
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 19 January 2017
Sec. Molecular Innate Immunity
Volume 8 - 2017 | https://doi.org/10.3389/fimmu.2017.00014
This article is part of the Research Topic Immunoregulatory mechanisms of Interferon View all 30 articles
 Sasha Silva-Barrios
Sasha Silva-Barrios Simona Stäger*
Simona Stäger*
For many years, the role of interferon (IFN)-I has been characterized primarily in the context of viral infections. However, regulatory functions mediated by IFN-I have also been described against bacterial infections and in tumor immunology. Only recently, the interest in understanding the immune functions mediated by IFN-I has dramatically increased in the field of protozoan infections. In this review, we discuss the discrete role of IFN-I in the immune response against major protozoan infections: Plasmodium, Leishmania, Trypanosoma, and Toxoplasma.
Innate and adaptive immune responses are key factors in the control of infectious and chronic diseases; the balance between these two systems is mainly orchestrated by cytokines. Interferons (IFNs) are a large family of cytokines that were first discovered in 1957 in the context of viral infections. The name IFN is due to the capacity of these antiviral factors to interfere with viral replication in mammalian cells (1). Numerous studies have been carried out since their discovery, which allowed the identification of several related molecules. Based on their structural characteristics and the restricted affinity by the receptor molecule with which they directly interact, IFNs are classified into three main groups: type I (IFN-I), type II (IFN-II), and the recently identified type III (IFN-III) (2).
The IFN-I family includes two main classes of related cytokines: IFN-α, which comprises 13 different subtypes encoded by 13/14 different genes; and IFN-β, a product encoded by a single gene and a group of other less studied IFNs (IFN-ϵ, IFNδ, IFNκ, IFNτ, IFNω) (2). The ability to produce and respond to IFN-I is distributed in a wide variety of cells. This confers several autocrine and paracrine effects that have been extensively characterized mainly in viral infections. IFN-I signaling is mediated through a common cell surface receptor, the IFN-I receptor (IFNAR) (3, 4).
The IFN-II family is represented by a single gene product, IFN-γ, and is mainly produced by T lymphocytes and natural killer (NK) cells. IFN-II responses are mediated by the binding of IFN-γ to a heterodimeric molecule, the IFN-γ receptor (IFNGR), ubiquitously expressed in a wide range of cells. IFNGR is involved in the modulation of different cell functions and is a key factor for host defence to intracellular pathogens in various infection models (5).
Finally, the IFN-III family, also known as IFN-λ, comprises four different subtypes: IFN-λ1, IFN-λ2, IFN-λ3, and IFN-λ4. The members of this novel IFN family interact through a unique receptor, the IFN-λ receptor (IFN-λR). In contrast to IFNAR and IFNGR, the expression of IFN-λR is mainly restricted to cells of epithelial origins. The role of IFN-III has yet to be better characterized; however, they appear to induce similar responses to IFN-I (6).
The crosstalk between IFNs and their specific receptors elicits an intracellular signaling cascade that mainly enhances inflammatory responses. The well-characterized signaling cascades of IFN-I and IFN-II are fairly similar. In both cases, Janus kinase 1 (JAK1) and tyrosine kinase 2, associated with IFNAR and IFNGR, are activated. This results in activation and following formation of a heterodimer complex comprised by the cytoplasmic transcription factor signal transducer and activator of transcription 1 and 2 (STAT1/STAT2). STAT1/STAT2 dimers can be translocated to the nucleus and interact with the IFN regulatory factor 9 to form the IFN-stimulated gene factor 3 complex, leading to the transcription of IFN-stimulated genes (ISGs). By contrast, IFN-II signaling through IFNGR activates the JAK/STAT pathway leading to the transcription of pro-inflammatory targets downstream of γ-activated sequences (2, 7).
Interferon (IFN)-I production is mainly induced in response to the activation of receptors on the membrane and/or cytosol, such as pattern recognition receptors (PRRs). PRRs can be activated by conserved pathogens component and endogenous molecules. In most of the cases, the production of IFN-I is related to the activation of PRRs that recognize xenogeneic or autologous nucleic acid, such as toll-like receptors (TLRs) (8).
Interferon (IFN)-I is historically best known for their capacity to elicit antiviral responses; however, they also play a role in bacterial infections and autoimmune diseases (4). The role of IFN-I in regulating the immune response against pathogens is fairly complicated. IFN-I can have enhancing or suppressive effects depending on the disease, the stage of infection, and the amount produced. For instance, IFN-I enhances the antigen-presenting capacity of DCs (9–11), favors the development of T cell responses (12–14), and promotes antibody responses (15, 16) during acute viral infections. By contrast, type I IFNs play an immunosuppressive role during chronic viral infections (17–19), reduce IFN-γ responsiveness in macrophages (20, 21), block B cell functions at high concentrations (22, 23), and can promote the expression of immunosuppressive factors such as IL-10 and PDL-1 (24–27). This duality is also observed in the context of autoimmune diseases, where IFN-I plays a pathogenic role in systemic lupus erythematosus and Sjogren’s syndrome (28, 29), whereas it has therapeutic effects in multiple sclerosis (30).
While IFN-γ has been widely characterized in the modulation of the immune response against protozoan infections, the contribution of IFN-I to host defence against parasites is less clear. In the past few years, a growing body of literature suggests an important role for IFN-I during protozoan infections, particularly in the innate immune response.
In this review, we provide a brief overview of IFN-I mediated effects on the host response in various protozoan infection models and the possible mechanisms involved.
Interferon (IFN)-I is involved in the modulation of innate immune responses promoting antigen presentation and NK cell functions. They are also known to play a role in the regulation of the adaptive immune system, promoting the development of antigen-specific T and B lymphocytes against numerous pathogens and inducing immunological memory (7). In most of the cases, these key features are important factors that limit pathogen proliferation; however, IFN-I may also lead to disease exacerbation. Protozoan parasites such as Plasmodium, Leishmania, Trypanosoma, and Toxoplasma are causing diseases that are among the most lethal and widespread around the world, primarily affecting populations of developing countries. The contribution of IFN-I in the host immune response to these pathogens will be discussed below.
Plasmodium parasites are the causative agents of malaria, one of the most widespread diseases in the world. The infection presents itself in a wide range of pathologies that can degenerate into severe anemia and the high-risk cerebral malaria (CM). Members of the Plasmodium genus have a complex life cycle between an invertebrate (female mosquitoes of the Anopheles genus), in which the sexual cycle occurs, and a mammalian host. During the mosquito blood meal, sporozoites are inoculated into the dermis of the mammalian host. In the initial phase of infection, circulating sporozoites can reach lymph nodes, where the priming of B and T cells occurs, or migrate to the liver (31, 32). Within the liver, sporozoites transform first into schizonts within hepatocytes and then into merozoites. This phase is asymptomatic and is known as the pre-erytocytic stage (33). Merozoites are then released into the blood stream. Once they reach the blood, merozoites invade red blood cells, where they undergo cyclic asexual replication initiating the typical symptomatic manifestations of blood-stage malaria, which are caused by the exponential growth of the parasite and massive destruction of erythroid cells (34).
Most of the current knowledge about the immune response to Plasmodium parasites has been derived from a combination of in vitro and in vivo observations in human patients (e.g., P. falciparum, P. vivax, P. malariae, P. knowlesi, and P. ovale) and murine models of infections (e.g., P. berghei, P. yoelii, P. chabaudi, and P. vinckei) (34).
During the pre-erythrocytic stage, sporozoite invasion of hepatocytes and subsequent development into merozoites can be blocked by sporozoite-specific antibodies generated by previous exposure to malaria or by immunization; however, this stage is not completely efficient because sporozoites remain in circulation for a short period of time. When T cell priming takes place, infected hepatocytes can be eliminated by cytotoxic CD8 T cells. CD8 T cells, IFNγ, and TNF are critical components required for elimination of infected hepatocytes in humans and the mouse model (35). However, the immune response at this stage is insufficient and released merozoites can reach erythrocytes giving rise to blood-stage malaria (35).
In the erythrocytic stage, early interaction between merozoites and innate immune cells such as dendritic cells, monocytes, macrophages, NK cells, NKT cells, and γδT cells is important for the control of parasite replication and the resolution of infection (33). This phase is characterized by a strong pro-inflammatory response, mediated by the activation of NK, NKT, CD8, and CD4 T cells that produce large amounts of IFNγ and other pro-inflammatory cytokines. IFNγ activates phagocytic cells, such as macrophages, enhancing the secretion of pro-inflammatory cytokines and promoting phagocytosis of circulating parasites and infected red blood cells, which results in the control of parasitemia (36). Polyreactive and specific antibodies against blood-stage malaria can limit parasite propagation between erythrocytes by opsonization and agglutination of parasites and infected erythrocytes; however, humoral responses during the infection are dependent on the presence of circulating merozoites (37). Infected erythrocytes on the surface express parasitic protein which allows them to bind to vascular endothelial cells and avoid clearance. This event induces obstructions in the blood flow and is associated with a strong inflammatory response and the development of CM (33).
Although IFN-γ is the most extensively studied IFN in malaria infection, part of the attention has now been diverted to type I IFNs. IFN-I can have a host-protective or detrimental effect, depending on the stage of the infection or the species of Plasmodium involved.
One of the first reports involving type I IFNs demonstrated that administration of mouse serum containing high levels of IFN-I protected mice from P. berghei infection by reducing blood parasitemia (38). Similar protective responses were observed after treatment with IFN-β, which prevented death related to CM in P. berghei-infected mice (39). By contrast, treatment with recombinant IFN-α during the hepatic cycle in mice infected P. yoelii sporozoites did not alter the hepatic parasite burden. However, mice showed reduced parasitemia and decreased signs of immunopathology (40).
Plasmodium parasites were reported to induce IFN-I responses. Transcriptomic analysis carried out in mice with blood-stage infection with P. berghei revealed that IFN regulatory factors were upregulated during the acute phase (41). Induction of a typical type I IFN signature was also observed in hepatocytes from mice infected with P. berghei and P. chabaudi sporozoites, where genes such as Mda, Irf3, Irf7, and Stat1 were upregulated (42–44). Similar results were observed in humans. Patients infected with P. vivax and P. falciparum showed a predominantly IFN-I transcriptional signature during the mild and the severe phase of infection (44, 45).
Recently, Liehl et al. showed that induction of IFN-I during liver stages of the infection is required for host defence against P. berghei. Recognition of P. berghei nucleic acids by Mda5 induced IFN-I and consequently, the recruitment of leukocytes necessary for parasite elimination in the liver (42). In P. yoelii-infected mice, recruitment and expansion of CD49b+CD3+NKT and CD8+T cells to the liver were mediated by IFN-I signaling (43). Migration of neutrophils to the liver is also modulated by IFN-I in mice infected with P. chabaudi (44). These studies suggest that functionality of the innate immune response in the liver relies on both IFN-I and IFN-II.
In contrast to the protective effects discussed above, a pathogenic role for IFN-I in Plasmodium infections has also been described. For instance, impaired IFN-I signaling has been linked to a protective effect in human patients. Polymorphism in the human gene encoding for IFNAR1 are strongly associated with protection against CM (46). This observation is in agreement with results obtained in a murine model, where the lack of IFN-I signaling led to strong resistance to CM and reduced parasite load during P. berghei infection (47, 48). Moreover, in P. chabaudi-infected mice, IFN-I appear to suppress Th1 responses that are crucial in the control of hyperparasitemia, by modulating dendritic cell functions (49). In addition, IFN-I and Myd88 signaling are responsible for a decreased recruitment of conventional DCs to the spleen during experimental P. berghei or P. yoelii infection (50).
Perhaps a better approach for truly understanding the role and function of IFN-I during malaria consists in the identification of modulator molecules that could act in the IFN-I signaling cascade. Recently, regulators of IFN-I response have been identified through genome-wide analysis (Trans-species expression quantitative trait locus, ts-eQLT) during P. yoelii infection. Eight genes (Ak3, Fcγr1, Fosl1, Havcr2, Sipr5, Parp14, Selenbp2, and Helb) had an effect on IFN-I activation. For example, Fcγr1−/− mice infected with P. yoelii showed significantly higher mRNA and protein levels of IFN-β than wild-type mice, suggesting a negative regulation in the IFN-β response (51).
Future experiments are granted to clarify the spatio-temporal role of IFN-I during malaria.
The role of IFN-I during Plasmodium infections is summarized in Table 1.
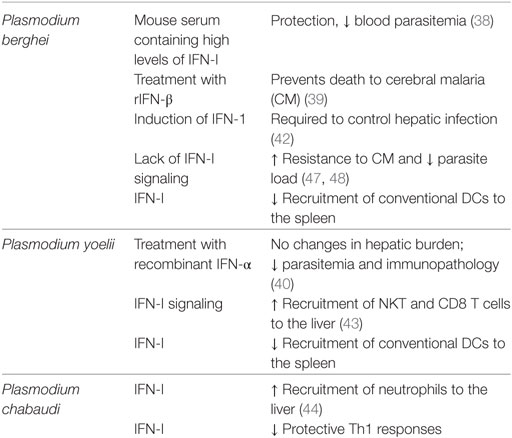
Table 1. Role of interferon (IFN)-I in Plasmodium infection.
Toxoplasma gondii is an obligate intracellular protozoan parasite that can infect a wide range of vertebrates and cause a zoonotic disease called toxoplasmosis. T. gondii could be considered one of the most successful parasites worldwide; at least 50% of the human population is infected with Toxoplasma. The parasite success is mainly due to its ability to invade any nucleated cell and to survive outside the mammalian host (52). T. gondii strains are classified in three main lineages, based on the virulence of the strain in the mouse model. This virulence profile does not necessarily correlate to the degree of human infection. Type I strains of T. gondii are the most virulent: less than 10 parasites are able to kill a mouse at the onset of infection. By contrast, type II and III strains are less virulent and lead to the establishment of chronic infection (53). T. gondii can undergo both asexual (schizogony) and sexual (gametogony) replication. Gametogony and oocyst formation is restricted to feline species that act as a definitive hosts; sexual reproduction of sporozoites occurs within intestinal epithelial cells. Asexual stages of T. gondii are not host-specific. Many mammals and birds can act as intermediate hosts. After ingestion of T. gondii oocysts by an intermediate host, the parasite transforms into tachyzoites that rapidly undergo multiplication within the parasitophorous vacuole inside various cell types. If the infection is controlled, parasites are retained in tissue cysts; if not, they can cause a systemic lethal disease (54, 55).
Humans are considered as an accidental intermediate host for Toxoplasma. In immune-competent individuals, the infection with T. gondii is mostly clinically silent, but cause severe diseases in immune-suppressed patients in particular with an impaired T cell and IFNγ response (55). Protective immunity is typically achieved by inducing an IL-12-driven Th1 immune response (56, 57).
In the mouse model, IFN-I can already be detected in the serum of T. gondii-infected animals during the acute phase (58–60); IFN-I levels gradually increase with the progression of the infection (60). IFN-I was also detected in the brain and spleen of infected mice (61). These results demonstrate that T. gondii not only induces IFN-γ, but also IFN-I.
Recently, inflammatory monocytes were identified as the major source of IFN-β in mesenteric lymph nodes. IFN-β production by inflammatory monocytes required three fundamental events: parasite internalization, TLR activation (mainly TLR4 and 2), and efficient MyD88 signaling. Interestingly, heat killed parasites induced higher levels of IFN-β in inflammatory monocytes (62), suggesting that Toxoplasma might limit IFN-I responses (62), possibly by blocking STAT1 (63).
As for many other infection models, the first studies carried out during the 1960s on the role of IFN-I in toxoplasmosis evaluated the impact of a treatment with IFN-I on infected cells in vitro. Pre-treatment of mouse fibroblast with IFNs conferred protection to T. gondii infection (64). In agreement with this observation, human neonatal and adult macrophages treated with IFN-I were able to control parasite multiplication, even if less effectively than IFN-γ treated cells (65). Moreover, human monocyte-derived macrophages treated with human IFN-β in combination with Escherichia coli lipopolysaccharides (LPS), but not with murine IFN-β (MuIFN-β) or rHuIFN-β alone (66), were more resistant to T. gondii infection (67).
In the mouse model of toxoplasmosis, treatment with HuIFN-β showed a protective effect, which was enhanced by the combination of rHuIFN-β and LPS and was IFN-γ dependent (66). In agreement with these results, it was shown that Ifnar−/− mice orally infected with T. gondii have an increased parasite load compared to wild-type mice; higher parasite burdens correlated with a decrease in survival (68).
These results suggest that IFN-β may be produced at the onset of infection to enhance the IFN-γ responses.
A study using human fibroblasts as host cells revealed that treatment of T. gondii-infected cells with IFN-I had no effect on parasite replication (69), suggesting that the protective effect of IFN-I depends on cell type and/or timing of exposure to the cytokine (prior to or after infection).
During T. gondii infection regulation of tryptophan metabolism is a key component for parasite survival. Indeed, tryptophan degradation inhibits parasite replication. In T. gondii-infected mice, indoleamine 2,3-dioxygenase (IDO), a tryptophan catalyzer (70, 71), is enhanced by IFN-II (72). However, it has also been reported that IFN-I can regulate IDO in human retinal pigment epithelial cells, inhibiting therefore T. gondii replication (73). Together, these results demonstrate that IFN-I also contribute to the regulation of protective immunity against T. gondii (Table 2).

Table 2. Role of interferon (IFN)-I in Toxoplasma infection.
Leishmania is a complex genus of obligate intracellular protozoan parasites that cause a widespread disease collectively known as Leishmaniasis. The life cycle of these parasites takes place between a mammalian host and a sandfly vector (genus Lutzomyia and Phlebotomus). Once in the hosts, the promastigote form of the parasite preferentially infects macrophages, but can also be found in other cell types, such as dendritic cells, neutrophils, and fibroblasts. Promastigotes then transform into the non-flagellated form called amastigotes within the host’s cell. The Leishmania spp. involved and the mammalian host immune status determine the clinical manifestation of the disease. Parasites can either reside in the skin and/or mucosal surfaces, which results in cutaneous (i.e., Leishmania major) or mucocutaneous (i.e., Leishmania braziliensis) Leishmaniasis; or disseminate to internal organs such as liver, spleen, and bone marrow, causing visceral Leishmaniasis (VL), the most severe form of the disease (i.e., Leishmania donovani) (74).
Leishmania immunity is mostly mediated by T lymphocytes. In experimental models, control of infection is mediated by a polarized Th1 response, induced by an initial production of IL-12 by DCs (75). IFN-γ secreting CD4 and CD8 T cells contribute to parasite control by enhancing the ability of phagocytic cells to kill intracellular Leishmania (74, 76).
As for many other protozoan models, IFN-II is the main mediators of the cellular immune response. However, IFN-I and IFN-I inducible genes are gradually gaining importance in the Leishmania field. One of the pioneer work on the role of IFN-I in Leishmaniasis described the prophylactic treatment with synthetic double-stranded RNA (Poly I:C) prior to L. donovani infection. Injection of Poly I:C triggered a burst of IFN-I and led to the control of the hepatic parasite burden (77). The role of endogenous IFN-I was studied for the first time using strains causing cutaneous Leishmaniasis. The induction of IFN-I was observed in macrophages infected in vitro with L. major promastigotes (78, 79) and in skin macrophages from infected animals (79), showing that promastigotes could enhance IFN-I expression in the host cell. The combination of exogenous IFN-I with L. major promastigotes was shown to activate macrophages, inducing type 2 nitric oxide synthase (NOS2). NOS2 is required for parasite elimination; mice deficient in this enzyme are more susceptible to L. major infection (80). As for T. gondii, the timing of the host cell’s exposure to IFN-I determines the effect on parasite control. Indeed, pre-treatment of macrophages with exogenous IFN-I failed to induce NOS2. Similar results were obtained with high doses of exogenous IFN-I, while a low IFN-I dose in combination with L. major enhanced leishmanicidal activity (78, 81). These results suggest that the design of in vitro experiments greatly influences the outcome of IFN-I treatment in infected macrophages and that the role of IFN-I should be better studied in in vivo models.
The protective role of endogenous IFN-I during infection was confirmed by neutralizing IFN-I in mice experimentally infected with L. major. In fact, IFN-I neutralization rendered L. major-infected mice more susceptible to infection and enhanced parasite multiplication. IFN-I blockade led to abolishment of NOS2 function and reduced cytotoxic activity and IFNγ production by NK cells at early stages of infection (79).
Opposite results were obtained in human macrophages infected in vitro with New World Leishmania spp. IFN-β treatment of L. braziliensis and Leishmania amazonensis-infected macrophages enhanced the parasite burden through a superoxide-dependent, NO-independent mechanism (82). In this model, it was shown that IFN-β can regulate the superoxide dismutase SOD1 activity. SOD1 is responsible for catalyzing the disproportionation of superoxide to hydrogen peroxide and dioxygen and is an important constituent in apoptotic signaling and oxidative stress. It has been observed that biopsies from cutaneous Leishmaniasis patients express high levels of SOD1 (82).
The importance of endogenous IFN-I during chronic infection has been investigated using IFNAR-deficient mice in the context of L. amazonensis infection. L. amazonensis infected Ifnar−/− mice developed attenuated cutaneous lesions and displayed a decreased parasite load. This effect appeared to be STAT1 independent, a key protein in the IFN signaling (83). Furthermore, L. amazonensis-infected Ifnar−/− mice exhibited high levels of neutrophils and lower inflammatory monocytes recruitment at early times post infection. This unique profile was also observed in L. major and L. braziliensis infections (83). In vitro coculture of infected WT macrophages with Ifnar−/− neutrophils revealed that IFNAR-deficient neutrophils promote parasite killing (83). This evidence supports the pathogenic role of IFN-I signaling in cutaneous Leishmaniasis caused by New World Leishmania species.
We also observed a negative role for IFN-I in an experimental model of VL. L. donovani amastigotes were shown to induce IFN-I expression in B cells in an endosomal TLR-dependent manner. This cytokine was involved in a positive regulatory loop that led to the upregulation of endosomal TLRs and to IL-10 production in B cells (84). B cell-derived IL-10 was shown to suppress protective T cells responses and increase disease susceptibility (85). B cells are known to play a detrimental role during VL (86), not only by secreting IL-10 but also for their excessive antibody production (87). Indeed, hypergammaglobulinemia is a hallmark of VL. Interestingly, IFN-I seems to be regulating antibody production during VL. Specific ablation of endosomal TLRs or IFN-I signaling in B cells was shown to severely reduce the Ig titer in the serum of L. donovani-infected mice, suggesting that parasite activation of B cells via endosomal TLRs and IFN-I are involved in the induction if hypergammaglobulinemia (84). Furthermore, mice with a B cell-specific deficiency in endosomal TLR or IFNAR were more resistant to L. donovani infection than their wild-type counterpart.
Very little is known about the function of IFN-I in VL patients. It was reported that human mononuclear phagocytes can be activated by IFN-β, but less efficiently than IFN-γ (88). Exogenous treatments with IFN-I and IFN-II but not IL-2, failed to restore the cytotoxic activity of NK isolated from VL patients (89). Also, treatment of the cutaneous lesion in patients with IFN-I did not improve healing, compared with IFN-γ treatment (72, 90).
Because dendritic cells can also be infected by Leishmania, it is important to consider the induction of IFN-I by the parasite and its possible effect in these cells as well. Transcriptomic analysis of human DCs infected in vitro with L. major or L. donovani showed a differential expression pattern for IL-12 associated genes, the NF-KB pathway, and IFN regulatory factors (91). IFN-β produced by L. major-infected DCs seems to be required for IL-12 secretion by the infected DC, suggesting that protective Th1 responses, which are IL-12 depended, may also depend on IFN-I (92).
A summary on the role of IFN-I during Leishmaniasis can be found in Table 3.
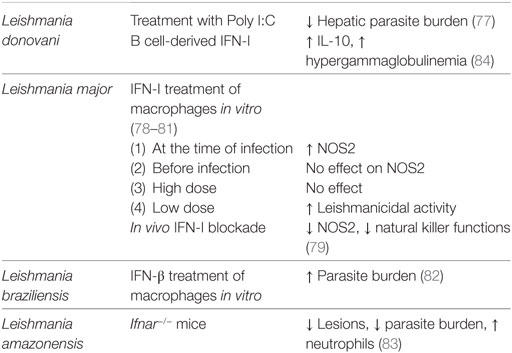
Table 3. Role of interferon (IFN)-I in Leishmania infection.
Trypanosomes are digenetic protozoan parasites that infect domestic and wild animals, as well humans. Although many species of trypanosomes cause important veterinary disease, mainly two species cause significant human morbidity: Trypanosoma brucei and Trypanosoma cruzi. These two species are responsible for causing the sleeping sickness (African trypanosomiasis) and the Chagas disease (American trypanosomiasis), respectively.
The life cycle of these parasites takes place between the invertebrate vector and the vertebrate host. T. brucei and other African’s trypanosomes are transmitted to the mammalian host by a tsetse fly bite. In the blood stream, metacyclic trypomastigotes differentiate into bloodstream trypomastigotes. In humans, trypanosomes proliferate in the blood and lymphatic system at early stages of the infection. This stage is associated with an anti-inflammatory response. At chronic stages, parasites can pass through the blood–brain barrier and enter the central nervous system. This stage is associated with inflammatory changes in the brain and is characterized by a neurological disturbance (93, 94).
In T. cruzi (American trypanosomiasis), metacyclic trypomastigotes are released in the feces/urine of the triatomine vector after a blood meal. Trypomastigotes can successfully infect the mammalian host if they are able to reach the mucosa or injured skin areas. In contrast to African trypanosomes, T. cruzi is an intracellular parasite that has the capacity to invade, differentiate into amastigotes, and replicate within a wide range of nucleated cells. This characteristic is one of the most important features of T. cruzi within the host. Amastigotes differentiate into infective bloodstream trypomastigotes, before being released upon cell lysis. The released parasites can then infect neighboring cells or enter the bloodstream (95).
During the acute phase, the innate immune response against T. cruzi is characterized by the induction of a cell-mediated response that involves the production of IFN-γ and TNF (by NK and T cells), required for enhancing iNOS activity by phagocytic cells and for priming the adaptive immune response. iNOS activation is critical for controlling parasite growth during the infection (95). T. cruzi elicits a prominent IFN-I response at early times of infection (96–99). As mentioned before for Plasmodium, the role of IFN-I in T. cruzi infection is controversial. Some studies ascribe a protective role to IFN-I; others demonstrate that IFN-I induces pathology. The effect of IFN-I mainly depends on the dose, amount of parasites, and the inoculation route used to set up the infection.
The first studies on the role of IFN-I investigated the outcome of exogenous IFN-I treatment in T. cruzi-infected mice. The results showed that administration of IFN-I increased resistance to infection by stimulating T and NK cell activities, which are essential for protection (100, 101).
In an intradermal model of infection, transcriptomic analysis of excised skin from the inoculation site revealed that T. cruzi upregulated the expression of ISGs as early as 24 h after infection. Induction of ISGs was dependent on IFN-I signaling, suggesting that IFN-I is an important component of the innate immune response to T. cruzi (99). In agreement with the above mentioned literature, studies carried out in Ifnar−/− mice infected with T. cruzi revealed that efficient IFN-I signaling was required for controlling parasites growth during the acute phase of infection (102, 103). IFN-I was necessary for enhancing NO production in phagocytic cells (102). NO is considered the major effector molecule for intracellular amastigotes elimination within infected cells, being important for the control of parasite multiplication (95).
By contrast, another group reported a potential pathogenic role for IFN-I. In this work, a lethal dose of parasites inoculated intradermally was used to set up the infection in WT and Ifnar−/− mice. Surprisingly, T. cruzi-infected Ifnar−/− mice survived the challenge and were able to control parasite replication (104). Besides the fact that splenocytes from Ifnar−/− mice produced higher levels of IFN-II, plasma cytokine profile in T. cruzi-infected Ifnar−/− mice were not different to control mice (104). Additionally, T cells populations were not inherently different compared with control mice (104), and IFN-γ production by CD8+T cells was not affected by impaired IFN-I signaling (105), suggesting that, in this model, endogenous IFN-I is not the only relevant signal in host defense against T. cruzi.
Taken together, the role of IFN-I in T. cruzii infection differs from one experimental model to the other, depending on the dose and the route of infection (106). This could explain the controversy about the observations on the role of IFN-I in the T. cruzi model of infection (Table 4).

Table 4. Role of interferon (IFN)-I in Trypanosoma infection.
The immune response to African trypanosomes is quite different than that to T. cruzi. First, parasites never enter the host cell at any stage of their development. The success of these parasites is mainly due to their ability to change the composition of the variant surface glycoprotein (VSG) by switching genes. This confers them the capacity to evade B- and T-cell-mediated immune responses and results in fluctuating waves of parasitemia that characterize African trypanosomiasis (94). VSG is a strong antigen that induces Th1 responses and promotes autoantibody and cytokines production, in particular TNF. Other trypanosome proteins and soluble factors, such as a trypanosome-released triggering factor, also trigger IFN-γ production by T and NK cells and are involved in macrophage activation toward an M1 phenotype, which is required for the control of parasite multiplication during the acute phase of infection. However, sustained activation of M1 macrophages is associated with disease exacerbation. The progression of the infection toward the development of an acute fatal disease or a prolonged chronic infection is determined by the balance between a type I and type II immune responses and the switch from the early type I immune response (dominated by M1 macrophage activation) from a type II (M2 macrophages) regulatory response that controls the inflammation (107).
The literature on the role of IFN-I in African trypanosomiasis is scarce. A study involving Trypanosoma brucei rhodesiense reported a beneficial effect of IFN-I during the acute phase of infection. Indeed, Ifnar−/− mice displayed delayed control of parasite burden during the first week of infection and died earlier than wild-type controls. Moreover, mice hyperresponsive for IFN-I (Ubp43−/−) exhibited a significant defect in Th1 responses and IFN-γ production, suggesting that IFN-I plays a role in the early stages of disease. Nevertheless, IFN-I contributes to the downregulation of IFN-γ production and loss of host resistance during chronic infection (108).
No effects of IFN-I signaling were observed in Trypanosoma brucei brucei-infected Ifnar−/− mice, which showed similar levels of parasitemia to wild-type mice, suggesting that in this model parasite control is independent of IFN-I (109). However, IFN-I regulates T cell infiltration to the brain parenchyma at chronic stages of the infection (109).
In conclusion, the contribution of IFN-I to protective immunity against several protozoan parasites is still unclear. Variations in parasite numbers used for infections, the site of inoculation, and the dose of IFN-I all seem to influence the outcome and the interpretation of the results. A spatio-temporal analysis of the role of IFN-I integrated with a more detailed investigation of cell-specific signaling pathways elicited by the cytokine could help to better dissect the involvement of IFN-I in the immune response.
All authors listed have made substantial, direct, and intellectual contribution to the work and approved it for publication.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
This work was supported by Canadian Institutes of Health Research grant MOP 123293 (to SS) and the Natural Science and Engineering Research Council of Canada (to SS). SS-B was partly supported by a studentship from the Fondation Armand-Frappier.
1. Isaacs A, Lindenmann J. Virus interference. I. The interferon. Proc R Soc B Biol Sci (1957) 147(927):258–67. doi:10.1098/rspb.1957.0049
2. Platanias LC. Mechanisms of type-I- and type-II-interferon-mediated signalling. Nat Rev Immunol (2005) 5(5):375–86. doi:10.1038/nri1604
3. de Weerd NA, Nguyen T. The interferons and their receptors – distribution and regulation. Immunol Cell Biol (2012) 90(5):483–91. doi:10.1038/icb.2012.9
4. Trinchieri G. Type I interferon: friend or foe? J Exp Med (2010) 207(10):2053–63. doi:10.1084/jem.20101664
5. Schoenborn JR, Wilson CB. Regulation of interferon-gamma during innate and adaptive immune responses. Adv Immunol (2007) 96:41–101. doi:10.1016/S0065-2776(07)96002-2
6. Egli A, Santer DM, O’Shea D, Tyrrell DL, Houghton M. The impact of the interferon-lambda family on the innate and adaptive immune response to viral infections. Emerg Microbes Infect (2014) 3(7):e51. doi:10.1038/emi.2014.51
7. Ivashkiv LB, Donlin LT. Regulation of type I interferon responses. Nat Rev Immunol (2014) 14(1):36–49. doi:10.1038/nri3581
8. Mogensen TH. Pathogen recognition and inflammatory signaling in innate immune defenses. Clin Microbiol Rev (2009) 22(2):240–73. doi:10.1128/CMR.00046-08
9. Montoya M, Schiavoni G, Mattei F, Gresser I, Belardelli F, Borrow P, et al. Type I interferons produced by dendritic cells promote their phenotypic and functional activation. Blood (2002) 99(9):3263–71. doi:10.1182/blood.V99.9.3263
10. Ito T, Amakawa R, Inaba M, Ikehara S, Inaba K, Fukuhara S. Differential regulation of human blood dendritic cell subsets by IFNs. J Immunol (2001) 166(5):2961–9. doi:10.4049/jimmunol.166.5.2961
11. Le Bon A, Etchart N, Rossmann C, Ashton M, Hou S, Gewert D, et al. Cross-priming of CD8+ T cells stimulated by virus-induced type I interferon. Nat Immunol (2003) 4(10):1009–15. doi:10.1038/ni978
12. Marrack P, Kappler J, Mitchell T. Type I interferons keep activated T cells alive. J Exp Med (1999) 189(3):521–30. doi:10.1084/jem.189.3.521
13. Aichele P, Unsoeld H, Koschella M, Schweier O, Kalinke U, Vucikuja S. CD8 T cells specific for lymphocytic choriomeningitis virus require type I IFN receptor for clonal expansion. J Immunol (2006) 176(8):4525–9. doi:10.4049/jimmunol.176.8.4525
14. Kolumam GA, Thomas S, Thompson LJ, Sprent J, Murali-Krishna K. Type I interferons act directly on CD8 T cells to allow clonal expansion and memory formation in response to viral infection. J Exp Med (2005) 202(5):637–50. doi:10.1084/jem.20050821
15. Lund JM, Alexopoulou L, Sato A, Karow M, Adams NC, Gale NW, et al. Recognition of single-stranded RNA viruses by toll-like receptor 7. Proc Natl Acad Sci U S A (2004) 101(15):5598–603. doi:10.1073/pnas.0400937101
16. Fink K, Lang KS, Manjarrez-Orduno N, Junt T, Senn BM, Holdener M, et al. Early type I interferon-mediated signals on B cells specifically enhance antiviral humoral responses. Eur J Immunol (2006) 36(8):2094–105. doi:10.1002/eji.200635993
17. Ng CT, Sullivan BM, Teijaro JR, Lee AM, Welch M, Rice S, et al. Blockade of interferon beta, but not interferon alpha, signaling controls persistent viral infection. Cell Host Microbe (2015) 17(5):653–61. doi:10.1016/j.chom.2015.04.005
18. Teijaro JR, Ng C, Lee AM, Sullivan BM, Sheehan KC, Welch M, et al. Persistent LCMV infection is controlled by blockade of type I interferon signaling. Science (2013) 340(6129):207–11. doi:10.1126/science.1235214
19. Wilson EB, Yamada DH, Elsaesser H, Herskovitz J, Deng J, Cheng G, et al. Blockade of chronic type I interferon signaling to control persistent LCMV infection. Science (2013) 340(6129):202–7. doi:10.1126/science.1235208
20. Rayamajhi M, Humann J, Penheiter K, Andreasen K, Lenz LL. Induction of IFN-alphabeta enables Listeria monocytogenes to suppress macrophage activation by IFN-gamma. J Exp Med (2010) 207(2):327–37. doi:10.1084/jem.20091746
21. Kearney SJ, Delgado C, Eshleman EM, Hill KK, O’Connor BP, Lenz LL. Type I IFNs downregulate myeloid cell IFN-gamma receptor by inducing recruitment of an early growth response 3/NGFI-A binding protein 1 complex that silences IFNGR1 transcription. J Immunol (2013) 191(6):3384–92. doi:10.4049/jimmunol.1203510
22. Moseman EA, Wu T, de la Torre JC, Schwartzberg PL, McGavern DB. Type I interferon suppresses virus-specific B cell responses by modulating CD8+ T cell differentiation. Sci Immunol (2016) 1(4).
23. Fallet B, Narr K, Ertuna YI, Remy M, Sommerstein R, Cornille K, et al. Interferon-driven deletion of antiviral B cells at the onset of chronic infection. Sci Immunol (2016) 1(4).
24. Carrero JA, Calderon B, Unanue ER. Lymphocytes are detrimental during the early innate immune response against Listeria monocytogenes. J Exp Med (2006) 203(4):933–40. doi:10.1084/jem.20060045
25. McNab FW, Ewbank J, Howes A, Moreira-Teixeira L, Martirosyan A, Ghilardi N, et al. Type I IFN induces IL-10 production in an IL-27-independent manner and blocks responsiveness to IFN-gamma for production of IL-12 and bacterial killing in Mycobacterium tuberculosis-infected macrophages. J Immunol (2014) 193(7):3600–12. doi:10.4049/jimmunol.1401088
26. Mayer-Barber KD, Andrade BB, Barber DL, Hieny S, Feng CG, Caspar P, et al. Innate and adaptive interferons suppress IL-1alpha and IL-1beta production by distinct pulmonary myeloid subsets during Mycobacterium tuberculosis infection. Immunity (2011) 35(6):1023–34. doi:10.1016/j.immuni.2011.12.002
27. McNab F, Mayer-Barber K, Sher A, Wack A, O’Garra A. Type I interferons in infectious disease. Nat Rev Immunol (2015) 15(2):87–103. doi:10.1038/nri3787
28. Emamian ES, Leon JM, Lessard CJ, Grandits M, Baechler EC, Gaffney PM, et al. Peripheral blood gene expression profiling in Sjogren’s syndrome. Genes Immun (2009) 10(4):285–96. doi:10.1038/gene.2009.20
29. Ko K, Franek BS, Marion M, Kaufman KM, Langefeld CD, Harley JB, et al. Genetic ancestry, serum interferon-alpha activity, and autoantibodies in systemic lupus erythematosus. J Rheumatol (2012) 39(6):1238–40. doi:10.3899/jrheum.111467
30. Javed A, Reder AT. Therapeutic role of beta-interferons in multiple sclerosis. Pharmacol Ther (2006) 110(1):35–56. doi:10.1016/j.pharmthera.2005.08.011
31. Chakravarty S, Cockburn IA, Kuk S, Overstreet MG, Sacci JB, Zavala F. CD8+ T lymphocytes protective against malaria liver stages are primed in skin-draining lymph nodes. Nat Med (2007) 13(9):1035–41. doi:10.1038/nm1628
32. Obeid M, Franetich JF, Lorthiois A, Gego A, Grüner AC, Tefit M, et al. Skin-draining lymph node priming is sufficient to induce sterile immunity against pre-erythrocytic malaria. EMBO Mol Med (2013) 5(2):250–63. doi:10.1002/emmm.201201677
33. Schofield L, Grau GE. Immunological processes in malaria pathogenesis. Nat Rev Immunol (2005) 5(9):722–35. doi:10.1038/nri1686
34. Stevenson MM, Riley EM. Innate immunity to malaria. Nat Rev Immunol (2004) 4(3):169–80. doi:10.1038/nri1311
35. Bertolino P, Bowen DG. Malaria and the liver: immunological hide-and-seek or subversion of immunity from within? Front Microbiol (2015) 6:41. doi:10.3389/fmicb.2015.00041
36. Riley EM, Stewart VA. Immune mechanisms in malaria: new insights in vaccine development. Nat Med (2013) 19(2):168–78. doi:10.1038/nm.3083
37. Urban BC, Ing R, Stevenson MM. Early interactions between blood-stage Plasmodium parasites and the immune system. Curr Top Microbiol Immunol (2005) 297:25–70.
38. Jahiel RI, Vilcek J, Nussenzweig RS. Exogenous interferon protects mice against Plasmodium berghei malaria. Nature (1970) 227(5265):1350–1. doi:10.1038/2271350a0
39. Morrell CN, Srivastava K, Swaim A, Lee MT, Chen J, Nagineni C, et al. Beta interferon suppresses the development of experimental cerebral malaria. Infect Immun (2011) 79(4):1750–8. doi:10.1128/IAI.00810-10
40. Vigario AM, Belnoue E, Grüner AC, Mauduit M, Kayibanda M, Deschemin JC, et al. Recombinant human IFN-alpha inhibits cerebral malaria and reduces parasite burden in mice. J Immunol (2007) 178(10):6416–25. doi:10.4049/jimmunol.178.10.6416
41. Portugal S, Carret C, Recker M, Armitage AE, Gonçalves LA, Epiphanio S, et al. Host-mediated regulation of superinfection in malaria. Nat Med (2011) 17(6):732–7. doi:10.1038/nm.2368
42. Liehl P, Zuzarte-Luís V, Chan J, Zillinger T, Baptista F, Carapau D, et al. Host-cell sensors for Plasmodium activate innate immunity against liver-stage infection. Nat Med (2014) 20(1):47–53. doi:10.1038/nm.3424
43. Miller JL, Sack BK, Baldwin M, Vaughan AM, Kappe SH. Interferon-mediated innate immune responses against malaria parasite liver stages. Cell Rep (2014) 7(2):436–47. doi:10.1016/j.celrep.2014.03.018
44. Rocha BC, Marques PE, Leoratti FM, Junqueira C, Pereira DB, Antonelli LR, et al. Type I interferon transcriptional signature in neutrophils and low-density granulocytes are associated with tissue damage in malaria. Cell Rep (2015) 13(12):2829–41. doi:10.1016/j.celrep.2015.11.055
45. Krupka M, Seydel K, Feintuch CM, Yee K, Kim R, Lin CY, et al. Mild Plasmodium falciparum malaria following an episode of severe malaria is associated with induction of the interferon pathway in Malawian children. Infect Immun (2012) 80(3):1150–5. doi:10.1128/IAI.06008-11
46. Aucan C, Walley AJ, Hennig BJ, Fitness J, Frodsham A, Zhang L, et al. Interferon-alpha receptor-1 (IFNAR1) variants are associated with protection against cerebral malaria in the Gambia. Genes Immun (2003) 4(4):275–82. doi:10.1038/sj.gene.6363962
47. Ball EA, Sambo MR, Martins M, Trovoada MJ, Benchimol C, Costa J, et al. IFNAR1 controls progression to cerebral malaria in children and CD8+ T cell brain pathology in Plasmodium berghei-infected mice. J Immunol (2013) 190(10):5118–27. doi:10.4049/jimmunol.1300114
48. Haque A, Best SE, Ammerdorffer A, Desbarrieres L, de Oca MM, Amante FH, et al. Type I interferons suppress CD4(+) T-cell-dependent parasite control during blood-stage Plasmodium infection. Eur J Immunol (2011) 41(9):2688–98. doi:10.1002/eji.201141539
49. Haque A, Best SE, Montes de Oca M, James KR, Ammerdorffer A, Edwards CL, et al. Type I IFN signaling in CD8- DCs impairs Th1-dependent malaria immunity. J Clin Invest (2014) 124(6):2483–96. doi:10.1172/JCI70698
50. Tamura T, Kimura K, Yui K, Yoshida S. Reduction of conventional dendritic cells during Plasmodium infection is dependent on activation induced cell death by type I and II interferons. Exp Parasitol (2015) 159:127–35. doi:10.1016/j.exppara.2015.09.010
51. Wu J, Cai B, Sun W, Huang R, Liu X, Lin M, et al. Genome-wide analysis of host-Plasmodium yoelii interactions reveals regulators of the type I interferon response. Cell Rep (2015) 12(4):661–72. doi:10.1016/j.celrep.2015.06.058
52. Elmore SA, Jones JL, Conrad PA, Patton S, Lindsay DS, Dubey JP. Toxoplasma gondii: epidemiology, feline clinical aspects, and prevention. Trends Parasitol (2010) 26(4):190–6. doi:10.1016/j.pt.2010.01.009
53. Darde ML. Toxoplasma gondii, “new” genotypes and virulence. Parasite (2008) 15(3):366–71. doi:10.1051/parasite/2008153366
54. Blader IJ, Coleman BI, Chen CT, Gubbels MJ. Lytic cycle of Toxoplasma gondii: 15 years later. Annu Rev Microbiol (2015) 69:463–85. doi:10.1146/annurev-micro-091014-104100
55. Dupont CD, Christian DA, Hunter CA. Immune response and immunopathology during toxoplasmosis. Semin Immunopathol (2012) 34(6):793–813. doi:10.1007/s00281-012-0339-3
56. Scharton-Kersten T, Caspar P, Sher A, Denkers EY. Toxoplasma gondii: evidence for interleukin-12-dependent and-independent pathways of interferon-gamma production induced by an attenuated parasite strain. Exp Parasitol (1996) 84(2):102–14. doi:10.1006/expr.1996.0096
57. Vossenkämper A, Struck D, Alvarado-Esquivel C, Went T, Takeda K, Akira S, et al. Both IL-12 and IL-18 contribute to small intestinal Th1-type immunopathology following oral infection with Toxoplasma gondii, but IL-12 is dominant over IL-18 in parasite control. Eur J Immunol (2004) 34(11):3197–207. doi:10.1002/eji.200424993
58. Omata Y, Yagami K, Sakurai H, Saito A, Suzuki N. Interferon induction in Toxoplasma gondii infected mice. Zentralbl Bakteriol Mikrobiol Hyg A (1983) 255(2–3):392–6.
59. Shirahata T, Mori A, Ishikawa H, Goto H. Strain differences of interferon-generating capacity and resistance in Toxoplasma-infected mice. Microbiol Immunol (1986) 30(12):1307–16. doi:10.1111/j.1348-0421.1986.tb03051.x
60. Diez B, Galdeano A, Nicolas R, Cisterna R. Relationship between the production of interferon-alpha/beta and interferon-gamma during acute toxoplasmosis. Parasitology (1989) 99(Pt 1):11–5. doi:10.1017/S0031182000060972
61. Mahmoud ME, Ui F, Salman D, Nishimura M, Nishikawa Y. Mechanisms of interferon-beta-induced inhibition of Toxoplasma gondii growth in murine macrophages and embryonic fibroblasts: role of immunity-related GTPase M1. Cell Microbiol (2015) 17(7):1069–83. doi:10.1111/cmi.12423
62. Han SJ, Melichar HJ, Coombes JL, Chan SW, Koshy AA, Boothroyd JC, et al. Internalization and TLR-dependent type I interferon production by monocytes in response to Toxoplasma gondii. Immunol Cell Biol (2014) 92(10):872–81. doi:10.1038/icb.2014.70
63. Rosowski EE, Nguyen QP, Camejo A, Spooner E, Saeij JP. Toxoplasma gondii inhibits gamma interferon (IFN-gamma)- and IFN-beta-induced host cell STAT1 transcriptional activity by increasing the association of STAT1 with DNA. Infect Immun (2014) 82(2):706–19. doi:10.1128/IAI.01291-13
64. Remington JS, Merigan TC. Interferon: protection of cells infected with an intracellular protozoan (Toxoplasma gondii). Science (1968) 161(3843):804–6. doi:10.1126/science.161.3843.804
65. Wilson CB, Westall J. Activation of neonatal and adult human macrophages by alpha, beta, and gamma interferons. Infect Immun (1985) 49(2):351–6.
66. Orellana MA, Suzuki Y, Araujo F, Remington JS. Role of beta interferon in resistance to Toxoplasma gondii infection. Infect Immun (1991) 59(9):3287–90.
67. Schmitz JL, Carlin JM, Borden EC, Byrne GI. Beta interferon inhibits Toxoplasma gondii growth in human monocyte-derived macrophages. Infect Immun (1989) 57(10):3254–6.
68. Han S. The role of type 1 IFN after oral Toxoplasma gondii infection. J Immunol (2011) 1486(Suppl).
69. Pfefferkorn ER, Guyre PM. Inhibition of growth of Toxoplasma gondii in cultured fibroblasts by human recombinant gamma interferon. Infect Immun (1984) 44(2):211–6.
70. Engin AB, Dogruman-Al F, Ercin U, Celebi B, Babur C, Bukan N. Oxidative stress and tryptophan degradation pattern of acute Toxoplasma gondii infection in mice. Parasitol Res (2012) 111(4):1725–30. doi:10.1007/s00436-012-3015-6
71. Sibley LD. Interactions between Toxoplasma gondii and its mammalian host cells. Semin Cell Biol (1993) 4(5):335–44. doi:10.1006/scel.1993.1040
72. Murray HW, Szuro-Sudol A, Wellner D, Oca MJ, Granger AM, Libby DM, et al. Role of tryptophan degradation in respiratory burst-independent antimicrobial activity of gamma interferon-stimulated human macrophages. Infect Immun (1989) 57(3):845–9.
73. Nagineni CN, Pardhasaradhi K, Martins MC, Detrick B, Hooks JJ. Mechanisms of interferon-induced inhibition of Toxoplasma gondii replication in human retinal pigment epithelial cells. Infect Immun (1996) 64(10):4188–96.
74. Kaye P, Scott P. Leishmaniasis: complexity at the host-pathogen interface. Nat Rev Microbiol (2011) 9(8):604–15. doi:10.1038/nrmicro2608
75. Engwerda CR, Murphy ML, Cotterell SE, Smelt SC, Kaye PM. Neutralization of IL-12 demonstrates the existence of discrete organ-specific phases in the control of Leishmania donovani. Eur J Immunol (1998) 28(2):669–80. doi:10.1002/(SICI)1521-4141(199802)28:02<669::AID-IMMU669>3.0.CO;2-N
76. Bankoti R, Stager S. Differential regulation of the immune response in the spleen and liver of mice infected with Leishmania donovani. J Trop Med (2012) 2012:639304. doi:10.1155/2012/639304
77. Herman R, Baron S. Effects of interferon inducers on the intracellular growth of the protozoan parasite, Leishmania donovani. Nature (1970) 226(5241):168–70. doi:10.1038/226168a0
78. Mattner J, Schindler H, Diefenbach A, Röllinghoff M, Gresser I, Bogdan C. Regulation of type 2 nitric oxide synthase by type 1 interferons in macrophages infected with Leishmania major. Eur J Immunol (2000) 30(8):2257–67. doi:10.1002/1521-4141(2000)30:8<2257::AID-IMMU2257>3.0.CO;2-U
79. Diefenbach A, Schindler H, Donhauser N, Lorenz E, Laskay T, MacMicking J, et al. Type 1 interferon (IFNalpha/beta) and type 2 nitric oxide synthase regulate the innate immune response to a protozoan parasite. Immunity (1998) 8(1):77–87. doi:10.1016/S1074-7613(00)80460-4
80. Olekhnovitch R, Ryffel B, Müller AJ, Bousso P. Collective nitric oxide production provides tissue-wide immunity during Leishmania infection. J Clin Invest (2014) 124(4):1711–22. doi:10.1172/JCI72058
81. Mattner J, Wandersee-Steinhäuser A, Pahl A, Röllinghoff M, Majeau GR, Hochman PS, et al. Protection against progressive leishmaniasis by IFN. J Immunol (2004) 172(12):7574–82. doi:10.4049/jimmunol.172.12.7574
82. Khouri R, Bafica A, Silva Mda P, Noronha A, Kolb JP, Wietzerbin J, et al. IFN-beta impairs superoxide-dependent parasite killing in human macrophages: evidence for a deleterious role of SOD1 in cutaneous leishmaniasis. J Immunol (2009) 182(4):2525–31. doi:10.4049/jimmunol.0802860
83. Xin L, Vargas-Inchaustegui DA, Raimer SS, Kelly BC, Hu J, Zhu L, et al. Type I IFN receptor regulates neutrophil functions and innate immunity to Leishmania parasites. J Immunol (2010) 184(12):7047–56. doi:10.4049/jimmunol.0903273
84. Silva-Barrios S, Smans M, Duerr CU, Qureshi ST, Fritz JH, Descoteaux A, et al. Innate immune B cell activation by Leishmania donovani exacerbates disease and mediates hypergammaglobulinemia. Cell Rep (2016) 15(11):2427–37. doi:10.1016/j.celrep.2016.05.028
85. Bankoti R, Gupta K, Levchenko A, Stäger S. Marginal zone B cells regulate antigen-specific T cell responses during infection. J Immunol (2012) 188(8):3961–71. doi:10.4049/jimmunol.1102880
86. Smelt SC, Cotterell SE, Engwerda CR, Kaye PM. B cell-deficient mice are highly resistant to Leishmania donovani infection, but develop neutrophil-mediated tissue pathology. J Immunol (2000) 164(7):3681–8. doi:10.4049/jimmunol.164.7.3681
87. Deak E, Jayakumar A, Cho KW, Goldsmith-Pestana K, Dondji B, Lambris JD, et al. Murine visceral leishmaniasis: IgM and polyclonal B-cell activation lead to disease exacerbation. Eur J Immunol (2010) 40(5):1355–68. doi:10.1002/eji.200939455
88. Passwell JH, Shor R, Shoham J. The enhancing effect of interferon-beta and -gamma on the killing of Leishmania tropica major in human mononuclear phagocytes in vitro. J Immunol (1986) 136(8):3062–6.
89. Manna PP, Bharadwaj D, Bhattacharya S, Chakrabarti G, Basu D, Mallik KK, et al. Impairment of natural killer cell activity in Indian kala-azar: restoration of activity by interleukin 2 but not by alpha or gamma interferon. Infect Immun (1993) 61(8):3565–9.
90. Trau H, Schewach-Millet M, Shoham J, Doerner T, Shor R, Passwell JH. Topical application of human fibroblast interferon (IFN) in cutaneous leishmaniasis. Isr J Med Sci (1987) 23(11):1125–7.
91. Chaussabel D, Semnani RT, McDowell MA, Sacks D, Sher A, Nutman TB. Unique gene expression profiles of human macrophages and dendritic cells to phylogenetically distinct parasites. Blood (2003) 102(2):672–81. doi:10.1182/blood-2002-10-3232
92. Favila MA, Geraci NS, Zeng E, Harker B, Condon D, Cotton RN, et al. Human dendritic cells exhibit a pronounced type I IFN signature following Leishmania major infection that is required for IL-12 induction. J Immunol (2014) 192(12):5863–72. doi:10.4049/jimmunol.1203230
93. MacGregor P, Szöőr B, Savill NJ, Matthews KR. Trypanosomal immune evasion, chronicity and transmission: an elegant balancing act. Nat Rev Microbiol (2012) 10(6):431–8. doi:10.1038/nrmicro2779
94. Namangala B. How the African trypanosomes evade host immune killing. Parasite Immunol (2011) 33(8):430–7. doi:10.1111/j.1365-3024.2011.01280.x
95. Cardoso MS, Reis-Cunha JL, Bartholomeu DC. Evasion of the immune response by Trypanosoma cruzi during acute infection. Front Immunol (2016) 6:659. doi:10.3389/fimmu.2015.00659
96. Chessler AD, Unnikrishnan M, Bei AK, Daily JP, Burleigh BA. Trypanosoma cruzi triggers an early type I IFN response in vivo at the site of intradermal infection. J Immunol (2009) 182(4):2288–96. doi:10.4049/jimmunol.0800621
97. Costales JA, Daily JP, Burleigh BA. Cytokine-dependent and-independent gene expression changes and cell cycle block revealed in Trypanosoma cruzi-infected host cells by comparative mRNA profiling. BMC Genomics (2009) 10:252. doi:10.1186/1471-2164-10-252
98. Vaena de Avalos S, Blader IJ, Fisher M, Boothroyd JC, Burleigh BA. Immediate/early response to Trypanosoma cruzi infection involves minimal modulation of host cell transcription. J Biol Chem (2002) 277(1):639–44. doi:10.1074/jbc.M109037200
99. Chessler AD, Ferreira LR, Chang TH, Fitzgerald KA, Burleigh BA. A novel IFN regulatory factor 3-dependent pathway activated by trypanosomes triggers IFN-beta in macrophages and fibroblasts. J Immunol (2008) 181(11):7917–24. doi:10.4049/jimmunol.181.11.7917
100. James SL, Kipnis TL, Sher A, Hoff R. Enhanced resistance to acute infection with Trypanosoma cruzi in mice treated with an interferon inducer. Infect Immun (1982) 35(2):588–93.
101. Rottenberg M, Cardoni RL, Andersson R, Segura EL, Orn A. Role of T helper/inducer cells as well as natural killer cells in resistance to Trypanosoma cruzi infection. Scand J Immunol (1988) 28(5):573–82. doi:10.1111/j.1365-3083.1988.tb01489.x
102. Costa VM, Torres KC, Mendonça RZ, Gresser I, Gollob KJ, Abrahamsohn IA. Type I IFNs stimulate nitric oxide production and resistance to Trypanosoma cruzi infection. J Immunol (2006) 177(5):3193–200. doi:10.4049/jimmunol.177.5.3193
103. Une C, Andersson J, Orn A. Role of IFN-alpha/beta and IL-12 in the activation of natural killer cells and interferon-gamma production during experimental infection with Trypanosoma cruzi. Clin Exp Immunol (2003) 134(2):195–201. doi:10.1046/j.1365-2249.2003.02294.x
104. Chessler AD, Caradonna KL, Da’dara A, Burleigh BA. Type I interferons increase host susceptibility to Trypanosoma cruzi infection. Infect Immun (2011) 79(5):2112–9. doi:10.1128/IAI.01176-10
105. Martin DL, Murali-Krishna K, Tarleton RL. Generation of Trypanosoma cruzi-specific CD8+ T-cell immunity is unaffected by the absence of type I interferon signaling. Infect Immun (2010) 78(7):3154–9. doi:10.1128/IAI.00275-10
106. Bogdan C, Mattner J, Schleicher U. The role of type I interferons in non-viral infections. Immunol Rev (2004) 202:33–48. doi:10.1111/j.0105-2896.2004.00207.x
107. Vincendeau P, Bouteille B. Immunology and immunopathology of African trypanosomiasis. An Acad Bras Cienc (2006) 78(4):645–65. doi:10.1590/S0001-37652006000400004
108. Lopez R, Demick KP, Mansfield JM, Paulnock DM. Type I IFNs play a role in early resistance, but subsequent susceptibility, to the African trypanosomes. J Immunol (2008) 181(7):4908–17. doi:10.4049/jimmunol.181.7.4908
109. Amin DN, Vodnala SK, Masocha W, Sun B, Kristensson K, Rottenberg ME, et al. Distinct toll-like receptor signals regulate cerebral parasite load and interferon alpha/beta and tumor necrosis factor alpha-dependent T-cell infiltration in the brains of Trypanosoma brucei-infected mice. J Infect Dis (2012) 205(2):320–32. doi:10.1093/infdis/jir734
Keywords: protozoan infections, IFN-I, Leishmania, Toxoplasma, Plasmodium, Trypanosoma
Citation: Silva-Barrios S and Stäger S (2017) Protozoan Parasites and Type I IFNs. Front. Immunol. 8:14. doi: 10.3389/fimmu.2017.00014
Received: 27 September 2016; Accepted: 05 January 2017;
Published: 19 January 2017
Edited by:
Joao P. B. Viola, Instituto Nacional de Cancer (INCA), BrazilReviewed by:
Claudia Ida Brodskyn, Centro de Pesquisas Gonçalo Moniz (Fiocruz Bahia), BrazilCopyright: © 2017 Silva-Barrios and Stäger. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Simona Stäger, c2ltb25hLnN0YWdlckBpYWYuaW5ycy5jYQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.