
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Immunol. , 28 August 2017
Sec. Microbial Immunology
Volume 8 - 2017 | https://doi.org/10.3389/fimmu.2017.01037
 Eleonora Ciarlo1
Eleonora Ciarlo1 Tytti Heinonen1
Tytti Heinonen1 Charlotte Théroude1
Charlotte Théroude1 Jacobus Herderschee1Matteo Mombelli1
Jacobus Herderschee1Matteo Mombelli1 Jérôme Lugrin1
Jérôme Lugrin1 Marc Pfefferlé1Beatrice Tyrrell1Sarah Lensch1
Marc Pfefferlé1Beatrice Tyrrell1Sarah Lensch1 Hans Acha-Orbea2
Hans Acha-Orbea2 Didier Le Roy1
Didier Le Roy1 Johan Auwerx3
Johan Auwerx3 Thierry Roger1*
Thierry Roger1*
Sirtuin 2 (SIRT2) is one of the seven members of the family of NAD+-dependent histone deacetylases. Sirtuins target histones and non-histone proteins according to their subcellular localization, influencing various biological processes. SIRT2 resides mainly in the cytoplasm and regulates cytoskeleton dynamics, cell cycle, and metabolic pathways. As such, SIRT2 has been implicated in the pathogenesis of neurodegenerative, metabolic, oncologic, and chronic inflammatory disorders. This motivated the development of SIRT2-directed therapies for clinical purposes. However, the impact of SIRT2 on antimicrobial host defense is largely unknown. Here, we address this question using SIRT2 knockout mice. We show that SIRT2 is the most highly expressed sirtuin in myeloid cells, especially macrophages. SIRT2 deficiency does not affect immune cell development and marginally impacts on intracellular signaling and cytokine production by splenocytes and macrophages. However, SIRT2 deficiency enhances bacterial phagocytosis by macrophages. In line with these observations, in preclinical models, SIRT2 deficiency increases survival of mice with chronic staphylococcal infection, while having no effect on the course of toxic shock syndrome toxin-1, LPS or TNF-induced shock, fulminant Escherichia coli peritonitis, sub-lethal Klebsiella pneumoniae pneumonia, and chronic candidiasis. Altogether, these data support the safety profile of SIRT2 inhibitors under clinical development in terms of susceptibility to infections.
Innate immune cells are at the vanguard of host defense against microbial infections. Monocytes/macrophages and dendritic cells (DCs) sense microbial or danger-associated molecular patterns (MAMPs and DAMPs released by injured or stressed cells) through pattern recognition receptors (PRRs) such as toll-like receptors (TLRs), NOD-like receptors, C-type lectins, scavenger receptors, RIG-I-like receptors, and intra-cytosolic DNA sensors (1, 2). The interaction of MAMPs or DAMPs with PRRs triggers the mitogen-activated protein kinases (MAPKs), nuclear factor-κB (NF-κB), and interferon (IFN) response factor signaling pathways that coordinate immune gene expression. The cellular and soluble mediators mobilized upon infection tightly regulate the development of the inflammatory response, the establishment of antimicrobial cellular and humoral responses, and the restoration of homeostasis once pathogens have been contained or eradicated.
The superfamily of histone deacetylases (HDACs) comprises eleven Zn-dependent HDACs (HDAC1-11) and seven NAD+-dependent sirtuins (SIRT1-7). HDACs are epigenetic erasers catalyzing histone deacetylation, chromatin compaction, and transcriptional repression. In addition, HDACs target thousands of non-histone proteins affecting many biological processes (3). The subfamily of sirtuins attracted much interest when sirtuins were proposed to promote longevity and represent attractive therapeutic targets for age-related pathologies, such as type 2 diabetes, as well as neurodegenerative, cardiovascular, and oncologic diseases (4–6). Our knowledge about the impact of sirtuins on innate immune responses is limited. Most studies have focused on SIRT1 and SIRT6, resulting in an overall complex picture attributing both proinflammatory and anti-inflammatory properties to sirtuins (7).
Sirtuin 2 (SIRT2) was originally reported to colocalize with the microtubule network and to deacetylate α-tubulin (8). Indeed, SIRT2 is mainly cytoplasmic, although it can translocate during the G2/M transition phase of cell cycle into the nucleus where it deacetylates histone H4 lysine 16 (9). Of note, SIRT2 possesses a proficient demyristoylation activity, the physiological relevance of which remains to be established (10). By targeting numerous proteins besides histones, SIRT2 regulates cytoskeleton dynamics, cell cycle, and metabolic pathways including inhibition of adipogenesis and promotion of lipolysis and gluconeogenesis. Additionally, SIRT2 can act as a tumor suppressor gene, and is involved in myelogenesis and other brain functions. Accordingly, SIRT2 has been implicated in tumorigenesis and neurodegeneration, and likely plays a role in metabolic disorders, such as obesity and type 2 diabetes (11–13).
With respect to inflammatory processes, SIRT2 was first shown to deacetylate NF-κB p65, resulting in the expression of a subset of p65-dependent genes in mouse embryonic fibroblasts (14). SIRT2 deficiency sustained brain inflammation in a model of traumatic brain injury and increased the severity of collagen-induced arthritis and colitis (15–17). However, SIRT2 deficiency was also reported to decrease NF-κB p65-mediated inflammatory response, renal tubular inflammation, and ischemia reperfusion-induced hepatocellular inflammation (18–20). Finally, in an experimental stroke model, SIRT2 deficiency preserved neurological functions without affecting inflammatory parameters (21). Overall, the function of SIRT2 in sterile and chronic inflammatory disorders appears to be context dependent.
Because sirtuins are pleiotropic and in consideration of the development of sirtuin-targeting drugs for clinical conditions, we sought to delineate the role of SIRT2 in the innate immune response. To this end, we used SIRT2 knockout mice to investigate the response of immune cells to immunological and microbial stimuli using in vitro experiments and in vivo preclinical models. Preclinical models included models of shock as well as Gram-negative and Gram-positive bacterial infections and fungal infection. Overall, SIRT2 deficiency does not modulate cytokine production by innate immune cells, but enhances bacterial phagocytosis by macrophages. SIRT2 deficiency protects from chronic staphylococcal infection, while having no impact on toxic shock, endotoxemia, fulminant peritonitis, non-lethal pneumonia, and chronic candidiasis. These data largely support the safety, in terms of susceptibility to infections, of SIRT2 inhibitors developed for clinical applications.
8- to 12-week-old female BALB/cByJ mice, C57BL/6J mice (Charles River Laboratories, Saint-Germain-sur-l’Arbresle, France), and SIRT2 knockout mice backcrossed 12 times on a C57BL/6J background (15) were used. Mice were housed under specific pathogen-free conditions and free of mouse norovirus. Splenocytes were cultured in RPMI 1640 medium containing 2 mM glutamine, 50 µM 2-ME, 100 IU/ml penicillin, 100 µg/ml streptomycin (Invitrogen, San Diego, CA) and 10% heat-inactivated fetal calf serum (FCS; Sigma-Aldrich, St. Louis, MO) (22). Bone marrow (BM) cells were cultured in IMDM (Invitrogen) containing 50 µM 2-ME, penicillin, streptomycin, and 10% FCS. Medium was supplemented with 20 ng/ml M-CSF, 20 ng/ml GM-CSF plus 20 ng/ml IL-4 (ProSpec, East Brunswick, NJ) or 200 ng/ml FMS-like tyrosine kinase 3 ligand (Flt3L, Shenandoah biotechnology, Warwick, PA) to generate BM-derived macrophages (BMDMs), BM-derived dendritic cells (BMDCs), or Flt3L-DCs, respectively. BMDCs were collected after 6 days. BMDMs and Flt3L-DCs were collected after 7 days of culture. Cells (1, 5, and 20 × 105) were seeded in 96-well, 24-well or 6-well plates in complete medium without growth factors and antibiotics unless specified.
Salmonella minnesota ultra pure lipopolysaccharide (LPS) was from List Biologicals Laboratories (Campbell, CA), Pam3CSK4 from EMC microcollections (Tübingen, Germany), CpG ODN 1826 (CpG) from InvivoGen (San Diego, CA, USA), toxic shock syndrome toxin-1 (TSST-1) and staphylococcal enterotoxin B (SEB) from Toxin Technology (Sarasota, FL, USA), concanavalin A and phytohemagglutinin (PHA) from Sigma-Aldrich, and anti-CD3ε and anti-CD28 antibodies (clones 145-2C11 and 37.51) from eBioscience (San Diego, CA, USA). Clinical strains of Escherichia coli (E. coli) O18, E. coli J5, E. coli O111, Salmonella enterica serovar Typhimurium C5 (Salmonella Typhimurium), Klebsiella pneumoniae caroli (K. pneumoniae), Neisseria meningitis, Streptococcus pneumoniae, Staphylococcus aureus AW7 (S. aureus), and Group B Streptococcus (GBS) were grown in brain heart infusion broth (BD Biosciences, Erembodegem, Belgium) (23–27). Candida albicans 5102 (C. albicans) (22) was cultured in yeast extract–peptone–dextrose (BD Biosciences). Microorganisms were washed in PBS and adjusted at 1010 CFU/ml. For in vitro stimulation, bacteria were heat-inactivated for 2 h at 56°C. Nocodazole and 2-deoxyglucose were from Sigma-Aldrich, cytochalasin D from Millipore (Billerica, MA, USA).
Total RNA was isolated, reverse transcribed (RNeasy and QuantiTect reverse transcription kits, Qiagen, Hilden, Germany), and used in real-time PCRs conducted with a QuantStudio™ 12K Flex system (Life Technologies, Carlsbad, CA, USA). Reactions consisted of 1.25 µl cDNA, 1.25 µl H2O, 0.62 µl 10 nM primers [Table S1 in Supplementary Material and Ref. (28, 29)], and 3.12 µl Fast SYBR® Green Master Mix (Life Technologies) and were tested in triplicate. Gene specific expression was normalized to hypoxanthine guanine phosphoribosyl transferase expression. Sirt2 expression levels in organs were extracted from the BioGPS resource (http://biogps.org).
Nuclear and total protein extracts were submitted to PAGE and transferred onto nitrocellulose membranes (30, 31). Membranes were incubated with antibodies directed against SIRT2, acetylated α-tubulin, total α-tubulin, total and phosphorylated ERK1/2, p38, JNK, and NF-κB p65 and TATA-box binding protein (used as a control of nuclear extracts) (see antibody description in Table S2 in Supplementary Material), then with a secondary horseradish peroxidase-conjugated antibody (Sigma-Aldrich) (32). Blots were imaged with the enhanced chemiluminescence Western blotting system (GE Healthcare, Little Chalfont, Royaume-Uni). Images were recorded using a Fusion Fx system (Viber Lourmat, Collégien, France).
Single cell suspensions from thymus and spleen, or BMDMs were enumerated and incubated with 2.4G2 monoclonal antibody (mAb). Immune cell subpopulations were tracked by staining performed using mAbs described in Table S2 in Supplementary Material. Splenic CD4+ CD25+ Foxp3+ cells were detected using The Mouse Regulatory T Cell Staining Kit (eBioscience). Data were acquired using a LSR II flow cytometer (BD Biosciences) and analyzed using FlowJo Version 10.2 software (FlowJo LLC, Ashland, OR, USA) (33).
The proliferation of 1.5 × 105 splenocytes cultured for 48 h in 96-well plates was quantified by measuring 3H-thymidine incorporation over 18 h (34).
Cytokine concentrations were quantified using DuoSet ELISA kits (R&D Systems, Abingdon, UK) or Luminex assays (Affimetrix eBioscience, Vienna, Austria) (35).
8- to 12-week-old female SIRT2+/+ and SIRT2−/− mice (6–13 mice per group) matched for age were used. To analyze the response to TSST-1, mice were challenged intraperitoneally (i.p.) with TSST-1 (0.5 mg/kg). Models of endotoxic shock were performed by challenging mice i.p. with LPS (10 and 25 mg/kg). To induce TNF shock, mice were sensitized with d-galactosamine (30 mg/kg i.p., Sigma-Aldrich) just before being challenged with TNF (25 mg/kg i.p., Preprotech, Rocky Hill, NJ, USA). Bacterial sepsis was induced by challenging mice i.p. with 105 CFU E. coli O18, intravenously (i.v.) with 107 CFU S. aureus or 105 CFU C. albicans or intranasally (i.n.) with 30 CFU K. pneumoniae. Blood and spleen were collected 0, 1, 6, 8, 24, or 48 h post-challenge to quantify cytokines and bacteria (28). Body weight loss, severity score, and survival were registered at least once daily. The severity score was graded from 1 to 5 (36). Animals were euthanized when they met a severity score of 4. Two to three operators performed animal follow-up.
Fluoresbrite® Yellow Green Microspheres (Polysciences Inc, Warrington, PA, USA) or FITC-labeled bacteria were added to cells at a ratio of 10 beads or bacteria/cell. After 1 h, cells were washed, incubated for 1 min with trypan blue (0.25 mg/ml) and analyzed by flow cytometry. When specified, beads were opsonized with serum for 30 min at 37°C. To assess phagocytosis of live bacteria, BMDMs (in quadruplicates or sextuplates) were incubated for 1 h with E. coli O18, S. aureus, and GBS (10 bacteria/cell). Non-adherent and extracellular bacteria were removed by washing and killed by a 30-min exposure to 100 µg/ml gentamicin (Essex Chemie, Luzern, Switzerland; for E. coli and GBS) or 10 µg/ml ciprofloxacin (Fresenius Kabi, Oberdorf, Switzerland; for S. aureus). Serial dilutions of cell lysates were plated on agar plates. Colonies were enumerated to calculate the number of phagocytosed bacteria.
The glycolytic activity of BMDMs was analyzed using a 96-well format Seahorse XFe® system and the Seahorse XF Glycolysis Stress Test Kit (Agilent Technologies, Santa Clara, CA, USA). Briefly, 4 × 104 BMDMs were plated in 96-well plates in IMDM medium. The next day, cells were incubated with or without 5 × 107 CFU/ml heat-killed S. aureus and rested 1 h in Seahorse medium without glucose. The glycolytic capacity was assessed by measuring the extracellular acidification rate following the sequentially addition of 10 mM glucose, 1 µM oligomycin, and 50 mM 2-deoxy-glucose (2-DG) according to manufacturer’s instructions.
Comparisons between the different groups were performed by analysis of variance followed by two-tailed unpaired Student’s t-test. The Kaplan–Meier method was used for building survival curves and differences were analyzed by the log-rank sum test. All analyses were performed using PRISM (GraphPad Software). P values were two-sided, and P < 0.05 was considered to indicate statistical significance.
SIRT1-7 mRNA expression was quantified in BM, BMDMs, and DCs (conventional BMDCs and Flt3L-derived DCs) (Figure 1A). SIRT2 was the most highly expressed sirtuin in all populations. SIRT2 was also the predominantly expressed sirtuin in RAW 264.7 macrophages and in the spleen, liver, and kidneys (Figure 1B and data not shown). Western blot analyses confirmed SIRT2 protein expression in BMDMs (see below). Primary osteoblasts, osteoclasts, macrophages, and mast cells expressed 2.6-fold to 7.1-fold higher levels of SIRT2 mRNA than granulocytes, NK cells, T cells, B cells, DCs, and thymocytes (Figure 1C). Overall, SIRT2 was highly expressed by myeloid-derived cells, suggesting that it could play a role in the control of immune responses. To address this question, SIRT2-deficient mice were used [Figures 1D,E and Ref. (15)].
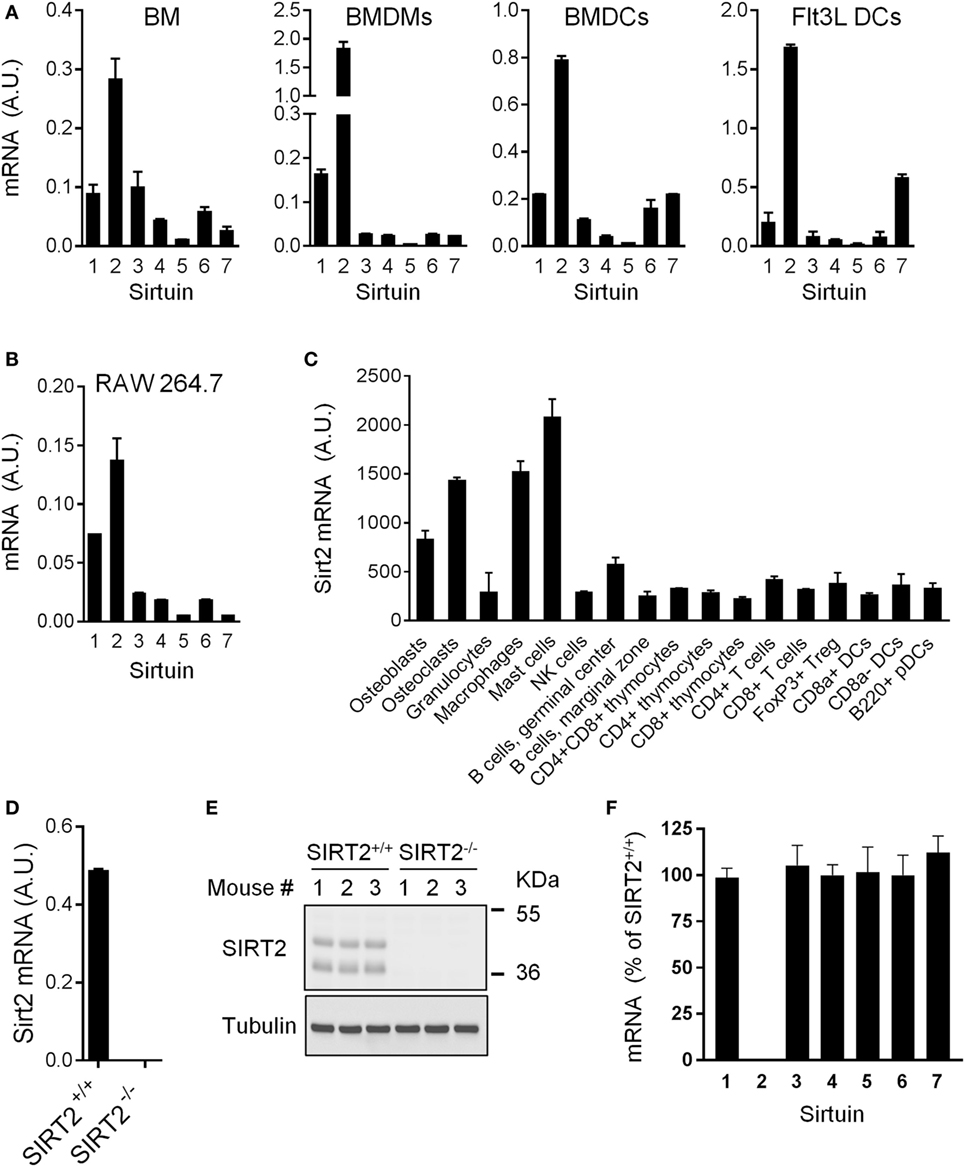
Figure 1. Sirt2 is strongly expressed in myeloid cells. (A,B) Sirt1-7 mRNA levels in bone marrow (BM), BM-derived macrophages (BMDMs) and dendritic cells (BMDCs), and Flt3L-derived DCs (A) and RAW 264.7 macrophages (B) analyzed by RT-PCR and normalized to Hprt mRNA levels. Data are means ± SD of triplicate samples from one experiment performed with four BALB/c mice (A) or three preparations of RAW 264.7 macrophages (B). (C) Sirt2 mRNA expression levels in a panel of cells (extracted from http://biogps.org). (D,E) Sirt2 mRNA (D) and Sirtuin 2 (SIRT2) protein (E) expression in SIRT2+/+ and SIRT2−/− BMDMs assessed by RT-PCR and western blotting, respectively. Data are means ± SD from one experiment performed with three mice (D). (F) Sirt1-7 mRNA expression levels in SIRT2−/− BMDMs, expressed relative to the mRNA levels in SIRT2+/+ BMDMs set at 100%. Data are means ± SD from one experiment performed with three mice. A.U., arbitrary units. Full-length blots of panel (E) are presented in Figure S1 in Supplementary Material.
SIRT2−/− mice were described previously (15). These mice were born at the expected Mendelian ratio and developed without abnormalities. SIRT2 mRNA and protein were undetectable in SIRT2−/− BMDMs (Figures 1D,E). Expression levels of Sirt1 and Sirt3-7 mRNA were unaltered in SIRT2−/− BMDMs (Figure 1F), suggesting that the lack of SIRT2 was not compensated by an increase in expression of other sirtuins.
Compared to SIRT2+/+ mice, SIRT2−/− mice expressed normal proportions and absolute numbers of CD4/CD8 double negative (DN1–4), double positive, and single positive (SP) thymocytes (Table 1). Additionally, SIRT2−/− mice had normal populations of splenic T cells (DN, SP, naïve, and memory), B cells (immature and mature B cells), DCs (B220− CD11c+ cDCs and B220+ CD11c+ pDCs), and Foxp3+ regulatory T cells (Table 2). Therefore, SIRT2 had no major impact on immune cell development.
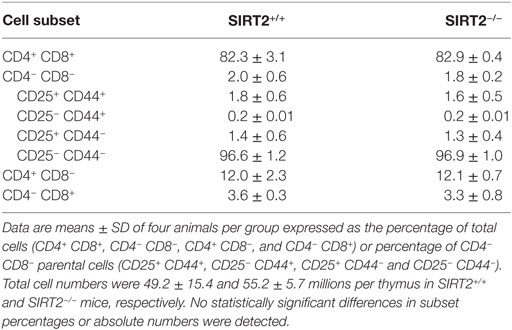
Table 1. Thymic cell subsets in SIRT2+/+ and SIRT2−/− mice.
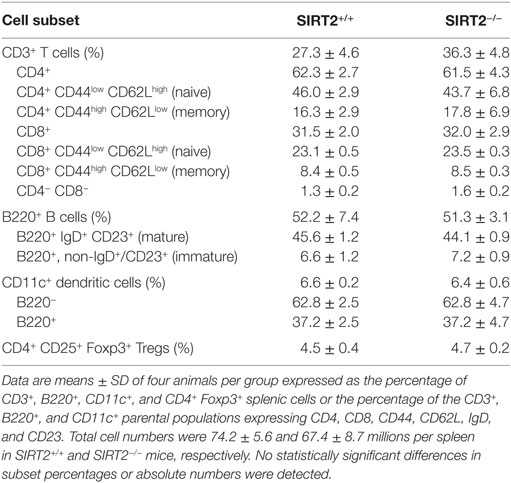
Table 2. Splenic cell subsets in SIRT2+/+ and SIRT2−/− mice.
As a first approach to evaluate whether SIRT2 influenced immune responses, SIRT2+/+ and SIRT2−/− splenocytes were exposed to microbial and immunological stimuli: LPS, CpG, concanavalin A, anti-CD3/CD28, PHA, TSST-1, and SEB. The proliferation and production of IL-2 and IFNγ (measured by ELISA) by splenocytes were not affected by SIRT2 deficiency (Figures 2A,B). In agreement, blood concentrations of IFNγ were similar in SIRT2+/+ and SIRT2−/− mice injected with TSST-1 (Figure 2C), a staphylococcal superantigen responsible of toxic shock syndrome. A Luminex assay was then used to quantify TNF, IL-6, IL-10, IL-12p70, CCL3/Mip1α, CCL4/Mip1β, and CCL5/Rantes secretion by splenocytes exposed to LPS, CpG, concanavalin A, anti-CD3/CD28, PHA, TSST-1, and SEB (Figure 2D). No differences were observed between SIRT2+/+ and SIRT2−/− splenocytes apart from a 20–28% reduction of LPS-induced IL-6, IL-10, and CCL5 and anti-CD3/CD28-induced CCL4, while the secretion of CCL5 was increased in response to anti-CD3/CD28.
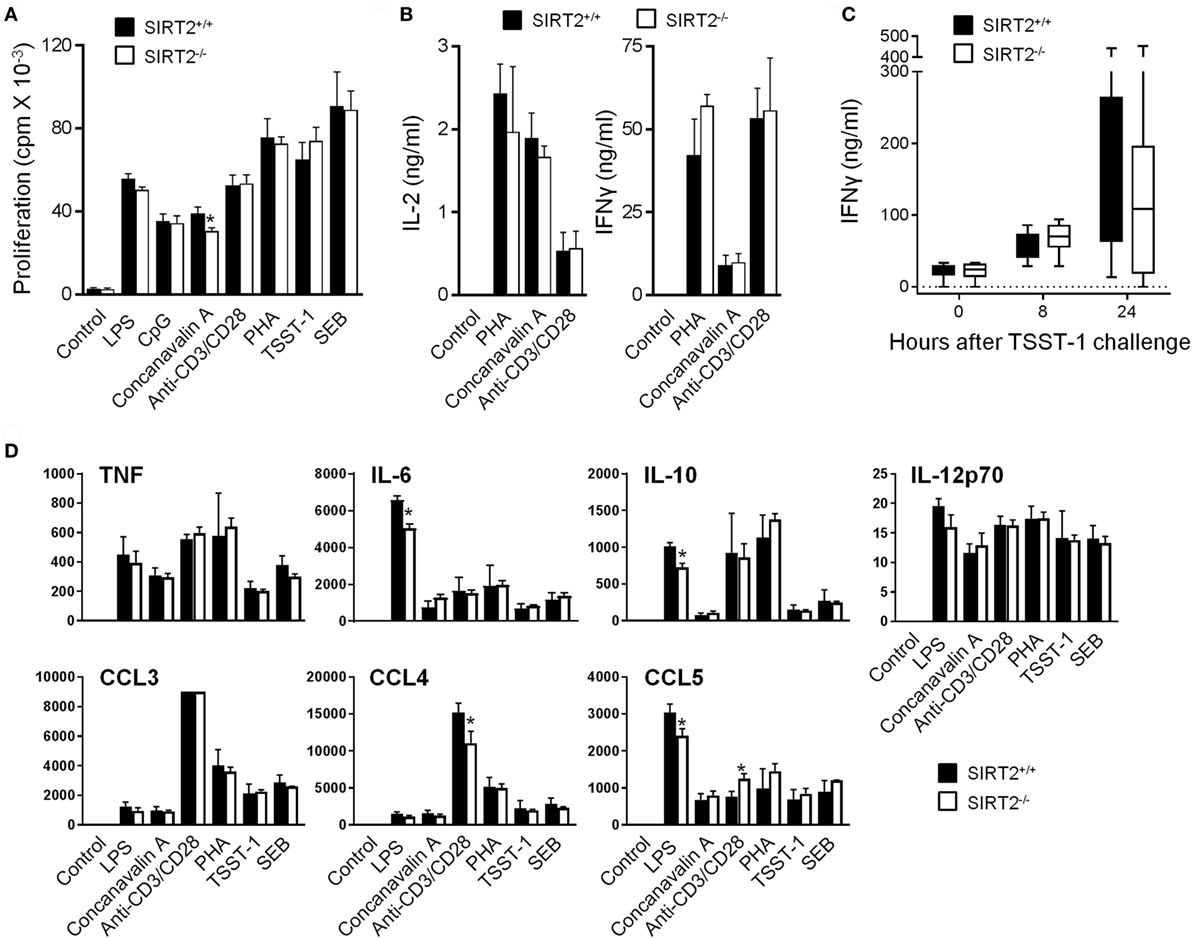
Figure 2. Sirtuin 2 (SIRT2) deficiency does not affect proliferation and cytokine response of splenocytes and IFNγ production in mice challenged with toxic shock syndrome toxin-1 (TSST-1). (A,B) SIRT2+/+ and SIRT2−/− splenocytes were incubated for 48 h with lipopolysaccharide (LPS) (5 µg/ml), CpG (2 µg/ml), concanavalin A (5 µg/ml), anti-CD3/CD28 antibodies (1 µg/ml), phytohemagglutinin (PHA) (10 µg/ml), TSST-1 (2 µg/ml), and staphylococcal enterotoxin B (SEB) (5 µg/ml). Proliferation was measured by 3H-thymidine incorporation (A) while IL-2 and IFNγ concentrations in cell culture supernatants were quantified by ELISA (B). Data are means ± SD of triplicate samples from one experiment performed with four mice and are representative of two experiments (*P < 0.05). (C) SIRT2+/+ and SIRT2−/− mice (n = 8 per group) were injected with TSST-1 (0.5 mg/kg i.p.). Blood was collected after 0, 8, and 24 h to quantify IFNγ concentrations. Data are means ± SD. P > 0.5 for all time points. (D) SIRT2+/+ and SIRT2−/− splenocytes were incubated for 48 h with LPS, CpG, concanavalin A, anti-CD3/CD28 antibodies, PHA, TSST-1, and SEB. TNF, IL-6, IL-10, IL-12p70, CCL3 (MIP-1α), CCL4 (MIP-1β), and CCL5 (RANTES) were quantified by Luminex. Data (in pg/ml) are means ± SD of one experiment performed with three mice. CCL3 values in response to anti-CD3/CD8 were over the upper limit of detection of the assay (*P < 0.05).
Macrophages are professional phagocytic cells that play a major role in antimicrobial host defenses. Therefore, we tested whether SIRT2 deficiency had an effect on phagocytosis by BMDMs. SIRT2+/+ and SIRT2−/− BMDMs were incubated with fluorescent beads and analyzed by flow cytometry (Figures 3A–C). A higher percentage of SIRT2−/− than SIRT2+/+ BMDMs phagocytosed beads (32.4 ± 1.9 vs 24.5 ± 1.2 percent positive cells, P = 0.002; Figures 3A,B), regardless of opsonization (Figure 3C). SIRT2−/− BMDMs also exhibited higher phagocytosis using a panel of fluorescently labeled heat-inactivated bacteria (% of SIRT2−/− vs SIRT2+/+ BMDMs ingesting bacteria: E. coli J5: 53.6 vs 43.6%, E. coli O111: 31.6 vs 23.0%, Salmonella Typhimurium: 24.1 vs 17.6%, Neisseria meningitis: 46.1 vs 37.2%, S. pneumoniae: 49.6 vs 34.7%). BMDMs were additionally exposed to live E. coli, S. aureus and GBS for 1 h before measuring the number of intracellular bacteria by plating cell lysates and enumerating colonies. Confirming the results obtained using inert beads and fluorescent bacteria, the numbers of phagocytosed E. coli, S. aureus and GBS were 1.3-fold to 1.6-fold higher using SIRT2−/− BMDMs (Figure 3D).
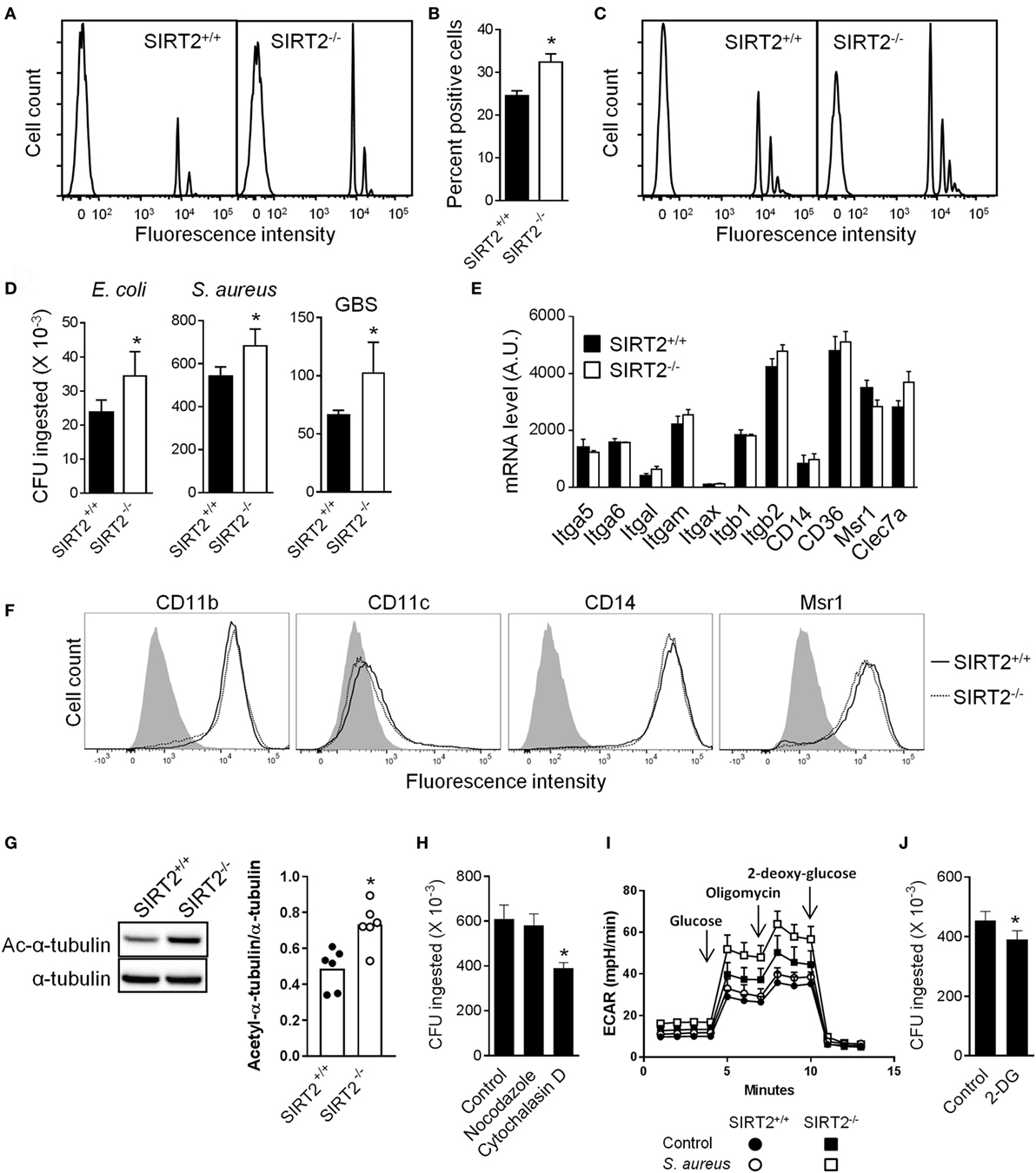
Figure 3. Sirtuin 2 (SIRT2) deficiency increases bacterial phagocytosis by macrophages. (A–C) SIRT2+/+ and SIRT2−/− BM-derived macrophages (BMDMs) were incubated with 10 fluorescent beads [opsonized in (C)] per cell. After 1 h, cells were analyzed by flow cytometry. Representative histograms are depicted in (A,C). The percentage of fluorescent cells was calculated (B). Data are means ± SD from an experiment performed with four mice (*P = 0.002). (D) SIRT2+/+ and SIRT2−/− BMDMs were incubated with live Escherchia coli, Staphylococcus aureus, and Group B Streptococcus (GBS) (10 bacteria/cell). Phagocytosis was assessed after 1 h. Data are means ± SD from one experiment performed with four (E. coli and GBS) or eight (S. aureus) mice. *P = 0.03, 0.006, and 0.03 for E. coli, S. aureus, and GBS, respectively. (E) Itga5, Itga6, Itgal, Itgam, Itgax, Itgb1, Itgb2, Cd14, Cd36, Msr1, and Clec7a mRNA expression levels in SIRT2+/+ and SIRT2−/− BMDMs. Data are means ± SD of one experiment performed with three mice. (F) CD11b, CD11c, CD14, and Msr1 expression levels were analyzed by flow cytometry. The gray histogram represents staining with an isotype control antibody. (G) Expression levels of acetylated (Ac) and total tubulin in BMDMs were analyzed by western blotting and quantified by imaging. Data are means ± SD from six mice (right panel) (*P = 0.015). Full-length blots are presented in Figure S2 in Supplementary Material. (H) Phagocytosis of S. aureus [performed as in (D)] by SIRT2−/− BMDMs preincubated for 1 h with or without nocodazole (10 µM) and cytochalasin D (10 µM). Data are means ± SD from one experiment performed with four mice (*P = 0.008). (I) Glycolytic capacity of BMDMs assessed by measuring the extracellular acidification rate (ECAR) using the Seahorse XF Glycolysis Stress Test Kit as described in Section “Materials and Methods.” Data are means ± SD from one experiment performed with three mice. (J) Phagocytosis of S. aureus [performed as in (D)] by SIRT2−/− BMDMs preincubated for 1 h with or without 25 mM 2-deoxy-glucose (2-DG). Data are means ± SD from one experiment performed with four mice (*P = 0.017).
Macrophages express phagocytic receptors, including macrophage scavenger receptor 1 (Msr1/SR-AI/CD204), CD14, CD36, C-type lectins such as dectin-1 (encoded by Clec7a), and members of the integrin superfamily (integrin α5/Itga5/CD49e, integrin αM/Itgam/CD11b, integrin αX/Itgax/CD11c, integrin β2/Itgb2/CD18). SIRT2−/− and SIRT2+/+ BMDMs expressed comparable mRNA levels of Itga5, Itga6, Itgal, Itgam, Itgax, Itgb1, Itgb2, Cd14, Cd36, Msr1, and Clec7a (Figure 3E). Moreover, SIRT2−/− and SIRT2+/+ BMDMs expressed similar levels of membrane-bound CD11b, CD11c, CD14, and Msr1 (Figure 3F). Hence, SIRT2 deficiency likely improved phagocytosis by BMDMs in a phagocytic receptor independent fashion.
Stabilization of microtubules and high glycolytic activity have been associated with efficient phagocytosis by macrophages (37–40). Since SIRT2 impacts on microtubules stabilization and glucose metabolism (8, 13, 41), we questioned whether these processes influenced phagocytosis by SIRT2−/− BMDMs. In BMDMs, SIRT2 deficiency increased 1.5-fold tubulin acetylation (Figure 3G), a hallmark of microtubule stabilization. However, the microtubule destabilizer nocodazole did not impair the phagocytosis of S. aureus by BMDMs (Figure 3H), while the actin depolymerization agent cytochalasin D efficiently inhibited phagocytosis. Interestingly, the glycolytic activity was higher in SIRT2−/− than SIRT2+/+ BMDMs exposed to S. aureus (Figure 3I). Moreover, 2-DG, which inhibits glycolysis, significantly reduced the phagocytosis of S. aureus by BMDMs (Figure 3J). Albeit preliminary, these results suggested that differences in the metabolic capacity of SIRT2+/+ and SIRT2−/− BMDMs may provide a mechanism by which SIRT2 impedes phagocytosis.
Sensing of microbial ligands through TLRs initiates MAPK and NF-κB signaling involved in the control of cytokine gene expression by innate immune cells (2). To address whether SIRT2 impacted intracellular signaling, the phosphorylation of ERK1/2, p38, and JNK MAPKs in BMDMs exposed to LPS for 0, 10, 30, and 60 min was analyzed by western blotting. The rate of LPS-induced phosphorylation of ERK1/2, p38 and JNK was very similar in SIRT2+/+ and SIRT2−/− BMDMs, with only a slight and late reduction of ERK1/2 phosphorylation in SIRT2−/− BMDMs (30% reduction at 1 hour) (Figure 4A). The nuclear translocation of phosphorylated NF-κB p65 was not different in SIRT2+/+ and SIRT2−/− BMDMs exposed to LPS for 0, 10, 30, and 60 min (P > 0.5 for all time points).
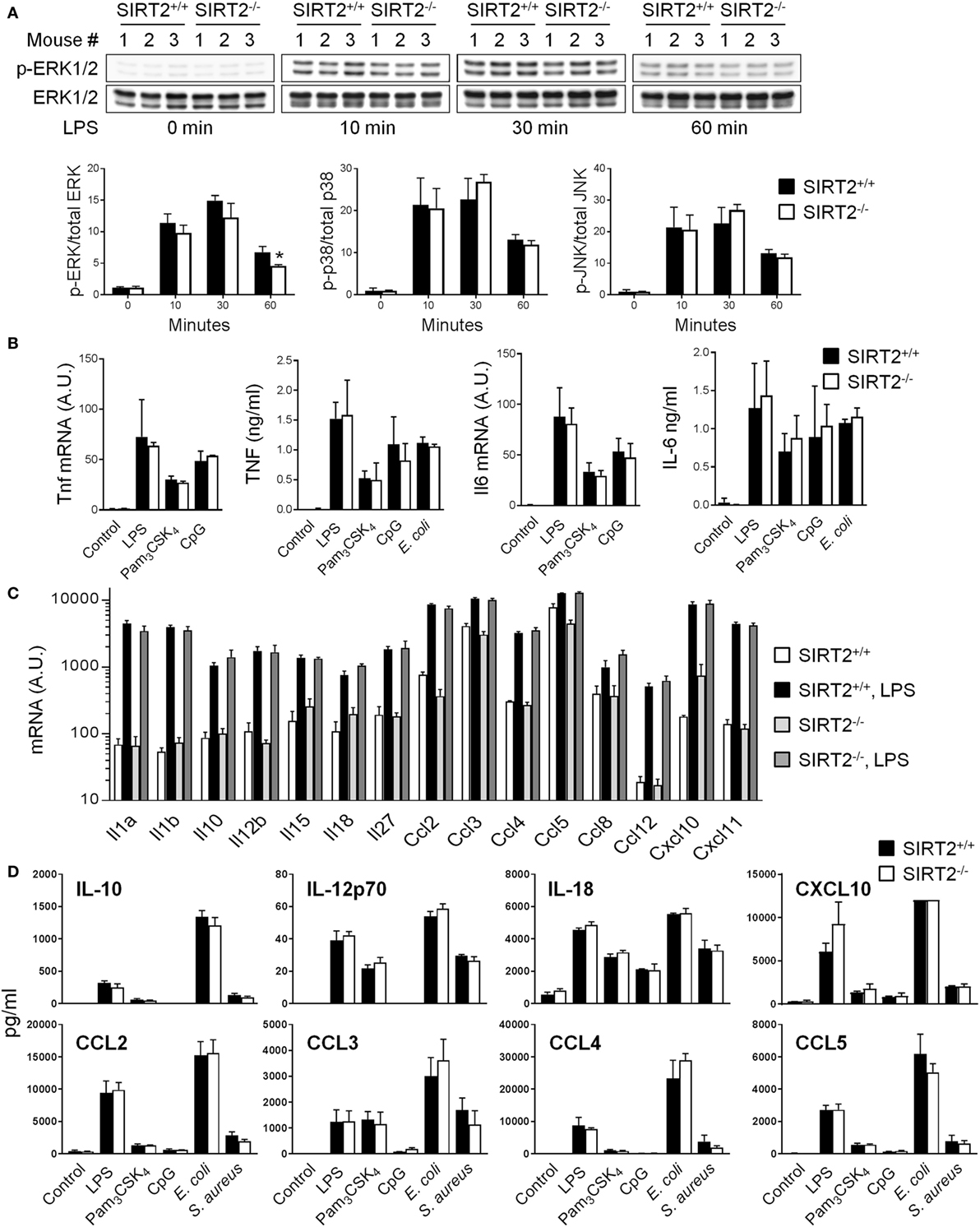
Figure 4. Sirtuin 2 (SIRT2) deficiency does not affect the response of macrophages to microbial stimulation. SIRT2+/+ and SIRT2−/− BM-derived macrophages were exposed to lipopolysaccharide (LPS) (10 ng/ml), Pam3CSK4 (10 ng/ml), CpG (2 µg/ml), Escherichia coli (106 CFU/ml), and Staphylococcus aureus (106 CFU/ml). (A) Expression levels of phosphorylated (p) and total ERK1/2 (upper panel), p38 and JNK were analyzed by western blotting and quantified by imaging. Data are means ± SD from one experiment performed with three mice (lower panel) (*P = 0.02). (B) Tnf and Il6 mRNA levels and TNF and IL-6 concentrations in cell culture supernatants 1 and 8 h after stimulation, respectively. (C) Il1a, Il1b, Il10, Il12b, Il15, Il18, Il27, Ccl2/Mcp1, Ccl3/Mip1a, Ccl4/Mip1b, Ccl5/Rantes, Ccl8/Mcp2, Ccl12/Mcp5, Cxcl10/Ip10, Cxcl11/Itac mRNA levels after 8 h of culture with or without LPS. (D) IL-10, IL-12p70, IL-18, CXCL10, CCL2, CCL3, CCL4, and CCL5 concentrations in cell culture supernatants 8 h after stimulation measured by Luminex. Data are means ± SD of triplicate samples from one experiment performed with three mice (mRNA analyses) or six mice (TNF and IL-6 secretion), or means ± SD of single measurements from one experiment performed with three mice (Luminex). CXCL10 values in response to E. coli were over the upper limit of detection of the assay. No statistically significant differences were detected in (B–D). A.U., arbitrary units. Full-length blots of panel (A) are presented in Figure S4 in Supplementary Material.
SIRT2+/+ and SIRT2−/− BMDMs exposed to LPS, Pam3CSK4, CpG (i.e., TLR4, TLR1/2 and TLR9 ligands, respectively), and E. coli upregulated Tnf and Il6 mRNA levels and secreted TNF and IL-6 to the same extend (Figure 4B). Additionally, Tlr1, Tlr2, Tlr4, and Tlr9 mRNA were modulated likewise in SIRT2+/+ and SIRT2−/− BMDMs (Figure S3 in Supplementary Material). SIRT2+/+ and SIRT2−/− BMDMs expressed also comparable mRNA levels of Il1a, Il1b, Il10, Il12b, Il15, Il18, Il27, Ccl2/Mcp1, Ccl3/Mip1a, Ccl4/Mip1b, Ccl5/Rantes, Ccl8/Mcp2, Ccl12/Mcp5, Cxcl10/Ip10, and Cxcl11/Itac at baseline and following LPS stimulation (Figure 4C), and secreted comparable levels of IL-10, IL-12p70, IL-18, CXCL10, CCL2, CCL3, CCL4, and CCL5 upon exposure to LPS, Pam3CSK4, CpG, E. coli, and S. aureus (Figure 4D). Altogether, these results argued against an important role of SIRT2 in controlling proinflammatory and anti-inflammatory cytokine response by macrophages exposed to TLR ligands.
To assess the relevance of these observations in vivo, we developed models of endotoxemia of different severity (Figures 5A–D). In a mild model of endotoxemia (induced by an i.p. challenge with 10 mg/kg LPS), TNF and IL-12p40 concentrations in blood and mortality rates (83 vs 100%, P = 0.3) were comparable in SIRT2+/+ and SIRT2−/− mice (Figures 5A,B). In a severe model of endotoxemia (induced by 25 mg/kg LPS), TNF, IL-6, and IL-12p40 concentrations in blood and mortality rates (88% in both groups, P = 0.69) were strongly increased, but remained similar in SIRT2+/+ and SIRT2−/− mice (Figures 5C,D). Furthermore, SIRT2+/+ and SIRT2−/− mice were equally sensitive to fulminant shock induced by TNF (25 mg/kg i.p. in d-galactosamine sensitized mice), the main driver of the lethal effect of endotoxemia (P = 0.6; Figure 5E). Overall, SIRT2 did not interfere with endotoxemia.
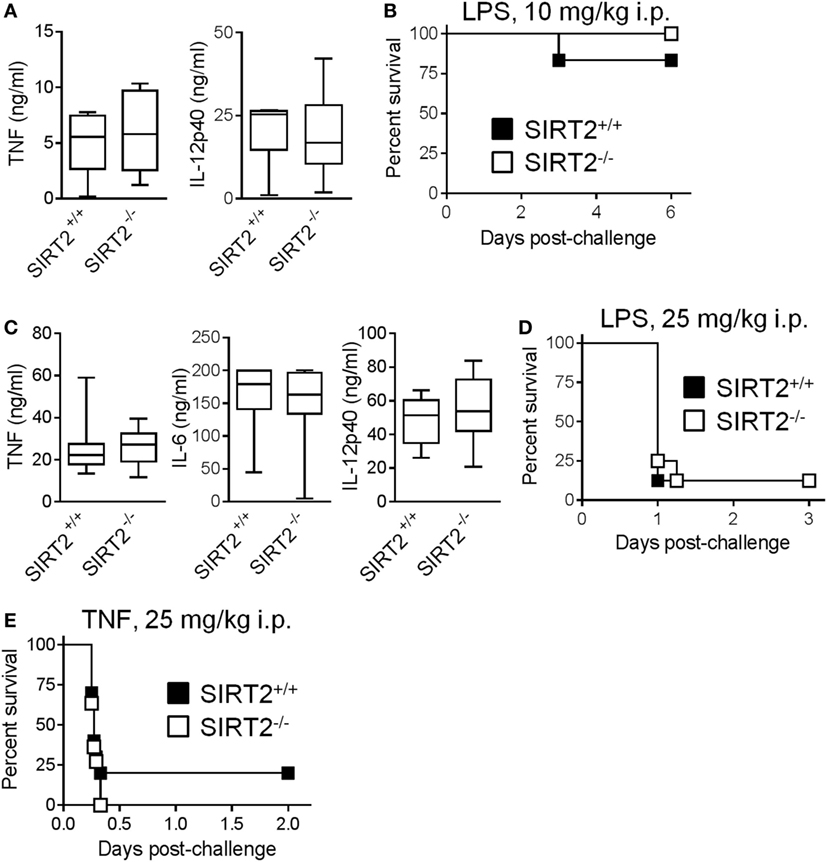
Figure 5. Sirtuin 2 (SIRT2) deficiency does not affect endotoxemia and TNF-induced shock. SIRT2+/+ and SIRT2−/− mice were injected i.p. with 10 mg/kg lipopolysaccharide (LPS) [(A,B), n = 6 per group] and 25 mg/kg LPS (C,D, n = 8 per group). (A,C) TNF, IL-6, and IL-12p40 concentrations in blood collected 1 h (TNF) and 6 h (IL-6 and IL-12p40) after LPS challenge. P > 0.5 for all conditions. (B,D) Survival of mice. P = 0.3 and 0.9. (E) Survival of SIRT2+/+ and SIRT2−/− mice (n = 11 per group) sensitized with 30 mg/kg d-galactosamine and challenged i.p. with 25 mg/kg TNF. P = 0.6.
Considering that SIRT2 impacted phagocytosis but not cytokine expression, we hypothesized that SIRT2 deficiency should provide some benefit during chronic lethal infection but not fulminant sepsis, and should not sensitize to benign infection. Therefore, we compared the impact of SIRT2 deficiency during rapidly lethal, sub-lethal, and chronic bacterial infections induced by E. coli, K. pneumonia, and S. aureus, three of the most frequent causes of bacterial sepsis in humans (42).
Supporting our working hypothesis, in a model of chronic infection by S. aureus in which mortality occurred 3 to 16 days post i.v. challenge with the bacteria, severity score, body weight loss, and survival (SIRT2+/+ vs SIRT2−/−: 33.3 vs 69.2%; P = 0.04) were all markedly improved in SIRT2−/− mice (Figure 6A). In agreement with these findings, 48 h postinfection, only 37.5% (3/8) of SIRT2−/− mice were bacteremic while 71.4% (5/7) of SIRT2+/+ mice were bacteremic (Figure 6B). Moreover, bacterial burden in the spleen was much lower in SIRT2−/− than in SIRT2+/+ mice (1.9 × 102 vs 3.3 × 103 mean CFU/organ; P = 0.04). TNF was not detected in blood, while IL-6 and IL-12p40 levels were not different between SIRT2−/− and SIRT2+/+ mice, although there was a trend toward lower IL-12p40 levels in SIRT2−/− mice (Figure 6C). In a model of fulminant, rapidly lethal peritonitis induced by E. coli, body weight loss, bacterial dissemination into the blood, and survival rate (12.5% in both groups, P = 0.7) were comparable in SIRT2+/+ and SIRT2−/− mice (Figure 6D). In a non-severe model of K. pneumoniae pneumonia, body weight loss and survival (85.7% in both groups, P = 0.9) were not affected by SIRT2 deficiency (Figure 6E). Finally, we questioned whether SIRT2 influenced host susceptibility to a non-bacterial chronic infection. Candidiasis was induced by i.v. inoculation of 105 CFU/ml C. albicans into SIRT2+/+ and SIRT2−/− mice (n = 14 and 16). Mice died 9–40 days postinfection, without survival differences between the SIRT2+/+ and SIRT2−/− groups (71 vs 56%; P = 0.4), suggesting that SIRT2 deficiency did not compromise host defenses to Candida infection.
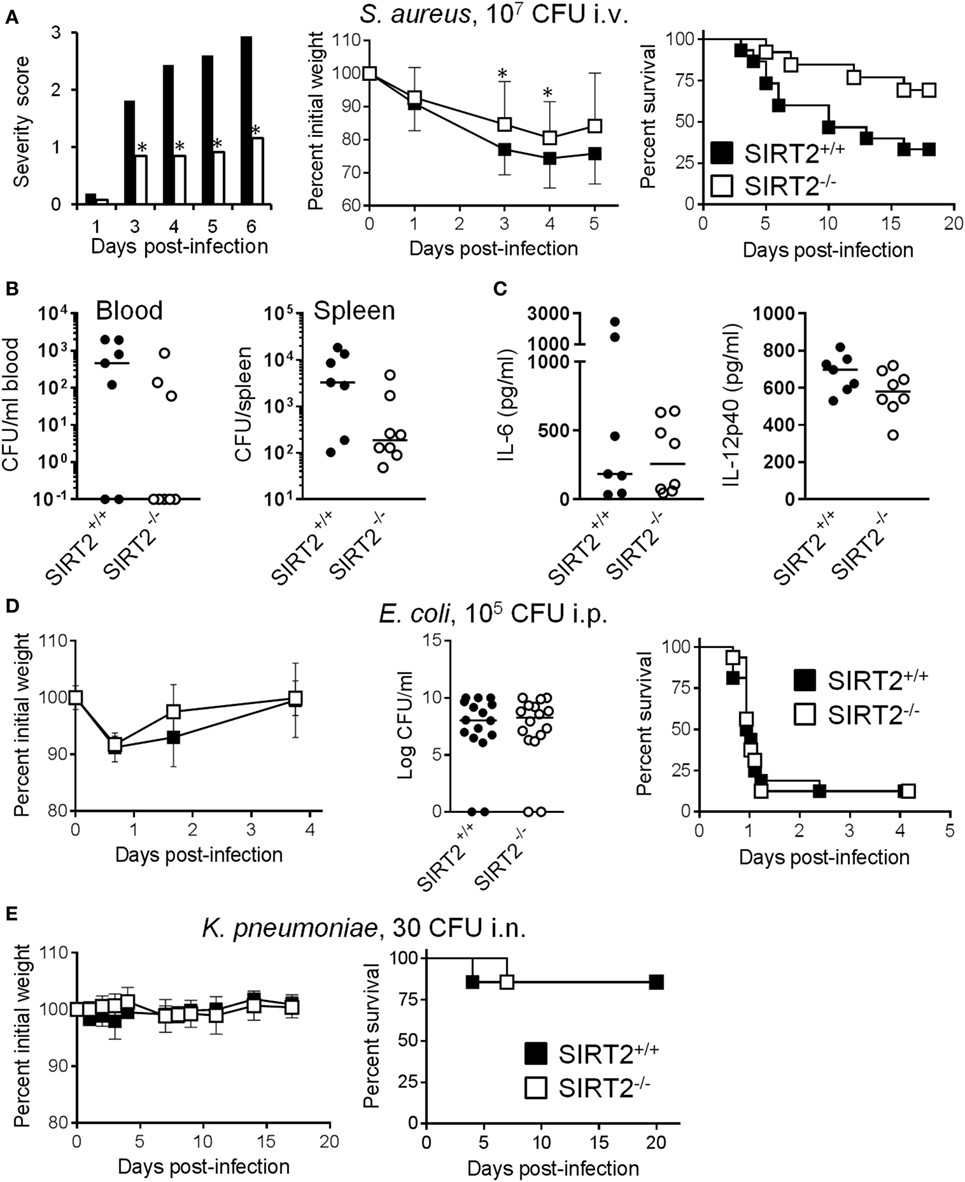
Figure 6. SIRT2 deficiency protects from chronic Staphytococcus aureus infection. (A) Severity score (*P ≤ 0.01), body weight (*P = 0.03 and 0.04) and survival (P = 0.04) of SIRT2+/+ and SIRT2−/− mice challenged i.v. with 107 CFU S. aureus (n = 13 and 9). (B,C) Blood and spleen were collected from SIRT2+/+ and SIRT2−/− mice (n = 7 and 8) 48 h after S. aureus challenge. Bacteria were quantified in blood and spleen (B) while IL-6 and IL-12p40 concentrations were quantified in blood (C). Horizontal bars represent the medians. P = 0.07 and 0.04 in (B), and P = 0.95 and 0.12 in (C). (D) Body weight, bacteria in blood 18 h postinfection (P = 0.9), and survival (P = 0.8) of SIRT2+/+ and SIRT2−/− mice challenged i.p. with 105 CFU E. coli (n = 16 per group). (E) Body weight and survival (P = 0.9) of SIRT2+/+ and SIRT2−/− mice challenged i.n. with 30 CFU K. pneumoniae (n = 7 per group).
In the present study, we identified a unique role for SIRT2 in host–pathogen interactions. SIRT2 deficiency promoted bacterial phagocytosis by macrophages but not cytokine production. In agreement with these findings, SIRT2 deficiency protected from chronic staphylococcal infection while having no influence on the course of endotoxemia, TNF-induced shock, fulminant bacterial peritonitis, non-severe bacterial pneumonia, and chronic candidiasis. These observations are particularly relevant in light of the development of pharmacological inhibitors of SIRT2 for clinical applications (43), as they suggest that their usage would not increase susceptibility to bacterial and candidal infections.
SIRT2 was the most highly expressed sirtuin in myeloid cells. Macrophages expressed elevated levels of SIRT2, in accordance previous reports describing SIRT2 expression in microglial cells in vivo (16, 17). Interestingly, SIRT2 deficiency had no major impact on LPS-induced MAPK activation, NF-κB nuclear translocation, and cytokine production in macrophages. Moreover, cytokine levels in blood were similar in SIRT2+/+ and SIRT2−/− endotoxemic mice. Likewise, inflammatory parameters were comparable in SIRT2+/+ and SIRT2−/− mice with experimental stroke and Mycobacterium tuberculosis infection (21, 44). However, contradictory findings have been reported in the literature. While SIRT2 deficiency promoted NF-κB p65 acetylation and p65-dependent gene expression, it was also reported to reduce NF-κB and p38 and JNK MAPKs activation through an increased stability of IκB and activity of MAPK phosphatase-1, respectively (14–16, 18, 20, 45). In these studies, SIRT2 deficiency sustained brain inflammation, colitis, and collagen-induced arthritis, but protected from renal and liver inflammation (15, 16, 18, 20, 45).
The discrepancy of the effects of SIRT2 on inflammatory responses mirrors conflicting results observed for other sirtuins. For example, SIRT1 protected from experimental autoimmune encephalomyelitis, arthritis, lung inflammation, hepatic steatosis, and insulin resistance, but promoted lupus, arthritis, allergic airway disease, and allograft rejection (46–54). SIRT6 has not only been reported to protect against liver fibrosis, atherosclerosis, osteoarthritis, and arthritis but has also been associated with increased TNF production and the development of autoimmune encephalomyelitis and cerebral ischemia (55–62). Different experimental conditions might explain these apparent conflicting results, for example qualitative and quantitative differences in caloric input or subtle variations of NAD+ availability influencing sirtuin activity indirectly. Moreover, SIRT1, SIRT3, and SIRT6 modulate circadian function and are affected by circadian oscillation in the abundance of NAD+ (63, 64). Furthermore, sirtuins might primarily play a role in the development of long-lasting, chronic metabolic, and/or inflammation-related disorders while having a modest impact during acute infectious processes. For instance, SIRT3 deficiency has been reported to increase insulin resistance, diabetic cardiac dysfunction, allograft graft injury, and lung fibrosis, but had no impact on innate immune responses and susceptibility to endotoxemia or bacterial and fungal sepsis (65–73).
Two prior studies examined SIRT2 in the context of infection by intracellular bacteria. SIRT2 deletion in the myeloid compartment had no noticeable impact on host defenses against M. tuberculosis infection as attested by cellular infiltrates, cytokine expression, and long-term bacterial burden in lungs (44). Listeria monocytogenes promoted SIRT2-dependent histone H3 deacetylation and redirected host gene expression to favor infection (74). Whether other microorganisms subvert SIRT2 or other sirtuins at their own benefit is unknown.
We analyzed host responses to extracellular bacteria most frequently isolated from septic patients. Strikingly, SIRT2 deficiency enhanced the engulfment of Gram-positive and Gram-negative bacteria by macrophages, an effect apparently unrelated with a differential expression of phagocytic receptors or microtubule polymerization. This observation was surprising considering that on the one hand SIRT2 deacetylates α-tubulin and destabilizes the microtubule network (8, 41) and on the other end microtubule depolymerizating agents were reported to inhibit phagocytosis (75, 76). Yet, the effects of microtubule depolymerizating agents were tested using an immortalized macrophage-like mouse cell line and human neutrophils that may behave differently than primary BMDMs. As a positive control (77), actin depolymerization efficiently inhibited phagocytosis by BMDMs. Increased glycolysis has been associated with efficient phagocytosis by macrophages (39, 40). SIRT2 deficiency reduced HIF-1α deacetylation and destablization (78), and augmented glycolysis in human fibroblasts (79). We observed that SIRT2 deficiency increased glycolysis and that glycolysis inhibition reduced phagocytosis in BMBMs. Albeit preliminary, these results suggest that SIRT2 may influence phagocytosis through metabolic constraints.
The improved control of bacterial burden during chronic staphylococcal infection might be related to improved phagocytosis but also to enhanced autophagy in SIRT2−/− mice. Autophagy facilitates the clearance of cytoplasmic bacteria (80) and has been involved in host defenses and tolerance to S. aureus infection (81, 82). Hyperacetylation of tubulin stimulated autophagy upon nutrient deprivation, and SIRT2 deficiency increased autophagy in a colorectal cancer cell line (83, 84). Thus, by regulating tubulin acetylation and metabolic activity, SIRT2 may contribute to modulate phagocytic and autophagy defense mechanisms, though the latter has not been formally demonstrated.
From a translational perspective, it was important to define the impact of SIRT2 in preclinical models of infection. A main observation of this study is that SIRT2 deficiency protected mice from chronic staphylococcal infection, while it neither protected nor sensitized mice to TNF-induced shock, endotoxemia, rapidly lethal E. coli peritonitis and mild K. pneumoniae pneumonia. Additionally, SIRT2 deficiency did not influence the development of chronic candidiasis as it did for chronic staphylococcal infection. This may not be surprising considering differences in host–pathogen interactions during fungal and bacterial infections. Whether SIRT2 protects from other chronic bacterial infections should be tested in the future. Nonetheless, these results support the clinical development of SIRT2 inhibitors regarding their infection-related safety profile. This contrasts with inhibitors of HDAC1-11 that impaired innate immune defenses, increased susceptibility to infection in preclinical mouse models and have been associated with severe infections in patients (29, 85–89).
Overall, SIRT2 has a subtle impact on host defense responses to bacterial infections. Considering that sirtuins are intricately linked with metabolism, age-associated dysfunctions and lifespan, it will be important to analyze the impact of SIRT2 on host defenses under metabolic stress conditions and according to age. To conclude, our results are encouraging with respect to developing inhibitors of SIRT2, which are safe in terms of susceptibility to infections, for treating metabolic and neurodegenerative diseases, such as Parkinson’s disease and Huntington’s disease (90, 91).
Animal experimentation was approved by the Service de la Consommation et des Affaires Vétérinaires (SCAV) du Canton de Vaud (Epalinges, Switzerland) under authorizations no. 876.8 and 877.8, and performed according to Swiss and ARRIVE guidelines.
EC, TH, CT, JH, MM, JL, MP, BT, and SL performed in vitro experiments. EC, TH, CT, and DLR performed in vivo experiments. HA-O and JA contributed to reagents. TR conceived the project, designed the experiments, and wrote the paper. All the authors revised the paper.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
This work was supported by grants from the Swiss National Science Foundation (SNSF) 145014, 149511, 173123 (TR), 132492 (HA-O), and 140780 (JA). TR received an interdisciplinary grant from the Faculty of Biology and Medicine of the University of Lausanne (Switzerland). CT was supported by the European Sepsis Academy Horizon 2020 Marie Skłodowska-Curie Action: Innovative Training Network (MSCA-ESA-ITN, grant number 676129). BT and SL were supported by the Summer Undergraduate Research (SUR) program of the University of Lausanne. The work in the JA laboratory was supported by the EPFL, NIH (R01AG043930), Systems X (SySX.ch 2013/153), and Velux Stiftung (1019).
The Supplementary Material for this article can be found online at http://journal.frontiersin.org/article/10.3389/fimmu.2017.01037/full#supplementary-material.
1. Broz P, Monack DM. Newly described pattern recognition receptors team up against intracellular pathogens. Nat Rev Immunol (2013) 13:551–65. doi:10.1038/nri3479
2. Savva A, Roger T. Targeting toll-like receptors: promising therapeutic strategies for the management of sepsis-associated pathology and infectious diseases. Front Immunol (2013) 4:387. doi:10.3389/fimmu.2013.00387
3. Falkenberg KJ, Johnstone RW. Histone deacetylases and their inhibitors in cancer, neurological diseases and immune disorders. Nat Rev Drug Discov (2014) 13:673–91. doi:10.1038/nrd4360
4. Haigis MC, Sinclair DA. Mammalian sirtuins: biological insights and disease relevance. Annu Rev Pathol (2010) 5:253–95. doi:10.1146/annurev.pathol.4.110807.092250
5. Guarente L. Franklin H. Epstein Lecture: sirtuins, aging, and medicine. N Engl J Med (2011) 364:2235–44. doi:10.1056/NEJMra1100831
6. Verdin E. NAD(+) in aging, metabolism, and neurodegeneration. Science (2015) 350:1208–13. doi:10.1126/science.aac4854
7. Preyat N, Leo O. Sirtuin deacylases: a molecular link between metabolism and immunity. J Leukoc Biol (2013) 93:669–80. doi:10.1189/jlb.1112557
8. North BJ, Marshall BL, Borra MT, Denu JM, Verdin E. The human Sir2 ortholog, SIRT2, is an NAD+-dependent tubulin deacetylase. Mol Cell (2003) 11:437–44. doi:10.1016/S1097-2765(03)00038-8
9. Herskovits AZ, Guarente L. Sirtuin deacetylases in neurodegenerative diseases of aging. Cell Res (2013) 23:746–58. doi:10.1038/cr.2013.70
10. Teng YB, Jing H, Aramsangtienchai P, He B, Khan S, Hu J, et al. Efficient demyristoylase activity of SIRT2 revealed by kinetic and structural studies. Sci Rep (2015) 5:8529. doi:10.1038/srep08529
11. Donmez G, Outeiro TF. SIRT1 and SIRT2: emerging targets in neurodegeneration. EMBO Mol Med (2013) 5:344–52. doi:10.1002/emmm.201302451
12. Kozako T, Suzuki T, Yoshimitsu M, Arima N, Honda S, Soeda S. Anticancer agents targeted to sirtuins. Molecules (2014) 19:20295–313. doi:10.3390/molecules191220295
13. Gomes P, Outeiro TF, Cavadas C. Emerging role of sirtuin 2 in the regulation of mammalian metabolism. Trends Pharmacol Sci (2015) 36:756–68. doi:10.1016/j.tips.2015.08.001
14. Rothgiesser KM, Erener S, Waibel S, Luscher B, Hottiger MO. SIRT2 regulates NF-kappaB dependent gene expression through deacetylation of p65 Lys310. J Cell Sci (2010) 123:4251–8. doi:10.1242/jcs.073783
15. Lo Sasso G, Menzies KJ, Mottis A, Piersigilli A, Perino A, Yamamoto H, et al. SIRT2 deficiency modulates macrophage polarization and susceptibility to experimental colitis. PLoS One (2014) 9:e103573. doi:10.1371/journal.pone.0103573
16. Pais TF, Szego EM, Marques O, Miller-Fleming L, Antas P, Guerreiro P, et al. The NAD-dependent deacetylase sirtuin 2 is a suppressor of microglial activation and brain inflammation. EMBO J (2013) 32:2603–16. doi:10.1038/emboj.2013.200
17. Yuan F, Xu ZM, Lu LY, Nie H, Ding J, Ying WH, et al. SIRT2 inhibition exacerbates neuroinflammation and blood-brain barrier disruption in experimental traumatic brain injury by enhancing NF-kappaB p65 acetylation and activation. J Neurochem (2016) 136:581–93. doi:10.1111/jnc.13423
18. Jung YJ, Lee AS, Nguyen-Thanh T, Kim D, Kang KP, Lee S, et al. SIRT2 regulates LPS-induced renal tubular CXCL2 and CCL2 expression. J Am Soc Nephrol (2015) 26:1549–60. doi:10.1681/ASN.2014030226
19. Lee AS, Jung YJ, Kim D, Nguyen-Thanh T, Kang KP, Lee S, et al. SIRT2 ameliorates lipopolysaccharide-induced inflammation in macrophages. Biochem Biophys Res Commun (2014) 450:1363–9. doi:10.1016/j.bbrc.2014.06.135
20. Wang J, Koh HW, Zhou L, Bae UJ, Lee HS, Bang IH, et al. Sirtuin 2 aggravates postischemic liver injury by deacetylating mitogen-activated protein kinase phosphatase-1. Hepatology (2017) 65:225–36. doi:10.1002/hep.28777
21. Krey L, Luhder F, Kusch K, Czech-Zechmeister B, Konnecke B, Outeiro TF, et al. Knockout of silent information regulator 2 (SIRT2) preserves neurological function after experimental stroke in mice. J Cereb Blood Flow Metab (2015) 35:2080–8. doi:10.1038/jcbfm.2015.178
22. Ciarlo E, Heinonen T, Herderschee J, Fenwick C, Mombelli M, Le Roy D, et al. Impact of the microbial derived short chain fatty acid propionate on host susceptibility to bacterial and fungal infections in vivo. Sci Rep (2016) 6:37944. doi:10.1038/srep37944
23. Renner P, Roger T, Bochud PY, Sprong T, Sweep FC, Bochud M, et al. A functional microsatellite of the macrophage migration inhibitory factor gene associated with meningococcal disease. FASEB J (2012) 26:907–16. doi:10.1096/fj.11-195065
24. Roger T, Froidevaux C, Le Roy D, Reymond MK, Chanson AL, Mauri D, et al. Protection from lethal Gram-negative bacterial sepsis by targeting toll-like receptor 4. Proc Natl Acad Sci U S A (2009) 106:2348–52. doi:10.1073/pnas.0808146106
25. Roger T, Schneider A, Weier M, Sweep FC, Le Roy D, Bernhagen J, et al. High expression levels of macrophage migration inhibitory factor sustain the innate immune responses of neonates. Proc Natl Acad Sci U S A (2016) 113:E997–1005. doi:10.1073/pnas.1514018113
26. Savva A, Brouwer MC, Roger T, Valls Seron M, Le Roy D, Ferwerda B, et al. Functional polymorphisms of macrophage migration inhibitory factor as predictors of morbidity and mortality of pneumococcal meningitis. Proc Natl Acad Sci U S A (2016) 113:3597–602. doi:10.1073/pnas.1520727113
27. Meller S, Di Domizio J, Voo KS, Friedrich HC, Chamilos G, Ganguly D, et al. T(H)17 cells promote microbial killing and innate immune sensing of DNA via interleukin 26. Nat Immunol (2015) 16:970–9. doi:10.1038/ni.3211
28. Ciarlo E, Roger T. Screening the impact of sirtuin inhibitors on inflammatory and innate immune responses of macrophages and in a mouse model of endotoxic shock. Methods Mol Biol (2016) 1436:313–34. doi:10.1007/978-1-4939-3667-0_21
29. Roger T, Lugrin J, Le Roy D, Goy G, Mombelli M, Koessler T, et al. Histone deacetylase inhibitors impair innate immune responses to toll-like receptor agonists and to infection. Blood (2011) 117:1205–17. doi:10.1182/blood-2010-05-284711
30. Roger T, Ding X, Chanson AL, Renner P, Calandra T. Regulation of constitutive and microbial pathogen-induced human macrophage migration inhibitory factor (MIF) gene expression. Eur J Immunol (2007) 37:3509–21. doi:10.1002/eji.200737357
31. Roger T, Out TA, Jansen HM, Lutter R. Superinduction of interleukin-6 mRNA in lung epithelial H292 cells depends on transiently increased C/EBP activity and durable increased mRNA stability. Biochim Biophys Acta (1998) 1398:275–84. doi:10.1016/S0167-4781(98)00075-X
32. Giannoni E, Guignard L, Knaup Reymond M, Perreau M, Roth-Kleiner M, Calandra T, et al. Estradiol and progesterone strongly inhibit the innate immune response of mononuclear cells in newborns. Infect Immun (2011) 79:2690–8. doi:10.1128/IAI.00076-11
33. Perreau M, Vigano S, Bellanger F, Pellaton C, Buss G, Comte D, et al. Exhaustion of bacteria-specific CD4 T cells and microbial translocation in common variable immunodeficiency disorders. J Exp Med (2014) 211:2033–45. doi:10.1084/jem.20140039
34. Lugrin J, Ciarlo E, Santos A, Grandmaison G, dos Santos I, Le Roy D, et al. The sirtuin inhibitor cambinol impairs MAPK signaling, inhibits inflammatory and innate immune responses and protects from septic shock. Biochim Biophys Acta (2013) 1833:1498–510. doi:10.1016/j.bbamcr.2013.03.004
35. Vacher G, Ciarlo E, Savova-Bianchi D, Le Roy D, Hantier G, Niculita-Hirzel H, et al. Innate immune sensing of Fusarium culmorum by mouse dendritic cells. J Toxicol Environ Health A (2015) 78:871–85. doi:10.1080/15287394.2015.1051201
36. Roger T, Delaloye J, Chanson AL, Giddey M, Le Roy D, Calandra T. Macrophage migration inhibitory factor deficiency is associated with impaired killing of Gram-negative bacteria by macrophages and increased susceptibility to Klebsiella pneumoniae sepsis. J Infect Dis (2013) 207:331–9. doi:10.1093/infdis/jis673
37. Niedergang F, Di Bartolo V, Alcover A. Comparative anatomy of phagocytic and immunological synapses. Front Immunol (2016) 7:18. doi:10.3389/fimmu.2016.00018
38. Westermann S, Weber K. Post-translational modifications regulate microtubule function. Nat Rev Mol Cell Biol (2003) 4:938–47. doi:10.1038/nrm1260
39. Fensterheim BA, Guo Y, Sherwood ER, Bohannon JK. The cytokine response to lipopolysaccharide does not predict the host response to infection. J Immunol (2017) 198:3264–73. doi:10.4049/jimmunol.1602106
40. Pavlou S, Wang L, Xu H, Chen M. Higher phagocytic activity of thioglycollate-elicited peritoneal macrophages is related to metabolic status of the cells. J Inflamm (Lond) (2017) 14:4. doi:10.1186/s12950-017-0151-x
41. Yuan Q, Zhan L, Zhou QY, Zhang LL, Chen XM, Hu XM, et al. SIRT2 regulates microtubule stabilization in diabetic cardiomyopathy. Eur J Pharmacol (2015) 764:554–61. doi:10.1016/j.ejphar.2015.07.045
42. Annane D, Bellissant E, Cavaillon JM. Septic shock. Lancet (2005) 365:63–78. doi:10.1016/S0140-6736(04)17667-8
43. Villalba JM, Alcain FJ. Sirtuin activators and inhibitors. Biofactors (2012) 38:349–59. doi:10.1002/biof.1032
44. Cardoso F, Castro F, Moreira-Teixeira L, Sousa J, Torrado E, Silvestre R, et al. Myeloid sirtuin 2 expression does not impact long-term Mycobacterium tuberculosis control. PLoS One (2015) 10:e0131904. doi:10.1371/journal.pone.0131904
45. Lin J, Sun B, Jiang C, Hong H, Zheng Y. Sirt2 suppresses inflammatory responses in collagen-induced arthritis. Biochem Biophys Res Commun (2013) 441:897–903. doi:10.1016/j.bbrc.2013.10.153
46. Hah YS, Cheon YH, Lim HS, Cho HY, Park BH, Ka SO, et al. Myeloid deletion of SIRT1 aggravates serum transfer arthritis in mice via nuclear factor-kappaB activation. PLoS One (2014) 9:e87733. doi:10.1371/journal.pone.0087733
47. Kim SR, Lee KS, Park SJ, Min KH, Choe YH, Moon H, et al. Involvement of sirtuin 1 in airway inflammation and hyperresponsiveness of allergic airway disease. J Allergy Clin Immunol (2010) 125:449–60.e14. doi:10.1016/j.jaci.2009.08.009
48. Schug TT, Xu Q, Gao H, Peres-da-Silva A, Draper DW, Fessler MB, et al. Myeloid deletion of SIRT1 induces inflammatory signaling in response to environmental stress. Mol Cell Biol (2010) 30:4712–21. doi:10.1128/MCB.00657-10
49. Zhang J, Lee SM, Shannon S, Gao B, Chen W, Chen A, et al. The type III histone deacetylase Sirt1 is essential for maintenance of T cell tolerance in mice. J Clin Invest (2009) 119:3048–58. doi:10.1172/JCI38902
50. Pfluger PT, Herranz D, Velasco-Miguel S, Serrano M, Tschop MH. Sirt1 protects against high-fat diet-induced metabolic damage. Proc Natl Acad Sci U S A (2008) 105:9793–8. doi:10.1073/pnas.0802917105
51. Legutko A, Marichal T, Fievez L, Bedoret D, Mayer A, de Vries H, et al. Sirtuin 1 promotes Th2 responses and airway allergy by repressing peroxisome proliferator-activated receptor-gamma activity in dendritic cells. J Immunol (2011) 187:4517–29. doi:10.4049/jimmunol.1101493
52. Niederer F, Ospelt C, Brentano F, Hottiger MO, Gay RE, Gay S, et al. SIRT1 overexpression in the rheumatoid arthritis synovium contributes to proinflammatory cytokine production and apoptosis resistance. Ann Rheum Dis (2011) 70:1866–73. doi:10.1136/ard.2010.148957
53. Hu N, Long H, Zhao M, Yin H, Lu Q. Aberrant expression pattern of histone acetylation modifiers and mitigation of lupus by SIRT1-siRNA in MRL/lpr mice. Scand J Rheumatol (2009) 38:464–71. doi:10.3109/03009740902895750
54. Beier UH, Wang L, Bhatti TR, Liu Y, Han R, Ge G, et al. Sirtuin-1 targeting promotes Foxp3+ T-regulatory cell function and prolongs allograft survival. Mol Cell Biol (2011) 31:1022–9. doi:10.1128/MCB.01206-10
55. Lee HS, Ka SO, Lee SM, Lee SI, Park JW, Park BH. Overexpression of sirtuin 6 suppresses inflammatory responses and bone destruction in mice with collagen-induced arthritis. Arthritis Rheum (2013) 65:1776–85. doi:10.1002/art.37963
56. Xiao C, Wang RH, Lahusen TJ, Park O, Bertola A, Maruyama T, et al. Progression of chronic liver inflammation and fibrosis driven by activation of c-JUN signaling in Sirt6 mutant mice. J Biol Chem (2012) 287:41903–13. doi:10.1074/jbc.M112.415182
57. Ailixiding M, Aibibula Z, Iwata M, Piao J, Hara Y, Koga D, et al. Pivotal role of Sirt6 in the crosstalk among ageing, metabolic syndrome and osteoarthritis. Biochem Biophys Res Commun (2015) 466:319–26. doi:10.1016/j.bbrc.2015.09.019
58. Balestrieri ML, Rizzo MR, Barbieri M, Paolisso P, D’Onofrio N, Giovane A, et al. Sirtuin 6 expression and inflammatory activity in diabetic atherosclerotic plaques: effects of incretin treatment. Diabetes (2015) 64:1395–406. doi:10.2337/db14-1149
59. Bruzzone S, Fruscione F, Morando S, Ferrando T, Poggi A, Garuti A, et al. Catastrophic NAD+ depletion in activated T lymphocytes through Nampt inhibition reduces demyelination and disability in EAE. PLoS One (2009) 4:e7897. doi:10.1371/journal.pone.0007897
60. Jiang H, Khan S, Wang Y, Charron G, He B, Sebastian C, et al. SIRT6 regulates TNF-alpha secretion through hydrolysis of long-chain fatty acyl lysine. Nature (2013) 496:110–3. doi:10.1038/nature12038
61. Lee OH, Kim J, Kim JM, Lee H, Kim EH, Bae SK, et al. Decreased expression of sirtuin 6 is associated with release of high mobility group box-1 after cerebral ischemia. Biochem Biophys Res Commun (2013) 438:388–94. doi:10.1016/j.bbrc.2013.07.085
62. Van Gool F, Galli M, Gueydan C, Kruys V, Prevot PP, Bedalov A, et al. Intracellular NAD levels regulate tumor necrosis factor protein synthesis in a sirtuin-dependent manner. Nat Med (2009) 15:206–10. doi:10.1038/nm.1906
63. Peek CB, Affinati AH, Ramsey KM, Kuo HY, Yu W, Sena LA, et al. Circadian clock NAD+ cycle drives mitochondrial oxidative metabolism in mice. Science (2013) 342:1243417. doi:10.1126/science.1243417
64. Masri S, Sassone-Corsi P. Sirtuins and the circadian clock: bridging chromatin and metabolism. Sci Signal (2014) 7:re6. doi:10.1126/scisignal.2005685
65. Akamata K, Wei J, Bhattacharyya M, Cheresh P, Bonner MY, Arbiser JL, et al. SIRT3 is attenuated in systemic sclerosis skin and lungs, and its pharmacologic activation mitigates organ fibrosis. Oncotarget (2016) 7:69321–36. doi:10.18632/oncotarget.12504
66. Beier UH, Angelin A, Akimova T, Wang L, Liu Y, Xiao H, et al. Essential role of mitochondrial energy metabolism in Foxp3(+) T-regulatory cell function and allograft survival. FASEB J (2015) 29:2315–26. doi:10.1096/fj.14-268409
67. Bindu S, Pillai VB, Kanwal A, Samant S, Mutlu GM, Verdin E, et al. SIRT3 blocks myofibroblast differentiation and pulmonary fibrosis by preventing mitochondrial DNA damage. Am J Physiol Lung Cell Mol Physiol (2017) 312:L68–78. doi:10.1152/ajplung.00188.2016
68. Gao J, Zheng Z, Gu Q, Chen X, Liu X, Xu X. Deacetylation of MnSOD by PARP-regulated SIRT3 protects retinal capillary endothelial cells from hyperglycemia-induced damage. Biochem Biophys Res Commun (2016) 472:425–31. doi:10.1016/j.bbrc.2015.12.037
69. Lantier L, Williams AS, Williams IM, Yang KK, Bracy DP, Goelzer M, et al. SIRT3 is crucial for maintaining skeletal muscle insulin action and protects against severe insulin resistance in high-fat-fed mice. Diabetes (2015) 64:3081–92. doi:10.2337/db14-1810
70. Morigi M, Perico L, Rota C, Longaretti L, Conti S, Rottoli D, et al. Sirtuin 3-dependent mitochondrial dynamic improvements protect against acute kidney injury. J Clin Invest (2015) 125:715–26. doi:10.1172/JCI77632
71. Sosulski ML, Gongora R, Feghali-Bostwick C, Lasky JA, Sanchez CG. Sirtuin 3 deregulation promotes pulmonary fibrosis. J Gerontol A Biol Sci Med Sci (2017) 72:595–602. doi:10.1093/gerona/glw151
72. Yu W, Gao B, Li N, Wang J, Qiu C, Zhang G, et al. Sirt3 deficiency exacerbates diabetic cardiac dysfunction: role of Foxo3A-Parkin-mediated mitophagy. Biochim Biophys Acta (2017) 1863:1973–83. doi:10.1016/j.bbadis.2016.10.021
73. Ciarlo E, Heinonen T, Lugrin J, Acha-Orbea H, Le Roy D, Auwerx J, et al. Sirtuin 3 deficiency does not alter host defenses against bacterial and fungal infections. Sci Rep (2017) 7:3853. doi:10.1038/s41598-017-04263-x
74. Eskandarian HA, Impens F, Nahori MA, Soubigou G, Coppee JY, Cossart P, et al. A role for SIRT2-dependent histone H3K18 deacetylation in bacterial infection. Science (2013) 341:1238858. doi:10.1126/science.1238858
75. Horwitz SB, Chia GH, Harracksingh C, Orlow S, Pifko-Hirst S, Schneck J, et al. Trifluoperazine inhibits phagocytosis in a macrophagelike cultured cell line. J Cell Biol (1981) 91:798–802. doi:10.1083/jcb.91.3.798
76. Roberts RL, Nath J, Friedman MM, Gallin JI. Effects of taxol on human neutrophils. J Immunol (1982) 129:2134–41.
77. Hartwig JH, Stossel TP. Interactions of actin, myosin, and an actin-binding protein of rabbit pulmonary macrophages. III. Effects of cytochalasin B. J Cell Biol (1976) 71:295–303. doi:10.1083/jcb.71.1.295
78. Seo KS, Park JH, Heo JY, Jing K, Han J, Min KN, et al. SIRT2 regulates tumour hypoxia response by promoting HIF-1alpha hydroxylation. Oncogene (2015) 34:1354–62. doi:10.1038/onc.2014.76
79. Cha Y, Han MJ, Cha HJ, Zoldan J, Burkart A, Jung JH, et al. Metabolic control of primed human pluripotent stem cell fate and function by the miR-200c-SIRT2 axis. Nat Cell Biol (2017) 19:445–56. doi:10.1038/ncb3517
80. Huang J, Brumell JH. Bacteria-autophagy interplay: a battle for survival. Nat Rev Microbiol (2014) 12:101–14. doi:10.1038/nrmicro3160
81. Maurer K, Reyes-Robles T, Alonzo F III, Durbin J, Torres VJ, Cadwell K. Autophagy mediates tolerance to Staphylococcus aureus alpha-toxin. Cell Host Microbe (2015) 17:429–40. doi:10.1016/j.chom.2015.03.001
82. Wu HM, Wang J, Zhang B, Fang L, Xu K, Liu RY. CpG-ODN promotes phagocytosis and autophagy through JNK/P38 signal pathway in Staphylococcus aureus-stimulated macrophage. Life Sci (2016) 161:51–9. doi:10.1016/j.lfs.2016.07.016
83. Geeraert C, Ratier A, Pfisterer SG, Perdiz D, Cantaloube I, Rouault A, et al. Starvation-induced hyperacetylation of tubulin is required for the stimulation of autophagy by nutrient deprivation. J Biol Chem (2010) 285:24184–94. doi:10.1074/jbc.M109.091553
84. Inoue T, Nakayama Y, Li Y, Matsumori H, Takahashi H, Kojima H, et al. SIRT2 knockdown increases basal autophagy and prevents postslippage death by abnormally prolonging the mitotic arrest that is induced by microtubule inhibitors. FEBS J (2014) 281:2623–37. doi:10.1111/febs.12810
85. Li Y, Liu B, Zhao H, Sailhamer EA, Fukudome EY, Zhang X, et al. Protective effect of suberoylanilide hydroxamic acid against LPS-induced septic shock in rodents. Shock (2009) 32:517–23. doi:10.1097/SHK.0b013e3181a44c79
86. Mombelli M, Lugrin J, Rubino I, Chanson AL, Giddey M, Calandra T, et al. Histone deacetylase inhibitors impair antibacterial defenses of macrophages. J Infect Dis (2011) 204:1367–74. doi:10.1093/infdis/jir553
87. Zhang LT, Yao YM, Lu JQ, Yan XJ, Yu Y, Sheng ZY. Sodium butyrate prevents lethality of severe sepsis in rats. Shock (2007) 27:672–7. doi:10.1097/SHK.0b013e31802e3f4c
88. Gojo I, Jiemjit A, Trepel JB, Sparreboom A, Figg WD, Rollins S, et al. Phase 1 and pharmacologic study of MS-275, a histone deacetylase inhibitor, in adults with refractory and relapsed acute leukemias. Blood (2007) 109:2781–90. doi:10.1182/blood-2006-05-021873
89. Ciarlo E, Savva A, Roger T. Epigenetics in sepsis: targeting histone deacetylases. Int J Antimicrob Agents (2013) 42(Suppl):S8–12. doi:10.1016/j.ijantimicag.2013.04.004
90. Outeiro TF, Kontopoulos E, Altmann SM, Kufareva I, Strathearn KE, Amore AM, et al. Sirtuin 2 inhibitors rescue alpha-synuclein-mediated toxicity in models of Parkinson’s disease. Science (2007) 317:516–9. doi:10.1126/science.1143780
Keywords: sirtuin, innate immunity, cytokine, macrophage, phagocytosis, sepsis, histone deacetylase, metabolism
Citation: Ciarlo E, Heinonen T, Théroude C, Herderschee J, Mombelli M, Lugrin J, Pfefferlé M, Tyrrell B, Lensch S, Acha-Orbea H, Le Roy D, Auwerx J and Roger T (2017) Sirtuin 2 Deficiency Increases Bacterial Phagocytosis by Macrophages and Protects from Chronic Staphylococcal Infection. Front. Immunol. 8:1037. doi: 10.3389/fimmu.2017.01037
Received: 08 April 2017; Accepted: 11 August 2017;
Published: 28 August 2017
Edited by:
Abhay Satoskar, The Ohio State University Columbus, United StatesReviewed by:
Ricardo Silvestre, Instituto de Pesquisa em Ciências da Vida e da Saúde (ICVS), PortugalCopyright: © 2017 Ciarlo, Heinonen, Théroude, Herderschee, Mombelli, Lugrin, Pfefferlé, Tyrrell, Lensch, Acha-Orbea, Le Roy, Auwerx and Roger. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Thierry Roger, dGhpZXJyeS5yb2dlckBjaHV2LmNo
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.