 Annette J. Theron
Annette J. Theron Ronald Anderson
Ronald Anderson Theresa M. Rossouw
Theresa M. Rossouw Helen C. Steel
Helen C. Steel
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 02 November 2017
Sec. HIV and AIDS
Volume 8 - 2017 | https://doi.org/10.3389/fimmu.2017.01461
Even after attainment of sustained viral suppression following implementation of highly active antiretroviral therapy, HIV-infected persons continue to experience persistent, low-grade, systemic inflammation. Among other mechanisms, this appears to result from ongoing microbial translocation from a damaged gastrointestinal tract. This HIV-related chronic inflammatory response is paralleled by counteracting, but only partially effective, biological anti-inflammatory processes. Paradoxically, however, this anti-inflammatory response not only exacerbates immunosuppression but also predisposes for development of non-AIDS-related, non-communicable disorders. With respect to the pathogenesis of both sustained immunosuppression and the increased frequency of non-AIDS-related disorders, the anti-inflammatory/profibrotic cytokine, transforming growth factor-β1 (TGF-β1), which remains persistently elevated in both untreated and virally suppressed HIV-infected persons, may provide a common link. In this context, the current review is focused on two different, albeit related, harmful activities of TGF-β1 in HIV infection. First, on the spectrum of anti-inflammatory/immunosuppressive activities of TGF-β1 and the involvement of this cytokine, derived predominantly from T regulatory cells, in driving disease progression in HIV-infected persons via both non-fibrotic and profibrotic mechanisms. Second, the possible involvement of sustained elevations in circulating and tissue TGF-β1 in the pathogenesis of non-AIDS-defining cardiovascular, hepatic, pulmonary and renal disorders, together with a brief comment on potential TGF-β1-targeted therapeutic strategies.
It is well established that the direct cytopathic effects of HIV, as well as destruction by cytotoxic CD8+ T cells, are the major contributors to chronic depletion of CD4+ T cells, both in the circulation and secondary lymphoid tissues, resulting in the progressive immunosuppression which culminates in the development of AIDS (1, 2). However, it is also now well recognized that HIV infection-related immunosuppression is exacerbated by several other, albeit indirect mechanisms, which may even precede significant loss of CD4+ T cells. Most noteworthy among these is secondary immunosuppression resulting from the sustained activation of the anti-inflammatory machinery of the host, mobilized to counteract not only HIV-mediated chronic immune activation but also chronic inflammation due to microbial translocation. Mechanistically, break down of the tight epithelial barrier of the gastrointestinal tract (GIT) results from a combination of factors, namely preferential infection and depletion of CCR5-expressing CD4+ T lymphocytes, accumulation of inflammatory cells and a concomitant decrease in cells that regulate epithelial homeostasis, alterations in the regulation of tight junction protein expression, and possibly epithelial and enterocyte apoptosis (3–5). This allows proinflammatory microbial products to translocate into the lamina propria of the GIT, and, eventually, into the systemic circulation (5).
With respect to HIV-related secondary immunosuppression, a number of studies has implicated chronic over-production of the anti-inflammatory, profibrotic cytokine, transforming growth factor beta-1 (TGF-β1), as a major cause of immunosuppression in HIV infection (6, 7). Not only does this cytokine promote immunosuppression directly by targeting cells of both the adaptive and innate immune systems (8, 9), but also indirectly via its profibrotic activity, resulting in the depletion of T cells in secondary lymphoid tissues, while also compromising repopulation of T cell-dependent zones following implementation of antiretroviral therapy (10).
Following brief descriptions of the subtypes of TGF-β, cellular origins and the intracellular signaling mechanisms utilized by this cytokine, as well as an overview of its spectrum of immunosuppressive activities, this review is focused primarily on the role of TGF-β, specifically TGF-β1, in driving HIV-associated, chronic, secondary immunosuppression. Particular emphasis is placed on: (i) T regulatory cells (Tregs) as a major source of the cytokine; (ii) profibrotic destruction of the architecture of secondary lymphoid organs/tissues; (iii) pro-oxidative interactions with macrophages, all three of which drive disease progression; and (iv) the involvement of sustained elevations of TGF-β1 in the pathogenesis of profibrotic, HIV-associated, non-AIDS-defining conditions, specifically those of cardiovascular, hepatic, pulmonary, and renal origin, as well as the possible role of highly active antiretroviral therapy (HAART).
Transforming growth factor beta belongs to a superfamily of cytokines that includes three TGF-β isoforms, as well as activins, bone morphogenetic proteins, growth and differentiation factors, and others (11). According to Blobe et al., “virtually every cell in the body, including epithelial, endothelial, hematopoietic, neuronal, and connective-tissue cells, produces TGF-β and has receptors for it” (12), while Prud’homme and Piccirillo state that TGF-βs are involved in a variety of different biological processes including embryonic development, fibrosis, wound healing, angiogenesis, hematopoesis, and regulation of the immune response (13).
All three isoforms (β1, β2, and β3) are produced by mammals with TGF-β1 being most common in the immune system, in which it functions as an important pleiotropic cytokine with potent immunoregulatory properties (14), the importance of which is underscored by observations that mice lacking TGF-β1 die of multiorgan inflammation early in life (15, 16).
The biological activities of TGF-β are achieved via binding of the cytokine to type II receptors (TGF-βRII) on target cells. This, in turn, causes activation of type 1 receptors (TGF-βRI), resulting in the phosphorylation and activation of the Sma- and Mad-related protein (Smad) transcription factors, Smad 2 and Smad3 (17). Phosphorylated receptor-regulated Smads then bind to the co-Smad, Smad 4, which translocates to the nucleus to modulate gene expression (17). TGF-β isoforms may also exert their effects through Smad-independent pathways (17, 18). Importantly, TGF-β is synthesized in an inactive form and must be converted to its active form prior to binding to TGF-βRII (19). Active TGF-β is produced by proteolytic processes that promote the dissociation of inactive TGF-β from latency-associated protein or latent-TGF-β binding protein, which maintains TGF-β in an inactive state (20).
Transforming growth factor beta-1 is encoded by a distinct gene located in the 19q13.2 chromosomal region, which comprises seven exons separated by six very large introns (21). Functional genetic variations of the TGF-β1 gene (gene polymorphisms) have been linked to variations of protein expression or functionality (22). According to Martelossi Cebinelli et al., eight single-nucleotide polymorphisms (SNPs) (rs2317130, rs11466313, rs1800468, rs1800469, rs11466314, rs1800471, rs1800470, and rs11466316) as well as one deletion/insertion polymorphism, have been shown to affect TGF-β1 expression (21). The authors state that these SNPs interfere with either transcriptional regulation of TGF-β1 or its synthesis (21). An earlier study by Kruit et al. alluded to the fact that TGF-β1 gene SNPs, which are present in codon 10 (Leu10Pro/rs1800470) and codon 25 (Arg25Pro/rs1800471), can contribute to variations in TGF-β1 production (22). The bi-allelic polymorphism changes the amino acid at codon 10 from leucine to proline, or at codon 25, from arginine to proline, which is associated with lower synthesis of TGF-β1 (23). The TGF-β1 high producer genotype may be a risk factor for certain conditions, e.g., those associated with fibrosis (23).
Transforming growth factor beta-1 utilizes various mechanisms to mediate suppression of the reactivity of T lymphocytes (8, 14, 20). In this context, the cytokine is a potent inhibitor of both T-helper (Th)1 and Th2 cell differentiation and proliferation in vitro, which is achieved via inhibition of production of the transcription factors, T bet, and GATA-3, respectively (24, 25). These mechanisms are likely to account for the suppressive effects of TGF-β1 on production of the cytokines interferon (IFN)-γ, interleukin (IL)-2, and IL-4 (14), and possibly the rapid loss of the β2 subunit of the IL-12 receptor (IL-12R) on CD4+ T cells, which then become unresponsive to IL-12 (26). In addition to these activities, TGF-β1 has also been reported to induce apoptosis of CD4+ T lymphocytes following macrophage-tropic (R5) HIV-1 infection, by reducing levels of Bcl-2 (survival signal), as well as by increasing those of the intracellular death signals, caspase-3, apoptosis-inducing factor, and BH3 interacting-domain death agonist (27).
Notwithstanding its suppressive effects on CD4+ T cells, TGF-β1 also regulates the proliferative and effector functions of CD8+ T cells. In the case of the latter, the synthesis of IFN-γ, the exocytic release of perforins and granzymes, and the expression of Fas ligand, which collectively contribute to the cytotoxicity of CD8+ T cells, are all downregulated by TGF-β1 (28, 29).
Transforming growth factor beta-1 also inhibits immune responses indirectly via regulation of CD4+, CD25+, Foxp3+ Tregs that potently suppress T cell functions. Contrary to its aforementioned proapoptotic effects on CD4+ T helper cells, TGF-β1 in this setting has been reported to protect Tregs from apoptosis during thymic development (30), as well as promoting the differentiation of induced Tregs. With regard to the latter, studies have shown that TGF-β1 is critically involved in mediating the transition of naive, peripheral CD4+, CD25− non-Tregs into functionally mature CD4+, CD25+, Foxp3+ Tregs (31, 32).
Transforming growth factor beta-1 also regulates humoral immune responses, with a very recent study having reported that both TGF-β1 and TGF-β3 suppress the survival and proliferation of B cells, as well as their differentiation into antibody-secreting cells (9).
Transforming growth factor beta-1 also regulates the reactivities of cells of the innate immune system. In this context, the cytokine was shown to inhibit the production of IFN-γ by human natural killer (NK) cells, as well as antibody-dependent cellular cytotoxicity (ADCC) induced by CD16 activation. The effect on ADCC was attributed to TGF-β1-mediated inhibition of synthesis of granzyme A and granzyme B protein, which was associated with decreased mRNA expression (33). TGF-β1 was also shown to decrease the expression of the activating receptors, natural killer protein 30 (NKp30), natural killer group 2D (NKG2D), and DNAX accessory molecule-1 (DNAM-1), leading to reduced synthesis and release of IFN-γ, as well as granule exocytosis, resulting in attenuation of tumor killing (34). A more recent study demonstrated that TGF-β1 inhibited the activation and functions of NK cells by repressing the mammalian target of rapamycin (mTOR) pathway, a central regulator of cellular metabolism and cytotoxic functions of these cells (35).
A number of studies [reviewed in Ref. (36–38)] have reported that TGF-β1 also has various regulatory effects on dendritic cells (DCs). In an earlier study, this cytokine was shown to inhibit the in vitro activation and maturation of DCs (39). In addition to inducing DC apoptosis (40), TGF-β1 has also been reported to impede critical functions of these cells, including migration (41), expression of costimulatory (CD40, CD80, and CD86) and HLA class II molecules (37, 38), antigen-presenting capacity (36), and production of tumor necrosis factor-α, IFN-α, and IL-12 (38).
The proinflammatory functions of macrophages are also negatively affected by TGF-β1. These include inhibition of expression of inducible nitric oxide synthase (iNOS) and matrix metalloproteinase (MMP)-12 (42). In addition, MyD88-dependent Toll-like receptor (TLR)-activated signaling pathways are also downregulated by this cytokine (43). In the case of neutrophils, an earlier study reported that TGF-β1 is a potent chemotactic and activating factor for these cells (44). In contrast, others have reported that TGF-β1 inhibits neutrophil transmigration through activated endothelium by down-regulating the expression of endothelial E-selectin and IL-8 (45), as well as suppressing neutrophil degranulation (46).
These immunosuppressive effects of TGF-β1 are summarized in Table 1.
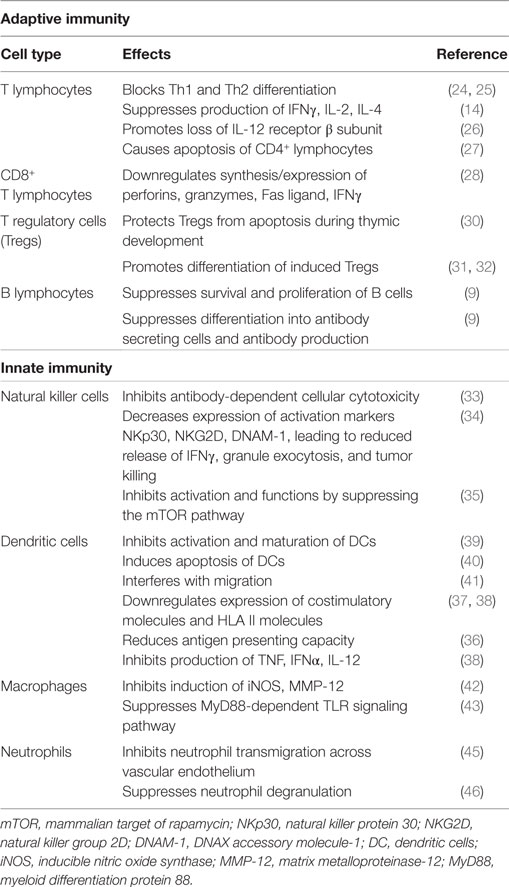
Table 1. Summary of the immunosuppressive effects of TGF-β1.
Possibly the first article documenting increases of plasma/serum TGF-β in treatment naive male patients with AIDS was published by Allen et al. (47). Several later reports also documented increased concentrations of this cytokine in the blood, lymphoid tissues and cerebrospinal fluid of HIV-infected individuals (6, 48–53). In addition, two other earlier studies reported that isolated peripheral blood mononuclear cells (PBMCs) from HIV-infected persons spontaneously released high levels of TGF-β, which was associated with selective upregulation of the TGF-β1 isoform (54, 55). In this setting, increased levels of this cytokine were associated with defective T cell proliferation to recall antigens, as well as B lymphocyte proliferative responses and immunoglobulin production, possibly related to the proapoptotic mechanisms described above (27), all of which were restored by the addition of a TGF-β1-neutralizing monoclonal antibody (54). Important caveats to bear in mind when measuring TGF-β1 in biological matrices are the requirement for stringent control of activation procedures, such as acidification, which precede measurement of this cytokine, as well as careful processing of blood specimens to avoid contamination with platelet-derived TGF-β1.
HIV-1 proteins may also contribute to the production of TGF-β1. The HIV-1 transactivator of transcription (Tat) protein has been shown to induce synthesis of TGF-β1 by human leukocytes (56), which may underpin the immunosuppressive effects of Tat (56). It has also been demonstrated that HIV-1 glycoprotein (gp)160 induces significant TGF-β mRNA expression and TGF-β1 secretion in PBMC from HIV-seronegative, healthy donors (57), which resulted from the CD4–gp160 interaction. In addition, Garba et al. demonstrated that HIV antigens induced TGF-β1 secretion by CD8+ T cells, which resulted in inhibition of production of IFN-β in response to HIV, as well as unrelated antigens (58).
The numbers of Tregs are highly increased in the mucosa and lymphoid tissues of untreated HIV patients and are associated with disease progression (49, 59, 60). Tregs exert their suppressive functions through various mechanisms including the production of TGF-β1, as well as another broadly -active anti-inflammatory cytokine, viz. IL-10 (61). TGF-β1 is normally a protective, anti-inflammatory cytokine, but its overproduction may have serious pathogenic effects. As mentioned above, increases in the concentrations of TGF-β1 in the circulation of HIV-1 infected patients have been reported in several studies, and, in one of these, were found to be highly correlated with increased numbers of circulating Tregs (r = 0.921, p = 0.001) (52), indicating that the cytokine is likely to originate predominantly from Tregs. This contention is supported by findings that elevated numbers of circulating Tregs and concentrations of TGF-β1 correlate with increased levels of circulating biomarkers of chronic immune activation (monocyte- and lymphocyte-derived activation markers) and bacterial translocation (51–53). As mentioned earlier, these are considered to be major drivers of immunosuppression and progression to AIDS (3–5, 62). This ongoing counterbalancing activity of Tregs and associated cytokines, which paradoxically also contributes to progressive immunosuppression, may underpin the prolonged asymptomatic phase of HIV infection. Worryingly, however, several studies have reported that even in the face of administration of virally suppressive HAART for periods of 6–12 months, circulating levels of both Tregs and TGF-β1 remained elevated. This appears to be consistent with ongoing chronic immune activation and its associated risks of persistent, albeit of lesser magnitude, immunosuppression, and development of HIV-associated, non-AIDS-defining conditions (6, 7, 51–53).
In this context, it is noteworthy that Chevalier and Weiss have proposed that Treg-mediated dampening of chronic immune activation may be less effective in untreated HIV-infected patients with high levels of immune activation as found in primary and chronic infection, compared with HAART-treated patients who have low levels of residual immune activation (63). These authors also proposed that HIV-driven Treg expansion may occur not only as a result of chronic immune activation, but may also be a consequence of both increased survival and enhanced production of these cells in the thymus (63). This contention is supported by the findings of two earlier studies. First, Chen et al., as alluded to above, reported that TGF-β1, as well as being produced by Tregs, also caused the induction of these cells by promoting the transition of CD4+, CD25− cells to CD4+, CD25+ Tregs that expressed the Foxp3 gene (31). These cells produced TGF-β1, but not Th1 or Th2 cytokines, and effectively suppressed T cell proliferation in vitro (31). Second, Ji and Cloyd have demonstrated that CD4+, CD25+ Treg cell binding to HIV-1 prolongs survival and enhances both the suppressive functions and accumulation of these cells in peripheral lymph nodes and mucosal lymphoid tissues (64).
According to Deeks, it remains an unanswered question as to why CD4+ T cells destroyed during HIV infection are not replaced by the immune system as only a “small fraction of cells are infected and killed on a daily basis after the acute phase of the disease” (65). Moreover, a substantial number of HIV-infected patients do not experience significant increases in their peripheral blood CD4 counts after initiation of HAART despite low HIV viral loads (2). These anomalies may be explained in part by the “damaged lymphoid tissue hypothesis” in which TGF-β1 plays a major role as a profibrotic agent in disrupting the fibroblastic reticular cell (FRC) network of secondary lymphoid structures.
The FRC network is located in the T cell zone of lymphoid tissues and provides a mechanical infrastructure that enables lymphocyte recruitment and organization, as well as promoting contact between T cells and antigen-presenting DCs. FRC-derived chemokines and cytokines, particularly IL-7, are critically involved in maintaining naive T cell recruitment and survival within lymphoid tissue (66). It has been demonstrated, however, that in HIV infection, as well as in simian immunodeficiency virus (SIV) infection of rhesus macaques, lymphoid tissue undergoes collagen deposition and fibrosis (2, 10), leading to the loss of the FRC network and reduced availability of IL-7 (2, 10). Consequently, there is an increased tendency for naive CD4+ and CD8+ T cell populations to undergo apoptosis, leading to a progressive reduction in the numbers of these cells (2). Depletion of T cells also leads to loss of lymphotoxin β, on which maintenance of the FRC depends, thereby exacerbating failure of production of IL-7, resulting in a vicious cycle of T cell and FRC depletion (10). Reversal of these effects was only found to be optimal when HAART was initiated in the acute stage of the infection (2).
These same authors also detected an increase in Tregs positive for TGF-β1, as well as an increase in the numbers of fibroblasts expressing the TGF-β1RII in lymphoid tissue, favoring chronic activation of the TGF-β1 signaling pathway, with consequent augmentation of production of procollagen by fibroblasts. Moreover, TGF-β1 also enhanced the expression of chitinase 3-like-1 activity in fibroblasts, thereby contributing to procollagen maturation and formation of collagen fibrils in the lymphoid tissue of HIV-infected patients (10). Taken together, these events strongly implicate potent and sustained Treg responses with production of profibrogenic “TGF-β which, in turn, causes collagen deposition, tissue fibrosis, the loss of the FRC network” and depletion of naive T cells (65).
The association of chronic systemic immune activation in HIV infection with persistently increased levels of TGF-β1 and the role of this cytokine in immunosuppression and disease progression are summarized in Figure 1.
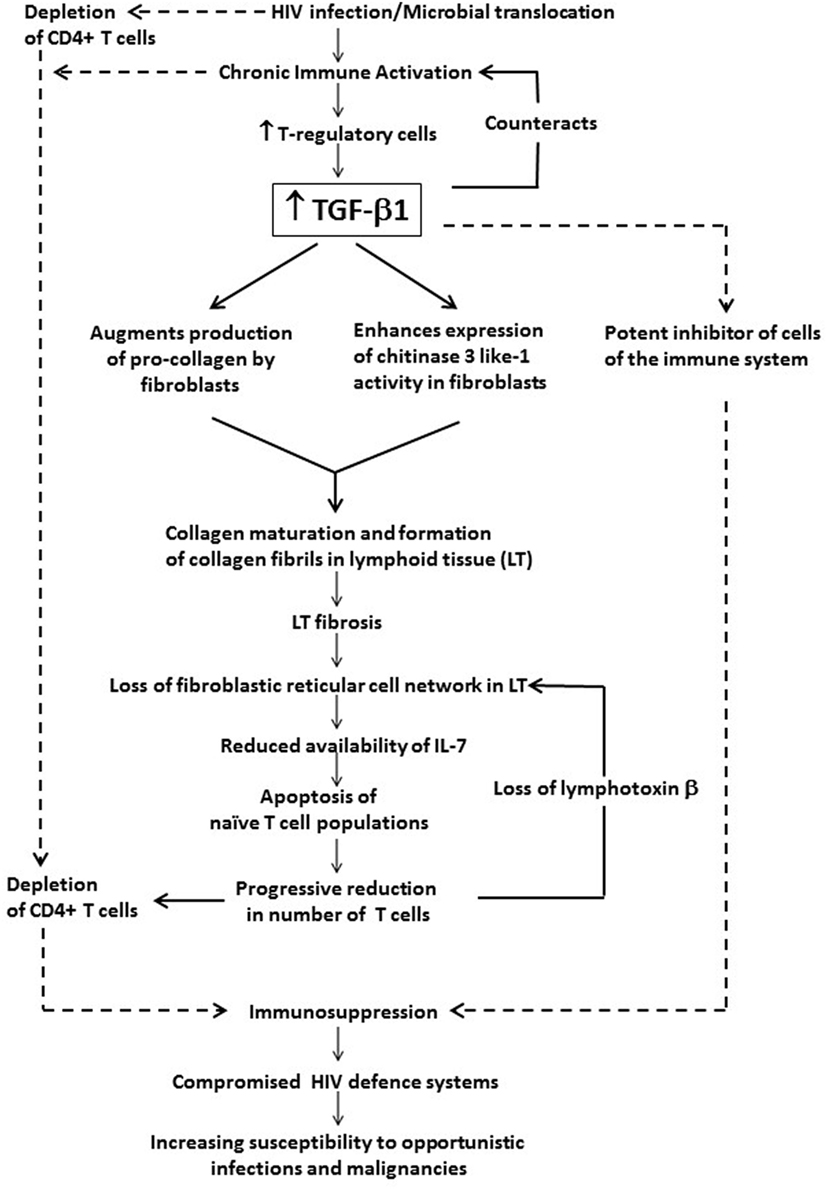
Figure 1. The potential contribution of chronic systemic immune activation in HIV infection to driving sustained increases in circulating and tissue concentrations of transforming growth factor beta-1 (TGF-β1). This cytokine, which appears to originate predominantly from T regulatory cells (Tregs), exacerbates immunosuppression and disease progression not only via its direct inhibitory effects on CD4+ T cells and other cells of the adaptive immune system, but also via prevention of T cell repopulation of secondary lymphoid tissue via profibrotic activity.
In addition to the immunosuppressive effects on immune cells, cytokines of the TGF-β family have also been reported to increase the production of reactive oxygen species (ROS) by various cell types, resulting in decreased concentrations of intracellular glutathione (GSH), the most abundant intracellular free thiol and an important antioxidant (67). ROS, in turn, augment the production and reactivity of TGF-β cytokines via induction of gene expression and activation of latent TGF-β (67).
In this context, it is noteworthy that monocyte-derived macrophages derived from monocytes isolated from the blood of HIV-infected individuals have been reported to have decreased ratios of GSH:oxidized glutathione (GSSG), consistent with increased intracellular oxidative stress (50). This contention was supported by several findings, including decreased activation of genes encoding enzymes involved in the synthesis of GSH, increased levels of lipid peroxidation products in macrophage lysates, and increased concentrations of the cytokines IL-1, IL-17, and TGF-β1 in cell lysates and plasma (50). In a follow-up study, administration of liposomal glutathione supplements (L-GSH) to HIV-infected individuals was found to alleviate oxidative stress and correct the Th1 cytokine imbalance, while decreasing the levels of TGF-β1 and IL-10 (68).
Although unproven, microbial translocation in HIV infection resulting in persistent macrophage activation, possibly due to chronic stimulation of TLRs by microbial products, may also drive the production of TGF-β1 by these cells, contributing to sustained immunosuppression and predisposition to development of HIV-associated, non-AIDS-defining conditions (69, 70).
Wiercińska-Drapalo et al. investigated the levels of circulating TGF-β1 and possible relationships with clinical parameters of HIV infection in 66 patients at different stages of HIV infection, 35 of whom were receiving HAART. A near twofold increase in plasma TGF-β1 was observed in HIV-infected patients compared to 20 healthy controls (6). Patients with a CD4 count below 200 cells/μL demonstrated the highest levels of TGF-β1, which were significantly and inversely correlated with circulating CD4 and CD8 counts, while no significant correlation was found with HIV viral load. Furthermore, patients in the symptomatic phase of the disease showed increased levels of TGF-β1 compared to those in the asymptomatic phase. No relationship was found between use of HAART and levels of TGF-β1. The authors proposed that TGF-β1 was associated with disease progression in HIV infection (6).
In a more recent study, TGF-β1 levels, as well as those of Th1 and Th2 cytokines, were also measured in HIV-infected patients stratified according to disease severity, i.e., non-progressive and progressive HIV infection (7). The latter group received HAART. The authors observed that TGF-β1 levels were increased in all HIV-infected patients, but even more so in patients with progressive disease when compared to those with non-progressive disease and healthy controls. These authors also observed that plasma levels of TGF-β1 and IL-10 in patients with non-progressive infection correlated inversely with CD4+ T cell counts and CD4:CD8 ratios, and proposed that these increases in TGF-β1 and IL-10 promote an immunosuppressive environment conducive to disease progression (7). In contrast, however, a study by Gaardbo et al. reported that Treg percentages and levels of TGF-β1 were comparable in healthy controls, HIV-infected viraemic controllers, elite controllers, long-term non-progressors, and progressors (71). The similar percentages of Tregs (and levels of TGF-β1) could possibly be attributed to the fact that in this study all patients had normal CD4+ T cell counts, as evidence suggests that increased Treg percentages are preferentially found in patients with low CD4+ T cell counts (71).
Although persistently elevated levels of circulating and tissue TGF-β1 have been implicated in sustained immunosuppression and progression to AIDS, it is also possible, albeit speculative, that this mechanism may also contribute to the pathogenesis of non-AIDS-related disorders, which is the topic of the remaining sections of this review.
The introduction of HAART in 1996, while resulting in significantly decreased mortality due to AIDS-related causes, has also been accompanied by an awareness of the emergence of a changing pattern of HIV/AIDS-related mortality. This is attributable to an increased prevalence of non-AIDS-defining malignancies and other non-communicable diseases (NCDs), particularly those of cardiovascular, hepatic, and renal origin (72–74). With the passage of time, this trend has become even more apparent (75–81), particularly in settings of advanced health care, most evident in developed countries, where mortality from HIV-related, non-AIDS-defining conditions may even exceed mortality due to opportunistic infections (82, 83). Among developed countries, however, the United Kingdom is an exception, where despite the ready accessibility of HAART, AIDS continues to account for the majority of deaths in HIV-infected persons (84). Persons at highest risk for development of HIV-related, non-AIDS-defining NCDs appear to be those who experience chronic immune activation associated with sustained, intense immunosuppression, even in the face of HAART-mediated viral suppression (75, 77–79).
An increasing body of evidence has implicated ongoing microbial translocation-associated chronic activation of monocytes/macrophages as a major contributor to the persistent systemic inflammation which underpins the pathogenesis of serious non-AIDS-defining morbidities and associated mortality (85–93). This contention is based on the frequency of reports that have consistently noted significant associations between non-AIDS-defining morbidities and/or mortality with elevated levels of circulating biomarkers of microbial translocation. These include intestinal fatty acid-binding protein and bacterial endotoxin (85, 86), as well as biomarkers indicative of monocyte/macrophage activation, particularly soluble CD14 (sCD14), IL-6 (85–91), and the macrophage scavenger receptor, sCD163, considered to be a more selective biomarker of macrophage activation and proliferation (91–93).
In addition, it is noteworthy that persistent systemic inflammation may also underpin the profibrotic mechanisms that have been implicated in the pathogenesis of HIV-related cardiovascular, hepatic, pulmonary, and renal disease (94–97). Surprisingly, however, with the notable exceptions of two earlier publications focused on HIV-related renal disease (98, 99), the involvement of persistently elevated levels of TGF-β1, in the pathogenesis of these non-AIDS-defining disorders remains largely unexplored. The remaining sections of this review are focused on the role of profibrotic mechanisms in the pathogenesis of HIV-related cardiovascular, renal, hepatic, and pulmonary disorders, and, by implication, the involvement of TGF-β1.
It is now well recognized that people living with HIV in the HAART era have a significantly increased risk for development of various types of CVD. These include coronary artery disease with its associated risk of types 1 and 2 myocardial infarction (MI), heart failure, arrhythmias, sudden cardiac death, cerebrovascular disease, pericardial diseases, and pulmonary hypertension (100–107). Several mechanisms have been proposed to contribute to the pathogenesis of CVD in HIV infection. Notwithstanding HIV-associated myocarditis and the existence of established risk factors such as older age, smoking and preexisting diabetes, hypertension, dyslipidaemia and renal dysfunction, as well as concomitant hepatitis C virus (HCV) infection (108), the most prominent contributors to CVD in HIV-infected patients are believed to be the adverse effects of certain components of HAART on the vasculature and heart, and, in particular, ongoing chronic immune activation encompassing both HAART-naive patients and even those who have achieved a significant degree of viral suppression (109).
An earlier study reported that exposure of rat ventricular myocytes to therapeutically relevant concentrations of the nucleoside reverse transcriptase inhibitor (NRTI), zidovudine, in vitro, resulted in damage to mitochondria (110). In the same study, exposure of human primary coronary artery endothelial cells and brain microvascular endothelial cells to zidovudine, or the protease inhibitor, indinavir, or, albeit to a lesser extent, the non-NTRI, efavirenz, caused disruption of intercellular gaps, as well as decreased transendothelial electrical resistance (110). In a subsequent study, administration of the NRTIs, zidovudine, and stavudine, but not lamivudine, to transgenic mice expressing the human mitochondrial deoxynucleotide carrier targeted to murine myocardium, resulted in increased left ventricular mass, damage to mitochondrial DNA and mitochondrial destruction (111). Other HAART-related mechanisms which predispose to development of CVD, particularly in the case of HIV protease inhibitors, include “elevations in serum levels of total cholesterol and triglycerides, decreased high-density lipoprotein cholesterol, lipodystrophy, insulin resistance and diabetes” reminiscent of the metabolic syndrome (112–114).
Protease inhibitors have also been proposed to promote myocardial oxidative stress, interfering, among other targets, with ion channel function (115). In the context of the current review, it is noteworthy that the protease inhibitor, ritonavir, has been implicated in the pathogenesis of HIV infection-associated myocardial fibrosis by TGF-β1-dependent mechanisms (116). These involve either direct activation of platelets resulting in the release of stored TGF-β1 by these cells, and/or by potentiation of production of TGF-β1 by other cell types via attenuation of the intracellular proteolysis of the signal-transducing adaptor protein, TNF receptor-associated factor 6 (116), which, in turn, is required for SMAD-independent synthesis of the cytokine (117). With respect to the possible involvement of these TGF-β1-associated mechanisms in the pathogenesis of HIV-related myocardial fibrosis, it is noteworthy that systemic activation of platelets has been described in both treatment-naive HIV-infected patients (n = 35) (118), as well as in patients on virally suppressive HAART (n = 73), which included a protease inhibitor in 76.7% of cases (119). However, possible associations between elevations in the concentrations of systemic biomarkers of platelet activation and types of antiretroviral agent were not mentioned in the latter study (119).
A systematic review and meta-analysis of eight studies, published in 2013, which complied with the authors’ inclusion criteria, described an “indication” of an increased risk of MI in the early stages of therapy with the NRTI, abacavir (RR 1.92, 95% CI 1.51–242), as well as with protease inhibitors (RR 2.13, 95% CI 1.06–4.28) (120). The authors conceded, however, that “our findings of increased cardiovascular risk from abacavir and protease inhibitors were in contrast to four published meta-analyses based on secondary analyses of randomized controlled trials, which found no increased risk from cardiovascular disease” (120). In the case of abacavir, Llibre and Hill in their recent review have proposed that the currently available evidence from clinical studies, together with lack of mechanistic data, is not supportive of the involvement of this agent in “causing a short/middle (but not cumulative) increase in the risk of acute MI or cardiovascular events” (121).
Myocardial fibrosis, of which TGF-β1 is a key mediator (122), is widely recognized as “a significant global health problem associated with nearly all forms of heart disease” (123) and has recently been identified as being a probable significant contributor to the excess incidence of HIV-related CV events (94, 124–126). In this context, Thiara et al., using magnetic resonance imaging (MRI), investigated the possible association of intramyocardial lipid accumulation and fibrosis with HIV-related myocardial dysfunction in a total of 95 adult patients, the majority of whom (93%) were receiving HAART (124). These authors observed that relative to matched healthy controls, systolic function was significantly impaired in HIV-infected participants and associated with significantly increased intramyocardial lipid accumulation and fibrosis (124). The following positive correlations were also noted: (i) intramyocardial lipid levels with duration of HAART and visceral adiposity, although the number of participants was too small to identify possible associations with individual antiretroviral agents and (ii) myocardial dysfunction with systemic biomarkers of inflammation, specifically lipopolysaccharide (LPS)-binding protein and monocyte chemoattractant protein 1. Taken together these findings are seemingly consistent with the involvement of both HAART and microbial translocation in the pathogenesis of HIV-related abnormal cardiac function (124). The authors concluded that “increased subclinical cardiac dysfunction is associated with cardiac steatosis and fibrosis in HIV-infected adults” (124).
The findings of the study reported by Thiara et al. (124) were confirmed and extended in two subsequent studies, which were also based on advanced MRI procedures, to which 22 and 92 HAART-treated, asymptomatic HIV-infected participants were recruited (125, 126). The authors documented the existence of subclinical myocardial inflammation, edema and fibrosis, which were associated with alterations in myocardial structure and function (125, 126). In one of these studies, myocardial fibrosis, which was detected “predominantly at the subepicardium of the midventricular and basal inferolateral wall,” was present in 82.1 and 27.3% of HIV-infected and healthy control participants, respectively (p < 0.001). The authors of both studies proposed chronic systemic inflammation involving the myocardium and pericardium to be the cause of myocardial fibrosis and associated CV morbidity and mortality (125, 126). However, conclusive support for this contention is dependent on the outcome of larger studies of this type which should also include extensive cardiac and inflammatory biomarker profiles (94), including TGF-β1 in particular.
From the early stages of the HIV pandemic, HIV-infected persons were recognized to have an increased risk of renal disease. Proteinuria and/or raised creatinine have also been associated with an increased risk of progression to AIDS and death in both the pre-HAART and HAART era (127). While it is possible that kidney disease may modulate HIV disease progression, these markers may merely be indicative of greater comorbidity (127). For instance, both acute kidney injury (AKI) and chronic kidney disease (CKD) are associated with CVD, which, as mentioned above, has become a leading cause of morbidity and mortality in HIV-infected populations (128).
Acute kidney injury can be attributed to prerenal, renal, and postrenal factors (129). The most common causes in the HAART era are intrinsic to the kidney, namely ischemic acute tubular necrosis, often in the context of an infection, or nephropathy secondary to nephrotoxic medication (130, 131). A study of a nationally representative database in the United States noted a twofold increase in the incidence of AKI requiring dialysis among hospitalized HIV-infected adults between 2002 and 2010, partially attributed to an increase in the prevalence of chronic comorbidities known to be risk factors for development of AKI (131).
Chronic kidney disease has also emerged as a major comorbid condition with HIV-infected persons estimated to have an almost fourfold increased risk. In addition to traditional risk factors, such as older age, hypertension, smoking and diabetes mellitus, late stage disease (i.e., low CD4 count and high HIV viral load) and coinfection with HCV have been identified as significant risk factors (132). Apart from glomerulonephritis secondary to hepatitis B virus (HBV) and HCV infection, HIV-related disorders, including HIV-associated nephropathy (HIVAN), HIV immune complex kidney disease (HIVICK), and less commonly, HIV-associated thrombotic microangiopathy, have emerged as important etiologies of CKD. CKD has a typical phenotype, characterized by a progressive reduction in nephrons associated with increased fibrosis and interstitial scarring, regardless of etiology (133). Many glomerular, tubular, and inflammatory processes involved in the development of this phenotype are thought to be mediated by TGF-β (134). Importantly, TGF-β increases expression of trophic and profibrotic factors, such as angiotensin II and connective tissue growth factor, as well as other pathological mediators, such as TNF-α and interleukins, that promote the formation of myofibroblasts (133, 135).
HIV-associated nephropathy is the pathognomonic form of HIV-related renal disease and is one of the leading causes of end-stage disease. This is especially true in patients of African descent due to high frequencies of apolipoprotein L1 risk alleles in this population (136, 137). HIVAN is characterized by a collapsing form of focal segmental glomerulosclerosis (FSGS), formation of tubular microcysts, inflammation in the tubulointerstitial space, and fibrosis (136, 138). FSGS has been proposed to have an inflammatory character similar to that of atherosclerosis, marked by the accumulation of cholesterol and cholesterol esters, activation of monocytes, production of lipid-laden macrophages, and expansion of contractile cells and matrix, leading to fibrosis (139). The pathogenesis of HIVAN is not fully understood, but seems to be triggered by direct viral infection and viral protein R-induced apoptosis of renal tubular epithelial cells, Nef-induced podocyte dysfunction, and upregulation of proinflammatory mediators, especially those induced by NF-κB (136).
The fibrogenic action of TGF-β, centered on its ability to induce extracellular matrix deposition in the glomeruli and tubular interstitium, is also believed to play an important role in the development of the pathological tissue fibrosis observed in HIVAN (140). Glomerular and tubulointerstitial expression of TGF-β has been found to be increased in experimental models and histological samples of patients with glomerulopathies that feature pathological extracellular matrix accumulation (141, 142). Recently, higher plasma levels of TGF-β have also been found to be associated with lower estimated glomerular filtration rate (143). In HIVAN, the mechanism underpinning the increase in TGF-β is proposed to be the HIV gene product, Tat protein, which has been shown to stimulate production of TGF-β by macrophages (144) and mesangial cells (140). TGF-β further stimulates HIV replication (145), thereby increasing Tat and inducing a vicious cycle of virus replication, TGF-β expression, and matrix deposition (140). Together with reduced expression of matrix proteins and overexpression of matrix remodeling enzymes, such as MMP-9, TGF-β is proposed to regulate the sclerosing and collapsing features of HIVAN (137). This is supported by the observation that the therapeutic effects of angiotensin II blockers in HIVAN correlate with decreased TGF-β-induced renal fibrosis (146).
HIV-associated nephropathy is principally seen in patients with advanced disease and has become less common in the HAART era (137). In contrast, diabetic nephropathy and arterionephrosclerosis have become more common, presumably secondary to chronic inflammation and dysmetabolism that persists in patients on virally suppressive treatment (137). Both pathologies have been associated with activation of TGF-β1 signaling with elevated levels of TGF-β1, mediated through increased levels of renin and angiotensin II (147, 148).
With more people on long-term treatment, HAART-induced nephrotoxicity is becoming an increasing problem with TDF-induced proximal tubular dysfunction and protease inhibitor-associated crystalluria the most common. TDF-associated nephrotoxicity mostly manifests as AKI, CKD, or proximal tubular injury secondary to mitochondrial damage caused by inhibition of polymerase γ (149). The protease inhibitor, indinavir, and to a lesser extent atazanavir, is poorly soluble in urine and can potentially cause crystalluria with subsequent interstitial nephritis and, rarely, parenchymal fibrosis (150). Although some medications, specifically those employed as immunosuppressants, have been associated with diffuse fibrosis and sclerosis secondary to increased expression of TGF-β (151), direct links between indinavir-mediated fibrosis and TGF-β1 have not been evaluated.
Liver disease is also increasingly being recognized as one of the leading causes of non-AIDS-related mortality and morbidity among HIV-infected individuals (73, 152). During HIV infection, a large number of cytokines and chemokines, including TGF-β1, as well as platelet-derived growth factor and endothelin I that promote the inflammatory response, fibrosis, contraction and mitosis, are released from Kupffer cells, lymphocytes, liver sinusoidal endothelial cells, platelets, and hepatic stellate cells (HSCs) (153, 154). HIV has been shown to modulate the activation and functioning of HSCs which are the main effector cells responsible for the fibrotic process (155–157). HSCs are non-parenchymal cells which, in their quiescent state, are responsible for the storage and metabolism of vitamin A. Following liver injury and/or viral infection, HSCs are transformed into proliferative, fibrogenic, proinflammatory, and contractile myofibroblasts that actively produce extracellular matrix components such as type I collagen (158). Accumulation of these proteins due to dysregulated release of the MMPs by HSCs leads to replacement of the liver parenchyma by scar tissue, resulting in liver fibrosis and its associated complications (95). Activated HSCs, in turn, perpetuate their own activation through several autocrine loops, including the secretion of additional TGF-β and upregulation of its receptors (159).
A number of other liver-related comorbidities with a profibrotic component are associated with HIV-infection, particularly in the setting of coinfection with HBV or HCV (5–30 and 15–40%, respectively, depending on the geographical region) (160–162). In this context, it is noteworthy that expression of TGF-β1 has been found to be significantly increased in both the serum and in the liver of individuals with HIV/HCV coinfection (163, 164). Other scenarios which predispose to and/or exacerbate HIV-associated chronic liver disease and hepatic fibrosis are alcohol/drug abuse, and the use of hepatotoxic traditional/herbal remedies (165–167), as well as metabolic abnormalities (insulin resistance, diabetes mellitus, and lipodystrophy syndrome) (166, 168).
Increased bacterial translocation, immune activation, and the presence of soluble proteins that alter the hepatic cytokine environment, producing a proinflammatory milieu, are also linked to direct and indirect profibrotic effects of HIV (152, 169). Although unproven, persistently elevated levels of circulating TGF-β1, consequent to chronic immune activation, may also contribute to the pathogenesis of HIV-related fibrotic disorders of the liver. In this context, it is noteworthy that microbial translocation promotes LPS entry into the peripheral circulation leading to increased TLR-4 signaling in Kupffer cells, with resultant generation of ROS and release of proinflammatory and profibrotic cytokines. This mechanism, in turn, exacerbates liver damage, compromising the ability of the liver to metabolize circulating LPS and promoting progression to liver fibrosis (170). Hepatic dysfunction, in turn, results in sustained elevations in the levels of circulating LPS due to reduced clearance by Kupffer cells (170).
Importantly, the introduction of HAART has also been associated with the emergence of a number of liver conditions including acute toxic hepatitis, steatosis, non-alcoholic steatohepatitis, non-alcoholic fatty liver disease and non-cirrhotic portal hypertension (157, 165, 171). However, the involvement of TGF-β1, if any, in the pathogenesis of these disorders remains to be established. Notwithstanding potential hepatotoxicity which has recently been extensively reviewed elsewhere (167), the short- and mid-term effects of HAART on the liver are, however, mostly considered to be beneficial, especially in HBV or HCV coinfected individuals, and are considered to outweigh the potential risks of long-term toxicity (172, 173).
With the notable exception of lung cancer, chronic obstructive lung disease (COPD) and pulmonary hypertension are the most common non-AIDS-defining pulmonary NCDs, occurring at higher frequencies than in the general population (174). Although compounded by a high prevalence of cigarette smoking among those living with the virus (175, 176), HIV/AIDS is now recognized as an independent risk factor for the development of COPD (174), a condition which has been identified as an emerging and serious threat to health in the HAART era (177, 178). With respect to the possible involvement of fibrotic mechanisms in disease pathogenesis, TGF-β1, via its profibrotic role in airway remodeling, has been reported to contribute to airflow limitation and disease severity in HIV-uninfected persons with COPD (179–181). Not surprisingly, albeit much less extensively characterized than in the clinical settings of HIV-related profibrotic disorders of CV, hepatic and renal origin, HIV infection has recently been reported to be associated with fibrosis-like changes in the lungs of a significant percentage (29%) of patients, correlating with viral load, but not with either HAART or CD4 count (182). In this context, the findings of an earlier study which reported excessive production of TGF-β by alveolar macrophages from HIV-infected persons are noteworthy (183). Nonetheless, the possible involvement of TGF-β1 in the pathogenesis of pulmonary fibrosis and COPD in the setting of HIV infection remains to be established.
Transforming growth factor beta has been described by Meng et al. in their recent review as being “the master regulator of fibrosis” (184). In their review, the authors have clearly described and depicted TGF-β-receptor-activated intracellular signaling mechanisms, identifying a range of potential therapeutic targets applicable to various inflammatory disorders, possibly including HIV/AIDS (184) a consideration of which is beyond the scope of this review. Of the many agents currently undergoing clinical evaluation in various fibrotic disorders (185), the anti-inflammatory/antioxidative drug, pirfenidone, and the αvβ6 integrin-targeted monoclonal antibody, STX-100X, have shown promise in the treatment of idiopathic pulmonary fibrosis (186, 187). With respect to pirfenidone, Estes et al. have reported that administration of this agent to SIV-infected rhesus macaques resulted in protection against lymphoid tissue, paracortical T cell zone, fibrosis (188). This, in turn, was associated with significant increases in the numbers of CD4+ T cells in peripheral blood and lymphoid tissues (188). The authors advocate antifibrotic drug/HAART combination therapy as a potential strategy to support recovery of immune function (188). Enthusiasm for this approach must, however, be countered by an awareness of the key protective biological activities of this cytokine. Alternative, and potentially safer strategies include the early implementation of efficacious antiretroviral therapy, reversal of increased intestinal permeability with probiotics, and the development of anti-inflammatory agents which effectively target intracellular signaling pathways common to those TLRs which are activated by bacterial LPS, bacterial DNA, and viral single-stranded RNA (TLRs-4, -7, -8, -9), as well as combinations of these.
Sustained elevations in circulating TGF-β1 are believed to contribute not only to immunosuppression and progression to AIDS, but also to residual immunosuppression in virally suppressed persons. In addition, the profibrotic activity of the cytokine, which also contributes to immunosuppression, may be linked to the pathogenesis of non-AIDS-defining cardiovascular, hepatic, renal, pulmonary and other disorders. Consequently, TGF-β1 (and possibly the other isoforms of this cytokine), represents an alternative, largely unexplored, therapeutic target in HIV infection which is dependent on the development of safe and efficacious strategies to counter the harmful activities of this cytokine.
All the authors contributed significantly to the planning and compilation of the manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. De Boer RJ. Time scales of CD4+ T cell depletion in HIV infection. PLoS Med (2007) 4:e193. doi:10.1371/journal.pmed.0040193
2. Zeng M, Southern PJ, Reilly CS, Beilman GJ, Chipman JG, Schacker TW, et al. Lymphoid tissue damage in HIV-infection depletes naïve T cells and limits T cell reconstitution after antiretroviral therapy. PLoS Pathog (2012) 8:e1002437. doi:10.1371/journal.ppat.1002437
3. Ipp H, Zemlin A. The paradox of the immune response in HIV infection: when inflammation becomes harmful. Clin Chim Acta (2013) 416:96–9. doi:10.1013/j.cca.2012.11.025
4. Klatt NR, Funderburg NT, Brenchley JM. Microbial translocation, immune activation, and HIV disease. Trends Microbiol (2013) 21:6–13. doi:10.1016/j.tim.2012.09.001
5. Doitsh G, Greene WC. Dissecting how CD4 T cells are lost during HIV infection. Cell Host Microbe (2016) 19:280–91. doi:10.1016/j.chom.2016.02.012
6. Wiercińska-Drapalo A, Flisiak R, Jaroszewicz J, Prokopowicz D. Increased plasma transforming growth factor-beta1 is associated with disease progression in HIV-1-infected patients. Viral Immunol (2004) 17:109–13. doi:10.1089/088282404322875502
7. Maina EK, Abana CZ, Bukusi EA, Sedegah M, Lartey M, Ampofo WK. Plasma concentrations of transforming growth factor beta 1 in non-progressive HIV-1 infection correlates with markers of disease progression. Cytokine (2016) 81:109–16. doi:10.1016/j.cyto.2016.02.006
8. Travis MA, Sheppard D. TGF-β activation and function in immunity. Annu Rev Immunol (2014) 32:51–82. doi:10.1146/annurev-immunol-032713-120257
9. Tsuchida Y, Sumitomo S, Ishigaki K, Suzuki A, Kochi Y, Tsuchiya H, et al. TGF-β3 inhibits antibody production by human B cells. PLoS One (2017) 12:e0169646. doi:10.1371/journal.pone.0169646
10. Zeng M, Smith AJ, Wietgrefe SW, Southern PJ, Schacker TW, Reilly CS, et al. Cumulative mechanisms of lymphoid tissue fibrosis and T cell depletion in HIV-1 and SIV infections. J Clin Invest (2011) 121:998–1008. doi:10.1172/JCI45157
11. Morikawa M, Derynck R, Miyazono K. TGF-β and the TGF-β family: context-dependent roles in cell and tissue physiology. Cold Spring Harb Perspect Biol (2016) 8:a021873. doi:10.1101/cshperspect.a021873
12. Blobe GC, Schiemann WP, Lodish F. Role of transforming growth factor β in human disease. N Engl J Med (2000) 342:1350–8. doi:10.1056/NEJM200005043421807
13. Prud’homme GJ, Piccirillo CA. The inhibitory effects of transforming growth factor-beta-1 (TGF-β1) in autoimmune diseases. J Autoimmun (2000) 14:23–42. doi:10.1006/jaut.1999.0339
14. Yoshimura A, Wakabayashi Y, Mori T. Cellular and molecular basis for the regulation of inflammation by TGF-β. J Biochem (2010) 147:781–92. doi:10.1093/jb/mvq043
15. Shull MM, Ormsby I, Kier AB, Pawlowski S, Diebold RJ, Yin M, et al. Targeted disruption of the mouse transforming growth factor-β1 gene results in multifocal inflammatory disease. Nature (1992) 359:693–9. doi:10.1038/359693a0
16. Kulkarni AB, Huh CG, Becker D, Geiser A, Lyght M, Flanders KC, et al. Transforming growth factor β1 null mutation in mice causes excessive inflammatory response and early death. Proc Natl Acad Sci U S A (1993) 90:770–4. doi:10.1073/pnas.90.2.770
17. Do T-V, Kubba LA, Du H, Sturgis CD, Woodruff TK. Transforming growth factor-β1, transforming growth factor-β2, and transforming growth factor-β3 enhance ovarian cancer metastatic potential by inducing a Smad3-dependent epithelial-to-mesenchymal transition. Mol Cancer Res (2008) 6:695–705. doi:10.1158/1541-7786.MCR-07-0294
18. Okamura T, Morita K, Iwasaki Y, Inoue M, Komai T, Fujio K, et al. Role of TGF-β3 in the regulation of immune responses. Clin Exp Rheumatol (2015) 33:S63–9.
19. Tran DQ. TGF-β: the sword, the wand, and the shield of FOXP3+ regulatory T cells. J Mol Cell Biol (2012) 4:29–37. doi:10.1093/jmcb/mjr033
20. Wan YY, Flavell RA. ‘Yin-Yang’ functions of TGF-β and Tregs in immune regulation. Immunol Rev (2007) 220:199–213. doi:10.1111/j.1600-065X.2007.00565.x
21. Martelossi Cebinelli GC, Paiva Trugilo K, Badaró Garcia S, Brajão de Oliveira K. TGF-β1 functional polymorphisms: a review. Eur Cytokine Netw (2016) 27:81–9. doi:10.1684/ecn.2016.0382
22. Kruit A, Grutters JC, Ruven HJT, Van Moorsel CHM, Weiskirchen R, Mengsteab S, et al. Transforming growth factor-beta gene polymorphisms in sarcoidosis patients with and without fibrosis. Chest (2006) 129:1584–91. doi:10.1378/chest.129.6.1584
23. Arkwright PD, Laurie S, Super M, Pravica V, Schwarz J, Webb AK, et al. TGF-β1 genotype and accelerated decline in lung function of patients with cystic fibrosis. Thorax (2000) 55:459–62. doi:10.1136/thorax.55.6.459
24. Gorelik L, Fields PE, Flavell RA. Cutting edge: TGF-beta inhibits Th type 2 development through inhibition of GATA-3 expression. J Immunol (2000) 165:4773–7. doi:10.4049/jimmunol.165.9.4773
25. Gorelik L, Constant S, Flavell RA. Mechanism of transforming growth factor beta-induced inhibition of T helper type 1 differentiation. J Exp Med (2002) 195:1499–505. doi:10.1084/jem.20012076
26. Gorham JD, Güler ML, Fenoglio D, Gubler U, Murphy KM. Low dose TGF-beta attenuates IL-12 responsiveness in murine Th cells. J Immunol (1998) 161:1664–70.
27. Wang J, Guan E, Roderiquez G, Norcross MA. Synergistic induction of apoptosis in primary CD4 (+) T cells by macrophage-tropic HIV-1 and TGF-beta1. J Immunol (2001) 167:3360–6. doi:10.4049/jimmunol.167.6.3360
28. Thomas DA, Massagué J. TGF-beta directly targets cytotoxic T cell functions during tumor evasion of immune surveillance. Cancer Cell (2005) 8:369–80. doi:10.1016/j.ccr.2005.10.012
29. Mempel TR, Pittet MJ, Khazaie K, Weninger W, Weissleder R, von Boehmer H, et al. Regulatory T cells reversibly suppress cytotoxic T cell function independent of effector differentiation. Immunity (2006) 25:129–41. doi:10.1016/j.immuni.2006.04.015
30. Ouyang W, Beckett O, Ma Q, Li MO. Transforming growth factor-β signalling curbs thymic negative selection promoting regulatory T cell development. Immunity (2010) 32:642–53. doi:10.1016/j.immuni.2010.04.012
31. Chen WJ, Jin WW, Hardegen N, Lei KJ, Li L, Marinos N, et al. Conversion of peripheral CD4+CD25- naïve T cells to CD4+CD25+ regulatory T cells by TGF-β induction of transcription factor Foxp3. J Exp Med (2003) 198:1875–86. doi:10.1084/jem.20030152
32. Zheng SG, Wang J, Wang P, Gray JD, Horwitz DA. IL-2 is essential for TGF-β to convert naïve CD4+CD25- cells to CD25+Foxp3+ regulatory T cells and for expansion of these cells. J Immunol (2007) 178:2018–27. doi:10.4049/jimmunol.178.4.2018
33. Trotta R, Dal Col J, Yu J, Ciarlariello D, Thomas B, Zhang X, et al. TGF-beta utilizes SMAD3 to inhibit CD16-mediated IFN-gamma production and antibody-dependent cellular cytotoxicity in human NK cells. J Immunol (2008) 181:3784–92. doi:10.4049/jimmunol.181.6.3784
34. Wilson EB, El-Jawhari JJ, Neilson AL, Hall GD, Melcher AA, Meade JL, et al. Human tumour immune evasion via TGF-β blocks NK cell activation but not survival allowing therapeutic restoration of anti-tumour activity. PLoS One (2011) 6:e22842. doi:10.1371/journal.pone.0022842
35. Viel S, Marçais A, Guimaraes FS, Loftus R, Rabilloud J, Grau M, et al. TGF-β inhibits the activation and functions of NK cells by repressing the mTOR pathway. Sci Signal (2016) 9:ra19. doi:10.1126/scisignal.aad1884
36. Strobl H, Knapp W. TGF-β1 regulation of dendritic cells. Microbes Infect (1999) 1:1283–90. doi:10.1016/S1286-4579(99)00256-7
37. Flavell RA, Sanjabi S, Wrzesinski SH, Licona-Limón P. The polarization of immune cells in the tumour environment by TGFβ. Nat Rev Immunol (2010) 10:554–67. doi:10.1038/nri2808
38. Seeger P, Musso T, Sozzani S. The TGF-β superfamily in dendritic cell biology. Cytokine Growth Factor Rev (2015) 26:647–57. doi:10.1016/j.cytogfr.2015.06.002
39. Yamaguchi Y, Tsumura H, Miwa M, Inaba K. Contrasting effects of TGF-beta 1 and TNF-alpha on the development of dendritic cells from progenitors in mouse bone marrow. Stem Cells (1997) 15:144–53. doi:10.1002/stem.150144
40. Ito M, Minamiya Y, Kawai H, Saito S, Saito H, Nakagawa T, et al. Tumor-derived TGFβ-1 induces dendritic cell apoptosis in the sentinel lymph node. J Immunol (2006) 176:5637–43. doi:10.4049/jimmunol.176.9.5637
41. Imai K, Minamiya Y, Koyota S, Ito M, Saito H, Sato Y, et al. Inhibition of dendritic cell migration by transforming growth factor-1 increases tumor-draining lymph node metastasis. J Exp Clin Cancer Res (2012) 31:3. doi:10.1186/1756-9966-31-3
42. Werner F, Jain MK, Feinberg MW, Sibinga NE, Pellacani A, Wiesel P, et al. Transforming growth factor-β1 inhibition of macrophage activation is mediated via Smad3. J Biol Chem (2000) 275:36653–8. doi:10.1074/jbc.M004536200
43. Naiki Y, Michelsen KS, Zhang W, Chen S, Doherty TM, Arditi M. Transforming growth factor-β differentially inhibits MyD88-dependent, but not TRAM- and TRIF-dependent, lipopolysaccharide-induced TLR4 signaling. J Biol Chem (2005) 280:5491–5. doi:10.1074/jbc.C400503200
44. Lagraoui M, Gagnon L. Enhancement of human neutrophil survival and activation by TGF-beta 1. Cell Mol Biol (1997) 43:313–8.
45. Smith WB, Noack L, Khew-Goodall Y, Isenmann S, Vadas MA, Gamble JR. Transforming growth factor-beta 1 inhibits the production of IL8 and the transmigration of neutrophils through activated endothelium. J Immunol (1996) 157:360–8.
46. Shen L, Smith JM, Shen Z, Erikkson M, Sentman C, Wira CR. Inhibition of human neutrophil degranulation by transforming growth factor-β1. Clin Exp Immunol (2007) 149:155–61. doi:10.1111/j.1365-2249.2007.03376.x
47. Allen JB, Wong HL, Guyre PM, Simon GL, Wahl SM. Association of circulating receptor FcγRIII-positive monocytes in AIDS patients with elevated levels of transforming growth factor-β. J Clin Invest (1991) 87:1773–9. doi:10.1170/JCI115196
48. Johnson MD, Kim P, Tourtellotte W, Federspiel CF. Transforming growth factor beta and monocyte chemotactic protein-1 are elevated in cerebrospinal fluid of immunocompromised patients with HIV-1 infection. J NeuroAIDS (2004) 2:33–43. doi:10.1300/J128v02n04_03
49. Nilsson J, Boasso A, Velilla PA, Zhang R, Vaccari M, Franchini G, et al. HIV-1-driven regulatory T-cell accumulation in lymphoid tissues is associated with disease progression in HIV/AIDS. Blood (2006) 108:3808–17. doi:10.1182/blood-2006-05-021576
50. Morris D, Guerra C, Donohue C, Oh H, Khurasany M, Venketaraman V. Unveiling of the mechanisms for decreased glutathione in individuals with HIV infection. Clin Dev Immunol (2012) 2012:734125. doi:10.1155/2012/734125
51. Malherbe G, Steel HC, Cassol S, de Oliveira T, Seebregts CJ, Anderson R, et al. Circulating biomarkers of immune activation distinguish viral suppression from nonsuppression in HAART-treated patients with advanced HIV-1 subtype C infection. Mediators Inflamm (2014) 2014:198413. doi:10.1155/2014/198413
52. Márquez M, Romero-Cores P, Montes-Oca M, Martin-Aspas A, Soto-Cárdenas M-J, Guerrero F, et al. Immune activation response in chronic HIV-infected patients: influence of hepatitis C virus coinfection. PLoS One (2015) 10:e0119568. doi:10.1371/journal.pone.0119568
53. Makhubele TG, Steel HC, Anderson R, van Dyk G, Theron AJ, Rossouw TM. Systemic immune activation profiles of HIV-1 subtype C-infected children and their mothers. Mediators Inflamm (2016) 2016:9026573. doi:10.1155/2016/9026573
54. Kekow J, Wachsman W, McCutchan AJ, Cronin M, Carson DA, Lotz M. Transforming growth factor β and noncytopathic mechanisms of immunodeficiency in human immunodeficiency virus infection. Proc Natl Acad Sci U S A (1990) 87:8321–5. doi:10.1073/pnas.87.21.8321
55. Kekow J, Wachsman W, McCutchan JA, Gross WL, Zachariah M, Carson DA, et al. Transforming growth factor-beta and suppression of humoral immune responses in HIV infection. J Clin Invest (1991) 87:1010–6. doi:10.1172/JCI115059
56. Reinhold D, Wrenger S, Kähne T, Ansorge S. HIV-1 Tat: immunosuppression via TGF-β1 induction. Immunol Today (1999) 20:384–5. doi:10.1016/S0167-5699(99)01497-8
57. Hu R, Oyaizu N, Than S, Kalyanaraman VS, Wang X-P, Pahwa S. HIV-1 gp160 induces transforming growth factor-β production in human PBMC. Clin Immunol Immunopathol (1996) 80:283–9. doi:10.1006/clin.1996.0125
58. Garba ML, Pilcher CD, Bingham AL, Eron J, Frelinger JA. HIV antigens can induce TGF-β1-producing immunoregulatory CD8+ T cells. J Immunol (2002) 168:2247–54. doi:10.4049/jimmunol.168.5.2247
59. Andersson J, Boasso A, Nilsson J, Zhang R, Shire NJ, Lindback S, et al. The prevalence of regulatory T cells in lymphoid tissue is correlated with viral load in HIV-infected patients. J Immunol (2005) 174:3143–7. doi:10.4049/jimmunol.174.6.3143
60. Epple HJ, Loddenkemper C, Kunkel D, Tröger H, Maul J, Moos V, et al. Mucosal but not peripheral FOXP3+ regulatory T cells are highly increased in untreated HIV-infection and normalize after suppressive HAART. Blood (2006) 108:3072–8. doi:10.1182/blood-2006-04-016923
61. Shalev I, Schmelzle M, Robson SC, Levy G. Making sense of regulatory T cell suppressive function. Semin Immunol (2011) 23:282–92. doi:10.1016/j.smim.2011.04.003
62. Paiardini M, Müller-Trutwin M. HIV-associated chronic immune activation. Immunol Rev (2013) 254:78–101. doi:10.1111/imr.12079
63. Chevalier MF, Weiss L. The split personality of regulatory T cells in HIV infection. Blood (2013) 121:29–37. doi:10.1182/blood-2012-07-409755
64. Ji J, Cloyd MW. HIV-1 binding to CD4 on CD4+CD25+ regulatory T cells enhances their suppressive function and induces them to home to, and accumulate in, peripheral and mucosal lymphoid tissues: an additional mechanism of suppression. Int Immunol (2009) 21:283–94. doi:10.1093/intimm/dxn146
65. Deeks SG. HIV infection, lymphoid fibrosis, and disease. Blood (2012) 120:1753–4. doi:10.1182/blood-2012-06-433987
66. Brown FD, Turley SJ. Fibroblastic reticular cells: organization and regulation of the T lymphocyte life cycle. J Immunol (2015) 194:1389–94. doi:10.4049/jimmunol.1402520
67. Liu RM, Gaston Pravia KA. Oxidative stress and glutathione in TGF-β-mediated fibrogenesis. Free Radic Biol Med (2010) 48:1–15. doi:10.1016/j.freeradbiomed.2009.09.026
68. Ly J, Lagman M, Saing T, Singh MK, Tudela EV, Morris D, et al. Liposomal glutathione supplementation restores Th1 cytokine response to Mycobacterium tuberculosis infection in HIV-infected individuals. J Interferon Cytokine Res (2015) 35:875–87. doi:10.1089/jir.2014.0210
69. Wallet MA, Rodriguez CA, Yin L, Saporta S, Chinratanapisit S, Hou W, et al. Microbial translocation induces persistent macrophage activation unrelated to HIV-1 levels or T cell activation following therapy. AIDS (2010) 24:1281–90. doi:10.1097/QAD.0b013e.328339e228
70. Stifano G, Affandi AJ, Mathes AL, Rice LM, Nakerakanti S, Nazari B, et al. Chronic Toll-like receptor 4 stimulation in skin induces inflammation, macrophage activation, transforming growth factor beta signature gene expression, and fibrosis. Arthritis Res Ther (2014) 16:R136. doi:10.1186/ar4598
71. Gaardbo JC, Ronit A, Harling HJ, Gjerdrum LMR, Springborg K, Ralfkiaer E, et al. Immunoregulatory T cells may be involved in preserving CD4 T cell counts in HIV-infected long-term nonprogressors and controllers. J Acquir Immune Defic Syndr (2014) 65:10–8. doi:10.1097/QAI.0b013e3182a7c932
72. Sackoff JE, Hanna DB, Pfeiffer MR, Torian LV. Causes of death among persons with AIDS in the era of highly active antiretroviral therapy: New York City. Ann Intern Med (2006) 145:397–406. doi:10.7326/0003-4819-145-6-200609190-00003
73. Weber R, Sabin CA, Friis-Møller N, Reiss P, El-Sadr WM, Kirk O, et al. Liver-related deaths in persons infected with the human immunodeficiency virus: the D:A:D study. Arch Intern Med (2006) 166:1632–41. doi:10.1001/archinte.166.15.1632
74. Strategies for Management of Antiretroviral Therapy (SMART) Study Group. CD4+ count-guided interruption of antiretroviral treatment. N Engl J Med (2006) 355:2283–96. doi:10.1056/NEJMoa062360
75. Wong CS, Lo FA, Cavailler P, Ng OT, Lee CC, Leo YS, et al. Causes of death in hospitalised human immunodeficiency virus (HIF)-infected patients at a national referral centre in Singapore: a retrospective review from 2008 to 2010. Ann Acad Med Singapore (2012) 41:571–6. doi:10.1111/j.1468-1293.2004.00223.x
76. Wang CJ, Silverberg MJ, Abrams DI. Non-AIDS-defining malignancies in the HIV-infected population. Curr Infect Dis Rep (2014) 16:406. doi:10.1007/s11908-014-0406-0
77. Althoff KN, McGinnis KA, Wyatt CM, Freiberg MS, Gilbert C, Oursler KK, et al. Comparison of risk and age at diagnosis of myocardial infarction, end-stage renal disease, and non-AIDS-defining cancer in HIV-infected versus uninfected adults. Clin Infect Dis (2015) 60:627–38. doi:10.1093/cid/ciu869
78. Pacheco YM, Jarrin I, Rosado I, Campins AA, Berenguer J, Iribarren JA, et al. Increased risk of non-AIDS-related events in HIV subjects with persistent low CD4 counts despite cART in the CoRIS cohort. Antiviral Res (2015) 117:69–74. doi:10.1016/j.antirival.2015.03.002
79. Shiels MS, Althoff KN, Pfeiffer RM, Achenbach CJ, Abraham AG, Castilho J, et al. HIV infection, immunosuppression, and age at diagnosis of non-AIDS-defining cancers. Clin Infect Dis (2017) 64:468–75. doi:10.1093/cid/ciw764
80. Langebeek N, Kooij KW, Wit FW, Stolte IG, Sprangers MAG, Reiss P, et al. Impact of co-morbidity and aging on health-related quality of life in HIV-positive and HIV-negative individuals. AIDS (2017) 31:1471–81. doi:10.1097/QAD.0000000000001511
81. Farahani M, Mulinder H, Farahani A, Marlink R. Prevalence and distribution of non-AIDS causes of death among HIV-infected individuals receiving antiretroviral therapy: a systematic review and meta-analysis. Int J STD AIDS (2017) 28:636–50. doi:10.1177/0956462416632428
82. Wester CW, Koethe JR, Shepherd BE, Stinnette SE, Rebeiro PF, Kipp AM, et al. Non-AIDS-defining events among HIV-1-infected adults receiving combination antiretroviral therapy in resource-replete versus resource-limited urban setting. AIDS (2011) 25:1471–9. doi:10.1097/QAD.0b013e.328347f9d4
83. Trickey A, May MT, Vehreschild J, Obel N, Gill MJ, Crane H, et al. Cause-specific mortality in HIV-positive patients who survived ten years after starting antiretroviral therapy. PLoS One (2016) 11:e0160460. doi:10.1371/journal.pone.0160460
84. Croxford S, Kitching A, Desai S, Kall M, Edelstein M, Skingsley A, et al. Mortality and causes of death in people diagnosed with HIV in the era of highly active antiretroviral therapy compared with the general population: an analysis of a national observational cohort. Lancet Public Health (2017) 2:e35–46. doi:10.1016/S2468-2667(16)30020-2
85. Sandler NG, Wand H, Roque A, Law M, Nason MC, Nixon DE, et al. Plasma levels of soluble CD14 independently predict mortality in HIV infection. J Infect Dis (2011) 203:780–90. doi:10.1093/infdis/jiq118
86. French AL, Evans CT, Agniel DM, Cohen MH, Peters M, Landay AL, et al. Microbial translocation and liver disease progression in women coinfected with HIV and hepatitis C virus. J Infect Dis (2013) 208:679–89. doi:10.1093/infdis/jit225
87. Tenorio AR, Zheng Y, Bosch RJ, Krishnan S, Rodriguez B, Hunt PW, et al. Soluble markers of inflammation and coagulation but not T-cell activation predict non-AIDS-defining morbid events during suppressive antiretroviral treatment. J Infect Dis (2014) 210:1248–59. doi:10.1093/infdis/jiu254
88. So-Armah KA, Tate JP, Chang CH, Butt AA, Gerschenson M, Gibert CL, et al. Do biomarkers of inflammation, monocyte activation, and altered coagulation explain excess mortality between HIV infected and uninfected people? J Acquir Immune Defic Syndr (2016) 72:206–13. doi:10.1097/QAI.0000000000000954
89. Borges ÁH, O’Connor JL, Phillips AN, Neaton JD, Grund B, Neuhaus J, et al. Interleukin 6 is a stronger predictor of clinical events than high-sensitivity C-reactive protein or D-dimer during HIV infection. J Infect Dis (2016) 214:408–16. doi:10.1093/infdis/jiw173
90. Sunil M, Nigalye M, Somasunderam A, Martinez ML, Yu X, Arduino RC, et al. Unchanged levels of soluble CD14 and IL-6 over time predict serious non-AIDS events in HIV-1-infected people. AIDS Res Hum Retroviruses (2016) 32:1205–9. doi:10.1089/AID.2016.0007
91. Imp BM, Rubin LH, Tien PC, Plankey MW, Golub ET, French AL, et al. Monocyte activation is associated with worse cognitive performance in HIV-infected women with virologic suppression. J Infect Dis (2017) 215:114–21. doi:10.1093/infdis/jiw506
92. Fitzpatrick ME, Nouraie M, Gingo MR, Camp D, Kessinger CJ, Sincebaugh JB, et al. Novel relationships of markers of monocyte activation and endothelial dysfunction with pulmonary dysfunction in HIV-infected persons. AIDS (2016) 30:1327–39. doi:10.1097/QAD.0000000000001092
93. Knudsen TB, Ertner G, Petersen J, Møller HJ, Moestrup SK, Eugen-Olsen J, et al. Plasma soluble CD163 level independently predicts all-cause mortality in HIV-1-infected individuals. J Infect Dis (2016) 214:1198–204. doi:10.1093/infdis/jiw263
94. Hsue PY, Tawakol A. Inflammation and fibrosis in HIV: getting to the heart of the matter. Circ Cardiovasc Imaging (2016) 9:e004427. doi:10.1161/CIRCIMAGING.116.004427
95. Debes JD, Bohjanen PR, Boonstra A. Mechanisms of accelerated liver fibrosis progression during HIV infection. J Clin Transl Hepatol (2016) 4:328–35. doi:10.14218/JCTH.2016.00034
96. Crothers K, Huang L, Goulet JL, Goetz MB, Brown ST, Rodriguez-Barradas MC, et al. HIV infection and risk for incident pulmonary diseases in the combination antiretroviral therapy era. Am J Respir Crit Care Med (2011) 183:388–95. doi:10.1164/rccm.201006-0836OC
97. Naicker S, Rahmanian S, Kopp JB. HIV and chronic kidney disease. Clin Nephrol (2015) 83:32–8. doi:10.5414/CNP835032
98. Bódi I, Kimmel PL, Abraham AA, Svetkey LP, Klotman PE, Kopp JB. Renal TGF-beta in HIV-associated kidney diseases. Kidney Int (1997) 51:1568–77. doi:10.1038/ki.1997.215
99. Cohen AH, Cohen GM. Distinguished scientists lecture series. HIV-associated nephropathy. Nephron (1999) 83:111–6. doi:10.1159/000045486
100. Tseng ZH, Secemsky EA, Dowdy D, Vittinghoff E, Moyers B, Wong JK, et al. Sudden cardiac death in patients with human immunodeficiency virus infection. J Am Coll Cardiol (2012) 59:1891–6. doi:10.1016/j.jacc.2012.02.024
101. Boccara F, Lang S, Meuleman C, Ederhy S, Mary-Krause M, Costagliola D, et al. HIV and coronary heart disease: time for a better understanding. J Am Coll Cardiol (2013) 61:511–23. doi:10.1016/j.jacc.2012.06.063
102. Freiberg MS, Chang CC, Kuller LH, Skanderson M, Lowy E, Kraemer KL, et al. HIV infection and the risk of acute myocardial infarction. JAMA Intern Med (2013) 173:614–22. doi:10.1001/jamainternmed.2013.3728
103. Calza L. HIV infection and myocardial infarction. Curr HIV Res (2016) 14:456–65. doi:10.2174/1570162X14666160803114651
104. Drozd DR, Kitahata MM, Althoff KN, Zhang J, Gange SJ, Napravnik S, et al. Increased risk of myocardial infarction in HIV-infected individuals in North America compared to the general population. J Acquir Immune Defic Syndr (2017) 75:568–76. doi:10.1097/QAI.0000000000001450
105. Crane HM, Paramsothy P, Drozd DR, Nance RM, Delaney JA, Heckbert SR, et al. Types of myocardial infarction among human immunodeficiency virus-infected individuals in the United States. JAMA Cardiol (2017) 2:260–7. doi:10.1001/jamacardio.2016.5139
106. Barnes RP, Lacson JC, Bahrami H. HIV infection and risk of cardiovascular diseases beyond coronary artery disease. Curr Atheroscler Rep (2017) 19:20. doi:10.1007/s11883-017-0652-3
107. Losina E, Hyle EP, Borre ED, Linas BP, Sax PE, Weinstein MC, et al. Projecting 10-yr, 20-yr and lifetime risks of cardiovascular disease in persons living with HIV in the US. Clin Infect Dis (2017) 65:1266–71. doi:10.1093/cid/cix547
108. Osibogun O, Ogunmoroti O, Michos ED, Spatz ES, Olubajo B, Nasir K, et al. HIV/HCV coinfection and the risk of cardiovascular disease: a meta-analysis. J Viral Hepat (2017) 24:998–1004. doi:10.1111/jvh.12725
109. Tawakol A, Lo J, Zanni M, Marmarelis E, Ihenachor E, MacNabb M, et al. Increased arterial inflammation relates to high-risk coronary plaque morphology in HIV-infected patients. J Acquir Immune Defic Syndr (2014) 66:164–71. doi:10.1097/QAI.0000000000000138
110. Fiala M, Murphy T, MacDougall J, Yang W, Luque A, Iruela-Arispe L, et al. HAART drugs induce mitochondrial damage and intercellular gaps and gp120 causes apoptosis. Cardiovasc Toxicol (2004) 4:327–37. doi:10.1385/CT:4:4:327
111. Lewis W, Kohler JJ, Hosseini SH, Haase CP, Copeland WC, Bienstock RJ, et al. Antiretroviral nucleosides, deoxynucleotide carrier and mitochondrial DNA: evidence supporting the DNA pol gamma hypothesis. AIDS (2006) 20:675–84. doi:10.1097/01.aids.0000216367.23325.58
112. Bozkurt B. Cardiovascular toxicity with highly active antiretroviral therapy: review of clinical studies. Cardiovasc Toxicol (2004) 4:243–60. doi:10.1385/CT:4:3:243
113. Dubé MP, Lipshultz SE, Fichtenbaum CJ, Greenberg R, Schecter AD, Fisher SD, et al. Effects of HIV infection and antiretroviral therapy on the heart and vasculature. Circulation (2008) 118:e36–40. doi:10.1161/CIRCULATIONAHA.107.189625
114. Melzi S, Carenzi L, Cossu MV, Passerini S, Capetti A, Rizzardini G. Lipid metabolism and cardiovascular risk in HIV-1 infection and HAART: present and future problems. Cholesterol (2010) 2010:271504. doi:10.1155/2010/271504
115. Reyskens KM, Essop MF. HIV protease inhibitors and onset of cardiovascular diseases: a central role for oxidative stress and dysregulation of the ubiquitin-proteasome system. Biochim Biophys Acta (2014) 1842:256–68. doi:10.1016/j.bbadis.2013.11.019
116. Ahamed J, Terry H, Choi ME, Laurence J. Transforming growth factor-β-mediated cardiac fibrosis: potential role in HIV and HIV/ART-linked cardiovascular disease. AIDS (2016) 30:535–42. doi:10.1097/QAD.0000000000000982
117. Zhang YE. Non-Smad pathways in TGF-β signaling. Cell Res (2009) 19:128–39. doi:10.1038/cr.2008.328
118. Nkambule BB, Davison G, Ipp H. Platelet leukocyte aggregates and markers of platelet aggregation, immune activation and disease progression in HIV infected treatment naïve asymptomatic individuals. J Thromb Thrombolysis (2015) 40:458–67. doi:10.1007/s11239-015-1212-8
119. Guzmán-Fulgencio M, Medrano J, Rallón N, Echeverria-Urabayen A, Benito JM, Restrepo C, et al. Soluble markers of inflammation are associated with Framingham scores in HIV-infected patients on suppressive antiretroviral therapy. J Infect (2011) 63:382–90. doi:10.1016/j.jinf.2011.08.006
120. Bavinger C, Bendavid E, Niehaus K, Olshen RA, Olkin I, Sundaram V, et al. Risk of cardiovascular disease from antiretroviral therapy for HIV: a systematic review. PLoS One (2013) 8:e59551. doi:10.1371/journal.pone.0059551
121. Llibre JM, Hill A. Abacavir and cardiovascular disease: a critical look at the data. Antiviral Res (2016) 132:116–21. doi:10.1016/j.antiviral.2016.05.015
122. Lijnen PJ, Petrov VV, Fagard RH. Induction of cardiac fibrosis by transforming growth factor-β1. Mol Genet Metab (2000) 71:418–35. doi:10.1006/mgme.2000.3032
123. Travers JG, Kamal FA, Robbins J, Yutzey KE, Blaxall BC. Cardiac fibrosis: the fibroblast awakens. Circ Res (2016) 118:1021–40. doi:10.1161/CIRCRESAHA.115.306565
124. Thiara DK, Liu CY, Raman F, Mangat S, Purdy JB, Duarte HA, et al. Abnormal myocardial function is related to myocardial steatosis and diffuse myocardial fibrosis in HIV-infected adults. J Infect Dis (2015) 212:1544–51. doi:10.1093/infdis/jiv274
125. Ntusi N, O’Dwyer E, Dorrell L, Wainwright E, Piechnik S, Clutton G, et al. HIV-1-related cardiovascular disease is associated with chronic inflammation, frequent pericardial effusions, and probable myocardial edema. Circ Cardiovasc Imaging (2016) 9:e004430. doi:10.1161/CIRCIMAGING.115.004430
126. Luetkens JA, Doerner J, Schwarze-Zander C, Wasmuth JC, Boesecke C, Sprinkart AM, et al. Cardiac magnetic resonance reveals signs of subclinical myocardial inflammation in asymptomatic HIV-infected patients. Circ Cardiovasc Imaging (2016) 9:e004091. doi:10.1161/CIRCIMAGING.115.004091
127. Szczech LA, Hoover DR, Feldman JG, Cohen MH, Gange SJ, Goozé L, et al. Association between renal disease and outcomes among HIV-infected women receiving or not receiving antiretroviral therapy. Clin Infect Dis (2004) 39:1199–206. doi:10.1086/424013
128. Chawla LS, Amdur RL, Shaw AD, Faselis C, Palant CE, Kimmel PL. Association between AKI and long-term renal and cardiovascular outcomes in United States veterans. Clin J Am Soc Nephrol (2014) 9:448–56. doi:10.2215/CJN.02440213
129. Mallipattu SK, Salem F, Wyatt CM. The changing epidemiology of HIV-related chronic kidney disease in the era of antiretroviral therapy. Kidney Int (2014) 86:259–65. doi:10.1038/ki.2014.44
130. Kalim S, Szczech LA, Wyatt CM. Acute kidney injury in HIV-infected patients. Semin Nephrol (2008) 28:556–62. doi:10.1016/j.semnephrol.2008.08.008
131. Nadkarni GN, Patel AA, Yacoub R, Benjo AM, Konstantinidis I, Annapureddy N, et al. The burden of dialysis-requiring acute kidney injury among hospitalized adults with HIV infection: a nationwide inpatient sample analysis. AIDS (2015) 29:1061–6. doi:10.1097/QAD.0000000000000653
132. Islam FM, Wu J, Jansson J, Wilson DP. Relative risk of renal disease among people living with HIV: a systematic review and meta-analysis. BMC Public Health (2012) 12:234. doi:10.1186/1471-2458-12-234
133. López-Hernández FJ, López-Novoa JM. Role of TGF-β in chronic kidney disease: an integration of tubular, glomerular and vascular effects. Cell Tissue Res (2012) 347:141–54. doi:10.1007/s00441-011-1275-6
134. Schnaper HW, Jandeska S, Runyan CE, Hubchak SC, Basu RK, Curley JF, et al. TGF-β signal transduction in chronic kidney disease. Front Biosci (2009) 14:2448–65. doi:10.2741/3389
135. Vega G, Alarcón S, San Martín R. The cellular and signalling alterations conducted by TGF-β contributing to renal fibrosis. Cytokine (2016) 88:115–25. doi:10.1013/j.cyto.2016.08.019
136. Medapalli RK, He JC, Klotman PE. HIV-associated nephropathy: pathogenesis. Curr Opin Nephrol Hypertens (2011) 20:306–11. doi:10.1097/MNH.0b013e328345359a
137. Rosenberg AZ, Naicker S, Winkler CA, Kopp JB. HIV-associated nephropathies: epidemiology, pathology, mechanisms and treatment. Nat Rev Nephrol (2015) 11:150–60. doi:10.1038/nrneph.2015.9
138. Wyatt CM, Klotman PE, D’Agati VD. HIV-associated nephropathy: clinical presentation, pathology, and epidemiology in the era of antiretroviral therapy. Semin Nephrol (2008) 28:513–22. doi:10.1016/j.semnephrol.2008.08.005
139. Rifkin DE, Sarnak MJ. Does inflammation fuel the fire in CKD? Am J Kidney Dis (2009) 53:572–5. doi:10.1053/j.ajkd.2009.01.001
140. Yamamoto T, Noble NA, Miller DE, Gold LI, Hishida A, Nagase M, et al. Increased levels of transforming growth factor-beta in HIV-associated nephropathy. Kidney Int (1999) 55:579–92. doi:10.1046/j.1523-1755.1999.00296.x
141. Yamamoto T, Nakamura T, Noble NA, Ruoslahti E, Border WA. Expression of transforming growth factor is elevated in human and experimental diabetic nephropathy. Proc Natl Acad Sci U S A (1993) 90:1814–8. doi:10.1073/pnas.90.5.1814
142. Border WA, Noble NA. Transforming growth factor beta in tissue fibrosis. N Engl J Med (1994) 331:1286–92. doi:10.1056/NEJM199411103311907
143. Mehta T, Buzkova P, Kizer JR, Diousse L, Chonchol M, Mukamal KJ, et al. Higher plasma transforming growth factor (TGF)-β is associated with kidney disease in older community dwelling adults. BMC Nephrol (2017) 18:98. doi:10.1186/s12882-017-0509-6
144. Zauli G, Davis BR, Re MC, Visani G, Furlini G, La Placa M. Tat protein stimulates production of transforming growth factor-beta 1 by marrow macrophages: a potential mechanism for human immunodeficiency virus-1-induced hematopoietic suppression. Blood (1992) 80:3036–43.
145. Shukla RR, Kumar A, Kimmel PL. Transforming growth factor beta increases the expression of HIV-1 gene in transfected human mesangial cells. Kidney Int (1993) 44:1022–9. doi:10.1038/ki.1993.344
146. Border WA, Noble NA. Interactions of transforming growth factor-beta and angiotensin II in renal fibrosis. Hypertension (1998) 31:181–8. doi:10.1161/01.HYP.31.1.181
147. Qiao YC, Chen YL, Pan YH, Ling W, Tian F, Zhang XX, et al. Changes of transforming growth factor beta 1 in patients with type 2 diabetes and diabetic nephropathy: a PRISMA-compliant systematic review and meta-analysis. Medicine (2017) 96:e6583. doi:10.1097/MD.0000000000006583
148. Meyrier A, Hill GS, Simon P. Ischemic renal diseases: new insights into old entities. Kidney Int (1998) 54:2–13. doi:10.1046/j.1523-1755.1998.00968.x
149. Lewis W, Day BJ, Copeland WC. Mitochondrial toxicity of NRTI antiviral drugs: an integrated cellular perspective. Nat Rev Drug Discov (2003) 2:812–22. doi:10.1038/nrd1201
150. Cooper RD, Tonelli M. Renal disease associated with antiretroviral therapy in the treatment of HIV. Nephron Clin Pract (2011) 118:c262–8. doi:10.1159/000321646
151. Bartoli E. Adverse effects of drugs on the kidney. Eur J Intern Med (2016) 28:1–8. doi:10.1016/j.ejim.2015.12.0001
152. Sacchi P, Cima S, Corbella M, Comolli G, Chiesa A, Baldanti F, et al. Liver fibrosis, microbial translocation and immune activation markers in HIV and HCV infections and in HIV/HCV co-infection. Dig Liver Dis (2015) 47:218–25. doi:10.1016/j.dld.2014.11.012
153. Hernandez-Gea V, Friedman SL. Pathogenesis of liver fibrosis. Annu Rev Pathol (2011) 6:425–56. doi:10.1146/annurev-pathol-011110-130246
154. Kawada N. Evolution of hepatic fibrosis research. Hepatol Res (2011) 41:199–208. doi:10.1111/j.1872-034X.2011.00776.x
155. Puoti M, Bonacini M, Spinetti A, Putzola V, Govindarajan S, Zaltron S, et al. Liver fibrosis progression is related to CD4 cell depletion in patients coinfected with hepatitis C virus and human immunodeficiency virus. J Infect Dis (2001) 183:134–7. doi:10.1086/317644
156. Gupta D, Rani M, Khan N, Jameel S. HIV-1 infected peripheral blood mononuclear cells modulate the fibrogenic activity of hepatic stellate cells through secreted TGF-β and JNK signaling. PLoS One (2014) 9:e91569. doi:10.1371/journal.pone.0091569
157. Mastroianni CM, Lichtner M, Mascia C, Zuccalà P, Vullo V. Molecular mechanisms of liver fibrosis in HIV/HCV coinfection. Int J Mol Sci (2014) 15:9184–208. doi:10.3390/ijms15069184
158. Friedman SL. Hepatic fibrosis – overview. Toxicology (2008) 254:120–9. doi:10.1016/j.tox.2008.06.013
159. Breitkopf K, Haas S, Wiercinska E, Singer MV, Dooley S. Anti-TGF-beta strategies for the treatment of chronic liver disease. Alcohol Clin Exp Res (2005) 29:121S–31S. doi:10.1097/01.alc.0000189284.98684.22
160. Soriano V, Puoti M, Sulkowski M, Cargnel A, Benhamou Y, Peters M, et al. Care of patients coinfected with HIV and hepatitis C virus: 2007 updated recommendations from the HCV-HIV International Panel. AIDS (2007) 21:1073–89. doi:10.1097/QAD.0b013e3281084e4d
161. Spradling PR, Richardson JT, Buchacz K, Moorman AC, Brooks JT, HIV Outpatient Study (HOPS) Investigators. Prevalence of chronic hepatitis B virus infection among patients in the HIV Outpatient Study, 1996-2007. J Viral Hepat (2010) 17:879–86. doi:10.1111/j.1365-2893.2009.01249.x
162. King J, Hagemeister DT. Hepatitis B co-infection in HIV-infected patients receiving antiretroviral therapy at the TC Newman Anti Retroviral Treatment Clinic in Paarl, Western Cape. S Afr J HIV Med (2016) 17:a336. doi:10.4102/sajhivmed.v17i1.336
163. Blackard JT, Kang M, Sherman KE, Koziel MJ, Peters MG, Chung RT. Effects of HCV treatment on cytokine expression during HCV/HIV coinfection. J Interferon Cytokine Res (2006) 26:834–8. doi:10.1089/jir.2006.26.834
164. Lin W, Weinberg EM, Tai AW, Peng LF, Brockman MA, Kim K-A, et al. HIV increases HCV replication in a TGF-β1-dependent manner. Gastroenterology (2008) 134:803–11. doi:10.1053/j.gastro.2008.01.005
165. Núñez M. Clinical syndromes and consequences of antiretroviral-related hepatotoxicity. Hepatology (2010) 52:1143–55. doi:10.1002/hep.23716
166. Rockstroh JK, Mohr R, Behrens G, Spengler U. Liver fibrosis in HIV: which role does HIV itself, long-term drug toxicities and metabolic changes play? Curr Opin HIV AIDS (2014) 9:365–70. doi:10.1097/COH.0000000000000064
167. Sonderup MW, Wainwright HC. Human immunodeficiency virus infection, antiretroviral therapy, and liver pathology. Gastroenterol Clin North Am (2017) 46:327–43. doi:10.1016/j.gtc.2017.01.007
168. Pedersen KK, Pedersen M, Trøseid M, Gaardbo JC, Lund TT, Thomsen C, et al. Microbial translocation in HIV infection is associated with dyslipidemia, insulin resistance and risk of myocardial infarction. J Acquir Immune Defic Syndr (2013) 64:425–33. doi:10.1097/QAI.0b013e312829f919d
169. Sherman KE, Thomas D, Chung RT. Human immunodeficiency virus and liver disease forum 2012. Hepatology (2014) 59:307–17. doi:10.1002/hep.26638
170. Marchetti G, Tincati C, Silvestri G. Microbial translocation in the pathogenesis of HIV infection and AIDS. Clin Microbiol Rev (2013) 26:2–18. doi:10.1128/CMR.00050-12
171. Lemoine M, Serfaty L, Capeau J. From nonalcoholic fatty liver to nonalcoholic steatohepatitis and cirrhosis in HIV-infected patients: diagnosis and management. Curr Opin Infect Dis (2012) 25:10–6. doi:10.1097/QCO.0b013e.0834ef599
172. Rohrbach J, Robinson N, Harcourt G, Hammond E, Gaudieri S, Gorgievski M, et al. Cellular immune responses to HCV core increase the HCV RNA levels decrease during successful antiretroviral therapy. Gut (2010) 59:1252–8. doi:10.1136/gut.2009.205971
173. Anderson JP, Tchetgen Tchetgen EJ, Lo Re V III, Tate JP, Williams PL, Seage GR III, et al. Antiretroviral therapy reduces the rate of hepatic decompensation among HIV- and hepatitis C virus-coinfected veterans. Clin Infect Dis (2014) 58:719–27. doi:10.1093/cid/cit779
174. Triplette M, Crothers K, Attia EF. Non-infectious pulmonary diseases and HIV. Curr HIV/AIDS Rep (2016) 13:140–8. doi:10.1007/s11904-016-0313-0
175. Rahmanian S, Wewers ME, Koletar S, Reynolds N, Ferketich A, Diaz P. Cigarette smoking in the HIV-infected population. Proc Am Thorac Soc (2011) 8:313–9. doi:10.1513/pats.201009-058WR
176. Rossouw TM, Anderson R, Feldman C. Impact of HIV infection and smoking on lung immunity and related disorders. Eur Respir J (2015) 46:1781–95. doi:10.1183/13993003.00353-2015
177. Scourfield AT, Doffman SR, Miller RF. Chronic obstructive pulmonary disease in patients with HIV: an emerging problem. Br J Hosp Med (2014) 75:678–84. doi:10.12968/hmed.2014.75.12.678
178. Ghadaki B, Kronfli N, Vanniyasingam T, Haider S. Chronic obstructive pulmonary disease and HIV: are we appropriately screening? AIDS Care (2016) 28:1338–43. doi:10.1080/09540121.2016.1189499
179. Yang YC, Zhang N, Van Crombruggen K, Hu GH, Hong SL, Bachert C. Transforming growth factor-beta1 in inflammatory airway disease: a key for understanding inflammation and remodeling. Allergy (2012) 67:1193–202. doi:10.1111/j.1398-9995.2012.02880.x
180. Chiang CH, Chuang CH, Liu SL. Transforming growth factor-β1 and tumor necrosis factor-α are associated with clinical severity and airflow limitation of COPD in an additive manner. Lung (2014) 192:95–102. doi:10.1007/s00408-013-9520-2
181. Aschner Y, Downey GP. Transforming growth factor-β: master regulator of the respiratory system in health and disease. Am J Respir Cell Mol Biol (2016) 54:647–55. doi:10.1165/rcmb.2015-0391TR
182. Leader JK, Crothers K, Huang L, King MA, Morris A, Thompson BW, et al. Risk factors associated with quantitative evidence of lung emphysema and fibrosis in an HIV-infected cohort. J Acquir Immune Defic Syndr (2016) 71:420–7. doi:10.1097/QAI.0000000000000894
183. Twigg HL III, Spain BA, Soliman DM, Bowen LK, Heidler KM, Wilkes DS. Impaired IgG production in the lungs of HIV-infected individuals. Cell Immunol (1996) 170:127–33. doi:10.1006/cimm.1996.0142
184. Meng X-M, Nikolic-Paterson DJ, Lan HY. TGF-β: the master regulator of fibrosis. Nat Rev Nephrol (2016) 12:325–38. doi:10.1038/nrneph.2016.48
185. Akhurst RJ, Hata A. Targeting the TGFβ signaling pathway in disease. Nat Rev Drug Discov (2012) 11:790–811. doi:10.1038/nrd3810
186. Woodcock HV, Maher TM. The treatment of idiopathic pulmonary fibrosis. F1000Prime Rep (2014) 6:16. doi:10.12703/P6-16
187. George PM, Wells AU. Pirfenidone for the treatment of idiopathic pulmonary fibrosis. Expert Rev Clin Pharmacol (2017) 10:483–91. doi:10.1080/17512433.2017
Keywords: highly active antiretroviral therapy, immunosuppression, lymphoid fibrosis, macrophages, non-AIDS-defining disorders, organ fibrosis, T regulatory cells, transforming growth factor-beta
Citation: Theron AJ, Anderson R, Rossouw TM and Steel HC (2017) The Role of Transforming Growth Factor Beta-1 in the Progression of HIV/AIDS and Development of Non-AIDS-Defining Fibrotic Disorders. Front. Immunol. 8:1461. doi: 10.3389/fimmu.2017.01461
Received: 15 September 2017; Accepted: 18 October 2017;
Published: 02 November 2017
Edited by:
Mario Clerici, Università degli Studi di Milano, ItalyReviewed by:
Nicholas Funderburg, The Ohio State University Columbus, United StatesCopyright: © 2017 Theron, Anderson, Rossouw and Steel. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Annette J. Theron, YXRoZXJvbkB1cC5hYy56YQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.