
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 06 September 2017
Sec. Inflammation
Volume 8 - 2017 | https://doi.org/10.3389/fimmu.2017.01085
This article is part of the Research Topic Neuro-immune Interactions in Inflammation and Autoimmunity View all 19 articles
 Takeshi Fujii1Masato Mashimo1
Takeshi Fujii1Masato Mashimo1 Yasuhiro Moriwaki2Hidemi Misawa2
Yasuhiro Moriwaki2Hidemi Misawa2 Shiro Ono3Kazuhide Horiguchi4
Shiro Ono3Kazuhide Horiguchi4 Koichiro Kawashima5*
Koichiro Kawashima5*
T and B cells express most cholinergic system components—e.g., acetylcholine (ACh), choline acetyltransferase (ChAT), acetylcholinesterase, and both muscarinic and nicotinic ACh receptors (mAChRs and nAChRs, respectively). Using ChATBAC-eGFP transgenic mice, ChAT expression has been confirmed in T and B cells, dendritic cells, and macrophages. Moreover, T cell activation via T-cell receptor/CD3-mediated pathways upregulates ChAT mRNA expression and ACh synthesis, suggesting that this lymphocytic cholinergic system contributes to the regulation of immune function. Immune cells express all five mAChRs (M1–M5). Combined M1/M5 mAChR-deficient (M1/M5-KO) mice produce less antigen-specific antibody than wild-type (WT) mice. Furthermore, spleen cells in M1/M5-KO mice produce less tumor necrosis factor (TNF)-α and interleukin (IL)-6, suggesting M1/M5 mAChRs are involved in regulating pro-inflammatory cytokine and antibody production. Immune cells also frequently express the α2, α5, α6, α7, α9, and α10 nAChR subunits. α7 nAChR-deficient (α7-KO) mice produce more antigen-specific antibody than WT mice, and spleen cells from α7-KO mice produce more TNF-α and IL-6 than WT cells. This suggests that α7 nAChRs are involved in regulating cytokine production and thus modulate antibody production. Evidence also indicates that nicotine modulates immune responses by altering cytokine production and that α7 nAChR signaling contributes to immunomodulation through modification of T cell differentiation. Together, these findings suggest the involvement of both mAChRs and nAChRs in the regulation of immune function. The observation that vagus nerve stimulation protects mice from lethal endotoxin shock led to the notion of a cholinergic anti-inflammatory reflex pathway, and the spleen is an essential component of this anti-inflammatory reflex. Because the spleen lacks direct vagus innervation, it has been postulated that ACh synthesized by a subset of CD4+ T cells relays vagal nerve signals to α7 nAChRs on splenic macrophages, which downregulates TNF-α synthesis and release, thereby modulating inflammatory responses. However, because the spleen is innervated solely by the noradrenergic splenic nerve, confirmation of an anti-inflammatory reflex pathway involving the spleen requires several more hypotheses to be addressed. We will review and discuss these issues in the context of the cholinergic system in immune cells.
Acetylcholine (ACh) is one of the old neurotransmitters identified in the central and peripheral nervous systems. First synthesized in 1867 by von Baeyer, who acetylated choline using acetylchloride, ACh was left on the chemical list for several decades without exploration of its biological activity [see a review by Burgen (1)]. In 1914, however, Ewins (2) identified ACh as the active principle in ergot that exerts an inhibitory effect on the heart but a stimulatory effect on intestinal muscle. This was the first discovery of ACh in a life form. Those findings prompted Dale (3) to extensively investigate the biological activities of choline derivatives, including ACh. A little later, Loewi (4) demonstrated that the effects of autonomic nerve impulses were transmitted through peripheral release of a specific chemical stimulant in isolated frog heart preparations; this was later proved pharmacologically to be ACh (5). Dale and Dudley (6) then successfully isolated ACh from the spleens of an ox and a horse, making them the first to isolate ACh from an animal organ. On the basis of these findings, and in conjunction with the resemblance between the effects of sympathetic nerves and those of adrenaline (7), Dale (8) suggested the term “cholinergic” to describe nerves that transmit their action through release of ACh, and “adrenergic” for those who employ a substance resembling adrenaline. In 1936, The Nobel Prize in Physiology or Medicine was awarded jointly to Sir Henry H. Dale and Otto Loewi “for their discoveries relating to chemical transmission of nerve impulses.” Since then, ACh has been widely recognized as a neurotransmitter.
The expression of muscarinic and nicotinic ACh receptors (mAChRs and nAChRs, respectively) in lymphocytes and thymocytes has been known since early 1970s, based on the various functional and biochemical changes elicited by ACh and agonists such as carbachol, oxotremorine (Oxo), and nicotine in these cells [see a review by Kawashima and Fujii (9)]. Moreover, expression of mAChRs and nAChRs in lymphocytes and thymocytes was confirmed in binding studies using radiolabeled mAChR and nAChR ligands such as [3H]quinuclinidyl benzilate, [3H]nicotine, and [125I]α-bungarotoxin (α-BTX) [see reviews in Ref. (9–12)]. At the time of its discovery, the origin of the splenic ACh isolated by Dale and Dudley (6) was left unexplored, and the findings summarized above were interpreted to show control of immune cells by the parasympathetic nervous system via ACh. Although the anatomy of immune system innervation has not yet been fully described, it is now generally agreed that the spleen receives innervation by sympathetic neurons but not by parasympathetic cholinergic neuron (10, 13–16). And the enigma of the origin of ACh that should act on the mAChRs and nAChRs on immune cells was ultimately solved based on the discovery of ACh in the blood and its localization to lymphocytes using a sensitive and specific radioimmunoassay for ACh (17, 18); also see reviews (9, 11, 12). Thereafter, data from a variety of investigations provided evidence that immune cells possess all the required components to constitute an independent cholinergic system, including choline acetyltransferase (ChAT, EC 2.3.2.6) and acetylcholinesterase (AChE, EC 3.1.1.7) as well as mAChRs and nAChRs [see reviews in Ref. (9, 11, 12)]. Furthermore, as reviewed by Fujii et al. (16), recent findings on the cholinergic system in immune cells suggest that ACh synthesized by immune cells plays a key role in the regulation of immune function by triggering signals that initiate and terminate cytokine production in immune cells.
In this review, we will discuss (1) the cholinergic components expressed in T and B cells, macrophages, and dendritic cells (DCs); (2) the functions of AChRs in the regulation of immune cell activity; and (3) the functions of the immune cell cholinergic system within an anti-inflammatory reflex.
We will first discuss the following major cholinergic system components: (1) ACh and ChAT, an ACh-synthesizing enzyme; (2) the ACh-degrading enzymes AChE and butyrylcholinesterase (BuChE, EC 3.1.1.8); (3) mAChRs and nAChRs; and (4) secreted lymphocyte antigen-6/urokinase-type plasminogen activator (SLURP)-1 and -2, two endogenous positive allosteric ligands for α7 and α3 nAChRs.
In immune cells and in the central and peripheral nervous systems, ACh is synthesized from choline and acetyl coenzyme A (acetyl-CoA) by ChAT.
After discovery of ACh in the peripheral blood and plasma of humans and animals, the presence of ACh in immune cells was first demonstrated in the human peripheral blood mononuclear leukocyte (MNL) fraction, which consists mainly of lymphocytes and a small monocyte fraction (18–23); also see a review (9). Later, the presence of ACh in immune cells was confirmed by detection of ACh in various human leukemic cell lines (24, 25) and rat lymphocytes, including T and B cells (26). These findings provided an explanation for the seemingly enigmatic observation of Dale and Dudley (6) that ACh was present in the spleen, though that organ is not cholinergically innervated.
In general, human leukemic T cell lines had higher ACh contents than B cell lines, prelymphoma cell lines, or a monocytic cell line (Table 1) (9, 25). Among rat lymphocytes, the ACh content in T cells was significantly higher than in B cells, and the ACh content in CD4+ T cells was significantly higher than in CD8+ T cells (26). The higher ACh content observed in rat T cells than B cells reflects the higher ChAT activity in T cells (27). Little information is available on the intracellular ACh contents in macrophages and DCs.
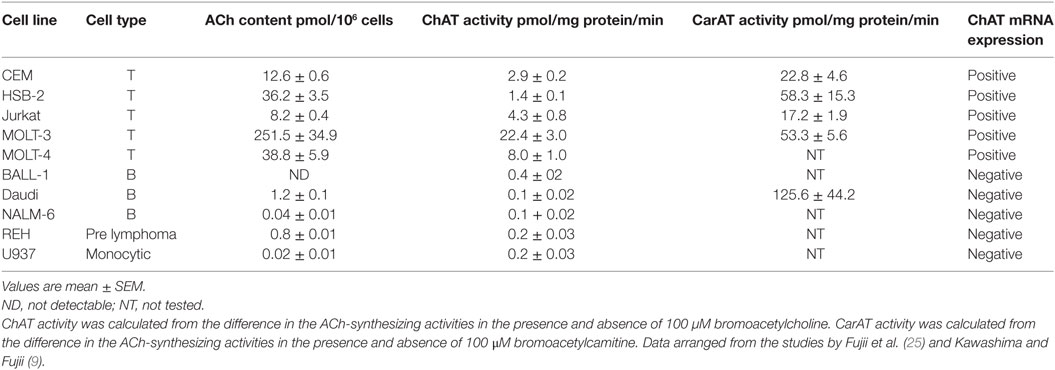
Table 1. ACh content, ChAT and CarAT activities, and ChAT mRNA expression in human leukemic cell lines.
It is important to note that, with the molecular weight of 146 Da, ACh is small, water soluble, and both physicochemically and enzymatically fragile. Furthermore, the chemical nature of ACh is quite different from that of catecholamines and serotonin, which are able to be fixed to a tissue using paraformaldehyde. At present, no technique is available to fix ACh to the tissue. It is therefore currently impossible to detect ACh in tissues or cells using immunohistochemical or immunocytochemical techniques. Although ACh may bind to an antibody, because it is not anchored to its site, it will be washed away with the antibody. Nonetheless, Takahashi et al. (28) reported a successful attempt to localize ACh in the mouse gut sections using a tandem imaging mass spectrometry.
Kajiyama et al. (29) observed that a major portion of rabbit blood ACh is present in the buffy coat along with ACh-synthesizing activity. On the basis of that observation, Rinner and Schauenstein (27) confirmed the expression of ACh-synthesizing activity in rat T and B cells in the thymus, spleen, and blood, suggesting the presence of ChAT in lymphocytes.
ChAT is primarily responsible for ACh synthesis within the nervous systems of mammalian species (30). However, in peripheral tissues and non-neuronal cells, the mitochondrial enzyme carnitine acetyltransferase (CarAT, EC 2.3.1.7) also contributes to ACh synthesis along with ChAT (30, 31). ACh-synthesizing activity determined in peripheral samples using the so-called Fonnum method (32) with [3H]acetyl coenzyme A and choline reflects the total activities of ChAT and CarAT. It is therefore recommended that one determines ChAT and CarAT activities in the presence of respective specific inhibitors, bromoacetylcholine and bromoacetylcarnitine. ChAT activity is proportional to the ACh content in cells of the T cell lines expressing ChAT mRNA (Table 1) (10, 25). However, Daudi B cells, which do not express ChAT mRNA, contained little ACh despite of a high CarAT activity. This suggests that ChAT is responsible for ACh synthesis in immune cells (10).
Fujii et al. (33) provided definitive evidence for the synthesis of ACh by ChAT in T cells by demonstrating expression of ChAT mRNA and the enzyme protein in MOLT-3 human leukemic T cells, using reverse transcription-polymerase chain reaction (RT-PCR) and western blot analysis (Figure 1A). Later, constitutive ChAT mRNA expression was detected in other human leukemic T cell lines (25), human blood CD4+ T cells (Figure 1B) (34), rat T and B cells (24, 26), and rat MNLs isolated from the renal vasculature (35). These findings support the idea that ACh production catalyzed by ChAT is occurring in lymphocytes, including T and B cells.
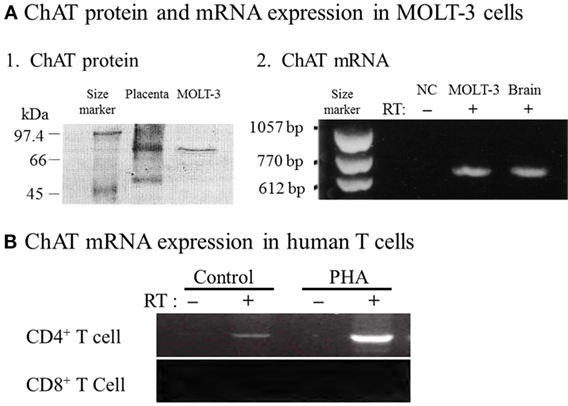
Figure 1. Expression of choline acetyltransferase (ChAT) mRNA and protein in human immune cells. (A1) Western blot analysis of ChAT protein expression in MOLT-3 human leukemic T cells. (A2) Expression of ChAT mRNA detected using reverse transcription-polymerase chain reaction (RT-PCR). NC, negative control of MOLT-3 without RT. Arranged from study by Fujii et al. (33). (B) Expression of ChAT mRNA in human CD4+ T cells and its potentiation by immunological activation with phytohemagglutinin (PHA). Note that CD8+ T cells do not express ChAT mRNA, even after immunological activation. RT, reverse transcriptase. Arranged from study by Fujii et al. (34).
Reverse transcription-polymerase chain reaction revealed ChAT mRNA expression in C57BL/6J mouse spleen-derived MNLs activated with concanavaline A (ConA) and bone marrow-derived DCs activated with lipopolysaccharide (LPS) (36). However, there was no detectable expression of ChAT mRNA in these cells under resting conditions, which suggests that immunological activation is required for ChAT transcription in these cells. While no ChAT mRNA was detected in peritoneal macrophages under either resting or LPS-activated conditions, even after an amplification protocol entailing 40 cycles (36), Koarai et al. (37) detected expression of ChAT mRNA in human lung and alveolar macrophages and monocytes using an RT-PCR protocol entailed 45 cycles, which suggests marginal ChAT mRNA expression in these cells. ChAT mRNA and protein were also detected in human mature and immature DCs using RT-PCR and immunocytochemistry (38). These findings confirm the expression of ChAT mRNA in T and B cells, DCs, and macrophages.
The recent development of ChATBAC-eGFP transgenic mice (39) and ChAT-Cre-tdTomato mice (40) provide the opportunity to detect ChAT-expressing cells using fluorescent reporter proteins. Tallini et al. (39) observed eGFP expression in a subset of lymphocytes in Peyer’s patches, leading to the detection of ChAT-GFP in splenic CD4+ T cells (15) and CD8+ T cells and B cells (41). In addition, Gautron et al. (40) observed expression of the reporter protein in T cells within Peyer’s patches in mice expressing tdTomato fluorescent protein in ChAT-expressing cells. These results confirm the earlier findings that peripheral blood CD4+ and CD8+ T cells and B cells express ChAT mRNA and contain ACh (26, 36).
ChAT expression in splenic DCs was confirmed using ChATBAC-eGFP mice (41). That finding along with the ChAT gene expression in DCs described above (36) suggests DCs are able to synthesize ACh using ChAT. So far, however, no additional data on ACh synthesis and release in DCs has been reported, and the physiological significance of ACh in DCs is yet to be determined.
ChAT expression was also observed in splenic macrophages using ChATBAC-eGFP transgenic mice (41). These findings are in line with those from Koarai et al. (37), who reported the expression of ChAT mRNA in human lung and alveolar macrophages and monocytes. However, Gautron et al. (40) did not find reporter expression in macrophages from gut-associated lymphoid tissue or the spleen of ChAT-Cre-tdTomato mice. As mentioned, we did not detect ChAT mRNA expression in either resting or activated mouse peritoneal macrophages (36). These findings suggest ChAT expression in macrophages may vary depending upon strain, species, tissue, cell processing procedure, and/or immunological status, or that the levels of ChAT expression are marginal in macrophages. Information on the physiological significance of ACh synthesis in macrophages is not available at present.
Lymphocytes
T Cells
Fujii et al. (24) first suggested a role for ACh synthesized by T cells in the regulation of immune system function by showing that phytohemagglutinin (PHA), a T cell activator, increased both intracellular ACh content and its release into the culture medium of HSB-2 and MOLT-3 human leukemic T cells used as models of T cells. In human MNLs, PHA activates protein kinase C (PKC) and mitogen-activated protein kinase (MAPK) pathways via the T-cell receptor (TCR)/CD3 complex, resulting in specific upregulation of ChAT mRNA expression, ChAT activity, and ACh synthesis (42). However, PHA does not upregulate CarAT activity, indicating that only ChAT is linked to T cell activity (42). Upregulation of ChAT mRNA expression by T cell activation with PHA or Con A was also confirmed in rat T cells (26) and mouse spleen cells (36). These findings support the notion that immunological activation of T cells upregulates ACh synthesis, leading to modulation of immune function.
Antithymocyte globulin (ATG)-Fresenius, an immunosuppressant that binds to cell surface molecules, including CD2, CD3, CD4/CD28, CD5, CD7, CD11a (lymphocyte function-associated antigen (LFA)-1), and intercellular adhesion molecule (ICAM)-1, increases ACh release but decreases intracellular ACh content in CCRF-CEM (CEM) human leukemic T cells in the short term (6 h), and increases both ACh release and intracellular ACh content in the long term (48 h) (43). Both anti-CD11a monoclonal antibody (mAb) and ATG-F upregulates ChAT mRNA expression after 48 h of culture, suggesting activation of T cell adhesion molecules facilitates ACh synthesis (43). By contrast, acting in a manner independent of hydroxymethylglutaryl-CoA reductase inhibition, statins, including simvastatin, are able to inhibit LFA-1 (CD11a/CD18)-mediated adhesion and co-stimulation of lymphocytes, leading to immune modulation (44). Simvastatin thus abolishes anti-CD11a mAb-induced increases in ChAT mRNA expression, ACh synthesis and release in MOLT-3 cells (45). These results confirm that cell adhesion molecules such as LFA-1 contribute to the regulation of lymphocytic cholinergic activity (45, 46).
Calcium ionophores such as A23187 and ionomycin upregulate expression of ChAT mRNA and its activity, and they increase the ACh content of MOLT-3 human leukemic T cells and their culture media (46, 47). By contrast, FK-506 (tacrolimus), a calcineurin inhibitor, suppresses PHA-induced upregulation of ChAT mRNA expression and ACh synthesis, which suggests that Ca2+ contributes to the regulation of T cell cholinergic activity through calcineurin-mediated pathways (46).
Phorbol 12-myristate 13-acetate (PMA), a non-specific PKC activator, and dibutyryl cAMP, a protein kinase A (PKA) activator, increased ChAT activity and ACh synthesis by upregulating ChAT gene expression in MOLT-3 human leukemic T cells (46). These data provide compelling evidence that T-cell activation via PKC–MAPK and/or adenylate cyclase-cAMP pathways during immune responses upregulates the synthesis and release of ACh, leading to the modulation of the T cell cholinergic activity.
B Cells
Staphylococcus aureus Cowan I (SAC) binds to B cells and triggers a signal transduction cascade involving tyrosine kinase-mediated activation of phospholipase C (PLC) and leads to activation of PKC–MAPK pathways (48). Incubation of human circulating MNLs consisting of mainly T and B cells and a small number of monocytes with SAC for 48 h induces a significant increase of the intracellular ACh content and upregulation of ChAT mRNA expression (47). These findings suggest that stimulation of B cells also facilitates cholinergic activity via upregulation of ChAT mRNA expression.
Murine B cells, but not human B cells, express toll-like receptor 4 (TLR-4) (49). LPS activates murine B cells, monocytes, DCs, and macrophages by binding to a CD14/TLR-4/MD2 complex, which leads to secretion of pro-inflammatory cytokines, nitric oxide, and eicosanoids (50). In splenic follicular B cells from ChATBAC-eGFP transgenic mice, LPS induces ChAT-GFP expression and increases ACh production (41). These results along with the aforementioned effects of SAC suggest that stimulation of TLRs on B cells activates cholinergic activity by enhancing ACh synthesis. Furthermore, ACh produced by ChAT+ B cells has been shown to reduce peritoneal neutrophil recruitment during sterile endotoxemia, suggesting the role for B cell-derived ACh in the regulation of innate immunity (41).
DCs and Macrophages
As described, LPS upregulates ChAT mRNA expression in bone marrow-derived DCs, but elicits no apparent effects on ChAT mRNA expression in peritoneal macrophages from C57BL/6J mice (36). Reardon et al. (41) found that induction of ChAT expression in murine macrophages and DCs by LPS elicits MyD88-dependent signal transduction in a cell-intrinsic manner.
Lipopolysaccharide
As described, among innate immune cells, DCs and macrophages have the potential for ChAT-catalyzed ACh synthesis upon activation with LPS (36–38, 41). LPS and TLR agonists induce expression of ChAT mRNA and enzyme protein in DCs (36, 41) and macrophages (41). MyD88-dependent TLRs are involved in LPS-induced ChAT expression in DCs and macrophages (41). Because DCs and macrophages express all five M1–M5 mAChR subtypes (36) and various nAChR subunits (Figure 2) (36), these findings suggest the possibility that activation of MyD88-dependent TLRs by LPS upregulates cholinergic activity in macrophages and DCs. Furthermore, ACh synthesized in these cells should act in autocrine/paracrine fashion on their own nAChRs and mAChRs and play a role in regulation of innate immune responses by modulating cytokine production, such as tumor necrosis factor (TNF)-α and interleukin (IL)-2.
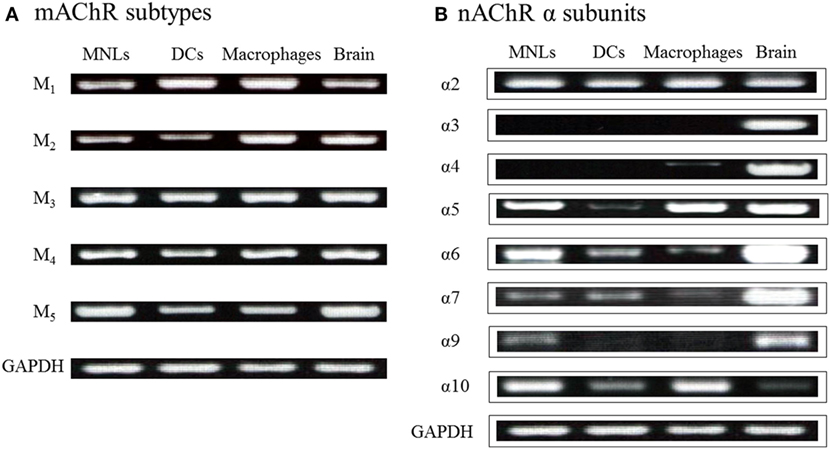
Figure 2. Expression of genes for mAChRs and nAChR α subunits in immune cells from C57BL/6J mice. (A) mRNA expression of mAChR subtypes detected using reverse transcription-polymerase chain reaction. MNLs, mononuclear leukocytes; DCs, dendritic cells. GAPDH, glyceraldehyde-3-phosphate dehydrogenase. (B) mRNA expression of nAChR α subunits. Arranged from study by Kawashima et al. (36).
Antigen Presentation
Phytohemagglutinin and ConA upregulate ChAT expression and ACh synthesis in T cells, which indicates that antigen presentation between naïve CD4+ T cells and antigen-presenting cells (APCs), including DCs and macrophages, enhances cholinergic activity in immune cells [see reviews in Ref. (11, 12, 16, 33, 47)]. Recognition by the TCR/CD3 complex of an antigen presented on major histocompatibility complex receptors on APCs triggers activation of PLCγ and Ca2+ release from endoplasmic reticulum (ER) in T cells (51, 52). Depletion of ER Ca2+ stores results in sustained Ca2+ influx through Ca2+-release activated Ca2+ (CRAC) channels, leading to activation of Ca2+-sensitive transcriptional factors, including nuclear factor of activated T cells, which promotes expression of cytokine genes critical for immune responses (53, 54). As described, PHA also upregulates AChE and M5 mAChR expression along with ChAT (33, 47, 55). This suggests that ACh released from T cells and APCs (DCs and macrophages) act on their own mAChRs and nAChRs affecting autocrine/paracrine pathways, leading to modification of immune function.
The functions of M5 mAChRs are not yet well defined at peripheral autonomic nerve effector junctions, the central nervous system, or immune cells. However, the observations that both antigen-specific IgG1 and pro-inflammatory cytokine production are decreased in the M1/M5-KO mice and that M5 mAChRs are upregulated by PHA and ConA suggest that M5 mAChRs in immune cells are involved in positive regulation of immune function (33, 47, 56). Consistent with that idea, antigen presentation between naïve CD4+ T cells and DCs upregulates T cells expression of ChAT mRNA, ChAT activity, ACh, AChE, and M5 mAChR, thereby enhancing cholinergic activity [see reviews in Ref. (11, 12, 33, 47)].
Both AChE and BuChE hydrolyze ACh into choline and acetate to terminate its activity at synapses. In the brain, AChE is mostly found within synaptic clefts between neurons (57, 58), while BuChE is mainly located outside the synaptic cleft and in glial cells (59, 60). Both AChE and BuChE are present at the mouse neuromuscular junction, but exhibit different localization patterns. AChE activity is present in both the primary cleft and in the secondary folds, while BuChE activity appears to be concentrated in structures resembling subsynaptic folds (61). AChE is also found within erythrocytes, but its physiological function there is unknown. BuChE is found primarily in plasma, liver, and the neuromuscular junction [see a review in Ref. (12)].
The decay time constants of focally recorded miniature endplate currents caused by ACh at the neuromuscular junction are 1.04 and 5.4 ms in wild-type (WT) and AChE-KO mice, respectively (62). It is important to note the differences in the inactivation rates and processes between ACh and other neurotransmitters, such as norepinephrine (NE) and epinephrine (EPI). Whereas the action of ACh is terminated within a few milliseconds through enzymatic breakdown, the actions of NE and EPI last much longer until reuptake into nerve terminals and surrounding tissues, and the diffusion decrease their concentrations within the synaptic cleft to subthreshold levels. As a consequence, to have a physiological action, non-neuronal ACh must be released into a microenvironment forming synapse-like structures, such as during antigen presentation and cell-to-cell interaction involving cell adhesion molecules.
AChE is expressed ubiquitously in mouse lymphocytes, DCs, and macrophages (36), while human blood MNLs, CEM human leukemic T cells, and Daudi B cells all express various types of AChE mRNA [see a review in Ref. (10)]. In addition, upregulation of AChE activity by PHA is detected in normal peripheral blood human lymphocytes and in leukemic T cell lines (55). These findings indicate that T cell activation via TCR/CD3-mediated pathways enhances expression of cholinergic elements, including ChAT and AChE, within T cells. However, the physiological function of AChE in immune cells has yet to be investigated.
Rivastigmine, which inhibits both AChE and ChE activities, relieves the clinical symptoms and spatial memory deficits in mice with autoimmune encephalomyelitis (EAE) (63). Rivastigmine also decreases the reactivity of encephalitogenic T cells and the production of TNF-α, interferon (IFN)-γ, and IL-17 cytokines in the EAE mouse. All of these effects are abolished by α-BTX, an α7 nAChR antagonist, which suggests that the effects are induced by ACh acting on α7 nAChRs after its levels were increased due to AChE and BuChE inhibition (63).
It has been suggested that functional defects in nAChRs on immune cells contribute to the etiology of inflammatory bowel diseases (IBD), as smoking modifies the development and progression of IBD (64, 65) and immunosuppressants are sometimes effective in patients with IBD (66–68). In addition, AChE-targeting microRNA-132 (miR-132) exhibited some potential to attenuate inflammation by reducing AChE levels in immune cells (69).
Early functional and binding studies revealed the presence of both mAChRs and nAChRs on immune cells [see reviews in Ref. (9, 11, 12, 16)].
The diversity of mAChR functions prompted investigation of their molecular basis using cloning techniques and led to the identification of five distinct mAChR subtypes (M1–M5) (70–72). These five mAChRs have been divided into two groups based on their functional coupling. The M1, M3, and M5 subtypes are coupled to pertussis toxin-insensitive Gq/11 proteins, which mediate activation of PLC activity. Upon activation of these mAChR subtypes, PLC hydrolyzes phosphatidylinositol 4,5-bisphosphate, leading to the formation of inositol 1,4,5-trisphosphate (IP3) and diacylglycerol. These products then act as second messengers by, respectively, mobilizing Ca2+ from intracellular stores and activating PKC (73). The M2 and M4 subtypes are coupled to pertussis toxin-sensitive Gi/o protein, which mediates inhibition of adenylate cyclase, and thus a decrease in cAMP formation, upon activation (74, 75).
mAChR Expression in Immune Cells
All five mAChRs subtypes have been detected in lymphocytes, macrophages, and DCs from humans, mice, and rats (Figure 2A) [see reviews in Ref. (16, 36, 76)]. As mentioned, PHA and SAC selectively upregulate expression of M5 mAChR mRNA in CEM human leukemic T cells and Daudi B cells, respectively (33, 47). Expression of other mAChR subtypes is not affected in either cell line. Stimulation with PMA plus ionomycin upregulates expression of both M3 and M5 mAChR mRNA in these cell lines (47). These findings show that immunological stimulation leads to M5 mAChR gene expression in lymphocytes and suggest a role for M5 mAChRs in the regulation of immune function.
Qian et al. (77) showed that activation of murine splenic T cells for differentiation using monoclonal antibodies against CD3 and CD28 modifies the expression patterns and intensities of mAChRs. These findings suggest the possibility that the expression patterns and intensities of mAChR expression in immune cells may vary among individuals, depending on their immunological status.
Nicotinic AChRs are located on the plasma membranes of skeletal muscle cells, neurons, and non-neuronal cells. Activation of nAChRs elicits membrane depolarization and excitation due to a rapid increase in membrane permeability to Na+, K+, and Ca2+. The molecular cloning of nAChR subunits (78) prompted a series of proteomic and genomic investigations of the various nAChR subunit proteins. At present, 10 α (α1–10), 4 β (β1–4), γ, δ, and ε nAChR subunits have been detected through molecular cloning. The α subunits can be subdivided according to their sensitivity to α-BTX: α1, α7, and α9 are α-BTX-sensitive, while α2–α6 are α-BTX-insensitive. nAChRs exist as pentamers composed of 1–5 different subunits. At least two copies of the α subunit are always present among these subunits, and multiple ACh binding sites are formed at the interface of each α subunit and a neighboring subunit.
nAChRs are classified into muscle and neuron types based on their expression sites. Muscle type nAChRs are expressed mainly in skeletal muscle and contain four different subunits within the pentameric complex: (α1)2/β1/δ/γ in embryonic and denervated muscle, and (α1)2/β1/δ/ε in innervated adult muscle. By contrast, neuron type nAChRs expressed in the nervous system and non-neuronal cells, including immune cells, are composed of only α (α2–α7, α9, and α10) and β (β2–β4) subunits. The α8 subunit is found only in the visual areas of the avian brain (79). nAChRs consisting of α3β2 and α3β4 are abundantly expressed in peripheral ganglia, while α4β2 subunits are expressed in the brain (80). Within the mammalian brain, about 80% of nAChRs are composed of α4β2 subunits, and 10–15% are composed of α7 (81). The α7 nAChR gene (CHRNA7) is expressed widely in the central and peripheral nervous system, and in non-neuronal cells, and the homomeric α7 nAChR exhibits uniquely high Ca2+ permeability upon activation (82, 83). The α9 and α10 subunits were first identified in the mechanosensory hair cells of the rat auditory system (84, 85). The α9 subunit was initially postulated to form a homomeric nAChR. However, subsequent studies identified expression of heteromeric α9α10 nAChR subtypes within the hair cells. Unlike other nAChRs, which mediate excitatory neurotransmission, α9α10 nAChR activation elicits hair cell hyperpolarization evoked by Ca2+ entry through the receptor, leading to activation of a small-conductance SK2 Ca2+-dependent potassium channel (85).
Specific Features Related to α7 nAChRs
Human-Specific dupα7 nAChRs
The α7 subunit gene CHRNA7 is composed of 10 exons, encoding 146 amino acids (exons 1–4) comprising an N-terminal extracellular domain and 384 amino acids (exons 5–10) comprising three transmembrane (M1–M3) domains, a large intracellular loop (M3–M4 loop), a fourth transmembrane (M4) domain, and a short C-terminal extracellular region (86). Gault et al. (87) found CHRNA7 exons 5–10 duplicated as the CHRFAM7A gene encoding dupα7 nAChR in the human genome and its expression in the brain. Although the α7 nAChR and dupα7 nAChR subunits share the same 384 amino acids comprising the four transmembrane domains of the ligand-gated ion channel transmembrane region and a short C-terminal extracellular region, the dupα7 subunit has a shorter N-terminal extracellular domain than the primal α7 nAChR subunit. Because the extracellular N-terminal region of the α7 nAChR contains multiple loops of agonist binding site (88), its structure suggests that the dupα7 nAChR subunit may lack the recognition sites for ACh and α-BTX. Experiments performed with Xenopus oocytes co-injected with various ratios of α7/dupα7 mRNA revealed a graded reduction in functional receptor generation proportional to the α7/dupα7 ratio, as measured based on nicotine-elicited α7 currents (89). These results were confirmed by measurements of α-BTX binding, which suggests that as the proportion of the dupα7 subunit increases, there is a reduction in the number of functional α7 receptors that reach the surface of the oocyte (89). These results are consistent with the findings of Araud et al. (86), who reported that CHRFAM7A functions as a dominant negative regulator of α7 nAChR.
Human-specific CHRFAM7A transcripts were first discovered in the brain (87), but were later detected in human peripheral blood leukocytes, including MNLs (90), macrophages (90, 91), and monocytic cell lines (90, 92). The role of the dupα7 subunit in the regulation of immune function remains to be determined.
Heteromeric α7β2 nAChRs
Although the α7 subunit has long been postulated to form a homomeric α7 nAChR, recent studies suggest the possibility that α7 and β2 subunits form heteromeric α7β2 nAChR subtypes in the brain (93–95). Differences in the pharmacological and functional properties between naturally occurring α7-containing nAChRs in the brain and those of recombinant homomeric α7 nAChRs prompted studies investigating whether the nAChR α7 and β2 subunits can co-assemble to form a functional heteromeric nAChR channel in Xenopus oocytes and cell lines (93–95). Detection of co-expressed rat α7 and β2 subunits in co-transfected TSA201 embryonal kidney cells and SH-EP1 human epithelial cells, and of α7 and β2 subunit mRNAs in the rat cholinergic neurons, confirmed their co-assembly in mammalian cells (93, 96, 97). Furthermore, the co-expression significantly slowed the rate of channel desensitization, compared to homomeric α7 channels, and altered the pharmacological properties of the channels. It thus appears that rat nAChR α7 and β2 subunits have the ability to co-assemble and form functional heteromeric nAChRs (94, 95, 98).
Expression of α7β2 nAChRs was confirmed in the human brain through purification of α7 subunit-containing proteins using α-BTX beads followed by western blotting using an anti-β2 subunit antibody (99). Thomsen et al. (99) also found that upon agonist stimulation, the currents carried by heteromeric α7β2 nAChRs show markedly slower rising and decay phases than homomeric α7 nAChRs in HEK293 cells. At present, no information is available on the expression of α7β2 nAChRs in immune cells. Because T and B cells, DCs, and macrophages all express α7 nAChRs (36, 98, 100), it is possible that α7β2 nAChRs are also expressed in immune cells and are involved in regulating immune function.
Ionotropic and Metabotropic Natures of α7 nAChRs
As described, activation of α7 nAChRs using ACh or nicotine elicits a transient increase of [Ca2+]i in neurons and immune cells, though in certain types of immune cells the receptor channel rapidly desensitizes, and no transient increase of [Ca2+]i is observed (101). Nevertheless, activation of α7 nAChRs can set into motion more prolonged signaling events operating downstream and leading to modulation of immune cell function. Prompted by these observations, the dual ionotropic/metabotropic nature of α7 receptors has been extensively explored to explain the role of α7 receptors (102).
(1) Ionotropic pathway: the Ca2+ influx through α7 nAChRs induced by stimulation with ACh and agonists activates a phosphorylation cascade via PKC. This in turn activates the PI3K/Akt pathway, which promotes nuclear factor erythroid 2-related factor 2 (Nrf2) translocation to the nucleus and overexpression of heme oxygenase 1, resulting in potent anti-inflammatory effects (67, 102–104).
(2) Metabolic pathway: evidence now suggests that α7 nAChRs may interact with G-proteins independently of G-protein coupled receptors (102, 105–108). By showing the binding of G-proteins to G-protein-binding sites located in the M3–M4 loop of α7 nAChRs, King et al. (108) demonstrated that direct coupling of α7 nAChRs to G-proteins makes it possible to elicit downstream Ca2+ signaling responses that can persist beyond the expected time course of channel activation. It has been suggested that α7 nAChRs coupled with Gs, Gq/11, and Gi/o proteins locating in the hippocampus and prefrontal cortex of C57BL/6J mice contribute to the regulation of neurite growth (101, 106). In addition, Razani-Boroujerdi et al. (101) found that a functional TCR/CD3 complex and leukocyte-specific tyrosine kinase are required for the nicotine-induced rise in the intracellular Ca2+ concentration ([Ca2+]i) evoked in the absence of extracellular Ca2+ via α7 nAChRs in T cells. This suggests an alternative metabotropic pathway leading to induction of Ca2+ signaling in human T cells. Agonist binding to α7 nAChRs also may activate Janus kinase 2/signal transducer and activator of transcription 3 signaling cascades independently of Ca2+ influx, leading to suppression of nuclear transcription factor kappaB-regulated transcriptional activity in macrophages (67, 102–104).
Specific Features Related to α9 and α10 nAChRs
Hecker et al. (109) reported that ACh, choline, phosphocholine, phosphocholine-modified LPS from Haemophilus influenzae, and phosphocholine-modified protein all inhibit ATP-mediated IL-1β release independently of the extracellular Ca2+ influx through the channel in human monocytic U937 cells expressing the α7, α9, and α10 nACR subunits, and in rat monocytes via nAChR-mediated pathways. Furthermore, using U937 cells, Richter et al. (110) found that choline and phosphocholine inhibit ATP-mediated P2X7 receptor activation and IL-1β release. While choline elicits ionotropic current responses at homomeric α9 nACRs, phosphocholine does not trigger ionotropic responses at either homomeric α9 or heteromeric α9/α10 nAChRs. These findings provide evidence that phosphocholine and their derivatives are able to function as metabotropic agonists for heteromeric α9/α10 nAChRs.
nAChR Expression in Immune Cells
Figure 2B shows the mRNA expression for nAChR α subunits in MNLs, DCs, and macrophages from C57BL/6J mice (36). Expression of the α2, α5, α6, α7, α9, and α10 subunits is frequently detected in immune cells, including T cells (16, 76, 111, 112). However, the specific patterns of nAChR subunit expression in particular immune cells are not yet settled. As mentioned, it appears that the patterns and the intensities of nAChR subunit expression vary depending on the immunological status of the cell in question (77). It is noteworthy, however, that data from immunohistochemistry and western blots must be interpreted cautiously due to the lack of the specificity of the commercially available antibodies (113, 114).
SLURP-1 and -2 are endogenous peptides that act as positive allosteric ligands on α7 and α3 nAChRs, respectively (115–119). The discovery of mutations in the gene encoding SLURP-1 in Mal de Meleda (MdM) patients with a characteristic transgressive palmoplantar keratoderma (120) is drawing major research attention to the capacity of SLURP-1 to serve as an epithelial growth modulator (112, 118–122). In addition, recent studies suggest the possibility that SLURP-1 and -2 are also involved in regulating immune cell function (36, 122–126).
SLURP-1 and -2 mRNAs are detected in nearly every organ in the C57BL/6J mouse (123). Gene expression for SLURP-1 and -2 was also detected in MNLs, DCs, and macrophages, but neither the T cell activator Con A nor the DC and macrophage activator LPS modified the levels of their expression (36, 123). This suggests that SLURP-1 and -2 are constitutively expressed in these cells, and their expression is independent of immunological stimulation.
Intense SLURP-1 immunoreactivity (SLURP-1+) was detected in DC-like cells residing mainly in the interfollicular zone surrounding the germinal center of human tonsils, and in a few cells scattered within the germinal center (Figure 3) (126). Some SLURP-1+ cells in the tonsil interfollicular zone also showed immunopositivity for CD205 (Figure 3A), a marker of mature DCs that mediates efficient antigen presentation (127), and these SLURP-1+ CD205+ DCs were surrounded by CD4+ T cells. These observations support the notion that ACh synthesized and released from T cells and activated macrophages during antigen presentation acts on α7 nAChRs expressed in immune cells, and that SLURP-1 potentiates the ACh activity at α7 nAChRs in both T cells and DCs (16, 126).
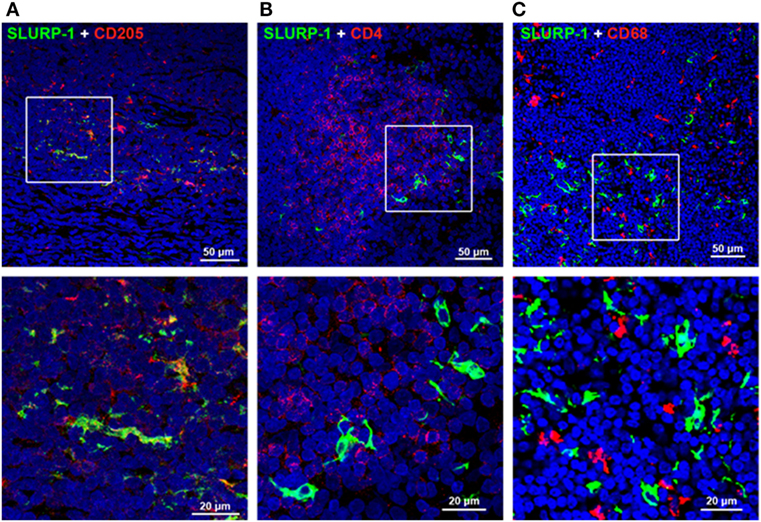
Figure 3. Immunohistochemical staining of SLURP-1, CD205, CD4, and CD68 in human tonsils. (A) Merged image showing SLURP-1 (green) and CD205 (red) immunoreactivities in the interfollicular zone. Note that some CD205+ cells were also SLURP-1+. An enlarged image of the boxed area is shown in the lower panel. (B) Merged image showing SLURP-1 (green) and CD4 (red) immunoreactivities in the interfollicular zone. Note that the SLURP-1+ cells are surrounded by CD4+ T cells. An enlarged image of the boxed area is shown in the lower panel. (C) Merged image showing SLURP-1 (green) and CD68 (red) immunoreactivities in the interfollicular zone. Note that the SLURP-1+ cells are located in close proximity to CD68+ macrophages. An enlarged image of the boxed area is shown in the lower panel. Arranged from data by Fujii et al. (126).
Agonist-evoked activation of mAChRs in immune cells evokes a variety of functional and biochemical effects, including enhanced cytotoxicity, increased cGMP and IP3 formation, and activation of cell proliferation [see reviews in Ref. (9, 12)]. Stimulation of mAChRs in CEM human leukemic T cells and Daudi human leukemic B cells using ACh, bethanechol, carbachol, or Oxo-M induced a transient rise in the intracellular Ca2+ concentration ([Ca2+]i) followed by extracellular Ca2+-dependent [Ca2+]i oscillations that persisted for about 10 min (128). In both cell lines, Oxo-M upregulated c-fos mRNA expression in an extracellular Ca2+-dependent manner. All these effects induced by mAChR agonists were abolished by atropine, a non-specific mAChR antagonist. This suggests that activation of mAChRs in immune cells triggers nuclear signaling, leading to modification of immune cell function. More recently, Mashimo et al. (54) showed that M3 and M5 mAChRs play a major role in Oxo-M-induced initial transient increases in [Ca2+]i and the following repetitive [Ca2+]i oscillations in CEM human leukemic T cells. The [Ca2+]i oscillations were blocked by removal of extracellular Ca2+ or YM-58483, a CRAC channel blocker, without affecting the Oxo-M-induced initial [Ca2+]i transient. Furthermore, CRAC channel blockade abolished Oxo-M-induced c-fos and IL-2 expression. These results suggest that activation of M3 or M5 mAChRs evokes IP3-mediated Ca2+ release from intracellular stores, leading to extracellular Ca2+ influx through CRAC channels, which generates repetitive [Ca2+]i oscillations and, in turn, enhances c-fos and IL-2 gene expression in T cells.
Upregulation of M5 mAChR mRNA expression in human T and B cells during immunological stimulation strongly suggests that mAChRs, including M5, are involved in regulating immune function (33, 47). Thus far, however, there have been few investigations of the roles played by mAChRs in regulation of immune function. The involvement of M1 mAChRs in the differentiation of CD8+ T cells into cytolytic T cells was first postulated by Zimring et al. (129), but it was later found that neither M1 nor M5 mAChRs are required for expansion of antigen-specific CD8+ T cells, in response to viral infection in C57BL/6 mice (130). On the basis of these observations, the authors concluded that the extent to which one can draw a generalized conclusion that M1 and M5 mAChRs are not involved in antiviral immunity mediated by CD8+ T cells, depends upon issues of antigen strength, genetic background, and receptor redundancy (130).
Fujii et al. (56) investigated the roles of M1 and M5 mAChRs in the regulation of immune function by immunizing combined M1 and M5 mAChR gene-knockout (M1/M5-KO) and WT mice with ovalbumin (OVA). One week after the immunization, serum concentrations of anti-OVA-specific IgG1 in the M1/M5-KO mice were significantly lower than in WT mice, though the serum concentrations of anti-OVA-specific IgM did not differ between the two genotypes. Spleen cells from M1/M5-KO mice activated with OVA secreted significantly lower amounts of TNF-α, IFN-γ, and IL-6 than those from WT mice. These observations suggest that M1 and/or M5 mAChRs contribute to the regulation of pro-inflammatory cytokine production related to adaptive immunity.
Among the various nAChRs, α7 nAChRs are drawing attention because their stimulation attenuates the synthesis and release of the pro-inflammatory cytokine TNF-α in LPS-activated macrophages, leading to modulation of inflammatory and immune responses (15, 131, 132).
Fujii et al. (133) observed a significantly higher serum anti-OVA-specific IgG1 levels in α7-KO than WT C57BL/6J mice 2 weeks after immunization with OVA. Moreover, antigen-stimulated spleen cells from α7-KO mice produced significantly greater amounts of TNF-α, IL-6, and IFN-γ responsible for antibody class switch induction to IgG1 than those from WT mice. These observations suggest the involvement of α7 nAChRs in regulating pro-inflammatory cytokine production, leading to modification of antibody production. In line with these findings, a recent study showed that nicotine attenuates production of TNF-α, IL-1β, and IL-12 in murine bone marrow-derived monocytes via α7 and α9 nAChR-mediated pathways (134). In addition, activation of B cells with anti-CD40 antibody elicited a greater proliferative response in α7-KO mice than in the WT, and the suppression of α7 nAChRs with methyllycaconitine (MLA) evoked a greater proliferative response in B cells stimulated either with anti-CD40 antibody or antibody against B cell receptor (135–138), suggesting a role for α7 nAChRs in downregulation of B cell proliferation. Taken together, the above findings suggest the possibility that a higher serum antigen-specific IgG1 concentration observed in α7-KO mice (133) can be ascribed to the upregulation of pro-inflammatory cytokine production and the increased number of B cells in α7-KO mice (135–138).
MOLT-3 human leukemic T cells and cultured spleen cells from C57BL/6J mice constitutively produce amounts of ACh sufficient to elicit autocrine changes in [Ca2+]i and upregulation of IL-2 mRNA and protein expressions (139). Mecamylamine, a nAChR inhibitor, suppressed the [Ca2+]i transients, IL-2 release, and cell proliferation. These findings indicate that T cells utilize ACh as a tool to interact with one another and that autocrine ACh-activated nAChRs are involved in regulating immune cell functions such as cytokine synthesis and cell proliferation.
Methyllycaconitine, a specific α7 nAChR antagonist, did not suppress the above described autocrine ACh-induced changes in [Ca2+]i, suggesting the involvement of nAChRs other than α7 nAChRs in the [Ca2+]i transients in freshly isolated spleen cells and resting MOLT-3 cells leading to upregulation of IL-2 production (139). In fact, Qian et al. (77) detected the expression of α7 nAChR mRNA in CD4+ and CD8+ T cells only after the activation via TCR/CD3 cross-linking, but not in freshly isolated CD4+ and CD8+ T cells. Taken together, these findings suggest the possibility that the pattern of nAChR subtype expression is variable depending on the immunological status. Furthermore, nicotine modified IFN-γ and IL-17 production in T cells activated with TCR/CD3 cross-linking (77, 140). These findings suggest that nAChRs including α7 nAChR contribute to immunomodulation through modification of T cell differentiation by altering cytokine production.
T cell activation with anti-CD3/anti-CD28 mAbs of peripheral blood MNLs isolated from MdM patients with SLURP-1 mutation showed a defect in their proliferative response (125). Moreover, addition of WT recombinant SLURP-1 (rSLURP-1) to cultures of T cells from MdM patients restored the normal T cell activation response, showing that SLURP-1 plays a key role during normal activation of T cells induced by immunological stimulation enhancing the actions of ACh via α7 nAChRs (125).
Recombinant SLURP-1 increases ChAT gene expression and the ACh content in MOLT-3 human leukemic T cells and human peripheral blood MNLs, and these effects are abolished by the α7 nAChR antagonist MLA. This suggests that, working as a positive allosteric ligand, SLURP-1 activates cholinergic transmission by potentiating ACh synthesis and its action on α7 nAChRs, thereby facilitating functional development of T cells (126). rSLURP-1 induces a slight but significant attenuation of cell growth in peripheral blood MNLs and MOLT-3 cells and that is abolished by MLA. These findings support the notion that SLURP-1 acts as a key modulator of T cell activity.
Chernyavsky et al. (122) found that rSLURP-1 decreases production of TNF-α in CEM human leukemic T cells, downregulates IL-1β and IL-6 secretion in U937 macrophages, and moderately upregulates IL-10 production in these immune cells. rSLURP-2 downregulates TNF-α and IFN-γ-receptors in CEM cells and reduces IL-6 production in U937 macrophages. These results demonstrate that SLURP-1 and -2 exert anti-inflammatory effects on T cells and macrophages.
As described, α7 nAChRs on immune cells are involved in regulating the synthesis of various cytokines (16, 131, 133). On the basis of those findings, Kawashima et al. (141) used α7-KO and WT mice to investigate the involvement of α7 nAChRs on naïve CD4+ T cells in the regulation of their differentiation to CD4+ CD25+ FoxP3+ regulatory T cells (Tregs). Non-specific activation of naïve CD4+ T cell differentiation in culture using anti-TCR and anti-CD28 mAbs in the presence GTS-21, a partial α7 nAChR agonist, upregulated the generation of Tregs from WT spleen cells, but not from α7-KO cells (Figure 4). On the other hand, assays of IFN-γ in the culture media suggested that Th1 differentiation was not affected by α7 nAChR activation. This suggests that when naïve CD4+ T cells are directly stimulated via their TCR/CD3 complex, activation of α7 nAChRs on the T cells leads to upregulation of Treg and that α7 nAChRs are also involved in regulating adaptive immunity.
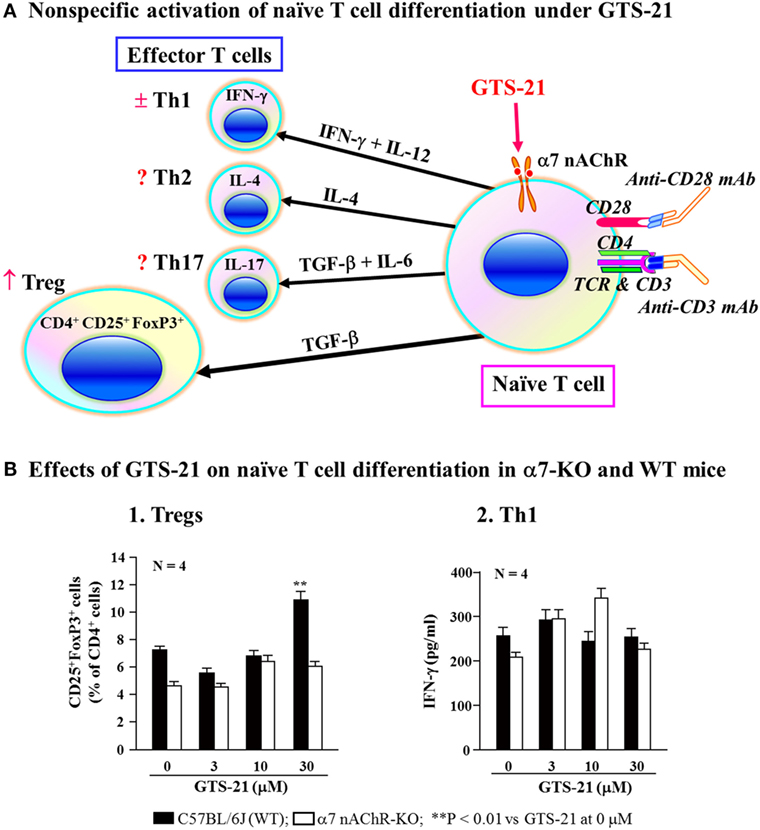
Figure 4. Role for α7 nAChRs during differentiation of naïve T cells. (A) Schematic drawing of non-specific activation of naïve T cell differentiation via T cell receptor and CD28 in the presence of GST-21, a partial agonist for α7 nAChRs. Differentiation of naïve CD4+ T cells into effector T cells (Th1, Th2, and Th17) and regulatory T cells (Tregs) is dictated by the presence of cytokines shown on the respective arrows (142, 143). GTS-21 may facilitate the expression of transcription factor FoxP3 or TGF-β leading to Treg expansion. IFN-γ, interferon-γ; IL-4, interleukin-4; IL-6, interleukin-6; IL-12, interleukin-12; IL-17, interleukin-17; TGF-β, transforming growth factor-β. (B) Effects of GST-21 on naïve T cell differentiation in α7 nAChR-deficient (α7-KO) and wild-type (WT) mice. Note that GTS-21 upregulated differentiation into Tregs in the WT, but not α7-KO mice. Th1 differentiation was not affected by GTS-21 in the both genotypes. Arranged from study by Kawashima et al. (141).
Studies examining the effects of adoptive transfer of antigen-specific Tregs have demonstrated their contribution to the protection and recovery of an animal model of autoimmune EAE (144). However, the availability of adequate numbers of antigen-specific Tregs for adoptive transfer is difficult to achieve and is one of the major limitations of its clinical application. For treatment of autoimmune diseases, it would be useful to develop procedures in which immune cell cholinergic activity was used to facilitate Treg differentiation from naïve CD4+ T cells. Conversely, suppression of Treg differentiation through manipulation of cholinergic activity in immune cells could potentially be of great help in cancers therapy.
The inflammatory reflex involves the nervous systems and humoral factors that operate to protect the body from infection and inflammation. Pro-inflammatory cytokines generated by immune cells in response to invading pathogens trigger a series of reflex responses. The inflammatory reflex loop consists of two major components: (1) a sensory afferent signaling pathway and (2) a motor efferent signaling pathway. The afferent signaling pathway conveys peripheral signals to the brain via the afferent sensory vagus nerve and humoral routes. After the signals are processed in the brainstem, the central nervous system transmits signals to the periphery via (1) humoral pathways that involve activation of the hypothalamic–pituitary–adrenal axis, leading to the release of glucocorticoids with anti-inflammatory actions (13), (2) neural pathways involving activation of the sympathetic nervous system (13, 14), and (3) the efferent vagus nerve (16, 145–147). Here, we will mainly touch on the efferent signaling pathways of the inflammatory reflex in the context of the immune cell cholinergic system.
Intraperitoneal injection of IL-1β, a pro-inflammatory cytokine, induces fever that is dependent on intact afferent vagus neurons (148). In a rat model of LPS-induced septic shock, electrical stimulation of the efferent vagus nerve protected animals from endotoxemia and attenuated the increase in serum and liver TNF-α levels (149). In addition, ACh and nicotine abolished the LPS-induced release of TNF-α from cultured human macrophages. Electrical stimulation of the vagus nerve also inhibited LPS-induced TNF-α synthesis in WT mice, but failed to inhibit TNF-α synthesis in α7-KO mice, suggesting that α7 nAChRs are essential for inhibiting cytokine synthesis via a cholinergic anti-inflammatory pathway (131, 150–152).
Following the observations that splenectomy and selective abdominal vagotomy abolished the anti-inflammatory effects of both vagus nerve stimulation and nicotine, the spleen was identified as an essential target of the cholinergic anti-inflammatory pathway involved not only in inflammatory signal generation in the afferent vagus nerve but also vagally mediated inhibition of cytokine production in macrophages (153–155).
Although the attenuation of LPS-induced septic shock by efferent vagal stimulation suggests the involvement of the vagus nerve in the anti-inflammatory reflex (150), the pathways and mechanisms that carry efferent inhibitory signals suppressing the synthesis and release of pro-inflammatory cytokines in the spleen are not yet settled. There is no neuroanatomical evidence for a direct parasympathetic or vagal nerve supply to any immune organ (156). The currently available evidence indicates that all primary and secondary immune organs receive innervation only by sympathetic postganglionic neurons [see a review by Nance and Sanders (14)]. Prevertebral sympathetic ganglia associated with the celiac-mesenteric plexus provide major sympathetic input to the spleen, and the splenic nerve is the final common pathway for neural input to the spleen (14, 157).
Because the efferent vagus nerve that should carry the efferent signals terminates in the celiac ganglion, Rosas-Ballina et al. (15) postulated that the efferent signals carried by the efferent vagus nerve are transmitted to the postganglionic sympathetic splenic nerve innervating the spleen in the celiac ganglion. This would activate the splenic nerve to release NE within the spleen, which would promote Ach release from a subset of CD4+ T cells that relay the neural signal to other immune cells—e.g., through activation of α7 nAChRs on macrophages, resulting in inhibition of the synthesis and release of pro-inflammatory cytokines, including TNF-α. Consistent with those ideas, they found that electrical vagus stimulation elevated ACh concentrations in the spleen, and attenuated serum TNF-α levels during endotoxemia. Their observations further confirmed that T cells mediate the inflammatory reflex. They found that vagus nerve stimulation did not diminish serum TNF-α concentrations during endotoxemia in nude mice lacking functional T cells, but that the transfusion of a subset of ChAT+ T cells from normal mice to the nude mice restored the ability of vagus nerve stimulation to suppress serum TNF-α concentrations. They, therefore, proposed that efferent vagus nerve signals are transferred to ChAT+ T cells via NE released from the splenic nerve in the spleen, leading to elevation of ACh synthesis through activation of β-adrenoceptors on the ChAT+ T cells. The elevated ACh in turn acted on α7 nAChRs to suppress synthesis and release of TNF-α in macrophages (15).
Based on the observation that electrical stimulation of the vagus nerve did not elicit action potentials in the splenic nerve (158), Martelli et al. (159) proposed that efferent vagal stimulation and/or antidromic vagal afferent activation elicits lymphocyte mobilization to the spleen from a deposit of lymphocytes (vagal target). Then within the spleen, ACh-synthesizing CD4+ T cells among the mobilized lymphocytes release ACh to activate α7 nAChRs on the sympathetic nerve terminals leading to NE release. NE released from sympathetic nerve terminals acts on β2-adrenoceptors on macrophages to suppress TNF-α synthesis and release [see reviews in Ref. (13, 14)]. However, considering the extreme enzymatic fragility of ACh, it is essential to confirm the formation of synapse-like structures between ACh-synthesizing CD4+ T cells and sympathetic nerve terminals in the spleen, and between vagus nerve terminals and the vagus target. It is also necessary to demonstrate α7 nAChR expression in the sympathetic nerve terminals in the spleen and their functional release of NE.
SLURP-1 acts as a positive allosteric ligand for α7 nAChRs (117, 118) and increases the potency and efficacy of ACh at α7 nAChRs more than 2.5 times (117). SLURP-1 co-localizes with subsets of sensory neurons containing substance P (SP) and calcitonin gene-related peptide (CGRP) (160), which have been detected among afferent vagus nerve fibers (161, 162). In the white pulp of the spleen, SP+ nerve fibers have been identified within the marginal zone and the outer regions of the periarteriolar lymphoid sheaths, which are filled with T cells (163). In mice, CGRP+ nerve fibers have been detected in the spleen and lymph nodes (164, 165). Furthermore, the significant increase in splenic CGRP levels seen during the initial swelling phase of antigen-induced arthritis in mice suggests inflammation-induced release of CGRP from the sensory neurons (166). These findings provide evidence for splenic innervation by sensory neurons containing both SP and CGRP, and suggest the possibility of co-localization of SLURP-1 in the SP and CGRP nerve fibers innervating the spleen.
A subset of CD205+ DCs located within the marginal zone of human tonsils contain immunoreactive SLURP-1 and are surrounded by CD4+ T cells and CD68+ macrophages (126) (Figure 3C). Detection of SLURP-1 in human plasma, urine, sweat, saliva, and tears is suggestive of the protein’s stability and mobility (167). Moreover, SLURP-1 potentiates ACh synthesis in T cells (126). It is therefore reasonable to suggest that by potentiating the action of ACh at α7 nAChRs, SLURP-1 released from CD205+ DCs during antigen presentation and from SP/CGRP-containing neurons may participate in the inflammatory reflex leading to suppression of TNF-α synthesis in splenic macrophages (Figure 5). Because CD4+ T cells can directly interact with splenic macrophages through antigen presentation or cell-to-cell adhesion via LFA-1 (CD11a), ACh released from CD4+ T cells should act effectively on α7 nAChRs on macrophages in the presence of SLURP-1, leading to the suppression of TNF-α synthesis and release.
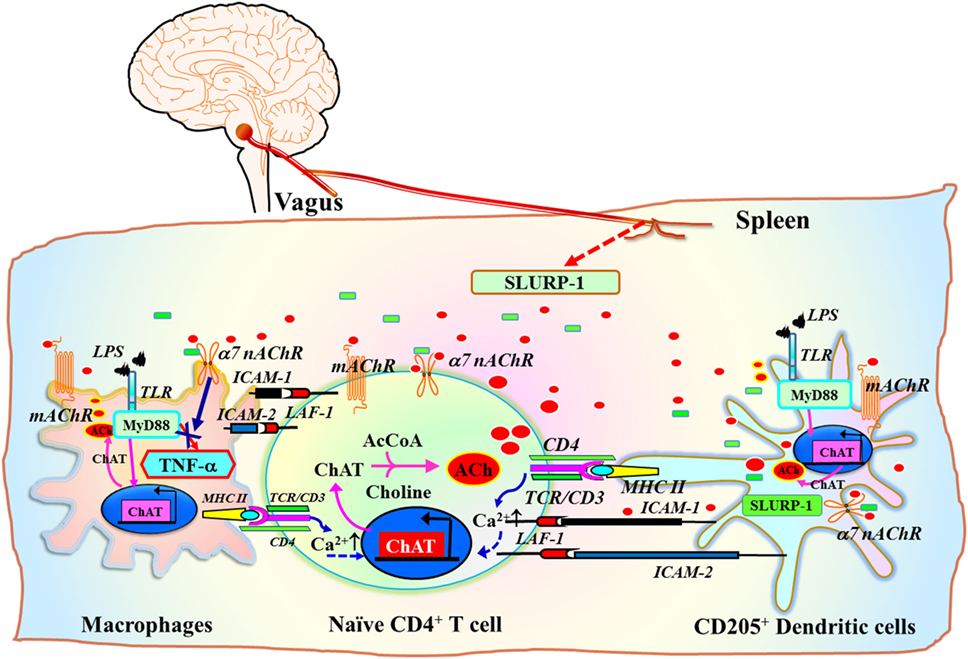
Figure 5. Schematic drawing of the efferent signaling pathway of the inflammatory reflex in the context of the cholinergic system in immune cells. ChAT+ T cell interaction with antigen peptides loaded on MHC class II on dendritic cells (DCs) or macrophages via the TCR/CD3 complex, CD80/CD86 (B7) co-stimulatory molecules with CD28, and ICAM-1/ICAM-2 with LFA-1, increases the synthesis and release of ACh from T cells [see a review by Fujii et al. (16)]. Inflammatory mediators induce the release of the positive allosteric α7 nAChR ligand SLURP-1 from SP/CGRP-containing sensory fibers. Efferent inflammatory reflex signaling via the vagus nerve may also induce released SLURP-1 from SP+/CGRP+ fibers in the spleen. This released SLURP-1, as well as SLURP-1 released from CD205+ mature DCs, potentiates the action of ACh from ChAT+ CD4+ T cells at α7 nAChRs on macrophages, thereby suppressing synthesis and release of tumor necrosis factor (TNF)-α. The model proposed here is from reviews by Kawashima et al. (111) and Fujii et al. (16). Green rectangles depict SLURP-1. Red ellipses depict acetylcholine. AcCoA, acetyl coenzyme A; ICAM-1, intercellular adhesion molecule-1; ICAM-2, intercellular adhesion molecule-2; LFA-1, lymphocyte function-associated antigen-1; LPS, lipopolysaccharide; MHC II, major histocompatibility complex class II; SLURP-1, secreted lymphocyte antigen-6/urokinase-type plasminogen activator receptor-related peptide-1; TCR, T cell receptor; TLR, toll-like receptor.
Currently available evidence suggests that the immune cell cholinergic system makes a critical contribution to the regulation of immune function. For example, the cholinergic signals generated by immune cells appear to be triggers of both the initiation and termination of cytokine synthesis (e.g., IL-2 in T cells and TNF-α in macrophages). Furthermore, the evidence raises the possibility that immune function can be modulated by manipulating the cholinergic activity of immune cells. Thus, a fuller understanding of the immune cell cholinergic system could be useful for the development of drugs and therapeutic strategies for the treatment of inflammation-related diseases and cancers.
TF, HM, SO, and KK conceived of the review. TF and KK drafted the initial version of the manuscript and refined the contents. MM, YM, SO, and KH contributed to reference analyses, prepared tables and figures, and revised the manuscript, which was progressively edited by TF, MM, SO, and KK. TF, MM, YM, HM, SO, KH, and KK reviewed and approved the final version of this review manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
This work was supported in part by Grant-in-Aid for Scientific Research (15K07969) from the Ministry of Education, Science, Sports and Culture (C) of Japan (KK, TF, MM, and KH) and funding from SRF (KK, TF, MM, KH, HM, and SO).
1. Burgen AS. The background of the muscarinic system. Life Sci (1995) 56(11–12):801–6. doi:10.1016/0024-3205(95)00013-V
2. Ewins AJ. Acetylcholine, a new active principle of ergot. Biochem J (1914) 8(1):44–9. doi:10.1042/bj0080044
3. Dale HH. The action of certain esters and ethers of choline, and their relation to muscarine. J Pharmacol Exp Ther (1914) 6:147–90.
4. Loewi O. Über humorale Übertragbarkeit der Herznervenwirkung. Pflugers Arch Gesamte Physiol Menschen Tiere (1921) 189:239–42. doi:10.1007/BF01741929
5. Loewi O, Navratil E. Über humorale Übertragbarkeit der Herznervenwirkung. X. Mitteilung: Über das Schicksal des Vagusstoffs. Pflugers Arch Gesamte Physiol Menschen Tiere (1926) 214:678–88. doi:10.1007/BF01741946
6. Dale HH, Dudley HW. The presence of histamine and acetylcholine in the spleen of the ox and the horse. J Physiol (1929) 68(2):97–123. doi:10.1113/jphysiol.1929.sp002598
7. Elliott TR. The action of adrenalin. J Physiol (1905) 32(5–6):401–67. doi:10.1113/jphysiol.1905.sp001093
8. Dale H. Pharmacology and nerve-endings (Walter Ernest Dixon Memorial Lecture): (Section of Therapeutics and Pharmacology). Proc R Soc Med (1935) 28(3):319–32.
9. Kawashima K, Fujii T. Extraneuronal cholinergic system in lymphocytes. Pharmacol Ther (2000) 86(1):29–48. doi:10.1016/S0163-7258(99)00071-6
10. Kawashima K, Fujii T. The lymphocytic cholinergic system and its biological function. Life Sci (2003) 72(18–19):2101–9. doi:10.1016/S0024-3205(03)00068-7
11. Kawashima K, Fujii T. The lymphocytic cholinergic system and its contribution to the regulation of immune activity. Life Sci (2003) 74(6):675–96. doi:10.1016/j.lfs.2003.09.037
12. Kawashima K, Fujii T. Expression of non-neuronal acetylcholine in lymphocytes and its contribution to the regulation of immune function. Front Biosci (2004) 9:2063–85. doi:10.2741/1390
13. Besedovsky HO, del Rey A. Immune-neuro-endocrine interactions: facts and hypotheses. Endocr Rev (1996) 17(1):64–102. doi:10.1210/edrv-17-1-64
14. Nance DM, Sanders VM. Autonomic innervation and regulation of the immune system (1987–2007). Brain Behav Immun (2007) 21(6):736–45. doi:10.1016/j.bbi.2007.03.008
15. Rosas-Ballina M, Olofsson PS, Ochani M, Valdes-Ferrer SI, Levine YA, Reardon C, et al. Acetylcholine-synthesizing T cells relay neural signals in a vagus nerve circuit. Science (2011) 334(6052):98–101. doi:10.1126/science.1209985
16. Fujii T, Mashimo M, Moriwaki Y, Misawa H, Ono S, Horiguchi K, et al. Physiological functions of the cholinergic system in immune cells. J Pharmacol Sci (2017) 134(1):1–21. doi:10.1016/j.jphs.2017.05.002
17. Kawashima K, Ishikawa H, Mochizuki M. Radioimmunoassay for acetylcholine in the rat brain. J Pharmacol Methods (1980) 3(2):115–23. doi:10.1016/0160-5402(80)90022-4
18. Kawashima K, Kajiyama K, Fujimoto K, Oohata H, Suzuki T. Presence of acetylcholine in human blood and its localization in circulating mononuclear leukocytes. Biog Amines (1993) 9:251–8.
19. Kawashima K, Oohata H, Fujimoto K, Suzuki T. Plasma concentration of acetylcholine in young women. Neurosci Lett (1987) 80(3):339–42. doi:10.1016/0304-3940(87)90478-2
20. Kawashima K, Oohata H, Fujimoto K, Suzuki T. Extraneuronal localization of acetylcholine and its release upon nicotinic stimulation in rabbits. Neurosci Lett (1989) 104(3):336–9. doi:10.1016/0304-3940(89)90599-5
21. Fujii T, Yamada S, Yamaguchi N, Fujimoto K, Suzuki T, Kawashima K. Species differences in the concentration of acetylcholine, a neurotransmitter, in whole blood and plasma. Neurosci Lett (1995) 201(3):207–10. doi:10.1016/0304-3940(95)12180-3
22. Fujii T, Mori Y, Tominaga T, Hayasaka I, Kawashima K. Maintenance of constant blood acetylcholine content before and after feeding in young chimpanzees. Neurosci Lett (1997) 227(1):21–4. doi:10.1016/S0304-3940(97)00287-5
23. Yamada S, Fujii T, Kawashima K. Oral administration of KW-5092, a novel gastroprokinetic agent with acetylcholinesterase inhibitory and acetylcholine release enhancing activities, causes a dose-dependent increase in the blood acetylcholine content of beagle dogs. Neurosci Lett (1997) 225(1):25–8. doi:10.1016/S0304-3940(97)00188-2
24. Fujii T, Tsuchiya T, Yamada S, Fujimoto K, Suzuki T, Kasahara T, et al. Localization and synthesis of acetylcholine in human leukemic T cell lines. J Neurosci Res (1996) 44(1):66–72. doi:10.1002/(SICI)1097-4547(19960401)44:1<66::AID-JNR9>3.0.CO;2-G
25. Fujii T, Tajima S, Yamada S, Watanabe Y, Sato KZ, Matsui M, et al. Constitutive expression of mRNA for the same choline acetyltransferase as that in the nervous system, an acetylcholine-synthesizing enzyme, in human leukemic T-cell lines. Neurosci Lett (1999) 259(2):71–4. doi:10.1016/S0304-3940(98)00921-5
26. Rinner I, Kawashima K, Schauenstein K. Rat lymphocytes produce and secrete acetylcholine in dependence of differentiation and activation. J Neuroimmunol (1998) 81(1–2):31–7. doi:10.1016/S0165-5728(97)00155-0
27. Rinner I, Schauenstein K. Detection of choline-acetyltransferase activity in lymphocytes. J Neurosci Res (1993) 35(2):188–91. doi:10.1002/jnr.490350209
28. Takahashi T, Ohnishi H, Sugiura Y, Honda K, Suematsu M, Kawasaki T, et al. Non-neuronal acetylcholine as an endogenous regulator of proliferation and differentiation of Lgr5-positive stem cells in mice. FEBS J (2014) 281(20):4672–90. doi:10.1111/febs.12974
29. Kajiyama K, Suzuki T, Fujimoto K, Kawashima K. Determination of acetylcholine content and choline acetyltransferase activity in rabbit blood cells obtained from the buffy coat layer. Jpn J Pharmacol (1991) 55(Suppl I):194.
30. Tucek S. The synthesis of acetylcholine in skeletal muscles of the rat. J Physiol (1982) 322:53–69. doi:10.1113/jphysiol.1982.sp014022
31. Tucek S. Choline acetyltransferase and the synthesis of acetylcholine. In: Whittaker VP, editor. Handbook of Experimental Pharmacology, the Cholinergic Synapse (Vol. 86), Berlin: Springer Verlag (1988). p. 125–65.
32. Fonnum F. A rapid radiochemical method for the determination of choline acetyltransferase. J Neurochem (1975) 24(2):407–9. doi:10.1111/j.1471-4159.1975.tb11895.x
33. Fujii T, Yamada S, Misawa H, Tajima S, Fujimoto K, Suzuki T, et al. Expression of choline acetyltransferase mRNA and protein in T-lymphocytes. Proc Jpn Acad (1995) 71B:231–5. doi:10.2183/pjab.71.231
34. Fujii T, Watanabe Y, Fujimoto K, Kawashima K. Expression of acetylcholine in lymphocytes and modulation of an independent lymphocytic cholinergic activity by immunological stimulation. Biog Amines (2003) 17:373–86. doi:10.1163/15693910260698320
35. Hecker A, Mikulski Z, Lips KS, Pfeil U, Zakrzewicz A, Wilker S, et al. Pivotal advance: up-regulation of acetylcholine synthesis and paracrine cholinergic signaling in intravascular transplant leukocytes during rejection of rat renal allografts. J Leukoc Biol (2009) 86(1):13–22. doi:10.1189/jlb.1107722
36. Kawashima K, Yoshikawa K, Fujii YX, Moriwaki Y, Misawa H. Expression and function of genes encoding cholinergic components in murine immune cells. Life Sci (2007) 80(24–25):2314–9. doi:10.1016/j.lfs.2007.02.036
37. Koarai A, Traves SL, Fenwick PS, Brown SM, Chana KK, Russell RE, et al. Expression of muscarinic receptors by human macrophages. Eur Respir J (2012) 39(3):698–704. doi:10.1183/09031936.00136710
38. Salamone G, Lombardi G, Gori S, Nahmod K, Jancic C, Amaral MM, et al. Cholinergic modulation of dendritic cell function. J Neuroimmunol (2011) 236(1–2):47–56. doi:10.1016/j.jneuroim.2011.05.007
39. Tallini YN, Shui B, Greene KS, Deng KY, Doran R, Fisher PJ, et al. BAC transgenic mice express enhanced green fluorescent protein in central and peripheral cholinergic neurons. Physiol Genomics (2006) 27(3):391–7. doi:10.1152/physiolgenomics.00092.2006
40. Gautron L, Rutkowski JM, Burton MD, Wei W, Wan Y, Elmquist JK. Neuronal and nonneuronal cholinergic structures in the mouse gastrointestinal tract and spleen. J Comp Neurol (2013) 521(16):3741–67. doi:10.1002/cne.23376
41. Reardon C, Duncan GS, Brustle A, Brenner D, Tusche MW, Olofsson PS, et al. Lymphocyte-derived ACh regulates local innate but not adaptive immunity. Proc Natl Acad Sci U S A (2013) 110(4):1410–5. doi:10.1073/pnas.1221655110
42. Fujii T, Yamada S, Watanabe Y, Misawa H, Tajima S, Fujimoto K, et al. Induction of choline acetyltransferase mRNA in human mononuclear leukocytes stimulated by phytohemagglutinin, a T-cell activator. J Neuroimmunol (1998) 82(1):101–7. doi:10.1016/S0165-5728(97)00195-1
43. Fujii T, Ushiyama N, Hosonuma K, Suenaga A, Kawashima K. Effects of human antithymocyte globulin on acetylcholine synthesis, its release and choline acetyltransferase transcription in a human leukemic T-cell line. J Neuroimmunol (2002) 128(1–2):1–8. doi:10.1016/S0165-5728(02)00111-X
44. Weitz-Schmidt G, Welzenbach K, Brinkmann V, Kamata T, Kallen J, Bruns C, et al. Statins selectively inhibit leukocyte function antigen-1 by binding to a novel regulatory integrin site. Nat Med (2001) 7(6):687–92. doi:10.1038/89058
45. Fujii T, Masuyama K, Kawashima K. Simvastatin regulates non-neuronal cholinergic activity in T lymphocytes via CD11a-mediated pathways. J Neuroimmunol (2006) 179(1–2):101–7. doi:10.1016/j.jneuroim.2006.05.029
46. Fujii T, Takada-Takatori Y, Kawashima K. Regulatory mechanisms of acetylcholine synthesis and release by T cells. Life Sci (2012) 91(21–22):981–5. doi:10.1016/j.lfs.2012.04.031
47. Fujii T, Watanabe Y, Inoue T, Kawashima K. Upregulation of mRNA encoding the M5 muscarinic acetylcholine receptor in human T- and B-lymphocytes during immunological responses. Neurochem Res (2003) 28(3–4):423–9. doi:10.1023/A:1022840416292
48. Das T, Sa G, Ray PK. Mechanisms of protein A superantigen-induced signal transduction for proliferation of mouse B cell. Immunol Lett (1999) 70(1):43–51. doi:10.1016/S0165-2478(99)00128-5
49. Browne EP. Regulation of B-cell responses by toll-like receptors. Immunology (2012) 136(4):370–9. doi:10.1111/j.1365-2567.2012.03587.x
50. Shimazu R, Akashi S, Ogata H, Nagai Y, Fukudome K, Miyake K, et al. MD-2, a molecule that confers lipopolysaccharide responsiveness on toll-like receptor 4. J Exp Med (1999) 189(11):1777–82. doi:10.1084/jem.189.11.1777
51. Lewis RS. Calcium signaling mechanisms in T lymphocytes. Annu Rev Immunol (2001) 19:497–521. doi:10.1146/annurev.immunol.19.1.497
52. Feske S, Gwack Y, Prakriya M, Srikanth S, Puppel SH, Tanasa B, et al. A mutation in Orai1 causes immune deficiency by abrogating CRAC channel function. Nature (2006) 441(7090):179–85. doi:10.1038/nature04702
53. Feske S, Giltnane J, Dolmetsch R, Staudt LM, Rao A. Gene regulation mediated by calcium signals in T lymphocytes. Nat Immunol (2001) 2(4):316–24. doi:10.1038/86318
54. Mashimo M, Yurie Y, Kawashima K, Fujii T. CRAC channels are required for [Ca2+]i oscillations and c-fos gene expression after muscarinic acetylcholine receptor activation in leukemic T cells. Life Sci (2016) 161:45–50. doi:10.1016/j.lfs.2016.07.014
55. Szelenyi J, Paldi-Haris P, Hollan S. Changes in the cholinergic system of lymphocytes due to mitogenic stimulation. Immunol Lett (1987) 16(1):49–54. doi:10.1016/0165-2478(87)90060-5
56. Fujii YX, Tashiro A, Arimoto K, Fujigaya H, Moriwaki Y, Misawa H, et al. Diminished antigen-specific IgG1 and interleukin-6 production and acetylcholinesterase expression in combined M1 and M5 muscarinic acetylcholine receptor knockout mice. J Neuroimmunol (2007) 188(1–2):80–5. doi:10.1016/j.jneuroim.2007.05.017
57. Schegg KM, Harrington LS, Neilsen S, Zweig RM, Peacock JH. Soluble and membrane-bound forms of brain acetylcholinesterase in Alzheimer’s disease. Neurobiol Aging (1992) 13(6):697–704. doi:10.1016/0197-4580(92)90092-C
58. Grossberg GT. Cholinesterase inhibitors for the treatment of Alzheimer’s disease: getting on and staying on. Curr Ther Res Clin Exp (2003) 64(4):216–35. doi:10.1016/S0011-393X(03)00059-6
59. Giacobini E. Selective inhibitors of butyrylcholinesterase: a valid alternative for therapy of Alzheimer’s disease? Drugs Aging (2001) 18(12):891–8. doi:10.2165/00002512-200118120-00001
60. Mizukami K, Akatsu H, Abrahamson EE, Mi Z, Ikonomovic MD. Immunohistochemical analysis of hippocampal butyrylcholinesterase: implications for regional vulnerability in Alzheimer’s disease. Neuropathology (2016) 36(2):135–45. doi:10.1111/neup.12241
61. Minic J, Chatonnet A, Krejci E, Molgo J. Butyrylcholinesterase and acetylcholinesterase activity and quantal transmitter release at normal and acetylcholinesterase knockout mouse neuromuscular junctions. Br J Pharmacol (2003) 138(1):177–87. doi:10.1038/sj.bjp.0705010
62. Girard E, Bernard V, Minic J, Chatonnet A, Krejci E, Molgo J. Butyrylcholinesterase and the control of synaptic responses in acetylcholinesterase knockout mice. Life Sci (2007) 80(24–25):2380–5. doi:10.1016/j.lfs.2007.03.011
63. Nizri E, Irony-Tur-Sinai M, Faranesh N, Lavon I, Lavi E, Weinstock M, et al. Suppression of neuroinflammation and immunomodulation by the acetylcholinesterase inhibitor rivastigmine. J Neuroimmunol (2008) 203(1):12–22. doi:10.1016/j.jneuroim.2008.06.018
64. Cosnes J. What is the link between the use of tobacco and IBD? Inflamm Bowel Dis (2008) 14(Suppl 2):S14–5. doi:10.1002/ibd.20555
65. Lunney PC, Leong RW. Review article: ulcerative colitis, smoking and nicotine therapy. Aliment Pharmacol Ther (2012) 36(11–12):997–1008. doi:10.1111/apt.12086
66. Siegel CA, Sands BE. Review article: practical management of inflammatory bowel disease patients taking immunomodulators. Aliment Pharmacol Ther (2005) 22(1):1–16. doi:10.1111/j.1365-2036.2005.02520.x
67. de Jonge WJ, Ulloa L. The alpha7 nicotinic acetylcholine receptor as a pharmacological target for inflammation. Br J Pharmacol (2007) 151(7):915–29. doi:10.1038/sj.bjp.0707264
68. Mehta SJ, Silver AR, Lindsay JO. Review article: strategies for the management of chronic unremitting ulcerative colitis. Aliment Pharmacol Ther (2013) 38(2):77–97. doi:10.1111/apt.12345
69. Shaked I, Meerson A, Wolf Y, Avni R, Greenberg D, Gilboa-Geffen A, et al. MicroRNA-132 potentiates cholinergic anti-inflammatory signaling by targeting acetylcholinesterase. Immunity (2009) 31(6):965–73. doi:10.1016/j.immuni.2009.09.019
70. Bonner TI, Buckley NJ, Young AC, Brann MR. Identification of a family of muscarinic acetylcholine receptor genes. Science (1987) 237(4814):527–32. doi:10.1126/science.3037705
71. Bonner TI, Young AC, Brann MR, Buckley NJ. Cloning and expression of the human and rat m5 muscarinic acetylcholine receptor genes. Neuron (1988) 1(5):403–10. doi:10.1016/0896-6273(88)90190-0
72. Bonner TI. New subtypes of muscarinic acetylcholine receptors. Trends Pharmacol Sci (1989) (Suppl):11–5.
73. Eglen RM, Nahorski SR. The muscarinic M5 receptor: a silent or emerging subtype? Br J Pharmacol (2000) 130(1):13–21. doi:10.1038/sj.bjp.0703276
74. Felder CC. Muscarinic acetylcholine receptors: signal transduction through multiple effectors. FASEB J (1995) 9(8):619–25.
75. Migeon JC, Thomas SL, Nathanson NM. Differential coupling of m2 and m4 muscarinic receptors to inhibition of adenylyl cyclase by Gi alpha and Goalpha subunits. J Biol Chem (1995) 270(27):16070–4. doi:10.1074/jbc.270.27.16070
76. Kawashima K, Fujii T, Moriwaki Y, Misawa H. Critical roles of acetylcholine and the muscarinic and nicotinic acetylcholine receptors in the regulation of immune function. Life Sci (2012) 91(21–22):1027–32. doi:10.1016/j.lfs.2012.05.006
77. Qian J, Galitovskiy V, Chernyavsky AI, Marchenko S, Grando SA. Plasticity of the murine spleen T-cell cholinergic receptors and their role in in vitro differentiation of naive CD4 T cells toward the Th1, Th2 and Th17 lineages. Genes Immun (2011) 12(3):222–30. doi:10.1038/gene.2010.72
78. Numa S, Noda M, Takahashi H, Tanabe T, Toyosato M, Furutani Y, et al. Molecular structure of the nicotinic acetylcholine receptor. Cold Spring Harb Symp Quant Biol (1983) 48(Pt 1):57–69. doi:10.1101/SQB.1983.048.01.008
79. Lohmann TH, Torrao AS, Britto LR, Lindstrom J, Hamassaki-Britto DE. A comparative non-radioactive in situ hybridization and immunohistochemical study of the distribution of alpha7 and alpha8 subunits of the nicotinic acetylcholine receptors in visual areas of the chick brain. Brain Res (2000) 852(2):463–9. doi:10.1016/S0006-8993(99)02082-X
80. Westfall TC, Westfall DP. Neurotransmission: the autonomic and somatic motor nervous system. 12th ed. In: Brunton L, Chabner BA, Knollmann BC, editors. Goodman and Gilman’s the Pharmacological Basisi of Therapeutics. New York: McGraw Hill (2011). p. 171–218.
81. Gotti C, Zoli M, Clementi F. Brain nicotinic acetylcholine receptors: native subtypes and their relevance. Trends Pharmacol Sci (2006) 27(9):482–91. doi:10.1016/j.tips.2006.07.004
82. Seguela P, Wadiche J, Dineley-Miller K, Dani JA, Patrick JW. Molecular cloning, functional properties, and distribution of rat brain alpha 7: a nicotinic cation channel highly permeable to calcium. J Neurosci (1993) 13(2):596–604.
83. Kalamida D, Poulas K, Avramopoulou V, Fostieri E, Lagoumintzis G, Lazaridis K, et al. Muscle and neuronal nicotinic acetylcholine receptors. Structure, function and pathogenicity. FEBS J (2007) 274(15):3799–845. doi:10.1111/j.1742-4658.2007.05935.x
84. Elgoyhen AB, Johnson DS, Boulter J, Vetter DE, Heinemann S. Alpha 9: an acetylcholine receptor with novel pharmacological properties expressed in rat cochlear hair cells. Cell (1994) 79(4):705–15. doi:10.1016/0092-8674(94)90555-X
85. Elgoyhen AB, Vetter DE, Katz E, Rothlin CV, Heinemann SF, Boulter J. alpha10: a determinant of nicotinic cholinergic receptor function in mammalian vestibular and cochlear mechanosensory hair cells. Proc Natl Acad Sci U S A (2001) 98(6):3501–6. doi:10.1073/pnas.051622798
86. Araud T, Graw S, Berger R, Lee M, Neveu E, Bertrand D, et al. The chimeric gene CHRFAM7A, a partial duplication of the CHRNA7 gene, is a dominant negative regulator of alpha7*nAChR function. Biochem Pharmacol (2011) 82(8):904–14. doi:10.1016/j.bcp.2011.06.018
87. Gault J, Robinson M, Berger R, Drebing C, Logel J, Hopkins J, et al. Genomic organization and partial duplication of the human alpha7 neuronal nicotinic acetylcholine receptor gene (CHRNA7). Genomics (1998) 52(2):173–85. doi:10.1006/geno.1998.5363
88. Changeux JP, Bertrand D, Corringer PJ, Dehaene S, Edelstein S, Lena C, et al. Brain nicotinic receptors: structure and regulation, role in learning and reinforcement. Brain Res Brain Res Rev (1998) 26(2–3):198–216. doi:10.1016/S0165-0173(97)00040-4
89. de Lucas-Cerrillo AM, Maldifassi MC, Arnalich F, Renart J, Atienza G, Serantes R, et al. Function of partially duplicated human alpha77 nicotinic receptor subunit CHRFAM7A gene: potential implications for the cholinergic anti-inflammatory response. J Biol Chem (2011) 286(1):594–606. doi:10.1074/jbc.M110.180067
90. Villiger Y, Szanto I, Jaconi S, Blanchet C, Buisson B, Krause KH, et al. Expression of an alpha7 duplicate nicotinic acetylcholine receptor-related protein in human leukocytes. J Neuroimmunol (2002) 126(1–2):86–98. doi:10.1016/S0165-5728(02)00057-7
91. van Maanen MA, Stoof SP, van der Zanden EP, de Jonge WJ, Janssen RA, Fischer DF, et al. The alpha7 nicotinic acetylcholine receptor on fibroblast-like synoviocytes and in synovial tissue from rheumatoid arthritis patients: a possible role for a key neurotransmitter in synovial inflammation. Arthritis Rheum (2009) 60(5):1272–81. doi:10.1002/art.24470
92. Benfante R, Antonini RA, De Pizzol M, Gotti C, Clementi F, Locati M, et al. Expression of the alpha7 nAChR subunit duplicate form (CHRFAM7A) is down-regulated in the monocytic cell line THP-1 on treatment with LPS. J Neuroimmunol (2011) 230(1–2):74–84. doi:10.1016/j.jneuroim.2010.09.008
93. Khiroug SS, Harkness PC, Lamb PW, Sudweeks SN, Khiroug L, Millar NS, et al. Rat nicotinic ACh receptor alpha7 and beta2 subunits co-assemble to form functional heteromeric nicotinic receptor channels. J Physiol (2002) 540(Pt 2):425–34. doi:10.1113/jphysiol.2001.013847
94. Moretti M, Zoli M, George AA, Lukas RJ, Pistillo F, Maskos U, et al. The novel alpha7beta2-nicotinic acetylcholine receptor subtype is expressed in mouse and human basal forebrain: biochemical and pharmacological characterization. Mol Pharmacol (2014) 86(3):306–17. doi:10.1124/mol.114.093377
95. Zwart R, Strotton M, Ching J, Astles PC, Sher E. Unique pharmacology of heteromeric alpha7beta2 nicotinic acetylcholine receptors expressed in Xenopus laevis oocytes. Eur J Pharmacol (2014) 726:77–86. doi:10.1016/j.ejphar.2014.01.031
96. Azam L, Winzer-Serhan U, Leslie FM. Co-expression of alpha7 and beta2 nicotinic acetylcholine receptor subunit mRNAs within rat brain cholinergic neurons. Neuroscience (2003) 119(4):965–77. doi:10.1016/S0306-4522(03)00220-3
97. Murray TA, Bertrand D, Papke RL, George AA, Pantoja R, Srinivasan R, et al. alpha7beta2 nicotinic acetylcholine receptors assemble, function, and are activated primarily via their alpha7-alpha7 interfaces. Mol Pharmacol (2012) 81(2):175–88. doi:10.1124/mol.111.074088
98. Wu J, Liu Q, Tang P, Mikkelsen JD, Shen J, Whiteaker P, et al. Heteromeric alpha7beta2 nicotinic acetylcholine receptors in the brain. Trends Pharmacol Sci (2016) 37(7):562–74. doi:10.1016/j.tips.2016.03.005
99. Thomsen MS, Zwart R, Ursu D, Jensen MM, Pinborg LH, Gilmour G, et al. alpha7 and beta2 nicotinic acetylcholine receptor subunits form heteromeric receptor complexes that are expressed in the human cortex and display distinct pharmacological properties. PLoS One (2015) 10(6):e0130572. doi:10.1371/journal.pone.0130572
100. Sato KZ, Fujii T, Watanabe Y, Yamada S, Ando T, Fujimoto F, et al. Diversity of mRNA expression for muscarinic acetylcholine receptor subtypes and neuronal nicotinic acetylcholine receptor subunits in human mononuclear leukocytes and leukemic cell lines. Neurosci Lett (1999) 266(1):17–20. doi:10.1016/S0304-3940(99)00259-1
101. Razani-Boroujerdi S, Boyd RT, Davila-Garcia MI, Nandi JS, Mishra NC, Singh SP, et al. T cells express alpha7-nicotinic acetylcholine receptor subunits that require a functional TCR and leukocyte-specific protein tyrosine kinase for nicotine-induced Ca2+ response. J Immunol (2007) 179(5):2889–98. doi:10.4049/jimmunol.179.5.2889
102. Corradi J, Bouzat C. Understanding the bases of function and modulation of alpha7 nicotinic receptors: implications for drug discovery. Mol Pharmacol (2016) 90(3):288–99. doi:10.1124/mol.116.104240
103. Báez-Pagán CA, Delgado-Vélez M, Lasalde-Dominicci JA. Activation of the macrophage alpha7 nicotinic acetylcholine receptor and control of inflammation. J Neuroimmune Pharmacol (2015) 10(3):468–76. doi:10.1007/s11481-015-9601-5
104. Egea J, Buendia I, Parada E, Navarro E, Leon R, Lopez MG. Anti-inflammatory role of microglial alpha7 nAChRs and its role in neuroprotection. Biochem Pharmacol (2015) 97(4):463–72. doi:10.1016/j.bcp.2015.07.032
105. Paulo JA, Brucker WJ, Hawrot E. Proteomic analysis of an alpha7 nicotinic acetylcholine receptor interactome. J Proteome Res (2009) 8(4):1849–58. doi:10.1021/pr800731z
106. Nordman JC, Kabbani N. An interaction between alpha7 nicotinic receptors and a G-protein pathway complex regulates neurite growth in neural cells. J Cell Sci (2012) 125(Pt 22):5502–13. doi:10.1242/jcs.110379
107. Kabbani N, Nordman JC, Corgiat BA, Veltri DP, Shehu A, Seymour VA, et al. Are nicotinic acetylcholine receptors coupled to G proteins? Bioessays (2013) 35(12):1025–34. doi:10.1002/bies.201300082
108. King JR, Nordman JC, Bridges SP, Lin MK, Kabbani N. Identification and characterization of a G protein-binding cluster in alpha7 nicotinic acetylcholine receptors. J Biol Chem (2015) 290(33):20060–70. doi:10.1074/jbc.M115.647040
109. Hecker A, Kullmar M, Wilker S, Richter K, Zakrzewicz A, Atanasova S, et al. Phosphocholine-modified macromolecules and canonical nicotinic agonists inhibit ATP-induced IL-1beta release. J Immunol (2015) 195(5):2325–34. doi:10.4049/jimmunol.1400974
110. Richter K, Mathes V, Fronius M, Althaus M, Hecker A, Krasteva-Christ G, et al. Phosphocholine – an agonist of metabotropic but not of ionotropic functions of alpha9-containing nicotinic acetylcholine receptors. Sci Rep (2016) 6:28660. doi:10.1038/srep28660
111. Kawashima K, Fujii T, Moriwaki Y, Misawa H, Horiguchi K. Reconciling neuronally and nonneuronally derived acetylcholine in the regulation of immune function. Ann N Y Acad Sci (2012) 1261:7–17. doi:10.1111/j.1749-6632.2012.06516.x
112. Grando SA, Kawashima K, Kirkpatrick CJ, Kummer W, Wessler I. Recent progress in revealing the biological and medical significance of the non-neuronal cholinergic system. Int Immunopharmacol (2015) 29(1):1–7. doi:10.1016/j.intimp.2015.08.023
113. Moser N, Mechawar N, Jones I, Gochberg-Sarver A, Orr-Urtreger A, Plomann M, et al. Evaluating the suitability of nicotinic acetylcholine receptor antibodies for standard immunodetection procedures. J Neurochem (2007) 102(2):479–92. doi:10.1111/j.1471-4159.2007.04498.x
114. Rommel FR, Raghavan B, Paddenberg R, Kummer W, Tumala S, Lochnit G, et al. Suitability of nicotinic acetylcholine receptor alpha7 and muscarinic acetylcholine receptor 3 antibodies for immune detection: evaluation in murine skin. J Histochem Cytochem (2015) 63(5):329–39. doi:10.1369/0022155415575028
115. Adermann K, Wattler F, Wattler S, Heine G, Meyer M, Forssmann WG, et al. Structural and phylogenetic characterization of human SLURP-1, the first secreted mammalian member of the Ly-6/uPAR protein superfamily. Protein Sci (1999) 8(4):810–9. doi:10.1110/ps.8.4.810
116. Tsuji H, Okamoto K, Matsuzaka Y, Iizuka H, Tamiya G, Inoko H. SLURP-2, a novel member of the human Ly-6 superfamily that is up-regulated in psoriasis vulgaris. Genomics (2003) 81(1):26–33. doi:10.1016/S0888-7543(02)00025-3
117. Chimienti F, Hogg RC, Plantard L, Lehmann C, Brakch N, Fischer J, et al. Identification of SLURP-1 as an epidermal neuromodulator explains the clinical phenotype of Mal de Meleda. Hum Mol Genet (2003) 12(22):3017–24. doi:10.1093/hmg/ddg320
118. Arredondo J, Chernyavsky AI, Webber RJ, Grando SA. Biological effects of SLURP-1 on human keratinocytes. J Invest Dermatol (2005) 125(6):1236–41. doi:10.1111/j.0022-202X.2005.23973.x
119. Arredondo J, Chernyavsky AI, Jolkovsky DL, Webber RJ, Grando SA. SLURP-2: a novel cholinergic signaling peptide in human mucocutaneous epithelium. J Cell Physiol (2006) 208(1):238–45. doi:10.1002/jcp.20661
120. Fischer J, Bouadjar B, Heilig R, Huber M, Lefevre C, Jobard F, et al. Mutations in the gene encoding SLURP-1 in Mal de Meleda. Hum Mol Genet (2001) 10(8):875–80. doi:10.1093/hmg/10.8.875
121. Grando SA. Basic and clinical aspects of non-neuronal acetylcholine: biological and clinical significance of non-canonical ligands of epithelial nicotinic acetylcholine receptors. J Pharmacol Sci (2008) 106(2):174–9. doi:10.1254/jphs.FM0070087
122. Chernyavsky AI, Galitovskiy V, Shchepotin IB, Grando SA. Anti-inflammatory effects of the nicotinergic peptides SLURP-1 and SLURP-2 on human intestinal epithelial cells and immunocytes. Biomed Res Int (2014) 2014:609086. doi:10.1155/2014/609086
123. Moriwaki Y, Yoshikawa K, Fukuda H, Fujii YX, Misawa H, Kawashima K. Immune system expression of SLURP-1 and SLURP-2, two endogenous nicotinic acetylcholine receptor ligands. Life Sci (2007) 80(24–25):2365–8. doi:10.1016/j.lfs.2006.12.028
124. Horiguchi K, Horiguchi S, Yamashita N, Irie K, Masuda J, Takano-Ohmuro H, et al. Expression of SLURP-1, an endogenous alpha7 nicotinic acetylcholine receptor allosteric ligand, in murine bronchial epithelial cells. J Neurosci Res (2009) 87(12):2740–7. doi:10.1002/jnr.22102
125. Tjiu JW, Lin PJ, Wu WH, Cheng YP, Chiu HC, Thong HY, et al. SLURP1 mutation-impaired T-cell activation in a family with mal de Meleda. Br J Dermatol (2011) 164(1):47–53. doi:10.1111/j.1365-2133.2010.10059.x
126. Fujii T, Horiguchi K, Sunaga H, Moriwaki Y, Misawa H, Kasahara T, et al. SLURP-1, an endogenous alpha7 nicotinic acetylcholine receptor allosteric ligand, is expressed in CD205+ dendritic cells in human tonsils and potentiates lymphocytic cholinergic activity. J Neuroimmunol (2014) 267(1–2):43–9. doi:10.1016/j.jneuroim.2013.12.003
127. Fukaya T, Murakami R, Takagi H, Sato K, Sato Y, Otsuka H, et al. Conditional ablation of CD205+ conventional dendritic cells impacts the regulation of T-cell immunity and homeostasis in vivo. Proc Natl Acad Sci U S A (2012) 109(28):11288–93. doi:10.1073/pnas.1202208109
128. Fujii T, Kawashima K. Ca2+ oscillation and c-fos gene expression induced via muscarinic acetylcholine receptor in human T- and B-cell lines. Naunyn Schmiedebergs Arch Pharmacol (2000) 362(1):14–21. doi:10.1007/s002100000251
129. Zimring JC, Kapp LM, Yamada M, Wess J, Kapp JA. Regulation of CD8+ cytolytic T lymphocyte differentiation by a cholinergic pathway. J Neuroimmunol (2005) 164(1–2):66–75. doi:10.1016/j.jneuroim.2005.03.018
130. Vezys V, Masopust D, Desmarets M, Wess J, Zimring JC. Analysis of CD8+ T cell-mediated anti-viral responses in mice with targeted deletions of the M1 or M5 muscarinic cholinergic receptors. Life Sci (2007) 80(24–25):2330–3. doi:10.1016/j.lfs.2007.01.001
131. Wang H, Yu M, Ochani M, Amella CA, Tanovic M, Susarla S, et al. Nicotinic acetylcholine receptor alpha7 subunit is an essential regulator of inflammation. Nature (2003) 421(6921):384–8. doi:10.1038/nature01339
132. Olofsson PS, Katz DA, Rosas-Ballina M, Levine YA, Ochani M, Valdes-Ferrer SI, et al. alpha7 nicotinic acetylcholine receptor (alpha7nAChR) expression in bone marrow-derived non-T cells is required for the inflammatory reflex. Mol Med (2012) 18:539–43. doi:10.2119/molmed.2011.00405
133. Fujii YX, Fujigaya H, Moriwaki Y, Misawa H, Kasahara T, Grando SA, et al. Enhanced serum antigen-specific IgG1 and proinflammatory cytokine production in nicotinic acetylcholine receptor alpha7 subunit gene knockout mice. J Neuroimmunol (2007) 189(1–2):69–74. doi:10.1016/j.jneuroim.2007.07.003
134. St-Pierre S, Jiang W, Roy P, Champigny C, LeBlanc E, Morley BJ, et al. Nicotinic acetylcholine receptors modulate bone marrow-derived pro-inflammatory monocyte production and survival. PLoS One (2016) 11(2):e0150230. doi:10.1371/journal.pone.0150230
135. Skok M, Grailhe R, Changeux JP. Nicotinic receptors regulate B lymphocyte activation and immune response. Eur J Pharmacol (2005) 517(3):246–51. doi:10.1016/j.ejphar.2005.05.011
136. Skok M, Grailhe R, Agenes F, Changeux JP. The role of nicotinic acetylcholine receptors in lymphocyte development. J Neuroimmunol (2006) 171(1–2):86–98. doi:10.1016/j.jneuroim.2005.09.011
137. Skok MV, Grailhe R, Agenes F, Changeux JP. The role of nicotinic receptors in B-lymphocyte development and activation. Life Sci (2007) 80(24–25):2334–6. doi:10.1016/j.lfs.2007.02.005
138. Koval L, Lykhmus O, Zhmak M, Khruschov A, Tsetlin V, Magrini E, et al. Differential involvement of alpha4beta2, alpha7 and alpha9alpha10 nicotinic acetylcholine receptors in B lymphocyte activation in vitro. Int J Biochem Cell Biol (2011) 43(4):516–24. doi:10.1016/j.biocel.2010.12.003
139. Mashimo M, Iwasaki Y, Inoue S, Saito S, Kawashima K, Fujii T. Acetylcholine released from T cells regulates intracellular Ca2+, IL-2 secretion and T cell proliferation through nicotinic acetylcholine receptor. Life Sci (2017) 172:13–8. doi:10.1016/j.lfs.2016.12.015
140. Nizri E, Irony-Tur-Sinai M, Lory O, Orr-Urtreger A, Lavi E, Brenner T. Activation of the cholinergic anti-inflammatory system by nicotine attenuates neuroinflammation via suppression of Th1 and Th17 responses. J Immunol (2009) 183(10):6681–8. doi:10.4049/jimmunol.0902212
141. Kawashima K, Mashimo M, Fujii T, Moriwaki M, Misawa H, Ono S. Role for alpha 7 nicotinic acetylcholine receptors in naïve T cell differentiation into regulatory T cells. Neuroscience Meeting Planner. San Diego, CA (2016).
142. Murphy KM, Reiner SL. The lineage decisions of helper T cells. Nat Rev Immunol (2002) 2(12):933–44. doi:10.1038/nri954
143. Murphy KM, Stockinger B. Effector T cell plasticity: flexibility in the face of changing circumstances. Nat Immunol (2010) 11(8):674–80. doi:10.1038/ni.1899
144. Fransson M, Piras E, Burman J, Nilsson B, Essand M, Lu B, et al. CAR/FoxP3-engineered T regulatory cells target the CNS and suppress EAE upon intranasal delivery. J Neuroinflammation (2012) 9:112. doi:10.1186/1742-2094-9-112
145. Andersson U, Tracey KJ. Neural reflexes in inflammation and immunity. J Exp Med (2012) 209(6):1057–68. doi:10.1084/jem.20120571
146. Andersson U, Tracey KJ. Reflex principles of immunological homeostasis. Annu Rev Immunol (2012) 30:313–35. doi:10.1146/annurev-immunol-020711-075015
147. Pavlov VA, Tracey KJ. Neural regulation of immunity: molecular mechanisms and clinical translation. Nat Neurosci (2017) 20(2):156–66. doi:10.1038/nn.4477
148. Watkins LR, Goehler LE, Relton JK, Tartaglia N, Silbert L, Martin D, et al. Blockade of interleukin-1 induced hyperthermia by subdiaphragmatic vagotomy: evidence for vagal mediation of immune-brain communication. Neurosci Lett (1995) 183(1–2):27–31. doi:10.1016/0304-3940(94)11105-R
149. Borovikova LV, Ivanova S, Zhang M, Yang H, Botchkina GI, Watkins LR, et al. Vagus nerve stimulation attenuates the systemic inflammatory response to endotoxin. Nature (2000) 405(6785):458–62. doi:10.1038/35013070
151. Pavlov VA, Tracey KJ. The cholinergic anti-inflammatory pathway. Brain Behav Immun (2005) 19(6):493–9. doi:10.1016/j.bbi.2005.03.015
152. Tracey KJ. Physiology and immunology of the cholinergic antiinflammatory pathway. J Clin Invest (2007) 117(2):289–96. doi:10.1172/JCI30555
153. Huston JM, Ochani M, Rosas-Ballina M, Liao H, Ochani K, Pavlov VA, et al. Splenectomy inactivates the cholinergic antiinflammatory pathway during lethal endotoxemia and polymicrobial sepsis. J Exp Med (2006) 203(7):1623–8. doi:10.1084/jem.20052362
154. Huston JM, Wang H, Ochani M, Ochani K, Rosas-Ballina M, Gallowitsch-Puerta M, et al. Splenectomy protects against sepsis lethality and reduces serum HMGB1 levels. J Immunol (2008) 181(5):3535–9. doi:10.4049/jimmunol.181.5.3535
155. Rosas-Ballina M, Ochani M, Parrish WR, Ochani K, Harris YT, Huston JM, et al. Splenic nerve is required for cholinergic antiinflammatory pathway control of TNF in endotoxemia. Proc Natl Acad Sci U S A (2008) 105(31):11008–13. doi:10.1073/pnas.0803237105
156. Berthoud HR, Powley TL. Characterization of vagal innervation to the rat celiac, suprarenal and mesenteric ganglia. J Auton Nerv Syst (1993) 42(2):153–69. doi:10.1016/0165-1838(93)90046-W
157. Bellinger DL, Felten SY, Lorton D, Felten DL. Origin of noradrenergic innervation of the spleen in rats. Brain Behav Immun (1989) 3(4):291–311. doi:10.1016/0889-1591(89)90029-9
158. Bratton BO, Martelli D, McKinley MJ, Trevaks D, Anderson CR, McAllen RM. Neural regulation of inflammation: no neural connection from the vagus to splenic sympathetic neurons. Exp Physiol (2012) 97(11):1180–5. doi:10.1113/expphysiol.2011.061531
159. Martelli D, McKinley MJ, McAllen RM. The cholinergic anti-inflammatory pathway: a critical review. Auton Neurosci (2014) 182:65–9. doi:10.1016/j.autneu.2013.12.007
160. Moriwaki Y, Watanabe Y, Shinagawa T, Kai M, Miyazawa M, Okuda T, et al. Primary sensory neuronal expression of SLURP-1, an endogenous nicotinic acetylcholine receptor ligand. Neurosci Res (2009) 64(4):403–12. doi:10.1016/j.neures.2009.04.014
161. Gamse R, Holzer P, Lembeck F. Decrease of substance P in primary afferent neurones and impairment of neurogenic plasma extravasation by capsaicin. Br J Pharmacol (1980) 68(2):207–13. doi:10.1111/j.1476-5381.1980.tb10409.x
162. Li Y, Jiang YC, Owyang C. Central CGRP inhibits pancreatic enzyme secretion by modulation of vagal parasympathetic outflow. Am J Physiol (1998) 275(5 Pt 1):G957–63.
163. Lorton D, Bellinger DL, Felten SY, Felten DL. Substance P innervation of spleen in rats: nerve fibers associate with lymphocytes and macrophages in specific compartments of the spleen. Brain Behav Immun (1991) 5(1):29–40. doi:10.1016/0889-1591(91)90005-U
164. Bulloch K, McEwen BS, Diwa A, Radojcic T, Hausman J, Baird S. The role of calcitonin gene-related peptide in the mouse thymus revisited. Ann N Y Acad Sci (1994) 741:129–36. doi:10.1111/j.1749-6632.1994.tb39653.x
165. Wang X, Fiscus RR, Tang Z, Yang L, Wu J, Fan S, et al. CGRP in the serum of endotoxin-treated rats suppresses lymphoproliferation. Brain Behav Immun (1994) 8(4):282–92. doi:10.1006/brbi.1994.1027
166. Ebbinghaus M, Segond von Banchet G, Massier J, Gajda M, Brauer R, Kress M, et al. Interleukin-6-dependent influence of nociceptive sensory neurons on antigen-induced arthritis. Arthritis Res Ther (2015) 17:334. doi:10.1186/s13075-015-0858-0
Keywords: dendritic cell, lymphocyte, macrophage, mAChR, nAChR, SLURP-1
Citation: Fujii T, Mashimo M, Moriwaki Y, Misawa H, Ono S, Horiguchi K and Kawashima K (2017) Expression and Function of the Cholinergic System in Immune Cells. Front. Immunol. 8:1085. doi: 10.3389/fimmu.2017.01085
Received: 20 June 2017; Accepted: 21 August 2017;
Published: 06 September 2017
Edited by:
Valentin A. Pavlov, Northwell Health, United StatesReviewed by:
Donald B. Hoover, East Tennessee State University, United StatesCopyright: © 2017 Fujii, Mashimo, Moriwaki, Misawa, Ono, Horiguchi and Kawashima. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Koichiro Kawashima, a29pY2hpcm8tamtAcGlhbm8ub2NuLm5lLmpw, a2F3YXNoaW1ha0BwaGFybS5raXRhc2F0by11LmFjLmpw
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.