
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 07 November 2017
Sec. Alloimmunity and Transplantation
Volume 8 - 2017 | https://doi.org/10.3389/fimmu.2017.01444
This article is part of the Research Topic Tailoring NK Cell Receptor-Ligand Interactions: an Art in Evolution View all 34 articles
 Gianfranco Pittari1
Gianfranco Pittari1 Luca Vago2,3
Luca Vago2,3 Moreno Festuccia4,5
Moreno Festuccia4,5 Chiara Bonini6,7
Chiara Bonini6,7 Deena Mudawi1
Deena Mudawi1 Luisa Giaccone4,5
Luisa Giaccone4,5 Benedetto Bruno4,5*
Benedetto Bruno4,5*
Transformed plasma cells in multiple myeloma (MM) are susceptible to natural killer (NK) cell-mediated killing via engagement of tumor ligands for NK activating receptors or “missing-self” recognition. Similar to other cancers, MM targets may elude NK cell immunosurveillance by reprogramming tumor microenvironment and editing cell surface antigen repertoire. Along disease continuum, these effects collectively result in a progressive decline of NK cell immunity, a phenomenon increasingly recognized as a critical determinant of MM progression. In recent years, unprecedented efforts in drug development and experimental research have brought about emergence of novel therapeutic interventions with the potential to override MM-induced NK cell immunosuppression. These NK-cell enhancing treatment strategies may be identified in two major groups: (1) immunomodulatory biologics and small molecules, namely, immune checkpoint inhibitors, therapeutic antibodies, lenalidomide, and indoleamine 2,3-dioxygenase inhibitors and (2) NK cell therapy, namely, adoptive transfer of unmanipulated and chimeric antigen receptor-engineered NK cells. Here, we summarize the mechanisms responsible for NK cell functional suppression in the context of cancer and, specifically, myeloma. Subsequently, contemporary strategies potentially able to reverse NK dysfunction in MM are discussed.
Multiple myeloma (MM) is a B-cell malignancy characterized by an abnormal growth of malignant plasma cells which derive from a post-germinal B-cell of the lymphoid cell lineage. The treatment paradigm for MM has undergone a dramatic evolution in the past decade given a considerable improvement in the understanding of disease pathogenesis. Despite the development of novel therapeutic agents such as proteasome inhibitors—bortezomib, carlfizomib—and immunomodulatory drugs—lenalidomide, pomalidomide—which target not only MM cells but also their interplay with the microenvironment, MM remains an incurable disease and the prognosis of patients with relapsed/refractory MM remains very poor. A number of factors concur to make MM a hard-to-treat hematologic malignancy. Drug resistance remains a major concern. MM is a highly heterogeneous disease with pathogenic processes that may greatly differ among newly diagnosed patients and others that may arise during the disease course. In recent years, several studies have focused on mechanisms of drug resistance even though many are not yet completely understood. It is widely assumed that cytogenetic and epigenetic abnormalities, deregulated signaling pathways, the MM bone marrow (BM) microenvironment, and the MM stem cell itself are all elements which play significant roles in drug resistance. Deletion 17p13 is one of the most relevant chromosomal abnormalities present in approximately 10–15% of newly diagnosed patients and observed more frequently in refractory-relapsed patients. It has been associated with resistance to new agents such as bortezomib and lenalidomide (1, 2). Aberrant drug transport processes and anti-apoptosis mechanisms have also been correlated with drug resistance (3, 4). Moreover, a pivotal role is played by the intense cell–cell crosstalk between the BM microenvironment and MM cells and their interplay with the extracellular matrix (5). All the abovementioned mechanisms make MM very challenging to eradicate with single-agent or combination modalities. Thus, an urgent need exists for new therapeutic strategies to overcome resistance to current therapies. MM is also characterized by a gradual and progressive immune dysregulation with impairs functions of B and T cell immunity, natural killer (NK) cells, and antigen-presenting/dendritic cells that allow malignant plasma cells to escape immunosurveillance. The combination of an “immunosuppressive” microenvironment and clonal evolution activate signaling pathways that invariably promote disease survival and progression. Several immunotherapies have recently been proposed and, among others, they have included monoclonal antibodies, antibody–drug conjugates, chimeric antigen receptor T cell therapy (CAR-T cells), tumor vaccines, and immune checkpoint inhibitors. This review provides an overview of the biological functions and potential clinical role of NK cells as a form of immunotherapy that may improve MM clinical outcomes.
In the early 1970s, immune effectors isolated from mice and humans were found to display in vitro antitumor cytotoxicity without prior immunization by tumor antigens in vivo (6–9). These cells were functionally defined as N-cells or NK cells and were believed to belong to the lymphoid lineage, but to be distinct from B and T cells (10–13). Mechanisms regulating NK cell-mediated target recognition and killing remained obscure for more than a decade after natural cytotoxicity was first described. In 1986, Karre et al. reported that resistance of mice lymphoma cells to NK cell-mediated rejection was dependent on major histocompatibility complex (MHC) class I antigen expression on cancer surface (14). This observation led to the assumption that NK cell would possess receptors able to transduce negative signals upon MHC class I engagement, thus sparing putative targets. Lack of MHC class I would instead trigger NK cell activation, a phenomenon known as missing-self recognition (15).
In humans, the NK cell inhibitory receptors able to recognize HLA class I are type I transmembrane structures belonging to the immunoglobulin (Ig) superfamily, known as killer immunoglobulin-like receptors (KIR). Inhibitory KIR share a long (L) cytoplasmic tail containing immunoreceptor tyrosine-based inhibitory motifs that can process signals through the recruitment and activation of the SH2-domain-containing tyrosine phosphatase 1 protein (16–20). Three inhibitory KIR engaging HLA class I ligand groups are critical regulators of NK cell function: KIR2DL1, specific for HLA-C2 group antigens (sharing Asn at position 77 and Lys at position 80 of the HLA-Cw heavy chain); KIR2DL2/3, specific for HLA-C1 group antigens (sharing Ser at position 77 and Asn at position 80 of the HLA-Cw heavy chain) (21, 22); and KIR3DL1, specific for the HLA-Bw4 epitope (located at position 77–83 of the heavy chain of certain HLA-B and HLA-A alleles) (23–25).
In the last two decades, multiple additional inhibitory NK cells receptors have been identified, leading to the currently accepted notion that NK cell effector function is dependent on the overall balance of signals transduced by multiple inhibitory and activating receptors recognizing cognate ligands on virally infected and cancer cells. Examples of non-KIR inhibitory NK receptors include the c-type lectin-like CD94/NKG2A (CD159a) heterodimer and ILT2 (LILRB1, CD85j), respectively, engaging HLA-E and various HLA class I antigens (26, 27); NKR-P1A (CD161) recognizing the lectin-like transcript 1 (28, 29); and the carcinoembryonic antigen-related cell adhesion molecule 1 (CD66a) recognizing the CD66 ligand (30–32).
Activating NK cell receptors are also described. Among them, NKG2D (CD314) has ligand specificity for a wide range of stress-induced cell surface ligands (NKG2D-L), including the MHC-related ligands MICA and MICB (33) and the human cytomegalovirus glycoprotein (UL16)-binding proteins ULBP1-6 (33, 34). Natural cytotoxicity receptors (NCRs) NKp46 (NCR1, CD335) (35, 36), NKp44 (NCR2, CD336) (37), and NKp30 (NCR3, CD337) (38) are potent activating receptors almost exclusively restricted to NK cells. Ligands for NCR are currently incompletely characterized. NKp46 and NKp44 are known to bind several viral hemagglutinins (39, 40), while NKp30 recognizes the HLA-B-associated transcript 3 (BAT3) (41) and B7-H6, a member of the B7 immunoreceptor family (42). CD94/NKG2C (CD159c) binds the non-classical HLA-E, similar to its inhibitory CD94/NKG2A counterpart (25). CD16 (FcγRIIIA) (43) is the low-affinity IgG receptor, strongly expressed on mature NK cells, mediating antibody-dependent cellular cytotoxicity (ADCC) (44). Other important activating receptors include the SLAM-related 2B4 (CD244) (45) engaging the pan-leukocyte surface antigen CD48 (46) and the adhesion molecule DNAM-1 (47) involved in recognition of PVR (CD155) and nectin-2 (CD112) (48).
Accumulating evidence indicates that microenvironment transformation may significantly impair NK cell effector function in MM (49). Plasma cells and T regulatory (Treg) cells from patients with MM secrete high levels of TGF-β (50, 51), a potent immunosuppressive cytokine known to downregulate multiple NK-activating receptors and to impair NK cytotoxicity (52–54). IL-10 and IL-6 are increased in MM (55–57) and independently act as powerful growth factors for malignant plasma cells (58, 59). IL-10 inhibits production of pro-inflammatory IFN-γ and TNF-α (60, 61) and promotes development of NK-resistant tumor phenotypes (62), although it may also enhance NK cytotoxicity in response to IL-15 exposure in vitro (63). IL-6 has been shown to impair NK cell activity in experimental models, human disease, and when administered to patients with advanced cancer (64–66). Altered levels of IFN-γ may also contribute to NK cell dysregulation in MM. In two studies, serum IFN-γ levels were found to be significantly lower in subjects with MM than in normal controls (55, 56), potentially affecting NK cell activity. Besides cytokines, other soluble factors are known to suppress NK-mediated antitumor capabilities. Prostaglandin E2 inhibits activating signals transduced by NCR, NKG2D, and CD16 (67) and has been shown to be actively produced in cultures of BM from patients with MM (68). Indoleamine 2,3-dioxygenase (IDO) promotes cancer cell immune escape through potent immunoregulatory effects on antigen-presenting cells via enzymatic degradation of l-tryptophan (69) (see IDO inhibitors). Della Chiesa et al. described that IDO-mediated immunosuppression also involves NK cells via l-kyreunine (Kyn), a l-tryptophan (Trp) degradation product impairing NKp46/NKG2D-specific lysis (70). Interestingly, interaction between CD28 on MM cells and CD80/86 stimulates IDO production by stromal dendritic cells (71), in agreement with the observation that CD28 expression on MM plasma cells is a marker correlating with poor disease outcome (72).
Additional microenvironmental factors may contribute to blunted NK cell cytotoxicity and cytokine production in MM. Among them, myeloid-derived suppressor cells (MDSCs) have been found to be expanded in MM (73, 74) and to directly contribute to downregulation of NK cell responsiveness via the NKp30-activating receptor (75), membrane-bound TGF-β (76), and TIGIT-mediated inhibitory signaling (77). Furthermore, reduced oxygenation described in MM BM (78, 79) may inhibit NK cell anti-myeloma responsiveness (80).
MICA and MICB (collectively named MIC) are stress-inducible NKG2D ligands frequently overexpressed in response to malignant transformation (81). When bound to tumor surface, they act as markers of “abnormal self” and may trigger NK cell cytotoxicity via NKG2D signaling. Conversely, cleavage of membrane-bound MIC is a strategy employed by MM and other tumors to evade NK cell immunosurveillance (82–85). In individuals with MIC+ tumors, soluble MIC (sMIC) ligands induce internalization of surface NKG2D (but also NCR and chemokine receptors) and substantial impairment of NK effector functions (86–88). In addition, sMIC has been shown to promote the accumulation of MDSC and macrophages with an immunosuppressive phenotype (89), potentially contributing to NK cell suppression. Not surprisingly, presence of sMIC is associated with poor cancer survival (90–92). In MM, shedding of MIC may result from exposure of MM cells to the genotoxic agents, doxorubicin and melphalan (93). Proteolytic cleavage by ADAMTS10 has been described to mediate this phenomenon, suggesting that the combination of metalloproteinase inhibitors with chemotherapy would exert a protective effect against escape of MM cells from NK-mediated recognition (93). Similar to NKG2D-L, NCR-specific soluble ligands may in some instances induce NK cell functional impairment. For example, circulating BAG6/BAT3 may inhibit NK cell cytotoxicity by inducing NKp30-specific hyporesponsiveness (94). Shedding of these ligands in the context of MM has not been investigated.
Derangement of NK cell effector functions may be further amplified by tumor ligand surface expression patterns favoring dominance of inhibitory NK signals. Ligands recognized by NK-activating receptors are often poorly expressed in cancer. Downregulation of membrane-bound NKG2D-L is common in multiple tumors, resulting in impaired NKG2D-dependent NK cell cytotoxicity (95–97) and unfavorable clinical outcomes (97). In the context of monoclonal gammopathy, expression of MICA is known to decrease upon transition from pre-cancerous monoclonal gammopathy of undetermined significance (MGUS) to MM (84). Of note, various pharmacological interventions may counter NKG2D-L downregulation in MM: vincristine, via p38 MAPK pathway activation (98); doxorubicin, melphalan, and bortezomib as a result of oxidative stress, DNA damage, and tumor senescence (99, 100) the heat shock protein-90 (HSP90) chaperone protein inhibitors 17-allylaminogeldanamycin and radicicol (101); and inhibition or degradation of bromodomain and extra-terminal proteins (102). Exposure to therapeutic agents with activity on MM has similarly been shown to induce upregulation of PVR (an activating ligand for DNAM-1) on malignant plasma cells (98, 100, 103). Besides NKG2D-L, surface expression of the B7-H6 ligand, engaging the NKp30 NCR, has been found to be downregulated in cell lines generated from multiple cancers, including MM, resulting in NKp30-dependent NK cell functional impairment (104).
Upregulation of tumor-bound HLA class I antigens is another mechanism of protection against NK cell immunosurveillance. Malignant plasma cells obtained from the BM of early-stage myeloma patients display low HLA class I expression potentially favoring NK-mediated killing (105). In contrast, high HLA class I levels are observed on plasma cells derived from pleural effusions of patients with advanced MM (105). HLA-E is a non-classical HLA class I antigen frequently upregulated on cancer cells, a phenomenon correlating with poor prognosis (106). In MM primary cells, high HLA-E expression results in restrained in vitro degranulation of NK cell subsets expressing the HLA-E-specific inhibitory NK receptor NKG2A (107).
Surface overexpression of ligands for inhibitory NK receptors is not restricted to HLA class I antigens. Notably, the CD200 glycoprotein is also commonly overexpressed on cancer surface, specifically in myeloid and lymphoid leukemias, where it is a marker of poor prognosis (108, 109). Leukemia blasts overexpressing CD200 escape NK-mediated immunosurveillance by dampening NK cell cytolytic capabilities and NKp44/NKp46 receptor expression (110), a phenomenon that can be reversed by CD200 blockade (111). CD200 is also frequently expressed in patients with MM, where it adversely affects clinical outcomes following stem cell transplantation (112).
Multiple reports describe numerical, phenotypic, and functional NK cell alterations in MM. Subjects with MGUS and untreated, early-stage MM have been generally found to have higher (113–115) or similar (116–118) numbers of circulating and BM NK cells than healthy donors. Upregulation of CD57 and CD16 on NK cell surface is also observed (119, 120), suggesting the emergence of terminally differentiated subsets with high-cytotoxic potential. While these findings suggest efficient response to malignant clones subject to NK-mediated immunosurveillance, several lines of evidence favor the view that such early anti-MM effects are rather to be interpreted as a sign of immunological stress resulting in poor disease control. In fact, the effector function of expanded NK cells from MM subjects has been unexpectedly found to be similar to that of NK cells obtained from healthy donors (114), and NK cells obtained from untreated or previously treated MM patients show a lower increase in cytotoxicity to the K562 cell line in response to pre-incubation with IFN-γ (121). Moreover, NK cell effector functions positively correlate with presence of adverse prognostic factors, including anemia, low albumin, high β2-microglobulin, and renal failure (115), suggesting a “stressed” immunoresponse under the pressure of an aggressive clonal expansion (115). Notably, NK cells from patients with MM display an “exhausted” phenotype signature that includes downregulation of multiple activating receptors and upregulation of programmed death receptor-1 (PD-1). Surface expression of activating 2B4 is reduced in both PB (122) and BM (123) NK cells obtained from untreated subjects with MM, potentially preventing killing of plasma cells despite low HLA class I expression (105). NKG2D and NCR are also downregulated in MM, but preferentially in the BM (122, 123), supporting the concept that downregulation of certain activating NK cell receptors is both dependent on soluble ligands and direct cell–cell contact. Negative signaling from PD-1 is a well-established marker of exhaustion on T cells, but can also disrupt NK cell cytotoxicity and cytokine production (124). In MM, both expression of PD-1 on NK cells and of its ligand PD-L1 on plasma cells has been described (125, 126). PD-1/PD-L1 interactions may therefore promote NK cell functional exhaustion in MM, a phenomenon potentially reversible by checkpoint blockade inhibition (see Inhibitors of the PD-1/PD-L1 Pathway; Figure 1).
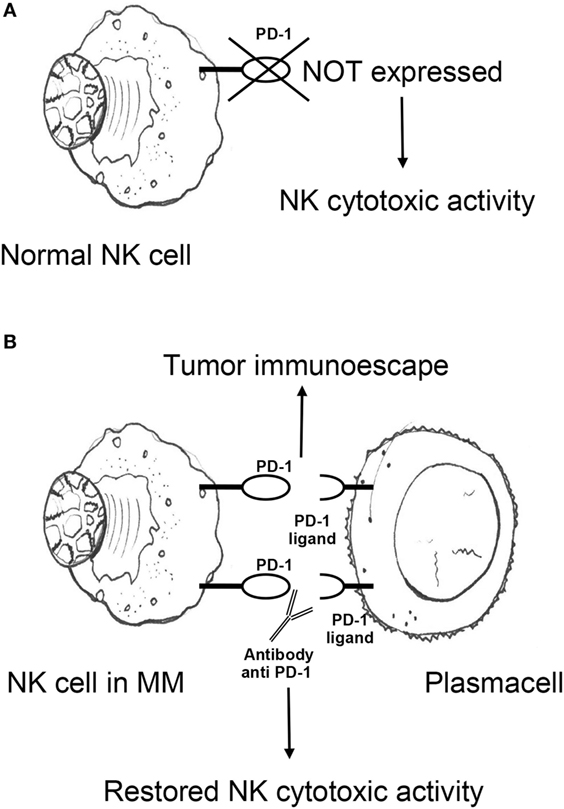
Figure 1. (A) Antitumor cytotoxic activity of NK cells in healthy individuals is not impaired by PD-1 expression. (B) NK cells from MM patients express PD-1, which promotes MM escape from NK cell-mediated immunosurveillance upon engagement with cognate ligand PD-L1 on plasma cells. PD-1/PD-L1 blocking monoclonal antibodies may potentiate NK cell effector functions against MM. NK, natural killer; PD-1, programmed death receptor-1; MM, multiple myeloma.
Natural killer cell-mediated immunity further deteriorates in advanced MM. Compared to MGUS and untreated MM, PB NK cell numbers are substantially reduced in advanced disease (113). Altered distribution of NK cell subsets in human BM may be likewise hypothesized based on studies in mice demonstrating selective decrease of KLRG1− NK cells during MM progression (127). Evolving phenotype editing further promotes tumor escape from NK cell-mediated immunosurveillance. Furthermore, the activating receptor DNAM-1, expressed on NK cells from healthy donors and with MM in complete remission, is downregulated on NK cells from patient with active MM (128). This phenomenon is particularly relevant for late-stage cancer immune escape, as killing of malignant plasma cells is in certain circumstances critically dependent on DNAM-1 engagement of PVR and nectin-2 (128, 129). In line with these findings, NK cell activity in advanced MM is significantly impaired (130). Taken together, these data indicate that NK cell immunity alterations, already detectable in early myeloma, progress in a clinical stage-dependent manner and that immunotherapy modalities based on efficient NK cell effector function such as (i.e., mAbs) are likely to exert a more effective anti-myeloma effect when used in early-stage disease.
Factors promoting NK immunity suppression in MM are summarized in Table 1.
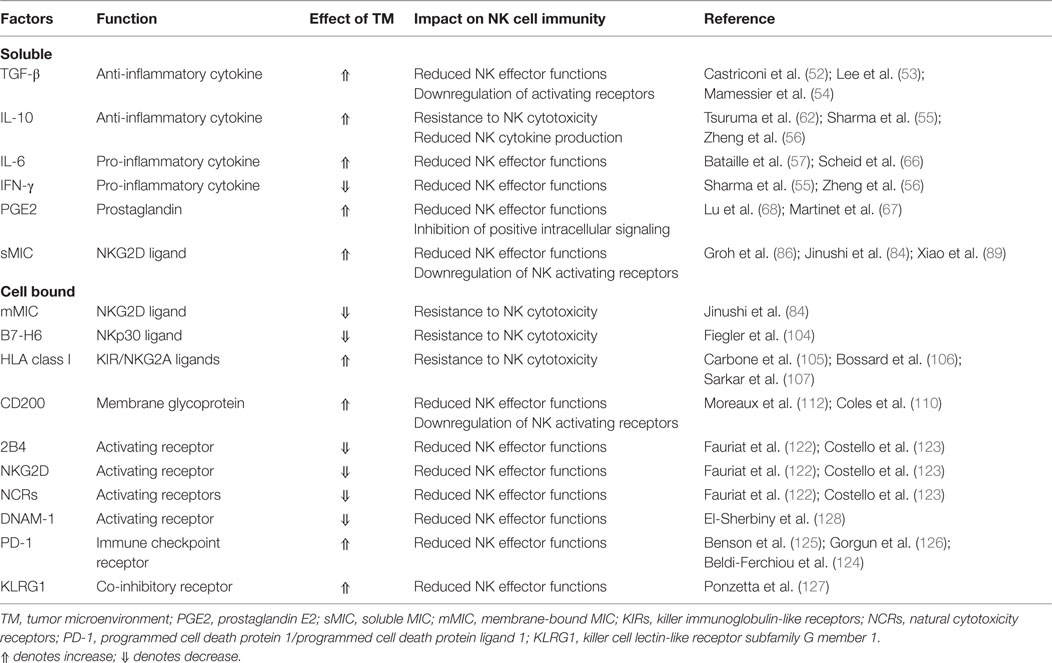
Table 1. Microenvironment alterations potentially promoting natural killer (NK) immunity suppression in multiple myeloma.
Programmed death receptor-1 is a transmembrane protein expressed on the surface of antigen-activated T and B cells. It has two ligands, PD-L1 and PD-L2. PD-L1 is expressed on both antigen-presenting cells/dendritic cells and a wide spectrum of non-hematopoietic cells. PD-1/PD-L1 interactions physiologically counter T cell stimulatory signals and allow T cell homeostasis and self-tolerance by suppressing activation and proliferation of autoreactive T cells. PD-1/PD-L1 binding delivers an inhibitory costimulatory signal that induces a state of T cell exhaustion that prevents activation and proliferation of T cells. Unlike NK cells from healthy donors, NK cells from MM patients express PD-1 (Figure 1A), suggesting that NK cells from healthy donors do not express PD-1 (Figure 1A), however, NK cells from MM patients do. This may show that a functional change in NK cells in response to MM may cause an immunosuppressive microenvironment for MM to grow. In the light of these observations and the broad expression of PD-1 and its ligands in the MM microenvironment, the PD-1/PD-L1 pathway may play a pivotal role in the immune evasion of MM cells (Figure 1B).
A role for the PD-1/PD-L1 signaling pathway in the NK cell immunoresponse against MM and of the anti-PD1 antibody CT-011 was first shown by Benson et al. (125). CT-011 was demonstrated to enhance human NK cell function against autologous, primary MM cells by affecting NK cell trafficking, immune complex formation with MM cells, and cytotoxicity toward MM cells expressing PD-L1 while sparing normal cells (Figure 1B).
It was also shown that lenalidomide had the ability to down regulate PD-L1 on primary MM cells and, by so doing, increase NK cell functions against MM. Thus, targeting the PD-1/PD-L1 pathway may become a feasible clinical strategy in MM, especially in patients with persistent residual disease (131).
One preliminary phase I study reported on 17 patients treated with pembrolizumab, a PD-1 inhibitor, in combination with lenalidomide and dexamethasone (132). Overall response and very good partial response rates were 76 and 23%, respectively. Some 75% of patients achieved stable disease. Many patients were heavily pretreated with other lines of therapy. Almost all patients, however, experienced at least one adverse event with anemia, neutropenia, thrombocytopenia, fatigue, hyperglycemia, and muscle spasms being the most common. Two other recent studies with nivolumab showed acceptable toxicity but no objective responses (133, 134). Efficacy assessment of nivolumab, alone or in combination, is ongoing.
More recently, a novel subpopulation of human NK cells expressing high levels of PD-1 have been identified in ovarian cancer, characterized by low proliferative responses, and impaired antitumor activity that can be partially restored by antibody-mediated disruption of PD-1/PD-L1 interaction (135).
Future studies to evaluate the real therapeutic role of anti-PD-1 antibodies, maybe in combination with other agents with potent anti-myeloma activity such as lenalidomide, are warranted.
The role of NK cells as graft-vs.-myeloma effectors was first investigated in preclinical models. Frohn et al. described for the first time the killing ability of NK cells against three different MM cell lines. The mean NK cell killing ability on MM samples ranged from 23 to 34.5% (136). Moreover, KIR-ligand mismatch in T cell-depleted allogeneic stem cell transplantation reduced the relapse incidence in MM recipients. The impact of KIR-ligand mismatch was assessed in a cohort of 73 MM patients who received reduced-intensity unrelated donor transplants. KIR-ligand mismatch in the graft-vs.-host disease direction was significantly associated with lower risk of relapse (HR: 0; p < 0.0001) (137).
To exploit this pathway, Romagné et al. generated an IgG monoclonal antibody, 1-7F9, against three different KIRs (KIR2DL-1, KIR2DL-2, and KIR2DL-3) to enhance the NK cells antitumor effect. This checkpoint inhibitor augmented NK cell-mediated lysis of HLA-C-expressing tumor cells without interfering with normal peripheral blood (PB) mononuclear cells (138) (Figure 2A). The therapeutic potential of 1-7F9 was then demonstrated in preclinical mouse models, providing the platform for translational studies in humans (139).
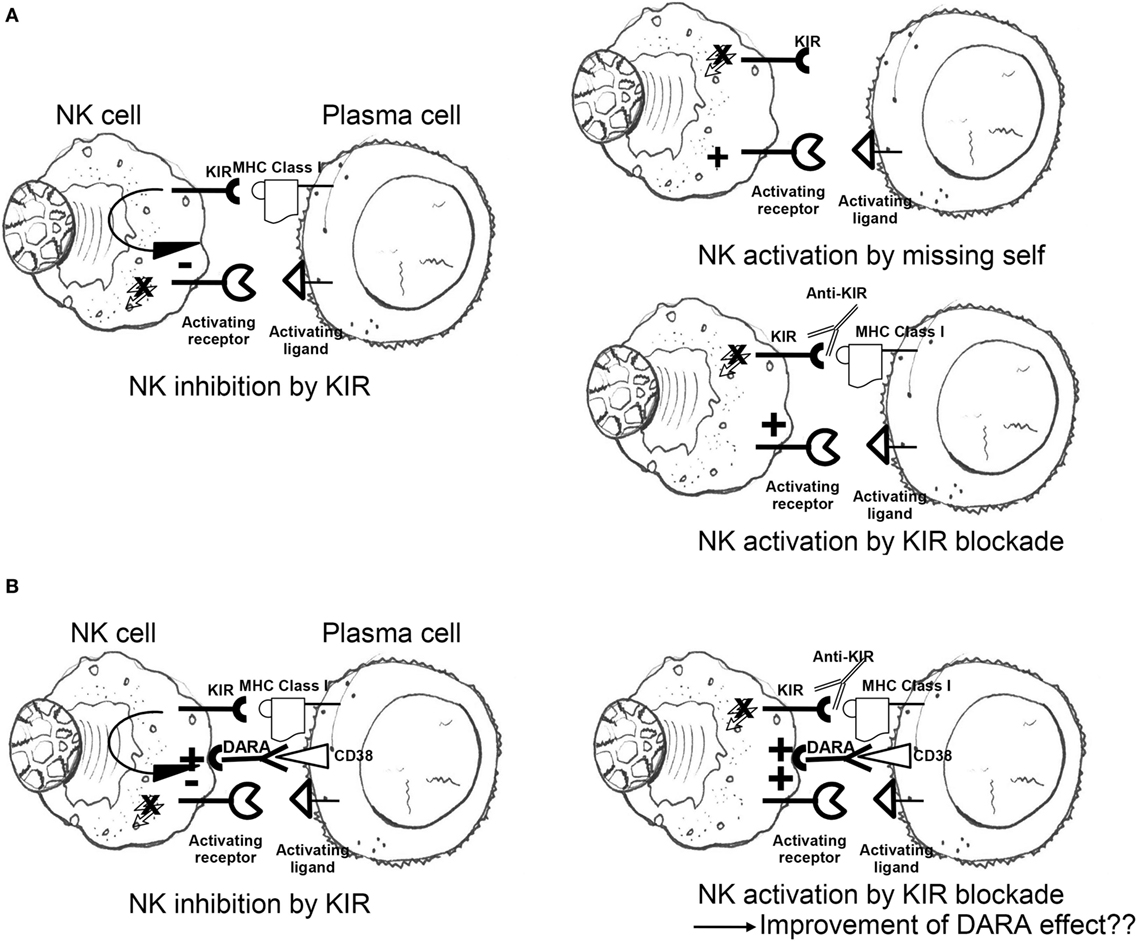
Figure 2. (A) Engagement of self-MHC class I by inhibitory KIR results in dominant-negative signals blocking competing activation responses; lack of MHC class I molecules triggers NK cell killing (missing-self recognition); inhibitory KIR blockade by anti-KIR mAbs abrogates KIR-mediated inhibition regardless of MHC class I ligand expression on target surface (“induced” missing self). (B) Negative signals transduced by inhibitory KIR antagonize anti-CD38 (DARA)-induced antibody-dependent cellular cytotoxicity, potentially dampening NK cytotoxicity to plasma cells; addition of KIR checkpoint inhibitors may potentiate the positive effects of DARA on NK cytotoxicity of malignant plasma cells (see also main text). NK, natural killer; KIR, killer cell immunoglobulin-like receptor; MHC, major histocompatibility complex; DARA, daratumumab.
The drug IPH2101, formerly 1-7F9, was tested in a phase I trial in 32 patients with relapsed/refractory MM. IPH2101 was administered for up to four 28-day cycles, in 7 dose-escalated cohorts (0.0003–3 mg/kg). Only one patient developed severe toxicity, characterized by grade 4 acute renal failure with hyperkalemia and hyperuricemia. From a biological point of view, the drug determined the full saturation of NK inhibitory KIRs (140). Furthermore, lenalidomide and IPH2101 were investigated as a novel, steroid-sparing, dual immunotherapy in 15 MM patients: the biological endpoint of full KIR occupancy was achieved, 5 patients had a response, and 5 severe adverse events were reported (141).
In an open-label, single arm two-stage phase II trial, IPH2101 was employed at the dose of 1 mg/kg every other month for six cycles in nine patients with smoldering MM. Despite the promising results from preclinical and phase I studies, the trial was terminated before planned second stage due to lack of patients meeting the primary objective (50% decline in M-protein) (142).
A recombinant version of IPH2101 was developed with a stabilized hinge (lirilumab). A phase I study of the safety and tolerability of lirilumab with elotuzumab in myeloma patients is currently in progress. Of note, lirilumab recognizes both the inhibitory KIR2DL1, -L2, and -L3 and the activating KIR2DS1-2. Therefore, lirilumab-mediated modulation of intracellular signals is expected to vary according to patient’s HLA class I genetic background and KIR receptor repertoire.
In vitro experiments showed that KIR2D molecules are removed from NK cells surface by trogocytosis. This phenomenon culminated in a strong reduction of NK cell cytotoxic function correlating with the loss of free KIR2D surface molecules (143). These data favor future protocol designs where lirilumab is administered in combination with other NK cell-activating agents, rather than as single agent.
Lenalidomide, a thalidomide analog, is an immunomodulatory drug with multiple mechanisms of action in MM. It is currently approved in both EU and USA in association with dexamethasone for the maintenance treatment of patients with newly diagnosed MM who have undergone an autograft. Four pivotal phase III studies have associated lenalidomide with improved progression-free survival and better overall response rates (144–147). Although lenalidomide has also been associated with increased risk of a second primary cancer, the overall survival benefits outweigh the risk (148).
Due to failure of single-agent anti-KIRs in phase II studies, researchers from multiple institutions investigated possible combined therapies. In vitro, the immunomodulatory agent lenalidomide was responsible of NK cell expansion and activation associated with malignant cells apoptosis (149). On this platform, Benson et al. tested the cytotoxicity of IPH2101 in combination to lenalidomide against MM cell lines U266 and K562 (139, 140). Healthy donor NK cells pretreated with lenalidomide or IPH2101 alone and combined showed increased IFN-γ production against primary MM cells compared to controls (p < 0.05). Furthermore, NK cells pretreated with both lenalidomide and IPH2101 led to the highest IFN-γ peak. The statistical interaction of p-value was 0.0182, suggesting a synergistic effect between the two drugs. Then, healthy donor PB mononuclear cells (PBMCs) incubated as control or with lenalidomide and/or with IPH2101 were used as effectors against U266 MM cell targets. Lenalidomide increased the specific release, a surrogate for cytotoxicity, by around 1.39-fold relative to control (p < 0.01). IPH2101 increased the specific release by 1.48-fold (p < 0.01). The two drugs combined increased the specific release by 2.09-fold relative to control (p < 0.001), which means a significantly higher cytotoxic effect than either lenalidomide or IPH2101 alone. Patient-derived NK cell cytotoxicity against autologous MM targets was enhanced by the combination of lenalidomide plus IPH2101 (128 ± 9 spots/well) compared with control conditions (81 ± 7 spots/well). Based on in vitro results, the authors evaluated the efficacy of the anti-KIR 5E6 in lenalidomide pretreated mice. The tumor burden was significantly reduced when the combination of 5E6 and lenalidomide was employed, in comparison to controls (p < 0.005). These data provide the basis for the translation of IPH2101 and lenalidomide combination in phase I and II studies.
Daratumumab is an IgGk monoclonal antibody targeting CD38, a cell surface protein that is overexpressed on MM cells (150, 151). Preclinical studies have shown that DARA induces MM cell death through several mechanisms, including complement-dependent cytotoxicity (152), ADCC (153), antibody-dependent cellular phagocytosis (154), and apoptosis (155). The drug showed efficacy as single agent in heavily pretreated MM patients or in combination with bortezomib and dexamethasone (156). When combined to lenalidomide, the DARA cell-mediated MM cell clearance was enhanced due to lenalidomide-dependent NK cell activation. In the light of preclinical results of lenalidomide in combination with anti-KIR agents, Nijhof et al. hypothesized that the NK cell-mediated cytotoxicity induced by DARA could be enhanced by anti-KIRs (Figure 2B). The effect could be further improved through the association with lenalidomide which stimulates the proliferation of NK cells and activates them (157), overcoming NK cells depletion induced by DARA itself (158).
Initially, Hsi et al. described a humanized antibody, HuLuc63, which specifically targeted CS1 (CCND3 subset 1, CRACC, and SLAMF7), a cell surface glycoprotein that had not previously been associated with MM cells. By flow-cytometry, HuLuc63 showed specific staining of CD138+ myeloma cells, NK cells, NK-like T cells, and CD8+ T cells. HuLuc63 showed significant in vitro ADCC against primary myeloma cells as targets and allogeneic or autologous NK cells as effectors. The authors concluded that HuLuc63 could eliminate MM partly through NK-mediated ADCC and targeting CS1 with HuLuc63 could become a novel treatment strategy (159). Tai et al. also showed that HuLuc63 was effective in inducing ADCC against primary MM cells resistant to novel therapies such as bortezomib and HSP90 inhibitor. Moreover, pre-treatment with conventional or novel anti-MM agents enhanced HuLuc63-induced MM cell lysis (160). Collins et al. also hypothesized that elotuzumab may have other mechanisms of action. A number of findings clearly suggested that elotuzumab may enhance NK cell function beyond ADCC. Elotuzumab was shown to induce NK cell activation by binding to CS1 which promotes cytotoxicity against CS1+ MM cells but not against autologous CS1+ NK cells. Moreover, NK cell activation was shown to be dependent on differential expression of the signaling intermediary EAT-2 which is present in NK cells but absent in primary, human MM cells (161). Therefore, HuLu63 enhances NK cell cytotoxicity to MM via a dual mechanism (Figure 3). The synergy between current anti-CS1 antibody elotuzumab, formerly known as HuLuc63, and bortezomib was also shown by van Rhee et al. (162). Elotuzumab was approved by FDA in 2015 for the treatment of MM, specific for signaling lymphocytic activation molecule-F7 (SLAMF7, or CS1) (163). As previously mentioned, SLAMF7 is a member of the Ig gene superfamily, almost universally expressed (>95%) on the surface of marrow MM cells, but not on normal tissues, with restricted expression on specific lymphocytes including NK cells. SLAMF7 determines activating or inhibitory effects on NK cells depending on the expression or not of EAT-2, an adapter protein (Figure 3). Given that MM cells lack EAT-2, the molecular mechanism by which SLAMF7 mediates inhibition in NK cells was investigated by Guo et al. It was shown that the inhibitory effects of SLAMF7 in EAT-2− NK cells was mediated by a mechanism implicating lipid phosphatase SHIP-1, Src kinases, and protein tyrosine phosphatase CD45. Coupling of SLAMF7 to SHIP-1 was highly compromised in MM cells. This correlated with a lack of CD45, which is required to activate Src family kinases in hematopoietic cells and was needed to initiate SLAMF7 inhibitory signals. This defect may explain why elotuzumab eliminates MM cells by an indirect mechanism that involves NK cells activation (164, 165). An elegant preclinical model clearly showed that elotuzumab activates NK cells and promotes myeloma cell death in healthy donor PB lymphocyte (PBL)/myeloma cell cocultures (166). Moreover, the combination of elotuzumab plus lenalidomide demonstrated higher anti-myeloma activity on established in vivo MM xenografts and in in vitro PBL/myeloma cell cocultures than either agent alone. In the same study, it was interestingly shown that the increased NK cell anti-myeloma functions were also due to increased secretion of IL-2 and production of TNF-α that combined to enhance NK cell activation and MM cell killing. All these findings supported the clinical application of combination strategies. Elotuzumab initially showed activity in combination with lenalidomide and dexamethasone in a phase I and a phase Ib-2 clinical studies in relapsed/refractory MM (167, 168). In a subsequet randomized study, patients with relapsed/refractory MM received either elotuzumab with lenalidomide and dexamethasone, or lenalidomide and dexamethasone alone. Patients who received a combination of elotuzumab, lenalidomide, and dexamethasone had a significant relative reduction of 30% in the risk of disease progression or death (169). Finally, Jakubowiak et al. reported on a phase II study in relapsed/refractory MM patients where combined elotuzumab/bortezomib/dexamethasone were compared with bortezomib/dexamethasone until progression or unacceptable toxicity. Overall, elotuzumab appeared to provide clinical benefit without clinically significant toxicity when combined with bortezomib (170).
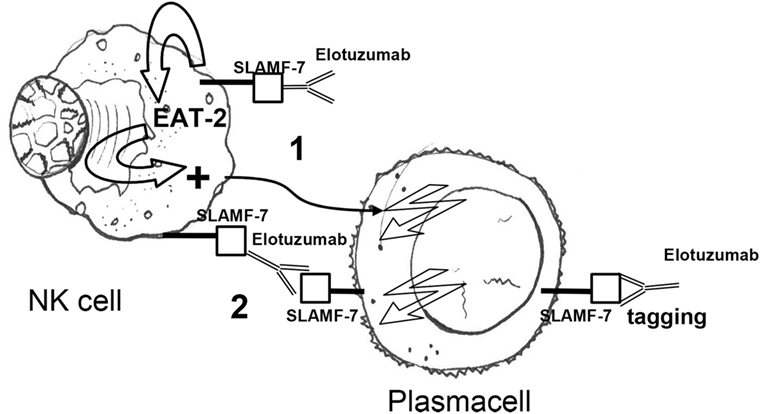
Figure 3. Elotuzumab activates NK cells via (1) an indirect mechanism, i.e., binding of the extracellular portion of SLAMF7 and recruitment of the EAT-2 adaptor protein and (2) a direct mechanism, i.e., antibody-dependent cellular cytotoxicity in response to SLAMF7 tagging on plasma cells. Owing to the absence of EAT-2 in plasma cells, elotuzumab engagement does not cause activation of plasma cells. NK, natural killer; EAT-2 Ewing’s sarcoma-associated transcript 2; Fc, fragment crystallizable; Fab, fragment antigen binding.
Indoleamine 2,3-dioxygenase plays a pivotal role in the metabolic cascade that converts the essential amino acid l-tryptophan (Trp) into l-kynurenine (Kyn). Moreover, IDO has also been shown to be involved in the establishment and maintenance of peripheral tolerance. This function may partly be due to IDO1 capacity to restrict the microenvironmental availability of Trp and to increase the accumulation of Kyn and derivatives. The expression of IDO on neoplastic lesions may help cancer to escape immunosurveillance. IDO inhibitors (i.e., 1-methyltryptophan) have therefore become a new class of anti-cancer agents. Current models imply that IDO limits both innate and adaptive immunoresponses by depleting immunoeffector cells of Trp (171, 172) and by promoting the accumulation of Kyn and its derivatives 3-dydroxykynurenine and 3-hydroxyanthranilic acid (173, 174). These derivatives have been shown not only to exert cytostatic and cytotoxic effects on several immunoeffectors including CD8 T cells, NK cells, and invariant NKT cells (173–175) but also to inhibit TH17 cells and to promote the differentiation of naıve CD4 T cells into Tregs and tolerogenic activity of dendritic cells (174–181). Interestingly, Bonanno et al. investigated IDO expression in 25 symptomatic MM and in 7 with either MGUS or smoldering MM (182). IDO-driven tryptophan breakdown was correlated with the release of hepatocyte growth factor (HGF) and with the frequency of Treg cells and NY-ESO-1-specific CD8 T cells. Kyn was increased in 75% of MM patients and correlated with the expansion of Treg cells and the contraction of NY-ESO-1-specific CD8 T cells. In vitro, primary MM cells promoted the differentiation of allogeneic CD4 T cells into Treg cells and suppressed IFN-γ/IL-2 secretion while preserving IL-4 and IL-10 production. Both Treg expansion and inhibition of Th1 differentiation were partly reverted by d,l-1-methyl-tryptophan, an inhibitor of IDO. Of note, HGF levels were higher within the marrow microenvironment of patients with IDO(+) MM as compared with patients with IDO(−) MM. The antagonism of MET receptor for HGF with SU11274, a MET inhibitor, prevented HGF-induced AKT phosphorylation and resulted in reduced IDO protein levels and functional activity. These findings suggest that IDO expression may contribute to immunosuppression in patients with MM. IDO inhibitors are currently being tested either as single agent or in combination with other anti-cancer drugs in a number of solid tumors even though this class has not yet been evaluated in MM.
Some of the most convincing proofs of the potential of NK cells as immunotherapeutic tools derive from evidences accumulated over the past two decades in the setting of allogeneic hematopoietic stem cell transplantation (allo-HSCT). The interest in NK cell immunobiology stemmed mainly from two observations: (a) NK cells are the first lymphocyte subset to recover after transplantation, often reaching percentages and absolute counts superior to those commonly observed in healthy subjects (183, 184), and (b) since KIRs and HLA ligands are encoded on different, independently inherited, chromosomes (chromosome 19 and 6, respectively), the KIR gene repertoire transferred from the donor into the host is often different posing the issue whether, and how, an efficient NK cell functionality can be achieved (185–187).
Two hallmark studies came from the Stanford group (188, 189) where it was demonstrated that after a variable number of months following HLA-matched, KIR-mismatched allo-HSCT, the NK cell repertoire is dominated by lymphocytes displaying an immature phenotype (CD56bright and CD94/NKG2A+) and then it stabilizes and becomes similar to that of donor origin. Moreover, these studies highlighted significant differences in the repertoire recovery kinetics among patients, and clearly correlated impaired NK cell reconstitution with occurrence of post-transplant complications. The development of a HSCT platform which allowed to safely infuse HLA-haploidentical grafts set the stage for some of the most exciting discoveries in transplant biology and translational potential of NK cells.
Starting from preclinical studies on the tolerogenic potential of “stem cell megadoses” (190), the Perugia team developed a protocol which combined a highly immunosuppressive myeloablative conditioning regimen with the infusion of high doses of extensively T cell-depleted HLA-haploidentical hematopoietic stem cells (HSCs). Full donor engraftment of the partially incompatible HSCs was successfully achieved, and despite the absence of post-transplant pharmacological prophylaxis, neither acute nor chronic GvHD ensued (191). This elegant and technologically advanced HSCT platform offered the opportunity to investigate the metrics of NK cell reconstitution in a partially HLA-mismatched host and in the absence of confounding factors such as alloreactive T cells or immunosuppressive drugs. This highly favorable environment further boosted the early expansion of NK cells that had been already described in other transplant settings, and, importantly, led to the appearance of donor-derived NK cells with alloreactivity against the host (192).
Velardi et al. brilliantly described the principles by which NK cell alloreactivity developed and designed an algorithm to easily predict it. Based on this model—later defined as “ligand-ligand” or “KIR-ligand mismatch” model—post-transplant alloreactivity is unleashed when the donor carried one or more KIR ligands (i.e., HLA class I alleles encompassing the Bw4, C1, or C2 motifs) absent in the host. In this setting, inhibitory KIRs expressed on the surface of donor-derived NK cells—which, in the host, are continuously engaged by their respective ligands in the pre-transplant phase—do not find their cognate HLA molecules on host cells and tissues leading to a perception of “missing self” that activates an alloreactive response (193, 194).
One of the most striking observations by the Perugia group was that NK alloreactivity did not result in clinical GvHD, but, conversely, led to a potentially eradicating mechanism of residual leukemic cells reducing relapse incidence and risk of graft failure and GvHD (195, 196). Over the following years, several studies confirmed and consolidated the evidence that in T cell-depleted haploidentical HSCT NK cell alloreactivity represents the main driver of the graft-vs.-leukemia effect and a major predictor of overall clinical outcomes in both adults (197–199) and children (200, 201). In addition, these studies were a major drive for the development of cell therapy protocols in which haploidentical KIR ligand-mismatched NK cells were infused in leukemic patients after lymphodepleting chemotherapy with highly promising results (202–204).
In more recent years, several new platforms of haploidentical HSCT have been developed, mainly with the aim at improving T cell immune reconstitution and at reducing post-transplant infectious complications (205). In this “new era,” studies on the impact of KIR-ligand mismatches on transplant outcomes reported some conflicting results. For instance, it was shown that infusion of unmanipulated BM grafts or of donor post-transplant T cell add-backs may mask, or blunt, the effectiveness of NK cell alloreactivity (206–208). By contrast, other recent haploidentical HSCT platforms based on the selective depletion of αβ T cells or on the infusion of balanced doses of conventional and regulatory T cells appeared to better preserve the positive effect of KIR ligand mismatches (209, 210).
In partially HLA-mismatched unrelated donor HSCT, either from adult volunteers or cord blood (CB) units, the potential role of NK alloreactivity has also been a matter of debate. Some studies supported a positive role of KIR ligand mismatches (211–213) and others found no significant advantage or even adverse effects (214–216).
To overcome these inconsistencies, several alternative immunogenetic models have been developed to better predict NK cell-driven effects on transplant outcomes. In particular, Cooley et al. focused on the donor genetic repertoire and demonstrated in a number of independent studies that donors with a KIR gene asset enriched in activating receptors—group B KIR haplotypes—can provide a superior relapse-free survival after unrelated HSCT for leukemias (217–219). Another model which takes into account both donor activating KIR asset and donor/recipient HLA typing has been proposed and validated by Venstrom et al. In an analysis on more than 1,200 unrelated HSCTs, the authors observed that the presence of donor-activating receptor KIR2DS1 and of HLA-C1 ligands provided a significant protection from relapse, further enhanced in case of recipient HLA-C1 positivity (220).
Despite the multiplicity of models proposed over the years in the setting of allo-HSCT, not all the immunogenetic mechanisms that regulate NK cell interactions and alloreactivity have fully been understood. However, it is widely assumed that NK cell alloreactivity is instrumental in control and eradication of hematological malignancies.
Expansion of NK cells from PBMC of patients with MM has been achieved using a culture system supplemented with IL-2 and OKT3 (221). NK cells could be extensively propagated (average 1,625-fold expansion in 20 days) and displayed increased levels of activating receptors as well as cytotoxicity to the NK-susceptible K562 line and to autologous MM cells (222). Another NK cell expansion strategy for MM immunotherapy is based on the artificial feeder K562 transfected with CD137L and membrane-bound IL-15. This technique allowed extensive in vitro NK cell propagation (average 447-fold, range 20–10,430 on harvest day, i.e., days 10–14). Transfer of these cells into a xenogeneic model of high-risk MM resulted in myeloma growth inhibition and protection against osteolysis (223). The same group tested the safety, persistence, and activity of expanded NK cells in seven heavily pretreated patients with high-risk relapsed myeloma: no serious adverse events related to NK cell infusion was observed. Moreover, the infusion of fresh, rather than cryopreserved, cells resulted of fundamental importance for their in vivo expansion. Two/seven patients showed some responses which lasted for at least 6 months (224). More recently, human studies were performed with allogeneic, KIR ligand-mismatched NK cells from haploidentical family donors. NK cells were cytotoxic to K562, the myeloma line U266, and recipient primary MM cells. Fifty percent of the patients with advanced MM achieved near complete remission when these cells were infused prior to autologous SCT (225). Another phase I clinical trial (NCT02481934) evaluated safety and efficacy of multiple infusions of activated and expanded NK cells in combination with lenalidomide- or bortezomib-based regimens (226). Five heavily pretreated refractory/relapsed patients were enrolled. NK cells were activated and expanded for 3 weeks with K562mb15-41BBL cells. Patients received four cycles of new drug-based treatment with two infusions of 7.5 × 106/kg NK cells. Four patients showed stable disease while on NK cell treatment, two showed a 50% reduction in BM plasma cell infiltration and one obtained a response >1 year. No major toxicities were reported. Expanded NK cells showed a highly cytotoxic phenotype and in vitro killing and were detected in both BM and PB of treated patients. While efficacy and safety of multiple NK cell infusions need further assessment, these data suggest that repeated transfer of in vitro activated and expanded NK cells into MM patients is feasible and may result in clinical benefit when combined with anti-myeloma drugs.
Umbilical CB represents a promising source of allogeneic NK cells. However, GMP-grade large scale ex vivo expansion is indispensable to generate CB-derived NK (CB-NK) cell doses that may be used in the clinical setting. Shah et al. recently described a strategy for the expansion of NK cells from cryopreserved CB units (227). By co-culturing for 14 days CB units using artificial antigen-presenting feeder cells (aAPC), a highly expanded cell product (average 1,848- and 2,389-fold in 14 days from fresh and cryopreserved samples, respectively) of 95% purity for CB-NK cells and less than 1% CD3+ cells was obtained. Despite differences in the expression of certain cytotoxicity receptors, aAPC-expanded CB-NK cells were phenotypically very similar to CB-NK cells expanded with IL-2 alone. Most importantly, aAPC-expanded CB-NK cells clearly showed cytotoxicity against both in vitro MM targets and in vivo anti-myeloma activity in a xenogenic mouse model. The same group investigated the mechanisms of CB-NK-mediated cytotoxicity against MM cells (228). Interestingly, a mechanism of transmissible cell death between cells induced by lipid–protein vesicles transferred from CB-NK to MM cells was described. Moreover, these vesicles were capable of migrating from recipient MM cells to neighboring MM cells enhancing cytotoxicity of CB-NK. Altogether, these findings supported the development of CB-NK-based cellular therapies for the treatment of MM. An encouraging first-in-human study of CB-NK cells for MM patients undergoing high dose chemotherapy and autologous transplantation was recently conducted (229). Patients received lenalidomide at a dose of 10 mg from day −8 through −2, standard melphalan at 200 mg/m2 on day −7. CB-NK cells were infused on day −5 and the autograft performed on day 0. Twelve patients were treated with different dose levels. Most patients were heavily pretreated and had high-risk cytogenetics. Overall CB-NK cells with an activated phenotype (NKG2D+/NKp30+) were detected in vivo in six patients. Importantly, no signs/symptoms of GVHD were observed. Eight patients achieved at least near complete remission and two additional patients a very good partial response. After a median follow-up of 21 months, four patients relapsed or experienced progressive disease.
The impressive clinical results obtained in patients with B cell malignancies with the infusion of T cells genetically modified to express synthetic chimeric antigen receptors (CARs) against the lineage-specific surface antigen CD19 represented a turning point in the history of cancer immunotherapy (230–236) Intriguingly, T cells engineered with an anti-CD19 CAR were capable to induce complete remission also in a patient with MM. However, given that the large majority of malignant plasma cells do not express CD19, studies to understand the mechanism that underlie this unexpected observation are currently in progress (237). More recently, a number of CARs have been developed to specifically target surface antigens expressed by pathological plasma cells, including CD38 (238, 239), CD138 (240), B cell maturation antigen (241, 242), κ light chains (243), SLAMF7 (244), and CD44v6 (245). However, despite their tremendous efficacy, CAR-T cells have also raised concerns on their short- and long-term toxicities, in particular the development of life-threatening cytokine release syndrome and the risks of prolonged aplasia of the healthy counterparts of the target tumor—“off tumor/on target toxicity”—and in case of allogeneic CAR-T cells the development of GvHD (246, 247).
To address these issues, genetic modifications with CARs of cells belonging to the innate immune system, and of NK cells in particular, may yield several potential advantages. For instance, most innate cells recognize and eliminate tumors by stereotyped patterns and have been infused into allogeneic recipient without excessive toxicities and with some promising intrinsic antitumor efficacy. Moreover, the short-lived persistence of innate immunocells in an allogeneic host, considered up to now one of the major limitations, may become an added value in case of CARs targeting antigens that are shared with mature cell types for which prolonged aplasia may be a concern (i.e., memory B cells, monocytes, or plasma cells) (248, 249).
Genetic modification of the human NK cell lines NKL and NK-92 by means of a lentiviral vector encoding for anti-SLAMF7 and anti-CD138 CARs has proven feasible. This did not substantially modify the expression profile of transduced cells and conferred selectivity for the target and the ability to kill human malignant plasma cells both in ex vivo and in an orthotopic xenograft models (250, 251). Overall, several steps to optimize and validate CAR-modified NK cells should be taken before their possible clinical use. In particular, the choice of the most appropriate NK cell source to be modified is a matter of intense debate (252). Whether freshly isolated NK cells may represent the most physiological choice to achieve sufficient cell doses and transduction efficiency remains unknown. Conversely, NK cells expanded from PB or from progenitor cells may be more easily modified even though their expression profile and functional competence may be negatively affected by prolonged ex vivo culture. Finally, immortalized human NK cell lines, such as NK-92, can be very efficiently transduced and expanded in desired numbers even for “off-the-shelf” use even though their cell surface expression of activating receptors is lower than in freshly isolated or expanded NK cells. Moreover, the need to irradiate the cell product before infusion would further limit their in vivo persistence (252, 253). A new modality that exploits the combination of the anti-CR38 monoclonal antibody DARA with CD38(−) NK cells armed with CS1 CAR has very recently been described by Wang et al. to treat relapsed MM (254). Given that both CS1 and CD38 are MM-associated antigens, their simultaneous targeting may prevent progression. The same authors previously showed that DARA induces apoptosis in CD38(+) NK cells but not in CD38(−) NK cells. It was then hypothesized that DARA in combination with CD38(−) CS1-CAR NK cells may show a synergistic effect and possibly lead to MM eradication. Long-term follow-up of clinical outcomes of this study are eagerly awaited.
The potent crosstalk between malignant plasma cells and their BM microenvironment plays a central role in MM progression and resistance to current therapies. Novel forms of immunotherapy against MM represent a rapidly developing area in cancer therapy. They include treatment strategies that may be delivered either alone or in combination with currently employed therapy lines such as IMiDs and proteasome inhibitors as well as newer agents (Table 2). Moreover, immunotherapy may attenuate the systemic toxicity of cytotoxic chemotherapy. A robust body of evidence has clearly shown that enhancing host anti-myeloma immunity within the BM microenvironment may lead to a more efficient disease control. NK cells play a pivotal role in the intricate network of cells and signaling pathways that may prevent immune escape mechanisms. NK cells were clearly shown to have potent in vivo antileukemia activity in patients undergoing allografting. Recent observations on NK cell functions in MM have become promising immunotherapeutic strategies. New avenues of research have included expansion of NK cells from PB as well as CB, and the generations of specific CAR-NK cells against myeloma-specific antigens. Moreover, MM NK cells express PD-1 whereas NK cells from healthy individuals do not. This phenotypic characteristic may indicate that immunocheckpoint blockade of NK cells may be an area to fully explore given the remarkable results obtained with anti-PD 1 inhibitors in cancer treatment. Altogether, the studies reported in this review show that NK cells hold promise in changing the natural course of MM and that may help restore immunity to MM and thereby improve survival outcomes.
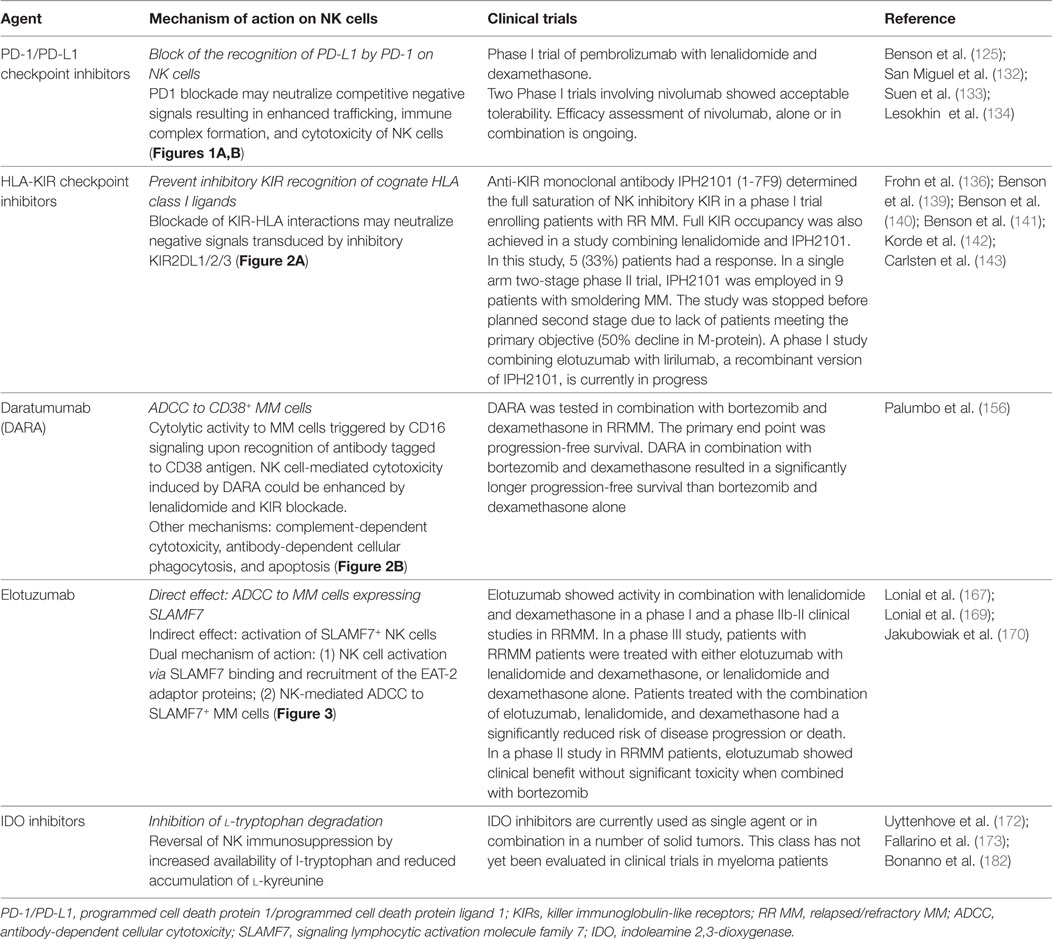
Table 2. Summary of current treatments with novel agents for multiple myeloma (MM) potentially affecting natural killer (NK) cell activity.
GP and BB contributed to the initial conception and designed of the manuscript. CB, MF, DM, and LG provided study materials and critically reviewed the manuscript. GP, LV, MF, and BB wrote the manuscript. All authors gave the final approval to the manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Fonseca R, Bergsagel PL, Drach J, Shaughnessy J, Gutierrez N, Stewart AK. International Myeloma Working Group molecular classification of multiple myeloma: spotlight review. Leukemia (2009) 23(12):2210–21. doi:10.1038/leu.2009.174
2. Reece D, Song KW, Fu T, Roland B, Chang H, Horsman DE, et al. Influence of cytogenetics in patients with relapsed or refractory multiple myeloma treated with lenalidomide plus dexamethasone: adverse effect of deletion 17p13. Blood (2009) 114(3):522–5. doi:10.1182/blood-2008-12-193458
3. Cornelissen JJ, Sonneveld P, Schoester M, Raaijmakers HG, Nieuwenhuis HK, Dekker AW, et al. MDR-1 expression and response to vincristine, doxorubicin, and dexamethasone chemotherapy in multiple myeloma refractory to alkylating agents. J Clin Oncol (1994) 12(1):115–9. doi:10.1200/JCO.1994.12.1.115
4. Yang HH, Ma MH, Vescio RA, Berenson JR. Overcoming drug resistance in multiple myeloma: the emergence of therapeutic approaches to induce apoptosis. J Clin Oncol (2003) 21(22):4239–47. doi:10.1200/JCO.2003.06.001
5. Meads MB, Gatenby RA, Dalton WS. Environment-mediated drug resistance: a major contributor to minimal residual disease. Nat Rev Cancer (2009) 9(9):665–74. doi:10.1038/nrc2714
6. Greenberg AH, Playfair JH. Spontaneously arising cytotoxicity to the P-815-Y mastocytoma in NZB mice. Clin Exp Immunol (1974) 16(1):99–109.
7. Herberman RB, Nunn ME, Lavrin DH, Asofsky R. Effect of antibody to theta antigen on cell-mediated immunity induced in syngeneic mice by murine sarcoma virus. J Natl Cancer Inst (1973) 51(5):1509–12. doi:10.1093/jnci/51.5.1509
8. Petranyi GG, Benczur M, Onody CE, Hollan SR. Letter: HL-A 3,7 and lymphocyte cytotoxic activity. Lancet (1974) 1(7860):736. doi:10.1016/S0140-6736(74)92943-2
9. Takasugi M, Mickey MR, Terasaki PI. Reactivity of lymphocytes from normal persons on cultured tumor cells. Cancer Res (1973) 33(11):2898–902.
10. Herberman RB, Nunn ME, Holden HT, Lavrin DH. Natural cytotoxic reactivity of mouse lymphoid cells against syngeneic and allogeneic tumors. II. Characterization of effector cells. Int J Cancer (1975) 16(2):230–9. doi:10.1002/ijc.2910160204
11. Herberman RB, Nunn ME, Lavrin DH. Natural cytotoxic reactivity of mouse lymphoid cells against syngeneic acid allogeneic tumors. I. Distribution of reactivity and specificity. Int J Cancer (1975) 16(2):216–29. doi:10.1002/ijc.2910160204
12. Kiessling R, Klein E, Pross H, Wigzell H. “Natural” killer cells in the mouse. II. Cytotoxic cells with specificity for mouse Moloney leukemia cells. Characteristics of the killer cell. Eur J Immunol (1975) 5(2):117–21. doi:10.1002/eji.1830050209
13. Kiessling R, Klein E, Wigzell H. “Natural” killer cells in the mouse. I. Cytotoxic cells with specificity for mouse Moloney leukemia cells. Specificity and distribution according to genotype. Eur J Immunol (1975) 5(2):112–7. doi:10.1002/eji.1830050208
14. Karre K, Ljunggren HG, Piontek G, Kiessling R. Selective rejection of H-2-deficient lymphoma variants suggests alternative immune defence strategy. Nature (1986) 319(6055):675–8. doi:10.1038/319675a0
15. Ljunggren HG, Karre K. In search of the ‘missing self’: MHC molecules and NK cell recognition. Immunol Today (1990) 11(7):237–44. doi:10.1016/0167-5699(90)90097-S
16. Burshtyn DN, Scharenberg AM, Wagtmann N, Rajagopalan S, Berrada K, Yi T, et al. Recruitment of tyrosine phosphatase HCP by the killer cell inhibitor receptor. Immunity (1996) 4(1):77–85. doi:10.1016/S1074-7613(00)80300-3
17. Campbell KS, Dessing M, Lopez-Botet M, Cella M, Colonna M. Tyrosine phosphorylation of a human killer inhibitory receptor recruits protein tyrosine phosphatase 1C. J Exp Med (1996) 184(1):93–100. doi:10.1084/jem.184.1.93
18. Fry AM, Lanier LL, Weiss A. Phosphotyrosines in the killer cell inhibitory receptor motif of NKB1 are required for negative signaling and for association with protein tyrosine phosphatase 1C. J Exp Med (1996) 184(1):295–300. doi:10.1084/jem.184.1.295
19. Burshtyn DN, Yang W, Yi T, Long EO. A novel phosphotyrosine motif with a critical amino acid at position-2 for the SH2 domain-mediated activation of the tyrosine phosphatase SHP-1. J Biol Chem (1997) 272(20):13066–72. doi:10.1074/jbc.272.20.13066
20. Yusa S, Campbell KS. Src homology region 2-containing protein tyrosine phosphatase-2 (SHP-2) can play a direct role in the inhibitory function of killer cell Ig-like receptors in human NK cells. J Immunol (2003) 170(9):4539–47. doi:10.4049/jimmunol.170.9.4539
21. Biassoni R, Falco M, Cambiaggi A, Costa P, Verdiani S, Pende D, et al. Amino acid substitutions can influence the natural killer (NK)-mediated recognition of HLA-C molecules. Role of serine-77 and lysine-80 in the target cell protection from lysis mediated by “group 2” or “group 1” NK clones. J Exp Med (1995) 182(2):605–9. doi:10.1084/jem.182.2.605
22. Winter CC, Long EO. A single amino acid in the p58 killer cell inhibitory receptor controls the ability of natural killer cells to discriminate between the two groups of HLA-C allotypes. J Immunol (1997) 158(9):4026–8.
23. Litwin V, Gumperz J, Parham P, Phillips JH, Lanier LL. NKB1: a natural killer cell receptor involved in the recognition of polymorphic HLA-B molecules. J Exp Med (1994) 180(2):537–43. doi:10.1084/jem.180.2.537
24. Wagtmann N, Rajagopalan S, Winter CC, Peruzzi M, Long EO. Killer cell inhibitory receptors specific for HLA-C and HLA-B identified by direct binding and by functional transfer. Immunity (1995) 3(6):801–9. doi:10.1016/1074-7613(95)90069-1
25. Gumperz JE, Litwin V, Phillips JH, Lanier LL, Parham P. The Bw4 public epitope of HLA-B molecules confers reactivity with natural killer cell clones that express NKB1, a putative HLA receptor. J Exp Med (1995) 181(3):1133–44. doi:10.1084/jem.181.3.1133
26. Braud VM, Allan DS, O’Callaghan CA, Soderstrom K, D’Andrea A, Ogg GS, et al. HLA-E binds to natural killer cell receptors CD94/NKG2A, B and C. Nature (1998) 391(6669):795–9.
27. Lee N, Llano M, Carretero M, Ishitani A, Navarro F, Lopez-Botet M, et al. HLA-E is a major ligand for the natural killer inhibitory receptor CD94/NKG2A. Proc Natl Acad Sci U S A (1998) 95(9):5199–204. doi:10.1073/pnas.95.9.5199
28. Aldemir H, Prod’homme V, Dumaurier MJ, Retiere C, Poupon G, Cazareth J, et al. Cutting edge: lectin-like transcript 1 is a ligand for the CD161 receptor. J Immunol (2005) 175(12):7791–5. doi:10.4049/jimmunol.175.12.7791
29. Lanier LL, Chang C, Phillips JH. Human NKR-P1A. A disulfide-linked homodimer of the C-type lectin superfamily expressed by a subset of NK and T lymphocytes. J Immunol (1994) 153(6):2417–28.
30. Teixeira AM, Fawcett J, Simmons DL, Watt SM. The N-domain of the biliary glycoprotein (BGP) adhesion molecule mediates homotypic binding: domain interactions and epitope analysis of BGPc. Blood (1994) 84(1):211–9.
31. Watt SM, Fawcett J, Murdoch SJ, Teixeira AM, Gschmeissner SE, Hajibagheri NM, et al. CD66 identifies the biliary glycoprotein (BGP) adhesion molecule: cloning, expression, and adhesion functions of the BGPc splice variant. Blood (1994) 84(1):200–10.
32. Markel G, Lieberman N, Katz G, Arnon TI, Lotem M, Drize O, et al. CD66a interactions between human melanoma and NK cells: a novel class I MHC-independent inhibitory mechanism of cytotoxicity. J Immunol (2002) 168(6):2803–10. doi:10.4049/jimmunol.168.6.2803
33. Bauer S, Groh V, Wu J, Steinle A, Phillips JH, Lanier LL, et al. Activation of NK cells and T cells by NKG2D, a receptor for stress-inducible MICA. Science (1999) 285(5428):727–9. doi:10.1126/science.285.5428.727
34. Cosman D, Mullberg J, Sutherland CL, Chin W, Armitage R, Fanslow W, et al. ULBPs, novel MHC class I-related molecules, bind to CMV glycoprotein UL16 and stimulate NK cytotoxicity through the NKG2D receptor. Immunity (2001) 14(2):123–33. doi:10.1016/S1074-7613(01)00095-4
35. Sivori S, Vitale M, Morelli L, Sanseverino L, Augugliaro R, Bottino C, et al. p46, a novel natural killer cell-specific surface molecule that mediates cell activation. J Exp Med (1997) 186(7):1129–36. doi:10.1084/jem.186.7.1129
36. Pessino A, Sivori S, Bottino C, Malaspina A, Morelli L, Moretta L, et al. Molecular cloning of NKp46: a novel member of the immunoglobulin superfamily involved in triggering of natural cytotoxicity. J Exp Med (1998) 188(5):953–60. doi:10.1084/jem.188.5.953
37. Vitale M, Bottino C, Sivori S, Sanseverino L, Castriconi R, Marcenaro E, et al. NKp44, a novel triggering surface molecule specifically expressed by activated natural killer cells, is involved in non-major histocompatibility complex-restricted tumor cell lysis. J Exp Med (1998) 187(12):2065–72. doi:10.1084/jem.187.12.2065
38. Pende D, Parolini S, Pessino A, Sivori S, Augugliaro R, Morelli L, et al. Identification and molecular characterization of NKp30, a novel triggering receptor involved in natural cytotoxicity mediated by human natural killer cells. J Exp Med (1999) 190(10):1505–16. doi:10.1084/jem.190.10.1505
39. Mandelboim O, Lieberman N, Lev M, Paul L, Arnon TI, Bushkin Y, et al. Recognition of haemagglutinins on virus-infected cells by NKp46 activates lysis by human NK cells. Nature (2001) 409(6823):1055–60. doi:10.1038/35059110
40. Arnon TI, Lev M, Katz G, Chernobrov Y, Porgador A, Mandelboim O. Recognition of viral hemagglutinins by NKp44 but not by NKp30. Eur J Immunol (2001) 31(9):2680–9. doi:10.1002/1521-4141(200109)31:9<2680::AID-IMMU2680>3.0.CO;2-A
41. Pogge von Strandmann E, Simhadri VR, von Tresckow B, Sasse S, Reiners KS, Hansen HP, et al. Human leukocyte antigen-B-associated transcript 3 is released from tumor cells and engages the NKp30 receptor on natural killer cells. Immunity (2007) 27(6):965–74. doi:10.1016/j.immuni.2007.10.010
42. Brandt CS, Baratin M, Yi EC, Kennedy J, Gao Z, Fox B, et al. The B7 family member B7-H6 is a tumor cell ligand for the activating natural killer cell receptor NKp30 in humans. J Exp Med (2009) 206(7):1495–503. doi:10.1084/jem.20090681
43. Perussia B, Acuto O, Terhorst C, Faust J, Lazarus R, Fanning V, et al. Human natural killer cells analyzed by B73.1, a monoclonal antibody blocking Fc receptor functions. II. Studies of B73.1 antibody-antigen interaction on the lymphocyte membrane. J Immunol (1983) 130(5):2142–8.
44. Mandelboim O, Malik P, Davis DM, Jo CH, Boyson JE, Strominger JL. Human CD16 as a lysis receptor mediating direct natural killer cell cytotoxicity. Proc Natl Acad Sci U S A (1999) 96(10):5640–4. doi:10.1073/pnas.96.10.5640
45. Garni-Wagner BA, Purohit A, Mathew PA, Bennett M, Kumar V. A novel function-associated molecule related to non-MHC-restricted cytotoxicity mediated by activated natural killer cells and T cells. J Immunol (1993) 151(1):60–70.
46. Brown MH, Boles K, van der Merwe PA, Kumar V, Mathew PA, Barclay AN. 2B4, the natural killer and T cell immunoglobulin superfamily surface protein, is a ligand for CD48. J Exp Med (1998) 188(11):2083–90. doi:10.1084/jem.188.11.2083
47. Shibuya A, Campbell D, Hannum C, Yssel H, Franz-Bacon K, McClanahan T, et al. DNAM-1, a novel adhesion molecule involved in the cytolytic function of T lymphocytes. Immunity (1996) 4(6):573–81. doi:10.1016/S1074-7613(00)70060-4
48. Bottino C, Castriconi R, Pende D, Rivera P, Nanni M, Carnemolla B, et al. Identification of PVR (CD155) and Nectin-2 (CD112) as cell surface ligands for the human DNAM-1 (CD226) activating molecule. J Exp Med (2003) 198(4):557–67. doi:10.1084/jem.20030788
49. Bianchi G, Munshi NC. Pathogenesis beyond the cancer clone(s) in multiple myeloma. Blood (2015) 125:3049–58. doi:10.1182/blood-2014-11-568881
50. Urashima M, Ogata A, Chauhan D, Hatziyanni M, Vidriales MB, Dedera DA, et al. Transforming growth factor-beta1: differential effects on multiple myeloma versus normal B cells. Blood (1996) 87:1928–38.
51. Beyer M, Kochanek M, Giese T, Endl E, Weihrauch MR, Knolle PA, et al. In vivo peripheral expansion of naive CD4+CD25high FoxP3+ regulatory T cells in patients with multiple myeloma. Blood (2006) 107:3940–9. doi:10.1182/blood-2005-09-3671
52. Castriconi R, Cantoni C, Della Chiesa M, Vitale M, Marcenaro E, Conte R, et al. Transforming growth factor beta 1 inhibits expression of NKp30 and NKG2D receptors: consequences for the NK-mediated killing of dendritic cells. Proc Natl Acad Sci U S A (2003) 100:4120–5. doi:10.1073/pnas.0730640100
53. Lee JC, Lee KM, Kim DW, Heo DS. Elevated TGF-beta1 secretion and down-modulation of NKG2D underlies impaired NK cytotoxicity in cancer patients. J Immunol (2004) 172:7335–40. doi:10.4049/jimmunol.172.12.7335
54. Mamessier E, Sylvain A, Thibult ML, Houvenaeghel G, Jacquemier J, Castellano R, et al. Human breast cancer cells enhance self tolerance by promoting evasion from NK cell antitumor immunity. J Clin Invest (2011) 121:3609–22. doi:10.1172/JCI45816
55. Sharma A, Khan R, Joshi S, Kumar L, Sharma M. Dysregulation in T helper 1/T helper 2 cytokine ratios in patients with multiple myeloma. Leuk Lymphoma (2010) 51:920–7. doi:10.3109/10428191003699563
56. Zheng MM, Zhang Z, Bemis K, Belch AR, Pilarski LM, Shively JE, et al. The systemic cytokine environment is permanently altered in multiple myeloma. PLoS One (2013) 8:e58504. doi:10.1371/journal.pone.0058504
57. Bataille R, Jourdan M, Zhang XG, Klein B. Serum levels of interleukin 6, a potent myeloma cell growth factor, as a reflect of disease severity in plasma cell dyscrasias. J Clin Invest (1989) 84:2008–11. doi:10.1172/JCI114392
58. Zhang XG, Klein B, Bataille R. Interleukin-6 is a potent myeloma-cell growth factor in patients with aggressive multiple myeloma. Blood (1989) 74:11–3.
59. Kovacs E. Interleukin-6 leads to interleukin-10 production in several human multiple myeloma cell lines. Does interleukin-10 enhance the proliferation of these cells? Leuk Res (2010) 34:912–6. doi:10.1016/j.leukres.2009.08.012
60. Fiorentino DF, Zlotnik A, Vieira P, Mosmann TR, Howard M, Moore KW, et al. IL-10 acts on the antigen-presenting cell to inhibit cytokine production by Th1 cells. J Immunol (1991) 146:3444–51.
61. Conti P, Kempuraj D, Kandere K, Di Gioacchino M, Barbacane RC, Castellani ML, et al. IL-10, an inflammatory/inhibitory cytokine, but not always. Immunol Lett (2003) 86:123–9. doi:10.1016/S0165-2478(03)00002-6
62. Tsuruma T, Yagihashi A, Hirata K, Torigoe T, Araya J, Watanabe N, et al. Interleukin-10 reduces natural killer (NK) sensitivity of tumor cells by downregulating NK target structure expression. Cell Immunol (1999) 198:103–10. doi:10.1006/cimm.1999.1586
63. Park JY, Lee SH, Yoon SR, Park YJ, Jung H, Kim TD, et al. IL-15-induced IL-10 increases the cytolytic activity of human natural killer cells. Mol Cells (2011) 32:265–72. doi:10.1007/s10059-011-1057-8
64. Vredevoe DL, Widawski M, Fonarow GC, Hamilton M, Martínez-Maza O, Gage JR. Interleukin-6 (IL-6) expression and natural killer (NK) cell dysfunction and anergy in heart failure. Am J Cardiol (2004) 93:1007–11. doi:10.1016/j.amjcard.2003.12.054
65. Cifaldi L, Prencipe G, Caiello I, Bracaglia C, Locatelli F, De Benedetti F, et al. Inhibition of natural killer cell cytotoxicity by interleukin-6: implications for the pathogenesis of macrophage activation syndrome. Arthritis Rheumatol (2015) 67:3037–46. doi:10.1002/art.39295
66. Scheid C, Prendiville J, Jayson G, Crowther D, Fox B, Pettit GR, et al. Immunomodulation in patients receiving intravenous Bryostatin 1 in a phase I clinical study: comparison with effects of Bryostatin 1 on lymphocyte function in vitro. Cancer Immunol Immunother (1994) 39:223–30. doi:10.1007/BF01525985
67. Martinet L, Jean C, Dietrich G, Fournié JJ, Poupot R. PGE2 inhibits natural killer and gamma delta T cell cytotoxicity triggered by NKR and TCR through a cAMP-mediated PKA type I-dependent signaling. Biochem Pharmacol (2010) 80:838–45. doi:10.1016/j.bcp.2010.05.002
68. Lu ZY, Bataille R, Poubelle P, Rapp MJ, Harousseau JL, Klein B. An interleukin 1 receptor antagonist blocks the IL-1-induced IL-6 paracrine production through a prostaglandin E2-related mechanism in multiple myeloma. Stem Cells (1995) 13(Suppl 2):28–34.
69. Mellor AL, Munn DH. Tryptophan catabolism and T-cell tolerance: immunosuppression by starvation? Immunol Today (1999) 20:469–73. doi:10.1016/S0167-5699(99)01520-0
70. Della Chiesa M, Carlomagno S, Frumento G, Balsamo M, Cantoni C, Conte R, et al. The tryptophan catabolite L-kynurenine inhibits the surface expression of NKp46- and NKG2D-activating receptors and regulates NK-cell function. Blood (2006) 108(13):4118–25. doi:10.1182/blood-2006-03-006700
71. Nair JR, Carlson LM, Koorella C, Rozanski CH, Byrne GE, Bergsagel PL, et al. CD28 expressed on malignant plasma cells induces a prosurvival and immunosuppressive microenvironment. J Immunol (2011) 187:1243–53. doi:10.4049/jimmunol.1100016
72. Mateo G, Montalbán MA, Vidriales MB, Lahuerta JJ, Mateos MV, Gutiérrez N, et al. Prognostic value of immunophenotyping in multiple myeloma: a study by the PETHEMA/GEM cooperative study groups on patients uniformly treated with high-dose therapy. J Clin Oncol (2008) 26:2737–44. doi:10.1200/JCO.2007.15.4120
73. Van Valckenborgh E, Schouppe E, Movahedi K, De Bruyne E, Menu E, De Baetselier P, et al. Multiple myeloma induces the immunosuppressive capacity of distinct myeloid-derived suppressor cell subpopulations in the bone marrow. Leukemia (2012) 26:2424–8. doi:10.1038/leu.2012.113
74. Zhuang J, Zhang J, Lwin ST, Edwards JR, Edwards CM, Mundy GR, et al. Osteoclasts in multiple myeloma are derived from Gr-1+CD11b+myeloid-derived suppressor cells. PLoS One (2012) 7:e48871. doi:10.1371/journal.pone.0048871
75. Hoechst B, Voigtlaender T, Ormandy L, Gamrekelashvili J, Zhao F, Wedemeyer H, et al. Myeloid derived suppressor cells inhibit natural killer cells in patients with hepatocellular carcinoma via the NKp30 receptor. Hepatology (2009) 50:799–807. doi:10.1002/hep.23054
76. Li H, Han Y, Guo Q, Zhang M, Cao X. Cancer-expanded myeloid-derived suppressor cells induce anergy of NK cells through membrane-bound TGF-beta 1. J Immunol (2009) 182:240–9. doi:10.4049/jimmunol.182.1.240
77. Sarhan D, Cichocki F, Zhang B, Yingst A, Spellman SR, Cooley S, et al. Adaptive NK cells with low TIGIT expression are inherently resistant to myeloid-derived suppressor cells. Cancer Res (2016) 76:5696–706. doi:10.1158/0008-5472.CAN-16-0839
78. Hu J, Handisides DR, Van Valckenborgh E, De Raeve H, Menu E, Vande Broek I, et al. Targeting the multiple myeloma hypoxic niche with TH-302, a hypoxia-activated prodrug. Blood (2010) 116(9):1524–7. doi:10.1182/blood-2010-02-269126
79. Martin SK, Diamond P, Gronthos S, Peet DJ, Zannettino AC. The emerging role of hypoxia, HIF-1 and HIF-2 in multiple myeloma. Leukemia (2011) 25(10):1533–42. doi:10.1038/leu.2011.122
80. Sarkar S, Germeraad WT, Rouschop KM, Steeghs EM, van Gelder M, Bos GM, et al. Hypoxia induced impairment of NK cell cytotoxicity against multiple myeloma can be overcome by IL-2 activation of the NK cells. PLoS One (2013) 8:e64835. doi:10.1371/journal.pone.0064835
81. Pende D, Rivera P, Marcenaro S, Chang CC, Biassoni R, Conte R, et al. Major histocompatibility complex class I-related chain A and UL16-binding protein expression on tumor cell lines of different histotypes: analysis of tumor susceptibility to NKG2D-dependent natural killer cell cytotoxicity. Cancer Res (2002) 62:6178–86.
82. Jinushi M, Takehara T, Tatsumi T, Hiramatsu N, Sakamori R, Yamaguchi S, et al. Impairment of natural killer cell and dendritic cell functions by the soluble form of MHC class I-related chain A in advanced human hepatocellular carcinomas. J Hepatol (2005) 43:1013–20. doi:10.1016/j.jhep.2005.05.026
83. Boissel N, Rea D, Tieng V, Dulphy N, Brun M, Cayuela JM, et al. BCR/ABL oncogene directly controls MHC class I chain-related molecule A expression in chronic myelogenous leukemia. J Immunol (2006) 176:5108–16. doi:10.4049/jimmunol.176.8.5108
84. Jinushi M, Vanneman M, Munshi NC, Tai YT, Prabhala RH, Ritz J, et al. MHC class I chain-related protein A antibodies and shedding are associated with the progression of multiple myeloma. Proc Natl Acad Sci U S A (2008) 105:1285–90. doi:10.1073/pnas.0711293105
85. Kohga K, Takehara T, Tatsumi T, Ohkawa K, Miyagi T, Hiramatsu N, et al. Serum levels of soluble major histocompatibility complex (MHC) class I-related chain A in patients with chronic liver diseases and changes during transcatheter arterial embolization for hepatocellular carcinoma. Cancer Sci (2008) 99:1643–9. doi:10.1111/j.1349-7006.2008.00859.x
86. Groh V, Wu J, Yee C, Spies T. Tumour-derived soluble MIC ligands impair expression of NKG2D and T-cell activation. Nature (2002) 419:734–8. doi:10.1038/nature01112
87. Doubrovina ES, Doubrovin MM, Vider E, Sisson RB, O’Reilly RJ, Dupont B, et al. Evasion from NK cell immunity by MHC class I chain-related molecules expressing colon adenocarcinoma. J Immunol (2003) 171:6891–9. doi:10.4049/jimmunol.171.12.6891
88. Wu JD, Higgins LM, Steinle A, Cosman D, Haugk K, Plymate SR. Prevalent expression of the immunostimulatory MHC class I chain-related molecule is counteracted by shedding in prostate cancer. J Clin Invest (2004) 114:560–8. doi:10.1172/JCI200422206
89. Xiao G, Wang X, Sheng J, Lu S, Yu X, Wu JD. Soluble NKG2D ligand promotes MDSC expansion and skews macrophage to the alternatively activated phenotype. J Hematol Oncol (2015) 8:13. doi:10.1186/s13045-015-0110-z
90. Paschen A, Sucker A, Hill B, Moll I, Zapatka M, Nguyen XD, et al. Differential clinical significance of individual NKG2D ligands in melanoma: soluble ULBP2 as an indicator of poor prognosis superior to S100B. Clin Cancer Res (2009) 15:5208–15. doi:10.1158/1078-0432.CCR-09-0886
91. Kumar V, Yi Lo PH, Sawai H, Kato N, Takahashi A, Deng Z, et al. Soluble MICA and a MICA variation as possible prognostic biomarkers for HBV-induced hepatocellular carcinoma. PLoS One (2012) 7:e44743. doi:10.1371/journal.pone.0044743
92. Liu G, Lu S, Wang X, Page ST, Higano CS, Plymate SR, et al. Perturbation of NK cell peripheral homeostasis accelerates prostate carcinoma metastasis. J Clin Invest (2013) 123:4410–22. doi:10.1172/JCI69369
93. Zingoni A, Cecere F, Vulpis E, Fionda C, Molfetta R, Soriani A, et al. Genotoxic stress induces senescence-associated ADAM10-dependent release of NKG2D MIC ligands in multiple myeloma cells. J Immunol (2015) 195:736–48. doi:10.4049/jimmunol.1402643
94. Reiners KS, Topolar D, Henke A, Simhadri VR, Kessler J, Sauer M, et al. Soluble ligands for NK cell receptors promote evasion of chronic lymphocytic leukemia cells from NK cell anti-tumor activity. Blood (2013) 121:3658–65. doi:10.1182/blood-2013-01-476606
95. Friese MA, Wischhusen J, Wick W, Weiler M, Eisele G, Steinle A, et al. RNA interference targeting transforming growth factor-beta enhances NKG2D-mediated antiglioma immune response, inhibits glioma cell migration and invasiveness, and abrogates tumorigenicity in vivo. Cancer Res (2004) 64:7596–603. doi:10.1158/0008-5472.CAN-04-1627
96. Eisele G, Wischhusen J, Mittelbronn M, Meyermann R, Waldhauer I, Steinle A, et al. TGF-beta and metalloproteinases differentially suppress NKG2D ligand surface expression on malignant glioma cells. Brain (2006) 129:2416–25. doi:10.1093/brain/awl205
97. Kamimura H, Yamagiwa S, Tsuchiya A, Takamura M, Matsuda Y, Ohkoshi S, et al. Reduced NKG2D ligand expression in hepatocellular carcinoma correlates with early recurrence. J Hepatol (2012) 56:381–8. doi:10.1016/j.jhep.2011.06.017
98. Soriani A, Borrelli C, Ricci B, Molfetta R, Zingoni A, Fionda C, et al. p38 MAPK differentially controls NK activating ligands at transcriptional and post-transcriptional level on multiple myeloma cells. Oncoimmunology (2017) 6:e1264564. doi:10.1080/2162402X.2016.1264564
99. Soriani A, Zingoni A, Cerboni C, Iannitto ML, Ricciardi MR, Di Gialleonardo V, et al. ATM-ATR-dependent up-regulation of DNAM-1 and NKG2D ligands on multiple myeloma cells by therapeutic agents results in enhanced NK-cell susceptibility and is associated with a senescent phenotype. Blood (2009) 113:3503–11. doi:10.1182/blood-2008-08-173914
100. Soriani A, Iannitto ML, Ricci B, Fionda C, Malgarini G, Morrone S, et al. Reactive oxygen species- and DNA damage response-dependent NK cell activating ligand upregulation occurs at transcriptional levels and requires the transcriptional factor E2F1. J Immunol (2014) 193:950–60. doi:10.4049/jimmunol.1400271
101. Fionda C, Soriani A, Malgarini G, Iannitto ML, Santoni A, Cippitelli M. Heat shock protein-90 inhibitors increase MHC class I-related chain A and B ligand expression on multiple myeloma cells and their ability to trigger NK cell degranulation. J Immunol (2009) 183:4385–94. doi:10.4049/jimmunol.0901797
102. Abruzzese MP, Bilotta MT, Fionda C, Zingoni A, Soriani A, Vulpis E, et al. Inhibition of bromodomain and extra-terminal (BET) proteins increases NKG2D ligand MICA expression and sensitivity to NK cell-mediated cytotoxicity in multiple myeloma cells: role of cMYC-IRF4-miR-125b interplay. J Hematol Oncol (2016) 9:134. doi:10.1186/s13045-016-0362-2
103. Fionda C, Abruzzese MP, Zingoni A, Cecere F, Vulpis E, Peruzzi G, et al. The IMiDs targets IKZF-1/3 and IRF4 as novel negative regulators of NK cell-activating ligands expression in multiple myeloma. Oncotarget (2015) 6:23609–30. doi:10.18632/oncotarget.4603
104. Fiegler N, Textor S, Arnold A, Rölle A, Oehme I, Breuhahn K, et al. Downregulation of the activating NKp30 ligand B7-H6 by HDAC inhibitors impairs tumor cell recognition by NK cells. Blood (2013) 122:684–93. doi:10.1182/blood-2013-02-482513
105. Carbone E, Neri P, Mesuraca M, Fulciniti MT, Otsuki T, Pende D, et al. HLA class I, NKG2D, and natural cytotoxicity receptors regulate multiple myeloma cell recognition by natural killer cells. Blood (2005) 105:251–8. doi:10.1182/blood-2004-04-1422
106. Bossard C, Bezieau S, Matysiak-Budnik T, Volteau C, Laboisse CL, Jotereau F, et al. HLA-E/beta2 microglobulin overexpression in colorectal cancer is associated with recruitment of inhibitory immune cells and tumor progression. Int J Cancer (2012) 131:855–63. doi:10.1002/ijc.26453
107. Sarkar S, van Gelder M, Noort W, Xu Y, Rouschop KM, Groen R, et al. Optimal selection of natural killer cells to kill myeloma: the role of HLA-E and NKG2A. Cancer Immunol Immunother (2015) 64:951–63. doi:10.1007/s00262-015-1694-4
108. Frey AB, Monu N. Effector-phase tolerance: another mechanism of how cancer escapes antitumor immune response. J Leukoc Biol (2006) 79:652–62. doi:10.1189/jlb.1105628
109. Tonks A, Hills R, White P, Rosie B, Mills KI, Burnett AK, et al. CD200 as a prognostic factor in acute myeloid leukaemia. Leukemia (2007) 21:566–8. doi:10.1038/sj.leu.2404559
110. Coles SJ, Wang EC, Man S, Hills RK, Burnett AK, Tonks A, et al. CD200 expression suppresses natural killer cell function and directly inhibits patient anti-tumor response in acute myeloid leukemia. Leukemia (2011) 25:792–9. doi:10.1038/leu.2011.1
111. Kretz-Rommel A, Qin F, Dakappagari N, Ravey EP, McWhirter J, Oltean D, et al. CD200 expression on tumor cells suppresses antitumor immunity: new approaches to cancer immunotherapy. J Immunol (2007) 178:5595–605. doi:10.4049/jimmunol.178.9.5595
112. Moreaux J, Hose D, Reme T, Jourdan E, Hundemer M, Legouffe E, et al. CD200 is a new prognostic factor in multiple myeloma. Blood (2006) 108:4194–7. doi:10.1182/blood-2006-06-029355
113. Osterborg A, Nilsson B, Björkholm M, Holm G, Mellstedt H. Natural killer cell activity in monoclonal gammopathies: relation to disease activity. Eur J Haematol (1990) 45:153–7. doi:10.1111/j.1600-0609.1990.tb00443.x
114. Famularo G, D’Ambrosio A, Quintieri F, Di Giovanni S, Parzanese I, Pizzuto F, et al. Natural killer cell frequency and function in patients with monoclonal gammopathies. J Clin Lab Immunol (1992) 37:99–109.
115. García-Sanz R, González M, Orfão A, Moro MJ, Hernández JM, Borrego D, et al. Analysis of natural killer-associated antigens in peripheral blood and bone marrow of multiple myeloma patients and prognostic implications. Br J Haematol (1996) 93:81–8. doi:10.1046/j.1365-2141.1996.4651006.x
116. Omedé P, Boccadoro M, Gallone G, Frieri R, Battaglio S, Redoglia V, et al. Multiple myeloma: increased circulating lymphocytes carrying plasma cell-associated antigens as an indicator of poor survival. Blood (1990) 76:1375–9.
117. King MA, Radicchi-Mastroianni MA. Natural killer cells and CD56+ T cells in the blood of multiple myeloma patients: analysis by 4-colour flow cytometry. Cytometry (1996) 26:121–4. doi:10.1002/(SICI)1097-0320(19960615)26:2<121::AID-CYTO4>3.0.CO;2-J
118. Paiva B, Mateos MV, Sanchez-Abarca LI, Puig N, Vidriales MB, López-Corral L, et al. Immune status of high-risk smoldering multiple myeloma patients and its therapeutic modulation under LenDex: a longitudinal analysis. Blood (2016) 127:1151–62. doi:10.1182/blood-2015-10-662320
119. Gonzalez M, San Miguel JF, Gascon A, Moro MJ, Hernandez JM, Ortega F, et al. Increased expression of natural-killer-associated and activation antigens in multiple myeloma. Am J Hematol (1992) 39:84–9. doi:10.1002/ajh.2830390203
120. Van den Hove LE, Meeus P, Derom A, Demuynck H, Verhoef GE, Vandenberghe P, et al. Lymphocyte profiles in multiple myeloma and monoclonal gammopathy of undetermined significance: flow-cytometric characterization and analysis in a two-dimensional correlation biplot. Ann Hematol (1998) 76:249–56. doi:10.1007/s002770050397
121. Nielsen H, Nielsen HJ, Tvede N, Klarlund K, Mansa B, Moesgaard F, et al. Immune dysfunction in multiple myeloma. Reduced natural killer cell activity and increased levels of soluble interleukin-2 receptors. APMIS (1991) 99:340–6. doi:10.1111/j.1699-0463.1991.tb05159.x
122. Fauriat C, Mallet F, Olive D, Costello RT. Impaired activating receptor expression pattern in natural killer cells from patients with multiple myeloma. Leukemia (2006) 20:732–3. doi:10.1038/sj.leu.2404096
123. Costello RT, Boehrer A, Sanchez C, Mercier D, Baier C, Le Treut T, et al. Differential expression of natural killer cell activating receptors in blood versus bone marrow in patients with monoclonal gammopathy. Immunology (2013) 139:338–41. doi:10.1111/imm.12082
124. Beldi-Ferchiou A, Lambert M, Dogniaux S, Vély F, Vivier E, Olive D, et al. PD-1 mediates functional exhaustion of activated NK cells in patients with Kaposi sarcoma. Oncotarget (2016) 7:72961–77. doi:10.18632/oncotarget.12150
125. Benson DM Jr, Bakan CE, Mishra A, Hofmeister CC, Efebera Y, Becknell B, et al. The PD-1/PD-L1 axis modulates the natural killer cell versus multiple myeloma effect: a therapeutic target for CT-011, a novel monoclonal anti-PD-1 antibody. Blood (2010) 116(13):2286–94. doi:10.1182/blood-2010-02-271874
126. Gorgun G, Samur MK, Cowens KB, Paula S, Bianchi G, Anderson JE, et al. Lenalidomide enhances immune checkpoint blockade-induced immune response in multiple myeloma. Clin Cancer Res (2015) 21:4607–18. doi:10.1158/1078-0432.CCR-15-0200
127. Ponzetta A, Benigni G, Antonangeli F, Sciumè G, Sanseviero E, Zingoni A, et al. Multiple myeloma impairs bone marrow localization of effector natural killer cells by altering the chemokine microenvironment. Cancer Res (2015) 75:4766–77. doi:10.1158/0008-5472.CAN-15-1320
128. El-Sherbiny YM, Meade JL, Holmes TD, McGonagle D, Mackie SL, Morgan AW, et al. The requirement for DNAM-1, NKG2D, and NKp46 in the natural killer cell-mediated killing of myeloma cells. Cancer Res (2007) 67:8444–9. doi:10.1158/0008-5472.CAN-06-4230
129. Guillerey C, Ferrari de Andrade L, Vuckovic S, Miles K, Ngiow SF, Yong MC, et al. Immunosurveillance and therapy of multiple myeloma are CD226 dependent. J Clin Invest (2015) 125:2077–89. doi:10.1172/JCI77181
130. Jurisic V, Srdic T, Konjevic G, Markovic O, Colovic M. Clinical stage-depending decrease of NK cell activity in multiple myeloma patients. Med Oncol (2007) 24:312–7. doi:10.1007/s12032-007-0007-y
131. Paiva B, Azpilikueta A, Puig N, Ocio EM, Sharma R, Oyajobi BO, et al. PD-L1/PD-1 presence in the tumor microenvironment and activity of PD-1 blockade in multiple myeloma. Leukemia (2015) 29(10):2110–3. doi:10.1038/leu.2015.79
132. San Miguel J, Mateos M-V, Shah JJ, Ocio EM, Rodriguez-Otero P, Reece D, et al. Pembrolizumab in combination with lenalidomide and low-dose dexamethasone for relapsed/refractory multiple myeloma (RRMM): keynote-023 [abstract 505]. Presented at the 57th ASH Annual Meeting and Exposition. (Vol. 73), Dec 5–8; Orlando, FL (2015).
133. Suen H, Brown R, Yang S, Ho PJ, Gibson J, Joshua D. The failure of immune checkpoint blockade in multiple myeloma with PD-1 inhibitors in a phase 1 study. Leukemia (2015) 29:1621–2. doi:10.1038/leu.2015.104
134. Lesokhin AM, Ansell SM, Armand P, Scott EC, Halwani A, Gutierrez M, et al. Preliminary results of a phase I study of nivolumab (BMS-936558) in patients with relapsed or refractory lymphoid malignancies [abstract 291]. Blood (2014) 124(Suppl).
135. Pesce S, Greppi M, Tabellini G, Rampinelli F, Parolini S, Olive D, et al. Identification of a subset of human natural killer cells expressing high levels of programmed death 1: a phenotypic and functional characterization. J Allergy Clin Immunol (2017) 139(1):335–46. doi:10.1016/j.jaci.2016.04.025
136. Frohn C, Hoppner M, Schlenke P, Kirchner H, Koritke P, Luhm J. Anti-myeloma activity of natural killer lymphocytes. Br J Haematol (2002) 119:660–4. doi:10.1046/j.1365-2141.2002.03879.x
137. Kroger N, Shaw B, Iacobelli S, Zabelina T, Peggs K, Shimoni A, et al. Comparison between antithymocyte globulin and alemtuzumab and the possible impact of KIR-ligand mismatch after dose-reduced conditioning and unrelated stem cell transplantation in patients with multiple myeloma. Br J Haematol (2005) 129:631–43. doi:10.1111/j.1365-2141.2005.05513.x
138. Romagné F, Andre P, Spee P, Zahn S, Anfossi N, Gauthier L, et al. Preclinical characterization of 1-7F9, a novel human anti-KIR receptor therapeutic antibody that augments natural killer-mediated killing of tumor cells. Blood (2009) 114:2667–77. doi:10.1182/blood-2009-02-206532
139. Benson DM Jr, Bakan CE, Zhang S, Collins SM, Liang J, Srivastava S, et al. IPH2101, a novel anti-inhibitory KIR antibody, and lenalidomide combine to enhance the natural killer cell versus multiple myeloma effect. Blood (2011) 118:6387–91. doi:10.1182/blood-2011-06-360255
140. Benson DM Jr, Hofmeister CC, Padmanabhan S, Suvannasankha A, Jagannath S, Abonour R, et al. A phase 1 trial of the anti-KIR antibody IPH2101 in patients with relapsed/refractory multiple myeloma. Blood (2012) 120:4324–33. doi:10.1182/blood-2012-06-438028
141. Benson DM Jr, Cohen AD, Jagannath S, Munshi NC, Spitzer G, Hofmeister CC, et al. A phase I trial of the anti-KIR antibody IPH2101 and lenalidomide in patients with relapsed/refractory multiple myeloma. Clin Cancer Res (2015) 21(18):4055–61. doi:10.1158/1078-0432.CCR-15-0304
142. Korde N, Carlsten M, Lee MJ, Minter A, Tan E, Kwok M, et al. A phase II trial of pan-KIR2D blockade with IPH2101 in smoldering multiple myeloma. Haematologica (2014) 99:e81–3. doi:10.3324/haematol.2013.103085
143. Carlsten M, Korde N, Kotecha R, Reger R, Bor S, Kazandjian D, et al. Checkpoint inhibition of KIR2D with the monoclonal antibody IPH2101 induces contraction and hyporesponsiveness of NK cells in patients with myeloma. Clin Cancer Res (2016) 22:5211–22. doi:10.1158/1078-0432.CCR-16-1108
144. Attal M, Lauwers-Cances V, Marit G, Caillot D, Moreau P, Facon T, et al. Lenalidomide maintenance after stem-cell transplantation for multiple myeloma. N Engl J Med (2012) 366(19):1782–91. doi:10.1056/NEJMoa1114138
145. McCarthy PL, Owzar K, Hofmeister CC, Hurd DD, Hassoun H, Richardson PG, et al. Lenalidomide after stem cell transplantation for multiple myeloma. N Engl J Med (2012) 366(19):1770–81. doi:10.1056/NEJMoa1114083
146. Palumbo A, Cavallo F, Gay F, Di Raimondo F, Ben Yehuda D, Petrucci MT, et al. Autologous transplantation and maintenance therapy in multiple myeloma. N Engl J Med (2014) 371(10):895–905. doi:10.1056/NEJMoa1402888
147. Jackson GH, Davies FE, Pawlyn C, Cairns DA, Striha A, Collett C, et al. Lenalidomide is a highly effective maintenance therapy inmyeloma patients of all ages; results of the phase III myeloma XI study [abstract no. 1143]. Blood (2016) 128(22).
148. Musto P, Anderson KC, Attal M, Richardson PG, Badros A, Hou J, et al. International Myeloma Working Group. Second primary malignancies in multiple myeloma: an overview and IMWG consensus. Ann Oncol (2017) 28:228–45. doi:10.1093/annonc/mdx160
149. Hayashi T, Hideshima T, Akiyama M, Podar K, Yasui H, Raje N, et al. Molecular mechanisms whereby immunomodulatory drugs activate natural killer cells: clinical application. Br J Haematol (2005) 128:192–203. doi:10.1111/j.1365-2141.2004.05286.x
150. Malavasi F, Funaro A, Roggero S, Horenstein A, Calosso L, Mehta K. Human CD38: a glycoprotein in search of a function. Immunol Today (1994) 15:95–7. doi:10.1016/0167-5699(94)90148-1
151. Lin P, Owens R, Tricot G, Wilson CS. Flow cytometric immunophenotypic analysis of 306 cases of multiple myeloma. Am J Clin Pathol (2004) 121:482–8. doi:10.1309/74R4TB90BUWH27JX
152. Deaglio S, Mehta K, Malavasi F. Human CD38: a (r)evolutionary story of enzymes and receptors. Leuk Res (2001) 25:1–12. doi:10.1016/S0145-2126(00)00093-X
153. Nijhof IS, Groen RW, Lokhorst HM, van Kessel B, Bloem AC, van Velzen J, et al. Upregulation of CD38 expression on multiple myeloma cells by all-trans retinoic acid improves the efficacy of daratumumab. Leukemia (2015) 29:2039–49. doi:10.1038/leu.2015.123
154. Overdijk MB, Verploegen S, Bögels M, van Egmond M, van Bueren JJL, Mutis T, et al. Antibody-mediated phagocytosis contributes to the anti-tumor activity of the therapeutic antibody daratumumab in lymphoma and multiple myeloma. MAbs (2015) 7:311–20. doi:10.1080/19420862.2015.1007813
155. Jansen JH, Boross P, Overdijk MB, van Bueren JJ, Parren PW, Leusen JH. Daratumumab, a human CD38 antibody induces apoptosis of myeloma tumor cells via Fc receptor-mediated crosslinking [abstract]. Blood (2012) 120:2974.
156. Palumbo A, Chanan-Khan A, Weisel K, Nooka AK, Masszi T, Beksac M, et al. Daratumumab, bortezomib, and dexamethasone for multiple myeloma. N Engl J Med (2016) 375:754–66. doi:10.1056/NEJMoa1606038
157. Nijhof IS, Lammerts van Bueren JJ, van Kessel B, Andre P, Morel Y, Lokhorst HM, et al. Daratumumab-mediated lysis of primary multiple myeloma cells is enhanced in combination with the human anti-KIR antibody IPH2102 and lenalidomide. Haematologica (2015) 100(2):263–8. doi:10.3324/haematol.2014.117531
158. Cherkasova E, Espinoza L, Kotecha R, Reger RN, Berg M, Aue G, et al. Treatment of ex vivo expanded NK cells with daratumumab F(ab’)2 fragments protects adoptively transferred NK cells from daratumumab-mediated killing and augments daratumumab-induced antibody dependent cellular toxicity (ADCC) of myeloma [abstract]. Blood (2015) 126:4244.
159. Hsi ED, Steinle R, Balasa B, Szmania S, Draksharapu A, Shum BP, et al. CS1, a potential new therapeutic antibody target for the treatment of multiple myeloma. Clin Cancer Res (2008) 14:2775–84. doi:10.1158/1078-0432.CCR-07-4246
160. Tai YT, Dillon M, Song W, Leiba M, Li XF, Burger P, et al. Anti-CS1 humanized monoclonal antibody HuLuc63 inhibits myeloma cell adhesion and induces antibody-dependent cellular cytotoxicity in the bone marrow milieu. Blood (2008) 112:1329–37. doi:10.1182/blood-2007-08-107292
161. Collins SM, Bakan CE, Swartzel GD, Hofmeister CC, Efebera YA, Kwon H, et al. Elotuzumab directly enhances NK cell cytotoxicity against myeloma via CS1 ligation: evidence for augmented NK cell function complementing ADCC. Cancer Immunol Immunother (2013) 62(12):1841–9. doi:10.1007/s00262-013-1493-8
162. van Rhee F, Szmania SM, Dillon M, van Abbema AM, Li X, Stone MK, et al. Combinatorial efficacy of anti-CS1 monoclonal antibody elotuzumab (HuLuc63) and bortezomib against multiple myeloma. Mol Cancer Ther (2009) 8:2616–24. doi:10.1158/1535-7163.MCT-09-0483
163. Magen H, Muchtar E. Elotuzumab: the first approved monoclonal antibody for multiple myeloma treatment. Ther Adv Hematol (2016) 7(4):187–95. doi:10.1177/2040620716652862
164. Guo H, Cruz-Munoz M-E, Wu N, Robbins M, Veillette A. Immune cell inhibition by SLAMF7 is mediated by a mechanism requiring src kinases, CD45, and SHIP-1 that is defective in multiple myeloma cells. Mol Cell Biol (2015) 35:41–51. doi:10.1128/MCB.01107-14
165. Pérez-Quintero LA, Roncagalli R, Guo H, Latour S, Davidson D, Veillette A. EAT-2, a SAP-like adaptor, controls NK cell activation through phospholipase Cγ, Ca++, and Erk, leading to granule polarization. J Exp Med (2014) 211:727–42. doi:10.1084/jem.20132038
166. Balasa B, Yun R, Belmar NA, Fox M, Chao DT, Robbins MD, et al. Elotuzumab enhances natural killer cell activation and myeloma cell killing through interleukin-2 and TNF-α pathways. Cancer Immunol Immunother (2015) 64(1):61–73. doi:10.1007/s00262-014-1610-3
167. Lonial S, Vij R, Harousseau JL, Facon T, Moreau P, Mazumder A, et al. Elotuzumab in combination with lenalidomide and low-dose dexamethasone in relapsed or refractory multiple myeloma. J Clin Oncol (2012) 30:1953–9. doi:10.1200/JCO.2011.37.2649
168. Richardson PG, Jagannath S, Moreau P, Jakubowiak A, Raab MS, Facon T, et al. Final results for the 1703 phase 1b/2 study of elotuzumab in combination with lenalidomide and dexamethasone in patients with relapsed/refractory multiple myeloma [abstract]. Blood (2014) 124:302.
169. Lonial S, Dimopoulos M, Palumbo A, White D, Grosicki S, Spicka I, et al. Elotuzumab therapy for relapsed or refractory multiple myeloma. ELOQUENT-2 investigators. N Engl J Med (2015) 373(7):621–31. doi:10.1056/NEJMoa1505654
170. Jakubowiak A, Offidani M, Pégourie B, De La Rubia J, Garderet L, Laribi K, et al. Randomized phase 2 study: elotuzumab plus bortezomib/dexamethasone vs bortezomib/dexamethasone for relapsed/refractory MM. Blood (2016) 127:2833–40. doi:10.1182/blood-2016-01-694604
171. Munn DH, Shafizadeh E, Attwood JT, Bondarev I, Pashine A, Mellor AL. Inhibition of T cell proliferation by macrophage tryptophan catabolism. J Exp Med (1999) 189:1363–72. doi:10.1084/jem.189.9.1363
172. Uyttenhove C, Pilotte L, Theate I, Stroobant V, Colau D, Parmentier N, et al. Evidence for a tumoral immune resistance mechanism based on tryptophan degradation by indoleamine 2,3-dioxygenase. Nat Med (2003) 9:1269–74. doi:10.1038/nm934
173. Fallarino F, Grohmann U, Vacca C, Orabona C, Spreca A, Fioretti MC, et al. T cell apoptosis by kynurenines. Adv Exp Med Biol (2003) 527:183–90. doi:10.1007/978-1-4615-0135-0_21
174. Hayashi T, Mo JH, Gong X, Rossetto C, Jang A, Beck L, et al. 3-Hydroxyanthranilic acid inhibits PDK1 activation and suppresses experimental asthma by inducing T cell apoptosis. Proc Natl Acad Sci U S A (2007) 104:18619–24. doi:10.1073/pnas.0709261104
175. Frumento G, Rotondo R, Tonetti M, Damonte G, Benatti U, Ferrara GB. Tryptophan-derived catabolites are responsible for inhibition of T and natural killer cell proliferation induced by indoleamine 2,3-dioxygenase. J Exp Med (2002) 196:459–68. doi:10.1084/jem.20020121
176. Sato N, Saga Y, Mizukami H, Wang D, Takahashi S, Nonaka H, et al. Downregulation of indoleamine-2,3-dioxygenase in cervical cancer cells suppresses tumor growth by promoting natural killer cell accumulation. Oncol Rep (2012) 28:1574–8. doi:10.3892/or.2012.1984
177. Molano A, Illarionov PA, Besra GS, Putterman C, Porcelli SA. Modulation of invariant natural killer T cell cytokine responses by indoleamine 2,3-dioxygenase. Immunol Lett (2008) 117:81–90. doi:10.1016/j.imlet.2007.12.013
178. Balachandran VP, Cavnar MJ, Zeng S, Bamboat ZM, Ocuin LM, Obaid H, et al. Imatinib potentiates antitumor T cell responses in gastrointestinal stromal tumor through the inhibition of IDO. Nat Med (2011) 17:1094–100. doi:10.1038/nm.2438
179. Nguyen NT, Kimura A, Nakahama T, Chinen I, Masuda K, Nohara K, et al. Aryl hydrocarbon receptor negatively regulates dendritic cell immunogenicity via a kynurenine dependent mechanism. Proc Natl Acad Sci U S A (2010) 107:19961–6. doi:10.1073/pnas.1014465107
180. Park MJ, Park KS, Park HS, Cho ML, Hwang SY, Min SY, et al. A distinct tolerogenic subset of splenic IDO(C)CD11b(C) dendritic cells from orally tolerized mice is responsible for induction of systemic immune tolerance and suppression of collagen-induced arthritis. Cell Immunol (2012) 278:45–54. doi:10.1016/j.cellimm.2012.06.009
181. Orabona C, Puccetti P, Vacca C, Bicciato S, Luchini A, Fallarino F, et al. Toward the identification of a tolerogenic signature in IDO-competent dendritic cells. Blood (2006) 107:2846–54. doi:10.1182/blood-2005-10-4077
182. Bonanno G, Mariotti A, Procoli A, Folgiero V, Natale D, De Rosa L, et al. Indoleamine 2,3-dioxygenase 1 (IDO1) activity correlates with immune system abnormalities in multiple myeloma. J Transl Med (2012) 10:247. doi:10.1186/1479-5876-10-247
183. Ault KA, Antin JH, Ginsburg D, Orkin SH, Rappeport JM, Keohan ML, et al. Phenotype of recovering lymphoid cell populations after marrow transplantation. J Exp Med (1985) 161(6):1483–502. doi:10.1084/jem.161.6.1483
184. Velardi A, Terenzi A, Cucciaioni S, Millo R, Grossi CE, Grignani F, et al. Imbalances within the peripheral blood T-helper (CD4+) and T-suppressor (CD8+) cell populations in the reconstitution phase after human bone marrow transplantation. Blood (1988) 71(5):1196–200.
185. Uhrberg M, Valiante NM, Shum BP, Shilling HG, Lienert-Weidenbach K, Corliss B, et al. Human diversity in killer cell inhibitory receptor genes. Immunity (1997) 7(6):753–63. doi:10.1016/S1074-7613(00)80394-5
186. Wende H, Colonna M, Ziegler A, Volz A. Organization of the leukocyte receptor cluster (LRC) on human chromosome 19q13.4. Mamm Genome (1999) 10(2):154–60. doi:10.1007/s003359900961
187. Shilling HG, Guethlein LA, Cheng NW, Gardiner CM, Rodriguez R, Tyan D, et al. Allelic polymorphism synergizes with variable gene content to individualize human KIR genotype. J Immunol (2002) 168(5):2307–15. doi:10.4049/jimmunol.168.5.2307
188. Shilling HG, Young N, Guethlein LA, Cheng NW, Gardiner CM, Tyan D, et al. Genetic control of human NK cell repertoire. J Immunol (2002) 169(1):239–47. doi:10.4049/jimmunol.169.1.239
189. Shilling HG, McQueen KL, Cheng NW, Shizuru JA, Negrin RS, Parham P. Reconstitution of NK cell receptor repertoire following HLA-matched hematopoietic cell transplantation. Blood (2003) 101(9):3730–40. doi:10.1182/blood-2002-08-2568
190. Bachar-Lustig E, Rachamim N, Li HW, Lan F, Reisner Y. Megadose of T cell-depleted bone marrow overcomes MHC barriers in sublethally irradiated mice. Nat Med (1995) 1(12):1268–73. doi:10.1038/nm1295-1268
191. Aversa F, Tabilio A, Velardi A, Cunningham I, Terenzi A, Falzetti F, et al. Treatment of high-risk acute leukemia with T-cell-depleted stem cells from related donors with one fully mismatched HLA haplotype. N Engl J Med (1998) 339(17):1186–93. doi:10.1056/NEJM199810223391702
192. Ruggeri L, Capanni M, Casucci M, Volpi I, Tosti A, Perruccio K, et al. Role of natural killer cell alloreactivity in HLA-mismatched hematopoietic stem cell transplantation. Blood (1999) 94(1):333–9.
193. Farag SS, Fehniger TA, Ruggeri L, Velardi A, Caligiuri MA. Natural killer cell receptors: new biology and insights into the graft-versus-leukemia effect. Blood (2002) 100(6):1935–47. doi:10.1182/blood-2002-02-0350
194. Velardi A, Ruggeri L, Alessandro M, Moretta L. NK cells: a lesson from mismatched hematopoietic transplantation. Trends Immunol (2002) 23(9):438–44. doi:10.1016/S1471-4906(02)02284-6
195. Ruggeri L, Capanni M, Urbani E, Perruccio K, Shlomchik WD, Tosti A, et al. Effectiveness of donor natural killer cell alloreactivity in mismatched hematopoietic transplants. Science (2002) 295(5562):2097–100. doi:10.1126/science.1068440
196. Parham P, McQueen KL. Alloreactive killer cells: hindrance and help for haematopoietic transplants. Nat Rev Immunol (2003) 3(2):108–22. doi:10.1038/nri999
197. Aversa F, Terenzi A, Tabilio A, Falzetti F, Carotti A, Ballanti S, et al. Full haplotype-mismatched hematopoietic stem-cell transplantation: a phase II study in patients with acute leukemia at high risk of relapse. J Clin Oncol (2005) 23(15):3447–54. doi:10.1200/JCO.2005.09.117
198. Ruggeri L, Mancusi A, Capanni M, Urbani E, Carotti A, Aloisi T, et al. Donor natural killer cell allorecognition of missing self in haploidentical hematopoietic transplantation for acute myeloid leukemia: challenging its predictive value. Blood (2007) 110(1):433–40. doi:10.1182/blood-2006-07-038687
199. Mancusi A, Ruggeri L, Urbani E, Pierini A, Massei MS, Carotti A, et al. Haploidentical hematopoietic transplantation from KIR ligand-mismatched donors with activating KIRs reduces nonrelapse mortality. Blood (2015) 125(20):3173–82. doi:10.1182/blood-2014-09-599993
200. Leung W, Iyengar R, Turner V, Lang P, Bader P, Conn P, et al. Determinants of antileukemia effects of allogeneic NK cells. J Immunol (2004) 172(1):644–50. doi:10.4049/jimmunol.172.1.644
201. Pende D, Marcenaro S, Falco M, Martini S, Bernardo ME, Montagna D, et al. Anti-leukemia activity of alloreactive NK cells in KIR ligand-mismatched haploidentical HSCT for pediatric patients: evaluation of the functional role of activating KIR and redefinition of inhibitory KIR specificity. Blood (2009) 113(13):3119–29. doi:10.1182/blood-2008-06-164103
202. Miller JS, Soignier Y, Panoskaltsis-Mortari A, McNearney SA, Yun GH, Fautsch SK, et al. Successful adoptive transfer and in vivo expansion of human haploidentical NK cells in patients with cancer. Blood (2005) 105(8):3051–7. doi:10.1182/blood-2004-07-2974
203. Rubnitz JE, Inaba H, Ribeiro RC, Pounds S, Rooney B, Bell T, et al. NKAML: a pilot study to determine the safety and feasibility of haploidentical natural killer cell transplantation in childhood acute myeloid leukemia. J Clin Oncol (2010) 28(6):955–9. doi:10.1200/JCO.2009.24.4590
204. Curti A, Ruggeri L, D’Addio A, Bontadini A, Dan E, Motta MR, et al. Successful transfer of alloreactive haploidentical KIR ligand-mismatched natural killer cells after infusion in elderly high risk acute myeloid leukemia patients. Blood (2011) 118(12):3273–9. doi:10.1182/blood-2011-01-329508
205. Kanakry CG, Fuchs EJ, Luznik L. Modern approaches to HLA-haploidentical blood or marrow transplantation. Nat Rev Clin Oncol (2016) 13(1):10–24. doi:10.1038/nrclinonc.2015.128
206. Huang XJ, Zhao XY, Liu DH, Liu KY, Xu LP. Deleterious effects of KIR ligand incompatibility on clinical outcomes in haploidentical hematopoietic stem cell transplantation without in vitro T-cell depletion. Leukemia (2007) 21(4):848–51. doi:10.1038/sj.leu.2404566
207. Vago L, Forno B, Sormani MP, Crocchiolo R, Zino E, Di Terlizzi S, et al. Temporal, quantitative, and functional characteristics of single-KIR-positive alloreactive natural killer cell recovery account for impaired graft-versus-leukemia activity after haploidentical hematopoietic stem cell transplantation. Blood (2008) 112(8):3488–99. doi:10.1182/blood-2007-07-103325
208. Symons HJ, Leffell MS, Rossiter ND, Zahurak M, Jones RJ, Fuchs EJ. Improved survival with inhibitory killer immunoglobulin receptor (KIR) gene mismatches and KIR haplotype B donors after nonmyeloablative, HLA-haploidentical bone marrow transplantation. Biol Blood Marrow Transplant (2010) 16(4):533–42. doi:10.1016/j.bbmt.2009.11.022
209. Locatelli F, Bauquet A, Palumbo G, Moretta F, Bertaina A. Negative depletion of α/β+ T cells and of CD19+ B lymphocytes: a novel frontier to optimize the effect of innate immunity in HLA-mismatched hematopoietic stem cell transplantation. Immunol Lett (2013) 155(1–2):21–3. doi:10.1016/j.imlet.2013.09.027
210. Martelli MF, Di Ianni M, Ruggeri L, Falzetti F, Carotti A, Terenzi A, et al. HLA-haploidentical transplantation with regulatory and conventional T-cell adoptive immunotherapy prevents acute leukemia relapse. Blood (2014) 124(4):638–44. doi:10.1182/blood-2014-03-564401
211. Giebel S, Locatelli F, Lamparelli T, Velardi A, Davies S, Frumento G, et al. Survival advantage with KIR ligand incompatibility in hematopoietic stem cell transplantation from unrelated donors. Blood (2003) 102(3):814–9. doi:10.1182/blood-2003-01-0091
212. Beelen DW, Ottinger HD, Ferencik S, Elmaagacli AH, Peceny R, Trenschel R, et al. Genotypic inhibitory killer immunoglobulin-like receptor ligand incompatibility enhances the long-term antileukemic effect of unmodified allogeneic hematopoietic stem cell transplantation in patients with myeloid leukemias. Blood (2005) 105(6):2594–600. doi:10.1182/blood-2004-04-1441
213. Willemze R, Rodrigues CA, Labopin M, Sanz G, Michel G, Socié G, et al. Eurocord-Netcord and Acute Leukaemia Working Party of the EBMT. KIR-ligand incompatibility in the graft-versus-host direction improves outcomes after umbilical cord blood transplantation for acute leukemia. Leukemia (2009) 23(3):492–500. Erratum in: Leukemia (2009) 23(3):630. doi:10.1038/leu.2008.365
214. Davies SM, Ruggieri L, DeFor T, Wagner JE, Weisdorf DJ, Miller JS, et al. Evaluation of KIR ligand incompatibility in mismatched unrelated donor hematopoietic transplants. Killer immunoglobulin-like receptor. Blood (2002) 100(10):3825–7. doi:10.1182/blood-2002-04-1197
215. Farag SS, Bacigalupo A, Eapen M, Hurley C, Dupont B, Caligiuri MA, et al. The effect of KIR ligand incompatibility on the outcome of unrelated donor transplantation: a report from the center for international blood and marrow transplant research, the European blood and marrow transplant registry, and the Dutch registry. Biol Blood Marrow Transplant (2006) 12(8):876–84. doi:10.1016/j.bbmt.2006.05.007
216. Brunstein CG, Wagner JE, Weisdorf DJ, Cooley S, Noreen H, Barker JN, et al. Negative effect of KIR alloreactivity in recipients of umbilical cord blood transplant depends on transplantation conditioning intensity. Blood (2009) 113(22):5628–34. doi:10.1182/blood-2008-12-197467
217. Cooley S, Trachtenberg E, Bergemann TL, Saeteurn K, Klein J, Le CT, et al. Donors with group B KIR haplotypes improve relapse-free survival after unrelated hematopoietic cell transplantation for acute myelogenous leukemia. Blood (2009) 113(3):726–32. doi:10.1182/blood-2008-07-171926
218. Cooley S, Weisdorf DJ, Guethlein LA, Klein JP, Wang T, Le CT, et al. Donor selection for natural killer cell receptor genes leads to superior survival after unrelated transplantation for acute myelogenous leukemia. Blood (2010) 116(14):2411–9. doi:10.1182/blood-2010-05-283051
219. Cooley S, Weisdorf DJ, Guethlein LA, Klein JP, Wang T, Marsh SG, et al. Donor killer cell Ig-like receptor B haplotypes, recipient HLA-C1, and HLA-C mismatch enhance the clinical benefit of unrelated transplantation for acute myelogenous leukemia. J Immunol (2014) 192(10):4592–600. doi:10.4049/jimmunol.1302517
220. Venstrom JM, Pittari G, Gooley TA, Chewning JH, Spellman S, Haagenson M, et al. HLA-C-dependent prevention of leukemia relapse by donor activating KIR2DS1. N Engl J Med (2012) 367(9):805–16. doi:10.1056/NEJMoa1200503
221. Carlens S, Gilljam M, Chambers BJ, Aschan J, Guven H, Ljunggren HG, et al. A new method for in vitro expansion of cytotoxic human CD3-CD56+ natural killer cells. Hum Immunol (2001) 62(10):1092–8. doi:10.1016/S0198-8859(01)00313-5
222. Alici E, Sutlu T, Bjorkstrand B, Gilljam M, Stellan B, Nahi H, et al. Autologous antitumor activity by NK cells expanded from myeloma patients using GMP-compliant components. Blood (2008) 111(6):3155–62. doi:10.1182/blood-2007-09-110312
223. Garg TK, Szmania SM, Khan JA, Hoering A, Malbrough PA, Moreno-Bost A, et al. Highly activated and expanded natural killer cells for multiple myeloma immunotherapy. Haematologica (2012) 97(9):1348–56. doi:10.3324/haematol.2011.056747
224. Szmania S, Lapteva N, Garg T, Greenway A, Lingo J, Nair B, et al. Ex vivo-expanded natural killer cells demonstrate robust proliferation in vivo in high-risk relapsed multiple myeloma patients. J Immunother (2015) 38(1):24–36. doi:10.1097/CJI.0000000000000059
225. Shi J, Tricot G, Szmania S, Rosen N, Garg TK, Malaviarachchi PA, et al. Infusion of haplo-identical killer immunoglobulin-like receptor ligand mismatched NK cells for relapsed myeloma in the setting of autologous stem cell transplantation. Br J Haematol (2008) 143(5):641–53. doi:10.1111/j.1365-2141.2008.07340.x
226. Leivas A, Perez-Martinez A, Blanchard MJ, Martín-Clavero E, Fernández L, Lahuerta JJ, et al. Novel treatment strategy with autologous activated and expanded natural killer cells plus anti-myeloma drugs for multiple myeloma. Oncoimmunology (2016) 5(12):e1250051. doi:10.1080/2162402X.2016.1250051
227. Shah N, Martin-Antonio B, Yang H, Ku S, Lee DA, Cooper LJ, et al. Antigen presenting cell-mediated expansion of human umbilical cord blood yields log-scale expansion of natural killer cells with anti-myeloma activity. PLoS One (2013) 8(10):e76781. doi:10.1371/journal.pone.0076781
228. Martin-Antonio B, Najjar A, Robinson SN, Chew C, Li S, Yvon E, et al. Transmissible cytotoxicity of multiple myeloma cells by cord blood-derived NK cells is mediated by vesicle trafficking. Cell Death Differ (2015) 22(1):96–107. doi:10.1038/cdd.2014.120
229. Shah N, Li L, McCarty J, Kaur I, Yvon E, Shaim H, et al. Phase I study of cord blood-derived natural killer cells combined with autologous stem cell transplantation in multiple myeloma. Br J Haematol (2017) 177:457–66. doi:10.1111/bjh.14570
230. Porter DL, Levine BL, Kalos M, Bagg A, June CH. Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. N Engl J Med (2011) 365(8):725–33. doi:10.1056/NEJMoa1103849
231. Grupp SA, Kalos M, Barrett D, Aplenc R, Porter DL, Rheingold SR, et al. Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. N Engl J Med (2013) 368(16):1509–18. doi:10.1056/NEJMoa1215134
232. Brentjens RJ, Davila ML, Riviere I, Park J, Wang X, Cowell LG, et al. CD19-targeted T cells rapidly induce molecular remissions in adults with chemotherapy-refractory acute lymphoblastic leukemia. Sci Transl Med (2013) 5(177):177ra38. doi:10.1126/scitranslmed.3005930
233. Kochenderfer JN, Wilson WH, Janik JE, Dudley ME, Stetler-Stevenson M, Feldman SA, et al. Eradication of B-lineage cells and regression of lymphoma in a patient treated with autologous T cells genetically engineered to recognize CD19. Blood (2010) 116(20):4099–102. doi:10.1182/blood-2010-04-281931
234. Maude SL, Frey N, Shaw PA, Aplenc R, Barrett DM, Bunin NJ, et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N Engl J Med (2014) 371(16):1507–17. doi:10.1056/NEJMoa1407222
235. Turtle CJ, Hanafi LA, Berger C, Gooley TA, Cherian S, Hudecek M, et al. CD19 CAR-T cells of defined CD4+:CD8+ composition in adult B cell ALL patients. J Clin Invest (2016) 126(6):2123–38. doi:10.1172/JCI85309
236. Jackson HJ, Rafiq S, Brentjens RJ. Driving CAR T-cells forward. Nat Rev Clin Oncol (2016) 13(6):370–83. doi:10.1038/nrclinonc.2016.36
237. Garfall AL, Maus MV, Hwang WT, Lacey SF, Mahnke YD, Melenhorst JJ, et al. Chimeric antigen receptor T cells against CD19 for multiple myeloma. N Engl J Med (2015) 373(11):1040–7. doi:10.1056/NEJMoa1504542
238. Mihara K, Bhattacharyya J, Kitanaka A, Yanagihara K, Kubo T, Takei Y, et al. T-cell immunotherapy with a chimeric receptor against CD38 is effective in eliminating myeloma cells. Leukemia (2012) 26(2):365–7. doi:10.1038/leu.2011.205
239. Drent E, Groen RW, Noort WA, Themeli M, Lammerts van Bueren JJ, Parren PW, et al. Pre-clinical evaluation of CD38 chimeric antigen receptor engineered T cells for the treatment of multiple myeloma. Haematologica (2016) 101(5):616–25. doi:10.3324/haematol.2015.137620
240. Guo B, Chen M, Han Q, Hui F, Dai H, Zhang W, et al. CD138-directed adoptive immunotherapy of chimeric antigen receptor (CAR)-modified T cells for multiple myeloma. J Cell Immunother (2016) 2(1):28–35. doi:10.1016/j.jocit.2014.11.001
241. Carpenter RO, Evbuomwan MO, Pittaluga S, Rose JJ, Raffeld M, Yang S, et al. B-cell maturation antigen is a promising target for adoptive T-cell therapy of multiple myeloma. Clin Cancer Res (2013) 19(8):2048–60. doi:10.1158/1078-0432.CCR-12-2422
242. Ali SA, Shi V, Maric I, Wang M, Stroncek DF, Rose JJ, et al. T cells expressing an anti-B-cell maturation antigen chimeric antigen receptor cause remissions of multiple myeloma. Blood (2016) 128(13):1688–700. doi:10.1182/blood-2016-04-711903
243. Ramos CA, Savoldo B, Torrano V, Ballard B, Zhang H, Dakhova O, et al. Clinical responses with T lymphocytes targeting malignancy-associated κ light chains. J Clin Invest (2016) 126(7):2588–96. doi:10.1172/JCI86000
244. Chu J, He S, Deng Y, Zhang J, Peng Y, Hughes T, et al. Genetic modification of T cells redirected toward CS1 enhances eradication of myeloma cells. Clin Cancer Res (2014) 20(15):3989–4000. doi:10.1158/1078-0432.CCR-13-2510
245. Casucci M, Nicolis di Robilant B, Falcone L, Camisa B, Norelli M, Genovese P, et al. CD44v6-targeted T cells mediate potent antitumor effects against acute myeloid leukemia and multiple myeloma. Blood (2013) 122(20):3461–72. doi:10.1182/blood-2013-04-493361
246. Casucci M, Hawkins RE, Dotti G, Bondanza A. Overcoming the toxicity hurdles of genetically targeted T cells. Cancer Immunol Immunother (2015) 64(1):123–30. doi:10.1007/s00262-014-1641-9
247. Brudno JN, Kochenderfer JN. Toxicities of chimeric antigen receptor T cells: recognition and management. Blood (2016) 127(26):3321–30. doi:10.1182/blood-2016-04-703751
248. Glienke W, Esser R, Priesner C, Suerth JD, Schambach A, Wels WS, et al. Advantages and applications of CAR-expressing natural killer cells. Front Pharmacol (2015) 6:21. doi:10.3389/fphar.2015.00021
249. Hermanson DL, Kaufman DS. Utilizing chimeric antigen receptors to direct natural killer cell activity. Front Immunol (2015) 6:195. doi:10.3389/fimmu.2015.00195
250. Chu J, Deng Y, Benson DM, He S, Hughes T, Zhang J, et al. CS1-specific chimeric antigen receptor (CAR)-engineered natural killer cells enhance in vitro and in vivo antitumor activity against human multiple myeloma. Leukemia (2014) 28(4):917–27. doi:10.1038/leu.2013.279
251. Jiang H, Zhang W, Shang P, Zhang H, Fu W, Ye F, et al. Transfection of chimeric anti-CD138 gene enhances natural killer cell activation and killing of multiple myeloma cells. Mol Oncol (2014) 8(2):297–310. doi:10.1016/j.molonc.2013.12.001
252. Tonn T, Schwabe D, Klingemann HG, Becker S, Esser R, Koehl U, et al. Treatment of patients with advanced cancer with the natural killer cell line NK-92. Cytotherapy (2013) 15(12):1563–70. doi:10.1016/j.jcyt.2013.06.017
253. Suck G, Odendahl M, Nowakowska P, Seidl C, Wels WS, Klingemann HG, et al. NK-92: an ‘off-the-shelf therapeutic’ for adoptive natural killer cell-based cancer immunotherapy. Cancer Immunol Immunother (2016) 65(4):485–92. doi:10.1007/s00262-015-1761-x
254. Wang Y, Yibo Zhang Y, Don Bnson D, Caligiuri M, Yu J. Daratumumab combined with CD38(-) natural killer cells armed with a CS1 chimeric antigen receptor for the treatment of relapsed multiple myeloma [abstract 4617]. Presented at American Association for Cancer Research (Vol. 77). Washington, DC (2017). doi:10.1158/1538-7445.AM2017-4617
Keywords: multiple myeloma, immunotherapy, natural killer cells, killer immunoglobulin-like receptors, cytokines, immune checkpoint inhibition, daratumumab, elotuzumab, IDO inhibitors, chimeric antigen receptor
Citation: Pittari G, Vago L, Festuccia M, Bonini C, Mudawi D, Giaccone L and Bruno B (2017) Restoring Natural Killer Cell Immunity against Multiple Myeloma in the Era of New Drugs. Front. Immunol. 8:1444. doi: 10.3389/fimmu.2017.01444
Received: 22 February 2017; Accepted: 17 October 2017;
Published: 07 November 2017
Edited by:
Hermann Einsele, University of Würzburg, GermanyReviewed by:
Daniel Olive, Institut national de la santé et de la recherche médicale, FranceCopyright: © 2017 Pittari, Vago, Festuccia, Bonini, Mudawi, Giaccone and Bruno. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Benedetto Bruno, YmVuZWRldHRvLmJydW5vQHVuaXRvLml0
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.