 Robert E. Brown
Robert E. Brown Robert L. Hunter
Robert L. Hunter Shen-An Hwang
Shen-An Hwang
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
PERSPECTIVE article
Front. Immunol., 02 February 2017
Sec. Vaccines and Molecular Therapeutics
Volume 8 - 2017 | https://doi.org/10.3389/fimmu.2017.00078
This article is part of the Research TopicTargeting host immunity for treatment of tuberculosisView all 8 articles
In an effort to develop more effective therapy for tuberculosis (TB), research efforts are looking toward host-directed therapy, reprograming the body’s natural defenses to better control the infection. While significant progress is being made, the efforts are limited by lack of understanding of the pathology and pathogenesis of adult type TB disease. We have recently published evidence that the developing lesions in human lungs are focal endogenous lipid pneumonia that constitutes a region of local susceptibility in a person with strong systemic immunity. Since most such lesions regress spontaneously, the ability to study them directly with immunohistochemistry provides means to investigate why some progress to clinical disease while others asymptomatically regress. Furthermore, this should enable us to develop more effective host-directed therapies. Morphoproteomics has proven to be an effective means of characterizing protein expression that can be used to identify metabolic pathways, which can lead to more effective therapies. The purpose of this perspective will argue that using morphoproteomics on human TB lung tissue is a particularly promising method to direct selection of host-directed therapeutics.
Progression of pulmonary tuberculosis (TB) in adults is a rare phenomenon in that at least 90% of cases regress spontaneously without producing clinical disease. Very little is known of why and how infection progresses to clinical disease in some people despite spontaneously regression in most. What is known is that TB disease is multifaceted, involving not just the actions of the pathogen Mycobacterium tuberculosis (MTB) on the host but also various immune mechanisms in response to bacterial antigens. TB disease is a chronic infection in immune competent hosts, displaying different pathologies, often simultaneously, in microenvironments in the same infected tissue, mostly in the lung (1–3). Protection from and progression to TB disease involves similar immune responses (4–6), and ongoing studies are trying to tease apart these differences.
There is no question that host immune responses play crucial roles in disease progression and transmission, but currently no therapeutic has been developed to suppress the immune induced pathology. Such host-directed therapy is routinely used and invested heavily in research in cancer (7–12), autoimmune (13–15), inflammatory (16), and other immune based diseases. Recently, immune directed therapy has been proposed and demonstrated to be potentially effective in TB disease (17–20). In order for this therapy to be effective, correct identification of critical host immune targets is paramount. This paper discusses newly developed means of studying host responses important for progression of pulmonary TB disease.
Host-directed therapy targets pathological mechanisms, either by shutting down pathways or manipulating immune responses to improve protection against the MTB pathogen. Proper identification of these pathological targets is crucial for the effectiveness of any host-directed therapy. Many pathological mechanisms of TB overlap with other immune-based diseases, providing TB researchers with a vast foundation of commercially available drugs (17) that have demonstrated protective responses in TB models. The use of in vitro and in vivo models to tease apart mechanistic parameters of diseases may be useful but may not adequately represent the human disease. Thus, targets identified through TB models may not be effective in the human patient. The best method to select effective targets for host-directed therapy for TB disease is by studying the human patient.
Mycobacterium tuberculosis is an obligate human pathogen since only humans develop cavities able to expel large numbers of organisms into the environment to infect new hosts (21–24). In order to eliminate TB disease, MTB transmission must be stopped by attenuating the caseation pathology. One key feature of caseation is that it occurs in localized pulmonary sites. Most people retain a high level of immunity in every part of their bodies except in localized pulmonary lesions. These lesions are areas of localized susceptibility that coexists with systemic immunity. Understanding the host mechanisms at these localized lesions that lead to susceptibility of MTB infection is hampered by the lack of access to appropriate clinical samples.
Since human tissues have not been available to most investigators since the introduction of antibiotics in the 1950s current descriptions of human pulmonary TB are based on animal models. While there are many animal models of TB, none of them develop pulmonary TB like humans. Consequently, some features of the pathology of human pulmonary TB have been largely forgotten. Through an extended study of human tuberculous tissues and relevant literature, we have formulated a corrected understanding of the pathology of human pulmonary TB and a new paradigm of its pathogenesis, reviewed extensively elsewhere (22, 25). The key finding is that pulmonary TB has a prolonged period of asymptomatic infection of alveolar macrophages in particular parts of the lung before the onset of clinical disease. This results from a localized susceptibility in parts of a lung in an otherwise immune person. A better understanding of how and why most of these lesions regress, while others progress to clinical disease might suggest ways to make them all regress and thereby eliminate TB.
Currently, most clinical samples from TB patients are either blood or lung. The former examine responses and mechanisms that are systemic, not at the site of infection, and may represent secondary effects of the primary response in the lung tissue. Alveolar lavages and lung biopsies are limited in the information they can provide: lavages are limited to immune responses in the alveolar and biopsies samples are too small to incorporate the surrounding parenchyma. Our group has taken the approach that in order to understand TB pathology we must study lung samples taken from untreated or under-treated TB individuals. Successful antibiotic treatment is known to significantly alter pathology (26–30). As MTB is killed by the antibiotic, the antigens that stimulate the host immune responses that generate the lung pathology are cleared. We propose that autopsy samples obtained from these untreated individuals may be the key to understanding the mechanisms of TB disease pathogenesis, especially the caseation process. Correct identification of key factors engaged in the caseation process will allow design of therapy directed toward controlling and ultimately stopping the pathology and arresting transmission of MTB.
There are several methods to identify pathological factors in lung tissue of TB patients. Our strategy has always focused on protein expression, as RNA expression may not necessarily result in changes in protein level due to post translational regulation. Additionally, we do not think that global proteomics will be useful due to the nature of MTB microenvironments. Within a single patient, MTB infection creates microenvironments with varying degree of pathology. In our samples, we often observed, in a single tissue section, areas of lipid pneumonia, matured cavities, developing cavities, fibrosis, caseation, and normal lung parenchyma. Each of these microenvironments has a different profile of immune responses, thus global proteomics will be unable to tease apart the critical targets of caseation. Our group proposes that the best method to identify effective targets for host-directed therapy is through the use of morphoproteomics, analyzing protein expression profile of specific pathological microenvironments.
Morphoproteomics is defined “as the identification by immunohistochemistry of the molecular circuitry of various proteins…by noting their state of activation (translocation and phosphorylation) and correlative expressions” (31). The method was originally developed for cancer patients, as tumors are often heterogeneous and was hypothesized to be more responsive to individualized guided therapy (32) as opposed to generalized standard protocols. Since this proposed method was developed in 2004 by Robert Brown, several publications have been peer reviewed and reported as to its effectiveness. A search in PubMed has yielded 37 publications. The majority of these used morphoproteomics to identify potential targets for adjuvant host-directed therapy (33–43) for an extensive list of cancers, such as prostate cancer, head and neck squamous cell carcinoma, Kaposi’s sarcoma, Hodgkin lymphoma, and others. Several publications have also indicated the clinical effectiveness of using morphoproteomics to guide host-directed therapy using commercially available drugs, including glioblastoma (44), osteosarcoma (45), pediatric brain tumors (46), and others. The success of morphoproteomic-guided therapy in cancer indicates that this method can be applied to other diseases where there is heterogeneous pathology and the host response directly causes the disease pathogenesis. Though morphoproteomics, we are able to identify cell types and characterize pathways in isolated lesions in human lungs. This manuscript reports recent findings and suggests future studies to investigate this key aspect of TB that takes place only in human lungs.
We propose that the heterogeneity of TB disease and the critical roles that the host response plays in the disease pathogenesis strongly indicate that morphoproteomic-guided host-directed therapy can be an effective tool to identify drugs with high possibility of ameliorating TB induced pathology. We believe that the future of host-directed therapy is to verify that pathology mechanisms identified in in vitro and/or animal models do occur in the human disease but also to demonstrate that the selected target(s) will affect critical pathology. From our extensive studies of human TB pathology, we hypothesize that foamy alveolar macrophages (obstructive lipid pneumonia) are the critical pathology directly responsible for the development of cavities (25). Thus, modulation of these pathologic macrophages may affect progression of pathology, the eventual cavitation, and stop the transmission process. As an example of how morphoproteomic-guided host-directed therapy can be applied, we decided to focus initially on two mechanisms of how MTB controls the host macrophage responses to promote its survival: mammalian target of rapamycin (mTOR) and cyclooxygenase 2 (COX-2) pathways.
Mycobacterium tuberculosis has evolved to escape host cell killing by preventing phagosome maturation into an acidic vesicle, the phagolysosome. Recent discoveries found that activation of autophagy through inhibition of mTOR can stimulate a double-membrane autophagosome that is capable of killing intracellular MTB (47, 48). The mTOR protein can bind other proteins to form two distinct complexes: mTORC1 (raptor-associated) that is sensitive to rapamycin and mTORC2 (rictor-associated) that is insensitive to rapamycin. In the context of MTB infection (both mouse and human studies), only mTORC1 has shown to be associated with TB disease. Since then, several animal studies have investigated the effectiveness of using rapamycin to inhibit mTOR as adjunct therapy (49–51) or for vaccination (52, 53). We completed preliminary staining of a clinical lung sample from a female who died suddenly at home. Diagnosis of TB disease was done at autopsy. No history of TB treatment was noted. Pathological analysis demonstrated the presence of foamy macrophages in alveolar spaces. We chose to stain for three markers of mTOR signaling (Figure 1). Foamy macrophages are heavily positive for expression of activated, pmTOR, phosphorylated (p) on serine 2448. Additionally, pmTOR is also positive in the alveolar walls, but to a lesser intensity. The second marker examined is the expression of insulin-like growth factor-1 receptor (IGF-1R), a strong inducer of mTOR via PI3K. Presence of IGF-1R is expressed not only in foamy macrophages but also in the surrounding parenchyma. The third marker is activated Akt (pAkt, on serine 473), the putative downstream effector of mTORC2 (54–57). We observed minimal presence of pAkt in foamy macrophages, suggesting that during MTB infection foamy macrophages are overexpressing mTORC1 and little to no activation of mTORC2. Activation of mTORC1 causes a negative feedback that decreases pAkt (58). This preliminary study suggests that foamy macrophages in this MTB-infected lung tissue over activate mTORC1, inhibiting autophagy of the infected cell and limiting MTB killing.
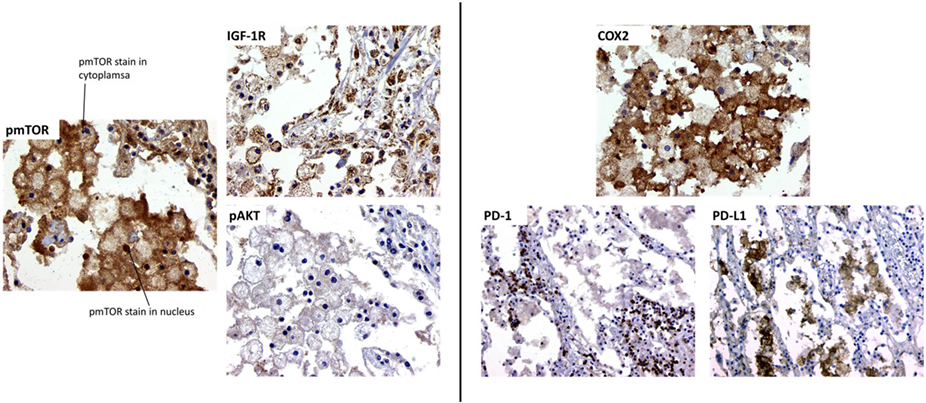
Figure 1. Morphoproteomic analysis of human TB lung sample. Left: stain for phosphorylated mTOR, insulin-like growth factor-1 receptor (IGF-1R), and phosphorylated Akt at 400× magnification. Right: sample stained with anti-human cyclooxygenase 2 (COX-2) and visualized at 400×. Programed death-1 (PD-1) and programed death-1 ligand (PD-L1) stain, magnification at 200×.
We also examined a second pathway of macrophage activity, COX-2. All studies of macrophage cultures suggest that MTB infection inhibit COX-2 activation and production of prostaglandin E2 (PGE2), leading to necrosis of the MTB-infected cell and MTB escape and spread of infection (59, 60). However, the effect of COX-2 activation in the in vivo lung local environment during MTB infection has not been well studied. Published reports on the cancer microenvironment often demonstrate that upregulation of COX-2 and PGE2 correlated to an increase in the presence and activity of T regulatory cells, which directly inhibited activity and function of effector T cells (61). Upregulation of T regulatory cells during active MTB infection blocks the ability of effector T cells to activate macrophages to control MTB infection (62), leading to loss of pathogen containment, uncontrolled proliferation, pathological inflammation, tissue necrosis, and spread of infection. Indeed, in the lungs of mice infected with MTB, COX-2 and PGE2 are overexpressed (63), suggesting that lung macrophage COX-2 activity may not reflect in vitro macrophage cultures studied. In this one MTB-infected lung sample, foamy macrophages varied in COX-2 intensity, indicating variability in the amount of COX-2 being expressed. Interestingly, COX-2 expression is mainly restricted to the foamy macrophage with nearly no COX-2 positivity in the alveolar walls (Figure 1).
In cancer studies, expression of COX-2 is associated with increase in T regulatory cells (64). T regulatory cell expansion in TB disease is associated with increases in expression of programed death-1 ligand (PD-L1) on antigen-presenting cells (65, 66). The expression of PD-L1 acts directly on programed death-1 (PD-1)-expressing T cells to inhibit their effector functions (67–70). In this MTB-infected lung microenvironment, PD-L1 is highly expressed in foamy macrophages, surrounded by PD-1-expressing lymphocytes in the interstitial (Figure 1). This suggests that foamy macrophages in this MTB-infected lung favor T effector cell suppression, possibly through macrophage COX-2 production. The increase in COX-2 in macrophages enhances surface expression of PD-L1, which ligates to PD-1, inhibiting activity of PD-1-expressing effector T cells. The increase in COX-2-producing macrophage may be due to increases in T regulatory cells in the MTB microenvironment, as previously observed (62). Thus, in this critical MTB microenvironment of foamy alveolar macrophages, two suppressor host response pathways are active (mTOR and COX-2), allowing TB disease progression.
Additional lung samples are currently undergoing the same morphoproteomic analysis. At the time of this manuscript preparation, four additional human TB lung samples have demonstrated the same pattern of mTOR and COX-2 staining in the alveolar macrophage pathological microenvironments (Hwang observations). With both mTOR and COX-2 mechanisms are potentially highly active in TB lungs, it seems logical to argue that designing host-directed therapy that would target both pathological pathways should offer the most success. There are several FDA-approved drugs that target mTOR and COX-2. Using commercially available products will enable clinical testing of the proposed therapy once proof-of-concept is established in appropriate animal models. We have identified two inhibitors that may offer the most effective outcome in reversing MTB-induced host pathological responses.
Metformin is an antidiabetic drug that activates adenosine monophosphate-activated kinase. In vitro analysis showed that Metformin directly reduced phosphorylation of mTOR and p70S6k, increasing apoptosis (71). Diabetic patients co-infected with MTB on metformin lived longer than those not taking metformin. In vivo mouse model studies demonstrated a significant decrease in lung bacterial load and pathology when treated with metformin (72). We believe metformin will inhibit overexpression of pmTOR and the expression of mTORC1 leading to decreased presence of foamy macrophages and increased autophagy and/or increased MTB-infected cell apoptosis, leading to observable decreases in bacterial load and lung pathology.
Celecoxib is a non-steroid anti-inflammatory drug that is a COX-2 inhibitor. However, celecoxib is capable of blocking several other proteins in the COX-2 signaling pathway and antiapoptotic proteins, such as Bcl-2 and Mcl-1 (73). We believe that using celecoxib during MTB disease will enhance apoptosis of foamy macrophages and increase effector T cell function, leading to decreased bacterial load and lung pathology, as observed by decreased clusters of foamy macrophages.
This is only one example of how morphoprotemics can aid in selection of host-targeted therapy. While human lung tissues have been previously investigated by immunohistochemistry, all these studies focused on MTB proteins (usually antigens) and/or host immune cell surface/secreted proteins (62, 74–77). Morphoproteomics is capable of identifying cell signaling pathways that are active in respect to specific pathological microenvironments, enabling understanding of the immunometabolism mechanisms that may be attractive targets for host-directed therapy (78). The application of routine morphoproteomic analysis for TB disease is still in its infancy due to the lack of appropriate human TB lung tissue and knowledgeable clinical pathologists. We are making progress toward creating a human TB pathology consortium that other researchers may access. Enabling researchers to verify their findings in the human patient is a must if we are to make significant breakthroughs in the future of TB research.
We offer the promise of an alternative strategy to developing new treatments for TB beyond just searching for effective antibiotics or choosing host-directed therapeutic targets from in vitro and/or animal models. Our approach is unique in that morphoproteomics directly analyzes pathological mechanisms in human tissue, allowing selection of targets for therapy that have been proven to be correlated with human disease. Additionally, morphoproteomics can also be used to tailor host-directed therapy to the individual patient, as it has been applied in cancer patients (32, 34, 45), if necessary. Since foamy alveolar macrophages are the lesion of TB that frequently undergoes spontaneous regression, we believe that studying it with morphoproteomics will identify the most promising targets for clinical testing and offer the highest chance of a positive outcome, to reduce or eliminate MTB transmission and reduce progression of the disease to the cavitary formation.
S-AH prepared the manuscript and did the final data analysis. RB performed the morphoproteomic analysis and help edit the manuscript. RH provided the pathology analysis and edit the manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer IO and handling Editor declared their shared affiliation, and the handling Editor states that the process nevertheless met the standards of a fair and objective review.
This work was supported by the UT Endowment Fund.
1. Chaves AS, Rodrigues MF, Mattos AM, Teixeira HC. Challenging Mycobacterium tuberculosis dormancy mechanisms and their immunodiagnostic potential. Braz J Infect Dis (2015) 19:636–42. doi: 10.1016/j.bjid.2015.08.004
2. Saunders BM, Britton WJ. Life and death in the granuloma: immunopathology of tuberculosis. Immunol Cell Biol (2007) 85:103–11. doi:10.1038/sj.icb.7100027
3. Vukmanovic-Stejic M, Reed JR, Lacy KE, Rustin MH, Akbar AN. Mantoux test as a model for a secondary immune response in humans. Immunol Lett (2006) 107:93–101. doi:10.1016/j.imlet.2006.08.002
4. Collins FM. Antituberculous immunity: new solutions to an old problem. Rev Infect Dis (1991) 13:940–50. doi:10.1093/clinids/13.5.940
5. Khan N, Vidyarthi A, Javed S, Agrewala JN. Innate immunity holding the flanks until reinforced by adaptive immunity against Mycobacterium tuberculosis infection. Front Microbiol (2016) 7:328. doi:10.3389/fmicb.2016.00328
6. Korb VC, Chuturgoon AA, Moodley D. Mycobacterium tuberculosis: manipulator of protective immunity. Int J Mol Sci (2016) 17:131. doi:10.3390/ijms17030131
7. Baxter D. Active and passive immunization for cancer. Hum Vaccin Immunother (2014) 10:2123–9. doi:10.4161/hv.29604
8. Hay MP, Hicks KO, Wang J. Hypoxia-directed drug strategies to target the tumor microenvironment. Adv Exp Med Biol (2014) 772:111–45. doi:10.1007/978-1-4614-5915-6_6
9. Jager K, Walter M. Therapeutic targeting of telomerase. Genes (Basel) (2016) 7:39–64. doi:10.3390/genes7070039.
10. Lechner MG, Russell SM, Bass RS, Epstein AL. Chemokines, costimulatory molecules and fusion proteins for the immunotherapy of solid tumors. Immunotherapy (2011) 3:1317–40. doi:10.2217/imt.11.115
11. Savas P, Hughes B, Solomon B. Targeted therapy in lung cancer: IPASS and beyond, keeping abreast of the explosion of targeted therapies for lung cancer. J Thorac Dis (2013) 5(Suppl 5):S579–92. doi:10.3978/j.issn.2072-1439.2013.08.52
12. Weitzenfeld P, Ben-Baruch A. The chemokine system, and its CCR5 and CXCR4 receptors, as potential targets for personalized therapy in cancer. Cancer Lett (2014) 352:36–53. doi:10.1016/j.canlet.2013.10.006
13. Chun J, Rosen H. Lysophospholipid receptors as potential drug targets in tissue transplantation and autoimmune diseases. Curr Pharm Des (2006) 12:161–71. doi:10.2174/138161206775193109
14. Dunne PJ, Fletcher JM. Recent advances in regulatory T cell therapy of autoimmunity, graft rejection and cancer. Recent Pat Inflamm Allergy Drug Discov (2010) 4:231–43. doi:10.2174/187221310793564182
15. Wang RF. Functional control of regulatory T cells and cancer immunotherapy. Semin Cancer Biol (2006) 16:106–14. doi:10.1016/j.semcancer.2005.11.004
16. Dinarello CA, van der Meer JW. Treating inflammation by blocking interleukin-1 in humans. Semin Immunol (2013) 25:469–84. doi:10.1016/j.smim.2013.10.008
17. Kaufmann SH, Lange C, Rao M, Balaji KN, Lotze M, Schito M, et al. Progress in tuberculosis vaccine development and host-directed therapies – a state of the art review. Lancet Respir Med (2014) 2:301–20. doi:10.1016/S2213-2600(14)70033-5
18. Zumla A, Rao M, Dodoo E, Maeurer M. Potential of immunomodulatory agents as adjunct host-directed therapies for multidrug-resistant tuberculosis. BMC Med (2016) 14:89. doi:10.1186/s12916-016-0635-1
19. Gupta A, Misra A, Deretic V. Targeted pulmonary delivery of inducers of host macrophage autophagy as a potential host-directed chemotherapy of tuberculosis. Adv Drug Deliv Rev (2016) 102:10–20. doi:10.1016/j.addr.2016.01.016
20. O’Connor G, Gleeson LE, Fagan-Murphy A, Cryan SA, O’Sullivan MP, Keane J. Sharpening nature’s tools for efficient tuberculosis control: a review of the potential role and development of host-directed therapies and strategies for targeted respiratory delivery. Adv Drug Deliv Rev (2016) 102:33–54. doi:10.1016/j.addr.2016.04.024
21. Hunter RL. Pathology of post primary tuberculosis of the lung: an illustrated critical review. Tuberculosis (Edinb) (2011) 91:497–509. doi:10.1016/j.tube.2011.03.007
22. Hunter RL, Actor JK, Hwang SA, Karev V, Jagannath C. Pathogenesis of post primary tuberculosis: immunity and hypersensitivity in the development of cavities. Ann Clin Lab Sci (2014) 44:365–87.
23. Kim MJ, Wainwright HC, Locketz M, Bekker LG, Walther GB, Dittrich C, et al. Caseation of human tuberculosis granulomas correlates with elevated host lipid metabolism. EMBO Mol Med (2010) 2:258–74. doi:10.1002/emmm.201000079
24. Mitchison DA, Davies GR. Assessment of the efficacy of new anti-tuberculosis drugs. Open Infect Dis J (2008) 2:59–76. doi:10.2174/1874279300802010059
25. Hunter RL. Tuberculosis as a three-act play: a new paradigm for the pathogenesis of pulmonary tuberculosis. Tuberculosis (Edinb) (2016) 97:8–17. doi:10.1016/j.tube.2015.11.010
26. Heo EY, Chun EJ, Lee CH, Kim YW, Han SK, Shim YS, et al. Radiographic improvement and its predictors in patients with pulmonary tuberculosis. Int J Infect Dis (2009) 13:e371–6. doi:10.1016/j.ijid.2009.01.007
27. Janols H, Abate E, Idh J, Senbeto M, Britton S, Alemu S, et al. Early treatment response evaluated by a clinical scoring system correlates with the prognosis of pulmonary tuberculosis patients in Ethiopia: a prospective follow-up study. Scand J Infect Dis (2012) 44:828–34. doi:10.3109/00365548.2012.694468
28. Jayasankar K, Shakila H, Umapathy KC, Ramanathan VD. Biochemical and histochemical changes pertaining to active and healed cutaneous tuberculosis. Br J Dermatol (2002) 146:977–82. doi:10.1046/j.1365-2133.2002.04784.x
29. Kim CH, Choi KJ, Yoo SS, Lee SY, Won DI, Lim JO, et al. Comparative analysis of whole-blood interferon-gamma and flow cytometry assays for detecting post-treatment immune responses in patients with active tuberculosis. Cytometry B Clin Cytom (2014) 86:236–43. doi:10.1002/cyto.b.21110
30. Qian X, Nguyen DT, Li Y, Lyu J, Graviss EA, Hu TY. Predictive value of serum bradykinin and desArg9-bradykinin levels for chemotherapeutic responses in active tuberculosis patients: a retrospective case series. Tuberculosis (Edinb) (2016) 101:S109–18. doi:10.1016/j.tube.2016.09.022
31. Brown RE, Bostrom B, Zhang PL. Morphoproteomics and bortezomib/dexamethasone-induced response in relapsed acute lymphoblastic leukemia. Ann Clin Lab Sci (2004) 34:203–5.
32. Brown RE. Morphogenomics and morphoproteomics: a role for anatomic pathology in personalized medicine. Arch Pathol Lab Med (2009) 133:568–79. doi:10.1043/1543-2165-133.4.568
33. Alexandrescu S, Tatevian N, Czerniak BA, Covinsky MH, Burns NK, Brown RE. Morphoproteomics provides support for TGF-beta pathway signaling in the osteoclastogenesis and immune dysregulation of osteolytic Langerhans cell histiocytosis. Int J Clin Exp Pathol (2012) 5:503–11.
34. Brown RE, Buryanek J, Katz AM, Paz K, Wolff JE. Alveolar rhabdomyosarcoma: morphoproteomics and personalized tumor graft testing further define the biology of PAX3-FKHR(FOXO1) subtype and provide targeted therapeutic options. Oncotarget (2016) 7:46263–72. doi:10.18632/oncotarget.10089
35. Brown RE, Buryanek J, Tammisetti VS, McGuire MF, Csencsits-Smith K. Morphoproteomics and biomedical analytics confirm the mTORC2/Akt pathway as a resistance signature and activated ERK and STAT3 as concomitant prosurvival/antiapoptotic pathways in metastatic renal cell carcinoma (RCC) progressing on rapalogs: pathogenesis. Oncotarget (2016) 7:41612–21. doi:10.18632/oncotarget.9508
36. Brown RE, Zhang PL, Lun M, Zhu S, Pellitteri PK, Riefkohl W, et al. Morphoproteomic and pharmacoproteomic rationale for mTOR effectors as therapeutic targets in head and neck squamous cell carcinoma. Ann Clin Lab Sci (2006) 36:273–82.
37. Brown RE, Zotalis G, Zhang PL, Zhao B. Morphoproteomic confirmation of a constitutively activated mTOR pathway in high grade prostatic intraepithelial neoplasia and prostate cancer. Int J Clin Exp Pathol (2008) 1:333–42.
38. Dhingra S, Rodriguez ME, Shen Q, Duan X, Stanton ML, Chen L, et al. Constitutive activation with overexpression of the mTORC2-phospholipase D1 pathway in uterine leiomyosarcoma and STUMP: morphoproteomic analysis with therapeutic implications. Int J Clin Exp Pathol (2010) 4:134–46.
39. Liu J, Brown RE. Morphoproteomics demonstrates activation of mTOR pathway in anaplastic thyroid carcinoma: a preliminary observation. Ann Clin Lab Sci (2010) 40:211–7.
40. Liu J, Brown RE. Morphoproteomic confirmation of an activated nuclear factor-small ka, CyrillicBp65 pathway in follicular thyroid carcinoma. Int J Clin Exp Pathol (2012) 5:216–23.
41. Quesada AE, Assylbekova B, Jabcuga CE, Zhang R, Covinsky M, Rios A, et al. Expression of Sirt1 and FoxP3 in classical Hodgkin lymphoma and tumor infiltrating lymphocytes: implications for immune dysregulation, prognosis and potential therapeutic targeting. Int J Clin Exp Pathol (2015) 8:13241–8.
42. Quesada AE, Nguyen ND, Rios A, Brown RE. Morphoproteomics identifies constitutive activation of the mTORC2/Akt and NF-kappaB pathways and expressions of IGF-1R, Sirt1, COX-2, and FASN in peripheral T-cell lymphomas: pathogenetic implications and therapeutic options. Int J Clin Exp Pathol (2014) 7:8732–9.
43. Shen Q, Stanton ML, Feng W, Rodriguez ME, Ramondetta L, Chen L, et al. Morphoproteomic analysis reveals an overexpressed and constitutively activated phospholipase D1-mTORC2 pathway in endometrial carcinoma. Int J Clin Exp Pathol (2010) 4:13–21.
44. Rios A, Hsu SH, Blanco A, Buryanek J, Day AL, McGuire MF, et al. Durable response of glioblastoma to adjuvant therapy consisting of temozolomide and a weekly dose of AMD3100 (plerixafor), a CXCR4 inhibitor, together with lapatinib, metformin and niacinamide. Oncoscience (2016) 3:156–63. doi:10.18632/oncoscience.311
45. Subbiah V, Wagner MJ, McGuire MF, Sarwari NM, Devarajan E, Lewis VO, et al. Personalized comprehensive molecular profiling of high risk osteosarcoma: implications and limitations for precision medicine. Oncotarget (2015) 6:40642–54. doi:10.18632/oncotarget.5841
46. Wolff JE, Brown RE, Buryanek J, Pfister S, Vats TS, Rytting ME. Preliminary experience with personalized and targeted therapy for pediatric brain tumors. Pediatr Blood Cancer (2012) 59:27–33. doi:10.1002/pbc.23402
47. Lam KK, Zheng X, Forestieri R, Balgi AD, Nodwell M, Vollett S, et al. Nitazoxanide stimulates autophagy and inhibits mTORC1 signaling and intracellular proliferation of Mycobacterium tuberculosis. PLoS Pathog (2012) 8:e1002691. doi:10.1371/journal.ppat.1002691
48. Petruccioli E, Romagnoli A, Corazzari M, Coccia EM, Butera O, Delogu G, et al. Specific T cells restore the autophagic flux inhibited by Mycobacterium tuberculosis in human primary macrophages. J Infect Dis (2012) 205:1425–35. doi:10.1093/infdis/jis226
49. Gupta A, Pant G, Mitra K, Madan J, Chourasia MK, Misra A. Inhalable particles containing rapamycin for induction of autophagy in macrophages infected with Mycobacterium tuberculosis. Mol Pharm (2014) 11:1201–7. doi:10.1021/mp4006563
50. Schiebler M, Brown K, Hegyi K, Newton SM, Renna M, Hepburn L, et al. Functional drug screening reveals anticonvulsants as enhancers of mTOR-independent autophagic killing of Mycobacterium tuberculosis through inositol depletion. EMBO Mol Med (2014) 7:127–39. doi:10.15252/emmm.201404137
51. Zullo AJ, Lee S. Mycobacterial induction of autophagy varies by species and occurs independently of mammalian target of rapamycin inhibition. J Biol Chem (2012) 287:12668–78. doi:10.1074/jbc.M111.320135
52. Jagannath C, Bakhru P. Rapamycin-induced enhancement of vaccine efficacy in mice. Methods Mol Biol (2012) 821:295–303. doi:10.1007/978-1-61779-430-8_18
53. Jagannath C, Lindsey DR, Dhandayuthapani S, Xu Y, Hunter RL Jr, Eissa NT. Autophagy enhances the efficacy of BCG vaccine by increasing peptide presentation in mouse dendritic cells. Nat Med (2009) 15:267–76. doi:10.1038/nm.1928
54. Harrington LS, Findlay GM, Gray A, Tolkacheva T, Wigfield S, Rebholz H, et al. The TSC1-2 tumor suppressor controls insulin-PI3K signaling via regulation of IRS proteins. J Cell Biol (2004) 166:213–23. doi:10.1083/jcb.200403069
55. Hsu PP, Kang SA, Rameseder J, Zhang Y, Ottina KA, Lim D, et al. The mTOR-regulated phosphoproteome reveals a mechanism of mTORC1-mediated inhibition of growth factor signaling. Science (2011) 332:1317–22. doi:10.1126/science.1199498
56. Yu Y, Yoon SO, Poulogiannis G, Yang Q, Ma XM, Villen J, et al. Phosphoproteomic analysis identifies Grb10 as an mTORC1 substrate that negatively regulates insulin signaling. Science (2011) 332:1322–6. doi:10.1126/science.1199484
57. Zhang H, Bajraszewski N, Wu E, Wang H, Moseman AP, Dabora SL, et al. PDGFRs are critical for PI3K/Akt activation and negatively regulated by mTOR. J Clin Invest (2007) 117:730–8. doi:10.1172/JCI28984
58. Sarbassov DD, Guertin DA, Ali SM, Sabatini DM. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science (2005) 307:1098–101. doi:10.1126/science.1106148
59. Nishimura T, Zhao X, Gan H, Koyasu S, Remold HG. The prostaglandin E2 receptor EP4 is integral to a positive feedback loop for prostaglandin E2 production in human macrophages infected with Mycobacterium tuberculosis. FASEB J (2013) 27:3827–36. doi:10.1096/fj.13-228858
60. Assis PA, Espindola MS, Paula-Silva FW, Rios WM, Pereira PA, Leao SC, et al. Mycobacterium tuberculosis expressing phospholipase C subverts PGE2 synthesis and induces necrosis in alveolar macrophages. BMC Microbiol (2014) 14:128. doi:10.1186/1471-2180-14-128
61. Whiteside TL. Disarming suppressor cells to improve immunotherapy. Cancer Immunol Immunother (2012) 61:283–8. doi:10.1007/s00262-011-1171-7
62. Welsh KJ, Risin SA, Actor JK, Hunter RL. Immunopathology of postprimary tuberculosis: increased T-regulatory cells and DEC-205-positive foamy macrophages in cavitary lesions. Clin Dev Immunol (2011) 2011:307631. doi:10.1155/2011/307631
63. Rangel Moreno J, Estrada Garcia I, De La Luz Garcia Hernandez M, Aguilar Leon D, Marquez R, Hernandez Pando R. The role of prostaglandin E2 in the immunopathogenesis of experimental pulmonary tuberculosis. Immunology (2002) 106:257–66. doi:10.1046/j.1365-2567.2002.01403.x
64. Jang TJ. Progressive increase of regulatory T cells and decrease of CD8+ T cells and CD8+ T cells/regulatory T cells ratio during colorectal cancer development. Korean J Pathol (2013) 47:443–51. doi:10.4132/KoreanJPathol.2013.47.5.443
65. McNab FW, Berry MP, Graham CM, Bloch SA, Oni T, Wilkinson KA, et al. Programmed death ligand 1 is over-expressed by neutrophils in the blood of patients with active tuberculosis. Eur J Immunol (2011) 41:1941–7. doi:10.1002/eji.201141421
66. Trinath J, Maddur MS, Kaveri SV, Balaji KN, Bayry J. Mycobacterium tuberculosis promotes regulatory T-cell expansion via induction of programmed death-1 ligand 1 (PD-L1, CD274) on dendritic cells. J Infect Dis (2012) 205:694–6. doi:10.1093/infdis/jir820
67. Jurado JO, Alvarez B, Pasquinelli V, Martinez GJ, Quiroga MF, Abbate E, et al. Programmed death (PD)-1:PD-ligand 1/PD-ligand 2 pathway inhibits T cell effector functions during human tuberculosis. J Immunol (2008) 181:116–25. doi:10.4049/jimmunol.181.1.116
68. Sakhno LV, Tikhonova MA, Tyrinova TV, Leplina OY, Shevela EY, Nikonov SD, et al. Cytotoxic activity of dendritic cells as a possible mechanism of negative regulation of T lymphocytes in pulmonary tuberculosis. Clin Dev Immunol (2012) 2012:628635. doi:10.1155/2012/628635
69. Singh A, Dey AB, Mohan A, Sharma PK, Mitra DK. Foxp3+ regulatory T cells among tuberculosis patients: impact on prognosis and restoration of antigen specific IFN-gamma producing T cells. PLoS One (2012) 7:e44728. doi:10.1371/journal.pone.0044728
70. Singh A, Mohan A, Dey AB, Mitra DK. Inhibiting the programmed death 1 pathway rescues Mycobacterium tuberculosis-specific interferon gamma-producing T cells from apoptosis in patients with pulmonary tuberculosis. J Infect Dis (2013) 208:603–15. doi:10.1093/infdis/jit206
71. Liu Y, Yang F, Ma W, Sun Q. Metformin inhibits proliferation and proinflammatory cytokines of human keratinocytes in vitro via mTOR-signaling pathway. Pharm Biol (2015) 54(7):1173–8. doi:10.1016/j.jpba.2014.11.001
72. Singhal A, Jie L, Kumar P, Hong GS, Leow MK, Paleja B, et al. Metformin as adjunct antituberculosis therapy. Sci Transl Med (2014) 6:263ra159. doi:10.1126/scitranslmed.3009885
73. Jendrossek V. Targeting apoptosis pathways by Celecoxib in cancer. Cancer Lett (2013) 332:313–24. doi:10.1016/j.canlet.2011.01.012
74. Ganbat D, Seehase S, Richter E, Vollmer E, Reiling N, Fellenberg K, et al. Mycobacteria infect different cell types in the human lung and cause species dependent cellular changes in infected cells. BMC Pulm Med (2016) 16:19. doi:10.1186/s12890-016-0185-5
75. Harriff MJ, Cansler ME, Toren KG, Canfield ET, Kwak S, Gold MC, et al. Human lung epithelial cells contain Mycobacterium tuberculosis in a late endosomal vacuole and are efficiently recognized by CD8(+) T cells. PLoS One (2014) 9:e97515. doi:10.1371/journal.pone.0097515
76. Mustafa T, Leversen NA, Sviland L, Wiker HG. Differential in vivo expression of mycobacterial antigens in Mycobacterium tuberculosis infected lungs and lymph node tissues. BMC Infect Dis (2014) 14:535. doi:10.1186/1471-2334-14-535
77. Purohit MR, Sviland L, Wiker H, Mustafa T. Rapid and specific diagnosis of extrapulmonary tuberculosis by immunostaining of tissues and aspirates with anti-MPT64. Appl Immunohistochem Mol Morphol (2016). doi:10.1097/PAI.0000000000000300
Keywords: morphoproteomics, tuberculosis, host-directed therapy, mTOR, COX-2
Citation: Brown RE, Hunter RL and Hwang S-A (2017) Morphoproteomic-Guided Host-Directed Therapy for Tuberculosis. Front. Immunol. 8:78. doi: 10.3389/fimmu.2017.00078
Received: 25 October 2016; Accepted: 17 January 2017;
Published: 02 February 2017
Edited by:
Jude Ezeh Uzonna, University of Manitoba, CanadaReviewed by:
Helen Alison Fletcher, London School of Hygiene & Tropical Medicine, UKCopyright: © 2017 Brown, Hunter and Hwang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Shen-An Hwang, c2hlbi1hbi5od2FuZ0B1dGgudG1jLmVkdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.