 Federica Ungaro
Federica Ungaro Federica Rubbino
Federica Rubbino Silvio Danese
Silvio Danese Silvia D’Alessio
Silvia D’Alessio- 1Department of Biomedical Sciences, Humanitas University, Rozzano, Italy
- 2Laboratory of Gastrointestinal Immunopathology, Humanitas Clinical and Research Center, IBD Center, Rozzano, Italy
In the last few decades, the pathogenesis of inflammatory bowel disease (IBD) in genetically predisposed subjects susceptible to specific environmental factors has been attributed to disturbance of both the immune and non-immune system and/or to the imbalanced interactions with microbes. However, increasing evidences support the idea that defects in pro-resolving pathways might strongly contribute to IBD onset. The resolution of inflammation is now recognized as a dynamic event coordinated by specialized pro-resolving lipid mediators (LMs), which dampen inflammation-sustaining events, such as angiogenesis, release of pro-inflammatory cytokines, clearance of apoptotic cells, and microorganisms. Among these pro-resolving molecules, those derived from essential polyunsaturated fatty acids (PUFAs) have been shown to induce favorable effects on a plethora of human inflammatory disorders, including IBD. Here, we offer a summary of mechanisms involving both cellular and molecular components of the immune response and underlying the anti-inflammatory and pro-resolving properties of PUFAs and their derivatives in the gut, focusing on both ω-3 and ω-6 LMs. These fatty acids may influence IBD progression by: reducing neutrophil transmigration across the intestinal vasculature and the epithelium, preventing the release of pro-inflammatory cytokines and the up-regulation of adhesion molecules, and finally by promoting the production of other pro-resolving molecules. We also discuss the numerous attempts in using pro-resolving PUFAs to ameliorate intestinal inflammation, both in patients with IBD and mouse models. Although their effects in reducing inflammation is incontestable, results from previous works describing the effects of PUFA administration to prevent or treat IBD are controversial. Therefore, more efforts are needed not only to identify and explain the physiological functions of PUFAs in the gut, but also to unveil novel biosynthetic pathways of these pro-resolving LMs that may be dysregulated in these gut-related disorders. We suppose that either PUFAs or new medications specifically promoting resolution-regulating mediators and pathways will be much better tolerated by patients with IBD, with the advantage of avoiding immune suppression.
Introduction
Inflammatory bowel diseases (IBDs), encompassing ulcerative colitis (UC), and Crohn’s disease (CD) are immunologically mediated inflammatory disorders of the gut, whose prevalence and incidence are dramatically increasing worldwide. Although clinical manifestations of these diseases are different, they share common features. In fact, both UC and CD are characterized by epithelial barrier damage that allows commensal bacteria and microbial products to translocate into and colonize the intestinal wall. This event triggers the release of cytokines, chemokines, and eicosanoids which thanks to regulatory mechanisms, mediate the physiological self-limiting immune-response (1, 2). Furthermore, both immune and non-immune components of the intestinal mucosa have been shown to exert a key role in IBD pathogenesis (3). In terms of immune components, the innate and the adaptive immune system are essential to chronic intestinal inflammation. In fact, innate immune cells (e.g., neutrophils, monocytes, and macrophages) hold the capability to remodel the response of adaptive T cells during the inflammatory process (4). Concomitantly, studies of the intestinal microbiota, environmental factors, and genetics have identified a significant contribution of non-immune components to the pathogenesis of IBD, which include: breach in the epithelial wall, that is, the first line of gut defense against bacteria and other microorganisms (5–7); defects in the biological activities of stromal cells, which hold immune-modulatory actions and the capability to clear chemokines and cytokines from the inflammatory milieu to re-establish mucosal homeostasis (8, 9); defective endothelial cell functions, crucial for the angiogenic process but also for the regulation of leukocyte adhesion, and trafficking across the hematic and lymphatic barriers (10–14). Activities of both immune and non-immune cells need to be finely modulated and constantly balanced, in order to avoid chronicity of inflammation and tissue damage.
Another key component of IBD pathogenesis is represented by the gut microbiota (15). In fact, the gastrointestinal tract hosts the largest microbial community of the organism that can be shaped by environmental factors, diet, and hygiene during childhood (16), whereas in adulthood this is more stable with a defined composition of bacteria (17, 18). In healthy subjects, homeostasis exists between the intestinal microbiome, mucosal barrier, and immune system. In IBD, this homeostasis is altered causing a “dysbiosis,” disrupted barrier function as well as immune system activation (15).
Although many efforts have been made to delineate the causes underlying the exact etiopathogenesis of IBD, so far our knowledge does not fully clarify what causes its onset. It is currently well accepted that at the basis of IBD pathogenesis (19, 20) there is an imbalance between pro- and anti-inflammatory signals (1). This suggests that defects in the proper release of pro-resolving molecules during the acute phase of inflammation may characterize IBD onset. For decades, the resolution of inflammation has been considered a passive event, in which pro-inflammatory signals simply dilute over time. This concept was overturned when Serhan and colleagues discovered for the first time that a specific class of lipids, known as eicosanoids and docosanoids, promotes, and orchestrates the resolution process (21, 22).
This discovery gave rise to a new field of research studying the mechanisms and the factors involved in the resolution phase of the inflammatory response, which is finely and temporally regulated by specialized pro-resolving mediators, named lipoxins (LXs), resolvins (Rv), protectins, and maresins. These resolving bioactive lipids are synthesized from ω-6 and ω-3 polyunsaturated fatty acids (PUFAs) and have been demonstrated to exert potent immune-resolving effects (2). However, this line of research is still at its infancy in the IBD field.
In fact, the vast majority of therapies currently in use for IBD aims at blocking key inflammatory mediators that are triggered during the early stages of acute inflammation. However, targeting infiltrating immune cells does not always lead to remission or stable resolution. Indeed, conventional anti-inflammatory agents do not alter the course of IBD because the naturally occurring resolution programs are likely to be subverted. For this reason, promotion and maintenance of the resolving milieu may represent a good alternative therapeutic approach to dampen chronic inflammation in IBD. In addition, defects in the production of resolving molecules may strongly contribute to IBD onset, thus expanding our understanding of what triggers these gut-related diseases.
This review aims to describe how the resolution process plays a fundamental role in the gut both at the physiological and pathological level. After a brief overview on IBD pathogenesis, we will emphasize which cellular and molecular components govern the resolving phase of intestinal inflammation and we will discuss the state of the art of preclinical and clinical studies employing PUFA-derived pro-resolving lipid mediators (LMs) in IBD.
Resolution of Inflammation and Pro-Resolving LMs: A Brief Overview
For decades, anti-inflammatory treatments have been used to treat chronic inflammatory conditions because of the concept that the chronically established inflammation was caused by an exaggerated immune response rather than a failure in the resolution of inflammation (23). Indeed, for years the resolution process has been considered a passive event where inflammatory signals progressively dissipate (2, 24). In contrast to this assumption, during the last decade resolution of inflammation has been conclusively recognized as an active and tightly regulated process orchestrated by pro-resolving LMs, which have been found to dampen inflammation-sustaining events such as cell proliferation, migration, and clearance of apoptotic cells and microorganisms (2, 25).
At tissue and cellular level, the key steps that characterize the resolution process are (i) clearance of the inciting stimuli, (ii) silencing of pro-inflammatory and local survival signals, including chemokine gradients, (iii) polymorphonuclear (PMN) efferocytosis and clearance by tissue and monocyte-derived macrophages, and (v) recirculation of macrophages via lymph flow. LMs represent the key signaling molecules in this process, which regulate the inflammatory profile and promote the return of affected tissues to homeostasis (26).
In this context, ω-3 and ω-6 PUFAs are specialized LMs that have the capability of influencing the inflammatory processes, such as those governing IBD. They are essential fatty acids that need to be obtained from the diet; in fact, since mammals lack of endogenous enzymes necessary for ω-3 PUFA desaturation, they cannot be synthesized by humans (27).
Polyunsaturated fatty acid metabolism is recognized as an important factor in immune regulation and disease control. In particular, the metabolic balance between ω-6 and ω-3 PUFAs is widely held to be important in human health and diseases (27–30). PUFA-derived bioactive metabolites are formed in vivo by enzymatic oxidation through the action of cyclooxygenases (COXs), lipoxygenases (LOXs), and cytochrome P450 (CYP450) monooxygenases. From ω-6 PUFAs, e.g., arachidonic acid (AA), the COX pathway leads to the formation of prostanoids, such as prostaglandins (PGs) and thromboxanes (TXs), the LOX pathway leads to leukotrienes (LTs) and LXs, and the CYP450 pathway gives rise to hydroxy-eicosatetraenoic acids (HETEs) and epoxy-eicosatrienoic acids (Figure 1A) (2, 24, 31, 32). Except for LXs (33), ω-6 PUFAs are conventionally involved in the initiation of inflammatory responses. On the contrary, ω-3 PUFAs seem to promote resolution of inflammation (34). α-linolenic acid (ALA) is an ω-3 PUFA and is categorized with the ω-6 linoleic acid (LA) as an essential fatty acid. As ω-6 LA can be metabolized into AA, ALA can be converted into precursors for long chain ω-3 PUFAs such as eicosapentaenoic acid (EPA) and docosahexaenoic acid (DHA). Both EPA and DHA, which can be found in some fish oils, are good substrates for LOX and CYP, thus being efficiently converted into bioactive metabolites such as E-series resolvins (RvEs), D-series resolvins (RvDs), protectins, and maresins that act as potent pro-resolving endogenous mediators in a wide range of human inflammatory disorders, including IBD (Figure 1B) (35–44). A large number of studies sustain the anti-inflammatory potential of EPA and DHA and their derivatives [for a recent review, see Ref. (35, 39)]. Nevertheless, the molecular mechanisms by which these essential fatty acids exert their anti-inflammatory effects remain controversial, particularly in the gut.
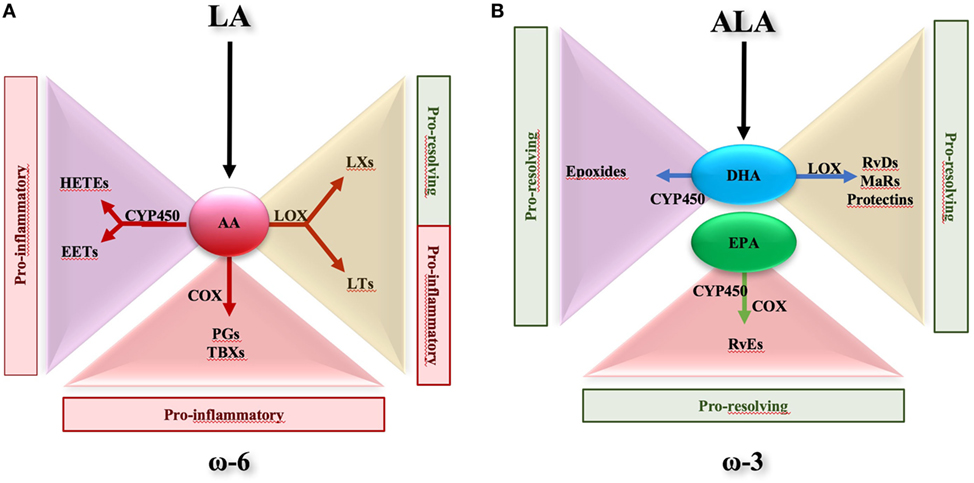
Figure 1. Metabolic route of ω-6- and ω-3-derived lipid mediators. (A) Essential fatty acid linoleic acid, classified as ω-6 polyunsaturated fatty acid, can be converted into arachidonic acid (AA). In turn, AA is metabolized in hydroxy-eicosatetraenoic acids (HETEs) and epoxy-eicosatrienoic acids (EETs) via cytochrome P450 (CYP450). Via lipoxygenase (LOX) pathway, AA is converted to lipoxins (LXs) and leukotriens (LTs), whereas via cyclooxygenase it is metabolized in prostaglandins (PGs) and tromboxanes (TBXs). HETEs, EETs, PGs, TBXs, and LTs are all pro-inflammatory, while LXs are considered pro-resolving mediators. (B) Essential fatty acid α-linolenic acid is converted to eicosapentaenoic acid (EPA) and docosahexaenoic acid (DHA). EPA and DHA may be substrate of CYP450, resulting into production of E-series resolvins (Rv) and epoxides, respectively. In addition, DHA is metabolized via LOX to D-series Rv, maresins, and protectins. All these EPA- and DHA-derived mediators are recognized to harbor pro-resolving properties.
Inflammatory bowel disease patients may display a deficiency in essential fatty acids and/or a defect in PUFA biosynthesis and metabolism. This is why the intake of ω-3 PUFAs may benefit patients with both UC and CD by a series of beneficial events such as inhibition of natural cytotoxicity, and improvement of oxidative stress (35, 45–47). This concept is strengthened by the fact that the intestinal mucosa seems to be highly responsive to ω-3 long-chain PUFAs (47–49).
Actions of Pro-Resolving PUFAs and Target Cell Types in the Gut
Active resolution of inflammation is characterized by a sequential series of events that starts from building an adequate inflammatory response against inciting agents, to minimizing local tissue damage. In this context, pro-resolving PUFAs act with various signals and mechanisms to different cell compartments, with the final purpose to remodel and clear healed tissues of unnecessary immune cells, thus bringing the inflamed organ to the original homeostasis.
The intestinal epithelium is a key coordinator of both inflammation and resolution. Thanks to tight junctions (TJs), intestinal epithelial cells form a dynamic barrier protected by a thick mucus layer which controls what can reach the lamina propria from the lumen (50, 51). In IBD pathogenesis, altered intestinal barrier functions, in terms of decreased mucous production (52) and reduced expression of TJs (53), have been associated with increased gut permeability, which facilitates the absorption of microbial products and triggers an excessive response, eventually leading to mucosa injury in both CD and UC (54, 55). In order to counteract pathogen infections, epithelial cells are able to produce and release in the luminal mucus antibacterial and endotoxin-neutralizing molecules called bactericidal permeability-increasing protein (BPI). BPI damages the membranes of Gram-negative bacteria, neutralizes endotoxin, and opsonizes bacteria for neutrophil phagocytosis (56). BPI is transcriptionally up-regulated by LXs and resolvin E1 (RvE1) (57). In addition, it was observed that RvE1 significantly upregulates the expression of intestinal alkaline phosphatase (ALPI), an enzyme whose activity is critical for the maintenance of bacterial homeostasis (57): for its luminal location, ALPI has been demonstrated to block Gram-negative growth such as Escherichia coli and strongly neutralizes LPS through dephosphorylation of moiety in lipid A (58). This was confirmed in the mouse model of dextran sodium sulfate (DSS)-induced colitis, during which the in vivo induction of ALPI by RvE1 positively correlated with the resolution process (57).
Lipoxins have also demonstrated to exert an ex vivo cytoprotective role on intestinal epithelial cells (59). Goh and colleagues showed that administration of LXs significantly ameliorates TNF-α-induced mucosal inflammation and reduces epithelial cell apoptosis. However, the mechanisms through which LXs exert these cytoprotective effects remain yet to be defined (33).
Polyunsaturated fatty acids have been shown to modulate other biological activities of intestinal epithelial cells. It is known that pro-resolving LMs exert their functions by binding with cell surface receptors, the majority of which belongs to G protein-coupled receptors (GPRs) (60). Among these receptors, GPR120 has been found to be the most abundantly expressed in the gut, particularly on epithelial cells and macrophages (59, 60). A study from Mobraten and colleagues shows that DHA, EPA, or AA are able to trigger GPR120 in Caco-2 cells, initiating multiple and independent signaling processes with different kinetics and intensity; these are (i) the activation of MAP kinases, (ii) the inhibition of IL-1β induced NF-κB activation, and (iii) the cytosolic accumulation of Ca2+ (61). Another group shows differential effects of activation of GPR120 by DHA in human intestinal Caco-2 and murine STC-1 cells, two different cell lines representing the mammalian intestinal epithelial layer. In this study, GPR120 stimulation by ω-3 PUFAs increased β-arrestin2 interaction with TAB 1 and attenuated TNFα-induced inflammatory effects by association of TAB 1 with TAK1, which resulted in reduced activation of NF-kB (59). Anti-inflammatory effects exerted by PUFAs through GPR120 were confirmed in vivo by Zhao et al., who demonstrated that triggering of GPR120 by DHA treatment ameliorate the experimental colitis in IL-10 deficient mice (62). Interestingly, transcription of GPR120 in intestinal epithelial cells was found tremendously increased by bacteria belonging to the Firmicutes, Bacteroides and Proteobacteria phyla (63), all classified as microorganisms harboring anti-inflammatory properties. This is intriguing, because the dysbiosis observed in patients with IBD is basically caused by a diminished diversity of Firmicutes (64). This suggests that reduced expression levels of GPR120 may be one of the causes underlining IBD pathogenesis, and that targeting this receptor may represent a new therapeutic strategy in IBD; however, to date there are no studies that deeply characterize and quantify GPR120 in the inflamed mucosa of IBD patients and further studies to elucidate this aspect are needed. The effects of PUFAs on intestinal epithelial cells are schematically summarized in Figure 2A.
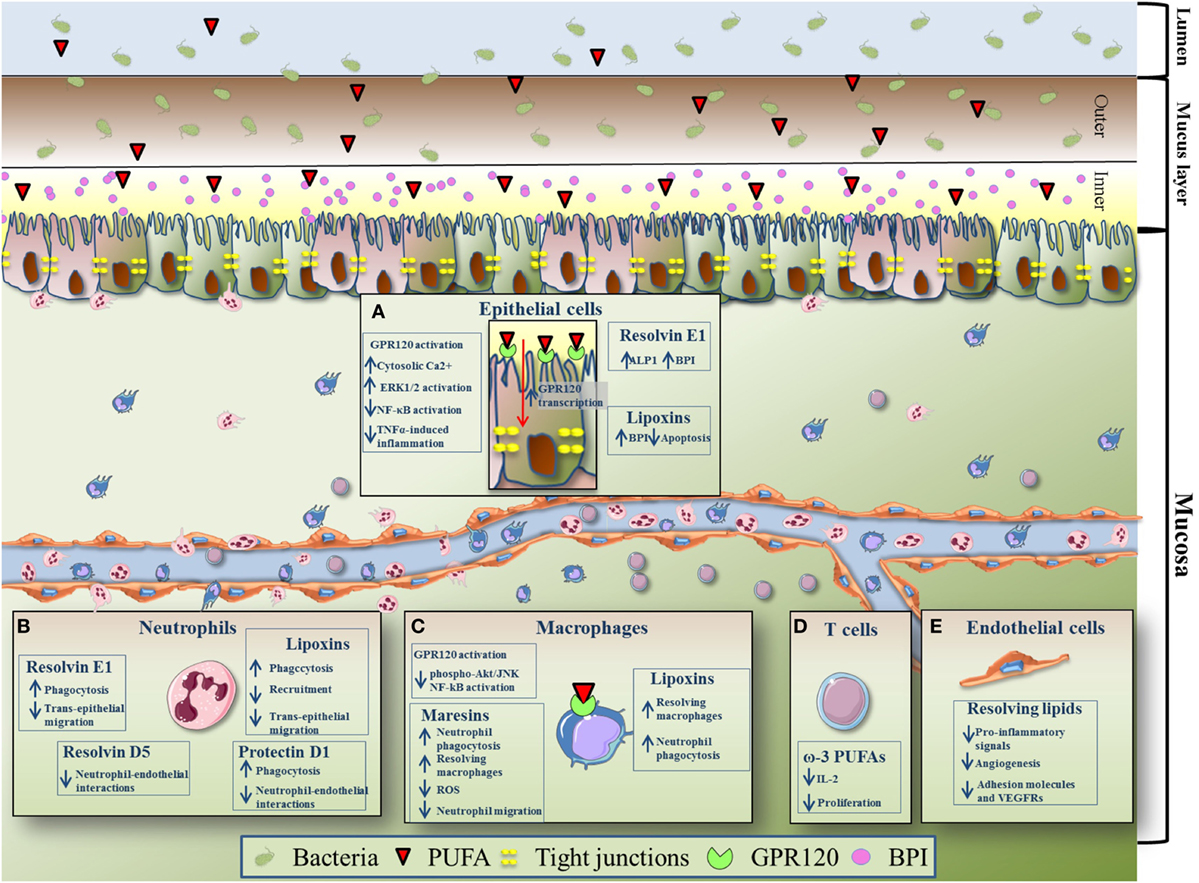
Figure 2. Effects of pro-resolving polyunsaturated fatty acids (PUFAs) on immune and non-immune intestinal components. (A) Thanks to tight junctions, intestinal epithelial cells form a dynamic barrier protected by a thick mucus layer (inner and outer) which controls what can reach the lamina propria from the lumen. In order to counteract pathogen infections, epithelial cells are able to produce and release in the luminal mucus antibacterial and endotoxin-neutralizing molecules called bactericidal permeability-increasing protein (BPI). BPI is transcriptionally up-regulated by lipoxins (LXs) and resolvin (Rv) E1. In addition, it was observed that resolvin E1 (RvE1) significantly upregulates the expression of intestinal alkaline phosphatase. Moreover, LXs inhibit epithelial cells apoptosis. G protein-coupled receptor (GPR)120 activation by PUFAs [eicosapentaenoic acid (EPA), docosahexaenoic acid (DHA), and arachidonic acid] leads to accumulation of cytosolic Ca2+, activation of MAP kinase ERK1/2, inhibition of IL-1β-induced NF-κB activation, and TNFα-induced inflammation. Transcription of GPR120 is increased by bacteria belonging to the bacteroides, proteobacteria, and firmicutes phyla. (B) Neutrophils (polymorphonuclear) are the first immune cells recruited to the site of inflammation, but are also important players in the first stages of the resolution program. LXs reduce neutrophil recruitment to the inflamed tissue, transepithelial migration, and phagocytosis. Protectin D1 promotes neutrophil phagocytosis. Similar to LXs, RvE1 reduces neutrophil transepithelial migration and induces neutrophil phagocytosis. Moreover, both protectin D1 and RvD5 have been shown to reduce neutrophil–endothelial interaction. (C) Macrophages, important for the resolution of intestinal inflammation, express high level of GPR120. EPA- and DHA-dependent activation of GPR120 has been shown to repress Akt/JNK phosphorylation and NF-kB induction. LXs enhance non-phlogistic phagocytosis of apoptotic neutrophils by macrophages. Treatment with LXs may also polarize intestinal macrophages into a resolving phenotype, thus promoting resolution of inflammation. Maresins exert potent pro-resolution and anti-inflammatory activities, ultimately leading to reduced neutrophil migration and increase macrophage phagocytic activities. Maresins induces also the resolving phenotype of macrophages and inhibit reactive oxygen species production. (D) EPA and DHA (ω-3 PUFAs) inhibit T cell proliferation and reduce IL-2 production. (E) Pro-resolving lipid mediators (DHA, α-linolenic acid-derived) exert anti-inflammatory and anti-angiogenic effects on the gut endothelium. They reduce the production of IL-6, IL-8, GM-CSF PGE-2, and LTB-4 (pro-inflammatory signals), decrease the levels of adhesion molecules (intercellular adhesion molecule 1 and vascular cell adhesion protein 1), and vascular endothelial growth factor receptor 2, thus suppressing the angiogenic component of inflammation.
Neutrophils (PMN leukocyte) are the first cell type of the innate immune system to reach inflamed areas and hold the essential role of limiting the invasion of microorganisms (65). In fact, upon transmigration through activated endothelial cells, PMNs infiltrate the intestinal epithelium, and once reached the apical portion of epithelial cells, they come into contact with tons of bacterial stimuli, which further sustain PMN activation. PMN accumulation within the intestinal crypts has been associated with transepithelial resistance (66, 67) and epithelial barrier integrity (68), and in IBD the persistent and prolonged PMN flux across the epithelium has been shown to cause mucosal ulceration and barrier disruption, ultimately facilitating microorganism entry into the submucosa (69), and contributing to the clinical syndrome of malabsorption and diarrhea in these patients (31, 68). However, PMNs are also recognized as important players in the first stages of the resolution program. For example, they release pro-inflammatory LMs (e.g., PGI2 and LTB4) during early inflammation, before producing pro-resolving molecules, such as LXs, Rvs, and protectins at the onset of resolution (21). Due to this dual role, PMN activity needs to be finely regulated in order to reduce tissue damage and avoid chronicity of diseases (70, 71). LXs deriving from the metabolic route of AA, have been shown to inhibit PMN flux across the epithelial barrier (21, 72). In line with this, patients with severe UC display colonic deficiency in LX biosynthesis, which causes low to absent production of lipoxin A4 (LXA4) (73). Accordingly, LXA4 analogs dampen colitis induced by 2,4,6-trinitrobenzene sulfonic acid (TNBS) or DSS (74, 75). RvE1 has been also shown to inhibit PMN transepithelial migration, and TNBS-induced colon damage (Figure 2B) (36, 57). These LMs, that include protectin D1, not only also support phagocytosis of apoptotic PMNs (76), but also mediate the overexpression of C–C chemokine receptor type 5 receptors on apoptotic neutrophils, thus sequestering inflammatory chemokines such as chemokine (C–C motif) ligand 3 and CCL5, and promoting PMN clearance at sites of inflammation (77).
During intestinal inflammation, PMNs not only represent the target cell type of many pro-resolving PUFAs, but they are also the main producers of many molecules. In fact, a number of recent studies (78–81) clearly indicate that activated PMNs generate crucial anti-inflammatory and pro-resolving mediators that characterize the onset of resolution (82, 83). This aspect has been convincingly demonstrated in vivo, by depletion of circulating PMNs with anti-Gr1 antibodies, which resulted in the exacerbation of colitis in various mouse models of IBD, implicating PMNs as a key protective factor in ongoing intestinal inflammation (84). This may justify the controversial role exerted by neutrophils to the pathogenesis of IBD, and why their contribution may differ between CD and UC (85). In fact, while in patients with active UC it has been observed a correlation between the extent of PMN infiltration and the severity of the disease (86), several other studies have reported PMN dysfunction in patients with CD (87–89).
Resident macrophages, located in the sub-epithelial layers of the gut, are designated for protecting the host against pathogens and for regulating mucosal responses to commensal bacteria. For this reason, they are considered important players in the resolution of inflammation (90). These cells of the innate immune system have the characteristic to express various GPRs, including GPR120 (19, 91). EPA- and DHA-dependent activation of GPR120 has been shown to have anti-inflammatory activities in both RAW 264.7 monocytes and primary intraperitoneal macrophages; these effects were abolished by GPR120 silencing (92). In another study, PUFA-dependent signaling cascade that follows GPR120 activation in the gut was observed also in macrophages, where the stimulation of this receptor led to the repression of Akt/JNK phosphorylation and NF-kB-mediated cyclooxygenase-2 (COX-2) induction (92–95). Blood-derived macrophages, that in chronic IBD are known to secrete inflammatory cytokines and tissue-degrading proteases (96, 97), and that well differentiate from resident macrophages, are recognized as either perpetuator of inflammation or effectors of the resolution process (33). Treatment of monocyte-derived macrophages with LXA4 and its analogs induced a strong enhancement in phagocytosis of apoptotic neutrophils (98), thus attributing to these PUFA derivatives an additional role in the resolution of intestinal inflammation.
Following studies on macrophages during the resolution process, a new pathway capable of producing potent mediators from DHA has been uncovered and the resulting metabolites have been coined maresins (MaR1 and MaR2), which exert potent pro-resolution and anti-inflammatory activities, ultimately leading to reduced neutrophil migration and increased macrophage phagoytic activities (99–102). Marcon and colleagues recently showed that MaR1 may cause a switch in the macrophage phenotype from the pro-inflammatory “classically activated M1” to the pro-resolving “alternatively activated M2,” as well as direct blockade of PMN transmigration and reactive oxygen species production, which could explain, at least in part, the beneficial actions of this LM in experimental colitis (103). The effects of PUFAs on macrophages are schematically summarized in Figure 2C.
Studies on the effects of PUFA derivatives on the adaptive immune system in the gut are still in their infancy. In general, both DHA and EPA were observed to reduce in vitro T cell proliferation and to decrease the expression of both Th1 and Th2 cytokine IL-2 (Figure 2D). Recent works have also unveiled the effects of ω-3 PUFAs on Th17 cells (104–106). However, only few in vivo studies have shown a real effect of pro-resolving LMs in T cell reactivity in the gut; these will be described in the paragraph on animal studies.
The excessive transfer of various immune cell types from the peripheral blood to the affected gastrointestinal tracts of IBD patients, depends not only on surface molecules expressed by activated leukocytes, but also on high levels of adhesion molecules expressed by endothelial cells (14). Endothelial cells are key regulators of the inflammatory response, not only providing in the steady state an anti-inflammatory and anti-coagulatory surface, but also controlling which cell type enters the site of inflammation (107). Thus, alterations of endothelial cells may cause an imbalance between initiation of pro-inflammatory mechanisms and those that promote resolution and restitution of tissue homeostasis, ultimately leading to chronic inflammatory disorders, such as IBD. Patients with IBD are indeed characterized by increased vascularization, and excessive release of angiogenic factors (108, 109). Resolving LMs were observed to exert anti-inflammatory and anti-angiogenic effects on the gut endothelium. For example, Ibrahim and colleagues demonstrated that DHA is able to decrease vascular cell adhesion protein 1 (VCAM-1), TLR4, COX-2, and vascular endothelial growth factor receptor 2 (VEGFR-2) expression and reduce the production of IL-6, IL-8, GM-CSF, and PGE-2 in intestinal microvascular endothelial cells (HIMEC) stimulated with IL-1β. Moreover, administration of ALA during the TNBS model resulted in the decrease of intercellular adhesion molecule 1 (ICAM-1), VCAM-1, and VEGFR-2 expression levels, thus leading to suppression of angiogenesis in the inflamed colon (Figure 2E) (110). Interestingly, Ungaro et al. demonstrated that the Major Facilitator Superfamily Domain containing 2A (MFSD2A) may act as a new player in the resolution of intestinal inflammation, likely promoting endothelial production of DHA-derived pro-resolving mediators (20). In this study, lentiviral induction of MFSD2A conferred anti-angiogenic properties to HIMEC, reducing in vitro capillary formation and proliferation, and significantly inhibited TNFα-triggered inflammatory machinery of NF-kB signaling, via production of pro-resolving DHA-derived metabolites. These findings suggest that stimulating MFSD2A activity in intestinal endothelial cells could be a novel and powerful therapeutic approach to treat IBD.
Overall we have reported that the main modes of action of PUFAs in the inflamed gut are: (i) inhibition of leukocyte chemotaxis, reduced expression of adhesion molecules, and diminished leukocyte-endothelial adhesive interactions, (ii) modulation of epithelial biological functions and interactions with PMN, (iii) suppression of macrophage phagocytic activities, (iv) production of inflammatory cytokines, and (v) modulation of endothelial functions and T-lymphocyte reactivity. However, there are other mechanisms of action that have not been described in the intestine, but that may be crucial for further studies in IBD. For example, it has been demonstrated that resolving ω-3 PUFAs, such as EPA and DHA, can compete with the enzymes that convert AA into pro-inflammatory eicosanoids, thus inhibiting the release of inflammatory cytokines (e.g., TNF-α and IL1-β) (111). Furthermore, administration of ω-3 PUFA derivatives may benefit IBD patients by change in the lipid composition of intestinal cell membranes, activation of anti-inflammatory proteins such as the transcription peroxisome proliferator activated receptor γ (PPAR-γ), and reduction in the gut production of pro-inflammatory molecules, like NF-κB, LTs, and PGs (35, 45–47, 112, 113).
Role of PUFAs in Animal Models of IBD
One of the first studies unveiling the contribution of PUFAs in IBD progression was done by Hudert et al., who exploited a transgenic mouse carrying Caenorhabditis elegans fat-1 gene, encoding for a specific desaturase capable of producing ω-3 PUFAs from ω-6 PUFAs (114). As a consequence, this transgenic mouse is characterized by a low ratio of ω-6/ω-3 fatty acids in its tissues and organs (115). They showed that fat-1 transgenic mice subjected to the DSS protocol of chemically induced experimental colitis, had significantly reduced signs of colon inflammation, in terms of both clinical manifestation and pathology, than wild-type littermates. Such amelioration was positively correlated with the production of anti-inflammatory ω-3 PUFA derivatives, reduced levels of pro-inflammatory cytokines and a concomitant increase of mucus-specific factors in their colons. Moreover, beside a reduced number of Th17 cells in lymphoid tissues, they also observed a reduced expression of Th17 cell type-specific cytokines and chemokine receptors specifically in the colonic mucosa, indicating a role for ω-3 PUFAs on T cell reactivity. The reduced susceptibility to chemically induced colitis in fat-1 mice is likely to result from reduced activation of the NF-κB pathway and decreased expression of TNFα, IL-1β, and inducible NO synthase. Conversely, the enhanced protection conferred by a thicker mucus layer in these mice was probably due to the concomitant up-regulation of trefoil factor 3, toll-interacting protein, and zonula occludens-1.
Initial studies on the efficacy of PUFAs in animal models of IBD considered the use of PUFA precursors instead of single metabolites. One of the first fatty acid used in experimental colitis was conjugated LA (CLA), a mixture of 28 isomers of LA (116); this has been tested in pig models of colitis. Animal treated with CLA showed reduced signs of intestinal inflammation, accompanied by decreased serum levels of TNF-α and NF-κB, and increased amount of transforming growth factor β and PPAR-γ (117). These findings were confirmed in two different experimental mouse models of colitis, either chemically (DSS)- or CD4-induced (118).
Other studies have focused their attention on the ω-6/ω-3 PUFA ratio. During the DSS-induced colitis model, mice administered with ALA-enriched diet, consequently resulting in a decreased uptake of LA, showed less severe colitis, with a markedly alleviated intestinal inflammation (119). The beneficial effects exerted by the ALA-enriched diet was probably due to the reduced PMN influx into the colonic mucosa, because of the decreased activity of both myeloperoxidase (MPO) and alkaline phosphatase. In addition, ALA supplementation blocked TNF-α and IL-1β up-regulation, by comparison with the control group.
Following studies were designed to use specific PUFA metabolites rather than precursors, with ω-3 EPA- and DHA-derived LMs as main candidates for both animal and clinical trials.
The first work involving ω-3 PUFA derivatives and IBD were conducted by using both TNBS- and DSS-induced colitis. Arita and colleagues demonstrated that RvE1 exerts protective effects in TNBS-induced intestinal inflammation, in terms of reduced body weight loss, colon shortening, and tissue damage, by reducing PMN flux into the colonic mucosa, and, at the same time, by limiting either the production of TNFα and IL-12, or the expression of pro-inflammatory enzymes, like COX-2. The authors also showed that the expression of the RvE1 receptor ChemR23 was up-regulated in colonic mucosa of TNBS-treated animals (36).
Similar effects were observed in the DSS-induced model of colitis by Ishida et al., who demonstrated that repeated administrations of RvE1 were able to dampen colitis severity in terms of body weight loss, colon shortening, and histological score (41). Concomitantly, they observed a reduction in NF-kB phosphorylation, TNFα, IL-1β, and IL-6 levels in colonic tissues, along with higher levels of ChemR23 mRNA, supporting a possible role for this receptor in the pathogenesis of intestinal inflammation (41). Other groups confirmed these findings additionally indicating that an interplay might exist between ALPI and RvE1 that ultimately leads to resolution of intestinal inflammation.
In 2011, Bento et al. showed that aspirin-triggered (AT)-RvD1 and RvD2 protect mice against both TNBS- and DSS-induced colitis (47). In this study, the preventive administration of these resolvins significantly ameliorated clinical manifestations, such as body weight loss, disease activity index, colonic damage, and colon shortening. Beside these clinical findings, they showed these mice to produce reduced levels of pro-inflammatory cytokines, and diminished activation of NF-kB pathway and expression of VCAM-1, ICAM-1, and leukocyte function-associated antigen-1. Finally, the authors demonstrated that blockage of LXA4 receptor (ALX), reversed the (AT)-RvD1protective effects in DSS-induced colitis, concluding that (AT)-RvD1 action may depend on ALX activation.
Other DHA-derived pro-resolving mediators, such as maresins, have also shown fundamental properties in experimental IBD. In fact, preventive or therapeutic administration of MaR1 (103) demonstrated for the first time that this DHA metabolite protects mice against both acute and chronic DSS-induced colitis, reducing disease activity index, colon shortening, body weight loss, and MPO activity. In addition, the authors demonstrated that MaR1 inhibited the production of pro-inflammatory cytokines like IL1-β, IL-6, TNF-α, and IFN-γ in colon tissue, together with down-regulation of NFk-B activation and diminished neutrophil transmigration in the inflamed mucosa (103). Similar results were obtained with the TNBS-induced model of colitis.
A very recent work from Gobbetti et al. shows that exogenous administration of LMs derived from ω-3 docosapentaenoic acid (ω-3 DPA), an intermediary product between EPA and DHA, named protectin D1n−3 DPA (PD1n−3 DPA) and resolvin D5n−3 DPA (RvD5n−3 DPA), was effective in preventing the acute model of DSS-induced colitis, in terms of reduced colon length, and microscopic damage score (120). These protective effects were partially linked to reduced granulocyte trafficking and PMN–endothelial interactions, which may occur downstream adhesion molecule activation. The translational impact of these data was determined not only by the ability of PD1n−3 DPA and RvD5n−3 DPA to reduce human neutrophil adhesion onto TNF-α-activated human endothelial monolayers, but also to the identification of ω-3 DPA metabolites in human colon biopsies. Using targeted LC-MS/MS-based LM metabololipidomics on colonic biopsies from controls and IBD patients they observed that LTB4, PGE2, and TX B2 were significantly increased in inflamed tissues in comparison with controls. Notably, they showed that RvD5n−3 DPA and PD1n−3 DPA were augmented in tissue biopsies from IBD patients compared with those from control. This finding on human IBD samples is in contrast with the fact that RvD5n−3 DPA and PD1n−3 DPA exert protective effects against chemically induced acute colitis, and warrants further investigation. There may be a dysfunctional susceptibility of cells targeted by these mediators in IBD. Moreover, it would be interesting to distinguish the effects of RvD5n−3 DPA and PD1n−3 DPA in patients with UC versus CD.
The last study that needs to be mentioned has been done by Meister and Ghosh, who treated IBD patient-derived biopsies with fish oil. They found reduced inflammation in terms of high IL-1a/IL-1b ratio in tissues derived from patients with UC, but not in tissues from patients with CD. These contrasting outcomes indicate that variations in diet composition may influence the success of a nutritional therapy for UC or CD patients (121). All mentioned animal studies are summarized in Table 1.
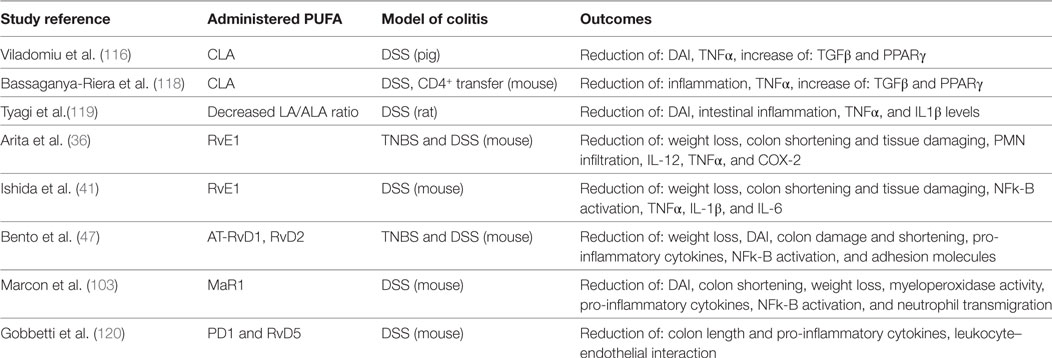
Table 1. Polyunsaturated fatty acid (PUFA) administration in animal models of IBD.
It is worth of note that although the majority of pre-clinical studies on animal model of IBD are promising and provide strong or mild anti-inflammatory properties of ω-3 PUFAs (122–131), other works revealed that an abundant intake of dietary ω-3 PUFAs could even worsen the clinical signs of colitis (132–135). This discrepancy may be explained by different treatment and dose regimen, by different animal facility conditions, and different racemic mixture that could have been used to treat mice. In any case, this must be taken into consideration when animal studies need to be translated into clinical management of IBD.
Clinical Application
As for animal models, many attempts have been done to prove ω-3 PUFA efficacy in human studies. The great therapeutic potential of ω-3 PUFAs has been also encouraged by some works reporting alterations in the production of pro-resolving LM. For example, Pearl and colleagues revealed the ω-6/ω-3 PUFA composition were altered in the inflamed gut mucosa of patients with active UC, in comparison with healthy samples (136). Additionally, Masoodi et al. reported that pro-inflammatory PUFA metabolites (PGD2, PGE2, TXB2, 5-HETE, 11-HETE, 12-HETE, and 15-HETE) not only were increased in the inflamed mucosa of patients with active UC, but their levels also correlated with the disease activity (137). Interestingly, our group recently characterized colonic biopsies isolated from patients with active UC showing that the production of pro-resolving DHA-derived metabolites was defective in inflamed mucosa in comparison with colon tissues from patients with UC in remission and healthy controls. This indicates that pro-resolving mechanisms are deficient in patients with active UC (20), suggesting that ω-3 PUFA administration can be exploited as a novel therapeutic approach to treat IBD.
The majority of studies that have been performed so far uses diet as way of delivery of ω-3 PUFAs, in combination or not with the conventional IBD therapies (138). John et al. found that the intake of dietary EPA and DHA was conversely correlated with the risk of developing incident UC (139). Similarly, in a cohort of patients with CD, the dietary DHA intake was conversely correlated with the development of incident CD, with statistical significance (140). Moreover, clinical trial for CD and UC revealed the beneficial effects of ω-3 enriched diet (141–151) in terms of clinical and histological parameters. Among these, Belluzzi et al. showed that in patients with CD in remission, fish-oil enriched diet is effective in decreasing relapse frequency (146). In another multicenter, randomized, double-blind, clinical trial the beneficial role of fish-oil administration in patients with UC was demonstrated. The positive clinical outcome was expressed in terms of reduced rectal leukotriene B4 (LTB4) levels, improvements in histological scores, and gain of weight.
Omega-3 PUFA administration may also be effective in pediatric patients. In children with CD treated with mesalazine, diet supplementation with ω-3 PUFAs significantly reduced the frequency of relapse within 1-year observation in comparison with patients receiving placebo, consisting in olive oil (145).
However, in a clinical trial (EPIC-1 and -2) conducted by Feagan et al. the efficacy of a mixture of ω-3 PUFA was revised; in fact, the treatment was not effective in preventing relapse and maintaining remission in CD patients (152). All clinical studies are summarized in Table 2.
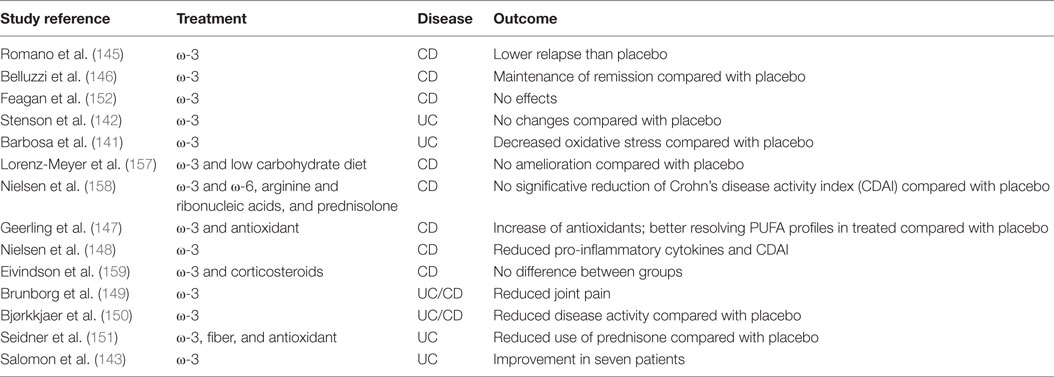
Table 2. Clinical studies with the use of polyunsaturated fatty acids (PUFAs) in inflammatory bowel disease.
The opportunity of clinical application for PUFAs has been evaluated by few systematic reviews and meta-analyses. For example, the study by Turner and colleagues found significant positive effects of ω-3 PUFA supplementation in CD patients. However, these conclusions derived from only six trials that are highly heterogeneous. Analysis of three clinical trials on ω-3 PUFA administration in patients with UC described no significant outcome. Thus, the authors concluded that data available were insufficient to prescribe the use of ω-3 PUFAs for the maintenance of remission in CD and UC (153, 154).
Overall, the studies conducted so far are elusive and displayed no real evidence of efficacy (138, 155–159). This might be due to different reasons: (a) the ω-3-based diet needs to be tightly controlled in IBD patients; (b) the administration of ω-3 PUFA (DHA or EPA) through diet is not effective because of insufficient intestinal absorption due to ulcers or because of biochemical modification of PUFAs when they are in the systemic circulation; (c) EPA and DHA are general precursors of a plethora of specific pro-resolving lipids, that, by definition, are locally and timely regulated. Therefore, the administration through the diet does not help to finely control such metabolism; (d) patients may harbor genetic predisposition impeding the correct DHA or EPA metabolism, thus leading to insufficient production of bioactive pro-resolving LMs.
Concluding Remarks
In IBD patients, diet and lifestyle changes, conventional or newly identified drugs, do not always resolve inflammation and relieve symptoms of the disease. One of theories formulated in the last few years is that anti-inflammatory agents do not alter the course of the disease, because naturally occurring resolution programs may have been subverted. Few studies, including findings from our group, showed that eventual dysfunctions in resolution pathways and/or deficits in precursors of pro-resolving mediators, such as ω-3 PUFAs, may lead to persistent inflammation and provoke alteration in gut mucosa homeostasis, thus being part of IBD pathogenesis. For this reason, the use of pro-resolving PUFAs, particularly the ω-3 ones, brings new possibilities to the treatment of IBD, and could be of great interest to pharmacological industry.
Although numerous pre-clinical and clinical studies employing the use of PUFAs, either as fatty acid precursors or single metabolites, showed controversial results, there is still much more to discover about the beneficial effects of these molecules, particularly in the IBD field. It would be important not only to uncover new cellular and molecular processes modulated by PUFAs under gut inflammatory conditions, but also to unveil novel biosynthetic pathways of these pro-resolving LMs that may likely be dysregulated in IBD. Ways of delivery, safety, dosage, and regimen treatment, and interaction with other drugs should also be further addressed in order to establish the most efficient replacement therapy. We suppose that either PUFAs or new medications specifically promoting resolution pathways will be much better tolerated by patients with IBD, mimicking the physiological processes through which inflammation naturally occurs in the organism, with the advantage of avoiding immune suppression.
Author Contributions
FU, FR, SD, and SDA conceived and wrote the manuscript, and realized the figure.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors would like to acknowledge the Broad Medical Research Program under grant agreement 510262 to SD.
References
1. Neurath MF. Cytokines in inflammatory bowel disease. Nat Rev Immunol (2014) 14:329–42. doi:10.1038/nri3661
2. Serhan CN, Chiang N, Dalli J. The resolution code of acute inflammation: novel pro-resolving lipid mediators in resolution. Semin Immunol (2015) 27:200–15. doi:10.1016/j.smim.2015.03.004
3. Danese S. Immune and nonimmune components orchestrate the pathogenesis of inflammatory bowel disease. Am J Physiol Gastrointest Liver Physiol (2011) 300:G716–22. doi:10.1152/ajpgi.00472.2010
4. Huang Y, Chen Z. Inflammatory bowel disease related innate immunity and adaptive immunity. Am J Transl Res (2016) 8:2490–7.
5. Viggiano D, Ianiro G, Vanella G, Bibbò S, Bruno G, Simeone G, et al. Gut barrier in health and disease: focus on childhood. Eur Rev Med Pharmacol Sci (2015) 19:1077–85.
6. Maloy KJ, Powrie F. Intestinal homeostasis and its breakdown in inflammatory bowel disease. Nature (2011) 474:298–306. doi:10.1038/nature10208
7. Blumberg RS. Inflammation in the intestinal tract: pathogenesis and treatment. Dig Dis (2009) 27:455–64. doi:10.1159/000235851
8. Messina V, Buccione C, Marotta G, Ziccheddu G, Signore M, Mattia G, et al. Gut mesenchymal stromal cells in immunity. Stem Cells Int (2017) 2017:8482326. doi:10.1155/2017/8482326
9. Sala E, Genua M, Petti L, Anselmo A, Arena V, Cibella J, et al. Mesenchymal stem cells reduce colitis in mice via release of TSG6, independently of their localization to the intestine. Gastroenterology (2015) 149:163–76.e20. doi:10.1053/j.gastro.2015.03.013
10. Scaldaferri F, Sans M, Vetrano S, Correale C, Arena V, Pagano N, et al. The role of MAPK in governing lymphocyte adhesion to and migration across the microvasculature in inflammatory bowel disease. Eur J Immunol (2009) 39:290–300. doi:10.1002/eji.200838316
11. Rutella S, Vetrano S, Correale C, Graziani C, Sturm A, Spinelli A, et al. Enhanced platelet adhesion induces angiogenesis in intestinal inflammation and inflammatory bowel disease microvasculature. J Cell Mol Med (2011) 15:625–34. doi:10.1111/j.1582-4934.2010.01033.x
12. Danese S, Sans M, de la Motte C, Graziani C, West G, Phillips MH, et al. Angiogenesis as a novel component of inflammatory bowel disease pathogenesis. Gastroenterology (2006) 130:2060–73. doi:10.1053/j.gastro.2006.03.054
13. D’Alessio S, Correale C, Tacconi C, Gandelli A, Pietrogrande G, Vetrano S, et al. VEGF-C-dependent stimulation of lymphatic function ameliorates experimental inflammatory bowel disease. J Clin Invest (2014) 124:3863–78. doi:10.1172/JCI72189
14. Danese S, Fiocchi C. Endothelial cell-immune cell interaction in IBD. Dig Dis (2016) 34:43–50. doi:10.1159/000442925
15. Vindigni SM, Zisman TL, Suskind DL, Damman CJ. The intestinal microbiome, barrier function, and immune system in inflammatory bowel disease: a tripartite pathophysiological circuit with implications for new therapeutic directions. Therap Adv Gastroenterol (2016) 9:606–25. doi:10.1177/1756283X16644242
16. Davis EC, Wang M, Donovan SM. The role of early life nutrition in the establishment of gastrointestinal microbial composition and function. Gut Microbes (2017) 8:143–71. doi:10.1080/19490976.2016.1278104
17. Carroll IM, Threadgill DW, Threadgill DS. The gastrointestinal microbiome: a malleable, third genome of mammals. Mamm Genome (2009) 20:395–403. doi:10.1007/s00335-009-9204-7
18. Spor A, Koren O, Ley R. Unravelling the effects of the environment and host genotype on the gut microbiome. Nat Rev Microbiol (2011) 9:279–90. doi:10.1038/nrmicro2540
19. Das UN. Inflammatory bowel disease as a disorder of an imbalance between pro- and anti-inflammatory molecules and deficiency of resolution bioactive lipids. Lipids Health Dis (2016) 15:11. doi:10.1186/s12944-015-0165-4
20. Ungaro F, Tacconi C, Massimino L, Corsetto PA, Correale C, Fonteyne P, et al. MFSD2A promotes endothelial generation of inflammation-resolving lipid mediators and reduces colitis in mice. Gastroenterology (2017). doi:10.1053/j.gastro.2017.07.048
21. Levy BD, Clish CB, Schmidt B, Gronert K, Serhan CN. Lipid mediator class switching during acute inflammation: signals in resolution. Nat Immunol (2001) 2:612–9. doi:10.1038/89759
22. Serhan CN. Novel eicosanoid and docosanoid mediators: resolvins, docosatrienes, and neuroprotectins. Curr Opin Clin Nutr Metab Care (2005) 8:115–21. doi:10.1097/00075197-200503000-00003
23. Perretti M. The resolution of inflammation: new mechanisms in patho-physiology open opportunities for pharmacology. Semin Immunol (2015) 27:145–8. doi:10.1016/j.smim.2015.06.001
24. Serhan CN, Savill J. Resolution of inflammation: the beginning programs the end. Nat Immunol (2005) 6:1191–7. doi:10.1038/ni1276
25. Headland SE, Norling LV. The resolution of inflammation: principles and challenges. Semin Immunol (2015) 27:149–60. doi:10.1016/j.smim.2015.03.014
26. Serhan CN. Novel lipid mediators and resolution mechanisms in acute inflammation: to resolve or not? Am J Pathol (2010) 177:1576–91. doi:10.2353/ajpath.2010.100322
27. Simopoulos A. An increase in the omega-6/omega-3 fatty acid ratio increases the risk for obesity. Nutrients (2016) 8:128. doi:10.3390/nu8030128
28. Shinohara M, Serhan CN. Novel endogenous proresolving molecules: essential fatty acid-derived and gaseous mediators in the resolution of inflammation. J Atheroscler Thromb (2016) 23:655–64. doi:10.5551/jat.33928
29. Simopoulos A. The importance of the ratio of omega-6/omega-3 essential fatty acids. Biomed Pharmacother (2002) 56:365–79. doi:10.1016/S0753-3322(02)00253-6
30. Skulas-Ray AC. Omega-3 fatty acids and inflammation: a perspective on the challenges of evaluating efficacy in clinical research. Prostaglandins Other Lipid Mediat (2015) 116–117C:104–11. doi:10.1016/j.prostaglandins.2015.02.001
31. Sugimoto MA, Sousa LP, Pinho V, Perretti M, Teixeira MM. Resolution of inflammation: what controls its onset? Front Immunol (2016) 7:160. doi:10.3389/fimmu.2016.00160
32. Serhan CN, Clish CB, Brannon J, Colgan SP, Chiang N, Gronert K. Novel functional sets of lipid-derived mediators with antiinflammatory actions generated from omega-3 fatty acids via cyclooxygenase 2-nonsteroidal antiinflammatory drugs and transcellular processing. J Exp Med (2000) 192:1197–204. doi:10.1084/jem.192.8.1197
33. Goh J, Godson C, Brady HR, Macmathuna P. Lipoxins: pro-resolution lipid mediators in intestinal inflammation. Gastroenterology (2003) 124:1043–54. doi:10.1053/gast.2003.50154
34. Schmitz G, Ecker J. The opposing effects of n-3 and n-6 fatty acids. Prog Lipid Res (2008) 47:147–55. doi:10.1016/j.plipres.2007.12.004
35. Calder PC. Marine omega-3 fatty acids and inflammatory processes: effects, mechanisms and clinical relevance. Biochim Biophys Acta (2015) 1851:469–84. doi:10.1016/j.bbalip.2014.08.010
36. Arita M, Yoshida M, Hong S, Tjonahen E, Glickman JN, Petasis NA, et al. Resolvin E1, an endogenous lipid mediator derived from omega-3 eicosapentaenoic acid, protects against 2,4,6-trinitrobenzene sulfonic acid-induced colitis. Proc Natl Acad Sci U S A (2005) 102:7671–6. doi:10.1073/pnas.0409271102
37. Buckley CD, Gilroy DW, Serhan CN. Proresolving lipid mediators and mechanisms in the resolution of acute inflammation. Immunity (2014) 40:315–27. doi:10.1016/j.immuni.2014.02.009
38. Randhawa PK, Singh K, Singh N, Jaggi AS. A review on chemical-induced inflammatory bowel disease models in rodents. Korean J Physiol Pharmacol (2014) 18:279. doi:10.4196/kjpp.2014.18.4.279
39. Calder PC. Omega-3 polyunsaturated fatty acids and inflammatory processes: nutrition or pharmacology? Br J Clin Pharmacol (2013) 75:645–62. doi:10.1111/j.1365-2125.2012.04374.x
40. Hong S, Gronert K, Devchand PR, Moussignac R-L, Serhan CN. Novel docosatrienes and 17S-resolvins generated from docosahexaenoic acid in murine brain, human blood, and glial cells. Autacoids in anti-inflammation. J Biol Chem (2003) 278:14677–87. doi:10.1074/jbc.M300218200
41. Ishida T, Yoshida M, Arita M, Nishitani Y, Nishiumi S, Masuda A, et al. Resolvin E1, an endogenous lipid mediator derived from eicosapentaenoic acid, prevents dextran sulfate sodium-induced colitis. Inflamm Bowel Dis (2010) 16:87–95. doi:10.1002/ibd.21029
42. Kremer JM. n-3 fatty acid supplements in rheumatoid arthritis. Am J Clin Nutr (2000) 71:349S–51S.
43. Marion-Letellier R, Savoye G, Beck PL, Panaccione R, Ghosh S. Polyunsaturated fatty acids in inflammatory bowel diseases: a reappraisal of effects and therapeutic approaches. Inflamm Bowel Dis (2013) 19:650–61. doi:10.1097/MIB.0b013e3182810122
44. Teitelbaum JE, Allan Walker W. Review: the role of omega 3 fatty acids in intestinal inflammation. J Nutr Biochem (2001) 12:21–32. doi:10.1016/S0955-2863(00)00141-8
45. Farrukh A, Mayberry JF. Is there a role for fish oil in inflammatory bowel disease? World J Clin cases (2014) 2:250–2. doi:10.12998/wjcc.v2.i7.250
46. Salem N, Eggersdorfer M. Is the world supply of omega-3 fatty acids adequate for optimal human nutrition? Curr Opin Clin Nutr Metab Care (2015) 18:147–54. doi:10.1097/MCO.0000000000000145
47. Bento AF, Claudino RF, Dutra RC, Marcon R, Calixto JB. Omega-3 fatty acid-derived mediators 17(R)-hydroxy docosahexaenoic acid, aspirin-triggered resolvin D1 and resolvin D2 prevent experimental colitis in mice. J Immunol (2011) 187:1957–69. doi:10.4049/jimmunol.1101305
48. de Vasconcelos Generoso S, Rodrigues NM, Trindade LM, Paiva NC, Cardoso VN, Carneiro CM, et al. Dietary supplementation with omega-3 fatty acid attenuates 5-fluorouracil induced mucositis in mice. Lipids Health Dis (2015) 14:54. doi:10.1186/s12944-015-0052-z
49. Arisue A, Shimojima N, Tomiya M, Shimizu T, Harada D, Nakayama M, et al. Effect of an omega-3 lipid emulsion in reducing oxidative stress in a rat model of intestinal ischemia-reperfusion injury. Pediatr Surg Int (2012) 28:913–8. doi:10.1007/s00383-012-3144-0
50. Koch S, Nusrat A. The life and death of epithelia during inflammation: lessons learned from the gut. Annu Rev Pathol (2012) 7:35–60. doi:10.1146/annurev-pathol-011811-120905
51. Ivanov AI, Parkos CA, Nusrat A. Cytoskeletal regulation of epithelial barrier function during inflammation. Am J Pathol (2010) 177:512–24. doi:10.2353/ajpath.2010.100168
52. Cornick S, Tawiah A, Chadee K. Roles and regulation of the mucus barrier in the gut. Tissue Barriers (2015) 3:e982426. doi:10.4161/21688370.2014.982426
53. Landy J, Ronde E, English N, Clark SK, Hart AL, Knight SC, et al. Tight junctions in inflammatory bowel diseases and inflammatory bowel disease associated colorectal cancer. World J Gastroenterol (2016) 22:3117. doi:10.3748/wjg.v22.i11.3117
54. Buisine MP, Desreumaux P, Debailleul V, Gambiez L, Geboes K, Ectors N, et al. Abnormalities in mucin gene expression in Crohn’s disease. Inflamm Bowel Dis (1999) 5:24–32. doi:10.1097/00054725-199902000-00004
55. Johansson MEV, Gustafsson JK, Holmén-Larsson J, Jabbar KS, Xia L, Xu H, et al. Bacteria penetrate the normally impenetrable inner colon mucus layer in both murine colitis models and patients with ulcerative colitis. Gut (2014) 63:281–91. doi:10.1136/gutjnl-2012-303207
56. Elsbach P, Weiss J. Role of the bactericidal/permeability-increasing protein in host defence. Curr Opin Immunol (1998) 10:45–9. doi:10.1016/S0952-7915(98)80030-7
57. Campbell EL, MacManus CF, Kominsky DJ, Keely S, Glover LE, Bowers BE, et al. Resolvin E1-induced intestinal alkaline phosphatase promotes resolution of inflammation through LPS detoxification. Proc Natl Acad Sci U S A (2010) 107:14298–303. doi:10.1073/pnas.0914730107
58. Bates JM, Akerlund J, Mittge E, Guillemin K. Intestinal alkaline phosphatase detoxifies lipopolysaccharide and prevents inflammation in zebrafish in response to the gut microbiota. Cell Host Microbe (2007) 2:371–82. doi:10.1016/j.chom.2007.10.010
59. Anbazhagan AN, Priyamvada S, Gujral T, Bhattacharyya S, Alrefai WA, Dudeja PK, et al. A novel anti-inflammatory role of GPR120 in intestinal epithelial cells. Am J Physiol Cell Physiol (2016) 310:C612–21. doi:10.1152/ajpcell.00123.2015
60. Marion-Letellier R, Savoye G, Ghosh S. Polyunsaturated fatty acids and inflammation. IUBMB Life (2015) 67:659–67. doi:10.1002/iub.1428
61. Mobraten K, Haug TM, Kleiveland CR, Lea T. Omega-3 and omega-6 PUFAs induce the same GPR120-mediated signalling events, but with different kinetics and intensity in Caco-2 cells. Lipids Health Dis (2013) 12:101. doi:10.1186/1476-511X-12-101
62. Zhao J, Wang H, Shi P, Wang W, Sun Y. GPR120, a potential therapeutic target for experimental colitis in IL-10 deficient mice. Oncotarget (2017) 8:8397–405. doi:10.18632/oncotarget.14210
63. Fredborg M, Theil PK, Jensen BB, Purup S. G protein-coupled receptor120 (GPR120) transcription in intestinal epithelial cells is significantly affected by bacteria belonging to the Bacteroides, Proteobacteria, and Firmicutes phyla. J Anim Sci (2012) 90(Suppl 4):10–2. doi:10.2527/jas.53792
64. Matsuoka K, Kanai T. The gut microbiota and inflammatory bowel disease. Semin Immunopathol (2015) 37:47–55. doi:10.1007/s00281-014-0454-4
65. Rosales C, Demaurex N, Lowell CA, Uribe-Querol E. Neutrophils: their role in innate and adaptive immunity. J Immunol Res (2016) 2016:1469780. doi:10.1155/2016/1469780
66. Nusrat A, Parkos CA, Liang TW, Carnes DK, Madara JL. Neutrophil migration across model intestinal epithelia: monolayer disruption and subsequent events in epithelial repair. Gastroenterology (1997) 113:1489–500. doi:10.1053/gast.1997.v113.pm9352851
67. Nash S, Stafford J, Madara JL. Effects of polymorphonuclear leukocyte transmigration on the barrier function of cultured intestinal epithelial monolayers. J Clin Invest (1987) 80:1104–13. doi:10.1172/JCI113167
68. Kucharzik T, Walsh SV, Chen J, Parkos CA, Nusrat A. Neutrophil transmigration in inflammatory bowel disease is associated with differential expression of epithelial intercellular junction proteins. Am J Pathol (2001) 159:2001–9. doi:10.1016/S0002-9440(10)63051-9
69. Danese S, Fiocchi C. Ulcerative colitis. N Engl J Med (2011) 365:1713–25. doi:10.1056/NEJMra1102942
70. El Kebir D, Filep JG. Targeting neutrophil apoptosis for enhancing the resolution of inflammation. Cells (2013) 2:330–48. doi:10.3390/cells2020330
71. Fullerton JN, O’Brien AJ, Gilroy DW. Pathways mediating resolution of inflammation: when enough is too much. J Pathol (2013) 231:8–20. doi:10.1002/path.4232
72. Colgan SP, Serhan CN, Parkos CA, Delp-Archer C, Madara JL. Lipoxin A4 modulates transmigration of human neutrophils across intestinal epithelial monolayers. J Clin Invest (1993) 92:75–82. doi:10.1172/JCI116601
73. Mangino MJ, Brounts L, Harms B, Heise C. Lipoxin biosynthesis in inflammatory bowel disease. Prostaglandins Other Lipid Mediat (2006) 79:84–92. doi:10.1016/j.prostaglandins.2005.10.004
74. Gewirtz AT, Collier-Hyams LS, Young AN, Kucharzik T, Guilford WJ, Parkinson JF, et al. Lipoxin A4 analogs attenuate induction of intestinal epithelial proinflammatory gene expression and reduce the severity of dextran sodium sulfate-induced colitis. J Immunol (2002) 168:5260–7. doi:10.4049/jimmunol.168.10.5260
75. Fiorucci S, Wallace JL, Mencarelli A, Distrutti E, Rizzo G, Farneti S, et al. A -oxidation-resistant lipoxin A4 analog treats hapten-induced colitis by attenuating inflammation and immune dysfunction. Proc Natl Acad Sci U S A (2004) 101:15736–41. doi:10.1073/pnas.0404722101
76. Schwab JM, Chiang N, Arita M, Serhan CN. Resolvin E1 and protectin D1 activate inflammation-resolution programmes. Nature (2007) 447:869–74. doi:10.1038/nature05877
77. Ariel A, Fredman G, Sun Y-P, Kantarci A, Van Dyke TE, Luster AD, et al. Apoptotic neutrophils and T cells sequester chemokines during immune response resolution through modulation of CCR5 expression. Nat Immunol (2006) 7:1209–16. doi:10.1038/ni1392
78. Colgan SP, Eltzschig HK, Eckle T, Thompson LF. Physiological roles for ecto-5’-nucleotidase (CD73). Purinergic Signal (2006) 2:351–60. doi:10.1007/s11302-005-5302-5
79. Khoury J, Ibla JC, Neish AS, Colgan SP. Antiinflammatory adaptation to hypoxia through adenosine-mediated cullin-1 deneddylation. J Clin Invest (2007) 117:703–11. doi:10.1172/JCI30049
80. Taylor CT, Colgan SP. Hypoxia and gastrointestinal disease. J Mol Med (Berl) (2007) 85:1295–300. doi:10.1007/s00109-007-0277-z
81. Eltzschig HK, Macmanus CF, Colgan SP. Neutrophils as sources of extracellular nucleotides: functional consequences at the vascular interface. Trends Cardiovasc Med (2008) 18:103–7. doi:10.1016/j.tcm.2008.01.006
82. Serhan CN, Chiang N, Van Dyke TE. Resolving inflammation: dual anti-inflammatory and pro-resolution lipid mediators. Nat Rev Immunol (2008) 8:349–61. doi:10.1038/nri2294
83. Bonnans C, Levy BD. Lipid mediators as agonists for the resolution of acute lung inflammation and injury. Am J Respir Cell Mol Biol (2007) 36:201–5. doi:10.1165/rcmb.2006-0269TR
84. Kühl AA, Kakirman H, Janotta M, Dreher S, Cremer P, Pawlowski NN, et al. Aggravation of different types of experimental colitis by depletion or adhesion blockade of neutrophils. Gastroenterology (2007) 133:1882–92. doi:10.1053/j.gastro.2007.08.073
85. Fournier BM, Parkos CA. The role of neutrophils during intestinal inflammation. Mucosal Immunol (2012) 5:354–66. doi:10.1038/mi.2012.24
86. Bressenot A, Salleron J, Bastien C, Danese S, Boulagnon-Rombi C, Peyrin-Biroulet L. Comparing histological activity indexes in UC. Gut (2015) 64:1412–8. doi:10.1136/gutjnl-2014-307477
87. Segal AW, Loewi G. Neutrophil dysfunction in Crohn’s disease. Lancet (1976) 2:219–21. doi:10.1016/S0140-6736(76)91024-2
88. Harbord MWN, Marks DJB, Forbes A, Bloom SL, Day RM, Segal AW. Impaired neutrophil chemotaxis in Crohn’s disease relates to reduced production of chemokines and can be augmented by granulocyte-colony stimulating factor. Aliment Pharmacol Ther (2006) 24:651–60. doi:10.1111/j.1365-2036.2006.03016.x
89. Vazeille E, Buisson A, Bringer M-A, Goutte M, Ouchchane L, Hugot J-P, et al. Monocyte-derived macrophages from Crohn’s disease patients are impaired in the ability to control intracellular adherent-invasive Escherichia coli and exhibit disordered cytokine secretion profile. J Crohns Colitis (2015) 9:410–20. doi:10.1093/ecco-jcc/jjv053
90. Smith PD, Smythies LE, Shen R, Greenwell-Wild T, Gliozzi M, Wahl SM. Intestinal macrophages and response to microbial encroachment. Mucosal Immunol (2011) 4:31–42. doi:10.1038/mi.2010.66
91. Lattin JE, Schroder K, Su AI, Walker JR, Zhang J, Wiltshire T, et al. Expression analysis of G protein-coupled receptors in mouse macrophages. Immunome Res (2008) 4:5. doi:10.1186/1745-7580-4-5
92. Oh DY, Talukdar S, Bae EJ, Imamura T, Morinaga H, Fan W, et al. GPR120 is an omega-3 fatty acid receptor mediating potent anti-inflammatory and insulin-sensitizing effects. Cell (2010) 142:687–98. doi:10.1016/j.cell.2010.07.041
93. Hirasawa A, Tsumaya K, Awaji T, Katsuma S, Adachi T, Yamada M, et al. Free fatty acids regulate gut incretin glucagon-like peptide-1 secretion through GPR120. Nat Med (2005) 11:90–4. doi:10.1038/nm1168
94. Li X, Yu Y, Funk CD. Cyclooxygenase-2 induction in macrophages is modulated by docosahexaenoic acid via interactions with free fatty acid receptor 4 (FFA4). FASEB J (2013) 27:4987–97. doi:10.1096/fj.13-235333
95. Im D-S. Functions of omega-3 fatty acids and FFA4 (GPR120) in macrophages. Eur J Pharmacol (2016) 785:36–43. doi:10.1016/j.ejphar.2015.03.094
96. Allison MC, Cornwall S, Poulter LW, Dhillon AP, Pounder RE. Macrophage heterogeneity in normal colonic mucosa and in inflammatory bowel disease. Gut (1988) 29:1531–8. doi:10.1136/gut.29.11.1531
97. Mahida YR, Wu KC, Jewell DP. Respiratory burst activity of intestinal macrophages in normal and inflammatory bowel disease. Gut (1989) 30:1362–70. doi:10.1136/gut.30.10.1362
98. Godson C, Mitchell S, Harvey K, Petasis NA, Hogg N, Brady HR. Cutting edge: lipoxins rapidly stimulate nonphlogistic phagocytosis of apoptotic neutrophils by monocyte-derived macrophages. J Immunol (2000) 164:1663–7. doi:10.4049/jimmunol.164.4.1663
99. Serhan CN, Yang R, Martinod K, Kasuga K, Pillai PS, Porter TF, et al. Maresins: novel macrophage mediators with potent antiinflammatory and proresolving actions. J Exp Med (2009) 206:15–23. doi:10.1084/jem.20081880
100. Dalli J, Zhu M, Vlasenko NA, Deng B, Haeggstrom JZ, Petasis NA, et al. The novel 13S,14S-epoxy-maresin is converted by human macrophages to maresin 1 (MaR1), inhibits leukotriene A4 hydrolase (LTA4H), and shifts macrophage phenotype. FASEB J (2013) 27:2573–83. doi:10.1096/fj.13-227728
101. Nordgren TM, Heires AJ, Wyatt TA, Poole JA, LeVan TD, Cerutis DR, et al. Maresin-1 reduces the pro-inflammatory response of bronchial epithelial cells to organic dust. Respir Res (2013) 14:51. doi:10.1186/1465-9921-14-51
102. Serhan CN, Dalli J, Karamnov S, Choi A, Park C-K, Xu Z-Z, et al. Macrophage proresolving mediator maresin 1 stimulates tissue regeneration and controls pain. FASEB J (2012) 26:1755–65. doi:10.1096/fj.11-201442
103. Marcon R, Bento AF, Dutra RC, Bicca MA, Leite DFP, Calixto JB. Maresin 1, a proresolving lipid mediator derived from omega-3 polyunsaturated fatty acids, exerts protective actions in murine models of colitis. J Immunol (2013) 191:4288–98. doi:10.4049/jimmunol.1202743
104. Monk JM, Jia Q, Callaway E, Weeks B, Alaniz RC, McMurray DN, et al. Th17 cell accumulation is decreased during chronic experimental colitis by (n-3) PUFA in fat-1 mice. J Nutr (2012) 142:117–24. doi:10.3945/jn.111.147058
105. Monk JM, Hou TY, Turk HF, Weeks B, Wu C, McMurray DN, et al. Dietary n-3 polyunsaturated fatty acids (PUFA) decrease obesity-associated Th17 cell-mediated inflammation during colitis. PLoS One (2012) 7:e49739. doi:10.1371/journal.pone.0049739
106. Allen MJ, Fan Y-Y, Monk JM, Hou TY, Barhoumi R, McMurray DN, et al. n-3 PUFAs reduce T-helper 17 cell differentiation by decreasing responsiveness to interleukin-6 in isolated mouse splenic CD4+ T cells. J Nutr (2014) 144:1306–13. doi:10.3945/jn.114.194407
107. Kadl A, Leitinger N. The role of endothelial cells in the resolution of acute inflammation. Antioxid Redox Signal (2005) 7:1744–54. doi:10.1089/ars.2005.7.1744
108. Deban L, Correale C, Vetrano S, Malesci A, Danese S. Multiple pathogenic roles of microvasculature in inflammatory bowel disease: a Jack of all trades. Am J Pathol (2008) 172:1457–66. doi:10.2353/ajpath.2008.070593
109. Harvey KA, Xu Z, Pavlina TM, Zaloga GP, Siddiqui RA. Modulation of endothelial cell integrity and inflammatory activation by commercial lipid emulsions. Lipids Health Dis (2015) 14:9. doi:10.1186/s12944-015-0005-6
110. Ibrahim A, Mbodji K, Hassan A, Aziz M, Boukhettala N, Coëffier M, et al. Anti-inflammatory and anti-angiogenic effect of long chain n-3 polyunsaturated fatty acids in intestinal microvascular endothelium. Clin Nutr (2011) 30:678–87. doi:10.1016/j.clnu.2011.05.002
111. Arnold C, Markovic M, Blossey K, Wallukat G, Fischer R, Dechend R, et al. Arachidonic acid-metabolizing cytochrome P450 enzymes are targets of {omega}-3 fatty acids. J Biol Chem (2010) 285:32720–33. doi:10.1074/jbc.M110.118406
112. Kris-Etherton PM, Innis S; American Dietetic Association, Dietitians of Canada. Position of the American Dietetic Association and Dietitians of Canada: dietary fatty acids. J Am Diet Assoc (2007) 107:1599–611.
113. Uchiyama K, Nakamura M, Odahara S, Koido S, Katahira K, Shiraishi H, et al. N-3 polyunsaturated fatty acid diet therapy for patients with inflammatory bowel disease. Inflamm Bowel Dis (2010) 16:1696–707. doi:10.1002/ibd.21251
114. Hudert CA, Weylandt KH, Lu Y, Wang J, Hong S, Dignass A, et al. Transgenic mice rich in endogenous omega-3 fatty acids are protected from colitis. Proc Natl Acad Sci U S A (2006) 103:11276–81. doi:10.1073/pnas.0601280103
115. Kang JX, Wang J, Wu L, Kang ZB. Transgenic mice: fat-1 mice convert n-6 to n-3 fatty acids. Nature (2004) 427:504. doi:10.1038/427504a
116. Viladomiu M, Hontecillas R, Yuan L, Lu P, Bassaganya-Riera J. Nutritional protective mechanisms against gut inflammation. J Nutr Biochem (2013) 24:929–39. doi:10.1016/j.jnutbio.2013.01.006
117. Hontecillas R, Wannemeulher MJ, Zimmerman DR, Hutto DL, Wilson JH, Ahn DU, et al. Nutritional regulation of porcine bacterial-induced colitis by conjugated linoleic acid. J Nutr (2002) 132:2019–27.
118. Bassaganya-Riera J, Reynolds K, Martino-Catt S, Cui Y, Hennighausen L, Gonzalez F, et al. Activation of PPAR gamma and delta by conjugated linoleic acid mediates protection from experimental inflammatory bowel disease. Gastroenterology (2004) 127:777–91. doi:10.1053/j.gastro.2004.06.049
119. Tyagi A, Kumar U, Reddy S, Santosh VS, Mohammed SB, Ehtesham NZ, et al. Attenuation of colonic inflammation by partial replacement of dietary linoleic acid with α-linolenic acid in a rat model of inflammatory bowel disease. Br J Nutr (2012) 108:1612–22. doi:10.1017/S0007114511007197
120. Gobbetti T, Dalli J, Colas RA, Federici Canova D, Aursnes M, Bonnet D, et al. Protectin D1n-3 DPA and resolvin D5n-3 DPA are effectors of intestinal protection. Proc Natl Acad Sci U S A (2017) 114:3963–8. doi:10.1073/pnas.1617290114
121. Meister D, Ghosh S. Effect of fish oil enriched enteral diet on inflammatory bowel disease tissues in organ culture: differential effects on ulcerative colitis and Crohn’s disease. World J Gastroenterol (2005) 11:7466–72. doi:10.3748/wjg.v11.i47.7466
122. Bassaganya-Riera J, Hontecillas R. CLA and n-3 PUFA differentially modulate clinical activity and colonic PPAR-responsive gene expression in a pig model of experimental IBD. Clin Nutr (2006) 25:454–65. doi:10.1016/j.clnu.2005.12.008
123. Grimstad T, Bjørndal B, Cacabelos D, Aasprong OG, Janssen EAM, Omdal R, et al. Dietary supplementation of krill oil attenuates inflammation and oxidative stress in experimental ulcerative colitis in rats. Scand J Gastroenterol (2012) 47:49–58. doi:10.3109/00365521.2011.634025
124. Camuesco D, Comalada M, Concha A, Nieto A, Sierra S, Xaus J, et al. Intestinal anti-inflammatory activity of combined quercitrin and dietary olive oil supplemented with fish oil, rich in EPA and DHA (n-3) polyunsaturated fatty acids, in rats with DSS-induced colitis. Clin Nutr (2006) 25:466–76. doi:10.1016/j.clnu.2005.12.009
125. Cho JY, Chi S-G, Chun HS. Oral administration of docosahexaenoic acid attenuates colitis induced by dextran sulfate sodium in mice. Mol Nutr Food Res (2011) 55:239–46. doi:10.1002/mnfr.201000070
126. Kitsukawa Y, Saito H, Suzuki Y, Kasanuki J, Tamura Y, Yoshida S. Effect of ingestion of eicosapentaenoic acid ethyl ester on carrageenan-induced colitis in guinea pigs. Gastroenterology (1992) 102:1859–66. doi:10.1016/0016-5085(92)90306-J
127. Kono H, Fujii H, Ogiku M, Tsuchiya M, Ishii K, Hara M. Enteral diets enriched with medium-chain triglycerides and N-3 fatty acids prevent chemically induced experimental colitis in rats. Transl Res (2010) 156:282–91. doi:10.1016/j.trsl.2010.07.012
128. Nieto N, Torres MI, Ríos A, Gil A. Dietary polyunsaturated fatty acids improve histological and biochemical alterations in rats with experimental ulcerative colitis. J Nutr (2002) 132:11–9.
129. Varnalidis I, Ioannidis O, Karamanavi E, Ampas Z, Poutahidis T, Taitzoglou I, et al. Omega 3 fatty acids supplementation has an ameliorative effect in experimental ulcerative colitis despite increased colonic neutrophil infiltration. Rev Esp Enferm Dig (2011) 103:511–8. doi:10.4321/S1130-01082011001000003
130. Vilaseca J, Salas A, Guarner F, Rodriguez R, Martinez M, Malagelada JR. Dietary fish oil reduces progression of chronic inflammatory lesions in a rat model of granulomatous colitis. Gut (1990) 31:539–44. doi:10.1136/gut.31.5.539
131. Whiting CV, Bland PW, Tarlton JF. Dietary N-3 polyunsaturated fatty acids reduce disease and colonic proinflammatory cytokines in a mouse model of colitis. Inflamm Bowel Dis (2005) 11:340–9. doi:10.1097/01.MIB.0000164016.98913.7c
132. Hegazi RAF, Saad RS, Mady H, Matarese LE, O’Keefe S, Kandil HM. Dietary fatty acids modulate chronic colitis, colitis-associated colon neoplasia and COX-2 expression in IL-10 knockout mice. Nutrition (2006) 22:275–82. doi:10.1016/j.nut.2005.06.006
133. Jia Q, Ivanov I, Zlatev ZZ, Alaniz RC, Weeks BR, Callaway ES, et al. Dietary fish oil and curcumin combine to modulate colonic cytokinetics and gene expression in dextran sodium sulphate-treated mice. Br J Nutr (2011) 106:519–29. doi:10.1017/S0007114511000390
134. Matsunaga H, Hokari R, Kurihara C, Okada Y, Takebayashi K, Okudaira K, et al. Omega-3 fatty acids exacerbate DSS-induced colitis through decreased adiponectin in colonic subepithelial myofibroblasts. Inflamm Bowel Dis (2008) 14:1348–57. doi:10.1002/ibd.20491
135. Woodworth HL, McCaskey SJ, Duriancik DM, Clinthorne JF, Langohr IM, Gardner EM, et al. Dietary fish oil alters T lymphocyte cell populations and exacerbates disease in a mouse model of inflammatory colitis. Cancer Res (2010) 70:7960–9. doi:10.1158/0008-5472.CAN-10-1396
136. Pearl DS, Masoodi M, Eiden M, Brümmer J, Gullick D, McKeever TM, et al. Altered colonic mucosal availability of n-3 and n-6 polyunsaturated fatty acids in ulcerative colitis and the relationship to disease activity. J Crohns Colitis (2014) 8:70–9. doi:10.1016/j.crohns.2013.03.013
137. Masoodi M, Pearl DS, Eiden M, Shute JK, Brown JF, Calder PC, et al. Altered colonic mucosal polyunsaturated fatty acid (PUFA) derived lipid mediators in ulcerative colitis: new insight into relationship with disease activity and pathophysiology. PLoS One (2013) 8:e76532. doi:10.1371/journal.pone.0076532
138. Khan I, Samson SE, Grover AK. Antioxidant supplements and gastrointestinal diseases: a critical appraisal. Med Princ Pract (2017) 26:201–17. doi:10.1159/000468988
139. John S, Luben R, Shrestha SS, Welch A, Khaw K-T, Hart AR. Dietary n-3 polyunsaturated fatty acids and the aetiology of ulcerative colitis: a UK prospective cohort study. Eur J Gastroenterol Hepatol (2010) 22:602–6. doi:10.1097/MEG.0b013e3283352d05
140. Chan SSM, Luben R, Olsen A, Tjonneland A, Kaaks R, Lindgren S, et al. Association between high dietary intake of the n-3 polyunsaturated fatty acid docosahexaenoic acid and reduced risk of Crohn’s disease. Aliment Pharmacol Ther (2014) 39:834–42. doi:10.1111/apt.12670
141. Barbosa DS, Cecchini R, El Kadri MZ, Rodríguez MAM, Burini RC, Dichi I. Decreased oxidative stress in patients with ulcerative colitis supplemented with fish oil omega-3 fatty acids. Nutrition (2003) 19:837–42. doi:10.1016/S0899-9007(03)00162-X
142. Stenson WF, Cort D, Rodgers J, Burakoff R, DeSchryver-Kecskemeti K, Gramlich TL, et al. Dietary supplementation with fish oil in ulcerative colitis. Ann Intern Med (1992) 116:609–14. doi:10.7326/0003-4819-116-8-609
143. Salomon P, Kornbluth AA, Janowitz HD. Treatment of ulcerative colitis with fish oil n-3-omega-fatty acid: an open trial. J Clin Gastroenterol (1990) 12:157–61. doi:10.1097/00004836-199004000-00009
144. Almallah YZ, Ewen SW, El-Tahir A, Mowat NA, Brunt PW, Sinclair TS, et al. Distal proctocolitis and n-3 polyunsaturated fatty acids (n-3 PUFAs): the mucosal effect in situ. J Clin Immunol (2000) 20:68–76. doi:10.1023/A:1006698728816
145. Romano C, Cucchiara S, Barabino A, Annese V, Sferlazzas C. Usefulness of omega-3 fatty acid supplementation in addition to mesalazine in maintaining remission in pediatric Crohn’s disease: a double-blind, randomized, placebo-controlled study. World J Gastroenterol (2005) 11:7118–21. doi:10.3748/wjg.v11.i45.7118
146. Belluzzi A, Brignola C, Campieri M, Pera A, Boschi S, Miglioli M. Effect of an enteric-coated fish-oil preparation on relapses in Crohn’s disease. N Engl J Med (1996) 334:1557–60. doi:10.1056/NEJM199606133342401
147. Geerling BJ, Badart-Smook A, van Deursen C, van Houwelingen AC, Russel MG, Stockbrügger RW, et al. Nutritional supplementation with N-3 fatty acids and antioxidants in patients with Crohn’s disease in remission: effects on antioxidant status and fatty acid profile. Inflamm Bowel Dis (2000) 6:77–84. doi:10.1002/ibd.3780060203
148. Nielsen AA, Jørgensen LGM, Nielsen JN, Eivindson M, Grønbaek H, Vind I, et al. Omega-3 fatty acids inhibit an increase of proinflammatory cytokines in patients with active Crohn’s disease compared with omega-6 fatty acids. Aliment Pharmacol Ther (2005) 22:1121–8. doi:10.1111/j.1365-2036.2005.02698.x
149. Brunborg LA, Madland TM, Lind RA, Arslan G, Berstad A, Frøyland L. Effects of short-term oral administration of dietary marine oils in patients with inflammatory bowel disease and joint pain: a pilot study comparing seal oil and cod liver oil. Clin Nutr (2008) 27:614–22. doi:10.1016/j.clnu.2008.01.017
150. Bjørkkjaer T, Brunborg LA, Arslan G, Lind RA, Brun JG, Valen M, et al. Reduced joint pain after short-term duodenal administration of seal oil in patients with inflammatory bowel disease: comparison with soy oil. Scand J Gastroenterol (2004) 39:1088–94. doi:10.1080/00365520410009429
151. Seidner DL, Lashner BA, Brzezinski A, Banks PLC, Goldblum J, Fiocchi C, et al. An oral supplement enriched with fish oil, soluble fiber, and antioxidants for corticosteroid sparing in ulcerative colitis: a randomized, controlled trial. Clin Gastroenterol Hepatol (2005) 3:358–69. doi:10.1016/S1542-3565(04)00672-X
152. Feagan BG, Sandborn WJ, Mittmann U, Bar-Meir S, D’Haens G, Bradette M, et al. Omega-3 free fatty acids for the maintenance of remission in Crohn disease. JAMA (2008) 299:1690. doi:10.1001/jama.299.14.1690
153. Lev-Tzion R, Griffiths AM, Leder O, Turner D. Omega 3 fatty acids (fish oil) for maintenance of remission in Crohn’s disease. Cochrane Database Syst Rev (2014) (2):CD006320. doi:10.1002/14651858.CD006320.pub4
154. Turner D, Shah PS, Steinhart AH, Zlotkin S, Griffiths AM. Maintenance of remission in inflammatory bowel disease using omega-3 fatty acids (fish oil): a systematic review and meta-analyses. Inflamm Bowel Dis (2011) 17:336–45. doi:10.1002/ibd.21374
155. Barbalho SM, de Alvares Goulart R, Quesada K, Bechara MD, de Cássio Alves de Carvalhoe A. Inflammatory bowel disease: can omega-3 fatty acids really help? Ann Gastroenterol (2016) 29:37–43.
156. MacLean CH, Mojica WA, Morton SC, Pencharz J, Hasenfeld Garland R, Tu W, et al. Effects of omega-3 fatty acids on lipids and glycemic control in type II diabetes and the metabolic syndrome and on inflammatory bowel disease, rheumatoid arthritis, renal disease, systemic lupus erythematosus, and osteoporosis. Evid Rep Technol Assess (Summ) (2004) (89):1–4.
157. Lorenz-Meyer H, Bauer P, Nicolay C, Schulz B, Purrmann J, Fleig WE, et al. Omega-3 fatty acids and low carbohydrate diet for maintenance of remission in Crohn’s disease. A randomized controlled multicenter trial. Study Group Members (German Crohn’s Disease Study Group). Scand J Gastroenterol (1996) 31:778–85. doi:10.3109/00365529609010352
158. Nielsen AA, Nielsen JN, Grønbaek H, Eivindson M, Vind I, Munkholm P, et al. Impact of enteral supplements enriched with omega-3 fatty acids and/or omega-6 fatty acids, arginine and ribonucleic acid compounds on leptin levels and nutritional status in active Crohn’s disease treated with prednisolone. Digestion (2007) 75:10–6. doi:10.1159/000101560
159. Eivindson M, Grønbaek H, Nielsen JN, Frystyk J, Flyvbjerg A, Jørgensen L, et al. Insulin-like growth factors (IGFs) and IGF binding proteins in active Crohn’s disease treated with omega-3 or omega-6 fatty acids and corticosteroids. Scand J Gastroenterol (2005) 40:1214–21. doi:10.1080/00365520510015728
Keywords: resolution of inflammation, pro-resolving lipid mediators, inflammatory bowel disease, polyunsatured fatty acids, pathogenesis, mucosal inflammation, tissue homeostasis
Citation: Ungaro F, Rubbino F, Danese S and D’Alessio S (2017) Actors and Factors in the Resolution of Intestinal Inflammation: Lipid Mediators As a New Approach to Therapy in Inflammatory Bowel Diseases. Front. Immunol. 8:1331. doi: 10.3389/fimmu.2017.01331
Received: 22 July 2017; Accepted: 29 September 2017;
Published: 23 October 2017
Edited by:
Christoph Becker, University of Erlangen-Nuremberg, GermanyCopyright: © 2017 Ungaro, Rubbino, Danese and D’Alessio. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Silvia D’Alessio, c2lsdmlhLmRhbGVzc2lvQGh1bmltZWQuZXU=