
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol., 30 June 2017
Sec. Inflammation
Volume 8 - 2017 | https://doi.org/10.3389/fimmu.2017.00763
This article is part of the Research TopicSphingolipids / PUFA and Respiratory SyndromeView all 10 articles
 Rituraj Niranjan*
Rituraj Niranjan* Ashwani Kumar Thakur*
Ashwani Kumar Thakur*
The environmental soot and carbon blacks (CBs) cause many diseases in humans, but their underlying mechanisms of toxicity are still poorly understood. Both are formed after the incomplete combustion of hydrocarbons but differ in their constituents and percent carbon contents. For the first time, “Sir Percival Pott” described soot as a carcinogen, which was subsequently confirmed by many others. The existing data suggest three main types of diseases due to soot and CB exposures: cancer, respiratory diseases, and cardiovascular dysfunctions. Experimental models revealed the involvement of oxidative stress, DNA methylation, formation of DNA adducts, and Aryl hydrocarbon receptor activation as the key mechanisms of soot- and CB-induced cancers. Metals including Si, Fe, Mn, Ti, and Co in soot also contribute in the reactive oxygen species (ROS)-mediated DNA damage. Mechanistically, ROS-induced DNA damage is further enhanced by eosinophils and neutrophils via halide (Cl− and Br−) dependent DNA adducts formation. The activation of pulmonary dendritic cells, T helper type 2 cells, and mast cells is crucial mediators in the pathology of soot- or CB-induced respiratory disease. Polyunsaturated fatty acids (PUFAs) were also found to modulate T cells functions in respiratory diseases. Particularly, telomerase reverse transcriptase was found to play the critical role in soot- and CB-induced cardiovascular dysfunctions. In this review, we propose integrated mechanisms of soot- and CB-induced toxicity emphasizing the role of inflammatory mediators and oxidative stress. We also suggest use of antioxidants and PUFAs as protective strategies against soot- and CB-induced disorders.
The environmental soots [black carbon (BC)] and carbon blacks (CBs) cause many health issues in humans and animals (1, 2). The terms soot and CB have been used interchangeably but, both are physically and chemically distinct entities (3–5). Soots are considered as unwanted byproducts derived from incomplete combustion of carbon-containing materials (3–5). In contrast, the CBs are manufactured under the controlled conditions in the rubber, printing and painting industries for commercial use (3–5). Soot is a powdery mass of fine black particles (6–8). It consists of impure carbon, formed after the incomplete combustion of hydrocarbons (9). The main source of environmental soot is the combustion of fossil-based fuels and biomass burning at the Earth’s surface (10). The other examples of soot may include coal, charred wood, petroleum coke, cenospheres, and tars (11, 12). To a smaller extent, quartz/halogen bulbs with settled dust, cooking, oil lamps, smoking of plant matter, fireplaces, candles, house fires, furnaces, and local field burning also contribute to the soot production (13). Soot particles range from about 10 nm to 1 mm in size (14–17). The relative amount of elemental carbon inside soot is considered to be less than 60% of the total mass of particle (4, 7, 18). Among hydrocarbons, the poly aromatic hydrocarbons (PAHs) are the main carcinogenic compound in the soot (19–21). At elemental level, the most characterized diesel soot contains carbon (as a main component), hydrogen, oxygen, sulfur, and trace amount of metals (22–24). The major component of soot, the BC, causes premature human mortality and disability (25). Furthermore, changes in the chemical composition of soot are accomplished due to heterogeneous oxidation reactions in the environment (26–28). From the regional point of view, developed nations were the biggest source of soot (BC) emissions but at the present scenario, soot emissions are majorly from developing countries (29, 30). It is noted that United States emits about 6.1% of the world’s soot (31). Notably, the biggest amount of soot comes from Latin America, Asia, and Africa (31, 32). India and China alone may account around 25–35% of total global soot emissions (31–34). The impact of soot on the human health and to the entire environment depends on its distribution and its distance from the source of origin (35–38). Soot from vehicle exhausts comes from combustion of diesel, gasoline, and other petroleum-based fuels materials that contains carbonaceous particles, having polycyclic aromatic hydrocarbons (PAHs) attached to it (21). Typically, diesel exhaust particles (DEP) are made up of carbon core with some volatile and semi-volatile (such as H2SO4 and organics) components adsorbed on it (9, 39). It has been believed that the vehicle exhaust contributes to approximately 50% of urban particulate matter (PM) (40). The special attention is given to the smaller fractions of PM (PM 2.5 and PM 0.1) because these particles can penetrate deep into the bronchiolar parts of the lungs and cause various health hazards (41).
In contrast to soot (BC), CB is generated by the partial combustion of heavy petroleum materials such as coal tar, ethylene cracking tar, and FCC tar (42). The common subtypes of CB are furnace black, acetylene black, lamp black, channel black, and thermal black (3, 43). CB is also known by the trade names such as Printex-90, Printex-140, Printex-G, and Lampblack-101 (3, 43). Around 95% of CB production is achieved by the oil furnace process (3, 44, 45). In this process, the heavy aromatic petroleum products are pyrolyzed at the very high temperatures (around 1,400–1,800°C) to make the CB particles and tail gas (e.g., carbon monoxide, hydrogen, and steam) (46, 47). This process is conducted as a continuous process in a closed reactor. It is interested to know that the production is highly controlled in the oil furnace process, such that various CBs with the differing properties can be made (3, 46). The amount of elemental carbon is greater than 97% in CB that is arranged as aciniform-like structures. CB has widely been used to produce both in vitro and animal models of soot toxicology and will be discussed in detail in latter sections of this article (3–5). CB is also dissimilar to environmental soot especially due to its higher surface area to volume ratio as well as very less (less bioavailable) polycyclic aromatic hydrocarbon contents (3). Importantly, both soot and CB mainly affect cardiovascular system, respiratory system, and cause different kinds of cancer (Figure 1) (41). Therefore, it is important to know soot- and CB-induced toxicity in these major disease areas.
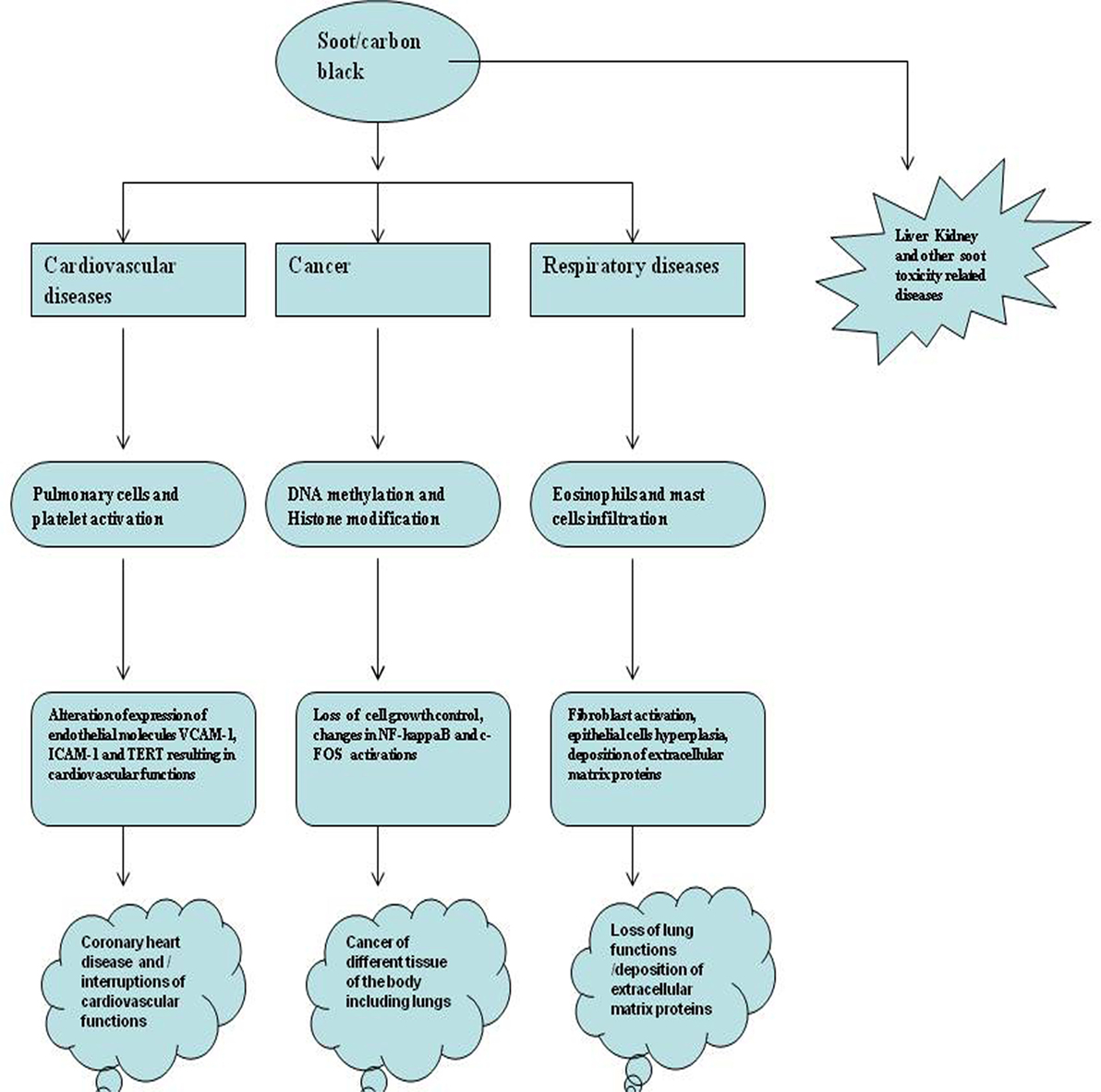
Figure 1. The major health problems due to soot and carbon black (CB). Figure shows soot- and CB-induced major health problems. The first hazard is cancer that is caused by DNA adducts formation, DNA strands breaks, or mutation in genes. Second is the respiratory toxicity caused by dysfunctional immune response involving activation of eosinophils and mast cells. The third is cardiovascular toxicology that also includes the coronary heart disease. Apart from these, soot also causes damage to the different organs of the body by some unknown mechanisms.
Historically for the first time, Sir Percival Pott, a London surgeon, in 1775 recognized that chimney sweeps were particularly susceptible to develop scrotal cancer. He attributed this disease to the soot exposure to workers (Figure 2) (48, 49). Later he described soot, as the first environmental factor to cause cancer. This linkage started the chain of events that led to the development of first experimental model of cancer and the synthesis of first carcinogen (49, 50). Later, Earle and Paget confirmed soot, as a general human skin carcinogen (48, 51). In 1936, the proof indicating soot as a carcinogen was first given by the findings of Kuroda and Kawahata (50, 51). In the year of 1969, Rosmanith et al. reported anthracofibrosis (characterized by the luminal narrowing and black pigmentation in the mucosa) in the workers of CB industry (52). Further in 1983, Riboli et al. have reported the mortality from lung cancer in the individuals working in the manufacturing plant of acetylene and phthalic anhydride, due to soot exposure (53). Subsequently, Snow found that the inhalation of CB leads to its accumulation into the larynx and trachea resulting into multiple disease situations (54). Kandt and Biendara (55) observed the appearance of chronic rhinitis more frequently in soot-exposed workers than in unexposed persons (55). Beck et al. (56) further confirmed that the soot exposure leads to causation of cancer (56). In 1987, Bourguet et al. considered soot as a major factor for cancer of skin in the persons working in the tire and rubber industry (57). Another study by Parent et al. was conducted in 1996 to find a relationship between exposure of CB and lung cancer risk assessment in a population-based study in Montreal, QC, Canada. This study provided additional support for the fact that exposure to CB leads toward the development of lung cancer (58). In 1994, Szozda described frequent occurrence of chronic bronchitis and ventilation disturbances in persons exposed to the BC (59). Recently, Parent et al. (60) demonstrated an association between esophageal cancer and in occupational exposures of sulfuric acid and CB (60). These evidences in the history clearly show the association of soot and its constituents to human health, but its exact mechanism of toxicity remains elusive and need further experimentation, both at epidemiological and animals levels (61). Nevertheless in the last decade, there is a significant increase in the number of soot and CB toxicity studies that will be discussed in detail (Figure 3).
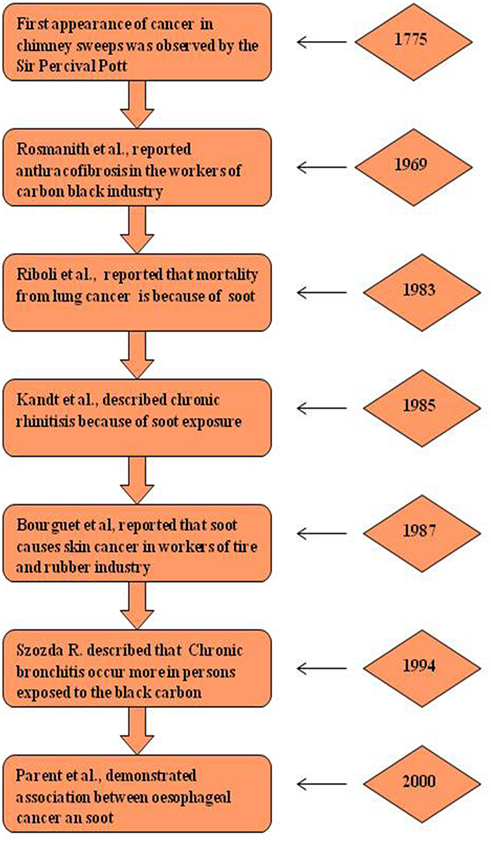
Figure 2. Historical perspective of soot-induced health hazards. Diagrammatic representation of major breakthrough studies due to soot and carbon black exposure (62). The left panel shows the pathological manifestation and right panel shows the corresponding year of study.
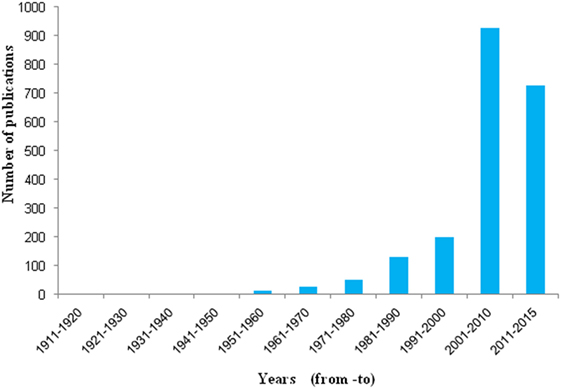
Figure 3. Number of publication found in PubMed related to soot. Histograms represent decade wise publications in the area of soot toxicity, collected from PubMed search using the word soot.
Over three centuries, the linkages of soot and CB with different diseases have been observed (25). Soot and CB cause many diseases but only three are understood to some details (Figure 1). The more complex disease associated with the soot and CB is the occurrence of cancer. Soot- and CB-induced cancers are localized and systemic in nature (34). The second major health issue with soot and CB is respiratory disorders, which sometimes can be very severe. The third one is the cardiovascular dysfunctions. Apart from these diseases, some unique pathological observations have also been seen in response to soot or CB exposures (Table 1). In a study, prenatal exposure of pritex-90 caused sexual and neuroinflammatory changes in mice (63). Surprisingly, lung exposure of diesel engine exhaust significantly influenced pro-inflammatory markers of the rat brain (64). In another study, Printex-90 lowered the sperm production (65). Similarly, carbon nanoparticles were found to adversely affect the male reproductive system of mice (66). Recently, it was known that CB exerts developmental toxicity by the immune activation in the male offspring of mice (67). Soot from a transformer fire was also seen to induce salivary gland duct metaplasia in guinea pigs (68). These studies show the involvement of systemic response of the body in the development of different pathologies that further need extensive exploration.
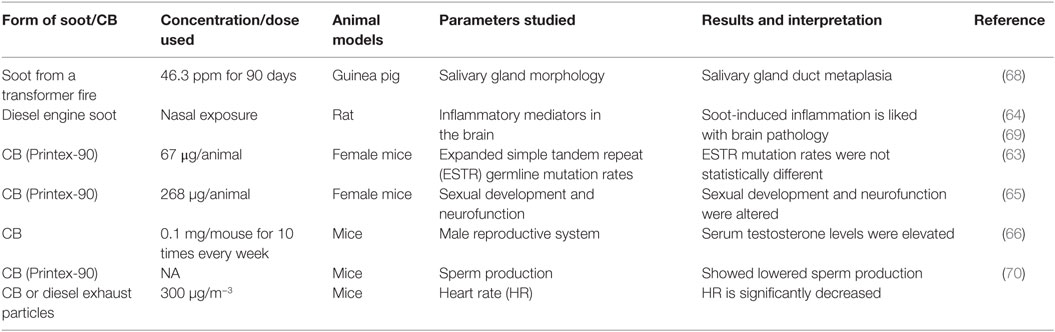
Table 1. Soot- and carbon black (CB)-induced special pathological manifestations in different experimental models.
Although soot and CB cause similar effects, but there is distinctions in their effects from the structure and composition point of view (71). It has been observed that the CB exposure in some studies has lees carcinogenic effect than the environmental soot exposure (Table 2) (21). It is believed that poly aromatic hydrocarbons exist in the CB are not biologically accessible as compared to the soot (BC) (46, 71, 72). As CB is formed under the controlled conditions, the bound poly aromatic hydrocarbons are less bioavailable compared to soot (BC). In the next sections of this article, three major diseases will be discussed in detail. At the beginning of each section, the epidemiologic studies and clinical manifestations will be discussed. Subsequently, findings from the animal models will be described.
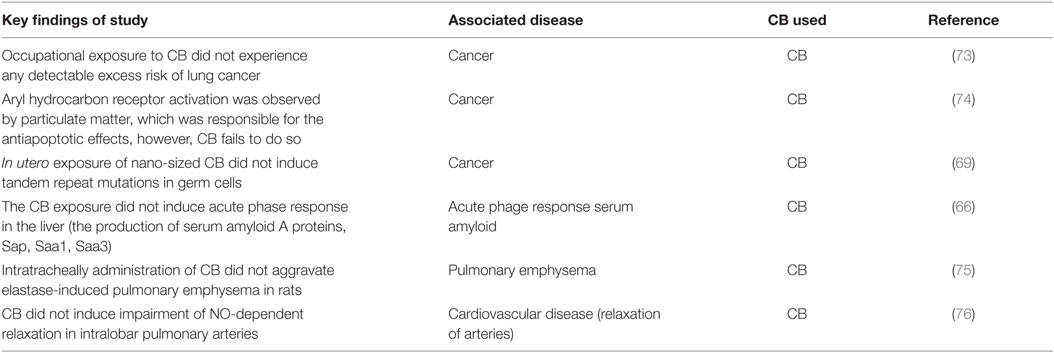
Table 2. Examples of studies with carbon black (CB) where it fails to cause pathology.
As introduced in the previous section, soot is the first known carcinogen responsible for the development of different types of cancer in humans and experimental models. These cancers may have local or distal appearance from the site of exposure (48, 50, 77). It was noted that despite the efforts of 200 years to control the safety in the soot-related work, chimney sweeps still show increased mortality from cancer (78). In line of this, a case report from Gerber described that the development of penis carcinoma in chimney sweeps was caused due to soot exposure (79). In Swedish chimney sweeps, the cancer excess was also reported due to soot and asbestos exposure (80). Soot is absorbed and transported to blood by airway epithelium and majority of the cancers in the distal body parts may be accompanied due to this mechanism of soot transportation (81). A population-based study showed that, occupational exposures to polycyclic aromatic hydrocarbons, a component of soot is responsible for respiratory and urinary tract cancers (82). Another case–control study in rubber manufacturing industry showed CB as a major contributor to the early cancer of skin (57). Subsequently, it became clear that exposure to polycyclic aromatic hydrocarbons (PAHs) in diesel soot are responsible in the development of prostate cancer (83). Furthermore, CB nanoparticle exposure-mediated human health risk was confirmed by gene expression profiling (84). Contradictory to the other studies, the International Agency for Research on Cancer (IARC) in Montreal, QC, Canada reported that the subjects with occupational exposure to titanium dioxide, industrial talc, CB, and cosmetic talc did not experience any detectable excess risk of lung cancer (85). However, this study was limited to the lung pathology alone, and no other organs were investigated (85).
In addition to the above described evidences, experimental models also provided data to further support the cancer causing properties of soot and CB (Figure 4). Study conducted on dogs demonstrated that the absorption of soot through alveolar epithelium is means of entry to the circulation of un-metabolized PAHs (86). It has been shown in rat model that soot particle interactions with lung tissue is responsible for morphological changes in the lungs (87). The diesel exhaust (DP) and CB when regularly inhaled by rats showed toxic and pulmonary carcinogenic properties (88, 89). In vitro study on the carcinogenic potency of CB confirmed the genotoxic basis of soot toxicity (90).
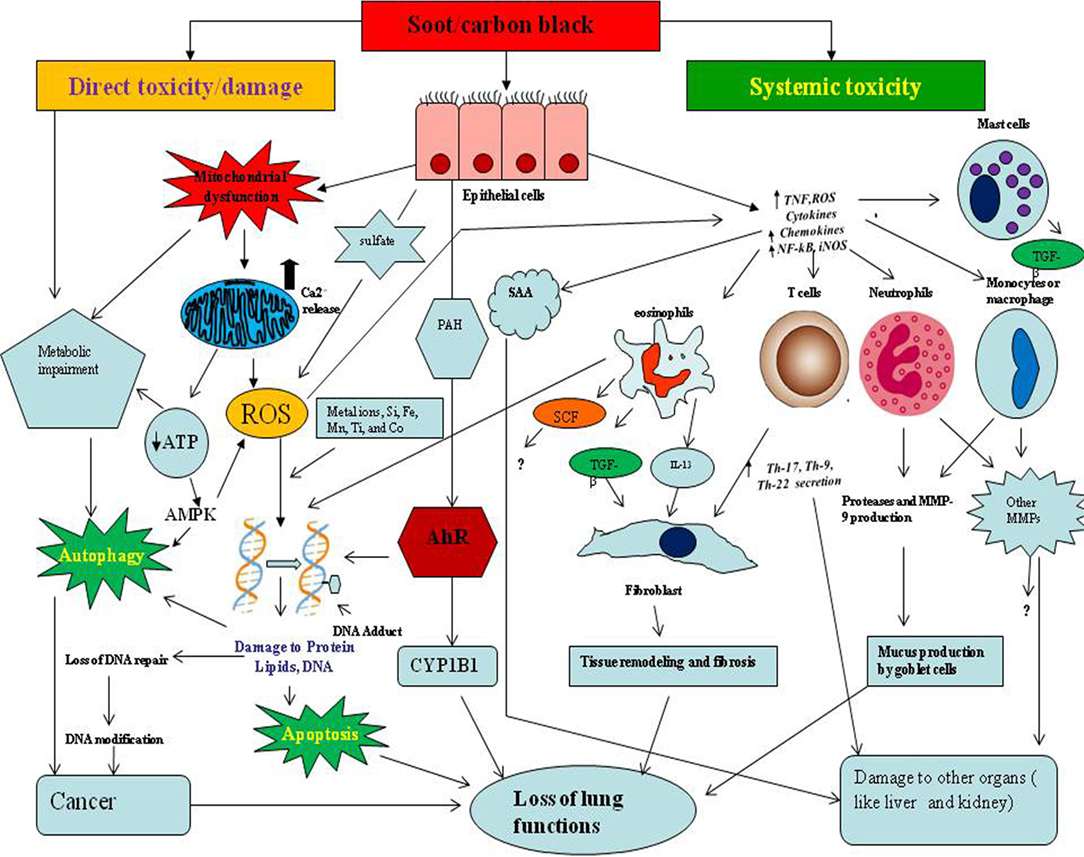
Figure 4. The proposed mechanism of soot- and carbon black (CB)-induced toxicity. The toxicological mechanisms of soot or CB can be theoretically classified into two types. The one is direct toxicity or localized damage, and the other is systemic toxicity. In direct toxicity, soot/CB comes into contact with lung epithelial cells, produces oxidative stress by affecting mitochondria, and upregulates calcium influx in the cells. The soot-induced oxidative stress initiates the cell survival or death mechanisms such as autophagy and apoptosis. Soot may lead to cancer development by interruption of autophagic and apoptotic cell death. Soot or CB also induces DNA methylation, DNA adducts formation, Aryl hydrocarbon receptor (AhR) activation, DNA double-strand breaks, and failure to DNA repair mechanisms, resulting in cancer. In systemic toxicity, soot triggers an inflammatory response in lungs and causes various symptoms. Due to soot or CB exposure, lung epithelial cells secrete inflammatory mediators (chemokine and cytokines), which further amplify the immune response. Immune cells produce interleukin-13 (IL-13) and transforming growth factor beta (TGF-beta) which activate fibroblast cells to acquire fibrotic phenotype (production and accumulations of fibrous proteins in extracellular space). Monocytes and macrophage produce a large number of matrix metalloproteases (MMPs) and other toxic mediators that alter many physiologic functions including brain and cardiovascular systems. Serum amyloid A (SAA) produced in response to soot causes problems to the liver and kidney.
At molecular level, DNA methylation changes occur after the exposure of ambient particulate pollutants (PM, BC) with aerodynamic diameter ≤2.5 µm [PM2.5], which in turn alter the expression profile of genes toward the development of cancer (Figure 4) (91). Interestingly, polycyclic aromatic hydrocarbons can alter the histone modification and therefore are responsible for epigenetic effects (92, 93). Another study reported that soot and CB cause genotoxic effects by making single- and double-stranded DNA breaks (92, 93). DEP enhanced nuclear factor kappa B (NF-kB) DNA-binding activity along with alteration profile of c-FOS proto-oncogene expression in cultured human epithelial cells, indicating the initiation of cancer (94). Notably, butadiene soot (a PAH-containing soot) exposure increases the of matrix metalloproteases-9 (MMP-9) protein expression in prostate cancer cell line. This indicates that hydrocarbons can also stimulate MMP-9 protein secretion that contributes in the development of cancer via inflammatory pathway (95). It was found that gene expression modulation occurs in response to combustion-derived nano-sized particles in A549 cells (96). This study demonstrated that several radiation-responsive genes such as GADD45beta and CDKN1A as well as NF-kappa B-dependent genes are altered due to the exposure of nano-sized combustion-derived particles (96).
The ability of soot particles to cause mutations has long been debated, but recent reports show that soot particles indeed can cause DNA mutations (97). Soot particles from the 1991 oil fires of Kuwait desert were studied for its property to induce genetic effects in in vitro conditions (98). In this study, dose-dependent increase was seen for both sister chromatids exchanges in peripheral blood lymphocytes. Also, a mutation at the hprt locus was observed in the metabolically competent AHH-1 cell line (human lymphoblast cell line) in response to soot (98). It was further confirmed in an in vitro study of cultured cells that the soot causes the mutation in the DNA and induces genotoxic effect (97). CBs also induced genotoxic effects by damaging DNA in presence of magnetite (Fe3O4) and polycyclic aromatic hydrocarbons (PAHs) (99). However, in utero exposure of nano-sized CB (Printex-90) did not cause mutations (tandem repeat) in germ cells (69). The one of the crucial mechanisms that causes DNA modification is the DNA adducts formation due to soot or CB exposure (100, 101). The chemical modification of DNA occurs when, polycyclic aromatic hydrocarbons (PAHs) reacts to the DNA molecule and form DNA adducts. In line of this, an important study described that chronic inhalation of diesel emissions and CB caused DNA adducts formation in the rat lungs (102). The formation of 8-oxo-7,8-dihydro-2′-deoxyguanosine in the DNA of rat lungs following sub-chronic exposure of CB (Printex-90) was also found (103–105). Subsequently, it was elucidated that soot and/or CB exposure induced oxidative stress in associated with the formation of DNA adducts via a failure of DNA repair mechanism (106). Mechanistically, it was observed that reactive oxygen species (ROS) enhance DNA damage by inflammatory cells such as eosinophils and neutrophils via the helide (Cl− and Br−)-dependent formation of DNA adducts and thus enhancement of cancer (107). It is already evident that sulfate (a semi-volatile component) present in the soot particles directly induces oxidative stress in the cells (108, 109). It is also found that sulfate in DEP may involve in the tissue remodeling and fibrosis (110). Another important mechanism that may play a key role at molecular level is the aryl hydrocarbon receptor (AhR) activation and DNA damage (101, 104, 111, 112). It was evident that soot nanoparticles significantly upregulate the AhR activation in the lungs, which may subsequently influence the oxidative damage and inflammation (113, 114). Soot also binds with the AhR receptor and may affect at cells and organ levels but the mechanism of its interaction to AhR receptor is not yet known (115). It was found that the activation of AhR via air pollutants can induce inflammation and subsequent allergic diseases (116). Activation of AhR induces the secretion of PDGF-BB by activated macrophages when exposed by DEP and stimulates lung fibroblast proliferation (117). Recently, it was confirmed that AhR activation and Nrf2 activation are the key mechanism in the induction of oxidative stress in response to air PM (soot) exposure (118). The AhR-mediated functions were also observed due to PM exposure, such as AhR-dependent cell proliferation and cytochrome P450 1A1/1B1 expression (119). The metals in PM2.5 were associated in the activation of AhR that subsequently influence the pathogenesis of infections in child (120). It was also observed that poly aromatic hydrocarbons found in PM are responsible for the AhR-mediated antiapoptotic effects where CB fails to do so (72, 74, 121).
It is also believed that trace amount of metal ions present in the soot are responsible for its toxic effects (122–124). A significant correlation in the metal content (Si, Fe, Mn, Ti, and Co) present in the ambient air samples was seen with the unregulated ROS formation in polymorphonuclear leukocytes (124–126). It was further proved that metal ions enhance the ROS formation capacity of ultrafine black particles both in vivo and in vitro (127). In has now been clear that metal ions present in the DEP are responsible for its genotoxic effects (128). A recent study described the estrogenic activity of soot is not related to the metal ion concentrations present in it (129). In soot, mainly BC and metal ions are responsible for the ROS generation in lung cells (130). Another study described that intracellular calcium ion formation and inflammation induced by ultrafine carbon black (ufCB) is not dependent on metal ions and other components. Collectively, sufficient amount of data support the claim that soot and CB are able to cause cancer via the genetic and molecular modifications.
The respiratory epithelium of the lungs is the first tissue to get constant exposure with different kinds of soots and CBs present in the environment. Soot or CB toxicity causes the interruption of respiratory process by alteration in lung functions (131). These toxicological mechanisms may be of two kinds. The first mechanism is the direct contact-mediated dysfunctions of lung cells that include ROS generation, cell hyperplasia, cell death, or apoptosis of lung airway epithelium and other adjacent cells (Figure 4) (132). The second mechanism includes the involvement of systemic immune response resulting in the development of tissue remodeling and fibrosis that causes problem in breathing and lung dysfunctions. In this section, we would discuss these two types of toxicities caused by soot or CB in the context of human clinical and animal studies.
The two respiratory diseases that are mainly reported in humans due to soot exposure are chronic obstructive pulmonary disease (COPD) and asthma (133). The pathophysiology of asthma involves the inflammation of airways, tissue remodeling and fibrosis, obstruction of airflow intermittently, and hyper responsiveness of bronchi (134, 135). The pathophysiology of COPD includes airway inflammation, mucociliary dysfunctions, and structural changes (136, 137). There are numerous evidences that support the linkage of soot or CB with asthma and COPD. A study reported that an early exposure to the air pollution leads to the development of childhood asthma (138). Ultrafine particles (UFPs) (soot) and carbon monoxide concentrations are associated with asthma enhancement in the urban children (139). DEP initiate the alveolar epithelial cell movement by alterations of polarity mechanisms (140). An epidemiological study reported that healthy subjects were affected by agriculture crop burning with their altered peak expiratory flow rate and pulmonary functions (1). The patients prone to COPD or asthma already exhibit preexisting oxidative stress and hence are more susceptible toward soot-mediated oxidative damage. Interestingly, it is known that ufCB causes adverse effects via ROS and may have worse manifestations in these susceptible persons (141).
The evidences from animal models also supported soot- and CB-mediated mechanisms of toxicity. A rat model of study described that the flame-generated ultrafine soot increased the ROS and upregulated Nrf2 antioxidants in the lungs (142). Similar studies found that the neonatal lungs are more susceptible to ultrafine soot as compared to adults (143). The ultrafine soot also generates ROS and induces DNA damage (144). Moreover, CB enhanced ROS in the rat alveolar macrophages, this is an example where non-biodegradable components of CB can generate an immune and oxidative stress response (145). Another study showed excessive generation of ROS by monocytes upon exposure of CB (146). It is already known that increased production of pro-inflammatory mediators are linked with the activation of specific transcription factors such as NF-kB, through the Ca2+ upregulation and ROS formation (146–148). A study suggested that ufCB triggers an increase in cytosolic Ca2+, possibly through entry of extracellular Ca2+ via the Ca2+ channels in the plasma membrane (146). Therefore, nanoparticles activate the opening of Ca2+ channels by means of ROS (146). It has now been unrevealed that alterations in the glutathione and superoxide dismutase activities are the key enzymatic mechanisms involved in the generation of oxidative stress by CB (149). CB induced ROS that involves lot of enzymatic reactions including ERK MAP kinase cascade pathway (145). NADPH quinone oxidoreductase-1 enzyme was also activated following DEP exposure and mediates activation of ROS (150). Interestingly, an antioxidant ceruloplasmin was found upregulated in epithelial cells of lung due to ufCB exposure (151). This emphasizes that antioxidant machinery is triggered in response to ufCB exposure in the lung (151). Increased nitrative stress has also been recently reported to cause DNA damage in response to ufCB particles (152).
Oxidative stress produced by soot or CB is subsequently linked with systemic immune response (inflammation) in the lungs, which results in the development of asthma and other diseases (Figure 4) (153–155). The existing literature supports that the inflammation causes serious damage to the lung functions by many mechanisms, most of which are not properly understood (156, 157). It was found that DEP were taken up by epithelial cells of human airway and altered cytokines production showing inflammation in lungs. These cytokines are known to cause damage to the lung functions (158). Notably, one of the key pathological manifestations of dysfunctional inflammatory response is the development of tissue remodeling and fibrosis. It was seen that soot triggers an inflammatory condition that leads to the accumulation of collagen fibers (56). This study provided the evidence that soot-associated lung inflammation leads to the tissue remodeling and fibrosis (56). In addition to the mediators of inflammation, immune cells also play a very critical role and modulate respiratory functions (33, 159). The Th2-type inflammatory responses and activation of pulmonary dendritic cells were seen on instillation of engineered DEP in vivo (160). Similarly, DE enhanced allergen-related eosinophils recruitment to airways and increased protein concentrations of granulocyte macrophage colony-stimulating factor and IL-5 in the lungs of mice (161).
Furthermore, exposure of DEP to rats by intratracheal instillation downregulated LPS-induced TNF-alpha and IL-1 release by alveolar macrophages (162). Similarly, exposure of DEP to rats downregulated the ability of alveolar macrophages to generate the antimicrobial reactive oxidant species in response to zymosan (a fungal component) (163). Moreover, ultrafine carbon particles downregulated cytochrome P450 1B1 expression in human monocytes (164). These data suggested that the Printex-90 decreased the expression profile of CYP gene that may interfere with the detoxification potential of inhaled toxic compounds (164). It is well established that TNF-alpha is a major cytokine responsible for cellular death and causes toxicity (165, 166). The 14-nm CB particles also synergize the ZnCl2 stimulated TNF-alpha release (167). Furthermore, zinc-induced morphological changes and cell death were altered by carbon nanoparticles treatment (167). However, there are also some examples where systemic response was not seen. In a study, CB exposure also lacks an acute phase response in the liver (the production of SAA proteins, Sap, Saa1, and Saa3) (66).
From nutritional point of view, polyunsaturated fatty acids (PUFAs) such as Omega-3 fatty acids or N3-fattyacids have shown protective against asthma risk development (168, 169). Some preclinical studies have shown omega-3 fatty acids as beneficial agents against asthma triggers, such as environmental allergens and viruses (170, 171). The connection with soot toxicity to PUFA is understood on the basis of its antioxidant and anti-inflammatory properties and on its T cell regulatory properties (171). As environmental soot produces plenty of oxidative stress and therefore antioxidant properties of PUFA can act as a protective agents in this area. In patients suffering from respiratory syndrome, a lower level of PUFA and other antioxidants were observed, which are related toward the development of various lung pathologies (172). A high-level PUFA also downregulates oxidative stress-induced chronic bronchopulmonary dysplasia (173). The antioxidant properties of PUFAs also help improve capacity the capacity of exercise in patients suffering from COPD (174). A pilot study described that there have not been any changes in the patients with stable asthma due to n-3 PUFAs dietary supplement (175). It is further important to note that there have been few reports that studied the effect of omega-3 fatty acids in the development of asthma pathophysiology (176, 177). Altogether, it can be said that soot and CB affects various biochemical and molecular mediators that in turn cause respiratory dysfunctions.
The cardiovascular diseases due to soot and CB exposure are of major concerns because of their distal appearance from the site of exposure and involvement of more systemic responses (Figure 4). A sufficient amount of clinical and epidemiological data linked soot and CB to cardiovascular dysfunctions. A case-crossover study showed that personal soot exposure is linked with acute myocardial infarction (178). Soot was also seen responsible in the incidence of myocardial infarction (179). In London, air pollution (BC) caused the activation of implantable cardioverter defibrillators (a device used to treat cardiovascular dysfunctions) (180). In Darwin, Australia, the risk of cardiovascular hospitalization was high in people exposed with bushfire particulates (181). Furthermore, air pollution was considered as a major risk factor to the ST-segment (a measure in electrocardiogram) depression in patients suffering from coronary artery disease (CAD) (182). Notably, traffic emission sources of primary organic carbon particles enhanced platelet activation, systemic inflammation, and potentially reduced antioxidant enzyme activity in old people, suffered from CAD (183). It was observed that ufCB particles were associated with accelerated cardiovascular changes, which may compromise “healthy aging” and may trigger cardiovascular diseases (2).
Many studies on experimental models demonstrated the mechanistic basis of soot toxicity leading to cardiovascular dysfunctions (Figure 4). A study revealed that CB affects cardiac autonomous nervous system functions in mice (184). This indicated that the CB can cause the cardiovascular dysfunctions independent of apparent myocardial and pulmonary injury (184). CB nanoparticles exposure also caused endothelial changes via modulating nitric oxide synthase expression when it is orally given to the rats (185). The long-term exposure of soot (fine particulate air pollution) was found associated with the adverse cardiovascular outcomes (186). The fact that biodiesel particles are more toxic to health and can cause more cumbersome cardiovascular health issues was shown in a mice model of study (187). In this study, heart rate (HR) and mean corpuscular volume were increased compared with control. Interestingly, leukocytes, reticulocytes, platelets, metamyelocytes, neutrophils, and macrophages were also increased compared with control (187). The involvement of a number of inflammatory mediators along with cells were upregulated in patients with cardiovascular dysfunctions, indicating a role of inflammation in diesel soot-mediated cardiovascular toxicity (187). The myth that UFPs go into the blood circulation was broken by a study showing translocation of UFPs in microcirculation of extrapulmonary organs after the inhalation (188). Moreover, when CB UFPs were infused into intra-arterially in C57BL/6 mice, significant enhancement in platelet on endothelium of post-sinusoidal venules and sinusoids was observed (188). It is already known that immune response is highly regulated by epigenetic mediators and its interactions with endothelial system may cause changes in the cardiovascular system. A study also described that pollution leads to endothelial dysfunctions through epigenetic associations (189). Importantly, ufCB particle changed the expression profile of endothelial nitric oxide synthase in abdominal aorta of animals (2).
Mechanistically, it is unveiled that ultrafine BC causes endothelial senescence and alters the cardiovascular functions at molecular levels (2). Telomerase reverse transcriptase, an enzyme that is required for telomere maintenance, is believed to be critical for proper endothelial cell functions and is inactivated by Src kinase in situations of excessive oxidative stress (2). ufCB increased Src kinase activation and decreased the telomerase activity in lung epithelial and endothelial cells (2). Consequently, ufCB increases senescence of endothelial cells and thus alters cardiovascular functions (2). The data from 642 elderly participated in the Veterans Administration study strongly emphasized the role of soot-mediated cardiovascular diseases. This study demonstrated that BC exposure affected soluble vascular cell adhesion molecule-1 (sVCAM-1), and soluble intercellular adhesion molecule-1 (sICAM-1) both molecule regulate endothelial and cardiovascular system (190). Particularly, diabetics were more sensitive to the BC for both sVCAM-1 and sICAM-1 (190). DEP and CB also altered expressions of cell adhesion molecules and caused oxidative damage in human endothelial cells (191). Proteomic analysis from bronchoalveolar lavage fluid unveiled the action of BC and demonstrated a strong relationship between albumin or alpha2-macroglobulin and vascular endothelial growth factor (151). A difference was also reported in the HR of the soot-exposed mice (70). The other supporting study linked the acute inflammation and cardiovascular functions due to inhalation of diesel and biodiesel exhaust particles (187). Collectively, all studies proved that soot and CB indeed are responsible for the adverse cardiovascular functions. Although these studies show a close association of soot or CB exposure to the cardiovascular symptoms, but the exact mechanism of soot-mediated cardiovascular diseases is not properly understood and needs further investigations.
Based on the existing literature discussed in the above sections, the mechanism of soot or CB toxicity can be proposed (Figure 4) (192, 193). Two main kinds of toxicities may exist due to soot or CB exposure: the localized toxicity and the systemic toxicity (194, 195). The localized or direct toxicity includes exposure of soot and its localized (contact mediated) effect by way of oxidative and/or necrotic damage to the lung epithelial cells (161). The second type of toxicity is more systemic in nature and produces toxicity beyond the site of exposure of soot or CB. In fact, systemic toxicity involves immune response that causes damage to the lungs and the other parts of the body (155, 161, 196). However, both types of toxic responses are interlinked and thus soot toxicity can be considered as a combined effect of them.
The localized response of soot is mainly caused by the oxidative stress and dysfunctions in the cellular machinery (Figure 4) (142, 197). One of the mechanisms by which soot or CB exert oxidative stress is the interruption of mitochondrial metabolism (142, 198). This oxidative stress may in turn increase the level of calcium ion release in the cytoplasm and thus affects the cellular signaling (199–202). It is already established that calcium signaling modulates the cell metabolic pathways and alters the cell survival or death pathways (200). Furthermore, it is interesting to know that excessive oxidative stress generated by soot or CB causes extensive DNA damage that leads to the development of cancer (203–205). In fact, oxidative stress produced by the soot interrupts DNA repair machinery and promote cancer like phenomenon (104). Apart from this soot also triggers the cell death probably through the autophagic and apoptotic pathways (132, 206). CB also induces DNA damage and genotoxicity in the in vitro cultured cells (99).
The systemic response of soot is more complex and affects both local and distal parts of the body by more than one mechanism (133). It is believed that the symptoms of the soot- or CB-induced immune response are similar to allergic diseases (207–209). The CB-induced IgE production in children confirms it as an allergen (210, 211). The subsequent pathologic response via IgE pathway is the development of tissue remodeling and fibrosis (209, 212). Importantly, it was noticed that toll-like receptors 2 and 4 are associated with traffic-related air pollutant (soot), which causes development of asthma in children (213). As illustrated in the Figure 4, when soot or CB comes in contact with the lung epithelial cells, various factors are released. These factors attract a huge number of immune cells that damage the lungs tissue in many ways (207, 214). Mainly, eosinophils, natural killer cells [NK and invariant natural killer cells (iNKT)], mast cells, T cells, and innate lymphocyte cells get activated due to the allergic responses (215–217). The blood monocytes and neutrophils may also participate in this initial inflammatory response (217–219). In sharp contrast to the diversity of cell types, mast cells and eosinophils play the critical role in the development of more cumbersome pathogenesis (Figure 5) (220, 221). Eosinophils secrete profibrotic cytokines such as IL-13, IL-4, transforming growth factor (TGF)-, and stem cell factor which in turn induce fibroblast and mast cells proliferation and activation (215, 222, 223). Activated mast cells promote tissue remodeling and fibrosis by secreting tryptase, chymase, and histamine, which work closely with fibroblasts (Figure 5) (215, 224–226). As seen in the figure, monocytes and macrophages become activated and secrete several inflammatory and toxic mediators including MMPs (200, 227–232). Importantly, it is seen that SAA protein is produced in the serum in response to soot (219, 233). It is therefore possible that SAA protein in response to soot exposure may lead to amyloidosis as observed in chronic inflammatory conditions (219, 233). The involvement of Th17 cells are postulated to play a pivotal role in the causation of soot-mediated toxicity (161, 227, 234).
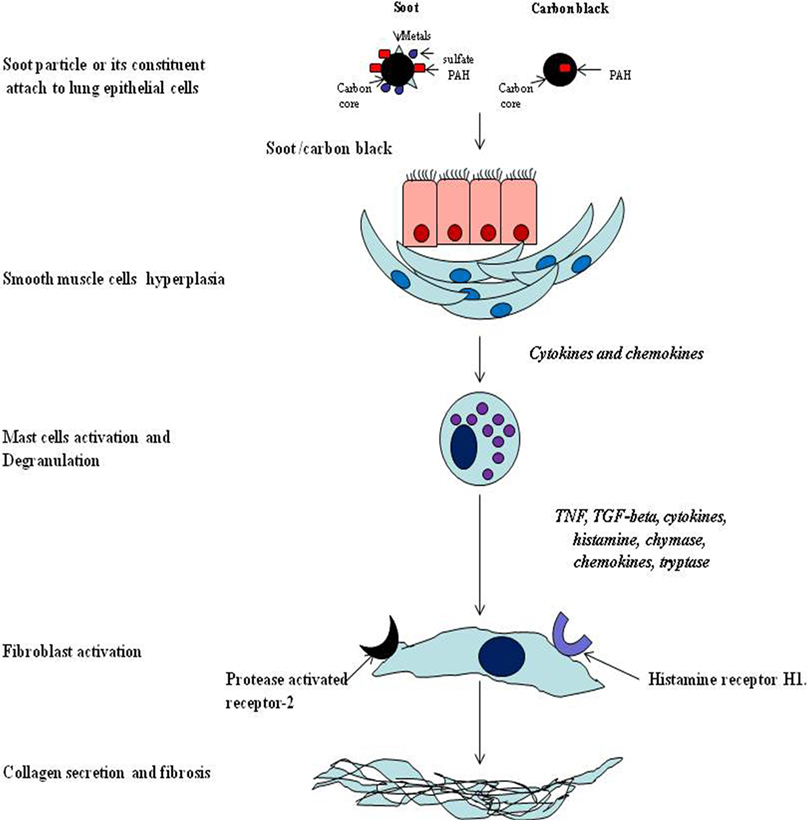
Figure 5. The mechanism of mast cells mediated allergic response due to soot and carbon black. Soot triggers activation of mast cells that in turn release the mediators of inflammation. These mediators subsequently activate fibroblast cells. The fibroblast dysfunctions by these mediators is caused by two receptors (heparin 1 and protease-activated receptor 2) leading to excess production of collagen and other fibrous proteins of the extracellular space.
As of now, a very small part of soot- or CB-induced toxicity is known and a lot more needs to be explored in this area (235). One of the main pathological manifestations of soot or CB is the development of tissue remodeling and fibrosis, the process of which is still largely unknown (161, 236). The fibroblast recruitment to the lung and its role in the soot-induced development of tissue remodeling and fibrosis still remain elusive (212). The role of NK and iNKT cells and eosinophils is not properly known (161, 217, 237, 238). The eosinophils interactions with mast cells are known to modulate the allergic responses in lungs and contribute to the development of tissue remodeling and fibrosis (239, 240). The involvement of chemotactic factors such as eotaxin-1 and eotaxin-2 is also not clear in response to soot or CB exposure (75). A recent break through study described the role of resistin-like molecule alpha and beta (RELM-α/β) in the lung pathology (241). How soot or CB toxicity is associated with the RELM-α/β is not clearly known (Figure 6). The amyloid formation due to inflammatory conditions in lung-related pathologies in response to soot or CB exposure needs exploration in future (242, 243).
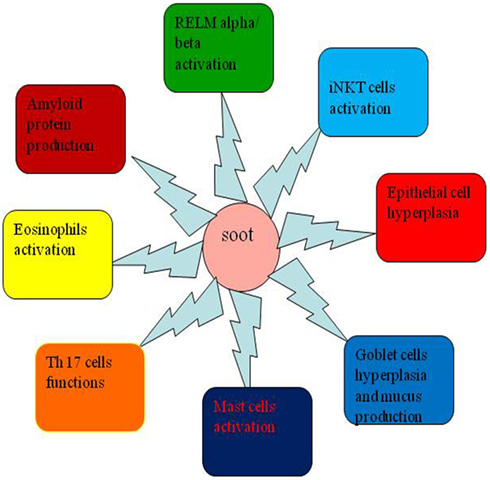
Figure 6. New aspects that need to be explored in the area of soot or carbon black (CB) toxicity. This figure shows new research areas, which need to be explored in soot or CB toxicity. The role of resistin-like molecule-α (RELM-alpha), role of eosinophils and mast cells, activation of invariant natural killer (iNKT) and Th17 cells, and involvement of serum amyloid protein leading to amyloidosis are some of the important areas that still need to be explored in detail.
In recent years, some therapeutic strategies have been suggested to combat the adverse effects of soot or CB (108, 244). As understood from the existing literature and from above discussions, the mechanism of soot toxicity involves immune cells, mediators of inflammation, and various molecules of oxidative stress responsive pathways (245, 246). Therefore, these all may contribute as important targets for the development of novel therapeutics (Figure 7) (247, 248). Here, we discuss some relevant strategies that have been already tested against immune dysfunctions and excessive oxidative stress. Therefore, these can possibly be used to treat soot- or CB-induced toxicities.
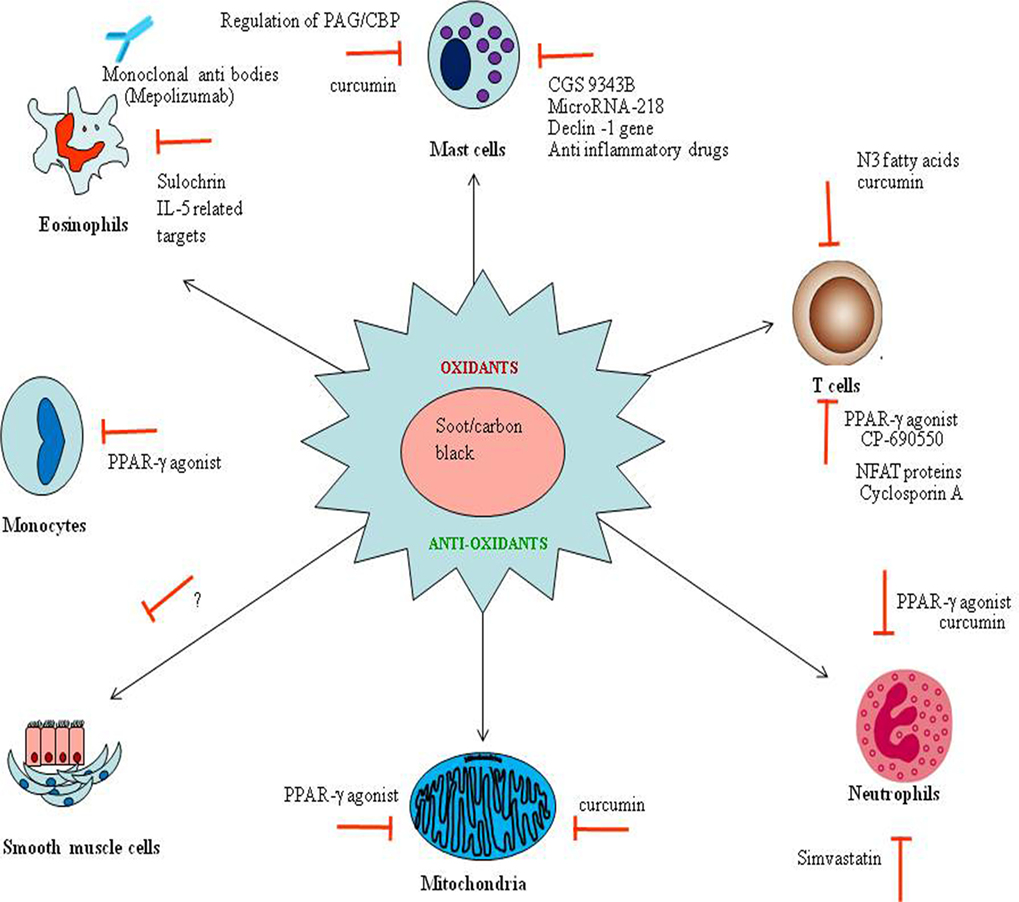
Figure 7. The possible therapeutic strategies to combat soot or carbon black (CB) toxicity. The therapeutic strategies are shown in the figure targeting oxidative stress (in the center) and immune cells (in the periphery) involved in soot or CB toxicity. Red signs show the inhibitors capable of arresting the appropriate response. Anti-eosinophils based antibodies to prevent eosinophils mediated toxic effects are shown. The microRNAs and some noble inhibitors can also be used to downregulate the mast-associated toxicities. N3 fatty acids (polyunsaturated fatty acids) based therapies may be useful to minimize the T cell-associated effects. Importantly, the PPR-γ-based therapeutic interventions become more interesting to target various cell-specific functions.
The antioxidant therapy can be an important way of treating soot and CB toxicity (249, 250). The existing literature already reported some examples of antioxidant therapy for the pulmonary toxicities (251). Zerumbone, an antioxidant, attenuated Th2 responses induced by ovalbumin and decreased airway inflammation in a mice model of study (252). Similarly, naringin, a flavinoid antioxidant, also attenuated airway inflammation in a mouse model of asthma (253). Allium cepa extract and quercetin also showed protective effect in a mice model of asthma (254). Crocus sativus, a natural antioxidant, and its main constituents, safranal and crocin, have shown the protective effects against the oxidative stress in the mice model of asthma (255). Resveratrol, a well-known antioxidant, has also shown its protective effects in a mice model of asthma (256–258). The clear example of antioxidant therapy to the soot or CB caused injury came from the effect of artesunate, which significantly decreased the levels of oxidative biomarkers, 3-nitrotyrosine, 8-isoprostane, and 8-OHdG in a mice model of lung injury (259). Similarly, melatonin, a natural antioxidant, was found to reduce airway inflammation in an asthma model (260). Recently, mitochondrial-based antioxidant therapy downregulates TGF-β-mediated deposition of collagen in a murine model of asthma (261). Overall, this can be considered that antioxidant-based therapeutics can be a promising approach against soot- and CB-associated disorders.
It is evidenced that immune cells such as mast cells, eosinophils, T cells, and neutrophils are the major culprit in soot and CB toxicity. Therefore, these may be targeted for the development of noble therapeutic approaches (Figure 7) (215, 226, 262). A monoclonal antibody mepolizumab against the eosinophils activation has been developed and is currently in clinical trials against severe eosinophilic asthma (DREAM) (263). Similar strategies can be used against eosinophils and other mediators of immune response in soot- and CB-mediated toxicity. The mast cells may be the next important target for which a number of therapeutic interventions have been developed (215, 264). Notably, CGS 9343B, a strong inhibitor of calmodulin family, has a potential to inhibit histamine release by mast cells as shown in rats (265, 266). Inhibitors of dectin-1 signaling (R406) downregulated mast cells activation, thus can also be used as a novel therapy to target soot-induced mast cell’s toxicity (267). Similarly, TGaseII/miR-218/-181 can also be used against mast cell activation (Figure 7) (268). Importantly, anti-inflammatory drugs can also interrupt mast cells degranulation and endothelial cells activation (269). Furthermore, a cross-talk between human mast cells and TGF-beta1 signaling has been shown. Therefore, it is postulated to use anti-TGF-beta1 signaling against mast cells associated soot toxicity (270). Recently, a novel, potent dual inhibitor (JNJ-10311795; RWJ-355871) of the leukocyte proteases cathepsin G and chymase has been discovered, which also has an anti-inflammatory activity in vivo (271). It has been already reported that cytochalasin B is able to inhibit the cytotoxic response of sensitized lymphoid cells and thus may attenuate mast cells responses (Figure 7) (272). The activation of transmembrane adaptor protein PAG/CBP, which is associated with the both positive and negative regulation cell signaling of mast cells, may also be used as a noble target against mast cells activation (273).
Inhibition of T-cell activation can also be a good idea against soot- or CB-triggered adverse responses (274). It has been reported that PPAR gamma is an important molecule and negatively regulates T cell activation (275). CP-690550 is a Janus kinase inhibitor that downregulate CD4+ T-cell-mediated diseases by inhibiting the interferon-gamma pathway (276). Cyclosporin A decreases surface antigen expression on the activated lymphocytes (277). A noble protein “NFAT” has also been reported as a key regulator of T-cell development and functions (278). A unique way of targeting T-cell activation can be the use of N3 fatty acids (omega-3 fatty acids), as they interrupts transcription of human IL-13, which indirectly inhibit the T-helper type 2 effecter immune responses (Figure 7) (279). More recently, the antagonism of noble microRNA-126 has been known to suppress the effecter functions of Th2 cells in the allergic airways disease (280).
Furthermore, inhibition of neutrophils can also be taken into consideration for the development of noble protective strategies. Simvastatin has been shown to affect many adverse functions of neutrophils. In a recent study, it has been shown that simvastatin downregulated secretion of interleukin-8 (IL-8) by neutrophils from the dyslipidemic patients (281). In addition to targeting specialized cells, some other miscellaneous strategies can also be used for the possible management of soot- and CB-associated toxicity. Study conducted in asthmatic rats showed that zinc suppressed inflammation in the airway with exerting effects on the level of eotaxin, IL-8, IL-4, monocyte chemotactic protein-1, and IFN-gamma (282). Curcumin suppresses ovalbumin-induced allergic disease (283). Inhibition of PPR-gamma can be a good strategy for combating the allergic response against soot (284).
Despite tremendous progress in management of air pollution throughout the world, it continues to harm people’s health and the environment. Nowadays, the problem of air pollution is intensified globally, and soot has been the key pollutant due to its effects on health of humans. The soot and CB toxicity is a broader area of research and should not be only limited to cancer, respiratory, and cardiovascular diseases but also include other disorders. Soot and CB induce cancers at the site of exposure and beyond due to DNA mutations, DNA adducts formation, AhR activation, DNA methylation, and altered oncogenes expressions. The soot- or CB-induced immune response in the lung play critical role in the development of cancer, cardiovascular dysfunctions, and respiratory diseases. The role of eosinophils and other immune cells is critically discussed pointing toward development of noble therapeutics. Based on the existing literature, a consensus mechanism has been proposed depicting the linkages between different cellular and metabolic pathways. As soot and CB can exert chronic inflammatory condition, it may elicit amyloid deposition in different tissues which is largely unknown. The effect of soot on cardiovascular and oxidative stress parameters need to be studied extensively. Altogether, this review provides a better understanding of soot- and CB-induced pathologies and strategies for the possible therapeutics.
RN wrote the manuscript. AT was involved in conceptualizing and suggestions and also helped to proof read and organize the final manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The “young scientist fellowship to RN by SERB (DST, India) on the project “To explore the role of eosinophiles in the development of tissue remodeling and fibrosis in the pathology of asthma” is gratefully acknowledged. AT thanks to Prof. Sachidanad Tripathi, Prof. Tarun Gupta [Civil Engineering], Prof. Debajyoti Paul [Earth Sciences], Prof. Ketan Rajawat [Electrical Engineering], and Indian Institute of Technology Kanpur in clarifying some of the points during the writing of this article.
The funding support from “young scientist fellowship grant No. SB/YS/LS-198/2014 to Dr. Rituraj Niranjan by SERB (DST, India.) is gratefully acknowledged. AKT also acknowledges the support by MHRD project No. MHRD/CESE/2013357 entitled “Building a noble system for soot: measurement, toxicity assessment and source identification”.
PM, particulate matter; DEP, diesel exhaust particles; ROS, reactive oxygen species; TNF-α, tumor necrosis factor-alpha; 8-OHdG, 8-hydroxy-2′-deoxyguanosine; iNKT, invariant natural killer T cells; IL-1, interleukin-1; AD, aerodynamic diameter; ufBC, ultrafine black carbon; PAH, polycyclic aromatic hydrocarbons; CAD, coronary artery disease; CB, carbon black; COPD, coronary occlusion pulmonary disease; Cp, ceruloplasmin; VEGF, vascular endothelial growth factor; ANS, autonomic nerves system; MMP-9, matrix metalloproteases-9; IARC, International Agency for Research on Cancer; UFPs, ultrafine particles; IL-5, interleukin-5; TERT, telomerase reverse transcriptase; sICAM-1, soluble intercellular adhesion molecule-1; Nrf2, nuclear factor erythroid 2; CYP1B1, cytochrome P450 1B1; DE, diesel exhaust; sVCAM-1, soluble vascular cell adhesion molecule-1; RELM, resistin-like molecule alpha and beta; IL-8, interleukin-8; IL-4, interleukin-4; TGF-β, transforming growth factor beta; IL-13, interleukin-13; SCF, stem cell factor; AMPK, adenosine monophosphate-activated protein kinase; SAA, serum amyloid A; MCP-1, monocyte chemotactic protein-1; eNOS, endothelial nitric oxide synthase; NF-kB, nuclear factor kappa B; GM-CSF, granulocyte macrophage colony-stimulating factor; VA, veteran administration; PUFAs, polyunsaturated fatty acids; NK, natural killer cells.
1. Agarwal R, Awasthi A, Singh N, Mittal SK, Gupta PK. Epidemiological study on healthy subjects affected by agriculture crop-residue burning episodes and its relation with their pulmonary function tests. Int J Environ Health Res (2013) 23(4):281–95. doi:10.1080/09603123.2012.733933
2. Buchner N, Ale-Agha N, Jakob S, Sydlik U, Kunze K, Unfried K, et al. Unhealthy diet and ultrafine carbon black particles induce senescence and disease associated phenotypic changes. Exp Gerontol (2013) 48(1):8–16. doi:10.1016/j.exger.2012.03.017
3. Long CM, Nascarella MA, Valberg PA. Carbon black vs. black carbon and other airborne materials containing elemental carbon: physical and chemical distinctions. Environ Pollut (2013) 181:271–86. doi:10.1016/j.envpol.2013.06.009
4. Watson AY, Valberg PA. Carbon black and soot: two different substances. AIHAJ (2001) 62(2):218–28. doi:10.1080/15298660108984625
5. Medalia AI, Rivin D, Sanders DR. A comparison of carbon black with soot. Sci Total Environ (1983) 31(1):1–22. doi:10.1016/0048-9697(83)90053-0
6. Lewis GP, Coughlin L. Lung “soot” accumulation in man. Atmos Environ (1973) 7(12):1249–55. doi:10.1016/0004-6981(73)90134-0
8. Chuang HC, Jones T, Chen Y, Bell J, Wenger J, BeruBe K. Characterisation of airborne particles and associated organic components produced from incense burning. Anal Bioanal Chem (2011) 401(10):3095–102. doi:10.1007/s00216-011-5209-7
9. Canagaratna MR, Onasch TB, Wood EC, Herndon SC, Jayne JT, Cross ES, et al. Evolution of vehicle exhaust particles in the atmosphere. J Air Waste Manag Assoc (2010) 60(10):1192–203. doi:10.3155/1047-3289.60.10.1192
10. Glaser B, Dreyer A, Bock M, Fiedler S, Mehring M, Heitmann T. Source apportionment of organic pollutants of a highway-traffic-influenced urban area in Bayreuth (Germany) using biomarker and stable carbon isotope signatures. Environ Sci Technol (2005) 39(11):3911–7. doi:10.1021/es050002p
11. Scheepers PT, Bos RP. Combustion of diesel fuel from a toxicological perspective. I. Origin of incomplete combustion products. Int Arch Occup Environ Health (1992) 64(3):149–61. doi:10.1007/BF00380904
12. Birky MM, Voorhees KJ. The use of soot analysis as an investigative tool in aircraft fires. Aviat Space Environ Med (1989) 60(10 Pt 2):B72–7.
13. Kamboures MA, Hu S, Yu Y, Sandoval J, Rieger P, Huang SM, et al. Black carbon emissions in gasoline vehicle exhaust: a measurement and instrument comparison. J Air Waste Manag Assoc (2013) 63(8):886–901. doi:10.1080/10962247.2013.787130
14. Niessner R. The many faces of soot: characterization of soot nanoparticles produced by engines. Angew Chem Int Ed Engl (2014) 53(46):12366–79. doi:10.1002/anie.201402812
15. Wang Q, Schwarz JP, Cao J, Gao R, Fahey DW, Hu T, et al. Black carbon aerosol characterization in a remote area of Qinghai-Tibetan Plateau, Western China. Sci Total Environ (2014) 47(9–480):151–8. doi:10.1016/j.scitotenv.2014.01.098
16. Rosen H, Hansen AD, Dod RL, Novakov T. Soot in urban atmospheres: determination by an optical absorption technique. Science (1980) 208(4445):741–4. doi:10.1126/science.208.4445.741
17. China S, Mazzoleni C, Gorkowski K, Aiken AC, Dubey MK. Morphology and mixing state of individual freshly emitted wildfire carbonaceous particles. Nat Commun (2013) 4:2122. doi:10.1038/ncomms3122
18. Palotas AB, Rainey LC, Feldermann CJ, Sarofim AF, Vander Sande JB. Soot morphology: an application of image analysis in high-resolution transmission electron microscopy. Microsc Res Tech (1996) 33(3):266–78. doi:10.1002/(SICI)1097-0029(19960215)33:3<266:AID-JEMT4>3.0.CO;2-O
19. Cain JP, Gassman PL, Wang H, Laskin A. Micro-FTIR study of soot chemical composition-evidence of aliphatic hydrocarbons on nascent soot surfaces. Phys Chem Chem Phys (2010) 12(20):5206–18. doi:10.1039/b924344e
20. Wang J, Levendis YA, Richter H, Howard JB, Carlson J. Polycyclic aromatic hydrocarbon and particulate emissions from two-stage combustion of polystyrene: the effect of the primary furnace temperature. Environ Sci Technol (2001) 35(17):3541–52. doi:10.1021/es0105109
21. Boffetta P, Jourenkova N, Gustavsson P. Cancer risk from occupational and environmental exposure to polycyclic aromatic hydrocarbons. Cancer Causes Control (1997) 8(3):444–72. doi:10.1023/A:1018465507029
22. Fernandes MB, Brooks P. Characterization of carbonaceous combustion residues: II. Nonpolar organic compounds. Chemosphere (2003) 53(5):447–58. doi:10.1016/S0045-6535(03)00452-1
23. Yang H, Jin H, Wang X, Liu Z, Yu M, Zhao F, et al. X-ray crystallographic characterization of new soluble endohedral fullerenes utilizing the popular C82 bucky cage. Isolation and structural characterization of Sm@C3v(7)-C82, Sm@C(s)(6)-C82, and Sm@C2(5)-C82. J Am Chem Soc (2012) 134(34):14127–36. doi:10.1021/ja304867j
24. Vedal S, Campen MJ, McDonald JD, Larson TV, Sampson PD, Sheppard L, et al. National particle component toxicity (NPACT) initiative report on cardiovascular effects. Res Rep Health Eff Inst (2013) 178:5–8.
25. Goto D. Modeling of black carbon in Asia using a global-to-regional seamless aerosol-transport model. Environ Pollut (2014) 195:330–5. doi:10.1016/j.envpol.2014.06.006
26. Browne EC, Franklin JP, Canagaratna MR, Massoli P, Kirchstetter TW, Worsnop DR, et al. Changes to the chemical composition of soot from heterogeneous oxidation reactions. J Phys Chem A (2015) 119(7):1154–63. doi:10.1021/jp511507d
27. Ivleva NP, Huckele S, Weinzierl B, Niessner R, Haisch C, Baumann T. Identification and characterization of individual airborne volcanic ash particles by Raman microspectroscopy. Anal Bioanal Chem (2013) 405(28):9071–84. doi:10.1007/s00216-013-7328-9
28. Zielinska B, Samy S, McDonald JD, Seagrave J. Atmospheric transformation of diesel emissions. Res Rep Health Eff Inst (2010) 147:5–60.
29. China’s environmental protection: challenges and countermeasures. China Popul Today (1993) 10(5):16–9.
30. Victor DG, Ramanathan V, Zaelke D. Air pollution: harmful soot spurs climate-policy action. Nature (2015) 517(7532):21. doi:10.1038/517021b
31. Wang R, Tao S, Shen H, Huang Y, Chen H, Balkanski Y, et al. Trend in global black carbon emissions from 1960 to 2007. Environ Sci Technol (2014) 48(12):6780–7. doi:10.1021/es5021422
32. Anenberg SC, Schwartz J, Shindell D, Amann M, Faluvegi G, Klimont Z, et al. Global air quality and health co-benefits of mitigating near-term climate change through methane and black carbon emission controls. Environ Health Perspect (2012) 120(6):831–9. doi:10.1289/ehp.1104301
34. Subramanian M. Global health: deadly dinners. Nature (2014) 509(7502):548–51. doi:10.1038/509548a
35. Liu S, Xia X, Zhai Y, Wang R, Liu T, Zhang S. Black carbon (BC) in urban and surrounding rural soils of Beijing, China: spatial distribution and relationship with polycyclic aromatic hydrocarbons (PAHs). Chemosphere (2011) 82(2):223–8. doi:10.1016/j.chemosphere.2010.10.017
36. Gramsch E, Reyes E, Oyola P, Ma R, Lopez G, Perez P, et al. Particle size distribution and its relationship to black carbon in two urban and one rural site in Santiago de Chile. J Air Waste Manag Assoc (2014) 64(7):785–96. doi:10.1080/10962247.2014.890141
37. Liggio J, Gordon M, Smallwood G, Li SM, Stroud C, Staebler R, et al. Are emissions of black carbon from gasoline vehicles underestimated? Insights from near and on-road measurements. Environ Sci Technol (2012) 46(9):4819–28. doi:10.1021/es2033845
38. Zhan C, Cao J, Han Y, Huang S, Tu X, Wang P, et al. Spatial distributions and sequestrations of organic carbon and black carbon in soils from the Chinese Loess Plateau. Sci Total Environ (2013) 465:255–66. doi:10.1016/j.scitotenv.2012.10.113
39. Alam MS, Zeraati-Rezaei S, Stark CP, Liang Z, Xu H, Harrison RM. The characterisation of diesel exhaust particles – composition, size distribution and partitioning. Faraday Discuss (2016) 189:69–84. doi:10.1039/c5fd00185d
40. Cassee FR, Heroux ME, Gerlofs-Nijland ME, Kelly FJ. Particulate matter beyond mass: recent health evidence on the role of fractions, chemical constituents and sources of emission. Inhal Toxicol (2013) 25(14):802–12. doi:10.3109/08958378.2013.850127
41. Valavanidis A, Vlachogianni T, Fiotakis K, Loridas S. Pulmonary oxidative stress, inflammation and cancer: respirable particulate matter, fibrous dusts and ozone as major causes of lung carcinogenesis through reactive oxygen species mechanisms. Int J Environ Res Public Health (2013) 10(9):3886–907. doi:10.3390/ijerph10093886
42. Apicella B, Barbella R, Ciajolo A, Tregrossi A. Comparative analysis of the structure of carbon materials relevant in combustion. Chemosphere (2003) 51(10):1063–9. doi:10.1016/S0045-6535(02)00715-4
43. Jeong BO, Kwon SW, Kim TJ, Lee EH, Jeong SH, Jung Y. Effect of carbon black materials on the electrochemical properties of sulfur-based composite cathode for lithium-sulfur cells. J Nanosci Nanotechnol (2013) 13(12):7870–4. doi:10.1166/jnn.2013.8111
44. Abbas K, Simonelli F, Holzwarth U, Cydzik I, Bulgheroni A, Gibson N, et al. Feasibility study of production of radioactive carbon black or carbon nanotubes in cyclotron facilities for nanobioscience applications. Appl Radiat Isot (2013) 73:44–8. doi:10.1016/j.apradiso.2012.11.012
45. Wang C, Yediler A, Lienert D, Wang Z, Kettrup A. Ozonation of an azo dye C.I. Remazol Black 5 and toxicological assessment of its oxidation products. Chemosphere (2003) 52(7):1225–32. doi:10.1016/S0045-6535(03)00331-X
46. Tsai PJ, Shieh HY, Lee WJ, Lai SO. Characterization of PAHs in the atmosphere of carbon black manufacturing workplaces. J Hazard Mater (2002) 91(1–3):25–42. doi:10.1016/S0304-3894(01)00384-3
47. Cadorim HR, Pereira ER, Carasek E, Welz B, de Andrade JB. Determination of sulfur in crude oil using high-resolution continuum source molecular absorption spectrometry of the SnS molecule in a graphite furnace. Talanta (2016) 146:203–8. doi:10.1016/j.talanta.2015.07.088
48. Denkler K. Sir Percivall Pott, Sir James Paget, and soot cancer of the hand. Lancet (2004) 364(9434):582. doi:10.1016/S0140-6736(04)16848-7
49. Androutsos G. The outstanding British surgeon Percivall Pott (1714–1789) and the first description of an occupational cancer. J BUON (2006) 11(4):533–9.
51. Novakov T, Rosen H. The black carbon story: early history and new perspectives. Ambio (2013) 42(7):840–51. doi:10.1007/s13280-013-0392-8
52. Rosmanith J, Kandus J, Holusa R. [Anthracofibrosis in the carbon black industry. (Carbon pneumoconiosis)]. Int Arch Arbeitsmed (1969) 25(4):292–8.
53. Riboli E, Bai E, Berrino F, Merisi A. Mortality from lung cancer in an acetylene and phthalic anhydride plant. A case-referent study. Scand J Work Environ Health (1983) 9(6):455–62. doi:10.5271/sjweh.2387
54. Snow JB Jr. Carbon black inhalation into the larynx and trachea. Laryngoscope (1970) 80(2):267–87. doi:10.1288/00005537-197002000-00012
55. Kandt D, Biendara E. [Clinical symptoms and findings in the upper airway of soot-exposed workers]. Z Erkr Atmungsorgane (1985) 165(1):13–24.
56. Beck B, Gohlke R, Sturm W, Bergmann L, Wolf E. [Soot lung as occupational disease]. Z Erkr Atmungsorgane (1985) 164(3):254–66.
57. Bourguet CC, Checkoway H, Hulka BS. A case-control study of skin cancer in the tire and rubber manufacturing industry. Am J Ind Med (1987) 11(4):461–73. doi:10.1002/ajim.4700110409
58. Parent ME, Siemiatycki J, Renaud G. Case-control study of exposure to carbon black in the occupational setting and risk of lung cancer. Am J Ind Med (1996) 30(3):285–92. doi:10.1002/(SICI)1097-0274(199609)30:3<285:AID-AJIM6>3.0.CO;2-Y
59. Szozda R. [Condition of the respiratory system in workers involved in carbon black production]. Med Pr (1994) 45(1):57–61.
60. Parent ME, Siemiatycki J, Fritschi L. Workplace exposures and oesophageal cancer. Occup Environ Med (2000) 57(5):325–34. doi:10.1136/oem.57.5.325
61. Agola JO, Jim PA, Ward HH, Basuray S, Wandinger-Ness A. Rab GTPases as regulators of endocytosis, targets of disease and therapeutic opportunities. Clin Genet (2011) 80(4):305–18. doi:10.1111/j.1399-0004.2011.01724.x
62. Raaschou-Nielsen O, Beelen R, Wang M, Hoek G, Andersen ZJ, Hoffmann B, et al. Particulate matter air pollution components and risk for lung cancer. Environ Int (2016) 87:66–73. doi:10.1016/j.envint.2015.11.007
63. Jackson P, Vogel U, Wallin H, Hougaard KS. Prenatal exposure to carbon black (Printex 90): effects on sexual development and neurofunction. Basic Clin Pharmacol Toxicol (2011) 109(6):434–7. doi:10.1111/j.1742-7843.2011.00745.x
64. Gerlofs-Nijland ME, van Berlo D, Cassee FR, Schins RP, Wang K, Campbell A. Effect of prolonged exposure to diesel engine exhaust on proinflammatory markers in different regions of the rat brain. Part Fibre Toxicol (2010) 7:12. doi:10.1186/1743-8977-7-12
65. Kyjovska ZO, Boisen AM, Jackson P, Wallin H, Vogel U, Hougaard KS. Daily sperm production: application in studies of prenatal exposure to nanoparticles in mice. Reprod Toxicol (2013) 36:88–97. doi:10.1016/j.reprotox.2012.12.005
66. Saber AT, Halappanavar S, Folkmann JK, Bornholdt J, Boisen AM, Moller P, et al. Lack of acute phase response in the livers of mice exposed to diesel exhaust particles or carbon black by inhalation. Part Fibre Toxicol (2009) 6:12. doi:10.1186/1743-8977-6-12
67. El-Sayed YS, Shimizu R, Onoda A, Takeda K, Umezawa M. Carbon black nanoparticle exposure during middle and late fetal development induces immune activation in male offspring mice. Toxicology (2015) 327:53–61. doi:10.1016/j.tox.2014.11.005
68. Burzynski NJ. Aging in guinea pig salivary gland. J Gerontol (1971) 26(2):204–7. doi:10.1093/geronj/26.2.204
69. Boisen AM, Shipley T, Jackson P, Wallin H, Nellemann C, Vogel U, et al. In utero exposure to nanosized carbon black (Printex90) does not induce tandem repeat mutations in female murine germ cells. Reprod Toxicol (2013) 41:45–8. doi:10.1016/j.reprotox.2013.06.068
70. Tankersley CG, Bierman A, Rabold R. Variation in heart rate regulation and the effects of particle exposure in inbred mice. Inhal Toxicol (2007) 19(8):621–9. doi:10.1080/08958370701353049
71. Locati G, Fantuzzi A, Consonni G, Gotti I. Li, Bonomi G. Identification of polycyclic aromatic hydrocarbons in carbon black with reference to cancerogenic risk in tire production. Am Ind Hyg Assoc J (1979) 40(7):644–52. doi:10.1080/15298667991430109
72. Goulaouic S, Foucaud L, Bennasroune A, Laval-Gilly P, Falla J. Effect of polycyclic aromatic hydrocarbons and carbon black particles on pro-inflammatory cytokine secretion: impact of PAH coating onto particles. J Immunotoxicol (2008) 5(3):337–45. doi:10.1080/15476910802371016
73. Mehta AJ, Kubzansky LD, Coull BA, Kloog I, Koutrakis P, Sparrow D, et al. Associations between air pollution and perceived stress: the Veterans Administration Normative Aging study. Environ Health (2015) 14(1):10. doi:10.1186/1476-069X-14-10
74. Ferecatu I, Borot MC, Bossard C, Leroux M, Boggetto N, Marano F, et al. Polycyclic aromatic hydrocarbon components contribute to the mitochondria-antiapoptotic effect of fine particulate matter on human bronchial epithelial cells via the aryl hydrocarbon receptor. Part Fibre Toxicol (2010) 7:18. doi:10.1186/1743-8977-7-18
75. Roulet A, Armand L, Dagouassat M, Rogerieux F, Simon-Deckers A, Belade E, et al. Intratracheally administered titanium dioxide or carbon black nanoparticles do not aggravate elastase-induced pulmonary emphysema in rats. BMC Pulm Med (2012) 12:38. doi:10.1186/1471-2466-12-38
76. Courtois A, Andujar P, Ladeiro Y, Baudrimont I, Delannoy E, Leblais V, et al. Impairment of NO-dependent relaxation in intralobar pulmonary arteries: comparison of urban particulate matter and manufactured nanoparticles. Environ Health Perspect (2008) 116(10):1294–9. doi:10.1289/ehp.11021
77. Laser diagnostic techniques used to measure soot formation. Appl Opt (1985) 24(8):1101. doi:10.1364/AO.24.001101
78. Evanoff BA, Gustavsson P, Hogstedt C. Mortality and incidence of cancer in a cohort of Swedish chimney sweeps: an extended follow up study. Br J Ind Med (1993) 50(5):450–9.
79. Gerber C, von Hochstetter AR, Schuler G, Hofmann V, Rosenthal C. [Penis carcinoma in a young chimney sweep. Case report 200 years following the description of the first occupational disease]. Schweiz Med Wochenschr (1995) 125(24):1201–5.
80. Hogstedt C, Jansson C, Hugosson M, Tinnerberg H, Gustavsson P. Cancer incidence in a cohort of Swedish chimney sweeps, 1958–2006. Am J Public Health (2013) 103(9):1708–14. doi:10.2105/AJPH.2012.300860
81. Gerde P, Muggenburg BA, Lundborg M, Tesfaigzi Y, Dahl AR. Respiratory epithelial penetration and clearance of particle-borne benzo[a]pyrene. Res Rep Health Eff Inst (2001) 101:5–25; discussion 27–32.
82. Bosetti C, Boffetta P, La Vecchia C. Occupational exposures to polycyclic aromatic hydrocarbons, and respiratory and urinary tract cancers: a quantitative review to 2005. Ann Oncol (2007) 18(3):431–46. doi:10.1093/annonc/mdl172
83. Seidler A, Heiskel H, Bickeboller R, Elsner G. Association between diesel exposure at work and prostate cancer. Scand J Work Environ Health (1998) 24(6):486–94. doi:10.5271/sjweh.373
84. Bourdon JA, Williams A, Kuo B, Moffat I, White PA, Halappanavar S, et al. Gene expression profiling to identify potentially relevant disease outcomes and support human health risk assessment for carbon black nanoparticle exposure. Toxicology (2013) 303:83–93. doi:10.1016/j.tox.2012.10.014
85. Ramanakumar AV, Parent ME, Latreille B, Siemiatycki J. Risk of lung cancer following exposure to carbon black, titanium dioxide and talc: results from two case-control studies in Montreal. Int J Cancer (2008) 122(1):183–9. doi:10.1002/ijc.23021
86. Gerde P, Muggenburg BA, Lundborg M, Dahl AR. The rapid alveolar absorption of diesel soot-adsorbed benzo[a]pyrene: bioavailability, metabolism and dosimetry of an inhaled particle-borne carcinogen. Carcinogenesis (2001) 22(5):741–9. doi:10.1093/carcin/22.5.741
87. Pylev LN. [Morphological changes in the lungs of rats as a result of administration of canal soot with 3,4-benzpyrene adsorbed on it]. Gig Sanit (1969) 34(2):102–4.
88. Mauderly JL, Jones RK, Griffith WC, Henderson RF, McClellan RO. Diesel exhaust is a pulmonary carcinogen in rats exposed chronically by inhalation. Fundam Appl Toxicol (1987) 9(2):208–21. doi:10.1016/0272-0590(87)90044-3
89. Mauderly JL, Snipes MB, Barr EB, Belinsky SA, Bond JA, Brooks AL, et al. Pulmonary toxicity of inhaled diesel exhaust and carbon black in chronically exposed rats. Part I: neoplastic and nonneoplastic lung lesions. Res Rep Health Eff Inst (1994) 68(Pt 1):1–75; discussion 77–97.
90. Roller M. In vitro genotoxicity data of nanomaterials compared to carcinogenic potency of inorganic substances after inhalational exposure. Mutat Res (2011) 727(3):72–85. doi:10.1016/j.mrrev.2011.03.002
91. Baccarelli A, Wright RO, Bollati V, Tarantini L, Litonjua AA, Suh HH, et al. Rapid DNA methylation changes after exposure to traffic particles. Am J Respir Crit Care Med (2009) 179(7):572–8. doi:10.1164/rccm.200807-1097OC
92. Mroz RM, Schins RP, Li H, Jimenez LA, Drost EM, Holownia A, et al. Nanoparticle-driven DNA damage mimics irradiation-related carcinogenesis pathways. Eur Respir J (2008) 31(2):241–51. doi:10.1183/09031936.00006707
93. Baccarelli A, Bollati V. Epigenetics and environmental chemicals. Curr Opin Pediatr (2009) 21(2):243–51. doi:10.1097/MOP.0b013e32832925cc
94. Baeza-Squiban A, Bonvallot V, Boland S, Marano F. Diesel exhaust particles increase NF-kappaB DNA binding activity and c-FOS proto-oncogene expression in human bronchial epithelial cells. Toxicol In Vitro (1999) 13(4–5):817–22. doi:10.1016/S0887-2333(99)00036-3
95. Haque M, Francis J, Sehgal I. Aryl hydrocarbon exposure induces expression of MMP-9 in human prostate cancer cell lines. Cancer Lett (2005) 225(1):159–66. doi:10.1016/j.canlet.2004.11.043
96. Arenz A, Hellweg CE, Stojicic N, Baumstark-Khan C, Grotheer HH. Gene expression modulation in A549 human lung cells in response to combustion-generated nano-sized particles. Ann N Y Acad Sci (2006) 1091:170–83. doi:10.1196/annals.1378.064
97. Jacobsen NR, Pojana G, White P, Moller P, Cohn CA, Korsholm KS, et al. Genotoxicity, cytotoxicity, and reactive oxygen species induced by single-walled carbon nanotubes and C(60) fullerenes in the FE1-mutatrade markMouse lung epithelial cells. Environ Mol Mutagen (2008) 49(6):476–87. doi:10.1002/em.20406
98. Kelsey KT, Xia F, Bodell WJ, Spengler JD, Christiani DC, Dockery DW, et al. Genotoxicity to human cells induced by air particulates isolated during the Kuwait oil fires. Environ Res (1994) 64(1):18–25. doi:10.1006/enrs.1994.1003
99. Gminski R, Decker K, Heinz C, Seidel A, Konczol M, Goldenberg E, et al. Genotoxic effects of three selected black toner powders and their dimethyl sulfoxide extracts in cultured human epithelial A549 lung cells in vitro. Environ Mol Mutagen (2011) 52(4):296–309. doi:10.1002/em.20621
100. Bond JA, Johnson NF, Snipes MB, Mauderly JL. DNA adduct formation in rat alveolar type II cells: cells potentially at risk for inhaled diesel exhaust. Environ Mol Mutagen (1990) 16(2):64–9. doi:10.1002/em.2850160203
101. Wolff RK, Bond JA, Sun JD, Henderson RF, Harkema JR, Griffith WC, et al. Effects of adsorption of benzo[a]pyrene onto carbon black particles on levels of DNA adducts in lungs of rats exposed by inhalation. Toxicol Appl Pharmacol (1989) 97(2):289–99. doi:10.1016/0041-008X(89)90334-7
102. Gallagher J, Heinrich U, George M, Hendee L, Phillips DH, Lewtas J. Formation of DNA adducts in rat lung following chronic inhalation of diesel emissions, carbon black and titanium dioxide particles. Carcinogenesis (1994) 15(7):1291–9. doi:10.1093/carcin/15.7.1291
103. Gallagher J, Sams R II, Inmon J, Gelein R, Elder A, Oberdorster G, et al. Formation of 8-oxo-7,8-dihydro-2’-deoxyguanosine in rat lung DNA following subchronic inhalation of carbon black. Toxicol Appl Pharmacol (2003) 190(3):224–31. doi:10.1016/S0041-008X(03)00187-X
104. Iwai K, Adachi S, Takahashi M, Moller L, Udagawa T, Mizuno S, et al. Early oxidative DNA damages and late development of lung cancer in diesel exhaust-exposed rats. Environ Res (2000) 84(3):255–64. doi:10.1006/enrs.2000.4072
106. Danielsen PH, Loft S, Jacobsen NR, Jensen KA, Autrup H, Ravanat JL, et al. Oxidative stress, inflammation, and DNA damage in rats after intratracheal instillation or oral exposure to ambient air and wood smoke particulate matter. Toxicol Sci (2010) 118(2):574–85. doi:10.1093/toxsci/kfq290
107. Shen Z, Wu W, Hazen SL. Activated leukocytes oxidatively damage DNA, RNA, and the nucleotide pool through halide-dependent formation of hydroxyl radical. Biochemistry (2000) 39(18):5474–82. doi:10.1021/bi992809y
108. Chuang HC, Cheng YL, Lei YC, Chang HH, Cheng TJ. Protective effects of pulmonary epithelial lining fluid on oxidative stress and DNA single-strand breaks caused by ultrafine carbon black, ferrous sulphate and organic extract of diesel exhaust particles. Toxicol Appl Pharmacol (2013) 266(3):329–34. doi:10.1016/j.taap.2012.12.004
109. Ball JC, Straccia AM, Young WC, Aust AE. The formation of reactive oxygen species catalyzed by neutral, aqueous extracts of NIST ambient particulate matter and diesel engine particles. J Air Waste Manag Assoc (2000) 50(11):1897–903. doi:10.1080/10473289.2000.10464231
110. Sato H, Onose J, Toyoda H, Toida T, Imanari T, Sagai M, et al. Quantitative changes in glycosaminoglycans in the lungs of rats exposed to diesel exhaust. Toxicology (2001) 166(3):119–28. doi:10.1016/S0300-483X(01)00453-X
111. Moller P, Folkmann JK, Danielsen PH, Jantzen K, Loft S. Oxidative stress generated damage to DNA by gastrointestinal exposure to insoluble particles. Curr Mol Med (2012) 12(6):732–45. doi:10.2174/156652412800792624
112. Wu J, Ramesh A, Nayyar T, Hood DB. Assessment of metabolites and AhR and CYP1A1 mRNA expression subsequent to prenatal exposure to inhaled benzo(a)pyrene. Int J Dev Neurosci (2003) 21(6):333–46. doi:10.1016/S0736-5748(03)00073-X
113. Rouse RL, Murphy G, Boudreaux MJ, Paulsen DB, Penn AL. Soot nanoparticles promote biotransformation, oxidative stress, and inflammation in murine lungs. Am J Respir Cell Mol Biol (2008) 39(2):198–207. doi:10.1165/rcmb.2008-0057OC
114. Reynolds LJ, Richards RJ. Can toxicogenomics provide information on the bioreactivity of diesel exhaust particles? Toxicology (2001) 165(2–3):145–52. doi:10.1016/S0300-483X(01)00417-6
115. Villalobos SA, Anderson MJ, Denison MS, Hinton DE, Tullis K, Kennedy IM, et al. Dioxinlike properties of a trichloroethylene combustion-generated aerosol. Environ Health Perspect (1996) 104(7):734–43. doi:10.1289/ehp.96104734
116. Hidaka T, Ogawa E, Kobayashi EH, Suzuki T, Funayama R, Nagashima T, et al. The aryl hydrocarbon receptor AhR links atopic dermatitis and air pollution via induction of the neurotrophic factor artemin. Nat Immunol (2017) 18(1):64–73. doi:10.1038/ni.3614
117. Jaguin M, Fardel O, Lecureur V. AhR-dependent secretion of PDGF-BB by human classically activated macrophages exposed to DEP extracts stimulates lung fibroblast proliferation. Toxicol Appl Pharmacol (2015) 285(3):170–8. doi:10.1016/j.taap.2015.04.007
118. Lawal AO. Air particulate matter induced oxidative stress and inflammation in cardiovascular disease and atherosclerosis: the role of Nrf2 and AhR-mediated pathways. Toxicol Lett (2017) 270:88–95. doi:10.1016/j.toxlet.2017.01.017
119. Andrysik Z, Vondracek J, Marvanova S, Ciganek M, Neca J, Pencikova K, et al. Activation of the aryl hydrocarbon receptor is the major toxic mode of action of an organic extract of a reference urban dust particulate matter mixture: the role of polycyclic aromatic hydrocarbons. Mutat Res (2011) 714(1–2):53–62. doi:10.1016/j.mrfmmm.2011.06.011
120. Hou W, Xu X, Lei Y, Cao J, Zhang Y, Chen L, et al. The role of the PM2.5-associated metals in pathogenesis of child Mycoplasma pneumoniae infections: a systematic review. Environ Sci Pollut Res Int (2016) 23(11):10604–14. doi:10.1007/s11356-016-6535-2
121. Agarwal T, Bucheli TD. Is black carbon a better predictor of polycyclic aromatic hydrocarbon distribution in soils than total organic carbon? Environ Pollut (2011) 159(1):64–70. doi:10.1016/j.envpol.2010.09.016
122. Cantor KP, Stewart PA, Brinton LA, Dosemeci M. Occupational exposures and female breast cancer mortality in the United States. J Occup Environ Med (1995) 37(3):336–48. doi:10.1097/00043764-199503000-00011
123. Schedle A, Samorapoompichit P, Rausch-Fan XH, Franz A, Fureder W, Sperr WR, et al. Response of L-929 fibroblasts, human gingival fibroblasts, and human tissue mast cells to various metal cations. J Dent Res (1995) 74(8):1513–20. doi:10.1177/00220345950740081301
124. Prahalad AK, Soukup JM, Inmon J, Willis R, Ghio AJ, Becker S, et al. Ambient air particles: effects on cellular oxidant radical generation in relation to particulate elemental chemistry. Toxicol Appl Pharmacol (1999) 158(2):81–91. doi:10.1006/taap.1999.8701
125. Kim SH, Fletcher RA, Zachariah MR. Understanding the difference in oxidative properties between flame and diesel soot nanoparticles: the role of metals. Environ Sci Technol (2005) 39(11):4021–6. doi:10.1021/es048828z
126. Brown DM, Stone V, Findlay P, MacNee W, Donaldson K. Increased inflammation and intracellular calcium caused by ultrafine carbon black is independent of transition metals or other soluble components. Occup Environ Med (2000) 57(10):685–91. doi:10.1136/oem.57.10.685
127. Wilson MR, Lightbody JH, Donaldson K, Sales J, Stone V. Interactions between ultrafine particles and transition metals in vivo and in vitro. Toxicol Appl Pharmacol (2002) 184(3):172–9. doi:10.1006/taap.2002.9501
128. Correa AX, Cotelle S, Millet M, Somensi CA, Wagner TM, Radetski CM. Genotoxicity assessment of particulate matter emitted from heavy-duty diesel-powered vehicles using the in vivo Vicia faba L. micronucleus test. Ecotoxicol Environ Saf (2016) 127:199–204. doi:10.1016/j.ecoenv.2016.01.026
129. Croes K, Debaillie P, Van den Bril B, Staelens J, Vandermarken T, Van Langenhove K, et al. Assessment of estrogenic activity in PM10 air samples with the ERE-CALUX bioassay: method optimization and implementation at an urban location in Flanders (Belgium). Chemosphere (2016) 144:392–8. doi:10.1016/j.chemosphere.2015.09.020
130. Dilger M, Orasche J, Zimmermann R, Paur HR, Diabate S, Weiss C. Toxicity of wood smoke particles in human A549 lung epithelial cells: the role of PAHs, soot and zinc. Arch Toxicol (2016) 90(12):3029–44. doi:10.1007/s00204-016-1659-1
131. Borm PJ, Driscoll K. Particles, inflammation and respiratory tract carcinogenesis. Toxicol Lett (1996) 88(1–3):109–13. doi:10.1016/0378-4274(96)03725-3
132. Hussain S, Thomassen LC, Ferecatu I, Borot MC, Andreau K, Martens JA, et al. Carbon black and titanium dioxide nanoparticles elicit distinct apoptotic pathways in bronchial epithelial cells. Part Fibre Toxicol (2010) 7:10. doi:10.1186/1743-8977-7-10
133. Brauer M, Hoek G, Van Vliet P, Meliefste K, Fischer PH, Wijga A, et al. Air pollution from traffic and the development of respiratory infections and asthmatic and allergic symptoms in children. Am J Respir Crit Care Med (2002) 166(8):1092–8. doi:10.1164/rccm.200108-007OC
134. Ozier A, Bara I, Girodet PO, Marthan R, Berger P. [Pathophysiology of asthma]. Rev Prat (2011) 61(3):339–45.
136. Margelidon-Cozzolino V, Chbini K, Freymond N, Devouassoux G, Belaaouaj A, Pacheco Y. [COPD: an early disease]. Rev Pneumol Clin (2016) 72(1):49–60. doi:10.1016/j.pneumo.2015.08.002
137. Schmeck B, Jerrentrup L, Bals R. [COPD update 2015: cell biology goes clinic? Important research findings for clinicians]. Pneumologie (2015) 69(12):704–10. doi:10.1055/s-0035-1563785
138. Clark NA, Demers PA, Karr CJ, Koehoorn M, Lencar C, Tamburic L, et al. Effect of early life exposure to air pollution on development of childhood asthma. Environ Health Perspect (2010) 118(2):284–90. doi:10.1289/ehp.0900916
139. Evans KA, Halterman JS, Hopke PK, Fagnano M, Rich DQ. Increased ultrafine particles and carbon monoxide concentrations are associated with asthma exacerbation among urban children. Environ Res (2014) 129:11–9. doi:10.1016/j.envres.2013.12.001
140. LaGier AJ, Manzo ND, Dye JA. Diesel exhaust particles induce aberrant alveolar epithelial directed cell movement by disruption of polarity mechanisms. J Toxicol Environ Health A (2013) 76(2):71–85. doi:10.1080/15287394.2013.738169
141. Dick CA, Brown DM, Donaldson K, Stone V. The role of free radicals in the toxic and inflammatory effects of four different ultrafine particle types. Inhal Toxicol (2003) 15(1):39–52. doi:10.1080/08958370304454
142. Chan JK, Charrier JG, Kodani SD, Vogel CF, Kado SY, Anderson DS, et al. Combustion-derived flame generated ultrafine soot generates reactive oxygen species and activates Nrf2 antioxidants differently in neonatal and adult rat lungs. Part Fibre Toxicol (2013) 10:34. doi:10.1186/1743-8977-10-34
143. Chan JK, Fanucchi MV, Anderson DS, Abid AD, Wallis CD, Dickinson DA, et al. Susceptibility to inhaled flame-generated ultrafine soot in neonatal and adult rat lungs. Toxicol Sci (2011) 124(2):472–86. doi:10.1093/toxsci/kfr233
144. Chuang HC, Jones TP, Lung SC, BeruBe KA. Soot-driven reactive oxygen species formation from incense burning. Sci Total Environ (2011) 409(22):4781–7. doi:10.1016/j.scitotenv.2011.07.041
145. Aam BB, Fonnum F. Carbon black particles increase reactive oxygen species formation in rat alveolar macrophages in vitro. Arch Toxicol (2007) 81(6):441–6. doi:10.1007/s00204-006-0164-3
146. Stone V, Tuinman M, Vamvakopoulos JE, Shaw J, Brown D, Petterson S, et al. Increased calcium influx in a monocytic cell line on exposure to ultrafine carbon black. Eur Respir J (2000) 15(2):297–303. doi:10.1034/j.1399-3003.2000.15b13.x
147. Choi J, Krushel LA, Crossin KL. NF-kappaB activation by N-CAM and cytokines in astrocytes is regulated by multiple protein kinases and redox modulation. Glia (2001) 33(1):45–56. doi:10.1002/1098-1136(20010101)33:1<45::AID-GLIA1005>3.0.CO;2-A
148. Lu T, Chai Q, Yu L, d’Uscio LV, Katusic ZS, He T, et al. Reactive oxygen species signaling facilitates FOXO-3a/FBXO-dependent vascular BK channel beta1 subunit degradation in diabetic mice. Diabetes (2012) 61(7):1860–8. doi:10.2337/db11-1658
149. Zhen X, Ng WC, Fendy, Tong YW, Dai Y, Neoh KG, et al. Toxicity assessment of carbon black waste: a by-product from oil refineries. J Hazard Mater (2017) 321:600–10. doi:10.1016/j.jhazmat.2016.09.043
150. Baulig A, Garlatti M, Bonvallot V, Marchand A, Barouki R, Marano F, et al. Involvement of reactive oxygen species in the metabolic pathways triggered by diesel exhaust particles in human airway epithelial cells. Am J Physiol Lung Cell Mol Physiol (2003) 285(3):L671–9. doi:10.1152/ajplung.00419.2002
151. Chang CC, Chen SH, Ho SH, Yang CY, Wang HD, Tsai ML. Proteomic analysis of proteins from bronchoalveolar lavage fluid reveals the action mechanism of ultrafine carbon black-induced lung injury in mice. Proteomics (2007) 7(23):4388–97. doi:10.1002/pmic.200700164
152. Hiraku Y, Nishikawa Y, Ma N, Afroz T, Mizobuchi K, Ishiyama R, et al. Nitrative DNA damage induced by carbon-black nanoparticles in macrophages and lung epithelial cells. Mutat Res (2017) 818:7–16. doi:10.1016/j.mrgentox.2017.04.002
153. Saber AT, Jensen KA, Jacobsen NR, Birkedal R, Mikkelsen L, Moller P, et al. Inflammatory and genotoxic effects of nanoparticles designed for inclusion in paints and lacquers. Nanotoxicology (2012) 6(5):453–71. doi:10.3109/17435390.2011.587900
154. Chan JK, Kodani SD, Charrier JG, Morin D, Edwards PC, Anderson DS, et al. Age-specific effects on rat lung glutathione and antioxidant enzymes after inhaling ultrafine soot. Am J Respir Cell Mol Biol (2013) 48(1):114–24. doi:10.1165/rcmb.2012-0108OC
155. Sarnat SE, Raysoni AU, Li WW, Holguin F, Johnson BA, Flores Luevano S, et al. Air pollution and acute respiratory response in a panel of asthmatic children along the U.S.-Mexico border. Environ Health Perspect (2012) 120(3):437–44. doi:10.1289/ehp.1003169
156. Morimoto Y, Oyabu T, Horie M, Kambara T, Izumi H, Kuroda E, et al. Pulmonary toxicity of printer toner following inhalation and intratracheal instillation. Inhal Toxicol (2013) 25(12):679–90. doi:10.3109/08958378.2013.835010
158. Boland S, Baeza-Squiban A, Fournier T, Houcine O, Gendron MC, Chevrier M, et al. Diesel exhaust particles are taken up by human airway epithelial cells in vitro and alter cytokine production. Am J Physiol (1999) 276(4 Pt 1):L604–13.
159. Canesi L, Ciacci C, Betti M, Fabbri R, Canonico B, Fantinati A, et al. Immunotoxicity of carbon black nanoparticles to blue mussel hemocytes. Environ Int (2008) 34(8):1114–9. doi:10.1016/j.envint.2008.04.002
160. Bezemer GF, Bauer SM, Oberdorster G, Breysse PN, Pieters RH, Georas SN, et al. Activation of pulmonary dendritic cells and Th2-type inflammatory responses on instillation of engineered, environmental diesel emission source or ambient air pollutant particles in vivo. J Innate Immun (2011) 3(2):150–66. doi:10.1159/000321725
161. Takano H, Ichinose T, Miyabara Y, Shibuya T, Lim HB, Yoshikawa T, et al. Inhalation of diesel exhaust enhances allergen-related eosinophil recruitment and airway hyperresponsiveness in mice. Toxicol Appl Pharmacol (1998) 150(2):328–37. doi:10.1006/taap.1998.8437
162. Siegel PD, Saxena RK, Saxena QB, Ma JK, Ma JY, Yin XJ, et al. Effect of diesel exhaust particulate (DEP) on immune responses: contributions of particulate versus organic soluble components. J Toxicol Environ Health A (2004) 67(3):221–31. doi:10.1080/15287390490266891
163. Castranova V, Ma JY, Yang HM, Antonini JM, Butterworth L, Barger MW, et al. Effect of exposure to diesel exhaust particles on the susceptibility of the lung to infection. Environ Health Perspect (2001) 109(Suppl 4):609–12. doi:10.1289/ehp.01109s4609
164. Eder C, Frankenberger M, Stanzel F, Seidel A, Schramm KW, Ziegler-Heitbrock L, et al. Ultrafine carbon particles down-regulate CYP1B1 expression in human monocytes. Part Fibre Toxicol (2009) 6:27. doi:10.1186/1743-8977-6-27
165. Nishanth RP, Jyotsna RG, Schlager JJ, Hussain SM, Reddanna P. Inflammatory responses of RAW 264.7 macrophages upon exposure to nanoparticles: role of ROS-NFkappaB signaling pathway. Nanotoxicology (2011) 5(4):502–16. doi:10.3109/17435390.2010.541604
166. Schmidt EP, Yang Y, Janssen WJ, Gandjeva A, Perez MJ, Barthel L, et al. The pulmonary endothelial glycocalyx regulates neutrophil adhesion and lung injury during experimental sepsis. Nat Med (2012) 18(8):1217–23. doi:10.1038/nm.2843
167. Wilson MR, Foucaud L, Barlow PG, Hutchison GR, Sales J, Simpson RJ, et al. Nanoparticle interactions with zinc and iron: implications for toxicology and inflammation. Toxicol Appl Pharmacol (2007) 225(1):80–9. doi:10.1016/j.taap.2007.07.012
168. Han YY, Forno E, Holguin F, Celedon JC. Diet and asthma: an update. Curr Opin Allergy Clin Immunol (2015) 15(4):369–74. doi:10.1097/ACI.0000000000000179
169. Varraso R. Nutrition and asthma. Curr Allergy Asthma Rep (2012) 12(3):201–10. doi:10.1007/s11882-012-0253-8
170. Kitz R, Rose MA, Schubert R, Beermann C, Kaufmann A, Bohles HJ, et al. Omega-3 polyunsaturated fatty acids and bronchial inflammation in grass pollen allergy after allergen challenge. Respir Med (2010) 104(12):1793–8. doi:10.1016/j.rmed.2010.06.019
171. Covar R, Gleason M, Macomber B, Stewart L, Szefler P, Engelhardt K, et al. Impact of a novel nutritional formula on asthma control and biomarkers of allergic airway inflammation in children. Clin Exp Allergy (2010) 40(8):1163–74. doi:10.1111/j.1365-2222.2010.03523.x
172. Schmidt R, Luboeinski T, Markart P, Ruppert C, Daum C, Grimminger F, et al. Alveolar antioxidant status in patients with acute respiratory distress syndrome. Eur Respir J (2004) 24(6):994–9. doi:10.1183/09031936.04.00120703
173. Rudiger M, von Baehr A, Haupt R, Wauer RR, Rustow B. Preterm infants with high polyunsaturated fatty acid and plasmalogen content in tracheal aspirates develop bronchopulmonary dysplasia less often. Crit Care Med (2000) 28(5):1572–7. doi:10.1097/00003246-200005000-00052
174. Broekhuizen R, Wouters EF, Creutzberg EC, Weling-Scheepers CA, Schols AM. Polyunsaturated fatty acids improve exercise capacity in chronic obstructive pulmonary disease. Thorax (2005) 60(5):376–82. doi:10.1136/thx.2004.030858
175. Moreira A, Moreira P, Delgado L, Fonseca J, Teixeira V, Padrao P, et al. Pilot study of the effects of n-3 polyunsaturated fatty acids on exhaled nitric oxide in patients with stable asthma. J Investig Allergol Clin Immunol (2007) 17(5):309–13.
176. Stanaland BE. Therapeutic measures for prevention of allergic rhinitis/asthma development. Allergy Asthma Proc (2004) 25(1):11–5.
177. Muley P, Shah M, Muley A. Omega-3 fatty acids supplementation in children to prevent asthma: is it worthy? A systematic review and meta-analysis. J Allergy (Cairo) (2015) 2015:312052. doi:10.1155/2015/312052
178. von Klot S, Cyrys J, Hoek G, Kuhnel B, Pitz M, Kuhn U, et al. Estimated personal soot exposure is associated with acute myocardial infarction onset in a case-crossover study. Prog Cardiovasc Dis (2011) 53(5):361–8. doi:10.1016/j.pcad.2011.01.002
179. Bhaskaran K, Hajat S, Haines A, Herrett E, Wilkinson P, Smeeth L. Effects of air pollution on the incidence of myocardial infarction. Heart (2009) 95(21):1746–59. doi:10.1136/hrt.2009.175018
180. Anderson HR, Armstrong B, Hajat S, Harrison R, Monk V, Poloniecki J, et al. Air pollution and activation of implantable cardioverter defibrillators in London. Epidemiology (2010) 21(3):405–13. doi:10.1097/EDE.0b013e3181d61600
181. Crabbe H. Risk of respiratory and cardiovascular hospitalisation with exposure to bushfire particulates: new evidence from Darwin, Australia. Environ Geochem Health (2012) 34(6):697–709. doi:10.1007/s10653-012-9489-4
182. Chuang KJ, Coull BA, Zanobetti A, Suh H, Schwartz J, Stone PH, et al. Particulate air pollution as a risk factor for ST-segment depression in patients with coronary artery disease. Circulation (2008) 118(13):1314–20. doi:10.1161/CIRCULATIONAHA.108.765669
183. Delfino RJ, Staimer N, Tjoa T, Polidori A, Arhami M, Gillen DL, et al. Circulating biomarkers of inflammation, antioxidant activity, and platelet activation are associated with primary combustion aerosols in subjects with coronary artery disease. Environ Health Perspect (2008) 116(7):898–906. doi:10.1289/ehp.11189
184. Jia X, Hao Y, Guo X. Ultrafine carbon black disturbs heart rate variability in mice. Toxicol Lett (2012) 211(3):274–80. doi:10.1016/j.toxlet.2012.04.007
185. Folkmann JK, Vesterdal LK, Sheykhzade M, Loft S, Moller P. Endothelial dysfunction in normal and prediabetic rats with metabolic syndrome exposed by oral gavage to carbon black nanoparticles. Toxicol Sci (2012) 129(1):98–107. doi:10.1093/toxsci/kfs180
186. Gan WQ, Koehoorn M, Davies HW, Demers PA, Tamburic L, Brauer M. Long-term exposure to traffic-related air pollution and the risk of coronary heart disease hospitalization and mortality. Environ Health Perspect (2011) 119(4):501–7. doi:10.1289/ehp.1002511
187. Brito JM, Belotti L, Toledo AC, Antonangelo L, Silva FS, Alvim DS, et al. Acute cardiovascular and inflammatory toxicity induced by inhalation of diesel and biodiesel exhaust particles. Toxicol Sci (2010) 116(1):67–78. doi:10.1093/toxsci/kfq107
188. Khandoga A, Stampfl A, Takenaka S, Schulz H, Radykewicz R, Kreyling W, et al. Ultrafine particles exert prothrombotic but not inflammatory effects on the hepatic microcirculation in healthy mice in vivo. Circulation (2004) 109(10):1320–5. doi:10.1161/01.CIR.0000118524.62298.E8
189. Bind MA, Baccarelli A, Zanobetti A, Tarantini L, Suh H, Vokonas P, et al. Air pollution and markers of coagulation, inflammation, and endothelial function: associations and epigene-environment interactions in an elderly cohort. Epidemiology (2012) 23(2):332–40. doi:10.1097/EDE.0b013e31824523f0
190. Alexeeff SE, Coull BA, Gryparis A, Suh H, Sparrow D, Vokonas PS, et al. Medium-term exposure to traffic-related air pollution and markers of inflammation and endothelial function. Environ Health Perspect (2011) 119(4):481–6. doi:10.1289/ehp.1002560
191. Frikke-Schmidt H, Roursgaard M, Lykkesfeldt J, Loft S, Nojgaard JK, Moller P. Effect of vitamin C and iron chelation on diesel exhaust particle and carbon black induced oxidative damage and cell adhesion molecule expression in human endothelial cells. Toxicol Lett (2011) 203(3):181–9. doi:10.1016/j.toxlet.2011.03.011
192. Eshel D, Ben-Arie R, Dinoor A, Prusky D. Resistance of gibberellin-treated persimmon fruit to Alternaria alternata arises from the reduced ability of the fungus to produce endo-1,4-beta-glucanase. Phytopathology (2000) 90(11):1256–62. doi:10.1094/PHYTO.2000.90.11.1256
193. Baulig A, Sourdeval M, Meyer M, Marano F, Baeza-Squiban A. Biological effects of atmospheric particles on human bronchial epithelial cells. Comparison with diesel exhaust particles. Toxicol In Vitro (2003) 17(5–6):567–73. doi:10.1016/S0887-2333(03)00115-2
194. Pedersen PA, Rung Weeke E. Seasonal variation of asthma and allergic rhinitis. Consultation pattern in general practice related to pollen and spore counts and to five indicators of air pollution. Allergy (1984) 39(3):165–70. doi:10.1111/j.1398-9995.1984.tb02620.x
195. Leuschner RM, Boehm G, Virchow C. [Pollen and inanimate dust particles as cause of obstructive airway diseases]. Z Erkr Atmungsorgane (1983) 161(2):141–52.
196. Patel MM, Chillrud SN, Deepti KC, Ross JM, Kinney PL. Traffic-related air pollutants and exhaled markers of airway inflammation and oxidative stress in New York City adolescents. Environ Res (2013) 121:71–8. doi:10.1016/j.envres.2012.10.012
197. Kumar RK, Shadie AM, Bucknall MP, Rutlidge H, Garthwaite L, Herbert C, et al. Differential injurious effects of ambient and traffic-derived particulate matter on airway epithelial cells. Respirology (2015) 20(1):73–9. doi:10.1111/resp.12381
198. Shiraiwa M, Selzle K, Poschl U. Hazardous components and health effects of atmospheric aerosol particles: reactive oxygen species, soot, polycyclic aromatic compounds and allergenic proteins. Free Radic Res (2012) 46(8):927–39. doi:10.3109/10715762.2012.663084
199. Hsu SI, Ito K, Kendall M, Lippmann M. Factors affecting personal exposure to thoracic and fine particles and their components. J Expo Sci Environ Epidemiol (2012) 22(5):439–47. doi:10.1038/jes.2012.23
200. Drumm K, Messner C, Kienast K. Reactive oxygen intermediate-release of fibre-exposed monocytes increases inflammatory cytokine-mRNA level, protein tyrosine kinase and NF-kappaB activity in co-cultured bronchial epithelial cells (BEAS-2B). Eur J Med Res (1999) 4(7):257–63.
201. Stone V, Johnston H, Clift MJ. Air pollution, ultrafine and nanoparticle toxicology: cellular and molecular interactions. IEEE Trans Nanobioscience (2007) 6(4):331–40. doi:10.1109/TNB.2007.909005
202. Stone V, Brown DM, Watt N, Wilson M, Donaldson K, Ritchie H, et al. Ultrafine particle-mediated activation of macrophages: intracellular calcium signaling and oxidative stress. Inhal Toxicol (2000) 12(Suppl 3):345–51. doi:10.1080/08958378.2000.11463244
203. Grant WB. Air pollution in relation to U.S. cancer mortality rates: an ecological study; likely role of carbonaceous aerosols and polycyclic aromatic hydrocarbons. Anticancer Res (2009) 29(9):3537–45.
204. Hodgson JT, Jones RD. A mortality study of carbon black workers employed at five United Kingdom factories between 1947 and 1980. Arch Environ Health (1985) 40(5):261–8. doi:10.1080/00039896.1985.10545929
205. Risom L, Moller P, Loft S. Oxidative stress-induced DNA damage by particulate air pollution. Mutat Res (2005) 592(1–2):119–37. doi:10.1016/j.mrfmmm.2005.06.012
206. Reisetter AC, Stebounova LV, Baltrusaitis J, Powers L, Gupta A, Grassian VH, et al. Induction of inflammasome-dependent pyroptosis by carbon black nanoparticles. J Biol Chem (2011) 286(24):21844–52. doi:10.1074/jbc.M111.238519
207. Delzell JE Jr. Common lung conditions: environmental pollutants and lung disease. FP Essent (2013) 409:32–42.
208. Myers TR, Tomasio L. Asthma: 2015 and beyond. Respir Care (2011) 56(9):1389–407; discussion 1407–10. doi:10.4187/respcare.01334
209. Franco Suglia S, Gryparis A, Schwartz J, Wright RJ. Association between traffic-related black carbon exposure and lung function among urban women. Environ Health Perspect (2008) 116(10):1333–7. doi:10.1289/ehp.11223
210. Jung KH, Hsu SI, Yan B, Moors K, Chillrud SN, Ross J, et al. Childhood exposure to fine particulate matter and black carbon and the development of new wheeze between ages 5 and 7 in an urban prospective cohort. Environ Int (2012) 45:44–50. doi:10.1016/j.envint.2012.03.012
211. Koinis-Mitchell D, Kopel SJ, Boergers J, Ramos K, LeBourgeois M, McQuaid EL, et al. Asthma, allergic rhinitis, and sleep problems in urban children. J Clin Sleep Med (2015) 11(1):101–10. doi:10.5664/jcsm.4450
212. Roth M. Airway and lung remodelling in chronic pulmonary obstructive disease: a role for muscarinic receptor antagonists? Drugs (2015) 75(1):1–8. doi:10.1007/s40265-014-0319-0
213. Kerkhof M, Postma DS, Brunekreef B, Reijmerink NE, Wijga AH, de Jongste JC, et al. Toll-like receptor 2 and 4 genes influence susceptibility to adverse effects of traffic-related air pollution on childhood asthma. Thorax (2010) 65(8):690–7. doi:10.1136/thx.2009.119636
214. Saric-Tanaskovic M, Nikolovski D, Belojevic G. [Air quality and respiratory health of school children in Pancevo]. Srp Arh Celok Lek (2006) 134(Suppl 2):108–12. doi:10.2298/SARH06S2108S
215. Landolina N, Gangwar RS, Levi-Schaffer F. Mast cells’ integrated actions with eosinophils and fibroblasts in allergic inflammation: implications for therapy. Adv Immunol (2015) 125:41–85. doi:10.1016/bs.ai.2014.09.002
216. Wegrzyn AS, Jakiela B, Ruckert B, Jutel M, Akdis M, Sanak M, et al. T-cell regulation during viral and nonviral asthma exacerbations. J Allergy Clin Immunol (2015) 136(1):194–7.e9. doi:10.1016/j.jaci.2014.12.1866
217. Mathias CB. Natural killer cells in the development of asthma. Curr Allergy Asthma Rep (2015) 15(2):500. doi:10.1007/s11882-014-0500-2
218. Oettinger R, Drumm K, Knorst M, Krinyak P, Smolarski R, Kienast K. Production of reactive oxygen intermediates by human macrophages exposed to soot particles and asbestos fibers and increase in NF-kappa B p50/p105 mRNA. Lung (1999) 177(6):343–54. doi:10.1007/PL00007652
219. Saber AT, Lamson JS, Jacobsen NR, Ravn-Haren G, Hougaard KS, Nyendi AN, et al. Particle-induced pulmonary acute phase response correlates with neutrophil influx linking inhaled particles and cardiovascular risk. PLoS One (2013) 8(7):e69020. doi:10.1371/journal.pone.0069020
220. White MJ, Galvis-Carvajal E, Gomer RH. A brief exposure to tryptase or thrombin potentiates fibrocyte differentiation in the presence of serum or serum amyloid p. J Immunol (2015) 194(1):142–50. doi:10.4049/jimmunol.1401777
221. Ben-Zimra M, Bachelet I, Seaf M, Gleich GJ, Levi-Schaffer F. Eosinophil major basic protein activates human cord blood mast cells primed with fibroblast membranes by integrin-beta1. Allergy (2013) 68(10):1259–68. doi:10.1111/all.12232
222. Corren J. Role of interleukin-13 in asthma. Curr Allergy Asthma Rep (2013) 13(5):415–20. doi:10.1007/s11882-013-0373-9
223. Zagai U, Dadfar E, Lundahl J, Venge P, Skold CM. Eosinophil cationic protein stimulates TGF-beta1 release by human lung fibroblasts in vitro. Inflammation (2007) 30(5):153–60. doi:10.1007/s10753-007-9032-4
224. Kazama I, Baba A, Endo Y, Toyama H, Ejima Y, Matsubara M, et al. Mast cell involvement in the progression of peritoneal fibrosis in rats with chronic renal failure. Nephrology (Carlton) (2015) 20(9):609–16. doi:10.1111/nep.12489
225. Pawelczyk T, Sakowicz-Burkiewicz M, Wesserling M, Grden M, Kuczkowski J. Altered response of fibroblasts from human tympanosclerotic membrane to interacting mast cells: implication for tissue remodeling. Int J Biochem Cell Biol (2014) 57:35–44. doi:10.1016/j.biocel.2014.10.002
226. Rydman EM, Ilves M, Koivisto AJ, Kinaret PA, Fortino V, Savinko TS, et al. Inhalation of rod-like carbon nanotubes causes unconventional allergic airway inflammation. Part Fibre Toxicol (2014) 11(1):48. doi:10.1186/s12989-014-0048-2
227. Yamaguchi A, Fujitani T, Ohyama K, Nakae D, Hirose A, Nishimura T, et al. Effects of sustained stimulation with multi-wall carbon nanotubes on immune and inflammatory responses in mice. J Toxicol Sci (2012) 37(1):177–89. doi:10.2131/jts.37.177
228. Foucaud L, Goulaouic S, Bennasroune A, Laval-Gilly P, Brown D, Stone V, et al. Oxidative stress induction by nanoparticles in THP-1 cells with 4-HNE production: stress biomarker or oxidative stress signalling molecule? Toxicol In Vitro (2010) 24(6):1512–20. doi:10.1016/j.tiv.2010.07.012
229. Drumm K, Attia DI, Kannt S, Micke P, Buhl R, Kienast K. Soot-exposed mononuclear cells increase inflammatory cytokine mRNA expression and protein secretion in cocultured bronchial epithelial cells. Respiration (2000) 67(3):291–7. doi:10.1159/000029513
230. Chen S, Weller MA, Barnhart MI. Effects of diesel engine exhaust on pulmonary alveolar macrophages. Scan Electron Microsc (1980) 3:327–38.
231. Kienast K, Bay M, Drumm K, Smolarski R, Ferlinz R. Modulation of TNF-alpha secretions by alveolar macrophages and blood monocytes after soot-particle or asbestos fibre exposure. Eur J Med Res (1996) 1(9):425–8.
232. Mastrofrancesco A, Alfe M, Rosato E, Gargiulo V, Beatrice C, Di Blasio G, et al. Proinflammatory effects of diesel exhaust nanoparticles on scleroderma skin cells. J Immunol Res (2014) 2014:138751. doi:10.1155/2014/138751
233. Bourdon JA, Saber AT, Jacobsen NR, Jensen KA, Madsen AM, Lamson JS, et al. Carbon black nanoparticle instillation induces sustained inflammation and genotoxicity in mouse lung and liver. Part Fibre Toxicol (2012) 9:5. doi:10.1186/1743-8977-9-5
234. Nygaard UC, Hansen JS, Samuelsen M, Alberg T, Marioara CD, Lovik M. Single-walled and multi-walled carbon nanotubes promote allergic immune responses in mice. Toxicol Sci (2009) 109(1):113–23. doi:10.1093/toxsci/kfp057
235. Yang H, Liu C, Yang D, Zhang H, Xi Z. Comparative study of cytotoxicity, oxidative stress and genotoxicity induced by four typical nanomaterials: the role of particle size, shape and composition. J Appl Toxicol (2009) 29(1):69–78. doi:10.1002/jat.1385
236. Miyabara Y, Ichinose T, Takano H, Lim HB, Sagai M. Effects of diesel exhaust on allergic airway inflammation in mice. J Allergy Clin Immunol (1998) 102(5):805–12. doi:10.1016/S0091-6749(98)70021-1
237. Penn A, Murphy G, Barker S, Henk W, Penn L. Combustion-derived ultrafine particles transport organic toxicants to target respiratory cells. Environ Health Perspect (2005) 113(8):956–63. doi:10.1289/ehp.7661
238. Ichinose T, Takano H, Miyabara Y, Sagai M. Long-term exposure to diesel exhaust enhances antigen-induced eosinophilic inflammation and epithelial damage in the murine airway. Toxicol Sci (1998) 44(1):70–9. doi:10.1006/toxs.1998.2459
239. Niranjan R, Mavi P, Rayapudi M, Dynda S, Mishra A. Pathogenic role of mast cells in experimental eosinophilic esophagitis. Am J Physiol Gastrointest Liver Physiol (2013) 304(12):G1087–94. doi:10.1152/ajpgi.00070.2013
240. Niranjan R, Rayapudi M, Mishra A, Dutt P, Dynda S, Mishra A. Pathogenesis of allergen-induced eosinophilic esophagitis is independent of interleukin (IL)-13. Immunol Cell Biol (2013) 91(6):408–15. doi:10.1038/icb.2013.21
241. Mavi P, Niranjan R, Dutt P, Zaidi A, Shukla JS, Korfhagen T, et al. Allergen-induced resistin-like molecule-alpha promotes esophageal epithelial cell hyperplasia in eosinophilic esophagitis. Am J Physiol Gastrointest Liver Physiol (2014) 307(5):G499–507. doi:10.1152/ajpgi.00141.2014
242. Hori Y, Takeda S, Cho H, Wegmann S, Shoup TM, Takahashi K, et al. A food and drug administration-approved asthma therapeutic agent impacts amyloid beta in the brain in a transgenic model of Alzheimer disease. J Biol Chem (2015) 290(4):1966–78. doi:10.1074/jbc.M114.586602
243. Zou J, Zhang R, Zhu F, Liu J, Madison V, Umland SP. ADAM33 enzyme properties and substrate specificity. Biochemistry (2005) 44(11):4247–56. doi:10.1021/bi0476230
244. Vivori E, Cudmore RE. Management of airway complications of burns in children. Br Med J (1977) 2(6100):1462–4. doi:10.1136/bmj.2.6100.1462
245. Patella V, Incorvaia C, Minciullo PL, Oricchio C, Saitta S, Florio G, et al. Oxidative stress markers in patients with hymenoptera venom allergy. Allergy Asthma Proc (2015) 36(1):9–13. doi:10.2500/aap.2015.36.3814
246. Allan K, Kelly FJ, Devereux G. Antioxidants and allergic disease: a case of too little or too much? Clin Exp Allergy (2010) 40(3):370–80. doi:10.1111/j.1365-2222.2009.03413.x
247. Hantson P, Butera R, Clemessy JL, Michel A, Baud FJ. Early complications and value of initial clinical and paraclinical observations in victims of smoke inhalation without burns. Chest (1997) 111(3):671–5. doi:10.1378/chest.111.3.671
248. Cho JY. Recent advances in mechanisms and treatments of airway remodeling in asthma: a message from the bench side to the clinic. Korean J Intern Med (2011) 26(4):367–83. doi:10.3904/kjim.2011.26.4.367
249. Hoffman S, Nolin J, McMillan D, Wouters E, Janssen-Heininger Y, Reynaert N. Thiol redox chemistry: role of protein cysteine oxidation and altered redox homeostasis in allergic inflammation and asthma. J Cell Biochem (2015) 116(6):884–92. doi:10.1002/jcb.25017
250. Provotorov VM, Budnevsky AV, Filatova YI, Perfil’eva MV. [Antioxidant therapy of bronchial asthma]. Klin Med (Mosk) (2015) 93(8):19–22.
251. Allen S, Britton JR, Leonardi-Bee JA. Association between antioxidant vitamins and asthma outcome measures: systematic review and meta-analysis. Thorax (2009) 64(7):610–9. doi:10.1136/thx.2008.101469
252. Shieh YH, Huang HM, Wang CC, Lee CC, Fan CK, Lee YL. Zerumbone enhances the Th1 response and ameliorates ovalbumin-induced Th2 responses and airway inflammation in mice. Int Immunopharmacol (2015) 24(2):383–91. doi:10.1016/j.intimp.2014.12.027
253. Guihua X, Shuyin L, Jinliang G, Wang S. Naringin protects ovalbumin-induced airway inflammation in a mouse model of asthma. Inflammation (2016) 39(2):891–9. doi:10.1007/s10753-016-0321-7
254. Oliveira TT, Campos KM, Cerqueira-Lima AT, Cana Brasil Carneiro T, da Silva Velozo E, Ribeiro Melo IC, et al. Potential therapeutic effect of Allium cepa L. and quercetin in a murine model of Blomia tropicalis induced asthma. Daru (2015) 23:18. doi:10.1186/s40199-015-0098-5
255. Bukhari SI, Pattnaik B, Rayees S, Kaul S, Dhar MK. Safranal of Crocus sativus L. inhibits inducible nitric oxide synthase and attenuates asthma in a mouse model of asthma. Phytother Res (2015) 29(4):617–27. doi:10.1002/ptr.5315
256. Chen G, Tang J, Ni Z, Chen Q, Li Z, Yang W, et al. Antiasthmatic effects of resveratrol in ovalbumin-induced asthma model mice involved in the upregulation of PTEN. Biol Pharm Bull (2015) 38(4):507–13. doi:10.1248/bpb.b14-00610
257. Chen J, Zhou H, Wang J, Zhang B, Liu F, Huang J, et al. Therapeutic effects of resveratrol in a mouse model of HDM-induced allergic asthma. Int Immunopharmacol (2015) 25(1):43–8. doi:10.1016/j.intimp.2015.01.013
258. Conte E, Fagone E, Fruciano M, Gili E, Iemmolo M, Vancheri C. Anti-inflammatory and antifibrotic effects of resveratrol in the lung. Histol Histopathol (2015) 30(5):523–9. doi:10.14670/HH-30.523
259. Ng DS, Liao W, Tan WS, Chan TK, Loh XY, Wong WS. Anti-malarial drug artesunate protects against cigarette smoke-induced lung injury in mice. Phytomedicine (2014) 21(12):1638–44. doi:10.1016/j.phymed.2014.07.018
260. Shin IS, Park JW, Shin NR, Jeon CM, Kwon OK, Kim JS, et al. Melatonin reduces airway inflammation in ovalbumin-induced asthma. Immunobiology (2014) 219(12):901–8. doi:10.1016/j.imbio.2014.08.004
261. Jaffer OA, Carter AB, Sanders PN, Dibbern ME, Winters CJ, Murthy S, et al. Mitochondrial-targeted antioxidant therapy decreases transforming growth factor-beta-mediated collagen production in a murine asthma model. Am J Respir Cell Mol Biol (2015) 52(1):106–15. doi:10.1165/rcmb.2013-0519OC
262. Kato A, Kyono H, Kuwabara N. [Electron-microscopic observations on rat lungs after long term inhalation of diesel emissions – non-neoplastic lesions]. Nihon Kyobu Shikkan Gakkai Zasshi (1992) 30(2):238–47.
263. Pavord ID, Korn S, Howarth P, Bleecker ER, Buhl R, Keene ON, et al. Mepolizumab for severe eosinophilic asthma (DREAM): a multicentre, double-blind, placebo-controlled trial. Lancet (2012) 380(9842):651–9. doi:10.1016/S0140-6736(12)60988-X
264. Hugle T. Beyond allergy: the role of mast cells in fibrosis. Swiss Med Wkly (2014) 144:w13999. doi:10.4414/smw.2014.13999
265. Grosman N. Influence of CGS 9343B, an inhibitor of calmodulin activity, on histamine release from isolated rat mast cells. Agents Actions (1992) 36(3–4):192–9.
266. Veerappan A, O’Connor NJ, Brazin J, Reid AC, Jung A, McGee D, et al. Mast cells: a pivotal role in pulmonary fibrosis. DNA Cell Biol (2013) 32(4):206–18. doi:10.1089/dna.2013.2005
267. Veraldi S, D’Agostino A, Pontini P, Guanziroli E, Gatt P. Nasal polyps: a predisposing factor for cutaneous leishmaniasis of the lips? Int J Dermatol (2016) 55(4):443–5. doi:10.1111/ijd.13118
268. Ding X, Wang X, Chen H, Zhao H, Chao Y, Yin W, et al. [A comparative study of different bedside lung ultrasound examination for the location and signs of diaphragmatic points]. Zhonghua Nei Ke Za Zhi (2015) 54(9):778–82.
269. Lopes-Ferreira M, Gomes EM, Bruni FM, Ferreira MJ, Charvet P, Lima C. First report of interruption of mast cell degranulation and endothelial cells activation by anti-inflammatory drugs controlling the acute response provoked by Pseudoplatystoma fasciatum fish venom. Toxicon (2014) 90:237–48. doi:10.1016/j.toxicon.2014.08.007
270. Green K, Ejlertsen JS, Madsen A, Buchvald FF, Kongstad T, Kobbernagel H, et al. Abbreviation modalities of nitrogen multiple-breath washout tests in school children with obstructed lung disease. Pediatr Pulmonol (2016) 51(6):624–32. doi:10.1002/ppul.23339
271. de Garavilla L, Greco MN, Sukumar N, Chen ZW, Pineda AO, Mathews FS, et al. A novel, potent dual inhibitor of the leukocyte proteases cathepsin G and chymase: molecular mechanisms and anti-inflammatory activity in vivo. J Biol Chem (2005) 280(18):18001–7. doi:10.1074/jbc.M501302200
272. Degiovanni G. [Cytochalasin B is an inhibitor of the cytotoxic activity of sensitized lymphoid cells]. C R Seances Soc Biol Fil (1972) 166(4):736–40.
273. Kusunoki Y, Nakamura T, Hattori K, Motegi T, Ishii T, Gemma A, et al. Atrial and ventricular arrhythmia-associated factors in stable patients with chronic obstructive pulmonary disease. Respiration (2016) 91(1):34–42. doi:10.1159/000442447
274. Heijink IH, Van Oosterhout AJ. Targeting T cells for asthma. Curr Opin Pharmacol (2005) 5(3):227–31. doi:10.1016/j.coph.2005.04.002
275. Park HJ, Kim DH, Choi JY, Kim WJ, Kim JY, Senejani AG, et al. PPARgamma negatively regulates T cell activation to prevent follicular helper T cells and germinal center formation. PLoS One (2014) 9(6):e99127. doi:10.1371/journal.pone.0099127
276. Park HB, Oh K, Garmaa N, Seo MW, Byoun OJ, Lee HY, et al. CP-690550, a Janus kinase inhibitor, suppresses CD4+ T-cell-mediated acute graft-versus-host disease by inhibiting the interferon-gamma pathway. Transplantation (2010) 90(8):825–35. doi:10.1097/TP.0b013e3181f24e59
277. Marder P, Schmidtke JR. Cyclosporin A inhibits helper/inducer surface antigen expression on activated human lymphocytes. Int J Immunopharmacol (1985) 7(2):165–75. doi:10.1016/0192-0561(85)90023-2
278. Macian F. NFAT proteins: key regulators of T-cell development and function. Nat Rev Immunol (2005) 5(6):472–84. doi:10.1038/nri1632
279. MacLean E, Madsen N, Vliagoftis H, Field C, Cameron L. n-3 fatty acids inhibit transcription of human IL-13: implications for development of T helper type 2 immune responses. Br J Nutr (2013) 109(6):990–1000. doi:10.1017/S0007114512002917
280. Mattes J, Collison A, Plank M, Phipps S, Foster PS. Antagonism of microRNA-126 suppresses the effector function of TH2 cells and the development of allergic airways disease. Proc Natl Acad Sci U S A (2009) 106(44):18704–9. doi:10.1073/pnas.0905063106
281. Marino F, Maresca AM, Castiglioni L, Cosentino M, Maio RC, Schembri L, et al. Simvastatin down-regulates the production of interleukin-8 by neutrophil leukocytes from dyslipidemic patients. BMC Cardiovasc Disord (2014) 14:37. doi:10.1186/1471-2261-14-37
282. Lu H, Xin Y, Tang Y, Shao G. Zinc suppressed the airway inflammation in asthmatic rats: effects of zinc on generation of eotaxin, MCP-1, IL-8, IL-4, and IFN-gamma. Biol Trace Elem Res (2012) 150(1–3):314–21. doi:10.1007/s12011-012-9493-7
283. Chung SH, Choi SH, Choi JA, Chuck RS, Joo CK. Curcumin suppresses ovalbumin-induced allergic conjunctivitis. Mol Vis (2012) 18:1966–72.
Keywords: soot (black carbon), carbon black, inflammation, oxidative stress, polyunsaturated fatty acids, air pollution
Citation: Niranjan R and Thakur AK (2017) The Toxicological Mechanisms of Environmental Soot (Black Carbon) and Carbon Black: Focus on Oxidative Stress and Inflammatory Pathways. Front. Immunol. 8:763. doi: 10.3389/fimmu.2017.00763
Received: 25 April 2017; Accepted: 16 June 2017;
Published: 30 June 2017
Edited by:
Hridayesh Prakash, University of Hyderabad, IndiaReviewed by:
Gabor Csanyi, Georgia Regents Medical Center, United StatesCopyright: © 2017 Niranjan and Thakur. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Rituraj Niranjan, cml0dXJham5pcmFuamFuQHJlZGlmZm1haWwuY29t;
Ashwani Kumar Thakur, YWt0aGFrdXJAaWl0ay5hYy5pbg==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.