 Andrea C. Lei-Leston
Andrea C. Lei-Leston Alison G. Murphy
Alison G. Murphy Kevin J. Maloy
Kevin J. Maloy- Sir William Dunn School of Pathology, University of Oxford, Oxford, United Kingdom
Pattern recognition receptors (PRR), such as NOD-like receptors (NLRs), sense conserved microbial signatures, and host danger signals leading to the coordination of appropriate immune responses. Upon activation, a subset of NLR initiate the assembly of a multimeric protein complex known as the inflammasome, which processes pro-inflammatory cytokines and mediates a specialized form of cell death known as pyroptosis. The identification of inflammasome-associated genes as inflammatory bowel disease susceptibility genes implicates a role for the inflammasome in intestinal inflammation. Despite the fact that the functional importance of inflammasomes within immune cells has been well established, the contribution of inflammasome expression in non-hematopoietic cells remains comparatively understudied. Given that intestinal epithelial cells (IEC) act as a barrier between the host and the intestinal microbiota, inflammasome expression by these cells is likely important for intestinal immune homeostasis. Accumulating evidence suggests that the inflammasome plays a key role in shaping epithelial responses at the host–lumen interface with many inflammasome components highly expressed by IEC. Recent studies have exposed functional roles of IEC inflammasomes in mucosal immune defense, inflammation, and tumorigenesis. In this review, we present the main features of the predominant inflammasomes and their effector mechanisms contributing to intestinal homeostasis and inflammation. We also discuss existing controversies in the field and open questions related to their implications in disease. A comprehensive understanding of the molecular basis of intestinal inflammasome signaling could hold therapeutic potential for clinical translation.
Introduction
Intestinal homeostasis is governed by complex interactions between the host immune system, the vast constitutive antigenic load in the lumen, and the epithelial barrier. Breakdown in this molecular dialog can lead to the development of chronic pathologies, such as inflammatory bowel diseases (IBD). The precise etiology of IBD remains unclear, although it is likely multifactorial involving a number of elements, such as host genetic susceptibility, environmental factors (e.g., smoking), and the composition of the microbiome (1). These factors contribute to the disturbance of homeostasis leading to the generation of chronic inflammation and development of IBD, including Crohn’s disease (CD) and ulcerative colitis (UC). IBD are debilitating, relapsing diseases affecting approximately 1:400 people. With no cure available, IBD patients are consigned to long-term anti-inflammatory and immune suppressive therapies, and surgery is often required. Thus, there is an urgent, unmet need to further understand the molecular mechanisms underlying IBD, to inform the development of new potential therapies. Genome-wide association studies (GWAS) revealed that inflammasome-associated genes were linked to IBD susceptibility (2), suggesting that this family of proteins is important for maintenance of intestinal homeostasis.
The inflammasome is a multimeric protein complex involved in inflammation. It comprised of an intracellular Pattern Recognition Receptors (PRR), usually a NOD-like receptor (NLR), and is activated in response to exogenous pattern-associated molecular patterns (PAMP) or endogenous danger-associated molecular patterns (DAMP) (3). NLR are highly conserved throughout evolution attesting to their important role in host defense (4). NLR possess three domains: the N-terminal effector domain that may be a caspase recruitment domain (CARD), a pyrin (PYD) domain, or a baculovirus inhibitor of apoptosis repeat (BIR) domain; the central nucleotide-binding oligomerization domain (NOD); and the C-terminal domain comprised of leucine rich repeat sequences (LRR) (5). Based on their N-terminal domains, NLR can be divided into four main families (Table 1). Different NLR have been linked to the detection of different signals, for example, NLRC4 recognizes bacterial flagellin (6, 7) whereas NLRP1 has been implicated in the sensing of anthrax lethal toxin (8), but the specific molecular ligands for a majority of NLRs remain uncharacterized. In some cases, the LRR of the C-terminal bind directly to the PAMP (5); however, the precise mechanism of agonist activation of NLR remains to be determined, as other reports have postulated an auto-inhibitory role for the LRR (9).
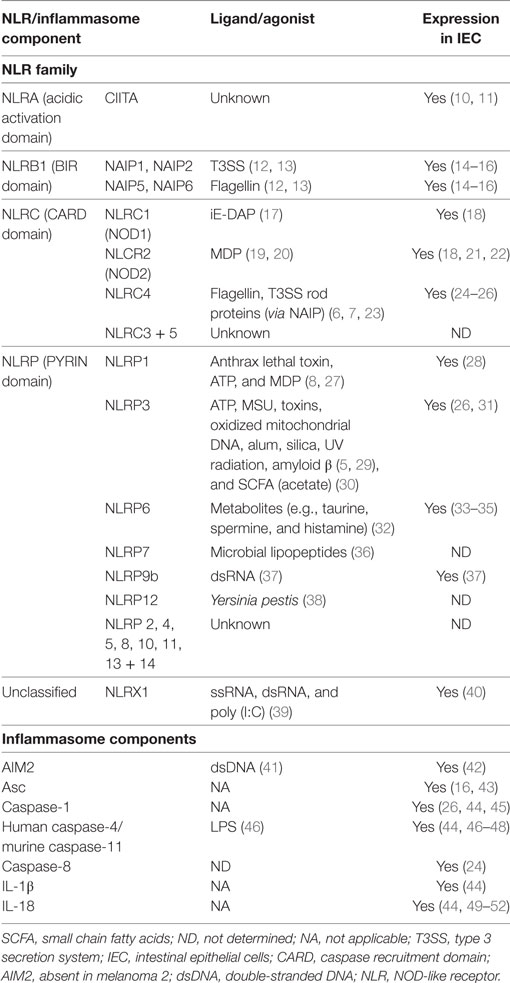
Table 1. NLR family members and other inflammasome components.
Upon sensing of endogenous or exogenous danger signals, some NLR oligomerize via their NOD domains. If the NLR contains a CARD domain this can facilitate the recruitment of the inactive enzyme pro-caspase-1, through direct CARD–CARD interactions. However, inflammasome-forming NLR lacking a CARD domain use their PYD domain to recruit the adaptor protein Apoptosis-associated speck-like protein containing CARD (Asc)—comprising a PYD and a CARD domain, and this serves as a scaffold, bridging the interactions between the NLR and pro-caspase-1. This “canonical” inflammasome formation results in the autocatalytic activation of caspase-1. Caspase-1 has two main functions, cleavage of pro-IL-1β and pro-IL-18 into their active forms for secretion (53, 54), and the induction of a specialized form of inflammatory cell death known as pyroptosis (55–57). Another form of inflammasome has been described which does not require a member of the NLR family, but instead contains members of the PYHIN family (PYD and HIN domain containing). For example, the PYHIN family member absent in melanoma 2 (AIM2) can directly bind to its stimulus, double-stranded DNA (dsDNA), which may be present in the cytosol during infection, to form a caspase-1 containing inflammasome (41).
Of emerging interest in the field is the formation of “non-canonical” inflammasomes by caspase-11 and caspase-8. Caspase-11 was originally discovered to be important in caspase-1 and -3 activation (58) and has been found to indirectly increase processing of pro-IL-1β and pro-IL-18 by promoting NLRP3 inflammasome activation (59). Indeed, it was shown that caspase-11 can detect intracellular LPS, and some intracellular bacteria, leading to cell death (60, 61). The human orthologs of murine caspase-11, namely, caspase-4 and -5, appear to serve similar functions (46, 62). Recently, an inflammasome formed by NLRC4, Asc, and potentially caspase-8 was described in a model of enteric Salmonella enterica serovar Typhimurium (S. Tm) infection, and this inflammasome was required for expulsion of infected intestinal epithelial cells (IEC) (Table 2) (24). There has also been a report of caspase-8 driving caspase-1 cleavage and downstream pro-IL-1β cleavage during Yersinia pestis infection of macrophages (63). Although immune cells and IEC express both “canonical” and “non-canonical” inflammasome components, how these complexes interact with one another upon stimulation and tailor their responses (e.g. pro-inflammatory cytokine secretion versus pyroptosis) remains to be elucidated (Figure 1).
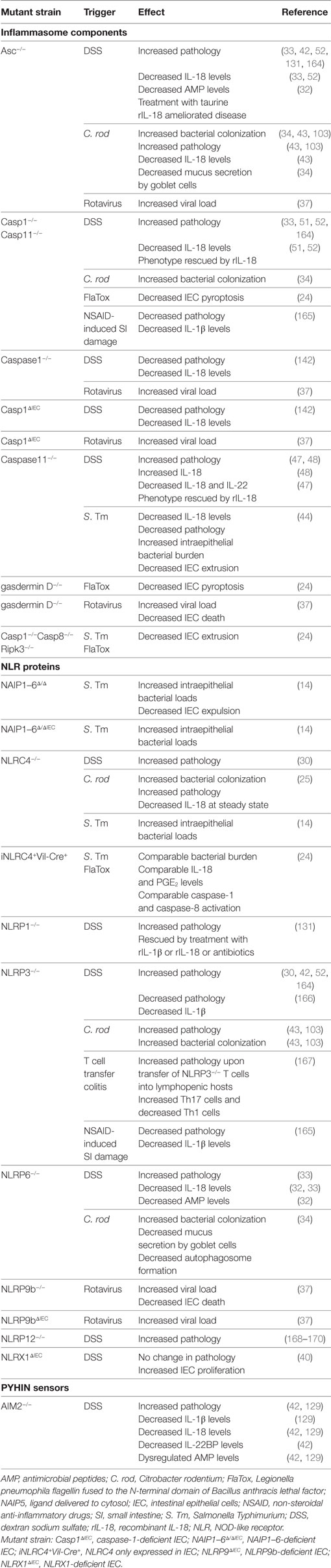
Table 2. Inflammasome components and intestinal inflammation.
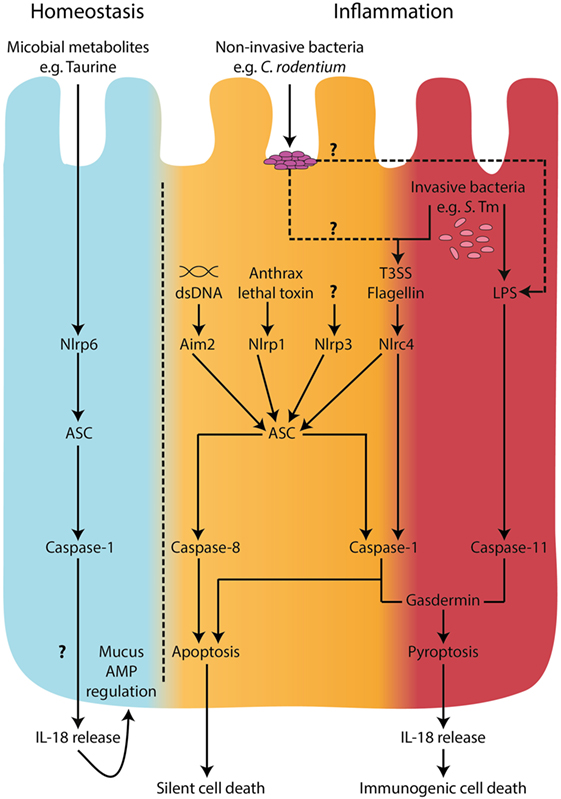
Figure 1. Inflammasomes in intestinal epithelial cells. During homeostatic conditions, in the absence of inflammation, IL-18 is released from epithelial cells and is involved in epithelial repair, proliferation, and maturation (33, 34). A metabolomics screen identified microbiome-derived metabolites, including taurine, that are capable of modulating NLRP6 inflammasome activation and subsequent IL-18 secretion (32). However, the mechanisms of release of IL-18 during homeostatic conditions are undefined. In the context of microbial invasion and pathogen-associated molecular pattern stimulation, inflammasome activation in intestinal epithelial cells has been described to engage both “canonical,” caspase-1-mediated and “non-canonical,” caspase-11 pathways (14, 24, 44). Recently, caspase-8 was also shown to be involved in inflammasome responses downstream of NLRC4 engagement with intracellular flagellin (24). Both caspase-1 and caspase-11 can lead to cell death by pyroptosis accompanied by IL-18 secretion; however, caspase-1 and caspase-8 were shown to lead to a non-lytic form of cell death upon NLRC4 sensing of intracellular flagellin (24). These observations raise the possibility of a distinction between a pro-immunogenic cell death signal driven by caspase-11 and GsdmD, a pro-silent cell death driven by caspase-8, and perhaps a threshold-dependent cellular decision between non-lytic and lytic forms of cell death involving caspase-1. Under low stress levels, it would be desirable to deal with the invading threat in an immunologically silent way. However, when the threat is high, an immunogenic cell death could recruit inflammatory cells to help clear the microbial insult.
Innate immune recognition at mucosal surfaces, in particular the intestine, is a critical mediator of homeostasis (64). Indeed, in the gut, PRR sensing has been implicated in several key processes, such as maintenance and repair of the epithelial barrier and production of antimicrobial peptides (AMP) (65–67). Aside from basal roles at steady state, effective PRR signaling also protects against enteric pathogens by initiating immune responses Tables 2 and 3 (68–70). To date, the majority of work has focused on the role of the hematopoietic compartment in microbial detection and inflammation, but non-hematopoietic cells, particularly IEC, are now appreciated to be important contributors to PRR sensing circuits in the gut (71).
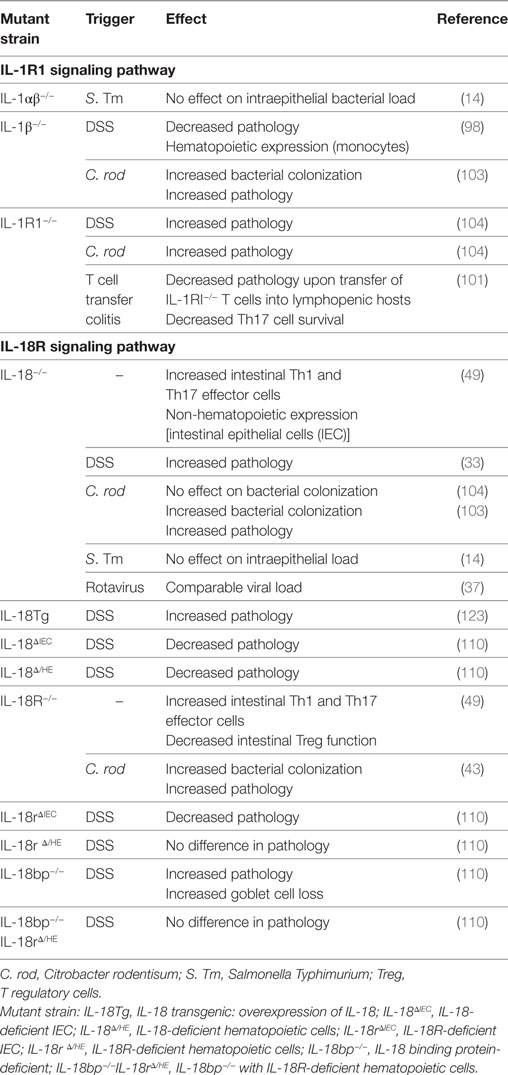
Table 3. Soluble mediators of inflammasome activation and intestinal inflammation.
Intestinal epithelial cells face a unique challenge as they constitute the first cellular border between the complex contents of the gut lumen and the largely sterile subepithelial compartment. This intestinal epithelial surface area is greatly increased by gland like invaginations called crypts, as well as projections of small finger like protrusions in the small intestine, known as villi. IEC are composed of various specialized cell types; enteroabsorptive cells, goblet cells, Paneth cells, neuroendrocrine cells, tuft cells, and stem cells. Due to the constant epithelial turnover, stem cells are responsible for replenishing any lost cells via Notch-mediated epithelial cell differentiation (72). Goblet cells secrete heavily glycosylated mucins which form a mucus matrix (73) into which Paneth cells secrete antimicrobial peptides (AMP) (74–76), together providing a physical and chemical barrier between the epithelial cell layer and the luminal contents. This barrier is further fortified by the secretion of IgA dimers into the mucus layer which act to sterically hinder any potential threats (77). In addition, goblet cells have been reported to deliver luminal antigens to subepithelial antigen-presenting cells enabling screening of the luminal contents (78). Thus, there are numerous antimicrobial mechanisms employed by the epithelium to limit access of potentially inflammatory stimuli.
During homeostasis, interactions with the microbial and dietary antigens induce a non-inflammatory IEC state that promotes immune tolerance. However, luminal content occasionally carries pathogenic microorganisms or toxic particles capable of causing mucosal damage and, in severe cases, systemic disease. Accumulating evidence suggests that the inflammasome plays a key role in modulating epithelial responses at the host–lumen interface. Data generated on purified IEC, in situ detection, or cell-specific ablation have revealed an expression of an array of inflammasome components within IEC including; NAIP, NLRC4, NLRP1, NLRP6, AIM2, caspase-1, caspase-4/5 (human), caspase-11 (mouse), Asc, and IL-18 (Tables 2 and 3) (79, 80). This review will discuss the functional importance of the inflammasome and its components within the context of epithelial cells and intestinal inflammation.
Inflammasomes and Their Soluble Mediators in Intestinal Homeostasis
Inflammasome formation and caspase-1 activation lead to cleavage and secretion of the active forms of IL-1 family member cytokines, such as IL-1β and IL-18. These cytokines play a central role in immunity due to their diverse array of biological functions and broad range of target cells. IL-1β is a potent pro-inflammatory cytokine exerting a plethora of systemic and local effects. IL-1β promotes the recruitment of immune cells to the site of inflammation via induction of adhesion molecules and chemoattractants (81, 82). Stimulation with IL-1β promotes the activation and effector functions of dendritic cells, macrophages, and neutrophils (83). In addition, IL-1β plays a role in adaptive immunity driving T cell activation and survival (84), and acting in concert with other cytokines to promote Th17 cell differentiation (85). Due to these highly pro-inflammatory properties, IL-1β release is tightly regulated via a two-step process, namely, TLR-induced production of an inactive ~31–34 kDa precursor pro-IL-1β, followed by caspase-1 dependent cleavage and secretion of the active form (86).
Several clinical studies reported high levels of IL-1β production by the lamina propria mononuclear cells from active colonic lesions of IBD patients (87–89). IL-1β levels in the colon correlated with disease activity suggesting an important role for this cytokine in driving local inflammation (90, 91). Elevated colonic IL-1β levels are also characteristic of many animal IBD models (92–94), and strategies blocking IL-1β signaling were beneficial in ameliorating acute models of intestinal inflammation (95–98). Moreover, genetic alterations of key innate immune molecules, such as NOD2 and Atg16l1, resulted in over production of IL-1β by macrophages and enhanced susceptibility to dextran sodium sulfate (DSS)-mediated intestinal injury (99, 100). In addition, IL-1β augmented the recruitment of granulocytes and the activation of innate lymphoid cells during Helicobacter hepaticus-triggered intestinal inflammation, and IL-1R signaling in T cells controlled the early accumulation and survival of pathogenic Th17 cells in the colon during T cell transfer colitis (101). The role of IL-1β in promoting intestinal inflammation has also been confirmed in infection studies, as blocking IL-1β ameliorated pathology in both Clostridium difficile-associated colitis and Salmonella Typhimurium-induced enteritis (68, 102). However, alternative studies suggest a protective role for IL-1β during Citrobacter rodentium induced intestinal inflammation, as IL-1R1−/− and IL-1β−/− animals suffered from increased bacterial loads and pathology (Table 3) (103, 104).
Although IL-1β signaling appears to play a predominant role in mediating intestinal inflammation, IEC do not produce significant levels of IL-1β themselves (44, 105). Interestingly, stratified epithelia at other sites produce considerable amounts of IL-1β upon activation of their NLRP3 inflammasome (106, 107). The significance of differential IL-1β expression between epithelial cell types in distinct tissues remains incompletely explored. In the gut, it appears that lamina propria phagocytes constitute the main source of IL-1β during intestinal inflammation (101, 108).
In contrast, there is substantial evidence for the expression and secretion of IL-18 by the intestinal epithelium. Notably, at steady state in the intestine IEC appear to be the primary source of IL-18 (44, 49, 50, 109). The inactive 24 kDa precursor pro-IL-18 is constitutively expressed by IEC, primed for release upon inflammasome activation (44, 49, 50, 109, 110). Akin to IL-1β, IL-18 has been shown to induce a diverse array of immune responses. Originally termed IFNγ-inducing factor, IL-18 is typically considered a Th1 promoting cytokine due to its ability to elicit IFNγ production by T cells (111). However, in the presence of the correct co-stimuli, IL-18 can also drive Th2 cytokine production (112), or IL-17 production by γδ T cells (113). In addition, IEC derived IL-18 can drive perforin production by NK cells during enteric infection with S. Typhimurium, revealing an important role for IEC in coordinating acute mucosal responses (114).
Genome-wide association studies have linked mutations within the IL-18R1-IL-18RAP locus with susceptibility to IBD (115–117). Furthermore, increased IL-18 levels were detected in the biopsies of CD patients (50, 118). Using immunohistochemical analysis, IL-18 localized to the epithelium of non-inflamed regions, whereas in involved regions IL-18 was detected in cells morphologically described as macrophages (50). However, this altered IL-18 distribution was specific to CD, as UC patients displayed an epithelial distribution of IL-18 regardless of severity (50). Moreover, the bioactivity of mature IL-18 is regulated by the production of IL-18 binding protein, levels of which are also elevated in CD patients (119, 120). Thus, although the contributions of IL-18 to clinical intestinal inflammation remain unclear, evidence suggests that dysregulated IL-18 signaling could influence intestinal inflammation.
In murine models, different studies have drawn conflicting conclusions on whether IL-18 plays a predominantly pathogenic or protective role in intestinal inflammation. Early studies using biochemical inhibition of IL-18 signaling revealed a detrimental role for the cytokine in intestinal inflammation mediated by DSS (121, 122). Furthermore, overexpression of IL-18 in IL-18 transgenic mice resulted in increased severity of DSS-mediated intestinal injury (Table 3) (123). Hyperproduction of IL-18 in mice deficient in Atg16l1, a key autophagy adaptor molecule, was also associated with increased susceptibility to DSS, a phenotype which was rescued by antibody-mediated blockade of IL-18 (100). This exacerbated inflammation associated with IL-18 may be due to its ability to induce pro-inflammatory effector T cell activation, even in the absence of T cell receptor engagement (111, 113, 124, 125). In fact, intestinal T cells express significantly greater amounts of IL-18R than those found in systemic lymphoid tissues, suggesting that they may be particularly sensitive to IL-18 signaling (49). Indeed, blocking IL-18 signaling protected mice against colitis mediated by transfer of naive T cells into lymphopenic hosts (126).
Conversely, independent studies using IL-18- and IL-18R-deficient mice revealed a beneficial role for IL-18 signaling during DSS colitis (Table 2) (127, 128). In addition, caspase-1−/− animals were more susceptible to DSS-mediated colitis, which was associated with decreased epithelial cell proliferation and IL-18 secretion (51). This was corroborated by Zaki et al., who also observed increased susceptibility to DSS colitis in the absence of caspase-1 (Table 2) (52). Interestingly, this exacerbated phenotype could be rescued through administration of recombinant IL-18 (rIL-18), but not by adoptive transfer of myeloid cells, suggesting that IL-18 expression in the non-hematopoietic compartment was essential for protection (51, 52). Similarly, non-hematopoietic NLRP6 expression was found to be necessary to protect against DSS colitis, an effect that was again associated with impaired IL-18 production (33). In addition, deficiencies in NLRP6 were associated with a dominant dysbiosis (33) and decreased microbiota diversity (32), with rIL-18 treatment ameliorating this effect by increasing AMP production by IEC (32). Furthermore, a metabolomics screen identified potential microbiome-derived metabolites capable of modulating NLRP6 inflammasome activation and subsequent IL-18 secretion (32). Thus, deficiencies in NLRP6 expression are associated with reduced IL-18 production and the emergence of a dysbiotic microbiome that sensitizes mice to exacerbated DSS-mediated intestinal inflammation. In addition, deficiency in the cytosolic dsDNA sensor AIM2 also led to increased pathology upon DSS administration, which was again associated with decreased IL-18 signaling (Table 2) (42, 129).
In fact, DSS colitis is ameliorated in antibiotic treated genetically susceptible mice (33, 42, 98, 129–131), or exacerbated in mice receiving transfers of pathobionts (98), signifying the importance of the microbiota composition in this model. Microbiota sensing may also mediate protective effects against DSS colitis as evidenced by reports of exacerbated disease in germ-free mice (132) and Myd88−/− mice (133). A key caveat of many studies using DSS colitis models in mice with genetic deficiencies is that they did not employ appropriate co-housing strategies to minimize any potential effects of the microbiota. As such, variations or “dysbiosis” in the microbiota may have occurred as a result of long-term microbial divergence due to extended isolation of breeding cohorts, as was reported for TLR-deficient mice (134). Therefore, studies in which inflammasome-deficient strains were compared to independent breeding cohorts of wild type mice must be interpreted with caution. In addition, these conflicting results emphasize the importance of using littermate controls to evaluate potential differences in susceptibility to experimental colitis in genetically modified mice.
Epithelium-derived IL-18 has also been implicated in protecting against infection-associated intestinal inflammation. For example, IL-18-deficient or IL-18R-deficient mice were more susceptible to colonization and inflammation upon infection with C. rodentium (Table 3) (43, 103, 109). Similarly, caspase1−/− animals suffered from increased susceptibility to C. rodentium infection which was associated with increased inflammatory responses and decreased IL-18 secretion, suggesting a protective role for IL-18 in this model (103). Consistent with these findings, mice deficient in NLRP3 or Asc also suffered from exacerbated C. rodentium infection and pathology (43). Furthermore, non-hematopoietic cells were the source of this protective NLRP3 and Asc circuit, with strong Asc expression evident in the IEC (43). However, although C. rodentium-infected Asc−/− animals almost completely lacked IL-18 in the intestine, the absence of NLRP3 did not affect IL-18 secretion (43). Thus, NLRP3 signaling may be mediating alternative protective pathways aside from IL-18 production (43). NLRC4 expression in IEC is also important for protection against C. rodentium induced intestinal inflammation, and NLRC4 deficiency was associated with decreased basal IL-18 levels and increased early pathogen colonization of the epithelium (25). Thus, the discrepancies in intestinal IL-18 production between the NLRP3- and Asc-deficient mice may be explained in part by compensation of the NLRC4 inflammasome in the absence of NLRP3 expression. Finally, NLRP6 inflammasome expression was also reported to protect against C. rodentium induced inflammation, and this was linked to effective mucin granule exocytosis by goblet cells (Table 2) (34). In addition, NLRP6 inflammasome formation and subsequent IL-18 secretion also enhanced AMP production by IEC (32). The non-redundant requirement for several NLR in protection from attaching and effacing pathogens like C. rodentium suggests that distinct NLR may mediate slightly different protective responses in IEC and/or that activation of NLR in additional cell types may contribute to epithelial protection. In addition, whether and how different inflammasomes interact during C. rodentium infection remains to be fully elucidated, although there is some evidence for the interaction of NLRP3 and NLRC4 inflammasomes during S. Typhimurium infection (135).
The epithelial protective effects of IL-18 may be explained by its roles in wound healing (127, 136) and in driving IL-22 (109), a cytokine important for AMP production and mucosal barrier integrity (137, 138). Of note, IL-22 expression has been shown to protect mice against several models of IBD (139, 140). In fact, administration of rIL-18 to IEC decreased their production of IL-22 binding protein allowing for greater amounts of IL-22 signaling (42). Interestingly, co-administration of IL-22 and IL-18 induced reprogramming of IEC gene expression, not observed with either cytokine alone, which correlated with protection against rotavirus infection, suggesting that these cytokines may act in concert in the intestine to promote antimicrobial responses (141). In addition, IL-18 has also been demonstrated to promote optimal T regulatory cells responses in the gut, with the lack of IL-18 associated with increased proinflammatory T effector cells (49).
Such studies have led to the conclusion that epithelial-derived IL-18 promotes barrier integrity and maintains a healthy microbiota, which contributes to protection against intestinal injury and inflammation. However, this function of IL-18 has been inferred from complete deletion of inflammasome components, as well as the cytokine itself, alongside bone marrow chimera studies. Recently, studies have been conducted using IEC-specific IL-18 knockouts (IL-18ΔIEC) (110) and IEC-specific caspase-1 knockouts (Casp1ΔIEC) (Tables 2 and 3) (142). These studies reported that caspase-1 activation and consequent IL-18 secretion by IEC during DSS colitis was associated with exacerbated inflammation and decreased goblet cell maturation (110, 142). These findings are somewhat surprising, as NLRP6 deficiencies were previously associated with both decreased IL-18 levels (33) and goblet cell mucus secretion (34), which led to increased susceptibility to DSS-mediated intestinal injury. In addition, several studies demonstrated that rIL-18 administration rescued inflammasome-deficient phenotypes from hypersusceptibility to DSS colitis (32, 47, 51, 52, 131). Considering these publications, the authors argue that extrapolation of IL-18 functions from mice fully deficient in inflammasome components should be interpreted with caution, as such deletions may affect the myeloid compartment beyond the scope of IL-18 production (i.e., there could be confounding effects on IL-1β production and pyroptosis). However, numerous bone marrow chimera experiments pointed to the importance of non-hematopoietic inflammasome expression in mediating protection against intestinal inflammation (25, 43, 51, 52, 129). As noted above, it is very likely that the microbiota is a key confounding factor, therefore repeating DSS colitis in IL-18ΔIEC mice housed in alternative vivariums could help clarify the contribution of genotype versus microbiota. Clearly, further studies using mice with cell-type specific ablation of inflammasome components (or effector molecules) need to be carried out to better understand their diverse functions in IEC.
In addition to IL-1 family cytokines, inflammasome activation affects the release of alternative bioactive factors by immune cells. The alarmin high-mobility group box 1 (HMGB1) was originally identified as a nuclear DNA-binding protein. Upon infection or injury, inflammasomes were shown to mediate extracellular release of HMGB1 from stimulated immune cells triggering inflammation (143, 144). In the context of epithelial cells, LPS transfection of IEC led to HMGB1 release (46) and infection of gingival epithelial cells with Fusobacterium nucleatum drove release of HMGB1 alongside Asc and IL-1β secretion (107), suggesting that inflammasomes may be involved in the active secretion of HMGB1 from IEC. Caspase-1 activation has also been hypothesized to play a role in unconventional protein secretion of leaderless peptides such as IL-1α and FGF2 from macrophages (145). Others have postulated that AMP may be regulated via post translation modification by an effector downstream of inflammasome activation (146). The lipid inflammatory mediators, eicosanoids, have also been linked to inflammasome-dependent unconventional secretion (147, 148). In fact, the eicosanoid prostaglandin PGE2 was secreted by murine IEC upon NLRC4 inflammasome activation (24). Examination of the downstream soluble mediators of inflammasome activation, aside from IL-1β and IL-18, remains comparatively understudied in IEC compared to classical immune cells. Future work will need to address this by systematically examining the inflammasome-dependent secretome of activated IEC, and its downstream activities.
Inflammasomes and Cell Death: Pyroptosis and Apoptosis
Inflammasome functional studies to date have largely focused on the secretion of downstream soluble mediators. However, there is much emerging interest in the role of inflammasome-dependent cell death, termed pyroptosis, an inflammatory form of cell death (149, 150). Pyroptosis takes place following engagement of “canonical” (caspase-1) or “non-canonical” (caspase-11) inflammasomes. “Non-canonical” triggering of pyroptosis occurs by intracellular LPS engagement with caspase-11 and has mainly been described in macrophages (151, 152). Identification of the “non-canonical” pathway followed from the finding that 129SvEv mice carried a passenger mutation that truncated the caspase-11 gene (59), meaning that the original caspase-1 knockout mice, which were generated on a 129SvEv background, were deficient in both caspase-1 and caspase-11. Using caspase-11 complementation, Kayagaki et al. showed that macrophages underwent caspase-1-independent “non-canonical” cell death in response to several inflammasome activating stimuli, including Gram-negative bacteria such as Escherichia coli, Vibrio cholerae, and C. rodentium, as well as LPS co-treatment with cholera toxin subunit B (59). Subsequently, it was found that macrophages that were loaded with intracellular LPS activated caspase-11 and died by pyroptosis, and that mice lacking caspase-11 were protected from LPS-induced endotoxemia and pyroptosis (59–61). Finally, two independent studies identified caspase-11 as the key intracellular receptor for LPS (46, 153).
Caspase-11-driven pyroptosis has been shown to be key for protection against intracellular pathogens, particularly those that can escape from phagocytic vacuoles, such as S. Tm (151, 154, 155). However, studies with phagocytes and embryonic fibroblasts reported that caspase-1 “canonical” inflammasomes were required for efficient processing of IL-1β and IL-18, even in the context of direct caspase-11 activation, which was only able to lead to cytokine cleavage via indirect activation of caspase-1 (156–159). Nevertheless, caspase-11-dependent activation of IL-18 has also been reported, for instance, cecal tissue explants from S. Tm-infected caspase-11-deficient mice were also defective in IL-18 but not IL-1β secretion (44). Furthermore, colonic tissue explants from C. rodentium-infected caspase-11-deficient mice also had decreased IL-18 secretion (160). This caspase-11-dependent IL-18 processing was proposed to occur in IEC, contrary to the caspase-1-dependent cleavage of IL-18 and IL-1β observed in myeloid cells (161).
The importance of “canonical” and “non-canonical” inflammasomes may vary depending on the nature and characteristics of the pathogenic threat and the cell types involved. For example, upon challenge with flagellin-deficient Salmonella, caspase-1-deficient macrophages died in a similar manner to WT macrophages, whereas caspase-11-deficient macrophages were resistant to cell death (158, 161). In contrast, both caspase-1 and caspase-11 were required for cell death in macrophages infected with WT Salmonella (158). This highlights the fact that Salmonella can activate both the “canonical” inflammasome, through flagellin–NAIP–NLRC4 interactions, and the “non-canonical” inflammasome, through direct LPS–caspase-11 interactions (Figure 1). The complementary roles of “canonical” and “non-canonical” inflammasomes are especially important in the context of bacterial infections. Bacterial evasion strategies can counteract inflammasome responses, such as inhibition of epithelial caspase-11 via NleF, a type 3 secretion system effector protein produced by E. coli and C. rodentium (160). In a caspase-11-deficient scenario, however, pyroptosis may still proceed due to intact caspase-1 activation, highlighting potential redundancy of these two caspases (162). It seems logical that the intestinal epithelium, as a first line of defense, would have intrinsic mechanisms to warn the immune system of an invading threat. Indeed, as noted above, caspase-11 in mice (an ortholog of human caspases-4/5) is important for the recognition and clearance of S. Tm, and mice lacking caspase-11 harbor increased loads of S. Tm in the intestinal epithelium (14, 44). Furthermore, siRNA knockdown of caspase-4 in human colonic IEC led to reduced cell death upon E. coli, S. Tm, and Shigella flexneri infection (44, 163), and this was accompanied by increased S. Tm intracellular load, and reduced IEC shedding (44, 161).
Recent studies, in addition to highlighting the importance of “non-canonical” inflammasomes in innate immune defense in IEC, have also shed some light on the mechanisms involved in IEC-intrinsic restriction of S. Tm invasion. The innate immune sensor NLRC4 and its NAIP adaptors were shown to be essential for the extrusion of infected IEC into the intestinal lumen following S. Tm challenge of streptomycin-treated mice (14). IEC extrusion may represent a cell-intrinsic defense mechanism that serves to limit the rate of pathogen colonization of the intestinal epithelium. In this study, it was unclear whether IEC extrusion was linked to pyroptosis, as plasma membrane integrity of extruded enterocytes seemed to be maintained (14). However, by using an inducible construct to drive the expression of NLRC4 specifically in the intestinal epithelium, Rauch et al. showed that IEC-specific NLRC4 activation by FlaTox (Legionella pneumophila flagellin fused to the N-terminal domain of Bacillus anthracis lethal factor to drive cytosolic delivery) was sufficient to drive pathology, IEC death and IL-18 release (24).
Moreover, in agreement with the findings of Sellin et al., FlaTox activation of NLRC4 in IEC also limited S. Tm colonization of intestinal tissues and drove IEC death and extrusion (14). However, FlaTox-induced expulsion of IEC was accompanied by lytic cell death with plasma membrane permeabilization, resembling pyroptosis (24) (see Table 4 for morphological features of pyroptosis). From these studies, it becomes clear that, upon NLRC4 activation, IEC can undergo cell death and expulsion from the intestinal epithelium. In parallel, experiments in which caspase-1 expression was selectively induced in IEC, it was found that caspase-1 could drive pyroptosis in response to NLRC4 activation by FlaTox. On the contrary, caspase-1-deficient IEC did not undergo lytic cell death but were expelled from the epithelial layer with intact plasma membranes, indicating that caspase-1 was required for pyroptosis but not for IEC extrusion (24). Furthermore, they also observed that caspase-1-independent IEC extrusion following NLRC4 activation was dependent on both Asc and caspase-8 (24). Taken together, these studies show that various inflammasome-dependent responses are triggered in IEC during S. Tm infection, and these encompass activation of NLRC4, caspase-1, caspase-11 and possibly caspase-8 (14, 24, 44) (see Figure 1). However, it is unclear how the different inflammasome responses are regulated in IEC and if they are redundant, complementary, or interdependent. In addition, further studies are required to better define the precise kinetics and interconnections between downstream responses, such as IEC expulsion and cell death.
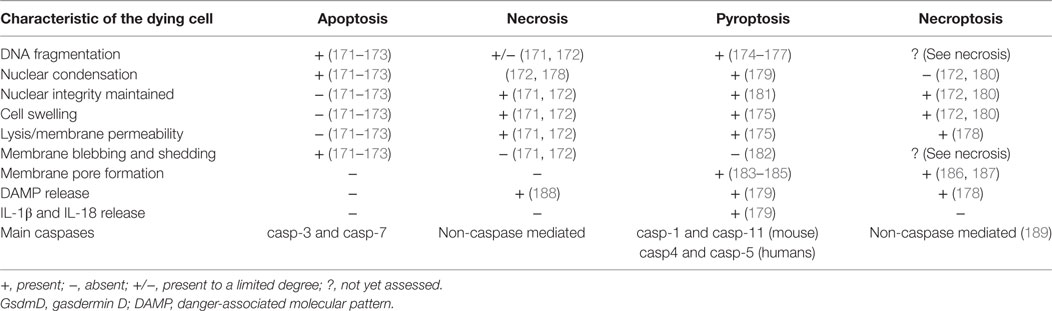
Table 4. Characteristic features of different cell death pathways.
The detailed role of pyroptosis in vivo remains largely unexplored due to limited knowledge of the downstream targets of caspase-1 and caspase-11 culminating in cell death. Recently, however, gasdermin D (GsdmD) was identified as a direct downstream target of caspase-1 and caspase-11 and was shown to be required for pyroptosis upon “canonical” and “non-canonical” inflammasome engagement (150, 183, 184, 190, 191). Indeed, upon GsdmD cleavage by caspase-1 or caspase-11, its ~30 kDa N-terminus embeds itself in the plasma membrane, forming 10–14 nm pores and ultimately leading to lytic cell death (184, 185, 192). Interestingly, GsdmD is highly expressed in the intestinal epithelium, suggesting that GsdmD may also be involved in pyroptosis in IEC (192, 193). Consistent with this hypothesis, IEC pyroptosis in response to in vivo administration FlaTox did not proceed in gasdermin D-deficient mice (24). A very recent study using a mouse model of rotavirus infection reported that activation of a novel NLR inflammasome that recognizes viral dsRNA, NLRP9b, contributed to the restriction of rotavirus replication in IEC organoids, at least partly through gasdermin D-induced pyroptosis (37). Furthermore, mice deficient in either GsdmD or NLRP9b displayed increased susceptibility to rotavirus infection in vivo (37). Collectively, these reports suggest that different IEC inflammasomes converge on GsdmD-induced pyroptosis to restrict pathogen load in infected IEC.
Pyroptosis shares a number of morphological features with both apoptotic and necrotic forms of cell death (Table 4). Akin to necrosis, in pyroptosis, nuclear integrity is maintained, and the cell undergoes cytoplasmic swelling due to membrane permeabilization that ultimately terminates in cell lysis (174, 179). Akin to apoptosis, pyroptotic cells exhibit DNA fragmentation and are TUNEL positive, as well as presenting nuclear condensation (174–176, 179). Before the acknowledgment of pyroptosis as a new form of cell death (57), its similarities to necrosis and apoptosis led researchers to attribute inflammasome-driven cell death to only apoptosis and/or necrosis (174, 176, 179). It is partly for this reason that the interconnections between the different types of cell death upon inflammasome activating stimuli remain poorly understood. The discovery of GsdmD as a key player in pyroptosis should help elucidate the molecular pathways involved (150, 191, 192).
In addition to pyroptosis, inflammasome responses in various cell types have also been linked to apoptotic cell death. For instance, ectopic expression of NLRC4 and Asc in HEK293T cells (which lack caspase-1) showed that these molecules can engage with caspase-8 to drive apoptosis (194). Furthermore, both apoptosis and pyroptosis have been observed in macrophages following NLRP3 or AIM2 activation (194, 195). Interestingly, macrophages lacking GsdmD were reported to undergo cell death upon LPS plus S. Tm or nigericin treatment, through a poorly defined mechanism that was independent of caspase-1, were delayed compared to pyroptosis, and had some features of apoptosis (192).
The literature also suggests some cross-regulation between pyroptosis and apoptosis as THP-1 cells treated with etoposide, an apoptosis inducing drug, resulted in the cleavage of GsdmD into a ~43 kDa fragment, different from the 30 kDa fragment observed in pyroptosis, that occurred independently of caspase-1 (196). The generation of the 43 kDa fragment was observed upon caspase-3 and -7 activation during apoptosis. This suggests that the apoptosis and pyroptosis pathways may compete for the same substrate and that cells may not be able to simultaneously undergo both forms of cell death. The authors speculated that the alternative cleavage of GsdmD by apoptotic caspases-3 and -7 may prevent apoptotic cells from becoming pyroptotic, thus maintaining and immunologically silent cell death (196).
Other studies suggest that differing thresholds may operate between the two cell death pathways following inflammasome activation. For example, in macrophages, for caspase-8 dependent apoptosis to occur upon AIM2 activation, the concentrations of DNA required were much lower than for pyroptosis (195). Under low stress levels, it would be desirable to deal with the invading threat in an immunologically silent way to avoid hyper inflammation, thus apoptosis would be favored. However, when the threat is high, an inflammatory response could help deal with the microbial insult, therefore pyroptosis may be beneficial. However, it is important to stress that it remains to be demonstrated if this threshold-dependent decision controls differential cell death pathways following inflammasome activation in vivo and in cells other than macrophages.
Linking Inflammasome Effector Mechanisms
Both IL-1β and IL-18 lack signal peptides and therefore are not secreted through the conventional ER–Golgi pathway (197–199). For IL-1β, the better described cytokine of the two, several routes of release have been proposed, including secretory lysosomes, exosomes, and microvesicles (200–204). The secretory exosome pathway was proposed through the observation that IL-1β in monocytes was localized in endosomal-like vesicles that are normally targeted for degradation, but can be redirected to the extracellular space (202, 205). In addition, microvesicle-mediated rapid secretion was proposed after observing vesicles associated with bioactive IL-1β as early as 2 min post-ATP stimulation in activated monocytes (203). However, studies on these secretory routes were often contradictory and employed different cell systems, thus these models of secretion remain controversial (206). The mechanisms of secretion of IL-18 are generally assumed to follow the mechanisms of IL-1β secretion but are much less investigated.
However, pyroptosis has now been proposed to be responsible for the release of IL-1β and IL-18 to alert the immune cells of the imminent danger, leading to the onset of inflammatory responses (207). This was first suggested by the observation of caspase-1-dependent pores in the plasma membrane of Salmonella-infected macrophages, ultimately leading to cell swelling and osmotic lysis (175). This was supported by more recent studies of ATP-stimulated BMDM, in which pharmacological inhibition of membrane permeabilization—a hallmark of pyroptosis—abolished IL-1β secretion, but not processing (200). The recent discovery of GsdmD and its requirement for pyroptosis offers a potential mechanistic explanation linking pyroptosis and cytokine secretion. Both caspase-1 and -11 are able to cleave GsdmD, releasing the active N-terminus that mediates pore formation and lytic cell death (184, 185, 191, 192) (Table 3). Consistent with the concept that pyroptosis facilitates cytokine secretion, macrophages lacking GsdmD exhibit defective IL-1β secretion in response to various “canonical” and “non-canonical” inflammasome activators, including intracellular LPS, Gram-negative bacteria and nigericin (150, 185, 191, 192). However, there is also evidence in the literature of IL-1β release in the absence of cell death in peritoneal macrophages, human monocytes, and neutrophils (208, 209). In particular, neutrophils were able to secrete IL-1β in response to Salmonella infection through a mechanism that was dependent on NLRC4 and caspase-1 but was independent of cell lysis (210). The mechanisms of secretion of inflammasome-processed cytokines may therefore be dependent on the cell type and the nature of the activatory signals.
It is again important to emphasize that inflammasome effector responses have largely been studied in leukocytes, particularly phagocytic cells. Whether the discoveries made in these cell types are applicable to tissue cells, including IEC, remains to be determined. For instance, classical activation of the inflammasome has long been viewed as a two-step process, starting with the transcriptional regulation of the inflammasome components. Thus, caspase-11 induced cell death in macrophages was dependent on priming by TLR4 ligands through TRIF, but not on Myd88 signals (157, 158, 211). Indeed, LPS administration in mice, rapidly induced caspase-11 expression in various tissues including thymus, spleen, and lung (161, 212). Conversely, IL-1β release in phagocytic cells depended on Myd88-mediated transcriptional priming (3, 213). These requirements appear to be somewhat different in the intestinal epithelium, for example, although TLR4 signaling is downregulated in IEC (214), caspase-11-dependent responses still occur. This suggests that caspase-11 is constitutively expressed in the intestinal epithelium and can be rapidly activated upon pathogen invasion (48). This “ready-to-go” phenotype of IEC inflammasome components is further supported by the observations that NLRC4 and pro-IL-18 are constitutively expressed by IEC and may not require priming (25, 49). Furthermore, the constitutive colonization of commensal Gram-negative bacteria in the intestine could explain the constitutive elevated expression of caspase-11 and IL-18 in the gut compared to other tissues (www.proteinatlas.org) (161).
It is also worth noting that during homeostatic conditions, and thus in the absence of inflammation, the inflammasome-dependent cytokine IL-18 is released from IEC and is believed to have functions in epithelial repair, proliferation and maturation (33, 34). The mechanisms of secretion of IL-18 by IEC during homeostatic conditions are not well understood and whether pyroptosis occurs in IEC under physiological conditions in vivo remains to be determined (215). Although there is increasing evidence that IEC-intrinsic inflammasome activation plays a key role in early innate defense against pathogens that target the intestinal epithelium (14, 24, 25, 43, 44), much remains to be learned on how inflammasomes and their downstream effector responses are regulated in IEC. Due to their constitutive exposure to microbial PAMP, inflammasome circuits and thresholds in IEC may be quite different to those primarily identified in macrophages and dendritic cells. Nevertheless, the constitutive secretion of IL-18 by IEC indicates that inflammasomes are active under homeostatic conditions in the intestinal epithelium. However, the precise signals or thresholds that determine when this may be superseded by the induction of pyroptosis or alternative cell death pathways remain to be determined. For example, it will be important to assess the role of GsdmD in IL-18 secretion and IEC turnover during steady-state conditions. Furthermore, it will also be vital to understand how inflammasome responses in IEC are modulated during pathogenic attack or during inflammatory conditions, where an optimal balance between apoptosis, pyroptosis, and cytokine release may be required to control potential pathogens and restore homeostasis.
Conclusion and Perspectives
High expression levels of many inflammasome proteins are enriched in the steady-state intestinal mucosa implicating their importance in barrier maintenance and immune monitoring. The spatial location of the IEC, directly facing the lumen, in combination with their primed phenotype, implies that inflammasomes are key sensors of intestinal insults. Indeed, as discussed throughout this review, deletion of these components is primarily associated with increased susceptibility to injury and infection. Thus, we can conclude that epithelial inflammasomes are critical for a healthy gut, both at steady state and during acute infection or injury. However, the molecular mechanisms orchestrating epithelial inflammasome activation remain incompletely understood, representing a key area for further research.
Frustratingly, the literature contains numerous examples of conflicting data pertaining to the functional impact and cellular sources of inflammasome components in various models of intestinal infection and inflammation. To better define these, the field needs to implement stringent lines of investigation that properly control for key environmental factors. Variation of the intestinal microbiome is likely responsible for most of the inconsistent findings reported the literature. For example, recent studies have identified protozoa (216) and microbial metabolites (32) as novel environmental factors capable of influencing inflammasome activation in the intestinal epithelium and in modulating susceptibility to intestinal inflammation. Therefore, standardized use of littermate controls for in vivo experiments should be implemented to circumvent misinterpretations resulting from differences in microbiota composition and baseline mucosal immune activation across distinct breeding cohorts. Furthermore, as different animal facilities will harbor their own distinct microbiotas, it would be advantageous if key experiments were reproduced in different vivariums.
To further assess the specific locations important for inflammasome function, tissue- and cell-specific deletion approaches represent an important approach, for example, the IL-18ΔIEC line specifically lacking IL-18 production in IEC (110). In addition, complementary studies using inducible knockouts will be useful for understanding acute responses while ruling out any developmental disadvantages. The increasing application of primary intestinal epithelial “organoid” cultures will complement the in vivo genetic approaches, enabling analysis of acute responses, as well as offering a tool for molecular manipulation of IEC (217). Moreover, transitioning from murine studies into humans will be bolstered by these new ex vivo techniques (218).
Murine bone marrow-derived macrophages have served as the gold standard for a majority of inflammasome research, contributing significantly to our understanding of inflammasome signaling and effector responses. However, it is likely that IEC inflammasomes are regulated differently to classic hematopoietic cells, due to the unique intestinal environment. Thus, we need to address how inflammasome activation and regulation in IEC differs from that described in myeloid cells and the resulting implications. For example, we can already surmise from the literature that IEC produce comparatively little IL-1β (44, 105) and constitutively express IL-18 (49). It is likely that within IEC there is a different composition of inflammasome machinery to tailor their immune responses. In addition, IEC could be capable of producing other potential secretory factors besides IL-18 upon inflammasome activation, for example, prostaglandin production by IEC was recently associated with NLRC4 activation (24). The signaling circuitry and relationship between different effector responses also needs to be elucidated. For example, are there distinct activation thresholds or can different inflammasome components work in concert, as has been described for NLRC4 and NLRP3 during S. Typhimurium infection of macrophages (219).
Our understanding of what specific agonists activate IEC inflammasomes is limited and warrants further investigation. Aside from microbial signals, how do dietary antigens interact with the intestinal epithelium? Evidence already exists for the capacity of dietary ligands to induce inflammasome activation [e.g., high fat and high cholesterol diets (79)] or dampen inflammasome activation [e.g., ketones (220)]. However, further investigation is required to delineate whether these dietary factors act directly and/or indirectly (e.g., through modulation of the microbiota) (30). Indeed, a recent study reported that a high fiber diet conferred protective effects in the DSS colitis model both by reshaping the gut microbiota and by increasing release of SCFAs that activated NLRP3 inflammasomes in a non-hematopoietic cell population.
Finally, inflammasome activation in IEC has been described to result in IEC extrusion and cell death (14, 24). Further investigation needs to be carried out into the role of pyroptotic cell death in mucosal immune responses. The regulation of different forms of cell death in IEC and the consequences for infection or inflammatory diseases also requires further characterization. For example, does too little IEC death result increased potential for invasive infection due to lack of cell extrusion and does too much IEC death perpetuate unnecessary inflammation? Finally, what function does dysregulated inflammasome activation and pyroptosis play in IBD? IBD patients are known to have necrotic lesions and increased levels of IL-18 and IL-1β in the inflamed intestine, but their relative contributions to chronic intestinal pathology remain incompletely understood.
Despite these challenges and limitations understanding gut-associated inflammasome signaling, its role in regulating dietary–microbiome–host immune interactions constitutes a critical component in maintaining homeostasis and mediating various immune-mediated disorders. Encouragingly, the identification of small molecules capable of targeting specific inflammasome components (44) could represent an opportunity for novel clinical interventions to tackle these currently incurable disorders.
Author Contributions
All authors listed have made a substantial, direct, and intellectual contribution to the work and approved it for publication.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
Our work is supported by a Wellcome Trust Investigator Award to KM (102972). AL-L is supported by funding from the Biotechnology and Biological Sciences Research Council (BBSRC) BB/J014427/1.
References
1. Xavier RJ, Podolsky DK. Unravelling the pathogenesis of inflammatory bowel disease. Nature (2007) 448(7152):427–34. doi:10.1038/nature06005
2. Khor B, Gardet A, Xavier RJ. Genetics and pathogenesis of inflammatory bowel disease. Nature (2011) 474(7351):307–17. doi:10.1038/nature10209
3. Latz E, Xiao TS, Stutz A. Activation and regulation of the inflammasomes. Nat Rev Immunol (2013) 13(6):397–411. doi:10.1038/nri3452
4. Ausubel FM. Are innate immune signaling pathways in plants and animals conserved? Nat Immunol (2005) 6(10):973–9. doi:10.1038/ni1253
5. von Moltke J, Ayres JS, Kofoed EM, Chavarria-Smith J, Vance RE. Recognition of bacteria by inflammasomes. Annu Rev Immunol (2013) 31:73–106. doi:10.1146/annurev-immunol-032712-095944
6. Franchi L, Amer A, Body-Malapel M, Kanneganti TD, Ozoren N, Jagirdar R, et al. Cytosolic flagellin requires Ipaf for activation of caspase-1 and interleukin 1beta in Salmonella-infected macrophages. Nat Immunol (2006) 7(6):576–82. doi:10.1038/ni1346
7. Miao EA, Alpuche-Aranda CM, Dors M, Clark AE, Bader MW, Miller SI, et al. Cytoplasmic flagellin activates caspase-1 and secretion of interleukin 1beta via Ipaf. Nat Immunol (2006) 7(6):569–75. doi:10.1038/ni1344
8. Boyden ED, Dietrich WF. Nalp1b controls mouse macrophage susceptibility to anthrax lethal toxin. Nat Genet (2006) 38(2):240–4. doi:10.1038/ng1724
9. Hu Z, Yan C, Liu P, Huang Z, Ma R, Zhang C, et al. Crystal structure of NLRC4 reveals its autoinhibition mechanism. Science (2013) 341(6142):172–5. doi:10.1126/science.1236381
10. Sanderson IR, Bustin SA, Dziennis S, Paraszczuk J, Stamm DS. Age and diet act through distinct isoforms of the class II transactivator gene in mouse intestinal epithelium. Gastroenterology (2004) 127(1):203–12. doi:10.1053/j.gastro.2004.04.014
11. Hershberg RM, Cho DH, Youakim A, Bradley MB, Lee JS, Framson PE, et al. Highly polarized HLA class II antigen processing and presentation by human intestinal epithelial cells. J Clin Invest (1998) 102(4):792–803. doi:10.1172/JCI3201
12. Kofoed EM, Vance RE. Innate immune recognition of bacterial ligands by NAIPs determines inflammasome specificity. Nature (2011) 477(7366):592–5. doi:10.1038/nature10394
13. Vance RE. The NAIP/NLRC4 inflammasomes. Curr Opin Immunol (2015) 32:84–9. doi:10.1016/j.coi.2015.01.010
14. Sellin ME, Muller AA, Felmy B, Dolowschiak T, Diard M, Tardivel A, et al. Epithelium-intrinsic NAIP/NLRC4 inflammasome drives infected enterocyte expulsion to restrict Salmonella replication in the intestinal mucosa. Cell Host Microbe (2014) 16(2):237–48. doi:10.1016/j.chom.2014.07.001
15. Maier JK, Balabanian S, Coffill CR, Stewart A, Pelletier L, Franks DJ, et al. Distribution of neuronal apoptosis inhibitory protein in human tissues. J Histochem Cytochem (2007) 55(9):911–23. doi:10.1369/jhc.6A7144.2007
16. Allam R, Maillard MH, Tardivel A, Chennupati V, Bega H, Yu CW, et al. Epithelial NAIPs protect against colonic tumorigenesis. J Exp Med (2015) 212(3):369–83. doi:10.1084/jem.20140474
17. Chamaillard M, Hashimoto M, Horie Y, Masumoto J, Qiu S, Saab L, et al. An essential role for NOD1 in host recognition of bacterial peptidoglycan containing diaminopimelic acid. Nat Immunol (2003) 4(7):702–7. doi:10.1038/ni945
18. Natividad JM, Petit V, Huang X, de Palma G, Jury J, Sanz Y, et al. Commensal and probiotic bacteria influence intestinal barrier function and susceptibility to colitis in Nod1-/-; Nod2-/- mice. Inflamm Bowel Dis (2012) 18(8):1434–46. doi:10.1002/ibd.22848
19. Girardin SE, Boneca IG, Viala J, Chamaillard M, Labigne A, Thomas G, et al. Nod2 is a general sensor of peptidoglycan through muramyl dipeptide (MDP) detection. J Biol Chem (2003) 278(11):8869–72. doi:10.1074/jbc.C200651200
20. Girardin SE, Travassos LH, Herve M, Blanot D, Boneca IG, Philpott DJ, et al. Peptidoglycan molecular requirements allowing detection by Nod1 and Nod2. J Biol Chem (2003) 278(43):41702–8. doi:10.1074/jbc.M307198200
21. Rosenstiel P, Fantini M, Brautigam K, Kuhbacher T, Waetzig GH, Seegert D, et al. TNF-alpha and IFN-gamma regulate the expression of the NOD2 (CARD15) gene in human intestinal epithelial cells. Gastroenterology (2003) 124(4):1001–9. doi:10.1053/gast.2003.50157
22. Petnicki-Ocwieja T, Hrncir T, Liu YJ, Biswas A, Hudcovic T, Tlaskalova-Hogenova H, et al. Nod2 is required for the regulation of commensal microbiota in the intestine. Proc Natl Acad Sci U S A (2009) 106(37):15813–8. doi:10.1073/pnas.0907722106
23. Miao EA, Mao DP, Yudkovsky N, Bonneau R, Lorang CG, Warren SE, et al. Innate immune detection of the type III secretion apparatus through the NLRC4 inflammasome. Proc Natl Acad Sci U S A (2010) 107(7):3076–80. doi:10.1073/pnas.0913087107
24. Rauch I, Deets KA, Ji DX, von Moltke J, Tenthorey JL, Lee AY, et al. NAIP-NLRC4 inflammasomes coordinate intestinal epithelial cell expulsion with eicosanoid and IL-18 release via activation of caspase-1 and -8. Immunity (2017) 46(4):649–59. doi:10.1016/j.immuni.2017.03.016
25. Nordlander S, Pott J, Maloy KJ. NLRC4 expression in intestinal epithelial cells mediates protection against an enteric pathogen. Mucosal Immunol (2014) 7(4):775–85. doi:10.1038/mi.2013.95
26. Hu B, Elinav E, Huber S, Booth CJ, Strowig T, Jin C, et al. Inflammation-induced tumorigenesis in the colon is regulated by caspase-1 and NLRC4. Proc Natl Acad Sci U S A (2010) 107(50):21635–40. doi:10.1073/pnas.1016814108
27. Faustin B, Lartigue L, Bruey JM, Luciano F, Sergienko E, Bailly-Maitre B, et al. Reconstituted NALP1 inflammasome reveals two-step mechanism of caspase-1 activation. Mol Cell (2007) 25(5):713–24. doi:10.1016/j.molcel.2007.01.032
28. Chen C, Wang B, Sun J, Na H, Chen Z, Zhu Z, et al. DAC can restore expression of NALP1 to suppress tumor growth in colon cancer. Cell Death Dis (2015) 6:e1602. doi:10.1038/cddis.2014.532
29. Schroder K, Tschopp J. The inflammasomes. Cell (2010) 140(6):821–32. doi:10.1016/j.cell.2010.01.040
30. Macia L, Tan J, Vieira AT, Leach K, Stanley D, Luong S, et al. Metabolite-sensing receptors GPR43 and GPR109A facilitate dietary fibre-induced gut homeostasis through regulation of the inflammasome. Nat Commun (2015) 6:6734. doi:10.1038/ncomms7734
31. Ruiz PA, Moron B, Becker HM, Lang S, Atrott K, Spalinger MR, et al. Titanium dioxide nanoparticles exacerbate DSS-induced colitis: role of the NLRP3 inflammasome. Gut (2017) 66(7):1216–24. doi:10.1136/gutjnl-2015-310297
32. Levy M, Thaiss CA, Zeevi D, Dohnalova L, Zilberman-Schapira G, Mahdi JA, et al. Microbiota-modulated metabolites shape the intestinal microenvironment by regulating NLRP6 inflammasome signaling. Cell (2015) 163(6):1428–43. doi:10.1016/j.cell.2015.10.048
33. Elinav E, Strowig T, Kau AL, Henao-Mejia J, Thaiss CA, Booth CJ, et al. NLRP6 inflammasome regulates colonic microbial ecology and risk for colitis. Cell (2011) 145(5):745–57. doi:10.1016/j.cell.2011.04.022
34. Wlodarska M, Thaiss CA, Nowarski R, Henao-Mejia J, Zhang JP, Brown EM, et al. NLRP6 inflammasome orchestrates the colonic host-microbial interface by regulating goblet cell mucus secretion. Cell (2014) 156(5):1045–59. doi:10.1016/j.cell.2014.01.026
35. Normand S, Delanoye-Crespin A, Bressenot A, Huot L, Grandjean T, Peyrin-Biroulet L, et al. Nod-like receptor pyrin domain-containing protein 6 (NLRP6) controls epithelial self-renewal and colorectal carcinogenesis upon injury. Proc Natl Acad Sci U S A (2011) 108(23):9601–6. doi:10.1073/pnas.1100981108
36. Khare S, Dorfleutner A, Bryan NB, Yun C, Radian AD, de Almeida L, et al. An NLRP7-containing inflammasome mediates recognition of microbial lipopeptides in human macrophages. Immunity (2012) 36(3):464–76. doi:10.1016/j.immuni.2012.02.001
37. Zhu S, Ding S, Wang P, Wei Z, Pan W, Palm NW, et al. Nlrp9b inflammasome restricts rotavirus infection in intestinal epithelial cells. Nature (2017) 546(7660):667–70. doi:10.1038/nature22967
38. Vladimer GI, Weng D, Paquette SW, Vanaja SK, Rathinam VA, Aune MH, et al. The NLRP12 inflammasome recognizes Yersinia pestis. Immunity (2012) 37(1):96–107. doi:10.1016/j.immuni.2012.07.006
39. Hong M, Yoon SI, Wilson IA. Structure and functional characterization of the RNA-binding element of the NLRX1 innate immune modulator. Immunity (2012) 36(3):337–47. doi:10.1016/j.immuni.2011.12.018
40. Tattoli I, Killackey SA, Foerster EG, Molinaro R, Maisonneuve C, Rahman MA, et al. NLRX1 acts as an epithelial-intrinsic tumor suppressor through the modulation of TNF-mediated proliferation. Cell Rep (2016) 14(11):2576–86. doi:10.1016/j.celrep.2016.02.065
41. Hornung V, Ablasser A, Charrel-Dennis M, Bauernfeind F, Horvath G, Caffrey DR, et al. AIM2 recognizes cytosolic dsDNA and forms a caspase-1-activating inflammasome with ASC. Nature (2009) 458(7237):514–8. doi:10.1038/nature07725
42. Ratsimandresy RA, Indramohan M, Dorfleutner A, Stehlik C. The AIM2 inflammasome is a central regulator of intestinal homeostasis through the IL-18/IL-22/STAT3 pathway. Cell Mol Immunol (2017) 14(1):127–42. doi:10.1038/cmi.2016.35
43. Song-Zhao GX, Srinivasan N, Pott J, Baban D, Frankel G, Maloy KJ. Nlrp3 activation in the intestinal epithelium protects against a mucosal pathogen. Mucosal Immunol (2014) 7(4):763–74. doi:10.1038/mi.2013.94
44. Knodler LA, Crowley SM, Sham HP, Yang H, Wrande M, Ma C, et al. Noncanonical inflammasome activation of caspase-4/caspase-11 mediates epithelial defenses against enteric bacterial pathogens. Cell Host Microbe (2014) 16(2):249–56. doi:10.1016/j.chom.2014.07.002
45. Hanby-Flarida MD, Trask OJ, Yang TJ, Baldwin CL. Modulation of WC1, a lineage-specific cell surface molecule of gamma/delta T cells augments cellular proliferation. Immunology (1996) 88(1):116–23. doi:10.1046/j.1365-2567.1996.d01-649.x
46. Shi J, Zhao Y, Wang Y, Gao W, Ding J, Li P, et al. Inflammatory caspases are innate immune receptors for intracellular LPS. Nature (2014) 514(7521):187–92. doi:10.1038/nature13683
47. Oficjalska K, Raverdeau M, Aviello G, Wade SC, Hickey A, Sheehan KM, et al. Protective role for caspase-11 during acute experimental murine colitis. J Immunol (2015) 194(3):1252–60. doi:10.4049/jimmunol.1400501
48. Demon D, Kuchmiy A, Fossoul A, Zhu Q, Kanneganti TD, Lamkanfi M. Caspase-11 is expressed in the colonic mucosa and protects against dextran sodium sulfate-induced colitis. Mucosal Immunol (2014) 7(6):1480–91. doi:10.1038/mi.2014.36
49. Harrison OJ, Srinivasan N, Pott J, Schiering C, Krausgruber T, Ilott NE, et al. Epithelial-derived IL-18 regulates Th17 cell differentiation and Foxp3(+) Treg cell function in the intestine. Mucosal Immunol (2015) 8(6):1226–36. doi:10.1038/mi.2015.13
50. Pizarro TT, Michie MH, Bentz M, Woraratanadharm J, Smith MF, Foley E, et al. IL-18, a novel immunoregulatory cytokine, is up-regulated in Crohn’s disease: expression and localization in intestinal mucosal cells. J Immunol (1999) 162(11):6829–35.
51. Dupaul-Chicoine J, Yeretssian G, Doiron K, Bergstrom KS, McIntire CR, LeBlanc PM, et al. Control of intestinal homeostasis, colitis, and colitis-associated colorectal cancer by the inflammatory caspases. Immunity (2010) 32(3):367–78. doi:10.1016/j.immuni.2010.02.012
52. Zaki MH, Boyd KL, Vogel P, Kastan MB, Lamkanfi M, Kanneganti TD. The NLRP3 inflammasome protects against loss of epithelial integrity and mortality during experimental colitis. Immunity (2010) 32(3):379–91. doi:10.1016/j.immuni.2010.03.003
53. Cerretti DP, Kozlosky CJ, Mosley B, Nelson N, Van Ness K, Greenstreet TA, et al. Molecular cloning of the interleukin-1 beta converting enzyme. Science (1992) 256(5053):97–100. doi:10.1126/science.1373520
54. Ghayur T, Banerjee S, Hugunin M, Butler D, Herzog L, Carter A, et al. Caspase-1 processes IFN-gamma-inducing factor and regulates LPS-induced IFN-gamma production. Nature (1997) 386(6625):619–23. doi:10.1038/386619a0
55. Hersh D, Monack DM, Smith MR, Ghori N, Falkow S, Zychlinsky A. The Salmonella invasin SipB induces macrophage apoptosis by binding to caspase-1. Proc Natl Acad Sci U S A (1999) 96(5):2396–401. doi:10.1073/pnas.96.5.2396
56. Hilbi H, Moss JE, Hersh D, Chen Y, Arondel J, Banerjee S, et al. Shigella-induced apoptosis is dependent on caspase-1 which binds to IpaB. J Biol Chem (1998) 273(49):32895–900. doi:10.1074/jbc.273.49.32895
57. Cookson BT, Brennan MA. Pro-inflammatory programmed cell death. Trends Microbiol (2001) 9(3):113–4. doi:10.1016/S0966-842X(00)01936-3
58. Kang SJ, Wang S, Hara H, Peterson EP, Namura S, Amin-Hanjani S, et al. Dual role of caspase-11 in mediating activation of caspase-1 and caspase-3 under pathological conditions. J Cell Biol (2000) 149(3):613–22. doi:10.1083/jcb.149.3.613
59. Kayagaki N, Warming S, Lamkanfi M, Vande Walle L, Louie S, Dong J, et al. Non-canonical inflammasome activation targets caspase-11. Nature (2011) 479(7371):117–21. doi:10.1038/nature10558
60. Hagar JA, Powell DA, Aachoui Y, Ernst RK, Miao EA. Cytoplasmic LPS activates caspase-11: implications in TLR4-independent endotoxic shock. Science (2013) 341(6151):1250–3. doi:10.1126/science.1240988
61. Kayagaki N, Wong MT, Stowe IB, Ramani SR, Gonzalez LC, Akashi-Takamura S, et al. Noncanonical inflammasome activation by intracellular LPS independent of TLR4. Science (2013) 341(6151):1246–9. doi:10.1126/science.1240248
62. Kajiwara Y, Schiff T, Voloudakis G, Gama Sosa MA, Elder G, Bozdagi O, et al. A critical role for human caspase-4 in endotoxin sensitivity. J Immunol (2014) 193(1):335–43. doi:10.4049/jimmunol.1303424
63. Philip NH, Dillon CP, Snyder AG, Fitzgerald P, Wynosky-Dolfi MA, Zwack EE, et al. Caspase-8 mediates caspase-1 processing and innate immune defense in response to bacterial blockade of NF-kappaB and MAPK signaling. Proc Natl Acad Sci U S A (2014) 111(20):7385–90. doi:10.1073/pnas.1403252111
64. Abreu MT. Toll-like receptor signalling in the intestinal epithelium: how bacterial recognition shapes intestinal function. Nat Rev Immunol (2010) 10(2):131–44. doi:10.1038/nri2707
65. Prabhakara R, Harro JM, Leid JG, Keegan AD, Prior ML, Shirtliff ME. Suppression of the inflammatory immune response prevents the development of chronic biofilm infection due to methicillin-resistant Staphylococcus aureus. Infect Immun (2011) 79(12):5010–8. doi:10.1128/IAI.05571-11
66. Joshi A, Pancari G, Cope L, Bowman EP, Cua D, Proctor RA, et al. Immunization with Staphylococcus aureus iron regulated surface determinant B (IsdB) confers protection via Th17/IL17 pathway in a murine sepsis model. Hum Vaccin Immunother (2012) 8(3):336–46. doi:10.4161/hv.18946
67. Taylor PR, Roy S, Leal SM Jr, Sun Y, Howell SJ, Cobb BA, et al. Activation of neutrophils by autocrine IL-17A-IL-17RC interactions during fungal infection is regulated by IL-6, IL-23, RORgammat and dectin-2. Nat Immunol (2014) 15(2):143–51. doi:10.1038/ni.2797
68. Muller AJ, Hoffmann C, Galle M, Van Den Broeke A, Heikenwalder M, Falter L, et al. The S. Typhimurium effector SopE induces caspase-1 activation in stromal cells to initiate gut inflammation. Cell Host Microbe (2009) 6(2):125–36. doi:10.1016/j.chom.2009.07.007
69. Kim YG, Kamada N, Shaw MH, Warner N, Chen GY, Franchi L, et al. The Nod2 sensor promotes intestinal pathogen eradication via the chemokine CCL2-dependent recruitment of inflammatory monocytes. Immunity (2011) 34(5):769–80. doi:10.1016/j.immuni.2011.04.013
70. Jarchum I, Liu M, Shi C, Equinda M, Pamer EG. Critical role for MyD88-mediated neutrophil recruitment during Clostridium difficile colitis. Infect Immun (2012) 80(9):2989–96. doi:10.1128/IAI.00448-12
71. Fritz JH, Le Bourhis L, Magalhaes JG, Philpott DJ. Innate immune recognition at the epithelial barrier drives adaptive immunity: APCs take the back seat. Trends Immunol (2008) 29(1):41–9. doi:10.1016/j.it.2007.10.002
72. Guilmeau S, Flandez M, Bancroft L, Sellers RS, Tear B, Stanley P, et al. Intestinal deletion of Pofut1 in the mouse inactivates notch signaling and causes enterocolitis. Gastroenterology (2008) 135(3):849–60, 60.e1–6. doi:10.1053/j.gastro.2008.05.050
73. Hansson GC. Role of mucus layers in gut infection and inflammation. Curr Opin Microbiol (2012) 15(1):57–62. doi:10.1016/j.mib.2011.11.002
74. Vaishnava S, Behrendt CL, Ismail AS, Eckmann L, Hooper LV. Paneth cells directly sense gut commensals and maintain homeostasis at the intestinal host-microbial interface. Proc Natl Acad Sci U S A (2008) 105(52):20858–63. doi:10.1073/pnas.0808723105
75. Bevins CL, Salzman NH. Paneth cells, antimicrobial peptides and maintenance of intestinal homeostasis. Nat Rev Microbiol (2011) 9(5):356–68. doi:10.1038/nrmicro2546
76. Vaishnava S, Yamamoto M, Severson KM, Ruhn KA, Yu X, Koren O, et al. The antibacterial lectin RegIIIgamma promotes the spatial segregation of microbiota and host in the intestine. Science (2011) 334(6053):255–8. doi:10.1126/science.1209791
77. Mantis NJ, Rol N, Corthesy B. Secretory IgA’s complex roles in immunity and mucosal homeostasis in the gut. Mucosal Immunol (2011) 4(6):603–11. doi:10.1038/mi.2011.41
78. McDole JR, Wheeler LW, McDonald KG, Wang B, Konjufca V, Knoop KA, et al. Goblet cells deliver luminal antigen to CD103+ dendritic cells in the small intestine. Nature (2012) 483(7389):345–9. doi:10.1038/nature10863
79. Progatzky F, Sangha NJ, Yoshida N, McBrien M, Cheung J, Shia A, et al. Dietary cholesterol directly induces acute inflammasome-dependent intestinal inflammation. Nat Commun (2014) 5:5864. doi:10.1038/ncomms6864
80. Sellin ME, Maslowski KM, Maloy KJ, Hardt WD. Inflammasomes of the intestinal epithelium. Trends Immunol (2015) 36(8):442–50. doi:10.1016/j.it.2015.06.002
81. Collins T, Read MA, Neish AS, Whitley MZ, Thanos D, Maniatis T. Transcriptional regulation of endothelial cell adhesion molecules: NF-kappa B and cytokine-inducible enhancers. FASEB J (1995) 9(10):899–909.
82. Chou RC, Kim ND, Sadik CD, Seung E, Lan Y, Byrne MH, et al. Lipid-cytokine-chemokine cascade drives neutrophil recruitment in a murine model of inflammatory arthritis. Immunity (2010) 33(2):266–78. doi:10.1016/j.immuni.2010.07.018
84. Ben-Sasson SZ, Hu-Li J, Quiel J, Cauchetaux S, Ratner M, Shapira I, et al. IL-1 acts directly on CD4 T cells to enhance their antigen-driven expansion and differentiation. Proc Natl Acad Sci U S A (2009) 106(17):7119–24. doi:10.1073/pnas.0902745106
85. Sutton CE, Lalor SJ, Sweeney CM, Brereton CF, Lavelle EC, Mills KH. Interleukin-1 and IL-23 induce innate IL-17 production from gammadelta T cells, amplifying Th17 responses and autoimmunity. Immunity (2009) 31(2):331–41. doi:10.1016/j.immuni.2009.08.001
86. Afonina IS, Muller C, Martin SJ, Beyaert R. Proteolytic processing of interleukin-1 family cytokines: variations on a common theme. Immunity (2015) 42(6):991–1004. doi:10.1016/j.immuni.2015.06.003
87. Satsangi J, Wolstencroft RA, Cason J, Ainley CC, Dumonde DC, Thompson RP. Interleukin 1 in Crohn’s disease. Clin Exp Immunol (1987) 67(3):594–605.
88. McAlindon ME, Hawkey CJ, Mahida YR. Expression of interleukin 1 beta and interleukin 1 beta converting enzyme by intestinal macrophages in health and inflammatory bowel disease. Gut (1998) 42(2):214–9. doi:10.1136/gut.42.2.214
89. Mahida YR, Wu K, Jewell DP. Enhanced production of interleukin 1-beta by mononuclear cells isolated from mucosa with active ulcerative colitis of Crohn’s disease. Gut (1989) 30(6):835–8. doi:10.1136/gut.30.6.835
90. Casini-Raggi V, Kam L, Chong YJ, Fiocchi C, Pizarro TT, Cominelli F. Mucosal imbalance of IL-1 and IL-1 receptor antagonist in inflammatory bowel disease. A novel mechanism of chronic intestinal inflammation. J Immunol (1995) 154(5):2434–40.
91. Ludwiczek O, Vannier E, Borggraefe I, Kaser A, Siegmund B, Dinarello CA, et al. Imbalance between interleukin-1 agonists and antagonists: relationship to severity of inflammatory bowel disease. Clin Exp Immunol (2004) 138(2):323–9. doi:10.1111/j.1365-2249.2004.02599.x
92. McCall RD, Haskill S, Zimmermann EM, Lund PK, Thompson RC, Sartor RB. Tissue interleukin 1 and interleukin-1 receptor antagonist expression in enterocolitis in resistant and susceptible rats. Gastroenterology (1994) 106(4):960–72. doi:10.1016/0016-5085(94)90755-2
93. Okayasu I, Hatakeyama S, Yamada M, Ohkusa T, Inagaki Y, Nakaya R. A novel method in the induction of reliable experimental acute and chronic ulcerative colitis in mice. Gastroenterology (1990) 98(3):694–702. doi:10.1016/0016-5085(90)90290-H
94. Cominelli F, Nast CC, Clark BD, Schindler R, Lierena R, Eysselein VE, et al. Interleukin 1 (IL-1) gene expression, synthesis, and effect of specific IL-1 receptor blockade in rabbit immune complex colitis. J Clin Invest (1990) 86(3):972–80. doi:10.1172/JCI114799
95. Cominelli F, Nast CC, Duchini A, Lee M. Recombinant interleukin-1 receptor antagonist blocks the proinflammatory activity of endogenous interleukin-1 in rabbit immune colitis. Gastroenterology (1992) 103(1):65–71. doi:10.1016/0016-5085(92)91096-M
96. Siegmund B, Lehr HA, Fantuzzi G, Dinarello CA. IL-1 beta-converting enzyme (caspase-1) in intestinal inflammation. Proc Natl Acad Sci U S A (2001) 98(23):13249–54. doi:10.1073/pnas.231473998
97. Thomas TK, Will PC, Srivastava A, Wilson CL, Harbison M, Little J, et al. Evaluation of an interleukin-1 receptor antagonist in the rat acetic acid-induced colitis model. Agents Actions (1991) 34(1–2):187–90. doi:10.1007/BF01993274
98. Seo SU, Kamada N, Munoz-Planillo R, Kim YG, Kim D, Koizumi Y, et al. Distinct commensals induce interleukin-1beta via NLRP3 inflammasome in inflammatory monocytes to promote intestinal inflammation in response to injury. Immunity (2015) 42(4):744–55. doi:10.1016/j.immuni.2015.03.004
99. Maeda S, Hsu LC, Liu H, Bankston LA, Iimura M, Kagnoff MF, et al. Nod2 mutation in Crohn’s disease potentiates NF-kappaB activity and IL-1beta processing. Science (2005) 307(5710):734–8. doi:10.1126/science.1103685
100. Saitoh T, Fujita N, Jang MH, Uematsu S, Yang BG, Satoh T, et al. Loss of the autophagy protein Atg16L1 enhances endotoxin-induced IL-1beta production. Nature (2008) 456(7219):264–8. doi:10.1038/nature07383
101. Coccia M, Harrison OJ, Schiering C, Asquith MJ, Becher B, Powrie F, et al. IL-1beta mediates chronic intestinal inflammation by promoting the accumulation of IL-17A secreting innate lymphoid cells and CD4(+) Th17 cells. J Exp Med (2012) 209(9):1595–609. doi:10.1084/jem.20111453
102. Ng J, Hirota SA, Gross O, Li Y, Ulke-Lemee A, Potentier MS, et al. Clostridium difficile toxin-induced inflammation and intestinal injury are mediated by the inflammasome. Gastroenterology (2010) 139(2):542–52, 52.e1–3. doi:10.1053/j.gastro.2010.04.005
103. Liu Z, Zaki MH, Vogel P, Gurung P, Finlay BB, Deng W, et al. Role of inflammasomes in host defense against Citrobacter rodentium infection. J Biol Chem (2012) 287(20):16955–64. doi:10.1074/jbc.M112.358705
104. Lebeis SL, Powell KR, Merlin D, Sherman MA, Kalman D. Interleukin-1 receptor signaling protects mice from lethal intestinal damage caused by the attaching and effacing pathogen Citrobacter rodentium. Infect Immun (2009) 77(2):604–14. doi:10.1128/IAI.00907-08
105. Thinwa J, Segovia JA, Bose S, Dube PH. Integrin-mediated first signal for inflammasome activation in intestinal epithelial cells. J Immunol (2014) 193(3):1373–82. doi:10.4049/jimmunol.1400145
106. Choi HW, Bowen SE, Miao Y, Chan CY, Miao EA, Abrink M, et al. Loss of bladder epithelium induced by cytolytic mast cell granules. Immunity (2016) 45(6):1258–69. doi:10.1016/j.immuni.2016.11.003
107. Bui FQ, Johnson L, Roberts J, Hung SC, Lee J, Atanasova KR, et al. Fusobacterium nucleatum infection of gingival epithelial cells leads to NLRP3 inflammasome-dependent secretion of IL-1beta and the danger signals ASC and HMGB1. Cell Microbiol (2016) 18(7):970–81. doi:10.1111/cmi.12560
108. Franchi L, Kamada N, Nakamura Y, Burberry A, Kuffa P, Suzuki S, et al. NLRC4-driven production of IL-1beta discriminates between pathogenic and commensal bacteria and promotes host intestinal defense. Nat Immunol (2012) 13(5):449–56. doi:10.1038/ni.2263
109. Munoz M, Eidenschenk C, Ota N, Wong K, Lohmann U, Kuhl AA, et al. Interleukin-22 induces interleukin-18 expression from epithelial cells during intestinal infection. Immunity (2015) 42(2):321–31. doi:10.1016/j.immuni.2015.01.011
110. Nowarski R, Jackson R, Gagliani N, de Zoete MR, Palm NW, Bailis W, et al. Epithelial IL-18 equilibrium controls barrier function in colitis. Cell (2015) 163(6):1444–56. doi:10.1016/j.cell.2015.10.072
111. Okamura H, Tsutsi H, Komatsu T, Yutsudo M, Hakura A, Tanimoto T, et al. Cloning of a new cytokine that induces IFN-gamma production by T cells. Nature (1995) 378(6552):88–91. doi:10.1038/378088a0
112. Greenfeder SA, Nunes P, Kwee L, Labow M, Chizzonite RA, Ju G. Molecular cloning and characterization of a second subunit of the interleukin 1 receptor complex. J Biol Chem (1995) 270(23):13757–65. doi:10.1074/jbc.270.23.13757
113. Lalor SJ, Dungan LS, Sutton CE, Basdeo SA, Fletcher JM, Mills KH. Caspase-1-processed cytokines IL-1beta and IL-18 promote IL-17 production by gammadelta and CD4 T cells that mediate autoimmunity. J Immunol (2011) 186(10):5738–48. doi:10.4049/jimmunol.1003597
114. Muller AA, Dolowschiak T, Sellin ME, Felmy B, Verbree C, Gadient S, et al. An NK cell perforin response elicited via IL-18 controls mucosal inflammation kinetics during Salmonella gut infection. PLoS Pathog (2016) 12(6):e1005723. doi:10.1371/journal.ppat.1005723
115. Barrett JC, Hansoul S, Nicolae DL, Cho JH, Duerr RH, Rioux JD, et al. Genome-wide association defines more than 30 distinct susceptibility loci for Crohn’s disease. Nat Genet (2008) 40(8):955–62. doi:10.1038/ng.175
116. Hedl M, Zheng S, Abraham C. The IL18RAP region disease polymorphism decreases IL-18RAP/IL-18R1/IL-1R1 expression and signaling through innate receptor-initiated pathways. J Immunol (2014) 192(12):5924–32. doi:10.4049/jimmunol.1302727
117. Imielinski M, Baldassano RN, Griffiths A, Russell RK, Annese V, Dubinsky M, et al. Common variants at five new loci associated with early-onset inflammatory bowel disease. Nat Genet (2009) 41(12):1335–40. doi:10.1038/ng.489
118. Monteleone G, Trapasso F, Parrello T, Biancone L, Stella A, Iuliano R, et al. Bioactive IL-18 expression is up-regulated in Crohn’s disease. J Immunol (1999) 163(1):143–7.
119. Corbaz A, ten Hove T, Herren S, Graber P, Schwartsburd B, Belzer I, et al. IL-18-binding protein expression by endothelial cells and macrophages is up-regulated during active Crohn’s disease. J Immunol (2002) 168(7):3608–16. doi:10.4049/jimmunol.168.7.3608
120. Naftali T, Novick D, Gabay G, Rubinstein M, Novis B. Interleukin-18 and its binding protein in patients with inflammatory bowel disease during remission and exacerbation. Isr Med Assoc J (2007) 9(7):504–8.
121. Sivakumar PV, Westrich GM, Kanaly S, Garka K, Born TL, Derry JM, et al. Interleukin 18 is a primary mediator of the inflammation associated with dextran sulphate sodium induced colitis: blocking interleukin 18 attenuates intestinal damage. Gut (2002) 50(6):812–20. doi:10.1136/gut.50.6.812
122. Siegmund B, Fantuzzi G, Rieder F, Gamboni-Robertson F, Lehr HA, Hartmann G, et al. Neutralization of interleukin-18 reduces severity in murine colitis and intestinal IFN-gamma and TNF-alpha production. Am J Physiol Regul Integr Comp Physiol (2001) 281(4):R1264–73.
123. Ishikura T, Kanai T, Uraushihara K, Iiyama R, Makita S, Totsuka T, et al. Interleukin-18 overproduction exacerbates the development of colitis with markedly infiltrated macrophages in interleukin-18 transgenic mice. J Gastroenterol Hepatol (2003) 18(8):960–9. doi:10.1046/j.1440-1746.2003.03097.x
124. Nakanishi K, Yoshimoto T, Tsutsui H, Okamura H. Interleukin-18 regulates both Th1 and Th2 responses. Annu Rev Immunol (2001) 19:423–74. doi:10.1146/annurev.immunol.19.1.423
125. Millward JM, Lobner M, Wheeler RD, Owens T. Inflammation in the central nervous system and Th17 responses are inhibited by IFN-gamma-induced IL-18 binding protein. J Immunol (2010) 185(4):2458–66. doi:10.4049/jimmunol.0902153
126. Wirtz S, Becker C, Blumberg R, Galle PR, Neurath MF. Treatment of T cell-dependent experimental colitis in SCID mice by local administration of an adenovirus expressing IL-18 antisense mRNA. J Immunol (2002) 168(1):411–20. doi:10.4049/jimmunol.168.1.411
127. Takagi H, Kanai T, Okazawa A, Kishi Y, Sato T, Takaishi H, et al. Contrasting action of IL-12 and IL-18 in the development of dextran sodium sulphate colitis in mice. Scand J Gastroenterol (2003) 38(8):837–44. doi:10.1080/00365520310004047
128. Salcedo R, Worschech A, Cardone M, Jones Y, Gyulai Z, Dai RM, et al. MyD88-mediated signaling prevents development of adenocarcinomas of the colon: role of interleukin 18. J Exp Med (2010) 207(8):1625–36. doi:10.1084/jem.20100199
129. Hu S, Peng L, Kwak YT, Tekippe EM, Pasare C, Malter JS, et al. The DNA sensor AIM2 maintains intestinal homeostasis via regulation of epithelial antimicrobial host defense. Cell Rep (2015) 13(9):1922–36. doi:10.1016/j.celrep.2015.10.040
130. Kitajima S, Morimoto M, Sagara E, Shimizu C, Ikeda Y. Dextran sodium sulfate-induced colitis in germ-free IQI/Jic mice. Exp Anim (2001) 50(5):387–95. doi:10.1538/expanim.50.387
131. Williams TM, Leeth RA, Rothschild DE, Coutermarsh-Ott SL, McDaniel DK, Simmons AE, et al. The NLRP1 inflammasome attenuates colitis and colitis-associated tumorigenesis. J Immunol (2015) 194(7):3369–80. doi:10.4049/jimmunol.1402098
132. Maslowski KM, Vieira AT, Ng A, Kranich J, Sierro F, Yu D, et al. Regulation of inflammatory responses by gut microbiota and chemoattractant receptor GPR43. Nature (2009) 461(7268):1282. doi:10.1038/nature08530
133. Rakoff-Nahoum S, Paglino J, Eslami-Varzaneh F, Edberg S, Medzhitov R. Recognition of commensal microflora by toll-like receptors is required for intestinal homeostasis. Cell (2004) 118(2):229–41. doi:10.1016/j.cell.2004.07.002
134. Ubeda C, Lipuma L, Gobourne A, Viale A, Leiner I, Equinda M, et al. Familial transmission rather than defective innate immunity shapes the distinct intestinal microbiota of TLR-deficient mice. J Exp Med (2012) 209(8):1445–56. doi:10.1084/jem.20120504
135. Qu Y, Misaghi S, Newton K, Maltzman A, Izrael-Tomasevic A, Arnott D, et al. NLRP3 recruitment by NLRC4 during Salmonella infection. J Exp Med (2016) 213(6):877–85. doi:10.1084/jem.20132234
136. Kampfer H, Kalina U, Muhl H, Pfeilschifter J, Frank S. Counterregulation of interleukin-18 mRNA and protein expression during cutaneous wound repair in mice. J Invest Dermatol (1999) 113(3):369–74. doi:10.1046/j.1523-1747.1999.00704.x
137. Ouyang W, Rutz S, Crellin NK, Valdez PA, Hymowitz SG. Regulation and functions of the IL-10 family of cytokines in inflammation and disease. Annu Rev Immunol (2011) 29:71–109. doi:10.1146/annurev-immunol-031210-101312
138. Sonnenberg GF, Fouser LA, Artis D. Border patrol: regulation of immunity, inflammation and tissue homeostasis at barrier surfaces by IL-22. Nat Immunol (2011) 12(5):383–90. doi:10.1038/ni.2025
139. Zheng Y, Valdez PA, Danilenko DM, Hu Y, Sa SM, Gong Q, et al. Interleukin-22 mediates early host defense against attaching and effacing bacterial pathogens. Nat Med (2008) 14(3):282–9. doi:10.1038/nm1720
140. Zenewicz LA, Yancopoulos GD, Valenzuela DM, Murphy AJ, Stevens S, Flavell RA. Innate and adaptive interleukin-22 protects mice from inflammatory bowel disease. Immunity (2008) 29(6):947–57. doi:10.1016/j.immuni.2008.11.003
141. Zhang B, Chassaing B, Shi Z, Uchiyama R, Zhang Z, Denning TL, et al. Viral infection. Prevention and cure of rotavirus infection via TLR5/NLRC4-mediated production of IL-22 and IL-18. Science (2014) 346(6211):861–5. doi:10.1126/science.1256999
142. Błażejewski AJ, Thiemann S, Schenk A, Pils MC, Gálvez EJ, Roy U, et al. Microbiota normalization reveals that canonical caspase-1 activation exacerbates chemically induced intestinal inflammation. Cell Rep (2017) 19(11):2319–30. doi:10.1016/j.celrep.2017.05.058
143. Lamkanfi M, Sarkar A, Vande Walle L, Vitari AC, Amer AO, Wewers MD, et al. Inflammasome-dependent release of the alarmin HMGB1 in endotoxemia. J Immunol (2010) 185(7):4385–92. doi:10.4049/jimmunol.1000803
144. Vande Walle L, Kanneganti TD, Lamkanfi M. HMGB1 release by inflammasomes. Virulence (2011) 2(2):162–5. doi:10.4161/viru.2.2.15480
145. Keller M, Ruegg A, Werner S, Beer HD. Active caspase-1 is a regulator of unconventional protein secretion. Cell (2008) 132(5):818–31. doi:10.1016/j.cell.2007.12.040
146. Lamkanfi M. Emerging inflammasome effector mechanisms. Nat Rev Immunol (2011) 11(3):213–20. doi:10.1038/nri2936
147. von Moltke J, Trinidad NJ, Moayeri M, Kintzer AF, Wang SB, van Rooijen N, et al. Rapid induction of inflammatory lipid mediators by the inflammasome in vivo. Nature (2012) 490(7418):107–11. doi:10.1038/nature11351
148. Jorgensen I, Lopez JP, Laufer SA, Miao EA. IL-1beta, IL-18, and eicosanoids promote neutrophil recruitment to pore-induced intracellular traps following pyroptosis. Eur J Immunol (2016) 46(12):2761–6. doi:10.1002/eji.201646647
149. Bergsbaken T, Fink SL, Cookson BT. Pyroptosis: host cell death and inflammation. Nat Rev Microbiol (2009) 7(2):99–109. doi:10.1038/nrmicro2070
150. Shi J, Zhao Y, Wang K, Shi X, Wang Y, Huang H, et al. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature (2015) 526(7575):660–5. doi:10.1038/nature15514
151. Aachoui Y, Leaf IA, Hagar JA, Fontana MF, Campos CG, Zak DE, et al. Caspase-11 protects against bacteria that escape the vacuole. Science (2013) 339(6122):975–8. doi:10.1126/science.1230751
152. Aachoui Y, Sagulenko V, Miao EA, Stacey KJ. Inflammasome-mediated pyroptotic and apoptotic cell death, and defense against infection. Curr Opin Microbiol (2013) 16(3):319–26. doi:10.1016/j.mib.2013.04.004
153. Yang J, Zhao Y, Shao F. Non-canonical activation of inflammatory caspases by cytosolic LPS in innate immunity. Curr Opin Immunol (2015) 32:78–83. doi:10.1016/j.coi.2015.01.007
154. Meunier E, Dick MS, Dreier RF, Schurmann N, Kenzelmann Broz D, Warming S, et al. Caspase-11 activation requires lysis of pathogen-containing vacuoles by IFN-induced GTPases. Nature (2014) 509(7500):366–70. doi:10.1038/nature13157
155. Miao EA, Leaf IA, Treuting PM, Mao DP, Dors M, Sarkar A, et al. Caspase-1-induced pyroptosis is an innate immune effector mechanism against intracellular bacteria. Nat Immunol (2010) 11(12):1136–42. doi:10.1038/ni.1960
156. Broz P, von Moltke J, Jones JW, Vance RE, Monack DM. Differential requirement for caspase-1 autoproteolysis in pathogen-induced cell death and cytokine processing. Cell Host Microbe (2010) 8(6):471–83. doi:10.1016/j.chom.2010.11.007
157. Rathinam VA, Vanaja SK, Waggoner L, Sokolovska A, Becker C, Stuart LM, et al. TRIF licenses caspase-11-dependent NLRP3 inflammasome activation by Gram-negative bacteria. Cell (2012) 150(3):606–19. doi:10.1016/j.cell.2012.07.007
158. Broz P, Ruby T, Belhocine K, Bouley DM, Kayagaki N, Dixit VM, et al. Caspase-11 increases susceptibility to Salmonella infection in the absence of caspase-1. Nature (2012) 490(7419):288–91. doi:10.1038/nature11419
159. Wang SY, Miura M, Jung YK, Zhu H, Li E, Yuan JY. Murine caspase-11, an ICE-interacting protease, is essential for the activation of ICE. Cell (1998) 92(4):501–9. doi:10.1016/S0092-8674(00)80943-5
160. Pallett MA, Crepin VF, Serafini N, Habibzay M, Kotik O, Sanchez-Garrido J, et al. Bacterial virulence factor inhibits caspase-4/11 activation in intestinal epithelial cells. Mucosal Immunol (2017) 10(3):602–12. doi:10.1038/mi.2016.77
161. Crowley SM, Vallance BA, Knodler LA. Noncanonical inflammasomes: antimicrobial defense that does not play by the rules. Cell Microbiol (2017) 19(4). doi:10.1111/cmi.12730
162. Man SM, Karki R, Briard B, Burton A, Gingras S, Pelletier S, et al. Differential roles of caspase-1 and caspase-11 in infection and inflammation. Sci Rep (2017) 7:45126. doi:10.1038/srep45126
163. Kobayashi T, Ogawa M, Sanada T, Mimuro H, Kim M, Ashida H, et al. The Shigella OspC3 effector inhibits caspase-4, antagonizes inflammatory cell death, and promotes epithelial infection. Cell Host Microbe (2013) 13(5):570–83. doi:10.1016/j.chom.2013.04.012
164. Allen IC, TeKippe EM, Woodford RM, Uronis JM, Holl EK, Rogers AB, et al. The NLRP3 inflammasome functions as a negative regulator of tumorigenesis during colitis-associated cancer. J Exp Med (2010) 207(5):1045–56. doi:10.1084/jem.20100050
165. Higashimori A, Watanabe T, Nadatani Y, Takeda S, Otani K, Tanigawa T, et al. Mechanisms of NLRP3 inflammasome activation and its role in NSAID-induced enteropathy. Mucosal Immunol (2016) 9(3):659–68. doi:10.1038/mi.2015.89
166. Bauer C, Duewell P, Mayer C, Lehr HA, Fitzgerald KA, Dauer M, et al. Colitis induced in mice with dextran sulfate sodium (DSS) is mediated by the NLRP3 inflammasome. Gut (2010) 59(9):1192–9. doi:10.1136/gut.2009.197822
167. Arbore G, West EE, Spolski R, Robertson AA, Klos A, Rheinheimer C, et al. T helper 1 immunity requires complement-driven NLRP3 inflammasome activity in CD4(+) T cells. Science (2016) 352(6292):aad1210. doi:10.1126/science.aad1210
168. Chen L, Wilson JE, Koenigsknecht MJ, Chou WC, Montgomery SA, Truax AD, et al. NLRP12 attenuates colon inflammation by maintaining colonic microbial diversity and promoting protective commensal bacterial growth. Nat Immunol (2017) 18(5):541–51. doi:10.1038/ni.3690
169. Zaki MH, Vogel P, Malireddi RK, Body-Malapel M, Anand PK, Bertin J, et al. The NOD-like receptor NLRP12 attenuates colon inflammation and tumorigenesis. Cancer Cell (2011) 20(5):649–60. doi:10.1016/j.ccr.2011.10.022
170. Allen IC, Wilson JE, Schneider M, Lich JD, Roberts RA, Arthur JC, et al. NLRP12 suppresses colon inflammation and tumorigenesis through the negative regulation of noncanonical NF-kappaB signaling. Immunity (2012) 36(5):742–54. doi:10.1016/j.immuni.2012.03.012
171. Galluzzi L, Vitale I, Abrams JM, Alnemri ES, Baehrecke EH, Blagosklonny MV, et al. Molecular definitions of cell death subroutines: recommendations of the Nomenclature Committee on Cell Death 2012. Cell Death Differ (2012) 19(1):107–20. doi:10.1038/cdd.2011.96
172. Kroemer G, Galluzzi L, Vandenabeele P, Abrams J, Alnemri ES, Baehrecke EH, et al. Classification of cell death: recommendations of the Nomenclature Committee on Cell Death 2009. Cell Death Differ (2009) 16(1):3–11. doi:10.1038/cdd.2008.150
173. Kerr JF, Wyllie AH, Currie AR. Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. Br J Cancer (1972) 26(4):239–57. doi:10.1038/bjc.1972.33
174. Labbe K, Saleh M. Cell death in the host response to infection. Cell Death Differ (2008) 15(9):1339–49. doi:10.1038/cdd.2008.91
175. Fink SL, Cookson BT. Caspase-1-dependent pore formation during pyroptosis leads to osmotic lysis of infected host macrophages. Cell Microbiol (2006) 8(11):1812–25. doi:10.1111/j.1462-5822.2006.00751.x
176. Monack DM, Raupach B, Hromockyj AE, Falkow S. Salmonella typhimurium invasion induces apoptosis in infected macrophages. Proc Natl Acad Sci U S A (1996) 93(18):9833–8. doi:10.1073/pnas.93.18.9833
177. Galluzzi L, Aaronson SA, Abrams J, Alnemri ES, Andrews DW, Baehrecke EH, et al. Guidelines for the use and interpretation of assays for monitoring cell death in higher eukaryotes. Cell Death Differ (2009) 16(8):1093–107. doi:10.1038/cdd.2009.44
178. Galluzzi L, Bravo-San Pedro JM, Vitale I, Aaronson SA, Abrams JM, Adam D, et al. Essential versus accessory aspects of cell death: recommendations of the NCCD 2015. Cell Death Differ (2015) 22(1):58–73. doi:10.1038/cdd.2014.137
179. Brennan MA, Cookson BT. Salmonella induces macrophage death by caspase-1-dependent necrosis. Mol Microbiol (2000) 38(1):31–40. doi:10.1046/j.1365-2958.2000.02103.x
180. Vandenabeele P, Galluzzi L, Vanden Berghe T, Kroemer G. Molecular mechanisms of necroptosis: an ordered cellular explosion. Nat Rev Mol Cell Biol (2010) 11(10):700–14. doi:10.1038/nrm2970
181. Aglietti RA, Dueber EC. Recent insights into the molecular mechanisms underlying pyroptosis and gasdermin family functions. Trends Immunol (2017) 38(4):261–71. doi:10.1016/j.it.2017.01.003
182. Fernandes-Alnemri T, Wu J, Yu JW, Datta P, Miller B, Jankowski W, et al. The pyroptosome: a supramolecular assembly of ASC dimers mediating inflammatory cell death via caspase-1 activation. Cell Death Differ (2007) 14(9):1590–604. doi:10.1038/sj.cdd.4402194
183. Aglietti RA, Estevez A, Gupta A, Ramirez MG, Liu PS, Kayagaki N, et al. GsdmD p30 elicited by caspase-11 during pyroptosis forms pores in membranes. Proc Natl Acad Sci U S A (2016) 113(28):7858–63. doi:10.1073/pnas.1607769113
184. Liu X, Zhang Z, Ruan J, Pan Y, Magupalli VG, Wu H, et al. Inflammasome-activated gasdermin D causes pyroptosis by forming membrane pores. Nature (2016) 535(7610):153–8. doi:10.1038/nature18629
185. Ding J, Wang K, Liu W, She Y, Sun Q, Shi J, et al. Pore-forming activity and structural autoinhibition of the gasdermin family. Nature (2016) 535(7610):111–6. doi:10.1038/nature18590
186. Chen X, He WT, Hu L, Li J, Fang Y, Wang X, et al. Pyroptosis is driven by non-selective gasdermin-D pore and its morphology is different from MLKL channel-mediated necroptosis. Cell Res (2016) 26(9):1007–20. doi:10.1038/cr.2016.100
187. Cai Z, Jitkaew S, Zhao J, Chiang H-C, Choksi S, Liu J, et al. Plasma membrane translocation of trimerized MLKL protein is required for TNF-induced necroptosis. Nat Cell Biol (2014) 16(1):55–65. doi:10.1038/ncb2883
188. Festjens N, Vanden Berghe T, Vandenabeele P. Necrosis, a well-orchestrated form of cell demise: signalling cascades, important mediators and concomitant immune response. Biochim Biophys Acta (2006) 1757(9–10):1371–87. doi:10.1016/j.bbabio.2006.06.014
189. Hitomi J, Christofferson DE, Ng A, Yao J, Degterev A, Xavier RJ, et al. Identification of a molecular signaling network that regulates a cellular necrotic cell death pathway. Cell (2008) 135(7):1311–23. doi:10.1016/j.cell.2008.10.044
190. Sborgi L, Ruhl S, Mulvihill E, Pipercevic J, Heilig R, Stahlberg H, et al. GSDMD membrane pore formation constitutes the mechanism of pyroptotic cell death. EMBO J (2016) 35(16):1766–78. doi:10.15252/embj.201694696
191. Kayagaki N, Stowe IB, Lee BL, O’Rourke K, Anderson K, Warming S, et al. Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature (2015) 526(7575):666–71. doi:10.1038/nature15541
192. He WT, Wan H, Hu L, Chen P, Wang X, Huang Z, et al. Gasdermin D is an executor of pyroptosis and required for interleukin-1beta secretion. Cell Res (2015) 25(12):1285–98. doi:10.1038/cr.2015.139
193. Saeki N, Sasaki H. Gasdermin superfamily: a novel gene family functioning in epithelial cells. In: Carrasco J, Mota M, editors. Endothelium and Epithelium: Composition, Functions and Pathology. New York: Nova Science Publishers, Inc (2012). p. 193–211.
194. Pierini R, Perret M, Djebali S, Juruj C, Michallet MC, Forster I, et al. ASC controls IFN-gamma levels in an IL-18-dependent manner in caspase-1-deficient mice infected with Francisella novicida. J Immunol (2013) 191(7):3847–57. doi:10.4049/jimmunol.1203326
195. Sagulenko V, Thygesen SJ, Sester DP, Idris A, Cridland JA, Vajjhala PR, et al. AIM2 and NLRP3 inflammasomes activate both apoptotic and pyroptotic death pathways via ASC. Cell Death Differ (2013) 20(9):1149–60. doi:10.1038/cdd.2013.37
196. Taabazuing CY, Okondo MC, Bachovchin DA. Pyroptosis and apoptosis pathways engage in bidirectional crosstalk in monocytes and macrophages. Cell Chem Biol (2017) 24(4):507–14.e4. doi:10.1016/j.chembiol.2017.03.009
197. Rothman JE. Mechanisms of intracellular protein transport. Nature (1994) 372(6501):55–63. doi:10.1038/372055a0
198. Auron PE, Webb AC, Rosenwasser LJ, Mucci SF, Rich A, Wolff SM, et al. Nucleotide sequence of human monocyte interleukin 1 precursor cDNA. Proc Natl Acad Sci U S A (1984) 81(24):7907–11. doi:10.1073/pnas.81.24.7907
199. Rubartelli A, Cozzolino F, Talio M, Sitia R. A novel secretory pathway for interleukin-1 beta, a protein lacking a signal sequence. EMBO J (1990) 9(5):1503–10.
200. Martin-Sanchez F, Diamond C, Zeitler M, Gomez AI, Baroja-Mazo A, Bagnall J, et al. Inflammasome-dependent IL-1beta release depends upon membrane permeabilisation. Cell Death Differ (2016) 23(7):1219–31. doi:10.1038/cdd.2015.176
201. Nickel W, Rabouille C. Mechanisms of regulated unconventional protein secretion. Nat Rev Mol Cell Biol (2009) 10(2):148–55. doi:10.1038/nrm2617
202. Andrei C, Dazzi C, Lotti L, Torrisi MR, Chimini G, Rubartelli A. The secretory route of the leaderless protein interleukin 1β involves exocytosis of endolysosome-related vesicles. Mol Biol Cell (1999) 10(5):1463–75. doi:10.1091/mbc.10.5.1463
203. MacKenzie A, Wilson HL, Kiss-Toth E, Dower SK, North RA, Surprenant A. Rapid secretion of interleukin-1β by microvesicle shedding. Immunity (2001) 15(5):825–35. doi:10.1016/S1074-7613(01)00229-1
204. Qu Y, Franchi L, Nunez G, Dubyak GR. Nonclassical IL-1β secretion stimulated by P2X7 receptors is dependent on inflammasome activation and correlated with exosome release in murine macrophages. J Immunol (2007) 179(3):1913–25. doi:10.4049/jimmunol.179.3.1913
205. Lopez-Castejon G, Brough D. Understanding the mechanism of IL-1β secretion. Cytokine Growth Factor Rev (2011) 22(4):189–95. doi:10.1016/j.cytogfr.2011.10.001
206. Monteleone M, Stow JL, Schroder K. Mechanisms of unconventional secretion of IL-1 family cytokines. Cytokine (2015) 74(2):213–8. doi:10.1016/j.cyto.2015.03.022
207. Alvarez-Errico D, Vento-Tormo R, Ballestar E. Genetic and epigenetic determinants in autoinflammatory diseases. Front Immunol (2017) 8:318. doi:10.3389/fimmu.2017.00318
208. Gaidt MM, Ebert TS, Chauhan D, Schmidt T, Schmid-Burgk JL, Rapino F, et al. Human monocytes engage an alternative inflammasome pathway. Immunity (2016) 44(4):833–46. doi:10.1016/j.immuni.2016.01.012
209. Pelegrin P, Barroso-Gutierrez C, Surprenant A. P2X7 receptor differentially couples to distinct release pathways for IL-1β in mouse macrophage. J Immunol (2008) 180(11):7147–57. doi:10.4049/jimmunol.180.11.7147
210. Chen KW, Gross CJ, Sotomayor FV, Stacey KJ, Tschopp J, Sweet MJ, et al. The neutrophil NLRC4 inflammasome selectively promotes IL-1beta maturation without pyroptosis during acute Salmonella challenge. Cell Rep (2014) 8(2):570–82. doi:10.1016/j.celrep.2014.06.028
211. Gurung P, Malireddi RK, Anand PK, Demon D, Vande Walle L, Liu Z, et al. Toll or interleukin-1 receptor (TIR) domain-containing adaptor inducing interferon-beta (TRIF)-mediated caspase-11 protease production integrates toll-like receptor 4 (TLR4) protein- and Nlrp3 inflammasome-mediated host defense against enteropathogens. J Biol Chem (2012) 287(41):34474–83. doi:10.1074/jbc.M112.401406
212. Wang S, Miura M, Jung Y-k, Zhu H, Gagliardini V, Shi L, et al. Identification and characterization of Ich-3, a member of the interleukin-1β converting enzyme (ICE)/Ced-3 family and an upstream regulator of ICE. J Biol Chem (1996) 271(34):20580–7. doi:10.1074/jbc.271.34.20580
213. Bauernfeind FG, Horvath G, Stutz A, Alnemri ES, MacDonald K, Speert D, et al. Cutting edge: NF-κB activating pattern recognition and cytokine receptors license NLRP3 inflammasome activation by regulating NLRP3 expression. J Immunol (2009) 183(2):787–91. doi:10.4049/jimmunol.0901363
214. Abreu MT, Arnold ET, Thomas LS, Gonsky R, Zhou YH, Hu B, et al. TLR4 and MD-2 expression is regulated by immune-mediated signals in human intestinal epithelial cells. J Biol Chem (2002) 277(23):20431–7. doi:10.1074/jbc.M110333200
215. Nagata S, Tanaka M. Programmed cell death and the immune system. Nat Rev Immunol (2017) 17(5):333–40. doi:10.1038/nri.2016.153
216. Chudnovskiy A, Mortha A, Kana V, Kennard A, Ramirez JD, Rahman A, et al. Host-protozoan interactions protect from mucosal infections through activation of the inflammasome. Cell (2016) 167(2):444–56.e14. doi:10.1016/j.cell.2016.08.076
217. Sato T, Clevers H. Growing self-organizing mini-guts from a single intestinal stem cell: mechanism and applications. Science (2013) 340(6137):1190–4. doi:10.1126/science.1234852
218. Fatehullah A, Tan SH, Barker N. Organoids as an in vitro model of human development and disease. Nat Cell Biol (2016) 18(3):246–54. doi:10.1038/ncb3312
219. Man SM, Hopkins LJ, Nugent E, Cox S, Gluck IM, Tourlomousis P, et al. Inflammasome activation causes dual recruitment of NLRC4 and NLRP3 to the same macromolecular complex. Proc Natl Acad Sci U S A (2014) 111(20):7403–8. doi:10.1073/pnas.1402911111
Keywords: inflammasome, IL-18, IL-1β, intestinal epithelial cells, NOD-like receptor, pyroptosis, inflammatory bowel disease
Citation: Lei-Leston AC, Murphy AG and Maloy KJ (2017) Epithelial Cell Inflammasomes in Intestinal Immunity and Inflammation. Front. Immunol. 8:1168. doi: 10.3389/fimmu.2017.01168
Received: 14 July 2017; Accepted: 04 September 2017;
Published: 20 September 2017
Edited by:
Moritz Leppkes, Universitätsklinikum Erlangen, GermanyReviewed by:
Laurence Macia, University of Sydney, AustraliaLuca Pastorelli, Università degli Studi di Milano, Italy
Copyright: © 2017 Lei-Leston, Murphy and Maloy. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kevin J. Maloy, a2V2aW4ubWFsb3lAcGF0aC5veC5hYy51aw==
†These authors have contributed equally to this work.