 Brandi M. Wynne
Brandi M. Wynne Li Zou2
Li Zou2 Valerie Linck
Valerie Linck Robert S. Hoover
Robert S. Hoover Douglas C. Eaton
Douglas C. Eaton
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 25 July 2017
Sec. Inflammation
Volume 8 - 2017 | https://doi.org/10.3389/fimmu.2017.00766
This article is part of the Research Topic Cytokine-ion channel interactions in pulmonary inflammation View all 16 articles
Acute lung injury leading to acute respiratory distress (ARDS) is a global health concern. ARDS patients have significant pulmonary inflammation leading to flooding of the pulmonary alveoli. This prevents normal gas exchange with consequent hypoxemia and causes mortality. A thin fluid layer in the alveoli is normal. The maintenance of this thin layer results from fluid movement out of the pulmonary capillaries into the alveolar interstitium driven by vascular hydrostatic pressure and then through alveolar tight junctions. This is then balanced by fluid reabsorption from the alveolar space mediated by transepithelial salt and water transport through alveolar cells. Reabsorption is a two-step process: first, sodium enters via sodium-permeable channels in the apical membranes of alveolar type 1 and 2 cells followed by active extrusion of sodium into the interstitium by the basolateral Na+, K+-ATPase. Anions follow the cationic charge gradient and water follows the salt-induced osmotic gradient. The proximate cause of alveolar flooding is the result of a failure to reabsorb sufficient salt and water or a failure of the tight junctions to prevent excessive movement of fluid from the interstitium to alveolar lumen. Cytokine- and chemokine-induced inflammation can have a particularly profound effect on lung sodium transport since they can alter both ion channel and barrier function. Cytokines and chemokines affect alveolar amiloride-sensitive epithelial sodium channels (ENaCs), which play a crucial role in sodium transport and fluid reabsorption in the lung. This review discusses the regulation of ENaC via local and systemic cytokines during inflammatory disease and the effect on lung fluid balance.
The maintenance of a thin fluid layer on the surface of the alveolar epithelium is critical for respiration. Two primary mechanisms regulate this fluid layer: Starling’s forces and active sodium (Na+) transport. Starling’s forces determine the movement of water from intravascular to extravascular or interstitial spaces caused by hydrostatic and oncotic pressures. An increase in pulmonary vascular pressure accounts for the increased alveolar flooding seen in cardiogenic pulmonary edema. However, the other regulator of the thickness of the alveolar fluid layer is the active transport of Na+, followed by potential-driven anion movement through cystic fibrosis transmembrane conductance regulator, and the aquaporin-mediated transport of water. The epithelial sodium channel (ENaC) is critical in the maintenance of the epithelial fluid layer. This review focuses on the primary physiological mechanisms required to maintain and regulate this layer and is an overview of the pathophysiological mechanisms of cytokine-mediated ENaC regulation in the lung (Figure 1).
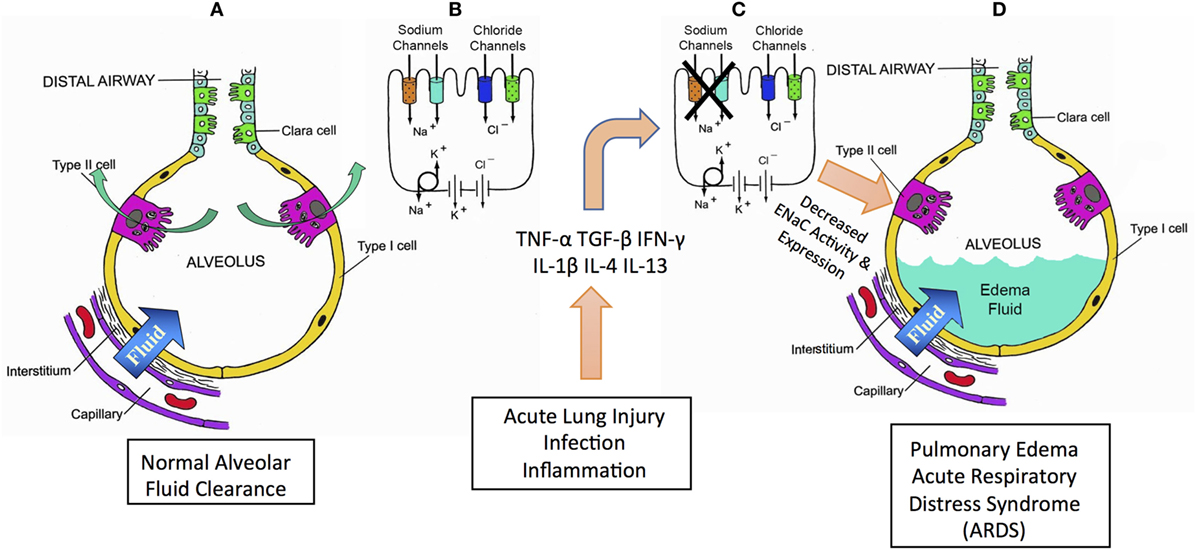
Figure 1. Summary schematic for role of cytokines in mediating pulmonary edema and acute respiratory distress syndrome (ARDS). Acute lung injury (ALI), ARDS, and pneumonia are all pathologies characterized by lung edema and alveolar flooding. Pneumonia mortality is typically caused by flooding of the pulmonary alveoli, preventing normal gas exchange and consequent hypoxemia. Airways normally have a critically regulated fluid layer essential for normal gas exchange and removal of foreign particulates from the airway (A). Maintaining this fluid layer in the alveoli depends critically on sodium reabsorption mediated by epithelial sodium channels (ENaCs) and CFTR chloride channels (B). During ALI, sepsis, inflammation or infection, inflammatory cytokines are produced that inhibit ENaC (C). A decrease in ENaC reabsorption allows fluid to accumulate in the alveoli causing alveolar flood in loss of normal gas exchange and consequent hypoxemia (D).
The primary function of the airways is exchange of gases; thus, both the anatomy and physiology of the lung have evolved to distribute gases efficiently. The diffusion of gases is facilitated in the alveoli by the large total surface area, coupled with thin, yet strong and elastic membranes (1). Human lungs are composed of a series of branched tubes, where conducting airways lead to the terminal respiratory units that are in close proximity to the vasculature (2–4).
The primary respiratory units, or alveoli, are composed of a single, polarized, epithelial cell layer that separates a gas-filled compartment and the pulmonary circulation (5). The two predominant cell types in this cell barrier are the squamous type I (AT1) and cuboidal type 2 (AT2) cells. The majority of the alveolar surface area consists of AT1 cells: the remainder of the area (≈2–5%) is AT2 cells. Both cell types contribute to alveolar fluid transport (6–10). These cells are responsible for Na+ transport from the apical to basolateral surface and maintenance of a thin layer of isotonic fluid on the alveolar surface. The AT2 cells have an additional function: they are also responsible for the secretion of surfactant, which is necessary to lower the surface tension at the interface of air and water and increase lung compliance. Overall, this anatomical structure and physiology ensures that the alveolar spaces remain open for gas exchange.
Regulation of the fluid interface occurs primarily through regulating Na+ uptake via ENaC in both AT1 and AT2 cells. After ENaC-mediated entry of Na+ across the apical membrane, Na+ leaves the cell across the basolateral membrane via the Na+–K+ ATPase and enters the interstitium where it is in equilibrium with vascular Na+. Some investigators have suggested that regulation of the ATPase also plays a role in controlling trans-epithelial Na+ transport (11–15); however, we will not consider ATPase regulation in this review. The paradigm in which vectorial Na+ transport is considered a primary drive for fluid transport from the alveolar surface has been established by numerous studies where pharmacological inhibitors of apical Na+ channels have been shown to reduce the rate at which fluid is cleared (16–21).
Epithelial sodium channel is composed of three homologous subunits, such as α, β, and γ. Together, these subunits assemble in the endoplasmic reticulum and traffic to the apical membrane and are highly selective for Na+ (22). Using ENaCα-subunit knock-out mice, investigators first showed the importance of ENaCα for proper lung function: neonates lacking ENaCα died within 40 h of birth (23). The α subunit is the ionophoric component of the heteromultimer and is required for the expression and assembly of functional ENaCs at the apical membrane. The importance of ENaC to normal lung function is underscored by the phenotype of several monogenetic disorders that affect ENaC. Patients with pseudohypoaldosteronism (PHA), a condition resulting from ENaC partial loss-of-function, were found to have twice the volume of airway surface liquid compared normal levels (24). Mice lacking the ubiquitin ligase, NEDD4-2, had increased levels of ENaC expression and increased ENaC-mediated current in AT2 cells (25). Additionally, overexpression of ENaCβ in an ENaCβ transgenic mouse model leads to airway dehydration and mucous obstruction, comparable to many features observed in cystic fibrosis (CF) (26). Together, these studies highlight the importance of proper ENaC expression and regulation for the airways.
Understanding the regulation of ENaC is significant for understanding lung fluid balance, as ENaC dysregulation is the source of pathological lung edema. In recent years, probably because monogenetic disorders often alter ENaC trafficking, much of the focus has examined how regulation of the number of channels at the apical membrane of alveolar epithelial cells can alter Na+ transport. However, since ENaC is an ion channel, regulating how much of the time the channel spends open (the open probability, Po) is also important. Both Liddle’s syndrome and PHA type 1 (PHA 1) are conventionally described as changes in channel density (an increase and decrease, respectively); however, examination of single ENaCs in these two syndromes shows that ENaC Po also changes. There are an observed increase in channel activity in Liddle’s (27) and a decrease in activity in PHA I (28). Steroid hormones increase Na+ transport and are often thought to do so by increasing subunit transcription and translation. Although Frindt and Palmer (29) have shown in Na+-transporting epithelial tissue that this is indeed true, the increase in subunit density accounts for less than 25% of the increase in trans-epithelial Na+ current implying that the remaining 75% is due to an increase in single channel Po. Single channel recordings show that acute application of steroids dramatically increases single channel Po (30–32).
Kleyman, Hughey, and their co-workers have shown that the α and γ subunits of ENaC must be proteolytically cleaved to be active loops (33–37). Some investigators have suggested that such cleavage might be a mechanism by which ENaC in the apical membrane could be regulated. In fact, proteolysis does appear to be required for ENaC to have any appreciable activity, and may be required for it to reach the membrane. As such, proteolysis appears to be, more or less, an all-or-none phenomenon: channels that are uncleaved are capable of little if any activity. However, under conditions of normal Na+ transport most channels are cleaved. Under these conditions, cleaved channels are capable of a wide range of activity by changing their Po.
Changes in membrane ENaC can occur by changing the rate of insertion into the membrane after transcription and translation (38). However, in any time frame less than 24 h, ENaC in the membrane is altered by recycling from intracellular pools into the membrane (22) or internalization of ENaC into recycling or degradative pools. Removal of ENaC occurs primarily via the ubiquitin ligase, NEDD 4-2, which targets ENaC for removal and proteosomal degradation (39, 40).
Therefore, in this review, we address both the regulation of Po in cleaved channels and change in membrane channel protein density.
The regulation of ENaC occurs via multiple, redundant systems to ensure that Na+ transport is not limited. ENaC is regulated by a many agents including transmitters interacting with G-protein-coupled receptors (GPCRs), circulating hormones, cytokines and chemokines, and reactive oxygen and nitrogen species. The regulation of ENaC via hormones and GPCRs is not a primary focus of this review, but we briefly review ENaC activation and regulation via steroids since their actions often interact with the activities of cytokines and chemokines.
In the lung, the glucocorticoid receptor (GR) is the primary receptor for corticosteroids (41–43). Once activated, the GR activates response elements inducing the transcription of signaling kinases, such as the serum- and glucocorticoid-regulated kinase 1 (30, 44, 45). Ligand-mediated activation of the GR via corticosteroids is used clinically as an anti-inflammatory treatment. The positive effects of corticosteroid therapy lie in the ability of the GR to bind to and inhibit nuclear factor kappa-light-chain-enhancer of activated B cells (Nf-κB) (46–48). Nf-κB is an important mediator of cytokine signaling. This transcription factor increases cyclooxygenase 2-induced prostaglandin production, as well as increases other proinflammatory factors (49). Corticosteroids reduce inflammation propagated via Nf-κB-mediated mechanisms but may not affect inflammation mediated by other signaling pathways. Indeed, GR activation may actually augment some downstream signaling pathways, such as those mediated through Smad proteins (50). This distinction is important because of the many heterogeneous pathways activated by each cytokine (51–54).
Regulation of the air/water interface is crucial for gas exchange, as the amount of the alveolar fluid layer must be precise. With injury, the inability of the lungs to clear this fluid can lead to pulmonary edema. Increased fluid accumulation can result from compromised ENaC function or when there is an asymmetrical hydrostatic force from the vasculature, pushing fluid from capillaries into the alveolar space (e.g., pulmonary hypertension) (5). In addition, tight junctions that maintain structural integrity and a tight epithelial layer can be disrupted, resulting in increased permeability. In fact, high levels of pulmonary inflammation causing airway tight junction damage that compromise alveolar barrier function is a primary cause of epithelial injury (55). And lastly, many proinflammatory and noxious factors cause changes in Na+ transport. Dysfunction in any single factor can lead to a dysregulation of the alveolar fluid (5). Because of the increased morbidity and mortality associated with alveolar fluid accumulation, understanding the mechanisms that regulate these factors are vital.
During inflammation most cells are capable of secreting a variety of small molecular weight proteins, called cytokines and chemokines, which communicate the inflammatory signals. In the airway, resident immune cells are mostly the alveolar macrophages; however, during infection or inflammation, other mononuclear and granular immune cells infiltrate (56). Several studies have proposed a role for the airway epithelium in propagating the immune response, especially as a “first responder” since the airway is the first to sense viral and bacterial pathogens as they enter the body. This layer can be an active participant in the immune response, producing a variety of cytokines and chemokines, as well as exclusive epithelial-derived cytokines (55, 57, 58).
Direct interaction with pathogens, such as influenza, reduces ENaC activity (59). However, other evidence suggests that some of the more chronic effects of pathogens may be via noxae-stimulated chronic cytokine production (60–62). Additionally, inflammatory activation of the airway epithelium can result in local nitric oxide (NO) production, most likely via increased cytokine production, further reducing ENaC activity and fluid transport (59, 63–67). Cytokines frequently increase local levels of reactive oxygen species (ROS) as well. Interestingly, ROS has been shown to activate ENaC at relatively low concentrations but to inhibit ENaC at higher concentrations often associated with massive pathogen-induced cytokine production (40, 68, 69). The overall redox environment of the alveoli is crucial and can rapidly change, often driven by high levels of Rac1-NADPH oxidase activity in AT1 cells (67, 70).
Some of the earliest studies revealed a correlation between large and sustained proinflammatory cytokine increases in bronchioalveolar lavage (BAL) fluid and an unfavorable outcome in acute respiratory distress syndrome (ARDS) (71). Overall, increased cytokine levels from lung injury can quickly lead to the accumulation of alveolar fluid, edema, and then acute respiratory distress. Thus, proinflammatory cytokines and chemokines produce a feed forward cycle decreasing lung Na+ transporter expression, as well as activity.
The TNF super family comprises 19 members and was originally named for its role in apoptosis (53). The best-studied member of this family is TNF-α, which plays a role in propagating the immune response and secretion of other cytokines. TNF-α was implicated in the pathogenesis of pulmonary edema, and increased levels were observed in patients with ARDS (72, 73). Monocytes and macrophages produce significant TNF-α, but it is also produced by alveolar epithelial cells following lipopolysaccharide stimulation (74).
Although TNF-α can bind to two different receptors that are linked to separate signaling pathways, much of the work in the airway has focused on TNF receptor 1. The effect that TNF-α elicits on ENaC function, and alveolar liquid clearance, seems to be critically dependent upon receptor activation or receptor-independent mechanisms and has been shown using both in vitro and in vivo models (75–78).
Tumor necrosis factor receptor 1-mediated activation of NF-κB increases cytokine (IL-1, IL-8, IL-6) and chemokine production. It also increases the expression of adhesion molecules including selectins, vascular cell adhesion molecules, and intercellular adhesion molecule (ICAM)-1 (53, 79, 80). In freshly isolated AT2 cells, TNF-α decreased α- and γ-ENaC mRNA and protein levels and reduced amiloride-sensitive trans-epithelial current (75, 78).
Tumor necrosis factor-α also plays an especially important role in endothelial activation, as well as disturbing the epithelial tight junction barrier. Disruption of the tight junctions not only leads to respiratory distress and increased exudate but also may reduce alveolar fluid clearance as well (81). TNF-α reduces the expression of tight junction proteins, including the claudins and zonula occludens protein 1, thus increasing alveolar permeability (55, 82). Consequently, TNF-α has a critical and multi-faceted role in the development of ARDS. TNF-α not only regulates Na+ and water clearance but also disrupts tight junction barriers and endothelial integrity and contributes to a pro-inflammatory environment.
Interestingly, TNF-α contains not only a receptor-binding domain but also a lectin-like domain (referred to as a TIP domain) that is spatially distinct from the receptor-binding site (83, 84). TNF-α produces an opposite response when there is binding of the lectin-like domain, or TIP, to certain oligosaccharides at high concentrations of TNF-α. This process increases Na+ uptake in AT2 cells and may account for the differential responses to TNF-α (85–87). In vivo, a peptide analog of TIP increased clearance in a murine flooded-lung model (85). Czikora and colleagues also demonstrated that this TIP domain directly binds to and, then, activates ENaC (83). This implies an endogenous mechanism to limit the effects of high TNF-α concentrations. Use of this may become a novel method in counteracting reduced alveolar clearance.
Transforming growth factor-β1 is a pathogenic cytokine, which has been implicated in the early phase of acute lung injury (ALI) prior to ARDS (72, 88). TGF-β1 levels were increased in ARDS patients compared to healthy controls (89). Furthermore, active TGF-β1 levels were more than doubled in the epithelial lining fluid from ARDS patients (90). As mentioned earlier, corticosteroids are a common tool to reduce inflammation and aid in lung clearance. Interestingly, TGF-β actually reduces the ability to produce multiple steroids, possibly leading to the inability for self-healing and furthering inflammatory damage, in addition to the activation of multiple Smad pathways (50, 52). Some of these pathways may be insensitive to corticosteroid treatment. However, there is still much to learn regarding which Smad-mediated pathways are downstream of TGF-β signaling during ALI and ARDS.
Other studies have specifically have explored the role of TGF-β in alveolar flooding. Using a bleomycin-induced lung injury model, TGF-β1-inducible genes were dramatically increased as early as 2 days, suggesting that TGF-β1 may precede alveolar flooding (91). Of interest, TGF-β may actually remain latent locally, covalently attached to a latency-associated peptide (LAP); pulmonary epithelial cells can activate and cause dissociation of TGF-β from LAP (92–94). One member of the integrin family, ανβ6, was recently shown to be a ligand for LAP (93). ανβ6 is expressed normally at lower levels, yet increased significantly with injury revealing a novel mechanism for rapid and local TGF-β activation (95). TGF-β is also redox sensitive, and in vitro models of increased ROS via ionizing radiation revealed another mechanism for TGF-β activation (96). Together, these studies show multiple, redundant possibilities for systemic and paracrine TGF-β activation during lung injury.
One of the first studies to directly implicate TGF-β1 in regulating ENaC was by Frank and colleagues. They showed that TGF-β1 reduced amiloride-sensitive Na+ transport in lung epithelial cells. Additionally, TGF-β1 reduced αENaC mRNA and protein expression via an ERK1/2 pathway in a model of ALI, thus promoting alveolar edema (97). In vivo studies then showed that TGF-β1 reduces vectorial Na+ and water transport and that this process occurs independently from increases in epithelial permeability (97, 98). Interestingly, TGF-β was also found to have an integral role in ENaC trafficking. Peters and colleagues were the first to demonstrate this acute regulation of ENaC in the lung; they found that TGF-β induces ENaC internalization via interaction with ENaCβ (99). In summary, TGF-β has been implicated in multiple mechanisms reducing ENaC expression and apical localization, thus contributing to the pathophysiology of ARDS and pulmonary edema (100).
The interferons (IFN) are a family of proteins originally classified by their ability to reduce viral replication. This family consists of both Type I and Type II IFNs; INF-γ is the only member of the Type II IFN family and is structurally unrelated to the other IFNs. During inflammation, INF-γ is secreted by multiple immune cells, but mostly by T lymphocytes. INF-γ increases ICAM-1 levels and increases NO production via inducible nitric oxide synthase. Little is known about the role of INF-γ in ENaC regulation; however, studies using human bronchial epithelial cells (BECs) showed that INF-γ treatment significantly reduced trans-epithelial Na+ transport in normal human BECs (101).
Several interleukins are correlated with the early stages of ALI; however, the best studied is IL-1β. This cytokine plays a diverse role in the pathogenesis of ALI and ARDS. IL-1β levels are increased in the BAL fluid, as well as the pulmonary edema fluid, of patients with ALI (102–106). IL-1β levels are higher in the pulmonary lavage fluids compared to serum suggesting that there is a local, pulmonary source for IL-1β similar to that of TGF-β (71, 104). An earlier study by Pugin and colleagues suggested that of the cytokines present in the BAL fluid, IL-1β is the most biologically active, and others have suggested that the source may be from early-infiltrating neutrophils (103, 107). BAL fluids from ARDS patients applied to AT2 cells increased ICAM-1 expression, while IL-1 inhibition reduced the increase in ICAM-1 (103).
IL-1β also seems to have significant effects on endothelial leakage and permeability. In vitro, IL-1β treatment significantly increased microvascular permeability (108). Several studies have also demonstrated that when given intratracheally, IL-1β increased endothelial permeability and lung leak (108–111). More recently, IL-1β has been shown to directly affect ENaC expression. Incubation with IL-1β reduced ENaC mRNA protein expression, possibly through promoter inhibition and a p38 MAPK-dependent mechanism. Additionally, IL-1β application reduced apical ENaC protein and amiloride-sensitive trans-epithelial current and Na+ flux (112).
Other studies have tried to reverse IL-1β effects. In vitro modeling suggests that the reduction of IL-1β, via suppressor of cytokine signaling-1, can rescue the IL-1β-mediated suppression of ENaCs (113). When investigating patients with ALI, those who had an increased activation of the stress protein response (SPR) positively correlated with preserved alveolar clearance rates (114). Thus, activation of this SPR during immune-related injury may ameliorate effects of IL-1β, if used as a “preconditioning” agent (115).
Classically, increases in IL-4 and IL-13 are associated with an increased goblet-cell hyperplasia and mucous secretion. These cytokines are implicated in allergic airway diseases and CF and contribute to reduced ciliary movement reducing the ability to clear the airways. These related cytokines frequently share signaling cascades and receptor subunits, such as the IL-4 receptor (116). However, studies in airway epithelial cells from human bronchi suggest that these cytokines may also alter ion transport. IL-4 significantly reduced ENaC subunits γ and β; interestingly, αENaC levels were not altered. IL-4 and IL-13 treatments reduced amiloride-sensitive short circuit current (using an Ussing chamber), which was reversed with an IL-4 receptor antagonist (116). Although these studies were investigating allergic diseases, one could infer a similar involvement in a variety of other inflammatory conditions where there are increased IL-4/IL-13 levels and reduced ENaC function.
Delineating the role of pro-inflammatory cytokines is important for the understanding of alveolar flooding and ALI; however, the lack of anti-inflammatory cytokines also plays a crucial role in mediating the “balance” necessary for regulating the epithelial fluid lining. Studies have shown that there is an increased mortality when there are reduced levels of “anti-inflammatory” cytokines, such as IL-10 and the IL-1 receptor antagonists (117). Much work is needed to understand the diverse and redundant roles of cytokines in disease progression. Nonetheless, pro-inflammatory cytokines seem to reduce the total expression, apical localization, and activity of ENaC in the lungs via multiple mechanisms (Figure 1). Given the prominent role for ENaC in maintaining alveolar fluid levels, understanding how inflammatory cytokines regulate ENaC will allow for the development of therapies to treat these complex diseases.
BW and DE conceived and wrote the manuscript. LZ, VL, H-PM, and RH edited and approved the manuscript.
The authors declare no commercial or financial relationships that could be construed as a potential conflict of interest.
We would like to thank Dr. Rudolph Lucas for extensive discussions and suggestions.
This study was funded by the National Institutes of Health T32 [DK07656] to BW, NIDDK R01 [DK100582] to H-PM, and R37 [DK030963] to DE, and Department of Veteran’s Affairs MERIT Award [I01BX002322-01] to RH.
1. West JB. Comparative physiology of the pulmonary circulation. Compr Physiol (2011) 1:1525–39. doi:10.1002/cphy.c090001
2. KH SNaA. The Structure of the Lungs Relative to Their Principle Function. Philadelphia, PA: Saunders (1988).
3. Matthay MA, Folkesson HG, Clerici C. Lung epithelial fluid transport and the resolution of pulmonary edema. Physiol Rev (2002) 82:569–600. doi:10.1152/physrev.00003.2002
4. Phalen RF, Oldham MJ. Tracheobronchial airway structure as revealed by casting techniques. Am Rev Respir Dis (1983) 128:S1–4.
5. Eaton DC, Helms MN, Koval M, Bao HF, Jain L. The contribution of epithelial sodium channels to alveolar function in health and disease. Annu Rev Physiol (2009) 71:403–23. doi:10.1146/annurev.physiol.010908.163250
6. Borok Z, Liebler JM, Lubman RL, Foster MJ, Zhou B, Li X, et al. Na transport proteins are expressed by rat alveolar epithelial type I cells. Am J Physiol Lung Cell Mol Physiol (2002) 282:L599–608. doi:10.1152/ajplung.00130.2000
7. Flodby P, Kim YH, Beard LL, Gao D, Ji Y, Kage H, et al. Knockout mice reveal a major role for alveolar epithelial type I cells in alveolar fluid clearance. Am J Respir Cell Mol Biol (2016) 55:395–406. doi:10.1165/rcmb.2016-0005OC
8. Helms MN, Chen XJ, Ramosevac S, Eaton DC, Jain L. Dopamine regulation of amiloride-sensitive sodium channels in lung cells. Am J Physiol Lung Cell Mol Physiol (2006) 290:L710–22. doi:10.1152/ajplung.00486.2004
9. Helms MN, Self J, Bao HF, Job LC, Jain L, Eaton DC. Dopamine activates amiloride-sensitive sodium channels in alveolar type I cells in lung slice preparations. Am J Physiol Lung Cell Mol Physiol (2006) 291:L610–8. doi:10.1152/ajplung.00426.2005
10. Johnson MD, Bao HF, Helms MN, Chen XJ, Tigue Z, Jain L, et al. Functional ion channels in pulmonary alveolar type I cells support a role for type I cells in lung ion transport. Proc Natl Acad Sci U S A (2006) 103:4964–9. doi:10.1073/pnas.0600855103
11. Adir Y, Welch LC, Dumasius V, Factor P, Sznajder JI, Ridge KM. Overexpression of the Na-K-ATPase alpha2-subunit improves lung liquid clearance during ventilation-induced lung injury. Am J Physiol Lung Cell Mol Physiol (2008) 294:L1233–7. doi:10.1152/ajplung.00076.2007
12. Comellas AP, Kelly AM, Trejo HE, Briva A, Lee J, Sznajder JI, et al. Insulin regulates alveolar epithelial function by inducing Na+/K+-ATPase translocation to the plasma membrane in a process mediated by the action of Akt. J Cell Sci (2010) 123:1343–51. doi:10.1242/jcs.066464
13. Gusarova GA, Trejo HE, Dada LA, Briva A, Welch LC, Hamanaka RB, et al. Hypoxia leads to Na,K-ATPase downregulation via Ca(2+) release-activated Ca(2+) channels and AMPK activation. Mol Cell Biol (2011) 31:3546–56. doi:10.1128/MCB.05114-11
14. Lecuona E, Sun H, Chen J, Trejo HE, Baker MA, Sznajder JI. Protein kinase A-Ialpha regulates Na,K-ATPase endocytosis in alveolar epithelial cells exposed to high CO(2) concentrations. Am J Respir Cell Mol Biol (2013) 48:626–34. doi:10.1165/rcmb.2012-0373OC
15. Lecuona E, Trejo HE, Sznajder JI. Regulation of Na,K-ATPase during acute lung injury. J Bioenerg Biomembr (2007) 39:391–5. doi:10.1007/s10863-007-9102-1
16. Berthiaume Y, Staub NC, Matthay MA. Beta-adrenergic agonists increase lung liquid clearance in anesthetized sheep. J Clin Invest (1987) 79:335–43. doi:10.1172/JCI112817
17. Finley N, Norlin A, Baines DL, Folkesson HG. Alveolar epithelial fluid clearance is mediated by endogenous catecholamines at birth in guinea pigs. J Clin Invest (1998) 101:972–81. doi:10.1172/JCI1478
18. Jain L, Chen XJ, Malik B, Al-Khalili O, Eaton DC. Antisense oligonucleotides against the alpha-subunit of ENaC decrease lung epithelial cation-channel activity. Am J Physiol (1999) 276:L1046–51.
19. Jayr C, Garat C, Meignan M, Pittet JF, Zelter M, Matthay MA. Alveolar liquid and protein clearance in anesthetized ventilated rats. J Appl Physiol (1985) (1994) 76:2636–42.
20. Matalon S. Mechanisms and regulation of ion transport in adult mammalian alveolar type II pneumocytes. Am J Physiol (1991) 261:C727–38.
21. Matalon S, Bridges RJ, Benos DJ. Amiloride-inhibitable Na+ conductive pathways in alveolar type II pneumocytes. Am J Physiol (1991) 260:L90–6.
22. Butterworth MB. Regulation of the epithelial sodium channel (ENaC) by membrane trafficking. Biochim Biophys Acta (2010) 1802:1166–77. doi:10.1016/j.bbadis.2010.03.010
23. Hummler E, Barker P, Gatzy J, Beermann F, Verdumo C, Schmidt A, et al. Early death due to defective neonatal lung liquid clearance in alpha-ENaC-deficient mice. Nat Genet (1996) 12:325–8. doi:10.1038/ng0396-325
24. Kerem E, Bistritzer T, Hanukoglu A, Hofmann T, Zhou Z, Bennett W, et al. Pulmonary epithelial sodium-channel dysfunction and excess airway liquid in pseudohypoaldosteronism. N Engl J Med (1999) 341:156–62. doi:10.1056/NEJM199907153410304
25. Boase NA, Rychkov GY, Townley SL, Dinudom A, Candi E, Voss AK, et al. Respiratory distress and perinatal lethality in Nedd4-2-deficient mice. Nat Commun (2011) 2:287. doi:10.1038/ncomms1284
26. Mall M, Grubb BR, Harkema JR, O’Neal WK, Boucher RC. Increased airway epithelial Na+ absorption produces cystic fibrosis-like lung disease in mice. Nat Med (2004) 10:487–93. doi:10.1038/nm1028
27. Auberson M, Hoffmann-Pochon N, Vandewalle A, Kellenberger S, Schild L. Epithelial Na+ channel mutants causing Liddle’s syndrome retain ability to respond to aldosterone and vasopressin. Am J Physiol Renal Physiol (2003) 285:F459–71. doi:10.1152/ajprenal.00071.2003
28. Kucher V, Boiko N, Pochynyuk O, Stockand JD. Voltage-dependent gating underlies loss of ENaC function in pseudohypoaldosteronism type 1. Biophys J (2011) 100:1930–9. doi:10.1016/j.bpj.2011.02.046
29. Frindt G, Palmer LG. Acute effects of aldosterone on the epithelial Na channel in rat kidney. Am J Physiol Renal Physiol (2015) 308:F572–8. doi:10.1152/ajprenal.00585.2014
30. Itani OA, Auerbach SD, Husted RF, Volk KA, Ageloff S, Knepper MA, et al. Glucocorticoid-stimulated lung epithelial Na(+) transport is associated with regulated ENaC and sgk1 expression. Am J Physiol Lung Cell Mol Physiol (2002) 282:L631–41. doi:10.1152/ajplung.00085.2001
31. Jain L, Chen XJ, Ramosevac S, Brown LA, Eaton DC. Expression of highly selective sodium channels in alveolar type II cells is determined by culture conditions. Am J Physiol Lung Cell Mol Physiol (2001) 280:L646–58.
32. Kemendy AE, Kleyman TR, Eaton DC. Aldosterone alters the open probability of amiloride-blockable sodium channels in A6 epithelia. Am J Physiol (1992) 263:C825–37.
33. Bruns JB, Carattino MD, Sheng S, Maarouf AB, Weisz OA, Pilewski JM, et al. Epithelial Na+ channels are fully activated by furin- and prostasin-dependent release of an inhibitory peptide from the gamma-subunit. J Biol Chem (2007) 282:6153–60. doi:10.1074/jbc.M610636200
34. Caldwell RA, Boucher RC, Stutts MJ. Serine protease activation of near-silent epithelial Na+ channels. Am J Physiol Cell Physiol (2004) 286:C190–4. doi:10.1152/ajpcell.00342.2003
35. Caldwell RA, Boucher RC, Stutts MJ. Neutrophil elastase activates near-silent epithelial Na+ channels and increases airway epithelial Na+ transport. Am J Physiol Lung Cell Mol Physiol (2005) 288:L813–9. doi:10.1152/ajplung.00435.2004
36. Hughey RP, Carattino MD, Kleyman TR. Role of proteolysis in the activation of epithelial sodium channels. Curr Opin Nephrol Hypertens (2007) 16:444–50. doi:10.1097/MNH.0b013e32821f6072
37. Sheng S, Maarouf AB, Bruns JB, Hughey RP, Kleyman TR. Functional role of extracellular loop cysteine residues of the epithelial Na+ channel in Na+ self-inhibition. J Biol Chem (2007) 282:20180–90. doi:10.1074/jbc.M611761200
38. Rossier BC, Baker ME, Studer RA. Epithelial sodium transport and its control by aldosterone: the story of our internal environment revisited. Physiol Rev (2015) 95:297–340. doi:10.1152/physrev.00011.2014
39. Loffing-Cueni D, Flores SY, Sauter D, Daidie D, Siegrist N, Meneton P, et al. Dietary sodium intake regulates the ubiquitin-protein ligase nedd4-2 in the renal collecting system. J Am Soc Nephrol (2006) 17:1264–74. doi:10.1681/ASN.2005060659
40. Snyder PM. Intoxicated Na(+) channels. Focus on “ethanol stimulates epithelial sodium channels by elevating reactive oxygen species”. Am J Physiol Cell Physiol (2012) 303:C1125–6. doi:10.1152/ajpcell.00301.2012
41. Folkesson HG, Norlin A, Wang Y, Abedinpour P, Matthay MA. Dexamethasone and thyroid hormone pretreatment upregulate alveolar epithelial fluid clearance in adult rats. J Appl Physiol (1985) (2000) 88:416–24.
42. McTavish N, Getty J, Burchell A, Wilson SM. Glucocorticoids can activate the alpha-ENaC gene promoter independently of SGK1. Biochem J (2009) 423:189–97. doi:10.1042/BJ20090366
43. Nakamura K, Stokes JB, McCray PB Jr. Endogenous and exogenous glucocorticoid regulation of ENaC mRNA expression in developing kidney and lung. Am J Physiol Cell Physiol (2002) 283:C762–72. doi:10.1152/ajpcell.00029.2002
44. Brennan FE, Fuller PJ. Rapid upregulation of serum and glucocorticoid-regulated kinase (sgk) gene expression by corticosteroids in vivo. Mol Cell Endocrinol (2000) 166:129–36. doi:10.1016/S0303-7207(00)00274-4
45. Chen SY, Bhargava A, Mastroberardino L, Meijer OC, Wang J, Buse P, et al. Epithelial sodium channel regulated by aldosterone-induced protein sgk. Proc Natl Acad Sci U S A (1999) 96:2514–9. doi:10.1073/pnas.96.5.2514
46. De Bosscher K, Vanden Berghe W, Haegeman G. The interplay between the glucocorticoid receptor and nuclear factor-kappaB or activator protein-1: molecular mechanisms for gene repression. Endocr Rev (2003) 24:488–522. doi:10.1210/er.2002-0006
47. McKay LI, Cidlowski JA. Molecular control of immune/inflammatory responses: interactions between nuclear factor-kappa B and steroid receptor-signaling pathways. Endocr Rev (1999) 20:435–59. doi:10.1210/er.20.4.435
48. Rhen T, Cidlowski JA. Antiinflammatory action of glucocorticoids – new mechanisms for old drugs. N Engl J Med (2005) 353:1711–23. doi:10.1056/NEJMra050541
49. Tanabe T, Tohnai N. Cyclooxygenase isozymes and their gene structures and expression. Prostaglandins Other Lipid Mediat (2002) 68-69:95–114. doi:10.1016/S0090-6980(02)00024-2
50. Schwartze JT, Becker S, Sakkas E, Wujak LA, Niess G, Usemann J, et al. Glucocorticoids recruit Tgfbr3 and Smad1 to shift transforming growth factor-beta signaling from the Tgfbr1/Smad2/3 axis to the Acvrl1/Smad1 axis in lung fibroblasts. J Biol Chem (2014) 289:3262–75. doi:10.1074/jbc.M113.541052
51. Halwani R, Al-Muhsen S, Al-Jahdali H, Hamid Q. Role of transforming growth factor-beta in airway remodeling in asthma. Am J Respir Cell Mol Biol (2011) 44:127–33. doi:10.1165/rcmb.2010-0027TR
52. Matsuki K, Hathaway CK, Lawrence MG, Smithies O, Kakoki M. The role of transforming growth factor beta1 in the regulation of blood pressure. Curr Hypertens Rev (2014) 10:223–38. doi:10.2174/157340211004150319123313
53. Thomas PS. Tumour necrosis factor-alpha: the role of this multifunctional cytokine in asthma. Immunol Cell Biol (2001) 79:132–40. doi:10.1046/j.1440-1711.2001.00980.x
54. Yanagisawa J, Yanagi Y, Masuhiro Y, Suzawa M, Watanabe M, Kashiwagi K, et al. Convergence of transforming growth factor-beta and vitamin D signaling pathways on SMAD transcriptional coactivators. Science (1999) 283:1317–21. doi:10.1126/science.283.5406.1317
55. Wittekindt OH. Tight junctions in pulmonary epithelia during lung inflammation. Pflugers Arch (2017) 469:135–47. doi:10.1007/s00424-016-1917-3
56. Martin TR, Frevert CW. Innate immunity in the lungs. Proc Am Thorac Soc (2005) 2:403–11. doi:10.1513/pats.200508-090JS
57. Cromwell O, Hamid Q, Corrigan CJ, Barkans J, Meng Q, Collins PD, et al. Expression and generation of interleukin-8, IL-6 and granulocyte-macrophage colony-stimulating factor by bronchial epithelial cells and enhancement by IL-1 beta and tumour necrosis factor-alpha. Immunology (1992) 77:330–7.
58. Kato A, Schleimer RP. Beyond inflammation: airway epithelial cells are at the interface of innate and adaptive immunity. Curr Opin Immunol (2007) 19:711–20. doi:10.1016/j.coi.2007.08.004
59. Chen XJ, Seth S, Yue G, Kamat P, Compans RW, Guidot D, et al. Influenza virus inhibits ENaC and lung fluid clearance. Am J Physiol Lung Cell Mol Physiol (2004) 287:L366–73. doi:10.1152/ajplung.00011.2004
60. Eisenhut M, Sidaras D, Barton P, Newland P, Southern KW. Elevated sweat sodium associated with pulmonary oedema in meningococcal sepsis. Eur J Clin Invest (2004) 34:576–9. doi:10.1111/j.1365-2362.2004.01386.x
61. Eisenhut M, Southern KW. Positive sweat test following meningococcal septicaemia. Acta Paediatr (2002) 91:361–2. doi:10.1111/j.1651-2227.2002.tb01731.x
62. Eisenhut M, Wallace H, Barton P, Gaillard E, Newland P, Diver M, et al. Pulmonary edema in meningococcal septicemia associated with reduced epithelial chloride transport. Pediatr Crit Care Med (2006) 7:119–24. doi:10.1097/01.PCC.0000200944.98424.E0
63. Belshe RB. Influenza prevention and treatment: current practices and new horizons. Ann Intern Med (1999) 131:621–4. doi:10.7326/0003-4819-131-8-199910190-00013
64. Doyle WJ, Skoner DP, Hayden F, Buchman CA, Seroky JT, Fireman P. Nasal and otologic effects of experimental influenza A virus infection. Ann Otol Rhinol Laryngol (1994) 103:59–69. doi:10.1177/000348949410300111
65. Greenberg SB. Respiratory viral infections in adults. Curr Opin Pulm Med (2002) 8:201–8. doi:10.1097/00063198-200205000-00009
66. Helms MN, Yu L, Malik B, Kleinhenz DJ, Hart CM, Eaton DC. Role of SGK1 in nitric oxide inhibition of ENaC in Na+-transporting epithelia. Am J Physiol Cell Physiol (2005) 289:C717–26. doi:10.1152/ajpcell.00006.2005
67. Matalon S, Hardiman KM, Jain L, Eaton DC, Kotlikoff M, Eu JP, et al. Regulation of ion channel structure and function by reactive oxygen-nitrogen species. Am J Physiol Lung Cell Mol Physiol (2003) 285:L1184–9. doi:10.1152/ajplung.00281.2003
68. Helms MN, Jain L, Self JL, Eaton DC. Redox regulation of epithelial sodium channels examined in alveolar type 1 and 2 cells patch-clamped in lung slice tissue. J Biol Chem (2008) 283:22875–83. doi:10.1074/jbc.M801363200
69. Yu L, Bao HF, Self JL, Eaton DC, Helms MN. Aldosterone-induced increases in superoxide production counters nitric oxide inhibition of epithelial Na channel activity in A6 distal nephron cells. Am J Physiol Renal Physiol (2007) 293:F1666–77. doi:10.1152/ajprenal.00444.2006
70. Takemura Y, Goodson P, Bao HF, Jain L, Helms MN. Rac1-mediated NADPH oxidase release of O2- regulates epithelial sodium channel activity in the alveolar epithelium. Am J Physiol Lung Cell Mol Physiol (2010) 298:L509–20. doi:10.1152/ajplung.00230.2009
71. Meduri GU, Kohler G, Headley S, Tolley E, Stentz F, Postlethwaite A. Inflammatory cytokines in the BAL of patients with ARDS. Persistent elevation over time predicts poor outcome. Chest (1995) 108:1303–14. doi:10.1378/chest.108.5.1303
72. Hamacher J, Lucas R, Lijnen HR, Buschke S, Dunant Y, Wendel A, et al. Tumor necrosis factor-alpha and angiostatin are mediators of endothelial cytotoxicity in bronchoalveolar lavages of patients with acute respiratory distress syndrome. Am J Respir Crit Care Med (2002) 166:651–6. doi:10.1164/rccm.2109004
73. Horgan MJ, Palace GP, Everitt JE, Malik AB. TNF-alpha release in endotoxemia contributes to neutrophil-dependent pulmonary edema. Am J Physiol (1993) 264:H1161–5.
74. McRitchie DI, Isowa N, Edelson JD, Xavier AM, Cai L, Man HY, et al. Production of tumour necrosis factor alpha by primary cultured rat alveolar epithelial cells. Cytokine (2000) 12:644–54. doi:10.1006/cyto.1999.0656
75. Dagenais A, Frechette R, Yamagata Y, Yamagata T, Carmel JF, Clermont ME, et al. Downregulation of ENaC activity and expression by TNF-alpha in alveolar epithelial cells. Am J Physiol Lung Cell Mol Physiol (2004) 286:L301–11. doi:10.1152/ajplung.00326.2002
76. Elia N, Tapponnier M, Matthay MA, Hamacher J, Pache JC, Brundler MA, et al. Functional identification of the alveolar edema reabsorption activity of murine tumor necrosis factor-alpha. Am J Respir Crit Care Med (2003) 168:1043–50. doi:10.1164/rccm.200206-618OC
77. Fukuda N, Jayr C, Lazrak A, Wang Y, Lucas R, Matalon S, et al. Mechanisms of TNF-alpha stimulation of amiloride-sensitive sodium transport across alveolar epithelium. Am J Physiol Lung Cell Mol Physiol (2001) 280:L1258–65.
78. Yamagata T, Yamagata Y, Nishimoto T, Hirano T, Nakanishi M, Minakata Y, et al. The regulation of amiloride-sensitive epithelial sodium channels by tumor necrosis factor-alpha in injured lungs and alveolar type II cells. Respir Physiol Neurobiol (2009) 166:16–23. doi:10.1016/j.resp.2008.12.008
79. Lassalle P, Gosset P, Delneste Y, Tsicopoulos A, Capron A, Joseph M, et al. Modulation of adhesion molecule expression on endothelial cells during the late asthmatic reaction: role of macrophage-derived tumour necrosis factor-alpha. Clin Exp Immunol (1993) 94:105–10. doi:10.1111/j.1365-2249.1993.tb05985.x
80. Pober JS, Gimbrone MA Jr, Lapierre LA, Mendrick DL, Fiers W, Rothlein R, et al. Overlapping patterns of activation of human endothelial cells by interleukin 1, tumor necrosis factor, and immune interferon. J Immunol (1986) 137:1893–6.
81. Rokkam D, Lafemina MJ, Lee JW, Matthay MA, Frank JA. Claudin-4 levels are associated with intact alveolar fluid clearance in human lungs. Am J Pathol (2011) 179:1081–7. doi:10.1016/j.ajpath.2011.05.017
82. Mazzon E, Cuzzocrea S. Role of TNF-alpha in lung tight junction alteration in mouse model of acute lung inflammation. Respir Res (2007) 8:75. doi:10.1186/1465-9921-8-75
83. Czikora I, Alli A, Bao HF, Kaftan D, Sridhar S, Apell HJ, et al. A novel tumor necrosis factor-mediated mechanism of direct epithelial sodium channel activation. Am J Respir Crit Care Med (2014) 190:522–32. doi:10.1164/rccm.201405-0833OC
84. Yang G, Hamacher J, Gorshkov B, White R, Sridhar S, Verin A, et al. The dual role of TNF in pulmonary edema. J Cardiovasc Dis Res (2010) 1:29–36. doi:10.4103/0975-3583.59983
85. Braun C, Hamacher J, Morel DR, Wendel A, Lucas R. Dichotomal role of TNF in experimental pulmonary edema reabsorption. J Immunol (2005) 175:3402–8. doi:10.4049/jimmunol.175.5.3402
86. Rezaiguia S, Garat C, Delclaux C, Meignan M, Fleury J, Legrand P, et al. Acute bacterial pneumonia in rats increases alveolar epithelial fluid clearance by a tumor necrosis factor-alpha-dependent mechanism. J Clin Invest (1997) 99:325–35. doi:10.1172/JCI119161
87. Tillie-Leblond I, Guery BP, Janin A, Leberre R, Just N, Pittet JF, et al. Chronic bronchial allergic inflammation increases alveolar liquid clearance by TNF-alpha-dependent mechanism. Am J Physiol Lung Cell Mol Physiol (2002) 283:L1303–9. doi:10.1152/ajplung.00147.2002
88. Wagener BM, Roux J, Carles M, Pittet JF. Synergistic inhibition of beta2-adrenergic receptor-mediated alveolar epithelial fluid transport by interleukin-8 and transforming growth factor-beta. Anesthesiology (2015) 122:1084–92. doi:10.1097/ALN.0000000000000595
89. Fahy RJ, Lichtenberger F, McKeegan CB, Nuovo GJ, Marsh CB, Wewers MD. The acute respiratory distress syndrome: a role for transforming growth factor-beta 1. Am J Respir Cell Mol Biol (2003) 28:499–503. doi:10.1165/rcmb.2002-0092OC
90. Wakefield LM, Letterio JJ, Chen T, Danielpour D, Allison RS, Pai LH, et al. Transforming growth factor-beta1 circulates in normal human plasma and is unchanged in advanced metastatic breast cancer. Clin Cancer Res (1995) 1:129–36.
91. Kaminski N, Allard JD, Pittet JF, Zuo F, Griffiths MJ, Morris D, et al. Global analysis of gene expression in pulmonary fibrosis reveals distinct programs regulating lung inflammation and fibrosis. Proc Natl Acad Sci U S A (2000) 97:1778–83. doi:10.1073/pnas.97.4.1778
92. Annes JP, Chen Y, Munger JS, Rifkin DB. Integrin alphaVbeta6-mediated activation of latent TGF-beta requires the latent TGF-beta binding protein-1. J Cell Biol (2004) 165:723–34. doi:10.1083/jcb.200312172
93. Annes JP, Rifkin DB, Munger JS. The integrin alphaVbeta6 binds and activates latent TGFbeta3. FEBS Lett (2002) 511:65–8. doi:10.1016/S0014-5793(01)03280-X
94. Munger JS, Huang X, Kawakatsu H, Griffiths MJ, Dalton SL, Wu J, et al. The integrin alpha v beta 6 binds and activates latent TGF beta 1: a mechanism for regulating pulmonary inflammation and fibrosis. Cell (1999) 96:319–28. doi:10.1016/S0092-8674(00)80545-0
95. Breuss JM, Gallo J, DeLisser HM, Klimanskaya IV, Folkesson HG, Pittet JF, et al. Expression of the beta 6 integrin subunit in development, neoplasia and tissue repair suggests a role in epithelial remodeling. J Cell Sci (1995) 108(Pt 6):2241–51.
96. Barcellos-Hoff MH, Dix TA. Redox-mediated activation of latent transforming growth factor-beta 1. Mol Endocrinol (1996) 10:1077–83. doi:10.1210/me.10.9.1077
97. Frank J, Roux J, Kawakatsu H, Su G, Dagenais A, Berthiaume Y, et al. Transforming growth factor-beta1 decreases expression of the epithelial sodium channel alphaENaC and alveolar epithelial vectorial sodium and fluid transport via an ERK1/2-dependent mechanism. J Biol Chem (2003) 278:43939–50. doi:10.1074/jbc.M304882200
98. Pittet JF, Griffiths MJ, Geiser T, Kaminski N, Dalton SL, Huang X, et al. TGF-beta is a critical mediator of acute lung injury. J Clin Invest (2001) 107:1537–44. doi:10.1172/JCI11963
99. Peters DM, Vadasz I, Wujak L, Wygrecka M, Olschewski A, Becker C, et al. TGF-beta directs trafficking of the epithelial sodium channel ENaC which has implications for ion and fluid transport in acute lung injury. Proc Natl Acad Sci U S A (2014) 111:E374–83. doi:10.1073/pnas.1306798111
100. Frank JA, Matthay MA. TGF-beta and lung fluid balance in ARDS. Proc Natl Acad Sci U S A (2014) 111:885–6. doi:10.1073/pnas.1322478111
101. Galietta LJ, Folli C, Marchetti C, Romano L, Carpani D, Conese M, et al. Modification of transepithelial ion transport in human cultured bronchial epithelial cells by interferon-gamma. Am J Physiol Lung Cell Mol Physiol (2000) 278:L1186–94.
102. Olman MA, White KE, Ware LB, Cross MT, Zhu S, Matthay MA. Microarray analysis indicates that pulmonary edema fluid from patients with acute lung injury mediates inflammation, mitogen gene expression, and fibroblast proliferation through bioactive interleukin-1. Chest (2002) 121:69S–70S. doi:10.1378/chest.121.3_suppl.69S
103. Pugin J, Ricou B, Steinberg KP, Suter PM, Martin TR. Proinflammatory activity in bronchoalveolar lavage fluids from patients with ARDS, a prominent role for interleukin-1. Am J Respir Crit Care Med (1996) 153:1850–6. doi:10.1164/ajrccm.153.6.8665045
104. Pugin J, Verghese G, Widmer MC, Matthay MA. The alveolar space is the site of intense inflammatory and profibrotic reactions in the early phase of acute respiratory distress syndrome. Crit Care Med (1999) 27:304–12. doi:10.1097/00003246-199902000-00036
105. Siler TM, Swierkosz JE, Hyers TM, Fowler AA, Webster RO. Immunoreactive interleukin-1 in bronchoalveolar lavage fluid of high-risk patients and patients with the adult respiratory distress syndrome. Exp Lung Res (1989) 15:881–94. doi:10.3109/01902148909069633
106. Suter PM, Suter S, Girardin E, Roux-Lombard P, Grau GE, Dayer JM. High bronchoalveolar levels of tumor necrosis factor and its inhibitors, interleukin-1, interferon, and elastase, in patients with adult respiratory distress syndrome after trauma, shock, or sepsis. Am Rev Respir Dis (1992) 145:1016–22. doi:10.1164/ajrccm/145.5.1016
107. Gonzales JN, Lucas R, Verin AD. The acute respiratory distress syndrome: mechanisms and perspective therapeutic approaches. Austin J Vasc Med (2015) 2:1009.
108. Lee YM, Hybertson BM, Cho HG, Terada LS, Cho O, Repine AJ, et al. Platelet-activating factor contributes to acute lung leak in rats given interleukin-1 intratracheally. Am J Physiol Lung Cell Mol Physiol (2000) 279:L75–80.
109. Hybertson BM, Lee YM, Cho HG, Cho OJ, Repine JE. Alveolar type II cell abnormalities and peroxide formation in lungs of rats given IL-1 intratracheally. Inflammation (2000) 24:289–303. doi:10.1023/A:1007092529261
110. Leff JA, Bodman ME, Cho OJ, Rohrbach S, Reiss OK, Vannice JL, et al. Post-insult treatment with interleukin-1 receptor antagonist decreases oxidative lung injury in rats given intratracheal interleukin-1. Am J Respir Crit Care Med (1994) 150:109–12. doi:10.1164/ajrccm.150.1.8025734
111. Repine JE. Interleukin-1-mediated acute lung injury and tolerance to oxidative injury. Environ Health Perspect (1994) 102(Suppl 10):75–8. doi:10.1289/ehp.94102s1075
112. Roux J, Kawakatsu H, Gartland B, Pespeni M, Sheppard D, Matthay MA, et al. Interleukin-1beta decreases expression of the epithelial sodium channel alpha-subunit in alveolar epithelial cells via a p38 MAPK-dependent signaling pathway. J Biol Chem (2005) 280:18579–89. doi:10.1074/jbc.M410561200
113. Galam L, Soundararajan R, Breitzig M, Rajan A, Yeruva RR, Czachor A, et al. SOCS-1 rescues IL-1beta-mediated suppression of epithelial sodium channel in mouse lung epithelial cells via ASK-1. Oncotarget (2016) 7:29081–91. doi:10.18632/oncotarget.8543
114. Ganter MT, Ware LB, Howard M, Roux J, Gartland B, Matthay MA, et al. Extracellular heat shock protein 72 is a marker of the stress protein response in acute lung injury. Am J Physiol Lung Cell Mol Physiol (2006) 291:L354–61. doi:10.1152/ajplung.00405.2005
115. Howard M, Roux J, Iles KE, Miyazawa B, Christiaans S, Anjum N, et al. Activation of the heat shock response attenuates the interleukin 1β-mediated inhibition of the amiloride-sensitive alveolar epithelial ion transport. Shock (2013) 39:189–96. doi:10.1097/SHK.0b013e31827e8ea3
116. Galietta LJ, Pagesy P, Folli C, Caci E, Romio L, Costes B, et al. IL-4 is a potent modulator of ion transport in the human bronchial epithelium in vitro. J Immunol (2002) 168:839–45. doi:10.4049/jimmunol.168.2.839
117. Donnelly SC, Strieter RM, Reid PT, Kunkel SL, Burdick MD, Armstrong I, et al. The association between mortality rates and decreased concentrations of interleukin-10 and interleukin-1 receptor antagonist in the lung fluids of patients with the adult respiratory distress syndrome. Ann Intern Med (1996) 125:191–6. doi:10.7326/0003-4819-125-3-199608010-00005
Keywords: lung, sodium channels, epithelial sodium channel, cytokines, physiology, inflammation, acute lung injury, acute respiratory distress syndrome
Citation: Wynne BM, Zou L, Linck V, Hoover RS, Ma H-P and Eaton DC (2017) Regulation of Lung Epithelial Sodium Channels by Cytokines and Chemokines. Front. Immunol. 8:766. doi: 10.3389/fimmu.2017.00766
Received: 18 March 2017; Accepted: 16 June 2017;
Published: 25 July 2017
Edited by:
Rudolf Lucas, Augusta University, United StatesReviewed by:
Ali Mobasheri, University of Surrey, United KingdomCopyright: © 2017 Wynne, Zou, Linck, Hoover, Ma and Eaton. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Brandi M. Wynne, Ynd5bm5lQGVtb3J5LmVkdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.