 Christin Peteranderl
Christin Peteranderl Susanne Herold
Susanne Herold
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol., 22 March 2017
Sec. Molecular Innate Immunity
Volume 8 - 2017 | https://doi.org/10.3389/fimmu.2017.00313
This article is part of the Research TopicImmunoregulatory mechanisms of InterferonView all 30 articles
Interferons (IFNs) are well described to be rapidly induced upon pathogen-associated pattern recognition. After binding to their respective IFN receptors and activation of the cellular JAK/signal transducer and activator of transcription signaling cascade, they stimulate the transcription of a plethora of IFN-stimulated genes (ISGs) in infected as well as bystander cells such as the non-infected epithelium and cells of the immune system. ISGs may directly act on the invading pathogen or can either positively or negatively regulate the innate and adaptive immune response. However, IFNs and ISGs do not only play a key role in the limitation of pathogen spread but have also been recently found to provoke an unbalanced, overshooting inflammatory response causing tissue injury and hampering repair processes. A prominent regulator of disease outcome, especially in—but not limited to—respiratory viral infection, is the IFN-dependent mediator TRAIL (TNF-related apoptosis-inducing ligand) produced by several cell types including immune cells such as macrophages or T cells. First described as an apoptosis-inducing agent in transformed cells, it is now also well established to rapidly evoke cellular stress pathways in epithelial cells, finally leading to caspase-dependent or -independent cell death. Hereby, pathogen spread is limited; however in some cases, also the surrounding tissue is severely harmed, thus augmenting disease severity. Interestingly, the lack of a strictly controlled and well balanced IFN/TRAIL signaling response has not only been implicated in viral infection but might furthermore be an important determinant of disease progression in bacterial superinfections and in chronic respiratory illness. Conclusively, the IFN/TRAIL signaling axis is subjected to a complex modulation and might be exploited for the evaluation of new therapeutic concepts aiming at attenuation of tissue injury.
In 1957, Isaacs and Lindenmann (1) first recognized the potential of a soluble and probably cell-derived factor to combat influenza virus infection and named this factor interferon [(IFN) from latin interferre, to interfere]. Since then, three subgroups of IFNs have been defined, primarily by their differential receptor usage. While the groups of type I IFN and type III IFN comprise largely agents directly limiting pathogen spread by improving cellular counter measurements, IFN-γ, the sole type II IFN, has been mainly implicated in the modulation of innate and also adaptive immune responses (2, 3). Accordingly, type I and III IFNs are key signaling molecules in viral control, and lack of both signaling pathways results in increased viral loads and disease severity. Still, there is accumulating evidence that not only lack of an antiviral response but that also an unbalanced overshooting activation of IFNs contributes to an exaggerated inflammatory reaction, tissue injury, reduced proliferative capacity, and thus enhanced disease severity (Table 1). Especially in viral infections, this effect has not only been tracked down to IFN signaling in general but specifically to the exaggerated production of key effector IFN-stimulated genes (ISGs) (4). A prominent example is the TNF-related apoptosis-inducing ligand (TRAIL) that displays an ambivalent role in viral infection (5–7) (Table 1). Whereas first identified as factor produced by immune cells in non-respiratory infection (8, 9), TRAIL is now especially well studied in influenza A virus (IAV) infection, where it is released in high amounts from bone marrow-derived macrophages upon pathogen-associated molecular patterns (PAMPs) recognition and type I IFN production (10). Macrophage-released soluble TRAIL, but also membrane-bound cell-associated TRAIL, acts via distinct receptors on infected but also on non-infected, neighboring cells. In viral infection, its preliminary role is to drive infected cells into apoptosis to limit virus spread. However, studies performed within the last decade demonstrate that TRAIL’s antiviral activity seems to be outweighed by the functional and structural damage it induces not only in infected but also in bystander cells such as uninfected cells of the alveolar epithelium (10, 11). This process is not only relevant in promoting viral disease progression but has further implications in bacterial superinfection and probably also in chronic diseases. The recognition of the ambivalent role of IFN-driven signaling in vivo is a first important step to better understand disease progression and to envision novel treatment options for primary viral respiratory infection targeting distinct host-derived signaling mediators such as TRAIL.
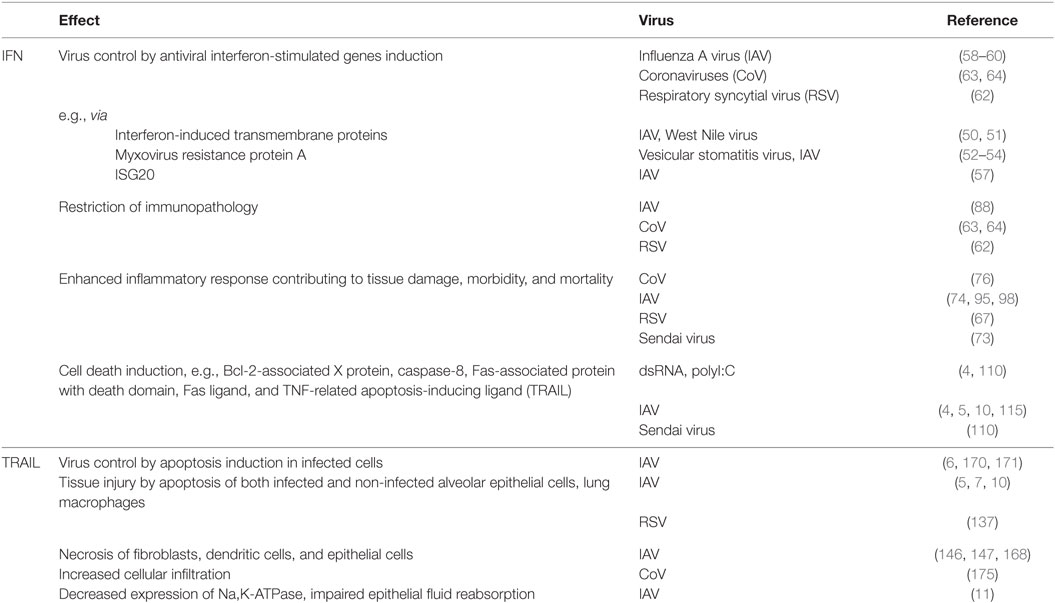
Table 1. Major effects of the interferon (IFN)/tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) signaling axis on host cells in respiratory viral infection.
It is a commonly accepted concept that—as Janeway (12) already proposed in 1989—immune activation toward invading pathogens is mounted upon recognition of PAMPs. PAMPs are evolutionary conserved biomolecules such as proteins, lipids, nitrogen bases, sugars, and complexed biomolecules such as lipoglycans that are essential to the survival of a given pathogen (13). PAMPs are recognized by distinct pattern recognition receptors (PRRs) that are germ-line encoded and—similar to PAMPs—usually show a high evolutionary conservation. The first recognized and probably most intensely studied family of PRRs are the toll-like receptors [TLRs; reviewed in Mogensen (14); Leifer and Medvedev (15)]. In viral infection, both host cell membrane-localized TLRs (TLR2, TLR4, detecting viral envelope proteins) and endosomal TLRs (TLR3, TLR7, TLR8, and TLR9, nucleic acid sensors) initiate signal transduction cascades leading to IFN production (Figure 1). TLR activation results in either myeloid differentiation factor 88 (MyD88) or TIR-domain-containing adaptor protein-inducing IFN-β (TRIF) recruitment that both trigger various downstream signaling events, eventually leading to IFN regulatory factor (IRF)3, IRF7, and NFκB nuclear translocation as well as MAP kinase and activator protein 1 (AP-1) activation (16, 17).
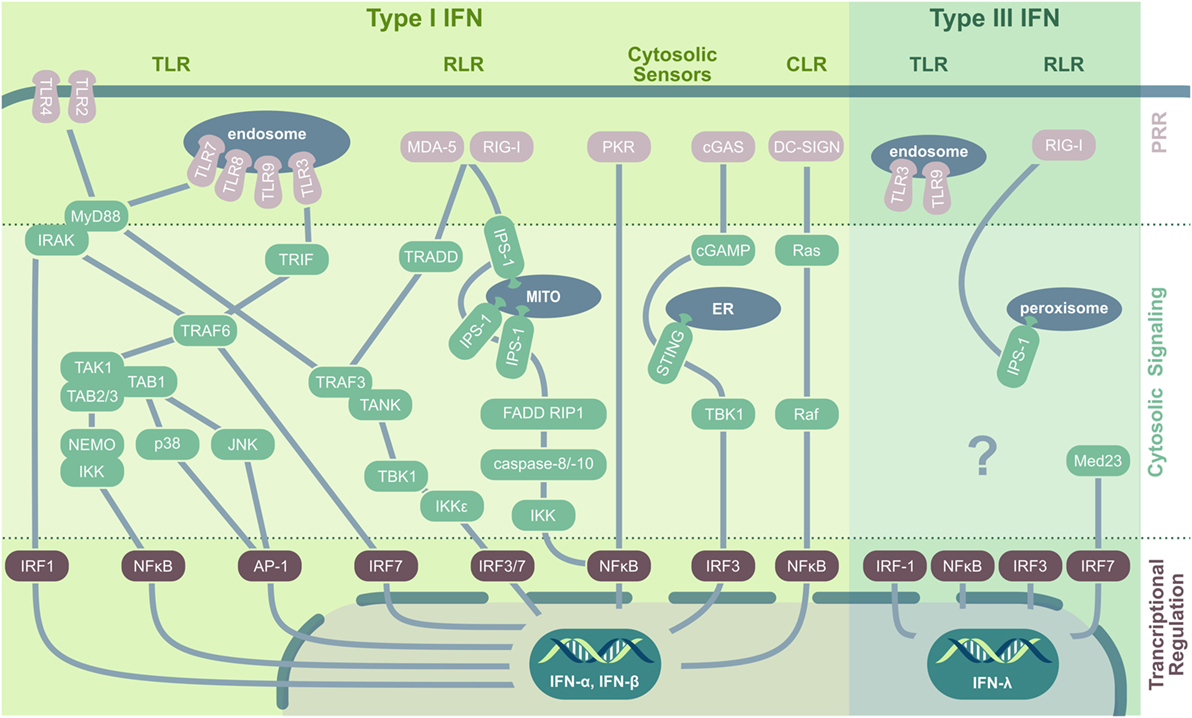
Figure 1. PRRs and their downstream signaling pathways in virus-induced IFN induction. In viral infection, type I IFNs are induced by TLR, RLR, CLR, and cytosolic nucleic acid sensors. Cell membrane-located TLRs ligate to viral envelope proteins (e.g., TLR2/herpes simplex virus), upon which they recruit MyD88. MyD88 interacts with IRAK kinase (IRAK-1, -2, or -4) that either directly activate IRF1 or interact with TRAF6, which induces IRF7 or assembles with TAK1. TAK1 forms a complex with TAB-1/-2 and -3 and subsequently either activates the MAPK kinases p38 and JNK, leading to AP-1 phosphorylation and nuclear translocation, or induces ubiquitination of NEMO followed by IκB degradation and NFκB activation. Endosomal TLRs recognizing viral nucleic acid and signal via the adaptor protein MyD88 (for TLR7/8/9) or interact with TRIF (for TLR3) followed by TRAF3, TANK, and TBK1 activation. TBK1 and IKKε then phosphorylate and activate IRF3 and IRF7. Additionally, TRIF can interact with TRAF6 to initiate TAK1 signaling. Both RLRs, RIG-I and MDA-5, recognize nucleic acid contents in the cytoplasm and stimulate the mitochondrial anchored IPS-1 for dimerization followed by TRADD recruitment that acts via TRAF3 on IRF3 and IRF7. Additionally, IPS-1 can interact with FADD and RIP1 to activate NFκB via IKK activated by caspase-8 and -10. Also PKR signaling results in NFκB activation and nuclear translocation. The dsDNA sensor cGAS produces cGAMP that activates ER-located STING that via TBK1 induces IRF3 translocation and type I IFN production. CLRs play a minor role in viral recognition; however, DC-SIGN activates the small GTPase Ras and Raf protein kinase, followed by NFκB activation. Type III IFN are induced by TLR3, TLR9, and via RIG-I and peroxisomal-resident IPS-1. Especially IRF1, but also NFκB, IRF3, and IRF7 are implicated in IFN-λ production, with the latter being stabilized by Med23. Abbreviations: TLR, toll-like receptor; RIG-I, retinoic acid-inducible gene; RLR, RIG-I-like receptors; CLR, C-type lectin receptors; PRR, pattern recognition receptor; MITO, mitochondrium; ER, endoplasmatic reticulum; IFN, interferon; MyD88, myeloid differentiation factor 88; IRAK, interleukin-1 receptor-associated kinase; IRF, IFN regulatory factor; TRIF, TIR-domain-containing adaptor protein-inducing IFN-β; TNF, tumor necrosis factor; TRAF, TNF receptor-associated factor; TAK, transforming growth factor β-activated kinase 1; TAB, TAK1-binding protein; NEMO, essential modulator; IKK, inhibitor-κB kinase; JNK, c-Jun N-terminal kinase; AP-1, activator protein 1; TANK, TRAF family member-associated NF-κB activator; TBK, TANK-binding kinase; MDA-5, melanoma differentiation antigen 5; TRADD, TNF receptor type 1-associated death protein; IPS-1, IFN-β promoter stimulation 1; FADD, Fas-associated protein with death domain; RIP1, receptor-interacting serine/threonine-protein kinase 1; PKR, protein kinase R; cGAS, cyclic GMP–AMP synthase; cGAMP, cyclic guanosine monophosphate–adenosine monophosphate; STING, stimulator of IFN genes; DC-SIGN, dendritic cell-specific ICAM3-grabbing non-integrin; AIM-2, absent in melanoma 2; Med23, mediator complex subunit 23; TRAF3, TNF receptor-associated factor 3.
Similar to endosomal TLRs, the cytosolic retinoic acid-inducible gene (RIG)-I-like receptors (RLRs) are specialized to recognize viral nucleic acid contents and are central PRRs relevant to mount an antiviral response, providing resistance to most RNA (e.g., orthomyxoviruses) and some DNA (e.g., reoviruses) viruses [reviewed in Ref. (18, 19)]. Both melanoma differentiation-associated gene 5 (MDA-5) and RIG-I recognize dsRNA, 5′-triphosphate RNA, or the synthetic analog to dsRNA, polyI:C (20, 21). Both drive the dimerization of the mitochondria-associated adaptor protein IFN-β promoter stimulation 1 (IPS-1) (also named MAVS, VISA, CARDIF). A subsequently activated cascade including TRADD (TNF receptor type 1-associated death domain protein), TRAF3 (TNF receptor-associated factor 3), and TANK (TRAF family member-associated NF-κB activator) induces the phosphorylation of IRF3 and IRF7, resulting in type I IFN production (22). The third RLR, LGP2, so far has primarily been implicated to regulate RIG-I or MDA-5 as a cofactor; however, a recent study by Stone et al. (23) demonstrated a novel, non-redundant, and independent role of LGP2 in West Nile virus infection. Another class of PRR, the nucleotide oligomerization domain (NOD)-like receptors (NLRs), has mainly been implicated in bacterial recognition (24), still several NLRs are activated as well upon virus infection. Especially, NLRP3 is known to recognize RNA of different viruses including hepatitis C virus, measles virus, influenza, and vesicular stomatitis virus (VSV) (25–28), resulting in inflammasome formation and caspase-1-dependent activation of IL-1β and IL-18 (29–31). In addition, virus infections are sensed also by a structurally diverse group of viral RNA and DNA sensors residing in the cytoplasm. These include the cyclic GMP–AMP synthase that synthesizes the second messenger cGAMP. cGAMP in turn activates stimulator of IFN genes (STING), TANK-binding kinase 1, and IRF3, triggering IFN production (32–34). Moreover, STING itself acts as a PRR and has been implicated in DNA virus recognition including HSV, adenovirus, vaccinia virus, and papilloma virus and in sensing of retroviral RNA–DNA hybrids (35) and RNA viruses after being activated by RIG-I (36, 37). Another cytosolic nucleic acid sensor, PKR, is well known for its phosphorylation of eukaryotic initiation factor 2 α (eIF2α) in response to viral dsRNA. Phosphorylation of eIF2α results in its deactivation, host translational shut-off, and the limitation of viral replication. Of note, the PKR–eIF2α-driven inhibition of protein synthesis can contribute to an IPS-1-dependent IFN-β induction (38). Furthermore, PKR has been implicated in efficient type I IFN activation by TLR3 in response to dsRNA (39) and can mediate—at least partially—activities of IRF1 (40).
In addition to type I IFNs, also type III IFNs exert antiviral activity and are widely expressed after viral recognition, being produced by most cell types including epithelial, endothelial, fibroblast, and polymorphonuclear cells [reviewed in Ref. (41, 42)]. Like type I IFNs, type III IFNs are induced in viral infection by the PRR RIG-I as well as TLR3 and TLR9 and rely on the activation of the same transcriptional activators, including IRF3, IRF7, and NFκB. These observations initially led to the conclusion that type I and type III IFN comprised two completely redundant systems to induce ISGs in response to PAMP recognition. However, more recent data suggest distinct selection mechanisms for either type I or type III IFN expression. As such, IPS-1 specifically induces IFN-λ, but not type I IFN, when located at the peroxisomal membrane instead of the mitochondrial membrane in response to RIG-I activation by reovirus, Sendai virus, or dengue virus challenge (43). Interestingly, type III IFN induction is largely independent toward AP-1 translocation, which facilitates an instantaneous induction of IFN-λ after viral recognition, highlighting it as an important immediate factor driving innate microbial defense mechanisms.
Release of IFNs upon pathogen recognition is a highly conserved mechanism—found from teleost fish to insects and mammals—to prepare the surrounding cells as well as the host defense against the invading threat (44, 45). Whereas often high-level IFN production relies on specialized sentinel cells such as macrophages or dendritic cells (DCs), mostly all cells of the multicellular organisms are able to respond to at least one type of IFN by expression of respective receptors. Receptor binding then induces a signal transduction cascade relying on the Janus kinase (JAK) and Signal Transducer and Activator of Transcription (STAT), which results in efficient transcription of a plethora of different ISGs in infected as well as bystander cells (46, 47). IFNs engage a classical canonical signal transduction cascade employing JAK/STAT molecules after binding to their respective receptors. Herein, type I IFNs ligate to their common heterodimeric receptor consisting of the IFN-α receptor (IFNAR)1 and IFNAR2 subunits, whereas type III IFNs act via a interleukin-10 receptor 2 (IL-10R2)/IFN-λ receptor 1 (IFNLR1) heterodimer that to date has been reported to be restricted in its expression to epithelial cells (48). In type I IFN signaling, IFNAR engagement leads to the activation of the receptor-associated protein tyrosine kinases JAK1 and tyrosine kinase 2 followed by the recruitment or repositioning of already associated but elsewise latent cytoplasmatic transcription factors STAT1 and STAT2. Consequently, STAT1/STAT2 are phosphorylated on conserved tyrosine residues, they disassemble, undergo conformational changes enabling their heterodimerization as well as the exposure of a nuclear localization sequence. Subsequently, the STAT1/STAT2 heterodimer translocates into the nucleus where it interacts with the IRF9 to form the trimeric IFN-stimulated factor 3 (ISGF3). ISGF3 binds cognate DNA sequences, the IFN-stimulated response elements (ISRE), finally leading to ISG induction. Also type III IFN interaction with the IL-10R2/IFNLR1 receptor complex triggers STAT1/STAT2 heterodimerization, nuclear translocation, and ISGF3 assembly (49).
Interferon signaling results in the induction of ISGs evoking different cellular responses against viral infection, both in infected as well as in non-infected cells, including direct antiviral, immune-modulatory, or cell death-inducing effects to enable an immediate and robust response to a pathogen challenge. Many ISGs directly interfere with viral replication on an intracellular level. Well-studied examples of antiviral ISGs comprise IFN-induced transmembrane proteins (IFITMs) effective in IAV, West Nile virus, and dengue virus infection (50, 51), the myxovirus resistance protein A (MxA) that interferes with VSV mRNA production and binds the IAV nucleocapsid to prevent nuclear translocation of viral genetic material (52–54), the 2-5-oligoadenylate synthase (OAS), which activates RNAse L triggering viral RNA degradation, or the PRR PKR, which besides activating the IFN response has a major impact on viral protein translation by inhibiting the eIF2α (55). More recently identified ISGs include the plasminogen activator inhibitor 1 that blocks IAV infection by inhibiting glycoprotein cleavage executed by extracellular airway proteases (56) or the antiviral ISG20 that limits IAV viral replication via its exonuclease activity most likely by interfering with the viral NP (57).
Accordingly, IFN pretreatment usually results in the establishment of an antiviral state that limits viral replication and spread from the start of infection and thus favors milder disease outcomes. IFN-α pretreatment has been demonstrated to limit viral spreading of seasonal IAV strains and thus decrease morbidity and mortality in mice, guinea pigs as well as ferrets (58–60). As shown by a study by Tumpey et al. (61), this effect can be attributed to the early induction of antiviral ISGs including MxA. Importantly, type I IFN pretreatment also dampens early replication of highly pathogenic avian influenza in ferrets (58). Also in respiratory syncytial virus (RSV) infection, treatment with recombinant IFN-α results in significantly decreased lung viral titers, alveolar inflammatory cell accumulation, and clinical disease in RSV-infected mice (62). In addition, respiratory infections caused by emerging coronaviruses (CoV) can be ameliorated by type I IFN pretreatment strategies. In an in vivo macaque model (Macaca fascicularis) of severe acute respiratory syndrome (SARS)-CoV infection, it could be demonstrated that pretreatment with pegylated IFN-α significantly diminished CoV replication and excretion and resulted in reduced pulmonary damage (63). Macaques also serve as a preclinical model for Middle East respiratory syndrome (MERS)-CoV, and similar to SARS-CoV, IFN-α in combination therapy with ribavirin reduces viral replication and severe histopathological changes (64).
In line, genetic alteration leading to an enhanced type I IFN signaling has been demonstrated to limit IAV-induced disease outcomes, as a recent study by Xing et al. (65) reported that deletion of TRIM29, a negative regulator of NEMO, which leads to NFκB induction and therefore enhanced type I IFN production, is protective in vivo in IAV-infected mice. Conversely, the genetic depletion of IFN signaling in IFN receptor-deficient mice can result in a lack of viral control, resulting in enhanced viral titers in different viral infections including RSV or IAV (66, 67). Still, it must be noted that this effect is often mild in IFNAR- or IFNLR-deficient animals, which is probably related to a certain redundancy between type I and type III IFN signaling in limiting viral spreading in epithelial cells (68). In contrast, IFNAR/IFNLR-double knockout or STAT1 knockout animals that are deficient in both type I and type III IFN signal transduction succumb more readily to infection due to excessive viral replication (69–71). Vice versa, mutations in key ISGs such as IFITM3 are associated with increased IAV disease severity in mice and humans (72).
However, IFN pretreatment and genetic loss-of-function approaches generally are not relevant to human respiratory virus-induced hospitalizations, where patients already present with ongoing respiratory infection and inflammation, and preclinical studies underline that type I IFN signaling in an already inflamed organ is rather detrimental and enhances tissue injury, and lack of type I IFN in vivo may even ameliorate disease outcome. Accordingly, in cases where the antiviral defense was not compromised (e.g., in animals with efficient type III IFN signaling) IFNAR-deficient mice infected with Sendai virus or IAV were reported to be more resistant to infection-induced morbidity and mortality (73, 74). Similarly, in Sendai virus in vivo infection, Wetzel et al. (75) showed that increased IFN-β levels in the lung homogenate correlates to increased morbidity and mortality, and also for SARS-CoV, a recent study demonstrates that high type I IFN induction in an already ongoing viral infection contributes to mortality in SARS-CoV-infected mice (76). Also for IAV infection, type I IFN application after infection has been proven to drive disease severity (74). Of note, the detrimental effects of type I IFNs were especially pronounced in mice lacking central antiviral factors, namely the IFIT protein in Sendai virus infection and MxA in IAV. Interestingly, Beilharz et al. (77) demonstrated that application of low doses of IFN-α reduces viral load, which to a certain degree led to attenuated disease progression, whereas high dose application of type I IFN contributed to morbidity (77). In line, high expression levels of ISGs have been shown to correlate to worse outcomes in ARDS patients (78). This observation corresponds to reports stating that the IFN threshold needed to induce antiviral ISGs—showing a beneficial effect in acute respiratory viral infection—is by at least 10-fold lower than the IFN dose necessary to trigger ISGs that show immunomodulatory, death-inducing, or anti-proliferative effects and thus can contribute to disease progression (79–82). Altogether, these data demonstrate that IFNs may significantly contribute to unbalanced inflammation and tissue injury during respiratory viral infection depending on expression levels and duration of IFN-related signaling events.
To date, the underlying mechanisms leading to the IFN-dependent enhanced disease progression are not fully understood but often result from a dysregulated IFN signaling response. One mode of action of IFN and IFN-stimulated ISGs is to stimulate negative feedback loops on IFN signaling. For example, suppression of JAK1 or STAT1 via specific phosphatases, expression of suppressor of cytokine signaling (SOCS)1 and SOCS3, or ubiquitination and endocytosis of the IFN receptors (83–86) desensitize cells to IFN signaling and allow recovery and the return to homeostasis after microbial challenge. As demonstrated by Bhattacharya et al. (87), the lack of IFNAR downregulation and thus the failure to initiate IFN-desensitization contributes to increased inflammatory signaling, extensive lung injury and, importantly, also impaired tissue regeneration (87). Moreover, IFNs are immunomodulatory and shape the specific responses of cells of the immune system, which has been implied to influence disease progression both positively and negatively. In a recent study, type I IFNs have been associated in the regulation of innate lymphoid immune cells (ILC)2 in IAV infection, where they—in concert with IFN-γ and IL-27—promote an ILC2-dependent restriction of immunopathology (88). Moreover, type I IFNs play an important role in stimulating the immune response driven by DCs; they stimulate the expression of MHC molecules as well as the co-stimulatory ligands CD80 and CD86 and thus activate T cell responses (89, 90). Additionally, ligand-driven activation of IFNAR enhances the proliferation of CD8 positive T cells, especially early in infection. However, late in infection, type I IFNs were also implied in decreasing T cell expansion upon SARS-CoV and arenavirus infection (76, 91), which might potentially be related to the above described desensitization upon prolonged IFN signaling and might be detrimental if initiated too early in infection. In line, Pinto et al. (92) reported an impairment of T cell responses upon type IFN induction in West Nile virus infection. In B cells, the lack of IFNAR has been demonstrated to result in enhanced release of neutralizing antibodies in IAV infection (93) implying a repressive role for type I IFN in B cell antibody production. However, immunization studies by Le Bon et al. (94) reported the necessity of IFNAR on B cells for efficient IgM and IgG production, underlining the need for further studies to understand the detailed effects of IFN-dosage and timing adaptive immunity activity upon respiratory viral infection.
Type I IFNs additionally induce the production of high levels of pro-inflammatory cytokines that have been closely linked to worsened outcomes of acute respiratory viral infection. Especially in IAV, disease severity and disease progression are linked with an overshooting, IFN-driven inflammatory response, in which further exogenous supplementation with type I IFN in fact correlates with increased morbidity and mortality (74, 95). In non-human primates, IAV infection with a highly pathogenic H5N1 isolate evokes a strong induction of type I IFN, resulting in severe lung injury by a necrotizing bronchiolitis and alveolotis (96). IFN levels in turn have been demonstrated to cause elevated pro-inflammatory cytokine levels after in vivo IAV infection and additionally, in human alveolar macrophages, the release of pro-inflammatory cytokines (e.g., MCP-1) are preceded by a robust type I IFN response (97). Importantly, also in human infection with H5N1, levels of pro-inflammatory cytokines are strongly elevated in bronchoalveolar lavage fluid, and cytokine levels have been associated with organ damage and worsened disease outcomes (98, 99). Still, it should be noted that due to strain differences in virus-elicited PRR activation and, importantly, IFN antagonism by the IAV non-structural (NS)1 protein, IFN levels and disease severity do not always directly correlate; actually, the extent to which NS1 can suppress the IFN response relates to prolonged viremia and thus can also be a determinant of virus pathogenicity both in human bronchial epithelial cells and in an in vivo model of IAV infection (100, 101). Alongside IAV, also in RSV infection the induction of high levels of pro-inflammatory cytokines has been directly related to type IFN, as RSV-infected but IFNAR-deficient mice presented with significantly diminished pro-inflammatory cytokine release, which translated into an attenuated disease course (67). Also in SARS-CoV, the late phase type I IFN induction relates to accumulation of inflammatory macrophage populations and elevated lung cytokine levels (76).
In addition to antiviral, immunomodulatory, and pro-inflammatory ISGs, IFN signaling results in the transcription and translation of cell death-inducing ISGs. In the context of viral infection, these factors provide a mode to block viral spreading and reinfection by killing those infected cells, in which the internal activation of antiviral ISGs is not sufficient to restrict viral replication. Thus, the infected cell is sacrificed to prevent the release of infectious progeny virions to limit viral spreading. However, especially in the lung, the disruption of the alveolar epithelial barrier by cell death of infected cells, but importantly also non-infected bystander cells induced by factors such as TRAIL, significantly contributes to worsened disease outcomes.
Controlled cell death or apoptosis can be induced by intrinsic and extrinsic signals. The intrinsic apoptosis pathway is initiated by diverse intracellular stimuli that influence the expression and activation of B cell lymphoma (Bcl)-2 family proteins that govern the permeabilization status of the outer mitochondrial membrane. Once cytochrome c is released from the mitochondria, it binds to the intracellular adaptor protein, apoptotic peptidase activating factor 1, forming the so-called apoptosome that in turn recruits pro-caspase-9 (102). Caspases (cysteine-aspartic proteases) exert their action by cleaving other proteins and substrates. Herein, initiator caspases such as caspase-8 and caspase-9 target other downstream caspases, whereas effector caspases, including caspase-3, -6, and -7, directly cause apoptosis by cleaving and thus inactivating or disassembling a vast array of cellular integral proteins and complexes (103). The extrinsic apoptosis pathway relies on an extracellular signal exerted by ligands of the tumor necrosis factor (TNF) receptor (TNFR) superfamily, including TRAIL, TNF-α, and Fas ligand (FasL) (104). Their ligation to their respective cell surface-expressed death receptors (DR) leads via the signal transmission by Fas-associated protein with death domain (FADD) to the activation of the initiator caspases-8 or -10, finally stimulating effector caspases including caspase-3 (105).
To date, several type I and type III IFN-induced, proapoptotic factors have been identified (106). Both caspase-4 and caspase-8 have been shown to be upregulated upon type I IFN signaling (4, 107); caspase-8 enhances the FADD-driven extrinsic apoptosis pathway, whereas the less-studied caspase-4 may promote pro-IL-1β cleavage and inflammasome-driven cell death (pyroptosis) in macrophages (108, 109). Chattopadhyay et al. (110) demonstrated that Sendai virus infection and polyI:C treatment resulted in Bcl-2-associated X protein (Bax) activation and apoptosis induction via one of the key transcription factors of IFN genes, IRF3. In addition IRF5 was reported to enhance TRAIL-dependent extrinsic apoptosis by nuclear translocation resulting in the translation of to date undefined factors that increase cell death upstream of caspase-8 activation (111). Furthermore, both RLRs, RIG-I and MDA-5, trigger the proteins Puma and Noxa that induce Bcl and thus activate the intrinsic mitochondrial apoptotic cascade (112). Also, PKR influences a cell’s susceptibility to apoptotic signals, as it was demonstrated to sensitize to the FADD/caspase-8 apoptosis pathway upon type I IFN signaling after challenge with IAV or dsRNA (4) and the OAS-RNAseL system has been suggested to contribute to IFN-α-related cell death induction, but the exact mechanisms remain to be elucidated (113). Finally, also two classical initiators of the extrinsic apoptosis cascade are induced as ISGs. Both FasL and its receptor Fas are upregulated on mRNA levels by IFN-α (114), and FasL was reported to be induced by type I IFN in IAV infection in the murine lung in vivo (115). Also, the proapoptotic factor TRAIL (or TNFSF10, Apo2L) is induced by IFN-mediated and ISGF3-executed transcriptional activation, as has been shown by Sato et al. (116), who revealed the presence of the ISRE sequence within the TRAIL promoter region (116). In IAV infection, TRAIL is released in high amounts from infected alveolar macrophages depending on a PKR- and IFN-β-driven autocrine signaling loop. Binding of IFN-β to macrophage-expressed IFNAR activates a JAK/STAT-dependent release of TRAIL, which then acts through its receptor DR5 on the alveolar epithelial cells (5, 10).
However, certain prerequisites may decrease the ability of a cell to undergo apoptosis, including a shortage in pro-caspase-8 availability, expression of cellular FADD-like IL-1β-converting enzyme-inhibitory proteins (c-FLIPs) that block FADD-driven caspase activation, inactivation, or degradation of FADD itself, or expression of CYLD, which acts as a receptor-interacting serine/threonine-protein (RIP)1 kinase de-ubiquitinase and thus stabilizes RIP1. However, in these cases IFN signaling can still promote a caspase-independent, programmed inflammatory cell death by activating the necroptosis pathway (117, 118). Necroptosis is induced by a complex formation by RIP1 and RIP3 kinases that activate both poly-ADP-ribose (PAR) polymerase 1 (PARP-1) and/or mixed lineage kinase domain-like (MLKL), leading to ATP depletion, calpain activation, PAR polymer accumulation or cell membrane permeabilization, and release of damage-associated molecular patterns, respectively [reviewed in Ref. (119, 120)]. Both a type I IFN-dependent JAK/STAT-driven activation of PKR as well as signaling by the PRR DAI (DNA-dependent activator of IRFs) initiates necroptosis via RIP1/RIP3 activation, respectively (117, 121).
Importantly, the activation of proapoptotic and pro-necroptotic pathways in respiratory infection can result in a structural disruption of the airway and the alveolar epithelial barrier, which is a major hallmark of respiratory disease and its progression to the acute respiratory distress syndrome (122, 123). In virus-induced lung injury, especially expression of TRAIL, which can initiate both apoptosis as well as necroptosis has been correlated with more severe outcomes.
As described earlier, TRAIL belongs to the superfamily of TNF ligands and has been reported to be inducible by both type I and type III IFNs. TRAIL has been found to be present in various cells of the immune system, among them natural killer (NK) cells, T cells, NK T cells, DC subsets such as IFN-γ-producing killer DCs and macrophages, and can be displayed in large amounts on the cell surface or be shed upon IFN- and/or pro-inflammatory cytokine signaling (124–126). In addition to cells of the immune system, fibroblasts have been shown to produce TRAIL after IFN-γ treatment or viral challenge. Also, club cells and the alveolar epithelium have been reported to produce TRAIL (127–130). Similar to other ligands of the TNF superfamily, TRAIL is a homotrimeric type II transmembrane protein with a conserved C-terminal extracellular domain that mediates receptor binding and can be cleaved by metalloproteinases to generate a soluble mediator (131). However, TRAIL can induce cell death also in its membrane-bound form, that is, similar to TRAIL expression levels and TRAIL shedding, upregulated by type I IFN (126). Direct cell-to-cell TRAIL–DR interactions have been demonstrated to play a role in macrophage, NK as well as CD4+ T cell-mediated induction of cellular death (132, 133).
In humans, five different binding partners for TRAIL are present: the membrane-bound DR4 (TRAIL-R1) and DR5 (TRAIL-R2) that both induce a proapoptotic signaling cascade, the membrane-bound anti-apoptotic decoy receptors (DcR)1 and DcR2, and the soluble interaction partner osteoprotegerin (134). In the murine system, only DR5 has been identified to ligate to TRAIL (135). In the human respiratory compartment, both DR4 and DR5 have been demonstrated to be present under steady-state conditions (136, 137). However, upon viral infection, cell-sensitivity to TRAIL-induced apoptosis is enhanced, which has been attributed to increased TRAIL receptor expression especially on infected cells, as DR levels are markedly increased in IAV-, adenovirus-, and paramyxovirus-infected cells in contrast to non-infected bystander cells (10, 138, 139). Of note, studies on the dependency of DR upregulation upon type I IFN signaling after IAV infection have yielded conflicting results in different strains of mice (10, 74), highlighting the complex interplay of IFN-induced cascades in a host- and tissue-specific context, whereas the exact virus- and host-specific mechanisms for DR regulation remain less well defined. Moreover, previous assumptions that also DcR expression would correlate with cell-sensitivity to TRAIL-induced cell death could not be experimentally verified (125).
Tumor necrosis factor-related apoptosis-inducing ligand ligation to the proapoptotic receptors DR4 or DR5 triggers a trimerization of the receptors. Subsequently, depending on additional stimuli, presence or absence of adaptor molecules or inhibitory proteins, different signaling pathways can be activated (Figure 2). In the classical TRAIL-dependent extrinsic apoptosis induction, the proteins RIP, TRADD, and FADD are subsequently recruited to the DR cytoplasmic domain upon TRAIL ligation (140, 141). These factors and the proapoptotic DRs all share a cytoplasmic death domain (DD), which is lacking or truncated and thus inactive in the DcR. The DD plays a central role in the concerted formation of the death-inducing signaling complex (DISC). DISC formation exposes a second functional domain of FADD, the death effector domain that is directly able to recruit pro-caspase-8 and pro-caspase-10. How exactly DISC formation induces caspase activation is still under debate. The most probable scenarios include either an autocatalytic cleavage of caspase-pro-domains enabled by the spatial proximity between pro-caspases (generated by their recruitment to DISC), by pro-caspase dimerization, or by pro-caspase conformational stabilization (125). Removal of the pro-domain of caspase-8 and caspase-10 results in the activation of the effector caspases-3 and -7, which cleave DNA fragmentation factor 45 and lead to apoptosis (142, 143). Moreover, TRAIL-binding to DR4 and DR5 can induce the JNK either via caspase-8 or recruitment of TNF receptor-associated protein 2 (TRAF2) to the DISC complex, which results in the activation of the intrinsic apoptotic cascade by Bax-dependent mitochondrial cytochrome c release (144). In addition, TRAIL signaling is also able to induce necroptosis by both activating the RIP1/RIP3 kinase downstream effectors PARP-1 and MLKL, contributing to epithelial cell death and tissue injury (145–147).
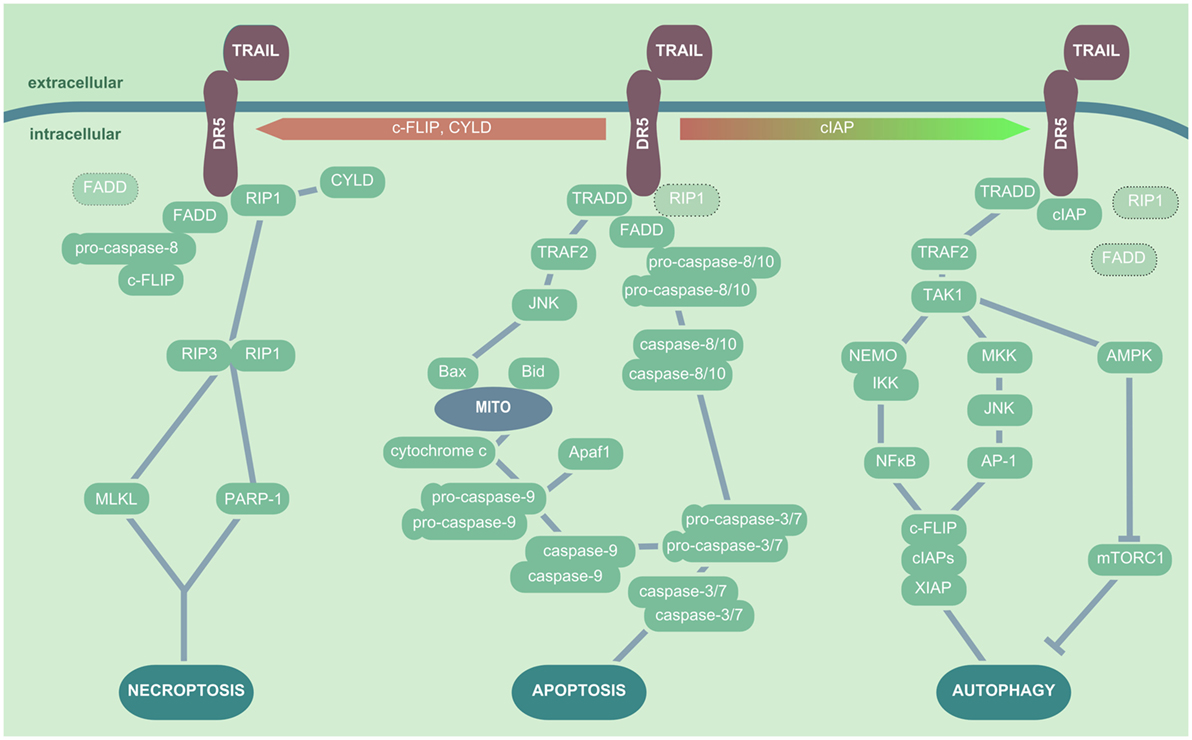
Figure 2. TRAIL/DR5-mediated cellular signaling pathways. In presence of RIP1, TRADD, and FADD, TRAIL ligation to DR5 results in apoptosis induction, which is initiated by recruitment of the pro-caspase-8 or -10 to FADD. These in turn activate the effector caspases-3 and -7, which leads to DNA fragmentation and apoptosis induction. In addition, TRADD can trigger a TRAF2- and JNK-dependent activation of Bax and subsequent release of mitochondrial cytochrome c, inducing the pro-caspase-9 activation. In the presence of CYLD, c-FLIP or absence of sufficient amounts of FADD or pro-caspase-8, TRAIL ligation to DR triggers the interaction of RIP1 and RIP3 kinase, which in turn cause cell death via induction of MLKL and/or PARP-1. In the presence of cIAPS, FADD is not recruited to DR5 upon TRAIL ligation, and TAK1 is activated by TRADD/TRAF2 interactions. TAK1 induces NEMO followed by IκB degradation and NFκB activation, as well as MKK and JNK activation leading to AP-1 nuclear translocation; both events promote the production of cytoprotective factors such as XIAP, cIAPs, and c-FLIP. Additionally, TAK1 triggers AMPK activation and thus mTORC inhibition, which results in enhanced autophagic activity. Abbreviations: AP-1, activator protein 1; TNF, tumor necrosis factor; TRAIL, TNF-related apoptosis-inducing ligand; DR5, death receptor 5; RIP1, receptor-interacting serine/threonine-protein kinase 1; TRADD, TNF receptor type 1-associated death protein; FADD, Fas-associated protein with death domain; TRAF, TNF receptor-associated protein; JNK, Janus kinase; Bax, Bcl-2-associated X protein; c-FLIP, cellular FADD-like IL-1β-converting enzyme-inhibitory proteins; RIP, receptor-interacting serine/threonine-protein; MLKL, mixed lineage kinase domain-like; PARP-1, poly-ADP-ribose (PAR) polymerase 1; cIAP, cytoprotective factors including inhibition of the autophagic machinery; XIAP, X-linked inhibitor of apoptosis protein; AMPK, AMP-activated protein kinase; mTORC, mammalian target of rapamycin complex; TRAF2, TNF receptor-associated protein 2; MKK, mitogen-activated protein kinase.
It has become apparent in recent years that TRAIL signaling is closely linked to induction of autophagy, a process generally associated with the blockade of apoptosis and necrosis. Indeed, autophagy has been reported to improve cellular survival in cell stress by catabolic removal of cytoplasmic long-lived proteins and damaged organelles. It also contributes to viral clearance and the transfer of viral material to endosomal-/lysosomal-located TLR7 or MHC class II compartments for the activation of adaptive immunity (148). Several studies outline that TRAIL ligation to DR5 can result in a TRAF2-dependent activation of TAK1 (MAP3K7) that has been attributed a central role in TRAIL-induced autophagy activation (149). TAK1 modulates the IKK-dependent translocation of NFκB, and it also induces JNK activation via mitogen-activated protein kinase. Both events lead to expression of autophagy-related factors including inhibition of the autophagic machinery (cIAP)1, cIAP2, X-linked inhibitor of apoptosis protein, and c-FLIP (150, 151). Especially, c-FLIP has been associated with desensitization of cells to TRAIL-induced apoptosis, favoring autophagy-related cascades (152). Another study revealed that upon TRAIL signaling the AMP-activated protein kinase (AMPK) is activated. AMPK in turn inhibits the mammalian target of rapamycin complex 1 that itself is an inhibitor of autophagy, thus the activation of the autophagic machinery is promoted (153). The decision if TRAIL signaling results rather in necroptotic or apoptotic cell death or in activation of autophagy seems to be dependent on the presence of cIAPs that promote RIP kinase ubiquitination and degradation (146), but also on the balance between active caspases and autophagic proteins such as Beclin-1 (154, 155). This suggests a scenario where autophagy is activated as cell protective mechanism until cell stress—as executed by enhanced TRAIL signaling or additional viral infection—increases over a threshold to favor cell death induction. Accordingly, as TRAIL signaling is not restricted to infected cells, excessive cell death activation might be limited by autophagy induction in non-infected bystander cells. However, autophagy is not only related to cell survival but can also positively affect apoptosis and induce—even if the exact mechanisms are still under debate—autosis, the autophagy-related cell death, another mode of TRAIL to trigger cell death (156, 157). Of note, autophagy activation needs to be placed into its virus-specific context, as some viruses, including Dengue virus, poliovirus, and Coxsackie B virus (158), can exploit autophagic pathways for their own replication and thus promote apoptosis and tissue injury.
As discussed above, TRAIL is a potent activator of cell death. However, its signaling outcomes can differ largely depending on its delivered form (e.g., membrane-bound versus soluble), the availability of DRs on the target cell membrane, alternate intracellular pathways that might be activated and finally the pathogen itself, as it might exploit TRAIL-induced pathways for its own survival and replication. In acute respiratory infection, TRAIL signaling is often part of an IFN-driven overshooting inflammatory reaction that promotes unspecific tissue injury and thus disease severity by increasing functional and structural changes in infected but also non-infected cells, as will be outlined below.
The release and effects of TRAIL have been especially well studied in IAV infection in the last decade. Earlier studies reported that within 3 days after infection, bronchial, bronchiolar, and alveolar epithelial cells undergo apoptosis (159). This early induction of cell death is mainly attributed to direct apoptosis induction by the virus itself, as IAV actively promotes apoptosis for efficient viral replication (160). Herein, the viral NS1 and PB-F2 proteins not only play a crucial role (161, 162) but also the viral M2 protein has been implicated in this process as it inhibits autophagy in infected cells (163). In addition, our own data revealed that later in IAV in vivo infection, the recruitment of bone marrow-derived macrophages via the CC chemokine receptor type 2 (CCR2)–CC-chemokine ligand 2 (CCL2) axis significantly contributes to alveolar cell apoptosis and structural damage of the alveolar epithelium (5). Studies by Wurzer et al. (164) had previously demonstrated that IAV promotes the production of proapoptotic factors in an auto- and paracrine fashion via NFκB transcriptional activation by IAV (164). Subsequently, Brincks et al. (6) elucidated that human peripheral blood mononuclear cell treated with IAV released TRAIL and that increased TRAIL levels correlated with type I as well as II IFN induction. Additionally, TRAIL sensitivity was increased in influenza virus-infected cells. In line, our investigations could elucidate that IAV triggers a PKR-dependent translocation of NFκB that results the production of type I IFNs. These in turn induce, via ligation to the IFNAR receptor complex, expression and shedding of TRAIL by bone marrow-derived macrophages (10). In addition, Davidson et al. (74) demonstrated that type I IFN application to IAV-infected mice increased morbidity and lung injury, which could be attributed to both DR5 and TRAIL upregulation inducing epithelial cell apoptosis. Importantly, Högner et al. also reported that the IAV strain used in these studies, A/PR8 (H1N1), which is highly pathogenic for mice, induced an approximately 800-fold induction in macrophage TRAIL expression, whereas the lower pathogenic virus A/X-31 (H3N2) only stimulated TRAIL by a factor of eight. Of note, the relation between TRAIL induction and IAV strain-specific pathogenicity also translates to the highly pathogenic avian H5N1 IAV, causing severe pneumonia in mice as well as in humans (165, 166). Moreover, human infection with both the highly pathogenic H5N1 as well as the pandemic 1918 H1N1 IAV strains are characterized by a massive influx of mononuclear phagocytes into the alveoli, which is correlated with extensive alveolar epithelial cell apoptosis (97, 167). Additionally, macrophages gained from bronchoalveolar lavages of patients presenting with ARDS caused by the pandemic H1N1/2009 virus strain showed high surface expression and release of TRAIL (10). Another recent report demonstrates that in highly pathogenic avian influenza, in addition to macrophages also the alveolar epithelium might be involved in causing elevated levels of TRAIL in the alveolar space (130). Besides its role in apoptosis, TRAIL signaling upon IAV infection has also been implicated in the induction of necroptosis in fibroblasts, DCs, and lung epithelial cells (146, 147, 168). Rodrigue-Gervais et al. (146) demonstrated that lack of cIPA2 promotes RIP3 kinase-mediated necroptosis in response to TRAIL—but also the proapoptotic factor FasL—released from hematopoietic cells. This contributed to severe lung epithelial degeneration and increased mortality, even though viral control was not compromised. Nogusa et al. (147) further elucidated that IAV-induced necroptosis depends on RIP3 kinase activation of MLKL, and that RIP3 kinase deficiency, similar to cIAP2-deficiency, increased IAV-susceptibility in vivo.
In IAV infection, as mentioned earlier, DR5 expression is elevated on infected alveolar epithelial cells, but not in non-infected cells in vivo, which might impact on TRAIL susceptibility to apoptosis induction (10). However, both infected as well as neighboring bystander cells were found to be targeted for apoptosis induction by macrophage-released TRAIL. Nonetheless, we could recently show that specifically in non-infected cells within the IAV-infected lung, TRAIL severely compromises the function of the ion channel Na,K-ATPase, which was mediated via induction of the stress kinase AMPK (11), thereby potentially revealing a cross-link to TRAIL-induced autophagic cell stress pathways in bystander cells both in vitro and in vivo. The TRAIL-induced and AMPK-mediated downregulation of the Na,K-ATPase, a major driver of vertical ion and fluid transport from the alveolar airspace toward the interstitium, resulted in a reduced capacity of IAV-infected mice to clear excessive fluid from the alveoli. Thus, TRAIL signaling contributes to intensive edema formation, a hallmark of disease in virus-induced ARDS (123). Notably, this effect of TRAIL on Na,K-ATPase expression was induced independently of cell death pathways elicited by caspases, as treatment of cells and mice with a specific caspase-3 inhibitor diminished apoptosis in alveolar epithelial cells but still allowed for the reduction of the Na,K-ATPase (11). Conclusively, treatment of IAV-infected mice with neutralizing antibodies directed against TRAIL or the abrogation of recruitment of TRAIL+ bone marrow-derived macrophages inhibited apoptosis of both non-infected and bystander cells. Thus, lung leakage due to loss of alveolar barrier function was reduced, whereas alveolar fluid clearance capacity was enhanced, resulting in reduced edema, improved survival, and outcome upon IAV challenge in vivo. However, TRAIL has also been shown to be upregulated on NK, DC, and on CD4+ and CD8+ T cells after IAV infection (169). Studies by Brincks et al. demonstrated that especially CD8+ T involved in cytotoxic T cell responses toward IAV and drive IAV-infected cells into apoptosis via TRAIL, thus contributing to efficient virus clearance (6, 170). In addition, both FasL and TRAIL are involved in DC-mediated CTL activation and cytotoxicity against IAV-infected cells (6, 171). Furthermore, studies showed delayed viral clearance upon neutralizing anti-TRAIL antibody administration (169, 172). Our data, however, demonstrate that the transfer of TRAIL-deficient bone marrow into irradiated wild-type mice, resulting in loss of TRAIL production by bone marrow-derived macrophages upon IAV infection, does not impact on the capacity to fully clear viral particles from the lung at day 7 after infection, suggesting that other compensatory mechanisms are recruited to guarantee viral clearance (10). Taken together, in IAV infection, TRAIL acts both as an important mediator of infected cell killing but particularly as a detrimental factor contributing to tissue injury and impaired inflammation resolution when released in excessive amounts by recruited immune cells.
Respiratory syncytial virus is an important cause of respiratory tract infections especially in children worldwide. Generally, there seem to be virus-elicited anti-apoptotic mechanisms active in the lung epithelium, as RSV-infected primary human airway cells show a minimal cytopathic effect (173). However, several cell lines including small airway cells, primary tracheal-bronchial cells, and A549 and HEp-2 showed increased expression of TRAIL and its ligands DR4 and DR5 in an in vitro RSV infection model (174). Moreover, soluble TRAIL released from leukocytes was elevated in the bronchoalveolar lavage fluid of patients with RSV-associated respiratory failure, suggesting that similar to IAV, TRAIL contributes to RSV-induced epithelial injury and disease progression (137).
Also in CoV respiratory tract infection, TRAIL levels, but less so FasL, have been reported to be markedly elevated (175, 176). For SARS-CoV that presents with a severe damage to both the upper and lower respiratory tract (177), especially DCs respond with a strong induction of TRAIL production, which was suggested to correlate to increased cellular lung infiltrations present in SARS-CoV patients (175). Interestingly, SARS-CoV infection drives cells into apoptosis by a PKR-driven but eIF2α-independent pathway (178), which might—similarly as seen in IAV infection—suggest a PKR-induced and autocrine/paracrine executed activation of apoptosis.
Also MERS-CoV, which causes pneumonia and respiratory failure, has been demonstrated to induce profound cell death within 24 h of infection, irrespective of viral titers produced by the infected cells. However, type I IFN expression is strongly reduced in MERS-CoV in comparison to seasonal human CoV in in vitro infection models, including human monocyte-derived macrophages, Calu-3, and human lung fibroblasts (179, 180), which might also dampen downstream TRAIL induction. Therefore, the exact mechanism by which MERS-CoV promotes cell death remains to be investigated.
Recurrently, viral infections of the respiratory tract are followed by outgrowth of colonizing Gram-positive bacteria that aggravates the course of illness. This is well documented for IAV, where “super” infections with Streptococcus pneumoniae and Staphylococcus aureus are the most frequent and increase viral pneumonia-associated morbidity and mortality (181). During the 1918 IAV pandemic, bacterial pneumonia was evident in most cases (182) and also during the recent 2009 H1N1 pandemic, coinfections were a relevant factor for severe disease in a young patient population without comorbidities (183). Interestingly, virus-induced elevation of the type I IFN response levels might promote secondary bacterial outgrowth by several mechanisms [reviewed in Ref. (184)]. In line, it has been repeatedly demonstrated that lack of type I IFN signaling results in better bacterial clearance and increased survival rates in IAV- and S. pneumoniae-superinfected mice (185–187). Herein, IFN-induced apoptosis induction as well as depletion or impaired recruitment of lymphocyte subsets necessary for bacterial control play a critical role (188, 189). Bacterial clearance from the lung has been reported to rely on sufficient phagocyte generation, recruitment, and survival. Type I IFN has been demonstrated to cause apoptosis in bone marrow-derived granulocytes, affecting the numbers of recruited neutrophils (189), but also to impair expression of the cytokines CXCL1 (or KC) and CXCL2 (or MIP-2), thus inhibiting neutrophil recruitment to the lungs with severe effects on survival of superinfected mice (185). A recent report by Schliehe et al. (190) elucidated the mechanistic background for impaired CXCL1 expression and secretion and demonstrated that type I IFNs activate the histone methyltransferase Setdb2, which in turn represses the Cxcl1 promoter and thus impairs neutrophil recruitment and bacterial clearance. Moreover, type I IFN production decreases CCL2 production, thus inhibiting macrophage recruitment, which as well has been reported to have detrimental effects on bacterial clearance and disease progression in bacterial superinfection after viral insult in vivo (186). In addition, type I IFNs also impair γδ T cell function and IL-17 release, which was shown to increase susceptibility to S. pneumoniae superinfection after IAV challenge (187). Also in S. aureus pneumonia, a robust type I IFN response is correlated to excessive morbidity and tissue injury (191). In a model of polyI:C, S. aureus (methicillin-resistant strain, MRSA) superinfection, polyI:C treatment prior to bacterial infection enhanced type I IFN levels and decreased bacterial clearance and survival (192). Furthermore, Shepardson et al. (193) demonstrated that late type I IFN induction rendered mice more susceptible to secondary bacterial pneumonia in a model of IAV–MRSA superinfection.
Only limited data are available on a direct role of TRAIL in respiratory disease progression due to bacterial superinfections. In a model of IAV–Haemophilus influenza infection, neither deficiency for CC chemokine receptor type 2, inhibiting bone marrow-derived macrophage recruitment, nor deficiency of Fas or TNFR1 impacted outcome (194). Yet, during S. pneumoniae single infection, early cell death of macrophages is thought to limit an exuberant inflammatory reaction and accordingly, a study by Steinwede et al. (195) revealed that neutrophil-derived TRAIL limits tissue injury by inducing cell death in DR5-epressing lung macrophages in bacterial mono-infection (195). In contrast, in the IAV–S. pneumoniae superinfection mouse model, IAV-induced TRAIL has a detrimental effect on overall mortality (7), as TRAIL-induced epithelial injury enhanced bacterial outgrowth of S. pneumoniae—administered at day 5 after IAV infection—markedly. Importantly, administration of anti-TRAIL neutralizing antibodies enhanced bacterial control by the host organism. Thus, the activation of IFN/TRAIL-mediated signaling in viral infection has detrimental implication for outcome of secondary bacterial infection following viral insult, rendering the IFN/TRAIL signaling axis an interesting therapeutic target not only in respiratory viral infections but also in complicating bacterial superinfection.
An increasing number of reports connect progression of chronic respiratory disease to acute respiratory virus infection or proapoptotic signaling events. In fact, TRAIL has been reported to be a critical determinant for promoting the development of chronic lung disease in early life (196); targeting TRAIL by genetic deletion or neutralizing antibody application in early-life respiratory infections ameliorated infection-induced histopathology, inflammation, as well as emphysema-like alveolar enlargement and lung function. Furthermore, TRAIL was also shown to play a role in the development of allergy and asthma. TRAIL is not only elevated in the sputum of asthmatic patients but has also been reported to be highly expressed in an experimental mouse model of asthma, where it induces CCL20 secretion by bronchial epithelial cells, thus promoting TH2 cell responses and airway hyperreactivity (197).
In COPD, acute exacerbations driven by viral and bacterial infection are a major factor increasing both mortality and morbidity, and both influenza and S. pneumoniae have been identified among the most common causes of COPD exacerbations (198). Indeed, primary bronchial epithelial cells isolated from subjects with COPD show an impaired production of type I IFN (199), which has been implied in the enhanced susceptibility of COPD patients to respiratory infections; however, even in absence of high IFN induction, both an abnormally elevated loss of alveolar epithelial cells due to apoptosis as well as elevated TRAIL and DR5 levels were reported (200), implying a possible link between viral/bacterial induction of TRAIL and acute exacerbations in COPD. TRAIL induction has also been directly linked to cigarette-smoke exposure, a common cause of COPD, and TRAIL deficiency resulted in decreased pulmonary inflammation and emphysema-like alveolar enlargement in vivo (201). Moreover, increased levels of both TRAIL and DR5 were associated to impaired lung function and increased systemic inflammation in human COPD patients (202). While alveolar epithelial cell death is closely connected to idiopathic pulmonary fibrosis (IPF), TRAIL and its receptors DR4 and DR5 in AEC were shown to be upregulated in IPF lungs (129). Also, in pulmonary arterial hypertension virus infection is considered to be a possible risk factor (203), and pulmonary hypertension has been reported to be a side effect of prolonged treatment with type I IFN (204, 205). In line, TRAIL has been closely linked to disease progression in pulmonary hypertension. TRAIL has been found to be increased within pulmonary vascular lesions of patients with pulmonary hypertension (206) and also in a mouse model of hypoxia-induced pulmonary hypertension, levels of soluble TRAIL correlated with right ventricular systolic pressure, right ventricular hypertrophy, and pathologic alterations (33, 34). Importantly, neutralizing antibody-treatment against TRAIL showed positive effects on survival while reducing pulmonary vascular remodeling (207). Notably, the extent to which infection-induced TRAIL release causes or exacerbates chronic lung disease or in how far TRAIL production in chronic lung diseases affects susceptibility to respiratory viral and complicating bacterial infection remains to be elucidated.
Respiratory viral infections are major causative agents for lung injury and ARDS; however, in many cases antivirals are not sufficient to limit disease (208). Besides the fact that most viruses are subject to strong selective pressures that favor quickly evolving, drug-resistant virus variants, recent advances in understanding the processes that contribute to tissue injury and ARDS highlight a crucial role of immune-related, IFN-driven events. Therefore, novel therapeutic strategies often aim to improve the outcome of severe respiratory infection by modulating host cell responses; however, to date, clinical trials trying to improve severe viral infections or ARDS outcomes by targeting host pathways have not resulted in approval of new drugs (122).
Of note, for establishment of such therapies it has to be considered that the timing and intensity of induction and amplification as well as of dampening and termination of the IFN-driven immune response needs to precisely match the pathogen- and organ-specific requirements of a given infection. A non-controlled regulation of these processes may lead to either an unrestricted pathogen spreading or, on the other extreme, to an overshooting inflammatory response, including the increased production of pro-inflammatory and proapoptotic mediators, elevated levels of recruited immune cells, and/or aberrant repair processes. Notably, both too low and too high levels of IFN-induced effects facilitate disease progression with a possible increase of fatal outcomes in ARDS patients (78). Accordingly, preclinical in vivo studies of IFN-directed therapies yielded seemingly adverse results, depending on the context, timing, and dosage of IFN modulation. However, in multiple settings of acute respiratory viral infection, studies demonstrate that an exaggerated signaling derived from type I IFN in an already inflamed tissue contributes to worsened outcomes, and importantly, might favor secondary bacterial superinfection [e.g., Ref. (75, 76, 209)]. Interestingly, Davidson et al. (209) demonstrated that type III IFN release upon influenza challenge—in contrast to type I IFN induction—does not trigger an unbalanced inflammatory response that critically contributes to respiratory disease progression in vivo, highlighting it as a possible therapeutic option in IAV-induced lung injury. Most likely, this effect derives from the lack of the IFN-λR1/IL-10R2 receptor complex, but presence of IFNAR, on immune cells, including bone marrow-derived macrophages. Nonetheless, other reports identify IFN-λ as a driver of macrophage polarization to an inflammatory M1 phenotype (41) that has been attributed to further promote an overshooting inflammatory response, highlighting the need for further studies of type III IFN biology in pathogen-associated disease progression.
As generally IFN-directed therapeutic approaches target various downstream signaling events that might both act beneficially as well as detrimentally on viral replication and pathogenesis, a further approach is to address specific ISGs that primarily show detrimental effects on disease progression. As outlined above, TRAIL or its downstream signaling events might comprise a suitable target for adjunct therapies in addition to antivirals. Accordingly, our own data in a preclinical mouse model of IAV infection demonstrate a clear benefit of the systemic application of neutralizing antibodies against TRAIL at days 3 and 5 postinfection for lung injury, morbidity, and mortality (10, 11). Targeting TRAIL as a major determinant of disease severity in respiratory viral infections including IAV, but also RSV and CoV, may yield therapeutic approaches that are superior to IFN-directed strategies, as they seemingly do not bear the risk of compromising host defense. Yet, it should be thoroughly excluded that blocking TRAIL-induced cell death of infected cells will not lead to an overwhelming viral spreading, especially as reports on viral loads upon TRAIL inhibition in preclinical models of IAV are controversial (6, 10, 170). Accordingly, additional studies are needed to understand how and to which extent virus-infected cells can be killed or viral spreading can be controlled by other means in the absence of TRAIL. Moreover, targeting pathways and signaling hubs downstream of TRAIL/DRs, such as AMPK (11), in a well-timed and lung compartment-specific way, may open new therapeutic avenues but requires more detailed preclinical studies on efficacies and side effects. A valid approach might be the use of a combination therapy of such a treatment together with a classical antiviral drug therapy limiting viral replication; however, exact dosage, timing, kinetics, and application routes remain to be defined.
CP and SH performed bibliographic research and drafted the manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
This work was supported by the German Research Foundation (SFB-TR84 B2, SFB1021 C05, KFO309 P2/P8, EXC147), by the German Center for Lung Research (DZL), and by the German Center for Infection Research (DZIF).
1. Isaacs A, Lindenmann J. Virus interference. I. The interferon. Proc R Soc London (1957) 7(5):429–38. doi:10.1098/rspb.1957.0048
2. Schoenborn JR, Wilson CB. Regulation of interferon-γ during innate and adaptive immune responses. Adv Immunol (2007) 96:41–101. doi:10.1016/S0065-2776(07)96002-2
3. Young HA, Hardy KJ. Role of interferon-gamma in immune cell regulation. J Leukoc Biol (1995) 58(4):373–81.
4. Balachandran S, Roberts PC, Kipperman T, Bhalla KN, Compans RW, Archer DR, et al. Alpha/beta interferons potentiate virus-induced apoptosis through activation of the FADD/caspase-8 death signaling pathway. J Virol (2000) 74(3):1513–23. doi:10.1128/JVI.74.3.1513-1523.2000
5. Herold S, Steinmueller M, Von Wulff W, Cakarova L, Pinto R, Pleschka S, et al. Lung epithelial apoptosis in influenza virus pneumonia: the role of macrophage-expressed TNF-related apoptosis-inducing ligand. J Exp Med (2008) 205(13):3065–77. doi:10.1084/jem.20080201
6. Brincks EL, Katewa A, Kucaba TA, Griffith TS, Legge KL. CD8 T cells utilize TRAIL to control influenza virus infection. J Immunol (2008) 181(7):4918–25. doi:10.4049/jimmunol.181.10.7428-a
7. Ellis GT, Davidson S, Crotta S, Branzk N, Papayannopoulos V, Wack A. TRAIL+ monocytes and monocyte-related cells cause lung damage and thereby increase susceptibility to influenza-Streptococcus pneumoniae coinfection. EMBO Rep (2015) 16(9):1203–18. doi:10.15252/embr.201540473
8. Miura Y, Koyanagi Y, Mizusawa H. TNF-related apoptosis-inducing ligand (TRAIL) induces neuronal apoptosis in HIV-encephalopathy. J Med Dental Sci (2003) 50(1):17–25.
9. Herbeuval JP, Grivel JC, Boasso A, Hardy AW, Chougnet C, Dolan MJ, et al. CD4+ T-cell death induced by infectious and noninfectious HIV-1: role of type 1 interferon-dependent, TRAIL/DR5-mediated apoptosis. Blood (2005) 106(10):3524–31. doi:10.1182/blood-2005-03-1243
10. Högner K, Wolff T, Pleschka S, Plog S, Gruber AD, Kalinke U, et al. Macrophage-expressed IFN-β contributes to apoptotic alveolar epithelial cell injury in severe influenza virus pneumonia. PLoS Pathog (2013) 9(2):e1003188. doi:10.1371/journal.ppat.1003188
11. Peteranderl C, Morales-Nebreda L, Selvakumar B, Lecuona E, Vadász I, Morty RE, et al. Macrophage-epithelial paracrine crosstalk inhibits lung edema clearance during influenza infection. J Clin Invest (2016) 126(4):1566–80. doi:10.1172/JCI83931
12. Janeway CA. Approaching the asymptote? Evolution and revolution in immunology. Cold Spring Harb Symp Quant Biol (1989) 54(0):1–13. doi:10.1101/SQB.1989.054.01.003
13. Mahla RS, Reddy MC, Raghava Prasad DV, Kumar H. Sweeten PAMPs: role of sugar complexed PAMPs in innate immunity and vaccine biology. Front Immunol (2013) 4:248. doi:10.3389/fimmu.2013.00248
14. Mogensen TH. Pathogen recognition and inflammatory signaling in innate immune defenses. Clin Microbiol Rev (2009) 22(2):240–73. doi:10.1128/CMR.00046-08
15. Leifer CA, Medvedev AE. Molecular mechanisms of regulation of toll-like receptor signaling. J Leukocy Biol (2016) 100(5):927–41. doi:10.1189/jlb.2MR0316-117RR
16. Wang C, Deng L, Hong M, Akkaraju GR, Inoue J, Chen ZJ. TAK1 is a ubiquitin-dependent kinase of MKK and IKK. Nature (2001) 412(6844):346–51. doi:10.1038/35085597
17. Oeckinghaus A, Ghosh S. The NF-kappaB family of transcription factors and its regulation. Cold Spring Harb Perspect Biol (2009) 1(4):a000034. doi:10.1101/cshperspect.a000034
18. Gack MU. Mechanisms of RIG-I-like receptor activation and manipulation by viral pathogens. J Virol (2014) 88(10):5213–6. doi:10.1128/JVI.03370-13
19. Kell AM, Gale M. RIG-I in RNA virus recognition. Virology (2015) 479–480:110–21. doi:10.1016/j.virol.2015.02.017
20. Kang D-C, Gopalkrishnan RV, Wu Q, Jankowsky E, Pyle AM, Fisher PB. MDA-5: an interferon-inducible putative RNA helicase with double-stranded RNA-dependent ATPase activity and melanoma growth-suppressive properties. Proc Natl Acad Sci U S A (2002) 99(2):637–42. doi:10.1073/pnas.022637199
21. Miyoshi K, Cui Y, Riedlinger G, Robinson P, Lehoczky J, Zon L, et al. Structure of the mouse Stat 3/5 Locus: evolution from Drosophila to zebrafish to mouse. Genomics (2001) 71(2):150–5. doi:10.1006/geno.2000.6433
22. Kawai T, Takahashi K, Sato S, Coban C, Kumar H, Kato H, et al. IPS-1, an adaptor triggering RIG-I- and MDA5-mediated type I interferon induction. Nat Immunol (2005) 6(10):981–8. doi:10.1038/ni1243
23. Stone AEL, Wilkins C, Green R, Gale MJ. RIG-I-like receptors control unique innate immune responses following West Nile virus infection. J Immunol (2016) 196(1 Suppl):203.9–203.9.
24. Chaput C, Sander LE, Suttorp N, Opitz B. NOD-like receptors in lung diseases. Front Immunol (2013) 4:393. doi:10.3389/fimmu.2013.00393
25. Chen W, Xu Y, Li H, Tao W, Xiang Y, Huang B, et al. HCV genomic RNA activates the NLRP3 inflammasome in human myeloid cells. PLoS One (2014) 9(1):e84953. doi:10.1371/journal.pone.0084953
26. Komune N, Ichinohe T, Ito M, Yanagi Y. Measles virus V protein inhibits NLRP3 inflammasome-mediated interleukin-1 secretion. J Virol (2011) 85(24):13019–26. doi:10.1128/JVI.05942-11
27. Ichinohe T, Pang IK, Iwasaki A. Influenza virus activates inflammasomes via its intracellular M2 ion channel. Nat Immunol (2010) 11(5):404–10. doi:10.1038/ni.1861
28. Rajan JV, Rodriguez D, Miao EA, Aderem A. The NLRP3 inflammasome detects encephalomyocarditis virus and vesicular stomatitis virus infection. J Virol (2011) 85(9):4167–72. doi:10.1128/JVI.01687-10
29. Allen IC, Scull MA, Moore CB, Holl EK, McElvania-TeKippe E, Taxman DJ, et al. The NLRP3 inflammasome mediates in vivo innate immunity to influenza A virus through recognition of viral RNA. Immunity (2009) 30(4):556–65. doi:10.1016/j.immuni.2009.02.005
30. Lawrence TM, Hudacek AW, de Zoete MR, Flavell RA, Schnell MJ. Rabies virus is recognized by the NLRP3 inflammasome and activates interleukin-1β release in murine dendritic cells. J Virol (2013) 87(10):5848–57. doi:10.1128/JVI.00203-13
31. Gram AM, Frenkel J, Ressing ME. Inflammasomes and viruses: cellular defence versus viral offence. J General Virol (2012) 93(Pt_10):2063–75. doi:10.1099/vir.0.042978-0
32. Wu J, Chen ZJ. Innate immune sensing and signaling of cytosolic nucleic acids. Annu Rev Immunol (2014) 32(1):461–88. doi:10.1146/annurev-immunol-032713-120156
33. Liu H, Yang E, Lu X, Zuo C, He Y, Jia D, et al. Serum levels of tumor necrosis factor-related apoptosis-inducing ligand correlate with the severity of pulmonary hypertension. Pulm Pharmacol Ther (2015) 33:39–46. doi:10.1016/j.pupt.2015.06.002
34. Liu S, Cai X, Wu J, Cong Q, Chen X, Li T, et al. Phosphorylation of innate immune adaptor proteins MAVS, STING, and TRIF induces IRF3 activation. Science (2015) 347(6227):aaa2630. doi:10.1126/science.aaa2630
35. Mankan AK, Schmidt T, Chauhan D, Goldeck M, Höning K, Gaidt M, et al. Cytosolic RNA:DNA hybrids activate the cGAS-STING axis. EMBO J (2014) 33(24):2937–46. doi:10.15252/embj.201488726
36. Liu Y, Goulet M-L, Sze A, Hadj SB, Belgnaoui SM, Lababidi RR, et al. RIG-I mediated STING up-regulation restricts HSV-1 infection. J Virol (2016) 90(20):9406–19. doi:10.1128/JVI.00748-16
37. Ran Y, Shu H-B, Wang Y-Y. MITA/STING: a central and multifaceted mediator in innate immune response. Cytokine Growth Factor Rev (2014) 25(6):631–9. doi:10.1016/j.cytogfr.2014.05.003
38. McAllister CS, Taghavi N, Samuel CE. Protein kinase PKR amplification of interferon β induction occurs through initiation factor eIF-2α-mediated translational control. J Biol Chem (2012) 287(43):36384–92. doi:10.1074/jbc.M112.390039
39. Jiang Z, Zamanian-Daryoush M, Nie H, Silva AM, Williams BRG, Li X. Poly(I-C)-induced toll-like receptor 3 (TLR3)-mediated activation of NFkappa B and MAP kinase is through an interleukin-1 receptor-associated kinase (IRAK)-independent pathway employing the signaling components TLR3-TRAF6-TAK1-TAB 2-PKR. J Biol Chem (2003) 278(19):16713–9. doi:10.1074/jbc.M300562200
40. Kirchhoff S, Koromilas AE, Schaper F, Grashoff M, Sonenberg N, Hauser H. IRF-1 induced cell growth inhibition and interferon induction requires the activity of the protein kinase PKR. Oncogene (1995) 11(3):439–45.
41. Egli A, Santer DM, O’Shea D, Tyrrell DL, Houghton M. The impact of the interferon-lambda family on the innate and adaptive immune response to viral infections. Emerg Microbes Infect (2014) 3(7):e51. doi:10.1038/emi.2014.51
42. Odendall C, Kagan JC. The unique regulation and functions of type III interferons in antiviral immunity. Curr Opin Virol (2015) 12(June):47–52. doi:10.1016/j.coviro.2015.02.003
43. Odendall C, Dixit E, Stavru F, Bierne H, Franz KM, Durbin AF, et al. Diverse intracellular pathogens activate type III interferon expression from peroxisomes. Nat Immunol (2014) 15(8):717–26. doi:10.1038/ni.2915
44. Zou J, Secombes CJ. Teleost fish interferons and their role in immunity. Dev Comp Immunol (2011) 35(12):1376–87. doi:10.1016/j.dci.2011.07.001
45. tenOever BR, Benjamin R, Aguado LC, Schmid S, Sachs D, Shim JV, et al. The evolution of antiviral defense systems. Cell Host Microbe (2016) 19(2):142–9. doi:10.1016/j.chom.2016.01.006
46. Schneider WM, Chevillotte MD, Rice CM. Interferon-stimulated genes: a complex web of host defenses. Annu Rev Immunol (2014) 32(1):513–45. doi:10.1146/annurev-immunol-032713-120231
47. von Recum-Knepper J, Sadewasser A, Weinheimer VK, Wolff T. Fluorescence-activated cell sorting-based analysis reveals an asymmetric induction of interferon-stimulated genes in response to seasonal influenza A virus. J Virol (2015) 89(14):6982–93. doi:10.1128/JVI.00857-15
48. Sommereyns C, Paul S, Staeheli P, Michiels T. IFN-lambda (IFN-λ) is expressed in a tissue-dependent fashion and primarily acts on epithelial cells in vivo. PLoS Pathog (2008) 4(3):e1000017. doi:10.1371/journal.ppat.1000017
49. Zhou Z, Hamming OJ, Ank N, Paludan SR, Nielsen AL, Hartmann R. Type III interferon (IFN) induces a type I IFN-like response in a restricted subset of cells through signaling pathways involving both the Jak-STAT pathway and the mitogen-activated protein kinases. J Virol (2007) 81(14):7749–58. doi:10.1128/JVI.02438-06
50. Brass AL, Huang I-C, Benita Y, John SP, Krishnan MN, Feeley EM, et al. The IFITM proteins mediate cellular resistance to influenza A H1N1 virus, West Nile virus, and dengue virus. Cell (2009) 139(7):1243–54. doi:10.1016/j.cell.2009.12.017
51. Gorman MJ, Poddar S, Farzan M, Diamond MS. The interferon-stimulated gene Ifitm3 restricts West Nile virus infection and pathogenesis. J Virol (2016) 90(18):8212–25. doi:10.1128/JVI.00581-16
52. Staeheli P, Pavlovic J. Inhibition of vesicular stomatitis virus mRNA synthesis by human MxA protein. J Virol (1991) 65(8):4498–501.
53. Matzinger SR, Carroll TD, Dutra JC, Ma Z-M, Miller CJ. Myxovirus resistance gene A (MxA) expression suppresses influenza A virus replication in alpha interferon-treated primate cells. J Virol (2013) 87(2):1150–8. doi:10.1128/JVI.02271-12
54. Xiao H, Killip MJ, Staeheli P, Randall RE, Jackson D. The human interferon-induced MxA protein inhibits early stages of influenza A virus infection by retaining the incoming viral genome in the cytoplasm. J Virol (2013) 87(23):13053–8. doi:10.1128/JVI.02220-13
55. Iwasaki A, Pillai PS. Innate immunity to influenza virus infection. Nat Rev Immunol (2014) 14(5):315–28. doi:10.1038/nri3665
56. Dittmann M, Hoffmann H-H, Scull MA, Gilmore RH, Bell KL, Ciancanelli M, et al. A serpin shapes the extracellular environment to prevent influenza A virus maturation. Cell (2015) 160(4):631–43. doi:10.1016/j.cell.2015.01.040
57. Qu H, Li J, Yang L, Sun L, Liu W, He H. Influenza A virus-induced expression of ISG20 inhibits viral replication by interacting with nucleoprotein. Virus Genes (2016) 52(6):759–67. doi:10.1007/s11262-016-1366-2
58. Kugel D, Kochs G, Obojes K, Roth J, Kobinger GP, Kobasa D, et al. Intranasal administration of alpha interferon reduces seasonal influenza A virus morbidity in ferrets. J Virol (2009) 83(8):3843–51. doi:10.1128/JVI.02453-08
59. Steel J, Staeheli P, Mubareka S, García-Sastre A, Palese P, Lowen AC. Transmission of pandemic H1N1 influenza virus and impact of prior exposure to seasonal strains or interferon treatment. J Virol (2010) 84(1):21–6. doi:10.1128/JVI.01732-09
60. Takano K, Jensen KE, Warren J. Passive interferon protection in mouse influenza. Proc Soc Exp Biol Med (1963) 114(2):472–5. doi:10.3181/00379727-114-28706
61. Tumpey TM, Szretter KJ, Van Hoeven N, Katz JM, Kochs G, Haller O, et al. The Mx1 gene protects mice against the pandemic 1918 and highly lethal human H5N1 influenza viruses. J Virol (2007) 81(19):10818–21. doi:10.1128/JVI.01116-07
62. Guerrero-Plata A, Baron S, Poast JS, Adegboyega PA, Casola A, Garofalo RP. Activity and regulation of alpha interferon in respiratory syncytial virus and human metapneumovirus experimental infections. J Virol (2005) 79(16):10190–9. doi:10.1128/JVI.79.16.10190-10199.2005
63. Haagmans BL, Kuiken T, Martina BE, Fouchier RAM, Rimmelzwaan GF, van Amerongen G, et al. Pegylated interferon-α protects type 1 pneumocytes against SARS coronavirus infection in macaques. Nat Med (2004) 10(3):290–3. doi:10.1038/nm1001
64. Falzarano D, de Wit E, Rasmussen AL, Feldmann F, Okumura A, Scott DP, et al. Treatment with interferon-α2b and ribavirin improves outcome in MERS-CoV-infected rhesus macaques. Nat Med (2013) 19(10):1313–7. doi:10.1038/nm.3362
65. Xing J, Weng L, Yuan B, Wang Z, Jia L, Jin R, et al. Identification of a role for TRIM29 in the control of innate immunity in the respiratory tract. Nat Immunol (2016) 17(12):1373–80. doi:10.1038/ni.3580
66. Müller U, Steinhoff U, Reis LF, Hemmi S, Pavlovic J, Zinkernagel RM, et al. Functional role of type I and type II interferons in antiviral defense. Science (1994) 264(5167):1918–21. doi:10.1126/science.8009221
67. Goritzka M, Durant LR, Pereira C, Salek-Ardakani S, Openshaw PJM, Johansson C. Alpha/Beta interferon receptor signaling amplifies early proinflammatory cytokine production in the lung during respiratory syncytial virus infection. J Virol (2014) 88(11):6128–36. doi:10.1128/JVI.00333-14
68. Davidson S, Maini MK, Wack A. Disease-promoting effects of type I interferons in viral, bacterial, and coinfections. J Interferon Cytokine Res (2015) 35(4):252–64. doi:10.1089/jir.2014.0227
69. Mordstein M, Kochs G, Dumoutier L, Renauld J-C, Paludan SR, Klucher K, et al. Interferon-λ contributes to innate immunity of mice against influenza A virus but not against hepatotropic viruses. PLoS Pathog (2008) 4(9):e1000151. doi:10.1371/journal.ppat.1000151
70. Mordstein M, Neugebauer E, Ditt V, Jessen B, Rieger T, Falcone V, et al. Lambda interferon renders epithelial cells of the respiratory and gastrointestinal tracts resistant to viral infections. J Virol (2010) 84(11):5670–7. doi:10.1128/JVI.00272-10
71. Crotta S, Davidson S, Mahlakoiv T, Desmet CJ, Buckwalter MR, Albert ML, et al. Type I and type III interferons drive redundant amplification loops to induce a transcriptional signature in influenza-infected airway epithelia. PLoS Pathog (2013) 9(11):e1003773. doi:10.1371/journal.ppat.1003773
72. Everitt AR, Clare S, Pertel T, John SP, Wash RS, Smith SE, et al. IFITM3 restricts the morbidity and mortality associated with influenza. Nature (2012) 484(7395):519–23. doi:10.1038/nature10921
73. Lopez CB, Yount JS, Hermesh T, Moran TM. Sendai virus infection induces efficient adaptive immunity independently of type I interferons. J Virol (2006) 80(9):4538–45. doi:10.1128/JVI.80.9.4538-4545.2006
74. Davidson S, Crotta S, McCabe TM, Wack A. Pathogenic potential of interferon Aβ in acute influenza infection. Nat Commun (2014) 5:3864. doi:10.1038/ncomms4864
75. Wetzel JL, Fensterl V, Sen GC. Sendai virus pathogenesis in mice is prevented by Ifit2 and exacerbated by interferon. J Virol (2014) 88(23):13593–601. doi:10.1128/JVI.02201-14
76. Channappanavar R, Fehr AR, Vijay R, Mack M, Zhao J, Meyerholz DK, et al. Dysregulated type I interferon and inflammatory monocyte-macrophage responses cause lethal pneumonia in SARS-CoV-infected mice. Cell Host Microbe (2016) 19(2):181–93. doi:10.1016/j.chom.2016.01.007
77. Beilharz MW, Cummins JM, Bennett AL. Protection from lethal influenza virus challenge by oral type 1 interferon. Biochem Biophys Res Commun (2007) 355(3):740–4. doi:10.1016/j.bbrc.2007.02.019
78. Nick JA, Caceres SM, Kret JE, Poch KR, Strand M, Faino AV, et al. Extremes of interferon-stimulated gene expression associate with worse outcomes in the acute respiratory distress syndrome. PLoS One (2016) 11(9):e0162490. doi:10.1371/journal.pone.0162490
79. Leaman DW, Chawla-Sarkar M, Jacobs B, Vyas K, Sun Y, Ozdemir A, et al. Novel growth and death related interferon-stimulated genes (ISGs) in melanoma: greater potency of IFN-beta compared with IFN-alpha2. J Interferon Cytokine Res (2003) 23(12):745–56. doi:10.1089/107999003772084860
80. Coelho LF, Magno de Freitas Almeida G, Mennechet FJ, Blangy A, Uzé G. Interferon-alpha and -beta differentially regulate osteoclastogenesis: role of differential induction of chemokine CXCL11 expression. Proc Natl Acad Sci U S A (2005) 102(33):11917–22. doi:10.1073/pnas.0502188102
81. Samarajiwa SA, Forster S, Auchettl K, Hertzog PJ. INTERFEROME: the database of interferon regulated genes. Nucleic Acids Res (2009) 37(Database):D852–7. doi:10.1093/nar/gkn732
82. Levin D, Harari D, Schreiber G. Stochastic receptor expression determines cell fate upon interferon treatment. Mol Cell Biol (2011) 31(16):3252–66. doi:10.1128/MCB.05251-11
83. ten Hoeve J, de Jesus Ibarra-Sanchez M, Fu Y, Zhu W, Tremblay M, David M, et al. Identification of a nuclear Stat1 protein tyrosine phosphatase. Mol Cell Biol (2002) 22(16):5662–8. doi:10.1128/MCB.22.16.5662-5668.2002
84. David M, Chen HE, Goelz S, Larner AC, Neel BG. Differential regulation of the alpha/beta interferon-stimulated Jak/Stat pathway by the SH2 domain-containing tyrosine phosphatase SHPTP1. Mol Cell Biol (1995) 15(12):7050–8. doi:10.1128/MCB.15.12.7050
85. Yoshimura A, Naka T, Kubo M. SOCS proteins, cytokine signalling and immune regulation. Nat Rev Immunol (2007) 7(6):454–65. doi:10.1038/nri2093
86. Kumar KG, Barriere H, Carbone CJ, Liu J, Swaminathan G, Xu P, et al. Site-specific ubiquitination exposes a linear motif to promote interferon-alpha receptor endocytosis. J Cell Biol (2007) 179(5):935–50. doi:10.1083/jcb.200706034
87. Bhattacharya S, Katlinski KV, Reichert M, Takano S, Brice A, Zhao B, et al. Triggering ubiquitination of IFNAR1 protects tissues from inflammatory injury. EMBO Mol Med (2014) 6(3):384–97. doi:10.1002/emmm.201303236
88. Duerr CU, McCarthy CDA, Mindt BC, Rubio M, Meli AP, Pothlichet J, et al. Type I interferon restricts type 2 immunopathology through the regulation of group 2 innate lymphoid cells. Nat Immunol (2015) 17(1):65–75. doi:10.1038/ni.3308
89. Ito T, Amakawa R, Inaba M, Ikehara S, Inaba K, Fukuhara S. Differential regulation of human blood dendritic cell subsets by IFNs. J Immunol (2001) 166(5):2961–9. doi:10.4049/JIMMUNOL.166.5.2961
90. Montoya M, Schiavoni G, Mattei F, Gresser I, Belardelli F, Borrow P, et al. Type I interferons produced by dendritic cells promote their phenotypic and functional activation. Blood (2002) 99(9):3263–71. doi:10.1182/blood.V99.9.3263
91. Marshall HD, Urban SL, Welsh RM. Virus-induced transient immune suppression and the inhibition of T cell proliferation by type I interferon. J Virol (2011) 85(12):5929–39. doi:10.1128/JVI.02516-10
92. Pinto AK, Daffis S, Brien JD, Gainey MD, Yokoyama WM, Sheehan KCF, et al. A temporal role of type I interferon signaling in CD8+ T cell maturation during acute West Nile virus infection. PLoS Pathog (2011) 7(12):e1002407. doi:10.1371/journal.ppat.1002407
93. Price GE, Gaszewska-Mastarlarz A, Moskophidis D. The role of alpha/beta and gamma interferons in development of immunity to influenza A virus in mice. J Virol (2000) 74(9):3996–4003. doi:10.1128/JVI.74.9.3996-4003.2000
94. Le Bon A, Thompson C, Kamphuis E, Durand V, Rossmann C, Kalinke U, et al. Cutting edge: enhancement of antibody responses through direct stimulation of B and T cells by type I IFN. J Immunol (2006) 176(4):2074–8. doi:10.4049/jimmunol.176.4.2074
95. Ramos I, Fernandez-Sesma A. Modulating the innate immune response to influenza A virus: potential therapeutic use of anti-inflammatory drugs. Front Immunol (2015) 6:361. doi:10.3389/fimmu.2015.00361
96. Baskin CR, Bielefeldt-Ohmann H, Tumpey TM, Sabourin PJ, Long JP, García-Sastre A, et al. Early and sustained innate immune response defines pathology and death in nonhuman primates infected by highly pathogenic influenza virus. Proc Natl Acad Sci U S A (2009) 106(9):3455–60. doi:10.1073/pnas.0813234106
97. Cheung CY, Poon LLM, Lau AS, Luk W, Lau YL, Shortridge KF, et al. Induction of proinflammatory cytokines in human macrophages by influenza A (H5N1) viruses: a mechanism for the unusual severity of human disease? Lancet (2002) 360(9348):1831–7. doi:10.1016/S0140-6736(02)11772-7
98. Peiris JS, Yu WC, Leung CW, Cheung CY, Ng WF, Nicholls JM, et al. Re-emergence of fatal human influenza A subtype H5N1 disease. Lancet (2004) 363(9409):617–9. doi:10.1016/S0140-6736(04)15595-5
99. de Jong MD, Simmons CP, Thanh TT, Hien VM, Smith GJ, Chau TN, et al. Fatal outcome of human influenza A (H5N1) is associated with high viral load and hypercytokinemia. Nat Med (2006) 12(10):1203–7. doi:10.1038/nm1477
100. Seo SH, Hoffmann S, Webster RG. Lethal H5N1 influenza viruses escape host anti-viral cytokine responses. Nat Med (2002) 8(9):950–4. doi:10.1038/nm757
101. Zeng H, Goldsmith C, Thawatsupha P, Chittaganpitch M, Waicharoen S, Zaki S, et al. Highly pathogenic avian influenza H5N1 viruses elicit an attenuated type I interferon response in polarized human bronchial epithelial cells. J Virol (2007) 81(22):12439–49. doi:10.1128/JVI.01134-07
102. Li P, Nijhawan D, Budihardjo I, Srinivasula SM, Ahmad M, Alnemri ES, et al. Cytochrome c and dATP-dependent formation of Apaf-1/Caspase-9 complex initiates an apoptotic protease cascade. Cell (1997) 91(4):479–89. doi:10.1016/S0092-8674(00)80434-1
103. Cohen GM. Caspases: the executioners of apoptosis. Biochem J (1997) 26(Pt 1):1–16. doi:10.1042/bj3260001
104. Locksley RM, Killeen N, Lenardo MJ. The TNF and TNF receptor superfamilies: integrating mammalian biology. Cell (2001) 104(4):487–501. doi:10.1016/S0092-8674(01)00237-9
105. McIlwain DR, Berger T, Mak TW. Caspase functions in cell death and disease. Cold Spring Harb Perspect Biol (2013) 5(4):a008656. doi:10.1101/cshperspect.a008656
106. Chawla-Sarkar M, Lindner DJ, Liu Y-F, Williams BR, Sen GC, Silverman RH, et al. Apoptosis and interferons: role of interferon-stimulated genes as mediators of apoptosis. Apoptosis (2003) 8(3):237–49. doi:10.1023/A:1023668705040
107. Stang MT, Armstrong M, Watson G, Liu Y, Sung K-Y, Yim J. IRF-1 mediated apoptosis is FADD and caspase 8 dependent. J Am Coll Surg (2006) 203(3):S82. doi:10.1016/j.jamcollsurg.2006.05.214
108. Sollberger G, Strittmatter GE, Kistowska M, French LE, Beer H-D. Caspase-4 is required for activation of inflammasomes. J Immunol (2012) 188(4):1992–2000. doi:10.4049/jimmunol.1101620
109. Trinchieri G. Type I interferon: friend or foe? J Exp Med (2010) 207(10):2053–63. doi:10.1084/jem.20101664
110. Chattopadhyay S, Marques JT, Yamashita M, Peters KL, Smith K, Desai A, et al. Viral apoptosis is induced by IRF-3-mediated activation of Bax. EMBO Journal (2010) 29(10):1762–73. doi:10.1038/emboj.2010.50
111. Hu G, Barnes BJ. IRF-5 is a mediator of the death receptor-induced apoptotic signaling pathway. J Biol Chem (2009) 284(5):2767–77. doi:10.1074/jbc.M804744200
112. Besch R, Poeck H, Hohenauer T, Senft D, Häcker G, Berking C, et al. Proapoptotic signaling induced by RIG-I and MDA-5 results in type I interferon-independent apoptosis in human melanoma cells. J Clin Invest (2009) 119(8):2399–411. doi:10.1172/JCI37155
113. Zhou A, Paranjape J, Brown TL, Nie H, Naik S, Dong B, et al. Interferon action and apoptosis are defective in mice devoid of 2’,5’-oligoadenylate-dependent RNase L. EMBO J (1997) 16(21):6355–63. doi:10.1093/emboj/16.21.6355
114. Kaser A, Nagata S, Tilg H. Interferon α augments activation-induced T cell death by upregulation of Fas (CD95/APO-1) and Fas ligand expression. Cytokine (1999) 11(10):736–43. doi:10.1006/cyto.1998.0484
115. Fujikura D, Chiba S, Muramatsu D, Kazumata M, Nakayama Y, Kawai T, et al. Type-I interferon is critical for FasL expression on lung cells to determine the severity of influenza. PLoS One (2013) 8(2):e55321. doi:10.1371/journal.pone.0055321
116. Sato K, Hida S, Takayanagi H, Yokochi T, Kayagaki N, Takeda K, et al. Antiviral response by natural killer cells through TRAIL gene induction by IFN-alpha/beta. Eur J Immunol (2001) 31(11):3138–46. doi:10.1002/1521-4141(200111)31:11<3138::AID-IMMU3138>3.0.CO;2-B
117. Thapa RJ, Nogusa S, Chen P, Maki JL, Lerro A, Andrake M, et al. Interferon-induced RIP1/RIP3-mediated necrosis requires PKR and is licensed by FADD and caspases. Proc Natl Acad Sci USA (2013) 110(33):E3109–18. doi:10.1073/pnas.1301218110
118. McComb S, Cessford E, Alturki NA, Joseph J, Shutinoski B, Startek JB, et al. Type-I interferon signaling through ISGF3 complex is required for sustained Rip3 activation and necroptosis in macrophages. Proc Natl Acad Sci U S A (2014) 111(31):E3206–13. doi:10.1073/pnas.1407068111
119. Baines CP. Role of the mitochondrion in programmed necrosis. Front Physiol (2010) 1:156. doi:10.3389/fphys.2010.00156
120. Pasparakis M, Vandenabeele P. Necroptosis and its role in inflammation. Nature (2015) 517(7534):311–20. doi:10.1038/nature14191
121. Thapa RJ, Ingram JP, Ragan KB, Nogusa S, Boyd DF, Benitez AA, et al. DAI senses influenza A virus genomic RNA and activates RIPK3-dependent cell death. Cell Host Microbe (2016) 20(5):674–81. doi:10.1016/j.chom.2016.09.014
122. Gonzales JN, Lucas R, Verin AD. The acute respiratory distress syndrome: mechanisms and perspective therapeutic approaches. Austin J Vasc Med (2015) 2(1):1009.
123. Matthay MA, Ware LB, Zimmerman GA. The acute respiratory distress syndrome. J Clin Invest (2012) 122(8):2731–40. doi:10.1172/JCI60331
124. Chan CW, Crafton E, Fan H-N, Flook J, Yoshimura K, Skarica M, et al. Interferon-producing killer dendritic cells provide a link between innate and adaptive immunity. Nat Med (2006) 12(2):207–13. doi:10.1038/nm1352
125. Shepard B, Badley A. The biology of TRAIL and the role of TRAIL-based therapeutics in infectious diseases. Antiinfect Agents Med Chem (2009) 8(2):87–101. doi:10.2174/187152109787846060
126. Ehrlich S, Infante-Duarte C, Seeger B, Zipp F. Regulation of soluble and surface-bound TRAIL in human T cells, B cells, and monocytes. Cytokine (2003) 24(6):244–53. doi:10.1016/S1043-4666(03)00094-2
127. Sedger LM, Shows DM, Blanton RA, Peschon JJ, Goodwin RG, Cosman D, et al. IFN-gamma mediates a novel antiviral activity through dynamic modulation of TRAIL and TRAIL receptor expression. J Immunol (1999) 163(2):920–6.
128. Akram KM, Lomas NJ, Spiteri MA, Forsyth NR. Club cells inhibit alveolar epithelial wound repair via TRAIL-dependent apoptosis. Eur Respir J (2013) 41(3):683–94. doi:10.1183/09031936.00213411
129. Akram KM, Lomas NJ, Forsyth NR, Spiteri MA. Alveolar epithelial cells in idiopathic pulmonary fibrosis display upregulation of TRAIL, DR4 and DR5 expression with simultaneous preferential over-expression of pro-apoptotic marker p53. Int J Clin Exp Pathol (2014) 7(2):552–64.
130. Hui KP, Li HS, Cheung MC, Chan RW, Yuen KM, Mok CK, et al. Highly pathogenic avian influenza H5N1 virus delays apoptotic responses via activation of STAT3. Sci Rep (2016) 6:28593. doi:10.1038/srep28593
131. Wiley SR, Schooley K, Smolak PJ, Din WS, Huang C-P, Nicholl JK, et al. Identification and characterization of a new member of the TNF Family that induces apoptosis. Immunity (1995) 3(6):673–82. doi:10.1016/1074-7613(95)90057-8
132. Sheard MA, Asgharzadeh S, Liu Y, Lin T-Y, Wu H-W, Ji L, et al. Membrane-bound TRAIL supplements natural killer cell cytotoxicity against neuroblastoma cells. J Immunother (2013) 36(5):319–29. doi:10.1097/CJI.0b013e31829b4493
133. Falschlehner C, Schaefer U, Walczak H. Following TRAIL’s path in the immune system. Immunology (2009) 127(2):145–54. doi:10.1111/j.1365-2567.2009.03058.x
134. LeBlanc HN, Ashkenazi A. Apo2L/TRAIL and its death and decoy receptors. Cell Death Differ (2003) 10(1):66–75. doi:10.1038/sj.cdd.4401187
135. Wu GS, Burns TF, Zhan Y, Alnemri ES, El-Deiry WS. Molecular cloning and functional analysis of the mouse homologue of the KILLER/DR5 tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) death receptor. Cancer Res (1999) 59(12):2770–5.
136. Spierings DC, de Vries EG, Vellenga E, van den Heuvel FA, Koornstra JJ, Wesseling J, et al. Tissue distribution of the death ligand TRAIL and its receptors. J Histochem Cytochem (2004) 52(6):821–31. doi:10.1369/jhc.3A6112.2004
137. Bem RA, Bos AP, Wösten-van Asperen RM, Bruijn M, Lutter R, Sprick MR, et al. Potential role of soluble TRAIL in epithelial injury in children with severe RSV infection. Am J Respir Cell Mol Biol (2010) 42(6):697–705. doi:10.1165/rcmb.2009-0100OC
138. Sträter J, Hinz U, Walczak H, Mechtersheimer G, Koretz K, Herfarth C, et al. Expression of TRAIL and TRAIL receptors in colon carcinoma: TRAIL-R1 is an independent prognostic parameter. Clin Cancer Res (2002) 8(12):3734–40.
139. Kirshner JR, Karpova AY, Kops M, Howley PM. Identification of TRAIL as an interferon regulatory factor 3 transcriptional target. J Virol (2005) 79(14):9320–4. doi:10.1128/JVI.79.14.9320-9324.2005
140. Schneider P, Thome M, Burns K, Bodmer JL, Hofmann K, Kataoka T, et al. TRAIL receptors 1 (DR4) and 2 (DR5) signal FADD-dependent apoptosis and activate NF-kappaB. Immunity (1997) 7(6):831–6. doi:10.1016/S1074-7613(00)80401-X
141. Chaudhary PM, Eby M, Jasmin A, Bookwalter A, Murray J, Hood L. Death receptor 5, a new member of the TNFR family, and DR4 induce FADD-dependent apoptosis and activate the NF-kappaB pathway. Immunity (1997) 7(6):821–30. doi:10.1016/S1074-7613(00)80400-8
142. Zhivotovsky B. Caspases: the enzymes of death. Essays Biochem (2003) 39(January):25–40. doi:10.1042/bse0390025
143. Liu X, Li P, Widlak P, Zou H, Luo X, Garrard WT, et al. The 40-kDa subunit of DNA fragmentation factor induces DNA fragmentation and chromatin condensation during apoptosis. Proc Natl Acad Sci U S A (1998) 95(15):8461–6. doi:10.1073/pnas.95.15.8461
144. Cummins N, Badley A. The TRAIL to viral pathogenesis: the good, the bad and the ugly. Curr Mol Med (2009) 9(4):495–505. doi:10.2174/156652409788167078
145. Jouan-Lanhouet S, Arshad MI, Piquet-Pellorce C, Martin-Chouly C, Le Moigne-Muller G, Van Herreweghe F, et al. TRAIL induces necroptosis involving RIPK1/RIPK3-dependent PARP-1 activation. Cell Death Differ (2012) 19(12):2003–14. doi:10.1038/cdd.2012.90
146. Rodrigue-Gervais IG, Labbé K, Dagenais M, Dupaul-Chicoine J, Champagne C, Morizot A, et al. Cellular inhibitor of apoptosis protein cIAP2 protects against pulmonary tissue necrosis during influenza virus infection to promote host survival. Cell Host Microbe (2014) 15(1):23–35. doi:10.1016/j.chom.2013.12.003
147. Nogusa S, Thapa RJ, Dillon CP, Liedmann S, Oguin TH, Ingram JP, et al. RIPK3 activates parallel pathways of MLKL-driven necroptosis and FADD-mediated apoptosis to protect against influenza A virus. Cell Host Microbe (2016) 20(1):13–24. doi:10.1016/j.chom.2016.05.011
148. Shoji-Kawata S, Levine B. Autophagy, antiviral immunity, and viral countermeasures. Biochim Biophys Acta (2009) 1793(9):1478–84. doi:10.1016/j.bbamcr.2009.02.008
149. Goodall ML, Fitzwalter BE, Zahedi S, Wu M, Rodriguez D, Mulcahy-Levy JM, et al. The autophagy machinery controls cell death switching between apoptosis and necroptosis. Dev Cell (2016) 37(4):337–49. doi:10.1016/j.devcel.2016.04.018
150. He W, Wang Q, Xu J, Xu X, Padilla MT, Ren G, et al. Attenuation of TNFSF10/TRAIL-induced apoptosis by an autophagic survival pathway involving TRAF2- and RIPK1/RIP1-mediated MAPK8/JNK activation. Autophagy (2012) 8(12):1811–21. doi:10.4161/auto.22145
151. Varfolomeev E, Maecker H, Sharp D, Lawrence D, Renz M, Vucic D, et al. Molecular determinants of kinase pathway activation by Apo2 ligand/tumor necrosis factor-related apoptosis-inducing ligand. J Biol Chem (2005) 280(49):40599–608. doi:10.1074/jbc.M509560200
152. Lluis JM, Nachbur U, Cook WD, Gentle IE, Moujalled D, Moulin M, et al. TAK1 Is required for survival of mouse fibroblasts treated with TRAIL, and does so by NF-κB dependent induction of cFLIPL. PLoS One (2010) 5(1):e8620. doi:10.1371/journal.pone.0008620
153. Herrero-Martín G, Høyer-Hansen M, García-García C, Fumarola C, Farkas T, López-Rivas A, et al. TAK1 activates AMPK-dependent cytoprotective autophagy in TRAIL-treated epithelial cells. EMBO J (2009) 28(6):677–85. doi:10.1038/emboj.2009.8
154. Hou W, Han J, Lu C, Goldstein LA, Rabinowich H. Autophagic degradation of active caspase-8. Autophagy (2010) 6(7):891–900. doi:10.4161/auto.6.7.13038
155. Bray K, Mathew R, Lau A, Kamphorst JJ, Fan J, Chen J, et al. Autophagy suppresses RIP kinase-dependent necrosis enabling survival to mTOR inhibition. PLoS One (2012) 7(7):e41831. doi:10.1371/journal.pone.0041831
156. Ojha R, Ishaq M, Singh S. Caspase-mediated crosstalk between autophagy and apoptosis: mutual adjustment or matter of dominance. J Cancer Res Ther (2015) 11(3):514. doi:10.4103/0973-1482.163695
157. Liu Y, Levine B. Autosis and autophagic cell death: the dark side of autophagy. Cell Death Differ (2015) 22(3):367–76. doi:10.1038/cdd.2014.143
158. Gyurkovska V, Ivanovska N. Distinct roles of TNF-related apoptosis-inducing ligand (TRAIL) in viral and bacterial infections: from pathogenesis to pathogen clearance. Inflamm Res (2016) 65(6):427–37. doi:10.1007/s00011-016-0934-1
159. Mori I, Komatsu T, Takeuchi K, Nakakuki K, Sudo M, Kimura Y. In vivo induction of apoptosis by influenza virus. J Gen Virol (1995) 76:2869–73. doi:10.1099/0022-1317-76-11-2869
160. Herold S, Ludwig S, Pleschka S, Wolff T. Apoptosis signaling in influenza virus propagation, innate host defense, and lung injury. J Leukoc Biol (2012) 92(1):75–82. doi:10.1189/jlb.1011530
161. Zamarin D, Ortigoza MB, Palese P. Influenza A virus PB1-F2 protein contributes to viral pathogenesis in mice. J Virol (2006) 80(16):7976–83. doi:10.1128/JVI.00415-06
162. Varga ZT, Ramos I, Hai R, Schmolke M, García-Sastre A, Fernandez-Sesma A, et al. The influenza virus protein PB1-F2 inhibits the induction of type I interferon at the level of the MAVS adaptor protein. PLoS Pathog (2011) 7(6):e1002067. doi:10.1371/journal.ppat.1002067
163. Gannagé M, Dormann D, Albrecht R, Dengjel J, Torossi T, Rämer PC, et al. Matrix protein 2 of influenza A virus blocks autophagosome fusion with lysosomes. Cell Host Microbe (2009) 6(4):367–80. doi:10.1016/j.chom.2009.09.005
164. Wurzer WJ, Ehrhardt C, Pleschka S, Berberich-Siebelt F, Wolff T, Walczak H, et al. NF-kappaB-dependent induction of tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) and Fas/FasL is crucial for efficient influenza virus propagation. J Biol Chem (2004) 279(30):30931–7. doi:10.1074/jbc.M403258200
165. Uiprasertkul M, Kitphati R, Puthavathana P, Kriwong R, Kongchanagul A, Ungchusak K, et al. Apoptosis and pathogenesis of avian influenza A (H5N1) virus in humans. Emerg Infect Dis (2007) 13(5):708–12. doi:10.3201/eid1305.060572
166. Szretter KJ, Gangappa S, Lu X, Smith C, Shieh W-J, Zaki SR, et al. Role of host cytokine responses in the pathogenesis of avian H5N1 influenza viruses in mice. J Virol (2007) 81(6):2736–44. doi:10.1128/JVI.02336-06
167. Perrone LA, Plowden JK, García-Sastre A, Katz JM, Tumpey TM. H5N1 and 1918 pandemic influenza virus infection results in early and excessive infiltration of macrophages and neutrophils in the lungs of mice. PLoS Pathog (2008) 4(8):e1000115. doi:10.1371/journal.ppat.1000115
168. Hartmann B, Albrecht R, Marjanovic N, Sealfon S. Regulation of necroptosis as a viral immune antagonistic mechanism (INM3P.361). J Immunol (2015) 194(1 Suppl):127.2–127.2.
169. Ishikawa E, Nakazawa M, Yoshinari M, Minami M. Role of tumor necrosis factor-related apoptosis-inducing ligand in immune response to influenza virus infection in mice. J Virol (2005) 79(12):7658–63. doi:10.1128/JVI.79.12.7658-7663.2005
170. Brincks EL, Gurung P, Langlois RA, Hemann EA, Legge KL, Griffith TS. The magnitude of the T cell response to a clinically significant dose of influenza virus is regulated by TRAIL. J Immunol (2011) 187(9):4581–8. doi:10.4049/jimmunol.1002241
171. Chaperot L, Blum A, Manches O, Lui G, Angel J, Molens J-P, et al. Virus or TLR agonists induce TRAIL-mediated cytotoxic activity of plasmacytoid dendritic cells. J Immunol (2006) 176(1):248–55. doi:10.4049/JIMMUNOL.176.1.248
172. Zhou J, Law HKW, Cheung CY, Ng IHY, Malik Peiris JS, Lau YL. Functional tumor necrosis factor-related apoptosis-inducing ligand production by avian influenza virus-infected macrophages. J Infect Dis (2006) 193(7):945–53. doi:10.1086/500954
173. Zhang L, Peeples ME, Boucher RC, Collins PL, Pickles RJ. Respiratory syncytial virus infection of human airway epithelial cells is polarized, specific to ciliated cells, and without obvious cytopathology. J Virol (2002) 76(11):5654–66. doi:10.1128/JVI.76.11.5654-5666.2002
174. Kotelkin A, Prikhod’ko EA, Cohen JI, Collins PL, Bukreyev A. Respiratory syncytial virus infection sensitizes cells to apoptosis mediated by tumor necrosis factor-related apoptosis-inducing ligand. J Virol (2003) 77(17):9156–72. doi:10.1128/JVI.77.17.9156-9172.2003
175. Law HKW, Cheung CY, Sia SF, Chan YO, Malik Peiris JS, Lau YL. Toll-like receptors, chemokine receptors and death receptor ligands responses in SARS coronavirus infected human monocyte derived dendritic cells. BMC Immunol (2009) 10:35. doi:10.1186/1471-2172-10-35
176. Marfè G, Tafani M, Fiorito F, Pagnini U, Iovane G, De Martino L. Involvement of FOXO transcription factors, TRAIL-FasL/Fas, and sirtuin proteins family in canine coronavirus type II-induced apoptosis. PLoS One (2011) 6(11):e27313. doi:10.1371/journal.pone.0027313
177. Chen J, Xie YQ, Zhang HT, Wan JW, Wang DT, Lu ZH, et al. Lung pathology of severe acute respiratory syndrome. Zhongguo Yi Xue Ke Xue Yuan Xue Bao (2003) 25(3):360–2.
178. Krähling V, Stein DA, Spiegel M, Weber F, Mühlberger E. Severe acute respiratory syndrome coronavirus triggers apoptosis via protein kinase r but is resistant to its antiviral activity. J Virol (2009) 83(5):2298–309. doi:10.1128/JVI.01245-08
179. Lau SK, Lau CC, Chan KH, Li CP, Chen H, Jin DY, et al. Delayed induction of proinflammatory cytokines and suppression of innate antiviral response by the novel Middle East respiratory syndrome coronavirus: implications for pathogenesis and treatment. J Gen Virol (2013) 94(Pt 12):2679–90. doi:10.1099/vir.0.055533-0
180. Zhou J, Chu H, Li C, Wong BH, Cheng ZS, Poon VK, et al. Active replication of Middle East respiratory syndrome coronavirus and aberrant induction of inflammatory cytokines and chemokines in human macrophages: implications for pathogenesis. J Infect Dis (2014) 209(9):1331–42. doi:10.1093/infdis/jit504
181. Rynda-Apple A, Robinson KM, Alcorn JF. Influenza and bacterial superinfection: illuminating the immunologic mechanisms of disease. Infect Immun (2015) 83(10):3764–70. doi:10.1128/IAI.00298-15
182. Sheng Z-M, Chertow DS, Ambroggio X, McCall S, Przygodzki RM, Cunningham RE, et al. Autopsy series of 68 cases dying before and during the 1918 influenza pandemic peak. Proc Natl Acad Sci U S A (2011) 108(39):16416–21. doi:10.1073/pnas.1111179108
183. Blyth CC, Webb SA, Kok J, Dwyer DE, van Hal SJ, Foo H, et al. The impact of bacterial and viral co-infection in severe influenza. Influenza Other Respir Viruses (2013) 7(2):168–76. doi:10.1111/j.1750-2659.2012.00360.x
184. McNab F, Mayer-Barber K, Sher A, Wack A, O’Garra A. Type I interferons in infectious disease. Nat Rev Immunol (2015) 15(2):87–103. doi:10.1038/nri3787
185. Shahangian A, Chow EK, Tian X, Kang JR, Ghaffari A, Liu SY, et al. Type I IFNs mediate development of postinfluenza bacterial pneumonia in mice. J Clin Invest (2009) 119(7):1910–20. doi:10.1172/JCI35412
186. Nakamura S, Davis KM, Weiser JN. Synergistic stimulation of type I interferons during influenza virus coinfection promotes Streptococcus pneumoniae colonization in mice. J Clin Invest (2011) 121(9):3657–65. doi:10.1172/JCI57762
187. Li W, Moltedo B, Moran TM. Type I interferon induction during influenza virus infection increases susceptibility to secondary Streptococcus pneumoniae infection by negative regulation of T cells. J Virol (2012) 86(22):12304–12. doi:10.1128/JVI.01269-12
188. Rayamajhi M, Humann J, Penheiter K, Andreasen K, Lenz LL. Induction of IFN-alphabeta enables Listeria monocytogenes to suppress macrophage activation by IFN-gamma. J Exp Med (2010) 207(2):327–37. doi:10.1084/jem.20091746
189. Navarini AA, Recher M, Lang KS, Georgiev P, Meury S, Bergthaler A, et al. Increased susceptibility to bacterial superinfection as a consequence of innate antiviral responses. Proc Natl Acad Sci USA (2006) 103(42):15535–9. doi:10.1073/pnas.0607325103
190. Schliehe C, Flynn EK, Vilagos B, Richson U, Swaminathan S, Bosnjak B, et al. The methyltransferase Setdb2 mediates virus-induced susceptibility to bacterial superinfection. Nat Immunol (2015) 16(1):67–74. doi:10.1038/ni.3046
191. Parker D, Planet PJ, Soong G, Narechania A, Prince A. Induction of type I interferon signaling determines the relative pathogenicity of Staphylococcus aureus strains. PLoS Pathog (2014) 10(2):e1003951. doi:10.1371/journal.ppat.1003951
192. Tian X, Xu F, Yi Lung W, Meyerson C, Ali A, Cheng G, et al. Poly I:C enhances susceptibility to secondary pulmonary infections by Gram-positive bacteria. PLoS One (2012) 7(9):e41879. doi:10.1371/journal.pone.0041879
193. Shepardson KM, Larson K, Morton RV, Prigge JR, Schmidt EE, Huber VC, et al. Differential type I interferon signaling is a master regulator of susceptibility to postinfluenza bacterial superinfection. MBio (2016) 7(3):e506–16. doi:10.1128/mBio.00506-16
194. Lee LN, Dias P, Han D, Yoon S, Shea A, Zakharov V, et al. A mouse model of lethal synergism between influenza virus and Haemophilus influenzae. Am J Pathol (2010) 176(2):800–11. doi:10.2353/ajpath.2010.090596
195. Steinwede K, Henken S, Bohling J, Maus R, Ueberberg B, Brumshagen C, et al. TNF-related apoptosis-inducing ligand (TRAIL) exerts therapeutic efficacy for the treatment of pneumococcal pneumonia in mice. J Exp Med (2012) 209(11):1937–52. doi:10.1084/jem.20120983
196. Starkey MR, Nguyen DH, Essilfie AT, Kim RY, Hatchwell LM, Collison AM, et al. Tumor necrosis factor-related apoptosis-inducing ligand translates neonatal respiratory infection into chronic lung disease. Mucosal Immunol (2014) 7(3):478–88. doi:10.1038/mi.2013.65
197. Weckmann M, Collison A, Simpson JL, Kopp MV, Wark PAB, Smyth MJ, et al. Critical link between TRAIL and CCL20 for the activation of TH2 cells and the expression of allergic airway disease. Nat Med (2007) 13(11):1308–15. doi:10.1038/nm1660
198. Shimizu K, Yoshii Y, Morozumi M, Chiba N, Ubukata K, Uruga H, et al. Viral and bacterial etiologies in acute exacerbation of COPD detected using a multiplex real-time polymerase chain reaction. Eur Respir J (2014) 44(Suppl):58.
199. Hsu AC-Y, Parsons K, Moheimani F, Knight DA, Hansbro PM, Fujita T, et al. Impaired antiviral stress granule and IFN-β enhanceosome formation enhances susceptibility to influenza infection in chronic obstructive pulmonary disease epithelium. Am J Respir Cell Mol Biol (2016) 55(1):117–27. doi:10.1165/rcmb.2015-0306OC
200. Morissette MC, Parent J, Milot J. Alveolar epithelial and endothelial cell apoptosis in emphysema: what we know and what we need to know. Int J Chron Obstruct Pulmon Dis (2009) 4:19–31. doi:10.2147/COPD.S4432
201. Haw TJ, Starkey MR, Nair PM, Pavlidis S, Liu G, Nguyen DH, et al. A pathogenic role for tumor necrosis factor-related apoptosis-inducing ligand in chronic obstructive pulmonary disease. Mucosal Immunol (2016) 9(4):859–72. doi:10.1038/mi.2015.111
202. Wen F, Wu Y, Shen Y, Zhang J, Wan C, Wang T, et al. Increased serum TRAIL and DR5 levels correlated with lung function and inflammation in stable COPD patients. Int J Chron Obstruct Pulmon Dis (2015) 10:2405–12. doi:10.2147/COPD.S92260
203. Cool CD, Voelkel NF, Bull T. Viral infection and pulmonary hypertension: is there an association? Expert Rev Respir Med (2011) 5(2):207–16. doi:10.1586/ers.11.17
204. Dhillon S, Kaker A, Dosanjh A, Japra D, VanThiel DH. Irreversible pulmonary hypertension associated with the use of interferon alpha for chronic hepatitis C. Dig Dis Sci (2010) 55(6):1785–90. doi:10.1007/s10620-010-1220-7
205. Savale L, Chaumais M-C, O’Connell C, Humbert M, Sitbon O. Interferon-induced pulmonary hypertension. Curr Opin Pulm Med (2016) 22(5):415–20. doi:10.1097/MCP.0000000000000307
206. Lawrie A. The role of the osteoprotegerin/tumor necrosis factor related apoptosis-inducing ligand axis in the pathogenesis of pulmonary arterial hypertension. Vascul Pharmacol (2014) 63(3):114–7. doi:10.1016/j.vph.2014.10.002
207. Hameed AG, Arnold ND, Chamberlain J, Pickworth JA, Paiva C, Dawson S, et al. Inhibition of tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) reverses experimental pulmonary hypertension. J Exp Med (2012) 209(11):1919–35. doi:10.1084/jem.20112716
208. Luks AM. Ventilatory strategies and supportive care in acute respiratory distress syndrome. Influenza Other Respir Viruses (2013) 7(Suppl 3):8–17. doi:10.1111/irv.12178
Keywords: interferon, interferon-stimulated genes, tumor necrosis factor-related apoptosis-inducing ligand, acute lung injury, innate immunity, influenza, respiratory syncytial virus, coronavirus
Citation: Peteranderl C and Herold S (2017) The Impact of the Interferon/TNF-Related Apoptosis-Inducing Ligand Signaling Axis on Disease Progression in Respiratory Viral Infection and Beyond. Front. Immunol. 8:313. doi: 10.3389/fimmu.2017.00313
Received: 15 November 2016; Accepted: 06 March 2017;
Published: 22 March 2017
Edited by:
Claudia U. Duerr, McGill University, CanadaReviewed by:
Gunnar Houen, Statens Serum Institut, DenmarkCopyright: © 2017 Peteranderl and Herold. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Christin Peteranderl, Y2hyaXN0aW4ucGV0ZXJhbmRlcmxAaW5uZXJlLm1lZC51bmktZ2llc3Nlbi5kZQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.