 Patrick Peschl
Patrick Peschl Monika Bradl
Monika Bradl Romana Höftberger
Romana Höftberger Thomas Berger
Thomas Berger Markus Reindl
Markus Reindl
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol., 08 May 2017
Sec. Multiple Sclerosis and Neuroimmunology
Volume 8 - 2017 | https://doi.org/10.3389/fimmu.2017.00529
This article is part of the Research TopicInduction of Central Nervous System Disease by the Adaptive Immune ResponseView all 12 articles
Myelin oligodendrocyte glycoprotein (MOG), a member of the immunoglobulin (Ig) superfamily, is a myelin protein solely expressed at the outermost surface of myelin sheaths and oligodendrocyte membranes. This makes MOG a potential target of cellular and humoral immune responses in inflammatory demyelinating diseases. Due to its late postnatal developmental expression, MOG is an important marker for oligodendrocyte maturation. Discovered about 30 years ago, it is one of the best-studied autoantigens for experimental autoimmune models for multiple sclerosis (MS). Human studies, however, have yielded controversial results on the role of MOG, especially MOG antibodies (Abs), as a biomarker in MS. But with improved detection methods using different expression systems to detect Abs in patients’ samples, this is meanwhile no longer the case. Using cell-based assays with recombinant full-length, conformationally intact MOG, several recent studies have revealed that MOG Abs can be found in a subset of predominantly pediatric patients with acute disseminated encephalomyelitis (ADEM), aquaporin-4 (AQP4) seronegative neuromyelitis optica spectrum disorders (NMOSD), monophasic or recurrent isolated optic neuritis (ON), or transverse myelitis, in atypical MS and in N-methyl-d-aspartate receptor-encephalitis with overlapping demyelinating syndromes. Whereas MOG Abs are only transiently observed in monophasic diseases such as ADEM and their decline is associated with a favorable outcome, they are persistent in multiphasic ADEM, NMOSD, recurrent ON, or myelitis. Due to distinct clinical features within these diseases it is controversially disputed to classify MOG Ab-positive cases as a new disease entity. Neuropathologically, the presence of MOG Abs is characterized by MS-typical demyelination and oligodendrocyte pathology associated with Abs and complement. However, it remains unclear whether MOG Abs are a mere inflammatory bystander effect or truly pathogenetic. This article provides deeper insight into recent developments, the clinical relevance of MOG Abs and their role in the immunpathogenesis of inflammatory demyelinating disorders.
Myelin oligodendrocyte glycoprotein is a minor myelin component, with a length of 245 amino acids (AA) and a molecular weight of 26–28 kDa. It is only present in mammals and has a highly conserved nucleotide and AA structure within different species (1). The human MOG gene is located at chromosome 6 within the human leukocyte antigen (HLA) gene locus, whereas the mouse MOG gene is located on chromosome 17 within the major histocompatibility complex (MHC) gene locus (2). MOG is exclusively expressed in the central nervous system (CNS) on the surface of myelin sheaths and oligodendrocyte processes (1–3). MOG expression starts at the onset of myelination and is therefore a potential differentiation marker for oligodendrocyte maturation (4). The function of MOG is not yet fully understood, but its molecular structure and its extracellular immunoglobulin (Ig) domain indicate a possible function as a cell surface receptor or cell adhesion molecule (5). MOG belongs to the Ig superfamily, with a single extracellular immunoglobuline variable (IgV) domain, one transmembrane domain, one cytoplasmic loop, a membrane-associated region, and a cytoplasmic tail (6). Fifteen different alternatively spliced isoforms have been detected in humans. Full-length variants alpha 1 and beta 1 are found in fetal stages, whereas alternative variants are expressed in later postnatal stages (1, 6). It has been shown, that these isoforms are localized on the cell surface, in the endoplasmic reticulum, in the endocytic system, or can be found in a secreted form. The secreted form could have important effects triggering autoimmunity if released into the cerebrospinal fluid (CSF) and then drained into the periphery. The cytoplasmic tail of MOG determines the intracellular localization of the various splice forms and could play a role in intracellular signaling (6). The cross-linking of antibodies (Abs) reactive with the extracellular domain of MOG resulted in the activation of intracellular signaling cascades resulting in survival signals, changes of cytoskeletal stability, and cellular stress responses (7). MOG is highly homologous to butyrophilins which are expressed in mammary glands (8) and might cause autoimmunity by molecular mimicry (9). Furthermore, a sequence homology of MOG AA 35–55 (MOG35–55) to medium-sized neurofilament leads to the activation of MOG35–55 specific T cells (10).
Myelin oligodendrocyte glycoprotein has been implicated to be the cellular receptor for Rubella virus (11), as a ligand for DC-SIGN on antigen-expressing cells (12), and as a receptor for nerve growth factor (13). The interaction of DC-SIGN and MOG along with its correct glycosylation might keep myeloid antigen-presenting cells (APC) in an immature and tolerogenic state and thereby prevent autoimmunity (12). However, the inactivation of mouse MOG by gene targeting resulted in no clinical or histological abnormalities (14, 15).
Whereas the biological function of MOG is still not clear, its topology at the surface of myelin and oligodendrocytes and its special characteristics predict MOG to be a very important target of autoantibodies and cell-mediated immune responses in inflammatory demyelinating diseases. Initially, MOG was discovered as a dominant target of autoantibodies (they so called it M2 antigen) after immunization of guinea pigs with CNS tissue (16, 17). Numerous studies have then established an important role of MOG as autoantigen for T and B cell responses in experimental models and inflammatory demyelinating diseases.
The first indications that humoral factors also contribute to demyelination have been described in 1947 by Kabat et al. who observed a demyelinating effect after immunization of rhesus monkeys with heterologous rabbit or homologous brain tissue (18). In 1968, it was noted that sera from guinea pigs sensitized with whole CNS preparations have a demyelinating effect in vitro (19). The first indication that MOG Abs might be pathogenic followed about 10 years later when it was observed that guinea pigs immunized with the M2 protein developed Abs with demyelinating activity in vitro (20, 21). Then, it was shown that the monoclonal MOG-specific Ab 8–18C5 induces demyelination in Lewis (LEW) rats with experimental autoimmune encephalomyelitis (EAE) (4, 22), that guinea pigs immunized with M2 show demyelinated lesions in their CNS, and that the M2 protein is identical to MOG (16). It soon became clear that MOG Abs may be pathogenic in a large number of additional species (Table 1) (23). Further characterizations of MOG revealed that this protein is found in the oligodendrocyte membrane with a large N-terminal extracellular IgG V-like domain (8) and that N-terminal domain (AA 1–125) is responsible for the formation of demyelinating Abs (23, 24). Studies in marmoset monkeys and mice clarified that pathogenic Abs recognize conformational epitopes on the extracellularly exposed MOG domain (25–27) and that strain specific differences in mounting such anti-conformational Ab responses correlate with exacerbation of diseases (28, 29). Epitopes for encephalitogenic T cells for many different strains of mice and for LEW rats are found on the extracellular domain of MOG (30–33), but also in its transmembrane region (34, 35) (Table 2).
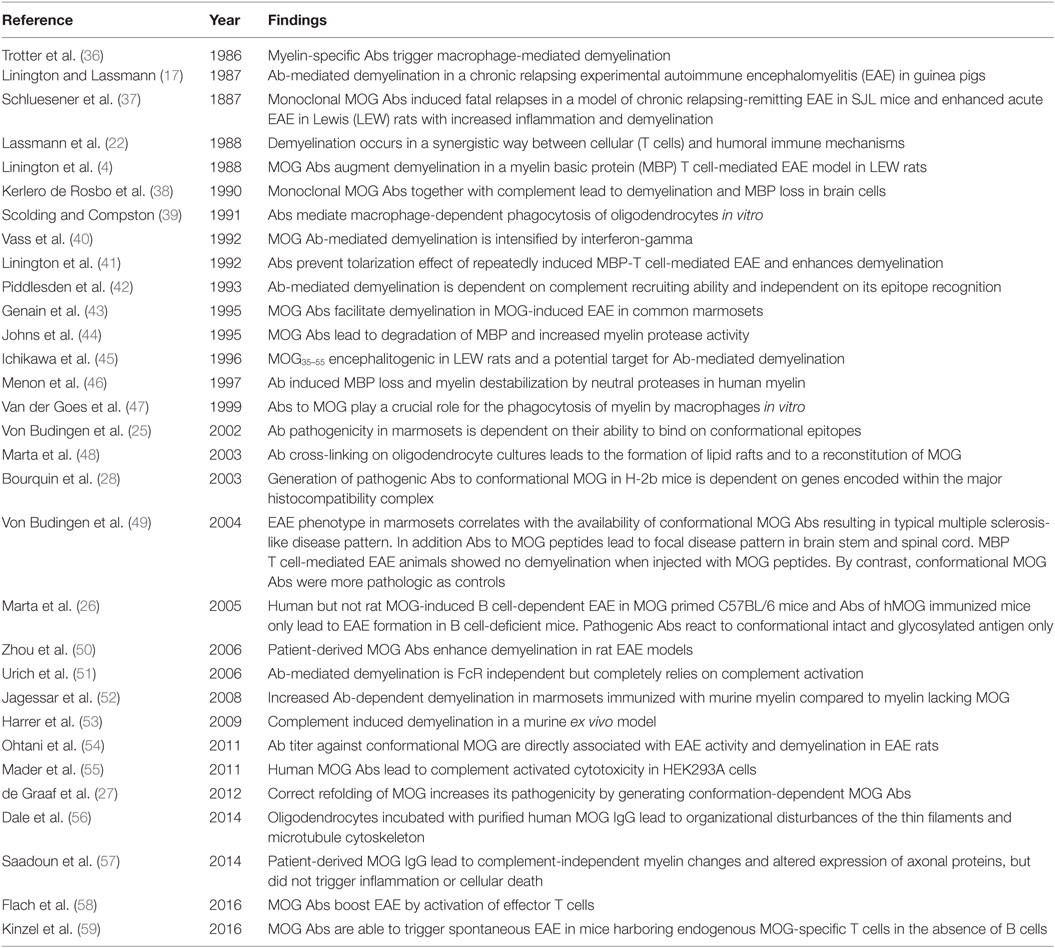
Table 1. The effects of myelin oligodendrocyte glycoprotein (MOG)-specific antibodies (Abs).

Table 2. T cell responses against myelin oligodendrocyte glycoprotein (MOG) in experimental autoimmune encephalomyelitis (EAE) animal models.
Immunizations of LEW rats with MOG activates MOG1–20- and MOG35–55- specific T cells which are only poorly encephalitogenic (24) and induces MOG-specific Abs which cause formation of focal small demyelinating lesions (24). In contrast to LEW rats are brown Norway and dark agouti rat strains highly susceptible to MOG-induced EAE (87). Different MHC haplotypes and non-MHC background genes modify the anti-MOG immune response (70, 75). This important information derived from EAE studies in MHC congenic LEW rats, i.e., in rats with different MHC class II alleles on the genetic background of LEW rats. Upon immunization with MOG, these animals either develop early onset acute lethal disease with extensive demyelinating plaques, chronic and/or relapsing types of disease, or do not show any evidence of clinical and histological disease, depending on the MHC class II haplotype present (62). Moreover, the MOG-induced T cell proliferation and interferon-gamma production, and the degree of MOG-specific B cell responses and Ab titers correlated with the severity of clinical disease (62). For further experiments, rats were selected which carried the most permissive MHC class II haplotype for the induction of MOG-specific autoimmune reactions, but differed in their non-MHC background genes. When these animals were sensitized with MOG, they mounted anti-MOG T cell and B cell responses, but showed differences in the maturation of these responses (62). Cumulatively, these data suggested that the MHC haplotype influences the degree of disease susceptibility, the clinical course, the recruitment of MOG-specific immunocompetent cells, and the CNS pathology, while non-MHC genes strongly influence the maturation of the anti-MOG response (62). A similar effect was also seen in human HLA DR4 transgenic mice which indicated that HLA DR shaped the anti-MOG response in both, humans and mice (88).
Further knowledge about the role of B cells in MOG-induced CNS inflammation derived from transgenic mice (Table 3). Mice were genetically engineered to express the heavy chain from the monoclonal anti-MOG Ab 8–18C5 described above, paired with endogenous Ig light chains (89). These animals had many MOG-reactive B cells in their immune repertoire and had titers of anti-MOG Abs in their circulation. And yet, they remained completely healthy until they were challenged with MOG. Then, they developed EAE with higher incidence, severity, and earlier onset compared to their non-transgenic counterparts (89). Further studies using B cell-deficient mice showed that B cells are required for EAE induction using the MOG protein, but are dispensable when the encephalitogenic MOG peptide is used for EAE induction (90–92). These studies also revealed that B cells are needed for the recovery from EAE, by the production of IL-10 and expression of CD40 (93). The role of B cells in promoting EAE was further confirmed by using transgenic mouse lines in which MHC class II products were knocked-out in B cells, or in which B cells were able to express MOG-specific B cell receptors on their surface, but were unable to secrete MOG-specific Abs (94). This and several other studies (see Table 3) revealed that B cells can act as APC, and that they can sufficiently promote pro-inflammatory T cell activation and spontaneous EAE onset (91, 94, 95). In another study, in which MOG-specific B cells and T cells were actively transferred into an intact immune repertoire of C57BL/6J mice, MOG-specific B cells were shown to aggravate CNS inflammation and EAE disease course. These results were further confirmed by using human MOG positive serum Abs, reproducing the same disease accelerating effects (58). Hence, both B cells and myelin-specific Abs can independently activate T cells and thus increase the risk of an autoimmune mediated inflammation of the CNS (59).

Table 3. The role of B cells in experimental autoimmune encephalomyelitis (EAE) animal models.
Also spontaneous models of MOG-induced CNS disease were highly informative for deciphering the role of anti-MOG responses in autoimmune disease. These models were based on the transgenic expression in mice of MOG-specific T cell receptors, either alone (71, 78) or in combination with MOG-specific B cell receptors (74, 97) and gave striking results:
The overexpression of MOG-specific T cell receptors in transgenic C57/BL6 (71) or SJL (78) mice lead to spontaneous optic neuritis (ON) in more than 30% of all animals and rendered the animals hyper-susceptible to the induction of ON in response to sensitization with suboptimal amounts of MOG (71), or to a severe spontaneous relapsing-remitting EAE with episodes often altering between different CNS compartments in more than 60% of all male, and more than 80% of all females within 160 days after birth (78). In these animals, the transgenic T cells expanded MOG-specific B cells from the endogenous immune repertoire, which produced pathogenic autoantibodies binding to a conformational epitope on native MOG protein (78). Overexpression of MOG-specific T cell receptors in NOD mice led to MOG-specific CD4+ and CD8+ T cell responses at the same time (79). These animals revealed that CD8+ MOG-specific T cells may be weakly encephalitogenic (79) and are able to regulate and attenuate CD4+ driven immune responses by modulating APC functions and reducing CD4+ T cell responses (80, 83).
Mice genetically engineered to express MOG-specific receptors on T and B cells (74, 97) showed a class switch of MOG Abs to an IgG1 subtype, and spontaneously developed inflammatory demyelinating CNS disease (74, 97). Most interestingly, spontaneous development of disease in these animals crucially depended on the presence of commensal microbiota in the gut (98).
Although many seminal observations on MOG-reactive T and B cell responses derive from murine EAE models, it is important to know that in these animals, large lesions with myelin loss are mainly caused by axonal degeneration with secondary demyelination, while primary demyelination is sparse (99, 100). Therefore, it is necessary to also study MOG autoreactivity in the marmoset (Callithrix jacchus), in which MOG-induced EAE resembles human demyelinating diseases more closely (100–102). When these animals are immunized with the recombinant IgV domain of rat MOG, they developed lesions which were very similar to chronic multiple sclerosis (MS) plaques with mononuclear cell infiltrates, primary demyelination, and astrogliosis (103), even at the ultrastructural level (65). Moreover, some animals developed a progressive form of EAE, which was triggered by cytotoxic effector memory T cells and further promoted demyelination in the gray matter (76, 82). As seen before in mice and rats, the marmoset CD4+ T cell response against MOG may cover several different epitopes, only one of which is highly encephalitogenic (104).
Cumulatively, these animal models revealed that
• autoimmune responses to MOG can be induced in many different species
• the susceptibility to MOG is determined by MHC- and non-MHC genes
• anti-MOG responses typically involve CD4+ T cells and complement-fixing Abs of the IgG1 subtype
• the MOG-specific T cell repertoire contains T cells specific for several different T cell epitopes which vary between different species and substrains dependent on the MHC haplotype
• not all MOG-specific T cells are encephalitogenic
• MOG-specific B cells have Ab-dependent and Ab-independent effects on tissue damage
• different types of anti-MOG Abs exist, but only those recognizing conformational epitopes on the extracellular domain of MOG are pathogenic
• MOG-specific autoantibodies in the circulation specific for such conformational epitopes are harmless, unless these Abs gain access to the CNS via an opened blood-brain barrier in an inflammatory environment
• MOG-specific Abs can cross-react with other proteins like butyrophilin
• the extent of demyelination caused by anti-MOG Abs depends on MHC-dependent and MHC-independent factors.
As outlined above, MOG is one of the best-studied autoantigens for experimental autoimmune models for MS. Attempts to translate these findings into the human disease have yielded controversial results, especially with regard to MOG Abs as a prognostic biomarker in MS (105, 106) [reviewed in Berger et al. (107)]. These results were caused by the use of inappropriate methods (e.g., immunoblotting, ELISA) and antigens (recombinant human MOG produced in Escherichia coli, MOG peptides) to determine disease-specific MOG Abs. However, with improved detection methods using correctly folded and glycosylated MOG protein expressed in mammalian cells for radioimmunoassays, flow cytometry, and immunofluorescence, MOG Abs were found in a subset of predominantly pediatric patients with acute disseminated encephalomyelitis (ADEM), aquaporin-4 (AQP4) seronegative neuromyelitis optica spectrum disorders (NMOSD), monophasic or recurrent isolated ON, or transverse myelitis (TM), in atypical MS, brainstem encephalitis, and N-methyl-d-aspartate receptor-encephalitis with overlapping demyelinating syndromes, but rarely in classical MS (50, 55, 56, 108–176). Since low-titer MOG Abs are often found in MS patients and controls, most of these studies have used either a “high-titer” cut-off or an IgG1 secondary Ab to increase specificity. Like many other autoantibodies, e.g., to AQP4, MOG Abs are therefore only present in rare diseases indicating widely established immunological tolerance to most autoantigens.
These findings, however, raise the important question whether MOG Abs are associated with a specific clinical phenotype like AQP4 Abs are associated with NMOSD (177). We have therefore reviewed the literature and compared all studies, which have analyzed the presence of MOG Abs in inflammatory demyelinating disorders (MS, ADEM, and AQP4 Ab seronegative and seropositive NMOSD) in comparison with a control group of patients with other neurological disorders or healthy controls. Results from these studies are shown in Table 4 and Figure 1. We have identified 26 studies which fulfilled these criteria (50, 55, 56, 109–116, 119, 121, 126, 130, 132, 134, 137, 141, 147, 148, 152, 156, 158, 165, 174). Only 13 of these studies included a control group with 50 or more individuals and only 5 studies included more than 100 controls (Table 4). Further, many patients and controls were repeatedly analyzed in some studies and therefore we decided not to include a statistical analysis of the reviewed publications. The specificity of these studies was calculated using the frequency of MOG Abs in other neurological disorders or healthy controls determined by the methods shown in Table 4. The overall specificity of these studies was 98.5% [95% confidence interval (CI) 97.8–99] and thus 1.5% (range 0–6%) of all controls were seropositive for MOG Abs (Table 4; Figure 1). The sensitivity of these studies was calculated using the frequency of MOG Abs in inflammatory demyelinating disorders determined by the methods shown in Table 4. The presence of MOG Abs in MS was analyzed in 23/26 studies and the overall sensitivity for MS was 5.1% (95% CI 4.2–6.1) and thus 5.1% (range 0–46.7%) of all MS patients were seropositive for MOG Abs. The highest frequency of MOG Abs within MS patients was found in pediatric MS patients and in one of the initial studies not using a high-titer cut-off. Therefore, it can be concluded that MOG Abs are rare in MS, particularly in adult MS, but are still found in a few patients in several studies. Since MOG Abs are associated with MS-like neuropathology (136, 149, 167, 172, 178, 179), they might play a role in pathophysiology in these patients and therefore the current practice to use MS as a negative control group for MOG Abs (141) should be regarded with caution. The presence of MOG Abs in ADEM was analyzed in 13/26 studies and the overall sensitivity for ADEM was 36.4% (95% CI 31.4–41.7; range 17.7–47.4%) and thus ADEM was the most frequent clinical presentation associated with MOG Abs. Again, the frequency of MOG Abs was highest in pediatric patients. Since the 26 studies used different clinical criteria for NMOSD, we reviewed the studies for the presence of MOG Abs in AQP4 seronegative patients with ON, TM, or NMOSD. The presence of MOG Abs in these conditions was analyzed in 15/26 studies and the overall sensitivity was 26.9% (95% CI 23.9–30.1; range 9.2–63.5%). Finally, the presence of MOG Abs in AQP4 seropositive NMOSD was analyzed in 13/26 studies and the overall sensitivity was 2% (95% CI 1.2–3.4; range 1.2–3.4%). Thus, the presence of MOG Abs in AQP4 Ab-positive NMOSD is in the range of the control group.
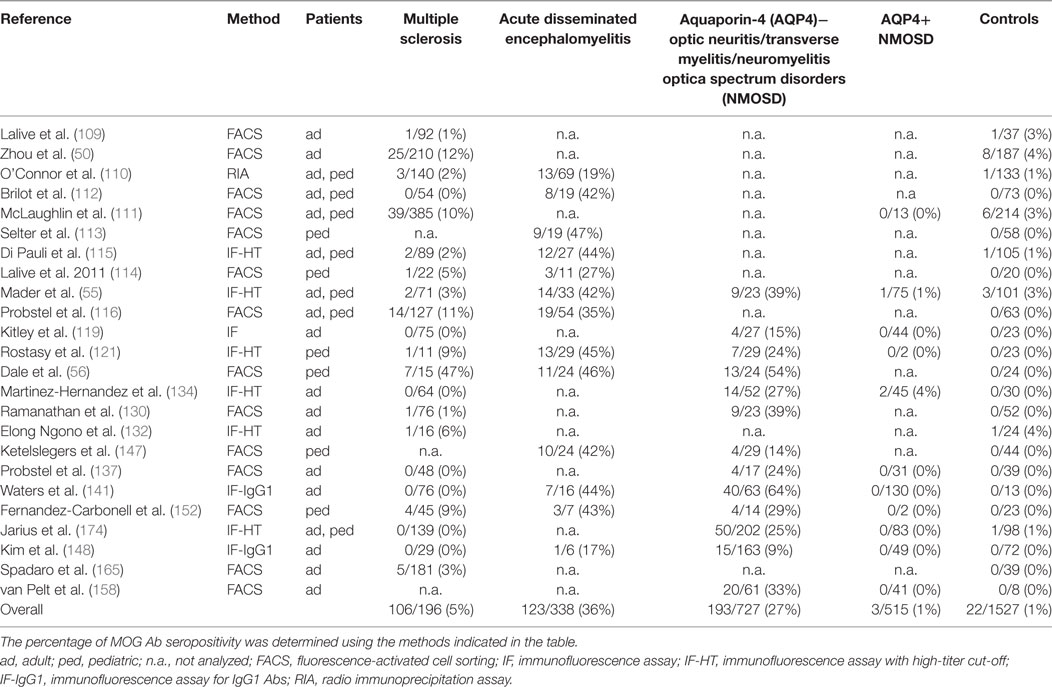
Table 4. Studies reporting the presence of myelin oligodendrocyte glycoprotein (MOG) antibodies (Abs) in patients with inflammatory demyelinating disorders in comparison to a control group of patients with other neurological disorders and/or healthy controls.
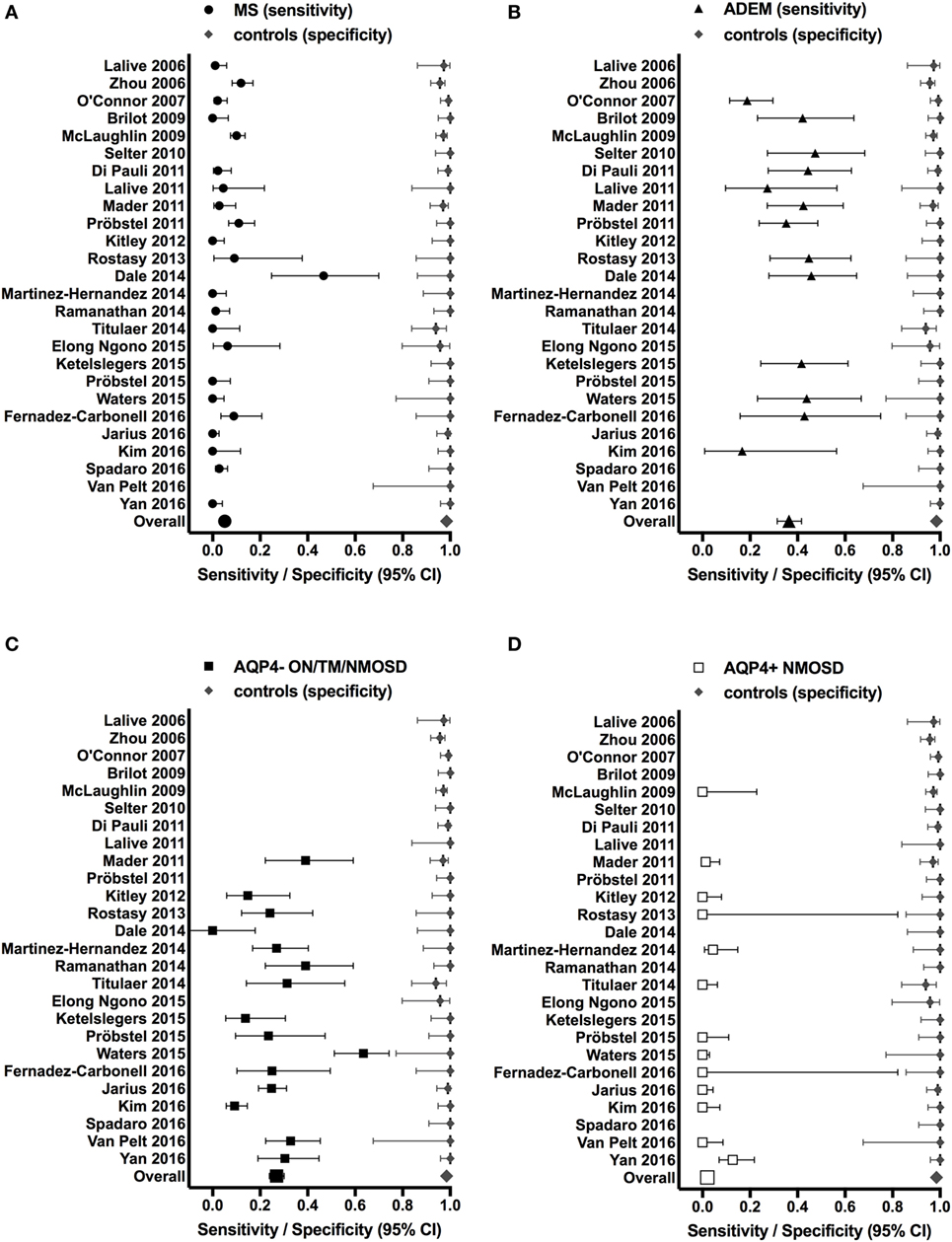
Figure 1. Studies reporting the presence of myelin oligodendrocyte glycoprotein (MOG) antibodies (Abs) in patients with inflammatory demyelinating disorders (A) MS; (B) ADEM; (C) AQP4− ON/TM/NMOSD; (D) AQP4+ NMOSD (sensitivity, left side of each graph) in comparison to a control group of patients with other neurological disorders and/or healthy controls (specificity, right side of each graph). Sensitivities and specificities are indicated by symbols with error bars (95% confidence intervals). Specificities were calculated using the frequency of MOG Abs in other neurological disorders or healthy controls determined by the methods shown in Table 4. Sensitivities were calculated using the frequency of MOG Abs in inflammatory demyelinating disorders determined by the methods shown in Table 4.
In conclusion, these studies revealed that MOG Abs are associated with heterogeneous clinical presentations and a younger age of onset in human inflammatory demyelinating diseases but a clear common clinical phenotype is missing.
The histopathology associated with MOG Abs has been described in few patients including NMOSD, atypical demyelination, CIS, and ADEM (136, 149, 167, 172, 178, 179) (Table 5). All cases showed demyelinating lesions with features of MS pattern II, with well-demarcated confluent plaques with loss of myelin, relative preservation of axons, well-preserved astrocytes, and numerous macrophages containing myelin debris. The inflammatory infiltrates were predominantly composed of perivascular and parenchymal T-cells and some perivascular B-cells. Moreover, the deposition of terminal complement complex C9neo was reported indicating complement-dependent cytotoxicity (136, 167). All lesions were characterized by well-preserved oligodendrocytes that were partly MOG-negative, most likely compatible with preoligodendrocytes. Demyelination associated with MOG Abs differs from AQP4 seropositive NMOSD that characteristically shows loss of astrocytes with deposition of IgG and terminal complement complex C9neo, inflammatory infiltrates including the presence of neutrophilic and eosinophilic granulocytes, and elevated glial fibrillary acidic protein levels in CSF (180).
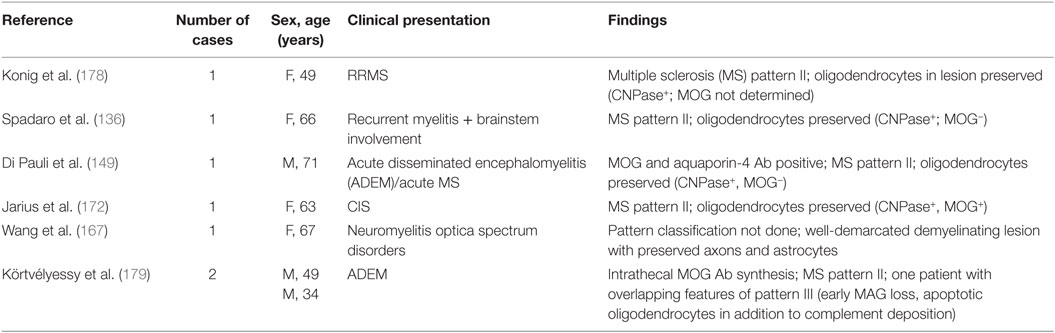
Table 5. Neuropathological findings in patients with myelin oligodendrocyte glycoprotein (MOG) antibody (Ab)-associated demyelination.
These similar immunopathological findings compatible with MS pattern II supports a humoral immune pathogenesis in patients with MOG Abs. Since the histopathological lesion type is independent from the clinical presentation the demyelinating lesions may be included under the term “MOG antibody syndrome.”
As MOG Ab binding has been shown to be dependent on the correct folding and glycosylation pattern of their antigen, studies were directed toward the binding motifs/epitopes of these Abs with the aim to identify specific binding patterns for diseases. Mayer and colleagues (122) performed epitope recognition studies of MOG Abs from several demyelinating diseases and seven distinct binding patterns were found. However, no clinical correlation between the binding patterns and different disease entities could be shown. Furthermore, these Abs were directed against only a single epitope or multiple epitopes and an association between glycosylation and an increased binding capacity could not be detected. The most frequent epitopes were found in the CC′-loop and FG-loop of the extracellular IgV domain of correctly folded human MOG protein. Within the CC′-loop, AA P42 was essential for binding and therefore human MOG Abs did not bind either to rodent MOG, which has a serine at position 42, or to mutated human MOG P42S (122). These findings were confirmed and extended by Sepulveda et al. (166) who demonstrated that only a subset of human MOG Abs is also reactive to rodent MOG epitopes as analyzed by cell-based assays and tissue immunohistochemistry and this reactivity to rodent MOG did not correlate with a specific clinical phenotype. Finally, it has been already demonstrated that species differences of MOG lead to the activation of different pathogenic mechanisms in EAE induced with rodent or human MOG35–55 or recombinant MOG (26, 92, 181).
The animal experiments described above clearly indicated that murine MOG Abs can be pathogenic. Furthermore, pathologic similarities to ADEM have been shown in transgenic MOG-IgG mice infected with several neurotrophic encephalitogenic viruses, exacerbating virus-induced CNS inflammation. These similarities were indicated by clinical defined extensive perivascular infiltrates (mixed inflammatory cell population, e.g., lymphocytes, neutrophils, NK cells, and blood born macrophages) and perivenous demyelination (182, 183).
By contrast, only four studies aimed to investigate the pathogenic role of human MOG Abs in vivo. Whereas several studies indicated that human MOG Abs can activate complement and cellular-dependent cytotoxicity (50, 55, 112) in vitro, these mechanisms were not observed after transfer of human MOG Abs to rodents in vivo: the injection of concentrated serum samples from MOG Ab-positive patients into LEW rats with EAE did not increase the clinical score of the disease, but led to a minor increase in demyelination and axonal loss (50). Intrathecal injection of purified human MOG IgG caused reversible brain edema and myelin loss with very little complement deposition at the lesion site (57).
A different pathogenic mechanism for MOG Abs was proposed in two recent studies (58, 59). In the first study (58), it was demonstrated that MOG-specific B cells and their products (MOG Abs) activate MOG-specific effector T cells via CNS resident APC. A similar effect was demonstrated for peripheral APC in the second study (59). Both studies emphasize an important role for Ab-mediated antigen opsonization and accumulation in Fc receptor expressing APCs and subsequent increased antigen presentation and activation of specific T cells.
The findings discussed in the previous chapter raise the important question whether human MOG Abs are pathogenic themselves or just a epiphenomenal bystander or a secondary immune reaction due to previous demyelination (184). An example for a secondary immune response was shown in a study using a transgenic myelin-specific T cell mice model, which developed spontaneous EAE (98). In this model, an interaction between MOG-specific T and B cells is necessary for inflammatory demyelination, resulting in the activation of native B cells by dendritic cells presenting MOG peptides in the cervical lymph nodes (78). In a gut germ free environment, autoreactive T cell activation failed, and therefore the signal cascade for producing autoantibodies producing B cells was significantly reduced, but increased after microbial re-colonization. One potential mechanism mediating the onset of spontaneous EAE is molecular mimicry, activating encephalitogenic T cells, with subsequent inflammation of the CNS and second, it leads to an activation of native MOG-specific B cells recruited to the CNS tissue via locally produced MOG material or drained into the CNS along peripheral lymph nodes.
But even if MOG Abs would only be a secondary immune reaction they still could be clinically relevant biomarkers such as seen in diabetes type I, an autoimmune disease affecting insulin producing β-cells in the pancreas. Four autoantibodies to insulin (185), glutamic acid decarboxylase (186), Islet antigen-2 (187), and zinc transporter 8 (188) have been identified as highly specific biomarkers to predict this disease. There is more than an 80% probability of developing diabetes in children and adolescents, if 2/4 autoantibodies are detected [reviewed in Bonifacio (189)]. However, these autoantibodies are not pathogenic itself, but rather indicate a disturbed immune activity or an underlying T cell-mediated autoimmune process (190). Similarly, it could be that human MOG Abs play only a minor role in the pathophysiology of inflammatory demyelination, but are highly specific markers for affected patients.
In the past years, autoantibodies emerged as important biomarkers in neurological autoimmune diseases. One of the best examples for these biomarkers is AQP4 Abs as diagnostic marker for NMOSD. Numerous studies have now established a possible similar role for MOG Abs that are associated with a very heterogeneous age-dependent clinical presentation and MS-like neuropathology. The exact pathologic effect of human MOG Abs is still unclear and needs to be critically investigated in order to clarify the immunopathological role of these Abs.
PP prepared the main body of the manuscript and tables. MB and TB participated in the preparation of the manuscript. RH participated in the preparation of the manuscript and prepared tables and figures. MR supervised the work and participated in the preparation of the manuscript and figures and tables. All authors approved the final version of the manuscript.
The Neurological Research Laboratory (Medical University of Innsbruck and Tirol Kliniken) receives payments for antibody assays (AQP4- and anti-neuronal antibodies) and for MOG and AQP4 antibody validation experiments organized by Euroimmun (Germany).
The authors are grateful to A. Navarro for proof-reading and comments on the manuscript.
PP and MR are supported by a research grant from the Fonds zur Förderung der wissenschaftlichen Forschung, Austria (FWF graduate program W1206 SPIN).
AA, amino acids; Ab, antibody; ADEM, acute disseminated encephalomyelitis; AQP4, aquaporin-4; BN, brown Norway; CNS, central nervous system; CSF, cerebrospinal fluid; DA, dark agouti; EAE, experimental autoimmune encephalomyelitis; HLA, human leukocyte antigen; Ig, immunoglobulin; MBP, myelin basic protein; MHC, major histocompatibility complex; MOG, myelin oligodendrocyte glycoprotein; MS, multiple sclerosis; NMOSD, neuromyelitis optica spectrum disorders; ON, optic neuritis; TM, transverse myelitis.
1. Delarasse C, Della Gaspera B, Lu CW, Lachapelle F, Gelot A, Rodriguez D, et al. Complex alternative splicing of the myelin oligodendrocyte glycoprotein gene is unique to human and non-human primates. J Neurochem (2006) 98(6):1707–17. doi: 10.1111/j.1471-4159.2006.04053.x
2. Pham-Dinh D, Mattei MG, Nussbaum JL, Roussel G, Pontarotti P, Roeckel N, et al. Myelin/oligodendrocyte glycoprotein is a member of a subset of the immunoglobulin superfamily encoded within the major histocompatibility complex. Proc Natl Acad Sci U S A (1993) 90(17):7990–4. doi:10.1073/pnas.90.17.7990
3. Brunner C, Lassmann H, Waehneldt TV, Matthieu JM, Linington C. Differential ultrastructural localization of myelin basic protein, myelin/oligodendroglial glycoprotein, and 2′,3′-cyclic nucleotide 3′-phosphodiesterase in the CNS of adult rats. J Neurochem (1989) 52(1):296–304. doi:10.1111/j.1471-4159.1989.tb10930.x
4. Linington C, Bradl M, Lassmann H, Brunner C, Vass K. Augmentation of demyelination in rat acute allergic encephalomyelitis by circulating mouse monoclonal antibodies directed against a myelin/oligodendrocyte glycoprotein. Am J Pathol (1988) 130(3):443–54.
5. Martini R, Schachner M. Immunoelectron microscopic localization of neural cell adhesion molecules (L1, N-CAM, and MAG) and their shared carbohydrate epitope and myelin basic protein in developing sciatic nerve. J Cell Biol (1986) 103(6 Pt 1):2439–48. doi:10.1083/jcb.103.6.2439
6. Boyle LH, Traherne JA, Plotnek G, Ward R, Trowsdale J. Splice variation in the cytoplasmic domains of myelin oligodendrocyte glycoprotein affects its cellular localisation and transport. J Neurochem (2007) 102(6):1853–62. doi:10.1111/j.1471-4159.2007.04687.x
7. Marta CB, Montano MB, Taylor CM, Taylor AL, Bansal R, Pfeiffer SE. Signaling cascades activated upon antibody cross-linking of myelin oligodendrocyte glycoprotein: potential implications for multiple sclerosis. J Biol Chem (2005) 280(10):8985–93. doi:10.1074/jbc.M413174200
8. Gardinier MV, Amiguet P, Linington C, Matthieu JM. Myelin/oligodendrocyte glycoprotein is a unique member of the immunoglobulin superfamily. J Neurosci Res (1992) 33(1):177–87. doi:10.1002/jnr.490330123
9. Guggenmos J, Schubart AS, Ogg S, Andersson M, Olsson T, Mather IH, et al. Antibody cross-reactivity between myelin oligodendrocyte glycoprotein and the milk protein butyrophilin in multiple sclerosis. J Immunol (2004) 172(1):661–8. doi:10.4049/jimmunol.172.1.661
10. Krishnamoorthy G, Saxena A, Mars LT, Domingues HS, Mentele R, Ben-Nun A, et al. Myelin-specific T cells also recognize neuronal autoantigen in a transgenic mouse model of multiple sclerosis. Nat Med (2009) 15(6):626–32. doi:10.1038/nm.1975
11. Cong H, Jiang Y, Tien P. Identification of the myelin oligodendrocyte glycoprotein as a cellular receptor for rubella virus. J Virol (2011) 85(21):11038–47. doi:10.1128/JVI.05398-11
12. Garcia-Vallejo JJ, Ilarregui JM, Kalay H, Chamorro S, Koning N, Unger WW, et al. CNS myelin induces regulatory functions of DC-SIGN-expressing, antigen-presenting cells via cognate interaction with MOG. J Exp Med (2014) 211(7):1465–83. doi:10.1084/jem.20122192
13. von Budingen HC, Mei F, Greenfield A, Jahn S, Shen YA, Reid HH, et al. The myelin oligodendrocyte glycoprotein directly binds nerve growth factor to modulate central axon circuitry. J Cell Biol (2015) 210(6):891–8. doi:10.1083/jcb.201504106
14. Delarasse C, Daubas P, Mars LT, Vizler C, Litzenburger T, Iglesias A, et al. Myelin/oligodendrocyte glycoprotein-deficient (MOG-deficient) mice reveal lack of immune tolerance to MOG in wild-type mice. J Clin Invest (2003) 112(4):544–53. doi:10.1172/JCI15861
15. Linares D, Mana P, Goodyear M, Chow AM, Clavarino C, Huntington ND, et al. The magnitude and encephalogenic potential of autoimmune response to MOG is enhanced in MOG deficient mice. J Autoimmun (2003) 21(4):339–51. doi:10.1016/j.jaut.2003.09.001
16. Lebar R, Baudrimont M, Vincent C. Chronic experimental autoimmune encephalomyelitis in the Guinea pig. Presence of anti-M2 antibodies in central nervous system tissue and the possible role of M2 autoantigen in the induction of the disease. J Autoimmun (1989) 2(2):115–32. doi:10.1016/0896-8411(89)90149-2
17. Linington C, Lassmann H. Antibody responses in chronic relapsing experimental allergic encephalomyelitis: correlation of serum demyelinating activity with antibody titre to the myelin/oligodendrocyte glycoprotein (MOG). J Neuroimmunol (1987) 17(1):61–9. doi:10.1016/0165-5728(87)90031-2
18. Kabat EA, Wolf A, Bezer AE. The rapid production of acute disseminated encephalomyelitis in rhesus monkeys by injection of heterologous and homologous brain tissue with adjuvants. J Exp Med (1947) 85(1):117–30. doi:10.1084/jem.85.1.117
19. Seil FJ, Falk GA, Kies MW, Alvord EC Jr. The in vitro demyelinating activity of sera from Guinea pigs sensitized with whole CNS and with purified encephalitogen. Exp Neurol (1968) 22(4):545–55. doi:10.1016/0014-4886(68)90148-9
20. Lebar R, Boutry JM, Vincent C, Robineaux R, Voisin GA. Studies on autoimmune encephalomyelitis in the Guinea pig. II. An in vitro investigation on the nature, properties, and specificity of the serum-demyelinating factor. J Immunol (1976) 116(5):1439–46.
21. Lebar R, Vincent C, Fischer-le Boubennec E. Studies on autoimmune encephalomyelitis in the Guinea pig – III. A comparative study of two autoantigens of central nervous system myelin. J Neurochem (1979) 32(5):1451–60. doi:10.1111/j.1471-4159.1979.tb11084.x
22. Lassmann H, Brunner C, Bradl M, Linington C. Experimental allergic encephalomyelitis: the balance between encephalitogenic T lymphocytes and demyelinating antibodies determines size and structure of demyelinated lesions. Acta Neuropathol (1988) 75(6):566–76. doi:10.1007/BF00686201
23. Bradl M, Linington C. Animal models of demyelination. Brain Pathol (1996) 6(3):303–11. doi:10.1111/j.1750-3639.1996.tb00857.x
24. Adelmann M, Wood J, Benzel I, Fiori P, Lassmann H, Matthieu JM, et al. The N-terminal domain of the myelin oligodendrocyte glycoprotein (MOG) induces acute demyelinating experimental autoimmune encephalomyelitis in the Lewis rat. J Neuroimmunol (1995) 63(1):17–27. doi:10.1016/0165-5728(95)00124-7
25. von Budingen HC, Hauser SL, Fuhrmann A, Nabavi CB, Lee JI, Genain CP. Molecular characterization of antibody specificities against myelin/oligodendrocyte glycoprotein in autoimmune demyelination. Proc Natl Acad Sci U S A (2002) 99(12):8207–12. doi:10.1073/pnas.122092499
26. Marta CB, Oliver AR, Sweet RA, Pfeiffer SE, Ruddle NH. Pathogenic myelin oligodendrocyte glycoprotein antibodies recognize glycosylated epitopes and perturb oligodendrocyte physiology. Proc Natl Acad Sci U S A (2005) 102(39):13992–7. doi:10.1073/pnas.0504979102
27. de Graaf KL, Albert M, Weissert R. Autoantigen conformation influences both B- and T-cell responses and encephalitogenicity. J Biol Chem (2012) 287(21):17206–13. doi:10.1074/jbc.M111.304246
28. Bourquin C, Schubart A, Tobollik S, Mather I, Ogg S, Liblau R, et al. Selective unresponsiveness to conformational B cell epitopes of the myelin oligodendrocyte glycoprotein in H-2b mice. J Immunol (2003) 171(1):455–61. doi:10.4049/jimmunol.171.1.455
29. Berer K, Wekerle H, Krishnamoorthy G. B cells in spontaneous autoimmune diseases of the central nervous system. Mol Immunol (2011) 48(11):1332–7. doi:10.1016/j.molimm.2010.10.025
30. Amor S, Groome N, Linington C, Morris MM, Dornmair K, Gardinier MV, et al. Identification of epitopes of myelin oligodendrocyte glycoprotein for the induction of experimental allergic encephalomyelitis in SJL and Biozzi AB/H mice. J Immunol (1994) 153(10):4349–56.
31. Kerlero de Rosbo N, Mendel I, Ben-Nun A. Chronic relapsing experimental autoimmune encephalomyelitis with a delayed onset and an atypical clinical course, induced in PL/J mice by myelin oligodendrocyte glycoprotein (MOG)-derived peptide: preliminary analysis of MOG T cell epitopes. Eur J Immunol (1995) 25(4):985–93. doi:10.1002/eji.1830250419
32. Mendel I, Kerlero de Rosbo N, Ben-Nun A. A myelin oligodendrocyte glycoprotein peptide induces typical chronic experimental autoimmune encephalomyelitis in H-2b mice: fine specificity and T cell receptor V beta expression of encephalitogenic T cells. Eur J Immunol (1995) 25(7):1951–9. doi:10.1002/eji.1830250723
33. Linington C, Berger T, Perry L, Weerth S, Hinze-Selch D, Zhang Y, et al. T cells specific for the myelin oligodendrocyte glycoprotein mediate an unusual autoimmune inflammatory response in the central nervous system. Eur J Immunol (1993) 23(6):1364–72. doi:10.1002/eji.1830230627
34. Delarasse C, Smith P, Baker D, Amor S. Novel pathogenic epitopes of myelin oligodendrocyte glycoprotein induce experimental autoimmune encephalomyelitis in C57BL/6 mice. Immunology (2013) 140(4):456–64. doi:10.1111/imm.12155
35. Shetty A, Gupta SG, Varrin-Doyer M, Weber MS, Prod’homme T, Molnarfi N, et al. Immunodominant T-cell epitopes of MOG reside in its transmembrane and cytoplasmic domains in EAE. Neurol Neuroimmunol Neuroinflamm (2014) 1(2):e22. doi:10.1212/NXI.0000000000000022
36. Trotter J, DeJong LJ, Smith ME. Opsonization with antimyelin antibody increases the uptake and intracellular metabolism of myelin in inflammatory macrophages. J Neurochem (1986) 47(3):779–89. doi:10.1111/j.1471-4159.1986.tb00679.x
37. Schluesener HJ, Sobel RA, Linington C, Weiner HL. A monoclonal antibody against a myelin oligodendrocyte glycoprotein induces relapses and demyelination in central nervous system autoimmune disease. J Immunol (1987) 139(12):4016–21.
38. Kerlero de Rosbo N, Honegger P, Lassmann H, Matthieu JM. Demyelination induced in aggregating brain cell cultures by a monoclonal antibody against myelin/oligodendrocyte glycoprotein. J Neurochem (1990) 55(2):583–7. doi:10.1111/j.1471-4159.1990.tb04173.x
39. Scolding NJ, Compston DA. Oligodendrocyte-macrophage interactions in vitro triggered by specific antibodies. Immunology (1991) 72(1):127–32.
40. Vass K, Heininger K, Schafer B, Linington C, Lassmann H. Interferon-gamma potentiates antibody-mediated demyelination in vivo. Ann Neurol (1992) 32(2):198–206. doi:10.1002/ana.410320212
41. Linington C, Engelhardt B, Kapocs G, Lassman H. Induction of persistently demyelinated lesions in the rat following the repeated adoptive transfer of encephalitogenic T cells and demyelinating antibody. J Neuroimmunol (1992) 40(2–3):219–24. doi:10.1016/0165-5728(92)90136-9
42. Piddlesden SJ, Lassmann H, Zimprich F, Morgan BP, Linington C. The demyelinating potential of antibodies to myelin oligodendrocyte glycoprotein is related to their ability to fix complement. Am J Pathol (1993) 143(2):555–64.
43. Genain CP, Nguyen MH, Letvin NL, Pearl R, Davis RL, Adelman M, et al. Antibody facilitation of multiple sclerosis-like lesions in a nonhuman primate. J Clin Invest (1995) 96(6):2966–74. doi:10.1172/JCI118368
44. Johns TG, Kerlero de Rosbo N, Menon KK, Abo S, Gonzales MF, Bernard CC. Myelin oligodendrocyte glycoprotein induces a demyelinating encephalomyelitis resembling multiple sclerosis. J Immunol (1995) 154(10):5536–41.
45. Ichikawa M, Johns TG, Adelmann M, Bernard CC. Antibody response in Lewis rats injected with myelin oligodendrocyte glycoprotein derived peptides. Int Immunol (1996) 8(11):1667–74. doi:10.1093/intimm/8.11.1667
46. Menon KK, Piddlesden SJ, Bernard CC. Demyelinating antibodies to myelin oligodendrocyte glycoprotein and galactocerebroside induce degradation of myelin basic protein in isolated human myelin. J Neurochem (1997) 69(1):214–22. doi:10.1046/j.1471-4159.1997.69010214.x
47. Van der Goes A, Kortekaas M, Hoekstra K, Dijkstra CD, Amor S. The role of anti-myelin (auto)-antibodies in the phagocytosis of myelin by macrophages. J Neuroimmunol (1999) 101(1):61–7. doi:10.1016/S0165-5728(99)00133-2
48. Marta CB, Taylor CM, Coetzee T, Kim T, Winkler S, Bansal R, et al. Antibody cross-linking of myelin oligodendrocyte glycoprotein leads to its rapid repartitioning into detergent-insoluble fractions, and altered protein phosphorylation and cell morphology. J Neurosci (2003) 23(13):5461–71.
49. von Budingen HC, Hauser SL, Ouallet JC, Tanuma N, Menge T, Genain CP. Frontline: epitope recognition on the myelin/oligodendrocyte glycoprotein differentially influences disease phenotype and antibody effector functions in autoimmune demyelination. Eur J Immunol (2004) 34(8):2072–83. doi:10.1002/eji.200425050
50. Zhou D, Srivastava R, Nessler S, Grummel V, Sommer N, Bruck W, et al. Identification of a pathogenic antibody response to native myelin oligodendrocyte glycoprotein in multiple sclerosis. Proc Natl Acad Sci U S A (2006) 103(50):19057–62. doi:10.1073/pnas.0607242103
51. Urich E, Gutcher I, Prinz M, Becher B. Autoantibody-mediated demyelination depends on complement activation but not activatory Fc-receptors. Proc Natl Acad Sci U S A (2006) 103(49):18697–702. doi:10.1073/pnas.0607283103
52. Jagessar SA, Smith PA, Blezer E, Delarasse C, Pham-Dinh D, Laman JD, et al. Autoimmunity against myelin oligodendrocyte glycoprotein is dispensable for the initiation although essential for the progression of chronic encephalomyelitis in common marmosets. J Neuropathol Exp Neurol (2008) 67(4):326–40. doi:10.1097/NEN.0b013e31816a6851
53. Harrer MD, von Budingen HC, Stoppini L, Alliod C, Pouly S, Fischer K, et al. Live imaging of remyelination after antibody-mediated demyelination in an ex-vivo model for immune mediated CNS damage. Exp Neurol (2009) 216(2):431–8. doi:10.1016/j.expneurol.2008.12.027
54. Ohtani S, Kohyama K, Matsumoto Y. Autoantibodies recognizing native MOG are closely associated with active demyelination but not with neuroinflammation in chronic EAE. Neuropathology (2011) 31(2):101–11. doi:10.1111/j.1440-1789.2010.01131.x
55. Mader S, Gredler V, Schanda K, Rostasy K, Dujmovic I, Pfaller K, et al. Complement activating antibodies to myelin oligodendrocyte glycoprotein in neuromyelitis optica and related disorders. J Neuroinflammation (2011) 8:184. doi:10.1186/1742-2094-8-184
56. Dale RC, Tantsis EM, Merheb V, Kumaran RY, Sinmaz N, Pathmanandavel K, et al. Antibodies to MOG have a demyelination phenotype and affect oligodendrocyte cytoskeleton. Neurol Neuroimmunol Neuroinflamm (2014) 1(1):e12. doi:10.1212/NXI.0000000000000012
57. Saadoun S, Waters P, Owens GP, Bennett JL, Vincent A, Papadopoulos MC. Neuromyelitis optica MOG-IgG causes reversible lesions in mouse brain. Acta Neuropathol Commun (2014) 2:35. doi:10.1186/2051-5960-2-35
58. Flach AC, Litke T, Strauss J, Haberl M, Gomez CC, Reindl M, et al. Autoantibody-boosted T-cell reactivation in the target organ triggers manifestation of autoimmune CNS disease. Proc Natl Acad Sci U S A (2016) 113(12):3323–8. doi:10.1073/pnas.1519608113
59. Kinzel S, Lehmann-Horn K, Torke S, Hausler D, Winkler A, Stadelmann C, et al. Myelin-reactive antibodies initiate T cell-mediated CNS autoimmune disease by opsonization of endogenous antigen. Acta Neuropathol (2016) 132(1):43–58. doi:10.1007/s00401-016-1559-8
60. Devaux B, Enderlin F, Wallner B, Smilek DE. Induction of EAE in mice with recombinant human MOG, and treatment of EAE with a MOG peptide. J Neuroimmunol (1997) 75(1–2):169–73. doi:10.1016/S0165-5728(97)00019-2
61. Slavin A, Ewing C, Liu J, Ichikawa M, Slavin J, Bernard CC. Induction of a multiple sclerosis-like disease in mice with an immunodominant epitope of myelin oligodendrocyte glycoprotein. Autoimmunity (1998) 28(2):109–20. doi:10.3109/08916939809003872
62. Weissert R, Wallstrom E, Storch MK, Stefferl A, Lorentzen J, Lassmann H, et al. MHC haplotype-dependent regulation of MOG-induced EAE in rats. J Clin Invest (1998) 102(6):1265–73. doi:10.1172/JCI3022
63. Storch MK, Stefferl A, Brehm U, Weissert R, Wallstrom E, Kerschensteiner M, et al. Autoimmunity to myelin oligodendrocyte glycoprotein in rats mimics the spectrum of multiple sclerosis pathology. Brain Pathol (1998) 8(4):681–94. doi:10.1111/j.1750-3639.1998.tb00194.x
64. Encinas JA, Wicker LS, Peterson LB, Mukasa A, Teuscher C, Sobel R, et al. QTL influencing autoimmune diabetes and encephalomyelitis map to a 0.15-cM region containing Il2. Nat Genet (1999) 21(2):158–60. doi:10.1038/5941
65. Raine CS, Cannella B, Hauser SL, Genain CP. Demyelination in primate autoimmune encephalomyelitis and acute multiple sclerosis lesions: a case for antigen-specific antibody mediation. Ann Neurol (1999) 46(2):144–60. doi:10.1002/1531-8249(199908)46:2<144::AID-ANA3>3.0.CO;2-K
66. Abdul-Majid KB, Jirholt J, Stadelmann C, Stefferl A, Kjellen P, Wallstrom E, et al. Screening of several H-2 congenic mouse strains identified H-2(q) mice as highly susceptible to MOG-induced EAE with minimal adjuvant requirement. J Neuroimmunol (2000) 111(1–2):23–33. doi:10.1016/S0165-5728(00)00360-X
67. Kerlero de Rosbo N, Brok HP, Bauer J, Kaye JF, ‘t Hart BA, Ben-Nun A. Rhesus monkeys are highly susceptible to experimental autoimmune encephalomyelitis induced by myelin oligodendrocyte glycoprotein: characterisation of immunodominant T- and B-cell epitopes. J Neuroimmunol (2000) 110(1–2):83–96. doi:10.1016/S0165-5728(00)00306-4
68. Bourquin C, Iglesias A, Berger T, Wekerle H, Linington C. Myelin oligodendrocyte glycoprotein-DNA vaccination induces antibody-mediated autoaggression in experimental autoimmune encephalomyelitis. Eur J Immunol (2000) 30(12):3663–71. doi:10.1002/1521-4141(200012)30:12<3663::AID-IMMU3663>3.0.CO;2-7
69. Brok HP, Uccelli A, Kerlero De Rosbo N, Bontrop RE, Roccatagliata L, de Groot NG, et al. Myelin/oligodendrocyte glycoprotein-induced autoimmune encephalomyelitis in common marmosets: the encephalitogenic T cell epitope pMOG24-36 is presented by a monomorphic MHC class II molecule. J Immunol (2000) 165(2):1093–101. doi:10.4049/jimmunol.165.2.1093
70. Weissert R, de Graaf KL, Storch MK, Barth S, Linington C, Lassmann H, et al. MHC class II-regulated central nervous system autoaggression and T cell responses in peripheral lymphoid tissues are dissociated in myelin oligodendrocyte glycoprotein-induced experimental autoimmune encephalomyelitis. J Immunol (2001) 166(12):7588–99. doi:10.4049/jimmunol.166.12.7588
71. Bettelli E, Pagany M, Weiner HL, Linington C, Sobel RA, Kuchroo VK. Myelin oligodendrocyte glycoprotein-specific T cell receptor transgenic mice develop spontaneous autoimmune optic neuritis. J Exp Med (2003) 197(9):1073–81. doi:10.1084/jem.20021603
72. Sun D, Zhang Y, Wei B, Peiper SC, Shao H, Kaplan HJ. Encephalitogenic activity of truncated myelin oligodendrocyte glycoprotein (MOG) peptides and their recognition by CD8+ MOG-specific T cells on oligomeric MHC class I molecules. Int Immunol (2003) 15(2):261–8. doi:10.1093/intimm/dxg023
73. Smith PA, Heijmans N, Ouwerling B, Breij EC, Evans N, van Noort JM, et al. Native myelin oligodendrocyte glycoprotein promotes severe chronic neurological disease and demyelination in Biozzi ABH mice. Eur J Immunol (2005) 35(4):1311–9. doi:10.1002/eji.200425842
74. Krishnamoorthy G, Lassmann H, Wekerle H, Holz A. Spontaneous opticospinal encephalomyelitis in a double-transgenic mouse model of autoimmune T cell/B cell cooperation. J Clin Invest (2006) 116(9):2385–92. doi:10.1172/JCI28330
75. de Graaf KL, Barth S, Herrmann MM, Storch MK, Wiesmuller KH, Weissert R. Characterization of the encephalitogenic immune response in a model of multiple sclerosis. Eur J Immunol (2008) 38(1):299–308. doi:10.1002/eji.200737475
76. Kap YS, Smith P, Jagessar SA, Remarque E, Blezer E, Strijkers GJ, et al. Fast progression of recombinant human myelin/oligodendrocyte glycoprotein (MOG)-induced experimental autoimmune encephalomyelitis in marmosets is associated with the activation of MOG34-56-specific cytotoxic T cells. J Immunol (2008) 180(3):1326–37. doi:10.4049/jimmunol.180.3.1326
77. Matsumoto Y, Park IK, Hiraki K, Ohtani S, Kohyama K. Role of pathogenic T cells and autoantibodies in relapse and progression of myelin oligodendrocyte glycoprotein-induced autoimmune encephalomyelitis in LEW.1AV1 rats. Immunology (2009) 128(1 Suppl):e250–61. doi:10.1111/j.1365-2567.2008.02955.x
78. Pollinger B, Krishnamoorthy G, Berer K, Lassmann H, Bosl MR, Dunn R, et al. Spontaneous relapsing-remitting EAE in the SJL/J mouse: MOG-reactive transgenic T cells recruit endogenous MOG-specific B cells. J Exp Med (2009) 206(6):1303–16. doi:10.1084/jem.20090299
79. Bettini M, Rosenthal K, Evavold BD. Pathogenic MOG-reactive CD8+ T cells require MOG-reactive CD4+ T cells for sustained CNS inflammation during chronic EAE. J Neuroimmunol (2009) 213(1–2):60–8. doi:10.1016/j.jneuroim.2009.05.017
80. York NR, Mendoza JP, Ortega SB, Benagh A, Tyler AF, Firan M, et al. Immune regulatory CNS-reactive CD8+T cells in experimental autoimmune encephalomyelitis. J Autoimmun (2010) 35(1):33–44. doi:10.1016/j.jaut.2010.01.003
81. Anderson AC, Chandwaskar R, Lee DH, Sullivan JM, Solomon A, Rodriguez-Manzanet R, et al. A transgenic model of central nervous system autoimmunity mediated by CD4+ and CD8+ T and B cells. J Immunol (2012) 188(5):2084–92. doi:10.4049/jimmunol.1102186
82. Jagessar SA, Heijmans N, Blezer EL, Bauer J, Blokhuis JH, Wubben JA, et al. Unravelling the T-cell-mediated autoimmune attack on CNS myelin in a new primate EAE model induced with MOG34-56 peptide in incomplete adjuvant. Eur J Immunol (2012) 42(1):217–27. doi:10.1002/eji.201141863
83. Ortega SB, Kashi VP, Tyler AF, Cunnusamy K, Mendoza JP, Karandikar NJ. The disease-ameliorating function of autoregulatory CD8 T cells is mediated by targeting of encephalitogenic CD4 T cells in experimental autoimmune encephalomyelitis. J Immunol (2013) 191(1):117–26. doi:10.4049/jimmunol.1300452
84. Haanstra KG, Jagessar SA, Bauchet AL, Doussau M, Fovet CM, Heijmans N, et al. Induction of experimental autoimmune encephalomyelitis with recombinant human myelin oligodendrocyte glycoprotein in incomplete Freund’s adjuvant in three non-human primate species. J Neuroimmune Pharmacol (2013) 8(5):1251–64. doi:10.1007/s11481-013-9487-z
85. Curtis AD II, Taslim N, Reece SP, Grebenciucova E, Ray RH, Rosenbaum MD, et al. The extracellular domain of myelin oligodendrocyte glycoprotein elicits atypical experimental autoimmune encephalomyelitis in rat and macaque species. PLoS One (2014) 9(10):e110048. doi:10.1371/journal.pone.0110048
86. Herrera SL, Palmer VL, Whittaker H, Smith BC, Kim A, Schellenberg AE, et al. Damage to the optic chiasm in myelin oligodendrocyte glycoprotein-experimental autoimmune encephalomyelitis mice. Magn Reson Insights (2014) 7:23–31. doi:10.4137/MRI.S19750
87. Stefferl A, Brehm U, Storch M, Lambracht-Washington D, Bourquin C, Wonigeit K, et al. Myelin oligodendrocyte glycoprotein induces experimental autoimmune encephalomyelitis in the “resistant” brown Norway rat: disease susceptibility is determined by MHC and MHC-linked effects on the B cell response. J Immunol (1999) 163(1):40–9.
88. Forsthuber TG, Shive CL, Wienhold W, de Graaf K, Spack EG, Sublett R, et al. T cell epitopes of human myelin oligodendrocyte glycoprotein identified in HLA-DR4 (DRB1*0401) transgenic mice are encephalitogenic and are presented by human B cells. J Immunol (2001) 167(12):7119–25. doi:10.4049/jimmunol.167.12.7119
89. Litzenburger T, Fassler R, Bauer J, Lassmann H, Linington C, Wekerle H, et al. B lymphocytes producing demyelinating autoantibodies: development and function in gene-targeted transgenic mice. J Exp Med (1998) 188(1):169–80. doi:10.1084/jem.188.1.169
90. Lyons JA, San M, Happ MP, Cross AH. B cells are critical to induction of experimental allergic encephalomyelitis by protein but not by a short encephalitogenic peptide. Eur J Immunol (1999) 29(11):3432–9. doi:10.1002/(SICI)1521-4141(199911)29:11<3432::AID-IMMU3432>3.0.CO;2-2
91. Lyons JA, Ramsbottom MJ, Cross AH. Critical role of antigen-specific antibody in experimental autoimmune encephalomyelitis induced by recombinant myelin oligodendrocyte glycoprotein. Eur J Immunol (2002) 32(7):1905–13. doi:10.1002/1521-4141(200207)32:7<1905::AID-IMMU1905>3.0.CO;2-L
92. Hjelmstrom P, Juedes AE, Fjell J, Ruddle NH. B-cell-deficient mice develop experimental allergic encephalomyelitis with demyelination after myelin oligodendrocyte glycoprotein sensitization. J Immunol (1998) 161(9):4480–3.
93. Fillatreau S, Sweenie CH, McGeachy MJ, Gray D, Anderton SM. B cells regulate autoimmunity by provision of IL-10. Nat Immunol (2002) 3(10):944–50. doi:10.1038/ni833
94. Molnarfi N, Schulze-Topphoff U, Weber MS, Patarroyo JC, Prod’homme T, Varrin-Doyer M, et al. MHC class II-dependent B cell APC function is required for induction of CNS autoimmunity independent of myelin-specific antibodies. J Exp Med (2013) 210(13):2921–37. doi:10.1084/jem.20130699
95. Parker Harp CR, Archambault AS, Sim J, Ferris ST, Mikesell RJ, Koni PA, et al. B cell antigen presentation is sufficient to drive neuroinflammation in an animal model of multiple sclerosis. J Immunol (2015) 194(11):5077–84. doi:10.4049/jimmunol.1402236
96. Svensson L, Abdul-Majid KB, Bauer J, Lassmann H, Harris RA, Holmdahl R. A comparative analysis of B cell-mediated myelin oligodendrocyte glycoprotein-experimental autoimmune encephalomyelitis pathogenesis in B cell-deficient mice reveals an effect on demyelination. Eur J Immunol (2002) 32(7):1939–46. doi:10.1002/1521-4141(200207)32:7<1939::AID-IMMU1939>3.0.CO;2-S
97. Bettelli E, Baeten D, Jager A, Sobel RA, Kuchroo VK. Myelin oligodendrocyte glycoprotein-specific T and B cells cooperate to induce a Devic-like disease in mice. J Clin Invest (2006) 116(9):2393–402. doi:10.1172/JCI28334
98. Berer K, Mues M, Koutrolos M, Rasbi ZA, Boziki M, Johner C, et al. Commensal microbiota and myelin autoantigen cooperate to trigger autoimmune demyelination. Nature (2011) 479(7374):538–41. doi:10.1038/nature10554
99. Lassmann H, Bradl M. Multiple sclerosis: experimental models and reality. Acta Neuropathol (2017) 133(2):223–44. doi:10.1007/s00401-016-1631-4
100. Hoftberger R, Leisser M, Bauer J, Lassmann H. Autoimmune encephalitis in humans: how closely does it reflect multiple sclerosis? Acta Neuropathol Commun (2015) 3(1):80. doi:10.1186/s40478-015-0260-9
101. ‘t Hart BA, Gran B, Weissert R. EAE: imperfect but useful models of multiple sclerosis. Trends Mol Med (2011) 17(3):119–25. doi:10.1016/j.molmed.2010.11.006
102. ‘t Hart BA, van Kooyk Y, Geurts JJ, Gran B. The primate autoimmune encephalomyelitis model; a bridge between mouse and man. Ann Clin Transl Neurol (2015) 2(5):581–93. doi:10.1002/acn3.194
103. Massacesi L, Genain CP, Lee-Parritz D, Letvin NL, Canfield D, Hauser SL. Active and passively induced experimental autoimmune encephalomyelitis in common marmosets: a new model for multiple sclerosis. Ann Neurol (1995) 37(4):519–30. doi:10.1002/ana.410370415
104. Genain CP, Hauser SL. Experimental allergic encephalomyelitis in the New World monkey Callithrix jacchus. Immunol Rev (2001) 183:159–72. doi:10.1034/j.1600-065x.2001.1830113.x
105. Berger T, Rubner P, Schautzer F, Egg R, Ulmer H, Mayringer I, et al. Antimyelin antibodies as a predictor of clinically definite multiple sclerosis after a first demyelinating event. N Engl J Med (2003) 349(2):139–45. doi:10.1056/NEJMoa022328
106. Kuhle J, Pohl C, Mehling M, Edan G, Freedman MS, Hartung HP, et al. Lack of association between antimyelin antibodies and progression to multiple sclerosis. N Engl J Med (2007) 356(4):371–8. doi:10.1056/NEJMoa063602
107. Berger T, Reindl M. Antibody biomarkers in CNS demyelinating diseases – a long and winding road. Eur J Neurol (2015) 22(8):1162–8. doi:10.1111/ene.12759
108. Pache F, Zimmermann H, Mikolajczak J, Schumacher S, Lacheta A, Oertel FC, et al. MOG-IgG in NMO and related disorders: a multicenter study of 50 patients. Part 4: afferent visual system damage after optic neuritis in MOG-IgG-seropositive versus AQP4-IgG-seropositive patients. J Neuroinflammation (2016) 13(1):282. doi:10.1186/s12974-016-0720-6
109. Lalive PH, Menge T, Delarasse C, Della Gaspera B, Pham-Dinh D, Villoslada P, et al. Antibodies to native myelin oligodendrocyte glycoprotein are serologic markers of early inflammation in multiple sclerosis. Proc Natl Acad Sci U S A (2006) 103(7):2280–5. doi:10.1073/pnas.0510672103
110. O’Connor KC, McLaughlin KA, De Jager PL, Chitnis T, Bettelli E, Xu C, et al. Self-antigen tetramers discriminate between myelin autoantibodies to native or denatured protein. Nat Med (2007) 13(2):211–7. doi:10.1038/nm1488
111. McLaughlin KA, Chitnis T, Newcombe J, Franz B, Kennedy J, McArdel S, et al. Age-dependent B cell autoimmunity to a myelin surface antigen in pediatric multiple sclerosis. J Immunol (2009) 183(6):4067–76. doi:10.4049/jimmunol.0801888
112. Brilot F, Dale RC, Selter RC, Grummel V, Kalluri SR, Aslam M, et al. Antibodies to native myelin oligodendrocyte glycoprotein in children with inflammatory demyelinating central nervous system disease. Ann Neurol (2009) 66(6):833–42. doi:10.1002/ana.21916
113. Selter RC, Brilot F, Grummel V, Kraus V, Cepok S, Dale RC, et al. Antibody responses to EBV and native MOG in pediatric inflammatory demyelinating CNS diseases. Neurology (2010) 74(21):1711–5. doi:10.1212/WNL.0b013e3181e04096
114. Lalive PH, Hausler MG, Maurey H, Mikaeloff Y, Tardieu M, Wiendl H, et al. Highly reactive anti-myelin oligodendrocyte glycoprotein antibodies differentiate demyelinating diseases from viral encephalitis in children. Mult Scler (2011) 17(3):297–302. doi:10.1177/1352458510389220
115. Di Pauli F, Mader S, Rostasy K, Schanda K, Bajer-Kornek B, Ehling R, et al. Temporal dynamics of anti-MOG antibodies in CNS demyelinating diseases. Clin Immunol (2011) 138(3):247–54. doi:10.1016/j.clim.2010.11.013
116. Probstel AK, Dornmair K, Bittner R, Sperl P, Jenne D, Magalhaes S, et al. Antibodies to MOG are transient in childhood acute disseminated encephalomyelitis. Neurology (2011) 77(6):580–8. doi:10.1212/WNL.0b013e318228c0b1
117. Kezuka T, Usui Y, Yamakawa N, Matsunaga Y, Matsuda R, Masuda M, et al. Relationship between NMO-antibody and anti-MOG antibody in optic neuritis. J Neuroophthalmol (2012) 32(2):107–10. doi:10.1097/WNO.0b013e31823c9b6c
118. Rostasy K, Mader S, Schanda K, Huppke P, Gartner J, Kraus V, et al. Anti-myelin oligodendrocyte glycoprotein antibodies in pediatric patients with optic neuritis. Arch Neurol (2012) 69(6):752–6. doi:10.1001/archneurol.2011.2956
119. Kitley J, Woodhall M, Waters P, Leite MI, Devenney E, Craig J, et al. Myelin-oligodendrocyte glycoprotein antibodies in adults with a neuromyelitis optica phenotype. Neurology (2012) 79(12):1273–7. doi:10.1212/WNL.0b013e31826aac4e
120. Huppke P, Rostasy K, Karenfort M, Huppke B, Seidl R, Leiz S, et al. Acute disseminated encephalomyelitis followed by recurrent or monophasic optic neuritis in pediatric patients. Mult Scler (2013) 19(7):941–6. doi:10.1177/1352458512466317
121. Rostasy K, Mader S, Hennes EM, Schanda K, Gredler V, Guenther A, et al. Persisting myelin oligodendrocyte glycoprotein antibodies in aquaporin-4 antibody negative pediatric neuromyelitis optica. Mult Scler (2013) 19(8):1052–9. doi:10.1177/1352458512470310
122. Mayer MC, Breithaupt C, Reindl M, Schanda K, Rostasy K, Berger T, et al. Distinction and temporal stability of conformational epitopes on myelin oligodendrocyte glycoprotein recognized by patients with different inflammatory central nervous system diseases. J Immunol (2013) 191(7):3594–604. doi:10.4049/jimmunol.1301296
123. Woodhall M, Coban A, Waters P, Ekizoglu E, Kurtuncu M, Shugaiv E, et al. Glycine receptor and myelin oligodendrocyte glycoprotein antibodies in Turkish patients with neuromyelitis optica. J Neurol Sci (2013) 335(1–2):221–3. doi:10.1016/j.jns.2013.08.034
124. Sato DK, Callegaro D, Lana-Peixoto MA, Waters PJ, de Haidar Jorge FM, Takahashi T, et al. Distinction between MOG antibody-positive and AQP4 antibody-positive NMO spectrum disorders. Neurology (2014) 82(6):474–81. doi:10.1212/WNL.0000000000000101
125. Kitley J, Waters P, Woodhall M, Leite MI, Murchison A, George J, et al. Neuromyelitis optica spectrum disorders with aquaporin-4 and myelin-oligodendrocyte glycoprotein antibodies: a comparative study. JAMA Neurol (2014) 71(3):276–83. doi:10.1001/jamaneurol.2013.5857
126. Titulaer MJ, Hoftberger R, Iizuka T, Leypoldt F, McCracken L, Cellucci T, et al. Overlapping demyelinating syndromes and anti-N-methyl-d-aspartate receptor encephalitis. Ann Neurol (2014) 75(3):411–28. doi:10.1002/ana.24117
127. Hacohen Y, Absoud M, Woodhall M, Cummins C, De Goede CG, Hemingway C, et al. Autoantibody biomarkers in childhood-acquired demyelinating syndromes: results from a national surveillance cohort. J Neurol Neurosurg Psychiatry (2014) 85(4):456–61. doi:10.1136/jnnp-2013-306411
128. Tanaka M, Tanaka K. Anti-MOG antibodies in adult patients with demyelinating disorders of the central nervous system. J Neuroimmunol (2014) 270(1–2):98–9. doi:10.1016/j.jneuroim.2014.03.001
129. Miyauchi A, Monden Y, Watanabe M, Sugie H, Morita M, Kezuka T, et al. Persistent presence of the anti-myelin oligodendrocyte glycoprotein autoantibody in a pediatric case of acute disseminated encephalomyelitis followed by optic neuritis. Neuropediatrics (2014) 45(3):196–9. doi:10.1055/s-0034-1371179
130. Ramanathan S, Reddel SW, Henderson A, Parratt JD, Barnett M, Gatt PN, et al. Antibodies to myelin oligodendrocyte glycoprotein in bilateral and recurrent optic neuritis. Neurol Neuroimmunol Neuroinflamm (2014) 1(4):e40. doi:10.1212/NXI.0000000000000040
131. Amano H, Miyamoto N, Shimura H, Sato DK, Fujihara K, Ueno S, et al. Influenza-associated MOG antibody-positive longitudinally extensive transverse myelitis: a case report. BMC Neurol (2014) 14:224. doi:10.1186/s12883-014-0224-x
132. Elong Ngono A, Lepetit M, Reindl M, Garcia A, Guillot F, Genty A, et al. Decreased frequency of circulating myelin oligodendrocyte glycoprotein B lymphocytes in patients with relapsing-remitting multiple sclerosis. J Immunol Res (2015) 2015:673503. doi:10.1155/2015/673503
133. Tsuburaya RS, Miki N, Tanaka K, Kageyama T, Irahara K, Mukaida S, et al. Anti-myelin oligodendrocyte glycoprotein (MOG) antibodies in a Japanese boy with recurrent optic neuritis. Brain Dev (2015) 37(1):145–8. doi:10.1016/j.braindev.2014.02.002
134. Martinez-Hernandez E, Sepulveda M, Rostasy K, Hoftberger R, Graus F, Harvey RJ, et al. Antibodies to aquaporin 4, myelin-oligodendrocyte glycoprotein, and the glycine receptor alpha1 subunit in patients with isolated optic neuritis. JAMA Neurol (2015) 72(2):187–93. doi:10.1001/jamaneurol.2014.3602
135. Baumann M, Sahin K, Lechner C, Hennes EM, Schanda K, Mader S, et al. Clinical and neuroradiological differences of paediatric acute disseminating encephalomyelitis with and without antibodies to the myelin oligodendrocyte glycoprotein. J Neurol Neurosurg Psychiatry (2015) 86(3):265–72. doi:10.1136/jnnp-2014-308346
136. Spadaro M, Gerdes LA, Mayer MC, Ertl-Wagner B, Laurent S, Krumbholz M, et al. Histopathology and clinical course of MOG-antibody-associated encephalomyelitis. Ann Clin Transl Neurol (2015) 2(3):295–301. doi:10.1002/acn3.164
137. Probstel AK, Rudolf G, Dornmair K, Collongues N, Chanson JB, Sanderson NS, et al. Anti-MOG antibodies are present in a subgroup of patients with a neuromyelitis optica phenotype. J Neuroinflammation (2015) 12:46. doi:10.1186/s12974-015-0256-1
138. Hacohen Y, Absoud M, Deiva K, Hemingway C, Nytrova P, Woodhall M, et al. Myelin oligodendrocyte glycoprotein antibodies are associated with a non-MS course in children. Neurol Neuroimmunol Neuroinflamm (2015) 2(2):e81. doi:10.1212/NXI.0000000000000081
139. Symmonds M, Waters PJ, Kuker W, Leite MI, Schulz UG. Anti-MOG antibodies with longitudinally extensive transverse myelitis preceded by CLIPPERS. Neurology (2015) 84(11):1177–9. doi:10.1212/WNL.0000000000001370
140. Morris KA, Waters P, Woodhall MR, Kuker W, Vincent A, Leite MI, et al. A 41-year-old woman with acute weakness and encephalopathy associated with MOG antibodies. Neurol Neuroimmunol Neuroinflamm (2015) 2(3):e88. doi:10.1212/NXI.0000000000000088
141. Waters P, Woodhall M, O’Connor KC, Reindl M, Lang B, Sato DK, et al. MOG cell-based assay detects non-MS patients with inflammatory neurologic disease. Neurol Neuroimmunol Neuroinflamm (2015) 2(3):e89. doi:10.1212/NXI.0000000000000089
142. Ikeda K, Kiyota N, Kuroda H, Sato DK, Nishiyama S, Takahashi T, et al. Severe demyelination but no astrocytopathy in clinically definite neuromyelitis optica with anti-myelin-oligodendrocyte glycoprotein antibody. Mult Scler (2015) 21(5):656–9. doi:10.1177/1352458514551455
143. Nakajima H, Motomura M, Tanaka K, Fujikawa A, Nakata R, Maeda Y, et al. Antibodies to myelin oligodendrocyte glycoprotein in idiopathic optic neuritis. BMJ Open (2015) 5(4):e007766. doi:10.1136/bmjopen-2015-007766
144. Hyun JW, Kim SH, Huh SY, Kim W, Yun J, Joung A, et al. Idiopathic aquaporin-4 antibody negative longitudinally extensive transverse myelitis. Mult Scler (2015) 21(6):710–7. doi:10.1177/1352458514551454
145. Hoftberger R, Sepulveda M, Armangue T, Blanco Y, Rostasy K, Cobo Calvo A, et al. Antibodies to MOG and AQP4 in adults with neuromyelitis optica and suspected limited forms of the disease. Mult Scler (2015) 21(7):866–74. doi:10.1177/1352458514555785
146. Chalmoukou K, Alexopoulos H, Akrivou S, Stathopoulos P, Reindl M, Dalakas MC. Anti-MOG antibodies are frequently associated with steroid-sensitive recurrent optic neuritis. Neurol Neuroimmunol Neuroinflamm (2015) 2(4):e131. doi:10.1212/NXI.0000000000000131
147. Ketelslegers IA, Van Pelt DE, Bryde S, Neuteboom RF, Catsman-Berrevoets CE, Hamann D, et al. Anti-MOG antibodies plead against MS diagnosis in an acquired demyelinating syndromes cohort. Mult Scler (2015) 21(12):1513–20. doi:10.1177/1352458514566666
148. Kim SM, Woodhall MR, Kim JS, Kim SJ, Park KS, Vincent A, et al. Antibodies to MOG in adults with inflammatory demyelinating disease of the CNS. Neurol Neuroimmunol Neuroinflamm (2015) 2(6):e163. doi:10.1212/NXI.0000000000000163
149. Di Pauli F, Hoftberger R, Reindl M, Beer R, Rhomberg P, Schanda K, et al. Fulminant demyelinating encephalomyelitis: insights from antibody studies and neuropathology. Neurol Neuroimmunol Neuroinflamm (2015) 2(6):e175. doi:10.1212/NXI.0000000000000175
150. Numa S, Kasai T, Kondo T, Kushimura Y, Kimura A, Takahashi H, et al. An adult case of anti-myelin oligodendrocyte glycoprotein (MOG) antibody-associated multiphasic acute disseminated encephalomyelitis at 33-year intervals. Intern Med (2016) 55(6):699–702. doi:10.2169/internalmedicine.55.5727
151. Adachi T, Yasui K, Takahashi T, Fujihara K, Watanabe Y, Nakashima K. Anti-myelin oligodendrocyte glycoprotein antibodies in a patient with recurrent optic neuritis involving the cerebral white matter and brainstem. Intern Med (2016) 55(10):1351–4. doi:10.2169/internalmedicine.55.6122
152. Fernandez-Carbonell C, Vargas-Lowy D, Musallam A, Healy B, McLaughlin K, Wucherpfennig KW, et al. Clinical and MRI phenotype of children with MOG antibodies. Mult Scler (2016) 22(2):174–84. doi:10.1177/1352458515587751
153. Piccolo L, Woodhall M, Tackley G, Jurynczyk M, Kong Y, Domingos J, et al. Isolated new onset ‘atypical’ optic neuritis in the NMO clinic: serum antibodies, prognoses and diagnoses at follow-up. J Neurol (2016) 263(2):370–9. doi:10.1007/s00415-015-7983-1
154. Baumann M, Hennes EM, Schanda K, Karenfort M, Kornek B, Seidl R, et al. Children with multiphasic disseminated encephalomyelitis and antibodies to the myelin oligodendrocyte glycoprotein (MOG): extending the spectrum of MOG antibody positive diseases. Mult Scler (2016) 22(14):1821–9. doi:10.1177/1352458516631038
155. Kakalacheva K, Regenass S, Wiesmayr S, Azzi T, Berger C, Dale RC, et al. Infectious mononucleosis triggers generation of IgG auto-antibodies against native myelin oligodendrocyte glycoprotein. Viruses (2016) 8(2):E51. doi:10.3390/v8020051
156. Yan Y, Li Y, Fu Y, Yang L, Su L, Shi K, et al. Autoantibody to MOG suggests two distinct clinical subtypes of NMOSD. Sci China Life Sci (2016) 59(12):1270–81. doi:10.1007/s11427-015-4997-y
157. Cobo-Calvo A, Sepulveda M, Bernard-Valnet R, Ruiz A, Brassat D, Martinez-Yelamos S, et al. Antibodies to myelin oligodendrocyte glycoprotein in aquaporin 4 antibody seronegative longitudinally extensive transverse myelitis: clinical and prognostic implications. Mult Scler (2016) 22(3):312–9. doi:10.1177/1352458515591071
158. van Pelt ED, Wong YY, Ketelslegers IA, Hamann D, Hintzen RQ. Neuromyelitis optica spectrum disorders: comparison of clinical and magnetic resonance imaging characteristics of AQP4-IgG versus MOG-IgG seropositive cases in the Netherlands. Eur J Neurol (2016) 23(3):580–7. doi:10.1111/ene.12898
159. Ramanathan S, Prelog K, Barnes EH, Tantsis EM, Reddel SW, Henderson AP, et al. Radiological differentiation of optic neuritis with myelin oligodendrocyte glycoprotein antibodies, aquaporin-4 antibodies, and multiple sclerosis. Mult Scler (2016) 22(4):470–82. doi:10.1177/1352458515593406
160. Akaishi T, Sato DK, Nakashima I, Takeshita T, Takahashi T, Doi H, et al. MRI and retinal abnormalities in isolated optic neuritis with myelin oligodendrocyte glycoprotein and aquaporin-4 antibodies: a comparative study. J Neurol Neurosurg Psychiatry (2016) 87(4):446–8. doi:10.1136/jnnp-2014-310206
161. Sepulveda M, Armangue T, Sola-Valls N, Arrambide G, Meca-Lallana JE, Oreja-Guevara C, et al. Neuromyelitis optica spectrum disorders: comparison according to the phenotype and serostatus. Neurol Neuroimmunol Neuroinflamm (2016) 3(3):e225. doi:10.1212/NXI.0000000000000225
162. Akaishi T, Nakashima I, Takeshita T, Mugikura S, Sato DK, Takahashi T, et al. Lesion length of optic neuritis impacts visual prognosis in neuromyelitis optica. J Neuroimmunol (2016) 293:28–33. doi:10.1016/j.jneuroim.2016.02.004
163. Polat I, Yis U, Karaoglu P, Ayanoglu M, Ozturk T, Guleryuz H, et al. Myelin oligodendrocyte glycoprotein antibody persistency in a steroid-dependent ADEM case. Pediatrics (2016) 137(5):e20151958. doi:10.1542/peds.2015-1958
164. Siritho S, Sato DK, Kaneko K, Fujihara K, Prayoonwiwat N. The clinical spectrum associated with myelin oligodendrocyte glycoprotein antibodies (anti-MOG-Ab) in Thai patients. Mult Scler (2016) 22(7):964–8. doi:10.1177/1352458515614093
165. Spadaro M, Gerdes LA, Krumbholz M, Ertl-Wagner B, Thaler FS, Schuh E, et al. Autoantibodies to MOG in a distinct subgroup of adult multiple sclerosis. Neurol Neuroimmunol Neuroinflamm (2016) 3(5):e257. doi:10.1212/NXI.0000000000000257
166. Sepulveda M, Armangue T, Martinez-Hernandez E, Arrambide G, Sola-Valls N, Sabater L, et al. Clinical spectrum associated with MOG autoimmunity in adults: significance of sharing rodent MOG epitopes. J Neurol (2016) 263(7):1349–60. doi:10.1007/s00415-016-8147-7
167. Wang JJ, Jaunmuktane Z, Mummery C, Brandner S, Leary S, Trip SA. Inflammatory demyelination without astrocyte loss in MOG antibody-positive NMOSD. Neurology (2016) 87(2):229–31. doi:10.1212/WNL.0000000000002844
168. Thulasirajah S, Pohl D, Davila-Acosta J, Venkateswaran S. Myelin oligodendrocyte glycoprotein-associated pediatric central nervous system demyelination: clinical course, neuroimaging findings, and response to therapy. Neuropediatrics (2016) 47(4):245–52. doi:10.1055/s-0036-1583184
169. Lechner C, Baumann M, Hennes EM, Schanda K, Marquard K, Karenfort M, et al. Antibodies to MOG and AQP4 in children with neuromyelitis optica and limited forms of the disease. J Neurol Neurosurg Psychiatry (2016) 87:897–905. doi:10.1136/jnnp-2015-311743
170. Hacohen Y, Nishimoto Y, Fukami Y, Lang B, Waters P, Lim MJ, et al. Paediatric brainstem encephalitis associated with glial and neuronal autoantibodies. Dev Med Child Neurol (2016) 58(8):836–41. doi:10.1111/dmcn.13090
171. Azumagawa K, Nomura S, Shigeri Y, Jones LS, Sato DK, Nakashima I, et al. Post-vaccination MDEM associated with MOG antibody in a subclinical Chlamydia infected boy. Brain Dev (2016) 38(7):690–3. doi:10.1016/j.braindev.2016.02.004
172. Jarius S, Metz I, Konig FB, Ruprecht K, Reindl M, Paul F, et al. Screening for MOG-IgG and 27 other anti-glial and anti-neuronal autoantibodies in ‘pattern II multiple sclerosis’ and brain biopsy findings in a MOG-IgG-positive case. Mult Scler (2016) 22(12):1541–9. doi:10.1177/1352458515622986
173. Kaneko K, Sato DK, Nakashima I, Nishiyama S, Tanaka S, Marignier R, et al. Myelin injury without astrocytopathy in neuroinflammatory disorders with MOG antibodies. J Neurol Neurosurg Psychiatry (2016) 87(11):1257–9. doi:10.1136/jnnp-2015-312676
174. Jarius S, Ruprecht K, Kleiter I, Borisow N, Asgari N, Pitarokoili K, et al. MOG-IgG in NMO and related disorders: a multicenter study of 50 patients. Part 1: frequency, syndrome specificity, influence of disease activity, long-term course, association with AQP4-IgG, and origin. J Neuroinflammation (2016) 13(1):279. doi:10.1186/s12974-016-0719-z
175. Jarius S, Ruprecht K, Kleiter I, Borisow N, Asgari N, Pitarokoili K, et al. MOG-IgG in NMO and related disorders: a multicenter study of 50 patients. Part 2: epidemiology, clinical presentation, radiological and laboratory features, treatment responses, and long-term outcome. J Neuroinflammation (2016) 13(1):280. doi:10.1186/s12974-016-0718-0
176. Jarius S, Kleiter I, Ruprecht K, Asgari N, Pitarokoili K, Borisow N, et al. MOG-IgG in NMO and related disorders: a multicenter study of 50 patients. Part 3: brainstem involvement – frequency, presentation and outcome. J Neuroinflammation (2016) 13(1):281. doi:10.1186/s12974-016-0717-1
177. Lennon VA, Wingerchuk DM, Kryzer TJ, Pittock SJ, Lucchinetti CF, Fujihara K, et al. A serum autoantibody marker of neuromyelitis optica: distinction from multiple sclerosis. Lancet (2004) 364(9451):2106–12. doi:10.1016/S0140-6736(04)17551-X
178. Konig FB, Wildemann B, Nessler S, Zhou D, Hemmer B, Metz I, et al. Persistence of immunopathological and radiological traits in multiple sclerosis. Arch Neurol (2008) 65(11):1527–32. doi:10.1001/archneur.65.11.1527
179. Körtvélyessy P, Breu M, Pawlitzki M, Metz I, Heinze H-J, Matzke M, et al. ADEM-like presentation, anti-MOG antibodies, and MS pathology: TWO case reports. Neurol Neuroimmunol Neuroinflamm (2017) 4(3):e335. doi:10.1212/NXI.0000000000000335
180. Misu T, Hoftberger R, Fujihara K, Wimmer I, Takai Y, Nishiyama S, et al. Presence of six different lesion types suggests diverse mechanisms of tissue injury in neuromyelitis optica. Acta Neuropathol (2013) 125(6):815–27. doi:10.1007/s00401-013-1116-7
181. Oliver AR, Lyon GM, Ruddle NH. Rat and human myelin oligodendrocyte glycoproteins induce experimental autoimmune encephalomyelitis by different mechanisms in C57BL/6 mice. J Immunol (2003) 171(1):462–8. doi:10.4049/jimmunol.171.9.4934
182. Burrer R, Buchmeier MJ, Wolfe T, Ting JP, Feuer R, Iglesias A, et al. Exacerbated pathology of viral encephalitis in mice with central nervous system-specific autoantibodies. Am J Pathol (2007) 170(2):557–66. doi:10.2353/ajpath.2007.060893
183. Young NP, Weinshenker BG, Parisi JE, Scheithauer B, Giannini C, Roemer SF, et al. Perivenous demyelination: association with clinically defined acute disseminated encephalomyelitis and comparison with pathologically confirmed multiple sclerosis. Brain (2010) 133(Pt 2):333–48. doi:10.1093/brain/awp321
184. Reindl M, Di Pauli F, Rostasy K, Berger T. The spectrum of MOG autoantibody-associated demyelinating diseases. Nat Rev Neurol (2013) 9(8):455–61. doi:10.1038/nrneurol.2013.118
185. Palmer JP, Asplin CM, Clemons P, Lyen K, Tatpati O, Raghu PK, et al. Insulin antibodies in insulin-dependent diabetics before insulin treatment. Science (1983) 222(4630):1337–9. doi:10.1126/science.6362005
186. Baekkeskov S, Aanstoot HJ, Christgau S, Reetz A, Solimena M, Cascalho M, et al. Identification of the 64K autoantigen in insulin-dependent diabetes as the GABA-synthesizing enzyme glutamic acid decarboxylase. Nature (1990) 347(6289):151–6. doi:10.1038/347151a0
187. Rabin DU, Pleasic SM, Shapiro JA, Yoo-Warren H, Oles J, Hicks JM, et al. Islet cell antigen 512 is a diabetes-specific islet autoantigen related to protein tyrosine phosphatases. J Immunol (1994) 152(6):3183–8.
188. Wenzlau JM, Juhl K, Yu L, Moua O, Sarkar SA, Gottlieb P, et al. The cation efflux transporter ZnT8 (Slc30A8) is a major autoantigen in human type 1 diabetes. Proc Natl Acad Sci U S A (2007) 104(43):17040–5. doi:10.1073/pnas.0705894104
189. Bonifacio E. Predicting type 1 diabetes using biomarkers. Diabetes Care (2015) 38(6):989–96. doi:10.2337/dc15-0101
Keywords: myelin oligodendrocyte glycoprotein, demyelination, autoantibodies, inflammation, MOG
Citation: Peschl P, Bradl M, Höftberger R, Berger T and Reindl M (2017) Myelin Oligodendrocyte Glycoprotein: Deciphering a Target in Inflammatory Demyelinating Diseases. Front. Immunol. 8:529. doi: 10.3389/fimmu.2017.00529
Received: 12 December 2016; Accepted: 19 April 2017;
Published: 08 May 2017
Edited by:
Robert Weissert, University of Regensburg, GermanyReviewed by:
Tomas Per Olsson, Karolinska Institutet, SwedenCopyright: © 2017 Peschl, Bradl, Höftberger, Berger and Reindl. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Markus Reindl, bWFya3VzLnJlaW5kbEBpLW1lZC5hYy5hdA==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.