 Jan Kempski
Jan Kempski Leonie Brockmann1†
Leonie Brockmann1† Samuel Huber
Samuel Huber
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Immunol., 25 October 2017
Sec. Mucosal Immunity
Volume 8 - 2017 | https://doi.org/10.3389/fimmu.2017.01373
This article is part of the Research TopicImmune-epithelial Crosstalk in Inflammatory Bowel Diseases and Mucosal Wound HealingView all 15 articles
The intestine is colonized by hundreds of different species of commensal bacteria, viruses, and fungi. Therefore, the intestinal immune system is constantly being challenged by foreign antigens. The immune system, the commensal microbiota, and the intestinal epithelial surface have to maintain a tight balance to guarantee defense against potential pathogens and to prevent chronic inflammatory conditions at the same time. Failure of these mechanisms can lead to a vicious cycle in which a perpetual tissue damage/repair process results in a pathological reorganization of the normal mucosal surface. This dysregulation of the intestine is considered to be one of the underlying causes for both inflammatory bowel disease (IBD) and colorectal cancer. TH17 cells have been associated with immune-mediated diseases, such as IBD, since their discovery in 2005. Upon mucosal damage, these cells are induced by a combination of different cytokines, such as IL-6, TGF-β, and IL-1β. TH17 cells are crucial players in the defense against extracellular pathogens and have various mechanisms to fulfill their function. They can activate and attract phagocytic cells. Additionally, TH17 cells can induce the release of anti-microbial peptides from non-immune cells, such as epithelial cells. The flip side of the coin is the strong potential of TH17 cells to be pro-inflammatory and promote pathogenicity. TH17 cells have been linked to both mucosal regeneration and inflammation. In turn, these cells and their cytokines emerged as potential therapeutic targets both for inflammatory diseases and cancer. This review will summarize the current knowledge regarding the TH17 cell-enterocyte crosstalk and give an overview of its clinical implications.
The gastrointestinal tract is essential for the absorption of nutrition and serves as a crucial barrier to protect the host against pathogens. It is colonized by up to 3.8 × 1013 microorganisms such as bacteria and fungi (1). The immune system of the intestine and commensal bacteria maintain a delicate and well-regulated homeostasis. However, when this balancing act is disrupted, chronic inflammatory conditions, such as IBD, can occur. The most common manifestations of IBD are Crohn’s disease (CD) and ulcerative colitis (UC). Their symptoms share common hallmarks such as diarrhea, abdominal pain, and relapsing inflammation in the intestine (2). The inflammation in UC patients is limited mostly to the colon with continuous inflammation of the mucosa and submucosa. CD patients suffer from patchy inflammation that causes deep ulcerations and can affect the whole gastrointestinal tract (3).
Even though the prevalence of IBD especially in western countries such as the USA is high (1.3%) and the resulting costs to the health systems are increasing (4), the underlying cause of IBD is still unknown. Twin studies revealed that genetic predispositions to develop IBD exist; however, the concordance rate for IBD in monozygotic twins does not exceed 50%, highlighting the importance of environmental factors, referred to as Exposome, for disease development (5–7). Furthermore, the intestinal microbiome in combination with the immune system seems to play a crucial role in IBD. Several studies describe a reduced diversity of commensal bacteria species in patients suffering from IBD (8, 9). Additionally, several bacterial pathogens have been implicated in the onset or progression of the disease (10). Furthermore, mouse studies indicate that changes in the microbiota composition can occur prior to colitis development (11, 12), suggesting that microbial changes might be involved in the development of IBD. However, whether dysbiosis is a cause or a consequence of IBD in humans remains to be solved. Nevertheless, barrier defects which are typically present in IBD seem to cause a dysregulated immune response against so far unknown components of the commensals. Especially the adaptive immunity, more specifically CD4+ T cells, seems to be inappropriately activated in response to commensal microorganisms in IBD. Classically, CD used to be associated with a chronic TH1 immunity, whereas during UC TH2 immunity has been thought to be implicated (13). However, after the discovery of the involvement of IL-23 and TH17 cells in autoimmune inflammation of the nervous system, further mouse studies revealed a prominent involvement of these cells during intestinal inflammation in CD and UC (14–16). Furthermore, already in 2003, TH17 cell-associated cytokines were reported to be upregulated in tissue biopsies and serum of patients with IBD (17, 18). Moreover, chronic inflammation predisposes IBD patients to the development of colorectal cancer (CRC) (19). The chronic inflammation and mucosal injury can trigger long-lasting healing responses that are not terminated, leading to tissue dysfunction and finally to carcinogenesis (20). TH17 cells and TH17 cell-associated cytokines are also involved during CRC in humans. TH17 cells were found elevated in tumors of CRC patients and an increased TH17 cell immune response correlates with advanced stages of CRC (21–23).
In summary, four main components lead to the pathology of IBD: genetic predisposition, environmental factors, intestinal microbiome, and a dysregulated immune system. So far, the main therapeutics broadly available to treat IBD are based on suppressing the immune response. But these therapies are unable to reset the intestinal homeostasis and do not directly treat the underlying cause of the chronic inflammation. Therefore, patients suffering from IBD mostly require lifelong treatment. TH17 cells could be the link between these four components. Thus, a better understanding of the interactions of these four components and TH17 cells is a main focus of current research, with the aim of developing more specific and efficient therapies. This review aims to summarize the current knowledge about the interactions of TH17 cells and TH17 cell-associated cytokines with the mucosal surface in the intestine and in the microbiota.
TH17 cell-mediated immunity is essential for the clearance of extracellular bacteria and fungi by attracting neutrophils and inducing the release of anti-microbial peptides from epithelial cells (24–28). TH17 cell cytokines include IL-17A, IL-17F, TNF-α, and IL-22 (15, 26). It has been demonstrated in mice that intestinal TH17 cells are induced by segmented filamentous bacteria (SFB), which are gram-positive, spore-forming bacteria located in the terminal ileum of the small intestine. Accordingly, TH17 cells are mainly located in this part of the intestine under physiological conditions (29). A critical feature of SFB is its ability to adhere to the intestinal epithelial cells (IEC). SFB and other bacteria with the same ability such as Citrobacter rodentium and Escherichia coli induce the production of serum amyloid A (SAA) from epithelial cells (30). Subsequently, SAA induces the release of IL-6, TGF-β, and IL-1β from intestinal cells, especially dendritic cells, which leads to the differentiation of TH17 cells (29–31). IL-6 signaling leads to the activation of STAT3 and subsequent induction of RORγt, one of the key transcription factors of TH17 cells, and of other TH17 cell-related factors such as IL-17A/F and IL-23R (25, 32–34). IL-1β is crucial for the differentiation of TH17 cells (35). Besides other effects, IL-1β induces the expression of the transcription factor IRF4, which is needed for the expression of RORγt (36). The role of TGF-β for TH17 cell differentiation is still controversial. TH17 cells can occur in the absence of TGF-β in the gut mucosa (37). However, TGF-β can negatively regulate TH1 and TH2 while promoting TH17 cell differentiation and therefore favors the contribution of TH17 cells (38). Due to the presence of microbiota in the intestine, the TH17 cell differentiation differs in comparison to sterile organs. One essential alteration is the activation of the transcription factor aryl hydrocarbon receptor (AHR) by ligands derived from food or intestinal microbiota (39, 40). AHR is highly expressed already in early stages of TH17 cell differentiation (41, 42). AHR expression is not essential for TH17 cell differentiation. However, it is nonetheless non-redundant for the secretion of IL-22 by TH17 cells, a cytokine vital for the anti-microbial properties of TH17 cells (42–44).
IL-23 is an important cytokine for TH17 cell biology. However, the IL-23 receptor is absent on naïve T cells. Accordingly, research led to the discovery that IL-23 is essential for the effector properties of TH17 cells rather than their induction (45, 46).
In the following sections, we want to outline the effects of TH17 cell-associated cytokines such as IL-17A, IL-22, and TNF-α on epithelial cells during IBD and carcinogenesis.
IL-17A is a member of the IL-17 family consisting of IL-17A, IL-17-B, IL-17-C, IL-17-D, IL-17E, and IL-17F (47). Both IL-17A and IL-17F signal through the IL-17RA–IL-17RC complex and activate the NF-κB and MAPK pathways (48). IL-17A is produced mainly by TH17 cells although production by many other cell types including CD8+ T cells, γδ T cells, NK cells, NKT cells, and innate lymphoid cells (ILCs) has been described. Initial studies have shown increased IL17A mRNA expression and increased numbers of TH17 cells in the inflamed tissue of IBD patients compared to healthy mucosa (18, 49, 50). Furthermore, the amount of IL-17A producing PBMCs correlates with disease severity in patients with UC (51). These results imply a pathogenic role of those cells in the intestine in IBD. IL-17A induces the recruitment and activation of granulocytes and locally promotes the production of other pro-inflammatory cytokines such as TNF-α, IL-6, and IL-1β (52, 53). In line with these findings, a blockade of IL-23 and IL-21 in murine models of colitis results in decreased numbers of TH17 cells and in a favorable disease outcome (54, 55). Surprisingly however, blockade or genetic deletion of IL-17A resulted in aggravated disease severity in the DSS-induced colitis, a mouse model of IBD (55, 56). Interestingly and in contrast with the data obtained using IL-17A knock-out mice, IL-17F knock-out mice show a less severe DSS-induced colitis (57). As IL-17F binds to the IL-17RA-IL-17RC complex with lower affinity than IL-17A, the different activation strength of the receptor complex might explain those opposing results. These findings do emphasize the need for strict distinction between the functions of TH17 cells and one of their signature cytokines, IL-17A. The protective function of IL-17A in mouse IBD models can be explained by the effect of this cytokine on enterocytes. Similar to IL-22, IL-17A induces their proliferation and tight-barrier formation and therefore promotes the integrity of the epithelial barrier (55, 58). The other side of the coin of those regenerative and physiological effects is the potential of IL-17A to promote carcinogenesis in the colon. Following a barrier defect, bacterial translocation and IL-23 production by innate immune cells, such as DCs and macrophages (Mφ), induce high levels of IL-17A which in turn can favor intestinal tumorigenesis (59). Importantly, an enterocyte-specific knock-out of the IL-17RA decreased tumor formation in mice, suggesting a direct tumorigenic function of IL-17A (60). Moreover, increased frequencies of IL-17A producing cells were found in human CRC and high IL17A mRNA expression correlates with a poor prognosis in these patients (60–62). Although these results do indicate a potential therapeutic benefit of targeting IL-17A, the heterogeneity of TH17 cells makes it difficult to conclude their exact function in human CRC and further research is needed before initiating clinical trials.
IL-22 is a member of the IL-10 family that has gained considerable attention in the last years due to its role in linking inflammation and regenerative processes. Although originally described as a TH1 cytokine (63), it can be produced by a variety of immune cells including CD4+ (TH1, TH17, TH22), CD8+ T cells, γδ T cells, NK cells, NKT cells, and group 3 ILCs. Innate lymphoid cells represent a newly described cell type which belongs to the lymphoid lineage but is characterized by the absence of antigen-specific T- or B-cell receptors. Group 3 ILCs represent a subgroup of innate lymphoid cells that are defined by the expression of the master transcriptional factor RORγt and the capacity to produce IL-17A and IL-22. They are therefore widely regarded as the innate counterpart of Th17 cells. Based on the expression of NKp46 (NCR1), group 3 ILCs can be further subdivided into ILC3s and lymphoid-tissue inducer cells. IL-22 signals through a heterodimeric receptor consisting of the ubiquitously expressed IL10R2 and the more specifically expressed IL22R1 (64, 65). Upon binding of IL22, the IL-22–IL-22R1–IL-10R2 complex signals mainly through STAT3 and the AKT-MAPK pathway, although activation of STAT1 and STAT5 has also been shown in certain cells (66, 67). The need for tight control of IL-22 activity is emphasized by the existence of an endogenous antagonist, called IL-22 binding protein (IL-22BP), which binds IL-22 and prevents binding to the membrane bound IL-22 receptor. In the intestine, IL-22 signaling elicits multiple responses that aim to maintain the integrity of the mucosal barrier, trigger antimicrobial responses, and promote wound healing (68, 69). In mice, levels of IL-22 increase upon chemically induced tissue damage and are crucial for tissue regeneration. Interestingly, levels of IL-22BP decrease in parallel, further increasing the activity of IL-22 (70). Similar results have been described in patients with acute diverticulitis (71). Surprisingly however, IL-22BP levels were recently shown to be elevated in patients with IBD and thus limit the tissue-healing effects of IL-22, suggesting that a disturbed IL-22–IL-22BP axis might be one of the mechanisms contributing to the chronification of inflammation (Figure 1) (71, 72). Although usually regarded as a tissue-protective cytokine, pro-inflammatory effects of IL-22 in the intestine have also been described. For instance, NCR-ILC-3, which coproduce IL-17A and IL-22, have been shown to be pathogenic in mouse models of IBD in a microbiota-dependent fashion and are increased in patients with IBD (73, 74). On the other hand, NCR-ILC-3 decreased in these patients and depletion of IL-22 producing ILC-3 makes mice more susceptible to C. rodentium infection (75–77). Similarly, T cell-derived IL-22 was shown to be both pathogenic and protective in murine models of IBD (78, 79). The distinct functions of ILC and T cell-derived IL-22 in IBD are still not completely understood. It can be hypothesized that ILC-derived IL-22 is crucial in the initial phases of inflammatory responses, whereas in chronically inflamed tissue, T cell become the major producers as suggested by Basu et al. (80).
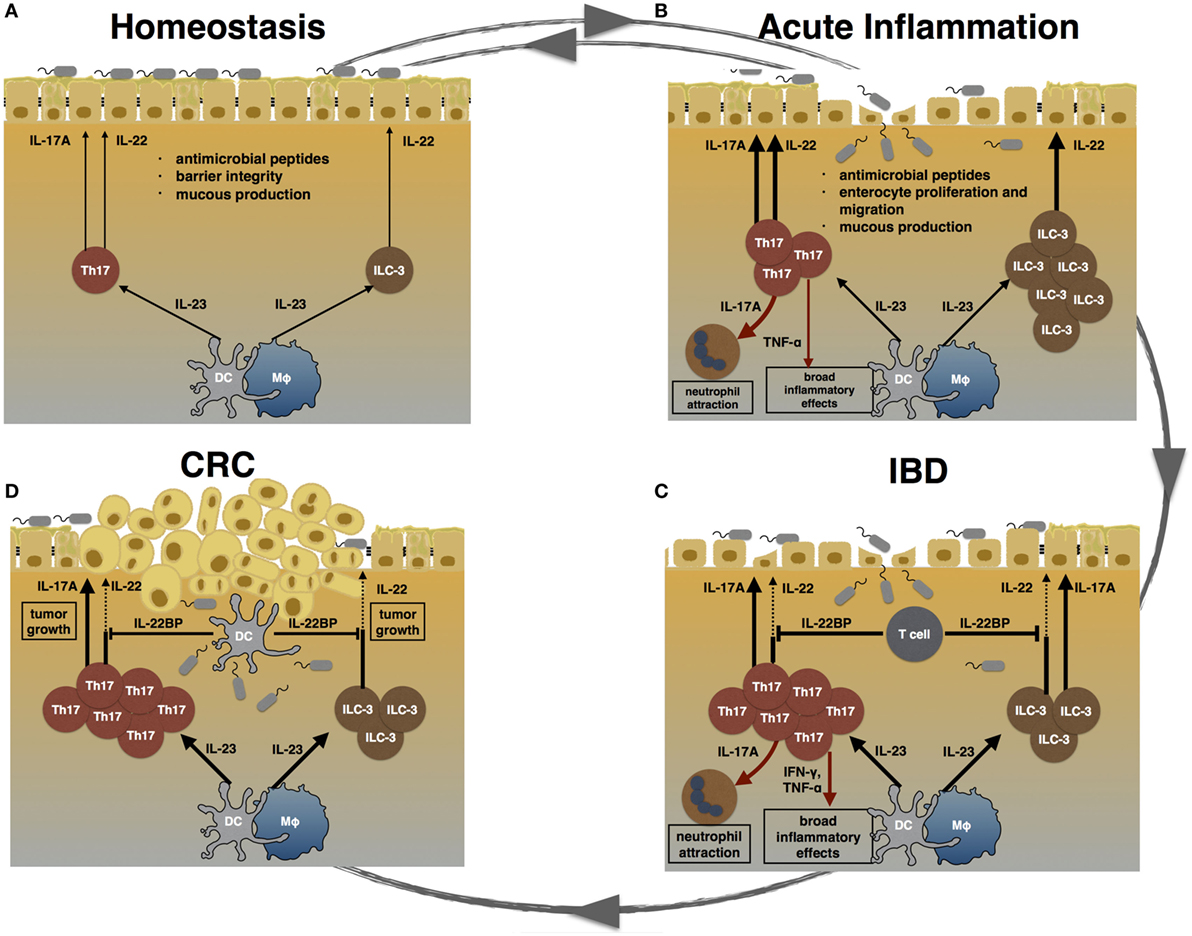
Figure 1. TH17 cells during homeostasis, inflammation, and carcinogenesis. (A) During homeostasis, TH17 cells and ILC3 are induced by certain species of the commensal microbiota. IL-17A and IL-22 promote epithelial barrier integrity, mucus production, and the release of anti-microbial peptides. (B) Acute inflammation can be caused by pathogenic bacteria. Invading pathogens induce the expansion of TH17 cells and ILC3. TH17 cell-associated cytokines attract neutrophils and trigger a pro-inflammatory response in order to clear the invading agent. Furthermore, IL-17A and IL-22 promote enterocyte proliferation and migration, thereby promoting mucosal healing. After clearance of the pathogen, the intestinal immune system returns to homeostasis. (C) Failure to terminate an intestinal immune response can lead to chronic inflammation. In inflammatory bowel disease (IBD), highly pathogenic TH17 cells expand and secret pro-inflammatory cytokines such as IL-17A, TNF-α, and IFN-γ. Especially, TNF-α and IFN-γ cause a broad inflammatory response. The regenerative effects of IL-22 are counter regulated by high levels of T cell-derived IL-22 binding protein (IL-22BP) in IBD patients. (D) Chronically elevated levels of IL-17A and IL-22 can promote carcinogenesis. Hereby, IL-22 can be controlled by DC-derived IL-22BP. However, whether this mechanism fails in human colorectal cancer (CRC) is currently unknown.
As mucosal healing is one of the major aims in the therapy of IBD, IL-22 is widely regarded as a potentially beneficial cytokine in those diseases. On the other hand, a relationship between regenerative agents and tumorigenesis has been known for a long time and patients with IBD are known to have an increased risk for the development of CRC. Indeed, prolonged or excessive IL-22 signaling promotes tumorigenesis in the intestine of both mice and humans. In murine models of colon cancer, knock-out of the IL-22BP resulted in accelerated tumor growth, which was IL-22 dependent (70). In line with these results, levels of IL-22 are increased in patients with CRC and IL-22 can directly promote growth of cancer cell lines in vitro (81). Although ILCs have been shown to promote tumor growth in mice through IL-22, data in humans indicate CCR6+ TH17 cells to be the major source (81, 82). Further research is required to understand the distinct effects of ILC vs. T cell derived IL-22 in human carcinogenesis.
The cytokine TNF-α has been linked to inflammatory responses for a long time. TNF-α can be produced by a multitude of immune cells such as macrophages, CD4+ lymphocytes, NK cells, neutrophils, mast cells, and eosinophils. A soluble form and transmembrane bound form of TNF-α exist (83). Both the soluble and the membrane-bound TNF-α can interact with two TNF receptors, TNF-R1 (TNFRSF1A) and TNF-R2 (TNFRSF1B), expressed on IEC (84). TNF-α signaling leads to the release of other pro-inflammatory molecules and can provoke both pro- and anti-apoptotic signals in the IEC (85). An involvement of TNF-α in the pathology of IBD was first assumed after assessing TNF-α levels in serum of children suffering from IBD (86). Further studies revealed elevated levels of TNF-α in stool and mucosal tissue also (87, 88). Based on the knowledge of the pro-inflammatory properties and these observations, TNF-α became an interesting target for new therapeutic approaches. The development of anti-TNF-α antibodies and their application in humans is one of the best examples of how the concept, “from bench to bedside” can be successfully employed. Already in 1995 Dullemen et al. reported the successful use of a monoclonal antibody cA2 (infliximab) in CD patients (89). Since then, anti-TNF-α therapy has greatly improved the management of IBD. Furthermore, it has been demonstrated that TNF-α can promote colitis-associated CRC, a long-term consequence of IBD. In a mouse model of colitis-induced carcinogenesis, the blockade of TNF-α led to reduced mucosal injury and in turn to decreased tumor formation (90). Accordingly, elevated expression of TNF-α in tumors of CRC patients is associated with advanced cancer stages in humans (91). However, the treatment with anti-TNF antibodies causes high costs to health care systems and can cause some severe side effects such as opportunistic infections (92). Finally, around 10–30% of patients with IBD do not respond to anti-TNF-α treatment and 20–40% of patients lose response over time (93). Moreover, a prognostic factor that can predict the response to this therapy is missing.
TH17 cells are highly enriched in IBD, and several genetic risk loci being associated with IBD are linked to TH17 cells. Thus, several drugs were developed with the intention to manipulate TH17 cell development or function in patients with IBD. First clinical trials using monoclonal antibodies targeting cytokines related to TH17 cells, such as IL-17A, show similar results to those obtained in murine IBD models and highlight the need to make a clear distinction between the biological functions of IL-17A and TH17 cells in general. As described above, despite the potential pro-inflammatory properties of TH17 cells, their signature cytokine IL-17A was shown to also have beneficial effects. IL-17A can promote enterocyte proliferation, tight-barrier formation and epithelial barrier integrity in the intestine (54, 55). A clinical trial with secukinumab, an anti-IL-17A antibody, further highlighted the importance of IL-17A for mucosal homeostasis. Although being highly effective in psoriasis, blockade of IL-17A resulted in aggravated disease course in IBD patients (94–96). In contrast to blockade of IL-17A, antibodies against IL-23 were effective in preventing colitis in mouse models (55). Similarly, ustekinumab proved to be effective in patients with CD (97). Ustekinumab targets the p40 subunit of IL-12, which is part of IL-12 and IL-23. Therefore, it affects both TH1 (together with ILC-1) and TH17 (together with ILC-3) lineages. Interestingly, a first trial involving the blockade of the p19 subunit of IL-23 (and therefore not affecting IL-12 signaling) using risankizumab also delivered promising results in patients with moderate-to-severe CD and might represent a future therapy in IBD (98). In contrast to the above-mentioned approaches, anti-TNF therapy is already well established in the therapy of both CD and UC. Surprisingly, we have recently shown that the effectiveness of this therapy is to some extent dependent on the IL-22–IL-22BP axis in both mice and humans (71). Direct therapeutic intervention into the IL-22–IL-22R1–IL-22BP system might therefore represent a novel strategy in patients with IBD. Importantly, as IL22R1 is expressed exclusively on non-hematopoietic cells, such a therapy would not lead to systemic immunosuppression, a common side-effect of most currently used medications. Nevertheless, the oncogenic properties of IL-22 could represent a major obstacle in the development of safe and effective drugs targeting the IL-22–IL-22R1–IL-22BP axis.
LB and JK wrote the manuscript and equally contributed to the manuscript; NG and SH supervised and revised the manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
This work was supported by the ERC (Stg 337251 to SH and Stg 715271 to NG) and the DFG (SFB841 to SH and NG).
1. Sender R, Fuchs S, Milo R. Revised estimates for the number of human and bacteria cells in the body. PLoS Biol (2016) 14(8):e1002533. doi:10.1371/journal.pbio.1002533
2. Podolsky DK. Inflammatory bowel disease (1). N Engl J Med (1991) 325(13):928–37. doi:10.1056/NEJM199109263251306
3. Kaser A, Zeissig S, Blumberg RS. Inflammatory bowel disease. Annu Rev Immunol (2010) 28:573–621. doi:10.1146/annurev-immunol-030409-101225
4. Voelker R. Inflammatory bowel disease is more common than earlier studies showed. JAMA (2016) 316(24):2590–2590. doi:10.1001/jama.2016.18375
5. Halfvarson J, Bodin L, Tysk C, Lindberg E, Järnerot G. Inflammatory bowel disease in a Swedish twin cohort: a long-term follow-up of concordance and clinical characteristics. Gastroenterology (2003) 124(7):1767–73. doi:10.1016/S0016-5085(03)00385-8
6. Orholm M, Binder V, Sørensen TI, Rasmussen LP, Kyvik KO. Concordance of inflammatory bowel disease among Danish twins. Results of a nationwide study. Scand J Gastroenterol (2000) 35(10):1075–81. doi:10.1080/003655200451207
7. Thompson NP, Driscoll R, Pounder RE, Wakefield AJ. Genetics versus environment in inflammatory bowel disease: results of a British twin study. BMJ (1996) 312(7023):95–6. doi:10.1136/bmj.312.7023.95
8. Lepage P, Colombet J, Marteau P, Sime-Ngando T, Dore J, Leclerc M. Dysbiosis in inflammatory bowel disease: a role for bacteriophages? Gut (2008) 57(3):424–5. doi:10.1136/gut.2007.134668
9. Qin J, Li R, Raes J, Arumugam M, Burgdorf KS, Manichanh C, et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature (2010) 464(7285):59–65. doi:10.1038/nature08821
10. Becker C, Neurath MF, Wirtz S. The intestinal microbiota in inflammatory bowel disease. ILAR J (2015) 56(2):192–204. doi:10.1093/ilar/ilv030
11. Glymenaki M, Singh G, Brass A, Warhurst G, McBain AJ, Else KJ, et al. Compositional changes in the gut mucus microbiota precede the onset of colitis-induced inflammation. Inflamm Bowel Dis (2017) 23(6):912–22. doi:10.1097/MIB.0000000000001118
12. Nagao-Kitamoto H, Shreiner AB, Gillilland MG 3rd, Kitamoto S, Ishii C, Hirayama A, et al. Functional characterization of inflammatory bowel disease-associated gut dysbiosis in gnotobiotic mice. Cell Mol Gastroenterol Hepatol (2016) 2(4):468–81. doi:10.1016/j.jcmgh.2016.02.003
13. Abraham C, Cho JH. Inflammatory bowel disease. N Engl J Med (2009) 361(21):2066–78. doi:10.1056/NEJMra0804647
14. Cua DJ, Sherlock J, Chen Y, Murphy CA, Joyce B, Seymour B, et al. Interleukin-23 rather than interleukin-12 is the critical cytokine for autoimmune inflammation of the brain. Nature (2003) 421(6924):744–8. doi:10.1038/nature01355
15. Langrish CL, Chen Y, Blumenschein WM, Mattson J, Basham B, Sedgwick JD, et al. IL-23 drives a pathogenic T cell population that induces autoimmune inflammation. J Exp Med (2005) 201(2):233–40. doi:10.1084/jem.20041257
16. Maloy KJ, Kullberg MC. IL-23 and Th17 cytokines in intestinal homeostasis. Mucosal Immunol (2008) 1(5):339–49. doi:10.1038/mi.2008.28
17. Andoh A, Zhang Z, Inatomi O, Fujino S, Deguchi Y, Araki Y, et al. Interleukin-22, a member of the IL-10 subfamily, induces inflammatory responses in colonic subepithelial myofibroblasts. Gastroenterology (2005) 129(3):969–84. doi:10.1053/j.gastro.2005.06.071
18. Fujino S, Andoh A, Bamba S, Ogawa A, Hata K, Araki Y, et al. Increased expression of interleukin 17 in inflammatory bowel disease. Gut (2003) 52(1):65–70. doi:10.1136/gut.52.1.65
19. Lasry A, Zinger A, Ben-Neriah Y. Inflammatory networks underlying colorectal cancer. Nat Immunol (2016) 17(3):230–40. doi:10.1038/ni.3384
20. Antonio N, Bonnelykke-Behrndtz ML, Ward LC, Collin J, Christensen IJ, Steiniche T, et al. The wound inflammatory response exacerbates growth of pre-neoplastic cells and progression to cancer. EMBO J (2015) 34(17):2219–36. doi:10.15252/embj.201490147
21. Amicarella F, Muraro MG, Hirt C, Cremonesi E, Padovan E, Mele V, et al. Dual role of tumour-infiltrating T helper 17 cells in human colorectal cancer. Gut (2017) 66(4):692–704. doi:10.1136/gutjnl-2015-310016
22. Mao H, Pan F, Guo H, Bu F, Xin T, Chen S, et al. Feedback mechanisms between M2 macrophages and Th17 cells in colorectal cancer patients. Tumour Biol (2016) 37(9):12223–30. doi:10.1007/s13277-016-5085-z
23. Sharp SP, Avram D, Stain SC, Lee EC. Local and systemic Th17 immune response associated with advanced stage colon cancer. J Surg Res (2017) 208:180–6. doi:10.1016/j.jss.2016.09.038
24. Archer NK, Adappa ND, Palmer JN, Cohen NA, Harro JM, Lee SK, et al. Interleukin-17A (IL-17A) and IL-17F are critical for antimicrobial peptide production and clearance of Staphylococcus aureus nasal colonization. Infect Immun (2016) 84(12):3575–83. doi:10.1128/IAI.00596-16
25. Ivanov II, McKenzie BS, Zhou L, Tadokoro CE, Lepelley A, Lafaille JJ, et al. The orphan nuclear receptor RORgammat directs the differentiation program of proinflammatory IL-17+ T helper cells. Cell (2006) 126(6):1121–33. doi:10.1016/j.cell.2006.07.035
26. Liang SC, Tan XY, Luxenberg DP, Karim R, Dunussi-Joannopoulos K, Collins M, et al. Interleukin (IL)-22 and IL-17 are coexpressed by Th17 cells and cooperatively enhance expression of antimicrobial peptides. J Exp Med (2006) 203(10):2271–9. doi:10.1084/jem.20061308
27. Meller S, Di Domizio J, Voo KS, Friedrich HC, Chamilos G, Ganguly D, et al. T(H)17 cells promote microbial killing and innate immune sensing of DNA via interleukin 26. Nat Immunol (2015) 16(9):970–9. doi:10.1038/ni.3211
28. Ouyang W, Kolls JK, Zheng Y. The biological functions of T helper 17 cell effector cytokines in inflammation. Immunity (2008) 28(4):454–67. doi:10.1016/j.immuni.2008.03.004
29. Ivanov II, Atarashi K, Manel N, Brodie EL, Shima T, Karaoz U, et al. Induction of intestinal Th17 cells by segmented filamentous bacteria. Cell (2009) 139(3):485–98. doi:10.1016/j.cell.2009.09.033
30. Atarashi K, Tanoue T, Ando M, Kamada N, Nagano Y, Narushima S, et al. Th17 cell induction by adhesion of microbes to intestinal epithelial cells. Cell (2015) 163(2):367–80. doi:10.1016/j.cell.2015.08.058
31. Shaw MH, Kamada N, Kim YG, Nunez G. Microbiota-induced IL-1beta, but not IL-6, is critical for the development of steady-state TH17 cells in the intestine. J Exp Med (2012) 209(2):251–8. doi:10.1084/jem.20111703
32. Bettelli E, Carrier Y, Gao W, Korn T, Strom TB, Oukka M, et al. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature (2006) 441(7090):235–8. doi:10.1038/nature04753
33. Mangan PR, Harrington LE, O’Quinn DB, Helms WS, Bullard DC, Elson CO, et al. Transforming growth factor-beta induces development of the T(H)17 lineage. Nature (2006) 441(7090):231–4. doi:10.1038/nature04754
34. Veldhoen M, Hocking RJ, Atkins CJ, Locksley RM, Stockinger B. TGFbeta in the context of an inflammatory cytokine milieu supports de novo differentiation of IL-17-producing T cells. Immunity (2006) 24(2):179–89. doi:10.1016/j.immuni.2006.01.001
35. Hu W, Troutman TD, Edukulla R, Pasare C. Priming microenvironments dictate cytokine requirements for T helper 17 cell lineage commitment. Immunity (2011) 35(6):1010–22. doi:10.1016/j.immuni.2011.10.013
36. Chung Y, Chang SH, Martinez GJ, Yang XO, Nurieva R, Kang HS, et al. Critical regulation of early Th17 cell differentiation by interleukin-1 signaling. Immunity (2009) 30(4):576–87. doi:10.1016/j.immuni.2009.02.007
37. Ghoreschi K, Laurence A, Yang XP, Tato CM, McGeachy MJ, Konkel JE, et al. Generation of pathogenic T(H)17 cells in the absence of TGF-beta signalling. Nature (2010) 467(7318):967–71. doi:10.1038/nature09447
38. Qin H, Wang L, Feng T, Elson CO, Niyongere SA, Lee SJ, et al. TGF-beta promotes Th17 cell development through inhibition of SOCS3. J Immunol (2009) 183(1):97–105. doi:10.4049/jimmunol.0801986
39. Zelante T, Iannitti RG, Cunha C, De Luca A, Giovannini G, Pieraccini G, et al. Tryptophan catabolites from microbiota engage aryl hydrocarbon receptor and balance mucosal reactivity via interleukin-22. Immunity (2013) 39(2):372–85. doi:10.1016/j.immuni.2013.08.003
40. Moura-Alves P, Faé K, Houthuys E, Dorhoi A, Kreuchwig A, Furkert J, et al. AhR sensing of bacterial pigments regulates antibacterial defence. Nature (2014) 512(7515):387–92. doi:10.1038/nature13684
41. Quintana FJ, Basso AS, Iglesias AH, Korn T, Farez MF, Bettelli E, et al. Control of T(reg) and T(H)17 cell differentiation by the aryl hydrocarbon receptor. Nature (2008) 453(7191):65–71. doi:10.1038/nature06880
42. Veldhoen M, Hirota K, Westendorf AM, Buer J, Dumoutier L, Renauld JC, et al. The aryl hydrocarbon receptor links TH17-cell-mediated autoimmunity to environmental toxins. Nature (2008) 453(7191):106–9. doi:10.1038/nature06881
43. Li Y, Innocentin S, Withers DR, Roberts NA, Gallagher AR, Grigorieva EF, et al. Exogenous stimuli maintain intraepithelial lymphocytes via aryl hydrocarbon receptor activation. Cell (2011) 147(3):629–40. doi:10.1016/j.cell.2011.09.025
44. Yeste A, Mascanfroni ID, Nadeau M, Burns EJ, Tukpah AM, Santiago A, et al. IL-21 induces IL-22 production in CD4+ T cells. Nat Commun (2014) 5:3753. doi:10.1038/ncomms4753
45. McGeachy MJ, Chen Y, Tato CM, Laurence A, Joyce-Shaikh B, Blumenschein WM, et al. The interleukin 23 receptor is essential for the terminal differentiation of interleukin 17-producing effector T helper cells in vivo. Nat Immunol (2009) 10(3):314–24. doi:10.1038/ni.1698
46. Zhou L, Lopes JE, Chong MM, Ivanov II, Min R, Victora GD, et al. TGF-beta-induced Foxp3 inhibits T(H)17 cell differentiation by antagonizing RORgammat function. Nature (2008) 453(7192):236–40. doi:10.1038/nature06878
47. Korn T, Bettelli E, Oukka M, Kuchroo VK. IL-17 and Th17 cells. Annu Rev Immunol (2009) 27:485–517. doi:10.1146/annurev.immunol.021908.132710
48. Gaffen SL. Structure and signalling in the IL-17 receptor family. Nat Rev Immunol (2009) 9(8):556–67. doi:10.1038/nri2586
49. Nielsen OH, Kirman I, Rudiger N, Hendel J, Vainer B. Upregulation of interleukin-12 and -17 in active inflammatory bowel disease. Scand J Gastroenterol (2003) 38(2):180–5. doi:10.1080/00365520310000672
50. Rovedatti L, Kudo T, Biancheri P, Sarra M, Knowles CH, Rampton DS, et al. Differential regulation of interleukin 17 and interferon gamma production in inflammatory bowel disease. Gut (2009) 58(12):1629–36. doi:10.1136/gut.2009.182170
51. Raza A, Shata MT. Letter: pathogenicity of Th17 cells may differ in ulcerative colitis compared with Crohn’s disease. Aliment Pharmacol Ther (2012) 36(2):204; author reply 205. doi:10.1111/j.1365-2036.2012.05124.x
52. Iwakura Y, Ishigame H, Saijo S, Nakae S. Functional specialization of interleukin-17 family members. Immunity (2011) 34(2):149–62. doi:10.1016/j.immuni.2011.02.012
53. Kasama T, Kobayashi K, Yajima N, Shiozawa F, Yoda Y, Takeuchi HT, et al. Expression of vascular endothelial growth factor by synovial fluid neutrophils in rheumatoid arthritis (RA). Clin Exp Immunol (2000) 121(3):533–8. doi:10.1046/j.1365-2249.2000.01272.x
54. Fina D, Sarra M, Fantini MC, Rizzo A, Caruso R, Caprioli F, et al. Regulation of gut inflammation and th17 cell response by interleukin-21. Gastroenterology (2008) 134(4):1038–48. doi:10.1053/j.gastro.2008.01.041
55. Maxwell JR, Zhang Y, Brown WA, Smith CL, Byrne FR, Fiorino M, et al. Differential roles for interleukin-23 and interleukin-17 in intestinal immunoregulation. Immunity (2015) 43(4):739–50. doi:10.1016/j.immuni.2015.08.019
56. Ogawa A, Andoh A, Araki Y, Bamba T, Fujiyama Y. Neutralization of interleukin-17 aggravates dextran sulfate sodium-induced colitis in mice. Clin Immunol (2004) 110(1):55–62. doi:10.1016/j.clim.2003.09.013
57. Yang XO, Chang SH, Park H, Nurieva R, Shah B, Acero L, et al. Regulation of inflammatory responses by IL-17F. J Exp Med (2008) 205(5):1063–75. doi:10.1084/jem.20071978
58. Kinugasa T, Sakaguchi T, Gu X, Reinecker HC. Claudins regulate the intestinal barrier in response to immune mediators. Gastroenterology (2000) 118(6):1001–11. doi:10.1016/S0016-5085(00)70351-9
59. Grivennikov SI, Wang K, Mucida D, Stewart CA, Schnabl B, Jauch D, et al. Adenoma-linked barrier defects and microbial products drive IL-23/IL-17-mediated tumour growth. Nature (2012) 491(7423):254–8. doi:10.1038/nature11465
60. Wang K, Kim MK, Di Caro G, Wong J, Shalapour S, Wan J, et al. Interleukin-17 receptor a signaling in transformed enterocytes promotes early colorectal tumorigenesis. Immunity (2014) 41(6):1052–63. doi:10.1016/j.immuni.2014.11.009
61. Dunne MR, Ryan C, Nolan B, Tosetto M, Geraghty R, Winter DC, et al. Enrichment of inflammatory IL-17 and TNF-alpha secreting CD4(+) T cells within colorectal tumors despite the presence of elevated CD39(+) T regulatory cells and increased expression of the immune checkpoint molecule, PD-1. Front Oncol (2016) 6:50. doi:10.3389/fonc.2016.00050
62. Mlecnik B, Bindea G, Angell HK, Maby P, Angelova M, Tougeron D, et al. Integrative analyses of colorectal cancer show immunoscore is a stronger predictor of patient survival than microsatellite instability. Immunity (2016) 44(3):698–711. doi:10.1016/j.immuni.2016.02.025
63. Wolk K, Kunz S, Asadullah K, Sabat R. Cutting edge: immune cells as sources and targets of the IL-10 family members? J Immunol (2002) 168(11):5397–402. doi:10.4049/jimmunol.168.11.5397
64. Dumoutier L, Van Roost E, Colau D, Renauld JC. Human interleukin-10-related T cell-derived inducible factor: molecular cloning and functional characterization as an hepatocyte-stimulating factor. Proc Natl Acad Sci U S A (2000) 97(18):10144–9. doi:10.1073/pnas.170291697
65. Xie MH, Aggarwal S, Ho WH, Foster J, Zhang Z, Stinson J, et al. Interleukin (IL)-22, a novel human cytokine that signals through the interferon receptor-related proteins CRF2-4 and IL-22R. J Biol Chem (2000) 275(40):31335–9. doi:10.1074/jbc.M005304200
66. Lejeune D, Dumoutier L, Constantinescu S, Kruijer W, Schuringa JJ, Renauld JC. Interleukin-22 (IL-22) activates the JAK/STAT, ERK, JNK, and p38 MAP kinase pathways in a rat hepatoma cell line. Pathways that are shared with and distinct from IL-10. J Biol Chem (2002) 277(37):33676–82. doi:10.1074/jbc.M204204200
67. Wolk K, Kunz S, Witte E, Friedrich M, Asadullah K, Sabat R. IL-22 increases the innate immunity of tissues. Immunity (2004) 21(2):241–54. doi:10.1016/j.immuni.2004.07.007
68. Brand S, Beigel F, Olszak T, Zitzmann K, Eichhorst ST, Otte JM, et al. IL-22 is increased in active Crohn’s disease and promotes proinflammatory gene expression and intestinal epithelial cell migration. Am J Physiol Gastrointest Liver Physiol (2006) 290(4):G827–38. doi:10.1152/ajpgi.00513.2005
69. Sugimoto K, Ogawa A, Mizoguchi E, Shimomura Y, Andoh A, Bhan AK, et al. IL-22 ameliorates intestinal inflammation in a mouse model of ulcerative colitis. J Clin Invest (2008) 118(2):534–44. doi:10.1172/JCI33194
70. Huber S, Gagliani N, Zenewicz LA, Huber FJ, Bosurgi L, Hu B, et al. IL-22BP is regulated by the inflammasome and modulates tumorigenesis in the intestine. Nature (2012) 491(7423):259–63. doi:10.1038/nature11535
71. Pelczar P, Witkowski M, Perez LG, Kempski J, Hammel AG, Brockmann L, et al. A pathogenic role for T cell-derived IL-22BP in inflammatory bowel disease. Science (2016) 354(6310):358–62. doi:10.1126/science.aah5903
72. Martin JC, Bériou G, Heslan M, Bossard C, Jarry A, Abidi A, et al. IL-22BP is produced by eosinophils in human gut and blocks IL-22 protective actions during colitis. Mucosal Immunol (2016) 9(2):539–49. doi:10.1038/mi.2015.83
73. Buonocore S, Ahern PP, Uhlig HH, Ivanov II, Littman DR, Maloy KJ, et al. Innate lymphoid cells drive interleukin-23-dependent innate intestinal pathology. Nature (2010) 464(7293):1371–5. doi:10.1038/nature08949
74. Geremia A, Arancibia-Cárcamo CV, Fleming MP, Rust N, Singh B, Mortensen NJ, et al. IL-23-responsive innate lymphoid cells are increased in inflammatory bowel disease. J Exp Med (2011) 208(6):1127–33. doi:10.1084/jem.20101712
75. Bernink JH, Peters CP, Munneke M, te Velde AA, Meijer SL, Weijer K, et al. Human type 1 innate lymphoid cells accumulate in inflamed mucosal tissues. Nat Immunol (2013) 14(3):221–9. doi:10.1038/ni.2534
76. Satoh-Takayama N, Vosshenrich CA, Lesjean-Pottier S, Sawa S, Lochner M, Rattis F, et al. Microbial flora drives interleukin 22 production in intestinal NKp46+ cells that provide innate mucosal immune defense. Immunity (2008) 29(6):958–70. doi:10.1016/j.immuni.2008.11.001
77. Sonnenberg GF, Monticelli LA, Elloso MM, Fouser LA, Artis D. CD4(+) lymphoid tissue-inducer cells promote innate immunity in the gut. Immunity (2011) 34(1):122–34. doi:10.1016/j.immuni.2010.12.009
78. Kamanaka M, Huber S, Zenewicz LA, Gagliani N, Rathinam C, O’Connor W Jr, et al. Memory/effector (CD45RB(lo)) CD4 T cells are controlled directly by IL-10 and cause IL-22-dependent intestinal pathology. J Exp Med (2011) 208(5):1027–40. doi:10.1084/jem.20102149
79. Zenewicz LA, Yancopoulos GD, Valenzuela DM, Murphy AJ, Stevens S, Flavell RA. Innate and adaptive interleukin-22 protects mice from inflammatory bowel disease. Immunity (2008) 29(6):947–57. doi:10.1016/j.immuni.2008.11.003
80. Basu R, O’Quinn DB, Silberger DJ, Schoeb TR, Fouser L, Ouyang W, et al. Th22 cells are an important source of IL-22 for host protection against enteropathogenic bacteria. Immunity (2012) 37(6):1061–75. doi:10.1016/j.immuni.2012.08.024
81. Kryczek I, Lin Y, Nagarsheth N, Peng D, Zhao L, Zhao E, et al. IL-22(+)CD4(+) T cells promote colorectal cancer stemness via STAT3 transcription factor activation and induction of the methyltransferase DOT1L. Immunity (2014) 40(5):772–84. doi:10.1016/j.immuni.2014.03.010
82. Kirchberger S, Royston DJ, Boulard O, Thornton E, Franchini F, Szabady RL, et al. Innate lymphoid cells sustain colon cancer through production of interleukin-22 in a mouse model. J Exp Med (2013) 210(5):917–31. doi:10.1084/jem.20122308
83. Perez C, Albert I, DeFay K, Zachariades N, Gooding L, Kriegler M. A nonsecretable cell surface mutant of tumor necrosis factor (TNF) kills by cell-to-cell contact. Cell (1990) 63(2):251–8. doi:10.1016/0092-8674(90)90158-B
84. Mizoguchi E, Mizoguchi A, Takedatsu H, Cario E, de Jong YP, Ooi CJ, et al. Role of tumor necrosis factor receptor 2 (TNFR2) in colonic epithelial hyperplasia and chronic intestinal inflammation in mice. Gastroenterology (2002) 122(1):134–44. doi:10.1053/gast.2002.30347
85. Janes KA, Gaudet S, Albeck JG, Nielsen UB, Lauffenburger DA, Sorger PK. The response of human epithelial cells to TNF involves an inducible autocrine cascade. Cell (2006) 124(6):1225–39. doi:10.1016/j.cell.2006.01.041
86. Murch SH, Lamkin VA, Savage MO, Walker-Smith JA, MacDonald TT. Serum concentrations of tumour necrosis factor alpha in childhood chronic inflammatory bowel disease. Gut (1991) 32(8):913–7. doi:10.1136/gut.32.8.913
87. Braegger CP, Nicholls S, Murch SH, Stephens S, MacDonald TT. Tumour necrosis factor alpha in stool as a marker of intestinal inflammation. Lancet (1992) 339(8785):89–91. doi:10.1016/0140-6736(92)90999-J
88. Breese EJ, Michie CA, Nicholls SW, Murch SH, Williams CB, Domizio P, et al. Tumor necrosis factor alpha-producing cells in the intestinal mucosa of children with inflammatory bowel disease. Gastroenterology (1994) 106(6):1455–66. doi:10.1016/0016-5085(94)90398-0
89. van Dullemen HM, van Deventer SJ, Hommes DW, Bijl HA, Jansen J, Tytgat GN, et al. Treatment of Crohn’s disease with anti-tumor necrosis factor chimeric monoclonal antibody (cA2). Gastroenterology (1995) 109(1):129–35. doi:10.1016/0016-5085(95)90277-5
90. Popivanova BK, Kitamura K, Wu Y, Kondo T, Kagaya T, Kaneko S, et al. Blocking TNF-alpha in mice reduces colorectal carcinogenesis associated with chronic colitis. J Clin Invest (2008) 118(2):560–70. doi:10.1172/JCI32453
91. Al Obeed OA, Alkhayal KA, Al Sheikh A, Zubaidi AM, Vaali-Mohammed M-A, Boushey B, et al. Increased expression of tumor necrosis factor-alpha is associated with advanced colorectal cancer stages. World J Gastroenterol (2014) 20(48):18390–6. doi:10.3748/wjg.v20.i48.18390
92. Sorrentino D, Nash P, Viladomiu M, Hontecillas R, Bassaganya-Riera J. Stopping anti-TNF agents in patients with Crohn’s disease in remission: is it a feasible long-term strategy? Inflamm Bowel Dis (2014) 20(4):757–66. doi:10.1097/01.MIB.0000442680.47427.bf
93. Roda G, Jharap B, Neeraj N, Colombel JF. Loss of response to anti-TNFs: definition, epidemiology, and management. Clin Transl Gastroenterol (2016) 7:e135. doi:10.1038/ctg.2015.63
94. Hueber W, Sands BE, Lewitzky S, Vandemeulebroecke M, Reinisch W, Higgins PD, et al. Secukinumab, a human anti-IL-17A monoclonal antibody, for moderate to severe Crohn’s disease: unexpected results of a randomised, double-blind placebo-controlled trial. Gut (2012) 61(12):1693–700. doi:10.1136/gutjnl-2011-301668
95. Langley RG, Elewski BE, Lebwohl M, Reich K, Griffiths CE, Papp K, et al. Secukinumab in plaque psoriasis – results of two phase 3 trials. N Engl J Med (2014) 371(4):326–38. doi:10.1056/NEJMoa1314258
96. Mease PJ, McInnes IB, Kirkham B, Kavanaugh A, Rahman P, van der Heijde D, et al. Secukinumab inhibition of interleukin-17a in patients with psoriatic arthritis. N Engl J Med (2015) 373(14):1329–39. doi:10.1056/NEJMoa1412679
97. Feagan BG, Sandborn WJ, Gasink C, Jacobstein D, Lang Y, Friedman JR, et al. Ustekinumab as induction and maintenance therapy for Crohn’s disease. N Engl J Med (2016) 375(20):1946–60. doi:10.1056/NEJMoa1602773
98. Feagan BG, Sandborn WJ, D’Haens G, Panés J, Kaser A, Ferrante M, et al. Induction therapy with the selective interleukin-23 inhibitor risankizumab in patients with moderate-to-severe Crohn’s disease: a randomised, double-blind, placebo-controlled phase 2 study. Lancet (2017) 389(10080):1699–709. doi:10.1016/S0140-6736(17)30570-6
Keywords: Th17 cells, inflammatory bowel diseases, colorectal cancer, enterocytes, cytokines
Citation: Kempski J, Brockmann L, Gagliani N and Huber S (2017) TH17 Cell and Epithelial Cell Crosstalk during Inflammatory Bowel Disease and Carcinogenesis. Front. Immunol. 8:1373. doi: 10.3389/fimmu.2017.01373
Received: 29 May 2017; Accepted: 05 October 2017;
Published: 25 October 2017
Edited by:
Christoph Becker, University of Erlangen-Nuremberg, GermanyReviewed by:
Wim Van Den Broeck, Ghent University, BelgiumCopyright: © 2017 Kempski, Brockmann, Gagliani and Huber. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Samuel Huber, c2h1YmVyQHVrZS5kZQ==
†These authors have contributed equally to this work.
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.