 Gwenoline Borhis
Gwenoline Borhis Maria Trovato
Maria Trovato Nada Chaoul4†
Nada Chaoul4† Hany M. Ibrahim
Hany M. Ibrahim Yolande Richard
Yolande Richard- 1INSERM u1016, Institut Cochin, Paris, France
- 2CNRS UMR 8104, Paris, France
- 3Université Paris-Descartes, Paris, France
- 4Commissariat à l’Energie Atomique, Institut des maladies Emergentes et Thérapies innovantes, Service d’Immuno-Virologie, Fontenay-aux Roses, France
With the goal to design effective HIV vaccines, intensive studies focused on broadly neutralizing antibodies, which arise in a fraction of HIV-infected people. Apart from identifying new vulnerability sites in the viral envelope proteins, these studies have shown that a fraction of these antibodies are produced by self/poly-reactive B-cells. These findings prompted us to revisit the B-cell differentiation and selection process during HIV/SIV infection and to consider B-cells as active players possibly shaping the helper T-cell program within germinal centers (GCs). In this context, we paid a particular attention to B-cell-activating factor (BAFF), a key cytokine in B-cell development and immune response that is overproduced during HIV/SIV infection. As it does in autoimmune diseases, BAFF excess might contribute to the abnormal rescue of self-reactive B-cells at several checkpoints of the B-cell development and impair memory B-cell generation and functions. In this review, we first point out what is known about the functions of BAFF/a proliferation-inducing ligand and their receptors [B-cell maturation, transmembrane activator and CAML interactor (TACI), and BAFF-R], in physiological and pathophysiological settings, in mice and humans. In particular, we highlight recent results on the previously underappreciated regulatory functions of TACI and on the highly regulated production of soluble TACI and BAFF-R that act as decoy receptors. In light of recent data on BAFF, TACI, and BAFF-R, we then revisit the altered phenotypes and functions of B-cell subsets during the acute and chronic phase of HIV/SIV infection. Given the atypical phenotype and reduced functions of memory B-cells in HIV/SIV infection, we particularly discuss the GC reaction, a key checkpoint where self-reactive B-cells are eliminated and pathogen-specific memory B-cells and plasmablasts/cells are generated in physiological settings. Through its capacity to differentially bind and process BAFF-R and TACI on GC B-cells and possibly on follicular helper T-cells, BAFF appears as a key regulator of the physiological GC reaction. Its local excess during HIV/SIV infection could play a key role in B-cell dysregulations.
Introduction
During pathogenic HIV/SIV infection, efficient antibody (Ab) protection hardly develops whereas immunoglobulin overproduction, germinal center (GC) hyperplasia (1), and increased recruitment of follicular helper T-cells (TFH) into GC occur concurrently from the acute phase of infection (2–6). In addition to several reports showing increased proportions of atypical memory B-cells in lymphoid organs and transitional B-cells in blood (7), recent molecular investigations established that a fraction of broadly neutralizing Abs (bNAbs) are produced by self/poly-reactive B-cells (8). In addition to direct B-cell activation by viral envelope proteins, inflammation is thought to play a major role in shaping these changes in B-cell phenotype and in virus-specific Ab responses (9–12). B-cell-activating factor (BAFF)/a proliferation-inducing ligand (APRIL) are instrumental cytokines for B-cell ontogeny and humoral responses in physiological settings (13), while their overproduction is detrimental in numerous autoimmune disorders (14, 15). During HIV/SIV or plasmodium infection, increased BAFF levels occur concurrently with expansion of atypical memory B-cells and inefficient Ab response (16–19). Thus, BAFF was thought to exert detrimental actions on pathogen-specific B-cells, and its overexpression has been associated with HIV/SIV disease progression (20–22). However, BAFF excess favors the expansion of immature-transitional B-cells and promotes self-Abs in mice and in patients with autoimmune diseases (23–25). Through a similar pathway, BAFF might be beneficial in expanding the pool of HIV cross-reactive B-cells, a potential source of bNAbs. Therefore, the role of BAFF excess in generating HIV/SIV-specific memory B-cells and neutralizing Abs needs to be further clarified. In this review, we first summarize what it is known about BAFF/APRIL and their receptors, with a special attention to transmembrane activator and CAML interactor (TACI), which might act as a key regulator of B-cell activation, BAFF-R shedding (26) and possibly self-reactivity. We then highlight data obtained in mice, humans, and macaques with the aim to better appreciate the role of BAFF and its receptors, BAFF-R and TACI, in HIV/SIV progression and in the expansion of HIV/SIV cross-reactive B-cells.
BAFF/APRIL and Their Receptors
The BAFF belonging to the tumor necrosis factor (TNF) superfamily (also called BLys) was first described as a key regulator of B-cell homeostasis and survival in mice and in humans (13). BAFF exerts its effects by binding to three different receptors: B-cell maturation (BCMA) (27, 28), TACI (29), and BAFF-R/BR3 (BLys receptor 3) (30). A highly similar homolog of BAFF (called APRIL) (31) also binds TACI and BCMA but not BAFF-R (32). APRIL only exists as a soluble form cleaved intracellularly, whereas BAFF can be found in both membrane-bound and soluble forms. In myeloid cells, BAFF is expressed on the cell surface as a membrane-bound form (mBAFF) and can then be released as a soluble form after cleavage by furin protease (33–35). Neutrophils directly release BAFF and APRIL as soluble cytokines (36, 37), whereas plasmacytoid dendritic cells (pDC) are unable to cleave mBAFF into its soluble form (19, 38, 39).
Through different expression and affinity for BAFF and APRIL, BAFF-R, TACI, and BCMA finely tune B-cell ontogeny and immune responses with species specificity (30, 40–44). Functional BAFF-R and TACI are expressed in B1 cells (45), and aging APRIL-transgenic mice develop B1 lymphoma (46, 47), whereas BAFF- and BAFF-R-deficient mice have normal proportions of B1 cells (48) (Table 1). This indicates that the TACI–APRIL pair likely plays a dominant role in murine B1 homeostasis. Absent from early transitional B-cells (T1, CD10+CD21lo), BAFF-R expression is acquired by transitional type-2 B-cells (T2, CD10+CD21+), and deficiency in BAFF-R inhibits B-cell ontogeny beyond the T1/T2 transition (49). However, this blockade is not absolute, and small proportions of mature B-cells are still present in BAFF-R-deficient mice and, to a lesser extent, in BAFF-deficient mice that mount residual responses to T-dependent (TD) antigens (50, 51). Consistently, BAFF- or BAFF-R-deficient mice form rudimentary GC in response to TD antigens (52, 53). Absent from naïve and memory B-cells, BCMA is dispensable for the survival of mature B-cells, spleen architecture, and GC development. Response to TD or T-independent (TI) antigens and isotype class switching are normal in BCMA-deficient mice (54–56). However, BCMA is important for long-term plasma cell biology (55, 57, 58) and antigen presentation (59). Upon binding to BCMA, APRIL and, to a lesser extent, BAFF promotes the survival of long-lived plasma cells in bone marrow (55). BCMA therefore constitutes one privileged target for the selective killing of malignant plasma cells, such as multiple myeloma cells (60, 61). Consistent with the recent description of constitutive BCMA shedding from the membrane of plasma cells by a γ-secretase (62), high serum BCMA level correlates with disease status and constitutes a valuable biomarker in multiple myeloma (63). Moreover, TACI expression distinguishes TACIlo from TACIhi myeloma, the latter with a signature of plasma cells, which are more dependent on bone marrow signals (64), likely osteoclast-derived BAFF/APRIL and IL6 (65). Accordingly, TACIhi myelomas are expected to be more responsive to BAFF-related immunotherapies. Based on these data in malignant cells, normal circulating plasmablasts are thought to be TACIlo in contrast to long-lived plasma cells present in bone marrow that would be TACIhi.
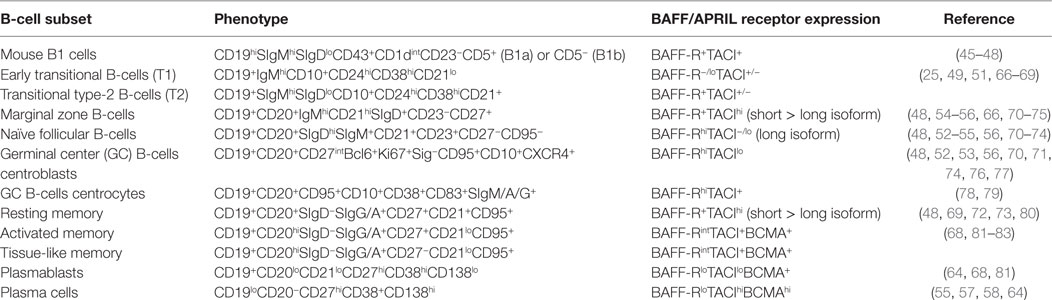
Table 1. Phenotype of B-cell subsets and expression of B-cell-activating factor (BAFF)/a proliferation-inducing ligand (APRIL) receptors.
TACI: A Multifaceted Receptor for BAFF/APRIL in Mice and Humans
Conventional and Regulatory Functions of TACI in Mice
BAFF-R is expressed by most follicular B-cells whereas TACI is absent (or very low) from naïve B-cells but highly present on marginal zone (MZ) and class-switched memory B-cells (48, 72, 73). TACI-deficient mice fail to respond to type-2 TI antigens (TI-2) but retain normal TD response (Table 2). However, they have reduced serum IgM and IgA levels, but normal IgG levels (48). In vitro, Castigli et al. have established that the murine TACI–APRIL pair is mandatory for IgA class switching and plays a dominant role over the BAFF-R–BAFF pair in IgG class switching (56). In another mouse model, TACI deficiency induces hyperplasia, enlarged MZ B-cell pool (66) and lupus-like autoimmune manifestations in aged mice (84). In agreement with TACI controlling exacerbated B-cell activation, knock-in mouse carrying a C76R mutation that impairs TACI-induced NF-κB activation develops splenomegaly with increased proportions of MZ and follicular B-cells (74). So, murine TACI that positively controls response to TI-2 antigens and IgA class switching can also deliver inhibitory signals that dampen abnormal B-cell activation and expansion. In vitro, Figgett et al. recently demonstrated that BAFF binding to TACI selectively limits TI innate response of TLR4-activated MZ B-cells by promoting FAS/FASL-mediated apoptosis (75). This process is thought to prevent inappropriate TI B-cell responses such as the expansion of self-reactive B-cells, and therefore to safeguard peripheral immune tolerance. Thus, membrane TACI controls excessive expansion/response of various mouse B-cell subsets.
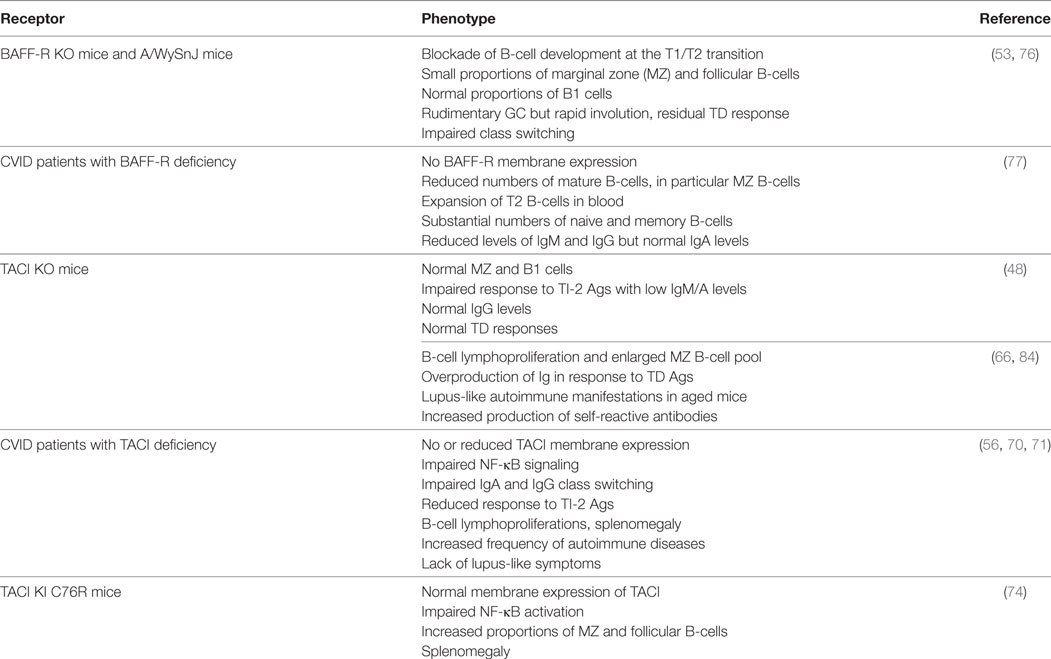
Table 2. Consequences of deficiency in BAFF-R and transmembrane activator and CAML interactor (TACI) in genetically modified mice and common variable immunodeficiency (CVID) patients.
BAFF-R and TACI in Humans: Lessons from Patients with Genetic Immunodeficiency
Spontaneous mutations occurring in individuals or families offer the opportunity to compare the biological importance of key molecules between mice and humans (Table 2). Studies in two patients with common variable immunodeficiency (CVID) carrying a homozygous deletion in BAFF-R gene, that precludes its membrane expression, confirm the key role of BAFF-R in human B-cell development. However, the phenotype of these patients is less severely compromised than that of BAFF-R-deficient mice, with significant numbers of circulating memory B-cells and normal IgA levels, despite B-cell lymphopenia and low levels of circulating IgM and IgG (77).
Similarly, the phenotype of CVID individuals with TACI deficiency differs from that of TACI-deficient mice (70, 71). These individuals combine Ab-deficiency syndrome, B-cell lymphoproliferation, and increased frequency of autoimmune manifestations without symptoms of lupus-like disease. Two homozygous mutations at positions C104R (the human equivalent of murine C76R) and S144X impair class switching to IgA but also to IgG, unlike TACI-deficient mice (71). Whereas TACI was expressed on B-cells from all individuals with heterozygous mutations (including C104R), its signaling was impaired leading to abnormal Ig production in vitro (70). Consistent with data in TACI-deficient mice, individuals with TACI deficiency have a strongly reduced response to TI-2 antigens with recurrent infections and more frequently develop splenomegaly. Thus, human TACI is mandatory for response to TI-2 antigens and IgA/G class switching. Splenomegaly and autoimmune manifestations in these patients clearly indicate that TACI also acts as negative regulator of B-cell expansion/response in humans.
Moreover, two recent studies evidenced the release of soluble TACI and BAFF-R, acting as soluble decoy receptors. Surface TACI is constitutively cleaved by ADAM17 from human and murine B-cells, producing a homotrimer acting as a soluble decoy receptor for BAFF and, to a lesser extent, for APRIL. Subsequent cleavage of its remaining membrane-bound C-terminal domain by γ − secretase prevents residual NF-κB activation (85). While ADAM17 cleaves BAFF-R from dark zone GC B-cells (centroblasts), BAFF-R cleavage by ADAM10, which depends on BAFF binding and TACI expression, occurs in memory and MZ B-cells as well as in light zone GC B-cells (centrocytes) (26). By amplifying BAFF-R cleavage from centrocytes, BAFF excess might impair B-cell selection and high affinity Ab maturation. Taken together, these results highlight a previously unexpected role for TACI as a key modulator of BAFF-mediated responses.
A supplementary level of complexity was introduced by the identification of two isoforms of human TACI produced by alternative splicing of the unique encoding gene. One isoform with two extracellular ligand-binding domains resembles murine TACI whereas the second isoform, which contains only one binding domain, was referred to as TACI-short by authors (80). In vitro studies have established that TACI-short binds APRIL and BAFF with higher affinity than the other isoform and that its triggering by either ligand leads to a more potent activation of canonical NF-κB pathway (86) and plasma cell differentiation (80). Consistent with previous data (87), intense NF-κB activation downstream TACI-short correlates with enhanced recruitment of MyD88. In particular, messengers of both TACI isoforms were found in isolated resting memory (RM, CD21+CD27+) and MZ B-cells, with TACI-short mRNA being present in higher amounts (80). It is therefore possible that the response to BAFF/APRIL is finely modulated through binding to TACI trimers containing various ratio of each isoform. Mechanisms favoring preferential TACI-short expression in vivo remain to be identified but, in vitro, TLR9 ligands strongly upregulate it in CD27+ B-cells. To what extent each TACI isoform contributes to the biology of memory B-cells and long-lived plasma cells remains to be studied. Since survival of memory B-cells is less dependent on BAFF in vivo than that of transitional and naïve B-cells, TACI-short expression might confer them an exceptional responsiveness to limited BAFF amounts. Whether TACI-short is released and whether it differently modulates BAFF-mediated BAFF-R cleavage on RM B-cells should be examined.
Evidence for Soluble and Membrane BAFF Overexpression During HIV/SIV Infection
Elevated circulating levels of BAFF and/or APRIL are associated with autoimmune diseases, chronic inflammation (14, 88), or occur after CD20 B-cell depleting therapy (89, 90). Because chronic inflammation and hypergammaglobulinemia are hallmarks of chronic HIV-1 infection, serum BAFF levels were first measured in chronically HIV-infected individuals (91). In this pioneer report, authors observed increased BAFF levels in most individuals, correlating with levels of self-Abs only in individuals with more than 200 CD4 T-cells per microliters. In these individuals, classical monocytes (CD14hi) overexpressing mBAFF were identified as a major source of soluble BAFF. Extending these first results, Fontaine et al. have evidenced increased levels of serum BAFF in HIV-infected people, with a sustained increase from the acute phase of infection in rapid and normal progressors (16). In these HIV-infected individuals, mBAFF expression was preferentially upregulated in blood myeloid dendritic cells (DC) (defined as HLA-DR+CD11c+) and their precursors (HLA-DR+CD14+CD11c+) (16). In a cohort of untreated individuals with primary HIV infection, we found that circulating BAFF levels were consistently increased at diagnosis (20–45 days after infection) but rapidly decreased toward baseline levels by 2–3 months of infection (1 month of follow-up) (19). Whereas mBAFF was mainly present in intermediate monocytes (CD14+CD16+) of healthy individuals, its expression was preferentially enhanced in CD1c+ DC and non-classical (CD14loCD16hi) monocytes in individuals with primary HIV infection (19). A similar trend was observed in BDCA-3+ DC and intermediate monocytes but did not reach significance. In vitro, the virus itself can directly drive mBAFF expression and its subsequent release in monocytes as well as in monocytes-derived DC and macrophages. In vivo, type I and II IFN could also contribute to BAFF increase. This virus-mediated effect is essentially independent on replication since it was observed with AT2-inactivated virus. Extending our results, Gomez et al. recently showed that HIV-1 does not induce BAFF expression in monocyte-derived macrophages displaying a M1 phenotype (92). Unexpectedly, our findings showed that mBAFF was expressed by a majority of pDC in healthy individuals, an expression that strongly decreased in patients with primary HIV infection. However, this loss was not due to BAFF release since pDC are unable to cleave mBAFF (19, 38, 39). Preferential cognate interactions of pDC with MZ and memory B-cells (93, 94) might relay on mBAFF binding to TACI-short, highly expressed by these B-cell subtypes (80).
In acutely SIV-infected macaques, we consistently observed a transient increase in BAFF plasma levels by 2 weeks of infection. BAFF levels correlate with total IgG levels, plasma viral loads and inversely with CD4 T-cell counts (21). However, steady BAFF overexpression was observed in spleen and intestinal mucosa (duodenum and terminal ileum) until 1 month post-infection. This BAFF signal was more intense in the spleen MZ, follicular mantle zone and within GC (21) but was also present all along the ileum villi in macrophages and in intraepithelial cells, likely CD8+ (Figure 1). According to previous data in humans, these latter cells might correspond to BAFF-expressing type-3 innate-lymphoid cells (ILC3) (95, 96). Retrospective measurement of blood BAFF levels in two groups of SIV-infected macaques treated or not by a 2-week antiretroviral therapy initiated at day 7 post-infection (97) showed a significant reduction of BAFF levels in treated animals at days 12 and 15 (42 and 56% reduction, respectively) (Figure 2). In these animals, the plasma viral load was concurrently reduced by 103-fold and the proportions of memory B-cells increased in blood and spleen. Median value of plasma IgM returned to pre-infection level and SIV-specific Abs were no longer detectable after treatment (97). Thus, early initiation of antiretroviral therapy dampens BAFF increase but inhibits early virus-specific Ab production. In agreement with our data, Poudrier et al. recently showed a transient BAFF increase during the first week of SIV infection and a progressive return to baseline values after 2 months before re-increasing by 3 months post-infection (early chronic infection) in progressor animals only. These authors established that granulocytes massively contribute to BAFF production during acute and chronic phases of infection (22). This observation fits well with increased proportions of activated neutrophils in the blood of chronically HIV-infected people (98, 99). Therefore, elevated BAFF levels might constitute a good predictor of disease progression at the early chronic phase (22). This conclusion is consistent with data of comparative transcriptomic analysis showing that upregulation of TNFSF13B (encoding BAFF) messenger is associated with disease progression during pathogenic HIV/SIV infections (20).
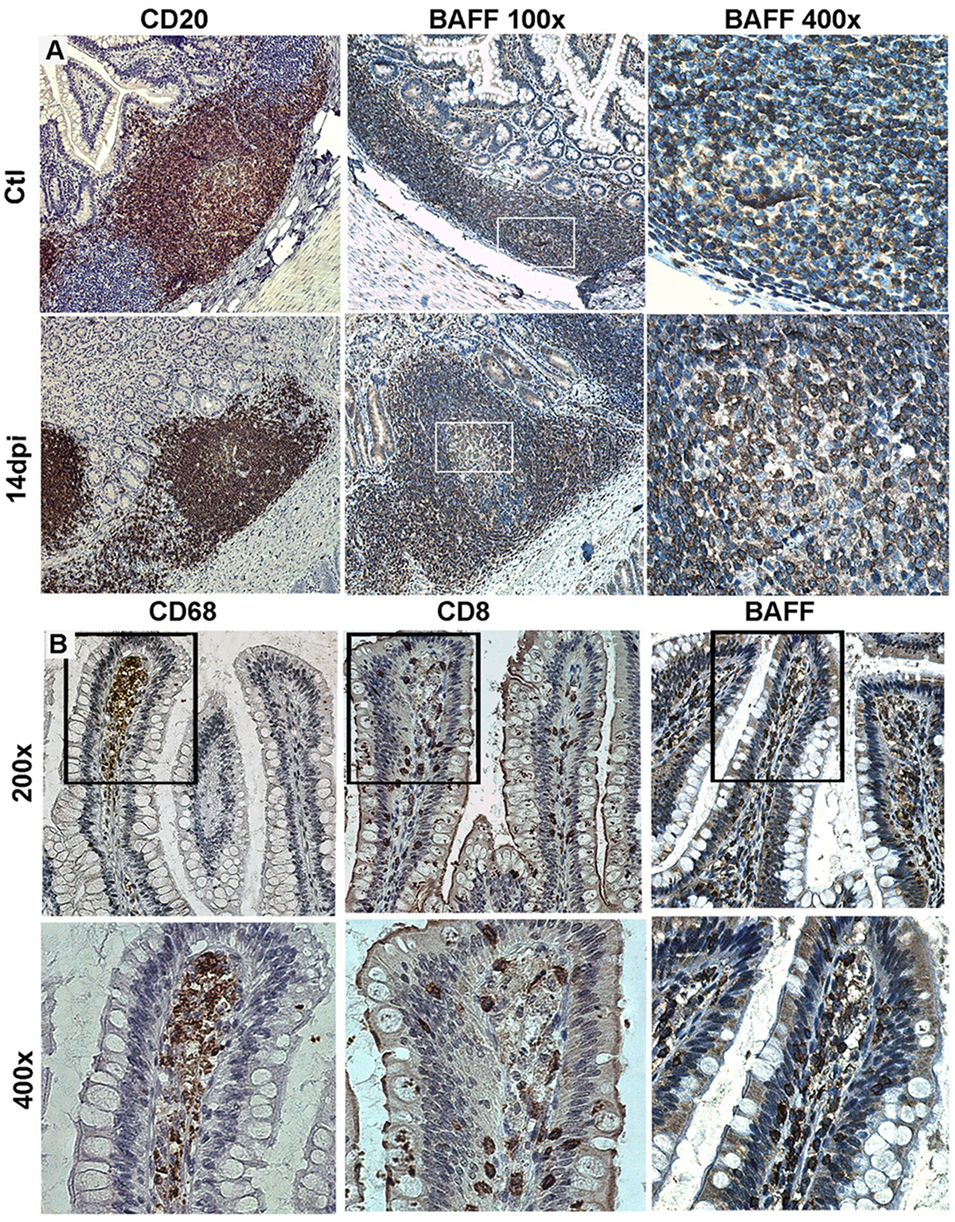
Figure 1. Tissue expression of B-cell-activating factor (BAFF) in SIV-infected macaques. (A) Terminal ileum sections from control macaques (upper panel) and macaques infected for 14 days (lower panel) were stained with anti-CD20 (B-cells, left panels) or anti-BAFF (clone Buffy 2, middle and right panels) antibodies (Abs). Original magnification: 200× for CD20, 100× and 400× for Buffy 2. (B) Terminal ileum sections with clear villi present were stained with CD68 (macrophages), CD8 (CD8+ and intraepithelial T-cells), and Buffy 2 (BAFF expression) Abs, respectively (original magnification 200×). Inserts from upper panels are shown in the lower panels (original magnification 400×). Reproduction authorized by SpringerNature.
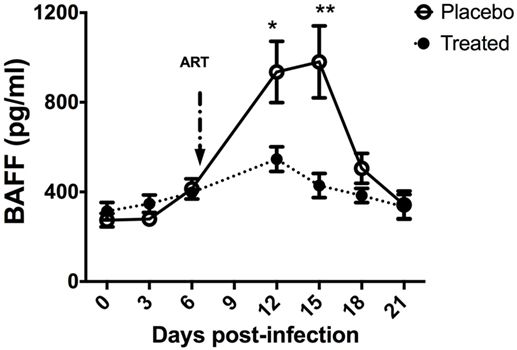
Figure 2. B-cell-activating factor (BAFF) levels in SIV-infected macaques upon antiretroviral therapy. Two groups of five macaques infected for 7 days by SIVmac 251 (50AID50) were treated or not (placebo) with antiretroviral therapy for 2 weeks and euthanized at day 21 post-infection (pi). Plasma BAFF concentration was determined using the BAFF Quantikine ELISA kit (R&D systems) in samples collected before infection and every 3 days pi. At each time point, mean value ± SEM is indicated for each group. At each time point, significant differences between treated and placebo groups were calculated by a Wilcoxon sign-ranked test (two-tailed, unpaired, and non-parametric t-test). The p values *(p < 0.05) and **(p < 0.01) were considered as significant.
In conclusion, non-classical monocytes and CD11c+ DC strongly contribute to elevated levels of soluble BAFF during HIV/SIV infection (16, 19, 91), but macrophages, granulocytes/neutrophils, epithelial cells, and ILC3 can also contribute to its local production in spleen and mucosa (21, 22, 100). Membrane BAFF-expressing pDC, which preferentially migrate into the vaginal mucosa and into the large intestine during pathogenic SIV infection (101, 102), might support TI B-cell response through cognate interaction with infiltrating B-cells. Through its binding to cell-type specific receptors, the virus can directly induce membrane/soluble BAFF overexpression but also the release of type I and II IFN that are keys inducers of BAFF expression. In our studies, IL1β, IL6, and TNFα are unable to modulate membrane and/or soluble BAFF overexpression by myeloid cells or pDC (19).
Preventing progression toward the chronic phase of virus infection generally requires the rapid production of potent neutralizing Abs that is rarely observed during acute HIV/SIV infection. That prompted us to interrogate the pathways of Ab production and the development of plasmablasts/cells as well as the nature of virus responsive B-cells.
Self-Reactive B-Cells: The Last Chance for Neutralizing HIV Abs?
Whereas GC hyperplasia is one the first sign of ongoing B-cell response described in HIV-infected patients (1), the virus-specific Ab production is delayed and globally inefficient in containing virus replication and in preventing the establishment of viral reservoirs (103). Even when present, most virus-specific Abs have limited and transient capacities to neutralize the virus. Whereas pioneers studies have evidenced that inactivated purified SIV or fixed SIV-infected cells can elicit protective virus-specific Abs during infection with autologous virus (104, 105), most candidate vaccines subsequently fail to clear HIV (8). Potent bNAbs are nevertheless produced by a minority of HIV-infected individuals, generally at low titers and only after years of infection. Analyses of bNAbs that target HIV-1 envelope trimer have considerably extended our knowledge on envelope epitopes susceptible to neutralization and therefore identified new targets for vaccine trials (106). The vulnerability sites include: the membrane-proximal external region (MPER) of gp41, the CD4-binding site of gp120, an exclusively glycan epitope on the outer domain of gp120, an extended region including residues from both gp120 and gp41 between the MPER and gp120 protomers, a gp120 V2-glycan site at the apex of the envelope trimer and a gp120 V3-glycan site centered at Asn332 and the fusion peptide of HIV-1 (106, 107). Whereas passive infusion of bNAbs in humans has limited impact on HIV-1 viral loads and disease progression, two recently identified bNAbs directed against the CD4-binding site (VRC01 and 3BNC117) have significant antiviral effects (108–110). Unexpected results have shown that a subset of bNAbs concurrently recognizes nuclear or cytoplasmic human (self) antigens or proteins of commensal pathogens. These self/poly-reactive Abs preferentially recognize the CD4-binding site and the MPER region (111–114). Rare poly-reactive Abs recognizing the gp120-V3 loop have been also cloned from memory B-cells of HIV-infected patients (115, 116). bNAbs have a high degree of somatic mutation, deletions and insertions and/or elongated highly hydrophobic heavy chain complementary-determining region 3 with development of breadth correlating with acquisition of self/poly-reactivity (8, 107). Whereas these features predict negative selection, current studies reveal that ancestors of B-cells producing bNAbs are frequently self-reactive (117). A clever study recently demonstrated that breaching tolerance in mice favors the generation of cross-reactive HIV-1 self-Abs (114). Early non-neutralizing Abs directed against HIV-1 gp41 subunit are also poly-reactive (118, 119) and derive from commensal bacteria-specific memory B-cells generated in terminal ileum before infection. These B-cells acquire cross-reactivity with HIV gp41 upon T-cell driven affinity maturation, which involves GC reaction in constitutive follicles (Peyer patches or mesenteric lymph nodes) or in virus-induced isolated follicles (118, 120). Therefore, HIV might preferentially interact with self/poly-reactive B-cells in different tissues.
In physiological settings, self-reactive B-cells are eliminated at the following three major checkpoints: (i) in the bone marrow before the surface IgM-positive immature B-cell stage; (ii) in spleen MZ (or peri-follicular zone in humans) when new emigrants mature into follicular or MZ B-cells, and finally (iii) within GC during Ab affinity maturation (121). In bone marrow, 50–75% of early B-cells are self-reactive, most of which are eliminated by central tolerance mechanisms before they reach the periphery. Despite this elimination based on “tonic” BCR signaling, a substantial proportion of self/poly-reactive B-cells are still present in the blood of healthy individuals and more frequent among immature and MZ B-cells than among naïve B-cells (122, 123). Given its capacity to support the survival of transitional and MZ B-cells through BAFF-R, BAFF overproduction might abnormally rescue self-reactive B-cells as shown in murine models (124, 125) or in patients with systemic lupus erythematosus (126). If BAFF-R can directly deliver survival signal to transitional B-cells, BAFF-R signaling also interferes with BCR signaling in mice and might thus abnormally rescue early B-cells expressing self-reactive BCR (127–129). More recently, self-reactive transitional B-cells (T1 and T2) abnormally expressing TACI have been identified in BAFF transgenic mice as a consequence of BAFF excess. These TACIhi transitional B-cells co-express AID (activation-induced cytidine deaminase), an enzyme mandatory for somatic hypermutation and isotype class switching, and T-bet, a transcriptional factor associated with IFNγ production and IgG class switching. Accordingly, binding of these TACI+ transitional B-cells by self-antigens promotes AID-mediated hyper-somatic mutations that spontaneously produce self-reactive IgG, ex vivo (25). Although less numerous, TACI+ transitional B-cells are present in wild-type mice with physiological BAFF settings. Increased proportions of T1-like (CD10+CD21lo) B-cells related to disrupted homeostasis have been reported in lymphopenic HIV-infected people with more advanced disease (67–69). Unfortunately, neither circulating BAFF level nor proportions of self-reactive B-cells, potentially HIV cross-reactive, have been estimated at the time of these studies. Whereas TACI+ transitional B-cells might also contribute to hypermutated Ab production during HIV infection, only rare transitional B-cells were found to express T-bet in healthy and chronically HIV-infected individuals (130). However, this might occur in highly lymphopenic HIV-infected individuals with more advanced disease.
Marginal zone B-cells that express diverse IgVH genes more frequently used by self/poly-reactive Abs including by bNAbs directed against CD4bs (131), might be a “natural reservoir” for HIV cross-reactive B-cells. As mentioned earlier, human MZ B-cells highly express TACI, in particular TACI-short, and are in close contact with different BAFF/APRIL-producing cells such as macrophages, DC, neutrophils, or ILC3 in the splenic peri-follicular zone (132, 133). Thus MZ B-cells likely produce a first pool of virus-specific Abs. Indeed, we showed that the frequency of spleen MZ B-cells decreased soon after the peak of plasma viral load whereas plasmablasts/cells, mainly expressing IgG or IgM, were more numerous in the MZ 1 month post-infection in SIV-infected macaques (97). Similarly, Fontaine et al. identified a circulating population with mixed features of transitional and MZ B-cells, thought to rapidly mature into MZ B-cells upon abnormal BAFF release by myeloid cells in viremic HIV-infected people (16). Together, these data suggest that HIV induces an early differentiation of MZ B-cells into plasmablasts/cells followed by a transient lymphopenia, which tends to be compensated by accelerated repopulation of the MZ B-cell pool in patients with higher levels of replication and/or inflammation (including high BAFF levels). Studying the expansion of early self-reactive B-cells, potentially expressing T-bet, in concert with BAFF levels during pathogenic SIV/HIV infection might be valuable. Whether this repopulation favors expansion of HIV/SIV cross-reactive B-cells or their deletion remains to be studied.
BAFF, B-Cells, and TFH in GCs: From Physiological Settings to HIV/SIV Infection
Memory B-cells and long-lived plasmablasts/cells are generated within the GC through a complex process including several cycles of somatic mutations/selection as elegantly described elsewhere (134, 135). Through somatic hypermutations of VH genes, an integrated process mandatory to Ab affinity maturation, the GC reaction constitutes an important stage where self-reactive B-cells are physiologically generated. Such self-reactive B-cells escaping peripheral tolerance and maturing into circulating memory IgG+ B-cells have been associated with autoimmunity (136) but might alternatively contribute to production of bNAbs (7). This directly questions the function of GC reaction with the generation of effectors B-cells (memory B-cells and long-lived plasmablasts/cells) in the context of chronic inflammation, where BAFF (and APRIL) can be overproduced.
Residual development of GC and efficient affinity maturation of Abs in response to TD antigen occur in BAFF or BAFF-R-deficient mice (50, 52, 53). However, GC more rapidly involute in these mice with reduced numbers of proliferating GC B-cells (centroblasts), impaired network of follicular dendritic cells (FDC) and reduced trapping of immune complexes (76). By contrast, BAFF overexpression in GC increases autoimmunity by reducing the competition between B-cell clones for T-cell help and survival signals, at least in mice (124, 137). Fibroblastic reticular cells throughout the body and FDC in GC are the main sources of BAFF in homeostatic settings (138) but also of CXCL13, which attracts B-cells to build B-cell follicles (139). During a TD response, TFH constitute not only the major source of BAFF, mandatory for the B-cell survival and the selection of high affinity B-cell clones (79) but also of CXCL13 as shown in vaccinated mice and primates (140). Consistent with ongoing TD response in HIV-infected patients, high blood CXCL13 levels have been reported with concomitant decrease of CXCR5 expression by circulating B-cells (141). Whether this decrease is due to B-cell activation or to an enhanced BAFF-mediated internalization of CXCR5, it likely perturbs the recruitment of B-cells into follicles. Given that BAFF enhances the CXCL13-mediated chemotactic response of CD27+ human B-cells, in vitro (142), it could potentiate the entry of recently antigen-activated B-cells (GC founders) or of memory B-cells into the follicle during a first or second exposure to antigen/pathogen, respectively. Within GC, BAFF overproduction might either increase the CXCL13-mediated response of B-clones in the light zone or accelerate the internalization of CXCR5 in centrocytes (light zone B-cells), favoring their rapid return to the dark zone. In both case, the asymmetric recycling of B-cell clones between dark and light zones and the selection process would be impaired leading to an abnormal pattern of mutation/selection of B-cell clones required for affinity maturation (143, 144). Alternatively, BAFF excess in GC might enhance BAFF-mediated BAFF-R cleavage on TACI+ GC B-cells (centrocytes). Decreased BAFF-R expression might consequently abolish BAFF effect on the CXCL13 chemotactic response of centrocytes or decrease the survival of high-affinity B-cell clones (26). At which step HIV cross-reactive B-cells clones appear and why they are not eliminated as self-reactive B-cells in the context of high amount of viral antigens is far from being clear. Vulnerability sites frequently buried in the envelope structure or masked by glycans are probably weakly accessible. This might favor their ignorance during the GC selection process.
In addition to its action on GC B-cells, two independent studies implied TACI in TFH expansion (Figure 3). In the first study, Ou et al. showed that BAFF preferentially binds BAFF-R and upregulates ICOSL expression by GC B-cells in physiological settings. When it is locally overproduced, BAFF also binds to TACI on GC B-cells leading to down modulation of BAFF-R-mediated ICOSL expression and subsequently TFH expansion (78). According to recent data on BAFF-R processing (26), one hypothesis could be that BAFF excess leads to TACI-mediated BAFF-R cleavage, which reduces BAFF-R signaling and thus down-modulates ICOSL expression. In a second study, IL21 produced by TFH, besides supporting the survival of both TFH and GC B-cells through IL21R, also diminishes TACI expression thus preventing premature loss of TFH (79). As TFH concurrently produce BAFF and IL21, a delicate balance is thought to control efficient GC reaction. The existence of two human TACI isoforms could further complicate our understanding of the role of BAFF and its receptors, BAFF-R and TACI, in GC reaction.
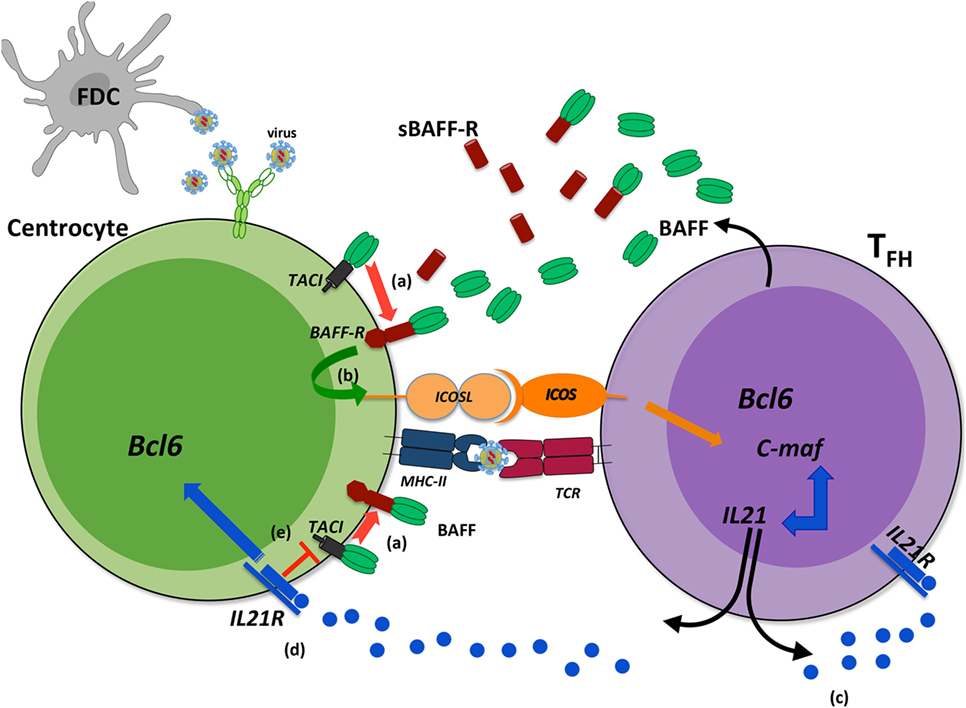
Figure 3. Transmembrane activator and CAML interactor (TACI) as a key regulator of B-cell-activating factor (BAFF)-dependent BAFF-R cleavage in germinal center (GC). During a T-dependent response, follicular helper T-cells (TFH) produce BAFF that can bind either to BAFF-R or to TACI. When BAFF is locally released in excess, its binding to BAFF-R can induce the cleavage of BAFF-R in a TACI-dependent manner from the surface of centrocytes (a). Reduced BAFF-R signaling leads to decreased ICOSL expression on B-cells (b) and therefore dampens ICOS signal, mandatory for TFH maintaining and IL21 production. This might constitute a physiological regulatory mechanism, exacerbated when high amounts of antigens are maintained within GC. IL21 is a key cytokine for TFH that ensures their survival (c) and that of light zone GC B-cells (d). In addition, IL21 decreases TACI expression that might prevent early TACI-dependent BAFF-R cleavage (e). Such regulatory roles would imply that IL21 and BAFF are produced sequentially during the GC reaction with possible consequences on recycling and differentiation of GC B-cells.
Impaired helper functions of TFH at the chronic phase of HIV/SIV infection (2, 6, 145) likely contribute to inefficient B-cell response to HIV/SIV. However, early functional TFH are present at elevated frequencies in nodal GC from the acute phase of HIV infection and their presence correlates with the breadth of bNAbs at the chronic phase (146). Thus, generation of bNAbs is dependent on the preservation of TFH functions, likely impaired in CXCR3+ TFH (147). As recently shown, human TFH express BAFF-R and release more IFNγ after culture with BAFF (148), thus BAFF excess might contribute to TFH1-expansion during HIV/SIV infection. Being produced by FDC and TFH in GC, BAFF likely exerts a physiological role on TFH, during response to TD natural or vaccine antigens. In conclusion, the overexpression of BAFF might impair GC reaction and even modulate TFH functions.
Memory B-Cells: The Weak Link in HIV/SIV Infection
It is now well established that chronically HIV-infected patients have an impaired memory B-cell compartment with lower frequency of HIV-specific and vaccine-specific memory B-cells as well as reduced anti-vaccine Abs (149–151). In addition to lower proportions of memory B-cells, viremic HIV-infected individuals also exhibited increased proportions of CD21lo mature B-cells (68, 81). This subset highly expressed BCMA and TACI but had decreased BAFF-R expression and BAFF binding. Based on this phenotype profile and on the concomitant increase in CD27, CD38, and CXCR3 expression, these CD21lo B-cells were first considered as circulating plasmablasts, prone to apoptosis and Ab production (67, 81) and expanded as a consequence of HIV-induced hyperactivation. After the identification of a subset of CD20hiCD21lo tissue memory cells in human tonsils exhibiting signs of exhaustion (82), the classification of this CD21lo population in HIV-infected patients has been revised. In addition to plasmablasts, the CD21lo B-cell subset comprised CD27+CD21lo and CD27loCD21lo cells often referred to as activated memory (ActM) and tissue-like memory (TLM) B-cells, respectively. These subsets differ from conventional RM B-cells by their expression of activation, inhibitory and/or apoptotic markers (69). In healthy donors, RM B-cells constitute the predominant fraction of blood memory B-cells with low percentages of CD21lo memory B-cells (152). By contrast, ActM and TLM are overrepresented in blood of chronically HIV-infected patients (153) and in rapidly progressing SIV-infected macaques (154). In contrast to influenza or tetanus-specific Abs enriched in RM B-cells, HIV-specific Abs are enriched in TLM B-cells in untreated individuals (155, 156). More recently, Muema et al. reported increased proportions of ActM, TLM, and plasmablasts but decreased proportions of naïve B-cells in vertically HIV-infected children in a viral-load-dependent manner (83). In agreement with other studies in children, lower IgG levels and proportions of switched memory B-cells against childhood vaccines were observed (150, 157, 158). In these HIV-infected children, circulating BAFF levels were elevated whereas BAFF-R and TACI expression were respectively decreased and increased in most B-cell subsets. B-cell interaction with viral proteins that can induce BCR- or TLR-mediated B-cell activation (9, 10, 159) might also increase TACI expression, possibly stabilized at the membrane by BAFF binding as shown in mice (79). By contrast, decreased BAFF-R expression might be due to potent receptor internalization in the presence of high BAFF levels as suggested during malaria infection (17, 160) or to enhanced BAFF-mediated BAFF-R processing (26).
It is not clear whether increase in TACI expression has any influence on ActM or TLM functions or survival, in vivo. Survival of human and simian RM B-cells (BAFF-R+ TACIhi) is less dependent on BAFF than that of naïve and transitional B-cells in physiological settings (161, 162). Moreover, BAFF levels correlate with proportions of MZ and RM B-cells, but not of CD21lo memory B-cells in HIV-infected children (83). By contrast, BAFF levels and proportions of CD21lo memory B-cells were concomitantly increased in individuals infected by Plasmodium (17, 160), an infection setting where the frequencies of TLM-like B-cells are increased (18, 163).
Similar to TLR9 ligands that elicit responses in TLM-like B-cells of malaria-exposed people (18, 164), BAFF/APRIL as TACI ligands might deliver differentiation signals to these B-cells through TACI and its downstream TLR-like signaling pathways (87) Whereas overrepresentation of CD21lo memory B-cells is consistently associated with chronic inflammation, the mechanisms leading to this phenotype are largely unknown. Studies in mice and more recently in HIV-infected people showing T-bet expression by CD21lo B-cells suggest simultaneous actions of pathogen-derived nucleic acids, through TLR9/7, and Th1-cytokines (IFNγ) (130, 165–167). According to high TACI expression in HIV-infected children, BAFF overexpression might directly or indirectly (for example, by upregulating IFNγ production by NK or Th1-cells) contribute to the generation or survival of these CD21lo memory B-cells and thus Ab-mediated protection against HIV-1.
Conclusion
B-cell-activating factor and its receptors (BAFF-R, TACI, and BCMA) are key actors for the B-cell survival and the immune responses of mature B-cells. Whereas BAFF-R is more widely expressed throughout the B-cell differentiation, TACI now appears as a key regulator of various BAFF-mediated responses. Indeed TACI is spontaneously released upon activation and orchestrates the cleavage of BAFF-R-BAFF complexes. This might have a major impact on memory and MZ B-cells that are TACIhi. Perturbations of these regulatory mechanisms likely impair the GC reaction: GC B-cell selection/survival or recycling between dark and light zones as well as the generation of appropriate effector B-cells during TD responses. Soluble but also membrane BAFF overexpression by key DC subsets during HIV/SIV infection might subsequently initiate the unexpected expansion of HIV cross-reactive B-cell clones and atypical memory B-cells. In this review, we pointed out previous data arguing for the involvement of BAFF in HIV-mediated B-cell dysfunctions and discussed more recent data on BAFF and TACI in physiological settings. Accordingly, we suggest BAFF-mediated mechanisms that could modulate B-cell response during pathogenic HIV/SIV infection. Our research around BAFF is part of a more global approach that aims to identify B-cell subset(s), which could constitute a reservoir of HIV cross-reactive B-cells, and to understand how to promote their expansion and/or prevent their elimination. This information is likely a prerequisite for the development of next-generation HIV vaccines.
Author Contributions
All authors contribute to the writing of this review.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by grants to YR from the “Agence Nationale de Recherches sur le SIDA et les Hépatites Virales” (ANRS). GB was supported by a postdoctoral fellowship from ANRS. HMI was a visiting researcher supported by a fellowship from the French Institute in Egypt. MT is supported by a postdoctoral fellowship from ANRS. NC was supported by doctoral fellowships from the Lebanese University St Joseph and the Fp6 Network of Excellence Europrise.
References
1. Amadori A, Chieco-Bianchi L. B-cell activation and HIV-1 infection: deeds and misdeeds. Immunol Today (1990) 11(10):374–9. doi:10.1016/0167-5699(90)90144-X
2. Lindqvist M, van Lunzen J, Soghoian DZ, Kuhl BD, Ranasinghe S, Kranias G, et al. Expansion of HIV-specific T follicular helper cells in chronic HIV infection. J Clin Invest (2012) 122(9):3271–80. doi:10.1172/JCI64314
3. Petrovas C, Yamamoto T, Gerner MY, Boswell KL, Wloka K, Smith EC, et al. CD4 T follicular helper cell dynamics during SIV infection. J Clin Invest (2012) 122(9):3281–94. doi:10.1172/JCI63039
4. Perreau M, Savoye AL, De Crignis E, Corpataux JM, Cubas R, Haddad EK, et al. Follicular helper T cells serve as the major CD4 T cell compartment for HIV-1 infection, replication, and production. J Exp Med (2013) 210(1):143–56. doi:10.1084/jem.20121932
5. Hong JJ, Amancha PK, Rogers KA, Courtney CL, Havenar-Daughton C, Crotty S, et al. Early lymphoid responses and germinal center formation correlate with lower viral load set points and better prognosis of simian immunodeficiency virus infection. J Immunol (2014) 193(2):797–806. doi:10.4049/jimmunol.1400749
6. Thornhill JP, Fidler S, Klenerman P, Frater J, Phetsouphanh C. The role of CD4+ T follicular helper cells in HIV infection: from the Germinal center to the periphery. Front Immunol (2017) 8:46. doi:10.3389/fimmu.2017.00046
7. Moir S, Fauci AS. B-cell responses to HIV infection. Immunol Rev (2017) 275(1):33–48. doi:10.1111/imr.12502
8. Borrow P, Moody MA. Immunologic characteristics of HIV-infected individuals who make broadly neutralizing antibodies. Immunol Rev (2017) 275(1):62–78. doi:10.1111/imr.12504
9. Badr G, Borhis G, Treton D, Moog C, Garraud O, Richard Y. HIV type 1 glycoprotein 120 inhibits human B cell chemotaxis to CXC chemokine ligand (CXCL) 12, CC chemokine ligand (CCL)20, and CCL21. J Immunol (2005) 175(1):302–10. doi:10.4049/jimmunol.175.1.302
10. He B, Qiao X, Klasse PJ, Chiu A, Chadburn A, Knowles DM, et al. HIV-1 envelope triggers polyclonal Ig class switch recombination through a CD40-independent mechanism involving BAFF and C-type lectin receptors. J Immunol (2006) 176(7):3931–41. doi:10.4049/jimmunol.176.7.3931
11. Ruffin N, Thang PH, Rethi B, Nilsson A, Chiodi F. The impact of inflammation and immune activation on B cell differentiation during HIV-1 infection. Front Immunol (2011) 2:90. doi:10.3389/fimmu.2011.00090
12. Jelicic K, Cimbro R, Nawaz F, Huang da W, Zheng X, Yang J, et al. The HIV-1 envelope protein gp120 impairs B cell proliferation by inducing TGF-beta1 production and FcRL4 expression. Nat Immunol (2013) 14(12):1256–65. doi:10.1038/ni.2746
13. Mackay F, Schneider P. Cracking the BAFF code. Nat Rev Immunol (2009) 9(7):491–502. doi:10.1038/nri2572
14. Mackay F, Sierro F, Grey ST, Gordon TP. The BAFF/APRIL system: an important player in systemic rheumatic diseases. Curr Dir Autoimmun (2005) 8:243–65. doi:10.1159/000082106
15. Tangye SG, Bryant VL, Cuss AK, Good KL. BAFF, APRIL and human B cell disorders. Semin Immunol (2006) 18(5):305–17. doi:10.1016/j.smim.2006.04.004
16. Fontaine J, Chagnon-Choquet J, Valcke HS, Poudrier J, Roger M, Montreal Primary HIVI, et al. High expression levels of B lymphocyte stimulator (BLyS) by dendritic cells correlate with HIV-related B-cell disease progression in humans. Blood (2011) 117(1):145–55. doi:10.1182/blood-2010-08-301887
17. Scholzen A, Teirlinck AC, Bijker EM, Roestenberg M, Hermsen CC, Hoffman SL, et al. BAFF and BAFF receptor levels correlate with B cell subset activation and redistribution in controlled human malaria infection. J Immunol (2014) 192(8):3719–29. doi:10.4049/jimmunol.1302960
18. Portugal S, Tipton CM, Sohn H, Kone Y, Wang J, Li S, et al. Malaria-associated atypical memory B cells exhibit markedly reduced B cell receptor signaling and effector function. Elife (2015) 4:07218. doi:10.7554/eLife.07218
19. Borhis G, Burelout C, Chaoul N, Smith N, Goujard C, Meyer L, et al. Plasmacytoid dendritic cells and myeloid cells differently contribute to B-cell-activating factor belonging to the tumor necrosis factor superfamily overexpression during primary HIV infection. AIDS (2016) 30(3):365–76. doi:10.1097/QAD.0000000000000965
20. Rotger M, Dalmau J, Rauch A, McLaren P, Bosinger SE, Martinez R, et al. Comparative transcriptomics of extreme phenotypes of human HIV-1 infection and SIV infection in sooty mangabey and rhesus macaque. J Clin Invest (2011) 121(6):2391–400. doi:10.1172/JCI45235
21. Chaoul N, Burelout C, Peruchon S, van Buu BN, Laurent P, Proust A, et al. Default in plasma and intestinal IgA responses during acute infection by simian immunodeficiency virus. Retrovirology (2012) 9:43. doi:10.1186/1742-4690-9-43
22. Poudrier J, Soulas C, Chagnon-Choquet J, Burdo T, Autissier P, Oskar K, et al. High expression levels of BLyS/BAFF by blood dendritic cells and granulocytes are associated with B-cell dysregulation in SIV-infected rhesus macaques. PLoS One (2015) 10(6):e0131513. doi:10.1371/journal.pone.0131513
23. Mackay F, Woodcock SA, Lawton P, Ambrose C, Baetscher M, Schneider P, et al. Mice transgenic for BAFF develop lymphocytic disorders along with autoimmune manifestations. J Exp Med (1999) 190(11):1697–710. doi:10.1084/jem.190.11.1697
24. Groom J, Kalled SL, Cutler AH, Olson C, Woodcock SA, Schneider P, et al. Association of BAFF/BLyS overexpression and altered B cell differentiation with Sjogren’s syndrome. J Clin Invest (2002) 109(1):59–68. doi:10.1172/JCI0214121
25. Jacobs HM, Thouvenel CD, Leach S, Arkatkar T, Metzler G, Scharping NE, et al. Cutting edge: BAFF promotes autoantibody production via TACI-dependent activation of transitional B cells. J Immunol (2016) 196(9):3525–31. doi:10.4049/jimmunol.1600017
26. Smulski CR, Kury P, Seidel LM, Staiger HS, Edinger AK, Willen L, et al. BAFF- and TACI-dependent processing of BAFFR by ADAM proteases regulates the survival of B cells. Cell Rep (2017) 18(9):2189–202. doi:10.1016/j.celrep.2017.02.005
27. Gras MP, Laabi Y, Linares-Cruz G, Blondel MO, Rigaut JP, Brouet JC, et al. BCMAp: an integral membrane protein in the Golgi apparatus of human mature B lymphocytes. Int Immunol (1995) 7(7):1093–106. doi:10.1093/intimm/7.7.1093
28. Shu HB, Johnson H. B cell maturation protein is a receptor for the tumor necrosis factor family member TALL-1. Proc Natl Acad Sci U S A (2000) 97(16):9156–61. doi:10.1073/pnas.160213497
29. Xia XZ, Treanor J, Senaldi G, Khare SD, Boone T, Kelley M, et al. TACI is a TRAF-interacting receptor for TALL-1, a tumor necrosis factor family member involved in B cell regulation. J Exp Med (2000) 192(1):137–43. doi:10.1084/jem.192.1.137
30. Thompson JS, Bixler SA, Qian F, Vora K, Scott ML, Cachero TG, et al. BAFF-R, a newly identified TNF receptor that specifically interacts with BAFF. Science (2001) 293(5537):2108–11. doi:10.1126/science.1061965
31. Kalled SL, Ambrose C, Hsu YM. The biochemistry and biology of BAFF, APRIL and their receptors. Curr Dir Autoimmun (2005) 8:206–42. doi:10.1159/000082105
32. Marsters SA, Yan M, Pitti RM, Haas PE, Dixit VM, Ashkenazi A. Interaction of the TNF homologues BLyS and APRIL with the TNF receptor homologues BCMA and TACI. Curr Biol (2000) 10(13):785–8. doi:10.1016/S0960-9822(00)00566-2
33. Schneider P, MacKay F, Steiner V, Hofmann K, Bodmer JL, Holler N, et al. BAFF, a novel ligand of the tumor necrosis factor family, stimulates B cell growth. J Exp Med (1999) 189(11):1747–56. doi:10.1084/jem.189.11.1747
34. Nardelli B, Belvedere O, Roschke V, Moore PA, Olsen HS, Migone TS, et al. Synthesis and release of B-lymphocyte stimulator from myeloid cells. Blood (2001) 97(1):198–204. doi:10.1182/blood.V97.1.198
35. Litinskiy MB, Nardelli B, Hilbert DM, He B, Schaffer A, Casali P, et al. DCs induce CD40-independent immunoglobulin class switching through BLyS and APRIL. Nat Immunol (2002) 3(9):822–9. doi:10.1038/ni829
36. Scapini P, Nardelli B, Nadali G, Calzetti F, Pizzolo G, Montecucco C, et al. G-CSF-stimulated neutrophils are a prominent source of functional BLyS. J Exp Med (2003) 197(3):297–302. doi:10.1084/jem.20021343
37. Huard B, McKee T, Bosshard C, Durual S, Matthes T, Myit S, et al. APRIL secreted by neutrophils binds to heparan sulfate proteoglycans to create plasma cell niches in human mucosa. J Clin Invest (2008) 118(8):2887–95. doi:10.1172/JCI33760
38. Deal EM, Lahl K, Narvaez CF, Butcher EC, Greenberg HB. Plasmacytoid dendritic cells promote rotavirus-induced human and murine B cell responses. J Clin Invest (2013) 123(6):2464–74. doi:10.1172/JCI60945
39. Gomez AM, Ouellet M, Tremblay MJ. HIV-1-triggered release of type I IFN by plasmacytoid dendritic cells induces BAFF production in monocytes. J Immunol (2015) 194(5):2300–8. doi:10.4049/jimmunol.1402147
40. Gross JA, Johnston J, Mudri S, Enselman R, Dillon SR, Madden K, et al. TACI and BCMA are receptors for a TNF homologue implicated in B-cell autoimmune disease. Nature (2000) 404(6781):995–9. doi:10.1038/35010115
41. Thompson JS, Schneider P, Kalled SL, Wang L, Lefevre EA, Cachero TG, et al. BAFF binds to the tumor necrosis factor receptor-like molecule B cell maturation antigen and is important for maintaining the peripheral B cell population. J Exp Med (2000) 192(1):129–35. doi:10.1084/jem.192.1.129
42. Wu Y, Bressette D, Carrell JA, Kaufman T, Feng P, Taylor K, et al. Tumor necrosis factor (TNF) receptor superfamily member TACI is a high affinity receptor for TNF family members APRIL and BLyS. J Biol Chem (2000) 275(45):35478–85. doi:10.1074/jbc.M005224200
43. Bossen C, Schneider P. BAFF, APRIL and their receptors: structure, function and signaling. Semin Immunol (2006) 18(5):263–75. doi:10.1016/j.smim.2006.04.006
44. Bossen C, Cachero TG, Tardivel A, Ingold K, Willen L, Dobles M, et al. TACI, unlike BAFF-R, is solely activated by oligomeric BAFF and APRIL to support survival of activated B cells and plasmablasts. Blood (2008) 111(3):1004–12. doi:10.1182/blood-2007-09-110874
45. Ng LG, Ng CH, Woehl B, Sutherland AP, Huo J, Xu S, et al. BAFF costimulation of Toll-like receptor-activated B-1 cells. Eur J Immunol (2006) 36(7):1837–46. doi:10.1002/eji.200635956
46. Planelles L, Carvalho-Pinto CE, Hardenberg G, Smaniotto S, Savino W, Gomez-Caro R, et al. APRIL promotes B-1 cell-associated neoplasm. Cancer Cell (2004) 6(4):399–408. doi:10.1016/j.ccr.2004.08.033
47. Dillon SR, Gross JA, Ansell SM, Novak AJ. An APRIL to remember: novel TNF ligands as therapeutic targets. Nat Rev Drug Discov (2006) 5(3):235–46. doi:10.1038/nrd1982
48. von Bulow GU, van Deursen JM, Bram RJ. Regulation of the T-independent humoral response by TACI. Immunity (2001) 14(5):573–82. doi:10.1016/S1074-7613(01)00130-3
49. Lentz VM, Hayes CE, Cancro MP. Bcmd decreases the life span of B-2 but not B-1 cells in A/WySnJ mice. J Immunol (1998) 160(8):3743–7.
50. Yan M, Marsters SA, Grewal IS, Wang H, Ashkenazi A, Dixit VM. Identification of a receptor for BLyS demonstrates a crucial role in humoral immunity. Nat Immunol (2000) 1(1):37–41. doi:10.1038/76889
51. Schiemann B, Gommerman JL, Vora K, Cachero TG, Shulga-Morskaya S, Dobles M, et al. An essential role for BAFF in the normal development of B cells through a BCMA-independent pathway. Science (2001) 293(5537):2111–4. doi:10.1126/science.1061964
52. Vora KA, Wang LC, Rao SP, Liu ZY, Majeau GR, Cutler AH, et al. Cutting edge: germinal centers formed in the absence of B cell-activating factor belonging to the TNF family exhibit impaired maturation and function. J Immunol (2003) 171(2):547–51. doi:10.4049/jimmunol.171.2.547
53. Sasaki Y, Casola S, Kutok JL, Rajewsky K, Schmidt-Supprian M. TNF family member B cell-activating factor (BAFF) receptor-dependent and -independent roles for BAFF in B cell physiology. J Immunol (2004) 173(4):2245–52. doi:10.4049/jimmunol.173.4.2245
54. Xu S, Lam KP. B-cell maturation protein, which binds the tumor necrosis factor family members BAFF and APRIL, is dispensable for humoral immune responses. Mol Cell Biol (2001) 21(12):4067–74. doi:10.1128/MCB.21.12.4067-4074.2001
55. O’Connor BP, Raman VS, Erickson LD, Cook WJ, Weaver LK, Ahonen C, et al. BCMA is essential for the survival of long-lived bone marrow plasma cells. J Exp Med (2004) 199(1):91–8. doi:10.1084/jem.20031330
56. Castigli E, Wilson SA, Scott S, Dedeoglu F, Xu S, Lam KP, et al. TACI and BAFF-R mediate isotype switching in B cells. J Exp Med (2005) 201(1):35–9. doi:10.1084/jem.20032000
57. Avery DT, Kalled SL, Ellyard JI, Ambrose C, Bixler SA, Thien M, et al. BAFF selectively enhances the survival of plasmablasts generated from human memory B cells. J Clin Invest (2003) 112(2):286–97. doi:10.1172/JCI18025
58. Zhang X, Park CS, Yoon SO, Li L, Hsu YM, Ambrose C, et al. BAFF supports human B cell differentiation in the lymphoid follicles through distinct receptors. Int Immunol (2005) 17(6):779–88. doi:10.1093/intimm/dxh259
59. Yang M, Hase H, Legarda-Addison D, Varughese L, Seed B, Ting AT. B cell maturation antigen, the receptor for a proliferation-inducing ligand and B cell-activating factor of the TNF family, induces antigen presentation in B cells. J Immunol (2005) 175(5):2814–24. doi:10.4049/jimmunol.175.5.2814
60. Tai YT, Mayes PA, Acharya C, Zhong MY, Cea M, Cagnetta A, et al. Novel anti-B-cell maturation antigen antibody-drug conjugate (GSK2857916) selectively induces killing of multiple myeloma. Blood (2014) 123(20):3128–38. doi:10.1182/blood-2013-10-535088
61. O’Donnell EK, Raje NS. New monoclonal antibodies on the horizon in multiple myeloma. Ther Adv Hematol (2017) 8(2):41–53. doi:10.1177/2040620716682490
62. Laurent SA, Hoffmann FS, Kuhn PH, Cheng Q, Chu Y, Schmidt-Supprian M, et al. Gamma-Secretase directly sheds the survival receptor BCMA from plasma cells. Nat Commun (2015) 6:7333. doi:10.1038/ncomms8333
63. Sanchez E, Li M, Kitto A, Li J, Wang CS, Kirk DT, et al. Serum B-cell maturation antigen is elevated in multiple myeloma and correlates with disease status and survival. Br J Haematol (2012) 158(6):727–38. doi:10.1111/j.1365-2141.2012.09241.x
64. Moreaux J, Cremer FW, Reme T, Raab M, Mahtouk K, Kaukel P, et al. The level of TACI gene expression in myeloma cells is associated with a signature of microenvironment dependence versus a plasmablastic signature. Blood (2005) 106(3):1021–30. doi:10.1182/blood-2004-11-4512
65. Yaccoby S, Pennisi A, Li X, Dillon SR, Zhan F, Barlogie B, et al. Atacicept (TACI-Ig) inhibits growth of TACI(high) primary myeloma cells in SCID-hu mice and in coculture with osteoclasts. Leukemia (2008) 22(2):406–13. doi:10.1038/sj.leu.2405048
66. Yan M, Wang H, Chan B, Roose-Girma M, Erickson S, Baker T, et al. Activation and accumulation of B cells in TACI-deficient mice. Nat Immunol (2001) 2(7):638–43. doi:10.1038/89790
67. Ho J, Moir S, Malaspina A, Howell ML, Wang W, DiPoto AC, et al. Two overrepresented B cell populations in HIV-infected individuals undergo apoptosis by different mechanisms. Proc Natl Acad Sci U S A (2006) 103(51):19436–41. doi:10.1073/pnas.0609515103
68. Malaspina A, Moir S, Ho J, Wang W, Howell ML, O’Shea MA, et al. Appearance of immature/transitional B cells in HIV-infected individuals with advanced disease: correlation with increased IL-7. Proc Natl Acad Sci U S A (2006) 103(7):2262–7. doi:10.1073/pnas.0511094103
69. Moir S, Fauci AS. Insights into B cells and HIV-specific B-cell responses in HIV-infected individuals. Immunol Rev (2013) 254(1):207–24. doi:10.1111/imr.12067
70. Castigli E, Wilson SA, Garibyan L, Rachid R, Bonilla F, Schneider L, et al. TACI is mutant in common variable immunodeficiency and IgA deficiency. Nat Genet (2005) 37(8):829–34. doi:10.1038/ng1601
71. Salzer U, Chapel HM, Webster AD, Pan-Hammarstrom Q, Schmitt-Graeff A, Schlesier M, et al. Mutations in TNFRSF13B encoding TACI are associated with common variable immunodeficiency in humans. Nat Genet (2005) 37(8):820–8. doi:10.1038/ng1600
72. Darce JR, Arendt BK, Chang SK, Jelinek DF. Divergent effects of BAFF on human memory B cell differentiation into Ig-secreting cells. J Immunol (2007) 178(9):5612–22. doi:10.4049/jimmunol.178.9.5612
73. Ettinger R, Sims GP, Robbins R, Withers D, Fischer RT, Grammer AC, et al. IL-21 and BAFF/BLyS synergize in stimulating plasma cell differentiation from a unique population of human splenic memory B cells. J Immunol (2007) 178(5):2872–82. doi:10.4049/jimmunol.178.5.2872
74. Bacchelli C, Buckland KF, Buckridge S, Salzer U, Schneider P, Thrasher AJ, et al. The C76R transmembrane activator and calcium modulator cyclophilin ligand interactor mutation disrupts antibody production and B-cell homeostasis in heterozygous and homozygous mice. J Allergy Clin Immunol (2011) 127(5):1253.e–9.e. doi:10.1016/j.jaci.2011.02.037
75. Figgett WA, Fairfax K, Vincent FB, Le Page MA, Katik I, Deliyanti D, et al. The TACI receptor regulates T-cell-independent marginal zone B cell responses through innate activation-induced cell death. Immunity (2013) 39(3):573–83. doi:10.1016/j.immuni.2013.05.019
76. Rahman ZS, Rao SP, Kalled SL, Manser T. Normal induction but attenuated progression of germinal center responses in BAFF and BAFF-R signaling-deficient mice. J Exp Med (2003) 198(8):1157–69. doi:10.1084/jem.20030495
77. Warnatz K, Salzer U, Rizzi M, Fischer B, Gutenberger S, Bohm J, et al. B-cell activating factor receptor deficiency is associated with an adult-onset antibody deficiency syndrome in humans. Proc Natl Acad Sci U S A (2009) 106(33):13945–50. doi:10.1073/pnas.0903543106
78. Ou X, Xu S, Lam KP. Deficiency in TNFRSF13B (TACI) expands T-follicular helper and germinal center B cells via increased ICOS-ligand expression but impairs plasma cell survival. Proc Natl Acad Sci U S A (2012) 109(38):15401–6. doi:10.1073/pnas.1200386109
79. Goenka R, Matthews AH, Zhang B, O’Neill PJ, Scholz JL, Migone TS, et al. Local BLyS production by T follicular cells mediates retention of high affinity B cells during affinity maturation. J Exp Med (2014) 211(1):45–56. doi:10.1084/jem.20130505
80. Garcia-Carmona Y, Cols M, Ting AT, Radigan L, Yuk FJ, Zhang L, et al. Differential induction of plasma cells by isoforms of human TACI. Blood (2015) 125(11):1749–58. doi:10.1182/blood-2014-05-575845
81. Moir S, Malaspina A, Pickeral OK, Donoghue ET, Vasquez J, Miller NJ, et al. Decreased survival of B cells of HIV-viremic patients mediated by altered expression of receptors of the TNF superfamily. J Exp Med (2004) 200(7):587–99. doi:10.1084/jem.20032236
82. Ehrhardt GR, Hsu JT, Gartland L, Leu CM, Zhang S, Davis RS, et al. Expression of the immunoregulatory molecule FcRH4 defines a distinctive tissue-based population of memory B cells. J Exp Med (2005) 202(6):783–91. doi:10.1084/jem.20050879
83. Muema DM, Macharia GN, Hassan AS, Mwaringa SM, Fegan GW, Berkley JA, et al. Control of viremia enables acquisition of resting memory B cells with age and normalization of activated B cell phenotypes in HIV-infected children. J Immunol (2015) 195(3):1082–91. doi:10.4049/jimmunol.1500491
84. Seshasayee D, Valdez P, Yan M, Dixit VM, Tumas D, Grewal IS. Loss of TACI causes fatal lymphoproliferation and autoimmunity, establishing TACI as an inhibitory BLyS receptor. Immunity (2003) 18(2):279–88. doi:10.1016/S1074-7613(03)00025-6
85. Hoffmann FS, Kuhn PH, Laurent SA, Hauck SM, Berer K, Wendlinger SA, et al. The immunoregulator soluble TACI is released by ADAM10 and reflects B cell activation in autoimmunity. J Immunol (2015) 194(2):542–52. doi:10.4049/jimmunol.1402070
86. Hymowitz SG, Patel DR, Wallweber HJ, Runyon S, Yan M, Yin J, et al. Structures of APRIL-receptor complexes: like BCMA, TACI employs only a single cysteine-rich domain for high affinity ligand binding. J Biol Chem (2005) 280(8):7218–27. doi:10.1074/jbc.M411714200
87. He B, Santamaria R, Xu W, Cols M, Chen K, Puga I, et al. The transmembrane activator TACI triggers immunoglobulin class switching by activating B cells through the adaptor MyD88. Nat Immunol (2010) 11(9):836–45. doi:10.1038/ni.1914
88. Mariette X, Roux S, Zhang J, Bengoufa D, Lavie F, Zhou T, et al. The level of BLyS (BAFF) correlates with the titre of autoantibodies in human Sjogren’s syndrome. Ann Rheum Dis (2003) 62(2):168–71. doi:10.1136/ard.62.2.168
89. Jacobson CA, Sun L, Kim HT, McDonough SM, Reynolds CG, Schowalter M, et al. Post-transplantation B cell activating factor and B cell recovery before onset of chronic graft-versus-host disease. Biol Blood Marrow Transplant (2014) 20(5):668–75. doi:10.1016/j.bbmt.2014.01.021
90. Min JW, Kim KW, Kim BM, Doh KC, Choi MS, Choi BS, et al. Clinical significance of pre- and post-transplant BAFF levels in kidney transplant recipients. PLoS One (2016) 11(9):e0162964. doi:10.1371/journal.pone.0162964
91. Stohl W, Cheema GS, Briggs WS, Xu D, Sosnovtseva S, Roschke V, et al. B lymphocyte stimulator protein-associated increase in circulating autoantibody levels may require CD4+ T cells: lessons from HIV-infected patients. Clin Immunol (2002) 104(2):115–22. doi:10.1006/clim.2002.5238
92. Gomez AM, Ouellet M, Deshiere A, Breton Y, Tremblay MJ. HIV-1-mediated BAFF secretion in macrophages does not require endosomal TLRs, type-I IFN, and Nef, but depends on the cellular phenotype status. J Immunol (2016) 196(9):3806–17. doi:10.4049/jimmunol.1501249
93. Jego G, Palucka AK, Blanck JP, Chalouni C, Pascual V, Banchereau J. Plasmacytoid dendritic cells induce plasma cell differentiation through type I interferon and interleukin 6. Immunity (2003) 19(2):225–34. doi:10.1016/S1074-7613(03)00208-5
94. Shaw J, Wang YH, Ito T, Arima K, Liu YJ. Plasmacytoid dendritic cells regulate B-cell growth and differentiation via CD70. Blood (2010) 115(15):3051–7. doi:10.1182/blood-2009-08-239145
95. Cella M, Otero K, Colonna M. Expansion of human NK-22 cells with IL-7, IL-2, and IL-1beta reveals intrinsic functional plasticity. Proc Natl Acad Sci U S A (2010) 107(24):10961–6. doi:10.1073/pnas.1005641107
96. Cella M, Miller H, Song C. Beyond NK cells: the expanding universe of innate lymphoid cells. Front Immunol (2014) 5:282. doi:10.3389/fimmu.2014.00282
97. Peruchon S, Chaoul N, Burelout C, Delache B, Brochard P, Laurent P, et al. Tissue-specific B-cell dysfunction and generalized memory B-cell loss during acute SIV infection. PLoS One (2009) 4(6):e5966. doi:10.1371/journal.pone.0005966
98. Cloke T, Munder M, Bergin P, Herath S, Modolell M, Taylor G, et al. Phenotypic alteration of neutrophils in the blood of HIV seropositive patients. PLoS One (2013) 8(9):e72034. doi:10.1371/journal.pone.0072034
99. Bowers NL, Helton ES, Huijbregts RP, Goepfert PA, Heath SL, Hel Z. Immune suppression by neutrophils in HIV-1 infection: role of PD-L1/PD-1 pathway. PLoS Pathog (2014) 10(3):e1003993. doi:10.1371/journal.ppat.1003993
100. Zeng M, Smith AJ, Shang L, Wietgrefe SW, Voss JE, Carlis JV, et al. Mucosal humoral immune response to SIVmac239nef vaccination and vaginal challenge. J Immunol (2016) 196(6):2809–18. doi:10.4049/jimmunol.1500156
101. Li Q, Estes JD, Schlievert PM, Duan L, Brosnahan AJ, Southern PJ, et al. Glycerol monolaurate prevents mucosal SIV transmission. Nature (2009) 458(7241):1034–8. doi:10.1038/nature07831
102. Kwa S, Kannanganat S, Nigam P, Siddiqui M, Shetty RD, Armstrong W, et al. Plasmacytoid dendritic cells are recruited to the colorectum and contribute to immune activation during pathogenic SIV infection in rhesus macaques. Blood (2011) 118(10):2763–73. doi:10.1182/blood-2011-02-339515
103. Cohen MS, Shaw GM, McMichael AJ, Haynes BF. Acute HIV-1 infection. N Engl J Med (2011) 364(20):1943–54. doi:10.1056/NEJMra1011874
104. Stott EJ, Chan WL, Mills KH, Page M, Taffs F, Cranage M, et al. Preliminary report: protection of cynomolgus macaques against simian immunodeficiency virus by fixed infected-cell vaccine. Lancet (1990) 336(8730):1538–41. doi:10.1016/0140-6736(90)93310-L
106. Pegu A, Hessell AJ, Mascola JR, Haigwood NL. Use of broadly neutralizing antibodies for HIV-1 prevention. Immunol Rev (2017) 275(1):296–312. doi:10.1111/imr.12511
107. Burton DR, Mascola JR. Antibody responses to envelope glycoproteins in HIV-1 infection. Nat Immunol (2015) 16(6):571–6. doi:10.1038/ni.3158
108. Caskey M, Klein F, Lorenzi JC, Seaman MS, West AP Jr, Buckley N, et al. Viraemia suppressed in HIV-1-infected humans by broadly neutralizing antibody 3BNC117. Nature (2015) 522(7557):487–91. doi:10.1038/nature14411
109. Lynch RM, Boritz E, Coates EE, DeZure A, Madden P, Costner P, et al. Virologic effects of broadly neutralizing antibody VRC01 administration during chronic HIV-1 infection. Sci Transl Med (2015) 7(319):319ra206. doi:10.1126/scitranslmed.aad5752
110. Scheid JF, Horwitz JA, Bar-On Y, Kreider EF, Lu CL, Lorenzi JC, et al. HIV-1 antibody 3BNC117 suppresses viral rebound in humans during treatment interruption. Nature (2016) 535(7613):556–60. doi:10.1038/nature18929
111. Yang G, Holl TM, Liu Y, Li Y, Lu X, Nicely NI, et al. Identification of autoantigens recognized by the 2F5 and 4E10 broadly neutralizing HIV-1 antibodies. J Exp Med (2013) 210(2):241–56. doi:10.1084/jem.20121977
112. Bonsignori M, Wiehe K, Grimm SK, Lynch R, Yang G, Kozink DM, et al. An autoreactive antibody from an SLE/HIV-1 individual broadly neutralizes HIV-1. J Clin Invest (2014) 124(4):1835–43. doi:10.1172/JCI73441
113. Liu M, Yang G, Wiehe K, Nicely NI, Vandergrift NA, Rountree W, et al. Polyreactivity and autoreactivity among HIV-1 antibodies. J Virol (2015) 89(1):784–98. doi:10.1128/JVI.02378-14
114. Schroeder KMS, Agazio A, Strauch PJ, Jones ST, Thompson SB, Harper MS, et al. Breaching peripheral tolerance promotes the production of HIV-1-neutralizing antibodies. J Exp Med (2017) 214(8):2283–302. doi:10.1084/jem.20161190
115. Mouquet H, Klein F, Scheid JF, Warncke M, Pietzsch J, Oliveira TY, et al. Memory B cell antibodies to HIV-1 gp140 cloned from individuals infected with clade A and B viruses. PLoS One (2011) 6(9):e24078. doi:10.1371/journal.pone.0024078
116. Mouquet H, Nussenzweig MC. Polyreactive antibodies in adaptive immune responses to viruses. Cell Mol Life Sci (2012) 69(9):1435–45. doi:10.1007/s00018-011-0872-6
117. Verkoczy L, Diaz M. Autoreactivity in HIV-1 broadly neutralizing antibodies: implications for their function and induction by vaccination. Curr Opin HIV AIDS (2014) 9(3):224–34. doi:10.1097/COH.0000000000000049
118. Tomaras GD, Yates NL, Liu P, Qin L, Fouda GG, Chavez LL, et al. Initial B-cell responses to transmitted human immunodeficiency virus type 1: virion-binding immunoglobulin M (IgM) and IgG antibodies followed by plasma anti-gp41 antibodies with ineffective control of initial viremia. J Virol (2008) 82(24):12449–63. doi:10.1128/JVI.01708-08
119. Liao HX, Chen X, Munshaw S, Zhang R, Marshall DJ, Vandergrift N, et al. Initial antibodies binding to HIV-1 gp41 in acutely infected subjects are polyreactive and highly mutated. J Exp Med (2011) 208(11):2237–49. doi:10.1084/jem.20110363
120. Trama AM, Moody MA, Alam SM, Jaeger FH, Lockwood B, Parks R, et al. HIV-1 envelope gp41 antibodies can originate from terminal ileum B cells that share cross-reactivity with commensal bacteria. Cell Host Microbe (2014) 16(2):215–26. doi:10.1016/j.chom.2014.07.003
121. Meffre E, Wardemann H. B-cell tolerance checkpoints in health and autoimmunity. Curr Opin Immunol (2008) 20(6):632–8. doi:10.1016/j.coi.2008.09.001
122. Wardemann H, Yurasov S, Schaefer A, Young JW, Meffre E, Nussenzweig MC. Predominant autoantibody production by early human B cell precursors. Science (2003) 301(5638):1374–7. doi:10.1126/science.1086907
123. Meffre E. The establishment of early B cell tolerance in humans: lessons from primary immunodeficiency diseases. Ann N Y Acad Sci (2011) 1246:1–10. doi:10.1111/j.1749-6632.2011.06347.x
124. Thien M, Phan TG, Gardam S, Amesbury M, Basten A, Mackay F, et al. Excess BAFF rescues self-reactive B cells from peripheral deletion and allows them to enter forbidden follicular and marginal zone niches. Immunity (2004) 20(6):785–98. doi:10.1016/j.immuni.2004.05.010
125. Hondowicz BD, Alexander ST, Quinn WJ III, Pagan AJ, Metzgar MH, Cancro MP, et al. The role of BLyS/BLyS receptors in anti-chromatin B cell regulation. Int Immunol (2007) 19(4):465–75. doi:10.1093/intimm/dxm011
126. Yurasov S, Wardemann H, Hammersen J, Tsuiji M, Meffre E, Pascual V, et al. Defective B cell tolerance checkpoints in systemic lupus erythematosus. J Exp Med (2005) 201(5):703–11. doi:10.1084/jem.20042251
127. Shinners NP, Carlesso G, Castro I, Hoek KL, Corn RA, Woodland RT, et al. Bruton’s tyrosine kinase mediates NF-kappa B activation and B cell survival by B cell-activating factor receptor of the TNF-R family. J Immunol (2007) 179(6):3872–80. doi:10.4049/jimmunol.179.6.3872
128. Jellusova J, Miletic AV, Cato MH, Lin WW, Hu Y, Bishop GA, et al. Context-specific BAFF-R signaling by the NF-kappaB and PI3K pathways. Cell Rep (2013) 5(4):1022–35. doi:10.1016/j.celrep.2013.10.022
129. Hobeika E, Levit-Zerdoun E, Anastasopoulou V, Pohlmeyer R, Altmeier S, Alsadeq A, et al. CD19 and BAFF-R can signal to promote B-cell survival in the absence of Syk. EMBO J (2015) 34(7):925–39. doi:10.15252/embj.201489732
130. Knox JJ, Buggert M, Kardava L, Seaton KE, Eller MA, Canaday DH, et al. T-bet+ B cells are induced by human viral infections and dominate the HIV gp140 response. JCI Insight (2017) 2(8):92943. doi:10.1172/jci.insight.92943
131. Zhou T, Georgiev I, Wu X, Yang ZY, Dai K, Finzi A, et al. Structural basis for broad and potent neutralization of HIV-1 by antibody VRC01. Science (2010) 329(5993):811–7. doi:10.1126/science.1192819
132. Puga I, Cols M, Barra CM, He B, Cassis L, Gentile M, et al. B cell-helper neutrophils stimulate the diversification and production of immunoglobulin in the marginal zone of the spleen. Nat Immunol (2012) 13(2):170–80. doi:10.1038/ni.2194
133. Magri G, Miyajima M, Bascones S, Mortha A, Puga I, Cassis L, et al. Innate lymphoid cells integrate stromal and immunological signals to enhance antibody production by splenic marginal zone B cells. Nat Immunol (2014) 15(4):354–64. doi:10.1038/ni.2830
134. Victora GD, Nussenzweig MC. Germinal centers. Annu Rev Immunol (2012) 30:429–57. doi:10.1146/annurev-immunol-020711-075032
135. Binder SC, Meyer-Hermann M. Implications of intravital imaging of murine germinal centers on the control of B cell selection and division. Front Immunol (2016) 7:593. doi:10.3389/fimmu.2016.00593
136. Tiller T, Tsuiji M, Yurasov S, Velinzon K, Nussenzweig MC, Wardemann H. Autoreactivity in human IgG+ memory B cells. Immunity (2007) 26(2):205–13. doi:10.1016/j.immuni.2007.01.009
137. Lesley R, Xu Y, Kalled SL, Hess DM, Schwab SR, Shu HB, et al. Reduced competitiveness of autoantigen-engaged B cells due to increased dependence on BAFF. Immunity (2004) 20(4):441–53. doi:10.1016/S1074-7613(04)00079-2
138. Cremasco V, Woodruff MC, Onder L, Cupovic J, Nieves-Bonilla JM, Schildberg FA, et al. B cell homeostasis and follicle confines are governed by fibroblastic reticular cells. Nat Immunol (2014) 15(10):973–81. doi:10.1038/ni.2965
139. Wang X, Cho B, Suzuki K, Xu Y, Green JA, An J, et al. Follicular dendritic cells help establish follicle identity and promote B cell retention in germinal centers. J Exp Med (2011) 208(12):2497–510. doi:10.1084/jem.20111449
140. Havenar-Daughton C, Lindqvist M, Heit A, Wu JE, Reiss SM, Kendric K, et al. CXCL13 is a plasma biomarker of germinal center activity. Proc Natl Acad Sci U S A (2016) 113(10):2702–7. doi:10.1073/pnas.1520112113
141. Cagigi A, Mowafi F, Phuong Dang LV, Tenner-Racz K, Atlas A, Grutzmeier S, et al. Altered expression of the receptor-ligand pair CXCR5/CXCL13 in B-cells during chronic HIV-1 infection. Blood (2008) 112(12):4401–10. doi:10.1182/blood-2008-02-140426
142. Badr G, Borhis G, Lefevre EA, Chaoul N, Deshayes F, Dessirier V, et al. BAFF enhances chemotaxis of primary human B cells: a particular synergy between BAFF and CXCL13 on memory B cells. Blood (2008) 111(5):2744–54. doi:10.1182/blood-2007-03-081232
143. Victora GD, Schwickert TA, Fooksman DR, Kamphorst AO, Meyer-Hermann M, Dustin ML, et al. Germinal center dynamics revealed by multiphoton microscopy with a photoactivatable fluorescent reporter. Cell (2010) 143(4):592–605. doi:10.1016/j.cell.2010.10.032
144. Meyer-Hermann M, Mohr E, Pelletier N, Zhang Y, Victora GD, Toellner KM. A theory of germinal center B cell selection, division, and exit. Cell Rep (2012) 2(1):162–74. doi:10.1016/j.celrep.2012.05.010
145. Cubas RA, Mudd JC, Savoye AL, Perreau M, van Grevenynghe J, Metcalf T, et al. Inadequate T follicular cell help impairs B cell immunity during HIV infection. Nat Med (2013) 19(4):494–9. doi:10.1038/nm.3109
146. Cohen K, Altfeld M, Alter G, Stamatatos L. Early preservation of CXCR5+ PD-1+ helper T cells and B cell activation predict the breadth of neutralizing antibody responses in chronic HIV-1 infection. J Virol (2014) 88(22):13310–21. doi:10.1128/JVI.02186-14
147. Locci M, Havenar-Daughton C, Landais E, Wu J, Kroenke MA, Arlehamn CL, et al. Human circulating PD-1+CXCR3-CXCR5+ memory Tfh cells are highly functional and correlate with broadly neutralizing HIV antibody responses. Immunity (2013) 39(4):758–69. doi:10.1016/j.immuni.2013.08.031
148. Coquery CM, Loo WM, Wade NS, Bederman AG, Tung KS, Lewis JE, et al. BAFF regulates follicular helper t cells and affects their accumulation and interferon-gamma production in autoimmunity. Arthritis Rheumatol (2015) 67(3):773–84. doi:10.1002/art.38950
149. Titanji K, Chiodi F, Bellocco R, Schepis D, Osorio L, Tassandin C, et al. Primary HIV-1 infection sets the stage for important B lymphocyte dysfunctions. AIDS (2005) 19(17):1947–55. doi:10.1097/01.aids.0000191231.54170.89
150. Pensieroso S, Cagigi A, Palma P, Nilsson A, Capponi C, Freda E, et al. Timing of HAART defines the integrity of memory B cells and the longevity of humoral responses in HIV-1 vertically-infected children. Proc Natl Acad Sci U S A (2009) 106(19):7939–44. doi:10.1073/pnas.0901702106
151. Moir S, Buckner CM, Ho J, Wang W, Chen J, Waldner AJ, et al. B cells in early and chronic HIV infection: evidence for preservation of immune function associated with early initiation of antiretroviral therapy. Blood (2010) 116(25):5571–9. doi:10.1182/blood-2010-05-285528
152. Good KL, Avery DT, Tangye SG. Resting human memory B cells are intrinsically programmed for enhanced survival and responsiveness to diverse stimuli compared to naive B cells. J Immunol (2009) 182(2):890–901. doi:10.4049/jimmunol.182.2.890
153. Moir S, Fauci AS. B cells in HIV infection and disease. Nat Rev Immunol (2009) 9(4):235–45. doi:10.1038/nri2524
154. Titanji K, Velu V, Chennareddi L, Vijay-Kumar M, Gewirtz AT, Freeman GJ, et al. Acute depletion of activated memory B cells involves the PD-1 pathway in rapidly progressing SIV-infected macaques. J Clin Invest (2010) 120(11):3878–90. doi:10.1172/JCI43271
155. Kardava L, Moir S, Shah N, Wang W, Wilson R, Buckner CM, et al. Abnormal B cell memory subsets dominate HIV-specific responses in infected individuals. J Clin Invest (2014) 124(7):3252–62. doi:10.1172/JCI74351
156. Buckner CM, Kardava L, Zhang X, Gittens K, Justement JS, Kovacs C, et al. Maintenance of HIV-specific memory B-cell responses in elite controllers despite low viral burdens. J Infect Dis (2016) 214(3):390–8. doi:10.1093/infdis/jiw163
157. Moss WJ, Scott S, Mugala N, Ndhlovu Z, Beeler JA, Audet SA, et al. Immunogenicity of standard-titer measles vaccine in HIV-1-infected and uninfected Zambian children: an observational study. J Infect Dis (2007) 196(3):347–55. doi:10.1086/519169
158. Nair N, Moss WJ, Scott S, Mugala N, Ndhlovu ZM, Lilo K, et al. HIV-1 infection in Zambian children impairs the development and avidity maturation of measles virus-specific immunoglobulin G after vaccination and infection. J Infect Dis (2009) 200(7):1031–8. doi:10.1086/605648
159. Lefevre EA, Krzysiek R, Loret EP, Galanaud P, Richard Y. Cutting edge: HIV-1 Tat protein differentially modulates the B cell response of naive, memory, and germinal center B cells. J Immunol (1999) 163(3):1119–22.
160. Nduati E, Gwela A, Karanja H, Mugyenyi C, Langhorne J, Marsh K, et al. The plasma concentration of the B cell activating factor is increased in children with acute malaria. J Infect Dis (2011) 204(6):962–70. doi:10.1093/infdis/jir438
161. Vugmeyster Y, Seshasayee D, Chang W, Storn A, Howell K, Sa S, et al. A soluble BAFF antagonist, BR3-Fc, decreases peripheral blood B cells and lymphoid tissue marginal zone and follicular B cells in cynomolgus monkeys. Am J Pathol (2006) 168(2):476–89. doi:10.2353/ajpath.2006.050600
162. Jacobi AM, Huang W, Wang T, Freimuth W, Sanz I, Furie R, et al. Effect of long-term belimumab treatment on B cells in systemic lupus erythematosus: extension of a phase II, double-blind, placebo-controlled, dose-ranging study. Arthritis Rheum (2010) 62(1):201–10. doi:10.1002/art.27189
163. Muellenbeck MF, Ueberheide B, Amulic B, Epp A, Fenyo D, Busse CE, et al. Atypical and classical memory B cells produce Plasmodium falciparum neutralizing antibodies. J Exp Med (2013) 210(2):389–99. doi:10.1084/jem.20121970
164. Sohn HW, Krueger PD, Davis RS, Pierce SK. FcRL4 acts as an adaptive to innate molecular switch dampening BCR signaling and enhancing TLR signaling. Blood (2011) 118(24):6332–41. doi:10.1182/blood-2011-05-353102
165. Rubtsov AV, Rubtsova K, Fischer A, Meehan RT, Gillis JZ, Kappler JW, et al. Toll-like receptor 7 (TLR7)-driven accumulation of a novel CD11c(+) B-cell population is important for the development of autoimmunity. Blood (2011) 118(5):1305–15. doi:10.1182/blood-2011-01-331462
166. Rubtsova K, Rubtsov AV, van Dyk LF, Kappler JW, Marrack P. T-box transcription factor T-bet, a key player in a unique type of B-cell activation essential for effective viral clearance. Proc Natl Acad Sci U S A (2013) 110(34):E3216–24. doi:10.1073/pnas.1312348110
Keywords: B-cell-activating factor, B-cells, dendritic cells, germinal center, HIV, memory B-cells, follicular helper T-cells, SIV
Citation: Borhis G, Trovato M, Chaoul N, Ibrahim HM and Richard Y (2017) B-Cell-Activating Factor and the B-Cell Compartment in HIV/SIV Infection. Front. Immunol. 8:1338. doi: 10.3389/fimmu.2017.01338
Received: 26 June 2017; Accepted: 03 October 2017;
Published: 27 October 2017
Edited by:
Francesca Chiodi, Karolinska Institute (KI), SwedenReviewed by:
Ivona Pandrea, University of Pittsburgh, United StatesLisa A. Chakrabarti, Institut Pasteur, France
Lucia Lopalco, San Raffaele Hospital (IRCCS), Italy
Copyright: © 2017 Borhis, Trovato, Chaoul, Ibrahim and Richard. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yolande Richard, eW9sYW5kZS5yaWNoYXJkJiN4MDAwNDA7aW5zZXJtLmZy
†Present address: Gwenoline Borhis, Kymab Ltd., Cambridge, United Kingdom;
Nada Chaoul, University of Bari, Bari, Italy;
Hany M. Ibrahim, Faculty of Science, Zoology Department, Menoufia University, Shibin El Kom, Egypt