
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Immunol., 13 September 2017
Sec. Microbial Immunology
Volume 8 - 2017 | https://doi.org/10.3389/fimmu.2017.01080
This article is part of the Research TopicImmune evasion strategies in protozoan-host interactionsView all 50 articles
 Thaís Rigueti Brasil1*
Thaís Rigueti Brasil1* Celio Geraldo Freire-de-Lima2
Celio Geraldo Freire-de-Lima2 Alexandre Morrot3,4
Alexandre Morrot3,4 Andrea Cristina Vetö Arnholdt1*
Andrea Cristina Vetö Arnholdt1*
Toxoplasma gondii has successfully developed strategies to evade host’s immune response and reach immune privileged sites, which remains in a controlled environment inside quiescent tissue cysts. In this review, we will approach several known mechanisms used by the parasite to modulate mainly the murine immune system at its favor. In what follows, we review recent findings revealing interference of host’s cell autonomous immunity and cell signaling, gene expression, apoptosis, and production of microbicide molecules such as nitric oxide and oxygen reactive species during parasite infection. Modulation of host’s metalloproteinases of extracellular matrix is also discussed. These immune evasion strategies are determinant to parasite dissemination throughout the host taking advantage of cells from the immune system to reach brain and retina, crossing crucial hosts’ barriers.
Toxoplasma gondii is a parasite acquired through food or water contamination, followed by gut invasion and systemic dissemination. The protozoan T. gondii is able to escape the immune system and cross the blood–brain and blood–retina barrier reaching immune privileged sites leading to long-term infection (1). Intracellular pathogen, T. gondii subverts innate immunitary system interfering with host signaling pathways according to virulence based on the parasite genotype and the cell type infected (2, 3). Moreover, distinct responses can be triggered depending on inflammatory cells recruited, parasite burden, and the parasites’ molecular arrangement (4). T. gondii is an example of host–parasite coadaptation, and several studies have unveiled molecular interactions that allow the parasite not to exterminate the host, evading from immune responses at different levels.
Host cell gene transcription is drastically affected by T. gondii including those genes involved in energy metabolism, immune responses, and signaling (5–7). Initially, pattern recognition receptors such as toll-like receptors (TLRs) are able to bind parasite molecules. In mice, TLR11 and TLR12 bind to TgPRF triggering a strong IL-12 response that most effective leads to interferon gamma-inducing response genes (IRGs) (8). In humans, those genes are not functional, and TLR2, 4, 8, and 9 are effective in inducing IL-12 (9). After active invasion of host cell, T. gondii surrounds itself with a combination of host membrane and it is able to exclude and to recruit host proteins to the resulting parasitophorous vacuole (PV) in which it develops (3).
Rhoptry organelle initiates vacuole formation by secretion of an array of proteins that are released directly into the host cell, collectively known as RONs, forming the moving junction (MJ) (10). RONs 2/4/5/8 anchor the MJ at host cell membrane during invasion and also function as a selective sieve to host cell proteins that will be incorporated to PV (11, 12). This process assures the formation of a PV devoided of host proteins required for recruitment of endosomes and lysosomes (13).
The non-fusogenic nature of the PV is critical since it inhibits one of the cell-autonomous immunity mechanisms, the autophagy (14). Autophagosomal compartments are generated in eukaryotic cells as part of a bulk degradation system, through the formation of an initial phagophorous derived of membrane cisterna where autophagy-related proteins (ATG) are orderly accumulated leading to the fusion with lysosomal pathway (15). In non-canonical autophagy, ATG proteins can build up from preformed membranes such as the PVM, and not all ATGs are required to participate in the process (16). Extensive experimental data indicate that the autophagy machinery can promote killing of a broad variety of pathogens (17, 18) including T. gondii, especially in mouse models, and the IFNγ produced early in infection is crucial for that (19–23). Upon the influence of IFNγ, infected host cells respond regulating nearly 2,000 genes that are called interferon-inducible genes (24). Among those, effector molecules such as the immunity-related p47 GTPases (IRGs) and guanylate-binding proteins (GBPs) rapidly accumulate on and around the PVM, leading to the disruption of the PVM and subsequent death of the parasite in mouse cells (25). In human cells, ubiquitination and recruitment of autophagy adaptors did not require GBPs (26).
The recruitment of IRGs and GBPs to PVM depends on autophagy-related (Atg) gene products (24, 27, 28). In mice, it has been demonstrated that Atg5 and Atg8 (LC3 in humans) are required for the proper targeting of the effectors onto the PVM of T. gondii (29–31). In addition, Atg12, Atg16L1, Atg3, and Atg7 are recruited to the PVM to promote parasite killing (26, 32). Infection of Atg5- and Atg3-deficient cells show decreased accumulation of immunity-related GTPase family member b10 (Irgb10) and guanylate-binding protein 2 (Gbp2) at T. gondii PVM (33, 34). Genotypes II and III are susceptible to the IRGs resistance system. On the other hand, infection with virulent strains (e.g., type I) has demonstrated that polymorphic T. gondii kinase proteins from rhoptries like ROP5, ROP17, and ROP18 phosphorylate IRG proteins in murine cells inactivating them in order to preserve PVM integrity (35–40), suggesting that in type I strain parasites can evade this cell autonomous immunity mechanism (Figure 1A).
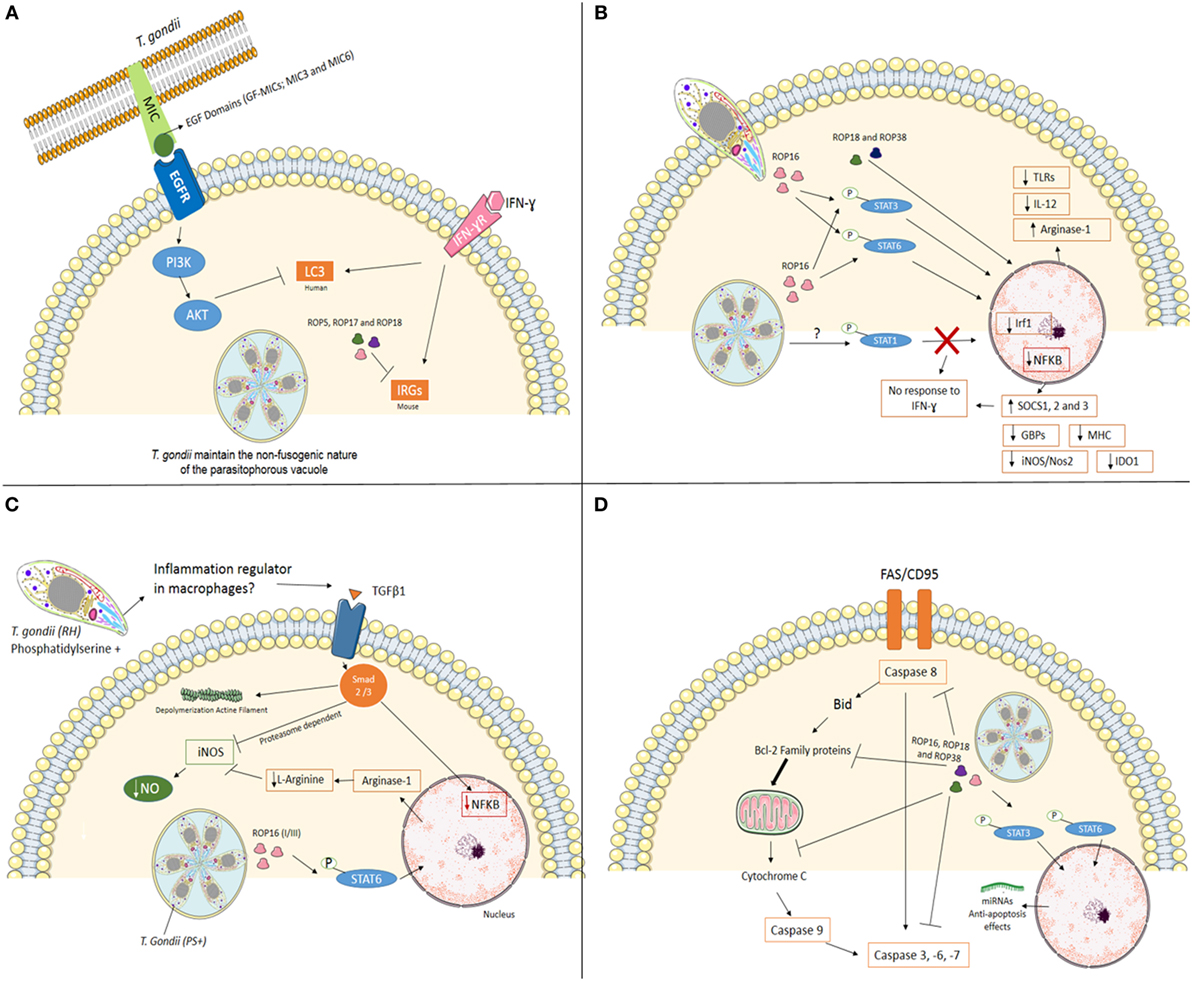
Figure 1. Mechanisms used by Toxoplasma gondii, type I strain, to modulate mainly immune system at its favor. (A) Interfering with cell-autonomous immunity. In infection with virulent strain (e.g., type I), the polymorphic effector proteins like ROP5, ROP17, and ROP18 cooperate to phosphorylate and inactivate mouse IRG proteins to preserve PVM integrity. In human cells (endothelial, retinal pigment epithelial, and microglia), T. gondii activates EGFR-Akt signaling to prevent targeting of the parasite by LC3 structures and pathogen killing dependent on autophagy proteins and lysosomal protease activity. (B) Cell signaling interference. The Toxoplasma rhoptry 16 kinase (ROP16), ROP18, and 38 can mediate the induction of arginase-1, suppress toll-like receptors (TLRs), IL-12, and nuclear factor-kappa B (NF-κB) through phosphorylation of signal transducer and activator of transcription (STAT) 3 and 6. T. gondii also interferes with STAT1 signaling, resulting in blockage of interferon regulatory factor 1 (Irf1), p65 guanylate-binding proteins (GBPs), inducible nitric oxide synthase (iNOS/Nos2), indoleamine 2,3-dioxygenase 1 (IDO1) and major histocompatibility complex (MHC). Furthermore, the infection upregulation SOCS1, 2 and 3. Together, T. gondii inhibiting pro-inflammatory response in different ways. (C) Silencing microbicide molecules. T. gondii [phosphatidylserine positive (PS+)] infection of murine blood monocyte-derived and peritoneal macrophages activated in vitro with IFN-γ and lipopolysaccharide (LPS) lead to a substantial decrease in NO production. Decreased mechanisms include phosphorylation of STAT6 by ROP16 resulting in arginine degradation and induction of TGFβ1 through Smad 2 and 3 leading to destruction of iNOS, actin filament (F-actin) depolymerization, and lack of NF-κB in the nucleus. (D) Maintaining the host cell alive. T. gondii has several strategies for inhibiting the initiation of the apoptotic cascade triggered by mitochondrial pathway or death receptor pathway in infected cells. The effector proteins like ROP16, ROP18, and ROP38 phosphorylate STAT3 and STAT6 and promotes mechanisms that include blocking of mitochondrial cytochrome c release, alterations of the balance between pro- and anti-apoptotic Bcl-2 proteins, degradation of caspase 8, blocking Fas/CD95-mediated apoptosis, and inactivation of effector caspases (-3, -6, -7) in infected cells.
Until recently, IFN inducible GTPases were thought to be non-functional in T. gondii response in human cells. Qin et al. showed that Gbp1 induced by IFNγ in mesenchymal stromal cells was responsible to decrease the number of parasites after 4 h of infection and showed that Gbp1 was found in association with at least 10% of PV (37). On the other hand, Johnston et al. (38) using A549 human epithelial cells showed that in the absence of Gbp1 parasite numbers increase rapidly. However, no Gbp1 was seen in association with PV at any moment. Clearly that are gaps in our knowledge of IRG system in the human autonomous immunity (38). Muniz-Feliciano et al. (22) showed that T. gondii micronemal proteins (MICs) containing epidermal growth factor (EGF) domains (MIC3 and MIC6) appeared to promote EGF receptor activation in endothelial cells, retinal pigment epithelial cells, and microglia in humans. These findings support the concept that T. gondii activates EGFR-Akt signaling in the host cell to prevent targeting of the PVM by LC3 (Atg8 orthologs in humans) and pathogen killing (22).
Moreover, T. gondii might also be killed by autophagy in mice macrophages independently of IFNγ, in a mechanism involving CD40, member of TNF receptor superfamily, and activation of ULK1, calcium/calmodulin-dependent kinase kinase b (CaMKKβ), AMP-activated kinase, and Jun-kinase (JKN are involved) (25).
Toxoplasma gondii modulates several signal transduction pathways once inside host cells. At the same time that an effective immune response is generated, intracellular survival strategies are adopted by the parasite. The equilibrium host-T. gondii is in the best interest of both allowing the establishment of long-lasting latent infection, increasing the chances of transmission to new hosts. Several T. gondii effector molecules have been identified that directly interact with signal transducer and activator of transcription (STAT) pathways, which influence the transcription of both pro and anti-inflammatory molecules such as IFN-γ and major histocompatibility complex class II (MHCII) (STAT1); IL-10 (STAT3); and IL-4 (STAT6) (41–44).
Rhoptry 16 kinase (ROP16) is secreted into the host cytosol during invasion and phosphorylates STAT6 in a rapid and sustained way (45). Phosphorylation of STAT6 by ROP16 mediates induction of arginase-1, resulting in arginine degradation, depriving the parasite from one important metabolite (42). Infection of mice with ROP16 knockout parasites shows that STAT3 is also phosphorylated by this kinase, suppressing TLRs and inhibiting pro-inflammatory responses at some level (42, 46, 47). ROP16 encoded by the type I/III strains, but not type II strains, maintains STAT3/6 activation for 24 h and suppresses IL-12 production from macrophages (45, 47) (Figure 1B). A recent report by Jensen et al. showed that ROP16, type II strain, induced the sustained phosphorylation and nuclear translocation of STAT5 in host infected cells, contributing to generation of protective immunity in murine gut mucosal system (48).
Toxoplasma gondii has also been shown to interfere with STAT1 signaling, resulting in blockage of interferon regulatory factor 1 (Irf1), p65 GBPs, inducible nitric oxide synthase (iNOS/Nos2), indoleamine 2,3-dioxygenase 1, and MHC (49–55) (Figure 1). Schneider et al. (56) showed that STAT1 is activated during infection of bone marrow-derived murine dendritic cells (BMDCs) through tyrosine 70 (Tyr70) and serine 727 (Ser727) phosphorylation with effective nuclear translocation in a ROP16 independent way. All clonal strains tested (Type I—RH, Type II—PTG, and Type III—M774.1) showed similar results, with a less effective and sustained phosphorylation induced by M774.1 (56). Besides its nuclear translocation, tyrosine-phosphorylated STAT-1 (pYSTAT1) was unable to bind to the Irf1 gene promoter and chromatin immunoprecipitation assays showed the presence of aberrant STAT1 complexes, as earlier described by Lang et al. (57) (Figure 1B).
One of the mechanisms of IFN-γ blockage is the dephosphorylation of STAT1 by SOCS1 (suppressor of cytokine signaling phosphatase) induced by positive feedback. T. gondii infection has been shown to induce both downregulation of SOCS1 in human fibroblasts (58) and upregulation in murine macrophages (48, 55). Infection of mice with target deletion of SOCS3 in neutrophils and macrophages results in death, as this molecule is upregulated during infection. Furthermore, the administration of anti-IL6 and IL-12 restored mice resistance to the infection (59).
Toxoplasma gondii induces the expression of SOCS2 in DCs through lipoxin A4 (LXA4), an arachidonic acid (Ah) with anti-inflammatory action that stimulates Ah and LXA receptors of the host cell, resulting in decreased expression of chemokine receptor type 5 (CCR5) and IL-12 secretion (60). However, enhancing transcriptions factors can also be an evasion strategy. Dense granule proteins 6 (GRA6) interferes with nuclear factor of activated T cells 4 (NFAT4), activating it via calcium-modulating ligand, which might lead to increased migration of inflammatory macrophages (61).
Release of dense granule protein GRA15 by type II strains, but not the type I/III strains, into the host cell cytoplasm mediates nuclear factor-κappaB (NF-κB) activation and initiates IL-12 synthesis (48, 62). On the other hand, type I strains inhibit NF-kB pathway through ROP18 and suppresses pro-inflammatory cytokine expression, resulting in the enhanced survival of the parasites in the hosts (63) (Figure 1B).
Inflammatory macrophages are able to contain dissemination of infection through microbicide molecules, such as nitric oxide (NO). However, T. gondii infection of murine blood monocytes and peritoneal macrophages activated in vitro with IFN-γ and lipopolysaccharide leads to a substantial decrease in NO production (51, 52, 54). Interestingly, pretreating T. gondii with annexin V, which binds to phosphatidylserine (PS) reverts NO inhibition (64). The authors demonstrated that infection induces TGFβ1 through Smad 2 and 3 leading to destruction of iNOS, actin filament (F-actin) depolymerization, and lack of NF-κB in the nucleus (64). Recently, the same group showed that degradation of iNOS is proteasome dependent (65). iNOS reduced expression is also observed in microglia infected with T. gondii, also involving TGFβ pathways, protecting neurons from death (54). PS positive (PS+) but not PS negative (PS−) subpopulations of T. gondii were capable of NO inhibition after infection of murine macrophages in vitro, and infection in vivo with PS+ subpopulations leads to high parasite burden and low inflammatory symptoms at peritoneal cavity, while low or absent infection observed when PS− parasites were used with active inflammatory response observed (66). Thus, PS expression at T. gondii cell surface seems to be an interesting regulator of exacerbated inflammation at the entry site (Figure 1C).
Toxoplasma gondii-infected cells are resistant to a series of apoptosis inducers (67), allowing intracellular survival and persistence within the host cells (68). In a recent study, He et al. (69) suggested that T. gondii (Type I—RH) targets transregulation factors in mouse spleen cells modulating host gene expression. The genes involved in apoptosis or anti-apoptosis were both targeted by differentially expressed miRNAs, which contributes to the fate of host apoptosis process (69). The same group revealed the T. gondii infection can alter the transcripts at mitochondria level that are involved in several biosynthetic and metabolic processes and also in apoptosis (70) (Figure 1D).
The initiation of the apoptotic cascades is disturbed by T. gondii at several key points. Blocking of mitochondrial cytochrome c release is one of the mechanisms affected (71–73). The balance of pro- and anti-apoptotic Bcl-2 proteins (71, 72, 74–76) and direct inhibition of cytochrome c-mediated activation of the caspase cascade (73) were also reported. Inhibition of caspase 8, blocking of Fas/CD95-mediated apoptosis (77–79), inactivation of caspase 3 and PARP (80), as well as abrogation of Granzyme B activity in infected cells (81) are also important in maintaining the host cell alive, in favor of parasite’s survival.
Inhibition of apoptosis is regulated also at the transcriptional level. Infection of mouse splenocytes induces activation of host’s NF-κB and the transcription of antiapoptotic genes (80). After cell invasion, cells increase levels of active serine threonine kinase/protein kinase B (Akt/PKB), exploiting PI3K in a Gi-dependent way to delay host cell apoptosis (82). Furthermore, T. gondii phosphorylates the pro-apoptotic Bad protein to prevent apoptosis (83). These findings suggest that during the early stages of infection T. gondii is able to evade induction of apoptosis remaining inside the cell allowing the spreading of infection. However, there are reports indicating that ROP18 from virulent T. gondii strains induces apoptosis of neurons via RE stress (84). On the other hand, the gp130 expressed by neuronal cells protects them through IL-6, TGFβ, and IL-27 (85).
Signal transducer and activator of transcription molecules are also exploited by T. gondii to prevent the apoptosis. Serine proteases, like SERPIN B3 and B4, are significantly expressed in macrophages infected by T. gondii via STAT6 activation. Extended parasite intracellular survival in THP-1 is gain through those enzymes that ultimately inhibit apoptosis (86). In a recent work, Cai et al. demonstrated that STAT3 mediates pro-survival by upregulating the miR-17–92 that in turn targets Bim, inhibiting apoptosis in infected macrophages (87).
Inflammatory cells attracted to the primary site of infection are targets of parasites that highjack the cell in order to circulate through the body inside the host cell, in a mechanism similar to a Trojan horse, delivering the parasite to deep tissues and immune privileged sites, such as central nervous system and eyes (88).
Our group showed that host extracellular matrix metalloproteases (MMPs) might be involved in infected macrophage dissemination (89, 90). Murine macrophages infected in vitro with T. gondii exhibit increased membrane type-1 matrix metalloproteinase (MT1-MMP) and disintegrin and metalloproteinase domain-containing protein 10 (ADAM10) while decreased levels of CD44 are observed at cell surface. On the other hand, augmented active MMP-9 is present at cell supernatant (89) resembling metastasis mechanisms used by invasive tumors (89). Upregulation of MMP-9 and -2 via an Erk1/2/NF-κB pathway was also observed in murine mast cells infected with T. gondii (91) and human macrophages infected with T. gondii showed increased levels of MT1-MMP, with decrease in pro-MMP-2 and pro-MMP-9, maintain the migratory capacity, although decrease some the costimulatory molecules (89).
Regulation of hosts’ MMPs processing involves extra- and intracellular mechanisms upon T. gondii infection. Urokinase-type PA/urokinase-type PA receptor (uPA/uPAR) pathway is known to be involved with MMPs processing at extracellular space and is regulated by endogenous inhibitor of plasminogen activator inhibitor (PAI-1) and protease nexin-1 (PN-1). We demonstrated that T. gondii-infected macrophages secrete a multiprotein complex containing MMP-9/TIMP1/uPAR, and incubation of infected cells with PAI-1 decreases the presence of this complex at cell supernatant (90) (Figure 2).
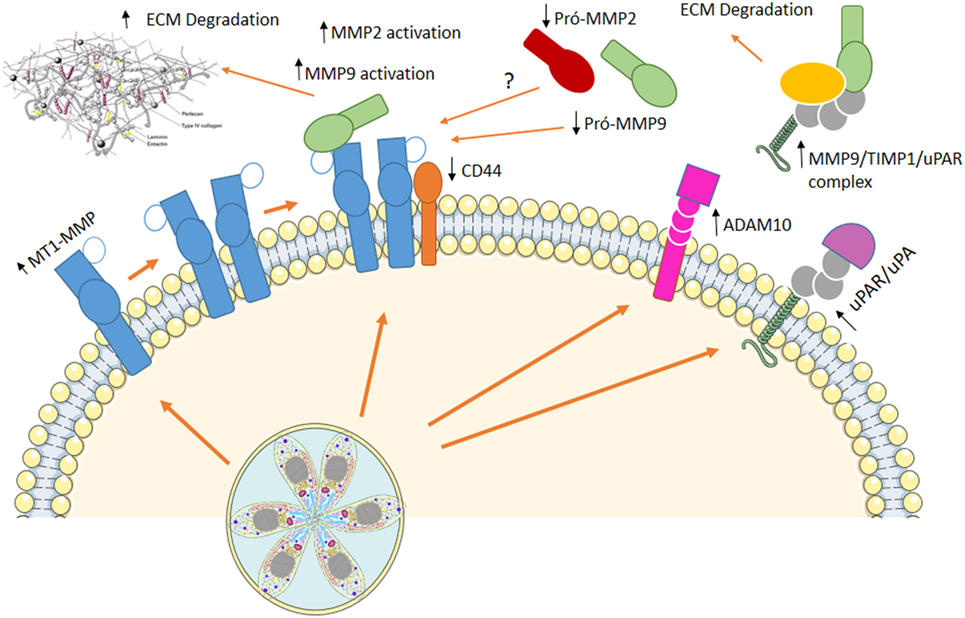
Figure 2. Using cells as Trojan horses. Host extracellular matrix metalloproteases (MMPs) are involved in infected macrophage dissemination. In vitro infection of murine macrophages induced an increase in membrane type-1 matrix metalloproteinase (MT1-MMP) and disintegrin and metalloproteinase domain-containing protein 10 (ADAM10), while decreased levels of CD44 are observed at cell surface. On the other hand, augmented active MMP-9, MMP-2, and a multiprotein complex containing MMP-9/TIMP1/urokinase-type PA receptor (uPAR) are present at cell supernatant. This mechanism resembling metastasis allows Toxoplasma gondii to disseminate throughout the host, reaching immune-privileged sites, where it remains in low proliferative state, with little damage to the host.
Toxoplasma gondii proteinases were identified (92) and might be involved in intracellular processing of MMPs zymogens, suggesting that hosts and T. gondii MMPs would work in favor of parasites’ dissemination to secondary organs and to immune privileged sites. Thus, dissemination through lymphatics and leukocytes could be the main form of dissemination, and cumulative information in this subject have been gathered (88, 93).
After oral infection, T. gondii is found in the blood inside CD11b+ monocytes and inside mouse CD11c+ DCs at lamina propria Peyer’s patches and mesenteric lymph nodes. Infected CD11b+ monocytes are observed at the extravascular space in the mouse brain after 7 days of infection (94). In human infected astroglia cells, the increase of MMP-2 and MMP-9 could promote leukocyte migration during toxoplasmic encephalitis (95). MMP-2 and -9 are higher in the sera and umbilical cord of pregnant women with T. gondii infection (96), suggesting that MMPs might be involved in the crossing of T. gondii through the placental barrier. Oral infection with T. gondii provokes small intestine inflammation as a result of Th1 responses, that depending of mice strain and/or parasite genotype is rapidly contained (3). Muñoz et al. (97) demonstrated that MMP-2 is involved in the development of T. gondii-induced immunopathology. The same paper shows that this gelatinase is regulated by IL-23 via IL-22 but independent of IL-17 (97).
T cells from CD4+ and CD8+ lineages are essential to control bradyzoites containing cysts at the brain, T cells expressing MMP-10 are present at the brain after 21 days of infection, while T cells expressing MMP-8 are observed at 28th day of infection. At this time, astrocytes express TIMP-1, probably in an attempt to control damage (98). The presence of TIMP-1 at the brain during chronicle stages of toxoplasmosis could control degradation/activation of cytokines, decreasing exacerbated inflammatory response at the brain.
Several effector molecules and mechanisms were presented here, which allow T. gondii to leave inside host, with little destructive effects to immunocompetent individuals. Both parasite and host have developed several strategies to decrease collateral damaging immediately after infection such as interfering with cell-autonomous immunity and cell signaling and also blocking apoptosis allowing infected host cell to remain alive. Also, controlling dissemination of parasites through metastatic-simile mechanisms, using host cell MMPs and migration, allows parasite to spread to immune-privileged sites, where it remains in low proliferative state, with little damage to the host. In order to successfully reach this semiequilibrium state between parasite–host, the initial events occurring at the parasite entrance site are crucial. Damaging control of ileitis by regulating levels of IFNγ, IL-23, and IL-17 and maintaining the fine tuning of MMPs and other enzymes and pro-zymogen enhancers, inducers, and/or converters are fundamental.
All author contributed equally for the manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank the technical support of Juliana de Azevedo Silva and Rita Mothé Escocard.
This work was supported by grants from CNPq 309481/2010-4 and 472133/2010; FAPERJ E-26 111.522/2010 and E-26 110.562/2010.
1. Harker KS, Ueno N, Lodoen MB. Toxoplasma gondii dissemination: a parasite’s journey through the infected host. Parasite Immunol (2015) 37(3):141–9. doi:10.1111/pim.12163
2. Denkers EY. From cells to signaling cascades: manipulation of innate immunity by Toxoplasma gondii. FEMS Immunol Med Microbiol (2003) 39(3):193–203. doi:10.1016/S0928-8244(03)00279-7
3. Hunter CA, Sibley LD. Modulation of innate immunity by Toxoplasma gondii virulence effectors. Nat Rev Microbiol (2012) 10:766–78. doi:10.1038/nrmicro2858
4. Pollard AM, Knoll LJ, Mordue DG. The role of specific Toxoplasma gondii molecules in manipulation of innate immunity. Trends Parasitol (2009) 25(11):491–4. doi:10.1016/j.pt.2009.07.009
5. Blader I, Manger ID, Boothroyd JC. Microarray analysis reveals previously unknown changes in Toxoplasma gondii infected human cells. J Biol Chem (2001) 276:24223–31. doi:10.1074/jbc.M100951200
6. Stutz A, Kessler H, Kaschel ME, Meissner M, Dalpke AH. Cell invasion and strain dependent induction of suppressor of cytokine signaling-1 by Toxoplasma gondii. Immunobiology (2012) 217:28–36. doi:10.1016/j.imbio.2011.08.008
7. Zhou CX, Elsheikha HM, Zhou DH, Liu Q, Zhu XQ, Suo X. Dual identification and analysis of differentially expressed transcripts of porcine PK-15 cells and Toxoplasma gondii during in vitro infection. Front Microbiol (2016) 13(7):721. doi:10.3389/fmicb.2016.00721
8. Gazzinelli RT, Mendonça-Neto R, Lilue J, Howard J, Sher A. Innate resistance against Toxoplasma gondii: an evolutionary tale of mice, cats and men. Cell Host Microbes (2014) 15(2):132–8. doi:10.1016/j.chom.2014.01.004
9. Morger J, Bajnok J, Boyce K, Craig PS, Rogan MT, Lun ZR, et al. Naturally occurring Toll-like receptor 11 (TLR11) and Toll-like receptor 12 (TLR12) polymorphisms are not associated with Toxoplasma gondii infection in wild wood mice. Infect Genet Evol (2014) 26:180–4. doi:10.1016/j.meegid.2014.05.032
10. Shen B, Sibley LD. The moving junction, a key portal to host cell invasion by apicomplexan parasites. Curr Opin Microbiol (2012) 15(4):449–55. doi:10.1016/j.mib.2012.02.007
11. Mordue DG, Desai N, Dustin M, Sibley LD. Invasion by Toxoplasma gondii establishes a moving junction that selectively excludes host cell plasma membrane proteins on the basis of their membrane anchoring. J Exp Med (1999) 190(12):1783–92. doi:10.1084/jem.190.12.1783
12. Charron AJ, Sibley LD. Molecular partitioning during host cell penetration by Toxoplasma gondii. Traffic (2004) 5(11):855–67. doi:10.1111/j.1600-0854.2004.00228.x
13. Mordue DG, Håkansson S, Niesman I, Sibley LD. Toxoplasma gondii resides in a vacuole that avoids fusion with host cell endocytic and exocytic vesicular trafficking pathways. Exp Parasitol (1999) 92(2):87–99. doi:10.1006/expr.1999.4412
14. Besteiro S. Which roles for autophagy in Toxoplasma gondii and related apicomplexan parasites? Mol Biochem Parasitol (2012) 184(1):1–8. doi:10.1016/j.molbiopara.2012.04.001
15. Strzyz P. Autophagy: membrane contacts lend a hand. Nat Rev Mol Cell Biol (2017) 18(7):404–5. doi:10.1038/nrm.2017.55
16. Latré de Laté P, Pineda M, Harnett M, Harnett W, Besteiro S, Langsley G. Apicomplexan autophagy and modulation of autophagy in parasite-infected host cells. Biomed J (2017) 40(1):23–30. doi:10.1016/j.bj.2017.01.001
17. Deretic V. Autophagy in immunity and cell-autonomous defense against intracellular microbes. Immunol Rev (2011) 240(1):92–104. doi:10.1111/j.1600-065X.2010.00995.x
18. Abdelaziz DH, Khalil H, Cormet-Boyaka E, Amer AO. The cooperation between the autophagy machinery and the inflammasome to implement an appropriate innate immune response: do they regulate each other? Immunol Rev (2015) 265(1):194–204. doi:10.1111/imr.12288
19. Andrade RM, Wessendarp M, Gubbels MJ, Striepen B, Subauste CS. CD40 induces macrophage anti-Toxoplasma gondii activity by triggering autophagy-dependent fusion of pathogen-containing vacuoles and lysosomes. J Clin Invest (2006) 116(9):2366–77. doi:10.1172/JCI28796
20. Portillo JA, Okenka G, Reed E, Subauste A, Van Grol J, Gentil K, et al. The CD40-autophagy pathway is needed for host protection despite IFN-Γ-dependent immunity and CD40 induces autophagy via control of P21 levels. PLoS One (2010) 5(12):e14472. doi:10.1371/journal.pone.0014472
21. Lee YJ, Song HO, Lee YH, Ryu JS, Ahn MH. Proliferation of Toxoplasma gondii suppresses host cell autophagy. Korean J Parasitol (2013) 51(3):279–87. doi:10.3347/kjp.2013.51.3.279
22. Muniz-Feliciano L, Van Grol J, Portillo JA, Liew L, Liu B, Carlin CR, et al. Toxoplasma gondii-induced activation of EGFR prevents autophagy protein-mediated killing of the parasite. PLoS Pathog (2013) 9(12):e1003809. doi:10.1371/journal.ppat.1003809
23. Souto XM, Barbosa HS, Menna-Barreto RF. The morphological analysis of autophagy in primary skeletal muscle cells infected with Toxoplasma gondii. Parasitol Res (2016) 115(7):2853–61. doi:10.1007/s00436-016-5040-3
24. Kim BH, Shenoy AR, Kumar P, Bradfield CJ, MacMicking JD. IFN-inducible GTPases in host cell defense. Cell Host Microbe (2012) 12(4):432–44. doi:10.1016/j.chom.2012.09.007
25. Liu E, Lopez Corcino Y, Portillo JA, Miao Y, Subauste CS. Identification of signaling pathways by which CD40 stimulates autophagy and antimicrobial activity against Toxoplasma gondii in macrophages. Infect Immun (2016) 84(9):2616–26. doi:10.1128/IAI.00101-16
26. Ohshima J, Lee Y, Sasai M, Saitoh T, Su Ma JJ, Kamiyama N, et al. Role of mouse and human autophagy proteins in IFN-gamma induced cell-autonomous responses against Toxoplasma gondii. J Immunol (2014) 192:3328–35. doi:10.4049/jimmunol.1302822
27. Khaminets A, Hunn JP, Könen-Waisman S, Zhao YO, Preukschat D, Coers J, et al. Coordinated loading of IRG resistance GTPases on to the Toxoplasma gondii parasitophorous vacuole. Cell Microbiol (2010) 12(7):939–61. doi:10.1111/j.1462-5822.2010.01443.x
28. Hunn JP, Feng CG, Sher A, Howard JC. The immunity-related GTPases in mammals: a fast-evolving cell-autonomous resistance system against intracellular pathogens. Mamm Genome (2011) 22(1–2):43–54. doi:10.1007/s00335-010-9293-3
29. Zhao Z, Fux B, Goodwin M, Dunay IR, Strong D, Miller BC, et al. Autophagosome-independent essential function for the autophagy protein Atg5 in cellular immunity to intracellular pathogens. Cell Host Microbe (2008) 4:458–69. doi:10.1016/j.chom.2008.10.003
30. Selleck EM, Fentress SJ, Beatty WL, Degrandi D, Pfeffer K, Virgin HW, et al. Guanylate-binding protein 1 (Gbp1) contributes to cell-autonomous immunity against Toxoplasma gondii. PLoS Pathog (2013) 9:e1003320. doi:10.1371/journal.ppat.1003320
31. Park S, Choi J, Biering SB, Dominici E, Williams LE, Hwang S. Targeting by autophagy proteins (TAG): targeting of IFNG-inducible GTPases to membranes by the LC3 conjugation system of autophagy. Autophagy (2016) 12(7):1153–67. doi:10.1080/15548627.2016.1178447
32. Choi J, Park S, Biering SB, Selleck E, Liu CY, Zhang X, et al. The parasitophorous vacuole membrane of Toxoplasma gondii is targeted for disruption by ubiquitin-like conjugation systems of autophagy. Immunity (2014) 40(6):924–35. doi:10.1016/j.immuni.2014.05.006
33. Haldar AK, Piro AS, Pilla DM, Yamamoto M, Coers J. The E2-like conjugation enzyme Atg3 promotes binding of IRG and Gbp proteins to chlamydia- and Toxoplasma-containing vacuoles and host resistance. PLoS One (2014) 9(1):e86684. doi:10.1371/journal.pone.0086684
34. Steinfeldt T, Könen-Waisman S, Tong L, Pawlowski N, Lamkemeyer T, Sibley LD, et al. Correction: phosphorylation of mouse immunity-related GTPase (IRG) resistance proteins is an evasion strategy for virulent Toxoplasma gondii. PLoS Biol (2015) 13(7):e1002199. doi:10.1371/journal.pbio.1002199
35. Reese ML, Shah N, Boothroyd JC. The Toxoplasma pseudokinase ROP5 is an allosteric inhibitor of the immunity-related GTPases. J Biol Chem (2014) 289(40):27849–58. doi:10.1074/jbc.M114.567057
36. Alaganan A, Fentress SJ, Tang K, Wang Q, Sibley LD. Toxoplasma GRA7 effector increases turnover of immunity-related GTPases and contributes to acute virulence in the mouse. Proc Natl Acad Sci U S A (2014) 111(3):1126–31. doi:10.1073/pnas.1313501111
37. Qin A, Lai DH, Liu Q, Huang W, Wu YP, Chen X, et al. Guanylate-binding protein 1 (GBP1) contributes to the immunity of human mesenchymal stromal cells against Toxoplasma gondii. Proc Natl Acad Sci U S A (2017) 114(6):1365–70. doi:10.1073/pnas.1619665114
38. Johnston AC, Piro A, Clough B, Siew M, Virreira Winter S, Coers J, et al. Human GBP1 does not localize to pathogen vacuoles but restricts Toxoplasma gondii. Cell Microbiol (2016) 18(8):1056–64. doi:10.1111/cmi.12579
39. Etheridge RD, Alaganan A, Tang K, Lou HJ, Turk BE, Sibley LD. The Toxoplasma pseudokinase ROP5 forms complexes with ROP18 and ROP17 kinases that synergize to control acute virulence in mice. Cell Host Microbe (2014) 15:537–50. doi:10.1016/j.chom.2014.04.00
40. Hermanns T, Müller UB, Könen-Waisman S, Howard JC, Steinfeldt T. The Toxoplasma gondii rhoptry protein ROP18 is an Irga6-specific kinase and regulated by the dense granule protein GRA7. Cell Microbiol (2016) 18(2):244–59. doi:10.1111/cmi.12499
41. Lüder CG, Walter W, Beuerle B, Maeurer MJ, Gross U. Toxoplasma gondii down-regulates MHC class II gene expression and antigen presentation by murine macrophages via interference with nuclear translocation of STAT1alpha. Eur J Immunol (2001) 31(5):1475–84. doi:10.1002/1521-4141(200105)31:5<1475::AID-IMMU1475>3.0.CO;2-C
42. Butcher BA, Kim L, Panopoulos AD, Watowich SS, Murray PJ, Denkers EY. IL-10-independent STAT3 activation by Toxoplasma gondii mediates suppression of IL-12 and TNF-alpha in host macrophages. J Immunol (2005) 174(6):3148–52. doi:10.4049/jimmunol.174.6.3148
43. Ahn HJ, Kim JY, Ryu KJ, Nam HW. STAT6 activation by Toxoplasma gondii infection induces the expression of Th2 C-C chemokine ligands and B clade serine protease inhibitors in macrophage. Parasitol Res (2009) 105(5):1445–53. doi:10.1007/s00436-009-1577-8
44. Barbosa BF, Lopes-Maria JB, Gomes AO, Angeloni MB, Castro AS, Franco PS, et al. IL10, TGF beta1, and IFN gamma modulate intracellular signaling pathways and cytokine production to control Toxoplasma gondii infection in BeWo trophoblast cells. Biol Reprod (2015) 92(3):82. doi:10.1095/biolreprod.114.124115
45. Ong YC, Reese ML, Boothroyd JC. Toxoplasma rhoptry protein 16 (ROP16) subverts host function by direct tyrosine phosphorylation of STAT6. J Biol Chem (2010) 285(37):28731–40. doi:10.1074/jbc.M110.112359
46. Saeij JP, Coller S, Boyle JP, Jerome ME, White MW, Boothroyd JC. Toxoplasma co-opts host gene expression by injection of a polymorphic kinase homologue. Nature (2007) 445(7125):324–7. doi:10.1038/nature05395
47. Yamamoto M, Standley DM, Takashima S, Saiga H, Okuyama M, Kayama H, et al. A single polymorphic amino acid on Toxoplasma gondii kinase ROP16 determines the direct and strain-specific activation of Stat3. J Exp Med (2009) 206(12):2747–60. doi:10.1084/jem.20091703
48. Jensen KD, Hu K, Whitmarsh RJ, Hassan MA, Julien L, Lu D, et al. Toxoplasma gondii rhoptry 16 kinase promotes host resistance to oral infection and intestinal inflammation only in the context of the dense granule protein GRA15. Infect Immun (2013) 81(6):2156–67. doi:10.1128/IAI.01185-12
49. Luder CG, Lang T, Beuerle B, Gross U. Down-regulation of MHC class II molecules and inability to up-regulate class I molecules in murine macrophages after infection with Toxoplasma gondii. Clin Exp Immunol (1998) 112:308–16. doi:10.1046/j.1365-2249.1998.00594.x
50. Lüder CGK, Gross U, Lopes MF. Intracellular protozoan parasites and apoptosis: diverse strategies to modulate parasite-host interactions. Trends Parasitol (2001) 17:480–6. doi:10.1016/S1471-4922(01)02016-5
51. Seabra SH, de Souza W, DaMatta RA. Toxoplasma gondii partially inhibits nitric oxide production of activated murine macrophages. Exp Parasitol (2002) 100(1):62–70. doi:10.1006/expr.2001.4675
52. Lüder CG, Algner M, Lang C, Bleicher N, Gross U. Reduced expression of the inducible nitric oxide synthase after infection with Toxoplasma gondii facilitates parasite replication in activated murine macrophages. Int J Parasitol (2003) 33(8):833–44. doi:10.1016/S0020-7519(03)00092-4
53. Lüder CG, Lang C, Giraldo-Velasquez M, Algner M, Gerdes J, Gross U. Toxoplasma gondii inhibits MHC class II expression in neural antigen-presenting cells by down-regulating the class II transactivator CIITA. J Neuroimmunol (2003) 134(1–2):12–24. doi:10.1016/S0165-5728(02)00320-X
54. Rozenfeld C, Martinez R, Seabra S, Sant’anna C, Gonçalves JG, Bozza M, et al. Toxoplasma gondii prevents neuron degeneration by interferon-gamma-activated microglia in a mechanism involving inhibition of inducible nitric oxide synthase and transforming growth factor-beta1 production by infected microglia. Am J Pathol (2005) 167(4):1021–31. doi:10.1016/S0002-9440(10)61191-1
55. Zimmermann S, Murray PJ, Heeg K, Dalpke AH. Induction of suppressor of cytokine signaling-1 by Toxoplasma gondii contributes to immune evasion in macrophages by blocking IFN-gamma signaling. J Immunol (2006) 176(3):1840–7. doi:10.4049/jimmunol.176.3.1840
56. Schneider AG, Abi Abdallah DS, Butcher BA, Denkers EY. Toxoplasma gondii triggers phosphorylation and nuclear translocation of dendritic cell STAT1 while simultaneously blocking IFNγ-induced STAT1 transcriptional activity. PLoS One (2013) 8(3):e60215. doi:10.1371/journal.pone.0060215
57. Lang C, Hildebrandt A, Brand F, Opitz L, Dihazi H, Lüder CG. Impaired chromatin remodelling at STAT1-regulated promoters leads to global unresponsiveness of Toxoplasma gondii-infected macrophages to IFN-γ. PLoS Pathog (2012) 8(1):e1002483. doi:10.1371/journal.ppat.1002483
58. Kim SK, Fouts AE, Boothroyd JC. Toxoplasma gondii dysregulates IFN-gamma-inducible gene expression in human fibroblasts: insights from a genome-wide transcriptional profiling. J Immunol (2007) 178(8):5154–65. doi:10.4049/jimmunol.178.8.5154
59. Whitmarsh RJ, Gray CM, Gregg B, Christian DA, May MJ, Murray PJ, et al. A critical role for SOCS3 in innate resistance to Toxoplasma gondii. Cell Host Microbe (2011) 10(3):224–36. doi:10.1016/j.chom.2011.07.009
60. Machado FS, Johndrow JE, Esper L, Dias A, Bafica A, Serhan CN, et al. Anti-inflammatory actions of lipoxin A4 and aspirin-triggered lipoxin are SOCS-2 dependent. Nat Med (2006) 12:330–4. doi:10.1038/nm1355
61. Ma JS, Sasai M, Ohshima J, Lee Y, Bando H, Takeda K, et al. Selective and strain-specific NFAT4 activation by the Toxoplasma gondii polymorphic dense granule protein GRA6. J Exp Med (2014) 211(10):2013–32. doi:10.1084/jem.20131272
62. Rosowski EE, Lu D, Julien L, Rodda L, Gaiser RA, Jensen KD, et al. Strain-specific activation of the NF-B pathway by GRA15, a novel Toxoplasma gondii dense granule protein. J Exp Med (2011) 208:195–212. doi:10.1084/jem.20100717
63. Du J, An R, Chen L, Shen Y, Chen Y, Cheng L, et al. Toxoplasma gondii virulence factor ROP18 inhibits the host NF-B pathway by promoting p65 degradation. J Biol Chem (2014) 289(18):12578–92. doi:10.1074/jbc.M113.544718
64. Seabra SH, de Souza W, Damatta RA. Toxoplasma gondii exposes phosphatidylserine inducing a TGF-beta1 autocrine effect orchestrating macrophage evasion. Biochem Biophys Res Commun (2004) 324(2):744–52. doi:10.1016/j.bbrc.2004.09.114
65. Padrão Jda C, Cabral GR, da Silva Mde F, Seabra SH, DaMatta RA. Toxoplasma gondii infection of activated J774-A1 macrophages causes inducible nitric oxide synthase degradation by the proteasome pathway. Parasitol Int (2014) 63(5):659–63. doi:10.1016/j.parint.2014.05.003
66. Santos TA, Portes Jde A, Damasceno-Sá JC, Caldas LA, Souza WD, Damatta RA, et al. Phosphatidylserine exposure by Toxoplasma gondii is fundamental to balance the immune response granting survival of the parasite and of the host. PLoS One (2011) 6(11):e27867. doi:10.1371/journal.pone.0027867
67. Nash PB, Purner MB, Leon RP, Clarke P, Duke RC, Curiel TJ. Toxoplasma gondii-infected cells are resistant to multiple inducers of apoptosis. J Immunol (1998) 160(4):1824–30.
68. Sinai AP, Payne TM, Carmen JC, Hardi L, Watson SJ, Molestina RE. Mechanisms underlying the manipulation of host apoptotic pathways by Toxoplasma gondii. Int J Parasitol (2004) 34(3):381–91. doi:10.1016/j.ijpara.2003.11.009
69. He JJ, Ma J, Li FC, Song HQ, Xu MJ, Zhu XQ. Transcriptional changes of mouse splenocyte organelle components following acute infection with Toxoplasma gondii. Exp Parasitol (2016) 167:7–16. doi:10.1016/j.exppara.2016.04.019
70. He JJ, Ma J, Wang JL, Xu MJ, Zhu XQ. Analysis of miRNA expression profiling in mouse spleen affected by acute Toxoplasma gondii infection. Infect Genet Evol (2016) 37:137–42. doi:10.1016/j.meegid.2015.11.005
71. Goebel S, Gross U, Lüder CG. Inhibition of host cell apoptosis by Toxoplasma gondii is accompanied by reduced activation of the caspase cascade and alterations of poly(ADP-ribose) polymerase expression. J Cell Sci (2001) 114(Pt 19):3495–505.
72. Carmen JC, Sinai AP. The differential effect of Toxoplasma gondii infection on the stability of BCL2-family members involves multiple activities. Front Microbiol (2011) 2:1. doi:10.3389/fmicb.2011.00001
73. Hippe D, Gais A, Gross U, Lüder CG. Modulation of caspase activation by Toxoplasma gondii. Methods Mol Biol (2009) 470:275–88. doi:10.1007/978-1-59745-204-5_19
74. Molestina RE, Payne TM, Coppens I, Sinai AP. Activation of NF-kappaB by Toxoplasma gondii correlates with increased expression of antiapoptotic genes and localization of phosphorylated IkappaB to the parasitophorous vacuole membrane. J Cell Sci (2003) 116(Pt 21):4359–71. doi:10.1242/jcs.00683
75. Hwang IY, Quan JH, Ahn MH, Ahmed HA, Cha GH, Shin DW, et al. Toxoplasma gondii infection inhibits the mitochondrial apoptosis through induction of Bcl-2 and HSP70. Parasitol Res (2010) 107(6):1313–21. doi:10.1007/s00436-010-1999-3
76. Choi SH, Park SJ, Cha GH, Quan JH, Chang NS, Ahn MH, et al. Toxoplasma gondii protects against H(2)O(2)-induced apoptosis in ARPE-19 cells through the transcriptional regulation of apoptotic elements and downregulation of the p38 MAPK pathway. Acta Ophthalmol (2011) 89(4):e350–6. doi:10.1111/j.1755-3768.2011.02113.x
77. Keller P, Schaumburg F, Fischer SF, Häcker G, Gross U, Lüder CG. Direct inhibition of cytochrome c-induced caspase activation in vitro by Toxoplasma gondii reveals novel mechanisms of interference with host cell apoptosis. FEMS Microbiol Lett (2006) 258(2):312–9. doi:10.1111/j.1574-6968.2006.00241.x
78. Vutova P, Wirth M, Hippe D, Gross U, Schulze-Osthoff K, Schmitz I, et al. Toxoplasma gondii inhibits Fas/CD95-triggered cell death by inducing aberrant processing and degradation of caspase 8. Cell Microbiol (2007) 6:1556e70. doi:10.1111/j.1462-5822.2007.00893.x
79. Hippe D, Lytovchenko O, Schmitz I, Lüder CG. Fas/CD95-mediated apoptosis of type II cells is blocked by Toxoplasma gondii primarily via interference with the mitochondrial amplification loop. Infect Immun (2008) 76(7):2905–12. doi:10.1128/IAI.01546-07
80. Kim JY, Ahn MH, Jun HS, Jung JW, Ryu JS, Min DY. Toxoplasma gondii inhibits apoptosis in infected cells by caspase inactivation and NF-kappaB activation. Yonsei Med J (2006) 47(6):862–9. doi:10.3349/ymj.2006.47.6.862
81. Yamada T, Tomita T, Weiss LM, Orlofsky A. Toxoplasma gondii inhibits granzyme B-mediated apoptosis by the inhibition of granzyme B function in host cells. Int J Parasitol (2011) 41(6):595–607. doi:10.1016/j.ijpara.2010.11.012
82. Kim L, Denkers EY. Toxoplasma gondii triggers Gi-dependent PI 3-kinase signaling required for inhibition of host cell apoptosis. J Cell Sci (2006) 119(Pt 10):2119–26. doi:10.1242/jcs.02934
83. Quan JH, Cha GH, Zhou W, Chu JQ, Nishikawa Y, Lee YH. Involvement of PI 3 kinase/Akt-dependent Bad phosphorylation in Toxoplasma gondii-mediated inhibition of host cell apoptosis. Exp Parasitol (2013) 133(4):462–71. doi:10.1016/j.exppara.2013.01.005
84. Wan L, Gong L, Wang W, An R, Zheng M, Jiang Z, et al. T. gondii rhoptry protein ROP18 induces apoptosis of neural cells via endoplasmic reticulum stress pathway. Parasit Vectors (2015) 21(8):554. doi:10.1186/s13071-015-1103-z
85. Händel U, Brunn A, Drögemüller K, Müller W, Deckert M, Schlüter D. Neuronal gp130 expression is crucial to prevent neuronal loss, hyperinflammation, and lethal course of murine Toxoplasma encephalitis. Am J Pathol (2012) 181(1):163–73. doi:10.1016/j.ajpath.2012.03.029
86. Song KJ, Ahn HJ, Nam HW. Anti-apoptotic effects of SERPIN B3 and B4 via STAT6 activation in macrophages after infection with Toxoplasma gondii. Korean J Parasitol (2012) 50(1):1–6. doi:10.3347/kjp.2012.50.1.1
87. Cai Y, Chen H, Mo X, Tang Y, Xu X, Zhang A, et al. Toxoplasma gondii inhibits apoptosis via a novel STAT3-miR-17-92-Bim pathway in macrophages. Cell Signal (2014) 26(6):1204–12. doi:10.1016/j.cellsig.2014.02.013
88. Da Gama LM, Ribeiro-Gomes FL, Guimarães U Jr, Arnholdt AC. Reduction in adhesiveness to extracellular matrix components, modulation of adhesion molecules and in vivo migration of murine macrophages infected with Toxoplasma gondii. Microbes Infect (2004) 6(14):1287–96. doi:10.1016/j.micinf.2004.07.008
89. Seipel D, Oliveira BC, Resende TL, Schuindt SH, Pimentel PM, Kanashiro MM, et al. Toxoplasma gondii infection positively modulates the macrophages migratory molecular complex by increasing matrix metalloproteinases, CD44 and alpha v beta 3 integrin. Vet Parasitol (2010) 169(3–4):312–9. doi:10.1016/j.vetpar.2009.12.042
90. Schuindt SH, Oliveira BC, Pimentel PM, Resende TL, Retamal CA, DaMatta RA, et al. Secretion of multi-protein migratory complex induced by Toxoplasma gondii infection in macrophages involves the uPA/uPAR activation system. Vet Parasitol (2012) 186(3–4):207–15. doi:10.1016/j.vetpar.2011.11.035
91. Wang MF, Lu CY, Lai SC. Up-regulation of matrix metalloproteinases-2 and -9 via an Erk1/2/NF-κB pathway in murine mast cells infected with Toxoplasma gondii. J Comp Pathol (2013) 149(2–3):146–55. doi:10.1016/j.jcpa.2013.03.002
92. Song KJ, Nam HW. Protease activity of 80 kDa protein secreted from the apicomplexan parasite Toxoplasma gondii. Korean J Parasitol (2003) 41(3):165–9. doi:10.3347/kjp.2003.41.3.165
93. JUnno A, Suzuki K, Xuan X, Nishikawa Y, Kitoh K, Takashima Y. Dissemination of extracellular and intracellular Toxoplasma gondii tachyzoites in the blood flow. Parasitol Int (2008) 57(4):515–8. doi:10.1016/j.parint.2008.06.004
94. Lambert H, Barragan A. Modelling parasite dissemination: host cell subversion and immune evasion by Toxoplasma gondii. Cell Microbiol (2010) 12(3):292–300. doi:10.1111/j.1462-5822.2009.01417.x
95. Courret N, Darche S, Sonigo P, Milon G, Buzoni-Gâtel D, Tardieux I. CD11c- and CD11b-expressing mouse leukocytes transport single Toxoplasma gondii tachyzoites to the brain. Blood (2006) 107(1):309–16. doi:10.1182/blood-2005-02-0666
96. Wang MF, Lai SC. Fibronectin degradation by MMP-2/MMP-9 in the serum of pregnant women and umbilical cord with Toxoplasma gondii infection. J Obstet Gynaecol (2013) 33(4):370–4. doi:10.3109/01443615.2013.769501
97. Muñoz M, Heimesaat MM, Danker K, Struck D, Lohmann U, Plickert R, et al. Interleukin (IL)-23 mediates Toxoplasma gondii-induced immunopathology in the gut via matrixmetalloproteinase-2 and IL-22 but independent of IL-17. J Exp Med (2009) 206(13):3047–59. doi:10.1084/jem.20090900
Keywords: Toxoplasma gondii, T cells, immunomodulation, cell signalling, immunity
Citation: Brasil TR, Freire-de-Lima CG, Morrot A and Vetö Arnholdt AC (2017) Host-Toxoplasma gondii Coadaptation Leads to Fine Tuning of the Immune Response. Front. Immunol. 8:1080. doi: 10.3389/fimmu.2017.01080
Received: 21 April 2017; Accepted: 21 August 2017;
Published: 13 September 2017
Edited by:
Christoph Hölscher, Forschungszentrum Borstel (LG), GermanyReviewed by:
Catherine Ropert, Universidade Federal de Minas Gerais, BrazilCopyright: © 2017 Brasil, Freire-de-Lima, Morrot and Vetö Arnholdt. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Thaís Rigueti Brasil, dGhhaXNyaWd1ZXRpQGdtYWlsLmNvbQ==;
Andrea Cristina Vetö Arnholdt, YWN2YXJuaG9sZHRAZ21haWwuY29t
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.