 Bas C. Mourik
Bas C. Mourik Erik Lubberts
Erik Lubberts Jurriaan E. M. de Steenwinkel
Jurriaan E. M. de Steenwinkel Tom H. M. Ottenhoff
Tom H. M. Ottenhoff Pieter J. M. Leenen
Pieter J. M. Leenen- 1Department of Medical Microbiology and Infectious Diseases, Erasmus University Medical Center, Rotterdam, Netherlands
- 2Department of Rheumatology, Erasmus University Medical Center, Rotterdam, Netherlands
- 3Department of Infectious Diseases, Leiden University Medical Center, Leiden, Netherlands
- 4Department of Immunology, Erasmus University Medical Center, Rotterdam, Netherlands
The classical paradigm of tuberculosis (TB) immunity, with a central protective role for Th1 responses and IFN-γ-stimulated cellular responses, has been challenged by unsatisfactory results of vaccine strategies aimed at enhancing Th1 immunity. Moreover, preclinical TB models have shown that increasing IFN-γ responses in the lungs is more damaging to the host than to the pathogen. Type 1 interferon signaling and altered Th17 responses have also been associated with active TB, but their functional roles in TB pathogenesis remain to be established. These two host responses have been studied in more detail in autoimmune diseases (AID) and show functional interactions that are of potential interest in TB immunity. In this review, we first identify the role of type 1 interferons and Th17 immunity in TB, followed by an overview of interactions between these responses observed in systemic AID. We discuss (i) the effects of GM-CSF-secreting Th17.1 cells and type 1 interferons on CCR2+ monocytes; (ii) convergence of IL-17 and type 1 interferon signaling on stimulating B-cell activating factor production and the central role of neutrophils in this process; and (iii) synergy between IL-17 and type 1 interferons in the generation and function of tertiary lymphoid structures and the associated follicular helper T-cell responses. Evaluation of these autoimmune-related pathways in TB pathogenesis provides a new perspective on recent developments in TB research.
1. Introduction
Tuberculosis (TB) has been responsible for an estimated one billion deaths worldwide over the last 200 years (1), which is more than any other infectious disease caused by a single pathogen. Given its global magnitude, it has been hypothesized that TB particularly contributed to the genetic selective pressure that predisposes for development of autoimmune diseases (AID) (2). This is supported by polymorphism studies of the TNF gene, which show an opposite association between susceptibility to TB vs. susceptibility to several AID (3). Additionally, a gender-dependent predisposition to either TB or AID exists with a male predominance among TB patients (4) opposed to increased AID incidences in women (5). The general concept of an inverse relation between infectious diseases and AID is best described by the hygiene hypothesis, which states that diminished exposure to infectious pathogens during childhood increases the chances of developing AID and allergies (6, 7). Also, epidemiologically, the decline in burden of infectious diseases over the last century in industrialized countries is accompanied by increasing rates of AID (8).
Despite support for an inverse relation, similarities between TB and AID have also been identified. TB is even hypothesized to be an infection-induced AID based on the observation that diverse clinical autoimmune phenomena frequently occur in TB patients (9, 10). Furthermore, up to 32% of patients with active TB have elevated autoantibody titers (11, 12). Rational explanations for these findings could be that either TB or AID activate common immunological pathways (10), or protective immunity in TB increases the chance to develop AID (2). In both scenarios, key findings in AID immunology could potentially contribute to our understanding of TB pathogenesis.
The current paradigm of the host response to Mtb infection is summarized in Figure 1. The indispensable role of IL-12/IFN-γ-mediated Th1 immunity against Mtb has long been recognized (13). However, stimulating Th1 immunity in TB can also result in excessive inflammation (see Box 1). More recently, the contributions of additional immune pathways have been explored, especially the role of type I interferons (T1-IFNs), Th17 immunity (14, 15), and unconventional T cell immunity (16–18). Little is known about the potential interaction between T1-IFNs and Th17 responses in TB, but interesting observations in this regard have been reported for multiple AID (19–21). To determine if these findings are relevant for the understanding of TB pathogenesis, we first review the separate involvements of T1-IFNs and Th17 responses in TB pathogenesis in Sections 2 and 3, respectively. Next, their known interactions in AID are discussed in Section 4. Finally, in Section 5, the potential relevance of these interacting pathways in TB is assessed and integrated into the current understanding of TB pathogenesis.
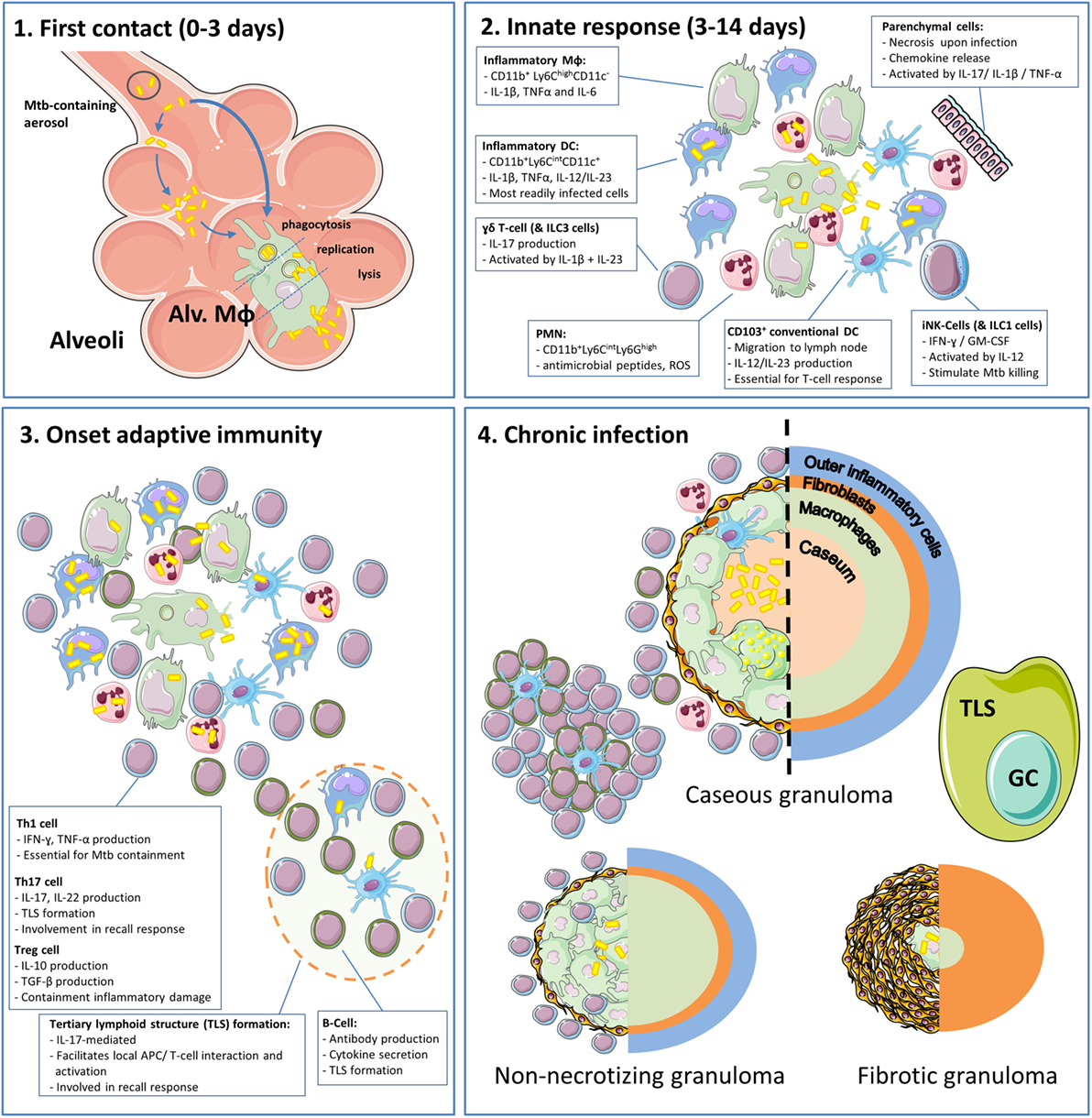
Figure 1. The phases and cell types involved in the immune response to tuberculosis (TB) in the lungs. (1) Inhaled Mtb-containing aerosols are deposited deep into the lung, reaching the alveoli (22). Within the alveoli, Mtb are phagocytosed by alveolar macrophages (Alv. MΦ) or infect alveolar epithelial cells prior to ending up in alveolar macrophages (23). Within Alv. MΦ, the bacteria are able to inhibit phagosome–lysosome fusion and replicate until cell lysis ensues, which takes approximately 3–5 days (24). (2) After the initial contact, Mtb encounters infiltrating myeloid cells of which inflammatory dendritic cells and PMN are most readily infected (13, 25). During these early phases, invariate natural killer (iNK) cells and type 1 innate lymphoid cells (ILC1) produce IFN-γ in response to IL-12 and stimulate myeloid cells to kill phagocytosed Mtb. In addition, γδ T-cells and ILC3 produce IL-17. There is increasing appreciation for the role of tertiary lymphoid structures (TLS) and their associated germinal centers (GC) that arise under influence of IL-17 and facilitate optimal activation of myeloid cells and efficient recall responses. During this process, loosely aggregated “innate granulomas” are already formed (26). It should be noted that the roles of ILC1s and ILC3s are based on their general function, which has not yet been formally demonstrated in TB (27). (3) Onset of adaptive immunity in Mtb infection is delayed to circa 14 days in mice and up to 6 weeks in humans (13, 22). At this point, distinct T-cell subsets and B-cells migrate to the site of infection and execute their different effector functions. (4) After onset of adaptive immunity, 90–97% of infected individuals develop sustained infection without clinical symptoms termed “latent TB infection” (LTBI) (13). LTBI was initially considered a static phase, but it is now known that this stage is hallmarked by the presence of granulomas in various stages (caseous, non-caseous, and fibrotic) and an ongoing balance between antimycobacterial activity and regulatory mechanisms to minimize immunopathology (13, 28). Cell phenotypes are as present in mouse TB models.
Box 1 The dual faces of IFN-γ in tuberculosis (TB) immunity.
In the current paradigm of a successful host response, lung DCs migrate to the draining lymph node after Mtb contact and induce a robust IL-12-mediated Th1 response (13). This results in migration of IFN-γ-producing CD4+ T-cells to the site of infection. Subsequently, activation of macrophages by IFN-γ results in killing of intracellular Mtb, while activated CD8+ T-cells lyse infected host cells. Conversely, unsuccessful clearance of infection is due to poor activation of adaptive immunity. This can result from insufficient antigen presentation (29), or from the action of regulatory factors that interfere with Th1 responses such as IL-10 or PDL1-PD1 interaction (13). Paradoxically, the current vaccine bacillus Calmette–Guérin (BCG) induces a strong Th1 response but is only partially effective in protecting against TB (30). Boosting the Th1-inducing potential of BCG by using a modified Ankara virus also has yielded disappointing results (31, 32). Thus, solely stimulating Th1 immunity might not be the solution in TB prevention. This is confirmed in a mouse TB study showing that increasing IFN-γ production by T-cells in the lungs is detrimental to the host due to hyper-inflammation that requires PD-1-mediated suppression to limit pathology (33). In line with this, Mtb-infected mice deficient in PD-1, or mice in which PD-1 is selectively inhibited, display excessive inflammation and disease progression (34, 35). Finally, ex vivo studies in human monocyte-derived macrophages show that protective effects of IFN-γ are dependent on multiple factors including time of contact, concentration, and the magnitude of the ensuing microbial challenge (36). Based on these observations, it can be concluded that boosting IFN-γ production and Th1 immunity in TB, besides potentially enhancing protection, can also result in unbalanced inflammation in the lungs that is more harmful to the host than to the pathogen. This emphasizes the need for involvement of additional immunological pathways for optimal protection.
2. T1-IFNs in TB
Type I interferons comprise a family of 13 IFN-α subtypes, IFN-β, IFN-ε, IFN-κ, and IFN-ω, which have the shared ability to bind to the IFN-α/β receptor (IFNAR) (37). Other interferons include the single type II interferon, interferon-γ, and the type III interferon family covering three IFN-λ types.
All nucleated cell types are capable of both producing T1-IFNs and responding to them, while type II/III interferons are mostly produced by leukocytes (37). The main function of T1-IFNs is to “interfere” with intracellular infections. Therefore, T1-IFN expression is primarily induced through cytoplasmic pattern recognition receptors (PRRs) and endosomal toll-like receptors (TLRs), which activate distinct interferon regulatory factors (IRFs) that act as transcription factors enabling expression of interferon-responsive genes (38). In contrast, extracellular pathogens trigger surface-bound TLRs that preferentially induce IL-1β and TNF-α through activation of NF-κB.
The role of T1-IFNs in infectious diseases is complex (15, 39–41). T1-IFNs boost the immune system upon pathogen encounter by activating dendritic cells and NK cells and by stimulating both B-cell responses and CD4+/CD8+ T-cell responses. However, T1-IFNs can also induce anti-inflammatory responses to control immune-mediated tissue damage during chronic infections. These contradictory effects of T1-IFNs in different situations can likely be ascribed to the heterogeneity of the T1-IFNs family, downstream activation of different STAT homo/heterodimers after binding to IFNAR (38, 42) and to differential priming of cells prior to induction of T1-IFN signaling (43).
2.1. T1-IFNs in Human TB
When recombinant or purified T1-IFNs became available as therapeutic agents in the 1980s, different applications have been established based on their antiviral, immune-stimulating, and suppressive effects. These include treatment of viral infections (e.g., IFN-α treatment of hepatitis B/C infections), AID [e.g., IFN-β treatment for multiple sclerosis (MS)], and various malignancies (44). Based on their well-described immune-stimulating effect, the use of T1-IFNs as adjuvant to antibiotic treatment for patients with active TB has also been explored (see Table 1). All studies found a positive influence of adjuvant T1-IFN therapy on clinical outcomes in active TB (45–49). Conversely, IFN-α treatment without concomitant antibiotic treatment, e.g., for hepatitis C, has been described to cause reactivation of latent TB (50–57). While reactivation of latent TB and treatment of active TB are two distinct clinical situations, the latter finding suggests an unfavorable role for T1-IFNs in TB pathogenesis.
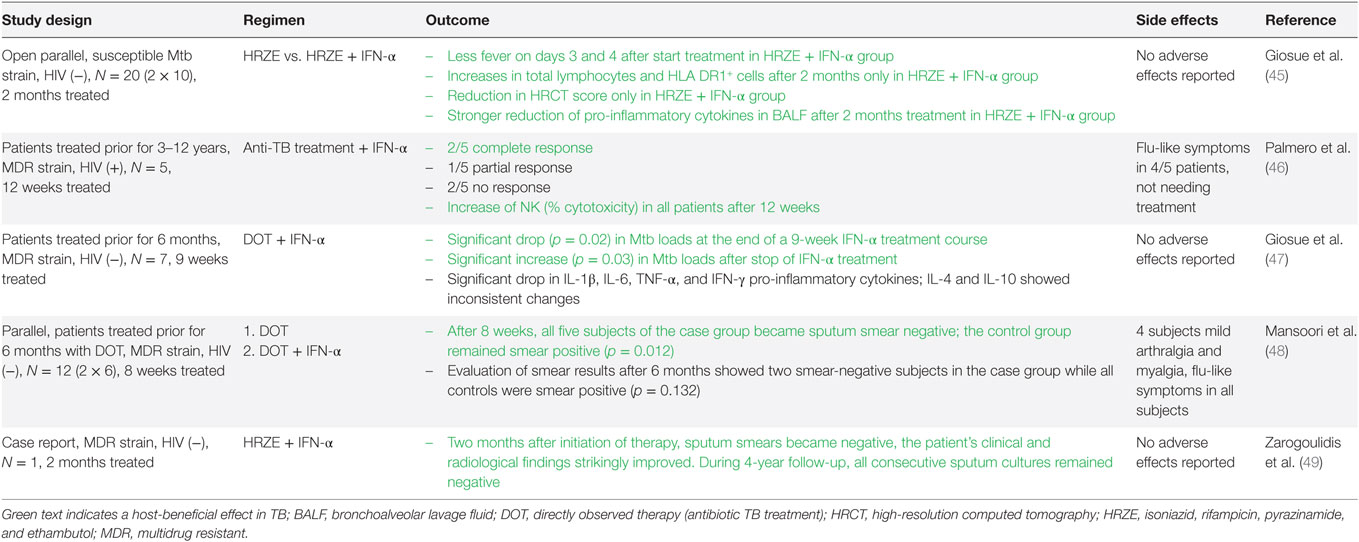
Table 1. Effect of type I interferons supplementation in human tuberculosis (TB).
In 2010, an interferon-inducible transcriptional signature was reported in circulating leukocytes of TB patients, thus linking increased T1-IFN signaling with active disease (58). This finding has been validated in several independent studies (59–62). A meta-analysis confirmed statistical significance but found a less dominant role for T1-IFN-related genes than expected (63). This is ascribed to the involvement of signaling components downstream of the T1-IFNs receptor in multiple overlapping intracellular pathways. Also, association studies do not necessarily implicate a causally detrimental effect of T1-IFNs in TB pathogenesis. In line with this, T1-IFN responses show potential as biomarkers or diagnostic tool for risk of active disease, but their functional involvement during TB progression in patients is not yet understood (62).
2.2. Preclinical Studies in Mice Support a Detrimental Role of T1-IFNs during Acute TB
A causal relationship between T1-IFN signaling and TB disease severity was first suggested in 2001 when IFN-α levels in the lungs of Mtb-infected mice were shown to be associated with Mtb strain virulence (64). Several approaches have been used to verify this relationship between increased T1-IFN signaling and unfavorable disease outcome. Blocking the T1-IFN signaling pathway through use of IFN-α/β receptor knockout (IFNAR−/−) improves survival, but only when applied on the background of mouse strains in which acute TB is lethal, such as the A129 strain (65). In IFNAR−/− mice with a relatively TB-resistant C57BL/6 background, survival rates were similar to wild-type mice, but mycobacterial loads in the lungs were lower (66–69). One study actually observed increased loads in the lungs (70) (Table 2).
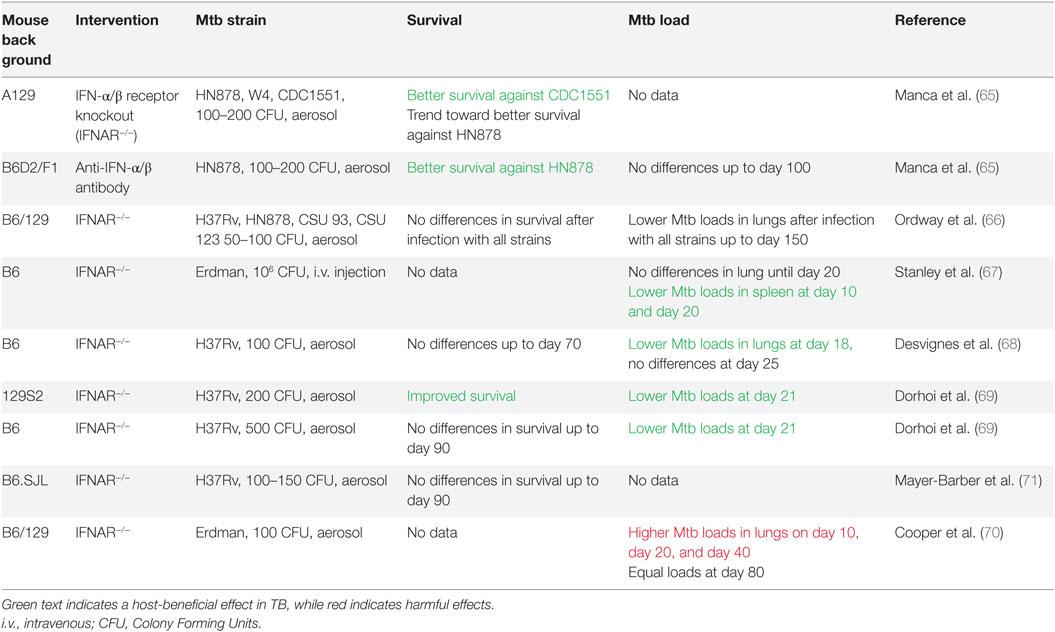
Table 2. Interference with T1-IFN signaling in preclinical tuberculosis (TB) studies.
In a second approach, Mtb-infected mice were supplemented with T1-IFNs after start of infection or treated with the TLR3-ligand poly-ICLC, which stimulates T1-IFN production and signaling (64, 72). Both studies showed increased mortality and higher mycobacterial loads in the supplemented groups, which were not observed when T1-IFNs or poly-ICLC were administered to Mtb-infected IFNAR−/− mice. Finally, in a third approach, mice were primed with a T1-IFN-inducing influenza virus prior to TB infection, which led to enhanced mycobacterial growth and reduced survival (73).
Enigmatically, reduced mycobacterial loads in IFNAR−/− mice are primarily observed in the acute phase of infection in which T1-IFNs are considered immune stimulating. No differences in survival or long-term control of infection were found in C57BL/6 IFNAR−/− mice compared to wild type. In support of this notion, T-cell analyses in several of the abovementioned studies convincingly excluded an effect of increased or decreased T1-IFN signaling on the adaptive immune response (68, 69, 72). Notably, none of these studies addressed the effect of T1-IFNs as adjunct treatment to antibiotics, which was shown to be beneficial in TB patients (Table 1).
2.3. Mtb Actively Induces T1-IFNs
Multiple studies indicate that Mtb employs both active and passive mechanisms to induce T1-IFNs (74–76). The mycobacterial ESAT-6 secretion system (ESX-1) and its 6 kDa early secretory antigenic target (ESAT-6) are essential in this process, as mycobacteria lacking ESX-1 fail to induce T1-IFN production (67, 77–81). ESAT-6 can disrupt the phagosomal membrane, which allows translocation of mycobacteria and mycobacterial products from the phagosome into the cytosol (78, 82).
Mycobacteria actively secrete several T1-IFN-inducing compounds, including double-stranded (ds)DNA and the bacterial second messenger cyclic-di-AMP (83). These compounds are recognized by different cytosolic PRRs, including cGAS (80), IFI-204 (78), AIM2 (84), and possibly NOD2 (77), although data on the latter are conflicting (67, 78). Activation of these cytosolic PRRs converges to activate “STimulator of INterferon Genes” (STING), which subsequently forms a complex with TANK-binding kinase 1 (79). This STING–TBK1 complex activates IRF3, leading to IFN-β production in mice (81) as well as human dendritic cells (74). IRF3−/− mice are poor producers of IFN-β and more resistant to Mtb infection, which supports a negative role for T1-IFNs in TB pathogenesis (78).
However, the overall picture is more complex. IRF3−/− mice are more resistant to Mtb infection, but mice deficient in the cytosolic PRR cGAS, upstream of IRF3, show diminished control of chronic Mtb infection (79). This can be traced back to a concomitant reduction in autophagy, which is also dependent on the cGAS-induced activation of the STING–TBK1 axis, but independent of IRF3. In line with this, mice infected with an Mtb strain that induces higher amounts of cyclic-di-AMP, thus stimulating both IRF3-mediated IFN-β production and STING–TBK1-mediated autophagy, show improved survival despite increased IFN-β levels (83). Taken together, this suggests that pro-mycobacterial effects of stimulating the cytosolic PRR/STING/IRF3/IFN-β axis by mycobacteria might be outweighed by the antimycobacterial effects of the PRR/STING/autophagy pathway.
Autocrine or paracrine IFN-β-signaling induces IRF7 and leads to the production of IFN-α in human dendritic cells (74). In line with this, injection of recombinant IFN-β in mice induces IFN-α production (85). Alternatively, myeloid cells and particularly plasmacytoid dendritic (pDC) cells are capable of directly activating IRF7-mediated IFN-α production after recognition of Mtb, particularly by endosomal TLR9 (86). In TB, this TLR9-IRF7 pathway is studied to lesser extent than the cytosolic PRR–IRF3 axis (87). This is possibly due to the dependence of T1-IFN-mediated pathogenic effects in mice on ESX-1, which induces IRF3 rather than IRF7 as explained above (67). However, IRF7 is recognized as commonly induced transcription factor by multiple clinical Mtb strains in alveolar epithelial cells (88). Moreover, TLR9−/− mice succumb earlier to high-dose Mtb infection than wild-type mice, which suggests a role for the TLR9/IRF7/IFN-α axis in TB as well (89).
2.4. T1-IFNs Drive the Influx of Mtb-Permissive Myeloid Cells during Acute Infection
Most studies in mouse TB models found significant functional effects of T1-IFNs specifically on CD11b+Gr1int myeloid cell populations (68, 69, 72). This population comprises monocyte-derived Ly6ChighCD11c−CCR2high inflammatory macrophages (iM) and Ly6CintCD11c+CCR2int inflammatory dendritic cells (iDC), but not CD11b+Gr1high PMN (90). This is an important distinction, as T1-IFNs actively inhibit PMN influx, as discussed in more detail in Section 2.4.3.
Inflammatory macrophages and iDC have been identified as major contributors to disease progression in mouse TB models (91–93). Several lines of evidence suggest that T1-IFNs regulate the influx of these cells and play a role in their functional impairment to resist Mtb. This interference with protective immunity is multifaceted and concerns four important interactions, which will be reviewed separately: (1) T1-IFNs mediate the influx of iM and iDC. (2) T1-IFNs inhibit IL-1β responses by these cells, which are essential in the initial host responses to Mtb. (3) Prolonged IL-1β signaling can also cause excessive inflammation and thus requires regulation during later phases. This can be mediated by T1-IFNs but also by IFN-γ through functionally different routes. (4) T1-IFNs and IFN-γ show a complex interplay in the activation of iM and iDC.
2.4.1. T1-IFNs Mediate the Influx of iM and iDC
Mtb-infected mice treated with the T1-IFN-inducing compound poly-ICLC show increased numbers of iM and iDC in the lungs, which are 10 times more permissive to Mtb infection than their counterparts in PBS-treated mice (72). Others confirmed that signaling through IFNAR indeed augments the recruitment of Mtb-permissive iM and iDC into the lungs (69). Mechanistically, IFNAR-dependent expression of the chemokine CCL2 mediates the influx of CCR2+ monocytes that differentiate into iM and iDC (72). Both myeloid and parenchymal cells can produce CCL2 in response to T1-IFNs, but parenchymal cells appear the main source of this chemokine (94–96). Expression of CCL2 is reduced in the lungs of IFNAR−/− mice, and the pathogenic effects of poly-ICLC treatment are absent in Mtb-infected CCR2−/− mice (72). Thus, preclinical TB studies indicate that T1-IFNs stimulate the influx of CCR2+ monocytes, but not PMN, to the site of infection in a CCR2-dependent way via the induction of CCL2 in parenchymal cells (74–76).
2.4.2. T1-IFNs Inhibit IL-1β Responses during Acute TB
Type I interferons not only stimulate the influx of CCR2+ monocytes but also stimulates their differentiation into Mtb-permissive iM and iDC (72, 75, 76). This can be traced back to a cross talk between T1-IFNs and IL-1β (71, 90). iM and iDC are the major sources of IL-1β in the lungs Mtb-infected mice, and IL-1β plays a crucial role in the acute host response to Mtb infection (71, 90). IL-1β augments TNF-α-stimulated Mtb killing and increases prostaglandin E2 (PGE2) production by upregulating cyclooxygenase-2 (COX2/PTGS2) (71, 97, 98). PGE2 is involved in control of intracellular Mtb replication but also prevents necrotic host cell death (99). In accordance, Ptgs2−/− mice, unable to produce PGE2, are more susceptible to Mtb infection than wild type mice, but to a lesser degree than IL1−/− mice. Further, information on PGE2 in TB is given in Box 2.
Box 2 The dual faces of prostaglandin E2 (PGE2) in tuberculosis (TB) immunity.
Prostaglandin E2 is generally considered a pro-inflammatory mediator and indispensable for the induction of fever, which is a hallmark symptom of active TB (100, 101). The anti-inflammatory effects of prostaglandin synthase (COX-) inhibitors such as NSAIDs underline this notion. However, high levels of PGE2 can also exert immunosuppressive effects as they stimulate alternative activation of macrophages (102), inhibit bactericidal activity (103), and promote production of IL-10 (104). Moreover, high PGE2 levels can stimulate the development of myeloid-derived suppressor cells with inhibitory effects on adaptive immune cells (104, 105). Finally, PGE2 inhibits IL-12 production by DCs and IFN-γ production by T-cells, thereby promoting Th2/Th17 immunity (106, 107).
In the serum and bronchoalveolar lavage fluid of TB patients, PGE2 levels were found to be elevated (71, 108, 109), and polymorphisms in the PGE2 receptor EP2 are associated with TB-susceptibility (110). Experimentally, one mouse study showed that low PGE2 levels in the acute phase of infection are essential for iNOS-mediated control of Mtb (111). Also, PGE2 plays an important role during acute TB since the PGE2-producing enzyme COX2 competes for arachidonic acid substrate with 5-lipoxygenase, which produces leukotrienes and lipoxins. Hereby, PGE2 prevents necrotic cell death thus benefiting the host (71). Opposed to the protective role of low PGE2 levels during acute disease, PGE2 levels are higher during the chronic phase of TB, and these concentrations contribute to disease by suppressing IFN-γ, TNF-α, and iNOS (111). Notably, the cellular source of PGE2 appears to differ between acute and chronic TB. During the acute phase of infection, inflammatory myeloid cells are the main source of PGE2, while foamy macrophages are strong producers of PGE2 during the chronic phase of disease (112). In line with a detrimental effect of high PGE2 levels in the chronic phase, foamy macrophages are typically associated with disease progression (113).
Type I interferons inhibit the expression and production of IL-1β and simultaneously increase the expression of 5-lipoxygenase (5-LO), which is a competitive enzyme for COX2 in the arachidonic acid metabolism (71, 90, 114, 115). As a result, IFNAR signaling causes a shift from COX2-mediated PGE2 production to an increase in the 5-LO products such as lipoxin A4 (LXA4) and leukotriene B4 (LTB4), which render cells more susceptible to necrotic cell death (71, 116). Pharmacological intervention in this process by administrating the 5-LO inhibitor Zileuton to Mtb-infected mice, improved disease outcomes during acute infection to similar extent as observed in IFNAR−/− mice (71). An overview on the balance between IL-1β and T1-IFNs is given in Figure 2.
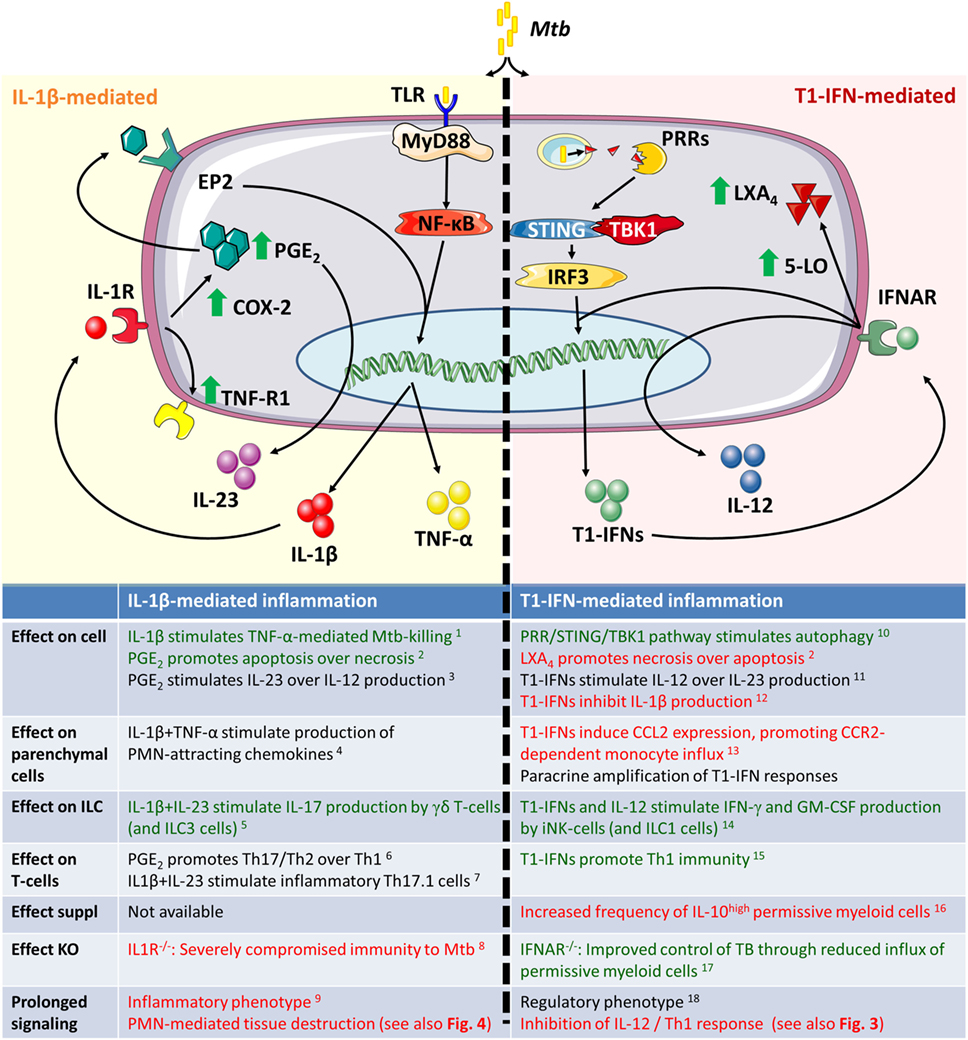
Figure 2. Inflammatory responses during acute infection in naïve inflammatory macrophages and dendritic cells. Green text indicates a beneficial host effect during Mtb infection and red indicates a detrimental effect. Mtb, Mycobacterium tuberculosis; PRR, pattern recognition receptor; STING, STimulator of INterferon Genes; TBK1, tank-binding kinase 1; IRF3, interferon regulatory factor 3; 5-LO, 5-lipoxygenase; COX-2, cyclooxygenase 2; PGE2, prostaglandin E2; EP2, prostaglandin E2 receptor 2; ILC3, innate lymphoid cells type 3. 1Jayaraman et al. (97), 2Chen et al. (116), 3Shi et al. (107), 4Di Paolo et al. (98), 5Lockhart et al. (117), 6Boniface et al. (106), 7El-Behi et al. (118), 8Fremond et al. (119), 9Mishra et al. (120), 10Watson et al. (81), 11Fleetwood et al. (94), 12Mayer-Barber et al. (71), 13Antonelli et al. (72), 14Une et al. (121), 15Longhi et al. (122), 16Manca et al. (64), 17Manca et al. (65), and 18Mayer-Barber et al. (90).
2.4.3. Prolonged IL-1β Signaling Causes PMN-Mediated Tissue Damage and Is Regulated by Both T1-IFNs and IFN-γ
The cross talk between T1-IFNs and IL-1β influences disease outcome in TB (71). However, this does not fully explain the harmful effects of T1-IFNs observed in TB. Most importantly, although IL-1β production is essential for protective immunity in the acute phase of disease in TB, it requires strict regulation as unchecked IL-1β signaling in TB can result in excessive PMN-mediated tissue damage (120, 123). Also, as explained in Box 2, IL-1β-mediated PGE2 production is protective during acute disease but appears to have a detrimental effect during chronic disease. Finally, inflammatory mediators associated with continuing infection, e.g., GM-CSF, predispose for IL-1β production over T1-IFNs by iM and iDC (36, 94, 124–126). This reflects an increasing need over time to limit IL-1β-mediated inflammatory responses.
To prevent PMN-mediated inflammation caused by excessive IL-1β signaling, the expression and production of IL-1β is inhibited not only by T1-IFNs but also by IFN-γ (90, 120). In line with this, both T1-IFNs and IFN-γ can inhibit PMN influx (127–131). T1-IFNs and IFN-γ can both reduce pro-IL-1β gene expression and increase the expression of soluble antagonists for the IL-1 receptor (114, 132).
Despite the abovementioned functional similarities between T1-IFNs and IFN-γ in IL-1β inhibition, mechanistic differences exist between these IFN types in mediating this effect.
Ex vivo studies in human iM and iDC demonstrate that IFN-β inhibits IL-1β production more potently than IFN-γ (90, 114). One explanation might be that IFN-γ inhibits IL-10, while T1-IFNs induce IL-10, which contributes to the inhibition of IL-1β production (90, 114, 115). Additionally, an IL-10-independent inhibition of IL-1β by T1-IFNs was recently identified (129). T1-IFNs induce cholesterol 25-hydroxylase, which potently reduces IL-1β transcription and broadly represses IL-1-activating inflammasomes. In contrast, IFN-γ inhibits IL-1β by increasing intracellular nitric oxide in an iNOS-dependent way (120). This prevents NLRP3 inflammasome activation and cleavage of pro-IL-1β into IL-1β. In contrast to the mechanisms exerted by T1-IFNs, IFN-γ-induced iNOS not only limits IL-1β-mediated inflammation but also markedly enhances the bactericidal potential of iM (120). Conversely, T1-IFNs suppress iNOS production (90). Based on the stimulation of iNOS by IFN-γ and the inhibition of iNOS by T1-IFNs, it appears that iDC are more sensitive to T1-IFN signaling and iM to IFN-γ when both types of interferon are present. T1-IFN-mediated inhibition of iNOS appears to occur primarily in iDC, since iDC only expressed iNOS in IFNAR−/− mice during viral infection, while iM appear more sensitive to IFN-γ and are the main source of iNOS in wild-type mice (131).
When taken together, these data suggest that IL-1β inhibition by either T1-IFNs or IFN-γ has strong implications on the bactericidal potential of iM and iDC. Furthermore, T1-IFNs interfere with the induction of iNOS by IFN-γ, particularly in iDC. This fits the observation that IFN-γ only inhibits IL-1β production by iM but not iDC in mouse TB models (90). Notably, iDC are the most readily infected cells in the lungs of Mtb-infected mice (25) and are present in larger numbers than iM during Mtb infection (72, 133).
2.4.4. The Interplay between T1-IFNs and IFN-γ
During direct contact with Mtb through TLRs, endogenous T1-IFN signaling through IRF3 promotes IL-12 production by iDC over IL-23 (see also Figure 2; Box 3) (94, 134, 135). This early IL-12 signaling is required to induce IFN-γ production by innate lymphoid cells (ILC) such as NK cells and possibly ILC1s (136, 137). However, exogeneous T1-IFNs or T1-IFN signaling in the absence of TLR stimulation can also inhibit IL-12 production by iDC (115, 138). This inhibition of IL-12 by T1-IFNs occurs particularly through induction of IL-10 (15). T1-IFNs also inhibit the responsiveness of iDC to IFN-γ-mediated activation, which is required for Mtb killing. This occurs partially by reducing the expression level of IFN-γ-receptor on the cell surface, but primarily through induction of an IL-10high regulatory phenotype in which antimicrobial pathways by IFN-γ are not readily activated, as discussed below (90, 115, 131, 139–141).
Box 3 IL-12 or IL-23 production by dendritic cells?
IL-12 and IL-23 are heterodimeric cytokines composed of a common p40 subunit, coupled with either a p35 subunit in IL-12 or a p19 subunit in IL-23. Both IL-12 and IL-23 are produced in particular by stimulated dendritic cells and to lesser degree by macrophages. The preferential production of IL-12 or IL-23 by these cells is multifactorial. Increased levels of PGE2 support IL-23 production over IL-12 (106, 107, 142). Activation of TLR2 and TLR4 also stimulates IL-23 production over IL-12, especially when NOD2 is simultaneously activated (143, 144). On the other hand, TLR9 and TLR3 agonists preferentially induce IL-12 (135, 145, 146). Downstream of PRRs, activation of IRF 4 and 5 favor induction of IL-23, while IRF 1, 3, and 7 induce IL-12 (135, 147). In line with this, T1-IFN-mediated IRF3 activation and IFN-γ-mediated IRF-1-activation both favor IL-12 production (148, 149).
IL-4 also favors IL-12 production and inhibits IL-23 production, especially in combination with IFN-γ or GM-CSF (150, 151). Finally, an important pathway that promotes IL-12 over IL-23 is ligation of the co-stimulatory molecule CD40 by CD40L on activated T-cells or by agonist antibodies (152). Taken together, IL-23 is induced in the presence of pathogens and innate signaling in the acute phase of infection, while onset of adaptive immunity with increased levels of IFN-γ and/or IL-4 shifts the balance toward IL-12 (153).
Recent findings might explain the mechanism behind this paradox where T1-IFNs initially support IL-12-mediated IFN-γ production by NK cells but can also induce an IL-10high phenotype in iDC, which interferes with IL-12 production and prevents IFN-γ-mediated activation. It has been observed in different mouse models, including Mtb-infected mice, that T1-IFNs can only induce an IL-10high regulatory phenotype in monocyte-derived DCs (iregDC) if these cells have been primed previously by IFN-γ (43). IFN-γ-primed DCs that did not receive T1-IFN signaling differentiated into iDC that stimulated robust T-cell responses. This phenomenon of monocyte priming by IFN-γ has been demonstrated to occur in the bone marrow (154). During gut infections, local production of IL-12 in mucosa-associated lymphoid tissue stimulates bone-marrow-resident NK cells to produce IFN-γ as early as 3 days post infection (154). This results in a uniform presence of an IFN-γ-primed signature of Ly6Chigh monocytes in the circulation at day 6. Furthermore, IFN-γ indeed primed these monocytes toward a regulatory phenotype, as they more effectively produced IL-10 in response to bacterial ligands (154). We speculate that a similar mechanism of IFN-γ priming is likely to be involved in pulmonary infections.
These data suggest interplay between T1-IFNs and IFN-γ as proposed in Figure 3. T1-IFNs initially induce IFN-γ responses by promoting IL-12 production in naïve cells as shown in Figure 2. These IL-12 responses stimulate IFN-γ production by ILC not only locally but also systemically, which results in IFN-γ priming of monocytes in the bone marrow. Once IFN-γ production is initiated, T1-IFNs mediate a regulatory function by inducing an IL-10high phenotype in IFN-γ-primed iDC. This prevents further production of IL-12 by these cells, inhibits their activation by IFN-γ, and results in an Mtb-permissive phenotype.
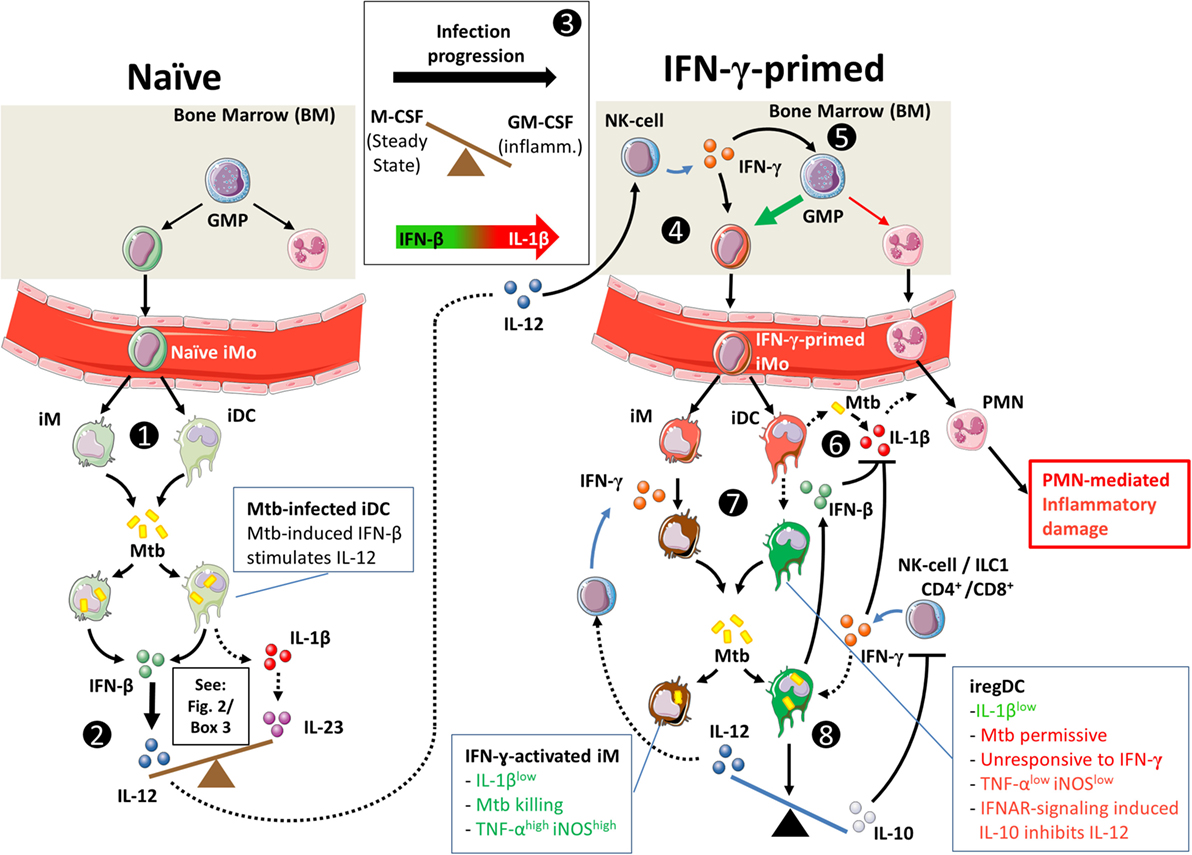
Figure 3. Hypothetical interplay between type I interferons (T1-IFNs) and IFN-γ in monocyte priming and shaping of the immune response in Mtb infection. Dashed lines indicate speculations in the context of pulmonary Mtb infection; solid lines indicate shown pathways in human and/or in animal models. (1) T1-IFN induces migration of CCR2+ monocytes (iMo) from the bone marrow to the lungs of Mtb-infected mice under influence of CCL2 (72). Locally, these cells develop into CD11b+Ly6Chigh inflammatory macrophages (iM) and CD11b+Ly6Cint inflammatory dendritic cells (iDC) (43). (2) As shown in Figure 1, naïve iM and iDC can initiate either IL-1β-mediated inflammation or T1-IFN-mediated inflammation. Mtb actively triggers intracellular pattern recognition receptors to induce a T1-IFN-mediated response. (3) Additionally, iM and iDC in the naïve situation have differentiated under influence of M-CSF, which makes them more responsive to T1-IFN signaling (94). During progression of Mtb infection, GM-CSF levels rise and increase the potential for IL-1β production by iM and iDC (94, 125, 155, 156). (4) Similar to the situation in gut infection, we propose that in tuberculosis (TB) IL-12 production in the lungs stimulates IFN-γ production by bone-marrow-resident NK cells, which locally primes monocytes (154). IFN-γ priming of monocyte-derived iDC is necessary for T1-IFNs to induce a regulatory (iregDC) phenotype in iDC in the lungs (43). (5) Additionally, IFN-γ stimulates monopoiesis over granulopoiesis by granulocyte/macrophage progenitor cells (128). (6) As Mtb infection progresses and GM-CSF levels increase, iM and iDC readily produce IL-1β [see also (3)] (124, 155), which can lead to PMN-mediated inflammatory damage in TB (120). (7) IL-1β production can be inhibited in response to either IFN-γ or IFN-β through mechanistically distinct pathways that differently affect Mtb killing. (8) Signaling through IFN-α/β receptor in IFN-γ-primed iDCs induces IL-10 production (43, 115, 131), inhibits IL-12 production (115), and makes these cells unresponsive to activation by IFN-γ (43, 115, 141), which together interfere with protective immunity during acute Mtb infection.
2.5. Summary: The Role of T1-IFNs in TB
Several modest clinical successes have been shown with IFN-α supplementation adjunct to antibiotic TB treatment (Table 1). However, case reports of TB reactivation under IFN-α treatment without concomitant antibiotics have put T1-IFNs in a negative spotlight (50–57). Furthermore, a T1-IFN transcriptional signature in circulating leukocytes is associated with active TB. Nevertheless, the functional role of T1-IFNs in TB patients remains to be determined (62).
Preclinical studies in mice support a detrimental role for T1-IFN in the acute phase of Mtb infection. T1-IFN signaling was associated with increased mortality in Mtb-susceptible mouse strains and higher Mtb loads in the lungs in most studies (Table 2).
However, it should be noted that most of these preclinical studies do not unequivocally support a harmful effect of T1-IFNs during the chronic phase of disease based on mortality, Mtb loads, or differences in adaptive immunity.
In support of a pathogenic role of T1-IFNs during acute infection, mycobacteria actively induce T1-IFNs by triggering cytosolic PRRs. This leads to IFN-β production in an IRF3-dependent way. Subsequently, T1-IFNs mediate the CCL2/CCR2-dependent migration of iM and iDC into the lungs (72). In these cells, interference of T1-IFNs with IL-1β and PGE2 as shown in Figure 2 can lead to an altered metabolism of arachidonic acids that leaves cells more vulnerable to necrotic cell death (71). However, sustained IL-1β signaling itself carries the risk of excessive inflammation in TB and not only T1-IFNs but also IFN-γ inhibits IL-1β to prevent excessive PMN-mediated inflammation (120). T1-IFNs inhibit IL-1β more effectively than IFN-γ but stimulate an IL-10high Mtb-permissive phenotype (72, 90).
Next to their shared ability to inhibit IL-1β, an interesting interplay between T1-IFNs and IFN-γ exists in TB as summarized in Figure 3. Two recent findings that are of particular interest include the observation that T1-IFNs can only induce an IL-10high phenotype in IFN-γ-primed cells (43) and the inductive role of T1-IFNs in early IL-12 signaling, which is required for IFN-γ priming in the bone marrow (154). Further, research into this complex interplay between T1-IFNs and IFN-γ during early host responses in TB would be highly interesting given the T1-IFN-inducing capacities of Mtb and the shaping effect of early T1-IFN or IFN-γ signaling on the ensuing immune response.
3. The Th17 Response in TB
As discussed in the previous paragraph, T1-IFNs induce IL-12 production by iDC, while IL-1β induces IL-23. Other factors also influence production of IL-12 or IL-23 (see Box 3). IL-12 is essential for the induction of IFN-γ responses in TB, but IL-1β is protective during acute TB despite inducing IL-23 over IL-12. Similar to the requirement of IL-12 for Th1 responses, IL-23 is essential for establishing Th17 immunity (157–159). Here, we review the effect of IL-23 signaling and the Th17 response in TB.
3.1. Introduction to the Th17 Response
The Th17 response is distinct from classical cell-mediated Th1 immunity or B-cell-stimulating Th2 responses and is often associated with a potent inflammatory response and tissue damage (159). Th17 cells display a high degree of plasticity and their ability to express signature markers of other T-helper lineages makes it difficult to establish their exact role in disease. Four different subsets of Th17 cells have been described to date with ranging inflammatory potential (160). On one side of the spectrum are highly inflammatory and often pathogenic IFN-γ/GM-CSF-producing Th17.1 cells that result from prolonged IL-1β and IL-23 signaling (161). On the other side are IL-10-producing Th17 cells, which can even transdifferentiate into regulatory T-cells and contribute to resolution of inflammation (162).
Despite the plasticity in cytokine production, IL-17 remains the hallmark cytokine of the Th17 response. Next to Th17 cells, γδ T-cells and ILC3 can also produce IL-17 in response to IL-23 and IL-1β (27, 117, 163). IL-17 exerts its effects primarily on nearby parenchymal cells and to lesser extent on hematopoietic cells, which is distinct from Th1 and Th2 cytokines like IFN-γ and IL-4 (159). In parenchymal cells, IL-17 primarily stimulates the production of the chemokines that attract PMN (164). However, it should be noted that IL-17 alone is a poor inducer of these chemokines and that synergistic activation by inflammatory ligands such as IL-1β, TNF-α, or GM-CSF markedly increases the effects of IL-17 (164, 165).
3.2. The Th17 Response in Human TB Infection
The exact role of the Th17 response in human TB remains a topic of debate (13, 14, 166). Polymorphisms in genes encoding IL-17 are associated with susceptibility to pulmonary TB, which indicates a role for this cytokine in TB (167–170). However, these findings could not be reproduced in different demographic settings (171, 172).
Analyses of Th17 responses in peripheral blood mononuclear cells (PBMC) from TB patients do not show uniform results either. Direct ex vivo analyses of unstimulated circulating CD4+ T-cells show that active TB (ATB) is associated with reduced frequencies of circulating Th17 cells compared to latent TB infection (LTBI) (173, 174). However, serum IL-17 levels do not differ between ATB and LTBI, and IL-17 is undetectable in the bronchoalveolar lavage fluid during both stages of disease (174, 175).
Different studies report PBMC stimulation assays with Mtb-specific antigens showing either increased or reduced Th17 responses in ATB compared to LTBI (Table 3). These diverse findings are similar to those observed in IFN-γ response assays (IGRA), in which the levels of IFN-γ often also cannot discriminate between ATB and LTBI (176, 177). Interestingly, both Th1 and Th17 cells appear functionally inhibited in ATB patients by a PD-1-mediated immunosuppressive state (178–181). In accord, reductions in PD-1 expression under TB treatment restored both Th1 and Th17 responses (182).
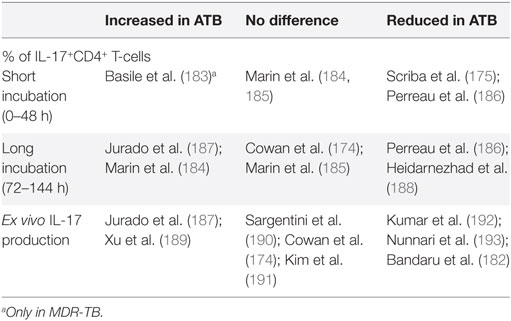
Table 3. IL-17 responses in patients with active TB (ATB) compared to latent tuberculosis infection (LTBI).
Taken together, systemic Th17 responses in TB patients demonstrate similar variability as observed for IGRA studies. Both are unable to distinguish ATB from LTBI. How these systemic responses relate to local host responses in the lungs has not been characterized in TB patients.
3.3. Preclinical Studies in Mice Support a Protective Role for IL-23 and IL-17 in TB
Based on mortality and mycobacterial loads, studies in Mtb-infected mice support a protective role for IL-23 and IL-17 in TB, but only during later stages of disease (Table 4).
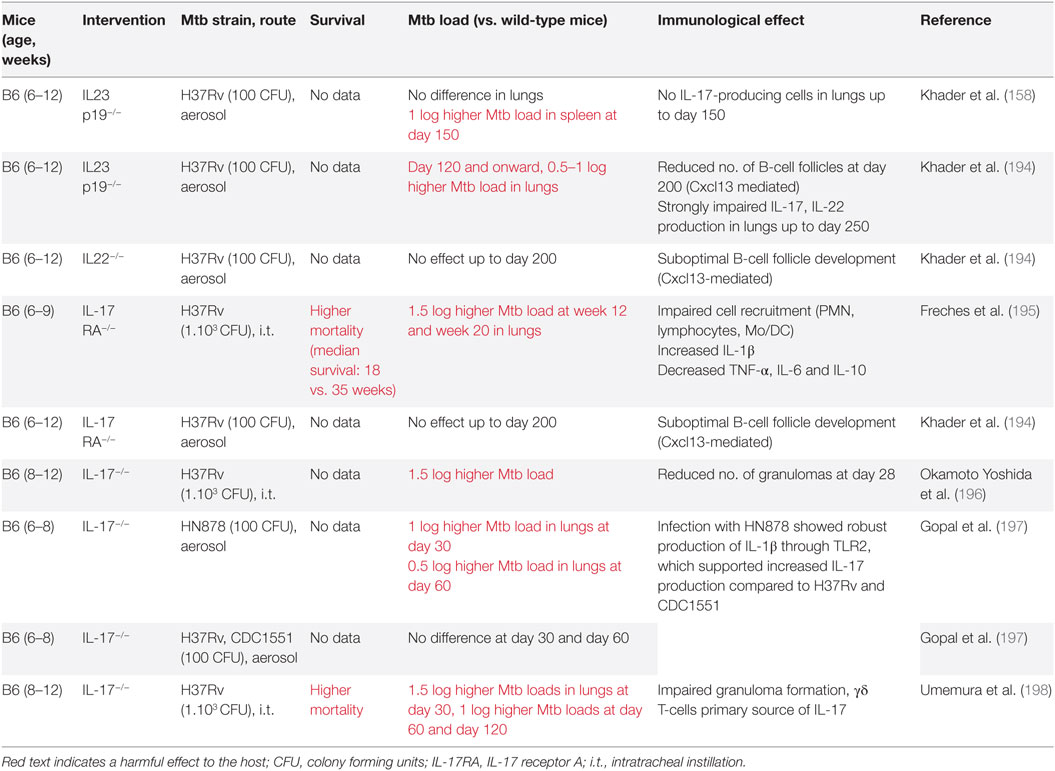
Table 4. Th17-related effects in preclinical tuberculosis (TB) studies in mice.
Interestingly, these late protective effects result from effects induced during the initial phase infection (142, 195). This is due to the essential roles of IL-23 and IL-17 in the local formation of tertiary lymphoid structures (TLS) (199, 200). These structures are formed during early infection but can persist for longer periods of time and are associated with protective immunity in Mtb-infected mice (199, 201) (Table 4; Figure 4). Furthermore, IL-17 and IL-23 increase the expression of the chemokine CXCL13 (194, 197). This chemokine stimulates the influx of TLS-associated CXCR5+ follicular helper (Tfh)-cell, which facilitate optimal localization of effector T-cell populations within the lung parenchyma, thereby promoting efficient T-cell-dependent macrophage activation and intracellular Mtb killing (194, 201).
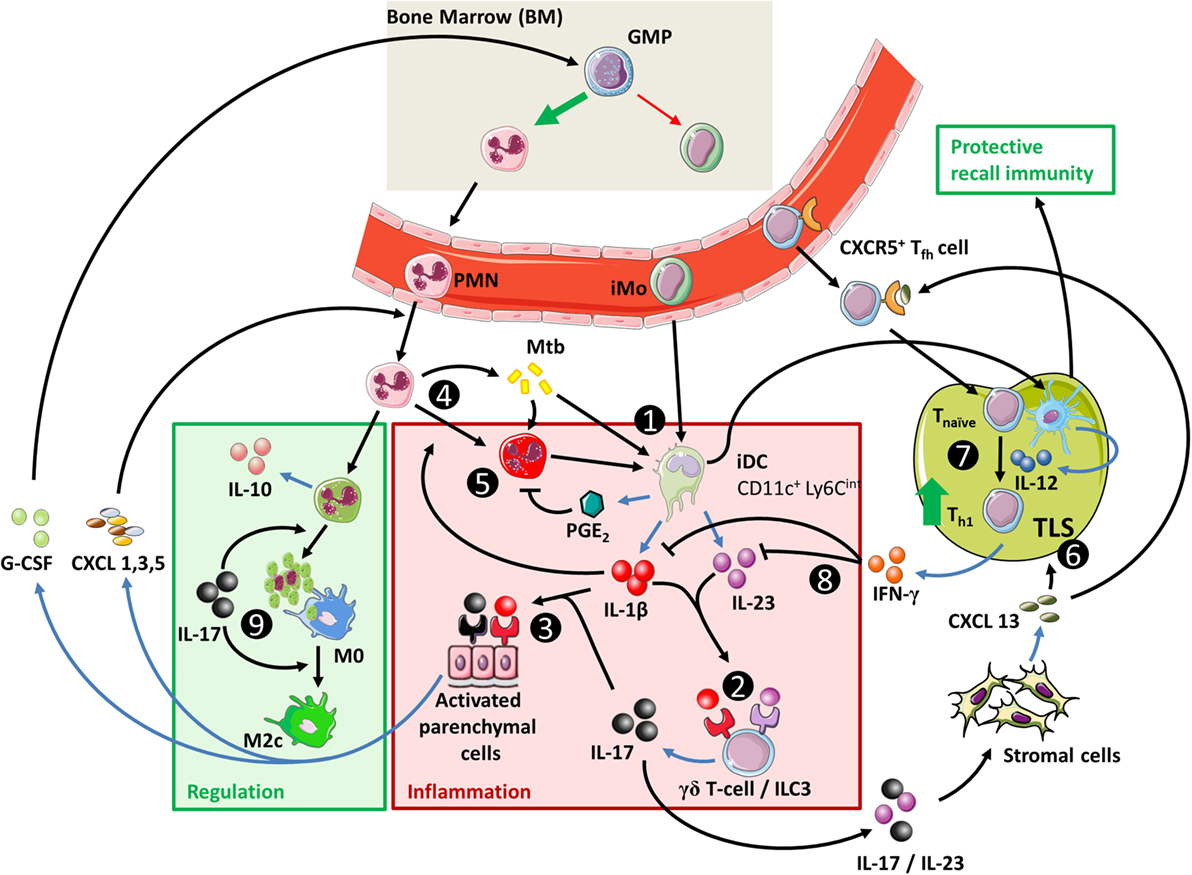
Figure 4. The IL-23/IL-17 axis in acute tuberculosis. (1) When inflammatory dendritic cells (iDC) recognize Mtb through membrane-bound toll-like receptors, they can secrete IL-1β, IL-23, and prostaglandin E2 (PGE2) (see Figure 1). This occurs more efficiently if iDC are activated through contact with Mtb-infected PMN, which also stimulates their migratory capacity to tertiary lymphoid structures (TLS) and promotes recall immunity (202–206). (2) The combination of IL-1β and IL-23 induces IL-17 production by γδ T-cells and possibly ILC3 (27, 117). (3) Activation of parenchymal cells by IL-17 in combination with IL-1β or other inflammatory mediators ultimately results in PMN influx. (4) PMN contribute to inflammation when stimulated by extracellular Mtb or inflammatory cytokines. (5) Activated PMN readily cause tissue damage through production of ROS and proteases; this effect is suppressed by activated iDC in a PGE2-dependent way (112, 207, 208). (6) IL-23 and IL-17 stimulate the local production of CXCL13 by stromal cells (194, 199). This promotes TLS formation and follicular helper T-cell migration to the site of infection. (7) CD40 ligation in the interaction between (i)DC and CD4+ T-cells is a strong stimulus for IL-12 production over IL-23 (Box 3) and leads to Th1 formation and IFN-γ production. (8) IFN-γ inhibits IL-1β production and shifts IL-23 production to IL-12, thus inhibiting IL-17 production and reinforcing the Th1 response (209). (9) In the absence of inflammatory stimuli, PMN can produce IL-10 and undergo apoptosis. Phagocytosis of apoptotic PMN induces an IL-10-producing regulatory M2c phenotype in macrophages and further contributes to resolution of inflammation.
On account of their ability to induce TLS formation, boosting IL-23 and IL-17 production is also an interesting strategy for vaccine-induced protection against TB. In this regard, IL-17 production by Th17 cells during recall responses is indeed dependent on IL-23 and could reduce mycobacterial loads in the lungs of Mtb-infected mice (210). Th17 cells preferentially migrate to the lungs and are better contained in the lungs compared to Th1 cells upon adoptive transfer to naïve mice (210, 211). The developmental flexibility of Th17 cells is illustrated in experiments where Mtb-antigen-primed Th17 cells have been adoptively transferred to naïve mice (210). Initially, these Th17 cells produce IL-17. However, upon recall immunity against Mtb, they primarily produce IFN-γ, with or without IL-17. Paradoxically, the latter switch results in a less effective reduction in bacterial loads compared to IL-17-producing Th17 cells that are adoptively transferred from IFN-γ−/− mice.
This tendency of Th17 cells to produce IFN-γ instead of IL-17 during recall responses might explain the observation that IL-17 production during later phases of Mtb infection is dominated by γδ T-cells rather than CD4+ cells (117, 198).
When taken together, initial shaping of the local inflammatory environment by IL-17 and IL-23 during acute infection stimulates local TLS formation. This facilitates the development of more robust Th1 responses by improving contact between antigen-presenting cells (APC) and lymphoid cells (Figure 4). Furthermore, Th17 cells confer protective immunity during recall responses by their enhanced capacity to migrate to the lungs and stimulate Tfh responses compared to other CD4+ T-helper cell populations.
3.4. The Th17 Response, PMN, and Inflammatory Damage
IL-17 stimulates granulopoiesis in the bone marrow and increases PMN influx to the site of infection by inducing G-CSF, CXCL1, CXCL3, and CXCL5 expression by parenchymal cells in mice or G-CSF and IL-8 in humans (159). These effects of IL-17 are markedly enhanced through synergistic activation by inflammatory mediators such as IL-1β, TNF-α, or GM-CSF (164, 212, 213). In this regard, IL-17 is not a strong inducer of inflammation by itself, but rather amplifies preexisting inflammation. This IL-17-mediated “inflammatory boost” can positively shape adaptive immunity, but prolonged or repeated antigen exposure can also lead to PMN-mediated pathological inflammation (214). Since IL-17 signaling is inevitably linked to PMN influx, the role of PMN in TB provides an additional perspective on the effects of IL-17 signaling in TB.
Review of available literature on the role of PMN in TB yields a complex picture with seemingly conflicting effects (14, 166, 215). In patients with active TB, PMN are the predominantly infected cells in the airways and provide a permissive site for a burst of active mycobacterial replication prior to transmission (216). On the other hand, PMN from healthy individuals, especially when stimulated with TNF-α, show a strong bactericidal effect (217). In preclinical TB models, highly susceptible mouse strains such as I/St, CBA/J, or DBA/2 show an enhanced influx of apoptosis-resistant, highly phagocytic neutrophils that negatively affect survival compared to more TB-resistant C57BL/6 and BALB/c mice (218–220). Moreover, PMN are poor producers of essential cytokines such as IL-1α/β and IL-12p40 in the anti-TB response (90, 221). These effects in preclinical models primarily suggest a negative contribution of PMN to acute disease. However, increasing evidence suggests a supportive role for PMN in protective immunity. PMN can indirectly augment IL-1β-mediated inflammatory responses in macrophages after contact with Mtb (202, 204, 205). Also, and consistent with Th17 responses, PMN play an essential role in generation of protective recall responses in Mtb-infected mice (195, 206, 222). Early, but not late PMN recruitment is essential for IL-17-mediated long-term control of Mtb infection (195). This can be explained by the finding that DCs that acquire Mtb through uptake of infected PMN are better able to activate T-cells (203, 222). The importance of this mechanism is recently highlighted in Mtb-infected mice, showing that PMN-depletion during vaccination prevented the generation of specific Th1 and Th17 responses (206).
A second emerging protective role of PMN is their contribution to initiating inflammation resolution (223). In mouse TB models, PMN are the main producers of IL-10 in the lungs and can dampen inflammatory damage (224). In this regulatory role, PMN inhibit Th17 responses but do not interfere with IFN-γ-mediated Th1 immunity due to relative insensitivity of Th1 cells to IL-10 (224, 225). Another regulatory effect of PMN concerns their apoptosis and subsequent phagocytosis by macrophages in the absence of extracellular Mtb. This inhibits IL-23 production by these macrophages and induces a regulatory IL-10high M2c phenotype under influence of IL-17 and IL-10 (see Figure 4) (226, 227). IL-17 can further contribute to this process by attenuating the anti-apoptotic effect of GM-CSF on PMN and by stimulating PMN apoptosis (228, 229).
Taken together, PMN recruitment to the site of infection is largely dependent on IL-17, but only in synergy with innate inflammatory cytokines such as IL-1β. Locally, these recruited PMN contribute to inflammation if pathogens are still present, improve dendritic cell function, and contribute to the formation of recall responses, or initiate resolution of inflammation in the absence of inflammatory or microbial stimuli.
3.5. Summary: The Role of Th17 Immunity in TB
The roles of IL-23 and IL-17 in TB are more subtle than the effects of Th1-related cytokines or T1-IFNs. Patient data are mostly limited to studies in PBMC. These show inconclusive results that are possibly confounded by the dynamics and heterogeneity of the Th17 response, which can range from highly pro-inflammatory IFN-γ/GM-CSF-producing Th17.1 cells to IL-10-producing regulatory Th17 cells.
Preclinical mouse TB models provide evidence for a protective role of the Th17 cytokines IL-23 and IL-17 in TB. These protective effects become apparent in the chronic phase of infection but result from IL-23/IL-17-mediated effects in the earlier, acute phase of infection. This is associated with early protective effects of IL-1β, which is a strong inducer of IL-23 and IL-17 (Figure 4). Mechanistically, evidence for the protective effects of IL-17 and IL-23 primarily points toward their role in the development of TLS during the acute phase of disease, which provides protective effects during later stages (199, 230). Additionally, IL-23 and IL-17 induce CXCL13 expression that mediates the influx of TLS-associated Tfh cells. TLS and Tfh responses facilitate optimal interactions between adaptive and innate immunity, contribute to granuloma formation, and improve the quality of T-cell recall responses in TB (201). In TB patients, TLS have also been associated with immune control, but more in-depth research is needed to establish their exact functional role and contribution to protective immunity (201).
Next to TLS formation and function, IL-23 and IL-17 mediate the influx of PMN into the lungs and the contribution of these cells to protective immunity in TB is increasingly recognized (206). Early, but not late PMN recruitment is essential for IL-17-mediated long-term control of Mtb infection (195) and DCs that acquire Mtb through uptake of infected PMN are better able to activate T-cells than DCs that directly interact with Mtb themselves (203, 222). The ability of IL-17 to induce the production of PMN-attracting chemokines in parenchymal cells is markedly improved when IL-17 signals in synergy with inflammatory mediators such as IL-1β, which again indicates synergy between IL-1β and IL-17 responses during acute TB. Prolonged activation of IL-1β and IL-17 responses can lead to massive accumulation of PMN, and their local necrotic death can also be damaging to the host. However, in the absence of inflammatory stimuli, PMN are an important source of IL-10 in the lungs and can initiate resolution of inflammation (Figure 4).
4. T1-IFNs, the Th17 Response and Their Interactions in Autoimmune Disease
Autoimmune diseases comprise a wide range of organ-specific and systemic disorders. Most systemic AID are considered classical B cell-mediated diseases, typified by circulating autoreactive antibodies against intracellular self-antigens. The clinical presentation of different AID varies, but evidence from genome-wide association studies points toward common immunogenetic mechanisms, as many systemic AID share disease-associated genes (231). Another trait particularly shared amongst different antibody-driven AID is the expression of a T1-IFN signature in both blood- and disease-affected tissue (232–234), the strength of which generally correlates with disease activity and severity (235–238). Vice versa, T1-IFN immunotherapy as treatment for other diseases is known to cause symptoms similar to those observed in AID, such as development of psoriatic lesions in MS or hepatitis C-infected patients (239, 240).
T-cells also have a major impact on the development and progression of AID and increasing evidence points toward crucial involvement of the Th17 response in the pathogenesis of multiple AID (160, 241). Th17 cells have been shown to be critical in the pathogenesis of MS and rheumatoid arthritis (RA) (19, 160). However, Th17 cells have also been associated with disease severity in AID characterized by a T1-IFN signature, such as systemic lupus erythematosus (SLE) (20, 233, 242, 243). Since T1-IFN signatures and Th17 responses are both associated with disease in AID, the question arises whether these two pathways act in concert to sustain and amplify autoimmune responses, or control each other (20, 21, 244). Therefore, we will discuss below the involvement of the T1-IFN and Th17 responses in AID individually as well as their interaction. We refer readers who are familiar with the contributions of T1-IFN and Th17 in AID to continue at Section 4.3 where we discuss the interaction between these pathways.
4.1. The Contribution of T1-IFNs to the Pathogenesis of AID
Most insight into the role of T1-IFNs in the pathogenesis of AID has been obtained in SLE, which was the first disease in which a T1-IFN transcriptional signature was identified in 2003 (235). Since then it has become clear that 60–80% of adult SLE patients and nearly 100% of pediatric SLE patients express a T1-IFN signature in their blood (245). Several mechanisms through which T1-IFNs contribute to disease in SLE, outlined below, have been elucidated.
4.1.1. Induction of T1-IFNs in AID
Specifically IFN-α appears to play a central role in SLE pathogenesis (245, 246). As mentioned in Section 2.3 IFN-α is produced in an IRF7-dependent way by pDC and other myeloid cell types. In accordance, pDC have been found to be a major source of T1-IFNs in SLE (247, 248). Immune complexes (IC), consisting of antibodies bound to self-DNA, are a major trigger for IFN-α production by pDC in AID (249). However, pDC are not activated by self-DNA under steady state conditions, which indicates that additional stimuli are required. One such stimulus is the PMN-derived antimicrobial peptide LL37 (249), which convert inert self-DNA into a potent activator of endosomal TLR9 (250). Another stimulus is the nuclear protein high mobility group box 1 (HMGB1) protein, which is secreted by activated myeloid cells and passively released by necrotic, but not apoptotic cells (251). HMGB1 binds DNA, and the formed complexes bind with high affinity to receptor for advanced glycation end-products, which facilitates internalization into the endosome where TLR9 can be activated (249). Extracellular HMGB1 also triggers the recruitment of PMN and stimulates their formation of neutrophil extracellular traps (NETs) (252). NETs contain large amounts of nucleic acids and LL37 and are also a major driving factor behind chronic pDC activation and IFN-α production in SLE (253).
It deserves mention that NET formation is driven by reactive oxygen species (ROS), which in PMN are particularly produced by NADPH oxidase and subsequently processed by myeloperoxidase (254). Paradoxically, despite the capacity of NETs to induce T1-IFNs and the pathogenic role of T1-IFNs in SLE, NADPH oxidase appears to be protective in SLE (255). Lupus-prone mice deficient in NADPH-oxidase develop more severe SLE (255). Moreover, autoimmunity with T1-IFN signatures can still develop in individuals with chronic granulomatous disease, who lack NADPH-oxidase activity (256). This seeming contradiction has been partially explained by the observation that IgG autoantibody-mediated NETosis, which is most relevant in SLE, is specifically reliant on mitochondrial ROS, while NETosis induced by, e.g., TLR4 signaling is NADPH dependent (256). In line with this, NETs from SLE patients have been shown to contain mitochondrial DNA (256). Thus, the way NETs are induced, and the type of DNA that is present on NETs probably also influences their ability to induce T1-IFNs and their role in disease.
Taken together, TLR9-mediated IFN-α production by pDC in response to IC and NETs appears the major driving factor behind T1-IFN production in autoantibody-mediated AID. Additionally, the way NETs are induced and the type of DNA present on NETs can influence disease outcomes.
4.1.2. Disease-Promoting Effects of T1-IFN in AID
Type I interferons exert a detrimental effect in AID through different pathways. In monocyte-derived cells, T1-IFNs stimulate maturation, increase phagocytic capacities (257), and increase the expression of co-stimulatory molecules (258). Also, T1-IFNs have a direct stimulating effect on T-cells. Together, these effects promote the generation of autoreactive T-cells, which support autoreactive B-cell responses (257, 259).
At cytokine level, T1-IFNs can induce the production of B-cell activating factor (BAFF) by myeloid cells (238, 260, 261). BAFF induction confers a significant proportion of T1-IFN-mediated damage in SLE as supported by the observation that IFN-α administration induces disease in SLE-prone mice but fails to do so in B-cell-deficient and BAFF-deficient mice on the same background (262). BAFF plays a central role in the development and selection of autoreactive B-cells (260). In line with this, increased BAFF expression correlates with disease severity in SLE (21, 260, 263). BAFF also induces class switch recombination in B-cells, leading to preferential expression of IgG and IgA over IgM, which is important for Fc-receptor-mediated NETosis induction in PMN (264). The clinical relevance of BAFF in SLE pathogenesis is illustrated by the current use of belimumab, a monoclonal antibody against BAFF, as treatment for SLE (265). Interestingly, targeting BAFF is effective in SLE patients, while B-cell depleting therapies using CD-20-targeting rituximab show disappointing results in phase III clinical trials (266, 267). This suggests effector functions of BAFF other than B-cell activation. In this regard, BAFF can act as a co-stimulatory molecule for T-cells and promote Th17 development (268, 269). BAFF can also directly activate plasma cells, which are not depleted by rituximab (270, 271).
4.2. The Contribution of Th17 in the Pathogenesis of AID
4.2.1. GM-CSF-Secreting Th17.1 Cells
Pathogenic effects of Th17-mediated immunity in AID have been studied most detailed in MS and RA and their respective mouse models, experimental autoimmune encephalitis (EAE), and collagen-induced arthritis (160, 242). MS was long believed to be primarily driven by an IL-12/Th1 response, but this concept was challenged by observations in the EAE mouse model for MS showing that the IL-23p19 subunit instead of IL-12p35 (see Box 3) caused disease (272). In addition, the classic cytokines of Th1 and Th17 immunity, i.e., IFN-γ and IL-17, respectively, were found dispensable in EAE and instead GM-CSF appeared to be the effector cytokine responsible for IL-23-induced encephalopathy (118). Notably, while most studies agree on a central pathogenic role for GM-CSF in MS, conflicting results are reported regarding its cellular source (19, 273–275). One study shows that GM-CSF expression in MS patients is promoted by the IL-12/T-bet/Th1 axis, instead of IL-23 as observed in mouse EAE (273). Other publications report that B-cells are a major source of GM-CSF and specifically act in concert with Th17 cells (274, 276). In accord with these discrepant results, MS is shown to be a heterogeneous disease that can be driven by either Th1 or Th17 immunity (242), which also has implications for therapy as will be discussed in Section 4.3.1.
One interesting observation in this regard is the development of “hybrid” Th17.1 cells that express markers of both Th17 cells and Th1 cells. Naïve CD4+ T-cells in both mice and man do not express the IL-23 receptor and can either differentiate into T-bet+ Th1 cells under influence of IL-12 or differentiate into CCR6+ Th17 cells under influence of IL-6 and TGF-β (161). These IL-6/TGF-β-differentiated Th17 cells have low inflammatory potential and are prone to adopt an IL-10-producing regulatory phenotype. However, IL-6 also induces STAT3-dependent upregulation of IL-23 receptor (277). Subsequent (re)activation of such IL-6-primed Th17 cells by IL-23 increases Th1-associated T-bet expression and generates inflammatory IFN-γ/GM-CSF-producing Th17.1 cells (161). These cells can also switch their chemokine receptor profile and become CCR2+ instead of CCR6+ (161). Expression of CCR2 by Th17.1 cells can contribute to their inflammatory potential as it can divert their migration to sites without concomitant influx of regulatory T-cells, which depend on CCR6 for their migration (278).
Mechanistically, it was shown in a mouse EAE model that GM-CSF exerted its pathogenic effector function by stimulating IL-1β production by monocyte-derived cells (279). This suggests a positive inflammatory feedback loop, since IL-1β in turn promotes IL-23 production and development of Th17.1 cells (118). A similar pathogenic Th17.1 response is observed in RA, which was the first AID in which IL-1β inhibition was approved for clinical use (280). Also, regarding the distinction between Th17 and Th17.1 responses in RA, it should be noted that anti-GM-CSF therapy shows more promise than anti-IL-17 in clinical phase I/II trials (160, 281).
4.2.2. The Contribution of IL-17-Producing Th17 Cells to AID Pathogenesis
Next to GM-CSF-secreting Th17.1 cells, regular IL-17-producing Th17 cells also have been identified as pathogenic in other AID. This is best exemplified by the clinical successes of targeting IL-17 in psoriasis (282). Th17-associated pathogenic effects in SLE also appear to be driven by IL-17 rather than GM-CSF (21, 283). This is further supported by the specific contribution of PMN to disease in SLE, which is dependent on IL-17, opposed to GM-CSF that primarily influences the inflammatory potential of monocytes in MS and RA.
4.3. Interactions between T1-IFNs and the Th17 Response in AID
Systemic lupus erythematosus and other autoantibody-mediated AID show a pathogenic role for T1-IFNs, while T-cell-mediated AID, such as MS, are driven primarily by GM-CSF-stimulated IL-1β production. With the functional dichotomy of IL-1β and T1-IFNs in mind, as shown in Figure 2, MS and SLE seem to be opposite ends of the disease spectrum in AID instead of demonstrating interactions between T1-IFNs and the Th17 response. However, the existence of different Th17 subsets might explain this seeming disparity and suggest roles for GM-CSF-producing Th17.1 cells in MS and regular IL-17-producing Th17 cells in SLE. Both Th17 responses interact differently with T1-IFNs as will be discussed here. We identify three relevant interactions: (1) Th17.1 responses are fueled by T1-IFN-stimulated influx of CCR2+ inflammatory monocytes; (2) a pathological IL-17/T1-IFN/BAFF axis driven by NET-forming PMN; and (3) Th17 immunity and T1-IFNs collaborate in the generation and function of TLS. An overview of these pathways is presented in Figure 5.
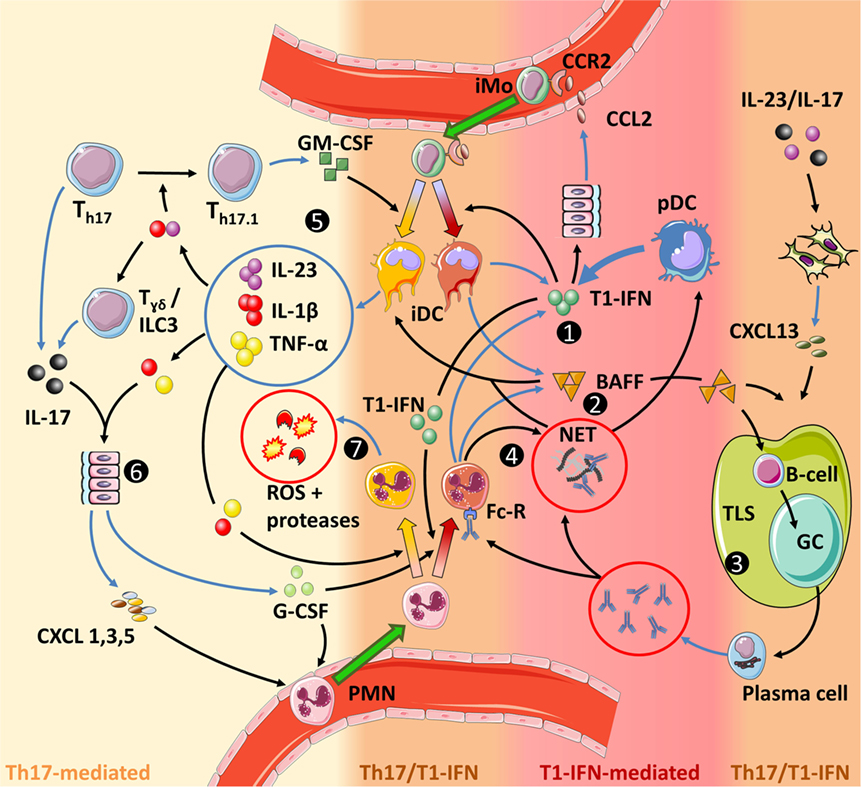
Figure 5. Interactions between type I interferons (T1-IFNs) and Th17 immunity in autoimmune diseases. The color grading in the figure indicates the level of involvement of either Th17 immunity or T1-IFN-associated signaling. (1) T1-IFNs, primarily produced by plasmacytoid dendritic (pDC) but also by inflammatory dendritic cells (iDC) and PMN, prime the latter cells toward a T1-IFN/B-cell activating factor (BAFF)-producing phenotype, promote NETosis by PMN and stimulate monocyte migration by inducing CCL2 production. (2) BAFF activates B-cells, stimulates tertiary lymphoid structures (TLS) formation together with CXCL13, directly promotes Th17 differentiation (not shown), and stimulates the release of IL-1β by iDC. (3) TLS facilitate optimal interaction between activated B-cells and antigen-presenting cells (APC), while necrosis, neutrophil extracellular traps, and T1-IFN increase the chance that these APC present self-antigens. Subsequent germinal center (GC) reactions within these TLS result in B-cells differentiating into plasma cells that produce large quantities of autoantibodies. These autoantibodies can mediate tissue damage and sustain a self-amplifying loop by inducing NETosis through binding the Fc-receptor on PMN. B-cells can also contribute to Th17 immunity by their ability to secrete IL-6 and GM-CSF (not shown and uncertain if this is BAFF dependent). (4) NETs trap antibodies. This facilitates their Fc-receptor-mediated internalization by pDC in which they stimulate T1-IFN production through endosomal TLR9 activation. Circulating NETs also stimulate IL-1β production by iDC and can mediate tissue damage. (5) In a pro-inflammatory feedback loop, IL-23 stimulates the development of GM-CSF-producing Th17 cells (Th17.1), which in turn, together with BAFF and/or NETs stimulate an inflammatory phenotype in iDC. (6) IL-1β and IL-23 stimulate IL-17 production by γδ T-cells, while concomitant stimulation with IL-1β and TNF-α is required for IL-17-induced G-CSF and chemokine production in parenchymal cells. (7) IL-1β and TNF-α activate PMN to release reactive oxygen species (ROS) and proteases that cause tissue damage. Furthermore, GM-CSF increases longevity of PMN (not shown). Finally, the priming of PMN and monocytes prior to entering the site of disease is important for their eventual effector function. For monocytes this is shown in more detail in Figure 3.
4.3.1. T1-IFNs Can Contribute to Th17.1-Mediated AID
Among MS patients treated with IFN-β, approximately 30–50% do not respond favorably to treatment (284). It was shown that IFN-β suppresses Th1-mediated inflammation in MS but is ineffective and may even exacerbate Th17-mediated inflammation (19). This is one of the first studies that report a detrimental interaction between T1-IFNs and Th17 responses. Given the importance of Th17.1 cells in MS, this negative outcome might be explained by the observation that IFN-β therapy in MS increases CCL2 production (285). Expression of this chemokine in the brain recruits inflammatory CCR2+ monocytes as well as Th17.1 cells, which switch their chemokine receptor profile from CCR6+ to CCR2+ upon terminal differentiation (161). Moreover, Th17.1 cells stimulate IL-1β production in CCR2+ monocytes (279, 286). Inflammatory monocytes may differentiate locally into dendritic cells further stimulating Th17 responses (287). Thus, a strongly pro-inflammatory condition is created in Th17.1-mediated MS. Since regulatory T lymphocytes rely on CCR6 rather than CCR2 (279), recruitment of these anti-inflammatory cells does not appear to hold pace with the influx of inflammatory monocytes and Th17.1 cells in MS.
4.3.2. A Pathological IL-17/T1-IFNs/BAFF Axis in AID
IL-17 induces PMN influx through induction of G-CSF and chemokines (see Section 3.4), which contribute to the production of IFN-α by pDC via the NETosis process (see Section 4.1.1). However, increasing evidence suggests a more prominent contribution of IL-17 and PMN to T1-IFN-mediated disease in SLE. First, besides being major inducers of IFN-α production by pDC upon NETosis, PMN also appear to be a significant source of IFN-α themselves (288, 289). This was related to their sheer numbers, as circulating pDC were 27 times more efficient in secreting IFN-α, but PMN were 100 times more frequent (289). Second, both T1-IFNs and IL-17-induced G-CSF prime PMN for NETosis (250, 290). In accord, circulating PMN of SLE patients are also the main cells expressing the transcriptional T1-IFN signature and release more NETs than PMN from healthy individuals (250, 253, 288, 289, 291, 292). Thirdly, T1-IFNs stimulate BAFF production, which is essential for T1-IFN-mediated pathogenic effects in mouse SLE (261, 262, 293). It is recently shown that IL-17 also induces BAFF production and that IL-17-driven, G-CSF-dependent PMN recruitment drives plasma cell responses during emergency granulopoiesis in a BAFF-dependent way (271). Also, therapeutically administered G-CSF, which is physiologically induced by IL-17, increases BAFF production by PMN (294).
These interactions indicate a prominent role for IL-17-mediated PMN influx in T1-IFN-production and induction in AID and synergistic induction of BAFF production by IL-17 and T1-IFNs. In support of this, IL-17 and Th17 cells are associated with disease severity in SLE to similar extent as T1-IFNs (20, 21, 241, 244). In turn, BAFF can promote Th17 responses (268, 269). This further suggests an inflammatory loop with a central role for PMN in which IL-17, T1-IFNs, and BAFF continuously increase each other’s production and contribute to autoantibody-mediated responses.
4.3.3. T1-IFNs, Th17 Responses, and TLS in AID
Finally, T1-IFNs and Th17 responses converge onto the development and functioning of TLS. In these structures, Tfh cells support germinal center (GC) reactions in which B-cells differentiate into antibody-producing plasma cells and memory cells (295). As expected from their function, TLS and Tfh cells are essential components in the pathogenesis of multiple autoantibody-mediated AID (296–303). The cytokines IL-17 and IL-22 secreted by ILC3, γδ T-cells and Th17 cells are required for local TLS formation (199, 230, 304). T1-IFN- and IL-17-induced BAFF promote the formation and integrity of GCs within TLS and stimulate Tfh development (305, 306). T1-IFNs directly induce the expression of the Tfh-markers CXCR5 and PD-1 on T cells (307, 308). Also, T1-IFNs promote the survival of aberrantly selected B-cells in the GC reactions during SLE directly and indirectly through BAFF induction as discussed in Section 4.2.2. Thus, it appears that by stimulating TLS development, the Th17 response facilitates an environment that promotes selection of autoreactive B-cells under influence of T1-IFNs and BAFF.
Taken together, several lines of evidence exist for interactions between the Th17 response and T1-IFNs in systemic AID. Current data support a scenario in which Th17 immunity fuels T1-IFN-related pathology by mediating PMN influx and driving TLS formation, which facilitates T1-IFN/BAFF-mediated plasma cell responses and autoantibody production. In turn, T1-IFNs can support pathogenic Th17.1 responses in AID by driving the influx of CCR2+ inflammatory monocytes and potentially CCR2+ Th17.1 cells themselves, which locally drive IL-1β mediated inflammation. An overview of these interacting pathways is shown in Figure 5.
5. Interactions Between T1-IFNs and Th17 Immunity in TB
In the previous section, we have outlined how T1-IFNs and Th17 immunity interact in AID (illustrated in Figure 5). These interactions primarily concern (1) Th17.1 responses fueled by T1-IFN-stimulated influx of CCR2+ monocytes; (2) The IL-17/T1-IFNs/BAFF axis driven by NET-forming PMN; and (3) synergism between Th17 immunity and T1-IFNs in TLS formation and function. In this section, we assess the relevance of these three pathways in TB based on cell types and effector molecules involved. Each subsection contains a part of Figure 5, supplemented with relevant finding and outstanding questions in TB.
5.1. Th17.1 Responses in TB
Studies in MS and RA emphasize the difference between GM-CSF/IFN-γ-producing Th17.1 cells and regular IL-17-producing Th17 cells. The former primarily increase the inflammatory potential of monocytes (Figure 6), while the latter are more closely associated with PMN. Data on subtypes of Th17 cells and particularly Th17.1 cells in human TB are limited. One study shows that circulating GM-CSF+ T-cells are not increased in ATB compared to LTBI, but it is unclear if this concerns Th17.1 cells or Th1 cells (309). Interestingly, GM-CSF production by both granuloma-associated T-cells and circulating CD4+ T-cells in TB patients only occurs after mycobacterial antigen stimulation (309, 310). In mice, adoptively transferred Mtb-primed Th17 cells that produce IL-17 upon transfer, predominantly produce IFN-γ upon subsequent contact with Mtb, which is suggestive of a Th17.1 phenotype (210).
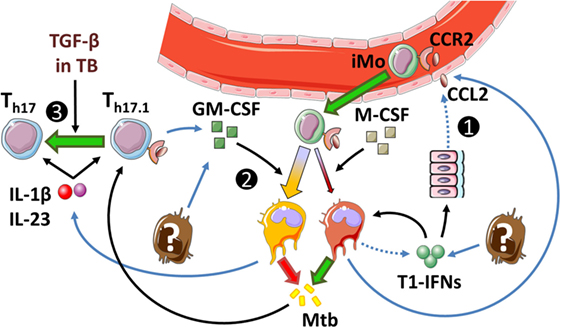
Figure 6. Th17.1 responses in tuberculosis (TB). (1) Type I interferons (T1-IFNs) induce CCL2 production in parenchymal cells and MDM, but not GMDM. This induces the influx of CCR2+ monocytes that mediate detrimental effects in TB as Mtb-permissive cells develop upon T1-IFN stimulation. (2) GM-CSF increases IL-1β production, limits responsiveness to T1-IFNs, and increases Mtb-killing potential. However, the exact cellular source of GM-CSF in TB is unknown. (3) Patients with active TB overexpress TGF-β, which may drive Th17 development over Th17.1 in the presence of IL-1β and IL-23. Dotted lines implicate mechanisms shown in autoimmune diseases that have not been confirmed in TB. Outstanding questions: (1) What is (are) the cellular source(s) of T1-IFNs in TB? (2) What is the ratio between different Th17 subsets in TB? (3) Do T-cells contribute to GM-CSF production in TB?
Th17.1 cells in AID result from prolonged innate IL-1β and IL-23 signaling. With regard to the role of IL-1β and IL-23 in human TB, IL-1β is essential for the expansion of both IFN-γ−IL-17+Th17 cells and IFN-γ+IL-17+Th17 cells (311, 312). IL-23 promotes the development of IFN-γ+IL-17+Th17 cells but promotes IFN-γ−IL-17+Th17 cells if TGF-β is concomitantly present (312). Since active TB is associated with elevated TGF-β levels (178, 313, 314), it is possible that Th17.1 cell differentiation does not play a major role, but this remains to be demonstrated.
Th17.1-derived GM-CSF exerts a pathogenic effect in AID by stimulating IL-1β production in CCR2+ monocytes. Although the role of Th17.1 cells in TB is uncertain, other cells such as NK cells and Th1 cells can also produce GM-CSF in TB, and during the course of infection, GM-CSF levels progressively increase in the lungs of Mtb-infected mice (125, 315). The functional role of GM-CSF is of interest in TB, because it importantly impacts on CCR2+ monocytes, which play a central role in T1-IFN-mediated pathogenic effects. T1-IFNs stimulate the influx of inflammatory CCR2+ monocytes but inhibit their IL-1β production and stimulate their differentiation into Mtb-permissive cells (see Figure 3). In contrast, GM-CSF is protective during acute TB, which is in line with the protective effects of IL-1β in this phase of disease. Mice deficient in GM-CSF succumb rapidly to infection due to their inability to mount Th1 responses (316, 317). Transgenic mice that overexpress GM-CSF in the lungs but are GM-CSF-deficient in all other organs can develop Th1 responses, but still succumb to infection more rapidly than wild-type mice due to their inability to develop a normal granulomatous response (316, 317). Evidence from in vitro studies suggests that GM-CSF exerts its protective effect in TB by countering the effects of T1-IFNs in CCR2+ monocytes (36, 94). Under physiological conditions, monocytes differentiate under influence of M-CSF into monocyte-derived macrophages (MDM). These MDM have a CCR2low phenotype, readily produce CCL2 and IL-10 in response to T1-IFNs, and have a low Mtb-killing capacity (94, 156, 318, 319). Conversely, monocytes that differentiate under influence of GM-CSF (GMDM) are CCR2high, relatively unresponsive to T1-IFN signaling, produce small amounts of CCL2 and IL-10, and have better Mtb-killing capacities than MDM in response to activation by IFN-γ (36, 126).
The relative unresponsiveness of GMDM to T1-IFNs might explain why preclinical studies primarily show effects of T1-IFNs during acute TB when the GM-CSF/M-CSF ratio in the lungs is relatively low, but less pronounced effects during later stages when GM-CSF-levels progressively increase (see Section 2.4.3; Figure 3) (125). However, similar to IL-1β, prolonged GM-CSF signaling also appears detrimental in TB. In particular, GM-CSF contributes to foamy macrophage development during later stages of infection, which can sustain persistent mycobacteria and contribute to inflammation (125, 320).
In summary, relatively few data are available on Th17.1 cells or T-cell-derived GM-CSF in TB. The requirement for antigen stimulation of T-cells to induce expression of GM-CSF is interesting. However, elevated TGF-β levels in TB patients suggest a limited contribution of Th17.1 cells to disease, as TGF-β favors the development of regular IL-17-producing Th17 cells. Regardless of its cellular source, preclinical TB studies support a protective role for GM-CSF during acute infection. GM-CSF causes monocytes to differentiate into cells with decreased T1-IFN responsiveness and increased Mtb-killing potential compared to their M-CSF-differentiated counterparts. However, during chronic Mtb infection, high GM-CSF levels appear detrimental as they stimulate foamy macrophage development and inflammation.
5.2. The IL-17/T1-IFNs/BAFF Axis in TB
In the previous paragraph, it was discussed that regular IL-17-producing T-cells are more likely to play a role in TB than Th17.1 cells. Opposed to Th17.1 cells, regular Th17 cells exert their effect primarily through PMN instead of CCR2+ monocytes in AID. Particularly in SLE, this was shown to be part of a pathogenic axis together with T1-IFNs and BAFF. The roles of T1-IFNs and IL-17 in TB have been discussed already in Sections 2 and 3. In this section, we assess the roles of the other components of the IL-17/T1-IFNs/BAFF axis in TB, which include PMN-derived NETs, pDC, and BAFF (Figure 7).
Figure 7. The IL-17/type I interferons (T1-IFNs)/B-cell activating factor (BAFF) axis in tuberculosis (TB). Mtb actively induces NET formation by PMN, but subsequent activation of IFN-α production by plasmacytoid dendritic (pDC) appears less relevant in TB than in autoimmune diseases (AID). IL-17 levels from TB patients vary (Table 3), but preclinical models support a protective role. Dotted lines implicate mechanisms present in AID that have not been confirmed in TB. Outstanding questions: (1) What are the specific contributions of IFN-α vs. IFN-β to disease in the different phases of TB? (2) Do BAFF and T1-IFNs promote the observed autoimmune phenomena in TB?
5.2.1. PMN, NETs, and pDC in TB
PMN isolated from SLE patients are the primary cells that express the transcriptional T1-IFN signature. Furthermore, a specific subclass of PMN, termed low-density granulocytes (LDG) have been identified in SLE that express a pro-inflammatory phenotype, has increased T1-IFN-production and more readily form NETs than PMN from healthy individuals (291, 292). Similar to SLE, the transcriptional T1-IFN signature in TB patients is mostly expressed in PMN (58). Moreover, LDG are also present in TB patients and correlate with disease severity, but it is unclear if these cells also have a similarly increased tendency for NETosis as their SLE counterparts (321). Nevertheless, NETosis does occurs in TB, as Mtb readily induces NETosis itself in PMN in an ESX-1-dependent way and can even stimulate extracellular trap formation in macrophages (204, 322–324).
Neutrophil extracellular traps are strong inducers of IFN-α production in pDC in SLE (256). Conversely, pDC produce only small amounts of IFN-α and appear of minor clinical significance in TB (325). In accord, circulating pDC are elevated in SLE (326) but reduced in TB patients (327).
Final support for a limited role of pDC in TB pathogenesis comes from the observation that pDC produce IFN-α after endosomal TLR-activation, while it is shown that Mtb primarily induces IFN-β through activation of cytoplasmic PRRs (see Section 2.3) (81, 250). While both IFN-α and IFN-β signal through IFNAR, this diversification in cellular source and type of T1-IFN that is induced can have important consequences for TB pathogenesis (see Box 4).
Box 4 IFN-α or IFN-β: which is relevant in tuberculosis (TB)?
IFN-α and IFN-β both exert their effect by binding to IFN-α/β receptor, but increasing evidence from autoimmune diseases (AID) and viral infections suggests divergent effector functions (328, 329). In AID, this is illustrated by the pathogenicity of IFN-α in systemic lupus erythematosus (SLE) opposed to the therapeutic application of IFN-β as immunosuppressive treatment in multiple sclerosis (MS). Recently, these different immunoregulatory roles of IFN-α and IFN-β in SLE and MS have been confirmed by more detailed analysis of blood transcriptional profiles in patients (330). The molecular explanation for the differential function of IFN-α and IFN-β traces back to subtle differences in receptor binding, signaling cascades, and feedback mechanisms initiated and has been reviewed in detail elsewhere (28, 331).
The specific contributions of IFN-α and IFN-β to the host response in infectious disease have been studied particularly in mice infected with lymphocytic choriomeningitis virus. This work supports an immune-stimulating, antiviral role for IFN-α as opposed to an immunosuppressive effect by IFN-β (328, 331, 332). IFN-β specifically inhibits antiviral T-cell responses and promotes viral persistence (331). In contrast, IFN-α-signaling associates with tissue damage and antiviral activity (331, 332).
In TB, evidence for the involvement of both type I interferons is present. Reactivation of TB has been reported specifically after treatment of patients with IFN-α, but not IFN-β (50–57). Also, mice infected with virulent Mtb strains specifically show higher IFN-α levels in the lungs compared to less virulent strains (64, 65). However, IFN-α-producing plasmacytoid dendritic cells seem to be of minor significance in TB patients (325, 327), and preclinical studies show that Mtb preferentially induces IFN-β through cytoplasmic pattern recognition receptors and IRF3 instead of IFN-α through endosomal toll-like receptors and IRF7 (79–81). Mycobacterial persistence in patients with TB is a major clinical problem and in line with the immunosuppressed state in active TB primarily supports a role for IFN-β (178, 333). However, exaggerated innate responses are also observed in TB where IFN-α might be involved. This is supported by recent evidence showing that IRF7 drives excessive innate inflammation during bacterial infections and provides an interesting therapeutic target (334). Taken together, little is known about the separate effects of IFN-α and IFN-β in TB, but clinical and preclinical studies support a role for both in different disease contexts. The diversification of IFN-α and IFN-β responses in transcriptional signatures observed in AID patients and the distinct effects of IFN-α and IFN-β in experimental LMCV infection therefore provide highly interesting perspectives for TB.
5.2.2. BAFF in TB
Both T1-IFNs and IL-17 can induce BAFF expression, which contributes to disease in SLE as illustrated by the clinical successes of BAFF-inhibition (261, 271, 335). BAFF increases B-cell numbers and antibody titers (293, 336) and treatment with anti-BAFF in SLE patients reduces serum IgG levels (335). The role of BAFF in TB has been explored to a much lesser extent, with currently one paper demonstrating BAFF levels to be elevated in patients with active TB without elaborating on its functional contribution to the host response (337).
The functional role of BAFF in TB might be of particular interest given its stimulation of humoral immunity and the recently demonstrated protective effects of antibody-mediated immunity in TB patients (338, 339). Next to antibody-mediated protection, B-cells also essentially support T-cell responses in TB, but circulating B-cells are dysfunctional and reduced in absolute numbers in patients with active TB (340). The protective effects of Mtb-specific antibodies and B cells in TB suggest that increased BAFF levels may supports host responses by stimulating antibody production and perhaps other B cell functions such as stimulating T cell responses (338). However, high BAFF levels also predispose for the development of autoreactive B-cells in AID (341). Thus, elevated BAFF levels in TB could relate to the observation that up to 32% of patients with active TB have elevated autoantibody levels (12). Such correlations between elevated BAFF levels and autoimmunity have been demonstrated in other chronic infections (342).
Additional support for a supposed protective role of BAFF in TB comes from its interaction with IL-17, which shows protective effects in preclinical early infection phase TB models, as discussed in Section 3. IL-17 stimulates the migration of PMN to lymphoid structures where they can produce large quantities of BAFF that directly drive plasma cell responses. Also, IL-17-induced G-CSF primes PMN for BAFF production upon activation (271, 294). Vice versa, elevated BAFF levels have been reported to increase Th17 immunity in AID and infection (268, 269, 343).
Taken together, preliminary pieces of evidence support the presence of interactions between IL-17, T1-IFNs, and BAFF in TB, similar to those demonstrated in AID. This primarily includes the presence of NETs and elevated BAFF levels. However, despite the T1-IFN signature observed in TB, NET-induced IFN-α production by pDC appears less relevant in TB than in AID, and the specific contributions of IFN-α and IFN-β are of high interest in TB, but currently largely unknown. Studies in TB patients show protective effects of antibody-mediated immunity but also elevated titers of autoantibodies. This supports a view in which BAFF is protective in TB, but excessive BAFF levels, driven by either T1-IFNs or IL-17 can also increase the chances of developing autoimmunity in TB patients.
5.3. TLS in TB
As a third place of interaction, IL-17, T1-IFNs, and BAFF converge in the local formation and functioning of TLS. In these structures, Tfh cells support GC reactions in which B-cells differentiate into plasma cells and memory cells (295). As discussed in Section 4.3.4, observations in AID suggest that Th17 responses drive TLS development and facilitate an environment that promotes development of autoreactive B-cells under influence of T1-IFNs and BAFF. Conversely, both TLS and Tfh cells are associated with immune control in TB patients and preclinical TB models, which is in line with the protective role of humoral immunity in TB discussed in the previous section (194, 201, 344, 345). Here, we discuss how TLS and Tfh responses are associated with protective immunity in TB and how interactions between IL-17, T1-IFNs, and BAFF may contribute to this immune response.
Migration of CXCR5+ Tfh cells into TLS is largely dependent on CXCL13, which is primarily induced by IL-23 and IL-17 in mouse TB models but can also be induced by T1-IFNs, as demonstrated in viral infections (194, 346). Mechanistically, CXCR5+ Tfh cells mediate their protective effect in Mtb-infected mice by facilitating optimal localization of effector T-cell populations within the lung parenchyma, thereby promoting efficient T-cell-dependent macrophage activation and intracellular Mtb killing (194, 201).
Another interesting observation regarding Tfh responses concerns the induction of PD-L1 expression on APC and PD1 on T-cells by T1-IFNs (43, 136, 307). In TB circulating PMN primarily express the T1-IFN signature but also overexpress PD-L1 (58, 347). The interaction of PD-L1 with PD1 on CD4+ T-cells is a key immunological checkpoint in TB that limits excessive T-helper responses (33, 35). In line with this, PD1-deficient mice are extraordinarily susceptible to TB (34). Tfh cells constitutively express PD1+, which distinguishes them from conventional CD4+ Th cells. Interestingly, while increased PD1/PD-L1 interaction suppresses conventional T-helper responses, the opposite is observed for Tfh responses (348). Interaction between PD1+ Tfh cells with PD-L1 has a stronger suppressive effect on the regulatory subset of Tfh cells than on stimulatory Tfh cells and results in a net increase of Tfh activity (348).
Taken together, IL-17, T1-IFNs, and BAFF act in concert to drive TLS formation and Tfh responses. These responses support the development of autoreactive B-cells and the subsequent production of autoantibodies in AID but confer protective immunity in TB by improving the interaction between adaptive and innate cells and facilitating antibody production, while simultaneously inhibiting excessive inflammation by conventional CD4+ T-cell responses.
6. Concluding Remarks
The notion that complex mechanisms beyond Th1 immunity are at play in TB immunity is supported by (1) the unsatisfactory results of vaccine strategies aimed at boosting Th1 immunity in TB patients (31); (2) the inflammatory damage associated with increasing IFN-γ production by T-cells in the lungs of Mtb-infected mice (33); and (3) the host-detrimental effect of targeting the Th1-inhibiting PD1/PD-L1 interaction in mice (34, 35).
Patients with active TB express a T1-IFN transcriptional signature in their circulating leukocytes, but the exact identity and functional role of T1-IFNs in patients remains to be elucidated (62). Others have speculated that deleterious effects of T1-IFN-signaling during bacterial infections are tolerated because of their ability to suppress myeloid cell responses (41). This review highlights two additional aspects of T1-IFNs that are of interest in TB. The first concerns the preconditioning of myeloid cells prior to their contact with T1-IFNs. IFN-γ priming appears essential for the induction of an Mtb-permissive phenotype, and monocytes that differentiate under GM-CSF are less responsive to T1-IFNs than their M-CSF-differentiated counterparts (Figure 3) (43, 94). The second aspect is the diversification of IFN-α and IFN-β responses on a transcriptional and functional level as explained in Box 4. We propose that the inflammatory effects of IRF7-mediated IFN-α might contribute to excessive innate inflammatory responses in TB, while the immunosuppressive effects of IFN-β are more likely to support mycobacterial persistence.
Determination of the role of the Th17 response in TB is impeded by its heterogeneity, reflected in the presence of different Th17 subsets with ranging inflammatory potentials. Observations in AID emphasize the difference between IFN-γ/GM-CSF-producing Th17.1 cells and regular IL-17-producing Th17 cells. The exact role of T-cell-derived GM-CSF in TB remains to be determined, but preclinical TB studies show a protective role for GM-CSF on monocyte differentiation in the acute phase of TB. In contrast, IL-17 and PMN appear more relevant in chronic control of Mtb infection and recall immunity.
Immunological similarities between TB and AID may result from commonly activated pathogenic pathways. Alternatively, compensatory mechanisms induced by one disease might predispose for the development of the other. Interactions between IL-17, T1-IFNs, and BAFF form a pathological axis in AID that promote autoantibody-mediated autoimmunity.
The newly appreciated functional roles of antibodies, B-cells, and Tfh cells in TB provide suggestive evidence that pathogenic mechanisms in AID confer protective immunity to TB. Further, insight into these mechanisms as discussed in Figures 6–8 may generate leads for immune-directed therapies adjunct to current and newly developed antimicrobial treatment protocols.
Figure 8. Tertiary lymphoid structures (TLS) in tuberculosis (TB). TLS, Tfh cells, B-cells, and antibodies are all associated with protective immunity in TB. Preclinical TB models show that TLS induction and CXCL13 production are driven by IL-17 and IL-23. Type I interferons (T1-IFNs) and B-cell activating factor (BAFF) support TLS function and Tfh responses in autoimmune diseases (AID). In TB, T1-IFNs and BAFF are associated with active disease, but their functional role remains to be identified. Dotted lines implicate mechanisms present in AID that have not been confirmed in TB. Outstanding question: (1) What are the functional roles of T1-IFNs and BAFF in TLS function and humoral immunity in Mtb infection?
Author Contributions
BM has written the manuscript and drafted the figures; EL has written the initial version of Section 4 and contributed to the conception of Figure 5; JS and TO have contributed substantially to Sections 1, 2, 3, and 5 and the conception of Figures 1–3; PL has contributed substantially to the design of the work, writing of the manuscript, conception of all figures, and final review of the manuscript. All authors have revised this manuscript for intellectual content, approved its final version for publication, and have agreed to be accountable for all aspects of the work and in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors thank Ko Hagoort for his critical reading of the manuscript and Servier Medical Art (http://servier.com/Powerpoint-image-bank) for providing base images for the figures. EL acknowledges the Dutch Arthritis Association (12-02-409; 13-3-403; 14-02-201; and 15-2-206). TO acknowledges EC FP7 ADITEC (Grant Agreement No. 280873); EC HORIZON2020 TBVAC2020 (Grant Agreement No. 643381); EC FP7 EURIPRED (FP7-INFRA-2012 Grant Agreement No. 312661); The Netherlands Organization for Scientific Research (NWO-TOP Grant Agreement No. 91214038); The Bill & Melinda Gates Foundation Grand Challenges in Global Health (Grant GC6-2013); and the National Institute of Allergy and Infectious Diseases of the National Institutes of Health under Award Number R21AI127133. Research for this manuscript was (in part) performed within the framework of the Erasmus post-graduate school Molecular Medicine. The content is solely the responsibility of the authors and does not necessarily represent the official views of any funder. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
2. Mobley JL. Is rheumatoid arthritis a consequence of natural selection for enhanced tuberculosis resistance? Med Hypotheses (2004) 62:839–43. doi:10.1016/j.mehy.2003.12.006
3. Correa PA, Gomez LM, Cadena J, Anaya JM. Autoimmunity and tuberculosis. Opposite association with TNF polymorphism. J Rheumatol (2005) 32:219–24.
4. Boum Y II, Atwine D, Orikiriza P, Assimwe J, Page AL, Mwanga-Amumpaire J, et al. Male Gender is independently associated with pulmonary tuberculosis among sputum and non-sputum producers people with presumptive tuberculosis in Southwestern Uganda. BMC Infect Dis (2014) 14:638. doi:10.1186/s12879-014-0638-5
5. Fairweather D, Frisancho-Kiss S, Rose NR. Sex differences in autoimmune disease from a pathological perspective. Am J Pathol (2008) 173:600–9. doi:10.2353/ajpath.2008.071008
6. Strachan DP. Hay fever, hygiene, and household size. BMJ (1989) 299:1259–60. doi:10.1136/bmj.299.6710.1259
7. Okada H, Kuhn C, Feillet H, Bach JF. The ‘hygiene hypothesis’ for autoimmune and allergic diseases: an update. Clin Exp Immunol (2010) 160:1–9. doi:10.1111/j.1365-2249.2010.04139.x
8. Bach JF. The effect of infections on susceptibility to autoimmune and allergic diseases. N Engl J Med (2002) 347:911–20. doi:10.1056/NEJMra020100
9. Shapira Y, Agmon-Levin N, Shoenfeld Y. Mycobacterium tuberculosis, autoimmunity, and vitamin D. Clin Rev Allergy Immunol (2010) 38:169–77. doi:10.1007/s12016-009-8150-1
10. Elkington P, Tebruegge M, Mansour S. Tuberculosis: an infection-initiated autoimmune disease? Trends Immunol (2016) 37:815–8. doi:10.1016/j.it.2016.09.007
11. Elkayam O, Caspi D, Lidgi M, Segal R. Auto-antibody profiles in patients with active pulmonary tuberculosis. Int J Tuberc Lung Dis (2007) 11:306–10.
12. Shen CY, Hsieh SC, Yu CL, Wang JY, Lee LN, Yu CJ. Autoantibody prevalence in active tuberculosis: reactive or pathognomonic? BMJ Open (2013) 3:e002665. doi:10.1136/bmjopen-2013-002665
13. O’Garra A, Redford PS, Mcnab FW, Bloom CI, Wilkinson RJ, Berry MP. The immune response in tuberculosis. Annu Rev Immunol (2013) 31:475–527. doi:10.1146/annurev-immunol-032712-095939
14. Lyadova IV, Panteleev AV. Th1 and Th17 cells in tuberculosis: protection, pathology, and biomarkers. Mediators Inflamm (2015) 2015:854507. doi:10.1155/2015/854507
15. McNab F, Mayer-Barber K, Sher A, Wack A, O’garra A. Type I interferons in infectious disease. Nat Rev Immunol (2015) 15:87–103. doi:10.1038/nri3787
16. Caccamo N, Pietra G, Sullivan LC, Brooks AG, Prezzemolo T, La Manna MP, et al. Human CD8 T lymphocytes recognize Mycobacterium tuberculosis antigens presented by HLA-E during active tuberculosis and express type 2 cytokines. Eur J Immunol (2015) 45:1069–81. doi:10.1002/eji.201445193
17. Godfrey DI, Uldrich AP, Mccluskey J, Rossjohn J, Moody DB. The burgeoning family of unconventional T cells. Nat Immunol (2015) 16:1114–23. doi:10.1038/ni.3298
18. van Meijgaarden KE, Haks MC, Caccamo N, Dieli F, Ottenhoff TH, Joosten SA. Human CD8+ T-cells recognizing peptides from Mycobacterium tuberculosis (Mtb) presented by HLA-E have an unorthodox Th2-like, multifunctional, Mtb inhibitory phenotype and represent a novel human T-cell subset. PLoS Pathog (2015) 11:e1004671. doi:10.1371/journal.ppat.1004671
19. Axtell RC, De Jong BA, Boniface K, Van Der Voort LF, Bhat R, De Sarno P, et al. T helper type 1 and 17 cells determine efficacy of interferon-beta in multiple sclerosis and experimental encephalomyelitis. Nat Med (2010) 16:406–12. doi:10.1038/nm.2110
20. Brkic Z, Corneth OB, Van Helden-Meeuwsen CG, Dolhain RJ, Maria NI, Paulissen SM, et al. T-helper 17 cell cytokines and interferon type I: partners in crime in systemic lupus erythematosus? Arthritis Res Ther (2014) 16:R62. doi:10.1186/ar4499
21. Lopez P, Rodriguez-Carrio J, Caminal-Montero L, Mozo L, Suarez A. A pathogenic IFNalpha, BLyS and IL-17 axis in systemic lupus erythematosus patients. Sci Rep (2016) 6:20651. doi:10.1038/srep20651
22. Grosset J. Mycobacterium tuberculosis in the extracellular compartment: an underestimated adversary. Antimicrob Agents Chemother (2003) 47:833–6. doi:10.1128/AAC.47.3.833-836.2003
23. Scordo JM, Knoell DL, Torrelles JB. Alveolar epithelial cells in Mycobacterium tuberculosis infection: active players or innocent bystanders? J Innate Immun (2016) 8:3–14. doi:10.1159/000439275
24. Repasy T, Lee J, Marino S, Martinez N, Kirschner DE, Hendricks G, et al. Intracellular bacillary burden reflects a burst size for Mycobacterium tuberculosis in vivo. PLoS Pathog (2013) 9:e1003190. doi:10.1371/journal.ppat.1003190
25. Wolf AJ, Linas B, Trevejo-Nunez GJ, Kincaid E, Tamura T, Takatsu K, et al. Mycobacterium tuberculosis infects dendritic cells with high frequency and impairs their function in vivo. J Immunol (2007) 179:2509–19. doi:10.4049/jimmunol.179.4.2509
26. Shaler CR, Horvath CN, Jeyanathan M, Xing Z. Within the Enemy’s Camp: contribution of the granuloma to the dissemination, persistence and transmission of Mycobacterium tuberculosis. Front Immunol (2013) 4:30. doi:10.3389/fimmu.2013.00030
27. Klose CS, Artis D. Innate lymphoid cells as regulators of immunity, inflammation and tissue homeostasis. Nat Immunol (2016) 17:765–74. doi:10.1038/ni.3489
28. Schreiber G, Piehler J. The molecular basis for functional plasticity in type I interferon signaling. Trends Immunol (2015) 36:139–49. doi:10.1016/j.it.2015.01.002
29. Coscolla M, Copin R, Sutherland J, Gehre F, De Jong B, Owolabi O, et al. M. tuberculosis T cell epitope analysis reveals paucity of antigenic variation and identifies rare variable TB antigens. Cell Host Microbe (2015) 18:538–48. doi:10.1016/j.chom.2015.10.008
30. Colditz GA, Brewer TF, Berkey CS, Wilson ME, Burdick E, Fineberg HV, et al. Efficacy of BCG vaccine in the prevention of tuberculosis. Meta-analysis of the published literature. JAMA (1994) 271:698–702. doi:10.1001/jama.271.9.698
31. Tameris MD, Hatherill M, Landry BS, Scriba TJ, Snowden MA, Lockhart S, et al. Safety and efficacy of MVA85A, a new tuberculosis vaccine, in infants previously vaccinated with BCG: a randomised, placebo-controlled phase 2b trial. Lancet (2013) 381:1021–8. doi:10.1016/S0140-6736(13)60177-4
32. Ndiaye BP, Thienemann F, Ota M, Landry BS, Camara M, Dieye S, et al. Safety, immunogenicity, and efficacy of the candidate tuberculosis vaccine MVA85A in healthy adults infected with HIV-1: a randomised, placebo-controlled, phase 2 trial. Lancet Respir Med (2015) 3:190–200. doi:10.1016/S2213-2600(15)00037-5
33. Sakai S, Kauffman KD, Sallin MA, Sharpe AH, Young HA, Ganusov VV, et al. CD4 T cell-derived IFN-gamma plays a minimal role in control of pulmonary Mycobacterium tuberculosis infection and must be actively repressed by PD-1 to prevent lethal disease. PLoS Pathog (2016) 12:e1005667. doi:10.1371/journal.ppat.1005667
34. Lazar-Molnar E, Chen B, Sweeney KA, Wang EJ, Liu W, Lin J, et al. Programmed death-1 (PD-1)-deficient mice are extraordinarily sensitive to tuberculosis. Proc Natl Acad Sci U S A (2010) 107:13402–7. doi:10.1073/pnas.1007394107
35. Barber DL, Mayer-Barber KD, Feng CG, Sharpe AH, Sher A. CD4 T cells promote rather than control tuberculosis in the absence of PD-1-mediated inhibition. J Immunol (2011) 186:1598–607. doi:10.4049/jimmunol.1003304
36. Vogt G, Nathan C. In vitro differentiation of human macrophages with enhanced antimycobacterial activity. J Clin Invest (2011) 121:3889–901. doi:10.1172/JCI57235
37. Manry J, Laval G, Patin E, Fornarino S, Itan Y, Fumagalli M, et al. Evolutionary genetic dissection of human interferons. J Exp Med (2011) 208:2747–59. doi:10.1084/jem.20111680
38. Ivashkiv LB, Donlin LT. Regulation of type I interferon responses. Nat Rev Immunol (2014) 14:36–49. doi:10.1038/nri3581
39. Trinchieri G. Type I interferon: friend or foe? J Exp Med (2010) 207:2053–63. doi:10.1084/jem.20101664
40. Kearney S, Delgado C, Lenz LL. Differential effects of type I and II interferons on myeloid cells and resistance to intracellular bacterial infections. Immunol Res (2013) 55:187–200. doi:10.1007/s12026-012-8362-y
41. Eshleman EM, Lenz LL. Type I interferons in bacterial infections: taming of myeloid cells and possible implications for autoimmunity. Front Immunol (2014) 5:431. doi:10.3389/fimmu.2014.00431
42. Divangahi M, King IL, Pernet E. Alveolar macrophages and type I IFN in airway homeostasis and immunity. Trends Immunol (2015) 36:307–14. doi:10.1016/j.it.2015.03.005
43. Cunningham CR, Champhekar A, Tullius MV, Dillon BJ, Zhen A, De La Fuente JR, et al. Type I and type II interferon coordinately regulate suppressive dendritic cell fate and function during viral persistence. PLoS Pathog (2016) 12:e1005356. doi:10.1371/journal.ppat.1005356
44. Zitvogel L, Galluzzi L, Kepp O, Smyth MJ, Kroemer G. Type I interferons in anticancer immunity. Nat Rev Immunol (2015) 15:405–14. doi:10.1038/nri3845
45. Giosue S, Casarini M, Alemanno L, Galluccio G, Mattia P, Pedicelli G, et al. Effects of aerosolized interferon-alpha in patients with pulmonary tuberculosis. Am J Respir Crit Care Med (1998) 158:1156–62. doi:10.1164/ajrccm.158.4.9803065
46. Palmero D, Eiguchi K, Rendo P, Castro Zorrilla L, Abbate E, Gonzalez Montaner LJ. Phase II trial of recombinant interferon-alpha2b in patients with advanced intractable multidrug-resistant pulmonary tuberculosis: long-term follow-up. Int J Tuberc Lung Dis (1999) 3:214–8.
47. Giosue S, Casarini M, Ameglio F, Zangrilli P, Palla M, Altieri AM, et al. Aerosolized interferon-alpha treatment in patients with multi-drug-resistant pulmonary tuberculosis. Eur Cytokine Netw (2000) 11:99–104.
48. Mansoori D, Tavana S, Mirsaeidi M, Yazdanpanah M, Sohrabpour H. The efficacy of interferon-α in the treatment of multidrug resistant tuberculosis. Tanaffos (2002) 1:29–34.
49. Zarogoulidis P, Kioumis I, Papanas N, Manika K, Kontakiotis T, Papagianis A, et al. The effect of combination IFN-alpha-2a with usual antituberculosis chemotherapy in non-responding tuberculosis and diabetes mellitus: a case report and review of the literature. J Chemother (2012) 24:173–7. doi:10.1179/1973947812Y.0000000005
50. Sabbatani S, Manfredi R, Marinacci G, Pavoni M, Cristoni L, Chiodo F. Reactivation of severe, acute pulmonary tuberculosis during treatment with pegylated interferon-alpha and ribavirin for chronic HCV hepatitis. Scand J Infect Dis (2006) 38:205–8. doi:10.1080/00365540500263268
51. Farah R, Awad J. The association of interferon with the development of pulmonary tuberculosis. Int J Clin Pharmacol Ther (2007) 45:598–600. doi:10.5414/CPP45598
52. Telesca C, Angelico M, Piccolo P, Nosotti L, Morrone A, Longhi C, et al. Interferon-alpha treatment of hepatitis D induces tuberculosis exacerbation in an immigrant. J Infect (2007) 54:e223–6. doi:10.1016/j.jinf.2006.12.009
53. Tsai MC, Lin MC, Hung CH. Successful antiviral and antituberculosis treatment with pegylated interferon-alfa and ribavirin in a chronic hepatitis C patient with pulmonary tuberculosis. J Formos Med Assoc (2009) 108:746–50. doi:10.1016/S0929-6646(09)60400-9
54. Belkahla N, Kchir H, Maamouri N, Ouerghi H, Hariz FB, Chouaib S, et al. [Reactivation of tuberculosis during dual therapy with pegylated interferon and ribavirin for chronic hepatitis C]. Rev Med Interne (2010) 31:e1–3. doi:10.1016/j.revmed.2009.11.017
55. Babudieri S, Soddu A, Murino M, Molicotti P, Muredda AA, Madeddu G, et al. Tuberculosis screening before anti-hepatitis C virus therapy in prisons. Emerg Infect Dis (2012) 18:689–91. doi:10.3201/eid1804.111016
56. Guardigni V, Fabbri G, Badia L, Grilli A, Contini C. Tuberculosis reactivation in a patient with chronic HBV infection undergoing PEG-interferon therapy: case report and literature review. J Infect Dis Ther (2013) 1:3–7.
57. Matsuoka S, Fujikawa H, Hasegawa H, Ochiai T, Watanabe Y, Moriyama M. Onset of tuberculosis from a pulmonary latent tuberculosis infection during antiviral triple therapy for chronic hepatitis C. Intern Med (2016) 55:2011–7. doi:10.2169/internalmedicine.55.6448
58. Berry MP, Graham CM, Mcnab FW, Xu Z, Bloch SA, Oni T, et al. An interferon-inducible neutrophil-driven blood transcriptional signature in human tuberculosis. Nature (2010) 466:973–7. doi:10.1038/nature09247
59. Maertzdorf J, Repsilber D, Parida SK, Stanley K, Roberts T, Black G, et al. Human gene expression profiles of susceptibility and resistance in tuberculosis. Genes Immun (2011) 12:15–22. doi:10.1038/gene.2010.51
60. Ottenhoff TH, Dass RH, Yang N, Zhang MM, Wong HE, Sahiratmadja E, et al. Genome-wide expression profiling identifies type 1 interferon response pathways in active tuberculosis. PLoS One (2012) 7:e45839. doi:10.1371/journal.pone.0045839
61. Cliff JM, Lee JS, Constantinou N, Cho JE, Clark TG, Ronacher K, et al. Distinct phases of blood gene expression pattern through tuberculosis treatment reflect modulation of the humoral immune response. J Infect Dis (2013) 207:18–29. doi:10.1093/infdis/jis499
62. Zak DE, Penn-Nicholson A, Scriba TJ, Thompson E, Suliman S, Amon LM, et al. A blood RNA signature for tuberculosis disease risk: a prospective cohort study. Lancet (2016) 387:2312–22. doi:10.1016/S0140-6736(15)01316-1
63. Joosten SA, Fletcher HA, Ottenhoff TH. A helicopter perspective on TB biomarkers: pathway and process based analysis of gene expression data provides new insight into TB pathogenesis. PLoS One (2013) 8:e73230. doi:10.1371/journal.pone.0073230
64. Manca C, Tsenova L, Bergtold A, Freeman S, Tovey M, Musser JM, et al. Virulence of a Mycobacterium tuberculosis clinical isolate in mice is determined by failure to induce Th1 type immunity and is associated with induction of IFN-alpha/beta. Proc Natl Acad Sci U S A (2001) 98:5752–7. doi:10.1073/pnas.091096998
65. Manca C, Tsenova L, Freeman S, Barczak AK, Tovey M, Murray PJ, et al. Hypervirulent M. tuberculosis W/Beijing strains upregulate type I IFNs and increase expression of negative regulators of the Jak-Stat pathway. J Interferon Cytokine Res (2005) 25:694–701. doi:10.1089/jir.2005.25.694
66. Ordway D, Henao-Tamayo M, Harton M, Palanisamy G, Troudt J, Shanley C, et al. The hypervirulent Mycobacterium tuberculosis strain HN878 induces a potent TH1 response followed by rapid down-regulation. J Immunol (2007) 179:522–31. doi:10.4049/jimmunol.179.1.522
67. Stanley SA, Johndrow JE, Manzanillo P, Cox JS. The Type I IFN response to infection with Mycobacterium tuberculosis requires ESX-1-mediated secretion and contributes to pathogenesis. J Immunol (2007) 178:3143–52. doi:10.4049/jimmunol.178.5.3143
68. Desvignes L, Wolf AJ, Ernst JD. Dynamic roles of type I and type II IFNs in early infection with Mycobacterium tuberculosis. J Immunol (2012) 188:6205–15. doi:10.4049/jimmunol.1200255
69. Dorhoi A, Yeremeev V, Nouailles G, Weiner J III, Jorg S, Heinemann E, et al. Type I IFN signaling triggers immunopathology in tuberculosis-susceptible mice by modulating lung phagocyte dynamics. Eur J Immunol (2014) 44:2380–93. doi:10.1002/eji.201344219
70. Cooper AM, Pearl JE, Brooks JV, Ehlers S, Orme IM. Expression of the nitric oxide synthase 2 gene is not essential for early control of Mycobacterium tuberculosis in the murine lung. Infect Immun (2000) 68:6879–82. doi:10.1128/IAI.68.12.6879-6882.2000
71. Mayer-Barber KD, Andrade BB, Oland SD, Amaral EP, Barber DL, Gonzales J, et al. Host-directed therapy of tuberculosis based on interleukin-1 and type I interferon crosstalk. Nature (2014) 511:99–103. doi:10.1038/nature13489
72. Antonelli LR, Gigliotti Rothfuchs A, Goncalves R, Roffe E, Cheever AW, Bafica A, et al. Intranasal Poly-IC treatment exacerbates tuberculosis in mice through the pulmonary recruitment of a pathogen-permissive monocyte/macrophage population. J Clin Invest (2010) 120:1674–82. doi:10.1172/JCI40817
73. Redford PS, Mayer-Barber KD, Mcnab FW, Stavropoulos E, Wack A, Sher A, et al. Influenza A virus impairs control of Mycobacterium tuberculosis coinfection through a type I interferon receptor-dependent pathway. J Infect Dis (2014) 209:270–4. doi:10.1093/infdis/jit424
74. Remoli ME, Giacomini E, Lutfalla G, Dondi E, Orefici G, Battistini A, et al. Selective expression of type I IFN genes in human dendritic cells infected with Mycobacterium tuberculosis. J Immunol (2002) 169:366–74. doi:10.4049/jimmunol.169.1.366
75. Mariotti S, Teloni R, Iona E, Fattorini L, Romagnoli G, Gagliardi MC, et al. Mycobacterium tuberculosis diverts alpha interferon-induced monocyte differentiation from dendritic cells into immunoprivileged macrophage-like host cells. Infect Immun (2004) 72:4385–92. doi:10.1128/IAI.72.8.4385-4392.2004
76. Lienard J, Movert E, Valfridsson C, Sturegard E, Carlsson F. ESX-1 exploits type I IFN-signalling to promote a regulatory macrophage phenotype refractory to IFNgamma-mediated autophagy and growth restriction of intracellular mycobacteria. Cell Microbiol (2016) 18:1471–85. doi:10.1111/cmi.12594
77. Pandey AK, Yang Y, Jiang Z, Fortune SM, Coulombe F, Behr MA, et al. NOD2, RIP2 and IRF5 play a critical role in the type I interferon response to Mycobacterium tuberculosis. PLoS Pathog (2009) 5:e1000500. doi:10.1371/journal.ppat.1000500
78. Manzanillo PS, Shiloh MU, Portnoy DA, Cox JS. Mycobacterium tuberculosis activates the DNA-dependent cytosolic surveillance pathway within macrophages. Cell Host Microbe (2012) 11:469–80. doi:10.1016/j.chom.2012.03.007
79. Collins AC, Cai H, Li T, Franco LH, Li XD, Nair VR, et al. Cyclic GMP-AMP synthase is an innate immune DNA sensor for Mycobacterium tuberculosis. Cell Host Microbe (2015) 17:820–8. doi:10.1016/j.chom.2015.05.005
80. Wassermann R, Gulen MF, Sala C, Perin SG, Lou Y, Rybniker J, et al. Mycobacterium tuberculosis differentially activates cGAS- and inflammasome-dependent intracellular immune responses through ESX-1. Cell Host Microbe (2015) 17:799–810. doi:10.1016/j.chom.2015.05.003
81. Watson RO, Bell SL, Macduff DA, Kimmey JM, Diner EJ, Olivas J, et al. The cytosolic sensor cGAS detects Mycobacterium tuberculosis DNA to induce type I interferons and activate autophagy. Cell Host Microbe (2015) 17:811–9. doi:10.1016/j.chom.2015.05.004
82. Houben D, Demangel C, Van Ingen J, Perez J, Baldeon L, Abdallah AM, et al. ESX-1-mediated translocation to the cytosol controls virulence of mycobacteria. Cell Microbiol (2012) 14:1287–98. doi:10.1111/j.1462-5822.2012.01799.x
83. Dey B, Dey RJ, Cheung LS, Pokkali S, Guo H, Lee JH, et al. A bacterial cyclic dinucleotide activates the cytosolic surveillance pathway and mediates innate resistance to tuberculosis. Nat Med (2015) 21:401–6. doi:10.1038/nm.3813
84. Saiga H, Kitada S, Shimada Y, Kamiyama N, Okuyama M, Makino M, et al. Critical role of AIM2 in Mycobacterium tuberculosis infection. Int Immunol (2012) 24:637–44. doi:10.1093/intimm/dxs062
85. Asano M, Hayashi M, Yoshida E, Kawade Y, Iwakura Y. Induction of interferon-alpha by interferon-beta, but not of interferon-beta by interferon-alpha, in the mouse. Virology (1990) 176:30–8. doi:10.1016/0042-6822(90)90227-I
86. Swiecki M, Colonna M. The multifaceted biology of plasmacytoid dendritic cells. Nat Rev Immunol (2015) 15:471–85. doi:10.1038/nri3865
87. Saraav I, Singh S, Sharma S. Outcome of Mycobacterium tuberculosis and Toll-like receptor interaction: immune response or immune evasion? Immunol Cell Biol (2014) 92:741–6. doi:10.1038/icb.2014.52
88. Mvubu NE, Pillay B, Gamieldien J, Bishai W, Pillay M. Canonical pathways, networks and transcriptional factor regulation by clinical strains of Mycobacterium tuberculosis in pulmonary alveolar epithelial cells. Tuberculosis (Edinb) (2016) 97:73–85. doi:10.1016/j.tube.2015.12.002
89. Bafica A, Scanga CA, Feng CG, Leifer C, Cheever A, Sher A. TLR9 regulates Th1 responses and cooperates with TLR2 in mediating optimal resistance to Mycobacterium tuberculosis. J Exp Med (2005) 202:1715–24. doi:10.1084/jem.20051782
90. Mayer-Barber KD, Andrade BB, Barber DL, Hieny S, Feng CG, Caspar P, et al. Innate and adaptive interferons suppress IL-1alpha and IL-1beta production by distinct pulmonary myeloid subsets during Mycobacterium tuberculosis infection. Immunity (2011) 35:1023–34. doi:10.1016/j.immuni.2011.12.002
91. Lyadova IV, Tsiganov EN, Kapina MA, Shepelkova GS, Sosunov VV, Radaeva TV, et al. In mice, tuberculosis progression is associated with intensive inflammatory response and the accumulation of Gr-1 cells in the lungs. PLoS One (2010) 5:e10469. doi:10.1371/journal.pone.0010469
92. Knaul JK, Jorg S, Oberbeck-Mueller D, Heinemann E, Scheuermann L, Brinkmann V, et al. Lung-residing myeloid-derived suppressors display dual functionality in murine pulmonary tuberculosis. Am J Respir Crit Care Med (2014) 190:1053–66. doi:10.1164/rccm.201405-0828OC
93. Tsiganov EN, Verbina EM, Radaeva TV, Sosunov VV, Kosmiadi GA, Nikitina IY, et al. Gr-1dimCD11b+ immature myeloid-derived suppressor cells but not neutrophils are markers of lethal tuberculosis infection in mice. J Immunol (2014) 192:4718–27. doi:10.4049/jimmunol.1301365
94. Fleetwood AJ, Dinh H, Cook AD, Hertzog PJ, Hamilton JA. GM-CSF- and M-CSF-dependent macrophage phenotypes display differential dependence on type I interferon signaling. J Leukoc Biol (2009) 86:411–21. doi:10.1189/jlb.1108702
95. Conrady CD, Zheng M, Mandal NA, Van Rooijen N, Carr DJ. IFN-alpha-driven CCL2 production recruits inflammatory monocytes to infection site in mice. Mucosal Immunol (2013) 6:45–55. doi:10.1038/mi.2012.46
96. Yoshimura T, Galligan C, Takahashi M, Chen K, Liu M, Tessarollo L, et al. Non-myeloid cells are major contributors to innate immune responses via production of monocyte chemoattractant protein-1/CCL2. Front Immunol (2014) 4:482. doi:10.3389/fimmu.2013.00482
97. Jayaraman P, Sada-Ovalle I, Nishimura T, Anderson AC, Kuchroo VK, Remold HG, et al. IL-1beta promotes antimicrobial immunity in macrophages by regulating TNFR signaling and caspase-3 activation. J Immunol (2013) 190:4196–204. doi:10.4049/jimmunol.1202688
98. Di Paolo NC, Shafiani S, Day T, Papayannopoulou T, Russell DW, Iwakura Y, et al. Interdependence between interleukin-1 and tumor necrosis factor regulates TNF-dependent control of Mycobacterium tuberculosis infection. Immunity (2015) 43:1125–36. doi:10.1016/j.immuni.2015.11.016
99. Behar SM, Martin CJ, Booty MG, Nishimura T, Zhao X, Gan HX, et al. Apoptosis is an innate defense function of macrophages against Mycobacterium tuberculosis. Mucosal Immunol (2011) 4:279–87. doi:10.1038/mi.2011.3
100. Barnes PF, Chan LS, Wong SF. The course of fever during treatment of pulmonary tuberculosis. Tubercle (1987) 68:255–60. doi:10.1016/0041-3879(87)90065-1
101. Lazarus M, Yoshida K, Coppari R, Bass CE, Mochizuki T, Lowell BB, et al. EP3 prostaglandin receptors in the median preoptic nucleus are critical for fever responses. Nat Neurosci (2007) 10:1131–3. doi:10.1038/nn1949
102. Luan B, Yoon YS, Le Lay J, Kaestner KH, Hedrick S, Montminy M. CREB pathway links PGE2 signaling with macrophage polarization. Proc Natl Acad Sci U S A (2015) 112:15642–7. doi:10.1073/pnas.1519644112
103. Serezani CH, Chung J, Ballinger MN, Moore BB, Aronoff DM, Peters-Golden M. Prostaglandin E2 suppresses bacterial killing in alveolar macrophages by inhibiting NADPH oxidase. Am J Respir Cell Mol Biol (2007) 37:562–70. doi:10.1165/rcmb.2007-0153OC
104. MacKenzie KF, Clark K, Naqvi S, Mcguire VA, Noehren G, Kristariyanto Y, et al. PGE(2) induces macrophage IL-10 production and a regulatory-like phenotype via a protein kinase A-SIK-CRTC3 pathway. J Immunol (2013) 190:565–77. doi:10.4049/jimmunol.1202462
105. Mao Y, Sarhan D, Poschke I, Lundqvist A, Kiessling R. Inhibiton of tumor-derived prostaglandin-e2 prevents the induction of human myeloid-derived suppressor cells (MDSCs) and rescues anti-tumor immunity. J Immunother Cancer (2014) 2:224–224. doi:10.1186/2051-1426-2-S3-P224
106. Boniface K, Bak-Jensen KS, Li Y, Blumenschein WM, Mcgeachy MJ, Mcclanahan TK, et al. Prostaglandin E2 regulates Th17 cell differentiation and function through cyclic AMP and EP2/EP4 receptor signaling. J Exp Med (2009) 206:535–48. doi:10.1084/jem.20082293
107. Shi Q, Yin Z, Zhao B, Sun F, Yu H, Yin X, et al. PGE2 elevates IL-23 production in human dendritic cells via a cAMP dependent pathway. Mediators Inflamm (2015) 2015:984690. doi:10.1155/2015/984690
108. Kaminskaia GO, Blonskaia G, Omarov TO, Lovacheva OV. [Level and ratio of prostaglandins group E and F2(alpha) in bronchoalveolar washes in pulmonary tuberculosis patients] Soderzhanie i sootnosheniia prostaglandinov grupp E i F2(alpha) v bronkhoal’veoliarnykh smyvakh u bol’nykh tuberkulezom legkikh. Vopr Med Khim (1991) 37:71–3.
109. Shu CC, Wu MF, Hsu CL, Huang CT, Wang JY, Hsieh SL, et al. Apoptosis-associated biomarkers in tuberculosis: promising for diagnosis and prognosis prediction. BMC Infect Dis (2013) 13:45. doi:10.1186/1471-2334-13-45
110. Liang L, Zhang Q, Luo LL, Yue J, Zhao YL, Han M, et al. Polymorphisms in the prostaglandin receptor EP2 gene confers susceptibility to tuberculosis. Infect Genet Evol (2016) 46:23–7. doi:10.1016/j.meegid.2016.10.016
111. Rangel Moreno J, Estrada Garcia I, De La Luz Garcia Hernandez M, Aguilar Leon D, Marquez R, Hernandez Pando R. The role of prostaglandin E2 in the immunopathogenesis of experimental pulmonary tuberculosis. Immunology (2002) 106:257–66. doi:10.1046/j.1365-2567.2002.01403.x
112. Kalinski P. Regulation of immune responses by prostaglandin E2. J Immunol (2012) 188:21–8. doi:10.4049/jimmunol.1101029
113. Russell DG, Cardona PJ, Kim MJ, Allain S, Altare F. Foamy macrophages and the progression of the human tuberculosis granuloma. Nat Immunol (2009) 10:943–8. doi:10.1038/ni.1781
114. Novikov A, Cardone M, Thompson R, Shenderov K, Kirschman KD, Mayer-Barber KD, et al. Mycobacterium tuberculosis triggers host type I IFN signaling to regulate IL-1beta production in human macrophages. J Immunol (2011) 187:2540–7. doi:10.4049/jimmunol.1100926
115. McNab FW, Ewbank J, Howes A, Moreira-Teixeira L, Martirosyan A, Ghilardi N, et al. Type I IFN induces IL-10 production in an IL-27-independent manner and blocks responsiveness to IFN-gamma for production of IL-12 and bacterial killing in Mycobacterium tuberculosis-infected macrophages. J Immunol (2014) 193:3600–12. doi:10.4049/jimmunol.1401088
116. Chen M, Divangahi M, Gan H, Shin DS, Hong S, Lee DM, et al. Lipid mediators in innate immunity against tuberculosis: opposing roles of PGE2 and LXA4 in the induction of macrophage death. J Exp Med (2008) 205:2791–801. doi:10.1084/jem.20080767
117. Lockhart E, Green AM, Flynn JL. IL-17 production is dominated by gammadelta T cells rather than CD4 T cells during Mycobacterium tuberculosis infection. J Immunol (2006) 177:4662–9. doi:10.4049/jimmunol.177.7.4662
118. El-Behi M, Ciric B, Dai H, Yan Y, Cullimore M, Safavi F, et al. The encephalitogenicity of T(H)17 cells is dependent on IL-1- and IL-23-induced production of the cytokine GM-CSF. Nat Immunol (2011) 12:568–75. doi:10.1038/ni.2031
119. Fremond CM, Togbe D, Doz E, Rose S, Vasseur V, Maillet I, et al. IL-1 receptor-mediated signal is an essential component of MyD88-dependent innate response to Mycobacterium tuberculosis infection. J Immunol (2007) 179:1178–89. doi:10.4049/jimmunol.179.2.1178
120. Mishra BB, Rathinam VA, Martens GW, Martinot AJ, Kornfeld H, Fitzgerald KA, et al. Nitric oxide controls the immunopathology of tuberculosis by inhibiting NLRP3 inflammasome-dependent processing of IL-1beta. Nat Immunol (2013) 14:52–60. doi:10.1038/ni.2474
121. Une C, Andersson J, Orn A. Role of IFN-alpha/beta and IL-12 in the activation of natural killer cells and interferon-gamma production during experimental infection with Trypanosoma cruzi. Clin Exp Immunol (2003) 134:195–201. doi:10.1046/j.1365-2249.2003.02294.x
122. Longhi MP, Trumpfheller C, Idoyaga J, Caskey M, Matos I, Kluger C, et al. Dendritic cells require a systemic type I interferon response to mature and induce CD4+ Th1 immunity with poly IC as adjuvant. J Exp Med (2009) 206:1589–602. doi:10.1084/jem.20090247
123. Zhang G, Zhou B, Li S, Yue J, Yang H, Wen Y, et al. Allele-specific induction of IL-1beta expression by C/EBPbeta and PU.1 contributes to increased tuberculosis susceptibility. PLoS Pathog (2014) 10:e1004426. doi:10.1371/journal.ppat.1004426
124. Berclaz PY, Carey B, Fillipi MD, Wernke-Dollries K, Geraci N, Cush S, et al. GM-CSF regulates a PU.1-dependent transcriptional program determining the pulmonary response to LPS. Am J Respir Cell Mol Biol (2007) 36:114–21. doi:10.1165/rcmb.2006-0174OC
125. Higgins DM, Sanchez-Campillo J, Rosas-Taraco AG, Higgins JR, Lee EJ, Orme IM, et al. Relative levels of M-CSF and GM-CSF influence the specific generation of macrophage populations during infection with Mycobacterium tuberculosis. J Immunol (2008) 180:4892–900. doi:10.4049/jimmunol.180.7.4892
126. Fejer G, Wegner MD, Gyory I, Cohen I, Engelhard P, Voronov E, et al. Nontransformed, GM-CSF-dependent macrophage lines are a unique model to study tissue macrophage functions. Proc Natl Acad Sci U S A (2013) 110:E2191–8. doi:10.1073/pnas.1302877110
127. Nandi B, Behar SM. Regulation of neutrophils by interferon-gamma limits lung inflammation during tuberculosis infection. J Exp Med (2011) 208:2251–62. doi:10.1084/jem.20110919
128. de Bruin AM, Voermans C, Nolte MA. Impact of interferon-gamma on hematopoiesis. Blood (2014) 124:2479–86. doi:10.1182/blood-2014-04-568451
129. Reboldi A, Dang EV, Mcdonald JG, Liang G, Russell DW, Cyster JG. Inflammation. 25-hydroxycholesterol suppresses interleukin-1-driven inflammation downstream of type I interferon. Science (2014) 345:679–84. doi:10.1126/science.1254790
130. Hoeksema MA, Scicluna BP, Boshuizen MC, Van Der Velden S, Neele AE, Van Den Bossche J, et al. IFN-gamma priming of macrophages represses a part of the inflammatory program and attenuates neutrophil recruitment. J Immunol (2015) 194:3909–16. doi:10.4049/jimmunol.1402077
131. Stifter SA, Bhattacharyya N, Pillay R, Florido M, Triccas JA, Britton WJ, et al. Functional interplay between type I and II interferons is essential to limit influenza A virus-induced tissue inflammation. PLoS Pathog (2016) 12:e1005378. doi:10.1371/journal.ppat.1005378
132. Guarda G, Braun M, Staehli F, Tardivel A, Mattmann C, Forster I, et al. Type I interferon inhibits interleukin-1 production and inflammasome activation. Immunity (2011) 34:213–23. doi:10.1016/j.immuni.2011.02.006
133. Mourik BC, Leenen PJ, De Knegt GJ, Huizinga R, Van Der Eerden BC, Wang J, et al. Immunotherapy added to antibiotic treatment reduces relapse of disease in a mouse model of tuberculosis. Am J Respir Cell Mol Biol (2017) 56:233–41. doi:10.1165/rcmb.2016-0185OC
134. Gautier G, Humbert M, Deauvieau F, Scuiller M, Hiscott J, Bates EE, et al. A type I interferon autocrine-paracrine loop is involved in Toll-like receptor-induced interleukin-12p70 secretion by dendritic cells. J Exp Med (2005) 201:1435–46. doi:10.1084/jem.20041964
135. Goriely S, Molle C, Nguyen M, Albarani V, Haddou NO, Lin R, et al. Interferon regulatory factor 3 is involved in Toll-like receptor 4 (TLR4)- and TLR3-induced IL-12p35 gene activation. Blood (2006) 107:1078–84. doi:10.1182/blood-2005-06-2416
136. Crouse J, Kalinke U, Oxenius A. Regulation of antiviral T cell responses by type I interferons. Nat Rev Immunol (2015) 15:231–42. doi:10.1038/nri3806
137. Spits H, Bernink JH, Lanier L. NK cells and type 1 innate lymphoid cells: partners in host defense. Nat Immunol (2016) 17:758–64. doi:10.1038/ni.3482
138. Dalod M, Salazar-Mather TP, Malmgaard L, Lewis C, Asselin-Paturel C, Briere F, et al. Interferon alpha/beta and interleukin 12 responses to viral infections: pathways regulating dendritic cell cytokine expression in vivo. J Exp Med (2002) 195:517–28. doi:10.1084/jem.20011672
139. Schreiber T, Ehlers S, Heitmann L, Rausch A, Mages J, Murray PJ, et al. Autocrine IL-10 induces hallmarks of alternative activation in macrophages and suppresses antituberculosis effector mechanisms without compromising T cell immunity. J Immunol (2009) 183:1301–12. doi:10.4049/jimmunol.0803567
140. de Paus RA, Van Wengen A, Schmidt I, Visser M, Verdegaal EM, Van Dissel JT, et al. Inhibition of the type I immune responses of human monocytes by IFN-alpha and IFN-beta. Cytokine (2013) 61:645–55. doi:10.1016/j.cyto.2012.12.005
141. Teles RM, Graeber TG, Krutzik SR, Montoya D, Schenk M, Lee DJ, et al. Type I interferon suppresses type II interferon-triggered human anti-mycobacterial responses. Science (2013) 339:1448–53. doi:10.1126/science.1233665
142. Gopal R, Lin Y, Obermajer N, Slight S, Nuthalapati N, Ahmed M, et al. IL-23-dependent IL-17 drives Th1-cell responses following Mycobacterium bovis BCG vaccination. Eur J Immunol (2012) 42:364–73. doi:10.1002/eji.201141569
143. Tada H, Aiba S, Shibata K, Ohteki T, Takada H. Synergistic effect of Nod1 and Nod2 agonists with toll-like receptor agonists on human dendritic cells to generate interleukin-12 and T helper type 1 cells. Infect Immun (2005) 73:7967–76. doi:10.1128/IAI.73.12.7967-7976.2005
144. Manni M, Granstein RD, Maestroni G. beta2-Adrenergic agonists bias TLR-2 and NOD2 activated dendritic cells towards inducing an IL-17 immune response. Cytokine (2011) 55:380–6. doi:10.1016/j.cyto.2011.05.013
145. Edwards AD, Manickasingham SP, Sporri R, Diebold SS, Schulz O, Sher A, et al. Microbial recognition via Toll-like receptor-dependent and -independent pathways determines the cytokine response of murine dendritic cell subsets to CD40 triggering. J Immunol (2002) 169:3652–60. doi:10.4049/jimmunol.169.7.3652
146. Napolitani G, Rinaldi A, Bertoni F, Sallusto F, Lanzavecchia A. Selected Toll-like receptor agonist combinations synergistically trigger a T helper type 1-polarizing program in dendritic cells. Nat Immunol (2005) 6:769–76. doi:10.1038/ni1223
147. Schlitzer A, Mcgovern N, Teo P, Zelante T, Atarashi K, Low D, et al. IRF4 transcription factor-dependent CD11b+ dendritic cells in human and mouse control mucosal IL-17 cytokine responses. Immunity (2013) 38:970–83. doi:10.1016/j.immuni.2013.04.011
148. Goriely S, Neurath MF, Goldman M. How microorganisms tip the balance between interleukin-12 family members. Nat Rev Immunol (2008) 8:81–6. doi:10.1038/nri2225
149. von Scheidt B, Leung PS, Yong MC, Zhang Y, Towne JE, Smyth MJ, et al. Combined anti-CD40 and anti-IL-23 monoclonal antibody therapy effectively suppresses tumor growth and metastases. Cancer Res (2014) 74:2412–21. doi:10.1158/0008-5472.CAN-13-1646
150. Hochrein H, O’keeffe M, Luft T, Vandenabeele S, Grumont RJ, Maraskovsky E, et al. Interleukin (IL)-4 is a major regulatory cytokine governing bioactive IL-12 production by mouse and human dendritic cells. J Exp Med (2000) 192:823–33. doi:10.1084/jem.192.6.823
151. Guenova E, Skabytska Y, Hoetzenecker W, Weindl G, Sauer K, Tham M, et al. IL-4 abrogates T(H)17 cell-mediated inflammation by selective silencing of IL-23 in antigen-presenting cells. Proc Natl Acad Sci U S A (2015) 112:2163–8. doi:10.1073/pnas.1416922112
152. Lyakh L, Trinchieri G, Provezza L, Carra G, Gerosa F. Regulation of interleukin-12/interleukin-23 production and the T-helper 17 response in humans. Immunol Rev (2008) 226:112–31. doi:10.1111/j.1600-065X.2008.00700.x
153. Roses RE, Xu S, Xu M, Koldovsky U, Koski G, Czerniecki BJ. Differential production of IL-23 and IL-12 by myeloid-derived dendritic cells in response to TLR agonists. J Immunol (2008) 181:5120–7. doi:10.4049/jimmunol.181.7.5120
154. Askenase MH, Han SJ, Byrd AL, Morais Da Fonseca D, Bouladoux N, Wilhelm C, et al. Bone-marrow-resident NK cells prime monocytes for regulatory function during infection. Immunity (2015) 42:1130–42. doi:10.1016/j.immuni.2015.05.011
155. Sorgi CA, Rose S, Court N, Carlos D, Paula-Silva FW, Assis PA, et al. GM-CSF priming drives bone marrow-derived macrophages to a pro-inflammatory pattern and downmodulates PGE2 in response to TLR2 ligands. PLoS One (2012) 7:e40523. doi:10.1371/journal.pone.0040523
156. Sierra-Filardi E, Nieto C, Dominguez-Soto A, Barroso R, Sanchez-Mateos P, Puig-Kroger A, et al. CCL2 shapes macrophage polarization by GM-CSF and M-CSF: identification of CCL2/CCR2-dependent gene expression profile. J Immunol (2014) 192:3858–67. doi:10.4049/jimmunol.1302821
157. Langrish CL, Mckenzie BS, Wilson NJ, De Waal Malefyt R, Kastelein RA, Cua DJ. IL-12 and IL-23: master regulators of innate and adaptive immunity. Immunol Rev (2004) 202:96–105. doi:10.1111/j.0105-2896.2004.00214.x
158. Khader SA, Pearl JE, Sakamoto K, Gilmartin L, Bell GK, Jelley-Gibbs DM, et al. IL-23 compensates for the absence of IL-12p70 and is essential for the IL-17 response during tuberculosis but is dispensable for protection and antigen-specific IFN-gamma responses if IL-12p70 is available. J Immunol (2005) 175:788–95. doi:10.4049/jimmunol.175.2.788
159. Korn T, Bettelli E, Oukka M, Kuchroo VK. IL-17 and Th17 cells. Annu Rev Immunol (2009) 27:485–517. doi:10.1146/annurev.immunol.021908.132710
160. Lubberts E. The IL-23-IL-17 axis in inflammatory arthritis. Nat Rev Rheumatol (2015) 11:415–29. doi:10.1038/nrrheum.2015.53
161. Kara EE, Mckenzie DR, Bastow CR, Gregor CE, Fenix KA, Ogunniyi AD, et al. CCR2 defines in vivo development and homing of IL-23-driven GM-CSF-producing Th17 cells. Nat Commun (2015) 6:8644. doi:10.1038/ncomms9644
162. Gagliani N, Amezcua Vesely MC, Iseppon A, Brockmann L, Xu H, Palm NW, et al. Th17 cells transdifferentiate into regulatory T cells during resolution of inflammation. Nature (2015) 523:221–5. doi:10.1038/nature14452
163. Sutton CE, Lalor SJ, Sweeney CM, Brereton CF, Lavelle EC, Mills KH. Interleukin-1 and IL-23 induce innate IL-17 production from gammadelta T cells, amplifying Th17 responses and autoimmunity. Immunity (2009) 31:331–41. doi:10.1016/j.immuni.2009.08.001
164. Griffin GK, Newton G, Tarrio ML, Bu DX, Maganto-Garcia E, Azcutia V, et al. IL-17 and TNF-alpha sustain neutrophil recruitment during inflammation through synergistic effects on endothelial activation. J Immunol (2012) 188:6287–99. doi:10.4049/jimmunol.1200385
165. Fischer JA, Hueber AJ, Wilson S, Galm M, Baum W, Kitson C, et al. Combined inhibition of tumor necrosis factor alpha and interleukin-17 as a therapeutic opportunity in rheumatoid arthritis: development and characterization of a novel bispecific antibody. Arthritis Rheumatol (2015) 67:51–62. doi:10.1002/art.38896
166. Torrado E, Cooper AM. IL-17 and Th17 cells in tuberculosis. Cytokine Growth Factor Rev (2010) 21:455–62. doi:10.1016/j.cytogfr.2010.10.004
167. Peng R, Yue J, Han M, Zhao Y, Liu L, Liang L. The IL-17F sequence variant is associated with susceptibility to tuberculosis. Gene (2013) 515:229–32. doi:10.1016/j.gene.2012.11.017
168. Du J, Han J, Li X, Zhang Y, Li H, Yang S. StIL-17 gene polymorphisms in the development of pulmonary tuberculosis. Int J Clin Exp Pathol (2015) 8:3225–9.
169. Milano M, Moraes MO, Rodenbusch R, Carvalho CX, Delcroix M, Mousquer G, et al. Single nucleotide polymorphisms in IL17A and IL6 are associated with decreased risk for pulmonary tuberculosis in Southern Brazilian population. PLoS One (2016) 11:e0147814. doi:10.1371/journal.pone.0147814
170. Wang M, Xu G, Lu L, Xu K, Chen Y, Pan H, et al. Genetic polymorphisms of IL-17A, IL-17F, TLR4 and miR-146a in association with the risk of pulmonary tuberculosis. Sci Rep (2016) 6:28586. doi:10.1038/srep28586
171. Abhimanyu, Bose M, Komal, Varma-Basil M. Lack of association between IL17A and IL17F polymorphisms and related serum levels in north Indians with tuberculosis. Gene (2013) 529:195–8. doi:10.1016/j.gene.2013.06.090
172. Bulat-Kardum LJ, Etokebe GE, Lederer P, Balen S, Dembic Z. Genetic polymorphisms in the toll-like receptor 10, interleukin (IL)17A and IL17F genes differently affect the risk for tuberculosis in croatian population. Scand J Immunol (2015) 82:63–9. doi:10.1111/sji.12300
173. Chen X, Zhang M, Liao M, Graner MW, Wu C, Yang Q, et al. Reduced Th17 response in patients with tuberculosis correlates with IL-6R expression on CD4+ T cells. Am J Respir Crit Care Med (2010) 181:734–42. doi:10.1164/rccm.200909-1463OC
174. Cowan J, Pandey S, Filion LG, Angel JB, Kumar A, Cameron DW. Comparison of interferon-gamma-, interleukin (IL)-17- and IL-22-expressing CD4 T cells, IL-22-expressing granulocytes and proinflammatory cytokines during latent and active tuberculosis infection. Clin Exp Immunol (2012) 167:317–29. doi:10.1111/j.1365-2249.2011.04520.x
175. Scriba TJ, Kalsdorf B, Abrahams DA, Isaacs F, Hofmeister J, Black G, et al. Distinct, specific IL-17- and IL-22-producing CD4+ T cell subsets contribute to the human anti-mycobacterial immune response. J Immunol (2008) 180:1962–70. doi:10.4049/jimmunol.180.3.1962
176. Metcalfe JZ, Everett CK, Steingart KR, Cattamanchi A, Huang L, Hopewell PC, et al. Interferon-gamma release assays for active pulmonary tuberculosis diagnosis in adults in low- and middle-income countries: systematic review and meta-analysis. J Infect Dis (2011) 204(Suppl 4):S1120–9. doi:10.1093/infdis/jir410
177. Pai M, Denkinger CM, Kik SV, Rangaka MX, Zwerling A, Oxlade O, et al. Gamma interferon release assays for detection of Mycobacterium tuberculosis infection. Clin Microbiol Rev (2014) 27:3–20. doi:10.1128/CMR.00034-13
178. Roberts T, Beyers N, Aguirre A, Walzl G. Immunosuppression during active tuberculosis is characterized by decreased interferon-gamma production and CD25 expression with elevated forkhead box P3, transforming growth factor-beta, and interleukin-4 mRNA levels. J Infect Dis (2007) 195:870–8. doi:10.1086/511277
179. Singh A, Mohan A, Dey AB, Mitra DK. Inhibiting the programmed death 1 pathway rescues Mycobacterium tuberculosis-specific interferon gamma-producing T cells from apoptosis in patients with pulmonary tuberculosis. J Infect Dis (2013) 208:603–15. doi:10.1093/infdis/jit206
180. Ashenafi S, Aderaye G, Bekele A, Zewdie M, Aseffa G, Hoang AT, et al. Progression of clinical tuberculosis is associated with a Th2 immune response signature in combination with elevated levels of SOCS3. Clin Immunol (2014) 151:84–99. doi:10.1016/j.clim.2014.01.010
181. Boer MC, Van Meijgaarden KE, Goletti D, Vanini V, Prins C, Ottenhoff TH, et al. KLRG1 and PD-1 expression are increased on T-cells following tuberculosis-treatment and identify cells with different proliferative capacities in BCG-vaccinated adults. Tuberculosis (Edinb) (2016) 97:163–71. doi:10.1016/j.tube.2015.11.008
182. Bandaru A, Devalraju KP, Paidipally P, Dhiman R, Venkatasubramanian S, Barnes PF, et al. Phosphorylated STAT3 and PD-1 regulate IL-17 production and IL-23 receptor expression in Mycobacterium tuberculosis infection. Eur J Immunol (2014) 44:2013–24. doi:10.1002/eji.201343680
183. Basile JI, Geffner LJ, Romero MM, Balboa L, Sabio YGC, Ritacco V, et al. Outbreaks of Mycobacterium tuberculosis MDR strains induce high IL-17 T-cell response in patients with MDR tuberculosis that is closely associated with high antigen load. J Infect Dis (2011) 204:1054–64. doi:10.1093/infdis/jir460
184. Marin ND, Paris SC, Rojas M, Garcia LF. Reduced frequency of memory T cells and increased Th17 responses in patients with active tuberculosis. Clin Vaccine Immunol (2012) 19:1667–76. doi:10.1128/CVI.00390-12
185. Marin ND, Paris SC, Rojas M, Garcia LF. Functional profile of CD4+ and CD8+ T cells in latently infected individuals and patients with active TB. Tuberculosis (Edinb) (2013) 93:155–66. doi:10.1016/j.tube.2012.12.002
186. Perreau M, Rozot V, Welles HC, Belluti-Enders F, Vigano S, Maillard M, et al. Lack of Mycobacterium tuberculosis-specific interleukin-17A-producing CD4+ T cells in active disease. Eur J Immunol (2013) 43:939–48. doi:10.1002/eji.201243090
187. Jurado JO, Pasquinelli V, Alvarez IB, Pena D, Rovetta AI, Tateosian NL, et al. IL-17 and IFN-gamma expression in lymphocytes from patients with active tuberculosis correlates with the severity of the disease. J Leukoc Biol (2012) 91:991–1002. doi:10.1189/jlb.1211619
188. Heidarnezhad F, Asnaashari A, Rezaee SA, Ghezelsofla R, Ghazvini K, Valizadeh N, et al. Evaluation of Interleukin 17 and Interleukin 23 expression in patients with active and latent tuberculosis infection. Iran J Basic Med Sci (2016) 19:844–50.
189. Xu L, Cui G, Jia H, Zhu Y, Ding Y, Chen J, et al. Decreased IL-17 during treatment of sputum smear-positive pulmonary tuberculosis due to increased regulatory T cells and IL-10. J Transl Med (2016) 14:179. doi:10.1186/s12967-016-0909-6
190. Sargentini V, Mariotti S, Carrara S, Gagliardi MC, Teloni R, Goletti D, et al. Cytometric detection of antigen-specific IFN-gamma/IL-2 secreting cells in the diagnosis of tuberculosis. BMC Infect Dis (2009) 9:99. doi:10.1186/1471-2334-9-99
191. Kim SY, Park MS, Kim YS, Kim SK, Chang J, Lee HJ, et al. The responses of multiple cytokines following incubation of whole blood from TB patients, latently infected individuals and controls with the TB antigens ESAT-6, CFP-10 and TB7.7. Scand J Immunol (2012) 76:580–6. doi:10.1111/j.1365-3083.2012.02776.x
192. Kumar NP, Anuradha R, Suresh R, Ganesh R, Shankar J, Kumaraswami V, et al. Suppressed type 1, type 2, and type 17 cytokine responses in active tuberculosis in children. Clin Vaccine Immunol (2011) 18:1856–64. doi:10.1128/CVI.05366-11
193. Nunnari G, Pinzone MR, Vancheri C, Palermo F, Cacopardo B. Interferon-gamma and interleukin-17 production from PPD-stimulated PBMCs of patients with pulmonary tuberculosis. Clin Invest Med (2013) 36:E64–71.
194. Khader SA, Guglani L, Rangel-Moreno J, Gopal R, Junecko BA, Fountain JJ, et al. IL-23 is required for long-term control of Mycobacterium tuberculosis and B cell follicle formation in the infected lung. J Immunol (2011) 187:5402–7. doi:10.4049/jimmunol.1101377
195. Freches D, Korf H, Denis O, Havaux X, Huygen K, Romano M. Mice genetically inactivated in interleukin-17A receptor are defective in long-term control of Mycobacterium tuberculosis infection. Immunology (2013) 140:220–31. doi:10.1111/imm.12130
196. Okamoto Yoshida Y, Umemura M, Yahagi A, O’brien RL, Ikuta K, Kishihara K, et al. Essential role of IL-17A in the formation of a mycobacterial infection-induced granuloma in the lung. J Immunol (2010) 184:4414–22. doi:10.4049/jimmunol.0903332
197. Gopal R, Monin L, Slight S, Uche U, Blanchard E, Fallert Junecko BA, et al. Unexpected role for IL-17 in protective immunity against hypervirulent Mycobacterium tuberculosis HN878 infection. PLoS Pathog (2014) 10:e1004099. doi:10.1371/journal.ppat.1004099
198. Umemura M, Okamoto-Yoshida Y, Yahagi A, Touyama S, Nakae S, Iwakura Y, et al. Involvement of IL-17A-producing TCR gammadelta T cells in late protective immunity against pulmonary Mycobacterium tuberculosis infection. Immun Inflamm Dis (2016) 4:401–12. doi:10.1002/iid3.121
199. Rangel-Moreno J, Carragher DM, De La Luz Garcia-Hernandez M, Hwang JY, Kusser K, Hartson L, et al. The development of inducible bronchus-associated lymphoid tissue depends on IL-17. Nat Immunol (2011) 12:639–46. doi:10.1038/ni.2053
200. Hwang JY, Randall TD, Silva-Sanchez A. Inducible bronchus-associated lymphoid tissue: taming inflammation in the lung. Front Immunol (2016) 7:258. doi:10.3389/fimmu.2016.00258
201. Slight SR, Rangel-Moreno J, Gopal R, Lin Y, Fallert Junecko BA, Mehra S, et al. CXCR5(+) T helper cells mediate protective immunity against tuberculosis. J Clin Invest (2013) 123:712–26. doi:10.1172/JCI65728
202. Persson YA, Blomgran-Julinder R, Rahman S, Zheng L, Stendahl O. Mycobacterium tuberculosis-induced apoptotic neutrophils trigger a pro-inflammatory response in macrophages through release of heat shock protein 72, acting in synergy with the bacteria. Microbes Infect (2008) 10:233–40. doi:10.1016/j.micinf.2007.11.007
203. Hedlund S, Persson A, Vujic A, Che KF, Stendahl O, Larsson M. Dendritic cell activation by sensing Mycobacterium tuberculosis-induced apoptotic neutrophils via DC-SIGN. Hum Immunol (2010) 71:535–40. doi:10.1016/j.humimm.2010.02.022
204. Braian C, Hogea V, Stendahl O. Mycobacterium tuberculosis-induced neutrophil extracellular traps activate human macrophages. J Innate Immun (2013) 5:591–602. doi:10.1159/000348676
205. Andersson H, Andersson B, Eklund D, Ngoh E, Persson A, Svensson K, et al. Apoptotic neutrophils augment the inflammatory response to Mycobacterium tuberculosis infection in human macrophages. PLoS One (2014) 9:e101514. doi:10.1371/journal.pone.0101514
206. Trentini MM, De Oliveira FM, Kipnis A, Junqueira-Kipnis AP. The role of neutrophils in the induction of specific Th1 and Th17 during vaccination against tuberculosis. Front Microbiol (2016) 7:898. doi:10.3389/fmicb.2016.00898
207. Grainger JR, Wohlfert EA, Fuss IJ, Bouladoux N, Askenase MH, Legrand F, et al. Inflammatory monocytes regulate pathologic responses to commensals during acute gastrointestinal infection. Nat Med (2013) 19:713–21. doi:10.1038/nm.3189
208. Shishikura K, Horiuchi T, Sakata N, Trinh DA, Shirakawa R, Kimura T, et al. Prostaglandin E2 inhibits neutrophil extracellular trap formation through production of cyclic AMP. Br J Pharmacol (2016) 173:319–31. doi:10.1111/bph.13373
209. Hoeve MA, Savage ND, De Boer T, Langenberg DM, De Waal Malefyt R, Ottenhoff TH, et al. Divergent effects of IL-12 and IL-23 on the production of IL-17 by human T cells. Eur J Immunol (2006) 36:661–70. doi:10.1002/eji.200535239
210. Monin L, Griffiths KL, Slight S, Lin Y, Rangel-Moreno J, Khader SA. Immune requirements for protective Th17 recall responses to Mycobacterium tuberculosis challenge. Mucosal Immunol (2015) 8:1099–109. doi:10.1038/mi.2014.136
211. Gopal R, Rangel-Moreno J, Slight S, Lin Y, Nawar HF, Fallert Junecko BA, et al. Interleukin-17-dependent CXCL13 mediates mucosal vaccine-induced immunity against tuberculosis. Mucosal Immunol (2013) 6:972–84. doi:10.1038/mi.2012.135
212. Maione F, Paschalidis N, Mascolo N, Dufton N, Perretti M, D’acquisto F. Interleukin 17 sustains rather than induces inflammation. Biochem Pharmacol (2009) 77:878–87. doi:10.1016/j.bcp.2008.11.011
213. van Nieuwenhuijze AEM, Van De Loo FA, Walgreen B, Bennink M, Helsen MMA, Van Den Bersselaar L, et al. 1.56 Synergism between GM-CSF and IL-17 causes enhanced joint pathology via the production of IL-6 and IL-23. Ann Rheum Dis (2014) 73:A24. doi:10.1136/annrheumdis-2014-eular.5430
214. Cruz A, Fraga AG, Fountain JJ, Rangel-Moreno J, Torrado E, Saraiva M, et al. Pathological role of interleukin 17 in mice subjected to repeated BCG vaccination after infection with Mycobacterium tuberculosis. J Exp Med (2010) 207:1609–16. doi:10.1084/jem.20100265
215. Rottenberg ME, Carow B. SOCS3 and STAT3, major controllers of the outcome of infection with Mycobacterium tuberculosis. Semin Immunol (2014) 26:518–32. doi:10.1016/j.smim.2014.10.004
216. Eum SY, Kong JH, Hong MS, Lee YJ, Kim JH, Hwang SH, et al. Neutrophils are the predominant infected phagocytic cells in the airways of patients with active pulmonary TB. Chest (2010) 137:122–8. doi:10.1378/chest.09-0903
217. Kisich KO, Higgins M, Diamond G, Heifets L. Tumor necrosis factor alpha stimulates killing of Mycobacterium tuberculosis by human neutrophils. Infect Immun (2002) 70:4591–9. doi:10.1128/IAI.70.8.4591-4599.2002
218. Eruslanov EB, Lyadova IV, Kondratieva TK, Majorov KB, Scheglov IV, Orlova MO, et al. Neutrophil responses to Mycobacterium tuberculosis infection in genetically susceptible and resistant mice. Infect Immun (2005) 73:1744–53. doi:10.1128/IAI.73.3.1744-1753.2005
219. Keller C, Hoffmann R, Lang R, Brandau S, Hermann C, Ehlers S. Genetically determined susceptibility to tuberculosis in mice causally involves accelerated and enhanced recruitment of granulocytes. Infect Immun (2006) 74:4295–309. doi:10.1128/IAI.00057-06
220. Yeremeev V, Linge I, Kondratieva T, Apt A. Neutrophils exacerbate tuberculosis infection in genetically susceptible mice. Tuberculosis (Edinb) (2015) 95:447–51. doi:10.1016/j.tube.2015.03.007
221. Feng CG, Kaviratne M, Rothfuchs AG, Cheever A, Hieny S, Young HA, et al. NK cell-derived IFN-gamma differentially regulates innate resistance and neutrophil response in T cell-deficient hosts infected with Mycobacterium tuberculosis. J Immunol (2006) 177:7086–93. doi:10.4049/jimmunol.177.10.7086
222. Blomgran R, Ernst JD. Lung neutrophils facilitate activation of naive antigen-specific CD4+ T cells during Mycobacterium tuberculosis infection. J Immunol (2011) 186:7110–9. doi:10.4049/jimmunol.1100001
223. Jones HR, Robb CT, Perretti M, Rossi AG. The role of neutrophils in inflammation resolution. Semin Immunol (2016) 28:137–45. doi:10.1016/j.smim.2016.03.007
224. Doz E, Lombard R, Carreras F, Buzoni-Gatel D, Winter N. Mycobacteria-infected dendritic cells attract neutrophils that produce IL-10 and specifically shut down Th17 CD4 T cells through their IL-10 receptor. J Immunol (2013) 191:3818–26. doi:10.4049/jimmunol.1300527
225. Zhang X, Majlessi L, Deriaud E, Leclerc C, Lo-Man R. Coactivation of Syk kinase and MyD88 adaptor protein pathways by bacteria promotes regulatory properties of neutrophils. Immunity (2009) 31:761–71. doi:10.1016/j.immuni.2009.09.016
226. Zizzo G, Cohen PL. IL-17 stimulates differentiation of human anti-inflammatory macrophages and phagocytosis of apoptotic neutrophils in response to IL-10 and glucocorticoids. J Immunol (2013) 190:5237–46. doi:10.4049/jimmunol.1203017
227. Tian F, Han Y, Song J, Lei J, Yan X, Xie N, et al. Pulmonary resident neutrophils regulate the production of GM-CSF and alveolar macrophages. FEBS J (2016) 283:1465–74. doi:10.1111/febs.13684
228. Dragon S, Saffar AS, Shan L, Gounni AS. IL-17 attenuates the anti-apoptotic effects of GM-CSF in human neutrophils. Mol Immunol (2008) 45:160–8. doi:10.1016/j.molimm.2007.04.027
229. Silverpil E, Glader P, Hansson M, Linden A. Impact of interleukin-17 on macrophage phagocytosis of apoptotic neutrophils and particles. Inflammation (2011) 34:1–9. doi:10.1007/s10753-010-9201-8
230. Grogan JL, Ouyang W. A role for Th17 cells in the regulation of tertiary lymphoid follicles. Eur J Immunol (2012) 42:2255–62. doi:10.1002/eji.201242656
231. Parkes M, Cortes A, Van Heel DA, Brown MA. Genetic insights into common pathways and complex relationships among immune-mediated diseases. Nat Rev Genet (2013) 14:661–73. doi:10.1038/nrg3502
232. Higgs BW, Liu Z, White B, Zhu W, White WI, Morehouse C, et al. Patients with systemic lupus erythematosus, myositis, rheumatoid arthritis and scleroderma share activation of a common type I interferon pathway. Ann Rheum Dis (2011) 70:2029–36. doi:10.1136/ard.2011.150326
233. Wahren-Herlenius M, Dorner T. Immunopathogenic mechanisms of systemic autoimmune disease. Lancet (2013) 382:819–31. doi:10.1016/S0140-6736(13)60954-X
234. Brkic Z, Versnel MA. Type I IFN signature in primary Sjogren’s syndrome patients. Expert Rev Clin Immunol (2014) 10:457–67. doi:10.1586/1744666X.2014.876364
235. Baechler EC, Batliwalla FM, Karypis G, Gaffney PM, Ortmann WA, Espe KJ, et al. Interferon-inducible gene expression signature in peripheral blood cells of patients with severe lupus. Proc Natl Acad Sci U S A (2003) 100:2610–5. doi:10.1073/pnas.0337679100
236. Bennett L, Palucka AK, Arce E, Cantrell V, Borvak J, Banchereau J, et al. Interferon and granulopoiesis signatures in systemic lupus erythematosus blood. J Exp Med (2003) 197:711–23. doi:10.1084/jem.20021553
237. Kirou KA, Lee C, George S, Louca K, Peterson MG, Crow MK. Activation of the interferon-alpha pathway identifies a subgroup of systemic lupus erythematosus patients with distinct serologic features and active disease. Arthritis Rheum (2005) 52:1491–503. doi:10.1002/art.21031
238. Brkic Z, Maria NI, Van Helden-Meeuwsen CG, Van De Merwe JP, Van Daele PL, Dalm VA, et al. Prevalence of interferon type I signature in CD14 monocytes of patients with Sjogren’s syndrome and association with disease activity and BAFF gene expression. Ann Rheum Dis (2013) 72:728–35. doi:10.1136/annrheumdis-2012-201381
239. Afshar M, Martinez AD, Gallo RL, Hata TR. Induction and exacerbation of psoriasis with Interferon-alpha therapy for hepatitis C: a review and analysis of 36 cases. J Eur Acad Dermatol Venereol (2013) 27:771–8. doi:10.1111/j.1468-3083.2012.04582.x
240. Kolb-Maurer A, Goebeler M, Maurer M. Cutaneous adverse events associated with interferon-beta treatment of multiple sclerosis. Int J Mol Sci (2015) 16:14951–60. doi:10.3390/ijms160714951
241. Martin JC, Baeten DL, Josien R. Emerging role of IL-17 and Th17 cells in systemic lupus erythematosus. Clin Immunol (2014) 154:1–12. doi:10.1016/j.clim.2014.05.004
242. Axtell RC, Raman C, Steinman L. Type I interferons: beneficial in Th1 and detrimental in Th17 autoimmunity. Clin Rev Allergy Immunol (2013) 44:114–20. doi:10.1007/s12016-011-8296-5
243. Raphael I, Nalawade S, Eagar TN, Forsthuber TG. T cell subsets and their signature cytokines in autoimmune and inflammatory diseases. Cytokine (2015) 74:5–17. doi:10.1016/j.cyto.2014.09.011
244. Ambrosi A, Espinosa A, Wahren-Herlenius M. IL-17: a new actor in IFN-driven systemic autoimmune diseases. Eur J Immunol (2012) 42:2274–84. doi:10.1002/eji.201242653
245. Crow MK. Type I interferon in the pathogenesis of lupus. J Immunol (2014) 192:5459–68. doi:10.4049/jimmunol.1002795
246. Niewold TB, Hua J, Lehman TJ, Harley JB, Crow MK. High serum IFN-alpha activity is a heritable risk factor for systemic lupus erythematosus. Genes Immun (2007) 8:492–502. doi:10.1038/sj.gene.6364408
247. Farkas L, Beiske K, Lund-Johansen F, Brandtzaeg P, Jahnsen FL. Plasmacytoid dendritic cells (natural interferon-alpha/beta-producing cells) accumulate in cutaneous lupus erythematosus lesions. Am J Pathol (2001) 159:237–43. doi:10.1016/S0002-9440(10)61689-6
248. Ronnblom L, Eloranta ML, Alm GV. Role of natural interferon-alpha producing cells (plasmacytoid dendritic cells) in autoimmunity. Autoimmunity (2003) 36:463–72. doi:10.1080/08916930310001602128
249. Tian J, Avalos AM, Mao SY, Chen B, Senthil K, Wu H, et al. Toll-like receptor 9-dependent activation by DNA-containing immune complexes is mediated by HMGB1 and RAGE. Nat Immunol (2007) 8:487–96. doi:10.1038/ni1457
250. Garcia-Romo GS, Caielli S, Vega B, Connolly J, Allantaz F, Xu Z, et al. Netting neutrophils are major inducers of type I IFN production in pediatric systemic lupus erythematosus. Sci Transl Med (2011) 3:73ra20. doi:10.1126/scitranslmed.3001201
251. Scaffidi P, Misteli T, Bianchi ME. Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature (2002) 418:191–5. doi:10.1038/nature00858
252. Huebener P, Pradere JP, Hernandez C, Gwak GY, Caviglia JM, Mu X, et al. The HMGB1/RAGE axis triggers neutrophil-mediated injury amplification following necrosis. J Clin Invest (2015) 125:539–50. doi:10.1172/JCI76887
253. Lande R, Ganguly D, Facchinetti V, Frasca L, Conrad C, Gregorio J, et al. Neutrophils activate plasmacytoid dendritic cells by releasing self-DNA-peptide complexes in systemic lupus erythematosus. Sci Transl Med (2011) 3:73ra19. doi:10.1126/scitranslmed.3001180
254. Bjornsdottir H, Welin A, Michaelsson E, Osla V, Berg S, Christenson K, et al. Neutrophil NET formation is regulated from the inside by myeloperoxidase-processed reactive oxygen species. Free Radic Biol Med (2015) 89:1024–35. doi:10.1016/j.freeradbiomed.2015.10.398
255. Campbell AM, Kashgarian M, Shlomchik MJ. NADPH oxidase inhibits the pathogenesis of systemic lupus erythematosus. Sci Transl Med (2012) 4:157ra141. doi:10.1126/scitranslmed.3004801
256. Lood C, Blanco LP, Purmalek MM, Carmona-Rivera C, De Ravin SS, Smith CK, et al. Neutrophil extracellular traps enriched in oxidized mitochondrial DNA are interferogenic and contribute to lupus-like disease. Nat Med (2016) 22:146–53. doi:10.1038/nm.4027
257. Ganguly D, Haak S, Sisirak V, Reizis B. The role of dendritic cells in autoimmunity. Nat Rev Immunol (2013) 13:566–77. doi:10.1038/nri3477
258. Blanco P, Palucka AK, Gill M, Pascual V, Banchereau J. Induction of dendritic cell differentiation by IFN-alpha in systemic lupus erythematosus. Science (2001) 294:1540–3. doi:10.1126/science.1064890
259. Ronnblom L, Pascual V. The innate immune system in SLE: type I interferons and dendritic cells. Lupus (2008) 17:394–9. doi:10.1177/0961203308090020
260. Vincent FB, Morand EF, Schneider P, Mackay F. The BAFF/APRIL system in SLE pathogenesis. Nat Rev Rheumatol (2014) 10:365–73. doi:10.1038/nrrheum.2014.33
261. Sjostrand M, Johansson A, Aqrawi L, Olsson T, Wahren-Herlenius M, Espinosa A. The expression of BAFF is controlled by IRF transcription factors. J Immunol (2016) 196:91–6. doi:10.4049/jimmunol.1501061
262. Jacob N, Guo S, Mathian A, Koss MN, Gindea S, Putterman C, et al. B cell and BAFF dependence of IFN-alpha-exaggerated disease in systemic lupus erythematosus-prone NZM 2328 mice. J Immunol (2011) 186:4984–93. doi:10.4049/jimmunol.1000466
263. Zollars E, Bienkowska J, Czerkowicz J, Allaire N, Ranger AM, Magder L, et al. BAFF (B cell activating factor) transcript level in peripheral blood of patients with SLE is associated with same-day disease activity as well as global activity over the next year. Lupus Sci Med (2015) 2:e000063. doi:10.1136/lupus-2014-000063
264. Litinskiy MB, Nardelli B, Hilbert DM, He B, Schaffer A, Casali P, et al. DCs induce CD40-independent immunoglobulin class switching through BLyS and APRIL. Nat Immunol (2002) 3:822–9. doi:10.1038/ni829
265. Navarra SV, Guzman RM, Gallacher AE, Hall S, Levy RA, Jimenez RE, et al. Efficacy and safety of belimumab in patients with active systemic lupus erythematosus: a randomised, placebo-controlled, phase 3 trial. Lancet (2011) 377:721–31. doi:10.1016/S0140-6736(10)61354-2
266. Merrill JT, Neuwelt CM, Wallace DJ, Shanahan JC, Latinis KM, Oates JC, et al. Efficacy and safety of rituximab in moderately-to-severely active systemic lupus erythematosus: the randomized, double-blind, phase II/III systemic lupus erythematosus evaluation of rituximab trial. Arthritis Rheum (2010) 62:222–33. doi:10.1002/art.27233
267. Rovin BH, Furie R, Latinis K, Looney RJ, Fervenza FC, Sanchez-Guerrero J, et al. Efficacy and safety of rituximab in patients with active proliferative lupus nephritis: the Lupus Nephritis Assessment with Rituximab study. Arthritis Rheum (2012) 64:1215–26. doi:10.1002/art.34359
268. Zhou X, Xia Z, Lan Q, Wang J, Su W, Han YP, et al. BAFF promotes Th17 cells and aggravates experimental autoimmune encephalomyelitis. PLoS One (2011) 6:e23629. doi:10.1371/journal.pone.0023629
269. Munari F, Fassan M, Capitani N, Codolo G, Vila-Caballer M, Pizzi M, et al. Cytokine BAFF released by Helicobacter pylori-infected macrophages triggers the Th17 response in human chronic gastritis. J Immunol (2014) 193:5584–94. doi:10.4049/jimmunol.1302865
270. Benson MJ, Dillon SR, Castigli E, Geha RS, Xu S, Lam KP, et al. Cutting edge: the dependence of plasma cells and independence of memory B cells on BAFF and APRIL. J Immunol (2008) 180:3655–9. doi:10.4049/jimmunol.180.6.3655
271. Parsa R, Lund H, Georgoudaki AM, Zhang XM, Ortlieb Guerreiro-Cacais A, Grommisch D, et al. BAFF-secreting neutrophils drive plasma cell responses during emergency granulopoiesis. J Exp Med (2016) 213:1537–53. doi:10.1084/jem.20150577
272. Langrish CL, Chen Y, Blumenschein WM, Mattson J, Basham B, Sedgwick JD, et al. IL-23 drives a pathogenic T cell population that induces autoimmune inflammation. J Exp Med (2005) 201:233–40. doi:10.1084/jem.20041257
273. Noster R, Riedel R, Mashreghi MF, Radbruch H, Harms L, Haftmann C, et al. IL-17 and GM-CSF expression are antagonistically regulated by human T helper cells. Sci Transl Med (2014) 6:241ra280. doi:10.1126/scitranslmed.3008706
274. Li R, Rezk A, Miyazaki Y, Hilgenberg E, Touil H, Shen P, et al. Proinflammatory GM-CSF-producing B cells in multiple sclerosis and B cell depletion therapy. Sci Transl Med (2015) 7:310ra166. doi:10.1126/scitranslmed.aab4176
275. Rasouli J, Ciric B, Imitola J, Gonnella P, Hwang D, Mahajan K, et al. Expression of GM-CSF in T Cells is increased in multiple sclerosis and suppressed by IFN-beta therapy. J Immunol (2015) 194:5085–93. doi:10.4049/jimmunol.1403243
276. Ireland SJ, Guzman AA, Frohman EM, Monson NL. B cells from relapsing remitting multiple sclerosis patients support neuro-antigen-specific Th17 responses. J Neuroimmunol (2016) 291:46–53. doi:10.1016/j.jneuroim.2015.11.022
277. Zhou L, Ivanov II, Spolski R, Min R, Shenderov K, Egawa T, et al. IL-6 programs T(H)-17 cell differentiation by promoting sequential engagement of the IL-21 and IL-23 pathways. Nat Immunol (2007) 8:967–74. doi:10.1038/ni1488
278. Yamazaki T, Yang XO, Chung Y, Fukunaga A, Nurieva R, Pappu B, et al. CCR6 regulates the migration of inflammatory and regulatory T cells. J Immunol (2008) 181:8391–401. doi:10.4049/jimmunol.181.12.8391
279. Croxford AL, Lanzinger M, Hartmann FJ, Schreiner B, Mair F, Pelczar P, et al. The cytokine GM-CSF drives the inflammatory signature of CCR2+ monocytes and licenses autoimmunity. Immunity (2015) 43:502–14. doi:10.1016/j.immuni.2015.08.010
280. Schett G, Dayer JM, Manger B. Interleukin-1 function and role in rheumatic disease. Nat Rev Rheumatol (2016) 12:14–24. doi:10.1038/nrrheum.2016.166
281. Avci AB, Feist E, Burmester GR. Targeting GM-CSF in rheumatoid arthritis. Clin Exp Rheumatol (2016) 34:39–44.
282. Canavan TN, Elmets CA, Cantrell WL, Evans JM, Elewski BE. Anti-IL-17 medications used in the treatment of plaque psoriasis and psoriatic arthritis: a comprehensive review. Am J Clin Dermatol (2016) 17:33–47. doi:10.1007/s40257-015-0162-4
283. Amarilyo G, Lourenco EV, Shi FD, La Cava A. IL-17 promotes murine lupus. J Immunol (2014) 193:540–3. doi:10.4049/jimmunol.1400931
284. Arnason BG. Immunologic therapy of multiple sclerosis. Annu Rev Med (1999) 50:291–302. doi:10.1146/annurev.med.50.1.291
285. Szczucinski A, Losy J. Long-term effect of IFN-beta 1a therapy on CCL2 (MCP-1) chemokine in patients with multiple sclerosis. Folia Neuropathol (2004) 42:15–8.
286. Shiomi A, Usui T. Pivotal roles of GM-CSF in autoimmunity and inflammation. Mediators Inflamm (2015) 2015:568543. doi:10.1155/2015/568543
287. Ifergan I, Kebir H, Bernard M, Wosik K, Dodelet-Devillers A, Cayrol R, et al. The blood-brain barrier induces differentiation of migrating monocytes into Th17-polarizing dendritic cells. Brain (2008) 131:785–99. doi:10.1093/brain/awm295
288. Decker P. Neutrophils and interferon-alpha-producing cells: who produces interferon in lupus? Arthritis Res Ther (2011) 13:118. doi:10.1186/ar3345
289. Lindau D, Mussard J, Rabsteyn A, Ribon M, Kotter I, Igney A, et al. TLR9 independent interferon alpha production by neutrophils on NETosis in response to circulating chromatin, a key lupus autoantigen. Ann Rheum Dis (2014) 73:2199–207. doi:10.1136/annrheumdis-2012-203041
290. Demers M, Wong SL, Martinod K, Gallant M, Cabral JE, Wang Y, et al. Priming of neutrophils toward NETosis promotes tumor growth. Oncoimmunology (2016) 5:e1134073. doi:10.1080/2162402X.2015.1134073
291. Carmona-Rivera C, Kaplan MJ. Low-density granulocytes: a distinct class of neutrophils in systemic autoimmunity. Semin Immunopathol (2013) 35:455–63. doi:10.1007/s00281-013-0375-7
292. Midgley A, Beresford MW. Increased expression of low density granulocytes in juvenile-onset systemic lupus erythematosus patients correlates with disease activity. Lupus (2016) 25:407–11. doi:10.1177/0961203315608959
293. Pers JO, Daridon C, Devauchelle V, Jousse S, Saraux A, Jamin C, et al. BAFF overexpression is associated with autoantibody production in autoimmune diseases. Ann N Y Acad Sci (2005) 1050:34–9. doi:10.1196/annals.1313.004
294. Scapini P, Bazzoni F, Cassatella MA. Regulation of B-cell-activating factor (BAFF)/B lymphocyte stimulator (BLyS) expression in human neutrophils. Immunol Lett (2008) 116:1–6. doi:10.1016/j.imlet.2007.11.009
295. Ueno H, Banchereau J, Vinuesa CG. Pathophysiology of T follicular helper cells in humans and mice. Nat Immunol (2015) 16:142–52. doi:10.1038/ni.3054
296. Linterman MA, Rigby RJ, Wong RK, Yu D, Brink R, Cannons JL, et al. Follicular helper T cells are required for systemic autoimmunity. J Exp Med (2009) 206:561–76. doi:10.1084/jem.20081886
297. Simpson N, Gatenby PA, Wilson A, Malik S, Fulcher DA, Tangye SG, et al. Expansion of circulating T cells resembling follicular helper T cells is a fixed phenotype that identifies a subset of severe systemic lupus erythematosus. Arthritis Rheum (2010) 62:234–44. doi:10.1002/art.25032
298. Craft JE. Follicular helper T cells in immunity and systemic autoimmunity. Nat Rev Rheumatol (2012) 8:337–47. doi:10.1038/nrrheum.2012.58
299. Dolff S, Quandt D, Feldkamp T, Jun C, Mitchell A, Hua F, et al. Increased percentages of PD-1 on CD4+ T cells is associated with higher INF-gamma production and altered IL-17 production in patients with systemic lupus erythematosus. Scand J Rheumatol (2014) 43:307–13. doi:10.3109/03009742.2013.869830
300. Jiao Q, Liu C, Yang Z, Ding Q, Wang M, Li M, et al. Upregulated PD-1 expression is associated with the development of systemic lupus erythematosus, but not the PD-1.1 allele of the PDCD1 gene. Int J Genomics (2014) 2014:950903. doi:10.1155/2014/950903
301. Choi JY, Ho JH, Pasoto SG, Bunin V, Kim ST, Carrasco S, et al. Circulating follicular helper-like T cells in systemic lupus erythematosus: association with disease activity. Arthritis Rheumatol (2015) 67:988–99. doi:10.1002/art.39020
302. Schiffer L, Worthmann K, Haller H, Schiffer M. CXCL13 as a new biomarker of systemic lupus erythematosus and lupus nephritis – from bench to bedside? Clin Exp Immunol (2015) 179:85–9. doi:10.1111/cei.12439
303. Blanco P, Ueno H, Schmitt N. T follicular helper (Tfh) cells in lupus: activation and involvement in SLE pathogenesis. Eur J Immunol (2016) 46:281–90. doi:10.1002/eji.201545760
304. Jones GW, Hill DG, Jones SA. Understanding immune cells in tertiary lymphoid organ development: it is all starting to come together. Front Immunol (2016) 7:401. doi:10.3389/fimmu.2016.00401
305. Rahman ZS, Rao SP, Kalled SL, Manser T. Normal induction but attenuated progression of germinal center responses in BAFF and BAFF-R signaling-deficient mice. J Exp Med (2003) 198:1157–69. doi:10.1084/jem.20030495
306. Coquery CM, Loo WM, Wade NS, Bederman AG, Tung KS, Lewis JE, et al. BAFF regulates follicular helper t cells and affects their accumulation and interferon-gamma production in autoimmunity. Arthritis Rheumatol (2015) 67:773–84. doi:10.1002/art.38950
307. Terawaki S, Chikuma S, Shibayama S, Hayashi T, Yoshida T, Okazaki T, et al. IFN-alpha directly promotes programmed cell death-1 transcription and limits the duration of T cell-mediated immunity. J Immunol (2011) 186:2772–9. doi:10.4049/jimmunol.1003208
308. Nakayamada S, Poholek AC, Lu KT, Takahashi H, Kato M, Iwata S, et al. Type I IFN induces binding of STAT1 to Bcl6: divergent roles of STAT family transcription factors in the T follicular helper cell genetic program. J Immunol (2014) 192:2156–66. doi:10.4049/jimmunol.1300675
309. Mueller H, Detjen AK, Schuck SD, Gutschmidt A, Wahn U, Magdorf K, et al. Mycobacterium tuberculosis-specific CD4+, IFNgamma+, and TNFalpha+ multifunctional memory T cells coexpress GM-CSF. Cytokine (2008) 43:143–8. doi:10.1016/j.cyto.2008.05.002
310. Tully G, Kortsik C, Hohn H, Zehbe I, Hitzler WE, Neukirch C, et al. Highly focused T cell responses in latent human pulmonary Mycobacterium tuberculosis infection. J Immunol (2005) 174:2174–84. doi:10.4049/jimmunol.174.4.2174
311. Stephen-Victor E, Sharma VK, Das M, Karnam A, Saha C, Lecerf M, et al. IL-1beta, but not programed death-1 and programed death ligand pathway, is critical for the human Th17 response to Mycobacterium tuberculosis. Front Immunol (2016) 7:465. doi:10.3389/fimmu.2016.00465
312. Basile JI, Kviatcovsky D, Romero MM, Balboa L, Monteserin J, Ritacco V, et al. Mycobacterium tuberculosis multi-drug-resistant strain M induces IL-17+ IFNgamma-CD4+ T cell expansion through an IL-23 and TGF-beta-dependent mechanism in patients with MDR-TB tuberculosis. Clin Exp Immunol (2017) 187:160–73. doi:10.1111/cei.12873
313. Toossi Z, Gogate P, Shiratsuchi H, Young T, Ellner JJ. Enhanced production of TGF-beta by blood monocytes from patients with active tuberculosis and presence of TGF-beta in tuberculous granulomatous lung lesions. J Immunol (1995) 154:465–73.
314. Olobo JO, Geletu M, Demissie A, Eguale T, Hiwot K, Aderaye G, et al. Circulating TNF-alpha, TGF-beta, and IL-10 in tuberculosis patients and healthy contacts. Scand J Immunol (2001) 53:85–91. doi:10.1046/j.1365-3083.2001.00844.x
315. Rothchild AC, Jayaraman P, Nunes-Alves C, Behar SM. iNKT cell production of GM-CSF controls Mycobacterium tuberculosis. PLoS Pathog (2014) 10:e1003805. doi:10.1371/journal.ppat.1003805
316. Gonzalez-Juarrero M, Hattle JM, Izzo A, Junqueira-Kipnis AP, Shim TS, Trapnell BC, et al. Disruption of granulocyte macrophage-colony stimulating factor production in the lungs severely affects the ability of mice to control Mycobacterium tuberculosis infection. J Leukoc Biol (2005) 77:914–22. doi:10.1189/jlb.1204723
317. Szeliga J, Daniel DS, Yang CH, Sever-Chroneos Z, Jagannath C, Chroneos ZC. Granulocyte-macrophage colony stimulating factor-mediated innate responses in tuberculosis. Tuberculosis (Edinb) (2008) 88:7–20. doi:10.1016/j.tube.2007.08.009
318. Verreck FA, De Boer T, Langenberg DM, Hoeve MA, Kramer M, Vaisberg E, et al. Human IL-23-producing type 1 macrophages promote but IL-10-producing type 2 macrophages subvert immunity to (myco)bacteria. Proc Natl Acad Sci U S A (2004) 101:4560–5. doi:10.1073/pnas.0400983101
319. Verreck FA, De Boer T, Langenberg DM, Van Der Zanden L, Ottenhoff TH. Phenotypic and functional profiling of human proinflammatory type-1 and anti-inflammatory type-2 macrophages in response to microbial antigens and IFN-gamma- and CD40L-mediated costimulation. J Leukoc Biol (2006) 79:285–93. doi:10.1189/jlb.0105015
320. Tall AR, Yvan-Charvet L. Cholesterol, inflammation and innate immunity. Nat Rev Immunol (2015) 15:104–16. doi:10.1038/nri3793
321. Deng Y, Ye J, Luo Q, Huang Z, Peng Y, Xiong G, et al. Low-density granulocytes are elevated in mycobacterial infection and associated with the severity of tuberculosis. PLoS One (2016) 11:e0153567. doi:10.1371/journal.pone.0153567
322. Ramos-Kichik V, Mondragon-Flores R, Mondragon-Castelan M, Gonzalez-Pozos S, Muniz-Hernandez S, Rojas-Espinosa O, et al. Neutrophil extracellular traps are induced by Mycobacterium tuberculosis. Tuberculosis (Edinb) (2009) 89:29–37. doi:10.1016/j.tube.2008.09.009
323. Wong KW, Jacobs WR Jr. Mycobacterium tuberculosis exploits human interferon gamma to stimulate macrophage extracellular trap formation and necrosis. J Infect Dis (2013) 208:109–19. doi:10.1093/infdis/jit097
324. Francis RJ, Butler RE, Stewart GR. Mycobacterium tuberculosis ESAT-6 is a leukocidin causing Ca2+ influx, necrosis and neutrophil extracellular trap formation. Cell Death Dis (2014) 5:e1474. doi:10.1038/cddis.2014.394
325. Lozza L, Farinacci M, Bechtle M, Staber M, Zedler U, Baiocchini A, et al. Communication between human dendritic cell subsets in tuberculosis: requirements for naive CD4(+) T cell stimulation. Front Immunol (2014) 5:324. doi:10.3389/fimmu.2014.00324
326. Jin O, Kavikondala S, Sun L, Fu R, Mok MY, Chan A, et al. Systemic lupus erythematosus patients have increased number of circulating plasmacytoid dendritic cells, but decreased myeloid dendritic cells with deficient CD83 expression. Lupus (2008) 17:654–62. doi:10.1177/0961203308089410
327. Lichtner M, Rossi R, Mengoni F, Vignoli S, Colacchia B, Massetti AP, et al. Circulating dendritic cells and interferon-alpha production in patients with tuberculosis: correlation with clinical outcome and treatment response. Clin Exp Immunol (2006) 143:329–37. doi:10.1111/j.1365-2249.2005.02994.x
328. Wilson EB, Yamada DH, Elsaesser H, Herskovitz J, Deng J, Cheng G, et al. Blockade of chronic type I interferon signaling to control persistent LCMV infection. Science (2013) 340:202–7. doi:10.1126/science.1235208
329. Crow MK. Autoimmunity: interferon alpha or beta: which is the culprit in autoimmune disease? Nat Rev Rheumatol (2016) 12:439–40. doi:10.1038/nrrheum.2016.117
330. de Jong TD, Vosslamber S, Mantel E, De Ridder S, Wesseling JG, Van Der Pouw Kraan TC, et al. Physiological evidence for diversification of IFNalpha- and IFNbeta-mediated response programs in different autoimmune diseases. Arthritis Res Ther (2016) 18:49. doi:10.1186/s13075-016-0946-9
331. Ng CT, Mendoza JL, Garcia KC, Oldstone MB. Alpha and beta type 1 interferon signaling: passage for diverse biologic outcomes. Cell (2016) 164:349–52. doi:10.1016/j.cell.2015.12.027
332. Baccala R, Welch MJ, Gonzalez-Quintial R, Walsh KB, Teijaro JR, Nguyen A, et al. Type I interferon is a therapeutic target for virus-induced lethal vascular damage. Proc Natl Acad Sci U S A (2014) 111:8925–30. doi:10.1073/pnas.1408148111
333. Kim K, Perera R, Tan DB, Fernandez S, Seddiki N, Waring J, et al. Circulating mycobacterial-reactive CD4+ T cells with an immunosuppressive phenotype are higher in active tuberculosis than latent tuberculosis infection. Tuberculosis (Edinb) (2014) 94:494–501. doi:10.1016/j.tube.2014.07.002
334. Puthia M, Ambite I, Cafaro C, Butler D, Huang Y, Lutay N, et al. IRF7 inhibition prevents destructive innate immunity-A target for nonantibiotic therapy of bacterial infections. Sci Transl Med (2016) 8:336ra359. doi:10.1126/scitranslmed.aaf1156
335. Stohl W, Hiepe F, Latinis KM, Thomas M, Scheinberg MA, Clarke A, et al. Belimumab reduces autoantibodies, normalizes low complement levels, and reduces select B cell populations in patients with systemic lupus erythematosus. Arthritis Rheum (2012) 64:2328–37. doi:10.1002/art.34400
336. Shah HB, Joshi SK, Rampuria P, Devera TS, Lang GA, Stohl W, et al. BAFF- and APRIL-dependent maintenance of antibody titers after immunization with T-dependent antigen and CD1d-binding ligand. J Immunol (2013) 191:1154–63. doi:10.4049/jimmunol.1300263
337. Liu K, Zhang Y, Hu S, Yu Y, Yang Q, Jin D, et al. Increased levels of BAFF and APRIL related to human active pulmonary tuberculosis. PLoS One (2012) 7:e38429. doi:10.1371/journal.pone.0038429
338. Achkar JM, Chan J, Casadevall A. B cells and antibodies in the defense against Mycobacterium tuberculosis infection. Immunol Rev (2015) 264:167–81. doi:10.1111/imr.12276
339. Lu LL, Chung AW, Rosebrock TR, Ghebremichael M, Yu WH, Grace PS, et al. A functional role for antibodies in tuberculosis. Cell (2016) 167:433–43.e14. doi:10.1016/j.cell.2016.08.072
340. Joosten SA, Van Meijgaarden KE, Del Nonno F, Baiocchini A, Petrone L, Vanini V, et al. Patients with tuberculosis have a dysfunctional circulating B-cell compartment, which normalizes following successful treatment. PLoS Pathog (2016) 12:e1005687. doi:10.1371/journal.ppat.1005687
341. Thien M, Phan TG, Gardam S, Amesbury M, Basten A, Mackay F, et al. Excess BAFF rescues self-reactive B cells from peripheral deletion and allows them to enter forbidden follicular and marginal zone niches. Immunity (2004) 20:785–98. doi:10.1016/j.immuni.2004.05.010
342. Toubi E, Gordon S, Kessel A, Rosner I, Rozenbaum M, Shoenfeld Y, et al. Elevated serum B-Lymphocyte activating factor (BAFF) in chronic hepatitis C virus infection: association with autoimmunity. J Autoimmun (2006) 27:134–9. doi:10.1016/j.jaut.2006.07.005
343. Francois A, Gombault A, Villeret B, Alsaleh G, Fanny M, Gasse P, et al. B cell activating factor is central to bleomycin- and IL-17-mediated experimental pulmonary fibrosis. J Autoimmun (2015) 56:1–11. doi:10.1016/j.jaut.2014.08.003
344. Ulrichs T, Kosmiadi GA, Jorg S, Pradl L, Titukhina M, Mishenko V, et al. Differential organization of the local immune response in patients with active cavitary tuberculosis or with nonprogressive tuberculoma. J Infect Dis (2005) 192:89–97. doi:10.1086/430621
345. Moguche AO, Shafiani S, Clemons C, Larson RP, Dinh C, Higdon LE, et al. ICOS and Bcl6-dependent pathways maintain a CD4 T cell population with memory-like properties during tuberculosis. J Exp Med (2015) 212:715–28. doi:10.1084/jem.20141518
346. Cohen KW, Dugast AS, Alter G, Mcelrath MJ, Stamatatos L. HIV-1 single-stranded RNA induces CXCL13 secretion in human monocytes via TLR7 activation and plasmacytoid dendritic cell-derived type I IFN. J Immunol (2015) 194:2769–75. doi:10.4049/jimmunol.1400952
347. McNab FW, Berry MP, Graham CM, Bloch SA, Oni T, Wilkinson KA, et al. Programmed death ligand 1 is over-expressed by neutrophils in the blood of patients with active tuberculosis. Eur J Immunol (2011) 41:1941–7. doi:10.1002/eji.201141421
Keywords: Mycobacterium tuberculosis, autoimmune diseases, neutrophils, inflammation, tertiary lymphoid structures, antibodies, B-cell-activating factor
Citation: Mourik BC, Lubberts E, de Steenwinkel JEM, Ottenhoff THM and Leenen PJM (2017) Interactions between Type 1 Interferons and the Th17 Response in Tuberculosis: Lessons Learned from Autoimmune Diseases. Front. Immunol. 8:294. doi: 10.3389/fimmu.2017.00294
Received: 21 December 2016; Accepted: 01 March 2017;
Published: 05 April 2017
Edited by:
Laurel L. Lenz, University of Colorado Denver School of Medicine, USAReviewed by:
Roland Lang, University Hospital Erlangen, GermanyRolf Billeskov, Statens Serum Institut, Denmark
Copyright: © 2017 Mourik, Lubberts, de Steenwinkel, Ottenhoff and Leenen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Pieter J. M. Leenen, cC5sZWVuZW5AZXJhc211c21jLm5s