 Sudan Prasad Neupane
Sudan Prasad Neupane- 1Norwegian National Advisory Unit on Concurrent Substance Abuse and Mental Health Disorders, Innlandet Hospital Trust, Brumunddal, Norway
- 2Norwegian Centre for Addiction Research (SERAF), University of Oslo, Oslo, Norway
Bidirectional communication links operate between the brain and the body. Afferent immune-to-brain signals are capable of inducing changes in mood and behavior. Chronic heavy alcohol drinking, typical of alcohol use disorder (AUD), is one such factor that provokes an immune response in the periphery that, by means of circulatory cytokines and other neuroimmune mediators, ultimately causes alterations in the brain function. Alcohol can also directly impact the immune functions of microglia, the resident immune cells of the central nervous system (CNS). Several lines of research have established the contribution of specific inflammatory mediators in the development and progression of depressive illness. Much of the available evidence in this field stems from cross-sectional data on the immune interactions between isolated AUD and major depression (MD). Given their heterogeneity as disease entities with overlapping symptoms and shared neuroimmune correlates, it is no surprise that systemic and CNS inflammation could be a critical determinant of the frequent comorbidity between AUD and MD. This review presents a summary and analysis of the extant literature on neuroimmune interface in the AUD–MD comorbidity.
Introduction
Alcohol consumption is responsible for 5.9% of global annual deaths and 5.1% of the global disease burden (1). Unipolar major depression (MD) was the second leading cause of years lived with disability worldwide, accounting for 8% of all global years lived with disabilities in 2013 (2). Together, alcohol use disorder (AUD) and MD disorders account for a half of the global disease burden attributable to mental and substance use disorders (3). An unequivocally high comorbidity exists between AUD and MD, with a lifetime comorbidity rate of 20.5% (4). About 30% of individuals with MD report lifetime AUD (5). Conversely, depressive symptoms are common in AUD to the extent that well over a third of AUD patients satisfies diagnostic criteria for MD at some point during their drinking career (6, 7). Compared to isolated disorder, patients with AUD–MD comorbidity carry higher risk of relapse to alcohol dependence, treatment dropout, suicide attempt, and poorer effect of antidepressant medication and have lower global functioning and less life satisfaction (4, 8, 9). Attempts to disentangle causal pathways between depression and AUD have resulted in the wider acceptance of bidirectional causality, with an estimate suggesting one disorder doubles the risk for the other (10). However, the mechanisms of such causality and the interfaces at which they interact remain unclear.
A colloquial understanding of the brain–body interaction is that the brain subjugates the body and pathogenic penetration of the blood–brain barrier is the only route by which bodily immune insults can reach the brain tissue. This view has changed with the demonstration of immune signals in the form of inflammatory cytokines that access the brain via afferent vagal fibers (11), by directly crossing leaky regions in the blood–brain barrier (e.g., area postrema), through cytokine-specific active transport molecules and through secondary messenger molecules within the CNS endothelia (12). Microglia and astrocytes can in turn accentuate CNS cytokine load. These cytokines and the relayed signals in the brain interact with various neurotransmitter systems as well as the hypothalamic–pituitary–adrenal (HPA) axis, the primary hormonal response system to stress (13). Furthermore, co-stimulatory signals that allow mast cells to interact with the immune cells and influence the integrity of the blood–brain barrier are important mediators of the cross talk between the peripheral and the central neuroimmune signaling (14). Thus, immune inflammatory signals in the brain are key to the translation of psychological and biological stressors into behavioral outcomes.
Several lines of research show both AUD and MD are, as isolated disorders, associated with various changes in immune function. There is, however, a paucity of knowledge on the role of neuroimmune function in the development and progression of comorbid AUD and MD. As an example, a binge pattern of drinking is particularly depressogenic (10), but the exact underlying neurobiological mechanism for this “alcoholic depression” awaits elucidation. The available evidence indicates that allostatic changes in the neuroimmune functioning could have significant impact on the development, progression, and outcome of AUD–MD comorbidity, and promising neuroimmune targets are being identified to address these issues. Several caveats remain before these developments in psychoneuroimmunology of comorbid psychiatric disorders could be capitalized.
AUD and Immunity
Alcohol is a potent modulator of the immune system and alters the expression of inflammatory mediators in the periphery as well as in the CNS. A well-described mechanistic explanation for this is that heavy alcohol consumption activates toll-like receptor (TLR) systems, including the TLR2 and TLR4 (15), through the danger-associated molecular pattern signaling, which renders the gut wall “leaky” then enabling the translocation of microbial products such as lipopolysaccharides (LPS) into circulation. This effect has been confirmed both in binge drinking (16) and chronic heavy drinking among humans (17, 18) and more widely in animal models (19, 20). The leaked LPS potentiates alcohol-induced liver inflammation and stimulates immune cells such as monocytes, macrophages, T lymphocytes, and dendritic cells to cause the release of pro-inflammatory cytokines, including interleukin (IL)-1β, IL-6, and tumor necrosis factor-alpha (TNF-α) (21). Peripherally produced cytokines and chemokines [e.g., monocyte chemoattractant protein-1 (MCP-1)] and/or their signals eventually relay to multiple brain regions, where they further activate brain microglia and astrocytes to produce CNS cytokines. The cytokine production in the brain is again dependent on TLR4 signaling and is propagated along the mitogen-activated protein kinase and NF-κB pathways. It appears that alcohol-induced cytokine upregulation follows the pattern of LPS but with less intensity. Within an hour of an intoxicating dose (5 g/kg) of ethanol, IL-10 levels were already significantly increased in rat hippocampus (22). Qin and colleagues demonstrated that comparable doses of ethanol in binge and chronic alcohol drinking paradigm in mice could induce IL-1β, TNF-α, and MCP-1 production in the liver, plasma, and brain tissues (23). In the liver and other peripheral organs, cytokine upregulation upon LPS or alcohol resolves within days to weeks. Remarkably though, brain immune activation induced by ethanol, or by LPS upon sensitization with ethanol, persisted for many months (23, 24). Using postmortem brain samples, the same group discovered that MCP-1 concentrations were increased in the ventral tegmental area, substantia nigra, hippocampus, and amygdala of alcoholic brains compared to the MCP-1 concentrations in those brain areas of moderate drinking controls (25). Since these areas are relevant to reward, emotion, and behavioral functions, MCP-1 is potentially involved in the neurodegenerative pathologies of alcohol. It is at this juncture that alcohol-induced neuroinflammation becomes clinically relevant because persistent neuroinflammation clearly precipitates cognitive and behavioral responses (26). It has recently been proposed that neuroimmune signaling is an important contributor to the development and maintenance of alcohol dependence (27). Thus, the enduring nature of the neuroimmune induction in the brain resonates with the chronicity of alcohol addiction and might represent a mechanism contributing to the development of closely comorbid conditions of alcohol dependence, such as depression (23, 24).
Alcohol modulation of the immune system involves a complex dynamic dependent on the dose and duration of exposure and chronicity of AUD (Figure 1). Acute heavy alcohol consumption (e.g., ≥3 g/kg), even in a single dose, inhibits inflammatory cell activation (28–30). Upon LPS challenge, alcohol-primed mice suppressed lung TNF-α activity, TNF-Rp55 mRNA expression, and soluble TNF-Rp55 levels (31). Ethanol suppressed LPS-induced expression of IL-1, IL-6, and their receptors while significantly upregulating IL-10 levels. In fact, acute ethanol blunted LPS-induced TNF-α secretion by 40%. This immune suppressing effect of alcohol drinking has long been appreciated. The exact molecular mechanism for the opposing immune effects of acute and chronic alcohol remains unclear. However, alcohol-induced tolerance and sensitization of TLRs depending on the length of exposure to alcohol may play a role. Through a series of experiments on human monocytes stimulated with LPS and on animal binge drinking models, Szabo and colleagues demonstrated that acute alcohol induces TLR4/LPS tolerance through activation of a nuclear protein Bcl-3, which interacts with the p50 subunit of the nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) (32). The Bcl-3–NF-κB p50 interaction results in the suppression of transcription of NF-κB-regulated genes, including that of pro-inflammatory cytokines (33). Furthermore, chronic alcohol switched the anti-inflammatory response to a pro-inflammatory response by human monocytic sensitization to LPS through decreased expression of interleukin-1 receptor-associated kinase-M, a negative regulator of TLR signaling, and subsequent activation of NF-κB, an effect opposite to acute alcohol (34).
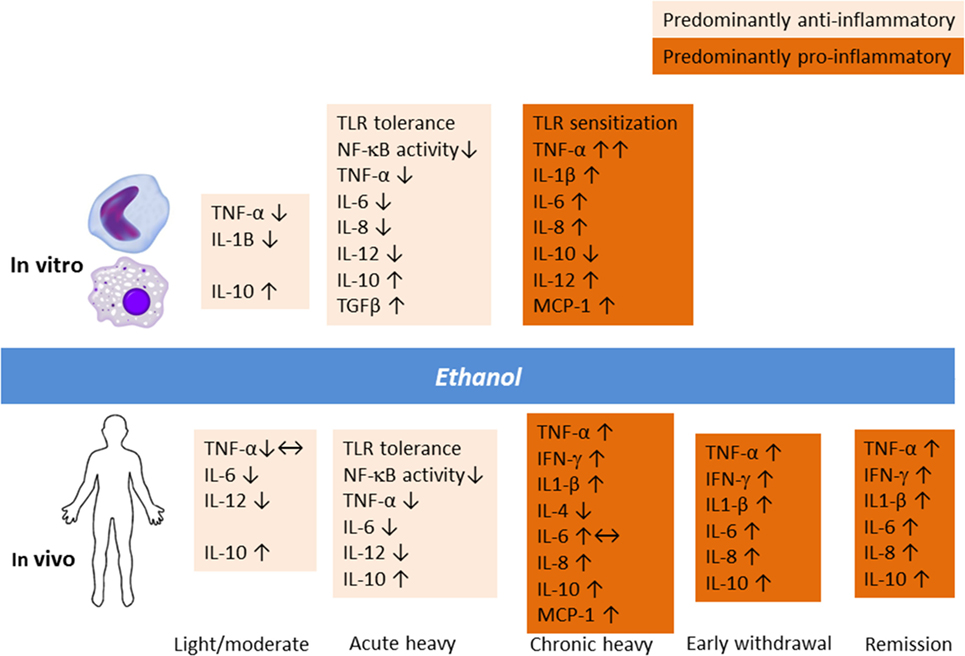
Figure 1. Alcohol modulation of the innate immune response in vitro and in vivo involves a complex dynamic depending upon the dose and duration of exposure and chronicity of alcohol use and alcohol use disorder.
Animal studies have demonstrated that an anti-inflammatory effect of acute binge drinking ensues already in the first hour and lasts beyond ethanol elimination from the body (22, 31). The same finding was replicated in humans 13 h after intake of 1.5 g/kg of alcohol (35). The experiment showed that stimulated peripheral blood mononuclear cell production of IL-10 and IL-12 as well as IFN-γ was increased upon early withdrawal. Thus, the continuum of heavy drinking, withdrawal, craving, and relapse to alcohol use potentially involves immune inflammatory signaling, an area that deserves further investigations. Recently, a study of rat models of acute alcohol intoxication suggested that the expression of inflammatory cytokines is elevated during the withdrawal phase, but changes in the central nervous system appeared to be site dependent (36). In particular, IL-6 levels were increased in multiple brain regions following alcohol exposure and lasted for up to 18 h. Thus, acute heavy drinking favors apoptotic and anti-inflammatory changes (37, 38), whereas chronic heavy drinking is known to induce monocytic TNF-α production as well as T- and B-cell activation (38, 39).
Clearly, the neuroimmune and endocrine modulatory function of alcohol varies depending on whether the individual is a social drinker or has a severe AUD (Figure 1). Gonzalez-Quintela et al. (40) reported that, among Spanish adult men and women, light-to-moderate alcohol drinking was not associated with altered levels of TNF-α. In clinical AUD populations, however, levels of cytokines are typically increased compared to non-drinking individuals (26, 41, 42). A recent study from Taiwan showed that the levels of inflammatory cytokines were elevated during the early withdrawal phase (up to 4 days of abstinence), which was considerably ameliorated upon 4 weeks of abstinence (43). Regarding alcohol effects on HPA axis, the findings have been controversial, but varying results probably indicate different vulnerability factors and the extent of familiarization with alcohol (44). Corticosterone levels surges following acute alcohol intake by social drinkers in a dose-dependent manner, but this response is dampened in chronic AUD (45). In clinical AUD populations even without liver disease, levels of cytokines are typically increased compared to non-drinking individuals (26, 41, 42). As I will elaborate later, pro-inflammatory responses seen in chronic alcohol misuse are akin to those seen in MD.
Recent studies have shown long-term negative health outcomes in animals exposed to ethanol prenatally. In adult rats prenatally exposed to ethanol, the corticosterone reservoir was depleted and the cytokine production upon immune challenge was exaggerated (46). The ensuing low-grade inflammation correlated with memory deficits, which implicated a microglial role in fetal alcohol spectrum disorders (47). Also, substantial neuroinflammation caused by traumatic brain injury induced escalation of drinking in ethanol-habituated animals (48). Thus, CNS inflammation in both low and high grades changes alcohol drinking behavior. This phenomenon is clinically relevant.
Human experimental studies on the immune effects of ethanol consumption in healthy individuals are rare for obvious ethical reasons. A few endeavors have confirmed an early recruitment of immune cells following ethanol intoxication. Afshar et al. (49) gave a binge dose of alcohol to healthy men (0.9 g ethanol/kg body weight) and women (0.8 g ethanol/kg body weight) and found a surge in the number of circulating monocytes, leukocytes, and natural killer cells—within 20 min of alcohol intake, which was followed by recovery toward baseline within 5 h. Thus, both innate and adaptive arms of the immune system are affected by alcohol. LPS induction of whole blood in the same sample showed fluctuations in inflammatory cytokines, and at 5 h, an anti-inflammatory state set in with elevated IL-10 and reduced IL-1β levels. No sex differences in immune response were reported, although animal studies indicate that females are more vulnerable to the neuroinflammatory effects of alcohol (50). For example, chronically ethanol-treated female mice expressed relatively greater levels of inflammatory mediators (iNOS and COX-2), cytokines (IL-1β, TNF-α), gliosis processes, caspase-3 activation, and neuronal loss in the cerebral cortex compared to their male counterparts (50). This finding was confirmed in postmortem brain specimens of AUD individuals who had higher MCP-1 levels and increased microglial activation markers compared to controls (25). Human pharmacogenetic studies on alcohol dependence have coincidentally discovered several immune-gene polymorphisms as underlying excessive drinking (51).
Neuroimmune Alterations in Depression
The identification of immune disturbance in depressive illness (52) led to the “macrophage hypothesis of depression,” the proposition that inflammatory products of macrophage were responsible for depression (53). Since then, a consistent body of literature has confirmed that inflammatory processes are involved in the development and progression of depressive illness. Numerous studies have consistently documented positive associations of MD with C-reactive protein (CRP) and IL-6 (54). Meta-analyses have also supported depression’s associations with IL-1β (55) and TNF-α (56, 57) as well as sIL-2 receptor (57). These associations held true for patient populations from the community as well as from clinical inpatient/outpatient settings. Patients with depression are found to have renormalized cytokine levels following treatment (58). Furthermore, several reports indicate longitudinal associations between CRP and subsequent development of depression (59), although an association was found in the opposite direction in a younger sample (60). Compelling evidence exists to suggest elevated levels of IL-6 as both a cause and a consequence of depression (61). In a 12-year prospective study of British civil servants, increased IL-6 levels at baseline predicted cognitive symptoms of depression at follow-up (62). These effects were reported to be consistent even after accounting for possible confounders such as, socio-demographics, behavioral and biological risk factors, health conditions, medication use, and baseline negative emotions. Recently, a population-based study from England (N = 5,909) showed positive associations between CRP and symptoms of fatigue, disturbed sleep, low energy, and low mood in a dose–response manner, a relationship that was absent in antidepressant medication users (63).
These novel findings quickly triggered drug trials using anti-inflammatory agents in depressive illness in humans. Notably, a proof-of-concept study examined infliximab, a TNF-α blocker in patients with treatment-resistant depression. Twelve weeks after the initiation of therapy, infliximab reduced depressive symptoms by at least a half among patients with baseline hs-CRP > 5 mg/L, but not among those with lower baseline hs-CRP levels (64). Yet, another trial showed that adjunctive celecoxib, a selective COX2 inhibitor, was more effective in reducing depressive symptoms than sertraline alone in MD (65). Again, a reduction of serum IL-6 levels correlated very well with a reduction in depression score. However, the observation period was only 6 weeks. Several other drug trials using non-steroidal anti-inflammatory drugs have been conducted (66), mostly without rigorous patient selection. Significant methodological heterogeneity and publication bias make the reported positive efficacy less tenable.
One mechanism by which activated inflammatory cytokines (mainly IFN-γ and TNF-α) can aggravate depressive symptoms is through their induction of indolamine 2,3-dioxygenase (IDO), an enzyme that metabolizes tryptophan along the neurotoxic kynurenine pathway (67, 68). IDO induction causes relative reduction in the availability of tryptophan, which is the amino acid precursor for serotonin synthesis. Tryptophan depletion and the neurotoxic metabolites produced downstream the kynurenine pathway may both trigger depression. In particular, peripheral macrophages and brain microglia preferentially metabolize kynurenine into anthranilic acid and quinolinic acid, both of which are NMDA receptor agonists and have potentially neurotoxic effects (69). Approximately, half of the cancer patients treated with IFN-α immunotherapy develops depression, and it was found that the severity of IFN-α-induced depression was related to the tryptophan degradation index (kynurenine to tryptophan ratio) along the kynurenine pathway (70). Studies also show a higher tryptophan degradation index ratio in individuals with MD compared to healthy controls (71, 72).
The failure of monoamine hypothesis to explain the delayed symptom relief in depression, despite early changes in brain monoamine neurotransmitter concentration following treatment, led to the emergence of the neurotrophic hypothesis of depression. It posits that chronic stress leads to reduced neurotrophic support to the brain limbic structures responsible for regulating mood and increases vulnerability to depression (73). Indeed, numerous studies have shown reduced serum levels of brain-derived neurotrophic factor (BDNF) in patients with MD compared to healthy controls (74–76), and evidence also exists to support renormalization of BDNF levels upon successful anti-depression interventions (74, 77). This process takes weeks to months. CNS and peripheral BDNF concentrations are altered in several mood and behavioral aberrations (74, 75, 78). Overexpressed pro-inflammatory cytokines in the brain and associated chronic neuroinflammation can lead to neurodegeneration and reduced neurogenesis, as indicated by decreased BDNF in multiple brain areas following LPS challenge (79). However, the cytokine network is rather complex, including pleiotropic effects that are sometimes paradoxical. For example, both neurotrophic and neurodestructive properties of IL-6 have been reported (80). Accordingly, circulating BDNF levels in depressed individuals were positively correlated with IL-6, but not with TNF-α (81). In recovering alcoholics, however, serum BDNF levels were positively correlated with IL-6 and TNF-α (82). Thus, the interaction between inflammatory cytokines and BDNF remains an active area of research.
While the search for neuroimmune targets in depression continues, alternative medicine has also contributed to the field. Salidroside, a traditional Tibetan herbal product, known for its antioxidative and immunotonic effects, was administered to mice that were later exposed to LPS (83). The study revealed that salidroside could effectively ameliorate LPS-induced depression-like behavior while also attenuating the inflammatory cytokine and NF-κB. Further investigations using polyphenolic compounds such as curcumin and resveratrol are underway to test the possible role of these agents in HPA axis modulation, hippocampal neurogenesis, and central monoamine homeostasis. Additionally, several other compounds related to immune regulation are of value: statins, polyunsaturated fatty acids, ketamine, TLR-inhibitors, glycogen synthase kinase-3 inhibitors, oleanolic acid analogs, and minocycline (84).
The most pressing caveat is that inflammation is neither necessary nor sufficient to cause depression, which means that activated inflammatory response would accompany only a subgroup of individuals with MD. Circulating and CNS levels of the inflammatory cytokines induced by alcohol are also modest, typically exceeding the levels in the healthy controls by a factor of 2–5 (24, 43). Unlike in purely inflammatory conditions, inflammatory markers in these low-inflammatory states rise only marginally, thus making interpersonal variations difficult to interpret (85). Nonetheless, a finding of sustained immune activation can connect depression as well as AUD with the often coexisting conditions of low-grade inflammation such as cardiovascular diseases, diabetes, fibromyalgia, multiple sclerosis, and cancer (86). A bulk of psychoneuoroimmunological literature stems from correlational evidence, which is clearly inadequate to explain the depression pathophysiology and to subsequently proffer clinical interventions. Thus, it is high time that the theoretically embraced entity of “inflammatory cytokine-associated depression” (87) be phenotype based on relevant biological and clinical characteristics. Omics-based approaches highlighting systems biomedicine could be beneficial (88). Only such progress would lead to an enhanced understanding of comorbid conditions of MD.
Neuroimmune Dysregulation in AUD–Depression Comorbidity
It should be noted that immune perturbations presented in the previous sections that focused on inflammatory cytokines are only parts of several interacting biological systems that are ascribed to AUD and MD. The proposed interrelated inflammatory and neurodegenerative mechanisms responsible for the neurobiological changes in depression involve activated central and peripheral pro-inflammatory cytokine response, lowered levels of zinc and ω3 polyunsaturated fatty acid overload, oxidative and nitrosative stress, tryptophan degradation along the kynurenine pathway, reduced neurogenesis, and increased neurodegeneration (89). A complex interaction between these processes produces neurobiology of depression and contributes to related brain disorders. For example, inflammatory cytokines in the brain are toxic to dopaminergic neurons and may precipitate Parkinson’s disease (90). Two main factors contributing to the development of alcohol addiction are reinforcement (positive and negative) and neuroadaptation, both of which seem closely related to alterations in these processes, as has been elaborated in previous sections. Given the high rates of comorbidity and overlapping pathophysiological changes in various aspects of the neuroimmune system that accompany each disorder, it will be no surprise if neuroimmune changes in AUD–MD comorbidity are somehow coordinated (Figure 2).
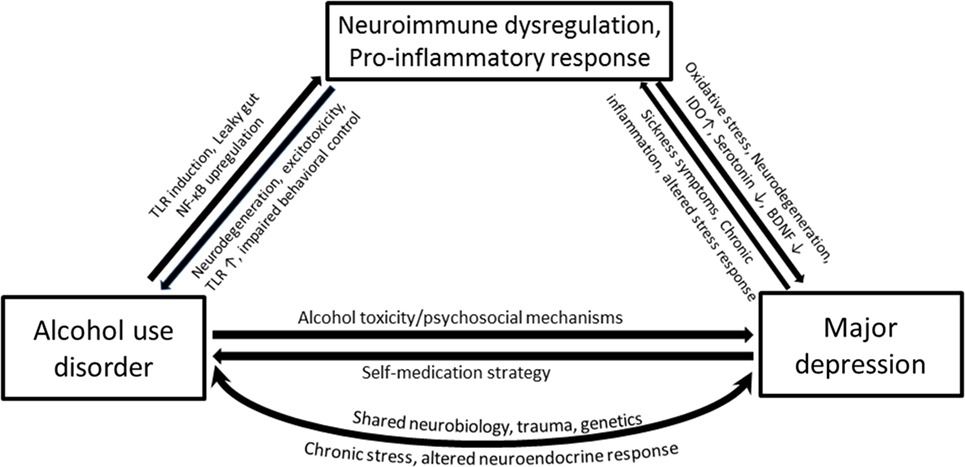
Figure 2. Simplified pathways illustrating the potential mechanisms that underlie associations between alcohol use disorder (AUD) and major depression (MD), with a compelling neuroimmune contribution. Heavy alcohol drinking may render gut wall permeable to bacterial proteins such as lipopolysaccharide (LPS) through the activation of toll-like receptors. Ethanol and LPS upregulate the transcription factor NF-κB and cause immune cells in the periphery as well as glial cells to produce pro-inflammatory cytokines. The pro-inflammatory cytokines within the brain activate the indolamine 2,3-dioxygenase enzyme, which metabolizes tryptophan away from serotonin production toward a potentially neurotoxic kynurenine pathway. MD accompanies altered monoamines, oxidative and nitrosative damage, and neurodegeneration. Depression is associated with chronic inflammatory conditions such as cancer and cardiovascular diseases, which together with sickness symptoms can feed the neuroimmune dysregulation causing further neurodegeneration, negative affect, and anxiety-like behavior and loss of behavioral control—all features characterized in AUD.
Hypothalamic–pituitary–adrenal-axis hyperactivity and glucocorticoid receptor impairment are reliable findings in depression (91), and altered HPA axis regulation is a hallmark of hormonal dysbalance in AUD (92). The nature of HPA axis abnormality upon ethanol depends on various stages of the disease and ethanol dose. In chronic AUD cases, basal ACTH levels are elevated and stress- and cue-induced corticotropin and cortisol responses are suppressed (93). Alcohol withdrawal syndrome is characterized by symptoms of autonomic hyperactivity such as tremor, sweating, anxiety, agitation, nausea, and malaise. Symptoms also include disturbed sleep and depressed mood. Interestingly, blocking the hypothalamic corticotropin-releasing factor (CRF) ameliorates the dysphoric symptoms of alcohol withdrawal (94) and the increased stress responsiveness and associated anxiety-like behavior during protracted abstinence (95). CRF blockade in depression-like behavior in a mouse model was shown to reduce those symptoms through modulation of neuronal plasticity (96). Taken together, involvement of brain stress systems in neuroadaptive changes accompanying addiction and emotional circuitry provides a common interface for AUD- and MD-related neuronal changes.
Inflammatory cytokines are potent inducers of CRF and, therefore, negative affect during withdrawal and negative reinforcement during long-term abstinence could potentially arise from immune-mediated CRF activation. Glucocorticoids thus produced cause tryptophan degradation by activating hepatic degradation of tryptophan 2,3-dioxygenase (TDO), which, along with cytokine-induced IDO in the brain, once again produces metabolites biased toward the neurotoxic edge (97, 98). TDO enzyme is activated upon acute alcohol consumption, subsequently inhibited with chronic alcohol drinking, and again surges during ethanol withdrawal (99–101). The altered tryptophan metabolism reportedly lasts for several months into abstention, as shown in a comparative study of 4 and 11 weeks of abstinence, wherein longer abstinence was related to increased kynurenine levels (102, 103). This could well be explained in terms of hyperactive stress response in concert with negative reinforcement, craving, and relapse. We reported increased tryptophan turnover with increased duration of abstinence (104). However, another study (105) showed that AUD individuals who abstained from alcohol for longer than two weeks, regardless of background variables, had much higher tryptophan levels compared to healthy controls. Literature also indicates a contradictory higher tryptophan and lower tryptophan degradation in depression, alongside activated pro-inflammatory pathway (104, 106, 107), but these findings are based on peripherally measured mediators and may not reflect brain levels. An overview of the few studies that have investigated neuroimmune mediators in the context of AUD–MD comorbidity are shown in Table 1. Further studies are needed to clarify these observations.
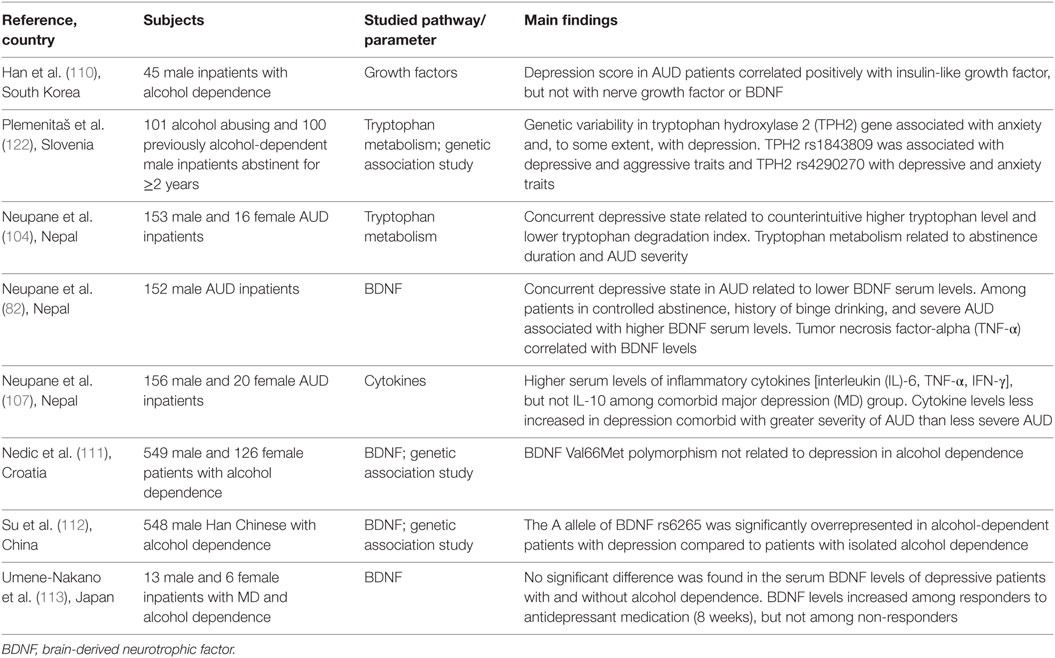
Table 1. Overview of studies investigating neuroimmune pathways in comorbid alcohol use disorder (AUD) and depression.
Neurotrophic changes in brain regions involved in depression and AUD are relevant considerations. As discussed in the previous section, depression is consistently associated with depleted BDNF. Indeed, neuroinflammation has an inhibitory effect on adult cortical and hippocampal neurogenesis, as evidenced by reduced BDNF expression concurrent to LPS-induced upregulation of inflammatory cytokines in rats (79). Similarly, chronic ethanol exposure in humans was accompanied by reduced BDNF expression in the hippocampus (108) as well as lower plasma BDNF protein levels (109). Reduced expression of BDNF in the hippocampus and cortical regions is a clear conjuncture for AUD and depression because these are critical target brain regions in both disorders. During the last decade, we (82) and others (110–113) have investigated BDNF in AUD patients. The findings of these studies indicate that neuronal repair initiates soon after the abstention commences, and BDNF levels continue to rise over several months (109, 113–117). Rat models of alcoholism showed that augmenting BDNF actions by the use of the selective BDNF tyrosine kinase B receptor agonist (7,8-dihydroflavone) removed withdrawal-induced depression-like behavior (118). Depressive symptoms are observed during various stages of AUD. In many cases of AUD, associated depressive symptoms do not disappear even after sustained abstinence. Against this backdrop, bidirectional causality between AUD and MD has been demonstrated with a more robust association seen from AUD leading toward MD (10). These evidences suggest the existence of what might be considered alcoholic depression and that a biological explanation for depression in AUD could be approached from the immune inflammatory and stress pathways. It remains to be shown how the neuroadaptive changes in recovering AUD individuals relate to depressive symptoms, and whether targeting key neuroimmune factors such as BDNF is a viable intervention option in AUD–MD comorbidity.
Immune signaling induces a range of physiological responses that are common to affective and behavioral disorders. Infection accompanies a TLR4-mediated pro-inflammatory response, indicated by raised IL-1β, IL-6, and TNF-α levels, which leads to “sickness behavior” (119, 120). Sickness behavior is also observed upon psychological stress and exogenous cytokine administration such as during cancer treatment with IFN-α and includes physiological responses (e.g., fever and disturbed sleep) as well as behavioral symptoms (e.g., anorexia, reduced mobility, disappearance of body care activities and reduced social interaction) (119). Many of these features overlap with those of depression. Compelling evidence also suggests activated TLR4 signaling to accentuate alcohol drinking but also negative affect and anxiety-like behavior, especially during the withdrawal phase (121). Sickness symptoms wane away over several days; however, cytokine induction of these behavioral changes may persist as MD. Thus, a better understanding of the loop between immunity, the brain, and behavioral outcomes holds promises to newer approaches to intervene AUD–MD pathologies.
Conclusion and Future Directions
The clinical realm of frequent comorbidity between AUD and MD requires an integrated psychobiological understanding that underlies these conditions. As presented in this review, mounting evidence supports neuroimmunological alterations, in particular activated immune responses (26), as a critical piece of the physiological link between AUD and MD. Neuroimmune gene induction in limbic brain regions increases negative affect, drug-seeking behavior, and loss of behavioral control (51). Diminished affect is a hallmark of depression, and anxiety-like behavior is pronounced in the withdrawal phase of alcohol addiction. Thus, mood symptoms as well as emotional and behavioral lability in AUD and MD appear to stem from neuroimmune mechanisms (21, 51, 87). The relative contribution of one phenomenon in the context of the other remains unclear. The current evidence is clearly inadequate to unravel the full scope of possible neuroimmune etiopathology of isolated AUD and MD, and an endeavor to attack their comorbidity may sound premature at this stage. However, this complexity should not be a hindrance to investigate these two disorders in their totality, because the relative neuroimmune contribution to each disorder may become clearer when one of the comorbid conditions vanishes. Prospective studies investigating longitudinal associations between changes in neuroimmune function and changes in depressive symptoms, drinking behavior, and treatment outcome are necessary. Furthermore, researchers in the field should be aware of the ethical obligation not to categorically exclude patients who have additional burden.
An extension of the research focus from isolated to comorbid disorders and from preclinical to clinical settings is conducive to appraising the significant overlaps in manifestation of AUD and MD, as well as common biological perturbations in their development and maintenance. It is important to note that the neuroimmune approach alone would not be sufficient to elucidate the underlying complex etiopathology of AUD and MD, which strongly involves other genetic, epigenetic, and environmental factors. However, the neuroimmune approach would constitute an essential component of the systems biomedicine and be applicable to a significant proportion of patient populations. Equally important is the identification of intermediary processes that may determine the ultimate neuroimmune allostasis. Taken together, an exploration of a neuroimmune model for AUD–MD comorbidity provides a foundation for the development of more effective immune-based pharmacotherapy against these burdensome disorders.
Author Contributions
The author confirms being the sole contributor of this work and approved it for publication.
Conflict of Interest Statement
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
This study was funded by the Norwegian South-East Regional Health Authority (Grant number 150348). The funder had no roles in the design of the study, interpretation of the reviewed data, or the decision to submit this manuscript.
References
1. World Health Organization. Global Status Report on Alcohol and Health-2014. Geneva: World Health Organization (2014).
2. Global Burden of Disease Study 2013 Collaborators. Global, regional, and national incidence, prevalence, and years lived with disability for 301 acute and chronic diseases and injuries in 188 countries, 1990-2013: a systematic analysis for the Global Burden of Disease Study 2013. Lancet (2015) 386:743–800. doi:10.1016/S0140-6736(15)60692-4
3. Whiteford HA, Degenhardt L, Rehm J, Baxter AJ, Ferrari AJ, Erskine HE, et al. Global burden of disease attributable to mental and substance use disorders: findings from the Global Burden of Disease Study 2010. Lancet (2013) 382:1575–86. doi:10.1016/S0140-6736(13)61611-6
4. Briere FN, Rohde P, Seeley JR, Klein D, Lewinsohn PM. Comorbidity between major depression and alcohol use disorder from adolescence to adulthood. Compr Psychiatry (2014) 55:526–33. doi:10.1016/j.comppsych.2013.10.007
5. Sullivan LE, Fiellin DA, O’Connor PG. The prevalence and impact of alcohol problems in major depression: a systematic review. Am J Med (2005) 118:330–41. doi:10.1016/j.amjmed.2005.01.007
6. Lynskey MT. The comorbidity of alcohol dependence and affective disorders: treatment implications. Drug Alcohol Depend (1998) 52:201–9. doi:10.1016/S0376-8716(98)00095-7
7. Schuckit MA. Comorbidity between substance use disorders and psychiatric conditions. Addiction (2006) 101:76–88. doi:10.1111/j.1360-0443.2006.01592.x
8. Nunes EV, Levin FR. Treatment of depression in patients with alcohol or other drug dependence. JAMA (2004) 291:1887–96. doi:10.1001/jama.291.15.1887
9. Hasin D, Liu X, Nunes E, McCloud S, Samet S, Endicott J. Effects of major depression on remission and relapse of substance dependence. Arch Gen Psychiatry (2002) 59:375–80. doi:10.1001/archpsyc.59.4.375
10. Boden JM, Fergusson DM. Alcohol and depression. Addiction (2011) 106:906–14. doi:10.1111/j.1360-0443.2010.03351.x
11. Watkins LR, Maier SF, Goehler LE. Cytokine-to-brain communication: a review & analysis of alternative mechanisms. Life Sci (1995) 57:1011–26. doi:10.1016/0024-3205(95)02047-M
12. Quan N, Banks WA. Brain-immune communication pathways. Brain Behav Immun (2007) 21:727–35. doi:10.1016/j.bbi.2007.05.005
13. Raison CL, Capuron L, Miller AH. Cytokines sing the blues: inflammation and the pathogenesis of depression. Trends Immunol (2006) 27:24–31. doi:10.1016/j.it.2005.11.006
14. Ribatti D. The crucial role of mast cells in blood-brain barrier alterations. Exp Cell Res (2015) 338:119–25. doi:10.1016/j.yexcr.2015.05.013
15. Pascual M, Balino P, Aragon CM, Guerri C. Cytokines and chemokines as biomarkers of ethanol-induced neuroinflammation and anxiety-related behavior: role of TLR4 and TLR2. Neuropharmacology (2015) 89:352–9. doi:10.1016/j.neuropharm.2014.10.014
16. Bala S, Marcos M, Gattu A, Catalano D, Szabo G. Acute binge drinking increases serum endotoxin and bacterial DNA levels in healthy individuals. PLoS One (2014) 9:e96864. doi:10.1371/journal.pone.0096864
17. Bjarnason I, Ward K, Peters T. The leaky gut of alcoholism: possible route of entry for toxic compounds. Lancet (1984) 323:179–82. doi:10.1016/S0140-6736(84)92109-3
18. Enomoto N, Ikejima K, Bradford BU, Rivera CA, Kono H, Goto M, et al. Role of Kupffer cells and gut-derived endotoxins in alcoholic liver injury. J Gastroenterol Hepatol (2000) 15(Suppl):D20–5. doi:10.1046/j.1440-1746.2000.02179.x
19. Keshavarzian A, Farhadi A, Forsyth CB, Rangan J, Jakate S, Shaikh M, et al. Evidence that chronic alcohol exposure promotes intestinal oxidative stress, intestinal hyperpermeability and endotoxemia prior to development of alcoholic steatohepatitis in rats. J Hepatol (2009) 50:538–47. doi:10.1016/j.jhep.2008.10.028
20. Szabo G. Gut-liver axis in alcoholic liver disease. Gastroenterology (2015) 148:30–6. doi:10.1053/j.gastro.2014.10.042
21. Crews FT, Vetreno RP. Mechanisms of neuroimmune gene induction in alcoholism. Psychopharmacology (Berl) (2016) 233:1543–57. doi:10.1007/s00213-015-3906-1
22. Suryanarayanan A, Carter JM, Landin JD, Morrow AL, Werner DF, Spigelman I. Role of interleukin-10 (IL-10) in regulation of GABAergic transmission and acute response to ethanol. Neuropharmacology (2016) 107:181–8. doi:10.1016/j.neuropharm.2016.03.027
23. Qin L, Wu X, Block ML, Liu Y, Breese GR, Hong JS, et al. Systemic LPS causes chronic neuroinflammation and progressive neurodegeneration. Glia (2007) 55:453–62. doi:10.1002/glia.20467
24. Qin L, He J, Hanes RN, Pluzarev O, Hong JS, Crews FT. Increased systemic and brain cytokine production and neuroinflammation by endotoxin following ethanol treatment. J Neuroinflammation (2008) 5:10. doi:10.1186/1742-2094-5-10
25. He J, Crews FT. Increased MCP-1 and microglia in various regions of the human alcoholic brain. Exp Neurol (2008) 210:349–58. doi:10.1016/j.expneurol.2007.11.017
26. Kelley KW, Dantzer R. Alcoholism and inflammation: neuroimmunology of behavioral and mood disorders. Brain Behav Immun (2011) 25(Suppl 1):S13–20. doi:10.1016/j.bbi.2010.12.013
27. Vetreno RP, Crews FT. Chapter 27 – current hypotheses on the mechanisms of alcoholism. In: Edith VS, Adolf P, editors. Handbook of Clinical Neurology. Amsterdam: Elsevier (2014). p. 477–97.
28. Pruett SB, Zheng Q, Fan R, Matthews K, Schwab C. Acute exposure to ethanol affects Toll-like receptor signaling and subsequent responses: an overview of recent studies. Alcohol (2004) 33:235–9. doi:10.1016/j.alcohol.2004.08.003
29. Szabo G, Catalano D, White B, Mandrekar P. Acute alcohol consumption inhibits accessory cell function of monocytes and dendritic cells. Alcohol Clin Exp Res (2004) 28:824–8. doi:10.1097/01.ALC.0000127104.80398.9B
30. Mandrekar P, Jeliazkova V, Catalano D, Szabo G. Acute alcohol exposure exerts anti-inflammatory effects by inhibiting IkappaB kinase activity and p65 phosphorylation in human monocytes. J Immunol (2007) 178:7686–93. doi:10.4049/jimmunol.178.12.7686
31. D’Souza El-Guindy NB, de Villiers WJ, Doherty DE. Acute alcohol intake impairs lung inflammation by changing pro- and anti-inflammatory mediator balance. Alcohol (2007) 41:335–45. doi:10.1016/j.alcohol.2007.07.002
32. Bala S, Tang A, Catalano D, Petrasek J, Taha O, Kodys K, et al. Induction of Bcl-3 by acute binge alcohol results in toll-like receptor 4/LPS tolerance. J Leukoc Biol (2012) 92:611–20. doi:10.1189/jlb.0112050
33. Kuwata H, Watanabe Y, Miyoshi H, Yamamoto M, Kaisho T, Takeda K, et al. IL-10-inducible Bcl-3 negatively regulates LPS-induced TNF-alpha production in macrophages. Blood (2003) 102:4123–9. doi:10.1182/blood-2003-04-1228
34. Mandrekar P, Bala S, Catalano D, Kodys K, Szabo G. The opposite effects of acute and chronic alcohol on lipopolysaccharide-induced inflammation are linked to IRAK-M in human monocytes. J Immunol (2009) 183:1320–7. doi:10.4049/jimmunol.0803206
35. Kim DJ, Kim W, Yoon SJ, Choi BM, Kim JS, Go HJ, et al. Effects of alcohol hangover on cytokine production in healthy subjects. Alcohol (2003) 31:167–70. doi:10.1016/j.alcohol.2003.09.003
36. Doremus-Fitzwater TL, Buck HM, Bordner K, Richey L, Jones ME, Deak T. Intoxication- and withdrawal-dependent expression of central and peripheral cytokines following initial ethanol exposure. Alcohol Clin Exp Res (2014) 38:2186–98. doi:10.1111/acer.12481
37. Deak T, Okrainets A, Doremus-Fitzwater TL. Mechanisms of stress-dependent neuroinflammation and their implications for understanding consequences of alcohol exposure. In: Cui C, Grandison L, Noronha A, editors. Neural-Immune Interactions in Brain Function and Alcohol Related Disorders. New York: Springer (2013). p. 133–66.
38. Barr T, Helms C, Grant K, Messaoudi I. Opposing effects of alcohol on the immune system. Prog Neuropsychopharmacol Biol Psychiatry (2016) 65:242–51. doi:10.1016/j.pnpbp.2015.09.001
39. Laso FJ, Vaquero JM, Almeida J, Marcos M, Orfao A. Production of inflammatory cytokines by peripheral blood monocytes in chronic alcoholism: relationship with ethanol intake and liver disease. Cytometry B Clin Cytom (2007) 72:408–15. doi:10.1002/cyto.b.20169
40. Gonzalez-Quintela A, Campos J, Loidi L, Quinteiro C, Perez LF, Gude F. Serum TNF-alpha levels in relation to alcohol consumption and common TNF gene polymorphisms. Alcohol (2008) 42:513–8. doi:10.1016/j.alcohol.2008.04.008
41. Achur RN, Freeman WM, Vrana KE. Circulating cytokines as biomarkers of alcohol abuse and alcoholism. J Neuroimmune Pharmacol (2010) 5:83–91. doi:10.1007/s11481-009-9185-z
42. Szabo G, Mandrekar P. A recent perspective on alcohol, immunity, and host defense. Alcohol Clin Exp Res (2009) 33:220–32. doi:10.1111/j.1530-0277.2008.00842.x
43. Yen C-H, Ho P-S, Yeh Y-W, Liang C-S, Kuo S-C, Huang C-C, et al. Differential cytokine levels between early withdrawal and remission states in patients with alcohol dependence. Psychoneuroendocrinology (2016) 76:183–91. doi:10.1016/j.psyneuen.2016.10.015
44. Mick I, Spring K, Uhr M, Zimmermann US. Alcohol administration attenuates hypothalamic–pituitary–adrenal (HPA) activity in healthy men at low genetic risk for alcoholism, but not in high-risk subjects. Addict Biol (2013) 18:863–71. doi:10.1111/j.1369-1600.2011.00420.x
45. King A, Munisamy G, de Wit H, Lin S. Attenuated cortisol response to alcohol in heavy social drinkers. Int J Psychophysiol (2006) 59:203–9. doi:10.1016/j.ijpsycho.2005.10.008
46. Bodnar TS, Hill LA, Weinberg J. Evidence for an immune signature of prenatal alcohol exposure in female rats. Brain Behav Immun (2016) 58:130–41. doi:10.1016/j.bbi.2016.05.022
47. Terasaki LS, Schwarz JM. Effects of moderate prenatal alcohol exposure during early gestation in rats on inflammation across the maternal-fetal-immune interface and later-life immune function in the offspring. J Neuroimmune Pharmacol (2016) 11(4):680–92. doi:10.1007/s11481-016-9691-8
48. Mayeux JP, Teng SX, Katz PS, Gilpin NW, Molina PE. Traumatic brain injury induces neuroinflammation and neuronal degeneration that is associated with escalated alcohol self-administration in rats. Behav Brain Res (2015) 279:22–30. doi:10.1016/j.bbr.2014.10.053
49. Afshar M, Richards S, Mann D, Cross A, Smith GB, Netzer G, et al. Acute immunomodulatory effects of binge alcohol ingestion. Alcohol (2015) 49:57–64. doi:10.1016/j.alcohol.2014.10.002
50. Alfonso-Loeches S, Pascual M, Guerri C. Gender differences in alcohol-induced neurotoxicity and brain damage. Toxicology (2013) 311:27–34. doi:10.1016/j.tox.2013.03.001
51. Crews FT, Zou J, Qin L. Induction of innate immune genes in brain create the neurobiology of addiction. Brain Behav Immun (2011) 25(Suppl 1):S4–12. doi:10.1016/j.bbi.2011.03.003
52. Maes M, Bosmans E, Suy E, Vandervorst C, De Jonckheere C, Raus J. Immune disturbances during major depression: upregulated expression of interleukin-2 receptors. Neuropsychobiology (1990) 24:115–20. doi:10.1159/000119472
53. Smith RS. The macrophage theory of depression. Med Hypotheses (1991) 35:298–306. doi:10.1016/0306-9877(91)90272-Z
54. Haapakoski R, Mathieu J, Ebmeier KP, Alenius H, Kivimaki M. Cumulative meta-analysis of interleukins 6 and 1beta, tumour necrosis factor alpha and C-reactive protein in patients with major depressive disorder. Brain Behav Immun (2015) 49:206–15. doi:10.1016/j.bbi.2015.06.001
55. Howren MB, Lamkin DM, Suls J. Associations of depression with C-reactive protein, IL-1, and IL-6: a meta-analysis. Psychosom Med (2009) 71:171–86. doi:10.1097/PSY.0b013e3181907c1b
56. Dowlati Y, Herrmann N, Swardfager W, Liu H, Sham L, Reim EK, et al. A meta-analysis of cytokines in major depression. Biol Psychiatry (2010) 67:446–57. doi:10.1016/j.biopsych.2009.09.033
57. Liu Y, Ho RC-M, Mak A. Interleukin (IL)-6, tumour necrosis factor alpha (TNF-α) and soluble interleukin-2 receptors (sIL-2R) are elevated in patients with major depressive disorder: a meta-analysis and meta-regression. J Affect Disord (2012) 139:230–9. doi:10.1016/j.jad.2011.08.003
58. Kim Y-K, Na K-S, Shin K-H, Jung H-Y, Choi S-H, Kim J-B. Cytokine imbalance in the pathophysiology of major depressive disorder. Prog Neuropsychopharmacol Biol Psychiatry (2007) 31:1044–53. doi:10.1016/j.pnpbp.2007.03.004
59. Valkanova V, Ebmeier KP, Allan CL. CRP, IL-6 and depression: a systematic review and meta-analysis of longitudinal studies. J Affect Disord (2013) 150:736–44. doi:10.1016/j.jad.2013.06.004
60. Copeland WE, Shanahan L, Worthman C, Angold A, Costello EJ. Cumulative depression episodes predict later C-reactive protein levels: a prospective analysis. Biol Psychiatry (2012) 71:15–21. doi:10.1016/j.biopsych.2011.09.023
61. Stewart JC, Rand KL, Muldoon MF, Kamarck TW. A prospective evaluation of the directionality of the depression-inflammation relationship. Brain Behav Immun (2009) 23:936–44. doi:10.1016/j.bbi.2009.04.011
62. Gimeno D, Kivimaki M, Brunner EJ, Elovainio M, De Vogli R, Steptoe A, et al. Associations of C-reactive protein and interleukin-6 with cognitive symptoms of depression: 12-year follow-up of the Whitehall II study. Psychol Med (2009) 39:413–23. doi:10.1017/S0033291708003723
63. White J, Kivimaki M, Jokela M, David Batty G. Association of inflammation with specific symptoms of depression in a general population of older people: the English Longitudinal Study of Ageing. Brain Behav Immun (2016). doi:10.1016/j.bbi.2016.08.012
64. Raison CL, Rutherford RE, Woolwine BJ, Shuo C, Schettler P, Drake DF, et al. A randomized controlled trial of the tumor necrosis factor antagonist infliximab for treatment-resistant depression: the role of baseline inflammatory biomarkers. JAMA Psychiatry (2013) 70:31–41. doi:10.1001/2013.jamapsychiatry.4
65. Abbasi SH, Hosseini F, Modabbernia A, Ashrafi M, Akhondzadeh S. Effect of celecoxib add-on treatment on symptoms and serum IL-6 concentrations in patients with major depressive disorder: randomized double-blind placebo-controlled study. J Affect Disord (2012) 141:308–14. doi:10.1016/j.jad.2012.03.033
66. Kohler O, Benros ME, Nordentoft M, Farkouh ME, Iyengar RL, Mors O, et al. Effect of anti-inflammatory treatment on depression, depressive symptoms, and adverse effects: a systematic review and meta-analysis of randomized clinical trials. JAMA Psychiatry (2014) 71:1381–91. doi:10.1001/jamapsychiatry.2014.1611
67. O’Connor JC, Andre C, Wang Y, Lawson MA, Szegedi SS, Lestage J, et al. Interferon-gamma and tumor necrosis factor-alpha mediate the upregulation of indoleamine 2,3-dioxygenase and the induction of depressive-like behavior in mice in response to bacillus Calmette-Guerin. J Neurosci (2009) 29:4200–9. doi:10.1523/JNEUROSCI.5032-08.2009
68. Dantzer R, O’Connor JC, Lawson MA, Kelley KW. Inflammation-associated depression: from serotonin to kynurenine. Psychoneuroendocrinology (2011) 36:426–36. doi:10.1016/j.psyneuen.2010.09.012
69. Walker AK, Dantzer R, Kelley KW. Mood Disorders and Immunity, Neural-Immune Interactions in Brain Function and Alcohol Related Disorders. New York: Springer (2013). p. 167–209.
70. Capuron L, Ravaud A, Neveu PJ, Miller AH, Maes M, Dantzer R. Association between decreased serum tryptophan concentrations and depressive symptoms in cancer patients undergoing cytokine therapy. Mol Psychiatry (2002) 7:468–73. doi:10.1038/sj.mp.4000995
71. Maes M, Leonard BE, Myint AM, Kubera M, Verkerk R. The new ‘5-HT’ hypothesis of depression: cell-mediated immune activation induces indoleamine 2,3-dioxygenase, which leads to lower plasma tryptophan and an increased synthesis of detrimental tryptophan catabolites (TRYCATs), both of which contribute to the onset of depression. Prog Neuropsychopharmacol Biol Psychiatry (2011) 35:702–21. doi:10.1016/j.pnpbp.2010.12.017
72. Myint AM, Kim YK, Verkerk R, Scharpe S, Steinbusch H, Leonard B. Kynurenine pathway in major depression: evidence of impaired neuroprotection. J Affect Disord (2007) 98:143–51. doi:10.1016/j.jad.2006.07.013
73. Duman RS, Monteggia LM. A neurotrophic model for stress-related mood disorders. Biol Psychiatry (2006) 59:1116–27. doi:10.1016/j.biopsych.2006.02.013
74. Brunoni AR, Lopes M, Fregni F. A systematic review and meta-analysis of clinical studies on major depression and BDNF levels: implications for the role of neuroplasticity in depression. Int J Neuropsychopharmacol (2008) 11:1169–80. doi:10.1017/S1461145708009309
75. Molendijk ML, Spinhoven P, Polak M, Bus BA, Penninx BW, Elzinga BM. Serum BDNF concentrations as peripheral manifestations of depression: evidence from a systematic review and meta-analyses on 179 associations (N=9484). Mol Psychiatry (2014) 19:791–800. doi:10.1038/mp.2013.105
76. Sen S, Duman R, Sanacora G. Serum brain-derived neurotrophic factor, depression, and antidepressant medications: meta-analyses and implications. Biol Psychiatry (2008) 64:527–32. doi:10.1016/j.biopsych.2008.05.005
77. Brunoni AR, Baeken C, Machado-Vieira R, Gattaz WF, Vanderhasselt MA. BDNF blood levels after electroconvulsive therapy in patients with mood disorders: a systematic review and meta-analysis. World J Biol Psychiatry (2014) 15:411–8. doi:10.3109/15622975.2014.892633
78. Autry AE, Monteggia LM. Brain-derived neurotrophic factor and neuropsychiatric disorders. Pharmacol Rev (2012) 64:238–58. doi:10.1124/pr.111.005108
79. Guan Z, Fang J. Peripheral immune activation by lipopolysaccharide decreases neurotrophins in the cortex and hippocampus in rats. Brain Behav Immun (2006) 20:64–71. doi:10.1016/j.bbi.2005.04.005
80. Spooren A, Kolmus K, Laureys G, Clinckers R, De Keyser J, Haegeman G, et al. Interleukin-6, a mental cytokine. Brain Res Rev (2011) 67:157–83. doi:10.1016/j.brainresrev.2011.01.002
81. Patas K, Penninx BW, Bus BA, Vogelzangs N, Molendijk ML, Elzinga BM, et al. Association between serum brain-derived neurotrophic factor and plasma interleukin-6 in major depressive disorder with melancholic features. Brain Behav Immun (2014) 36:71–9. doi:10.1016/j.bbi.2013.10.007
82. Neupane SP, Lien L, Ueland T, Mollnes TE, Aukrust P, Bramness JG. Serum brain-derived neurotrophic factor levels in relation to comorbid depression and cytokine levels in Nepalese men with alcohol-use disorders. Alcohol (2015) 49:471–8. doi:10.1016/j.alcohol.2015.01.012
83. Zhu L, Wei T, Gao J, Chang X, He H, Miao M, et al. Salidroside attenuates lipopolysaccharide (LPS) induced serum cytokines and depressive-like behavior in mice. Neurosci Lett (2015) 606:1–6. doi:10.1016/j.neulet.2015.08.025
84. Schmidt FM, Kirkby KC, Lichtblau N. Inflammation and immune regulation as potential drug targets in antidepressant treatment. Curr Neuropharmacol (2016) 14:674–87. doi:10.2174/1570159X14666160115130414
85. Raison CL, Miller AH. Is depression an inflammatory disorder? Curr Psychiatry Rep (2011) 13:467–75. doi:10.1007/s11920-011-0232-0
86. Maes M, Kubera M, Obuchowiczwa E, Goehler L, Brzeszcz J. Depression’s multiple comorbidities explained by (neuro) inflammatory and oxidative & nitrosative stress pathways. Neuroendocrinol Lett (2011) 32:7–24.
87. Lotrich FE. Inflammatory cytokine-associated depression. Brain Res (2014) 1617:113–25. doi:10.1016/j.brainres.2014.06.032
88. Maes M, Nowak G, Caso JR, Leza JC, Song C, Kubera M, et al. Toward omics-based, systems biomedicine, and path and drug discovery methodologies for depression-inflammation research. Mol Neurobiol (2016) 53:2927–35. doi:10.1007/s12035-015-9183-5
89. Maes M, Yirmyia R, Noraberg J, Brene S, Hibbeln J, Perini G, et al. The inflammatory & neurodegenerative (I&ND) hypothesis of depression: leads for future research and new drug developments in depression. Metab Brain Dis (2009) 24:27–53. doi:10.1007/s11011-008-9118-1
90. Hernandez-Romero MC, Arguelles S, Villaran RF, de Pablos RM, Delgado-Cortes MJ, Santiago M, et al. Simvastatin prevents the inflammatory process and the dopaminergic degeneration induced by the intranigral injection of lipopolysaccharide. J Neurochem (2008) 105:445–59. doi:10.1111/j.1471-4159.2007.05148.x
91. Stetler C, Miller GE. Depression and hypothalamic-pituitary-adrenal activation: a quantitative summary of four decades of research. Psychosom Med (2011) 73:114–26. doi:10.1097/PSY.0b013e31820ad12b
92. Burke HM, Davis MC, Otte C, Mohr DC. Depression and cortisol responses to psychological stress: a meta-analysis. Psychoneuroendocrinology (2005) 30:846–56. doi:10.1016/j.psyneuen.2005.02.010
93. Sinha R, Fox HC, Hong KI, Hansen J, Tuit K, Kreek MJ. Effects of adrenal sensitivity, stress- and cue-induced craving, and anxiety on subsequent alcohol relapse and treatment outcomes. Arch Gen Psychiatry (2011) 68:942–52. doi:10.1001/archgenpsychiatry.2011.49
94. Overstreet DH, Knapp DJ, Breese GR. Modulation of multiple ethanol withdrawal-induced anxiety-like behavior by CRF and CRF1 receptors. Pharmacol Biochem Behav (2004) 77:405–13. doi:10.1016/j.pbb.2003.11.010
95. Breese GR, Overstreet DH, Knapp DJ, Navarro M. Prior multiple ethanol withdrawals enhance stress-induced anxiety-like behavior: inhibition by CRF1- and benzodiazepine-receptor antagonists and a 5-HT1a-receptor agonist. Neuropsychopharmacology (2005) 30:1662–9. doi:10.1038/sj.npp.1300706
96. Alonso R, Griebel G, Pavone G, Stemmelin J, Le Fur G, Soubrie P. Blockade of CRF(1) or V(1b) receptors reverses stress-induced suppression of neurogenesis in a mouse model of depression. Mol Psychiatry (2004) 9:278–86. doi:10.1038/sj.mp.4001484
97. Oxenkrug GF. Tryptophan kynurenine metabolism as a common mediator of genetic and environmental impacts in major depressive disorder: the serotonin hypothesis revisited 40 years later. Isr J Psychiatry Relat Sci (2010) 47:56–63.
98. Badawy AA. Tryptophan: the key to boosting brain serotonin synthesis in depressive illness. J Psychopharmacol (2013) 27:878–93. doi:10.1177/0269881113499209
99. Branchey L, Branchey M, Shaw S, Lieber CS. Relationship between changes in plasma amino acids and depression in alcoholic patients. Am J Psychiatry (1984) 141:1212–5. doi:10.1176/ajp.141.10.1212
100. Badawy AA. Tryptophan metabolism in alcoholism. Nutr Res Rev (2002) 15:123–52. doi:10.1079/NRR200133
101. Badawy AA, Doughrty DM, Marsh-Richard DM, Steptoe A. Activation of liver tryptophan pyrrolase mediates the decrease in tryptophan availability to the brain after acute alcohol consumption by normal subjects. Alcohol Alcohol (2009) 44:267–71. doi:10.1093/alcalc/agp005
102. Gleissenthall GV, Geisler S, Malik P, Kemmler G, Benicke H, Fuchs D, et al. Tryptophan metabolism in post-withdrawal alcohol-dependent patients. Alcohol Alcohol (2014) 49:251–5. doi:10.1093/alcalc/agu011
103. Sinha R, Fox HC, Hong KA, Bergquist K, Bhagwagar Z, Siedlarz KM. Enhanced negative emotion and alcohol craving, and altered physiological responses following stress and cue exposure in alcohol dependent individuals. Neuropsychopharmacology (2009) 34:1198–208. doi:10.1038/npp.2008.78
104. Neupane SP, Lien L, Martinez P, Hestad K, Bramness JG. The relationship of alcohol use disorders and depressive symptoms to tryptophan metabolism: cross-sectional data from a Nepalese alcohol treatment sample. Alcohol Clin Exp Res (2015) 39:514–21. doi:10.1111/acer.12651
105. Farren CK, Dinan TG. Elevated tryptophan levels in post-withdrawal alcoholics. Acta Psychiatr Scand (1996) 94:465–70. doi:10.1111/j.1600-0447.1996.tb09892.x
106. Quak J, Doornbos B, Roest AM, Duivis HE, Vogelzangs N, Nolen WA, et al. Does tryptophan degradation along the kynurenine pathway mediate the association between pro-inflammatory immune activity and depressive symptoms? Psychoneuroendocrinology (2014) 45:202–10. doi:10.1016/j.psyneuen.2014.03.013
107. Neupane SP, Lien L, Martinez P, Aukrust P, Ueland T, Mollnes TE, et al. High frequency and intensity of drinking may attenuate increased inflammatory cytokine levels of major depression in alcohol-use disorders. CNS Neurosci Ther (2014) 20:898–904. doi:10.1111/cns.12303
108. Davis MI. Ethanol-BDNF interactions: still more questions than answers. Pharmacol Ther (2008) 118:36–57. doi:10.1016/j.pharmthera.2008.01.003
109. Joe KH, Kim YK, Kim TS, Roh SW, Choi SW, Kim YB, et al. Decreased plasma brain-derived neurotrophic factor levels in patients with alcohol dependence. Alcohol Clin Exp Res (2007) 31:1833–8. doi:10.1111/j.1530-0277.2007.00507.x
110. Han C, Ahn D, Hahm W, Nam J, Park Y, Lim S, et al. Serum levels of growth factors in alcohol-dependent patients according to comorbid depressive symptoms. Clin Psychopharmacol Neurosci (2016) 14:43. doi:10.9758/cpn.2016.14.1.43
111. Nedic G, Perkovic MN, Sviglin KN, Muck-Seler D, Borovecki F, Pivac N. Brain-derived neurotrophic factor Val 66 Met polymorphism and alcohol-related phenotypes. Prog Neuropsychopharmacol Biol Psychiatry (2013) 40:193–8. doi:10.1016/j.pnpbp.2012.09.005
112. Su N, Zhang L, Fei F, Hu H, Wang K, Hui H, et al. The brain-derived neurotrophic factor is associated with alcohol dependence-related depression and antidepressant response. Brain Res (2011) 1415:119–26. doi:10.1016/j.brainres.2011.08.005
113. Umene-Nakano W, Yoshimura R, Ikenouchi-Sugita A, Hori H, Hayashi K, Ueda N, et al. Serum levels of brain-derived neurotrophic factor in comorbidity of depression and alcohol dependence. Hum Psychopharmacol (2009) 24:409–13. doi:10.1002/hup.1035
114. Huang MC, Chen CH, Chen CH, Liu SC, Ho CJ, Shen WW, et al. Alterations of serum brain-derived neurotrophic factor levels in early alcohol withdrawal. Alcohol Alcohol (2008) 43:241–5. doi:10.1093/alcalc/agm172
115. Lee BC, Choi IG, Kim YK, Ham BJ, Yang BH, Roh S, et al. Relation between plasma brain-derived neurotrophic factor and nerve growth factor in the male patients with alcohol dependence. Alcohol (2009) 43:265–9. doi:10.1016/j.alcohol.2009.04.003
116. D’Sa C, Dileone RJ, Anderson GM, Sinha R. Serum and plasma brain-derived neurotrophic factor (BDNF) in abstinent alcoholics and social drinkers. Alcohol (2012) 46:253–9. doi:10.1016/j.alcohol.2011.12.001
117. Costa MA, Girard M, Dalmay F, Malauzat D. Brain-derived neurotrophic factor serum levels in alcohol-dependent subjects 6 months after alcohol withdrawal. Alcohol Clin Exp Res (2011) 35:1966–73. doi:10.1111/j.1530-0277.2011.01548.x
118. Briones TL, Woods J. Chronic binge-like alcohol consumption in adolescence causes depression-like symptoms possibly mediated by the effects of BDNF on neurogenesis. Neuroscience (2013) 254:324–34. doi:10.1016/j.neuroscience.2013.09.031
119. Dantzer R. Cytokine-induced sickness behaviour: a neuroimmune response to activation of innate immunity. Eur J Pharmacol (2004) 500:399–411. doi:10.1016/j.ejphar.2004.07.040
120. Kelley KW, Bluthe RM, Dantzer R, Zhou JH, Shen WH, Johnson RW, et al. Cytokine-induced sickness behavior. Brain Behav Immun (2003) 17(Suppl 1):S112–8. doi:10.1016/S0889-1591(02)00077-6
121. Blednov YA, Benavidez JM, Geil C, Perra S, Morikawa H, Harris RA. Activation of inflammatory signaling by lipopolysaccharide produces a prolonged increase of voluntary alcohol intake in mice. Brain Behav Immun (2011) 25(Suppl 1):S92–105. doi:10.1016/j.bbi.2011.01.008
Keywords: alcohol use disorder, depression, comorbidity, neuroimmune interface, neuroinflammation, alcohol drinking, cytokines
Citation: Neupane SP (2016) Neuroimmune Interface in the Comorbidity between Alcohol Use Disorder and Major Depression. Front. Immunol. 7:655. doi: 10.3389/fimmu.2016.00655
Received: 29 September 2016; Accepted: 15 December 2016;
Published: 27 December 2016
Edited by:
Ihssane Zouikr, RIKEN Brain Science Institute, JapanReviewed by:
Anna Fogdell-Hahn, Karolinska Institutet, SwedenJulie Lasselin, University Hospital Essen, Germany
Copyright: © 2016 Neupane. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sudan Prasad Neupane, c3VkYW4ucHJhc2FkLm5ldXBhbmVAc3lrZWh1c2V0LWlubmxhbmRldC5ubw==, dGhlbG9uZWx5cGxhbmV0QGdtYWlsLmNvbQ==