 Lena Müller
Lena Müller Petra Aigner1
Petra Aigner1 Dagmar Stoiber
Dagmar Stoiber
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 31 March 2017
Sec. Molecular Innate Immunity
Volume 8 - 2017 | https://doi.org/10.3389/fimmu.2017.00304
This article is part of the Research Topic Immunoregulatory mechanisms of Interferon View all 30 articles
Type I interferons (IFNs) are known to mediate antitumor effects against several tumor types and have therefore been commonly used in clinical anticancer treatment. However, how IFN signaling exerts its beneficial effects is only partially understood. The clinically relevant activity of type I IFNs has been mainly attributed to their role in tumor immune surveillance. Different mechanisms have been postulated to explain how type I IFNs stimulate the immune system. On the one hand, they modulate innate immune cell subsets such as natural killer (NK) cells. On the other hand, type I IFNs also influence adaptive immune responses. Here, we review evidence for the impact of type I IFNs on immune surveillance against cancer and highlight the role of NK cells therein.
Type I interferons (IFNs) have been initially identified 60 years ago as antiviral substances (1). They are a family of monomeric cytokines consisting of 14 IFNα subtypes, IFNβ, IFNε, IFNκ, and IFNω. While IFNα and IFNβ have been extensively studied during the past decades, the functions of IFNε, IFNκ, and IFNω remain poorly understood (2, 3). The term type I IFNs in this review therefore refers to the well-characterized forms IFNα and IFNβ, whereas the other type I IFN subtypes have been reviewed elsewhere (4, 5).
Type I IFNs can be secreted by most cell types in the body in response to activation of host pattern recognition receptors such as toll-like receptors (TLRs) and retinoic acid inducible gene-I-like RNA helicases that are triggered by bacterial or viral components (6–8). IFNα and IFNβ signal through the interferon α/β receptor (IFNAR), a heterodimeric transmembrane receptor that is composed of the two subunits IFNAR1 and IFNAR2. Following receptor binding, downstream signals lead to the phosphorylation and translocation of signal transducer and activator of transcription (STAT) factors to the nucleus to drive the expression of IFN-regulated genes (IRGs). For type I IFNs, the main STAT signaling complex is formed by IFN-stimulated gene factor 3 consisting of STAT1, STAT2, and IFN regulatory factor (IRF)-9 (3, 9, 10) (Figure 1), however, alternative pathways of IRG stimulation have been described as well (11–13).
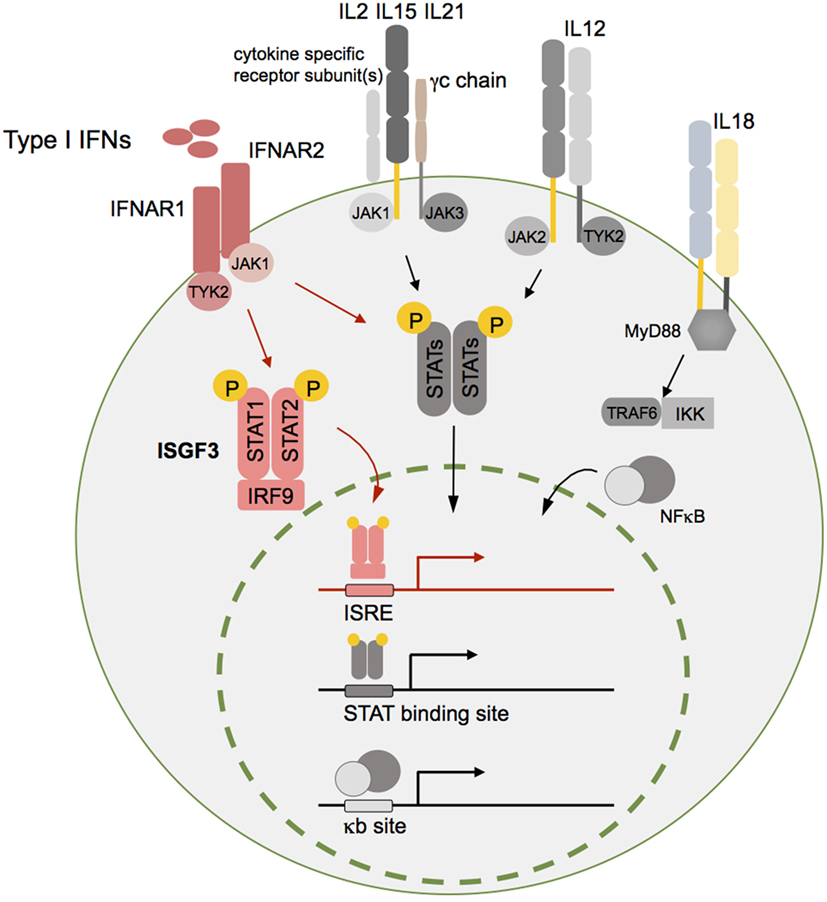
Figure 1. Type I interferons (IFNs) and different other cytokines are essential for natural killer (NK) cell homeostasis and function. Although type I IFNs are in focus of this review, additional cytokine pathways such as interleukin (IL)-2, IL12, IL15, IL18, and IL21 are schematically indicated here as important mediators of NK cell function. Cytokine receptor binding triggers downstream signaling pathways such as the Janus kinase (JAK)/signal transducer and activator of transcription (STAT) or nuclear factor kappa B (NFκB) pathway. The respective activated transcription factor complex—IFN-stimulated gene factor 3 (ISGF3) (type I IFNs), STAT dimers (IL2, IL15, IL21, and IL12), and NFκB (IL18)—translocates into the nucleus and induces target gene transcription leading to expression of genes that are crucial for survival, proliferation, differentiation, and cytotoxic function of NK cells. For reasons of simplicity, IL2R, IL15R, and IL21R were summarized in this graph. The receptor-specific subunit(s) in case of IL2R and IL15R refer to the β- and high-affinity α chain and for IL21R only to one specific subunit. Of note, IL15Rα chain is mainly expressed by other cells such as DCs, which is not displayed here. Abbreviations: ISRE, interferon stimulated response element; γc, common gamma chain; MyD88, myeloid differentiation primary response 88; TRAF6, TNF receptor associated factor 6; IKK, I kappa B kinase; NFκB, nuclear factor kappa B.
It has become well-accepted that functions of IFNα and β reach far beyond antiviral and microbial defense and include the regulation of physiological processes such as cell survival (12), immune cell homeostasis and functions (14), cell cycle, and differentiation (15–17). Many years back, it came as a surprise that constitutively released endogenous IFNα and IFNβ contribute to tissue homeostasis and inhibit malignant cellular transformation (14, 18, 19). Consequently, the finding that type I IFNs have antineoplastic functions stimulated the clinical development of type I IFN anticancer therapies for certain neoplasias.
However, unraveling molecular key mechanisms underlying the antitumor function of type I IFNs remained very challenging for a long time. Recent advances in the development of genetically engineered mouse models have provided useful tools for investigating these mechanisms and continuously improved our understanding of how IFN signaling interferes with tumor development.
Type I IFNs have been shown to prevent cellular transformation in premalignant cells in vitro by sustaining the expression of the tumor suppressor gene p53 (20). Moreover, cell-intrinsic roles for type I IFN signaling in negatively regulating tumor cell proliferation and in triggering apoptosis in different human cancer cell lines have been suggested as well (21). In vitro generated findings on direct antineoplastic effects of type I IFNs were substantiated by more recently performed in vivo studies, where tissue-specific deletion of IFNAR1 from intestinal epithelial cells increased tumor formation in mice treated with dextran sodium sulfate and the carcinogen azoxymethane to induce colitis (22).
However, a growing number of studies during the past decades provided solid evidence that type I IFNs execute antitumor functions mainly indirectly via stimulating immune cells to rapidly eliminate malignant cells. Owing to the ubiquitous IFNAR expression, type I IFNs have been shown to have crucial regulatory effects on immune cells in the context of inflammatory and viral diseases (2, 23). Thus, cellular mediators of the innate as well as the adaptive immune response may be regulated by type I IFNs in the protection of the host against malignant diseases. Indeed, an increasing number of studies performed during the past decades have supported the idea of an anticancer immune response analogous to the reaction of the host against pathogens.
A study performed by Dunn and colleagues elegantly demonstrated for the first time an essential role of endogenously produced type I IFNs in a process widely known as tumor immune surveillance (24). Unexpectedly and in contrast to IFNγ, type I IFNs were found in bone marrow transfer experiments to act on host hematopoietic cells and not on the tumor cell itself during the formation of a protective antitumor immune response.
The knowledge on how type I IFNs impact on cells of the innate and adaptive immune system in the context of tumor surveillance has been refined in numerous subsequent studies [reviewed in Ref. (21, 25)]. Some of the earliest studies identified an essential role of type I IFNs, particularly, for the function of host antigen presenting cells (26–28). Early produced type I IFNs act on the level of CD8α+ dendritic cells (DCs) that are required for the successful activation of tumor antigen-specific cytotoxic CD8+ T lymphocytes (CTLs). Based on in vitro data, it was demonstrated that type I IFN signaling specifically enhances the ability of CD8α+ DCs to cross-present antigens (27), most likely by promoting survival of DCs and enhancing antigen persistence on the cell surface during cross-presentation (21, 29, 30). Moreover, type I IFNs have been shown to promote DC maturation, differentiation, and migration (28).
Finally, type I IFNs induce the release of interleukin 15 (IL15) by DCs (31), thus promoting the survival of CD8+ memory cells and NK cells (32), which will be discussed in more detail later on. In response to type I IFNs, CTLs have also been shown to acquire full effector functions (26, 33). Also by impacting on other innate immune cell subsets such as neutrophils (34–38), NKT, and γδ T cells (39), type I IFNs exhibit tumor-growth limiting properties.
In addition, type I IFNs promote a protective antitumor response by inhibiting cells of the tolerogenic tumor microenvironment such as myeloid-derived suppressor cells (MDSCs) (40, 41) and regulatory T cells (Tregs) (42–45) that might interfere with the host tumor immune response.
Type I IFNs are released very early during infections (46), thus it was not surprising that they are important regulators specifically of innate immune cell subsets such as DCs and NK cells in anticancer host responses. For NK cells, type I IFNs have already been demonstrated in viral infection to be critical for early responses and are thought to enhance NK cell cytotoxicity and cytokine production (47, 48). However, how type I IFNs regulate NK cell function in the context of tumor development will be outlined in detail in the following sections.
The importance of NK cells in tumor immune surveillance was initially demonstrated via depletion of NK cells from mice rendering them more susceptible to transplanted tumor cells or methylcholanthrene (MCA)-induced sarcomas (49, 50). Furthermore, NK cells have been shown to control the development of B cell lymphomas that arise in mice deficient for perforin, and NK cells were able to recognize and eliminate some of the tumors in the absence of major histocompatibility complex class I (MHC I) (49, 51, 52). Importantly, impaired type I IFN signaling in NK cells leads to a substantial loss of mature NK cell functions that are essential for efficient tumor cell killing. Initially, the effect of type I IFNs on NK cell homeostasis and development has been studied in mice deficient for IFNAR1 or IFNAR2. In the spleens of those mice, NK cell proportions were significantly decreased and mature NK cells of both genotypes expressed lower levels of the surface molecules CD122, CD11b, and Ly49 C + I (53). Thus, IFNAR-deficient NK cells are reduced in numbers and exhibit impaired cytotoxic capacity (24, 53). The cellular and molecular mechanisms of how type I IFN signaling impacts on NK cells and their effector functions are discussed in detail in this and the following section.
Murine NK cells develop in the bone marrow and at alternative sites such as thymus and liver (54–57). However, the majority of NK cells detected in the periphery is likely to have developed in the bone marrow. There, common lymphoid progenitor cells lose their potential to develop into precursor cells of other lineages and differentiate toward an NK cell-restricted precursor cell (NKP) via intermediate stages (58–60). Based on the expression of cell-specific markers and the acquisition of functional competence, NK cell differentiation is subdivided into distinct developmental stages. Natural killer cell-restricted precursor cells express CD122 that enables the cell to respond to IL15, which is the hallmark cytokine of NK cell lineage specification. Natural killer cell-restricted precursor cells progress to a transitory immature NK cell (iNK) stage that is characterized by the upregulation of the pan-NK cell marker NK1.1. The terminal maturation step from iNK cells to mature NK cells (mNK) involves the upregulation of Ly49 receptor family members together with CD11b and DX5. Following their complete maturation, mNK cells egress from the bone marrow and reside in the blood, spleen, liver, lung, and various other organs, where they continue to mature to tissue-specific and functionally distinct NK cell subsets (54). In the periphery, classical stages of NK cell maturation are described based on the expression of CD11b and killer cell lectin-like receptor subfamily G, member 1 as well as loss of CD27 and TNF-related apoptosis inducing ligand (TRAIL) expression (61–64).
We have previously identified an unexpected role for type I IFNs in NK cell development. In IFNAR-deficient mice, type I IFN signaling was dispensable for NK cell maturation in the bone marrow, but lack of IFNAR1 expression on NK cells significantly abrogated peripheral maturation in the spleen. Of note, late stage deletion of Ifnar1 in mature NK cells (Ifnar1f/f Ncr1-iCre mice) did not interfere with splenic NK cell maturation indicating that type I IFNs are required at an earlier stage or by other cells for full NK cell maturation in the spleen (65). The impact on NK cell maturation by systemic type I IFNs was also evidenced by Guan and colleagues (66). By generating mixed bone marrow chimeric mice from Ifnar−/− and wild-type animals, they showed an intrinsic effect of IFNAR signaling on early NK cell maturation in the bone marrow and also in the liver. In line with results from our study (65), mature NK cell numbers remained unchanged in spleen and blood.
Similar to T cells, NK cells as part of the innate immune system are also able to form an immunological memory and terminally differentiate into memory NK cells. Different educational routes have been described that lead to the formation of NK cell memory by antigen-dependent (hapten- and virus-induced) or antigen-independent (cytokine-induced) mechanisms (67, 68).
Sensitization of mice with haptens in the presence of the pro-inflammatory cytokines IL12, IFNγ, and IFNα leads to hapten-specific memory NK cells in the liver (67, 68). Type I IFNs play an important role herein as hepatic NK cells in hapten-sensitized Ifnar1−/− (and Il12−/−, Ifng−/−) mice failed to induce contact hypersensitivity after adoptive transfer to the challenged host (69).
Interestingly, in a murine cytomegalovirus (MCMV) infection model, type I IFNs have been proposed to play a role in the differentiation of antigen-dependent memory NK cells. Acute MCMV infection stimulates the production of type I IFNs and other pro-inflammatory cytokines (IL12, IL18, IFNγ, IL21) (70, 71). These pro-inflammatory signals drive the expression of the BTB-ZF transcription factor Zbtb32 (also known as ROG, FAZF, TZFP, PLZP) in antigen-specific NK cells, which is essential for their proliferation and protective function during MCMV infection (72). By using NK cells deficient for IFNAR1 in mixed bone marrow chimeric mice, Madera et al. demonstrated that direct type I IFN signaling in NK cells promotes their optimal activation and function during MCMV infection. However, type I IFNs were shown to be dispensable for the survival of NK cells and NK memory formation (73).
Also in other virus infection models, type I IFNs and NK cells play important roles. In mice, lytic infection in macrophages with gammaherpesvirus was restricted by NK cells independently of type I IFNs, but spreading of virions to the spleen was only possible in the absence of both, type I IFNs and NK cells (74).
Of note, NK cell memory against tumors has not been observed under physiological conditions. Receptors such as NKG2D that are involved in the recognition of tumor cells by NK cells may not be capable of efficiently generating memory. Moreover, it is also conceivable that host-derived factors such as cytokines in addition to specific ligands for activating NK receptors are needed for the generation of memory NK cells against tumors and that these factors are under-represented in the tumor microenvironment (68). Still, memory NK cells bear the potential to be further manipulated to target tumor cells (see section “Type I IFNs and Anticancer Therapies—A Role for NK Cells Therein?”).
As mentioned above, IFNAR1 as well as IFNAR2-deficient NK cells are diminished in numbers and exhibit considerably reduced cytotoxic capacity. These defects finally translate into severely impaired tumor surveillance in Ifnar1−/− and Ifnar2−/− mice, which succumb earlier to carcinogen-induced fibrosarcoma and RMA-S lymphoma (24, 53). These findings were substantiated by the importance of type I IFN signaling on NK cell-mediated v-Abl oncogene-driven B cell leukemogenesis (65). In this context, mice with impaired type I IFN signaling (i.e., Ifnar1−/− and Ifnb−/− mice) had an increased susceptibility to v-Abl-induced leukemia/lymphoma and B16F10 melanoma. Increased tumor incidence in these models is linked to defects in NK cell-mediated tumor surveillance, which is dependent on their reduced cytotoxic capacity. Indeed, NK cells derived from Ifnar1−/− and Ifnb−/− animals display impaired cytotoxic effector function against their target cells in vitro (24, 53, 65).
In line with reduced cytotoxicity observed in NK cells lacking either IFNAR1 or IFNβ expression, a similar effect has been reported for NK cells deficient for downstream components of the type I IFN pathway, such as TYK2 (75) or STAT1 (47, 76).
Similar to NK cells derived from IFNAR1-deficient mice, NK cells isolated from mice lacking type I IFN signaling only at the mature NK cell stage (Ifnar1f/f Ncr1-iCre mice) (65, 77) display a substantial defect in cytolytic capacity against hematopoietic tumor cell lines (YAC-1, RMA-S) in vitro. However, challenging these Ifnar1f/f Ncr1-iCre mice with the v-Abl oncogene revealed that IFN signaling in mature NK cells is dispensable for the surveillance of leukemia (65). This result might be explainable by the complex cytokine milieu in vivo compensating for the obvious defects under IL2-dependent in vitro culturing. Previous studies showed that Ifnar1 deficiency severely curtails NK cell cytotoxicity even in the presence of high doses of IL2 (53). Interestingly, exogenous IL12 stimulation significantly enhances the cytotoxicity of Ifnar1−/− and Stat1−/− NK cells. Moreover, IL15 stimulation completely restores cytotoxic activity of Stat1−/− NK cells in vitro (47). These findings clearly show that NK cell defects in Ifnar1−/− or Stat1−/− animals cannot be overcome by IL2 stimulation, but might be partially compensated by other cytokines in vivo. This underscores the importance of other cytokines in NK cell biology such as IL15 and IL21 that are known to increase the cytolytic activity of NK cells in vivo (52, 78, 79) (Figure 1). An additional possible explanation is that in Ifnar1−/− mice other cell types that do require type I IFNs are critically involved in tumor surveillance.
Natural killer cells do not possess immediate and permanent effector functions. A process called “priming” is required to induce the establishment of the entire NK cell competence (80, 81). Natural killer cell priming is dominated by type I IFNs, which provide essential signals for DCs to produce IL15, the master cytokine for promoting NK cell development, proliferation, and function (54, 80, 82–84).
The activation of NK cells can be induced by DCs through pathways that require cell–cell contact (NKG2D-MICA and/or MICB) and cytokines such as IFNα, IFNβ, IL2, IL12, IL15, and IL18 (82) (Figure 1). Dendritic cell-derived signals elicit both NK-cell-mediated cytolysis as well as cytokine production. Resting and activated DCs are capable of activating NK cells, however, the latter far more potently. The interaction between activated DCs and NK cells has been shown to augment the efficiency of NK cell antitumor effector function in different in vitro and in vivo models (85, 86). Upon type I IFN signal recognition, DCs produce IL15 and trans-present IL15 to resting NK cells (80). Thus, the interaction with DCs equips NK cells for full effector function. In turn, NK cells are also capable of affecting DC functions through their involvement in DC maturation and DC elimination (82).
More recently, myeloid cells came again into focus as a mechanism was proposed on how cells such as DCs and macrophages could assist NK cell-mediated tumor control (87). Dectin-1 expressed on myeloid cells is critical for NK cell-mediated killing of tumor cells that express high levels of N-glycan structures. Receptor recognition of such tumor cells led to activation of IRF5, an IRF best known for its function in pathogen-induced immunity via activation of MyD88-dependent TLR pathway. This Dectin-1-IRF5 pathway activation in myeloid cells led to activation and efficient tumoricidal function of NK cells. The interaction of myeloid cells and NK cells here may be partially dependent on the expression of the IRF3-dependent NK activating molecule, a membrane-bound protein known to activate NK cells via its homophilic interaction (88).
Apart from effects elicited by type I IFNs on myeloid cells, the following mechanisms could also affect NK cell-mediated tumor surveillance. Although most of those mechanisms have been identified in the context of viral infections, they might be of significant importance in the tumor setting.
The interaction of NK and T cells is also influenced by type I IFN signaling. Type I IFNs keep NK cells from eliminating antigen-activated CTLs by modulating the expression of NK cell receptor ligands (89, 90). In the context of lymphocytic choriomeningitis virus infection, Crouse et al. demonstrated that direct sensing of type I IFNs by T cells prevents them from NK cell-mediated killing by keeping the expression of NCR1 ligands on the CTLs low (89). With the same viral infection setting, Xu et al. showed that the elimination of virus-activated T cells by NK cells was inhibited by type I IFN-induced expression of selected inhibitory NK cell receptor ligands, i.e., classical and nonclassical MHC molecules (MHC I and Qa-1b) (90). An effect of type I IFN signaling on MHC I expression and therefore antigen presentation was reported already earlier, however, the differences in MHC I expression on IFNAR1-deficient cells appeared to be of minor extent (91–93).
Another NK cell surface molecule, TRAIL, was reported to be critical for NK antitumor function in mice and humans (94–96). For example, murine liver NK cells contribute to natural antimetastatic function against TRAIL sensitive tumor cells and constitutive TRAIL expression on these NK cells is IFNγ dependent (96, 97). During viral infection, type I IFNs were also described to enhance antiviral response by NK cell cytotoxicity through induction of TRAIL on NK cells (98).
Another aspect of tumorigenesis influenced by type I IFN signaling is oncogene-induced senescence. In this context, DNA-damage-induced production of type I IFNs enhances cellular senescence (99). In addition, type I IFNs produced by senescent cells indirectly stimulate NKG2D ligand expression on senescent malignant cells, thus promoting the elimination by NK cells (100). However, IFNα has been shown to downregulate the expression of NKG2D ligand H60 in MCA-induced tumors in 129/Sv mice resulting in reduced effectiveness of NK target recognition and NK-dependent killing (101). This indicates that depending on the tumor model, type I IFNs differentially regulate NKG2DL expression.
Finally, ligands for receptors of immune checkpoints such as those of the programmed cell death protein 1 (PD1) family are induced by type I IFNs (102, 103). This immunoregulatory function of type I IFNs is of great relevance and needs to be taken into consideration for the design of clinical anticancer treatments. Recently, targeting of PD1-ligand (PDL1), which is recognized by its inhibitory receptor PD1 expressed on NK cells and other immune cell subsets gained a lot of attention in oncology and will be discussed in more detail later on.
The mechanisms involving NK cells and type I IFN signaling in tumor surveillance are summarized in Figure 2.
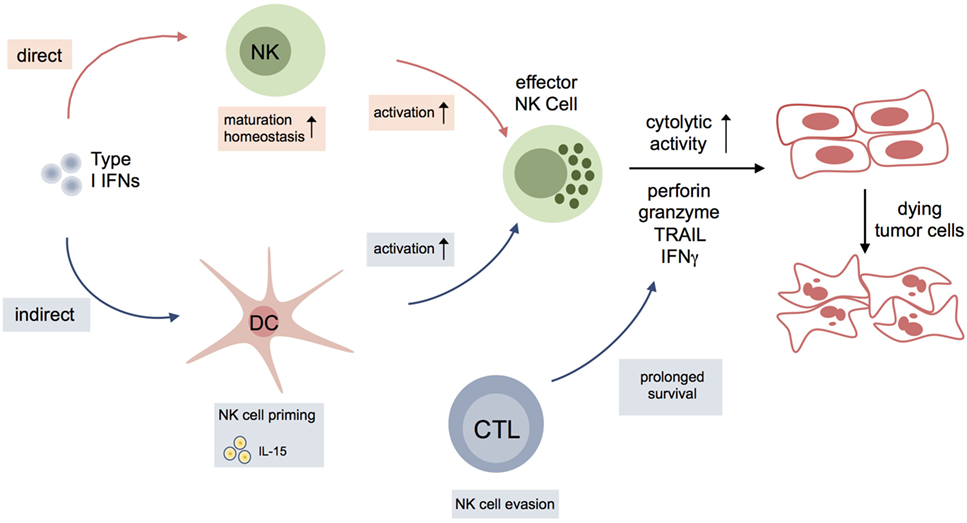
Figure 2. Interplay of type I interferons (IFNs) and natural killer (NK) cell activation during antitumor response. Type I IFNs either impact on maturation, homeostasis, and activation of NK cells, or indirectly influence NK cells to kill tumor cells via other immune cells or cells of the tumor microenvironment. Dendritic cells (DCs), in particular, are essential for NK cell priming via production of IL15. Another indirect effect of type I IFNs on NK cell function in cancer might result from modulation of surface molecules on CD8+ cytotoxic T lymphocytes (CTLs) [NCR1 ligands; classical and nonclassical major histocompatibility complex class I (MHC I)] leading to evasion of CTLs from NK cell-mediated elimination.
Metastasis as the dreadful consequence of tumorigenesis has recently been shown to be controlled by antitumor immune responses. In this context, NK and CD8+ T cells as the main cellular mediators of tumor immune surveillance have been described to be capable of restricting metastatic tumor growth. Therefore, depletion of CD8+ T cells or NK cells increased metastasis formation in a breast cancer mouse model without affecting primary tumor growth (104). One mechanism proposed for the metastasis surveillance function of NK cells relies on the inhibition of the MERTK (also known as TAM; TYR3, AXL, and MER) family tyrosine kinase receptors that suppress NK cell activation (105, 106). Of note, the protective function of NK cells against metastases can be also linked to and is partially dependent on type I IFN signaling. In a syngeneic mouse model of mammary tumor metastasis using 4T1.2 cells, Bidwell and coworkers identified a number of IRF7 target genes that are suppressed in bone metastases (104). Consequently, metastasis formation in spontaneous (MMTV-PyMT) and orthotopic mammary tumorigenesis models was accelerated in mice deficient for IFNAR1, NK cell, or CD8+ T cell responses (104, 107). Conversely, enforced expression of IRF7 in tumor cells or treatment with type I IFNs enhanced the immune activity and suppressed bone metastasis, thus prolonging survival of the diseased mice. Of note, depletion of both CD8+ T and NK cells significantly accelerated metastasis and shortened survival time in mice harboring 4T1.2 tumors ectopically expressing IRF7. This indicates that IRF7-induced and type I IFN-dependent inhibition of bone metastasis was mediated by CD8+ T and NK cells (104). In line with the data obtained from metastasis studies in mice, loss of IRF7-associated gene signature in primary tumors of breast cancer patients predicted an increased risk of bone metastasis and also additional studies suggest a suppressive role for type I IFN signaling on breast cancer progression (25).
However, tumor cells use different immune evasive strategies to survive at distinct metastatic sites. The recruitment of immunosuppressive cells is one major mechanism to overcome the immune surveillance system (108). For example, systemic factors from hypoxic breast cancer cells increase myeloid CD11b+ cell accumulation and reduce the cytotoxic functions of NK cells in the premetastatic lung (109). Myeloid cells, especially MDSCs, have the capacity to suppress immune responses, thus it is conceivable that recruited myeloid cells establish a premetastatic immune-suppressive niche to promote tumor metastasis. Moreover, platelet activation and the resulting fibrin clot formation support survival of tumor cells that are nested at metastatic sites by protecting them from NK cells (108, 110).
In mice engrafted with mammary tumor cell lines, type I IFN treatment has been shown to reduce metastasis to bone. Interestingly, while MDSC accumulation was substantially decreased, there was an increase in numbers of NK cells present in the bone marrow of these mice (104). Hence, the authors proposed that type I IFNs specifically inhibit bone metastases of mammary cancer by a selective modulation of MDSCs and NK effector cells in the bone marrow (104).
A consecutive study demonstrated that endogenous type I IFN signaling in the host hematopoietic system is indispensable for the responsiveness of circulating NK cells and therefore essential for metastasis-free survival. Consistently, in vivo stimulated NK cells derived from Ifnar1−/− mice but not from wild-type counterparts failed to eliminate the 4T1 and 66cl4 mammary tumor cell lines in vitro (107).
In summary, these studies clearly highlight an essential role for IFN signaling and NK cells during metastasis formation and could pave the way for type I IFNs for new therapeutic means in metastatic cancer.
As outlined above, ample evidence substantiates the importance of type I IFN signaling in NK cell-mediated tumor surveillance. Interferons mainly function by modulating the immune system rather than executing direct anticancer effects. In the clinics, type I IFNs have been used for decades as anticancer therapy, however, the exact mechanism of action of type I IFNs has not been clarified yet (111, 112). IFNα has been and is still used mainly for the treatment of hematopoietic neoplasms. Especially, before the advent and breakthrough of the BCR-ABL inhibitor imatinib as therapy for chronic myeloid leukemia (CML), IFNα was the treatment of choice for patients not suitable for bone marrow transplantation. Interestingly, in chronic myeloproliferative neoplasms the positive effect of IFNα coincided with a substantially higher frequency of circulating CD56bright NK cells that produced increased levels of IFNγ (113). Recently, IFNα has gained attention for further use as therapeutic option in CML, preferably in combination with imatinib or its next generation inhibitors (114).
Trials with IFN therapies in solid malignancies have met with varied success. However, besides virus-related cancers at least in melanoma as one type of solid tumors, IFNα is clinically used (21). In high-risk melanoma patients, high-dose IFNα treatment leads to an extension of relapse-free survival and is therefore considered a valid therapeutic option. Interestingly, IFN therapy is more effective at targeting disseminated cancer cells and minimal residual disease before they form large proliferative metastases, emphasizing again that promotion of antitumor immunity rather than direct antiproliferative effects is the predominant mechanism of action (25).
Data obtained mainly from tumor studies in mice strongly suggest that the success of conventional chemotherapeutics (such as anthracyclines, cyclophosphamide), targeted anticancer agents, radiotherapy, and immunotherapy depends on type I IFN signaling (21, 115). Under certain circumstances, this mode of action of IFN signaling involves NK cells. For example, some immunogenic chemotherapeutics lead to the activation of TLR3 in malignant cells by cancer-cell derived RNA which results in type I IFN production. Subsequently, IRGs such as CXC-chemokine ligand 10 (CXCL10) are expressed, which in turn are crucial for recruitment in NK cell-mediated tumor control (116, 117).
The concept of tumor immune surveillance has triggered an increasing interest in immunomodulatory treatment strategies. However, immune-activating therapies are likely to induce the expression of immunosuppressive ligands and receptors such as PDL1, PD1, and CTLA4. Since type I IFNs have immunostimulatory functions, they can promote the upregulation of such surface molecules (102, 118), thus preventing prolonged antitumor immune responses. In this case, a sustained therapeutic antitumor response could be achieved by the combination of type I IFN therapy with other therapeutic means targeting the PD1–PDL1 axis to block secondary immune suppression. Programmed cell death protein 1- and CTLA4-targeted therapeutics have been proven in some cancers to significantly prolong survival of the patients. Combining these agents with type I IFNs could be a suitable strategy to overcome immunosuppression and raise patient responsiveness. Programmed cell death protein 1 is well documented in the context of T-cell responses and has recently been shown to be upregulated on NK cells, which leads to downregulation of anticancer function (110, 119, 120).
On the contrary, IFN signaling seems to be also important for the success of checkpoint immunotherapy, which is illustrated by a recent study on late relapses of PD1 blockade treatment in metastatic melanoma. Here, a loss-of-function mutation in the Janus kinase 1 has been identified in one patient, suggesting that disruption of type I and type II IFN signaling might be involved in preventing the success of checkpoint immunotherapies (121). If this turns out to be a more frequent observation, the combination of type I IFNs with checkpoint inhibitors would be desirable for an improved treatment outcome. Furthermore, in anticancer virotherapy, type I IFNs play a key role, as intratumoral injection of the oncolytic Newcastle disease virus combined with systemic CTLA4 blockade leads to regression of murine B16 melanomas. Interestingly, this effect has been reported to be dependent on CTLs, NK cells, and IFNAR signaling (122).
As outlined above, there are a number of reasons pleading for type I IFNs as tools in anticancer treatment. However, one big disadvantage are dose-limiting side effects, including influenza-like symptoms (fatigue, fever, headache, and muscle aches), nausea, anorexia, dizziness, depression, and leukopenia. To avoid these side effects of IFN therapy, strategies are now being developed to deliver type I IFNs directly to the tumor microenvironment (21). Different types of cells can be manipulated to express type I IFNs to augment their own antitumor activity or to promote the activity of other immune effector cells of the host. This has been also assessed with NK cells: a genetically engineered NK cell line expressing human IFNα displayed improved cytotoxicity functions against hepatocellular carcinoma cells in vitro, as well as in xenograft tumor models (123). Moreover, mesenchymal stem cells modified to express mouse IFNα efficiently decreased the growth of murine B16 melanomas in vivo, an effect that was shown to be dependent on NK and T cells (90). However, translating this strategy to the clinics might be difficult and other means, such as the usage of modulators of specific immune cell subtypes and/or pathways might be preferred.
As described above, memory NK cells against tumors have not been observed yet, but would be highly appreciable if those could be generated in vitro by different manipulations such as transduction of proliferating NK cells with chimeric antigen receptors, or enhanced antibody dependent cellular cytotoxicity (ADCC) using newly identified human FcεRIγ-deficient adaptive NK cells (68). As antigen-dependent memory NK cell formation relies also on type I IFN signaling, this could be another strategy for type I IFNs and NK cells in cancer control.
The studies on type I IFNs and breast cancer metastasis (see section “Type I IFNs, NK cells and metastasis”) may provide a rationale for targeting the endogenous type I IFN pathway as an antimetastatic strategy. As IFN signaling modulates the tumor immune response, targeting type I IFNs to a specific cellular compartment of the tumor mass may mediate optimal therapeutic effects for some cancer types. Type I IFN signaling within tumors is essential for both natural and therapy-induced immune surveillance. Thus, downstream effectors of type I IFN signaling would be suitable candidates for further investigation as prognostic and predictive biomarkers in cancer diagnosis and progression (21).
The high potential and importance of type I IFNs and NK cells in cancer is also illustrated by a glimpse on current clinical trials. Searching for IFNα, NK cells, and cancer at ClinicalTrials.gov resulted in 16 studies, half of them dealing with type I IFNs and NK cells for cancer patients (https://clinicaltrials.gov; November 2016). Already in 1997, Nagler et al. combined type I IFNs with NK cell-stimulating molecules such as IL2 and indeed showed increased survival in lymphoma patients after stem cell transplantation (124). Recently, a more specific approach using adoptive transfer of autologous or allogeneic NK cells is frequently tested for cancer treatment (125). Here, even synergistic or additive effects of type I IFNs applied in this context could be imagined.
The combination of type I IFNs with other immunostimulatory agents such as immune checkpoint blockers, cytokines, or other inhibitors that target different immunosuppressive circuits is likely to result in optimal NK cell anticancer function and tumor control.
Type I IFNs are essential in antitumor control and execute their function predominantly by modulating the activity of other immune cells. Although type I IFNs affect various immune cell subsets, the impact of type I IFNs on NK cells is especially crucial for efficient tumor immune surveillance. Type I IFNs not only positively regulate NK cell maturation and memory, but also NK cell priming and NK cell-mediated tumor surveillance by various mechanisms. Detailed knowledge about underlying mechanisms of immunoregulatory cell recruitment and their suppressive functions in primary tumors and at metastatic sites should lead to more effective immunotherapies.
Thus, therapeutic approaches will need to include the evaluation of immune cell profiles in individual cancers, so that drug targeting can be precisely tailored to maximize the response. In addition, tumor-type specific treatments of type I IFNs and other therapeutic concepts might extend the pharmacological armament to combat diverse cancer types.
LM, PA, and DS have written the review article. LM has compiled graphics for this review article.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Financial support was provided by the St. Anna Kinderkrebsforschung, Children’s Cancer Research Institute, Vienna, Austria, financing the position of PA. The authors thank Silvia Stockinger, Eva-Maria Zebedin-Brandl, and Emilio Casanova for critical reading of the manuscript.
1. Isaacs A, Lindenmann J. Virus interference. I. The interferon. Proc R Soc Lond B Biol Sci (1957) 147:258–67. doi:10.1098/rspb.1957.0048
2. Decker T, Müller M, Stockinger S. The yin and yang of type I interferon activity in bacterial infection. Nat Rev Immunol (2005) 5:675–87. doi:10.1038/nri1684
3. Ivashkiv LB, Donlin LT. Regulation of type I interferon responses. Nat Rev Immunol (2014) 14:36–49. doi:10.1038/nri3581
4. Pestka S, Krause CD, Walter MR. Interferons, interferon-like cytokines, and their receptors. Immunol Rev (2004) 202:8–32. doi:10.1111/j.0105-2896.2004.00204.x
5. Hertzog PJ, Williams BRG. Fine tuning type I interferon responses. Cytokine Growth Factor Rev (2013) 24:217–25. doi:10.1016/j.cytogfr.2013.04.002
6. Akira S, Uematsu S, Takeuchi O. Pathogen recognition and innate immunity. Cell (2006) 124:783–801. doi:10.1016/j.cell.2006.02.015
7. Uematsu S, Akira S. Toll-like receptors and type I interferons. J Biol Chem (2007) 282:15319–23. doi:10.1074/jbc.R700009200
8. Cavlar T, Ablasser A, Hornung V. Induction of type I IFNs by intracellular DNA-sensing pathways. Immunol Cell Biol (2012) 90:474–82. doi:10.1038/icb.2012.11
9. Levy DE, Darnell JE. Stats: transcriptional control and biological impact. Nat Rev Mol Cell Biol (2002) 3:651–62. doi:10.1038/nrm909
10. Stark GR, Darnell JE. The JAK-STAT pathway at twenty. Immunity (2012) 36:503–14. doi:10.1016/j.immuni.2012.03.013
11. Silvennoinen O, Ihle JN, Schlessinger J, Levy DE. Interferon-induced nuclear signalling by Jak protein tyrosine kinases. Nature (1993) 366:583–5. doi:10.1038/366583a0
12. Platanias LC. Mechanisms of type-I- and type-II-interferon-mediated signalling. Nat Rev Immunol (2005) 5:375–86. doi:10.1038/nri1604
13. Du Z, Wei L, Murti A, Pfeffer SR, Fan M, Yang CH, et al. Non-conventional signal transduction by type 1 interferons: the NF-κB pathway. J Cell Biochem (2007) 102:1087–94. doi:10.1002/jcb.21535
14. Gough DJ, Messina NL, Clarke CJP, Johnstone RW, Levy DE. Constitutive type I interferon modulates homeostatic balance through tonic signaling. Immunity (2012) 36:166–74. doi:10.1016/j.immuni.2012.01.011
15. Essers MAG, Offner S, Blanco-Bose WE, Waibler Z, Kalinke U, Duchosal MA, et al. IFNalpha activates dormant haematopoietic stem cells in vivo. Nature (2009) 458:904–8. doi:10.1038/nature07815
16. Sato T, Onai N, Yoshihara H, Arai F, Suda T, Ohteki T. Interferon regulatory factor-2 protects quiescent hematopoietic stem cells from type I interferon-dependent exhaustion. Nat Med (2009) 15:696–700. doi:10.1038/nm.1973
17. Takayanagi H, Kim S, Matsuo K, Suzuki H, Suzuki T, Sato K, et al. RANKL maintains bone homeostasis through c-Fos-dependent induction of interferon-beta. Nature (2002) 416:744–9. doi:10.1038/416744a
18. Tovey MG, Streuli M, Gresser I, Gugenheim J, Blanchard B, Guymarho J, et al. Interferon messenger RNA is produced constitutively in the organs of normal individuals. Proc Natl Acad Sci U S A (1987) 84:5038–42. doi:10.1073/pnas.84.14.5038
19. Gresser I. Biologic effects of interferons. J Invest Dermatol (1990) 95:66S–71S. doi:10.1111/1523-1747.ep12874776
20. Takaoka A, Hayakawa S, Yanai H, Stoiber D, Negishi H, Kikuchi H, et al. Integration of interferon-α/β signalling to p53 responses in tumour suppression and antiviral defence. Nature (2003) 424:516–23. doi:10.1038/nature01850
21. Zitvogel L, Galluzzi L, Kepp O, Smyth MJ, Kroemer G. Type I interferons in anticancer immunity. Nat Rev Immunol (2015) 15:405–14. doi:10.1038/nri3845
22. Tschurtschenthaler M, Wang J, Fricke C, Fritz TMJ, Niederreiter L, Adolph TE, et al. Type I interferon signalling in the intestinal epithelium affects Paneth cells, microbial ecology and epithelial regeneration. Gut (2014) 63:1921–31. doi:10.1136/gutjnl-2013-305863
23. Stetson DB, Medzhitov R. Type I interferons in host defense. Immunity (2006) 25:373–81. doi:10.1016/j.immuni.2006.08.007
24. Dunn GP, Bruce AT, Sheehan KCF, Shankaran V, Uppaluri R, Bui JD, et al. A critical function for type I interferons in cancer immunoediting. Nat Immunol (2005) 6:722–9. doi:10.1038/ni1213
25. Parker BS, Rautela J, Hertzog PJ. Antitumour actions of interferons: implications for cancer therapy. Nat Rev Cancer (2016) 16:131–44. doi:10.1038/nrc.2016.14
26. Fuertes MB, Kacha AK, Kline J, Woo S-R, Kranz DM, Murphy KM, et al. Host type I IFN signals are required for antitumor CD8+ T cell responses through CD8α+ dendritic cells. J Exp Med (2011) 208:2005–16. doi:10.1084/jem.20101159
27. Diamond MS, Kinder M, Matsushita H, Mashayekhi M, Dunn GP, Archambault JM, et al. Type I interferon is selectively required by dendritic cells for immune rejection of tumors. J Exp Med (2011) 208:1989–2003. doi:10.1084/jem.20101158
28. Fuertes MB, Woo S-R, Burnett B, Fu Y-X, Gajewski TF. Type I interferon response and innate immune sensing of cancer. Trends Immunol (2013) 34:67–73. doi:10.1016/j.it.2012.10.004
29. Lorenzi S, Mattei F, Sistigu A, Bracci L, Spadaro F, Sanchez M, et al. Type I IFNs control antigen retention and survival of CD8α(+) dendritic cells after uptake of tumor apoptotic cells leading to cross-priming. J Immunol (2011) 186:5142–50. doi:10.4049/jimmunol.1004163
30. Schiavoni G, Mattei F, Gabriele L. Type I interferons as stimulators of DC-mediated cross-priming: impact on anti-tumor response. Front Immunol (2013) 4:483. doi:10.3389/fimmu.2013.00483
31. Mattei F, Schiavoni G, Belardelli F, Tough DF. IL-15 is expressed by dendritic cells in response to type I IFN, double-stranded RNA, or lipopolysaccharide and promotes dendritic cell activation. J Immunol (2001) 167:1179–87. doi:10.4049/jimmunol.167.3.1179
32. Huntington ND. The unconventional expression of IL-15 and its role in NK cell homeostasis. Immunol Cell Biol (2014) 92:210–3. doi:10.1038/icb.2014.1
33. Curtsinger JM, Mescher MF. Inflammatory cytokines as a third signal for T cell activation. Curr Opin Immunol (2010) 22:333–40. doi:10.1016/j.coi.2010.02.013
34. Granot Z, Jablonska J. Distinct functions of neutrophil in cancer and its regulation. Mediators Inflamm (2015) 2015:701067. doi:10.1155/2015/701067
35. Jablonska J, Leschner S, Westphal K, Lienenklaus S, Weiss S. Neutrophils responsive to endogenous IFN-β regulate tumor angiogenesis and growth in a mouse tumor model. J Clin Invest (2010) 120:1151–64. doi:10.1172/JCI37223
36. Andzinski L, Wu C-F, Lienenklaus S, Kröger A, Weiss S, Jablonska J. Delayed apoptosis of tumor associated neutrophils in the absence of endogenous IFN-β. Int J Cancer (2015) 136:572–83. doi:10.1002/ijc.28957
37. Jablonska J, Wu C-F, Andzinski L, Leschner S, Weiss S. CXCR2-mediated tumor-associated neutrophil recruitment is regulated by IFN-β. Int J Cancer (2014) 134:1346–58. doi:10.1002/ijc.28551
38. Wu C-F, Andzinski L, Kasnitz N, Kröger A, Klawonn F, Lienenklaus S, et al. The lack of type I interferon induces neutrophil-mediated pre-metastatic niche formation in the mouse lung. Int J Cancer (2015) 137:837–47. doi:10.1002/ijc.29444
39. Woo S-R, Corrales L, Gajewski TF. Innate immune recognition of cancer. Annu Rev Immunol (2015) 33:445–74. doi:10.1146/annurev-immunol-032414-112043
40. Yu J, Du W, Yan F, Wang Y, Li H, Cao S, et al. Myeloid-derived suppressor cells suppress antitumor immune responses through IDO expression and correlate with lymph node metastasis in patients with breast cancer. J Immunol (2013) 190:3783–97. doi:10.4049/jimmunol.1201449
41. Zoglmeier C, Bauer H, Nörenberg D, Wedekind G, Bittner P, Sandholzer N, et al. CpG blocks immunosuppression by myeloid-derived suppressor cells in tumor-bearing mice. Clin Cancer Res (2011) 17:1765–75. doi:10.1158/1078-0432.CCR-10-2672
42. Pace L, Vitale S, Dettori B, Palombi C, La Sorsa V, Belardelli F, et al. APC activation by IFN-alpha decreases regulatory T cell and enhances Th cell functions. J Immunol (2010) 184:5969–79. doi:10.4049/jimmunol.0900526
43. Sisirak V, Faget J, Gobert M, Goutagny N, Vey N, Treilleux I, et al. Impaired IFN-α production by plasmacytoid dendritic cells favors regulatory T-cell expansion that may contribute to breast cancer progression. Cancer Res (2012) 72:5188–97. doi:10.1158/0008-5472.CAN-11-3468
44. Bacher N, Raker V, Hofmann C, Graulich E, Schwenk M, Baumgrass R, et al. Interferon-α suppresses cAMP to disarm human regulatory T cells. Cancer Res (2013) 73:5647–56. doi:10.1158/0008-5472.CAN-12-3788
45. Hashimoto H, Ueda R, Narumi K, Heike Y, Yoshida T, Aoki K. Type I IFN gene delivery suppresses regulatory T cells within tumors. Cancer Gene Ther (2014) 21:532–41. doi:10.1038/cgt.2014.60
46. Biron CA. Initial and innate responses to viral infections – pattern setting in immunity or disease. Curr Opin Microbiol (1999) 2:374–81. doi:10.1016/S1369-5274(99)80066-6
47. Lee CK, Rao DT, Gertner R, Gimeno R, Frey AB, Levy DE. Distinct requirements for IFNs and STAT1 in NK cell function. J Immunol (2000) 165:3571–7. doi:10.4049/jimmunol.165.7.3571
48. Nguyen KB, Salazar-Mather TP, Dalod MY, Van Deusen JB, Wei X, Liew FY, et al. Coordinated and distinct roles for IFN-alpha beta, IL-12, and IL-15 regulation of NK cell responses to viral infection. J Immunol (2002) 169:4279–87. doi:10.4049/jimmunol.169.8.4279
49. Smyth MJ, Taniguchi M, Street SE. The anti-tumor activity of IL-12: mechanisms of innate immunity that are model and dose dependent. J Immunol (2000) 165:2665–70. doi:10.4049/jimmunol.165.5.2665
50. Smyth MJ, Crowe NY, Godfrey DI. NK cells and NKT cells collaborate in host protection from methylcholanthrene-induced fibrosarcoma. Int Immunol (2001) 13:459–63. doi:10.1093/intimm/13.4.459
51. Smyth MJ, Swann J, Kelly JM, Cretney E, Yokoyama WM, Diefenbach A, et al. NKG2D recognition and perforin effector function mediate effective cytokine immunotherapy of cancer. J Exp Med (2004) 200:1325–35. doi:10.1084/jem.20041522
52. Brady J, Hayakawa Y, Smyth MJ, Nutt SL. IL-21 induces the functional maturation of murine NK cells. J Immunol (2004) 172:2048–58. doi:10.4049/jimmunol.172.4.2048
53. Swann JB, Hayakawa Y, Zerafa N, Sheehan KCF, Scott B, Schreiber RD, et al. Type I IFN contributes to NK cell homeostasis, activation, and antitumor function. J Immunol (2007) 178:7540–9. doi:10.4049/jimmunol.178.12.7540
54. Sun JC, Lanier LL. NK cell development, homeostasis and function: parallels with CD8+ T cells. Nat Rev Immunol (2011) 11:645–57. doi:10.1038/nri3044
55. Huntington ND, Vosshenrich CAJ, Di Santo JP. Developmental pathways that generate natural-killer-cell diversity in mice and humans. Nat Rev Immunol (2007) 7:703–14. doi:10.1038/nri2154
56. Carotta S, Pang SHM, Nutt SL, Belz GT. Identification of the earliest NK-cell precursor in the mouse BM. Blood (2011) 117:5449–52. doi:10.1182/blood-2010-11-318956
57. Yu J, Freud AG, Caligiuri MA. Location and cellular stages of natural killer cell development. Trends Immunol (2013) 34:573–82. doi:10.1016/j.it.2013.07.005
58. Spits H, Artis D, Colonna M, Diefenbach A, Di Santo JP, Eberl G, et al. Innate lymphoid cells – a proposal for uniform nomenclature. Nat Rev Immunol (2013) 13:145–9. doi:10.1038/nri3365
59. Huntington ND, Nutt SL, Carotta S. Regulation of murine natural killer cell commitment. Front Immunol (2013) 4:14. doi:10.3389/fimmu.2013.00014
60. Geiger TL, Sun JC. Development and maturation of natural killer cells. Curr Opin Immunol (2016) 39:82–9. doi:10.1016/j.coi.2016.01.007
61. Chiossone L, Chaix J, Fuseri N, Roth C, Vivier E, Walzer T. Maturation of mouse NK cells is a 4-stage developmental program. Blood (2009) 113:5488–96. doi:10.1182/blood-2008-10-187179
62. Hayakawa Y, Smyth MJ. CD27 dissects mature NK cells into two subsets with distinct responsiveness and migratory capacity. J Immunol (2006) 176:1517–24. doi:10.4049/jimmunol.176.3.1517
63. Huntington ND, Tabarias H, Fairfax K, Brady J, Hayakawa Y, Degli-Esposti MA, et al. NK cell maturation and peripheral homeostasis is associated with KLRG1 up-regulation. J Immunol (2007) 178:4764–70. doi:10.4049/jimmunol.178.8.4764
64. Takeda K, Cretney E, Hayakawa Y, Ota T, Akiba H, Ogasawara K, et al. TRAIL identifies immature natural killer cells in newborn mice and adult mouse liver. Blood (2005) 105:2082–9. doi:10.1182/blood-2004-08-3262
65. Mizutani T, Neugebauer N, Putz EM, Moritz N, Simma O, Zebedin-Brandl E, et al. Conditional IFNAR1 ablation reveals distinct requirements of type I IFN signaling for NK cell maturation and tumor surveillance. Oncoimmunology (2012) 1:1027–37. doi:10.4161/onci.21284
66. Guan J, Miah SMS, Wilson ZS, Erick TK, Banh C, Brossay L. Role of type I interferon receptor signaling on NK cell development and functions. PLoS One (2014) 9:e111302. doi:10.1371/journal.pone.0111302
67. O’Sullivan TE, Sun JC, Lanier LL. Natural killer cell memory. Immunity (2015) 43:634–45. doi:10.1016/j.immuni.2015.09.013
68. Cerwenka A, Lanier LL. Natural killer cell memory in infection, inflammation and cancer. Nat Rev Immunol (2016) 16:112–23. doi:10.1038/nri.2015.9
69. Majewska-Szczepanik M, Paust S, von Andrian UH, Askenase PW, Szczepanik M. Natural killer cell-mediated contact sensitivity develops rapidly and depends on interferon-α, interferon-γ and interleukin-12. Immunology (2013) 140:98–110. doi:10.1111/imm.12120
70. Biron CA, Tarrio ML. Immunoregulatory cytokine networks: 60 years of learning from murine cytomegalovirus. Med Microbiol Immunol (2015) 204:345–54. doi:10.1007/s00430-015-0412-3
71. Beverley PCL, Ruzsics Z, Hey A, Hutchings C, Boos S, Bolinger B, et al. A novel murine cytomegalovirus vaccine vector protects against Mycobacterium tuberculosis. J Immunol (2014) 193:2306–16. doi:10.4049/jimmunol.1302523
72. Beaulieu AM, Zawislak CL, Nakayama T, Sun JC. The transcription factor Zbtb32 controls the proliferative burst of virus-specific natural killer cells responding to infection. Nat Immunol (2014) 15:546–53. doi:10.1038/ni.2876
73. Madera S, Rapp M, Firth MA, Beilke JN, Lanier LL, Sun JC. Type I IFN promotes NK cell expansion during viral infection by protecting NK cells against fratricide. J Exp Med (2016) 213:225–33. doi:10.1084/jem.20150712
74. Lawler C, Tan CSE, Simas JP, Stevenson PG. Type I interferons and NK cells restrict gammaherpesvirus lymph node infection. J Virol (2016) 90:9046–57. doi:10.1128/JVI.01108-16
75. Stoiber D, Kovacic B, Schuster C, Schellack C, Karaghiosoff M, Kreibich R, et al. TYK2 is a key regulator of the surveillance of B lymphoid tumors. J Clin Invest (2004) 114:1650–8. doi:10.1172/JCI22315
76. Kovacic B, Stoiber D, Moriggl R, Weisz E, Ott RG, Kreibich R, et al. STAT1 acts as a tumor promoter for leukemia development. Cancer Cell (2006) 10:77–87. doi:10.1016/j.ccr.2006.05.025
77. Eckelhart E, Warsch W, Zebedin E, Simma O, Stoiber D, Kolbe T, et al. A novel Ncr1-Cre mouse reveals the essential role of STAT5 for NK-cell survival and development. Blood (2011) 117:1565–73. doi:10.1182/blood-2010-06-291633
78. Marçais A, Viel S, Grau M, Henry T, Marvel J, Walzer T. Regulation of mouse NK cell development and function by cytokines. Front Immunol (2013) 4:450. doi:10.3389/fimmu.2013.00450
79. Croce M, Rigo V, Ferrini S. IL-21: a pleiotropic cytokine with potential applications in oncology. J Immunol Res (2015) 2015:1–15. doi:10.1155/2015/696578
80. Lucas M, Schachterle W, Oberle K, Aichele P, Diefenbach A. Dendritic cells prime natural killer cells by trans-presenting interleukin 15. Immunity (2007) 26:503–17. doi:10.1016/j.immuni.2007.03.006
81. Fehniger TA, Cai SF, Cao X, Bredemeyer AJ, Presti RM, French AR, et al. Acquisition of murine NK cell cytotoxicity requires the translation of a pre-existing pool of granzyme B and perforin mRNAs. Immunity (2007) 26:798–811. doi:10.1016/j.immuni.2007.04.010
82. Degli-Esposti MA, Smyth MJ. Close encounters of different kinds: dendritic cells and NK cells take centre stage. Nat Rev Immunol (2005) 5:112–24. doi:10.1038/nri1549
83. Verbist KC, Klonowski KD. Functions of IL-15 in anti-viral immunity: multiplicity and variety. Cytokine (2012) 59:467–78. doi:10.1016/j.cyto.2012.05.020
84. Lodolce JP, Boone DL, Chai S, Swain RE, Dassopoulos T, Trettin S, et al. IL-15 receptor maintains lymphoid homeostasis by supporting lymphocyte homing and proliferation. Immunity (1998) 9:669–76. doi:10.1016/S1074-7613(00)80664-0
85. Fernandez NC, Lozier A, Flament C, Ricciardi-Castagnoli P, Bellet D, Suter M, et al. Dendritic cells directly trigger NK cell functions: cross-talk relevant in innate anti-tumor immune responses in vivo. Nat Med (1999) 5:405–11. doi:10.1038/7403
86. van den Broeke LT, Daschbach E, Thomas EK, Andringa G, Berzofsky JA. Dendritic cell-induced activation of adaptive and innate antitumor immunity. J Immunol (2003) 171:5842–52. doi:10.4049/jimmunol.171.11.5842
87. Chiba S, Ikushima H, Ueki H, Yanai H, Kimura Y, Hangai S, et al. Recognition of tumor cells by dectin-1 orchestrates innate immune cells for anti-tumor responses. Elife (2014) 3:e04177. doi:10.7554/eLife.04177
88. Ebihara T, Azuma M, Oshiumi H, Kasamatsu J, Iwabuchi K, Matsumoto K, et al. Identification of a polyI:C-inducible membrane protein that participates in dendritic cell-mediated natural killer cell activation. J Exp Med (2010) 207:2675–87. doi:10.1084/jem.20091573
89. Crouse J, Bedenikovic G, Wiesel M, Ibberson M, Xenarios I, Von Laer D, et al. Type I interferons protect T cells against NK cell attack mediated by the activating receptor NCR1. Immunity (2014) 40:961–73. doi:10.1016/j.immuni.2014.05.003
90. Xu HC, Grusdat M, Pandyra AA, Polz R, Huang J, Sharma P, et al. Type I interferon protects antiviral CD8+ T cells from NK cell cytotoxicity. Immunity (2014) 40:949–60. doi:10.1016/j.immuni.2014.05.004
91. Moro H, Otero DC, Tanabe Y, David M. T cell-intrinsic and -extrinsic contributions of the IFNAR/STAT1-axis to thymocyte survival. PLoS One (2011) 6:e24972. doi:10.1371/journal.pone.0024972
92. Zietara N, Łyszkiewicz M, Gekara N, Puchałka J, Dos Santos VAPM, Hunt CR, et al. Absence of IFN-beta impairs antigen presentation capacity of splenic dendritic cells via down-regulation of heat shock protein 70. J Immunol (2009) 183:1099–109. doi:10.4049/jimmunol.0803214
93. Lee CK, Gimeno R, Levy DE. Differential regulation of constitutive major histocompatibility complex class I expression in T and B lymphocytes. J Exp Med (1999) 190:1451–64. doi:10.1084/jem.190.10.1451
94. Zamai L, Ahmad M, Bennett IM, Azzoni L, Alnemri ES, Perussia B. Natural killer (NK) cell-mediated cytotoxicity: differential use of TRAIL and Fas ligand by immature and mature primary human NK cells. J Exp Med (1998) 188:2375–80. doi:10.1084/jem.188.12.2375
95. Kayagaki N, Yamaguchi N, Nakayama M, Takeda K, Akiba H, Tsutsui H, et al. Expression and function of TNF-related apoptosis-inducing ligand on murine activated NK cells. J Immunol (1999) 163:1906–13.
96. Takeda K, Hayakawa Y, Smyth MJ, Kayagaki N, Yamaguchi N, Kakuta S, et al. Involvement of tumor necrosis factor-related apoptosis-inducing ligand in surveillance of tumor metastasis by liver natural killer cells. Nat Med (2001) 7:94–100. doi:10.1038/83416
97. Cretney E, Takeda K, Yagita H, Glaccum M, Peschon JJ, Smyth MJ. Increased susceptibility to tumor initiation and metastasis in TNF-related apoptosis-inducing ligand-deficient mice. J Immunol (2002) 168:1356–61. doi:10.4049/jimmunol.168.3.1356
98. Sato K, Hida S, Takayanagi H, Yokochi T, Kayagaki N, Takeda K, et al. Antiviral response by natural killer cells through TRAIL gene induction by IFN-alpha/beta. Eur J Immunol (2001) 31:3138–46. doi:10.1002/1521-4141(200111)31:11<3138::AID-IMMU3138>3.0.CO;2-B
99. Yu Q, Katlinskaya YV, Carbone CJ, Zhao B, Katlinski KV, Zheng H, et al. DNA-damage-induced type I interferon promotes senescence and inhibits stem cell function. Cell Rep (2015) 11:785–97. doi:10.1016/j.celrep.2015.03.069
100. Katlinskaya YV, Carbone CJ, Yu Q, Fuchs SY. Type 1 interferons contribute to the clearance of senescent cell. Cancer Biol Ther (2015) 16:1214–9. doi:10.1080/15384047.2015.1056419
101. Bui JD, Carayannopoulos LN, Lanier LL, Yokoyama WM, Schreiber RD. IFN-dependent down-regulation of the NKG2D ligand H60 on tumors. J Immunol (2006) 176:905–13. doi:10.4049/jimmunol.176.2.905
102. Schreiner B, Mitsdoerffer M, Kieseier BC, Chen L, Hartung H-P, Weller M, et al. Interferon-beta enhances monocyte and dendritic cell expression of B7-H1 (PD-L1), a strong inhibitor of autologous T-cell activation: relevance for the immune modulatory effect in multiple sclerosis. J Neuroimmunol (2004) 155:172–82. doi:10.1016/j.jneuroim.2004.06.013
103. Eppihimer MJ, Gunn J, Freeman GJ, Greenfield EA, Chernova T, Erickson J, et al. Expression and regulation of the PD-L1 immunoinhibitory molecule on microvascular endothelial cells. Microcirculation (2002) 9:133–45. doi:10.1038/sj/mn/7800123
104. Bidwell BN, Slaney CY, Withana NP, Forster S, Cao Y, Loi S, et al. Silencing of IRF7 pathways in breast cancer cells promotes bone metastasis through immune escape. Nat Med (2012) 18:1224–31. doi:10.1038/nm.2830
105. Eyles J, Puaux A-L, Wang X, Toh B, Prakash C, Hong M, et al. Tumor cells disseminate early, but immunosurveillance limits metastatic outgrowth, in a mouse model of melanoma. J Clin Invest (2010) 120:2030–9. doi:10.1172/JCI42002
106. Paolino M, Choidas A, Wallner S, Pranjic B, Uribesalgo I, Loeser S, et al. The E3 ligase Cbl-b and TAM receptors regulate cancer metastasis via natural killer cells. Nature (2014) 507:508–12. doi:10.1038/nature12998
107. Rautela J, Baschuk N, Slaney CY, Jayatilleke KM, Xiao K, Bidwell BN, et al. Loss of host type-I IFN signaling accelerates metastasis and impairs NK-cell antitumor function in multiple models of breast cancer. Cancer Immunol Res (2015) 3:1207–17. doi:10.1158/2326-6066.CIR-15-0065
108. Kitamura T, Qian B-Z, Pollard JW. Immune cell promotion of metastasis. Nat Rev Immunol (2015) 15:73–86. doi:10.1038/nri3789
109. Sceneay J, Chow MT, Chen A, Halse HM, Wong CSF, Andrews DM, et al. Primary tumor hypoxia recruits CD11b+/Ly6Cmed/Ly6G+ immune suppressor cells and compromises NK cell cytotoxicity in the premetastatic niche. Cancer Res (2012) 72:3906–11. doi:10.1158/0008-5472.CAN-11-3873
110. Morvan MG, Lanier LL. NK cells and cancer: you can teach innate cells new tricks. Nat Rev Cancer (2016) 16:7–19. doi:10.1038/nrc.2015.5
111. Ferrantini M, Capone I, Belardelli F. Interferon-α and cancer: mechanisms of action and new perspectives of clinical use. Biochimie (2007) 89:884–93. doi:10.1016/j.biochi.2007.04.006
112. Rizza P, Moretti F, Belardelli F. Recent advances on the immunomodulatory effects of IFN-alpha: implications for cancer immunotherapy and autoimmunity. Autoimmunity (2010) 43:204–9. doi:10.3109/08916930903510880
113. Riley CH, Hansen M, Brimnes MK, Hasselbalch HC, Bjerrum OW, Straten PT, et al. Expansion of circulating CD56bright natural killer cells in patients with JAK2-positive chronic myeloproliferative neoplasms during treatment with interferon-α. Eur J Haematol (2015) 94:227–34. doi:10.1111/ejh.12420
114. Talpaz M, Hehlmann R, Quintás-Cardama A, Mercer J, Cortes J. Re-emergence of interferon-α in the treatment of chronic myeloid leukemia. Leukemia (2013) 27:803–12. doi:10.1038/leu.2012.313
115. Zitvogel L, Galluzzi L, Smyth MJ, Kroemer G. Mechanism of action of conventional and targeted anticancer therapies: reinstating immunosurveillance. Immunity (2013) 39:74–88. doi:10.1016/j.immuni.2013.06.014
116. Saudemont A, Jouy N, Hetuin D, Quesnel B. NK cells that are activated by CXCL10 can kill dormant tumor cells that resist CTL-mediated lysis and can express B7-H1 that stimulates T cells. Blood (2005) 105:2428–35. doi:10.1182/blood-2004-09-3458
117. Wennerberg E, Kremer V, Childs R, Lundqvist A. CXCL10-induced migration of adoptively transferred human natural killer cells toward solid tumors causes regression of tumor growth in vivo. Cancer Immunol Immunother (2015) 64:225–35. doi:10.1007/s00262-014-1629-5
118. Terawaki S, Chikuma S, Shibayama S, Hayashi T, Yoshida T, Okazaki T, et al. IFN-α directly promotes programmed cell death-1 transcription and limits the duration of T cell-mediated immunity. J Immunol (2011) 186:2772–9. doi:10.4049/jimmunol.1003208
119. Benson DM, Bakan CE, Mishra A, Hofmeister CC, Efebera Y, Becknell B, et al. The PD-1/PD-L1 axis modulates the natural killer cell versus multiple myeloma effect: a therapeutic target for CT-011, a novel monoclonal anti-PD-1 antibody. Blood (2010) 116:2286–94. doi:10.1182/blood-2010-02-271874
120. Concha-Benavente F, Srivastava RM, Trivedi S, Lei Y, Chandran U, Seethala RR, et al. Identification of the cell-intrinsic and -extrinsic pathways downstream of EGFR and IFNγ that induce PD-L1 expression in head and neck cancer. Cancer Res (2016) 76:1031–43. doi:10.1158/0008-5472.CAN-15-2001
121. Zaretsky JM, Garcia-Diaz A, Shin DS, Escuin-Ordinas H, Hugo W, Hu-Lieskovan S, et al. Mutations associated with acquired resistance to PD-1 blockade in melanoma. N Engl J Med (2016) 375:819–29. doi:10.1056/NEJMoa1604958
122. Zamarin D, Holmgaard RB, Subudhi SK, Park JS, Mansour M, Palese P, et al. Localized oncolytic virotherapy overcomes systemic tumor resistance to immune checkpoint blockade immunotherapy. Sci Transl Med (2014) 6:226ra32. doi:10.1126/scitranslmed.3008095
123. Jiang W, Zhang C, Tian Z, Zhang J. hIFN-α gene modification augments human natural killer cell line anti-human hepatocellular carcinoma function. Gene Ther (2013) 20:1062–9. doi:10.1038/gt.2013.31
124. Nagler A, Ackerstein A, Or R, Naparstek E, Slavin S. Immunotherapy with recombinant human interleukin-2 and recombinant interferon-alpha in lymphoma patients postautologous marrow or stem cell transplantation. Blood (1997) 89:3951–9.
Keywords: type I interferon, interferon signaling, natural killer cells, tumor surveillance, innate immunity, tumor microenvironment, anticancer therapy
Citation: Müller L, Aigner P and Stoiber D (2017) Type I Interferons and Natural Killer Cell Regulation in Cancer. Front. Immunol. 8:304. doi: 10.3389/fimmu.2017.00304
Received: 15 December 2016; Accepted: 03 March 2017;
Published: 31 March 2017
Edited by:
Jorg Hermann Fritz, McGill University, CanadaCopyright: © 2017 Müller, Aigner and Stoiber. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Dagmar Stoiber, ZGFnbWFyLnN0b2liZXJAbWVkdW5pd2llbi5hYy5hdA==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.