 Alessandro Borghesi
Alessandro Borghesi Mauro Stronati
Mauro Stronati Jacques Fellay
Jacques Fellay
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
HYPOTHESIS AND THEORY article
Front. Immunol. , 07 March 2017
Sec. Vaccines and Molecular Therapeutics
Volume 8 - 2017 | https://doi.org/10.3389/fimmu.2017.00215
This article is part of the Research Topic Reducing Neonatal Infectious Morbidity and Mortality: Joining Up Our Thinking View all 15 articles
Only a small proportion of newborn infants exposed to a pathogenic microorganism develop overt infection. Susceptibility to infection in preterm infants and infants with known comorbidities has a likely multifactorial origin and can be often attributed to the concurrence of iatrogenic factors, environmental determinants, underlying pathogenic processes, and probably genetic predisposition. Conversely, infection occurring in otherwise healthy full-term newborn infants is unexplained in most cases. Microbial virulence factors and the unique characteristics of the neonatal immune system only partially account for the interindividual variability in the neonatal immune responses to pathogens. We here suggest that neonatal infection occurring in otherwise healthy infants is caused by a failure of the specific protective immunity to the microorganism. To explain infection in term and preterm infants, we propose an extension of the previously proposed model of the genetic architecture of infectious diseases in humans. We then focus on group B streptococcus (GBS) disease, the best characterized neonatal infection, and outline the potential molecular mechanisms underlying the selective failure of the immune responses against GBS. In light of the recent discoveries of pathogen-specific primary immunodeficiencies and of the role of anticytokine autoantibodies in increasing susceptibility to specific infections, we hypothesize that GBS disease occurring in otherwise healthy infants could reflect an immunodeficiency caused either by rare genetic defects in the infant or by transmitted maternal neutralizing antibodies. These hypotheses are consistent with available epidemiological data, with clinical and epidemiological observations, and with the state of the art of neonatal physiology and disease. Studies should now be designed to comprehensively search for genetic or immunological factors involved in susceptibility to severe neonatal infections.
Neonates are commonly thought to be vulnerable to pathogens because of neonatal immaturity, immune tolerance, or immune deviation, a developmentally regulated transitional state (1–4). These concepts, while useful to describe the highest incidence of infection during the neonatal age at the population level, do not take into account interindividual variability. Even if the highest incidence of infection is observed during the first 28 days of life, the majority of newborn infants are resistant to common pathogens, and only a small proportion of infants exposed to a given microorganism develop overt disease.
It is very clear from epidemiological studies that multiple risk factors contribute to the individual risk of developing neonatal infections. Based on them, neonates can be classified into high- and low-risk groups; individual risks can be estimated; and preventive protocols can be designed for infants who are at high risk of suffering from severe infections (5–7).
Despite their proven clinical utility, most preventive protocols are only partially effective. This can be explained in part by incomplete adherence by healthcare practitioners and missed opportunities (8–10). However, another critical limitation is the inability of current protocols to accurately predict susceptibility to severe infection at the individual level. Furthermore, infections that occur in the absence of any recognizable factors are currently unpredictable.
Many research groups are focusing on the mechanisms of host susceptibility and resistance to pediatric and adult infections (11, 12). Conversely, neonatal infections have been much less studied from a host susceptibility perspective. Several layers of complexity have indeed prevented researchers from fully understanding the neonatal-specific protective immunity, beyond maternal protection of the neonate through transplancentally transmitted antibodies (Abs). Considering the global burden of neonatal infectious diseases, this looks like a missed opportunity to address a critical public health problem.
The study of neonatal infections raises several practical and ethical issues and is challenging from a scientific perspective. First, the neonatal immune system is a rapidly evolving entity, as is every other organ and system soon after birth (13). Second, and possibly more importantly, there is a complex immune interplay between the mother and the child. The maternal environment (the maternal immune system and microbiome) is intertwined with physiological and pathological processes occurring in the fetal and neonatal tissues (e.g., the maturation of fetal and neonatal immune responses, the composition of the neonatal microbiome) (13–17).
To date, little is known about the mechanisms leading to individual vulnerability and resistance to specific pathogens in the neonatal age. We here propose novel, testable hypotheses that could explain the interindividual differences in pathogen susceptibility and help dissect the molecular and cellular bases of severe neonatal infections.
The Global Burden of Disease Study 2015 reports that “sepsis and other neonatal infections” account for 336,300 neonatal deaths each year worldwide (18).
The distribution of infecting microorganisms varies between term and preterm infants and is different in the neonatal period compared to other age groups. Early-onset and late-onset infections are defined as infection occurring during and after the first 6 days of life, respectively. According to other definitions, 48–96 h of life could be used as cutoff (19).
Group B streptococcus (GBS), or Streptococcus agalactiae, is one of the leading pathogens in neonatal infections occurring in full-term newborn infants during the first week of life (9). It is also the most frequent cause of sepsis and meningitis in young infants after the first week of life (20, 21). Recent reports show an increase in the proportion of Escherichia coli infection, mostly associated with urinary tract infection, in previously healthy, full-term infants aged 1 week to 3 months (22, 23). Other pathogens responsible for invasive infection in full-term infants include Gram-positive (Staphylococcus aureus, Streptococcus spp., Enterococcus spp., and, less frequently, Listeria monocytogenes) and Gram-negative microorganisms (Klebsiella spp., Citrobacter spp., Serratia marcescens, Salmonella spp., Haemophilus influenzae) (22, 23). Deep organ infections by Candida spp. and other fungal microorganisms are exceedingly rare in full-term infants.
In very low-birth-weight (VLBW; <1,500 g at birth) infants, Gram-negative pathogens are the most frequently isolated microorganism in early-onset infections, while Gram-positive bacteria are the most frequently isolated pathogens in late-onset infections in the neonatal intensive care units (NICUs), followed by Gram-negative bacteria and fungal organisms (24–29).
The neonatal immune responses differ in many aspects from immune responses in other age groups. A fine-tuning is required to balance the need for tolerance to beneficial antigens (microbial flora and nutrients) and the need for defense against harmful microorganisms.
The cord blood is enriched in CD4+CD25+ T regulatory cells with potent suppressor activity (30, 31) and other immunosuppressive cell populations including some B cell populations and CD71+ erythroid cells (32, 33).
Despite this strong immunosuppressive component, the neonatal immune system has been demonstrated to be able to mount pro-inflammatory responses that are appropriate for the protection against common pathogens in most infants (34). The two main components of the adaptive immune system, the T and B cell compartments, undergo maturation during human fetal life, with progressive and regulated acquisition of B and T cell repertoire diversity and complexity (35). In addition, the human cord blood possesses several pro-inflammatory cell populations, including newborn-specific interleukin (IL)8-producing T cells (36) and a population of CD4+ T cells with a memory-like phenotype and a variety of effector functions (37).
Cells of the neonatal adaptive immune system are capable of mounting a wide range of responses, from poor or “deviant” T helper 2 (Th2)-skewed antiinflammatory responses to balanced Th1/Th2 responses, and even strong adult-like pro-inflammatory responses (2–4, 38). A series of elegant experiments have shown that neonatal T cells, unlike adult cells, are able to produce large amounts of the Th2 cytokines, IL4 and IL13, upon polyclonal stimulation (39, 40). This phenomenon is linked to extensive epigenetic modifications at the Th2 locus (IL5, IL13, and IL4 genes) and in particular to hypomethylation of the conserved non-coding sequence 1 locus, an enhancer and coordinate regulator of Th2 cytokine production (38, 41). Despite this Th2 bias, neonatal adaptive immune responses can shift toward a dominant Th1 and pro-inflammatory response depending on the type of innate responses and the conditions of antigen exposure (38, 42).
Adaptive immune responses require, however, several days to take place. Neonates cannot rely on preexistent immunological memory because exposure to foreign antigens and pathogens is limited during intrauterine life (34, 42). Furthermore, humoral immunity largely depends on maternally transmitted antimicrobial IgG Abs during the first days of life. The rapid decline of maternal IgG in the neonatal plasma after birth (with a half-life of 21–30 days) is accompanied by a relatively slow maturation of both T-dependent and T-independent B-cell responses throughout the first months of life (13, 43).
Infections occurring in the neonatal period are, by definition, primary infections, and neonates mostly rely on the innate immune responses that provide a first line of defense against invading pathogenic microorganisms (34, 44, 45).
A number of studies demonstrated that the neonatal innate immune responses are characterized by dampened Th1-polarizing and pro-inflammatory responses [low amounts of tumor necrosis factor (TNF) upon toll-like receptor (TLR) stimulation] and by increased production of Th2-polarizing and antiinflammatory cytokines (higher IL6/TNF ratio compared to adult responses) (45–48). Furthermore, decreased phosphorylation of signal transducer and activation of transcription 1 in response to interferon gamma (IFN-γ) (49) and developmental maturation of specific dendritic cell subsets (50) contribute to the neonatal-specific Th2-polarizing innate immunity.
Interestingly, full-term healthy newborn infants do not appear specifically vulnerable to deep infection by microorganisms typically causing disease in immunodeficient patients, most notably Nocardia and fungi-like Aspergillus, Candida, Cryptococcus, Pneumocystis, and other opportunistic pathogens, suggesting a substantial maturation of the specific antifungal protective responses in most full-term newborn infants.
Altogether, the characterization of the neonatal immune responses over the past two decades has shown profound differences with adult immunity that in part explain the overall increased susceptibility to life-threatening infection of newborn infants. However, little is known so far about the interindividual differences in the immune protection against pathogens in the neonatal age.
There is controversy over the heritability of susceptibility to neonatal sepsis. In one study, comparing the concordance of late-onset sepsis in same-sex vs. unlike-sex twin pairs, no evidence was found of a genetic component of susceptibility to late-onset sepsis among VLBW infants (51). Conversely, another study compared sepsis concordance rates between monozygotic and dizygotic twins; the authors found that 49% of the variance in liability to late-onset sepsis could be explained by genetic factors alone and 51% by residual environmental factors (52). Both studies focused on cohorts of very preterm/VLBW infants. No study so far addressed the question of heritability of neonatal sepsis in late-preterm and full-term newborn infants.
The question of the role of the genetic background on neonatal host susceptibility to infection has been addressed by several studies. All published studies, included in a recent meta-analysis, used a candidate gene approach on cohorts of preterm infants (53). One genome-wide association study (GWAS) is ongoing (54). Such studies are useful to investigate the contribution of host genetics in the setting of a likely multifactorial pathogenesis, as it is probably the case for most infections occurring in preterm infants. Different approaches are needed to find the genetic determinants of susceptibility to life-threatening infections occurring in full-term infants with no underlying medical conditions in which susceptibility to infection is largely unexplained.
Inborn errors of immunity or primary immunodeficiencies (PIDs) are a group of genetic disorders characterized by increased susceptibility to infection. Historically, the so-called conventional PIDs have been the first PIDs described and dissected from a molecular perspective (55). They are typically Mendelian diseases, caused by highly penetrant single-gene defects. They often occur in families or in the presence of consanguinity and are characterized by a profound defect in one or more arms of the immune system leading to susceptibility to recurrent infections by a broad range of microorganisms (56).
Over the past two decades, it has become clear that infectious diseases previously thought to be due to the sole virulence of the pathogen may be the expression of a monogenic disorder underlying a PID. Inborn errors of immunity resulting from single-gene defects have been shown to underlie multiple bacterial infections [myeloid differentiation primary response 88 (MYD88) and interleukin 1 receptor-associated kinase 4 (IRAK4) deficiency], monogenic susceptibility to mycobacterial disease (deficiency of genes in the IL12/IFN-γ loop), herpes simplex encephalitis (defect in TLR3-dependent immune responses), and severe primary Influenza virus infection (interferon regulatory factor 7 deficiency) (56–60).
These “non-conventional” PIDs are distinguished from conventional PIDs as they often occur in sporadic cases without any family history of severe infection. Individuals affected by non-conventional PIDs are often otherwise healthy. The immunological phenotype is not detectable with first-line immunological studies, and the disease might manifest as a single episode of severe and potentially lethal infection caused by a common or opportunistic pathogen, mostly during primary infection (56, 58, 59).
The discovery of non-conventional PIDs suggested that monogenic conditions might underlie infectious diseases of infancy and childhood more frequently than previously thought (11). The model of the genetic architecture of human infectious diseases that has been proposed based on these observations suggests that infections occurring early in life are more likely to be caused by single-gene disorders (61).
The proportion of neonatal infections that can be explained by known PIDs is unknown. However, there is evidence from case reports or small case studies that life-threatening infections occurring early in life may represent the first phenotypic manifestation of an inborn error of immunity.
The role of conventional PIDs in conferring susceptibility to infection in the neonatal age has been recently reviewed by Walkovich and Connelly (62). It is important here to remember that a high index of suspicion is required, given that the infectious and potential extraimmunological phenotypes may be only partially expressed during the neonatal period.
Non-conventional PIDs have also been shown to underlie life-threatening neonatal infections. Pyogenic infections occurring during the first few weeks of life have been described as the first phenotypic manifestation of IRAK4 and MYD88 deficiencies (63–65). Klebsiella pneumoniae infection often striking in neonatal units as a fulminant and fatal disease, has been linked in some pediatric patients to IL12 receptor subunit beta 1 deficiency (66).
Loss-of-function mutations in interferon induced with helicase c domain 1 (IFIH1), a cytosolic sensor of the viral RNA, have been implicated as causative factors in lower respiratory tract infections (pneumonitis, bronchiolitis) caused by RNA viruses (67). Interestingly, the phenotype of IFIH1 deficiency is narrow (restricted to few related RNA viruses), transient (recurrence was found in one of eight patients), and organ specific (only affects the lungs).
Variants in single Ig And TIR domain containing (SIGIRR) have been implicated as a possible causative or facilitating factor of necrotizing enterocolitis (NEC) (68), but fulminant and infection-associated NEC (69) in infants with no other identifiable facilitating iatrogenic factor or medical condition has not been linked yet to a genetic condition.
From a clinical perspective, newborn infants suffering from life-threatening infections may be divided in two major groups:
(1) Newborn infants with a known medical condition. This group includes all infants admitted to a NICU (therefore exposed to nosocomial pathogens) and specifically very preterm (<32 weeks gestational age) and extremely preterm (<28 weeks gestational age) infants, infants undergoing surgery, infants with organ disease (e.g., urinary tract malformations, neurological conditions), and infants receiving medical procedures or treatments that are per se sufficient to explain an increased vulnerability to colonizing microorganisms. Infections in this group are multifactorial or linked to one specific known factor of vulnerability, and only a small proportion of the risk is probably explained by individual genetic variation.
(2) Otherwise healthy, full-term, or late-preterm newborn infants with no identifiable medical conditions. Severe infections in these infants occur without any apparent risk or facilitating factor and, from a host perspective, can be considered idiopathic diseases.
Most of these infections occur as isolated events (the spectrum of susceptibility is extremely narrow, in most cases restricted to a single microorganism) and rarely recur.
Some infections are almost never observed in healthy children after the first year of life or in adults. These include neonatal GBS disease, viral bronchiolitis, and rare cases of infection-related NEC in late preterm and full-term infants. Conversely, other infections are not age specific, but may occur with particular frequency and severity in the neonatal period and infancy. These include infections by E. coli, Klebsiella spp., Listeria monocytogenes, and other Gram-negative and Gram-positive pathogens.
The biological underpinnings of the interindividual differences in resistance and vulnerability to specific pathogens in otherwise healthy infants are currently unknown.
A general model to explain susceptibility to neonatal infections in full-term and preterm infants is lacking.
Single factors with high effect size explain some of the most severe diseases occurring in infants without known comorbidities. A prime example in neonatal medicine is the rare occurrence of rapidly progressive neonatal jaundice and kernicterus in otherwise healthy, full-term babies, which is due to neonatal hemolysis resulting from either monogenic defects (e.g., spherocytosis, G6PD deficiency) or alloimmune maternal Abs (anti-Rh, anti-ABO) (70). Conversely, hemolysis leading to kernicterus in extremely preterm infants is more likely to be multifactorial, depending on the combined contribution of common genetic polymorphisms, underlying medical conditions, iatrogenic factors, and other environmental determinants (71).
As a general observation, single-gene or single-factor disorders are more likely to underlie severe neonatal disease phenotypes in otherwise healthy full-term infants, while a multifactorial pathogenesis is more likely to explain mild-to-severe neonatal disease in the presence of comorbidities or iatrogenic factors, with severity depending on the underlying pathogenic process (Table 1).
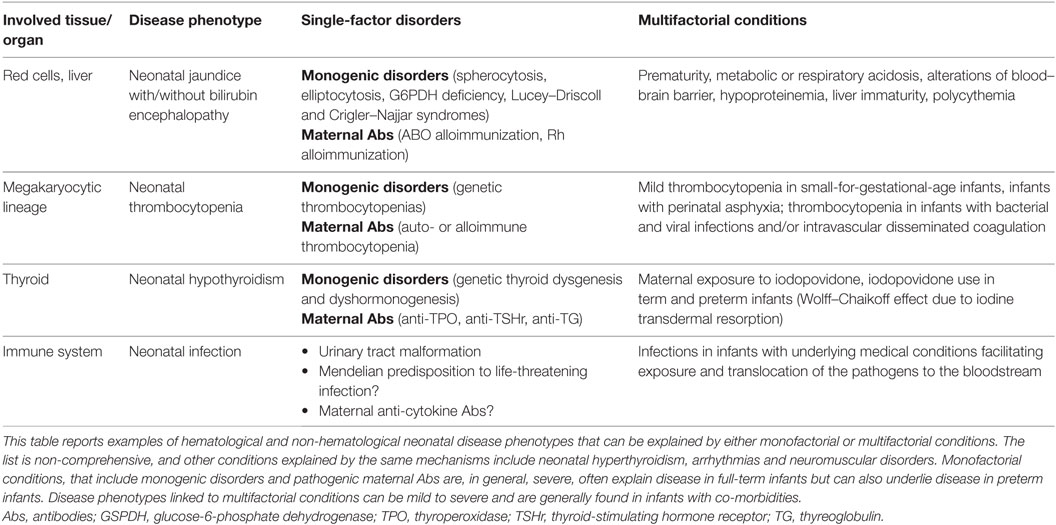
Table 1. Mechanisms of disease in term and preterm infants.
Along the same lines, we here suggest that single factors with high effect size may underlie life-threatening infections in otherwise healthy, full-term, or late-preterm babies, while a polygenic/multifactorial model may better explain the occurrence and severity of infections in very and extremely preterm infants.
Accordingly, we propose an extension of the model of the genetic architecture of infectious diseases proposed by Alcais et al. (61) to include full-term and preterm infants (Figure 1).
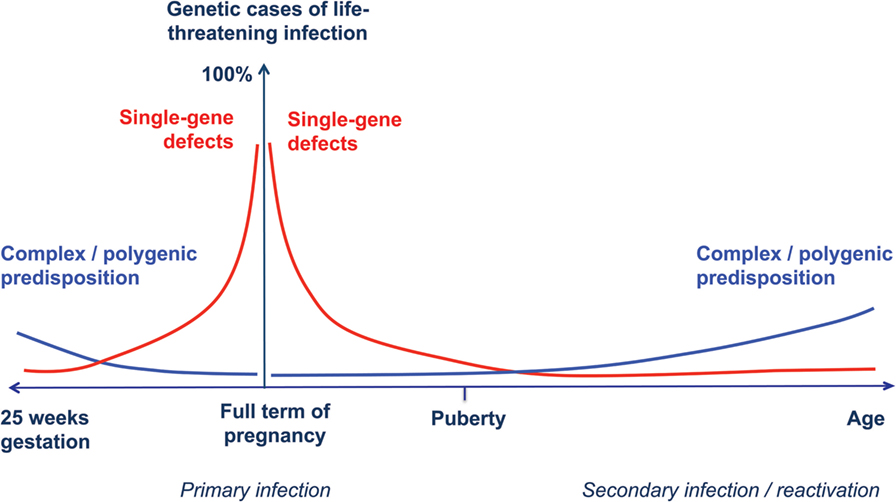
Figure 1. Human genetic architecture of infections. Modified from Alcais et al. (61). The contribution of Mendelian genetic defects (red lines) to life-threatening infectious diseases is mostly observed during childhood, while complex interactions between environmental influences and polygenic susceptibility (blue lines) play a more important role for infections occurring later in life. We propose a specular trend for the contribution of human genetic variants to infection susceptibility with decreasing gestational age. In newborn infants at extremely low gestational ages, exogenous factors play a major role, while host genetic defects are more likely to explain life-threatening infection in full-term, otherwise healthy babies.
Additional host factors (maternal antimicrobial protective Abs, vaginal and breast microbiome, epigenetic, and maturational changes in the immune system) and determinants of microbial virulence may also modulate disease severity (17).
In preterm infants, multiple factors are well known to contribute to both the occurrence and the severity of infections. Colonization of deep mucosal tissues by hospital-acquired microorganisms and translocation to the bloodstream is facilitated by several factors: biomedical devices (endotracheal or nasogastric tubes), invasive procedures, thin skin and mucosal layers, central catheters, total parenteral nutrition, drugs (histamine type 2 receptor-antagonists, steroids, antibiotics), delayed initiation of enteral nutrition with formula milk, associated diseases, male gender, an incomplete maturation of the preterm immune system (6, 35, 72–74), and, possibly, a weak polygenic predisposition (52).
In full-term infants, supporting evidence for a role of single host factors in determining susceptibility to infection is provided by the example of urinary tract malformation as one single, high effect-size factor, in determining susceptibility to urosepsis (75) independently of other protective or risk factors. In infants with urinary tract malformation, the effect of the alterations in urinary flow on the facilitation of urosepsis exceeds by far the effect of other potentially modulating factors.
Where no apparent determinant of higher susceptibility to infection is identified, a failure of the individual specific protective innate immune responses can be hypothesized. The failure of specific arms of the immune system that are non-redundant in the neonatal defense against a given microorganism would exceed in effect size the modulating potential of other protective factors.
The view of single-gene defects contributing to the burden of neonatal infections in otherwise healthy infants is supported by the growing body of evidence in the literature describing neonatal infections as the first phenotypic manifestation of a known conventional or non-conventional PID (62–65, 67, 76). However, the great majority of neonatal infections still need to be characterized from a host molecular perspective.
In the past years, given its predominant role among neonatal infections, neonatal GBS disease has been extensively characterized from an epidemiological standpoint. The elucidation of the mechanisms underlying neonatal vulnerability to GBS may serve as a model to understand the pathogenesis of other neonatal infectious diseases. In the following paragraphs, we discuss the unique susceptibility to GBS infection of some young infants and propose that it could be due to genetic or immune factors.
Group B streptococcus is a Gram-positive, β-hemolytic bacterium frequently colonizing the human gastrointestinal and genitourinary tracts. Invasive GBS disease is extremely rare in healthy adults, with a reported incidence of 10/100,000 non-pregnant individuals (20, 21). Young infants, pregnant and post-partum women, and older adults with underlying medical conditions display higher rates of invasive disease (77).
The global incidence of neonatal GBS disease is estimated to be as high as 0.53/1,000 live births (78). The incidence is highest in infants during the first 3 months of life and dramatically declines afterward (7). Early-onset GBS disease (EOD, onset during the first 6 days of life) occurs after vertical transmission of the bacterium through ascending infection or during delivery through a GBS-colonized birth canal. Risk of EOD can be reduced by administration of antibiotics to the mother during labor. Late-onset GBS disease (LOD) (onset between 7 and 89 days of life) is thought to result from horizontal transmission in most cases. The source of GBS can been identified in some cases. Potential routes of transmission include persistent mucous membrane and skin colonization from acquisition of GBS at birth or after birth from mothers with vaginal colonization; gut colonization through ingestion of infected breast milk from mothers with or without mastitis; or the community or hospital environment (15, 79–81). No preventive strategy exists for LOD. After the introduction of intrapartum antibiotic prophylaxis in clinical practice, the incidence of EOD has dropped in the United States from 1.7/1,000 live births in 1993 to ~0.3/1,000 live births, but the incidence of LOD remained stable (7, 82). Clinically, neonatal GBS disease has the features of a severe, life-threatening bacterial infection with systemic disease (sepsis), often associated with organ involvement (meningitis, osteoarthritis, NEC), requiring admission to a NICU. Untreated, it is almost always fatal with multiorgan failure due to septic shock and disseminated intravascular coagulopathy. Case-fatality ratio was as high as 50% in the 1970s (7) and has now dropped to <10% (78, 82), thanks to improvements in neonatal intensive care techniques and the prompt detection of clinical signs of infection and immediate initiation of antibiotic treatment.
Approximately 50% of infants born to GBS-colonized mothers (10–30% of all pregnancies) are in turn colonized. Of these, only 1–2% develops overt EOD (7). Data on the proportion of GBS-exposed infants developing LOD are lacking, but it is probably low, given a likely increase in the cumulative exposure/colonization rate with age and a concurrent decline in the incidence of GBS disease.
During the past decades, epidemiological studies led to the identification of several risk factors for EOD, including maternal colonization with GBS and bacteriuria, prematurity, chorioamnionitis, and/or intrapartum fever, prolonged (>18 h) premature rupture of membranes (PROM), low maternal anticapsular polysaccharide GBS Abs (29, 83–88), and GBS disease in an older sibling (89). Established risk factors for LOD include prematurity and gut colonization by the pathogen (80, 90).
In many cases, invasive GBS infection develops in otherwise healthy, full-term newborn infants, with as many as 42% of early-onset cases (91) and most late-onset cases occurring in the absence of any established risk factor. Known risk factors are therefore unable to reliably predict the occurrence of GBS disease at the individual level. Rather, they identify groups of infants enriched for determinants of susceptibility, but the nature of such determinants has remained elusive.
Fetal and neonatal exposure to the microorganism is the sine qua non-for neonatal colonization and subsequent infection. Heavy maternal vaginal colonization has since long been recognized as a risk factor for EOD, possibly due to greater bacterial inoculum to the lungs (7). High bacterial load in maternal milk has been linked to neonatal gut colonization and subsequent invasive LOD (80). The determinants of maternal carriage and the maternal bacterial overgrowth are poorly understood. Mild maternal disease may accompany heavy maternal colonization: maternal GBS urinary tract infection in pregnancy is considered a sign of heavy colonization (7), and maternal mastitis may be responsible for high bacterial load in maternal milk (80).
Additional microbial factors, beyond bacterial load, contribute to the development of invasive disease. Ten different GBS serotypes have been described (Ia, Ib, II–IX), based on the capsular polysaccharide antigen. Serotypes Ia, Ib, II, III, and V are most frequently found in EOD; serotype III is the most frequently isolated serotype in LOD and meningitis, but all serotypes can cause neonatal infection (19, 82, 92, 93). The capsular polysaccharide is thought to contribute to the virulence of the microorganism by aiding to escape the host immune responses. Deeper investigation on GBS isolates through multilocus sequence typing and grouping of genetically related sequence types (STs) into clonal complexes (CCs) has shown that most human isolates belong to few CCs (CC1, CC10, CC17, CC19, CC23, and CC26) (94–98) (http://pubmlst.org/sagalactiae/). The hypervirulent CC17 strains (including the hypervirulent ST-17 strain) are newborn specific. They possess the adhesin HvgA and other surface proteins conferring the ability to invade the neonatal central nervous system and are responsible for most LOD with meningitis, but are usually not responsible for adult disease (99). Strains belonging to all the six CCs have been reported in EOD (82, 98, 99).
The neonatal-specific hypervirulence of some bacterial strains and the bacterial load may explain in part the occurrence of neonatal disease. Nonetheless, individual susceptibility is not fully explained by bacterial virulence, especially in cases in which infection is caused by non-hypervirulent strains.
One fundamental and yet-unanswered question in the field is which are the non-redundant pathways of the innate immune system conferring neonatal protection to GBS.
Several different methodologies in in vitro and animal models have been used to attempt to answer this question.
Both knockout mouse and in vitro models of GBS infection identified a critical role for TLR and IL1 receptor signaling and/or signaling through MYD88 in bacterial clearance, TNF-mediated inflammation, septic shock, and microglia activation and neurodegeneration (100–109). Specifically, TLR2 and IL1R signaling have been shown to be both beneficial and harmful, depending on the experimental conditions (101, 110–112). A role for IL6, IL10, IL12, and IL18 has been demonstrated in mouse models of GBS infection through administration of anticytokine specific Abs (113–116).
The relevance of the studied pathways in the experimental settings may largely depend on the experimental conditions. Conversely, the non-redundant role of the studied signaling pathways in the human model in natural (as opposed to experimental) conditions still needs to be elucidated (117).
One human study suggested that a null polymorphism in sialic acid-binding immunoglobulin-like lectin 14 (SIGLEC14) influences human inflammatory responses to GBS in neutrophils and amniotic membranes and is possibly correlated with GBS-related preterm birth (118), but no data are available on the possible role of SIGLEC proteins in the pathogenesis of GBS infection.
Despite advances in the understanding of both the host and the microbial sides of neonatal GBS infection, currently available data are not able to fully explain neonatal susceptibility to infection at the individual level.
We hypothesize that susceptibility to neonatal GBS disease in otherwise healthy infants is due to a failure of the specific neonatal protective innate immune responses to GBS. This neonatal immunodeficiency could be either intrinsic (genetic defect in the infant) or extrinsic/environmental (interference of maternal Abs). In the next paragraphs, we present the genetic and the “maternal antibody” hypotheses of GBS disease and explain how these fit with current evidence.
Several reports, recently reviewed (76), demonstrate that adult and neonatal GBS infection may be a phenotypic expression of both conventional (Kostmann disease, transient hypogammaglobulinemia of infancy, chronic granulomatous disease, activated phosphatidylinositol 3-kinase δ syndrome—like immunodeficiency, C2 and IgG4 subclass deficiency, and isolated congenital asplenia) and non-conventional (IRAK4 and MYD88 deficiency) PIDs. Even when occurring in the context of a non-conventional PID, neonatal GBS infection may be one of the several manifestations of a broader phenotype that, for MYD88 and IRAK4 deficiency, includes susceptibility to multiple pyogenic bacteria. Conversely, most cases of neonatal GBS disease occur as an isolated infection, indicating that the susceptibility to GBS is pathogen specific and not linked to a more general state of immunosuppression.
We hypothesize that inborn errors of the primary innate immune responses to GBS, i.e., monogenic susceptibility to GBS disease, underlie some cases of isolated neonatal GBS infection occurring in otherwise healthy neonates. The clinical and immunological phenotypes of isolated neonatal GBS disease may indeed be consistent with those of non-conventional PIDs (57): (i) GBS disease is a potentially lethal infection striking early in life; (ii) the infecting strain/serotype and its virulence factors, while accounting for some variability in the occurrence and severity of infection (92, 119, 120), are not sufficient to explain susceptibility and resistance at the individual level; (iii) the spectrum of susceptibility is extremely narrow, restricted to GBS; and (iv) in most cases, there are no immunological defects at first-line immunological studies that would be consistent with conventional PIDs. In addition, GBS infection usually strikes once in life and only rarely recurs (~1% of cases) (121). This observation is consistent with a low recurrence rate in the subset of non-conventional PIDs characterized by immunodeficiency of the protective immunity to primary infections (57).
The highest incidence of GBS disease during the first 3 months of life would be explained by the high likelihood of being exposed to GBS in the perinatal period and/or by the full penetrance of the genetic defects in this age group.
Recurrence of GBS infection concerns only a small percentage of cases, both singletons and twins, and has been linked to re-exposure to GBS through maternal milk or other sources, to inappropriate treatment, or to persistence of GBS on skin and mucosal surfaces after the first infectious episode (122–125). A genetic explanation for recurrence is also plausible. In some PIDs of protective immunity to primary infection, the genotype has been shown to influence the recurrence rate (126). Therefore, recurrence of neonatal GBS disease may indicate a more severe phenotype or represent the phenotypic manifestation of a specific genetic defect.
The occurrence of GBS disease in siblings (89), as well as the recurrence described in a consanguineous family (76), suggests that the genetic hypothesis may be a plausible explanation for some cases. Infection by poorly virulent strains, the presence of other cases with overlapping phenotypes in the family, consanguinity in the parents, recurrence and severity of the clinical signs, and slow or absent response to antimicrobials despite appropriate treatment strengthen (although their absence does not exclude) the hypothesis of a PID underlying GBS infection.
The highest incidence of GBS disease is registered during the first 3 months of life, with most cases (77–78%) occurring during the first week of life (20, 21). This observation, together with a known role of GBS in prenatal disease, both prematurely and at full term of pregnancy (GBS-related stillbirth, term or preterm PROM, chorioaminionitis), suggest that a maternal factor might be particularly important for perinatal infection.
Recently, neutralizing anticytokine auto-Abs have been found in adult and pediatric patients suffering from life-threatening infections, revealing novel mechanisms of unusual susceptibility to specific pathogens (127–130). Auto-Abs against IL17 and/or IL22 have been associated with chronic mucocutaneous candidiasis; anti-IFN-γ auto-Abs with adult-onset immunodeficiency; anti-IL6 auto-Abs with recurrent skin infection; and auto-Abs against GM-CSF with pulmonary alveolar proteinosis (131).
The clinical phenotypes resulting from anticytokine auto-Abs partially (anti-IFN-γ, anti-IL6) or completely (anti-IL17, anti-GM-CSF) overlap with known monogenic conditions affecting the same pathways, demonstrating that Ab-mediated diseases may be immunophenocopies of monogenic immune disorders.
In neonates, autoimmunity is an exceedingly rare condition, but Ab-mediated disease due to transplacental crossing of maternal auto- or allo-Abs is a well-recognized and relatively frequent mechanism of organ dysfunction. This has been shown in the thyroid (congenital hypothyroidism), the blood (fetal and neonatal hemolytic disease and fetal and neonatal auto- and alloimmune thrombocytopenia), the neuromuscular junction (transient neonatal myasthenia gravis), the heart (congenital heart block due to SSA/Ro Abs), and other organs and tissues (132–137). We therefore hypothesize that neonatal GBS disease may be caused by yet-undiscovered neutralizing maternal auto-Abs or allo-Abs against components of the fetal and neonatal immune system that are non-redundant in conferring neonatal protection against GBS. The progressive decay of circulating maternal Abs in the infant plasma might then explain the decreasing incidence of infection over the first 3 months of life. Furthermore, the presence of pathogenic circulating Abs in the maternal blood would be consistent with the occurrence of mild disease in the mother (GBS-related urinary tract infection or mastitis) that is often associated with neonatal GBS disease, as well as with the well-documented higher risk of GBS-EOD in infants with a previous sibling with GBS disease (7). Finally the removal, with exchange transfusion, of pathogenic Abs from neonatal plasma could be an additional explanation to the efficacy of the procedure in infants with septic shock (138).
The proposed mechanism could in part explain neonatal GBS disease in full-term infants. Despite transplacental transfer of Abs is reduced at low gestational ages, allo- or autoimmune pathogenic maternal Abs have been demonstrated to be able to cause disease in the preterm infant or during gestation (71, 134). Therefore, the “maternal antibody” hypothesis could also explain some cases of neonatal GBS disease occurring in preterm infants.
Previous studies that addressed the role of genetics in the susceptibility to neonatal infection focused on the associations between selected common single-nucleotide polymorphisms and infectious outcomes (139). A more integrated approach including genomics, transcriptomics, proteomics, and functional studies is required to uncover the precise molecular determinants of susceptibility to specific pathogens causing neonatal infections.
Ad hoc studies should be designed depending on the phenotype under investigation.
Multicenter GWASs may provide some insight into the pathogenesis of suspected multifactorial infections as, for instance, those occurring in preterm infants. GWAS are currently ongoing on neonatal cohorts (54).
Exome or genome sequencing studies have potential to uncover the cause of suspected monogenic disorders. Cases should be prioritized based on the clinical profile most suggestive of a monogenic etiology, including extreme severity, consanguinity, recurrence of infection, and familial presentation. Depending on the design of the study, analysis of the trio (proband and parents) and of the family or cohort studies should be carried out to uncover the individual, rare (<1% in the general population), and functionally deleterious genetic variants that best fit the most likely genetic model (de novo, autosomal dominant with complete or incomplete penetrance, autosomal recessive with mono- or biallelic mutations). This approach could shed light on the pathways that are non-redundant in neonatal protection against GBS.
Functional follow-up will be required to validate candidate variants and confirm their causative role. These studies should be designed to assess the integrity of the molecular pathways affected by the mutations and determine how they are relevant to the neonatal immune responses in primary cells and/or immortalized cell lines.
Laboratory experiments will be also needed to investigate the possible interfering effect of maternal plasma on the neonatal immune responses. The laboratory tests could include cytokine production assays, detailed analyses of RNA (transcriptome analysis) and protein expression in ex vivo samples (blood collected during sepsis), and in vitro experiments (stimulation of patient and control cells with different ligands, cell differentiation assays) in the presence of maternal or control plasma. Specific assays should be used for the detection of specific Abs in the maternal and in the perinatal plasma.
Ultimately, these experiments should aim at demonstrating a causative link between the molecular findings, the observed cellular phenotypes, and the patient’s clinical phenotype.
Transient susceptibility to a narrow range of infections during the neonatal age may be explained by inborn errors of immunity, in the context of a relatively immature, non-redundant immune system. The early recognition of a PID as an essential contributing factor to a severe neonatal infection is clinically very relevant, as it may change the management and allow the referral of the patient to the clinical immunologist for specific follow-up and family counseling.
In parallel fields, the discovery of concurrent genetic and auto-/alloimmune mechanisms for several neonatal diseases has dramatically changed practice, as exemplified by the development of highly effective screening and diagnostic procedures for neonatal hemolysis, which reduced the incidence of fetal erythroblastosis and neonatal bilirubin encephalopathy by two orders of magnitude, from ~1/1,000 to ~1/100,000 live births (Table 2) (140). Similar observations can be made for congenital hypothyroidism and other common and rare neonatal diseases (Table 1).
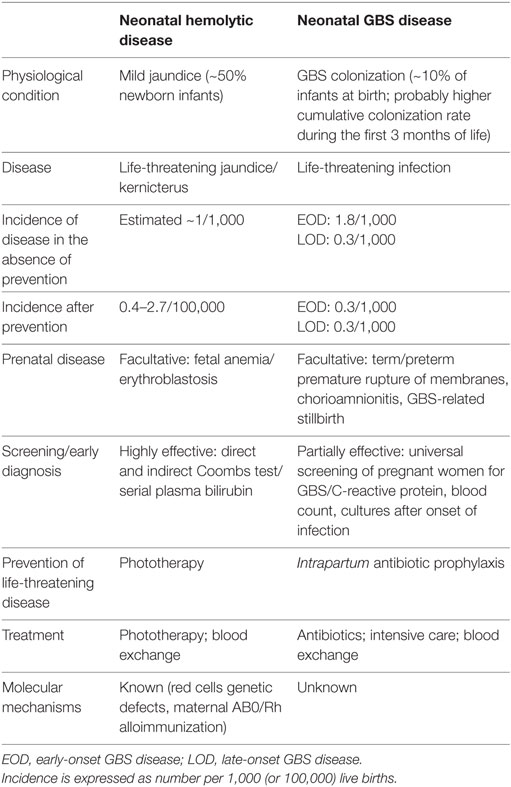
Table 2. Comparison of neonatal hemolytic disease and neonatal group B streptococcus (GBS) disease.
Current prevention efforts, although invaluable for neonatal health, only had a limited impact on the global incidence of neonatal infections (9, 141) (Table 2). A more complete understanding of the mechanisms underlying the interindividual variability in the neonatal innate immune responses to pathogens is required to develop highly effective, pathogen-specific and individual-tailored preventive protocols.
AB conceived the manuscript; conducted the literature search; and drafted, edited, and approved the final version of the paper. MS participated in the discussion of ideas, helped with the writing, revised critically, and approved the final version of the manuscript. JF participated in the discussion of ideas, edited, revised critically, and approved the final version of the manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The authors wish to thank the colleagues who collaborate with them on ongoing projects on GBS disease and supported them with suggestions and critical discussions. From San Matteo Hospital, Pavia (Italy), Neonatal Intensive Care Unit: Stefania Longo and Iolanda Mazzucchelli. From Ecole Polytechnique Fédérale de Lausanne (Switzerland), Fellay lab: Samira Asgari, Nimisha Chaturvedi, Yann Dubois, Christian Hammer, Thomas Junier, Olivier Naret, Petar Scepanovic, and Christian Thorball. From the Swiss Pediatric Sepsis Study (SPSS): Luregn Schlapbach (Mater Children’s Hospital, Brisbane, Australia) and Eric Giannoni (Centre hospitalier universitaire vaudois, Lausanne, Switzerland). From the Rockefeller University, New York (USA): Janet Markle. The authors are obliged to Jean-Laurent Casanova (Laboratory of Human Genetics of Infectious Diseases, Rockefeller University, New York and Institut Imagine, Paris, France) for his precious advices and hints on human genetics of infectious diseases. The authors are grateful to all the colleagues from neonatal and pediatric units who struggle with them in the attempt to understand the molecular bases of neonatal infections. The authors’ most heart-felt thanks are due to the little patients and their families, who are their most important collaborators.
The authors did not receive any specific financial support for the writing of the submitted work. JF is the recipient of an SNF Professorship from the Swiss National Science Foundation (PP00P3_133703).
1. Adkins B, Chun K, Hamilton K, Nassiri M. Naive murine neonatal T cells undergo apoptosis in response to primary stimulation. J Immunol (1996) 157(4):1343–9.
2. Forsthuber T, Yip HC, Lehmann PV. Induction of TH1 and TH2 immunity in neonatal mice. Science (1996) 271(5256):1728–30. doi:10.1126/science.271.5256.1728
3. Ridge JP, Fuchs EJ, Matzinger P. Neonatal tolerance revisited: turning on newborn T cells with dendritic cells. Science (1996) 271(5256):1723–6. doi:10.1126/science.271.5256.1723
4. Sarzotti M, Robbins DS, Hoffman PM. Induction of protective CTL responses in newborn mice by a murine retrovirus. Science (1996) 271(5256):1726–8. doi:10.1126/science.271.5256.1726
5. Royal College of Obstetricians and Gynaecologists. (2012). Available from: https://www.rcog.org.uk/globalassets/documents/guidelines/gtg_36.pdf
6. Borghesi A, Stronati M. Strategies for the prevention of hospital-acquired infections in the neonatal intensive care unit. J Hosp Infect (2008) 68(4):293–300. doi:10.1016/j.jhin.2008.01.011
7. Centers for Disease Control and Prevention. 2010 guidelines for the prevention of perinatal group B streptococcal disease. Morb Mortal Wkly Rep (2010) 59(RR–10):1–32.
8. Vergnano S, Embleton N, Collinson A, Menson E, Russell AB, Heath P. Missed opportunities for preventing group B Streptococcus infection. Arch Dis Child Fetal Neonatal Ed (2010) 95(1):F72–3. doi:10.1136/adc.2009.160333
9. Stoll BJ, Hansen NI, Sanchez PJ, Faix RG, Poindexter BB, Van Meurs KP, et al. Early onset neonatal sepsis: the burden of group B Streptococcal and E. coli disease continues. Pediatrics (2011) 127(5):817–26. doi:10.1542/peds.2010-2217
10. Stoll BJ. Early-onset neonatal sepsis: a continuing problem in need of novel prevention strategies. Pediatrics (2016) 138(6):e20163038. doi:10.1542/peds.2016-3038
11. Casanova JL. Severe infectious diseases of childhood as monogenic inborn errors of immunity. Proc Natl Acad Sci U S A (2015) 112(51):E7128–37. doi:10.1073/pnas.1521651112
12. Casanova JL. Human genetic basis of interindividual variability in the course of infection. Proc Natl Acad Sci U S A (2015) 112(51):E7118–27. doi:10.1073/pnas.1521644112
13. Siegrist CA, Aspinall R. B-cell responses to vaccination at the extremes of age. Nat Rev Immunol (2009) 9(3):185–94. doi:10.1038/nri2508
14. Ardeshir A, Narayan NR, Mendez-Lagares G, Lu D, Rauch M, Huang Y, et al. Breast-fed and bottle-fed infant rhesus macaques develop distinct gut microbiotas and immune systems. Sci Transl Med (2014) 6(252):252ra120. doi:10.1126/scitranslmed.3008791
15. Le Doare K, Kampmann B. Breast milk and Group B streptococcal infection: vector of transmission or vehicle for protection? Vaccine (2014) 32(26):3128–32. doi:10.1016/j.vaccine.2014.04.020
16. Le Doare K, Allen L, Kampmann B, Heath PT, Taylor S, Hesseling AC, et al. Anti-group B Streptococcus antibody in infants born to mothers with human immunodeficiency virus (HIV) infection. Vaccine (2015) 33(5):621–7. doi:10.1016/j.vaccine.2014.12.025
17. Gomez de Aguero M, Ganal-Vonarburg SC, Fuhrer T, Rupp S, Uchimura Y, Li H, et al. The maternal microbiota drives early postnatal innate immune development. Science (2016) 351(6279):1296–302. doi:10.1126/science.aad2571
18. GBD 2015 Mortality and Causes of Death Collaborators. Global, regional, and national life expectancy, all-cause mortality, and cause-specific mortality for 249 causes of death, 1980-2015: a systematic analysis for the Global Burden of Disease Study 2015. Lancet (2016) 388(10053):1459–544. doi:10.1016/S0140-6736(16)31012-1
19. Heath PT, Schuchat A. Perinatal group B streptococcal disease. Best Pract Res Clin Obstet Gynaecol (2007) 21(3):411–24. doi:10.1016/j.bpobgyn.2007.01.003
20. Centers for Disease Control and Prevention. (2010). Available from: http://www.cdc.gov/groupbstrep/about/adults.html
21. Centers for Disease Control and Prevention. (2010). Available from: http://www.cdc.gov/groupbstrep/guidelines/slidesets.html
22. Byington CL, Rittichier KK, Bassett KE, Castillo H, Glasgow TS, Daly J, et al. Serious bacterial infections in febrile infants younger than 90 days of age: the importance of ampicillin-resistant pathogens. Pediatrics (2003) 111(5 Pt 1):964–8. doi:10.1542/peds.111.5.964
23. Greenhow TL, Hung YY, Herz AM. Changing epidemiology of bacteremia in infants aged 1 week to 3 months. Pediatrics (2012) 129(3):e590–6. doi:10.1542/peds.2011-1546
24. Stoll BJ, Hansen N, Fanaroff AA, Wright LL, Carlo WA, Ehrenkranz RA, et al. Changes in pathogens causing early-onset sepsis in very-low-birth-weight infants. N Engl J Med (2002) 347(4):240–7. doi:10.1056/NEJMoa012657
25. Stoll BJ, Hansen N, Fanaroff AA, Wright LL, Carlo WA, Ehrenkranz RA, et al. Late-onset sepsis in very low birth weight neonates: the experience of the NICHD Neonatal Research Network. Pediatrics (2002) 110(2 Pt 1):285–91. doi:10.1542/peds.110.2.285
26. Clark R, Powers R, White R, Bloom B, Sanchez P, Benjamin DK Jr. Nosocomial infection in the NICU: a medical complication or unavoidable problem? J Perinatol (2004) 24(6):382–8. doi:10.1038/sj.jp.7211120
27. Lachassine E, Letamendia-Richard E, Gaudelus E. Epidemiology of nosocomial infections in neonates. Archives de Pediatrie (2004) 11:229–33. doi:10.1016/j.arcped.2003.10.016
28. Vergnano S, Menson E, Kennea N, Embleton N, Russell AB, Watts T, et al. Neonatal infections in England: the NeonIN surveillance network. Arch Dis Child Fetal Neonatal Ed (2011) 96(1):F9–14. doi:10.1136/adc.2009.178798
29. Shane AL, Stoll BJ. Neonatal sepsis: progress towards improved outcomes. J Infect (2014) 68(Suppl 1):S24–32. doi:10.1016/j.jinf.2013.09.011
30. Godfrey WR, Spoden DJ, Ge YG, Baker SR, Liu B, Levine BL, et al. Cord blood CD4(+)CD25(+)-derived T regulatory cell lines express FoxP3 protein and manifest potent suppressor function. Blood (2005) 105(2):750–8. doi:10.1182/blood-2004-06-2467
31. Li L, Godfrey WR, Porter SB, Ge Y, June CH, Blazar BR, et al. CD4+CD25+ regulatory T-cell lines from human cord blood have functional and molecular properties of T-cell anergy. Blood (2005) 106(9):3068–73. doi:10.1182/blood-2005-04-1531
32. Walker WE, Goldstein DR. Neonatal B cells suppress innate toll-like receptor immune responses and modulate alloimmunity. J Immunol (2007) 179(3):1700–10. doi:10.4049/jimmunol.179.3.1700
33. Elahi S, Ertelt JM, Kinder JM, Jiang TT, Zhang X, Xin L, et al. Immunosuppressive CD71+ erythroid cells compromise neonatal host defence against infection. Nature (2013) 504(7478):158–62. doi:10.1038/nature12675
34. Wynn JL, Levy O. Role of innate host defenses in susceptibility to early-onset neonatal sepsis. Clin Perinatol (2010) 37(2):307–37. doi:10.1016/j.clp.2010.04.001
35. Rechavi E, Lev A, Lee YN, Simon AJ, Yinon Y, Lipitz S, et al. Timely and spatially regulated maturation of B and T cell repertoire during human fetal development. Sci Transl Med (2015) 7(276):276ra225. doi:10.1126/scitranslmed.aaa0072
36. Gibbons D, Fleming P, Virasami A, Michel ML, Sebire NJ, Costeloe K, et al. Interleukin-8 (CXCL8) production is a signatory T cell effector function of human newborn infants. Nat Med (2014) 20(10):1206–10. doi:10.1038/nm.3670
37. Zhang X, Mozeleski B, Lemoine S, Deriaud E, Lim A, Zhivaki D, et al. CD4 T cells with effector memory phenotype and function develop in the sterile environment of the fetus. Sci Transl Med (2014) 6(238):238ra272. doi:10.1126/scitranslmed.3008748
38. Adkins B. Heterogeneity in the CD4 T cell compartment and the variability of neonatal immune responsiveness. Curr Immunol Rev (2007) 3(3):151–9. doi:10.2174/157339507781483496
39. Adkins B, Bu Y, Guevara P. The generation of Th memory in neonates versus adults: prolonged primary Th2 effector function and impaired development of Th1 memory effector function in murine neonates. J Immunol (2001) 166(2):918–25. doi:10.4049/jimmunol.166.2.918
40. Adkins B. Peripheral CD4+ lymphocytes derived from fetal versus adult thymic precursors differ phenotypically and functionally. J Immunol (2003) 171(10):5157–64. doi:10.4049/jimmunol.171.10.5157
41. Rose S, Lichtenheld M, Foote MR, Adkins B. Murine neonatal CD4+ cells are poised for rapid Th2 effector-like function. J Immunol (2007) 178(5):2667–78. doi:10.4049/jimmunol.178.5.2667
42. Levy O. Innate immunity of the newborn: basic mechanisms and clinical correlates. Nat Rev Immunol (2007) 7(5):379–90. doi:10.1038/nri2075
43. Kruetzmann S, Rosado MM, Weber H, Germing U, Tournilhac O, Peter HH, et al. Human immunoglobulin M memory B cells controlling Streptococcus pneumoniae infections are generated in the spleen. J Exp Med (2003) 197(7):939–45. doi:10.1084/jem.20022020
44. Marodi L. Innate cellular immune responses in newborns. Clin Immunol (2006) 118(2–3):137–44. doi:10.1016/j.clim.2005.10.012
45. Dowling DJ, Levy O. Ontogeny of early life immunity. Trends Immunol (2014) 35(7):299–310. doi:10.1016/j.it.2014.04.007
46. Levy O, Zarember KA, Roy RM, Cywes C, Godowski PJ, Wessels MR. Selective impairment of TLR-mediated innate immunity in human newborns: neonatal blood plasma reduces monocyte TNF-alpha induction by bacterial lipopeptides, lipopolysaccharide, and imiquimod, but preserves the response to R-848. J Immunol (2004) 173(7):4627–34. doi:10.4049/jimmunol.173.7.4627
47. Angelone DF, Wessels MR, Coughlin M, Suter EE, Valentini P, Kalish LA, et al. Innate immunity of the human newborn is polarized toward a high ratio of IL-6/TNF-alpha production in vitro and in vivo. Pediatr Res (2006) 60(2):205–9. doi:10.1203/01.pdr.0000228319.10481.ea
48. Cuenca AG, Wynn JL, Moldawer LL, Levy O. Role of innate immunity in neonatal infection. Am J Perinatol (2013) 30(2):105–12. doi:10.1055/s-0032-1333412
49. Marodi L, Goda K, Palicz A, Szabo G. Cytokine receptor signalling in neonatal macrophages: defective STAT-1 phosphorylation in response to stimulation with IFN-gamma. Clin Exp Immunol (2001) 126(3):456–60. doi:10.1046/j.1365-2249.2001.01693.x
50. Lee HH, Hoeman CM, Hardaway JC, Guloglu FB, Ellis JS, Jain R, et al. Delayed maturation of an IL-12-producing dendritic cell subset explains the early Th2 bias in neonatal immunity. J Exp Med (2008) 205(10):2269–80. doi:10.1084/jem.20071371
51. Boghossian NS, Page GP, Bell EF, Stoll BJ, Murray JC, Cotten CM, et al. Late-onset sepsis in very low birth weight infants from singleton and multiple-gestation births. J Pediatr (2013) 162(6):1120–4. doi:10.1016/j.jpeds.2012.11.089
52. Bizzarro MJ, Jiang Y, Hussain N, Gruen JR, Bhandari V, Zhang H. The impact of environmental and genetic factors on neonatal late-onset sepsis. J Pediatr (2011) 158(2):234–8. doi:10.1016/j.jpeds.2010.07.060
53. Srinivasan L, Swarr DT, Sharma M, Cotten CM, Kirpalani H. Systematic review and meta-analysis: gene association studies in neonatal sepsis. Am J Perinatol (2016). doi:10.1055/s-0036-1597132
54. Srinivasan L, Kirpalani H, Cotten CM. Elucidating the role of genomics in neonatal sepsis. Semin Perinatol (2015) 39(8):611–6. doi:10.1053/j.semperi.2015.09.008
55. Picard C, Al-Herz W, Bousfiha A, Casanova JL, Chatila T, Conley ME, et al. Primary immunodeficiency diseases: an update on the classification from the International Union of Immunological Societies Expert Committee for Primary Immunodeficiency 2015. J Clin Immunol (2015) 35(8):696–726. doi:10.1007/s10875-015-0201-1
56. Casanova JL, Abel L. Primary immunodeficiencies: a field in its infancy. Science (2007) 317(5838):617–9. doi:10.1126/science.1142963
57. Bousfiha A, Picard C, Boisson-Dupuis S, Zhang SY, Bustamante J, Puel A, et al. Primary immunodeficiencies of protective immunity to primary infections. Clin Immunol (2010) 135(2):204–9. doi:10.1016/j.clim.2010.02.001
58. Casanova JL, Fieschi C, Bustamante J, Reichenbach J, Remus N, von Bernuth H, et al. From idiopathic infectious diseases to novel primary immunodeficiencies. J Allergy Clin Immunol (2005) 116(2):426–30. doi:10.1016/j.jaci.2005.03.053
59. Bustamante J, Boisson-Dupuis S, Jouanguy E, Picard C, Puel A, Abel L, et al. Novel primary immunodeficiencies revealed by the investigation of paediatric infectious diseases. Curr Opin Immunol (2008) 20(1):39–48. doi:10.1016/j.coi.2007.10.005
60. Ciancanelli MJ, Huang SX, Luthra P, Garner H, Itan Y, Volpi S, et al. Infectious disease. Life-threatening influenza and impaired interferon amplification in human IRF7 deficiency. Science (2015) 348(6233):448–53. doi:10.1126/science.aaa1578
61. Alcais A, Quintana-Murci L, Thaler DS, Schurr E, Abel L, Casanova JL. Life-threatening infectious diseases of childhood: single-gene inborn errors of immunity? Ann N Y Acad Sci (2010) 1214:18–33. doi:10.1111/j.1749-6632.2010.05834.x
62. Walkovich K, Connelly JA. Primary immunodeficiency in the neonate: early diagnosis and management. Semin Fetal Neonatal Med (2016) 21(1):35–43. doi:10.1016/j.siny.2015.12.005
63. Chapel H, Puel A, von Bernuth H, Picard C, Casanova JL. Shigella sonnei meningitis due to interleukin-1 receptor-associated kinase-4 deficiency: first association with a primary immune deficiency. Clin Infect Dis (2005) 40(9):1227–31. doi:10.1086/428733
64. von Bernuth H, Picard C, Jin Z, Pankla R, Xiao H, Ku CL, et al. Pyogenic bacterial infections in humans with MyD88 deficiency. Science (2008) 321(5889):691–6. doi:10.1126/science.1158298
65. Krause JC, Ghandil P, Chrabieh M, Casanova JL, Picard C, Puel A, et al. Very late-onset group B Streptococcus meningitis, sepsis, and systemic shigellosis due to interleukin-1 receptor-associated kinase-4 deficiency. Clin Infect Dis (2009) 49(9):1393–6. doi:10.1086/630206
66. Pedraza S, Lezana JL, Samarina A, Aldana R, Herrera MT, Boisson-Dupuis S, et al. Clinical disease caused by Klebsiella in 2 unrelated patients with interleukin 12 receptor beta1 deficiency. Pediatrics (2010) 126(4):e971–6. doi:10.1542/peds.2009-2504
67. Asgari S, Fellay J. Host and Pathogen Genomics of Severe Pediatric Infections. PhD thesis 6656. Lausanne: École Polytechnique Fédérale de Lausanne, EPFL (2016).
68. Sampath V, Menden H, Helbling D, Li K, Gastonguay A, Ramchandran R, et al. SIGIRR genetic variants in premature infants with necrotizing enterocolitis. Pediatrics (2015) 135(6):e1530–4. doi:10.1542/peds.2014-3386
69. Gordon PV, Swanson JR, MacQueen BC, Christensen RD. A critical question for NEC researchers: can we create a consensus definition of NEC that facilitates research progress? Semin Perinatol (2016). doi:10.1053/j.semperi.2016.09.013
70. Christensen RD, Yaish HM. hemolytic disorders causing severe neonatal hyperbilirubinemia. Clin Perinatol (2015) 42(3):515–27. doi:10.1016/j.clp.2015.04.007
71. Christensen RD, Yaish HM. Hemolysis in preterm neonates. Clin Perinatol (2016) 43(2):233–40. doi:10.1016/j.clp.2016.01.002
72. Borghesi A, Tzialla C, Decembrino L, Manzoni P, Stronati M. New possibilities of prevention of infection in the newborn. J Matern Fetal Neonatal Med (2011) 24(sup2):28–30. doi:10.3109/14767058.2011.604934
73. Currie AJ, Curtis S, Strunk T, Riley K, Liyanage K, Prescott S, et al. Preterm infants have deficient monocyte and lymphocyte cytokine responses to group B Streptococcus. Infect Immun (2011) 79(4):1588–96. doi:10.1128/IAI.00535-10
74. Schelonka RL, Maheshwari A, Carlo WA, Taylor S, Hansen NI, Schendel DE, et al. T cell cytokines and the risk of blood stream infection in extremely low birth weight infants. Cytokine (2011) 53(2):249–55. doi:10.1016/j.cyto.2010.11.003
75. Beetz R. Evaluation and management of urinary tract infections in the neonate. Curr Opin Pediatr (2012) 24(2):205–11. doi:10.1097/MOP.0b013e32834f0423
76. Licciardi F, Montin D, Versace A, Migliore G, Tzialla C, Fellay J, et al. Familial segregation of group B streptococcal infection in a consanguineous kindred. Int J Infect Dis (2016) 51:22–4. doi:10.1016/j.ijid.2016.08.010
77. Phares CR, Lynfield R, Farley MM, Mohle-Boetani J, Harrison LH, Petit S, et al. Epidemiology of invasive group B streptococcal disease in the United States, 1999-2005. JAMA (2008) 299(17):2056–65. doi:10.1001/jama.299.17.2056
78. Edmond KM, Kortsalioudaki C, Scott S, Schrag SJ, Zaidi AK, Cousens S, et al. Group B streptococcal disease in infants aged younger than 3 months: systematic review and meta-analysis. Lancet (2012) 379(9815):547–56. doi:10.1016/S0140-6736(11)61651-6
79. Morinis J, Shah J, Murthy P, Fulford M. Horizontal transmission of group B Streptococcus in a neonatal intensive care unit. Paediatr Child Health (2011) 16(6):e48–50.
80. Berardi A, Rossi C, Lugli L, Creti R, Bacchi Reggiani ML, Lanari M, et al. Group B Streptococcus late-onset disease: 2003-2010. Pediatrics (2013) 131(2):e361–8. doi:10.1542/peds.2012-1231
81. Filleron A, Lombard F, Jacquot A, Jumas-Bilak E, Rodiere M, Cambonie G, et al. Group B streptococci in milk and late neonatal infections: an analysis of cases in the literature. Arch Dis Child Fetal Neonatal Ed (2014) 99(1):F41–7. doi:10.1136/archdischild-2013-304362
82. Le Doare K, Heath PT. An overview of global GBS epidemiology. Vaccine (2013) 31(Suppl 4):D7–12. doi:10.1016/j.vaccine.2013.01.009
83. Baker CJ, Edwards MS, Kasper DL. Role of antibody to native type III polysaccharide of group B Streptococcus in infant infection. Pediatrics (1981) 68(4):544–9.
84. Schuchat A, Deaver-Robinson K, Plikaytis BD, Zangwill KM, Mohle-Boetani J, Wenger JD. Multistate case-control study of maternal risk factors for neonatal group B streptococcal disease. The Active Surveillance Study Group. Pediatr Infect Dis J (1994) 13(7):623–9. doi:10.1097/00006454-199407000-00008
85. Schuchat A. Group B Streptococcus. Lancet (1999) 353(9146):51–6. doi:10.1016/S0140-6736(98)07128-1
86. Schuchat A, Zywicki SS, Dinsmoor MJ, Mercer B, Romaguera J, O’Sullivan MJ, et al. Risk factors and opportunities for prevention of early-onset neonatal sepsis: a multicenter case-control study. Pediatrics (2000) 105(1 Pt 1):21–6. doi:10.1542/peds.105.1.21
87. Oddie S, Embleton ND. Risk factors for early onset neonatal group B streptococcal sepsis: case-control study. BMJ (2002) 325(7359):308. doi:10.1136/bmj.325.7359.308
88. Baker CJ, Carey VJ, Rench MA, Edwards MS, Hillier SL, Kasper DL, et al. Maternal antibody at delivery protects neonates from early onset group B streptococcal disease. J Infect Dis (2014) 209(5):781–8. doi:10.1093/infdis/jit549
89. Carstensen H, Christensen KK, Grennert L, Persson K, Polberger S. Early-onset neonatal group B streptococcal septicaemia in siblings. J Infect (1988) 17(3):201–4. doi:10.1016/S0163-4453(88)96426-2
90. Carl MA, Ndao IM, Springman AC, Manning SD, Johnson JR, Johnston BD, et al. Sepsis from the gut: the enteric habitat of bacteria that cause late-onset neonatal bloodstream infections. Clin Infect Dis (2014) 58(9):1211–8. doi:10.1093/cid/ciu084
91. Heath PT, Balfour G, Weisner AM, Efstratiou A, Lamagni TL, Tighe H, et al. Group B streptococcal disease in UK and Irish infants younger than 90 days. Lancet (2004) 363(9405):292–4. doi:10.1016/S0140-6736(03)15389-5
92. Baker CJ, Barrett FF. Group B streptococcal infections in infants. The importance of the various serotypes. JAMA (1974) 230(8):1158–60. doi:10.1001/jama.230.8.1158
93. Alhhazmi A, Hurteau D, Tyrrell GJ. Epidemiology of invasive group B streptococcal disease in Alberta, Canada, from 2003 to 2013. J Clin Microbiol (2016) 54(7):1774–81. doi:10.1128/JCM.00355-16
94. Davies HD, Miller MA, Faro S, Gregson D, Kehl SC, Jordan JA. Multicenter study of a rapid molecular-based assay for the diagnosis of group B Streptococcus colonization in pregnant women. Clin Infect Dis (2004) 39(8):1129–35. doi:10.1086/424518
95. Jolley KA, Maiden MC. BIGSdb: scalable analysis of bacterial genome variation at the population level. BMC Bioinformatics (2010) 11:595. doi:10.1186/1471-2105-11-595
96. Imperi M, Gherardi G, Berardi A, Baldassarri L, Pataracchia M, Dicuonzo G, et al. Invasive neonatal GBS infections from an area-based surveillance study in Italy. Clin Microbiol Infect (2011) 17(12):1834–9. doi:10.1111/j.1469-0691.2011.03479.x
97. Da Cunha V, Davies MR, Douarre PE, Rosinski-Chupin I, Margarit I, Spinali S, et al. Streptococcus agalactiae clones infecting humans were selected and fixed through the extensive use of tetracycline. Nat Commun (2014) 5:4544. doi:10.1038/ncomms5544
98. Teatero S, Ramoutar E, McGeer A, Li A, Melano RG, Wasserscheid J, et al. Clonal Complex 17 Group B Streptococcus strains causing invasive disease in neonates and adults originate from the same genetic pool. Sci Rep (2016) 6:20047. doi:10.1038/srep20047
99. Tazi A, Bellais S, Tardieux I, Dramsi S, Trieu-Cuot P, Poyart C. Group B Streptococcus surface proteins as major determinants for meningeal tropism. Curr Opin Microbiol (2012) 15(1):44–9. doi:10.1016/j.mib.2011.12.002
100. Henneke P, Takeuchi O, Malley R, Lien E, Ingalls RR, Freeman MW, et al. Cellular activation, phagocytosis, and bactericidal activity against group B Streptococcus involve parallel myeloid differentiation factor 88-dependent and independent signaling pathways. J Immunol (2002) 169(7):3970–7. doi:10.4049/jimmunol.169.7.3970
101. Mancuso G, Midiri A, Beninati C, Biondo C, Galbo R, Akira S, et al. Dual role of TLR2 and myeloid differentiation factor 88 in a mouse model of invasive group B streptococcal disease. J Immunol (2004) 172(10):6324–9. doi:10.4049/jimmunol.172.10.6324
102. Henneke P, Morath S, Uematsu S, Weichert S, Pfitzenmaier M, Takeuchi O, et al. Role of lipoteichoic acid in the phagocyte response to group B Streptococcus. J Immunol (2005) 174(10):6449–55. doi:10.4049/jimmunol.174.10.6449
103. Draper DW, Bethea HN, He YW. Toll-like receptor 2-dependent and -independent activation of macrophages by group B streptococci. Immunol Lett (2006) 102(2):202–14. doi:10.1016/j.imlet.2005.09.005
104. Lehnardt S, Henneke P, Lien E, Kasper DL, Volpe JJ, Bechmann I, et al. A mechanism for neurodegeneration induced by group B streptococci through activation of the TLR2/MyD88 pathway in microglia. J Immunol (2006) 177(1):583–92. doi:10.4049/jimmunol.177.1.583
105. Asplin IR, Carl DJ, Way SS, Jones AL. Role of Toll-like receptor 2 in innate resistance to group B Streptococcus. Microb Pathog (2008) 44(1):43–51. doi:10.1016/j.micpath.2007.08.001
106. Puliti M, Uematsu S, Akira S, Bistoni F, Tissi L. Toll-like receptor 2 deficiency is associated with enhanced severity of group B streptococcal disease. Infect Immun (2009) 77(4):1524–31. doi:10.1128/IAI.00965-08
107. Xiao N, Eidenschenk C, Krebs P, Brandl K, Blasius AL, Xia Y, et al. The Tpl2 mutation Sluggish impairs type I IFN production and increases susceptibility to group B streptococcal disease. J Immunol (2009) 183(12):7975–83. doi:10.4049/jimmunol.0902718
108. Costa A, Gupta R, Signorino G, Malara A, Cardile F, Biondo C, et al. Activation of the NLRP3 inflammasome by group B streptococci. J Immunol (2012) 188(4):1953–60. doi:10.4049/jimmunol.1102543
109. Biondo C, Mancuso G, Midiri A, Signorino G, Domina M, Lanza Cariccio V, et al. Essential role of interleukin-1 signaling in host defenses against group B Streptococcus. MBio (2014) 5(5):e1428–1414. doi:10.1128/mBio.01428-14
110. Teti G, Mancuso G, Tomasello F. Cytokine appearance and effects of anti-tumor necrosis factor alpha antibodies in a neonatal rat model of group B streptococcal infection. Infect Immun (1993) 61(1):227–35.
111. Vallette JD Jr, Goldberg RN, Suguihara C, Del Moral T, Martinez O, Lin J, et al. Effect of an interleukin-1 receptor antagonist on the hemodynamic manifestations of group B streptococcal sepsis. Pediatr Res (1995) 38(5):704–8. doi:10.1203/00006450-199511000-00012
112. Andrade EB, Alves J, Madureira P, Oliveira L, Ribeiro A, Cordeiro-da-Silva A, et al. TLR2-induced IL-10 production impairs neutrophil recruitment to infected tissues during neonatal bacterial sepsis. J Immunol (2013) 191(9):4759–68. doi:10.4049/jimmunol.1301752
113. Mancuso G, Tomasello F, Migliardo M, Delfino D, Cochran J, Cook JA, et al. Beneficial effects of interleukin-6 in neonatal mouse models of group B streptococcal disease. Infect Immun (1994) 62(11):4997–5002.
114. Cusumano V, Genovese F, Mancuso G, Carbone M, Fera MT, Teti G. Interleukin-10 protects neonatal mice from lethal group B streptococcal infection. Infect Immun (1996) 64(7):2850–2.
115. Mancuso G, Cusumano V, Genovese F, Gambuzza M, Beninati C, Teti G. Role of interleukin 12 in experimental neonatal sepsis caused by group B streptococci. Infect Immun (1997) 65(9):3731–5.
116. Cusumano V, Midiri A, Cusumano VV, Bellantoni A, De Sossi G, Teti G, et al. Interleukin-18 is an essential element in host resistance to experimental group B streptococcal disease in neonates. Infect Immun (2004) 72(1):295–300. doi:10.1128/IAI.72.1.295-300.2004
117. Zhang SY, Jouanguy E, Ugolini S, Smahi A, Elain G, Romero P, et al. TLR3 deficiency in patients with herpes simplex encephalitis. Science (2007) 317(5844):1522–7. doi:10.1126/science.1139522
118. Ali SR, Fong JJ, Carlin AF, Busch TD, Linden R, Angata T, et al. Siglec-5 and Siglec-14 are polymorphic paired receptors that modulate neutrophil and amnion signaling responses to group B Streptococcus. J Exp Med (2014) 211(6):1231–42. doi:10.1084/jem.20131853
119. Davies HD, Adair C, McGeer A, Ma D, Robertson S, Mucenski M, et al. Antibodies to capsular polysaccharides of group B Streptococcus in pregnant Canadian women: relationship to colonization status and infection in the neonate. J Infect Dis (2001) 184(3):285–91. doi:10.1086/322029
120. Dangor Z, Kwatra G, Izu A, Lala SG, Madhi SA. Review on the association of Group B Streptococcus capsular antibody and protection against invasive disease in infants. Expert Rev Vaccines (2015) 14(1):135–49. doi:10.1586/14760584.2014.953939
121. Ekelund K, Konradsen HB. Invasive group B streptococcal disease in infants: a 19-year nationwide study. Serotype distribution, incidence and recurrent infection. Epidemiol Infect (2004) 132(6):1083–90. doi:10.1017/S0950268804002808
122. Green PA, Singh KV, Murray BE, Baker CJ. Recurrent group B streptococcal infections in infants: clinical and microbiologic aspects. J Pediatr (1994) 125(6 Pt 1):931–8. doi:10.1016/S0022-3476(05)82012-8
123. Moylett EH, Fernandez M, Rench MA, Hickman ME, Baker CJ. A 5-year review of recurrent group B streptococcal disease: lessons from twin infants. Clin Infect Dis (2000) 30(2):282–7. doi:10.1086/313655
124. Kotiw M, Zhang GW, Daggard G, Reiss-Levy E, Tapsall JW, Numa A. Late-onset and recurrent neonatal Group B streptococcal disease associated with breast-milk transmission. Pediatr Dev Pathol (2003) 6(3):251–6. doi:10.1007/s10024-001-0276-y
125. Jawa G, Hussain Z, da Silva O. Recurrent late-onset group B Streptococcus sepsis in a preterm infant acquired by expressed breastmilk transmission: a case report. Breastfeed Med (2013) 8(1):134–6. doi:10.1089/bfm.2012.0016
126. Lim HK, Seppanen M, Hautala T, Ciancanelli MJ, Itan Y, Lafaille FG, et al. TLR3 deficiency in herpes simplex encephalitis: high allelic heterogeneity and recurrence risk. Neurology (2014) 83(21):1888–97. doi:10.1212/WNL.0000000000000999
127. Doffinger R, Helbert MR, Barcenas-Morales G, Yang K, Dupuis S, Ceron-Gutierrez L, et al. Autoantibodies to interferon-gamma in a patient with selective susceptibility to mycobacterial infection and organ-specific autoimmunity. Clin Infect Dis (2004) 38(1):e10–4. doi:10.1086/380453
128. Puel A, Picard C, Lorrot M, Pons C, Chrabieh M, Lorenzo L, et al. Recurrent staphylococcal cellulitis and subcutaneous abscesses in a child with autoantibodies against IL-6. J Immunol (2008) 180(1):647–54. doi:10.4049/jimmunol.180.1.647
129. Browne SK, Holland SM. Anticytokine autoantibodies in infectious diseases: pathogenesis and mechanisms. Lancet Infect Dis (2010) 10(12):875–85. doi:10.1016/S1473-3099(10)70196-1
130. Puel A, Doffinger R, Natividad A, Chrabieh M, Barcenas-Morales G, Picard C, et al. Autoantibodies against IL-17A, IL-17F, and IL-22 in patients with chronic mucocutaneous candidiasis and autoimmune polyendocrine syndrome type I. J Exp Med (2010) 207(2):291–7. doi:10.1084/jem.20091983
131. Bousfiha A, Jeddane L, Al-Herz W, Ailal F, Casanova JL, Chatila T, et al. The 2015 IUIS phenotypic classification for primary immunodeficiencies. J Clin Immunol (2015) 35(8):727–38. doi:10.1007/s10875-015-0198-5
132. Saint-Faust M, Perelman S, Dupont D, Velin P, Chatel M. Transient neonatal myasthenia gravis revealing a myasthenia gravis and a systemic lupus erythematosus in the mother: case report and review of the literature. Am J Perinatol (2010) 27(2):107–10. doi:10.1055/s-0029-1224873
133. Gruters A, Krude H. Detection and treatment of congenital hypothyroidism. Nat Rev Endocrinol (2012) 8(2):104–13. doi:10.1038/nrendo.2011.160
134. Peterson JA, McFarland JG, Curtis BR, Aster RH. Neonatal alloimmune thrombocytopenia: pathogenesis, diagnosis and management. Br J Haematol (2013) 161(1):3–14. doi:10.1111/bjh.12235
135. Peart E, Clowse ME. Systemic lupus erythematosus and pregnancy outcomes: an update and review of the literature. Curr Opin Rheumatol (2014) 26(2):118–23. doi:10.1097/BOR.0000000000000030
136. Lewin S, Bussel JB. Review of fetal and neonatal immune cytopenias. Clin Adv Hematol Oncol (2015) 13(1):35–43.
137. Fasano RM. Hemolytic disease of the fetus and newborn in the molecular era. Semin Fetal Neonatal Med (2016) 21(1):28–34. doi:10.1016/j.siny.2015.10.006
138. Pugni L, Ronchi A, Bizzarri B, Consonni D, Pietrasanta C, Ghirardi B, et al. Exchange transfusion in the treatment of neonatal septic shock: a ten-year experience in a neonatal intensive care unit. Int J Mol Sci (2016) 17(5):695. doi:10.3390/ijms17050695
139. Esposito S, Zampiero A, Pugni L, Tabano S, Pelucchi C, Ghirardi B, et al. Genetic polymorphisms and sepsis in premature neonates. PLoS One (2014) 9(7):e101248. doi:10.1371/journal.pone.0101248
140. Watchko JF, Tiribelli C. Bilirubin-induced neurologic damage – mechanisms and management approaches. N Engl J Med (2013) 369(21):2021–30. doi:10.1056/NEJMra1308124
141. Bekker V, Bijlsma MW, van de Beek D, Kuijpers TW, van der Ende A. Incidence of invasive group B streptococcal disease and pathogen genotype distribution in newborn babies in the Netherlands over 25 years: a nationwide surveillance study. Lancet Infect Dis (2014) 14(11):1083–9. doi:10.1016/S1473-3099(14)70919-3
Keywords: newborn infant, life-threatening, primary immunodeficiency, genetic predisposition to disease, Mendelian diseases, monogenic, infection, group B streptococcus
Citation: Borghesi A, Stronati M and Fellay J (2017) Neonatal Group B Streptococcal Disease in Otherwise Healthy Infants: Failure of Specific Neonatal Immune Responses. Front. Immunol. 8:215. doi: 10.3389/fimmu.2017.00215
Received: 29 November 2016; Accepted: 15 February 2017;
Published: 07 March 2017
Edited by:
Kirsty Le Doare, Imperial College London, UKReviewed by:
Manish Sadarangani, University of British Columbia, CanadaCopyright: © 2017 Borghesi, Stronati and Fellay. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Alessandro Borghesi, YWxlc3NhbmRyby5ib3JnaGVzaUBlcGZsLmNo
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.