 Wendy Dankers
Wendy Dankers Edgar M. Colin
Edgar M. Colin Jan Piet van Hamburg
Jan Piet van Hamburg Erik Lubberts
Erik Lubberts
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 20 January 2017
Sec. Inflammation
Volume 7 - 2016 | https://doi.org/10.3389/fimmu.2016.00697
Over the last three decades, it has become clear that the role of vitamin D goes beyond the regulation of calcium homeostasis and bone health. An important extraskeletal effect of vitamin D is the modulation of the immune system. In the context of autoimmune diseases, this is illustrated by correlations of vitamin D status and genetic polymorphisms in the vitamin D receptor with the incidence and severity of the disease. These correlations warrant investigation into the potential use of vitamin D in the treatment of patients with autoimmune diseases. In recent years, several clinical trials have been performed to investigate the therapeutic value of vitamin D in multiple sclerosis, rheumatoid arthritis, Crohn’s disease, type I diabetes, and systemic lupus erythematosus. Additionally, a second angle of investigation has focused on unraveling the molecular pathways used by vitamin D in order to find new potential therapeutic targets. This review will not only provide an overview of the clinical trials that have been performed but also discuss the current knowledge about the molecular mechanisms underlying the immunomodulatory effects of vitamin D and how these advances can be used in the treatment of autoimmune diseases.
Autoimmune diseases, including rheumatoid arthritis (RA), multiple sclerosis (MS), and Crohn’s disease (CD), result from an aberrant activation of the immune system, whereby the immune response is directed against harmless self-antigens. This results in inflammation, tissue damage, and loss of function of the affected organs or joints. With the increasing prevalence of autoimmunity in the Western countries (1), the societal burden of these diseases also increases. Although the treatment of autoimmune diseases has improved due to the development of so-called biologics, like tumor necrosis factor alpha (TNFα) inhibitors, a large proportion of patients are still not adequately responding to these treatments (2). Therefore, it is still important to improve current therapies or to uncover new treatment options.
In this context, the immunomodulatory effects of vitamin D provide opportunities to enhance the treatment of autoimmune diseases. First, given the high prevalence of vitamin D deficiency in patients suffering from autoimmunity, vitamin D supplementation might decrease disease severity or augment the therapeutic effect of current medication. Second, knowing the molecular mechanisms underlying the immunomodulatory effects could lead to the discovery of new potential therapeutic targets. Therefore, this review will explore the advances that have been made in both clinical trials and molecular studies. In addition, it will give an overview of the challenges that still remain before the immunomodulatory effects of vitamin D can be utilized in clinical practice.
Vitamin D, or cholecalciferol, is a secosteroid hormone that can be obtained from dietary sources, but that is predominantly synthesized in the skin from 7-dehydroxycholesterol in response to UV light (Figure 1). Cholecalciferol is bound by vitamin D-binding protein (DBP) and transported to the liver. In the liver, various cytochrome p450 (Cyp) vitamin D hydroxylases convert cholecalciferol into 25(OH)D3. Cyp2R1 is considered to be the primary 25-hydroxylase responsible for this process. Subsequently, DBP transports 25(OH)D3 to the kidneys, where the 1α-hydroxylase Cyp27B1 converts 25(OH)D3 into 1,25(OH)2D3. 1,25(OH)2D3, also called calcitriol, is the active vitamin D metabolite. To control calcitriol concentrations, the 24-hydroxylase Cyp24A1 hydroxylates 25(OH)D3 or 1,25(OH)2D3 at C-24, yielding the less active metabolites 24,25(OH)2D3 and 1,24,25(OH)3D3, respectively (3). The level of 1,25(OH)2D3 is therefore mainly determined by the balance between Cyp27B1 and Cyp24A1. Two proteins that are important for regulating this balance are fibroblast growth factor 23 (FGF23) and parathyroid hormone (PTH). FGF23 shifts the balance toward Cyp24A1 and therefore inactivation of vitamin D signaling, and is induced by high concentrations of 1,25(OH)2D3 and low serum phosphate. On the other hand, PTH favors the balance toward Cyp27B1 and activation of vitamin D signaling. PTH is inhibited by high concentrations of 1,25(OH)2D3 and induced by low serum calcium (3) (Figure 1).
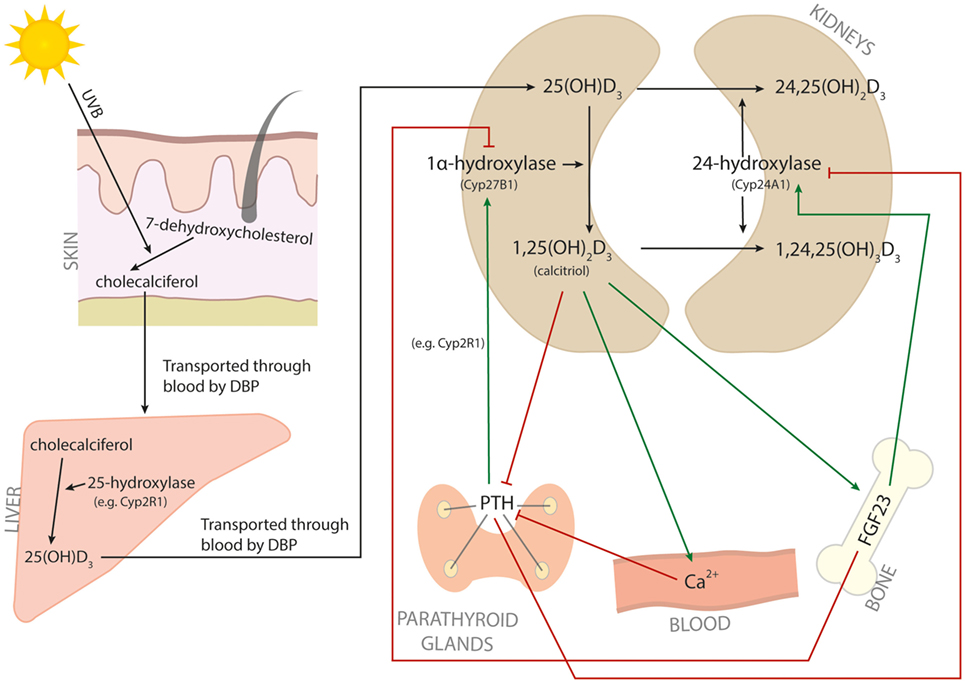
Figure 1. Vitamin D metabolism. The metabolic pathway of vitamin D. Red arrows indicate inhibition, and green arrows indicate induction.
1,25(OH)2D3 initiates its signaling cascade by binding to the vitamin D receptor (VDR), which is a nuclear receptor that acts as a transcription factor. VDR binds to vitamin D responsive elements (VDREs) in the DNA, mostly to so-called DR3-type VDREs that are characterized by two hexameric core binding motifs separated by three nucleotides. In the absence of ligand, VDR is mostly bound to non-DR3-type VDREs and is associated with corepressor proteins. When 1,25(OH)2D3 binds to VDR, this induces a conformational change leading to the formation of two new protein interaction surfaces. One is for binding with heterodimeric partners to facilitate specific DNA binding, such as retinoid X receptor (RXR), and the other is for recruitment of co-regulatory complexes that will exert the genomic effects of VDR (4). Furthermore, there is a shift in binding to primarily DR3-type VDREs (5). Interestingly, although RXR has multiple binding partners, specifically with VDR it will bind to the DR3-type elements. This indicates that the heterodimerization of VDR and RXR is important for functioning of the VDR (6). However, research in colorectal cancer cells has shown that 25% of the VDR binding sites are not enriched for RXR (7). No direct data on colocalization of VDR and RXR in immune cells have been reported, although Handel et al. found a significant overlap between VDR in CD4+ T cells and RXR in a promyelocytic leukemia cell line (8). Therefore, it is currently unknown whether the rate of VDR/RXR colocalization differs between cell types. Also, the functional consequence of VDR binding with or without RXR remains to be understood.
The best known function of 1,25(OH)2D3 is the maintenance of calcium homeostasis by facilitating the absorption of calcium in the intestine. However, in the presence of low 1,25(OH)2D3 levels, calcium will be mobilized from the bone rather than the intestine. If these conditions are prolonged, this may lead to osteomalacia and rickets, both well-known clinical signs of vitamin D deficiency. An overview of the current knowledge on the role of vitamin D signaling in calcium homeostasis was recently given by Carmeliet et al. and will not be discussed here (9). The first hint that vitamin D might also be important for extraskeletal health came from mycobacterial infections such as tuberculosis, in which vitamin D was used as a treatment before antibiotics were discovered (10). The discovery that the VDR is expressed in almost all human cells has further increased the attention for the extraskeletal effects of vitamin D. As a result, vitamin D deficiency has now been linked to not only bone health but also, for example, cancer, cardiovascular diseases, and autoimmune diseases (9).
Since the discovery of the VDR on blood lymphocytes (11, 12), the effects of vitamin D on the immune system and immune-related diseases became the subject of a large number of studies. In this context, it was discovered that supplementation with 1,25(OH)2D3 could prevent both the initiation and progression of experimental autoimmune encephalomyelitis (EAE) and collagen-induced arthritis (CIA), experimental models of MS and RA, respectively (13–15). In addition, VDR deficiency aggravated arthritis severity in human TNFα transgenic mice (16). Similarly, vitamin D deficiency increased enterocolitis severity in IL-10 knock-out (KO) mice, which are used as a model system for inflammatory bowel diseases (IBDs). Treatment with 1,25(OH)2D3 decreased disease symptoms in both the IL-10 KO mice and in the dextran sulfate sodium (DSS)-induced colitis model (17, 18). Finally, treatment with 1,25(OH)2D3 reduced the incidence of diabetes in non-obese diabetic (NOD) mice (19, 20) and the severity of systemic lupus erythematosus (SLE) in MRL/1 mice (21).
These studies in experimental autoimmune models underscore the need to examine whether there is a protective role for vitamin D in human autoimmune diseases. In the last few decades, numerous studies have investigated the link between vitamin D and the incidence and severity of autoimmune diseases. One of the first indications was the correlation between increasing MS prevalence and increasing latitude, and consequently with decreasing sunlight exposure. Exceptions to this gradient can at least partially be explained by genetic variants (like the HLA-DRB1 allele) or lifestyle differences, such as high fish consumption (22). The relation between latitude and disease prevalence was also found for other autoimmune diseases such as type I diabetes mellitus (T1D) and IBD (23, 24). Further strengthening the link between sun exposure and autoimmunity is the finding that the risk of developing MS is correlated with the month of birth, with for the northern hemisphere a higher risk in April and a lower risk in October and November (25, 26). Importantly, this correlation can only be found in areas where the UV exposure changes during the year (25).
Next to UV exposure, vitamin D can also be obtained from dietary sources and supplements. A meta-analysis by Song et al. found that the incidence of RA is inversely correlated with vitamin D intake, both when considering dietary intake and supplements or supplements alone (27). In addition, vitamin D supplementation in early childhood might reduce the risk of developing T1D up to 30% depending on the supplementation frequency (28, 29). Also the effect of maternal vitamin D intake on the risk of T1D in the offspring has been investigated, but due to the limited amount of studies there is currently not sufficient evidence to prove a correlation (29).
Investigating the correlation between vitamin D intake and prevalence of autoimmunity is challenging because the measurements of dietary intake and UV exposure are often based on estimations. Therefore, it might be more useful to analyze the correlation between the serum 25(OH)D3 level and autoimmunity. Indeed, in many autoimmune diseases, patients have a lower serum 25(OH)D3 than healthy controls (30–36). In addition, patients with a lower 25(OH)D3 level are implicated to have higher disease activity (32, 35, 37). Although it is not clear whether the lower 25(OH)D3 level also increases the risk of autoimmunity, the study by Hiraki et al. suggested that there is a strong correlation between the risk of developing RA and the 25(OH)D3 level between 3 months and 4 years before diagnosis (38). It should be noted that all these studies merely demonstrate correlations, so it is still under debate whether the low 25(OH)D3 level is the cause or the result of the autoimmune disease.
Another line of evidence that indicates a role for vitamin D in human autoimmunity is the correlation with polymorphisms in the VDR. There are four well-known VDR polymorphisms that have been extensively studied for their potential role in autoimmunity: ApaI, BsmI, TaqI, and FokI. All of these polymorphisms have been associated with the risk of developing an autoimmune disease, although it differs between diseases and polymorphisms whether it is protective or a risk factor. Also, ethnicity plays a role in the correlation between the polymorphisms and autoimmune diseases (39–47).
In summary, autoimmune diseases are correlated with 25(OH)D3 serum levels, vitamin D intake, UV exposure, and VDR polymorphisms. Furthermore, 1,25(OH)2D3 suppresses disease in experimental autoimmune models. Although these data do not prove a causal relationship between vitamin D and autoimmune diseases, they warrant further investigation into whether at-risk individuals and patients could benefit from vitamin D supplementation.
Despite the beneficial effects of 1,25(OH)2D3 supplementation in experimental autoimmune models, the application of vitamin D derivatives in clinical practice is currently limited to topical use for the treatment of psoriasis (48). The systemic use of vitamin D in the treatment of other autoimmune diseases is still under investigation. Table 1 gives an overview of the placebo-controlled clinical trials investigating the effect of vitamin D supplementation in autoimmune diseases other than psoriasis. Here, we discuss these trials and what this means for the therapeutic potential of vitamin D in each of these autoimmune diseases.
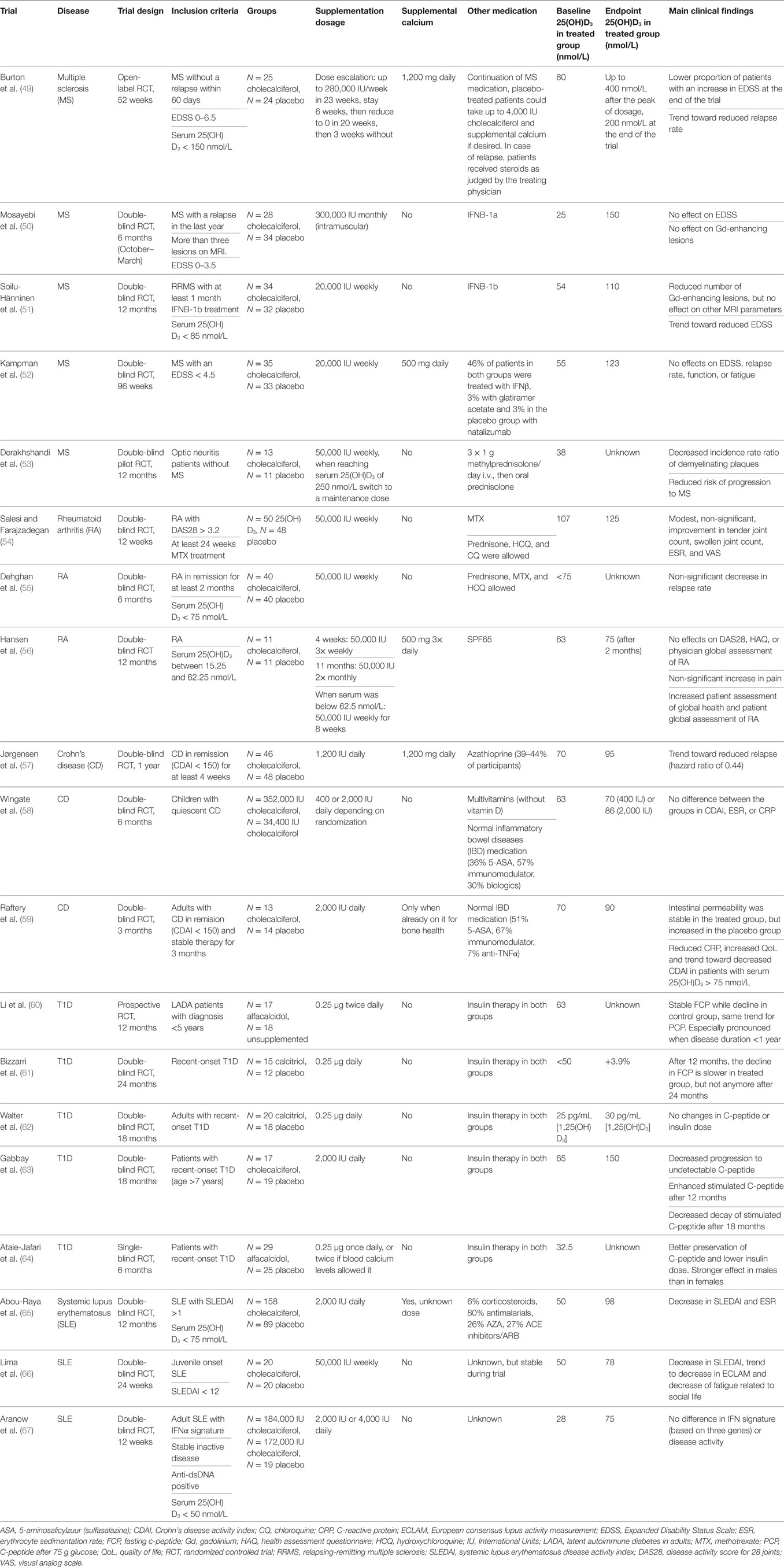
Table 1. Overview of randomized controlled trials with vitamin D supplementation in autoimmune diseases.
In the field of MS, several trials have been performed in which cholecalciferol was given to the patients, but the results are contradictory. Beneficial effects of cholecalciferol supplementation that have been reported include decrease in Expanded Disability Status Scale (EDSS), decrease in MRI lesions, increased functionality, and reduced relapse rates (49, 51). Importantly, cholecalciferol has an added effect when used as a supplement to interferon β (IFNβ) treatment (51). On the other hand, two other trials reported no difference in any of these parameters (50, 52). Vitamin D supplementation might also be important in the pre-MS stage, since cholecalciferol supplementation decreased the conversion rate of optic neuritis to chronic MS (53).
Due to the small sample size (no more than 35 patients/group) of these trials, it is difficult to draw conclusions from these data. Although the effect of cholecalciferol on conversion to chronic effect appears promising, this was only one study with 13 treated patients and 11 placebo controls. Therefore, more research is necessary to determine whether therapy with cholecalciferol is beneficial for MS patients.
Despite the beneficial effect of 1,25(OH)2D3 supplementation on experimental arthritis (15), there are to date only three randomized trials investigating the effect of supplementation on disease activity in RA. Although the studies performed by Salesi and Farajzadegan and Dehghan et al. suggested a beneficial effect on disease activity and relapse rate, respectively, neither results reach statistical significance (54, 55). However, Dehghan et al. pointed out that for every 10 patients treated with cholecalciferol, relapse would be prevented in one patient. Considering the costs and safety profile of cholecalciferol supplementation, this might be worth following up. Ergocalciferol, the less potent fungal equivalent of human cholecalciferol, had no effect on disease activity and was associated with worse patient-related health assessments (56). Similar to studies in MS, the major limitation in the three RA studies is the group size, which limits the power of the analyses. Therefore no definitive conclusion can be drawn yet whether vitamin D can be used as a therapeutic agent in RA.
Crohn’s disease is a subtype of the IBDs and is investigated intensively for the effect of vitamin D supplementation. However, the difficulty with this disease is that the intestinal inflammation may lead to decreased absorption of the supplemented vitamin D. Nevertheless, for adult patients, cholecalciferol supplementation might reduce the risk of relapses, although the difference does not reach statistical significance (p = 0.06) (57). Correspondingly, cholecalciferol prevented further increase of intestinal permeability, which may be an early marker of relapse (59). This is even more pronounced when the patients are stratified based on their serum 25(OH)D3 level. Additionally, patients with a serum level above 75 nmol/L have significantly lower serum levels of C-reactive protein (CRP, a marker of inflammation) and a non-significant decrease in disease activity as measured with Crohn’s Disease Activity Index (59). These studies used 1,200–2,000 IU cholecalciferol daily in adults, but in children there is no difference in disease activity between supplementing 400 and 2,000 IU daily despite a serum 25(OH)D3 level that is 25 nmol/L higher in the latter group (58).
When compared to RA and MS, the results for adult CD are more consistently showing a beneficial effect of cholecalciferol treatment. Since group sizes are again small, more research is required to confirm these data.
In contrast to the other autoimmune diseases where cholecalciferol supplementation is investigated, in T1D almost all trials use 1,25(OH)2D3 or an analog. Both forms appear to delay, but not prevent, the progression of β cell destruction in three studies (60, 63, 64). On the other hand, no effect of 1,25(OH)2D3 on T1D was observed in studies performed by Bizzarri et al. (61) and Walter et al. (62). This lack of effect could be due to the low level of remaining β cell function at the start of the study, suggesting that the therapeutic window for vitamin D supplementation is in the earliest phases of the disease. The study by Li et al. found that the protective effect is only visible when the disease duration was less than 1 year, supporting this hypothesis (60). In T1D, the beneficial effects of 1,25(OH)2D3 may lie more in the prevention of disease onset (28, 29) than in the treatment of disease, since the destruction of β cells cannot be reversed.
Vitamin D supplementation in SLE might even be more relevant than in the other autoimmune diseases, since 80% of the patients is sensitive for sunlight and therefore protect themselves against UV exposure (68). Two studies supplementing either 2,000 IU daily or 50,000 IU weekly demonstrated decreasing disease activity score, auto-antibody levels, and fatigue (65, 66). Conversely, the type I interferon (IFN) signature was unchanged after 12 weeks of 2,000 or 4,000 IU cholecalciferol in another study (67). Since this study was performed in patients with inactive disease, had a short supplementation period, and the signature was based on the expression of only three genes, it remains to be determined whether cholecalciferol supplementation truly does not affect the complete IFN signature in patients with active disease.
Systemic lupus erythematosus is the only autoimmune disease is which a larger study was done, with 158 cholecalciferol-treated patients and 89 placebo controls (65). The promising results in this clinical trial await further confirmation before vitamin D can be used therapeutically in these patients.
In addition to exploring the potential of therapeutic vitamin D supplementation, there has been a great deal of research toward the working mechanisms of 1,25(OH)2D3 in cells of the immune system. Since autoimmune diseases are characterized by an overactive immune response, it seems logical that the beneficial effects of vitamin D on autoimmunity are due to effects on the immune system. Furthermore, virtually all immune cells express the VDR, making them susceptible to 1,25(OH)2D3-mediated modulation (11, 12, 69, 70). Various immune cells, including monocytes, dendritic cells, macrophages, B cells, and T cells, also have the capability to convert 25(OH)D3 into 1,25(OH)2D3 (71–78). This allows for local regulation of the concentration of 1,25(OH)2D3 at the site of inflammation and illustrates an important role for the cells of the immune system in the systemic effects of vitamin D.
Therefore, insight into how 1,25(OH)2D3 modulates the immune system could uncover new therapeutic targets in autoimmune diseases. Here, we discuss the effects of vitamin D on various cell types involved in the immune response, the current knowledge about the underlying mechanisms, and what this means for the therapeutic potential of vitamin D in autoimmunity (Figure 2).
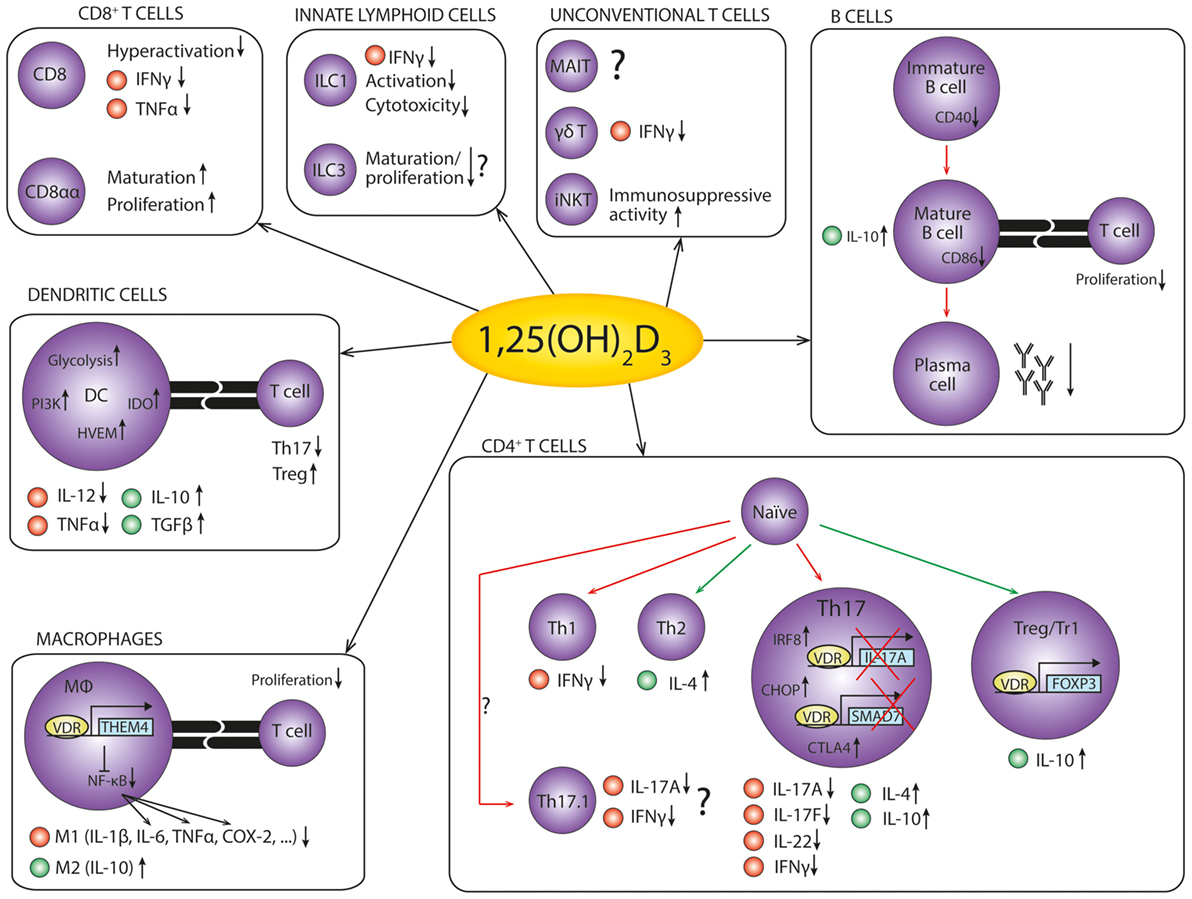
Figure 2. The anti-inflammatory effects of 1,25(OH)2D3 on cells of the immune system. An overview of the anti-inflammatory effects of 1,25(OH)2D3 on the cells of the immune system in autoimmunity. Red dots represent pro-inflammatory cytokines, while green dots represent anti-inflammatory cytokines. Red arrows indicate decreased differentiation, and green arrows indicate increased differentiation. References: CD8+ T cells (79–81); innate lymphoid cells (82–86); unconventional T cells (87–89); B cells (75, 90–96); dendritic cells (97–103); macrophages (104–108); CD4+ T cells (109–125).
Dendritic cells are antigen-presenting cells (APCs), which means that their main function is to take up foreign antigens and present them as peptides to T cells on the human leukocyte antigen (HLA) molecules. DCs are predominantly found in an immature state in peripheral tissues such as the skin, gut, and lungs, where they probe the surroundings for potential pathogens. Upon encountering a foreign antigen, they mature and migrate to the lymphoid tissues to stimulate antigen-specific T cells. Depending on the cytokines secreted by the DC, the T cell will differentiate into an effector cell with appropriate pro- or anti-inflammatory properties. Through these actions, APCs are crucial in initiating effective adaptive immune responses against pathogens, and also for maintaining self-tolerance and immune homeostasis.
The important role of DCs in autoimmune pathogenesis is illustrated in experimental autoimmune models, where deletion of specific DC subtypes ameliorates, or even prevents disease onset (126–129). In addition, APCs, including DCs and also macrophages and B cells, are associated with human autoimmunity through the correlation between specific HLA alleles and the risk of developing an autoimmune disease. For example, HLA-DRB1*15:01 is associated with an increased risk for MS (130), while HLA-DRB1*04:01 confers a greater susceptibility to RA (131).
Dendritic cells differentiated in vitro from monocytes or bone marrow cells in the presence of 1,25(OH)2D3 will remain in an immature-like tolerogenic state. This is characterized by decreased production of pro-inflammatory factors like IL-12 and TNFα and increased anti-inflammatory IL-10 production. These tolerogenic DCs (tDCs) are less capable of promoting proliferation and cytokine production of pro-inflammatory T cells, while they induce the differentiation of T regulatory (Treg) cells (97–99). Furthermore, they specifically induce apoptosis in autoreactive T cells, while not affecting proliferation of other T cells (132). Of note, 1,25(OH)2D3 can only induce this tolerogenic phenotype in DCs when it is added before their maturation. Once a maturation stimulus like lipopolysaccharide (LPS) is present or when the cells have already matured, the effects of 1,25(OH)2D3 on DCs are minimal (133). Aside from in vitro differentiated DCs, 1,25(OH)2D3 also induces a tolerogenic phenotype in dermal DCs, Langerhans cells, and plasmacytoid DCs, even though there are subtle differences between the effects on these subsets (100, 134, 135).
While the tolerizing effects of 1,25(OH)2D3 on DCs are well described, the underlying mechanisms are less clear. Recently, Ferreira et al. suggested that a metabolic switch toward glycolysis and activation of the PI3K-Akt-mTOR pathway are the first steps for the generation of tDCs by 1,25(OH)2D3 (101). Also the induction of indoleamine 2,3-dioxygenase (IDO) on DCs has been reported to be essential for the induction of a tDC phenotype and thereby for the beneficial effect of 1,25(OH)2D3 on EAE (102). Although all tDCs promote regulatory T cells (Tregs), the mechanism by which they do this depends on the type of DC. While tDC derived in vitro from bone marrow cells promote Tregs via induction of herpesvirus entry mediator (HVEM), tolerized Langerhans cells use TGFβ for this (100, 103). Dermal DCs induce the differentiation of T regulatory 1 (Tr1) cells, another type of Treg, via IL-10 (100). So in recent years, advances have been made to fully understand how 1,25(OH)2D3 modulates DCs, but the picture is not yet complete.
Despite the incomplete understanding of the molecular mechanism behind the effects of 1,25(OH)2D3 on DCs, tDCs generated with 1,25(OH)2D3 alone or in combination with dexamethasone are considered for therapy in autoimmune diseases (136). Their persistent tolerogenic state and the possibility to pulse them with tissue-specific antigens have made them valuable candidates to treat various diseases, including autoimmune diseases (99, 132, 137). This is illustrated in experimental disease models for T1D, MS, and RA, where administered antigen-specific tDCs migrate to inflammatory sites and reduce disease activity upon administration (102, 138–140). Importantly, DCs with an increased activation status from patients with autoimmune diseases can become equally tolerogenic in response to 1,25(OH)2D3 as healthy DCs (141–145). Because they can also be pulsed with auto-antigens and they can be generated under current Good Manufacturing Practice conditions, this opens up the way for the use of autologous tDCs in the treatment of human autoimmune diseases (141, 146). Currently, the use of tDCs generated with 1,25(OH)2D3 has not been clinically tested. However, tDCs generated using antisense oligonucleotides or Bay11-7082 were found to be safe upon administration in patients with T1D or RA, respectively (147, 148).
It remains to be determined whether these tDCs also have effects on disease activity and whether tDCs generated using 1,25(OH)2D3 could also be used in this context. Increased understanding on how 1,25(OH)2D3, with or without dexamethasone, modulates the DCs can provide insights in how to further optimize the tolerogenic potential of the DCs.
Macrophages are known for their supreme phagocytic capacities, but they are also important APCs. In a normal immune response, an infection activates tissue-resident macrophages after which they produce inflammatory mediators and recruit other immune cells to eradicate the pathogen. Macrophages can roughly be divided into two categories: the M1 and M2 macrophages. M1 macrophages produce pro-inflammatory mediators like nitric oxide, TNFα, IL-23, IL-12, and IL-1β, whereby they kill pathogens and promote the polarization of T helper (Th) cells to T helper 1 (Th1) and Th17 cells to assist in the immune response. On the other hand, M2 macrophages produce the anti-inflammatory cytokine IL-10 and are important in wound repair and restoring tissue homeostasis (149).
The role of macrophages in the pathogenesis of autoimmune diseases is illustrated by an increase in macrophages at inflammatory sites (150–153). In addition, macrophages are hyperactivated and produce more pro-inflammatory cytokines, suggesting a dysregulated balance between M1 and M2 cells (104, 151, 154). As a result of their hyper-inflammatory state, they are essential for the development and activation of β-cell specific cytotoxic T cells, which leads to insulitis in NOD mice (155). Interestingly, the suppression of EAE by 1,25(OH)2D3 is preceded by a rapid reduction of macrophages in the CNS. This suggests that macrophages are another important target for vitamin D in the suppression of autoimmunity (156).
Notably, 1,25(OH)2D3 has dual roles in macrophage differentiation and activation. In the early stages of infection, 1,25(OH)2D3 stimulates differentiation of monocytes into macrophages (157). Furthermore, toll-like receptor triggering or IFNγ-induced activation activates Cyp27B1 and thereby potentiates the conversion of 25(OH)D3 into 1,25(OH)2D3 (158, 159). 1,25(OH)2D3 obtained via this pathway is then required for producing cathelicidin and for the antimicrobial activity of human monocytes and macrophages (160, 161). In addition, 1,25(OH)2D3 induces IL-1β, either directly or via upregulation of C/EBPβ or Erk1/2 (162, 163). So initially, 1,25(OH)2D3 is essential for effective pathogen clearance.
The hyperresponsiveness of VDR−/− mice to LPS stimulation indicates that in the later stages of infection, 1,25(OH)2D3 plays a role in the contraction of the immune response (105). The anti-inflammatory effect of 1,25(OH)2D3 on macrophages is characterized by decreased production of pro-inflammatory factors such as IL-1β, IL-6, TNFα, RANKL, COX-2, and nitric oxide and increased anti-inflammatory IL-10 (104–108). These changes suggest that 1,25(OH)2D3 promotes the M2 phenotype while inhibiting the M1 phenotype, thereby restoring the balance between these subsets. Finally, 1,25(OH)2D3-treated macrophages have reduced T cell stimulatory capacity (108).
In recent years, some advances were made with unraveling the mechanism behind this anti-inflammatory effect of 1,25(OH)2D3 on macrophages. An important target of 1,25(OH)2D3 is thioesterase superfamily member 4 (THEM4), an inhibitor of the NFκB signaling pathway. THEM4 inhibits the direct binding of NFκB to the COX-2 locus and thereby prevents COX-2 transcription (106). Furthermore, THEM4 inhibits IL-6 and TNFα expression by preventing the signaling cascade in which NFκB induces miR-155 to suppress SOCS (105). Whether this THEM4-dependent pathway also inhibits the other pro-inflammatory mediators is not yet clear (104).
The balancing effect of 1,25(OH)2D3 between the pro- and anti-inflammatory status of macrophages is of particular interest in the treatment of autoimmune diseases. Currently, many inflammatory mediators secreted by M1 macrophages, like IL-1β, COX-2, IL-6, and especially TNFα, are already successful therapeutic targets in various autoimmune diseases. However, since current therapies result in systemic reduction of these mediators, patients may become prone to infections. Therefore, it is of interest to understand the mechanism by which 1,25(OH)2D3 balances between pro- and anti-inflammatory actions. This may provide insights in how to suppress the pro-inflammatory cytokines only in case of hyperactivation, without affecting the normal immune response.
B cells are mostly known for their crucial role in the immune response via the differentiation toward plasma cells and the production of antibodies. However, they also modulate the immune response via antigen presentation and cytokine secretion. In the context of autoimmunity, B cells play a crucial role by the production of autoreactive antibodies. These auto-antibodies, like anti-nuclear antibodies (ANAs) in SLE and anti-citrullinated peptide antibodies (ACPA) in RA, can be found in >95% and 70% of patients, respectively (164, 165).
Interestingly, the VDR binds to the promoter region of genes involved in the immune system in lymphoblastoid B cell lines, suggesting a role for B cells in the effect of vitamin D on autoimmune diseases (166). Here, we discuss what is known about the direct effects of 1,25(OH)2D3 on B cell differentiation and the three B cell functions of antibody production, cytokine secretion, and antigen presentation.
Before B cells become plasma cells that secrete high-affinity antibodies, they have to go through various stages of differentiation, class-switch recombination and somatic hypermutation (167). Various reports indicate that 1,25(OH)2D3 reduces the proliferation of B cells, induces their apoptosis and inhibits immunoglobulin class switching (90–92). This inhibition of differentiation may involve preventing nuclear translocation of NF-κB p65 and thereby inhibiting the signaling pathway downstream of CD40 costimulation (93). On the other hand, 1,25(OH)2D3 stimulates plasma cell development when added to terminally differentiating B cells. Furthermore, it induces the chemokine receptor CCR10 on these plasma cells, promoting their migration toward mucosal sites of inflammation (168). Therefore, it appears that the effect of 1,25(OH)2D3 depends on the activation and differentiation status of the B cells.
Independent of the effect of 1,25(OH)2D3 on B cell differentiation, there is ample evidence that it decreases the antibody production (90–92, 94, 95). Interestingly, the presence of ANA is correlated with a lower serum 25(OH)D3 level even in healthy people without SLE (169), while cholecalciferol supplementation decreases auto-antibody titers (65, 109).
Next to antibody production, B cells also secrete cytokines to influence the inflammatory milieu. Interestingly, VDR binds directly to the promoter region of IL-10 in B cells, thereby inducing the expression of IL-10 (75). However, in a cohort of healthy controls and relapsing-remitting MS patients, there was no correlation between IL-10 producing B cells and serum 25(OH)D3 levels (170).
There has been limited research toward the effect of 1,25(OH)2D3 on the APC function of B cells. However one study suggested that B cells primed with 1,25(OH)2D3 have decreased CD86 surface expression. Thereby, these B cells are less potent stimulators of naïve T cell proliferation and cytokine production (96).
Altogether, the effect of 1,25(OH)2D3 on B cells is still not completely clear. Currently, it is hypothesized that 1,25(OH)2D3 inhibits the pathogenic function of B cells in autoimmunity by preventing plasma cell differentiation and thereby auto-antibody production, by inducing IL-10 production and by inhibiting the antigen presentation capabilities. However, the limited amount of studies warrants further research to support this hypothesis and what role these effects play in the suppression of autoimmunity by 1,25(OH)2D3.
Historically, it was thought that DCs were the main target of vitamin D and that effects observed on T cells were mediated via DCs. However, it has now become clear that upon activation, various T cell populations express the VDR, including CD4+ Th cells, CD8+ cytotoxic T cells, and TCRγδ cells (12, 87, 171). This makes the T cell another direct immunological target for 1,25(OH)2D3. The effects of 1,25(OH)2D3 on T cells include modulation of cytokine secretion and differentiation, but VDR is also required for the activation of T cell by propagating TCR signaling (77). Since T cells are proposed to play an important role in the pathogenesis of autoimmunity, we will discuss the effects of 1,25(OH)2D3 on the various T cell populations.
CD4+ T cells are a heterogeneous group of cells, including Th1, Th2, Th17, and Treg cells. In the normal immune response, Th1 cells are important for fighting intracellular pathogens, Th2 cells for helminth infections and Th17 cells for extracellular pathogens and fungi. On the other hand, Tregs mediate immunological tolerance against self-antigens and harmless foreign antigens such as food and intestinal microbiota. Furthermore, they control the immune response via various mechanisms, including the secretion of anti-inflammatory mediators such as IL-10 and TGF-β (172). However, in autoimmune diseases, T cells mediate an immune response against the body itself, suggesting either hyperactivation of the pro-inflammatory T cells or insufficient control by Treg cells, or both.
The importance of the T cells as a target of 1,25(OH)2D3 in experimental autoimmune diseases is illustrated by Mayne et al., who showed that 1,25(OH)2D3 is not able to suppress EAE when the VDR is absent in T cells (173). For these studies, they used the CD4-Cre system, resulting in VDR deficiency in both CD4+ and CD8+ T cells. However, in this disease model, CD4+ T cells are likely the prime 1,25(OH)2D3 target cells, since other studies show that in this model CD8+ T cells are dispensable for the effects of 1,25(OH)2D3 (174). Further strengthening the hypothesis that the suppression of EAE by 1,25(OH)2D3 is driven by modulation of CD4+ T cells, is the finding that 1,25(OH)2D3 prevents CD4+ Th cell migration into the CNS (175). Finally, VDR binding is enriched near SNPs associated with autoimmune diseases in human CD4+ T cells, suggesting that these cells are also important in the effects of 1,25(OH)2D3 in human autoimmunity (8).
Because the effects of 1,25(OH)2D3 differ between the various CD4+ Th cell subsets (110), we will give an overview of the current knowledge on how these individual subsets are modulated by 1,25(OH)2D3 to suppress the autoimmune response.
Th1 and Th2 Cells
Classically, CD4+ T cells were subdivided into two classes: Th1 and Th2 cells. Th1 cells are characterized by the expression of IFNγ and T-bet, while Th2 cells produce IL-4, IL-5, and IL-13 and express the transcription factor GATA3. In the context of autoimmunity, it was long thought that Th1 cells mediate the disease pathogenesis, since mice lacking the transcription factor T-bet are protected against EAE (176). However, the discovery of Th17 cells, which will be discussed in the next section, and the finding that IFNγ is not required for induction of autoimmunity have led to a debate as to whether Th1 cells are important for autoimmune pathogenesis (177, 178). However, since adoptive transfer of myelin-specific IFNγ+ cells induces EAE (179), Th1 cells may still play a role in the disease pathogenesis.
Within Th1 cells, some studies suggest that 1,25(OH)2D3 inhibits IFNγ production when added at the first phases of differentiation (111, 180). On the other hand, another study found no effects on IFNγ (110). This contradiction could be explained by the addition of exogenous IL-2 in the first two studies. Since 1,25(OH)2D3 directly downregulates IL-2, exogenous IL-2 might be required for the inhibition of IFNγ by 1,25(OH)2D3 (181, 182). Although these studies indicate that 1,25(OH)2D3 modulates Th1 cells under certain circumstances, given their relatively small role in autoimmune pathogenesis and the low expression of VDR compared to other CD4+ T cell subsets, it is unlikely that they play an important role in the suppression of autoimmunity by 1,25(OH)2D3 (110, 112).
In contrast to Th1 cells, Th2 cells might be protective in Th17-driven autoimmune diseases even though they are pathogenic in the development of asthma and allergies. Studies in experimental arthritis demonstrate that T cell-specific overexpression of GATA3 is protective in autoimmunity due to suppression of Th17 responses (183). Interestingly, IL-4 is required for 1,25(OH)2D3 to inhibit EAE, suggesting an important role for this cytokine in the effect of 1,25(OH)2D3 (88). In the same model, 1,25(OH)2D3 induces GATA3 and its regulator STAT6. The functional relevance of this upregulation is demonstrated in STAT6-KO mice, where 1,25(OH)2D3 is unable to inhibit EAE development (184). Altogether these studies suggest a role for Th2 induction in the immune suppression by 1,25(OH)2D3.
However, the data on the effect of 1,25(OH)2D3 on Th2 cytokines like IL-4 seems contradictory. When naïve CD4+ T cells or the entire CD4+ T cell population are cultured without polarizing cytokines, 1,25(OH)2D3 induces IL-4 and GATA3 (113, 114). Also, in PBMC of treatment-naïve early RA patients, where IL-4 production is diminished, 1,25(OH)2D3 restores the IL-4 levels to the levels of healthy controls (115). However, when naïve CD4+ T cells, effector CD4+ T cells, or total CD4+ T cells are cultured in the presence of IL-4 to induce Th2 polarization, cellular IL-4 production is unaffected or even inhibited by 1,25(OH)2D3 (111, 180). Also when patients are supplemented with cholecalciferol, there is no increased IL-4 production by their T cells (109, 116, 117). Combining these data leads to the hypothesis that 1,25(OH)2D3 promotes Th2 differentiation and IL-4 production to assist in suppression of autoimmunity, but only when no sufficient IL-4 is present. The mechanism behind the precise regulation of IL-4 is of interest not only for treatment of autoimmunity but also of allergies and asthma where Th2 cytokines play an important pathogenic role.
Th17 Cells
In most autoimmune diseases, Th17 cells are considered to be important drivers of disease pathogenesis. Th17 cells are characterized by production of cytokines such as IL-17A, IL-17F, TNFα, and GM-CSF and the transcription factor RORC2 (RORγt in mice). They can also be distinguished based on the expression of the chemokine receptor CCR6, which directs migration toward the chemokine CCL20. Their differentiation can be driven by TGFβ, IL-6, and IL-1β, but they require IL-23 to become pathogenic Th17 cells (185). In 2003, two hallmark studies showed that IL-23, and not IL-12, is required for the induction of EAE and CIA (186, 187), suggesting an important role for the IL-23/IL-17 immune pathway in the pathogenesis of autoimmune diseases. Indeed, local IL-17A overexpression in mouse knee joints induces an arthritis-like phenotype with inflammation, bone erosions, and damaged cartilage (188). In EAE, the pathogenic cells appear to be the ex-Th17 cells, which now express IFNγ and T-bet, indicating the importance of Th17 plasticity in autoimmune diseases (189). In human autoimmunity, for example, in RA and SLE, levels of Th17 cells are elevated in the peripheral blood and synovial fluid of patients and correlate with disease activity (190–192). Furthermore, specifically the CCR6+ memory Th cells, which include Th17 cells, are potent activators of synovial fibroblasts (190). We have previously shown that this interaction leads to a pro-inflammatory feedback loop with increased production of IL-17A, IL-6, IL-8, and tissue-destructive enzymes. Via this mechanism, Th17 cells may contribute to local joint inflammation in RA (190). Combining the important role of Th17 cells in autoimmunity and the beneficial effect of 1,25(OH)2D3 on autoimmune diseases, it is hypothesized that 1,25(OH)2D3 suppresses autoimmunity at least partially via the inhibition of Th17 activity.
In support of this hypothesis, the effect of 1,25(OH)2D3 on an experimental model for antiretinal autoimmunity depends on inhibiting Th17 activity (193). Also in vitro 1,25(OH)2D3 decreases expression of pro-inflammatory cytokines like IL-17A, IL-17F, and IL-22 in CD4+ T cells, CD4+ memory cells, or CD4+CCR6+ memory cells (115, 118–120). Functionally, this decrease in Th17 activity diminishes activation of synovial fibroblasts, thereby inhibiting the pro-inflammatory loop between these cell types (120). Interestingly, 1,25(OH)2D3 also inhibits the secretion of IL-17A and other Th17 cytokines in the presence of Th17-polarizing cytokines (119, 121).
1,25(OH)2D3 not only inhibits the activity of Th17 cells but also Th17 differentiation. When naïve CD4+ T cells are differentiated toward the Th17 lineage in vitro, the presence of 1,25(OH)2D3 inhibits Th17-related cytokines and transcription factors such as IL-17A, IL-17F, RORC, and CCR6 (110, 112, 122). Functionally, MOG-specific Th17 cells differentiated in the presence of 1,25(OH)2D3 are less capable of inducing EAE upon adoptive transfer (119). Aside from the decreased pathogenicity of the cells, this effect may also be due to a decrease in CCR6, the chemokine receptor required for migration to the CNS (123).
Although the inhibitory effect on Th17 activity is well described, the mechanisms behind it are less clear. First of all, Joshi et al. showed that the regulation of IL-17A can be mediated via direct binding of the VDR to the IL-17A promoter. VDR–RXR complexes compete with NFAT for the binding sites in the promoter, after which they recruit RUNX1 and HDAC (histone deacetylase) to inhibit IL-17A gene expression (119). This competition for the NFAT binding site also occurs at the promoter of IL-2, a known primary 1,25(OH)2D3 target gene, suggesting that this may be a general mechanism that also applies to other NFAT-regulated genes (181). Recruitment of HDAC indicates that epigenetic regulation is also important in the inhibition of IL-17A by 1,25(OH)2D3, especially given the relative epigenetic instability of the IL-17A gene locus (194). Aside from this direct regulation of IL-17A, other mechanisms have also been proposed. One study showed that CHOP is crucial for the inhibitory effect of 1,25(OH)2D3, while a second study indicated IRF8 to be important (112, 122). Yet another study indicated that VDR forms a complex with VDR, RXR, HDAC2, and Smad3 to inhibit Smad7 transcription, thereby preventing IL-17A production (124). Of note, TGFβ is the cytokine that induces Smad3 and Erk, leading to this inhibition of IL-17A, but it is also the cytokine responsible for inducing the VDR (121). How these mechanisms relate to each other remains to be investigated.
Th17.1 Cells
Before the discovery of Th17 cells, it was thought that Th1 cells, characterized by expression of IFNγ, T-bet, and CXCR3, were the major drivers of the autoimmune response. The finding that IL-23, and not IL-12, was required for experimental autoimmunity, at first completely shifted the viewpoint toward Th17 cells as the pathogenic drivers of autoimmunity. However, lately more and more studies indicate that the subdivision into Th17 and Th1 is not as linear as previously assumed. Upon stimulation by IL-12 or TNFα, Th17 cells can become double producers of IL-17A and IFNγ or even shift toward high IFNγ production with little or no IL-17A. Since these latter cells still express CCR6 and RORC, together with T-bet and CXCR3, they are called non-classic Th1 or Th17.1 cells (195). Currently, it is hypothesized that the Th17.1 cells are more pathogenic than Th17 cells in autoimmune diseases, because they are enriched at the sites of inflammation in several diseases (196, 197).
Interestingly, we have shown that in CCR6+ cells, which includes Th17 and Th17.1 cells, 1,25(OH)2D3 reduces the frequency of IFNγ+, IL-17A+, and IFNγ+ IL-17A+ cells (120). This suggests that 1,25(OH)2D3 can inhibit Th cell pathogenicity in autoimmunity via the inhibition of Th17 and Th17.1 cells. A similar effect was found in the CD4+ T cells of SLE patients supplemented with 10,400 IU cholecalciferol for 6 months (198). Other supplementation studies have not addressed the combined or single expression of IFNγ and IL-17A, but the results on total IL-17A+ or total IFNγ+ cells are ambiguous (109, 116, 117).
Regulatory T Cells
In contrast to the pro-inflammatory Th subsets mentioned above, regulatory T cells, or Tregs, suppress the immune response. Tregs express FoxP3, the anti-inflammatory cytokines IL-10 and TGFβ, the inhibitory co-receptor CTLA4, and a high level of CD25. They exert immunomodulatory effects on other immune cells such as macrophages, DCs, CD8+ T cells, and also other CD4+ T cells, thereby maintaining immune homeostasis. Their essential role in preventing autoimmunity is demonstrated in patients with a mutation in FoxP3. These patients are suffering from the IPEX syndrome, which is characterized by massive autoimmunity (199). In the autoimmune diseases discussed here, it is hypothesized that an imbalance between pro-inflammatory T cells, such as Th17 or Th17.1, and Tregs underlies the immune pathogenesis. 1,25(OH)2D3 may act by restoring this balance and thereby restoring immune homeostasis.
Indeed, 1,25(OH)2D3 induces FoxP3+ Tregs in the spleen, lymph nodes, and spinal cord of EAE mice (119, 124). Additionally, without IL-10 or IL-10-mediated signaling, 1,25(OH)2D3 cannot inhibit EAE (200). In in vitro cultures of Tregs, either obtained via in vitro polarization or sorted from peripheral blood, 1,25(OH)2D3 induces the production of IL-10, but not FoxP3 (114, 201, 202). Polarized Tregs express a higher level of Treg-associated markers such as CTLA4, PD1, and CD25 and their suppressive capacity is enhanced by 1,25(OH)2D3 (202). Also, the suppressive capacity of Tregs is positively correlated with the serum 25(OH)D3 level in MS patients (203). However, when sorted Tregs are used, 1,25(OH)2D3 does not further enhance their suppressive capacity (114, 201). This suggests that 1,25(OH)2D3 optimizes Treg function in order to suppress autoimmunity.
Interestingly, 1,25(OH)2D3 also induces IL-10 production when CD4+ cells are cultured under neutral conditions, and even further in the presence of Th17 polarizing cytokines. Furthermore, in these cultures, 1,25(OH)2D3 also induces FoxP3 and CTLA4, while enhancing the suppressive capacity of the cells (113, 118, 119, 121, 122, 124, 125). Because 1,25(OH)2D3 inhibits Th17 polarization while inducing IL-10 in these cultures, it was postulated that 1,25(OH)2D3 may inhibit Th17 activity via IL-10 induction. However, IL-10 is dispensable for the inhibition of IL-17A, suggesting that Th17 inhibition and Treg induction are two independent mechanisms of 1,25(OH)2D3 (110).
On a molecular level, three mechanisms have been proposed by which 1,25(OH)2D3 can stimulate a Treg-like phenotype even under Th17 polarizing conditions. First, the VDR can bind to three VDREs in the conserved non-coding sequence of the FoxP3 promoter, thereby directly controlling FoxP3 transcription (119, 125). The second mechanism is by reversing the inhibitory effect of Th17 polarizing cytokines on CTLA4, leading to upregulation of CTLA4 (121). Finally, 1,25(OH)2D3 induces the expression of IDO, which increases the number of Tregs (76). The latter finding is interesting, since IDO was also reported to be important for the induction of tDCs (see Dendritic Cells) (102), suggesting it might be a general target of 1,25(OH)2D3 in the immune system.
Although the in vitro data demonstrate that 1,25(OH)2D3 induces Treg cells, not all cholecalciferol supplementation studies find an effect on Tregs. Several studies suggest an increase in the proportion or number of Treg cells based on surface marker expression (109, 116, 204) or based on IL-10 production (50, 117). However, another study did not find this induction in Treg cells (63), and Treg suppressive function is unaffected by cholecalciferol supplementation (117).
Overall, in CD4+ T cells, 1,25(OH)2D3 inhibits the pro-inflammatory Th cell functions while stimulating Treg activity. These effects are observed under both healthy and pathogenic conditions, such as in patients with autoimmune diseases (201). Therefore, restoring the disturbed balance between effector T cells and Treg cells may underlie the beneficial effects of 1,25(OH)2D3 on autoimmunity.
In addition to CD4+ T cells, cytotoxic CD8+ T cells comprise the second important class within the T cells. These cells contribute to the immune response by inducing apoptosis in abnormal cells, for example, in case of infection or uncontrolled growth in cancer. In addition, they modulate other immune cells by secreting cytokines (205). Although the role of CD8+ T cells in autoimmune diseases is not as well characterized as the role of CD4+ T cells, various studies indicate that they play a role in disease pathogenesis. For example, myelin-specific CD8+ T cells induce EAE in mice, with characteristics of human MS that are not conferred by myelin-specific CD4+ T cells (206, 207). Similarly, hsp60-specific CD8+ T cells induce autoimmune intestinal inflammation (208). More recently, it was shown that IL-17A+ CD8+ T cells are enriched in the synovial fluid of psoriatic arthritis patients. These cells do not express cytolytic markers, but their levels are positively correlated with markers of disease activity (209). Since CD8+ T cells have a higher expression of VDR than CD4+ T cells (171), CD8+ T cells may also be a target for 1,25(OH)2D3 in the suppression of autoimmunity.
Indeed, adoptive transfer of VDR−/− CD8+ T cells in Rag-deficient mice induces intestinal inflammation. When VDR−/− IL-10−/− CD8+ T cells are transferred, the intestinal inflammation is even worse and leads to wasting disease (79). The increased proliferation of VDR−/− CD8+ T cells, even in the naive state, suggests that VDR-induced signaling is required for maintaining quiescence of these cells. Thereby 1,25(OH)2D3 prevented hyperactivation of CD8+ T cells and subsequent autoimmune pathology in diseases such as CD (79). In addition to maintaining quiescence, 1,25(OH)2D3 also inhibits the secretion of IFNγ and TNFα by activated CD8+ T cells (80). Finally, topical treatment with calcipotriol decreases the frequency of IL-17A+ CD8+ cells in psoriatic lesions, which is interesting in light of the correlations between these cells and disease activity in psoriatic arthritis (82, 209).
Aside from modulating the activity of the classical CD8+ T cells to reduce autoimmunity, 1,25(OH)2D3 is also important in the development of CD8αα+ T cells. CD8αα+ T cells are self-reactive cells that have a regulatory function by maintaining homeostasis in the gut. In VDR−/− mice, the number of these cells is reduced, which may explain the susceptibility of these animals to intestinal inflammation (81).
It is important to note that the effect of 1,25(OH)2D3 is not mediated via the CD8+ T cells in every autoimmune disease, since they were dispensable for the attenuation of EAE by 1,25(OH)2D3 (174). However, it seems that in IBD and psoriatic arthritis, the CD8+ T cells are target for 1,25(OH)2D3. It will be of great interest to determine what the role of the CD8+ T cells is in the effect of 1,25(OH)2D3 on other autoimmune diseases. This will not only provide insight into the mechanisms behind the effect of vitamin D but also about the differences in pathogenesis in the various autoimmune diseases.
Next to the traditional CD4+ and CD8+ T cells, there are also cells expressing the TCR but lacking both CD4 and CD8. These so-called unconventional T cells have a less diverse TCR repertoire and they are not restricted to MHC class I or II. The unconventional T cells include mucosal-associated invariant T (MAIT) cells, TCRγδ T cells and natural killer T (NKT) cells.
Although MAIT cells have been implicated to be suppressive in autoimmunity, as reviewed by Godfrey et al. (210), there is currently no data available on the effect of 1,25(OH)2D3 on these cells.
TCRγδ T cells are rapid responders in the event of an infection with intracellular pathogens, due to their recognition of phosphoantigens. Interestingly, they are pathogenic in autoimmune models like EAE and CIA and they produce a wide range of pro-inflammatory cytokines like IL-17A, IL-17F, GM-CSF, TNFα, and IFNγ (211). There is only one study that investigated the effect of 1,25(OH)2D3 on the pro-inflammatory activity of these cells. They demonstrated that TCRγδ T cells express the VDR upon activation. In response to 1,25(OH)2D3, the production of IFNγ and the proliferation of these cells was inhibited (87). Currently, it is thought that the main pathogenic action of the TCRγδ T cells in autoimmunity is the secretion of IL-17A (211). Unfortunately, there are no data available yet that describe the effect of 1,25(OH)2D3 on this cytokine, or any of the other cytokines secreted by the TCRγδ T cells.
The last subset of unconventional T cells that will be discussed here are the NKT cells. They recognize glycolipid antigens and are thereby involved in the protection against a wide range of pathogens. Upon TCR stimulation, NKT cells can rapidly secrete various pro-inflammatory cytokines, including IL-4, IFNγ, and IL-17A. NKT cells can be divided into type I and type II NKT cells. Type I NKT cells are also called invariant NKT (iNKT) cells due to their invariant TCR. Type II NKT cells have a variable TCR and are therefore called the variant NKT cells. The exact role of NKT cells in the pathogenesis of autoimmune disease is not yet completely clear. They are pathogenic in CIA, but they are protective in EAE, T1D, and SLE (88, 212).
Interestingly, VDR is required in the thymus for the development of functionally mature iNKT cells. Furthermore, the iNKT cells in VDR−/− mice are hyporesponsive to TCR stimulation (89). In addition, the protective effect of 1,25(OH)2D3 in EAE is partially dependent on iNKT cells, possibly via inducing IL-4 in these cells (88). These data suggest that 1,25(OH)2D3 promotes a suppressive function of iNKT cells. However, given the two-sided effect of iNKT cells in the different autoimmune diseases, further research is needed to fully examine the effect of 1,25(OH)2D3 on iNKT cell activity and what this means for each individual disease.
Recently, a new group of cells became the center of attention in the field of immunology; the innate lymphoid cells (ILC). ILCs play an important role in tissue repair, tissue homeostasis, and the immune response against bacteria, viruses, and fungi. ILCs can be grouped into three classes as follows: (i) the group 1 ILCs (ILC1) that secrete IFNγ and depend on T-bet expression, (ii) the group 2 ILCs (ILC2) that secrete type 2 cytokines such as IL-5 and IL-13 and depend on GATA3, and (iii) the group 3 ILCs (ILC3) that secrete IL-17A and/or IL-22 and depend on RORC (213).
The ILC1s include natural killer cells, which have been known for a longer time and play a role in the clearance of viruses. Since viral triggers are thought to play a role in the initiation of some autoimmune diseases, the NK cells have been investigated for their role in this context. However, under some circumstances, NK cells are protective, while in others they can be pathogenic as recently reviewed by Poggi and Zocchi (214). Also the data on the effect of 1,25(OH)2D3 on NK cells are somewhat contradictory. In an NK cell line, 1,25(OH)2D3 induces the cytolytic killing capacity of NK cells (83), but this effect has not been found in healthy control peripheral blood (84, 85). However, when 1,25(OH)2D3 is added during the in vitro differentiation of NK cells from hematopoietic stem cells, the development of NK cells is impaired and their cytotoxicity and IFNγ production are reduced (84). Interestingly, 1,25(OH)2D3 specifically inhibits activation, cytotoxic capacity and pro-inflammatory cytokine production in overactivated NK cells in women with recurrent pregnancy losses (85). This supports a hypothesis in which 1,25(OH)2D3 is not a general inhibitor of the immune response, but rather a regulator of immune homeostasis. Therefore, it is of interest whether this abnormal NK activation is also seen in autoimmune diseases and can be modulated by 1,25(OH)2D3.
Based on their cytokine signature, it can be hypothesized that in the context of autoimmunity ILC3 cells play a role in disease pathogenesis. Indeed, an increase in ILC3 cells has been demonstrated in the lesional skin of psoriasis patients (215, 216), in the inflamed intestine of CD patients (217), in the peripheral blood of MS patients (218), and in the gut, peripheral blood, bone marrow, and synovial fluid of patients with ankylosing spondylitis (219). Furthermore, ILC3 were shown to be responsible for experimental innate-induced colitis (220). Interestingly, in VDR-KO mice, which are susceptible for colitis, the levels of ILC1 and ILC3 are increased (86). On the other hand, calcipotriol treatment did not affect the frequencies of ILC subsets in psoriatic skin lesions after 2 weeks (82).
Since the research into ILC has only started to expand in recent years, the effects of 1,25(OH)2D3 on these cells have not been investigated extensively. Current data suggest that 1,25(OH)2D3 may also have anti-inflammatory effects on these cells, but more studies are required to distinguish the effects on the different subsets and its role in the protective effect of vitamin D in autoimmunity.
In the previous sections, we discussed the direct modulatory effects of 1,25(OH)2D3 on various cells of the immune system. However, 1,25(OH)2D3 and the VDR also affect tissue-resident cells, such as hepatic and pancreatic stellate cells, and the inflammatory mediators that they secrete (221, 222). This indirect mechanism of immune modulation by 1,25(OH)2D3 is also relevant in autoimmune diseases. For example, in RA, the interaction between T cells and synovial fibroblasts contributes to disease pathogenesis (190). Therefore, it is also of interest to study the effect of 1,25(OH)2D3 on the tissue-resident cells in the context of autoimmunity.
Similar to the tissue-resident tissue cells in liver and pancreas, 1,25(OH)2D3 also directly affects RA synovial fibroblasts. Not only is the IL-1β-induced production of tissue-degrading matrix metalloprotease 1 inhibited, also the infiltration capacity of RA fibroblasts is reduced upon treatment with 1,25(OH)2D3 (223). But this effect on tissue-resident cells is not only found in the synovial cells. It was also shown that the VDR is required for intestinal homeostasis by limiting the production of IL-6 by epithelial cells through inhibition of the NFκB pathway (224). Finally, 1,25(OH)2D3 also affects brain pericytes, which may be relevant for MS. The pericytes line the epithelial cells of blood vessels, and in the brain, they are important for maintaining the blood–brain barrier and neuron functioning. Brain pericytes cells produce less pro-inflammatory genes when exposed to 1,25(OH)2D3 while upregulating anti-inflammatory genes. Interestingly, brain pericytes express Cyp27B1 upon stimulation with TNFα and IFNγ. This indicates that an inflammatory environment promotes the conversion of 25(OH)D3 into 1,25(OH)2D3, which then can dampen the inflammation by modulating the pericytes (225).
Overall, the indirect effects of vitamin D and the VDR on immune cells via tissue-resident cells have been underexposed in the past years. However, if we truly want to understand the molecular mechanisms by which 1,25(OH)2D3 acts in autoimmune diseases, these effects are very important for future studies.
In this review, we have discussed the advancements that have been made regarding the clinical effects of vitamin D and the molecular mechanisms that underlie these effects. However, there is still a lot that is unclear at the moment, which will be subject of investigation in the coming years.
Based on the current data on the effect of vitamin D supplementation, it is still not possible to draw conclusions about the added value for the treatment of autoimmunity. This is due to the low number of trials, small patient numbers and heterogeneity in trial setup. In order to determine the therapeutic value of vitamin D supplementation, there are two big open questions that need to be addressed.
First, it is important to assess what serum 25(OH)D3 level is required for a beneficial effect of vitamin D in autoimmune diseases. Based on the requirements for calcium homeostasis, current guidelines indicate that a level below 50 nmol/L corresponds with deficiency, between 50 and 74 nmol/L as insufficiency and above 75 nmol/L as a sufficient 25(OH)D3 level (226, 227). However, in the context of autoimmunity, it is not known whether it is enough to correct deficiency or whether we should strive for an even higher serum 25(OH)D3 level. Using 75 nmol/L as a cut-off point, Raftery et al. showed that CD patients with sufficient serum 25(OH)D3 have significantly higher quality of life and less severe disease as measured by intestinal permeability, LL-37 expression, and CDAI (59). Furthermore, in healthy individuals, the serum 25(OH)D3 level is correlated with number of VDR binding sites in CD4+ T cells. When they have a level above 75 nmol/L, the VDR binding is enriched near genes associated with autoimmune diseases and Tregs (8). However, clinical trials, either with or without placebo controls, do not consistently find immune modulation regardless of the baseline and endpoint serum 25(OH)D3 level (Table 2). It should be noted that these measurements have been done in the peripheral blood or in cells from the peripheral blood, which is not the site of inflammation and therefore may not be the most relevant place to look for immunological effects.
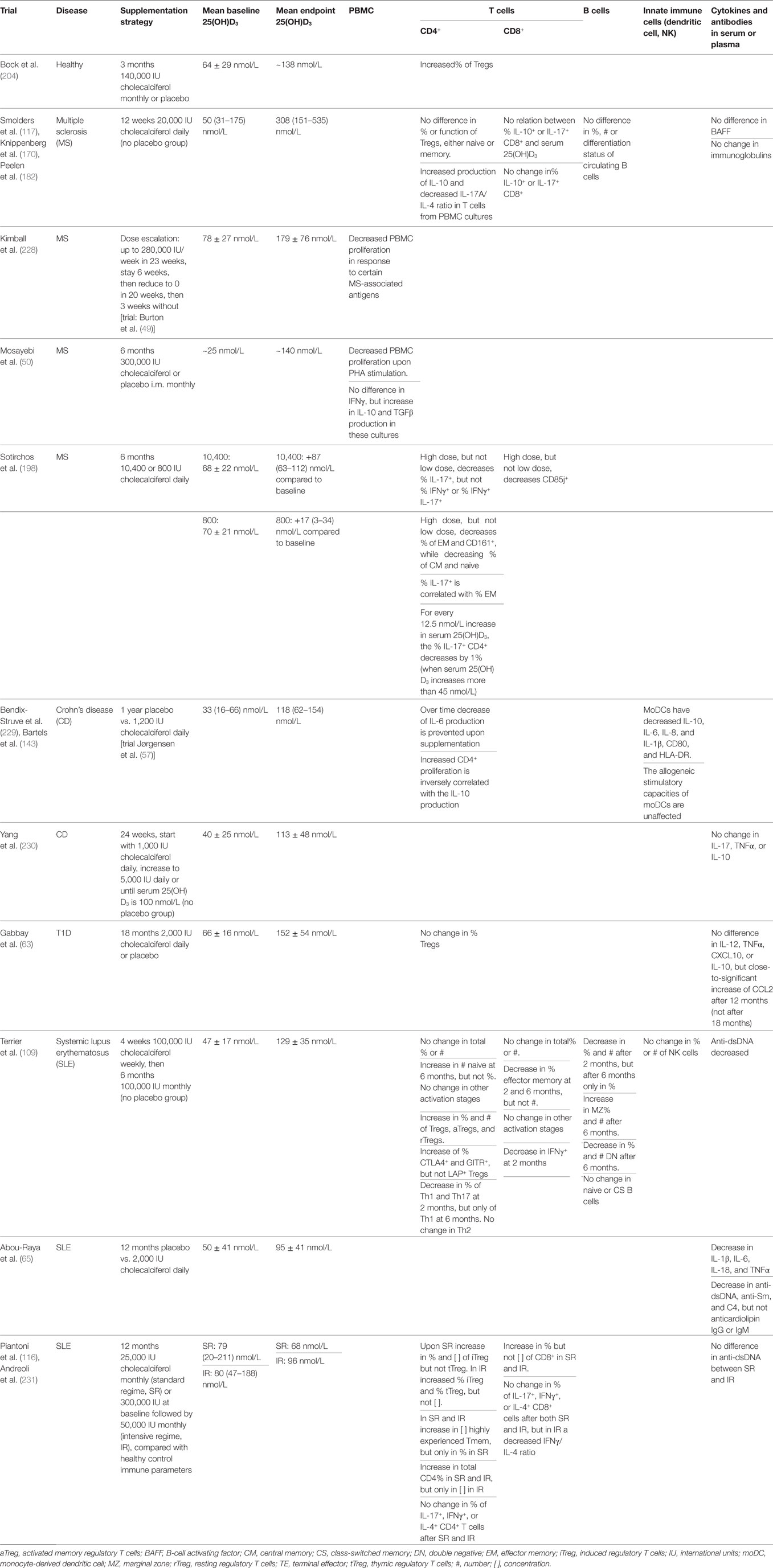
Table 2. Overview of clinical trials looking at immunological parameters after vitamin D supplementation.
The second question that is still matter of debate is in what form and dosage vitamin D should be supplemented. In the experimental autoimmune models, animals are mostly supplemented with a high dose of 1,25(OH)2D3, but in humans, this strategy may lead to hypercalcemia. Therefore, most clinical trials use cholecalciferol as the form of choice, although some use 1,25(OH)2D3 or less calcemic analogs like alfacalcidol. Of note, a study comparing the effects of alfacalcidol [analog for 1,25(OH)2D3] with colecalciferol (analog for cholecalciferol) indicates that in the short term alfacalcidol might be more effective, but this effect disappears after 12 months (232). Analogs like calcipotriol that are used in the topical treatment of psoriasis have not been tested in the other autoimmune diseases that were discussed here. Other analogs have been developed, which show equal or better immunomodulatory potential and have been successfully used in experimental autoimmune diseases (201, 233–237). The only analog that was used in clinical trials was alfacalcidol, mainly in type 1 diabetes patients (Table 1). However, the effects of alfacalcidol do not seem better than calcitriol, and at the same dosage, there were no severe side effects from either alfacalcidol or calcitriol (61, 62, 64). More research into the actual effects of vitamin D analogs on human autoimmune disease is required for establishing whether these analogs can be used safely and effectively. Furthermore, in the clinical trials performed so far, there were no serious adverse events after cholecalciferol supplementation. Therefore, it is important to establish the added value of the vitamin D analogs compared to cholecalciferol supplementation. Currently, cholecalciferol is the most used supplementation form in clinical practice. Vitamin D supplementation guidelines indicate a maximum safe dose of 4,000 IU cholecalciferol/day for healthy adults (226). However, no adverse effects were found with dosages of up to 50,000 IU cholecalciferol weekly for 12 weeks, or 100,000 IU weekly for 1 month followed by 100,000 IU monthly for 5 months (54, 109, 117). Interestingly, the dose-escalation regime used by Burton et al. and 20,000 IU weekly by Smolders et al. did not elicit hypercalcemia despite reaching a serum 25(OH)D3 level of 400 and 380 nmol/L, respectively (49, 117).
In considering the best strategy for cholecalciferol supplementation, it should also not be forgotten that 1,25(OH)2D3 may have a synergistic effect with other treatments. For example, in vitro studies have shown that 1,25(OH)2D3 synergizes with retinoic acid (an active vitamin A metabolite) or dexamethason in the inhibition of Th17 pathogenicity (115, 238). Also in monocytes, the combination of dexamethasone and 1,25(OH)2D3 has added effects over the compounds separately, partially because 1,25(OH)2D3 enhances the effects of the glucocorticoid receptor (239, 240). Furthermore, we have previously shown that 1,25(OH)2D3 has an added effect on TNFα blockade in inhibiting the pro-inflammatory loop between Th17 cells and RASF in RA, suggesting that vitamin D combined with anti-TNFα could yield a better treatment response in the treatment of RA patients (120). Finally, combining 1,25(OH)2D3 with lovastatin has an added therapeutic effect on EAE. This is due to the inhibition of RhoA-ROCK signaling in autoreactive T cells, leading to decreased expression of Cyp24A1 and thereby less inactivation of 1,25(OH)2D3 (241). Altogether, these data indicate that it may be worthwhile to investigate the addition of cholecalciferol to current treatments like anti-TNFα, or to combine cholecalciferol with, for example, retinoic acid or statins. Due to the synergy between 1,25(OH)2D3 and these already approved drugs, a lower dose of cholecalciferol may be sufficient for achieving beneficial clinical effects.
Currently, several clinical trials are ongoing and recruiting patients in MS (http://clinicaltrials.gov identifier NCT01490502), RA (NCT02243800), and IBD (NCT02704624, NCT01046773, NCT02208310) for which the results are expected in the coming 3–5 years. Hopefully, they can provide more insight into the answers on these remaining questions. However, to firmly establish the added value of cholecalciferol supplementation, large multicenter trials are required. Ideally, in these trials, the patients should be randomized into different treat-to-target arms, in which every arm has a target 25(OH)D3 serum level, such as 75, 100, and 150 nmol/L. Since the effect of cholecalciferol alone is probably not sufficient to control disease activity, patients should receive standard care following pre-defined, harmonized treatment protocols in addition to the cholecalciferol supplementation.
In addition to the studies where cholecalciferol has been supplemented, attention has also focused on understanding the immunomodulatory effects of 1,25(OH)2D3 on a cellular level. Based on the current knowledge, 1,25(OH)2D3 reduced the pathogenicity of DCs, macrophages, CD4+ T cells, CD8+ T cells, and B cells. Similar effects have been observed in γδ T cells, iNKT cells, and ILCs, but more research is necessary to confirm these data (see section 5). It should be noted that 1,25(OH)2D3 does not merely work as an anti-inflammatory agent. Instead, 1,25(OH)2D3 assists in maintaining the balance between a pro- and anti-inflammatory state and is thereby able to restore the disturbed balance that is associated with autoimmunity.
This balancing effect of 1,25(OH)2D3 is best illustrated in monocytes and macrophages, where it has pro-inflammatory effects in the early stages of activation but later shifts to an anti-inflammatory state (242). Therefore, it is interesting to study the effects of 1,25(OH)2D3 in more detail in the various stages of differentiation and activation from monocyte to macrophage. The Carlberg lab has performed ChIP-seq experiments in the monocytic THP-1 cell line at early time points (5). Detailed studies have revealed several primary target genes such as ASAP2 and THBD (243–245), but also identified Bcl6 as a primary target that mediates important secondary responses (246). Next to the primary target genes, combining the ChIP-seq dataset with publically available ChIA-PET and FAIRE-seq datasets has improved the knowledge on VDR binding kinetics (247, 248).
This is just an example of how next-generation sequencing techniques can be combined to yield more understanding of the molecular mechanisms behind the effects of 1,25(OH)2D3. Since it has already been shown that 1,25(OH)2D3 has different effects on every cell type, even closely related cell types such as Th1 and Th17 (110), it will be interesting to study VDR DNA binding and identify primary target genes in separate cell types. This will give insight into the similarities and differences between the effects of 1,25(OH)2D3 on each cell, and what will be important to balance the immune response in patients with autoimmune diseases.
Although various studies have shown a beneficial effect of cholecalciferol supplementation in autoimmune diseases, there are also studies that do not find any effect on disease parameters. This might be due to the supplementation strategy or the subjects included in the study, which are issues that should be addressed in properly designed multicenter clinical trials.
However, it is also possible that systemic cholecalciferol supplementation is not sufficient to establish effects in every patient. Therefore, another way to use the immunomodulatory effects of vitamin D to the advantage of patients with autoimmune diseases is to mimic the effects by targeting important pathways within immune cells. In order to do this, it is crucial to understand the working mechanisms of 1,25(OH)2D3. In the coming years, attention should be paid toward unraveling these molecular mechanisms to optimize the therapeutic potential of vitamin D.
WD has performed literature research, designed the review layout, and written the review. EC has designed the review layout, contributed to the clinical section, and revised the manuscript. JH has designed the review layout and revised the manuscript. EL has designed the review layout, contributed to the molecular section, and revised the manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
This project was funded by the Dutch Arthritis Foundation (Reumafonds, grant no DAA 10-1-407 to E.L.).
1. Lerner A, Jeremias P, Matthias T. The world incidence and prevalence of autoimmune diseases is increasing. Int J Celiac Dis (2015) 3(4):151–5. doi: 10.12691/ijcd-3-4-8
2. Chang C. Unmet needs in the treatment of autoimmunity: from aspirin to stem cells. Autoimmun Rev (2014) 13(4–5):331–46. doi:10.1016/j.autrev.2014.01.052
3. Christakos S, Ajibade DV, Dhawan P, Fechner AJ, Mady LJ. Vitamin D: metabolism. Endocrinol Metab Clin North Am (2010) 39(2):243–53. doi:10.1016/j.ecl.2010.02.002
4. Pike JW, Meyer MB. The vitamin D receptor: new paradigms for the regulation of gene expression by 1,25-dihydroxyvitamin D(3). Endocrinol Metab Clin North Am (2010) 39(2):255–69. doi:10.1016/j.ecl.2010.02.007
5. Heikkinen S, Vaisanen S, Pehkonen P, Seuter S, Benes V, Carlberg C. Nuclear hormone 1alpha,25-dihydroxyvitamin D3 elicits a genome-wide shift in the locations of VDR chromatin occupancy. Nucleic Acids Res (2011) 39(21):9181–93. doi:10.1093/nar/gkr654
6. Evans RM, Mangelsdorf DJ. Nuclear receptors, RXR, and the big bang. Cell (2014) 157(1):255–66. doi:10.1016/j.cell.2014.03.012
7. Meyer MB, Goetsch PD, Pike JW. VDR/RXR and TCF4/beta-catenin cistromes in colonic cells of colorectal tumor origin: impact on c-FOS and c-MYC gene expression. Mol Endocrinol (2012) 26(1):37–51. doi:10.1210/me.2011-1109
8. Handel AE, Sandve GK, Disanto G, Berlanga-Taylor AJ, Gallone G, Hanwell H, et al. Vitamin D receptor ChIP-seq in primary CD4+ cells: relationship to serum 25-hydroxyvitamin D levels and autoimmune disease. BMC Med (2013) 11:163. doi:10.1186/1741-7015-11-163
9. Carmeliet G, Dermauw V, Bouillon R. Vitamin D signaling in calcium and bone homeostasis: a delicate balance. Best Pract Res Clin Endocrinol Metab (2015) 29(4):621–31. doi:10.1016/j.beem.2015.06.001
10. Ellman P, Anderson KH. Calciferol in tuberculous peritonitis with disseminated tuberculosis. Br Med J (1948) 1(4547):394. doi:10.1136/bmj.1.4547.394
11. Bhalla AK, Amento EP, Clemens TL, Holick MF, Krane SM. Specific high-affinity receptors for 1,25-dihydroxyvitamin D3 in human peripheral blood mononuclear cells: presence in monocytes and induction in T lymphocytes following activation. J Clin Endocrinol Metab (1983) 57(6):1308–10. doi:10.1210/jcem-57-6-1308
12. Provvedini DM, Tsoukas CD, Deftos LJ, Manolagas SC. 1,25-dihydroxyvitamin D3 receptors in human leukocytes. Science (1983) 221(4616):1181–3. doi:10.1126/science.6310748
13. Lemire JM, Archer DC. 1,25-dihydroxyvitamin D3 prevents the in vivo induction of murine experimental autoimmune encephalomyelitis. J Clin Invest (1991) 87(3):1103–7. doi:10.1172/JCI115072
14. Cantorna MT, Hayes CE, DeLuca HF. 1,25-Dihydroxyvitamin D3 reversibly blocks the progression of relapsing encephalomyelitis, a model of multiple sclerosis. Proc Natl Acad Sci U S A (1996) 93(15):7861–4. doi:10.1073/pnas.93.15.7861
15. Cantorna MT, Hayes CE, DeLuca HF. 1,25-Dihydroxycholecalciferol inhibits the progression of arthritis in murine models of human arthritis. J Nutr (1998) 128(1):68–72.
16. Zwerina K, Baum W, Axmann R, Heiland GR, Distler JH, Smolen J, et al. Vitamin D receptor regulates TNF-mediated arthritis. Ann Rheum Dis (2011) 70(6):1122–9. doi:10.1136/ard.2010.142331
17. Zhang H, Wu H, Liu L, Li H, Shih DQ, Zhang X. 1,25-dihydroxyvitamin D3 regulates the development of chronic colitis by modulating both T helper (Th)1 and Th17 activation. APMIS (2015) 123(6):490–501. doi:10.1111/apm.12378
18. Cantorna MT, Munsick C, Bemiss C, Mahon BD. 1,25-Dihydroxycholecalciferol prevents and ameliorates symptoms of experimental murine inflammatory bowel disease. J Nutr (2000) 130(11):2648–52.
19. Mathieu C, Laureys J, Sobis H, Vandeputte M, Waer M, Bouillon R. 1,25-Dihydroxyvitamin D3 prevents insulitis in NOD mice. Diabetes (1992) 41(11):1491–5. doi:10.2337/diabetes.41.11.1491
20. Mathieu C, Waer M, Laureys J, Rutgeerts O, Bouillon R. Prevention of autoimmune diabetes in NOD mice by 1,25 dihydroxyvitamin D3. Diabetologia (1994) 37(6):552–8. doi:10.1007/BF00403372
21. Lemire JM, Ince A, Takashima M. 1,25-Dihydroxyvitamin D3 attenuates the expression of experimental murine lupus of MRL/l mice. Autoimmunity (1992) 12(2):143–8. doi:10.3109/08916939209150321
22. Simpson S Jr, Blizzard L, Otahal P, Van der Mei I, Taylor B. Latitude is significantly associated with the prevalence of multiple sclerosis: a meta-analysis. J Neurol Neurosurg Psychiatry (2011) 82(10):1132–41. doi:10.1136/jnnp.2011.240432
23. Mohr SB, Garland CF, Gorham ED, Garland FC. The association between ultraviolet B irradiance, vitamin D status and incidence rates of type 1 diabetes in 51 regions worldwide. Diabetologia (2008) 51(8):1391–8. doi:10.1007/s00125-008-1061-5
24. Szilagyi A, Leighton H, Burstein B, Xue X. Latitude, sunshine, and human lactase phenotype distributions may contribute to geographic patterns of modern disease: the inflammatory bowel disease model. Clin Epidemiol (2014) 6:183–98. doi:10.2147/CLEP.S59838
25. Dobson R, Giovannoni G, Ramagopalan S. The month of birth effect in multiple sclerosis: systematic review, meta-analysis and effect of latitude. J Neurol Neurosurg Psychiatry (2013) 84(4):427–32. doi:10.1136/jnnp-2012-303934
26. Torkildsen O, Grytten N, Aarseth J, Myhr KM, Kampman MT. Month of birth as a risk factor for multiple sclerosis: an update. Acta Neurol Scand (2012) 126(Supp. 195):58–62. doi:10.1111/ane.12040
27. Song GG, Bae SC, Lee YH. Association between vitamin D intake and the risk of rheumatoid arthritis: a meta-analysis. Clin Rheumatol (2012) 31(12):1733–9. doi:10.1007/s10067-012-2080-7
28. Zipitis CS, Akobeng AK. Vitamin D supplementation in early childhood and risk of type 1 diabetes: a systematic review and meta-analysis. Arch Dis Child (2008) 93(6):512–7. doi:10.1136/adc.2007.128579
29. Dong JY, Zhang WG, Chen JJ, Zhang ZL, Han SF, Qin LQ. Vitamin D intake and risk of type 1 diabetes: a meta-analysis of observational studies. Nutrients (2013) 5(9):3551–62. doi:10.3390/nu5093551
30. Shen L, Zhuang QS, Ji HF. Assessment of vitamin D levels in type 1 and type 2 diabetes patients: results from meta-analysis. Mol Nutr Food Res (2016) 60(5):1059–67. doi:10.1002/mnfr.201500937
31. Duan S, Lv Z, Fan X, Wang L, Han F, Wang H, et al. Vitamin D status and the risk of multiple sclerosis: a systematic review and meta-analysis. Neurosci Lett (2014) 570:108–13. doi:10.1016/j.neulet.2014.04.021
32. Lin J, Liu J, Davies ML, Chen W. Serum vitamin D level and rheumatoid arthritis disease activity: review and meta-analysis. PLoS One (2016) 11(1):e0146351. doi:10.1371/journal.pone.0146351
33. Del Pinto R, Pietropaoli D, Chandar AK, Ferri C, Cominelli F. Association between inflammatory bowel disease and vitamin D deficiency: a systematic review and meta-analysis. Inflamm Bowel Dis (2015) 21(11):2708–17. doi:10.1097/MIB.0000000000000546
34. Lu C, Yang J, Yu W, Li D, Xiang Z, Lin Y, et al. Association between 25(OH)D level, ultraviolet exposure, geographical location, and inflammatory bowel disease activity: a systematic review and meta-analysis. PLoS One (2015) 10(7):e0132036. doi:10.1371/journal.pone.0132036
35. Sadeghian M, Saneei P, Siassi F, Esmaillzadeh A. Vitamin D status in relation to Crohn’s disease: meta-analysis of observational studies. Nutrition (2016) 32(5):505–14. doi:10.1016/j.nut.2015.11.008
36. Feng R, Li Y, Li G, Li Z, Zhang Y, Li Q, et al. Lower serum 25 (OH) D concentrations in type 1 diabetes: a meta-analysis. Diabetes Res Clin Pract (2015) 108(3):e71–5. doi:10.1016/j.diabres.2014.12.008
37. Sahebari M, Nabavi N, Salehi M. Correlation between serum 25(OH)D values and lupus disease activity: an original article and a systematic review with meta-analysis focusing on serum VitD confounders. Lupus (2014) 23(11):1164–77. doi:10.1177/0961203314540966
38. Hiraki LT, Arkema EV, Cui J, Malspeis S, Costenbader KH, Karlson EW. Circulating 25-hydroxyvitamin D level and risk of developing rheumatoid arthritis. Rheumatology (Oxford) (2014) 53(12):2243–8. doi:10.1093/rheumatology/keu276
39. Tizaoui K, Kaabachi W, Hamzaoui A, Hamzaoui K. Association between vitamin D receptor polymorphisms and multiple sclerosis: systematic review and meta-analysis of case-control studies. Cell Mol Immunol (2015) 12(2):243–52. doi:10.1038/cmi.2014.47
40. Tizaoui K, Hamzaoui K. Association between VDR polymorphisms and rheumatoid arthritis disease: systematic review and updated meta-analysis of case-control studies. Immunobiology (2015) 220(6):807–16. doi:10.1016/j.imbio.2014.12.013
41. Lee YH, Bae SC, Choi SJ, Ji JD, Song GG. Associations between vitamin D receptor polymorphisms and susceptibility to rheumatoid arthritis and systemic lupus erythematosus: a meta-analysis. Mol Biol Rep (2011) 38(6):3643–51. doi:10.1007/s11033-010-0477-4
42. Xue LN, Xu KQ, Zhang W, Wang Q, Wu J, Wang XY. Associations between vitamin D receptor polymorphisms and susceptibility to ulcerative colitis and Crohn’s disease: a meta-analysis. Inflamm Bowel Dis (2013) 19(1):54–60. doi:10.1002/ibd.22966
43. Wang L, Wang ZT, Hu JJ, Fan R, Zhou J, Zhong J. Polymorphisms of the vitamin D receptor gene and the risk of inflammatory bowel disease: a meta-analysis. Genet Mol Res (2014) 13(2):2598–610. doi:10.4238/2014.April.8.2
44. Tizaoui K, Kaabachi W, Hamzaoui A, Hamzaoui K. Contribution of VDR polymorphisms to type 1 diabetes susceptibility: systematic review of case-control studies and meta-analysis. J Steroid Biochem Mol Biol (2014) 143:240–9. doi:10.1016/j.jsbmb.2014.03.011
45. Wang G, Zhang Q, Xu N, Xu K, Wang J, He W, et al. Associations between two polymorphisms (FokI and BsmI) of vitamin D receptor gene and type 1 diabetes mellitus in Asian population: a meta-analysis. PLoS One (2014) 9(3):e89325. doi:10.1371/journal.pone.0089325
46. Zhang J, Li W, Liu J, Wu W, Ouyang H, Zhang Q, et al. Polymorphisms in the vitamin D receptor gene and type 1 diabetes mellitus risk: an update by meta-analysis. Mol Cell Endocrinol (2012) 355(1):135–42. doi:10.1016/j.mce.2012.02.003
47. Zhou TB, Jiang ZP, Lin ZJ, Su N. Association of vitamin D receptor gene polymorphism with the risk of systemic lupus erythematosus. J Recept Signal Transduct Res (2015) 35(1):8–14. doi:10.3109/10799893.2014.926927
48. Mason AR, Mason J, Cork M, Dooley G, Hancock H. Topical treatments for chronic plaque psoriasis. Cochrane Database Syst Rev (2013) (3). doi:10.1002/14651858.CD005028.pub3
49. Burton JM, Kimball S, Vieth R, Bar-Or A, Dosch HM, Cheung R, et al. A phase I/II dose-escalation trial of vitamin D3 and calcium in multiple sclerosis. Neurology (2010) 74(23):1852–9. doi:10.1212/WNL.0b013e3181e1cec2
50. Mosayebi G, Ghazavi A, Ghasami K, Jand Y, Kokhaei P. Therapeutic effect of vitamin D3 in multiple sclerosis patients. Immunol Invest (2011) 40(6):627–39. doi:10.3109/08820139.2011.573041
51. Soilu-Hänninen M, Aivo J, Lindstrom BM, Elovaara I, Sumelahti ML, Farkkila M, et al. A randomised, double blind, placebo controlled trial with vitamin D3 as an add on treatment to interferon beta-1b in patients with multiple sclerosis. J Neurol Neurosurg Psychiatry (2012) 83(5):565–71. doi:10.1136/jnnp-2011-301876
52. Kampman MT, Steffensen LH, Mellgren SI, Jørgensen L. Effect of vitamin D3 supplementation on relapses, disease progression, and measures of function in persons with multiple sclerosis: exploratory outcomes from a double-blind randomised controlled trial. Mult Scler (2012) 18(8):1144–51. doi:10.1177/1352458511434607
53. Derakhshandi H, Etemadifar M, Feizi A, Abtahi SH, Minagar A, Abtahi MA, et al. Preventive effect of vitamin D3 supplementation on conversion of optic neuritis to clinically definite multiple sclerosis: a double blind, randomized, placebo-controlled pilot clinical trial. Acta Neurol Belg (2013) 113(3):257–63. doi:10.1007/s13760-012-0166-2
54. Salesi M, Farajzadegan Z. Efficacy of vitamin D in patients with active rheumatoid arthritis receiving methotrexate therapy. Rheumatol Int (2012) 32(7):2129–33. doi:10.1007/s00296-011-1944-5
55. Dehghan A, Rahimpour S, Soleymani-Salehabadi H, Owlia MB. Role of vitamin D in flare ups of rheumatoid arthritis. Z Rheumatol (2014) 73(5):461–4. doi:10.1007/s00393-013-1297-4
56. Hansen KE, Bartels CM, Gangnon RE, Jones AN, Gogineni J. An evaluation of high-dose vitamin D for rheumatoid arthritis. J Clin Rheumatol (2014) 20(2):112–4. doi:10.1097/RHU.0000000000000072
57. Jørgensen SP, Agnholt J, Glerup H, Lyhne S, Villadsen GE, Hvas CL, et al. Clinical trial: vitamin D3 treatment in Crohn’s disease – a randomized double-blind placebo-controlled study. Aliment Pharmacol Ther (2010) 32(3):377–83. doi:10.1111/j.1365-2036.2010.04355.x
58. Wingate KE, Jacobson K, Issenman R, Carroll M, Barker C, Israel D, et al. 25-Hydroxyvitamin D concentrations in children with Crohn’s disease supplemented with either 2000 or 400 IU daily for 6 months: a randomized controlled study. J Pediatr (2014) 164(4):860–5. doi:10.1016/j.jpeds.2013.11.071
59. Raftery T, Martineau AR, Greiller CL, Ghosh S, McNamara D, Bennett K, et al. Effects of vitamin D supplementation on intestinal permeability, cathelicidin and disease markers in Crohn’s disease: results from a randomised double-blind placebo-controlled study. United European Gastroenterol J (2015) 3(3):294–302. doi:10.1177/2050640615572176
60. Li X, Liao L, Yan X, Huang G, Lin J, Lei M, et al. Protective effects of 1-alpha-hydroxyvitamin D3 on residual beta-cell function in patients with adult-onset latent autoimmune diabetes (LADA). Diabetes Metab Res Rev (2009) 25(5):411–6. doi:10.1002/dmrr.977
61. Bizzarri C, Pitocco D, Napoli N, Di Stasio E, Maggi D, Manfrini S, et al. No protective effect of calcitriol on beta-cell function in recent-onset type 1 diabetes: the IMDIAB XIII trial. Diabetes Care (2010) 33(9):1962–3. doi:10.2337/dc10-0814
62. Walter M, Kaupper T, Adler K, Foersch J, Bonifacio E, Ziegler AG. No effect of the 1alpha,25-dihydroxyvitamin D3 on beta-cell residual function and insulin requirement in adults with new-onset type 1 diabetes. Diabetes Care (2010) 33(7):1443–8. doi:10.2337/dc09-2297
63. Gabbay MA, Sato MN, Finazzo C, Duarte AJ, Dib SA. Effect of cholecalciferol as adjunctive therapy with insulin on protective immunologic profile and decline of residual beta-cell function in new-onset type 1 diabetes mellitus. Arch Pediatr Adolesc Med (2012) 166(7):601–7. doi:10.1001/archpediatrics.2012.164
64. Ataie-Jafari A, Loke SC, Rahmat AB, Larijani B, Abbasi F, Leow MK, et al. A randomized placebo-controlled trial of alphacalcidol on the preservation of beta cell function in children with recent onset type 1 diabetes. Clin Nutr (2013) 32(6):911–7. doi:10.1016/j.clnu.2013.01.012
65. Abou-Raya A, Abou-Raya S, Helmii M. The effect of vitamin D supplementation on inflammatory and hemostatic markers and disease activity in patients with systemic lupus erythematosus: a randomized placebo-controlled trial. J Rheumatol (2013) 40(3):265–72. doi:10.3899/jrheum.111594
66. Lima GL, Paupitz J, Aikawa NE, Takayama L, Bonfa E, Pereira RM. Vitamin D supplementation in adolescents and young adults with juvenile systemic lupus erythematosus for improvement in disease activity and fatigue scores: a randomized, double-blind, placebo-controlled trial. Arthritis Care Res (Hoboken) (2016) 68(1):91–8. doi:10.1002/acr.22621
67. Aranow C, Kamen DL, Dall’Era M, Massarotti EM, Mackay MC, Koumpouras F, et al. Randomized, double-blind, placebo-controlled trial of the effect of vitamin D3 on the interferon signature in patients with systemic lupus erythematosus. Arthritis Rheumatol (2015) 67(7):1848–57. doi:10.1002/art.39108
68. Foering K, Chang AY, Piette EW, Cucchiara A, Okawa J, Werth VP. Characterization of clinical photosensitivity in cutaneous lupus erythematosus. J Am Acad Dermatol (2013) 69(2):205–13. doi:10.1016/j.jaad.2013.03.015
69. Brennan A, Katz DR, Nunn JD, Barker S, Hewison M, Fraher LJ, et al. Dendritic cells from human tissues express receptors for the immunoregulatory vitamin D3 metabolite, dihydroxycholecalciferol. Immunology (1987) 61(4):457–61.
70. Morgan JW, Kouttab N, Ford D, Maizel AL. Vitamin D-mediated gene regulation in phenotypically defined human B cell subpopulations. Endocrinology (2000) 141(9):3225–34. doi:10.1210/endo.141.9.7666
71. Kongsbak M, von Essen MR, Levring TB, Schjerling P, Woetmann A, Odum N, et al. Vitamin D-binding protein controls T cell responses to vitamin D. BMC Immunol (2014) 15(1):35. doi:10.1186/s12865-014-0035-2
72. Morgan JW, Reddy GS, Uskokovic MR, May BK, Omdahl JL, Maizel AL, et al. Functional block for 1 alpha,25-dihydroxyvitamin D3-mediated gene regulation in human B lymphocytes. J Biol Chem (1994) 269(18):13437–43.
73. Sigmundsdottir H, Pan J, Debes GF, Alt C, Habtezion A, Soler D, et al. DCs metabolize sunlight-induced vitamin D3 to ‘program’ T cell attraction to the epidermal chemokine CCL27. Nat Immunol (2007) 8(3):285–93. doi:10.1038/ni1433
74. Jeffery LE, Wood AM, Qureshi OS, Hou TZ, Gardner D, Briggs Z, et al. Availability of 25-hydroxyvitamin D(3) to APCs controls the balance between regulatory and inflammatory T cell responses. J Immunol (2012) 189(11):5155–64. doi:10.4049/jimmunol.1200786
75. Heine G, Niesner U, Chang HD, Steinmeyer A, Zugel U, Zuberbier T, et al. 1,25-dihydroxyvitamin D(3) promotes IL-10 production in human B cells. Eur J Immunol (2008) 38(8):2210–8. doi:10.1002/eji.200838216
76. Correale J, Ysrraelit MC, Gaitan MI. Immunomodulatory effects of Vitamin D in multiple sclerosis. Brain (2009) 132(Pt 5):1146–60. doi:10.1093/brain/awp033
77. von Essen MR, Kongsbak M, Schjerling P, Olgaard K, Odum N, Geisler C. Vitamin D controls T cell antigen receptor signaling and activation of human T cells. Nat Immunol (2010) 11(4):344–9. doi:10.1038/ni.1851
78. Bakdash G, van Capel TM, Mason LM, Kapsenberg ML, de Jong EC. Vitamin D3 metabolite calcidiol primes human dendritic cells to promote the development of immunomodulatory IL-10-producing T cells. Vaccine (2014) 32(47):6294–302. doi:10.1016/j.vaccine.2014.08.075
79. Chen J, Bruce D, Cantorna MT. Vitamin D receptor expression controls proliferation of naive CD8+ T cells and development of CD8 mediated gastrointestinal inflammation. BMC Immunol (2014) 15:6. doi:10.1186/1471-2172-15-6
80. Lysandropoulos AP, Jaquiery E, Jilek S, Pantaleo G, Schluep M, Du Pasquier RA. Vitamin D has a direct immunomodulatory effect on CD8+ T cells of patients with early multiple sclerosis and healthy control subjects. J Neuroimmunol (2011) 233(1–2):240–4. doi:10.1016/j.jneuroim.2010.11.008
81. Bruce D, Cantorna MT. Intrinsic requirement for the vitamin D receptor in the development of CD8alphaalpha-expressing T cells. J Immunol (2011) 186(5):2819–25. doi:10.4049/jimmunol.1003444
82. Dyring-Andersen B, Bonefeld CM, Bzorek M, Lovendorf MB, Lauritsen JP, Skov L, et al. The vitamin D analogue calcipotriol reduces the frequency of CD8+ IL-17+ T cells in psoriasis lesions. Scand J Immunol (2015) 82(1):84–91. doi:10.1111/sji.12304
83. Balogh G, de Boland AR, Boland R, Barja P. Effect of 1,25(OH)(2)-vitamin D(3) on the activation of natural killer cells: role of protein kinase C and extracellular calcium. Exp Mol Pathol (1999) 67(2):63–74. doi:10.1006/exmp.1999.2264
84. Weeres MA, Robien K, Ahn YO, Neulen ML, Bergerson R, Miller JS, et al. The effects of 1,25-dihydroxyvitamin D3 on in vitro human NK cell development from hematopoietic stem cells. J Immunol (2014) 193(7):3456–62. doi:10.4049/jimmunol.1400698
85. Ota K, Dambaeva S, Kim MW, Han AR, Fukui A, Gilman-Sachs A, et al. 1,25-Dihydroxy-vitamin D3 regulates NK-cell cytotoxicity, cytokine secretion, and degranulation in women with recurrent pregnancy losses. Eur J Immunol (2015) 45(11):3188–99. doi:10.1002/eji.201545541
86. Chen J, Waddell A, Lin YD, Cantorna MT. Dysbiosis caused by vitamin D receptor deficiency confers colonization resistance to Citrobacter rodentium through modulation of innate lymphoid cells. Mucosal Immunol (2015) 8(3):618–26. doi:10.1038/mi.2014.94
87. Chen L, Cencioni MT, Angelini DF, Borsellino G, Battistini L, Brosnan CF. Transcriptional profiling of gamma delta T cells identifies a role for vitamin D in the immunoregulation of the V gamma 9V delta 2 response to phosphate-containing ligands. J Immunol (2005) 174(10):6144–52. doi:10.4049/jimmunol.174.10.6144
88. Waddell A, Zhao J, Cantorna MT. NKT cells can help mediate the protective effects of 1,25-dihydroxyvitamin D3 in experimental autoimmune encephalomyelitis in mice. Int Immunol (2015) 27(5):237–44. doi:10.1093/intimm/dxu147
89. Yu S, Cantorna MT. The vitamin D receptor is required for iNKT cell development. Proc Natl Acad Sci U S A (2008) 105(13):5207–12. doi:10.1073/pnas.0711558105
90. Chen S, Sims GP, Chen XX, Gu YY, Chen S, Lipsky PE. Modulatory effects of 1,25-dihydroxyvitamin D3 on human B cell differentiation. J Immunol (2007) 179(3):1634–47. doi:10.4049/jimmunol.179.3.1634
91. Lemire JM, Adams JS, Sakai R, Jordan SC. 1 alpha,25-dihydroxyvitamin D3 suppresses proliferation and immunoglobulin production by normal human peripheral blood mononuclear cells. J Clin Invest (1984) 74(2):657–61. doi:10.1172/JCI111465
92. Iho S, Takahashi T, Kura F, Sugiyama H, Hoshino T. The effect of 1,25-dihydroxyvitamin D3 on in vitro immunoglobulin production in human B cells. J Immunol (1986) 136(12):4427–31.
93. Geldmeyer-Hilt K, Heine G, Hartmann B, Baumgrass R, Radbruch A, Worm M. 1,25-dihydroxyvitamin D3 impairs NF-kappaB activation in human naive B cells. Biochem Biophys Res Commun (2011) 407(4):699–702. doi:10.1016/j.bbrc.2011.03.078
94. Chen WC, Vayuvegula B, Gupta S. 1,25-Dihydroxyvitamin D3-mediated inhibition of human B cell differentiation. Clin Exp Immunol (1987) 69(3):639–46.
95. Heine G, Anton K, Henz BM, Worm M. 1alpha,25-dihydroxyvitamin D3 inhibits anti-CD40 plus IL-4-mediated IgE production in vitro. Eur J Immunol (2002) 32(12):3395–404. doi:10.1002/1521-4141(2002012)32:12<3395::AID-IMMU3395>3.0.CO;2-I
96. Drozdenko G, Scheel T, Heine G, Baumgrass R, Worm M. Impaired T cell activation and cytokine production by calcitriol-primed human B cells. Clin Exp Immunol (2014) 178(2):364–72. doi:10.1111/cei.12406
97. Penna G, Adorini L. 1 Alpha,25-dihydroxyvitamin D3 inhibits differentiation, maturation, activation, and survival of dendritic cells leading to impaired alloreactive T cell activation. J Immunol (2000) 164(5):2405–11. doi:10.4049/jimmunol.164.5.2405
98. Piemonti L, Monti P, Sironi M, Fraticelli P, Leone BE, Dal Cin E, et al. Vitamin D3 affects differentiation, maturation, and function of human monocyte-derived dendritic cells. J Immunol (2000) 164(9):4443–51. doi:10.4049/jimmunol.164.9.4443
99. Unger WW, Laban S, Kleijwegt FS, van der Slik AR, Roep BO. Induction of Treg by monocyte-derived DC modulated by vitamin D3 or dexamethasone: differential role for PD-L1. Eur J Immunol (2009) 39(11):3147–59. doi:10.1002/eji.200839103
100. van der Aar AM, Sibiryak DS, Bakdash G, van Capel TM, van der Kleij HP, Opstelten DJ, et al. Vitamin D3 targets epidermal and dermal dendritic cells for induction of distinct regulatory T cells. J Allergy Clin Immunol (2011) 127(6):1532.e–40.e. doi:10.1016/j.jaci.2011.01.068
101. Ferreira GB, Vanherwegen AS, Eelen G, Gutierrez AC, Van Lommel L, Marchal K, et al. Vitamin D3 induces tolerance in human dendritic cells by activation of intracellular metabolic pathways. Cell Rep (2015) 10:711–25. doi:10.1016/j.celrep.2015.01.013
102. Farias AS, Spagnol GS, Bordeaux-Rego P, Oliveira CO, Fontana AG, de Paula RF, et al. Vitamin D3 induces IDO+ tolerogenic DCs and enhances Treg, reducing the severity of EAE. CNS Neurosci Ther (2013) 19(4):269–77. doi:10.1111/cns.12071
103. Huang Y, Zhao Y, Ran X, Wang C. Increased expression of herpesvirus entry mediator in 1,25-dihydroxyvitamin D3-treated mouse bone marrow-derived dendritic cells promotes the generation of CD4(+)CD25(+)Foxp3(+) regulatory T cells. Mol Med Rep (2014) 9(3):813–8. doi:10.3892/mmr.2013.1874
104. Neve A, Corrado A, Cantatore FP. Immunomodulatory effects of vitamin D in peripheral blood monocyte-derived macrophages from patients with rheumatoid arthritis. Clin Exp Med (2014) 14(3):275–83. doi:10.1007/s10238-013-0249-2
105. Chen Y, Liu W, Sun T, Huang Y, Wang Y, Deb DK, et al. 1,25-Dihydroxyvitamin D promotes negative feedback regulation of TLR signaling via targeting microRNA-155-SOCS1 in macrophages. J Immunol (2013) 190(7):3687–95. doi:10.4049/jimmunol.1203273
106. Wang Q, He Y, Shen Y, Zhang Q, Chen D, Zuo C, et al. Vitamin D inhibits COX-2 expression and inflammatory response by targeting thioesterase superfamily member 4. J Biol Chem (2014) 289(17):11681–94. doi:10.1074/jbc.M113.517581
107. Zhang X, Zhou M, Guo Y, Song Z, Liu B. 1,25-dihydroxyvitamin D(3) promotes high glucose-induced M1 macrophage switching to M2 via the VDR-PPARgamma signaling pathway. Biomed Res Int (2015) 2015:157834. doi:10.1155/2015/157834
108. Korf H, Wenes M, Stijlemans B, Takiishi T, Robert S, Miani M, et al. 1,25-dihydroxyvitamin D3 curtails the inflammatory and T cell stimulatory capacity of macrophages through an IL-10-dependent mechanism. Immunobiology (2012) 217(12):1292–300. doi:10.1016/j.imbio.2012.07.018
109. Terrier B, Derian N, Schoindre Y, Chaara W, Geri G, Zahr N, et al. Restoration of regulatory and effector T cell balance and B cell homeostasis in systemic lupus erythematosus patients through vitamin D supplementation. Arthritis Res Ther (2012) 14(5):R221. doi:10.1186/ar4060
110. Palmer MT, Lee YK, Maynard CL, Oliver JR, Bikle DD, Jetten AM, et al. Lineage-specific effects of 1,25-dihydroxyvitamin D(3) on the development of effector CD4 T cells. J Biol Chem (2011) 286(2):997–1004. doi:10.1074/jbc.M110.163790
111. Pichler J, Gerstmayr M, Szepfalusi Z, Urbanek R, Peterlik M, Willheim M. 1 alpha,25(OH)2D3 inhibits not only Th1 but also Th2 differentiation in human cord blood T cells. Pediatr Res (2002) 52(1):12–8. doi:10.1203/01.PDR.0000017267.23950.48
112. Chang SH, Chung Y, Dong C. Vitamin D suppresses Th17 cytokine production by inducing C/EBP homologous protein (CHOP) expression. J Biol Chem (2010) 285(50):38751–5. doi:10.1074/jbc.C110.185777
113. Boonstra A, Barrat FJ, Crain C, Heath VL, Savelkoul HF, O’Garra A. 1alpha,25-Dihydroxyvitamin d3 has a direct effect on naive CD4(+) T cells to enhance the development of Th2 cells. J Immunol (2001) 167(9):4974–80. doi:10.4049/jimmunol.167.9.4974
114. Khoo AL, Joosten I, Michels M, Woestenenk R, Preijers F, He XH, et al. 1,25-dihydroxyvitamin D3 inhibits proliferation but not the suppressive function of regulatory T cells in the absence of antigen-presenting cells. Immunology (2011) 134(4):459–68. doi:10.1111/j.1365-2567.2011.03507.x
115. Colin EM, Asmawidjaja PS, van Hamburg JP, Mus AM, van Driel M, Hazes JM, et al. 1,25-dihydroxyvitamin D3 modulates Th17 polarization and interleukin-22 expression by memory T cells from patients with early rheumatoid arthritis. Arthritis Rheum (2010) 62(1):132–42. doi:10.1002/art.25043
116. Piantoni S, Andreoli L, Scarsi M, Zanola A, Dall’Ara F, Pizzorni C, et al. Phenotype modifications of T-cells and their shift toward a Th2 response in patients with systemic lupus erythematosus supplemented with different monthly regimens of vitamin D. Lupus (2015) 24(4–5):490–8. doi:10.1177/0961203314559090
117. Smolders J, Peelen E, Thewissen M, Cohen Tervaert JW, Menheere P, Hupperts R, et al. Safety and T cell modulating effects of high dose vitamin D3 supplementation in multiple sclerosis. PLoS One (2010) 5(12):e15235. doi:10.1371/journal.pone.0015235
118. Jeffery LE, Burke F, Mura M, Zheng Y, Qureshi OS, Hewison M, et al. 1,25-Dihydroxyvitamin D3 and IL-2 combine to inhibit T cell production of inflammatory cytokines and promote development of regulatory T cells expressing CTLA-4 and FoxP3. J Immunol (2009) 183(9):5458–67. doi:10.4049/jimmunol.0803217
119. Joshi S, Pantalena LC, Liu XK, Gaffen SL, Liu H, Rohowsky-Kochan C, et al. 1,25-dihydroxyvitamin D(3) ameliorates Th17 autoimmunity via transcriptional modulation of interleukin-17A. Mol Cell Biol (2011) 31(17):3653–69. doi:10.1128/MCB.05020-11
120. van Hamburg JP, Asmawidjaja PS, Davelaar N, Mus AM, Cornelissen F, van Leeuwen JP, et al. TNF blockade requires 1,25(OH)2D3 to control human Th17-mediated synovial inflammation. Ann Rheum Dis (2012) 71(4):606–12. doi:10.1136/annrheumdis-2011-200424
121. Jeffery LE, Qureshi OS, Gardner D, Hou TZ, Briggs Z, Soskic B, et al. Vitamin D antagonises the suppressive effect of inflammatory cytokines on CTLA-4 expression and regulatory function. PLoS One (2015) 10(7):e0131539. doi:10.1371/journal.pone.0131539
122. Tian Y, Wang C, Ye Z, Xiao X, Kijlstra A, Yang P. Effect of 1,25-dihydroxyvitamin D3 on Th17 and Th1 response in patients with Behcet’s disease. Invest Ophthalmol Vis Sci (2012) 53(10):6434–41. doi:10.1167/iovs.12-10398
123. Chang JH, Cha HR, Lee DS, Seo KY, Kweon MN. 1,25-dihydroxyvitamin D3 inhibits the differentiation and migration of T(H)17 cells to protect against experimental autoimmune encephalomyelitis. PLoS One (2010) 5(9):e12925. doi:10.1371/journal.pone.0012925
124. Nanduri R, Mahajan S, Bhagyaraj E, Sethi K, Kalra R, Chandra V, et al. The active form of vitamin D transcriptionally represses Smad7 signaling and activates extracellular signal-regulated kinase (ERK) to inhibit the differentiation of a inflammatory T helper cell subset and suppress experimental autoimmune encephalomyelitis. J Biol Chem (2015) 290(19):12222–36. doi:10.1074/jbc.M114.621839
125. Kang SW, Kim SH, Lee N, Lee WW, Hwang KA, Shin MS, et al. 1,25-dihyroxyvitamin D3 promotes FOXP3 expression via binding to vitamin D response elements in its conserved noncoding sequence region. J Immunol (2012) 188(11):5276–82. doi:10.4049/jimmunol.1101211
126. Teichmann LL, Ols ML, Kashgarian M, Reizis B, Kaplan DH, Shlomchik MJ. Dendritic cells in lupus are not required for activation of T and B cells but promote their expansion, resulting in tissue damage. Immunity (2010) 33(6):967–78. doi:10.1016/j.immuni.2010.11.025
127. Wohn C, Ober-Blobaum JL, Haak S, Pantelyushin S, Cheong C, Zahner SP, et al. Langerin(neg) conventional dendritic cells produce IL-23 to drive psoriatic plaque formation in mice. Proc Natl Acad Sci U S A (2013) 110(26):10723–8. doi:10.1073/pnas.1307569110
128. Sisirak V, Ganguly D, Lewis KL, Couillault C, Tanaka L, Bolland S, et al. Genetic evidence for the role of plasmacytoid dendritic cells in systemic lupus erythematosus. J Exp Med (2014) 211(10):1969–76. doi:10.1084/jem.20132522
129. Ferris ST, Carrero JA, Mohan JF, Calderon B, Murphy KM, Unanue ER. A minor subset of Batf3-dependent antigen-presenting cells in islets of Langerhans is essential for the development of autoimmune diabetes. Immunity (2014) 41(4):657–69. doi:10.1016/j.immuni.2014.09.012
130. Schmidt H, Williamson D, Ashley-Koch A. HLA-DR15 haplotype and multiple sclerosis: a HuGE review. Am J Epidemiol (2007) 165(10):1097–109. doi:10.1093/aje/kwk118
131. Mackie SL, Taylor JC, Martin SG, Consortium Y, Consortium U, Wordsworth P, et al. A spectrum of susceptibility to rheumatoid arthritis within HLA-DRB1: stratification by autoantibody status in a large UK population. Genes Immun (2012) 13(2):120–8. doi:10.1038/gene.2011.60
132. van Halteren AG, Tysma OM, van Etten E, Mathieu C, Roep BO. 1alpha,25-dihydroxyvitamin D3 or analogue treated dendritic cells modulate human autoreactive T cells via the selective induction of apoptosis. J Autoimmun (2004) 23(3):233–9. doi:10.1016/j.jaut.2004.06.004
133. Gambhir V, Kim J, Siddiqui S, Taylor M, Byford V, Petrof EO, et al. Influence of 1,25-dihydroxy vitamin D3 on TLR4-induced activation of antigen presenting cells is dependent on the order of receptor engagement. Immunobiology (2011) 216(9):988–96. doi:10.1016/j.imbio.2011.03.011
134. Penna G, Amuchastegui S, Giarratana N, Daniel KC, Vulcano M, Sozzani S, et al. 1,25-Dihydroxyvitamin D3 selectively modulates tolerogenic properties in myeloid but not plasmacytoid dendritic cells. J Immunol (2007) 178(1):145–53. doi:10.4049/jimmunol.178.1.145
135. Karthaus N, van Spriel AB, Looman MW, Chen S, Spilgies LM, Lieben L, et al. Vitamin D controls murine and human plasmacytoid dendritic cell function. J Invest Dermatol (2014) 134(5):1255–64. doi:10.1038/jid.2013.501
136. Hu J, Wan Y. Tolerogenic dendritic cells and their potential applications. Immunology (2011) 132(3):307–14. doi:10.1111/j.1365-2567.2010.03396.x
137. Griffin MD, Lutz W, Phan VA, Bachman LA, McKean DJ, Kumar R. Dendritic cell modulation by 1alpha,25 dihydroxyvitamin D3 and its analogs: a vitamin D receptor-dependent pathway that promotes a persistent state of immaturity in vitro and in vivo. Proc Natl Acad Sci U S A (2001) 98(12):6800–5. doi:10.1073/pnas.121172198
138. Stoop JN, Harry RA, von Delwig A, Isaacs JD, Robinson JH, Hilkens CM. Therapeutic effect of tolerogenic dendritic cells in established collagen-induced arthritis is associated with a reduction in Th17 responses. Arthritis Rheum (2010) 62(12):3656–65. doi:10.1002/art.27756
139. Ferreira GB, Gysemans CA, Demengeot J, da Cunha JP, Vanherwegen AS, Overbergh L, et al. 1,25-Dihydroxyvitamin D3 promotes tolerogenic dendritic cells with functional migratory properties in NOD mice. J Immunol (2014) 192(9):4210–20. doi:10.4049/jimmunol.1302350
140. Mansilla MJ, Selles-Moreno C, Fabregas-Puig S, Amoedo J, Navarro-Barriuso J, Teniente-Serra A, et al. Beneficial effect of tolerogenic dendritic cells pulsed with MOG autoantigen in experimental autoimmune encephalomyelitis. CNS Neurosci Ther (2015) 21(3):222–30. doi:10.1111/cns.12342
141. Harry RA, Anderson AE, Isaacs JD, Hilkens CM. Generation and characterisation of therapeutic tolerogenic dendritic cells for rheumatoid arthritis. Ann Rheum Dis (2010) 69(11):2042–50. doi:10.1136/ard.2009.126383
142. Bartosik-Psujek H, Tabarkiewicz J, Pocinska K, Stelmasiak Z, Rolinski J. Immunomodulatory effects of vitamin D on monocyte-derived dendritic cells in multiple sclerosis. Mult Scler (2010) 16(12):1513–6. doi:10.1177/1352458510379611
143. Bartels LE, Jørgensen SP, Bendix M, Hvas CL, Agnholt J, Agger R, et al. 25-Hydroxy vitamin D3 modulates dendritic cell phenotype and function in Crohn’s disease. Inflammopharmacology (2013) 21(2):177–86. doi:10.1007/s10787-012-0168-y
144. Volchenkov R, Brun JG, Jonsson R, Appel S. In vitro suppression of immune responses using monocyte-derived tolerogenic dendritic cells from patients with primary Sjogren’s syndrome. Arthritis Res Ther (2013) 15(5):R114. doi:10.1186/ar4294
145. Wahono CS, Rusmini H, Soelistyoningsih D, Hakim R, Handono K, Endharti AT, et al. Effects of 1,25(OH)2D3 in immune response regulation of systemic lupus erythematosus (SLE) patient with hypovitamin D. Int J Clin Exp Med (2014) 7(1):22–31.
146. Raiotach-Regue D, Grau-Lopez L, Naranjo-Gomez M, Ramo-Tello C, Pujol-Borrell R, Martinez-Caceres E, et al. Stable antigen-specific T-cell hyporesponsiveness induced by tolerogenic dendritic cells from multiple sclerosis patients. Eur J Immunol (2012) 42(3):771–82. doi:10.1002/eji.201141835
147. Giannoukakis N, Phillips B, Finegold D, Harnaha J, Trucco M. Phase I (safety) study of autologous tolerogenic dendritic cells in type 1 diabetic patients. Diabetes Care (2011) 34(9):2026–32. doi:10.2337/dc11-0472
148. Benham H, Nel HJ, Law SC, Mehdi AM, Street S, Ramnoruth N, et al. Citrullinated peptide dendritic cell immunotherapy in HLA risk genotype-positive rheumatoid arthritis patients. Sci Transl Med (2015) 7(290):290ra87. doi:10.1126/scitranslmed.aaa9301
149. Murray PJ, Wynn TA. Protective and pathogenic functions of macrophage subsets. Nat Rev Immunol (2011) 11(11):723–37. doi:10.1038/nri3073
150. Tak PP, Smeets TJ, Daha MR, Kluin PM, Meijers KA, Brand R, et al. Analysis of the synovial cell infiltrate in early rheumatoid synovial tissue in relation to local disease activity. Arthritis Rheum (1997) 40(2):217–25. doi:10.1002/art.1780400206
151. Kamada N, Hisamatsu T, Okamoto S, Chinen H, Kobayashi T, Sato T, et al. Unique CD14 intestinal macrophages contribute to the pathogenesis of Crohn disease via IL-23/IFN-gamma axis. J Clin Invest (2008) 118(6):2269–80. doi:10.1172/JCI34610
152. van Horssen J, Singh S, van der Pol S, Kipp M, Lim JL, Peferoen L, et al. Clusters of activated microglia in normal-appearing white matter show signs of innate immune activation. J Neuroinflammation (2012) 9:156. doi:10.1186/1742-2094-9-156
153. Singh S, Metz I, Amor S, van der Valk P, Stadelmann C, Bruck W. Microglial nodules in early multiple sclerosis white matter are associated with degenerating axons. Acta Neuropathol (2013) 125(4):595–608. doi:10.1007/s00401-013-1082-0
154. Orme J, Mohan C. Macrophage subpopulations in systemic lupus erythematosus. Discov Med (2012) 13(69):151–8.
155. Jun HS, Yoon CS, Zbytnuik L, van Rooijen N, Yoon JW. The role of macrophages in T cell-mediated autoimmune diabetes in nonobese diabetic mice. J Exp Med (1999) 189(2):347–58. doi:10.1084/jem.189.2.347
156. Nashold FE, Miller DJ, Hayes CE. 1,25-dihydroxyvitamin D3 treatment decreases macrophage accumulation in the CNS of mice with experimental autoimmune encephalomyelitis. J Neuroimmunol (2000) 103(2):171–9. doi:10.1016/S0165-5728(99)00247-7
157. Xu H, Soruri A, Gieseler RK, Peters JH. 1,25-Dihydroxyvitamin D3 exerts opposing effects to IL-4 on MHC class-II antigen expression, accessory activity, and phagocytosis of human monocytes. Scand J Immunol (1993) 38(6):535–40. doi:10.1111/j.1365-3083.1993.tb03237.x
158. Krutzik SR, Hewison M, Liu PT, Robles JA, Stenger S, Adams JS, et al. IL-15 links TLR2/1-induced macrophage differentiation to the vitamin D-dependent antimicrobial pathway. J Immunol (2008) 181(10):7115–20. doi:10.4049/jimmunol.181.10.7115
159. Fabri M, Stenger S, Shin DM, Yuk JM, Liu PT, Realegeno S, et al. Vitamin D is required for IFN-gamma-mediated antimicrobial activity of human macrophages. Sci Transl Med (2011) 3(104):104ra2. doi:10.1126/scitranslmed.3003045
160. Liu PT, Stenger S, Li H, Wenzel L, Tan BH, Krutzik SR, et al. Toll-like receptor triggering of a vitamin D-mediated human antimicrobial response. Science (2006) 311(5768):1770–3. doi:10.1126/science.1123933
161. Liu PT, Stenger S, Tang DH, Modlin RL. Cutting edge: vitamin D-mediated human antimicrobial activity against Mycobacterium tuberculosis is dependent on the induction of cathelicidin. J Immunol (2007) 179(4):2060–3. doi:10.4049/jimmunol.179.4.2060
162. Lee BN, Kim TH, Jun JB, Yoo DH, Woo JH, Choi SJ, et al. Upregulation of interleukin-1beta production by 1,25-dihydroxyvitamin D(3) in activated human macrophages. Mol Biol Rep (2011) 38(3):2193–201. doi:10.1007/s11033-010-0348-z
163. Verway M, Bouttier M, Wang TT, Carrier M, Calderon M, An BS, et al. Vitamin D induces interleukin-1beta expression: paracrine macrophage epithelial signaling controls M. tuberculosis infection. PLoS Pathog (2013) 9(6):e1003407. doi:10.1371/journal.ppat.1003407
164. Kumar V, Abbas AK, Fausto N, Mitchell R. Robbins Basic Pathology. 8 ed. Philadelphia: Elsevier Health Sciences (2007).
165. van Venrooij WJ, van Beers JJ, Pruijn GJ. Anti-CCP antibodies: the past, the present and the future. Nat Rev Rheumatol (2011) 7(7):391–8. doi:10.1038/nrrheum.2011.76
166. Ramagopalan SV, Heger A, Berlanga AJ, Maugeri NJ, Lincoln MR, Burrell A, et al. A ChIP-seq defined genome-wide map of vitamin D receptor binding: associations with disease and evolution. Genome Res (2010) 20(10):1352–60. doi:10.1101/gr.107920.110
167. Pieper K, Grimbacher B, Eibel H. B-cell biology and development. J Allergy Clin Immunol (2013) 131(4):959–71. doi:10.1016/j.jaci.2013.01.046
168. Shirakawa AK, Nagakubo D, Hieshima K, Nakayama T, Jin Z, Yoshie O. 1,25-dihydroxyvitamin D3 induces CCR10 expression in terminally differentiating human B cells. J Immunol (2008) 180(5):2786–95. doi:10.4049/jimmunol.180.5.2786
169. Ritterhouse LL, Crowe SR, Niewold TB, Kamen DL, Macwana SR, Roberts VC, et al. Vitamin D deficiency is associated with an increased autoimmune response in healthy individuals and in patients with systemic lupus erythematosus. Ann Rheum Dis (2011) 70(9):1569–74. doi:10.1136/ard.2010.148494
170. Knippenberg S, Peelen E, Smolders J, Thewissen M, Menheere P, Cohen Tervaert JW, et al. Reduction in IL-10 producing B cells (Breg) in multiple sclerosis is accompanied by a reduced naive/memory Breg ratio during a relapse but not in remission. J Neuroimmunol (2011) 239(1–2):80–6. doi:10.1016/j.jneuroim.2011.08.019
171. Veldman CM, Cantorna MT, DeLuca HF. Expression of 1,25-dihydroxyvitamin D(3) receptor in the immune system. Arch Biochem Biophys (2000) 374(2):334–8. doi:10.1006/abbi.1999.1605
172. Hirahara K, Nakayama T. CD4+ T-cell subsets in inflammatory diseases: beyond the Th1/Th2 paradigm. Int Immunol (2016) 28(4):163–71. doi:10.1093/intimm/dxw006
173. Mayne CG, Spanier JA, Relland LM, Williams CB, Hayes CE. 1,25-Dihydroxyvitamin D3 acts directly on the T lymphocyte vitamin D receptor to inhibit experimental autoimmune encephalomyelitis. Eur J Immunol (2011) 41(3):822–32. doi:10.1002/eji.201040632
174. Meehan TF, DeLuca HF. CD8(+) T cells are not necessary for 1 alpha,25-dihydroxyvitamin D(3) to suppress experimental autoimmune encephalomyelitis in mice. Proc Natl Acad Sci U S A (2002) 99(8):5557–60. doi:10.1073/pnas.082100699
175. Grishkan IV, Fairchild AN, Calabresi PA, Gocke AR. 1,25-Dihydroxyvitamin D3 selectively and reversibly impairs T helper-cell CNS localization. Proc Natl Acad Sci U S A (2013) 110(52):21101–6. doi:10.1073/pnas.1306072110
176. Nath N, Prasad R, Giri S, Singh AK, Singh I. T-bet is essential for the progression of experimental autoimmune encephalomyelitis. Immunology (2006) 118(3):384–91. doi:10.1111/j.1365-2567.2006.02385.x
177. Ferber IA, Brocke S, Taylor-Edwards C, Ridgway W, Dinisco C, Steinman L, et al. Mice with a disrupted IFN-gamma gene are susceptible to the induction of experimental autoimmune encephalomyelitis (EAE). J Immunol (1996) 156(1):5–7.
178. Damsker JM, Hansen AM, Caspi RR. Th1 and Th17 cells: adversaries and collaborators. Ann N Y Acad Sci (2010) 1183:211–21. doi:10.1111/j.1749-6632.2009.05133.x
179. Waldburger KE, Hastings RC, Schaub RG, Goldman SJ, Leonard JP. Adoptive transfer of experimental allergic encephalomyelitis after in vitro treatment with recombinant murine interleukin-12. Preferential expansion of interferon-gamma-producing cells and increased expression of macrophage-associated inducible nitric oxide synthase as immunomodulatory mechanisms. Am J Pathol (1996) 148(2):375–82.
180. Staeva-Vieira TP, Freedman LP. 1,25-dihydroxyvitamin D3 inhibits IFN-gamma and IL-4 levels during in vitro polarization of primary murine CD4+ T cells. J Immunol (2002) 168(3):1181–9. doi:10.4049/jimmunol.168.3.1181
181. Takeuchi A, Reddy GS, Kobayashi T, Okano T, Park J, Sharma S. Nuclear factor of activated T cells (NFAT) as a molecular target for 1alpha,25-dihydroxyvitamin D3-mediated effects. J Immunol (1998) 160(1):209–18.
182. Peelen E, Thewissen M, Knippenberg S, Smolders J, Muris AH, Menheere P, et al. Fraction of IL-10+ and IL-17+ CD8 T cells is increased in MS patients in remission and during a relapse, but is not influenced by immune modulators. J Neuroimmunol (2013) 258(1–2):77–84. doi:10.1016/j.jneuroim.2013.02.014
183. van Hamburg JP, Mus AM, de Bruijn MJ, de Vogel L, Boon L, Cornelissen F, et al. GATA-3 protects against severe joint inflammation and bone erosion and reduces differentiation of Th17 cells during experimental arthritis. Arthritis Rheum (2009) 60(3):750–9. doi:10.1002/art.24329
184. Sloka S, Silva C, Wang J, Yong VW. Predominance of Th2 polarization by vitamin D through a STAT6-dependent mechanism. J Neuroinflammation (2011) 8:56. doi:10.1186/1742-2094-8-56
185. McGeachy MJ, Bak-Jensen KS, Chen Y, Tato CM, Blumenschein W, McClanahan T, et al. TGF-beta and IL-6 drive the production of IL-17 and IL-10 by T cells and restrain T(H)-17 cell-mediated pathology. Nat Immunol (2007) 8(12):1390–7. doi:10.1038/ni1539
186. Cua DJ, Sherlock J, Chen Y, Murphy CA, Joyce B, Seymour B, et al. Interleukin-23 rather than interleukin-12 is the critical cytokine for autoimmune inflammation of the brain. Nature (2003) 421(6924):744–8. doi:10.1038/nature01355
187. Murphy CA, Langrish CL, Chen Y, Blumenschein W, McClanahan T, Kastelein RA, et al. Divergent pro- and antiinflammatory roles for IL-23 and IL-12 in joint autoimmune inflammation. J Exp Med (2003) 198(12):1951–7. doi:10.1084/jem.20030896
188. Lubberts E, Joosten LA, van de Loo FA, Schwarzenberger P, Kolls J, van den Berg WB. Overexpression of IL-17 in the knee joint of collagen type II immunized mice promotes collagen arthritis and aggravates joint destruction. Inflamm Res (2002) 51(2):102–4. doi:10.1007/BF02684010
189. Hirota K, Duarte JH, Veldhoen M, Hornsby E, Li Y, Cua DJ, et al. Fate mapping of IL-17-producing T cells in inflammatory responses. Nat Immunol (2011) 12(3):255–63. doi:10.1038/ni.1993
190. van Hamburg JP, Asmawidjaja PS, Davelaar N, Mus AM, Colin EM, Hazes JM, et al. Th17 cells, but not Th1 cells, from patients with early rheumatoid arthritis are potent inducers of matrix metalloproteinases and proinflammatory cytokines upon synovial fibroblast interaction, including autocrine interleukin-17A production. Arthritis Rheum (2011) 63(1):73–83. doi:10.1002/art.30093
191. Yang J, Chu Y, Yang X, Gao D, Zhu L, Yang X, et al. Th17 and natural Treg cell population dynamics in systemic lupus erythematosus. Arthritis Rheum (2009) 60(5):1472–83. doi:10.1002/art.24499
192. Leipe J, Grunke M, Dechant C, Reindl C, Kerzendorf U, Schulze-Koops H, et al. Role of Th17 cells in human autoimmune arthritis. Arthritis Rheum (2010) 62(10):2876–85. doi:10.1002/art.27622
193. Tang J, Zhou R, Luger D, Zhu W, Silver PB, Grajewski RS, et al. Calcitriol suppresses antiretinal autoimmunity through inhibitory effects on the Th17 effector response. J Immunol (2009) 182(8):4624–32. doi:10.4049/jimmunol.0801543
194. Mukasa R, Balasubramani A, Lee YK, Whitley SK, Weaver BT, Shibata Y, et al. Epigenetic instability of cytokine and transcription factor gene loci underlies plasticity of the T helper 17 cell lineage. Immunity (2010) 32(5):616–27. doi:10.1016/j.immuni.2010.04.016
195. Maggi L, Santarlasci V, Capone M, Rossi MC, Querci V, Mazzoni A, et al. Distinctive features of classic and nonclassic (Th17 derived) human Th1 cells. Eur J Immunol (2012) 42(12):3180–8. doi:10.1002/eji.201242648
196. Cosmi L, Liotta F, Maggi E, Romagnani S, Annunziato F. Th17 and non-classic Th1 cells in chronic inflammatory disorders: two sides of the same coin. Int Arch Allergy Immunol (2014) 164(3):171–7. doi:10.1159/000363502
197. Paulissen SM, van Hamburg JP, Dankers W, Lubberts E. The role and modulation of CCR6+ Th17 cell populations in rheumatoid arthritis. Cytokine (2015) 74(1):43–53. doi:10.1016/j.cyto.2015.02.002
198. Sotirchos ES, Bhargava P, Eckstein C, Van Haren K, Baynes M, Ntranos A, et al. Safety and immunologic effects of high- vs low-dose cholecalciferol in multiple sclerosis. Neurology (2016) 86(4):382–90. doi:10.1212/WNL.0000000000002316
199. Bennett CL, Christie J, Ramsdell F, Brunkow ME, Ferguson PJ, Whitesell L, et al. The immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome (IPEX) is caused by mutations of FOXP3. Nat Genet (2001) 27(1):20–1. doi:10.1038/83713
200. Spach KM, Nashold FE, Dittel BN, Hayes CE. IL-10 signaling is essential for 1,25-dihydroxyvitamin D3-mediated inhibition of experimental autoimmune encephalomyelitis. J Immunol (2006) 177(9):6030–7. doi:10.4049/jimmunol.177.9.6030
201. Van Belle TL, Vanherwegen AS, Feyaerts D, De Clercq P, Verstuyf A, Korf H, et al. 1,25-dihydroxyvitamin D3 and its analog TX527 promote a stable regulatory T cell phenotype in T cells from type 1 diabetes patients. PLoS One (2014) 9(10):e109194. doi:10.1371/journal.pone.0109194
202. Urry Z, Chambers ES, Xystrakis E, Dimeloe S, Richards DF, Gabrysova L, et al. The role of 1alpha,25-dihydroxyvitamin D3 and cytokines in the promotion of distinct Foxp3+ and IL-10+ CD4+ T cells. Eur J Immunol (2012) 42(10):2697–708. doi:10.1002/eji.201242370
203. Smolders J, Thewissen M, Peelen E, Menheere P, Tervaert JW, Damoiseaux J, et al. Vitamin D status is positively correlated with regulatory T cell function in patients with multiple sclerosis. PLoS One (2009) 4(8):e6635. doi:10.1371/journal.pone.0006635
204. Bock G, Prietl B, Mader JK, Holler E, Wolf M, Pilz S, et al. The effect of vitamin D supplementation on peripheral regulatory T cells and beta cell function in healthy humans: a randomized controlled trial. Diabetes Metab Res Rev (2011) 27(8):942–5. doi:10.1002/dmrr.1276
205. Zhang N, Bevan MJ. CD8(+) T cells: foot soldiers of the immune system. Immunity (2011) 35(2):161–8. doi:10.1016/j.immuni.2011.07.010
206. Huseby ES, Liggitt D, Brabb T, Schnabel B, Ohlen C, Goverman J. A pathogenic role for myelin-specific CD8(+) T cells in a model for multiple sclerosis. J Exp Med (2001) 194(5):669–76. doi:10.1084/jem.194.5.669
207. Sun D, Whitaker JN, Huang Z, Liu D, Coleclough C, Wekerle H, et al. Myelin antigen-specific CD8+ T cells are encephalitogenic and produce severe disease in C57BL/6 mice. J Immunol (2001) 166(12):7579–87. doi:10.4049/jimmunol.166.12.7579
208. Steinhoff U, Brinkmann V, Klemm U, Aichele P, Seiler P, Brandt U, et al. Autoimmune intestinal pathology induced by hsp60-specific CD8 T cells. Immunity (1999) 11(3):349–58. doi:10.1016/S1074-7613(00)80110-7
209. Menon B, Gullick NJ, Walter GJ, Rajasekhar M, Garrood T, Evans HG, et al. Interleukin-17+CD8+ T cells are enriched in the joints of patients with psoriatic arthritis and correlate with disease activity and joint damage progression. Arthritis Rheumatol (2014) 66(5):1272–81. doi:10.1002/art.38376
210. Godfrey DI, Uldrich AP, McCluskey J, Rossjohn J, Moody DB. The burgeoning family of unconventional T cells. Nat Immunol (2015) 16(11):1114–23. doi:10.1038/ni.3298
211. Edwards SC, McGinley AM, McGuinness NC, Mills KH. gammadelta T cells and NK cells – distinct pathogenic roles as innate-like immune cells in CNS autoimmunity. Front Immunol (2015) 6:455. doi:10.3389/fimmu.2015.00455
212. Wu L, Van Kaer L. Natural killer T cells in health and disease. Front Biosci (Schol Ed) (2011) 3:236–51. doi:10.2741/148
213. Spits H, Artis D, Colonna M, Diefenbach A, Di Santo JP, Eberl G, et al. Innate lymphoid cells – a proposal for uniform nomenclature. Nat Rev Immunol (2013) 13(2):145–9. doi:10.1038/nri3365
214. Poggi A, Zocchi MR. NK cell autoreactivity and autoimmune diseases. Front Immunol (2014) 5:27. doi:10.3389/fimmu.2014.00027
215. Villanova F, Flutter B, Tosi I, Grys K, Sreeneebus H, Perera GK, et al. Characterization of innate lymphoid cells in human skin and blood demonstrates increase of NKp44+ ILC3 in psoriasis. J Invest Dermatol (2014) 134(4):984–91. doi:10.1038/jid.2013.477
216. Teunissen MB, Munneke JM, Bernink JH, Spuls PI, Res PC, Te Velde A, et al. Composition of innate lymphoid cell subsets in the human skin: enrichment of NCR(+) ILC3 in lesional skin and blood of psoriasis patients. J Invest Dermatol (2014) 134(9):2351–60. doi:10.1038/jid.2014.146
217. Geremia A, Arancibia-Carcamo CV, Fleming MP, Rust N, Singh B, Mortensen NJ, et al. IL-23-responsive innate lymphoid cells are increased in inflammatory bowel disease. J Exp Med (2011) 208(6):1127–33. doi:10.1084/jem.20101712
218. Perry JS, Han S, Xu Q, Herman ML, Kennedy LB, Csako G, et al. Inhibition of LTi cell development by CD25 blockade is associated with decreased intrathecal inflammation in multiple sclerosis. Sci Transl Med (2012) 4(145):145ra06. doi:10.1126/scitranslmed.3004140
219. Ciccia F, Guggino G, Giardina A, Ferrante A, Carrubbi F, Giacomelli R, et al. The role of innate and lymphoid IL-22-producing cells in the immunopathology of primary Sjogren’s syndrome. Expert Rev Clin Immunol (2014) 10(4):533–41. doi:10.1586/1744666X.2014.884461
220. Buonocore S, Ahern PP, Uhlig HH, Ivanov II, Littman DR, Maloy KJ, et al. Innate lymphoid cells drive interleukin-23-dependent innate intestinal pathology. Nature (2010) 464(7293):1371–5. doi:10.1038/nature08949
221. Ding N, Yu RT, Subramaniam N, Sherman MH, Wilson C, Rao R, et al. A vitamin D receptor/SMAD genomic circuit gates hepatic fibrotic response. Cell (2013) 153(3):601–13. doi:10.1016/j.cell.2013.03.028
222. Sherman MH, Yu RT, Engle DD, Ding N, Atkins AR, Tiriac H, et al. Vitamin D receptor-mediated stromal reprogramming suppresses pancreatitis and enhances pancreatic cancer therapy. Cell (2014) 159(1):80–93. doi:10.1016/j.cell.2014.08.007
223. Laragione T, Shah A, Gulko PS. The vitamin D receptor regulates rheumatoid arthritis synovial fibroblast invasion and morphology. Mol Med (2012) 18:194–200. doi:10.2119/molmed.2011.00410
224. Wu S, Liao AP, Xia Y, Li YC, Li JD, Sartor RB, et al. Vitamin D receptor negatively regulates bacterial-stimulated NF-kappaB activity in intestine. Am J Pathol (2010) 177(2):686–97. doi:10.2353/ajpath.2010.090998
225. Nissou MF, Guttin A, Zenga C, Berger F, Issartel JP, Wion D. Additional clues for a protective role of vitamin D in neurodegenerative diseases: 1,25-dihydroxyvitamin D3 triggers an anti-inflammatory response in brain pericytes. J Alzheimers Dis (2014) 42(3):789–99. doi:10.3233/JAD-140411
226. Holick MF, Binkley NC, Bischoff-Ferrari HA, Gordon CM, Hanley DA, Heaney RP, et al. Evaluation, treatment, and prevention of vitamin D deficiency: an endocrine society clinical practice guideline. J Clin Endocrinol Metab (2011) 96(7):1911–30. doi:10.1210/jc.2011-0385
227. Aspray TJ, Bowring C, Fraser W, Gittoes N, Javaid MK, Macdonald H, et al. National Osteoporosis Society vitamin D guideline summary. Age Ageing (2014) 43(5):592–5. doi:10.1093/ageing/afu093
228. Kimball S, Vieth R, Dosch HM, Bar-Or A, Cheung R, Gagne D, et al. Cholecalciferol plus calcium suppresses abnormal PBMC reactivity in patients with multiple sclerosis. J Clin Endocrinol Metab (2011) 96(9):2826–34. doi:10.1210/jc.2011-0325
229. Bendix-Struve M, Bartels LE, Agnholt J, Dige A, Jørgensen SP, Dahlerup JF. Vitamin D3 treatment of Crohn’s disease patients increases stimulated T cell IL-6 production and proliferation. Aliment Pharmacol Ther (2010) 32(11–12):1364–72. doi:10.1111/j.1365-2036.2010.04463.x
230. Yang L, Weaver V, Smith JP, Bingaman S, Hartman TJ, Cantorna MT. Therapeutic effect of vitamin d supplementation in a pilot study of Crohn’s patients. Clin Transl Gastroenterol (2013) 4:e33. doi:10.1038/ctg.2013.1
231. Andreoli L, Dall’Ara F, Piantoni S, Zanola A, Piva N, Cutolo M, et al. A 24-month prospective study on the efficacy and safety of two different monthly regimens of vitamin D supplementation in pre-menopausal women with systemic lupus erythematosus. Lupus (2015) 24(4–5):499–506. doi:10.1177/0961203314559089
232. Miheller P, Muzes G, Hritz I, Lakatos G, Pregun I, Lakatos PL, et al. Comparison of the effects of 1,25 dihydroxyvitamin D and 25 hydroxyvitamin D on bone pathology and disease activity in Crohn’s disease patients. Inflamm Bowel Dis (2009) 15(11):1656–62. doi:10.1002/ibd.20947
233. Ferreira GB, Overbergh L, Verstuyf A, Mathieu C. 1alpha,25-Dihydroxyvitamin D3 and its analogs as modulators of human dendritic cells: a comparison dose-titration study. J Steroid Biochem Mol Biol (2013) 136:160–5. doi:10.1016/j.jsbmb.2012.10.009
234. Verlinden L, Leyssens C, Beullens I, Marcelis S, Mathieu C, De Clercq P, et al. The vitamin D analog TX527 ameliorates disease symptoms in a chemically induced model of inflammatory bowel disease. J Steroid Biochem Mol Biol (2013) 136:107–11. doi:10.1016/j.jsbmb.2012.09.017
235. Kiekhaefer CM, Weber B, Huggins M, Gorichanaz C, Nehring JA, DeLuca HF. 2alpha-Methyl-19-nor-(20S)-1,25-dihydroxyvitamin D(3) protects the insulin 2 knockout non-obese diabetic mouse from developing type 1 diabetes without hypercalcaemia. Clin Exp Immunol (2011) 166(3):325–32. doi:10.1111/j.1365-2249.2011.04481.x
236. Sochorova K, Budinsky V, Rozkova D, Tobiasova Z, Dusilova-Sulkova S, Spisek R, et al. Paricalcitol (19-nor-1,25-dihydroxyvitamin D2) and calcitriol (1,25-dihydroxyvitamin D3) exert potent immunomodulatory effects on dendritic cells and inhibit induction of antigen-specific T cells. Clin Immunol (2009) 133(1):69–77. doi:10.1016/j.clim.2009.06.011
237. Larsson P, Mattsson L, Klareskog L, Johnsson C. A vitamin D analogue (MC 1288) has immunomodulatory properties and suppresses collagen-induced arthritis (CIA) without causing hypercalcaemia. Clin Exp Immunol (1998) 114(2):277–83. doi:10.1046/j.1365-2249.1998.00706.x
238. Ikeda U, Wakita D, Ohkuri T, Chamoto K, Kitamura H, Iwakura Y, et al. 1alpha,25-dihydroxyvitamin D3 and all-trans retinoic acid synergistically inhibit the differentiation and expansion of Th17 cells. Immunol Lett (2010) 134(1):7–16. doi:10.1016/j.imlet.2010.07.002
239. Ferreira GB, Kleijwegt FS, Waelkens E, Lage K, Nikolic T, Hansen DA, et al. Differential protein pathways in 1,25-dihydroxyvitamin d(3) and dexamethasone modulated tolerogenic human dendritic cells. J Proteome Res (2012) 11(2):941–71. doi:10.1021/pr200724e
240. Zhang Y, Leung DY, Goleva E. Vitamin D enhances glucocorticoid action in human monocytes: involvement of granulocyte-macrophage colony-stimulating factor and mediator complex subunit 14. J Biol Chem (2013) 288(20):14544–53. doi:10.1074/jbc.M112.427054
241. Paintlia AS, Paintlia MK, Hollis BW, Singh AK, Singh I. Interference with RhoA-ROCK signaling mechanism in autoreactive CD4+ T cells enhances the bioavailability of 1,25-dihydroxyvitamin D3 in experimental autoimmune encephalomyelitis. Am J Pathol (2012) 181(3):993–1006. doi:10.1016/j.ajpath.2012.05.028
242. Matilainen JM, Husso T, Toropainen S, Seuter S, Turunen MP, Gynther P, et al. Primary effect of 1alpha,25(OH)(2)D(3) on IL-10 expression in monocytes is short-term down-regulation. Biochim Biophys Acta (2010) 1803(11):1276–86. doi:10.1016/j.bbamcr.2010.07.009
243. Seuter S, Heikkinen S, Carlberg C. Chromatin acetylation at transcription start sites and vitamin D receptor binding regions relates to effects of 1alpha,25-dihydroxyvitamin D3 and histone deacetylase inhibitors on gene expression. Nucleic Acids Res (2013) 41(1):110–24. doi:10.1093/nar/gks959
244. Seuter S, Ryynanen J, Carlberg C. The ASAP2 gene is a primary target of 1,25-dihydroxyvitamin D3 in human monocytes and macrophages. J Steroid Biochem Mol Biol (2014) 144:12–8. doi:10.1016/j.jsbmb.2013.08.014
245. Ryynanen J, Carlberg C. Primary 1,25-dihydroxyvitamin D3 response of the interleukin 8 gene cluster in human monocyte- and macrophage-like cells. PLoS One (2013) 8(10):e78170. doi:10.1371/journal.pone.0078170
246. Nurminen V, Neme A, Ryynanen J, Heikkinen S, Seuter S, Carlberg C. The transcriptional regulator BCL6 participates in the secondary gene regulatory response to vitamin D. Biochim Biophys Acta (2015) 1849(3):300–8. doi:10.1016/j.bbagrm.2014.12.001
247. Seuter S, Pehkonen P, Heikkinen S, Carlberg C. Dynamics of 1alpha,25-dihydroxyvitamin D3-dependent chromatin accessibility of early vitamin D receptor target genes. Biochim Biophys Acta (2013) 1829(12):1266–75. doi:10.1016/j.bbagrm.2013.10.003
Keywords: vitamin D, autoimmune disease, supplementation, T cells, B cells, dendritic cells, macrophages
Citation: Dankers W, Colin EM, van Hamburg JP and Lubberts E (2017) Vitamin D in Autoimmunity: Molecular Mechanisms and Therapeutic Potential. Front. Immunol. 7:697. doi: 10.3389/fimmu.2016.00697
Received: 12 October 2016; Accepted: 29 December 2016;
Published: 20 January 2017
Edited by:
Junji Yodoi, Kyoto University, JapanReviewed by:
Kiyoshi Hirahara, Chiba University, JapanCopyright: © 2017 Dankers, Colin, van Hamburg and Lubberts. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Erik Lubberts, ZS5sdWJiZXJ0c0BlcmFzbXVzbWMubmw=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.