 Alessandra Geremia
Alessandra Geremia Carolina V. Arancibia-Cárcamo
Carolina V. Arancibia-Cárcamo- Translational Gastroenterology Unit, Nuffield Department of Experimental Medicine, University of Oxford, Oxford, United Kingdom
Inflammatory bowel disease (IBD) is a chronic inflammatory disorder of the intestine that encompasses Crohn’s disease (CD) and ulcerative colitis. The cause of IBD is unknown, but the evidence suggests that an aberrant immune response toward the commensal bacterial flora is responsible for disease in genetically susceptible individuals. Results from animal models of colitis and human studies indicate a role for innate lymphoid cells (ILC) in the pathogenesis of chronic intestinal inflammation in IBD. ILC are a population of lymphocytes that are enriched at mucosal sites, where they play a protective role against pathogens including extracellular bacteria, helminthes, and viruses. ILC lack an antigen-specific receptor, but can respond to environmental stress signals contributing to the rapid orchestration of an early immune response. Several subsets of ILC reflecting functional characteristics of T helper subsets have been described. ILC1 express the transcription factor T-bet and are characterized by secretion of IFNγ, ILC2 are GATA3+ and secrete IL5 and IL13 and ILC3 depend on expression of RORγt and secrete IL17 and IL22. However, ILC retain a degree of plasticity depending on exposure to cytokines and environmental factors. IL23 responsive ILC have been implicated in the pathogenesis of colitis in several innate murine models through the production of IL17, IFNγ, and GM-CSF. We have previously identified IL23 responsive ILC in the human intestine and found that they accumulate in the inflamed colon and small bowel of patients with CD. Other studies have confirmed accumulation of ILC in CD with increased frequencies of IFNγ-secreting ILC1 in both the intestinal lamina propria and the epithelium. Moreover, IL23 driven IL22 producing ILC have been shown to drive bacteria-induced colitis-associated cancer in mice. Interestingly, our data show increased ILC accumulation in patients with IBD and primary sclerosing cholangitis, who carry an increased risk of developing colorectal cancer. ILC may play an important amplifying role in IBD and IBD-associated cancer, through secretion of inflammatory cytokines and interaction with other immune and non-immune cells. Here, we will review the evidence indicating a role for ILC in the pathogenesis of chronic intestinal inflammation.
Introduction
Innate lymphoid cells (ILC) belong to a family of innate immune cells that share similarities with the phenotype and functions of T lymphocytes. They present classical lymphocyte morphology, but lack a rearranged antigen-specific receptor. ILC do not express T, B, or myeloid cell markers and, therefore, they are lineage negative (Lin−), but express the IL2 receptor (CD25) and the IL7 receptor (IL7R or CD127) (1). ILC also include cytotoxic NK cells and lymphoid tissue inducer (LTi) cells that contribute to lymph node organogenesis during embryonic development (2).
Innate lymphoid cells are crucial players mediating immunity against pathogens and maintenance of tissue homeostasis. They bridge the innate and adaptive immune systems, sense environmental changes, and are able to mediate these processes mainly by secreting and responding to a finely tuned cytokine crosstalk between various cell types. ILC are able to swiftly react to microbial and inflammatory challenges with cytokine production, limiting pathogen spread, and tissue injury. They are strategically located at sites where there is the highest exposure to the outside world and infections are more likely to first arise, such as the intestinal mucosa, the skin, and the lungs. They respond to environmental stress signals contributing to the rapid orchestration of an early immune response to pathogens, such as extracellular bacteria, helminthes, and viruses (Figure 1). However, recent evidence suggests that ILC are also involved in the pathogenesis of chronic inflammatory disorders, including allergic reactions, inflammatory bowel disease (IBD) and cancer (1).
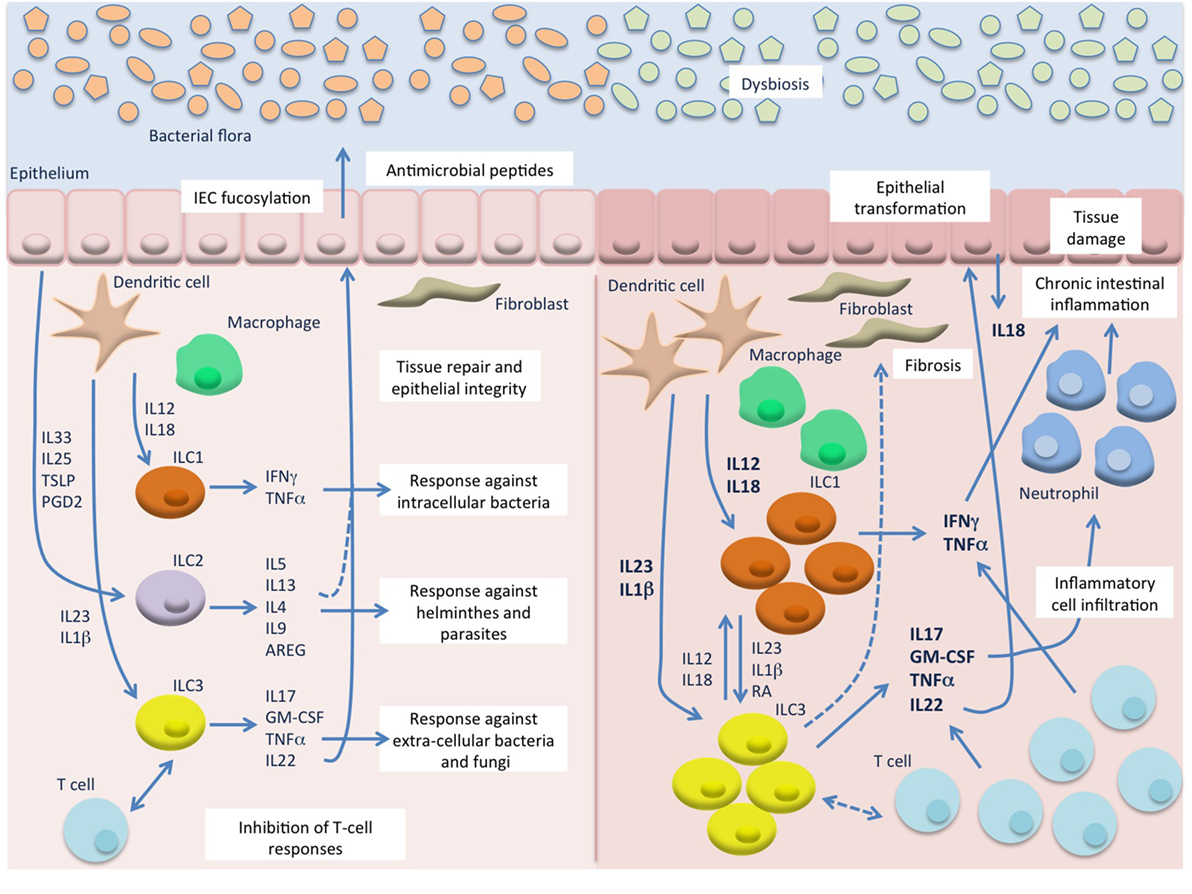
Figure 1. Innate lymphoid cells (ILC) in intestinal homeostasis and chronic inflammation. In the healthy mucosa (left) ILC contribute to the maintenance of intestinal homeostasis through induction of protective immune responses against pathogens and promotion of tissue integrity. In response to microbial stimuli, innate cells, such as dendritic cells (DC) and macrophages, secrete IL12 and IL18 that stimulate ILC1 responses against intracellular pathogens, and IL23 and IL1β that induce ILC3 responses against extracellular bacteria and fungi. On the other hand, epithelial derived factors, such as IL33, IL25, TSLP, and prostaglandin D2 (PGD2), induce secretion of type 2 cytokines from ILC2 that contribute to the expulsion of helminthes. ILC3 and possibly ILC2 also promote epithelial integrity and tissue repair, particularly through IL22 production, and can inhibit T cell activation and proliferation. During chronic intestinal inflammation, such as Crohn’s disease (CD) (right), in the presence of an altered bacterial flora, excessive levels of IL12, IL18, IL23, and IL1β are secreted by inflammatory DC and macrophages and lead to accumulation and activation of ILC1 and ILC3 that secrete high amount of type 1 and type 17 cytokines. These, also produced by accumulating T cells, induce chemotaxis of more inflammatory cells, such as neutrophils, and result in chronic inflammation and tissue damage. Besides the production of cytokines, ILC may also interact with immune (T cells) and non-immune cells (epithelial cells and fibroblasts) leading to secretion of more inflammatory mediators, such as IL18 from the epithelium, and to tissue reorganization (epithelial proliferation and fibrosis). A possible role for IL22 in epithelial transformation in colitis-associated cancer has also emerged from murine studies.
Although it has been reported that human ILC express toll-like receptors (3), they mostly respond to cytokines by making more cytokines. Unlike innate cells that respond directly to the pathogens and adaptive cells that respond through more specific antigen recognition, ILC act like messengers that in response to innate cell produced cytokines secrete more cytokines to orchestrate the local immune response. A small population, through an impressive per cell ability to release cytokines, can have an important amplifying role and the combination of different cytokines can lead to a dramatic shift in responses. ILC also go further by interacting with other cells, such as non-immune cells like epithelial and stromal cells, and may induce tissue reorganization. Moreover, they can influence T cell responses by promoting T helper (Th) cell differentiation and effector functions, have been shown to express MHCII and are able to present antigen to Th cells and interact with regulatory T cells (Tregs) (4–7) (Figure 1).
Innate lymphoid cells arise from the common lymphoid progenitor and their development is dependent on the transcription factors Id2, the common γ-chain, GATA3, and PLZF (8–10). Three main subsets of ILC have been described reflecting functional characteristics of the Th subsets (2). Hence, like Th1, ILC1 express the transcription factor T-bet and are characterized by secretion of IFNγ and TNFα and are involved in the immune response against intracellular pathogens. ILC2, mirroring Th2 cells, express GATA3, secrete IL4, IL5, IL9, and IL13 and are involved in protection against helminthes and in allergic reactions. Finally, ILC3 depend on expression of RORγt and secrete IL17, IL22, GM-CSF, and TNFα, resembling their Th17 counterpart, and participate in the response against extracellular pathogens at mucosal sites (Figure 1).
However, it is becoming apparent that, similarly to what is observed for Th subsets, ILC retain a degree of plasticity depending on exposure to environmental stimuli, such as cytokines or metabolites. It has been shown that ILC1 can differentiate into ILC3 in presence of IL1β and IL23, an effect that is enhanced by the vitamin A metabolite retinoic acid (RA) (11), and to ILC2 in the presence of IL4 (12). On the other hand, ILC3 when exposed to IL12, IL18, and/or IL1β up-regulate T-bet and can therefore express IFNγ, while repressing RORγt and the secretion of IL17 and IL22 (13). In mice, a gradient of expression of RORγt and T-bet by ILC3 is associated with functional plasticity (14). In particular, NKp46− ILC3 can develop into NKp46+ “ex-ILC3” through induction of T-bet and repression of RORγt. The “ex-ILC3” down-regulate the IL23 receptor, up-regulate the IL12 receptor, and mainly produce IFNγ in response to IL12. Similarly ILC2 can also trans-differentiate into ILC1 after exposure to IL12 and IL18 (12, 15).
Recent studies have particularly challenged the existence of a defined ILC1 subset, distinct from conventional NK cells, and other ILC subsets. Simoni et al. did not find human ILC1 in various healthy and disease-derived tissues using CyTOF mass-cytometry to assess the expression of an extensive range of cell surface and intracellular parameters. The t-SNE analysis showed clear clustering of ILC2, ILC3, and conventional NK cells, but was unable to identify a distinct subset of ILC1 that appeared instead to be scattered throughout other clusters (16). Furthermore, transcriptomic analysis of murine ILC and NK cell populations has shown overlapping profiles for ILC1 cells, NKp46+ “ex-ILC3” cells, and NK cells (17). On the other hand, in a human study using single cell RNA sequencing of tonsil derived CD127+ ILC and NK cells, unbiased clustering of cellular transcriptomes revealed four distinct cell populations, corresponding to ILC1, ILC2, ILC3, and NK cells (18). These observations suggest that a dogmatic classification of ILC in rigidly defined subtypes is likely to represent an over-simplification of the real scenario. It appears more plausible to conceive that ILC, like their T cell counterpart, can exert flexible and adaptable functional properties in response to the evolving environment at mucosal sites.
ILC are key contributors to the already complex immune system playing a critical role in health and disease. In this review, we focus on the role of non-cytotoxic or helper-like ILC (called ILC from now on for simplicity) in intestinal homeostasis and inflammation.
ILC in Intestinal Homeostasis
The maintenance and regulation of the intestinal microenvironment to keep a stable and functional healthy gut are not a small task and involve constant tweaking of physiological, biochemical, and immune pathways. The intestinal environment has been described as the Wild West of the immune system and represents the epitome of the complexity of immune organization. In this environment, several factors, such as commensal microbes, foods, and metabolites, can modify the immunological balance of the region and the gut needs to distinguish unharmful from pernicious agents. The process of homeostasis in the intestine is not a passive process, but it actually involves several active mechanisms on the part of local cells especially from ILC.
The distribution of ILC varies across the different parts of the human gastrointestinal (GI) tract with a progressive increase of total ILC from the proximal to the distal intestine (19). Moreover, distinct subsets are represented at different sites. Indeed, ILC1 are enriched in the upper GI tract, while ILC2 are only present at very low frequencies in the entire intestine, and ILC3 increase toward the colon and this correlates with higher distal expression of IL7. Interestingly, the functional properties of these cells were also found to differ across different parts of the intestine, with highest secretion of IL22 being observed in the ileum and in the esophagus (19).
ILC play a role in the maintenance of intestinal homeostasis by contributing to the protective immune response toward intestinal pathogens, as it has emerged by murine studies. In particular, T-bet expressing ILC participate in the defense against Helicobacter typhlonius, a commensal in the murine microbiota that closely resembles Helicobacter pylori, the frequent colonizer of the human stomach associated with gastritis, peptic ulcer, and gastric cancer. Mice lacking T-bet in the innate immune compartment develop colitis spontaneously triggered by H. typhlonius through induction of TNFα secretion by dendritic cells (DC). In turn, TNFα synergizes with IL23 to induce IL17 secretion by ILC in this animal model (20). These findings are particularly interesting in light of the high representation of ILC1 in the human upper GI tract where they may be involved in the inflammatory and pro-carcinogenic responses induced by H. pylori. ILC1 were also shown to contribute to protection against intracellular pathogens, such as Salmonella enterica. During Salmonella infection T-bet expressing ILC (including ILC1 and “ex-ILC3”) are the main source of IFNγ, which drives the secretion of mucus-forming glycoproteins required to protect the epithelial barrier (14). Similarly, ILC1 are the main producers of both IFNγ and TNFα during Toxoplasma gondii infection and T-bet deficient mice fail to control parasite replication (21).
On the other hand, ILC2 were found to play an important role in mounting protective innate responses against parasites and helminthes through induction of eosinophilia and goblet cell hyperplasia. In particular, IL25 and IL33 responsive ILC2 are critical for Nippostrongylus brasiliensis expulsion in mice that lack adaptive immune cells (6, 22, 23). As for all other ILC, ILC2 development depends on the common γ-chain cytokine receptor, IL7 and the transcription factor Id2 and GATA3 (24, 25), and they also require RORα (26, 27) and TCF-1 (28). ILC2 respond to stimulation with epithelial derived cytokines, such as IL33, IL25, and TSLP, but also eicosanoids, such as prostaglandin D2 (PGD2) (29, 30) and leukotriene D4 (31), and secrete type 2 cytokines, mainly IL5 and IL13, but also IL4, IL9 (31–34), and the epidermal growth family member amphiregulin that is responsible for lung epithelial repair after murine infection with the H1N1 influenza virus (35).
Human ILC2 have been identified in the peripheral blood and in fetal and adult tissues, including the gut, even if at low frequencies (19, 36). These cells express the IL33R (ST2) and the IL25R, and are positive for the chemoattractant receptor-homologous molecule expressed on Th2 lymphocytes (CRTH2) (36). CRTH2 is a G-protein coupled receptor for PGD2, which is released by activated mast cells during allergic reactions. PGD2 binding to CRTH2 has been shown to induce ILC2 migration, production of type 2 cytokines, and up-regulation of IL33R and IL25R possibly creating a positive feedback loop that amplifies type 2 responses during allergy (30).
Three independent groups initially identified ILC2 in mice and applied different denominations to the groups, “natural helper cells,” “nuocytes,” and “innate helper 2 cells” (6, 22, 23). “Natural helper cells” were identified in lymphoid clusters associated with the adipose tissue in the peritoneal cavity. Interestingly, similar fat-associate lymphoid clusters (FALC) were also found in the human mesentery. These cells are Lin−, but express c-Kit, Sca-1, the IL7R and the IL33R, produce large amounts of Th2 cytokines, such as IL5 and IL13 and also induce B cell proliferation. After helminthic infection and in response to IL33, FALC Lin−c-Kit+Sca-1+ cells produce large amounts of IL13, which leads to goblet cell hyperplasia and contribute to the expulsion of helminthes. The location of these cells in the visceral fat, and not at the classical barrier surface, is interesting. Recent studies indicate that ILC2 play a role in adipose tissue deposition and metabolism. In fact, they were shown to promote weight loss and glucose tolerance, through induction of caloric expenditure and browning of the adipose tissue (37–40). “Nuocytes” were identified in the IL13 reporter mice as non-B, non-T lymphocytes that expand in vivo in response to IL25 and IL33, and mediate the early protective immune response to helminthes through secretion of IL13. Similarly, “innate helper 2 cells” were identified as Lin−, IL25, and IL33 responsive cells in IL4 and IL13 reporter mice. These cells are widely distributed and expand after helminthic infection and secrete IL13 resulting in worm clearance. More recently, it has been shown that IL33-mediated induction of IL13 by ILC2 is necessary to mediate protection against helminthes (41).
Around the same time, an IL25 responsive Lin− multipotent progenitor precursor called MPPtype2 was also identified in the gut-associated lymphoid tissue (GALT). These cells are also able to elicit protective type 2 responses during parasitic infection (42). Compared with the other three populations, however, MPPtype2 are Id2 independent, show a distinct transcriptional profile and are able to differentiate into multiple monocyte/macrophage and granulocyte populations and are therefore classically not included in the ILC family (43).
ILC2 were shown to be critical for epithelial repair in the lung following influenza virus infection through secretion of amphiregulin (35). Murine gut-associated ILC2 can also secrete amphiregulin in response to IL33 and were shown to have a protective role in the dextran sodium sulfate model of intestinal damage and inflammation (44). These data suggest that intestinal ILC2 may play a role in the maintenance of epithelial integrity in the gut (Figure 1).
Together with fetal LTi cells, ILC3 belong to the Group 3 of ILC. Both types of cells depend on RORγt and IL7 for their development and function (2). The classical Group 3 LTi, in conjunction with stromal cells, are responsible for the induction of secondary lymphoid tissues during embryonic development (45). On the other hand, ILC3 expand postnatally in response to microbiota derived signals (46–48). Phenotypically, fetal LTi cells and adult ILC3 populations can be distinguished on the basis of CCR6 expression, with LTi cells being of CCR6hi phenotype and adult ILC3 populations of CCR6low/− phenotype (14).
ILC3 are (arguably) the most interesting ILC in the intestine and are the most abundant ILC subset at mucosal tissues, particularly in the intestinal tract. In fact, ILC3 constitute the majority of ILC in the ileum (13, 49) and in the colon (19). Interestingly, it has been shown that the composition of the different ILC subsets correlates with localized IL7 expression. IL7 stimulation increases RORγt expression and this is a striking result, as the implication would be that IL7 does not just control ILC survival but can modulate which ILC subsets you have, i.e., the more IL7, the more ILC3.
In the healthy gut, ILC3 exhibit a range of cytokine dependent and cell surface receptor mediated mechanisms to apply homeostatic control of intestinal immunity. Indeed, one of the major functions of ILC3 is to maintain barrier integrity in an environment where a tug of war is constant between the enhancement and inhibition of immune responses against the microbiota (50).
Once they sense bacterial antigens, CX3CR1+ mononuclear cells and DC are major sources of IL23 or IL1β in the gut (Figure 1). In turn, these cytokines stimulate ILC3 to produce the effector cytokines IL22, GM-CSF, and IL17. The IL22 cytokine has a key role in homeostatic control and is secreted by Th17, Th22, and ILC3 (51–54). IL22 expression by ILC3 is also controlled by the aryl hydrocarbon receptor (Ahr), a ligand dependent transcription factor that is activated by xenobiotic and endogenous ligands (55). Ahr controls the development of adult, but not fetal RORγt+ ILC in mice, and RORγt+ ILC that lack Ahr were shown to produce less IL22 through both direct inhibition of Il22 gene transcription and reduced responsiveness to IL23 (47, 48).
In the intestine, the IL22 dependent crosstalk between ILC3 and epithelial cells has been shown to influence the composition of the intestinal microbiota in mice, resulting in augmented host resistance to colonization with pathogenic organisms. The production of IL22 by ILC3 is critical for innate responses against intestinal bacterial pathogens such as Citrobacter rodentium (56), while their production of IL17 is central for antifungal responses against the opportunistic pathogen Candida albicans in the oral mucosa (57).
IL22 derived from ILC3 can also prevent bacterial translocation and keep opportunistic pathogens in check. Indeed, mice lacking IL22 or ILC3 show disrupted barrier integrity and hence cannot contain commensal bacteria resulting in translocation. ILC3 derived IL22 has been shown to protect mice against systemic dissemination of pathogenic bacteria following Clostridium difficile infection and infection-induced damage to the intestinal epithelium. ILC3 derived IL22 was required for the induction of complement factor C3 in the liver and intestine, which enhanced bacterial phagocytosis and allowed effective control of antigen load in peripheral organs (58). Additionally, IL22–IL22R interactions on intestinal epithelial cells (IEC) induce STAT3 phosphorylation and stimulate production and secretion of anti-microbial peptides and proteins such as β-defensins, RegIIIβ and RegIIIγ, calgranulins S100A8 and S100A9, and lipocalin-2 and mucins like Muc1, Muc3, Muc10, and Muc13 (59–63). In mice, it has been shown that suboptimal expression of anti-microbial factors by IEC results in systemic dissemination of commensal bacteria of the genus Alcaligenes spp, which is normally contained in GALT and Peyer’s patches. Peripheral Alcaligenes spp. dissemination was associated with low-grade systemic inflammation, which could be reversed by treating mice with recombinant IL22 (64).
IL22 binding to IL22R in IEC triggers a signaling cascade necessary for the production and maintenance of IL18 mRNA and pro-IL18 induction in epithelial cells (65). In addition, activation of the inflammasome in these cells leads to the downregulation of IL22 binding protein and hence increases the effects of IL22 (65, 66). Therefore, IL18 and IL22 production is tightly and mutually controlled contributing to the maintenance of gut homeostasis. When this delicate balance is altered under inflammatory conditions, intestinal pathology ensues. In mice, it has been shown that ILC3 responses are indirectly downregulated via IL25, which is secreted by microbiota-stimulated epithelial cells and likely acts via DC, which in turn limit ILC3 activation through a, so far, unknown mechanism (67).
In addition to being regulated by immune cell subsets such as DC (68), ILC3 have the capacity to regulate the activity of other immune cells via direct cell–cell interactions with important implications for gut homeostasis. Moreover, ILC3 respond to IL1β with the secretion of GM-CSF, a crucial regulator of mononuclear phagocytes, which induce intestinal Tregs. In fact, IL1β stimulation elicits production of pro-inflammatory cytokines by ILC3, including TNFα, IL6, and GM-CSF as well as regulating the expression of MHCII and co-stimulatory ligand expression in both ex vivo and in vitro generated mouse ILC3 (4). Hepworth et al. have demonstrated a requirement for class II transactivator, a master transcriptional regulator of MHCII, in driving MHCII expression by ILC3 (69). ILC3 are indeed capable of processing ingested protein antigens and load them onto MHCII for presentation to CD4 T cells, although the efficiency at which ILC were shown to do this was expectedly much lower than that of professional antigen presenting cells (5, 7).
ILC3 inhibit commensal-specific T cells via the MHCII receptor together with a withdrawal of IL2. Under steady-state conditions, IL2 and MHCII dependent interactions between intestinal ILC3 and effector CD4 T cells can inhibit CD4 T cell responses (5, 69). In humans, HLA-DR has been shown to be expressed on intestinal ILC3 as well as in ILC2. ILC3 have also been reported to limit commensal-specific CD4 T cell responses, through direct inhibitory interactions with CD4 T cells (5, 69) and through induction of tolerogenic RA and IL10 secreting intestinal DC and macrophages (70).
Data from our unit (Anna-Lena Schaupp et al.) have shown that ILC populations in human blood and the intestinal lamina propria express HLA-DR and co-stimulatory ligands, such as CD80, CD86, OX40, and PD-L1. In the colon, ILC3 were found to contain the highest proportion of HLA-DR expressing cells, with intermediate expression by ILC1 and lowest expression by ILC2. A similar distribution of HLA-DR expression among mouse ILC subsets in the small intestine has been reported with ILC3 populations containing a significantly higher percentage of HLA-DR+ cells compared with ILC2 or ILC1 (5).
Furthermore, ILC3 can also constrain commensal dissemination through effects on B cell responses. Thanks to their expression of B cell activating factors, such as BAFF, APRIL, and CD40L, as well as through lymphotoxin-dependent pathways, ILC3 promote the production of T cell-dependent and -independent IgA and IgG antibodies by mucosal and splenic B cell populations (71, 72). In turn, mucosal IgA and low-affinity systemic IgG antibodies contribute to the maintenance of mucosal homeostasis by regulating the composition, size, and anatomical containment of the intestinal commensal microbiota (73).
ILC3 express high levels of CD1d on their surface, therefore, they can acquire and present lipids to iNKT cells and this engagement stimulates them to produce IL22 (74). IL22 plays a central role in epithelial barrier function and tissue repair (75), and thus, ILC3 integration of signals obtained through CD1d, along with those received from cytokines in the local inflammatory milieu, such as IL23, may contribute to maintenance of homeostasis and to regulation of immune responses. By sensing the environment via the interaction microbial lipids-CD1d, ILC3 make an important contribution to the maintenance of barrier function.
The function of TLR on ILC3 is still an open question. TLR1, 2, 5, 6, 7, and 9 transcripts in human ILC3 have been shown to be expressed, albeit at low levels, however, only TLR2 engagement on ILC3 induces cytokine production in the presence of IL2, IL15, and IL23 (3). These results imply that ILC3 may have a role during C. albicans infection.
Another mechanism through which intestinal ILC3 have been shown to control commensal homeostasis is the regulation of IEC fucosylation (Figure 1). Commensal bacteria induce IEC fucosylation and epithelial fucose is used as a dietary carbohydrate. Fucosylated carbohydrates on IEC are thought to be important for the generation of an intestinal niche for commensal microbes (76). IL22 engagement with its receptor and secretion of lymphotoxin by ILC3 triggers fucosylation of epithelial cells (77). Indeed, IEC fucosylation is mediated by the fucosyltransferase enzymes Fut1 and Fut2 (78). ILC3 have been shown to induce intestinal epithelial Fut2 expression and fucosylation in mice. Abrogation of IEC fucosylation in mice lacking Fut2 has been linked to intestinal commensal dysbiosis and led to increased susceptibility to infection with the pathogens Salmonella typhimurium and C. rodentium (76). Thus, by regulating IEC anti-microbial functions, IL22 producing ILC3 plays an important role in preventing the systemic dissemination of microbial commensals.
ILC in Intestinal Pathology
Recent evidence from murine and human studies indicates that ILC participate in the pathogenesis of chronic intestinal inflammation and colitis-associated cancer in patients with IBD. On the other hand, a protective role for ILC has emerged in murine models of graft versus host disease (GVHD), a major complication of allogeneic hematopoietic stem cell transplant (A-HSCT). These findings identify ILC as potential therapeutic target for patients’ treatment and suggest that modulation of ILC function and activity may be beneficial in pathological conditions.
Inflammatory Bowel Disease
Inflammatory bowel disease is a chronic inflammatory disorder of the GI tract that encompasses two main forms, such as Crohn’s disease (CD) and ulcerative colitis (UC). In UC, inflammation is confined to the colonic mucosa while in CD it involves all layers of the gut wall and can affect any part of the GI tract, often leading to stricturing or fistulisation. Extra-intestinal manifestations of the disease are frequent with possible involvement of joints, skin, eyes, and kidneys. Moreover, the risk of developing other chronic immune disorders, such as psoriasis, ankylosing spondylitis, or primary sclerosing cholangitis (PSC), is also increased. Patients with long standing IBD colitis also have increased risk of colon cancer, and this risk is five times higher in patients with PSC-associated IBD (79). Patients are typically treated with immune suppressive treatment (corticosteroids, immunosuppressant agents, and biologic therapies, such as anti-TNFα or anti α4β7 antibodies), but surgery is often required during the course of the disease (80). The etiology of IBD remains unknown, but the available evidence suggests that IBD is caused by an abnormal immune response against the microorganisms of the intestinal flora in genetically susceptible individuals (81). Both dysregulated innate and adaptive immune pathways have been implicated in the pathogenesis of intestinal inflammation in these patients (82).
Early IBD research mainly focused on understanding the contribution of abnormal adaptive T cell responses to disease pathogenesis. CD has long been considered to be driven by an IL12 dependent Th1 response, while UC has been rather associated with a non-conventional Th2 response. However, an important role for IL23 driven Th17 responses in the pathogenesis of IBD has emerged by GWAS studies, and was confirmed in animal models and in translational studies in patients (83). Furthermore, advances arisen from genetic studies contributed to moving the focus of IBD pathogenesis on to mucosal innate immune responses. It became apparent that effector cytokines traditionally attributed to CD4 or CD8 T cells could also be secreted by unconventional innate lymphocytes, including ILC. This prompted an extensive research into the role of ILC in intestinal inflammation.
Buonocore et al. have shown that IL23 responsive ILC3 are required for induction of innate bacterial driven colitis in mice lacking B or T cells following infection with the pathogenic bacterium Helicobacter hepaticus. Colitis development was dependent on IL23 induced IL17 and IFNγ production by ILC3 that accumulated in the gut in response to IL1β. RORγt+ ILC3 were critical for colitis development, since intestinal inflammation was abrogated in mice that lack ILC3 (84). As previously mentioned, pathogenic IL17 production by intestinal ILC3 has also been shown to underlie the development of spontaneous colitis in mice lacking the type 1 transcription factor T-bet. In this model, ILC3 accumulate in the intestinal lamina propria and are activated in response to TNFα, IL23, and IL6 produced by activated DC (20).
Pearson et al. set out to investigate whether the movement of ILC within tissues contributed to immune and inflammatory responses (85). Using the anti-CD40 animal model of colitis, they observed ILC movement within cryptopatches. The authors concluded that by accumulating, clustering, and moving within the cryptopatches, ILC might initiate an inflammatory immune cascade resulting in intestinal inflammation. Interestingly, ILC appeared to wriggle their way directly through the tissue, possibly following a chemokine gradient.
Commensal dysbiosis secondary to ILC3 dysfunction can promote systemic inflammatory responses in mice. Outgrowth of commensal segmented filamentous bacteria, which are implicated in the development of pro-inflammatory Th17 cells (86), has been observed in mice lacking Ahr with lower levels of intestinal ILC3 derived IL22. Further reduction of ILC3 numbers and IL22 production in Ahr−/−RorcγtGFP/− mice promoted development of spontaneous, Th17-driven colitis, indicating that ILC3/IL22 mediated regulation of the commensal flora plays a role in mitigating the induction of potentially pathogenic Th17 responses to commensal antigens (48).
Furthermore, as already discussed, IL22 induces production of IL18 by IEC in mice and uncontrolled IL18 can amplify intestinal inflammation hence disrupting the mucosal barrier and creating a feedback loop of injury (65). Moreover, IL18 is up-regulated in intestinal lesions and peripheral blood in patients with CD (87–89), suggesting the presence of an altered IL22-IL18 regulation (Figure 1).
Interestingly, in both mice and humans, ILC3 were also found to secrete GM-CSF, which can recruit myeloid cells and further promote intestinal inflammation (85). In the same study, it was shown that ILC3 move within the intestinal tissue when activated. These two mechanisms can contribute to the induction and progression of inflammation throughout the gut.
In intestinal samples from IBD patients, we observed accumulation of Lin−CD56−CD127+ ILC in the inflamed ileum and colon of patients with CD, but not UC. These cells were able to respond to IL23 in vitro and they expressed IL17 and IFNγ (90). More, recently, we also found increased ILC frequencies in patients with PSC-associated IBD, but again no accumulation was observed in UC patients (91). Follow-up studies have confirmed that ILC contribute to inflammation in CD and suggested that IFNγ secreting ILC1 are especially increased in the intestinal lesions in these patients. Two populations of ILC1 have been described in the human intestine. Bernink et al. identified a population of CD127+NKp44−c-Kit− cells that express high levels of T-bet and IFNγ compared to c-Kit+ ILC3 (13). Similarly to NK cells, these cells respond to IL12 and IL18 stimulation in vitro with induction of IFNγ, however, they do not express granzyme B or perforin, which are classically associated to NK cell cytotoxic ability. Another population of IFNγ secreting intraepithelial ILC1, which are CD127−/low and express the epithelial homing integrin CD103, was identified in the human gut (92). Like CD127+ ILC1 and CD127−/lowCD103+ ILC1 are T-bet+, but they express the transcription factor Eomes and perforin and granzyme B granules, similarly to conventional cytotoxic NK cells. IFNγ secreting CD127+NKp44−C-Kit− ILC1 were found to accumulate in the inflamed intestine of patients with CD (13). The frequency of intraepithelial CD127−/lowCD103+ ILC1 was also found to be increased in the inflamed tissue of patients with CD (92). These data are in agreement with our first observation of increased frequencies of ILC in the inflamed lamina propria of patients with CD and may reflect ILC functional flexibility and their exposure to high levels of IL12 in the mucosa (90, 93) (Figure 1).
A role for ILC2 in pathologic intestinal inflammation has also been suggested by results in the oxazolone induced model of colitis, where ILC2 participate in intestinal inflammation in an IL25-dependent manner (94). Interestingly, increased frequencies of ILC2, which are able to secrete IL13 and IFNγ, have been detected in intestinal samples from CD patients (15). In the same study, stimulation of ILC2 with IL12 in vitro induced the expression of T-bet and IFNγ. IL12 is classically increased in the mucosa of patients with CD (93) and the finding of increased frequency of IFNγ secreting ILC2 may, therefore, reflect the functional plasticity of ILC depending on the mucosal microenvironment. CRTH2+ ILC2 were found to accumulate in esophageal tissues of patients with the allergic disease eosinophilic esophagitis (95), in nasal polyps of patients with chronic rhinosinusitis (36), an allergic condition characterized by high levels of IL5 and IL13, and in the inflamed skin of patients with atopic dermatitis (96), suggesting they can contribute to type 2 cell-mediated disease. In these allergic conditions, CRTH2+ ILC2 can be activated by mast cell released PGD2, up-regulate IL33R and IL25R and secrete IL4, IL5, and IL13 leading to the amplification of the type 2 response (30). On the other hand, while IL13 induction has been associated to mucosal inflammation in patients with UC (97, 98), there are no reports of increased ILC2 frequency in UC to date. Very low frequencies of ILC2 are found in the human colon and they may not represent major players in the colonic immune response, at least in the absence of parasitic infections (19). Interestingly, frequencies of CD127+ ILC were similar between UC patients and controls in our studies (90, 91), suggesting these cells may not be involved in intestinal inflammation in these patients.
Colorectal Cancer
Inflammation is one of the 10 hallmarks of cancer (99). The immune system can profoundly influence carcinogenesis with both favorable anti-tumorigenic effects and detrimental pro-tumorigenic activities. Despite the protective role of mucosal inflammation to limit damage and promote tissue repair, a dysregulated, chronic and extensive intestinal inflammation is regarded as a promoter of carcinogenesis. The exact mechanisms remain unknown, but inflammatory cytokines can act on IEC to promote many processes involved in neoplastic transformation, including proliferation, inhibition of apoptosis, invasion, angiogenesis, epithelial to mesenchymal transition, and metastasis (100).
Evidence from animal models suggests that ILC and especially IL23 responsive ILC3 may contribute to intestinal carcinogenesis. In particular, Kirchberger et al. identified a role for IL23 driven IL22 producing ILC in bacteria-induced colitis-associated cancer in T and B cell deficient mice infected with H. hepaticus long-term or co-treated with 2-azoxymethane (a cancer inducing agent) (101). In this setting, IL22 producing CD4−Nkp46−Thy-1+ ILC accumulate at the expense of Nkp46+ and CD4+ ILC populations. Depletion of Thy-1+ ILC or IL22 blockade can reverse established invasive carcinoma and inflammation. Importantly, IL22+ T cells and non-T cells are present in human colorectal cancer, and IL22 gene expression is increased in cancer tissue relative to matched adjacent normal tissue in these patients. In another model, transgenic expression of IL23 was shown to drive the de novo development of duodenal adenomatous tumors that was dependent on IL17 secreting Thy-1+ Sca-1+ ILC (102). However, one must be careful to translate results from T cell depleted animals into clinical settings, especially in light of the beneficial effects of IL22 in promoting barrier protection and epithelial regeneration. It is evident that IL22 can act as a “double edged sword” with both protective and pathogenic activities depending on the context.
Patients with IBD are at increased risk of developing intestinal cancer. This risk is further increased in patients co-diagnosed with PSC, a chronic progressive disorder of the hepato-biliary system, and strongly associated to IBD. PSC is characterized by inflammation, fibrosis, and stricturing of intrahepatic and extrahepatic bile ducts, leading to liver cirrhosis (103). Interestingly, patients with UC and PSC have a fivefold increased risk to develop colon cancer than those with UC alone (104). Patients with PSC are also at increased risk of other malignancies, including cholangiocarcinoma, gall-bladder adenoma, dysplasia, and carcinoma (105).
Our data show that secretion of Th17-cytokines from colonic T cells did not differ between patients with PSC–IBD, patients with UC and healthy controls, while we observed enhanced Th1 responses in PSC–IBD. On the other hand, ILC accumulate in the colon of PSC–IBD patients suggesting that ILC may play a role in intestinal inflammation and increased cancer risk in PSC and IBD (91). However, further studies are needed to evaluate the functional activity of ILC in these patients, and whether they contribute to pro-carcinogenic responses, including cytokine secretion and/or cell–cell interaction, that can promote epithelial transformation in PSC–IBD.
Graft Versus Host Disease
Allogeneic hematopoietic stem cell transplant is a potentially curative treatment for patients with leukemia and lymphoid malignancies (106). However, A-HSCT is frequently complicated by potentially fatal GVHD (107), caused by allo-reactive donor T cells that are activated against recipient antigens and lead to severe tissue damage, most frequently involving the GI tract, liver, and skin, where ILC are particularly represented.
Murine studies showed that IL23 responsive ILC3 secrete IL22 after bone marrow transplant, but are decreased in mice with acute GVHD. The reduction of ILC3 is associated with decreased secretion of IL22, loss of intestinal stem cells, impaired epithelial barrier function, and increased intestinal inflammation (108). A recent study showed that ILC2 are highly reduced after radiation and chemotherapy before A-HSCT in mice, and only limited repopulation with donor bone marrow derived ILC2 is observed in the GI tract. Interestingly, infusion of IL33–activated donor ILC2 significantly reduced the severity of GI GVHD in two separate murine models and this effect was dependent on the recruitment of donor myeloid-derived suppressor cells. Furthermore, ILC2 treatment of recipients with established GVHD reduced disease severity and increased survival (109).
Accordingly, ILC are depleted by pre-conditioning therapy in patients with acute leukemia and the reconstitution of ILC1, ILC2, and NKp44− ILC3 is slow compared with neutrophils and monocytes. On the other hand, NKp44+ ILC3 that are not present in the peripheral blood of healthy individuals, appear after conditioning and A-HSCT. A significant proportion of NKp44+ ILC3 expresses the gut-homing and skin-homing receptors, cutaneous lymphocyte-associated antigen and CCR10. The frequency of activated NKp44+ ILC3 expressing skin- and gut-homing receptors were significantly higher in patients who did not develop therapy induced mucositis and acute or chronic GVHD. These data suggest that NKp44+ ILC3, similarly to their murine counterpart, are involved in intestinal homeostasis and tissue repair contributing to GVHD prevention possibly through the secretion of IL22 (110).
On the other hand, ILC were shown to be dispensable in patients with severe combined immune deficiency that received A-HSCT. In these patients, while T cell and B cell reconstitution was obtained after A-HSCT, ILC were still deficient after transplant. However, no increased risk of infections or other disease was observed in the medium-long term in these subjects (111).
Besides their protective role in maintaining intestinal homeostasis, ILC have been shown to participate in chronic intestinal inflammation in both animal models and human diseases. In the presence of pro-inflammatory mediators in the microenvironment a shift from protective ILC populations toward pro-inflammatory and possibly pathogenic ILC subsets may occur. While IL22 mediated induction of epithelial proliferation and tissue repair may play a protective role in intestinal inflammation in GVHD, chronic exposure to IL22 may lead to acquisition of epithelial stemness and neoplastic transformation contributing to the increased risk of colon cancer that is associated to longstanding colitis.
Future Perspectives and Therapeutic Applications
The identification of ILC as a heterogeneous group of innate lymphocytes particularly abundant at mucosal sites has prompted both animal and human studies aimed to evaluate their role in intestinal pathophysiology. They are present at low frequencies, compared to other lymphocytic or myeloid populations, however, results from murine studies have highlighted their important contribution to maintaining intestinal homeostasis and to promoting chronic intestinal inflammation. As discussed, ILC can exert protective activities in the gut, through regulation of the microbial niche and contribution to epithelial and tissue repair. On the other hand, they can induce inflammation through the production of inflammatory cytokines and the regulation of innate and adaptive immune responses. It is becoming apparent that ILC can exert their function, not only through the secretion of soluble mediators, but also through their interaction with other cells, such as immune cells, including myeloid and T cell populations, and non-immune cells, such as epithelial cells and stromal cells, as suggested by the role of LTi-stromal cell crosstalk during lymphoid tissue embryogenesis. Targeting ILC activities, through cytokine blocking or modulation of ILC activation and their interaction with other cells, may result in beneficial effects in order to maintain homeostasis and prevent or treat inflammation. However, due to the lack of ILC specific markers, mediators or functions, at present any therapeutic approach will also have effects on other immune cells, and particularly on T cell mediated responses.
IL22 is emerging as a central mediator in intestinal homeostasis and tissue repair (75), while IL17 and IFNγ preferentially secreted by ILC3 and ILC1 have been associated to more pathogenic effects. A differential accumulation of IL22 secreting and IFNγ− and IL17A-secreting ILC has been found in patients with IBD and particularly in CD. Our own data suggest that ILC populations that respond to IL23 stimulation with secretion of IL17A and IFNγ accumulate in the inflamed intestine of patients with CD, but not in UC. Further studies have confirmed accumulation of IFNγ secreting ILC populations in both the lamina propria and the epithelium in CD (13, 90). Moreover, a reduction in the frequency of IL22 secreting ILC3, associated to IFNγ secreting ILC accumulation, has been described in CD (112, 113). Interestingly, no study to date has described accumulation of ILC in the gut of patients with UC and our studies may suggest they are not implicated in UC inflammation, even if larger studies are needed. On the contrary accumulation of ILC is present in patients with PSC–IBD, who classically exhibit mild colitis but significantly increased risk of colon cancer compared to other patients with IBD (91).
ILC1 secreted IFNγ may contribute to propagation of inflammation and tissue damage in CD. Both Th1 and ILC1 responses are increased in CD with higher levels of IFNγ expressed in the intestinal mucosa of these patients. However, Fontolizumab, a humanized murine anti-IFNγ antibody, did not show efficacy in clinical trials, even if some concerns about the design of these studies have been raised and Fontolizumab may induce some partial benefit in CD (114).
ILC3 derived IL17 promotes the recruitment of inflammatory neutrophils by inducing the release of chemokines from epithelial and endothelial cells. In this context, IL17 promotes epithelial barrier function (115). However, IL17 also presents cytokine functional dichotomy, and in the presence of IL23, IL17 and pathogenic Th17 cells have been implicated in intestinal inflammation and tissue damage (116). Hence, IL23 dependent IL17 production might be counter-indicative. Based on the results of genetic studies and immunological studies models and human tissue, blocking Th17- and unconventional lymphocyte-secreted IL17 seemed a reasonable therapeutic approach for patient with IBD. However, specific targeting of IL17 with Secukinumab, a human anti-IL17A monoclonal antibody, led to disappointing results in a multicentre phase IIa study (117). Secukinumab was not only ineffective in CD patients, but also actually resulted in disease exacerbation, likely interfering with the more protective IL17 functions or resulting in increased Th1 and IFNγ-mediated responses. The results of this study highlighted the complexity of the IL23/IL17 axis, with IL17 being only one of the many IL23 induced mediators with often redundant and dichotomic effects (118).
It is conceivable that insufficient levels of intestinal ILC derived IL22 in CD patients, resulting in impaired epithelial regeneration, innate anti-microbial defense and containment of commensal microbes, may contribute to disease pathogenesis. Selective enhancement of IL22 production by ILC may have protective effects in these patients, although this strategy would have to be carefully balanced against a potentially increased risk of colorectal cancer. In fact, chronic IL22 exposure promotes intestinal epithelial proliferation and may result in increased risk of colitis-associated cancer as suggested by murine studies (101). Targeting of IL22 or more specifically IL22 secreting ILC may be beneficial in the prevention or treatment of longstanding IBD-associated increase risk of colon cancer.
Studies in murine models also suggested that blocking ILC3 secreted GM-CSF or altering ILC3 movement or activity might help reduce intestinal inflammation (85). However, blocking GM-CSF activity could also result in the inhibition of important protective mechanisms (119, 120). Furthermore, anti-GM-CSF autoantibodies were found to be increased in IBD (121) and recombinant GM-CSF (Sargramostim) showed some efficacy in patients with CD (122). However, these results were not confirmed in further studies (123). In any case, clinical trials are underway to investigate the use of anti-GM-CSF in other inflammatory conditions, such as rheumatoid arthritis or multiple sclerosis and these studies could offer insight into whether this approach could have beneficial effect on intestinal inflammation in patients with concomitant IBD.
ILC3 might well be divided into several subsets and an understanding of the different functions of each one might help us in the design of specific therapies according to the specific roles of ILC3 in intestinal inflammation and homeostasis. A crucial open question of ILC biology is how these rare cells impact homeostatic or, in the case of IBD, pathogenic immune responses in the presence of a functional adaptive immune system. This is evidenced by the existence of functionally discrete ILC3 subsets, including those that present antigen and inhibit microbiota-directed T cell immunity and those that promote anti-microbial T cell responses via IL22 production.
Elevated levels of pro-inflammatory cytokines, such as IL12, IL23, or IL1β, in the intestine of patients with IBD may be responsible for the acquisition of pathogenic ILC phenotypes. IL12 and IL23, can be targeted by Ustekinumab and Briakinumab, monoclonal antibodies against their shared p40 subunit. This strategy has proven to be effective for patients with CD (124–126) and it remains to be elucidated how ILC and T cell populations are affected by IL23 and IL12 blockade. Interestingly, the efficacy of anti-IL12/IL23 treatment in CD is not comparable to the striking results obtained in patients with psoriasis (127, 128) highlighting the complexity of the immune mechanisms involved in intestinal inflammation in these patients (118). More recently Risankizumab, a monoclonal antibody against the IL23 specific subunit p19, showed higher efficacy than placebo at inducing clinical remission in CD patients in a phase 2 study (129).
It is conceivable that future therapeutic strategies could include interfering with ILC interaction with immune and non-immune cells. As discussed, ILC3 inhibit commensal-specific T cells via the MHCII receptor together with a withdrawal of IL2 (5, 69). However, in IBD an increase in the availability of IL2 (130) due to widespread immune activation could mean that IL2 uptake by ILC may no longer be a limiting factor for effector CD4 T cells. In this context, T cell-derived IL2 in conjunction with other pro-inflammatory cytokines (IL1β, IL6, IL12, and IL23) could furthermore drive inflammatory ILC3 activation. ILC3 derived effector cytokines (IL17A, IFNγ, TNFα, and GM-CSF) could then feedback to potentiate pathogenic effector CD4 T cell responses, either directly or indirectly via antigen presenting cells.
Taken together, a better understanding of the regulation of cytokine expression by ILC and their interaction with other immune and non-immune cells will help to develop new strategies to treat inflammatory diseases in humans.
Author Contributions
AG and CVA-C contributed equally to this work.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to thank Fiona Powrie, FRS, for fostering our interest in mucosal immunology, Claire Pearson, Ph.D. for critically reading this review and Anna-Lena Schaupp, D. Phil. for her enthusiasm in exploring the biology of ILC for her doctoral thesis.
Funding
Our research is funded by Crohn’s and Colitis UK [grant number M/13/3] and the Wellcome Trust [grant number 101734/Z/13/Z] (AG) and by the National Institute for Health Research (NIHR) Oxford Biomedical Research Centre (BRC) (CVA-C). The views expressed are those of the authors and not necessarily those of the NHS, the NIHR or the Department of Health.
References
1. Artis D, Spits H. The biology of innate lymphoid cells. Nature (2015) 517:293–301. doi:10.1038/nature14189
2. Spits H, Artis D, Colonna M, Diefenbach A, Di Santo JP, Eberl G, et al. Innate lymphoid cells – a proposal for uniform nomenclature. Nat Rev Immunol (2013) 13:145–9. doi:10.1038/nri3365
3. Crellin NK, Trifari S, Kaplan CD, Satoh-Takayama N, Di Santo JP, Spits H. Regulation of cytokine secretion in human CD127(+) LTi-like innate lymphoid cells by toll-like receptor 2. Immunity (2010) 33:752–64. doi:10.1016/j.immuni.2010.10.012
4. Burg von N, Turchinovich G, Finke D. Maintenance of immune homeostasis through ILC/T cell interactions. Front Immunol (2015) 6:416. doi:10.3389/fimmu.2015.00416
5. Hepworth MR, Monticelli LA, Fung TC, Ziegler CGK, Grunberg S, Sinha R, et al. Innate lymphoid cells regulate CD4+ T-cell responses to intestinal commensal bacteria. Nature (2013) 498:113–7. doi:10.1038/nature12240
6. Neill DR, Wong SH, Bellosi A, Flynn RJ, Daly M, Langford TKA, et al. Nuocytes represent a new innate effector leukocyte that mediates type-2 immunity. Nature (2010) 464:1367–70. doi:10.1038/nature08900
7. Oliphant CJ, Hwang YY, Walker JA, Salimi M, Wong SH, Brewer JM, et al. MHCII-mediated dialog between group 2 innate lymphoid cells and CD4(+) T cells potentiates type 2 immunity and promotes parasitic helminth expulsion. Immunity (2014) 41:283–95. doi:10.1016/j.immuni.2014.06.016
8. Yokota Y, Mansouri A, Mori S, Sugawara S, Adachi S, Nishikawa S, et al. Development of peripheral lymphoid organs and natural killer cells depends on the helix-loop-helix inhibitor Id2. Nature (1999) 397:702–6. doi:10.1038/17812
9. Spits H, Cupedo T. Innate lymphoid cells: emerging insights in development, lineage relationships, and function. Annu Rev Immunol (2012) 30:647–75. doi:10.1146/annurev-immunol-020711-075053
10. Yagi R, Zhong C, Northrup DL, Yu F, Bouladoux N, Spencer S, et al. The transcription factor GATA3 is critical for the development of all IL-7Rα-expressing innate lymphoid cells. Immunity (2014) 40:378–88. doi:10.1016/j.immuni.2014.01.012
11. Bernink JH, Krabbendam L, Germar K, de Jong E, Gronke K, Kofoed-Nielsen M, et al. Interleukin-12 and -23 control plasticity of CD127(+) group 1 and group 3 innate lymphoid cells in the intestinal lamina propria. Immunity (2015) 43:146–60. doi:10.1016/j.immuni.2015.06.019
12. Bal SM, Bernink JH, Nagasawa M, Groot J, Shikhagaie MM, Golebski K, et al. IL-1β, IL-4 and IL-12 control the fate of group 2 innate lymphoid cells in human airway inflammation in the lungs. Nat Immunol (2016) 17:636–45. doi:10.1038/ni.3444
13. Bernink JH, Peters CP, Munneke M, Velde te AA, Meijer SL, Weijer K, et al. Human type 1 innate lymphoid cells accumulate in inflamed mucosal tissues. Nat Immunol (2013) 14:221–9. doi:10.1038/ni.2534
14. Klose CSN, Kiss EA, Schwierzeck V, Ebert K, Hoyler T, d’Hargues Y, et al. A T-bet gradient controls the fate and function of CCR6-RORγt+ innate lymphoid cells. Nature (2013) 494:261–5. doi:10.1038/nature11813
15. Lim AI, Menegatti S, Bustamante J, Le Bourhis L, Allez M, Rogge L, et al. IL-12 drives functional plasticity of human group 2 innate lymphoid cells. J Exp Med (2016) 213:569–83. doi:10.1084/jem.20151750
16. Simoni Y, Fehlings M, Kløverpris HN, McGovern N, Koo S-L, Loh CY, et al. Human innate lymphoid cell subsets possess tissue-type based heterogeneity in phenotype and frequency. Immunity (2017) 46:148–61. doi:10.1016/j.immuni.2016.11.005
17. Robinette ML, Fuchs A, Cortez VS, Lee JS, Wang Y, Durum SK, et al. Transcriptional programs define molecular characteristics of innate lymphoid cell classes and subsets. Nat Immunol (2015) 16:306–17. doi:10.1038/ni.3094
18. Björklund ÅK, Forkel M, Picelli S, Konya V, Theorell J, Friberg D, et al. The heterogeneity of human CD127(+) innate lymphoid cells revealed by single-cell RNA sequencing. Nat Immunol (2016) 17:451–60. doi:10.1038/ni.3368
19. Krämer B, Goeser F, Lutz P, Glässner A, Boesecke C, Schwarze-Zander C, et al. Compartment-specific distribution of human intestinal innate lymphoid cells is altered in HIV patients under effective therapy. PLoS Pathog (2017) 13:e1006373. doi:10.1371/journal.ppat.1006373
20. Powell N, Walker AW, Stolarczyk E, Canavan JB, Gökmen MR, Marks E, et al. The transcription factor T-bet regulates intestinal inflammation mediated by interleukin-7 receptor+ innate lymphoid cells. Immunity (2012) 37:674–84. doi:10.1016/j.immuni.2012.09.008
21. Klose CSN, Flach M, Möhle L, Rogell L, Hoyler T, Ebert K, et al. Differentiation of type 1 ILCs from a common progenitor to all helper-like innate lymphoid cell lineages. Cell (2014) 157:340–56. doi:10.1016/j.cell.2014.03.030
22. Moro K, Yamada T, Tanabe M, Takeuchi T, Ikawa T, Kawamoto H, et al. Innate production of T(H)2 cytokines by adipose tissue-associated c-Kit(+)Sca-1(+) lymphoid cells. Nature (2010) 463:540–4. doi:10.1038/nature08636
23. Price AE, Liang H-E, Sullivan BM, Reinhardt RL, Eisley CJ, Erle DJ, et al. Systemically dispersed innate IL-13-expressing cells in type 2 immunity. Proc Natl Acad Sci U S A (2010) 107:11489–94. doi:10.1073/pnas.1003988107
24. Hoyler T, Klose CSN, Souabni A, Turqueti-Neves A, Pfeifer D, Rawlins EL, et al. The transcription factor GATA-3 controls cell fate and maintenance of type 2 innate lymphoid cells. Immunity (2012) 37:634–48. doi:10.1016/j.immuni.2012.06.020
25. Klein Wolterink RGJ, Serafini N, van Nimwegen M, Vosshenrich CAJ, de Bruijn MJW, Fonseca-Pereira D, et al. Essential, dose-dependent role for the transcription factor Gata3 in the development of IL-5+ and IL-13+ type 2 innate lymphoid cells. Proc Natl Acad Sci U S A (2013) 110:10240–5. doi:10.1073/pnas.1217158110
26. Halim TYF, MacLaren A, Romanish MT, Gold MJ, McNagny KM, Takei F. Retinoic-acid-receptor-related orphan nuclear receptor alpha is required for natural helper cell development and allergic inflammation. Immunity (2012) 37:463–74. doi:10.1016/j.immuni.2012.06.012
27. Wong SH, Walker JA, Jolin HE, Drynan LF, Hams E, Camelo A, et al. Transcription factor RORα is critical for nuocyte development. Nat Immunol (2012) 13:229–36. doi:10.1038/ni.2208
28. Yang Q, Monticelli LA, Saenz SA, Chi AW-S, Sonnenberg GF, Tang J, et al. T cell factor 1 is required for group 2 innate lymphoid cell generation. Immunity (2013) 38:694–704. doi:10.1016/j.immuni.2012.12.003
29. Wojno EDT, Monticelli LA, Tran SV, Alenghat T, Osborne LC, Thome JJ, et al. The prostaglandin D2 receptor CRTH2 regulates accumulation of group 2 innate lymphoid cells in the inflamed lung. Mucosal Immunol (2015) 8:1313–23. doi:10.1038/mi.2015.21
30. Xue L, Salimi M, Panse I, Mjösberg JM, McKenzie ANJ, Spits H, et al. Prostaglandin D2 activates group 2 innate lymphoid cells through chemoattractant receptor-homologous molecule expressed on TH2 cells. J Allergy Clin Immunol (2014) 133:1184–94. doi:10.1016/j.jaci.2013.10.056
31. Doherty TA, Khorram N, Lund S, Mehta AK, Croft M, Broide DH. Lung type 2 innate lymphoid cells express cysteinyl leukotriene receptor 1, which regulates TH2 cytokine production. J Allergy Clin Immunol (2013) 132:205–13. doi:10.1016/j.jaci.2013.03.048
32. Wilhelm C, Hirota K, Stieglitz B, Van Snick J, Tolaini M, Lahl K, et al. An IL-9 fate reporter demonstrates the induction of an innate IL-9 response in lung inflammation. Nat Immunol (2011) 12:1071–7. doi:10.1038/ni.2133
33. Mjösberg J, Bernink J, Golebski K, Karrich JJ, Peters CP, Blom B, et al. The transcription factor GATA3 is essential for the function of human type 2 innate lymphoid cells. Immunity (2012) 37:649–59. doi:10.1016/j.immuni.2012.08.015
34. KleinJan A, Klein Wolterink RGJ, Levani Y, de Bruijn MJW, Hoogsteden HC, van Nimwegen M, et al. Enforced expression of Gata3 in T cells and group 2 innate lymphoid cells increases susceptibility to allergic airway inflammation in mice. J Immunol (2014) 192:1385–94. doi:10.4049/jimmunol.1301888
35. Monticelli LA, Sonnenberg GF, Abt MC, Alenghat T, Ziegler CGK, Doering TA, et al. Innate lymphoid cells promote lung-tissue homeostasis after infection with influenza virus. Nat Immunol (2011) 12:1045–54. doi:10.1031/ni.2131
36. Mjösberg JM, Trifari S, Crellin NK, Peters CP, van Drunen CM, Piet B, et al. Human IL-25- and IL-33-responsive type 2 innate lymphoid cells are defined by expression of CRTH2 and CD161. Nat Immunol (2011) 12:1055–62. doi:10.1038/ni.2104
37. Molofsky AB, Nussbaum JC, Liang H-E, Van Dyken SJ, Cheng LE, Mohapatra A, et al. Innate lymphoid type 2 cells sustain visceral adipose tissue eosinophils and alternatively activated macrophages. J Exp Med (2013) 210:535–49. doi:10.1084/jem.20121964
38. Hams E, Locksley RM, McKenzie ANJ, Fallon PG. Cutting edge: IL-25 elicits innate lymphoid type 2 and type II NKT cells that regulate obesity in mice. J Immunol (2013) 191:5349–53. doi:10.4049/jimmunol.1301176
39. Brestoff JR, Kim BS, Saenz SA, Stine RR, Monticelli LA, Sonnenberg GF, et al. Group 2 innate lymphoid cells promote beiging of white adipose tissue and limit obesity. Nature (2015) 519:242–6. doi:10.1038/nature14115
40. Lee M-W, Odegaard JI, Mukundan L, Qiu Y, Molofsky AB, Nussbaum JC, et al. Activated type 2 innate lymphoid cells regulate beige fat biogenesis. Cell (2015) 160:74–87. doi:10.1016/j.cell.2014.12.011
41. Hung L-Y, Lewkowich IP, Dawson LA, Downey J, Yang Y, Smith DE, et al. IL-33 drives biphasic IL-13 production for noncanonical Type 2 immunity against hookworms. Proc Natl Acad Sci U S A (2013) 110:282–7. doi:10.1073/pnas.1206587110
42. Saenz SA, Siracusa MC, Perrigoue JG, Spencer SP, Urban JF, Tocker JE, et al. IL25 elicits a multipotent progenitor cell population that promotes T(H)2 cytokine responses. Nature (2010) 464:1362–6. doi:10.1038/nature08901
43. Saenz SA, Siracusa MC, Monticelli LA, Ziegler CGK, Kim BS, Brestoff JR, et al. IL-25 simultaneously elicits distinct populations of innate lymphoid cells and multipotent progenitor type 2 (MPPtype2) cells. J Exp Med (2013) 210:1823–37. doi:10.1084/jem.20122332
44. Monticelli LA, Osborne LC, Noti M, Tran SV, Zaiss DMW, Artis D. IL-33 promotes an innate immune pathway of intestinal tissue protection dependent on amphiregulin-EGFR interactions. Proc Natl Acad Sci U S A (2015) 112:10762–7. doi:10.1073/pnas.1509070112
45. Bar-Ephraïm YE, Mebius RE. Innate lymphoid cells in secondary lymphoid organs. Immunol Rev (2016) 271:185–99. doi:10.1111/imr.12407
46. Lee JS, Cella M, McDonald KG, Garlanda C, Kennedy GD, Nukaya M, et al. AHR drives the development of gut ILC22 cells and postnatal lymphoid tissues via pathways dependent on and independent of Notch. Nat Immunol (2011) 13:144–51. doi:10.1038/ni.2187
47. Kiss EA, Vonarbourg C, Kopfmann S, Hobeika E, Finke D, Esser C, et al. Natural aryl hydrocarbon receptor ligands control organogenesis of intestinal lymphoid follicles. Science (2011) 334:1561–5. doi:10.1126/science.1214914
48. Qiu J, Heller JJ, Guo X, Chen Z-ME, Fish K, Fu Y-X, et al. The aryl hydrocarbon receptor regulates gut immunity through modulation of innate lymphoid cells. Immunity (2012) 36:92–104. doi:10.1016/j.immuni.2011.11.011
49. Forkel M, Mjösberg J. Dysregulation of group 3 innate lymphoid cells in the pathogenesis of inflammatory bowel disease. Curr Allergy Asthma Rep (2016) 16:73. doi:10.1007/s11882-016-0652-3
50. Neill DR, Flynn RJ. Origins and evolution of innate lymphoid cells: Wardens of barrier immunity. Parasite Immunol (2017) 5:e12436. doi:10.1111/pim.12436
51. McKenzie ANJ, Spits H, Eberl G. Innate lymphoid cells in inflammation and immunity. Immunity (2014) 41:366–74. doi:10.1016/j.immuni.2014.09.006
52. Diefenbach A, Colonna M, Koyasu S. Development, differentiation, and diversity of innate lymphoid cells. Immunity (2014) 41:354–65. doi:10.1016/j.immuni.2014.09.005
53. Montaldo E, Juelke K, Romagnani C. Group 3 innate lymphoid cells (ILC3s): origin, differentiation, and plasticity in humans and mice. Eur J Immunol (2015) 45:2171–82. doi:10.1002/eji.201545598
54. Eberl G, Colonna M, Di Santo JP, McKenzie ANJ. Innate lymphoid cells. Innate lymphoid cells: a new paradigm in immunology. Science (2015) 348:aaa6566. doi:10.1126/science.aaa6566
55. Qiu J, Zhou L. Aryl hydrocarbon receptor promotes RORγt+ group 3 ILCs and controls intestinal immunity and inflammation. Semin Immunopathol (2013) 35:657–70. doi:10.1007/s00281-013-0393-5
56. Sonnenberg GF, Monticelli LA, Elloso MM, Fouser LA, Artis D. CD4(+) lymphoid tissue-inducer cells promote innate immunity in the gut. Immunity (2011) 34:122–34. doi:10.1016/j.immuni.2010.12.009
57. Gladiator A, Wangler N, Trautwein-Weidner K, LeibundGut-Landmann S. Cutting edge: IL-17-secreting innate lymphoid cells are essential for host defense against fungal infection. J Immunol (2013) 190:521–5. doi:10.4049/jimmunol.1202924
58. Hasegawa M, Yada S, Liu MZ, Kamada N, Muñoz-Planillo R, Do N, et al. Interleukin-22 regulates the complement system to promote resistance against pathobionts after pathogen-induced intestinal damage. Immunity (2014) 41:620–32. doi:10.1016/j.immuni.2014.09.010
59. Killig M, Glatzer T, Romagnani C. Recognition strategies of group 3 innate lymphoid cells. Front Immunol (2014) 5:142. doi:10.3389/fimmu.2014.00142
60. Wolk K, Kunz S, Witte E, Friedrich M, Asadullah K, Sabat R. IL-22 increases the innate immunity of tissues. Immunity (2004) 21:241–54. doi:10.1016/j.immuni.2004.07.007
61. Aujla SJ, Chan YR, Zheng M, Fei M, Askew DJ, Pociask DA, et al. IL-22 mediates mucosal host defense against Gram-negative bacterial pneumonia. Nat Med (2008) 14:275–81. doi:10.1038/nm1710
62. Liang SC, Tan X-Y, Luxenberg DP, Karim R, Dunussi-Joannopoulos K, Collins M, et al. Interleukin (IL)-22 and IL-17 are coexpressed by Th17 cells and cooperatively enhance expression of antimicrobial peptides. J Exp Med (2006) 203:2271–9. doi:10.1084/jem.20061308
63. Zheng Y, Valdez PA, Danilenko DM, Hu Y, Sa SM, Gong Q, et al. Interleukin-22 mediates early host defense against attaching and effacing bacterial pathogens. Nat Med (2008) 14:282–9. doi:10.1038/nm1720
64. Sonnenberg GF, Monticelli LA, Alenghat T, Fung TC, Hutnick NA, Kunisawa J, et al. Innate lymphoid cells promote anatomical containment of lymphoid-resident commensal bacteria. Science (2012) 336:1321–5. doi:10.1126/science.1222551
65. Muñoz M, Eidenschenk C, Ota N, Wong K, Lohmann U, Kühl AA, et al. Interleukin-22 induces interleukin-18 expression from epithelial cells during intestinal infection. Immunity (2015) 42:321–31. doi:10.1016/j.immuni.2015.01.011
66. Huber S, Gagliani N, Zenewicz LA, Huber FJ, Bosurgi L, Hu B, et al. IL-22BP is regulated by the inflammasome and modulates tumorigenesis in the intestine. Nature (2012) 491:259–63. doi:10.1038/nature11535
67. Sawa S, Lochner M, Satoh-Takayama N, Dulauroy S, Bérard M, Kleinschek M, et al. RORγt+ innate lymphoid cells regulate intestinal homeostasis by integrating negative signals from the symbiotic microbiota. Nat Immunol (2011) 12:320–6. doi:10.1038/ni.2002
68. Cording S, Medvedovic J, Cherrier M, Eberl G. Development and regulation of RORγt(+) innate lymphoid cells. FEBS Lett (2014) 588:4176–81. doi:10.1016/j.febslet.2014.03.034
69. Hepworth MR, Fung TC, Masur SH, Kelsen JR, McConnell FM, Dubrot J, et al. Immune tolerance. Group 3 innate lymphoid cells mediate intestinal selection of commensal bacteria-specific CD4+ T cells. Science (2015) 348:1031–5. doi:10.1126/science.aaa4812
70. Mortha A, Chudnovskiy A, Hashimoto D, Bogunovic M, Spencer SP, Belkaid Y, et al. Microbiota-dependent crosstalk between macrophages and ILC3 promotes intestinal homeostasis. Science (2014) 343:1249288–1249288. doi:10.1126/science.1249288
71. Magri G, Miyajima M, Bascones S, Mortha A, Puga I, Cassis L, et al. Innate lymphoid cells integrate stromal and immunological signals to enhance antibody production by splenic marginal zone B cells. Nat Immunol (2014) 15:354–64. doi:10.1038/ni.2830
72. Tsuji M, Suzuki K, Kitamura H, Maruya M, Kinoshita K, Ivanov II, et al. Requirement for lymphoid tissue-inducer cells in isolated follicle formation and T cell-independent immunoglobulin A generation in the gut. Immunity (2008) 29:261–71. doi:10.1016/j.immuni.2008.05.014
73. Mantis NJ, Rol N, Corthésy B. Secretory IgA’s complex roles in immunity and mucosal homeostasis in the gut. Mucosal Immunol (2011) 4:603–11. doi:10.1038/mi.2011.41
74. Saez de Guinoa J, Jimeno R, Farhadi N, Jervis PJ, Cox LR, Besra GS, et al. CD1d-mediated activation of group 3 innate lymphoid cells drives IL-22 production. EMBO Rep (2017) 18:39–47. doi:10.15252/embr.201642412
75. Sonnenberg GF, Fouser LA, Artis D. Border patrol: regulation of immunity, inflammation and tissue homeostasis at barrier surfaces by IL-22. Nat Immunol (2011) 12:383–90. doi:10.1038/ni.2025
76. Pickard JM, Maurice CF, Kinnebrew MA, Abt MC, Schenten D, Golovkina TV, et al. Rapid fucosylation of intestinal epithelium sustains host-commensal symbiosis in sickness. Nature (2014) 514:638–41. doi:10.1038/nature13823
77. Goto Y, Obata T, Kunisawa J, Sato S, Ivanov II, Lamichhane A, et al. Innate lymphoid cells regulate intestinal epithelial cell glycosylation. Science (2014) 345:1254009–1254009. doi:10.1126/science.1254009
78. Goto Y, Uematsu S, Kiyono H. Epithelial glycosylation in gut homeostasis and inflammation. Nat Immunol (2016) 17:1244–51. doi:10.1038/ni.3587
79. Beaugerie L, Itzkowitz SH. Cancers complicating inflammatory bowel disease. N Engl J Med (2015) 372:1441–52. doi:10.1056/NEJMra1403718
80. Baumgart DC, Sandborn WJ. Inflammatory bowel disease: clinical aspects and established and evolving therapies. Lancet (2007) 369:1641–57. doi:10.1016/S0140-6736(07)60751-X
81. Abraham C, Cho JH. Inflammatory bowel disease. N Engl J Med (2009) 361:2066–78. doi:10.1056/NEJMra0804647
82. Geremia A, Biancheri P, Allan P, Corazza GR, Di Sabatino A. Innate and adaptive immunity in inflammatory bowel disease. Autoimmun Rev (2014) 13:3–10. doi:10.1016/j.autrev.2013.06.004
83. Geremia A, Jewell DP. The IL-23/IL-17 pathway in inflammatory bowel disease. Expert Rev Gastroenterol Hepatol (2012) 6:223–37. doi:10.1586/egh.11.107
84. Buonocore S, Ahern PP, Uhlig HH, Ivanov II, Littman DR, Maloy KJ, et al. Innate lymphoid cells drive interleukin-23-dependent innate intestinal pathology. Nature (2010) 464:1371–5. doi:10.1038/nature08949
85. Pearson C, Thornton EE, McKenzie B, Schaupp A-L, Huskens N, Griseri T, et al. ILC3 GM-CSF production and mobilisation orchestrate acute intestinal inflammation. Elife (2016) 5:e10066. doi:10.7554/eLife.10066
86. Ivanov II, McKenzie BS, Zhou L, Tadokoro CE, Lepelley A, Lafaille JJ, et al. The orphan nuclear receptor RORgammat directs the differentiation program of proinflammatory IL-17+ T helper cells. Cell (2006) 126:1121–33. doi:10.1016/j.cell.2006.07.035
87. Monteleone G, Trapasso F, Parrello T, Biancone L, Stella A, Iuliano R, et al. Bioactive IL-18 expression is up-regulated in Crohn’s disease. J Immunol (1999) 163:143–7.
88. Pizarro TT, Michie MH, Bentz M, Woraratanadharm J, Smith MF, Foley E, et al. IL-18, a novel immunoregulatory cytokine, is up-regulated in Crohn’s disease: expression and localization in intestinal mucosal cells. J Immunol (1999) 162:6829–35.
89. Furuya D, Yagihashi A, Komatsu M, Masashi N, Tsuji N, Kobayashi D, et al. Serum interleukin-18 concentrations in patients with inflammatory bowel disease. J Immunother (2002) 25(Suppl 1):S65–7. doi:10.1097/00002371-200203001-00010
90. Geremia A, Arancibia-Cárcamo CV, Fleming MPP, Rust N, Singh B, Mortensen NJ, et al. IL-23-responsive innate lymphoid cells are increased in inflammatory bowel disease. J Exp Med (2011) 208:1127–33. doi:10.1084/jem.20101712
91. Gwela A, Siddhanathi P, Oxford IBD Cohort InvestigatorsChapman RW, Travis S, Powrie F, et al. Th1 and innate lymphoid cells accumulate in primary sclerosing cholangitis-associated inflammatory bowel disease. J Crohns Colitis (2017) 11(9):1124–34. doi:10.1093/ecco-jcc/jjx050
92. Fuchs A, Vermi W, Lee JS, Lonardi S, Gilfillan S, Newberry RD, et al. Intraepithelial type 1 innate lymphoid cells are a unique subset of IL-12- and IL-15-responsive IFN-γ-producing cells. Immunity (2013) 38:769–81. doi:10.1016/j.immuni.2013.02.010
93. Monteleone G, Biancone L, Marasco R, Morrone G, Marasco O, Luzza F, et al. Interleukin 12 is expressed and actively released by Crohn’s disease intestinal lamina propria mononuclear cells. Gastroenterology (1997) 112:1169–78. doi:10.1016/S0016-5085(97)70128-8
94. Camelo A, Barlow JL, Drynan LF, Neill DR, Ballantyne SJ, Wong SH, et al. Blocking IL-25 signalling protects against gut inflammation in a type-2 model of colitis by suppressing nuocyte and NKT derived IL-13. J Gastroenterol (2012) 47:1198–211. doi:10.1007/s00535-012-0591-2
95. Doherty TA, Baum R, Newbury RO, Yang T, Dohil R, Aquino M, et al. Group 2 innate lymphocytes (ILC2) are enriched in active eosinophilic esophagitis. J Allergy Clin Immunol (2015) 136:792–4.e3. doi:10.1016/j.jaci.2015.05.048
96. Salimi M, Barlow JL, Saunders SP, Xue L, Gutowska-Owsiak D, Wang X, et al. A role for IL-25 and IL-33-driven type-2 innate lymphoid cells in atopic dermatitis. J Exp Med (2013) 210:2939–50. doi:10.1084/jem.20130351
97. Fuss IJ, Neurath M, Boirivant M, Klein JS, la Motte de C, Strong SA, et al. Disparate CD4+ lamina propria (LP) lymphokine secretion profiles in inflammatory bowel disease. Crohn’s disease LP cells manifest increased secretion of IFN-gamma, whereas ulcerative colitis LP cells manifest increased secretion of IL-5. J Immunol (1996) 157:1261–70.
98. Heller F, Florian P, Bojarski C, Richter J, Christ M, Hillenbrand B, et al. Interleukin-13 is the key effector Th2 cytokine in ulcerative colitis that affects epithelial tight junctions, apoptosis, and cell restitution. Gastroenterology (2005) 129:550–64. doi:10.1016/j.gastro.2005.05.002
99. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell (2011) 144:646–74. doi:10.1016/j.cell.2011.02.013
100. West NR, McCuaig S, Franchini F, Powrie F. Emerging cytokine networks in colorectal cancer. Nat Rev Immunol (2015) 15:615–29. doi:10.1038/nri3896
101. Kirchberger S, Royston DJ, Boulard O, Thornton E, Franchini F, Szabady RL, et al. Innate lymphoid cells sustain colon cancer through production of interleukin-22 in a mouse model. J Exp Med (2013) 210:917–31. doi:10.1084/jem.20122308
102. Chan IH, Jain R, Tessmer MS, Gorman D, Mangadu R, Sathe M, et al. Interleukin-23 is sufficient to induce rapid de novo gut tumorigenesis, independent of carcinogens, through activation of innate lymphoid cells. Mucosal Immunol (2014) 7:842–56. doi:10.1038/mi.2013.101
103. Lazaridis KN, LaRusso NF. Primary sclerosing cholangitis. N Engl J Med (2016) 375:1161–70. doi:10.1056/NEJMra1506330
104. Loftus EV, Harewood GC, Loftus CG, Tremaine WJ, Harmsen WS, Zinsmeister AR, et al. PSC-IBD: a unique form of inflammatory bowel disease associated with primary sclerosing cholangitis. Gut (2005) 54:91–6. doi:10.1136/gut.2004.046615
105. Ananthakrishnan AN, Cagan A, Gainer VS, Cheng S-C, Cai T, Szolovits P, et al. Mortality and extraintestinal cancers in patients with primary sclerosing cholangitis and inflammatory bowel disease. J Crohns Colitis (2014) 8:956–63. doi:10.1016/j.crohns.2014.01.019
106. Copelan EA. Hematopoietic stem-cell transplantation. N Engl J Med (2006) 354:1813–26. doi:10.1056/NEJMra052638
107. Ferrara JLM, Levine JE, Reddy P, Holler E. Graft-versus-host disease. Lancet (2009) 373:1550–61. doi:10.1016/S0140-6736(09)60237-3
108. Hanash AM, Dudakov JA, Hua G, O’Connor MH, Young LF, Singer NV, et al. Interleukin-22 protects intestinal stem cells from immune-mediated tissue damage and regulates sensitivity to graft versus host disease. Immunity (2012) 37:339–50. doi:10.1016/j.immuni.2012.05.028
109. Bruce DW, Stefanski HE, Vincent BG, Dant TA, Reisdorf S, Bommiasamy H, et al. Type 2 innate lymphoid cells treat and prevent acute gastrointestinal graft-versus-host disease. J Clin Invest (2017) 127:1813–25. doi:10.1172/JCI91816
110. Munneke JM, Björklund AT, Mjösberg JM, Garming-Legert K, Bernink JH, Blom B, et al. Activated innate lymphoid cells are associated with a reduced susceptibility to graft-versus-host disease. Blood (2014) 124:812–21. doi:10.1182/blood-2013-11-536888
111. Vély F, Barlogis V, Vallentin B, Neven B, Piperoglou C, Ebbo M, et al. Evidence of innate lymphoid cell redundancy in humans. Nat Immunol (2016) 17:1291–9. doi:10.1038/ni.3553
112. Takayama T, Kamada N, Chinen H, Okamoto S, Kitazume MT, Chang J, et al. Imbalance of NKp44(+)NKp46(-) and NKp44(-)NKp46(+) natural killer cells in the intestinal mucosa of patients with Crohn’s disease. Gastroenterology (2010) 139:882–92.e1–3. doi:10.1053/j.gastro.2010.05.040
113. Li J, Doty AL, Iqbal A, Glover SC. The differential frequency of Lineage(-)CRTH2(-)CD45(+)NKp44(-)CD117(-)CD127(+)ILC subset in the inflamed terminal ileum of patients with Crohn’s disease. Cell Immunol (2016) 30(4–305):63–8. doi:10.1016/j.cellimm.2016.05.001
114. Kaser A. Not all monoclonals are created equal – lessons from failed drug trials in Crohn’s disease. Best Pract Res Clin Gastroenterol (2014) 28:437–49. doi:10.1016/j.bpg.2014.04.005
115. Maloy KJ, Kullberg MC. IL-23 and Th17 cytokines in intestinal homeostasis. Mucosal Immunol (2008) 1:339–49. doi:10.1038/mi.2008.28
116. Stockinger B, Omenetti S. The dichotomous nature of T helper 17 cells. Nat Rev Immunol (2017) 7:13416. doi:10.1038/nri.2017.50
117. Hueber W, Sands BE, Lewitzky S, Vandemeulebroecke M, Reinisch W, Higgins PDR, et al. Secukinumab, a human anti-IL-17A monoclonal antibody, for moderate to severe Crohn’s disease: unexpected results of a randomised, double-blind placebo-controlled trial. Gut (2012) 61:1693–700. doi:10.1136/gutjnl-2011-301668
118. Raine T, Kaser A. Seventeen in Crohn’s disease: less prime than we thought? Gut (2012) 61:1653–4. doi:10.1136/gutjnl-2012-302525
119. Hirata Y, Egea L, Dann SM, Eckmann L, Kagnoff MF. GM-CSF-facilitated dendritic cell recruitment and survival govern the intestinal mucosal response to a mouse enteric bacterial pathogen. Cell Host Microbe (2010) 7:151–63. doi:10.1016/j.chom.2010.01.006
120. Sainathan SK, Hanna EM, Gong Q, Bishnupuri KS, Luo Q, Colonna M, et al. Granulocyte macrophage colony-stimulating factor ameliorates DSS-induced experimental colitis. Inflamm Bowel Dis (2008) 14:88–99. doi:10.1002/ibd.20279
121. Han X, Uchida K, Jurickova I, Koch D, Willson T, Samson C, et al. Granulocyte-macrophage colony-stimulating factor autoantibodies in murine ileitis and progressive ileal Crohn’s disease. Gastroenterology (2009) 136:1261–71.e1–3. doi:10.1053/j.gastro.2008.12.046
122. Korzenik JR, Dieckgraefe BK, Valentine JF, Hausman DF, Gilbert MJ, Sargramostim in Crohn’s Disease Study Group. Sargramostim for active Crohn’s disease. N Engl J Med (2005) 352:2193–201. doi:10.1056/NEJMoa041109
123. Roth L, MacDonald JK, McDonald JWD, Chande N. Sargramostim (GM-CSF) for induction of remission in Crohn’s disease: a cochrane inflammatory bowel disease and functional bowel disorders systematic review of randomized trials. Inflamm Bowel Dis (2012) 18:1333–9. doi:10.1002/ibd.22973
124. Mannon PJ, Fuss IJ, Mayer L, Elson CO, Sandborn WJ, Present D, et al. Anti-interleukin-12 antibody for active Crohn’s disease. N Engl J Med (2004) 351:2069–79. doi:10.1056/NEJMoa033402
125. Sandborn WJ, Feagan BG, Fedorak RN, Scherl E, Fleisher MR, Katz S, et al. A randomized trial of Ustekinumab, a human interleukin-12/23 monoclonal antibody, in patients with moderate-to-severe Crohn’s disease. Gastroenterology (2008) 135:1130–41. doi:10.1053/j.gastro.2008.07.014
126. Sandborn WJ, Gasink C, Gao L-L, Blank MA, Johanns J, Guzzo C, et al. Ustekinumab induction and maintenance therapy in refractory Crohn’s disease. N Engl J Med (2012) 367:1519–28. doi:10.1056/NEJMoa1203572
127. Reich K, Langley RG, Papp KA, Ortonne J-P, Unnebrink K, Kaul M, et al. A 52-week trial comparing briakinumab with methotrexate in patients with psoriasis. N Engl J Med (2011) 365:1586–96. doi:10.1056/NEJMoa1010858
128. Griffiths CEM, Strober BE, van de Kerkhof P, Ho V, Fidelus-Gort R, Yeilding N, et al. Comparison of ustekinumab and etanercept for moderate-to-severe psoriasis. N Engl J Med (2010) 362:118–28. doi:10.1056/NEJMoa0810652
129. Feagan BG, Sandborn WJ, D’Haens G, Panés J, Kaser A, Ferrante M, et al. Induction therapy with the selective interleukin-23 inhibitor risankizumab in patients with moderate-to-severe Crohn’s disease: a randomised, double-blind, placebo-controlled phase 2 study. Lancet (2017) 389:1699–709. doi:10.1016/S0140-6736(17)30570-6
Keywords: innate lymphocytes, epithelial barrier integrity, mucosal immune response, microbial flora, cytokines, inflammatory bowel disease, cancer
Citation: Geremia A and Arancibia-Cárcamo CV (2017) Innate Lymphoid Cells in Intestinal Inflammation. Front. Immunol. 8:1296. doi: 10.3389/fimmu.2017.01296
Received: 24 July 2017; Accepted: 27 September 2017;
Published: 13 October 2017
Edited by:
Marina Cella, Washington University School of Medicine, United StatesReviewed by:
Giuseppe Sciume, Sapienza Università di Roma, ItalyAharon Freud, The Ohio State University Columbus, United States
Copyright: © 2017 Geremia and Arancibia-Cárcamo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Alessandra Geremia, alessandra.geremia@ndm.ox.ac.uk