 Shona A. Hendry1,2
Shona A. Hendry1,2 Rae H. Farnsworth3Benjamin Solomon4Marc G. Achen2,3
Rae H. Farnsworth3Benjamin Solomon4Marc G. Achen2,3 Steven A. Stacker2,3
Steven A. Stacker2,3 Stephen B. Fox1*
Stephen B. Fox1*
- 1Department of Pathology, Peter MacCallum Cancer Centre, Melbourne, VIC, Australia
- 2The Sir Peter MacCallum Department of Oncology, University of Melbourne, Parkville, VIC, Australia
- 3Tumour Angiogenesis and Microenvironment Program, Peter MacCallum Cancer Centre, Melbourne, VIC, Australia
- 4Department of Medical Oncology, Peter MacCallum Cancer Centre, Melbourne, VIC, Australia
Recently developed cancer immunotherapy approaches including immune checkpoint inhibitors and chimeric antigen receptor T cell transfer are showing promising results both in trials and in clinical practice. These approaches reflect increasing recognition of the crucial role of the tumor microenvironment in cancer development and progression. Cancer cells do not act alone, but develop a complex relationship with the environment in which they reside. The host immune response to tumors is critical to the success of immunotherapy; however, the determinants of this response are incompletely understood. The immune cell infiltrate in tumors varies widely in density, composition, and clinical significance. The tumor vasculature is a key component of the microenvironment that can influence tumor behavior and treatment response and can be targeted through the use of antiangiogenic drugs. Blood vascular and lymphatic endothelial cells have important roles in the trafficking of immune cells, controlling the microenvironment, and modulating the immune response. Improving access to the tumor through vascular alteration with antiangiogenic drugs may prove an effective combinatorial strategy with immunotherapy approaches and might be applicable to many tumor types. In this review, we briefly discuss the host’s immune response to cancer and the treatment strategies utilizing this response, before focusing on the pathological features of tumor blood and lymphatic vessels and the contribution these might make to tumor immune evasion.
Introduction
The interaction between tumor cells and the microenvironment in which they exist is increasingly recognized as a key player in the development and progression of cancer. The microenvironment of a tumor includes the blood and lymphatic vasculatures, stroma, nerves, and cells of the immune system, which may be resident in the involved tissue or recruited from the periphery. The hallmarks of cancer include features of the tumor cells themselves, such as replicative immortality and resistance to cell death, as well as features relating to the microenvironment, such as induction of angiogenesis and evasion of the immune response (1). Successful reversal of this immune evasion by checkpoint inhibitors is now a clinical reality, with inhibitors of cytotoxic T lymphocyte-associated protein-4 (CTLA-4) as well as programed cell death protein-1 (PD-1) and programed death ligand-1 (PD-L1) delivering durable responses in a subset of patients with a range of cancer types including melanoma (2, 3), urothelial carcinoma (4), Hodgkin lymphoma (5), non-small cell lung carcinoma (6–8), Merkel cell carcinoma (9), and squamous cell carcinoma of the head and neck (10). In addition, decades of research into the use of adoptive cell transfer and genetic engineering of tumor killing T cells has resulted in breakthrough therapy designation of anti-CD19 chimeric antigen receptor (CAR) T cell transfer for use in B-acute lymphoblastic leukemia (11). However, there is marked variability in patient response to immune checkpoint blockade (12), and the use of CAR T cells against solid tumors has seen little success in the clinic (13).
Immunotherapy, particularly checkpoint inhibitors, differs from conventional cancer therapies. A complex intermediate step is introduced by activating the host’s immune system, instead of a direct toxic effect on tumor cells or targeting of a tumor cell-specific mutation. Understanding the tumor microenvironment is critical to understanding the exact mechanisms of actions of these therapies and predicting response. There is a clear need for robust microenvironmental biomarkers to direct therapeutic strategies. The presence of tumor-infiltrating lymphocytes (TILs) is correlated with improved prognosis in many tumor types, as well as improved response to some conventional therapies and most immunotherapies (14). Tumors can exert direct effects to adapt to, escape, and suppress antitumor immunity, which is reviewed in Ref. (15). The access of immune cells to the tumor is a critical factor in the efficacy of both adoptive cell transfer and immune checkpoint inhibition, and the role of the tumor vasculature in providing or blocking access to the tumor is likely to prove an important consideration in immunotherapeutic strategies. In addition, blood vessels, lymphatic vessels, and the hypoxic tumor environment have important immunomodulatory roles, which contribute to the immune evasion of tumors. In this review, we provide a brief overview of factors affecting the host immune response to tumors and current immunotherapy approaches, which show exciting clinical results. We then focus on the molecular and mechanical features of the tumor vasculature that modulate the host antitumor immune response and consider the implications of these interactions for potential therapeutic approaches to enhance immunotherapy.
The Host Immune Response to Tumors
For an effective host immune response, the tumor must be recognized as foreign and the immune effector cells must be able to access the tumor to destroy it. It is well established that tumors are antigenic and able to induce a systemic, tumor-specific immune response (16, 17). Unstable tumor genomes contain many mutations that generate altered protein products, which have the potential to be recognized as foreign by the host immune system during surveillance. The tumors must therefore develop mechanisms of evading this immune response in order to establish, grow, and eventually metastasize. For example, circulating T cells specific to tumor antigens can be demonstrated in patients with metastatic melanoma, yet the tumor progresses (18, 19).
There is wide variation in the immune cell infiltrate seen in solid tumors, both within and between different tumor types, which is illustrated in Figure 1. This can provide important prognostic and predictive information. The density of TILs correlates with improved survival in many tumors ranging from melanoma to colorectal cancer, renal cell carcinoma, and non-small cell lung carcinoma (20). However, specific immune cell subsets modify this association, including regulatory T cells (Tregs), myeloid-derived suppressor cells (MDSCs), and tumor-associated macrophages (TAMs) (20, 21). The presence of TILs has also been shown to be predictive of response to conventional anticancer treatment, for example, anti-HER2/neu therapy and trastuzumab and anthracycline chemotherapy in breast cancer (22). A classification of tumors based on their immune phenotypes has been proposed, both as a broad conceptual approach (23, 24) and as specific quantitative scoring (21). Broadly, tumors can be classified as “T-cell inflamed” or “non-inflamed” based on the presence or absence of CD8+ cytotoxic T cells within the tumor (23). For example, Figure 1A shows a basal phenotype breast carcinoma with a florid lymphocytic infiltrate, whereas Figure 1B, which is also a basal phenotype breast carcinoma, shows very few TILs. Even in melanoma, widely accepted as an immunogenic tumor and the solid tumor in which immunotherapy has had the most success, approximately 40% of tumors display a non-inflamed phenotype (24). The existence of an inflamed phenotype is supported by gene expression profiling of tumors, through which a subset of tumors rich in immune-related gene transcripts has been identified in pancreatic ductal adenocarcinoma, colorectal carcinoma, and melanoma (25–27). A multitude of different scoring systems and methodologies have been proposed to describe the immune infiltrate in tumors, with variable reproducibility and practicality (21, 28, 29). As such, use of these scoring systems is limited in routine pathology practice, despite the valuable information they could convey.
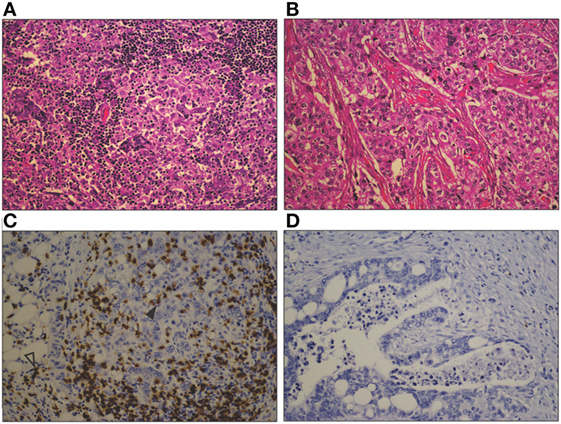
Figure 1. Photomicrographs comparing a heavy lymphocytic infiltrate in a basal phenotype breast carcinoma (A), with a sparse infiltrate in a different basal phenotype breast carcinoma (B) (H&E, original magnification 200×). A similar contrast is seen between a marked CD8+ T cell infiltrate in a mismatch repair-deficient colon cancer (C), and the sparse infiltrate in a mismatch repair proficient colon cancer (D). CD8+ T cells are seen both within the tumor epithelium (closed arrowhead) and in the tumor stroma (open arrowhead) (CD8 immunohistochemical stain, original magnification 200×).
It is hypothesized that the mutational load of the tumor correlates with the presence of an immune infiltrate, due to the greater potential for neoantigen formation. In support of this hypothesis is the evidence that mismatch repair deficient tumors with vast mutational loads show higher immune cell infiltrates than mismatch repair proficient tumors (30) (for example, see Figures 1C,D, respectively). The tumor types showing high levels of response to immune checkpoint blockade—melanoma, smoking associated lung cancer, and urothelial cancer—are the tumor types with the highest overall mutational loads (31). However, this correlation is weak at an individual tumor level, as the presence of mutations does not necessarily result in neoantigen formation, and multiple factors are involved in the presentation of antigens to elicit an immune response (32, 33). In addition, the extent and composition of the immune infiltrate varies widely between individual tumors within these highly mutated types (29, 34). Features of the microenvironment, including blood and lymphatic vessel structure, stromal fibroblasts, and extracellular matrix, may contribute to this variation by modulating the access of immune cells to the tumor and their activation and function in the tumor microenvironment.
Trafficking of effector T cells to tumors is complex and tightly regulated. T cell migration, activation, and differentiation are intricately linked processes. Following activation by antigen-presenting cells (APCs), T cells upregulate chemokine receptors and ligands for endothelial adhesion molecules. Binding of inflammatory chemokines enhances adhesion and extravasation, allowing effector T cells to enter the tumor microenvironment (35, 36). Levels of chemokines within tumors, particularly the CXCR3 ligands CXCL9 and CXCL10, have been shown to correlate with T cell infiltration into tumors and enhanced antitumor responses (37, 38). Chemokine/chemokine receptor mismatching is postulated as an important mechanism of reduced T cell trafficking into tumors (35). Post-translational modification of chemokines can also affect immune cell infiltration. For example, nitration of CCL2 as a result of the intratumoral production of reactive nitrogen species can reduce T cell infiltration into tumors, while macrophages and MDSCs can still be attracted by nitrated CCL2 (39).
Once arriving within the tumor microenvironment, T cells must also proliferate locally, as evidenced by the enrichment of cancer-specific T cells in the tumor compared to the peripheral blood (40). A range of cellular, metabolic, and molecular features of the tumor microenvironment contribute to limit the proliferation and activation of antitumor immune effector cells. Activation of CD8+ T cells requires APCs that can efficiently cross-present antigen. However, hypoxia in the tumor microenvironment can impair the maturation and differentiation of dendritic cells (DCs) and polarize macrophages to an immunosuppressive phenotype (41). Nutritional depletion, hypoxia, and reactive nitrogen species, features characteristic of the abnormal metabolic environment of tumors, can limit the activation of T cells and induce apoptosis [reviewed in Ref. (42)]. Enzymes contributing to immunosuppression are also found in the tumor microenvironment. Indoleamine 2,3-dioxygenase (IDO) is an intracellular enzyme preferentially expressed by subsets of APCs, which functions to catalyze catabolism of tryptophan to kynurenine (43). Depletion of tryptophan and accumulation of kynurenine in the tumor microenvironment impairs DC function and limits the clonal expansion of T cells (44), induces CD8+ T cell anergy (45), and promotes Treg induction and activation (46, 47). IDO has been implicated in resistance to immune checkpoint inhibitors (48), and blockade of the IDO pathway is under investigation in clinical trials (49). Depletion of l-arginine in the microenvironment can also result in the impairment of T cell function. Enzymes of the arginase and nitric oxide synthase (NOS) families control the metabolism of l-arginine [reviewed in Ref. (50)]. Expression of inducible NOS and arginase-1 has been demonstrated to limit T cell responses and promote the immunosuppressive microenvironment in different tumor types (51–53). These metabolic features of the tumor microenvironment combine with cellular mechanisms such as the expression of co-inhibitory immune checkpoint molecules [reviewed elsewhere (54)] to control the activity and proliferation of immune cells in the tumor microenvironment. Both exclusion of immune cells and inhibition of their function clearly contribute to the creation of an immunosuppressive microenvironment, which allows tumor immune evasion. The contribution of the tumor vasculature to T cell trafficking, the regulation of endothelial adhesion molecule expression, and the creation of an immunosuppressive microenvironment are discussed in the following sections.
Current Therapies Utilizing the Host Immune Response
Tumors that do support T cell trafficking and show high levels of immune cell infiltration appear to use a range of immunosuppressive pathways to evade the host response. An important immune evasion strategy is the use of inhibitory signaling pathways, known as immune checkpoints, which are part of the physiological process of peripheral tolerance, designed to protect against autoimmunity (55). In this process, self-antigens taken up by APCs will be presented to T cells without the appropriate coactivation signals such as the binding of CD80 or CD86 to CD28, or in the presence of co-inhibitory signals such as the binding of PD-1 to PD-L1. This results in anergy or deletion of the self-reactive T cell. Tumors can co-opt these signaling pathways to evade the immune response, by expressing high levels of co-inhibitory molecules such as PD-L1 (54). Release of these immune checkpoints through the use of inhibitory monoclonal antibodies targeting CTLA-4, PD-1, or PD-L1 can result in durable antitumor responses in a subset of patients (2–7, 56, 57). Responses have been demonstrated across multiple tumor types; however, the selection of patients likely to respond remains problematic (12). The presence of TILs is critical to the success of these immune checkpoint inhibitors (58).
An alternative approach that utilizes the host immune response to fight tumors is termed adoptive cell transfer. Here, TILs are isolated from the patient’s tumor tissue, expanded ex vivo and reintroduced into the patient’s blood stream. This approach has a number of limitations and to date has seen minimal success in the clinic (59). Genetic modification of the T cells can improve tumor cell specificity and enhance activation (59). CARs include a specific antigen-binding domain and an intracellular signaling domain, which allow MHC-independent activation of T cells. Limited success has been seen in the use of CAR T cell and adoptive cell transfer against solid tumors compared to impressive results in hematological malignancies (13).
A limiting factor in the efficacy of CAR T cells in solid tumors is the lack of infiltration into the tumor itself. This therapeutic approach has seen the most success in B cell leukemia, in which the tumor cells express a common and specific antigen (CD19) and are easily accessible, as they are circulating in the peripheral blood (11). Infiltration of solid tumors by the transferred T cells is required for efficacy (60); however, it has been demonstrated in both humans and mice that only a small fraction of transferred T cells reach the tumor tissue (35). Following transfer, CAR T cells may be readily identifiable in peripheral blood, but scant in the tumor tissue (61). It has also been shown that mesothelin-targeted CAR T cells demonstrated markedly superior efficacy in an orthotopic mouse model of mesothelioma when delivered regionally rather than systemically (62). Current clinical trials are investigating methods to overcome this suboptimal trafficking of CAR T cells, including altering the chemokine milieu of the tumor and expressing matched chemokine receptors on the engineered T cells (35, 63). Investigations into local delivery approaches are also ongoing (13).
Is There an Access Issue?
The existence of the non-inflamed tumor phenotype and the lack of success of CAR T cell therapy in solid tumors support the concept that exclusion of immune cells from the microenvironment plays an important role in the immune escape of tumors. It has been recognized that the tumor vasculature is part of the permissive microenvironment that prevents the immune rejection of tumors (64). Understanding the impact of the tumor vasculature’s role in this exclusion will be important in selecting appropriate therapeutic strategies to enhance the potential of immunotherapy. The immunomodulatory effects of tumor blood vessels and lymphatics are also important targets in understanding and manipulating the tumor microenvironment.
Role of the Tumor Vasculature in Immune Cell Exclusion
Molecular Mechanisms
Specialized endothelial cells line the blood and lymphatic vessels of the body and act in a variety of ways to control the delivery and removal of oxygen, nutrients, and circulating cells to the tissues. Endothelial cells are active participants in the immune response to inflammation (65), through their role in regulating the trafficking and activation of immune cells. A summary of the alterations in leukocyte–endothelium interactions seen in tumors is provided in Figure 2. Migration of leukocytes (lymphocytes, monocytes, and granulocytes) from the blood vessels into peripheral tissues is a multistep process involving rolling, slow rolling, activation, firm adhesion, adhesion strengthening, intraluminal crawling, and transcellular and paracellular migration (66). E-selectin and P-selectin on endothelial cells and L-selectin on granulocytes, monocytes, and most lymphocytes mediate rolling through interaction with P-selectin glycoprotein ligand-1 and other glycosylated ligands (66). Selectins require shear stress resulting from the flow of blood to support adhesion (67). Intercellular adhesion molecule-1 (ICAM-1) is a member of the immunoglobulin superfamily that plays an important role in the adhesion cascade, participating in rolling, firm adhesion, and transcellular migration (68). ICAM-1 and vascular cell adhesion molecule-1 (VCAM-1), another immunoglobulin superfamily member (69), are located on the luminal surfaces of endothelial cells and bind to the integrins such as lymphocyte function-associated antigen-1 (LFA-1) and very late antigen-4 (VLA-4), respectively (70, 71). LFA-1 is expressed on lymphocytes, monocytes, and neutrophils, whereas VLA-4 is expressed on lymphocytes and monocytes (72). Clustering of ICAM-1 and VCAM-1 is also a critical step in transendothelial migration, and blocking this clustering is sufficient to prevent migration of leukocytes expressing LFA-1 or VLA-4 (73). Expression of vascular adhesion molecules in intratumoral blood vessels is correlated with the number of TILs. E-selectin is required for T cell extravasation in skin, and expression of E-selectin in cutaneous squamous cell carcinoma and Merkel cell carcinoma correlates with infiltration by CD8+ T cells and better prognosis (74, 75). Medullary breast carcinomas are defined in part by a florid lymphocytic infiltrate and showed a higher expression of ICAM-1 on intratumoral blood vessels than ductal breast carcinomas of no special type (76).
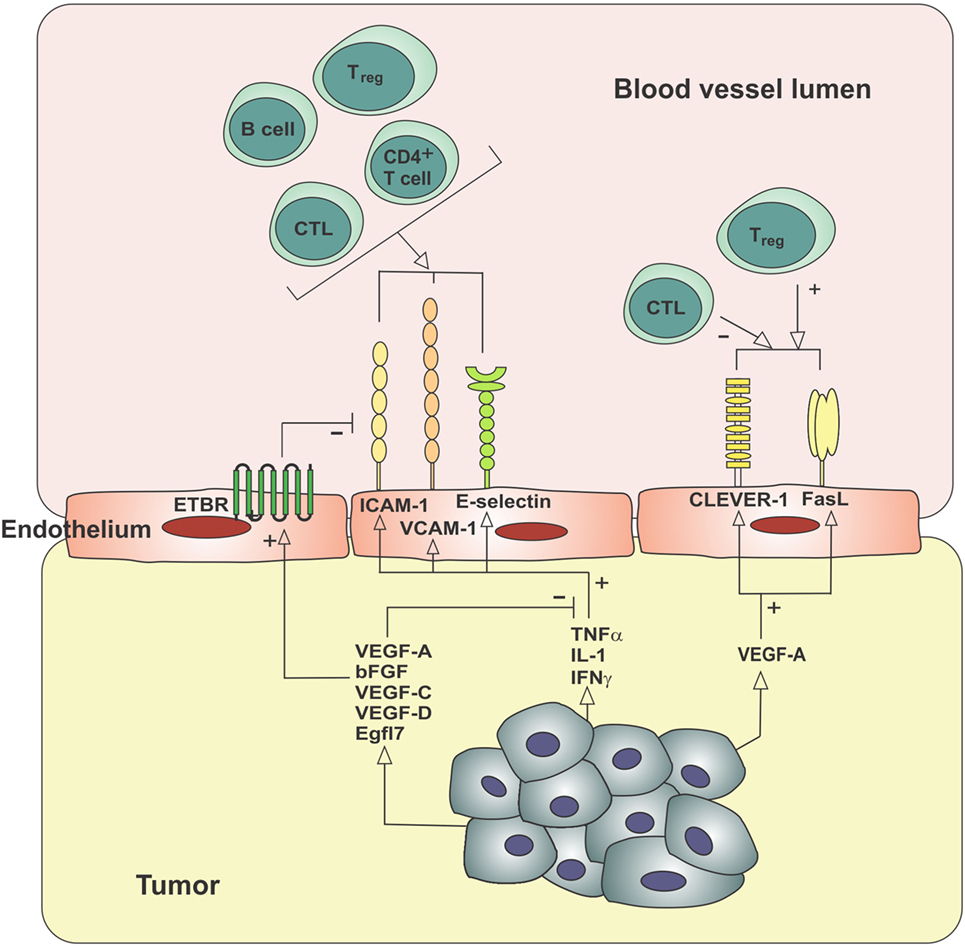
Figure 2. Molecular mechanisms contributing to the exclusion of immune cells from the tumor microenvironment. Tumor-derived angiogenic factors can block the usual upregulation of cell adhesion molecules in response to inflammatory mediators (endothelial anergy), increase the expression of ETBR, which blocks the clustering of ICAM-1 required for lymphocyte adhesion, and increase expression of cell surface receptors, which selectively decrease CTL extravasation while increasing Treg extravasation. bFGF, basic fibroblast growth factor; CLEVER-1, common lymphatic endothelial and vascular endothelial receptor-1; CTL, cytotoxic T lymphocyte; EGFL7, epidermal growth factor-like domain 7; ETBR, endothelin B receptor; FasL, Fas ligand; ICAM-1, intercellular adhesion molecule-1; IFNγ, interferon-gamma; IL-1, interleukin-1; TNFα, tumor necrosis factor-α; Treg, regulatory T cell; VCAM-1, vascular cell adhesion molecule-1; VEGF, vascular endothelial growth factor.
Inflammatory signals are required to upregulate expression of ICAM-1, which can be expressed by a range of cells in addition to endothelial cells, including fibroblasts, thymic epithelial cells, macrophages, and follicular DCs (70). In addition to mediating the adhesion of leukocytes to endothelial cells, ICAM-1:LFA-1 interactions also participate in the formation of an immune synapse between T cells and APCs (77). A mature immune synapse requires molecular interactions mediating adhesion, antigen presentation, and costimulation or inhibition. A synapse may also form within the docking structure forming the adhesion between endothelial cells and lymphocytes (78). Inflammatory cytokines IL-1, TNFα and, to a lesser degree, IFNγ, cause a rapid rise in the expression of ICAM-1 on cultured endothelial cells (79). Different cell types may vary as to which inflammatory signals are capable of inducing ICAM-1 expression (77).
Adhesion molecules including ICAM-1, VCAM-1, and E-selectin may be absent or expressed at low levels on tumor vasculature, despite the inflammatory microenvironment of the tumor. Pro-inflammatory pathways are induced in tumor cells by oncogenic activation of transcription factors such as HIF-1α and NFκB, resulting in the high levels of inflammatory mediators detected in most solid tumors (80). However, this inflammatory environment appears to fail to induce the expression of vascular adhesion molecules on intratumoral vessels. This has been demonstrated in experimental models of melanoma and carcinoma (81), as well as in human cutaneous squamous cell carcinoma, Merkel cell carcinoma, and metastatic melanoma tissue (74, 75, 82). This lack of responsiveness to inflammatory signals has been termed endothelial anergy (83) and may play an important role in the exclusion of antitumor immune effector cells from the tumor microenvironment.
Evidence suggests that endothelial anergy is due at least in part to angiogenic factors (84, 85), a range of molecules including vascular endothelial growth factor-A (VEGF-A), VEGF-C, VEGF-D, and basic fibroblast growth factor (bFGF), some of which are produced in response to tissue hypoxia. The tumor microenvironment is characteristically hypoxic due to disordered and loosely regulated angiogenesis that fails to adequately supply the expanding tumor mass (86). This hypoxia leads to stabilization and nuclear accumulation of hypoxia-inducible factors (HIF-1α and HIF-2α), transcription factors that lead to upregulation of angiogenic factors, and other molecules that act to improve tissue oxygenation. VEGF-A can be secreted by tumor cells and TAMs and is overexpressed in the majority of solid tumors (87, 88). VEGF-A and bFGF, also a strong mitogenic factor for endothelium produced by tumor cells, contribute to the suppression of ICAM-1 in tumors (84). This downregulation of adhesion molecules in response to angiogenic factors has been demonstrated in vitro (83, 84, 89, 90) and in mouse tumor models (85, 91, 92). As described above, tumor vasculature appears unresponsive to inflammatory signals that mediate the expression of adhesion molecules through the NFκB signaling pathway. bFGF can block this stimulation by preventing the degradation of pathway inhibitor Iκβα, thus stopping the translocation of NFκB to the nucleus and activation of target gene transcription (93).
The concept of endothelial anergy and the downregulation of adhesion molecules mediated by angiogenic factors is supported by the evidence that antiangiogenic therapy results in increased expression of adhesion molecules on tumor vasculature (94). Angiostatic therapy using platelet factor 4, anginex, angiostatin, or endostatin results in upregulation of ICAM-1, VCAM-1, and E-selectin in animal models and in vitro (94, 95) and also reinstates the responsiveness of the endothelium to inflammatory signals (94). These anti-angiogenic peptides showed promising anti-tumor effects in initial pre-clinical trials, however have failed to demonstrate efficacy in human cancers and are no longer being clinically investigated (96). Multi-target tyrosine kinase inhibitors such as SU6668, sunitinib, and sorafenib are a more promising antiangiogenic treatment approach and are approved for the treatment of some human cancers such as the highly angiogenic renal cell carcinoma (96). These small molecules inhibit the activation of a range of tyrosine kinase receptors, including vascular endothelial growth factor receptor-1 (VEGFR-1), VEGFR-2, and fibroblast growth factor receptor (FGFR-1), receptors for angiogenic factors VEGF-A, VEGF-C, and VEGF-D, and bFGF, as well as growth factor receptors such as platelet-derived growth factor receptor-β (PDGFRβ) and c-kit. Use of SU6668, a small molecule inhibitor of VEGFR-2, FGFR-1, and PDGFRβ, blocked the actions of bFGF and showed reversal of adhesion molecule downregulation in a mouse model of metastatic breast cancer (89). A number of pre-clinical studies have shown that various antiangiogenic therapies, including tyrosine kinase inhibitors and inhibitory monoclonal antibodies against VEGF-A and VEGFR-2, may help to increase tumor infiltration by lymphocytes (97–108). These are summarized in Table 1 and discussed further in Section “Implications for Treatment Strategies”. It would be of interest to delineate the extent to which this increased infiltration is due to reversal of endothelial anergy or alternatively due to blockade of the direct effects of VEGF-A on tumor cells, stromal cells, or immune cells, or alteration of the hypoxic microenvironment. Initial clinical studies also support an increase in tumor infiltration by immune cells with the combination of immunotherapies and antiangiogenic agents, summarized in Table 2 and discussed further in Section “Implications for Treatment Strategies” (109, 110). To the best of our knowledge, reversal of endothelial anergy in human tumors by antiangiogenic agents remains to be conclusively demonstrated. Further investigations of changes in adhesion molecule expression and lymphocyte infiltration resulting from antiangiogenic drugs currently approved for use in the clinic, which largely target the VEGF-VEGFR signaling pathway, may provide useful information and should be a high priority.
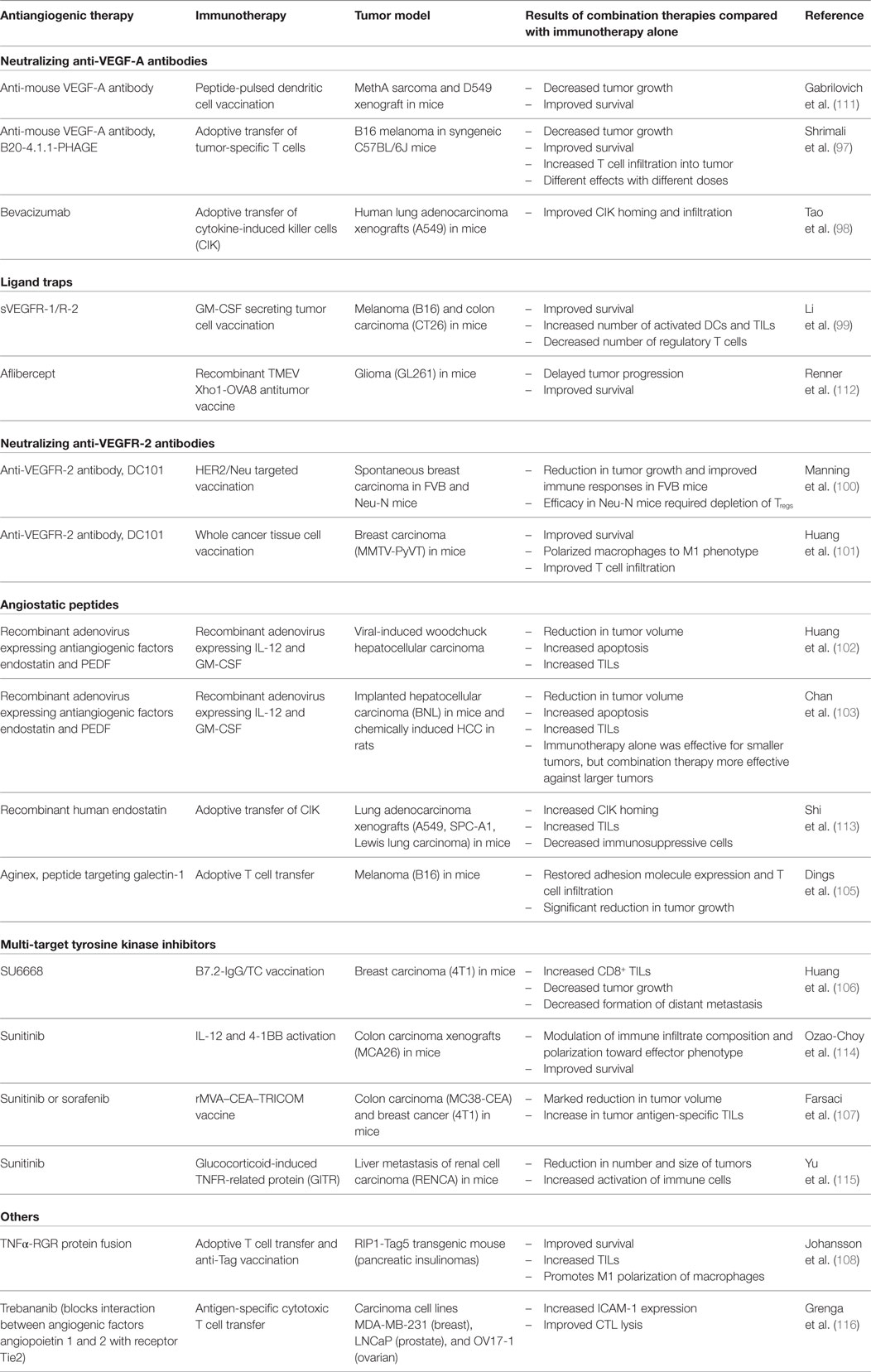
Table 1. Summary of pre-clinical studies combining antiangiogenic therapies and immunotherapy.
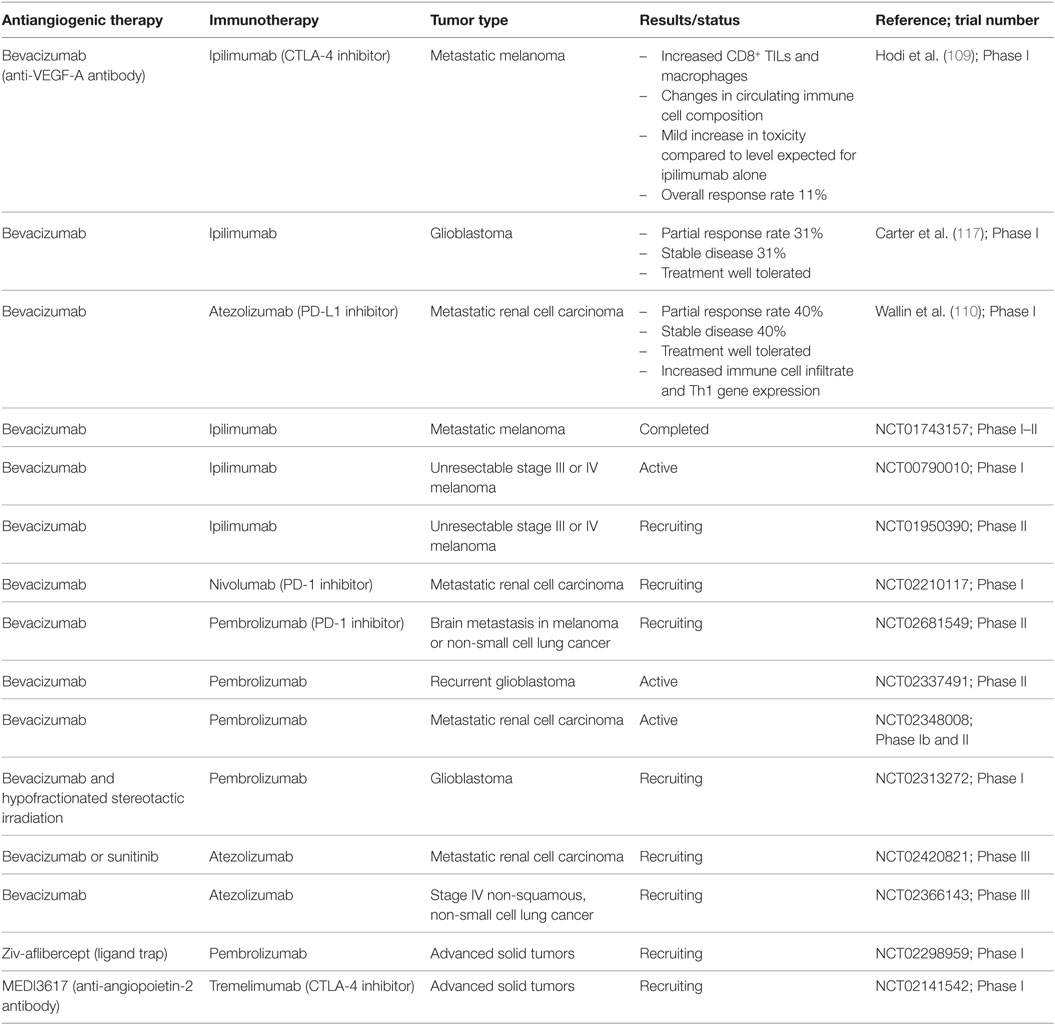
Table 2. Summary of published and ongoing clinical trials combining antiangiogenic therapies and immunotherapy.
In addition to VEGF-A and bFGF, other angiogenic and tumor-associated factors may also contribute to the exclusion of TILs. VEGF-C and VEGF-D are closely related members of the VEGF family that promote angiogenesis, lymphangiogenesis, and cancer metastasis (118–122). These factors can be secreted by tumor cells, immune cells, and tumor-associated fibroblasts (123–125). In human breast carcinoma, higher levels of VEGF-C and VEGF-D were seen in ductal carcinomas compared to medullary carcinomas and correlated with decreased ICAM-1 expression and lower numbers of infiltrating lymphocytes (76). Other growth factors including placenta growth factor (PlGF) and epidermal growth factor have also been shown to downregulate ICAM-1 expression in vitro (126). Epidermal growth factor-like domain 7 (EGFL7) is secreted by normal blood endothelial cells, at sites of pathological angiogenesis, and by tumor cells (127, 128). Higher levels of EGFL7 have been correlated with poor prognosis in some tumor types such as colorectal cancer (127). Delfortrie et al. have shown that EGFL7 also functions to decrease levels of adhesion molecules ICAM-1 and VCAM-1, resulting in a reduction in TILs (128).
Endothelin-1 (ET-1) is a molecule that plays a role in both angiogenesis and controlling the trafficking of immune cells. ET-1 acts through two receptors, the endothelin A receptor (ETAR) and the endothelin B receptor (ETBR) (129). ET-1, ETAR, and ETBR expression is correlated with VEGF-A expression and microvessel density in breast and ovarian carcinoma (130). Messenger RNA profiling of microdissected endothelial cells from ovarian cancer showed overexpression of ETBR in tumors lacking infiltrating lymphocytes (131). The binding of ET-1 to ETBR prevented T cell adhesion to endothelium, even in the presence of the inflammatory cytokine TNFα, an additional mechanism of endothelial anergy (131). Findings suggesting selectivity in lymphocyte extravasation due to ETBR expression were reported for glial tumors (132). Glioblastomas with higher numbers of ETBR-expressing vessels showed lower infiltration by cytotoxic T cells and higher numbers of regulatory T cells. Cytotoxic T cells infiltrated around ETBR-negative blood vessels, but were absent around vessels expressing ETBR (132). Similar findings were seen in primary central nervous system lymphoma, in which both endothelial and tumor cells expressed ETBR (133). However, no correlation between ETBR expression and TILs was seen in oral squamous cell carcinoma (134). Blockade of ETBR increased T cell adhesion to endothelium through the upregulation and clustering of ICAM-1 (131). Blockade of ETBR was also shown to increase T cell homing to tumors and increase the effectiveness of cancer vaccines in mice (131).
Selective extravasation of different leukocyte subsets may also be mediated by additional molecules including common lymphatic endothelial and vascular endothelial receptor-1 (CLEVER-1) (135) and Fas ligand (FasL) (136). CLEVER-1, also known as stabilin-1 and FEEL-1, is a multifunctional scavenging receptor expressed constitutively on lymphatic endothelial cells (LECs) and type 2 macrophages and induced by inflammation on blood endothelial cells (137, 138). Functions have been demonstrated to include both lymphocyte trafficking and adherence of cancer cells to lymphatic endothelium (139, 140). In a mouse model of melanoma, levels of CLEVER-1 correlated with increased infiltration by FoxP3+ Tregs and type II macrophages. Following administration of anti-CLEVER-1 antibody, numbers of Tregs and type II macrophages were reduced, and there was increased immune activation and decreased tumor growth (135). FasL mediates T cell apoptosis and can be induced on blood vascular endothelial cells in solid tumors by tumor-derived VEGF-A, prostaglandin E2, and IL-10 (136). Endothelial FasL is able to kill activated T lymphocytes, but CD4+CD25+ regulatory T cells are resistant to FasL-mediated killing due to high levels of antiapoptotic protein c-FLIP (136). Endothelial FasL expression correlated with lower numbers of CD8+ T cells in a range of cancer types. Blockade of VEGF-A, prostaglandins, or FasL resulted in increased CD8+ T cell infiltration and impaired tumor growth (136).
In addition to effects on the tumor vasculature, hypoxia and angiogenic factors such as VEGF-A also have direct immunomodulatory effects, which are summarized in Figure 3. As mentioned above, hypoxia-inducible factors are transcription factors activated by low tissue oxygen levels sensed by hydroxylase enzymes (141). HIFs control the transcription of various genes involved in the adaptation to hypoxic conditions, and also have a number of direct effects on immune cells. In hypoxic tumors, macrophages are polarized toward an immunosuppressive M2 phenotype, MDSCs accumulate and DC maturation and differentiation is impaired, inhibiting the activation of T cells (41). Cytotoxic T cells show increased lytic capacity under hypoxic conditions, but decreased proliferation and differentiation (41). Hypoxic stress increases secretion of CCL28 and CXCL12 by tumor cells, thereby attracting regulatory T cells (142, 143). HIF-1α also directly binds to a hypoxia response element in the promoter of the gene encoding immune checkpoint molecule PD-L1, and hypoxia thereby increases expression of PD-L1 on MDSCs, tumor cells, DCs, and macrophages (144). VEGF-A also directly enhances the expression of PD-1, TIM-3, and CTLA-4 on intratumoral CD8+ T cells, contributing to T cell anergy (145). These data suggest an important role for hypoxia, angiogenesis, and the endothelium in creating a permissive microenvironment to prevent the immune rejection of tumors.
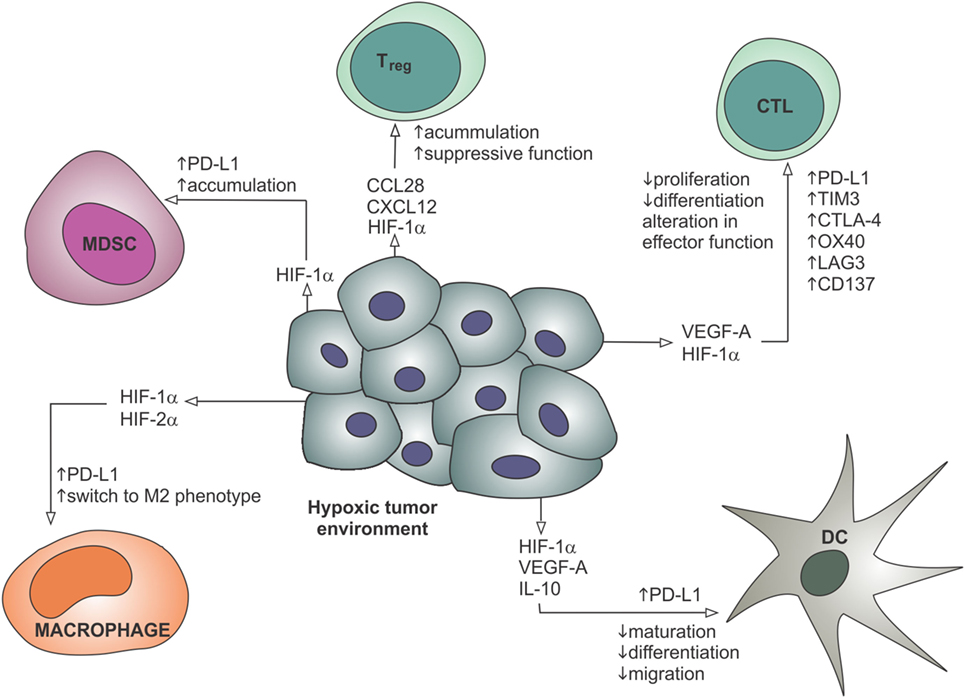
Figure 3. Hypoxia contributes to the recruitment of suppressive immune cells, restricts the maturation and migration of dendritic cells, reduces proliferation and differentiation of effector CTLs, and leads to the upregulation of immune checkpoint molecules such as PD-L1. These effects are mediated through gene regulation by hypoxia-inducible factors and secreted factors such as VEGF-A. CTL, cytotoxic T lymphocyte; DC, dendritic cell; HIF, hypoxia-inducible factor; IL-10, interleukin-10; MDSC, myeloid-derived suppressor cell; Treg, regulatory T cell; VEGF-A, vascular endothelial growth factor-A.
Mechanical Properties
The tumor vasculature may also contribute to the exclusion of effector lymphocytes from the tumor microenvironment by physical means. In normal immune responses, T cells exit the vasculature predominantly in the post-capillary venule, a site of low shear stress where adhesion molecules are preferentially expressed (78, 146). Newly formed blood vessels within tumors, however, are structurally and functionally abnormal, lacking the specialized organization of normal tissue vasculature (147). Tumor vessels are heterogeneous, tortuous, and irregularly branched (148, 149). The vessel walls are leaky with wide junctions between endothelial cells, increased fenestrations and loss, or abnormalities of the surrounding pericytes and basement membranes. Tumor endothelial cells lose polarity, can detach, and stratify (149). The normal laminar flow of blood is disrupted, and with it, the margination, rolling, and adhesion of lymphocytes. Areas of stagnation and increased interstitial fluid pressure are also present, resulting in heterogeneous tumor perfusion (150). The delivery of chemotherapeutic agents is hampered by this chaotic and inefficient tumor blood flow (149, 151), and access of antitumor lymphocytes may also be impaired.
Shear stress, the parallel force applied to the endothelial lining of blood vessels by laminar blood flow in normal vasculature, is a key regulator of vascular physiology (152). Endothelial cells respond to shear stress through mechanosensory molecules including CD31 (platelet endothelial adhesion molecule) and VE-cadherin, which can activate various signaling pathways leading to complex and context-dependent effects on endothelial adhesion molecule expression (153). In tumors, the disrupted and sluggish blood flow in tumors due to abnormal vasculature results in lower levels of shear stress (154). A threshold level of shear stress is required for the expression of E-selectin, P-selectin, and L-selectin, which mediate leukocyte rolling (67). Low shear stress can enhance expression of adhesion molecules on endothelial cells, particularly ICAM-1, but can also decrease the responsiveness of the endothelium to inflammatory signals such as TNFα, thus becoming an additional promoter of endothelial anergy (155). Low shear can also upregulate VEGF-A expression by tumor cells (154), which may modulate adhesion molecule expression and perpetuate angiogenesis. The direct effects of the mechanical properties of abnormal tumor blood vessels on immune cell extravasation remain to be fully elucidated.
Pericytes and vascular smooth muscle cells are contractile cells that surround and interact with the endothelial cell layer of blood vessels. Pericytes are required for vessel stabilization and maturation, and in tumor vessels they are often immature, less abundant, and loosely attached (156). Recruitment of pericytes to immature and proliferating blood vessels involves, among others, the PDGF/PDGFRβ and angiopoietin (Ang)-1/Tie2 signaling pathways (157). Disrupting pericyte coverage through targeting of the PDGF/PDGFRβ pathway results in increased vessel leakiness, decreased tumor vascularity, and decreased tumor growth, particularly when combined with anti-VEGF-A treatment (158–160). Conversely, promotion of pericyte coverage and pericyte–endothelial cell interactions through activation of VEGFR and PDGFRβ has been proposed to enhance vessel stabilization and normalization (160). During changes in oxygen availability, Ang2 can bind to Tie2 on endothelial cells, thus blocking the binding of Ang1, releasing the pericyte, and destabilizing the vessel (161). Inhibition of Ang2 can improve pericyte coverage and normalize tumor vessels in mouse models (162). Clinical trials of pericyte modulation by PDGFRβ inhibition alone have been largely disappointing (163, 164). Other approaches to modulate pericyte coverage require further investigation in the clinic. To the best of our knowledge, no clinical trials have yet examined the effect of vascular normalization due to pericyte modulation on lymphocyte infiltration. However, pericytes may however have additional immunomodulatory effects. Hong et al. demonstrated an increase in MDSCs in tumors grown in a pericyte deficient mouse model, due to IL-6 production in the hypoxic tumor microenvironment (165). MDSC levels decreased when pericyte coverage was restored (165). In human breast cancers, MDSC gene expression correlated with decreased pericyte gene expression and poor prognosis (165). Pericyte coverage is thus an important consideration in vascular normalization studies and may play a role in creation of the immunosuppressive tumor microenvironment. Rgs5, one of a family of molecules that inhibits signaling by G protein-coupled receptors, is expressed by pericytes and hypoxic endothelial cells and has been shown to be overexpressed in tumor vasculature (166, 167). Loss of Rgs5 in mice results in pericyte maturation, vascular normalization, improved oxygenation, and reduced vessel leakiness (166). Importantly, it was also found that tumor infiltration by both endogenous and adoptively transferred lymphocytes was increased in Rgs5-deficient mice (166). This finding supports the hypothesis that physical normalization of the blood vessels and their supporting cells improves immune cell extravasation. Human RGS5 shows high homology to the mouse gene and appears to perform similar functions (168), although data describing its role in human tumors are limited.
The abnormal, poorly organized structure of tumor blood vessel walls results in leakiness and extravasation of fluid into the tumor microenvironment (169). Angiogenic factors also contribute to this leakiness. VEGF-A was initially described as vascular permeability factor (170) due to its marked enhancement of vessel permeability and is found in high levels in malignant effusions (171). However, data appear to suggest that this permeability of tumor blood vessels does not result in increased lymphocyte extravasation. As discussed above, expression of angiogenic factors instead correlates with reduced TILs (76, 172). Use of antiangiogenic therapy and vascular normalization can improve lymphocyte infiltration into tumors, discussed further below. Lymphocyte extravasation requires controlled molecular regulation and as such increased vessel wall permeability, and fluid extravasation alone may not increase the lymphocyte infiltration in the tumor.
High Endothelial Venules and the Recruitment of Naïve T Cells
High endothelial venules (HEVs) are specialized post-capillary venules normally found in secondary lymphoid organs including lymph nodes and Peyer’s patches, characterized histologically by their cuboidal “high” endothelial lining. They are adapted to promote trafficking of naïve lymphocytes into the lymphoid organ, expressing specific addressins including peripheral node addressin (PNAd) and mucosal addressin (MAdCAM-1). Activated lymphocytes, including effector T cells and memory T cells, can also be recruited by HEVs into lymph nodes under inflammatory conditions through the upregulation of VCAM-1, E-selectin, and P-selectin (173). Blood vessels with morphological and immunohistochemical features of HEVs have been identified in a range of human tumors, including breast, ovarian, colorectal, and lung cancers (174). The presence of HEVs correlates strongly with the presence of CD8+ effector T cells as well as B cells and Th1 cells (174), often organized as tertiary lymphoid structures, that is, ectopic lymphoid structures with all the characteristics of lymph nodes (175). Evidence suggests that these local tertiary lymphoid structures may play a role in recruitment and priming of naïve T cells and promote differentiation into tumor-specific effector T cells, within the tumor microenvironment itself (176). Interestingly, both positive and negative effects on antitumor immunity have been associated with tertiary lymphoid structures and lymph node-like vasculature (177, 178). The recruitment of naïve T cells and differentiation into effector T cells seen in some settings (177) contrasts with the recruitment of MDSCs and differentiation of Tregs seen in others (178). The inflammatory context in which these tertiary lymphoid structures develop may help to explain these findings.
Lymphangiogenesis, Interstitial Fluid Pressure, and Immune Evasion
Recent work has established a key role of LECs in inducing immune tolerance, both in peripheral tissues and the draining lymph node. Tumors and their microenvironments promote lymphangiogenesis and lymphatic remodeling through both molecular and mechanical means. VEGF-C and VEGF-D signaling via interactions with VEGFR-2 and VEGFR-3 are important drivers of tumor lymphangiogenesis, promoting intratumoral and peritumoral lymphatic growth and metastasis (179). These growth factors may be secreted by tumor cells, immune cells, and stromal cells (123–125).
As described in previous sections, loosely regulated angiogenesis in tumors results in abnormal, leaky blood vessels. In conjunction with alterations in the stroma and extracellular matrix surrounding the tumor, this results in increased interstitial fluid pressure within the tumor (180). Interstitial fluid pressure within tumors can measure up to 60 mmHg, whereas normal tissue has a range of −3 to +3 mmHg (180). This pressure gradient causes an increase in interstitial flow at the tumor margin, and increased lymphatic drainage by peritumoral lymphatics (181). Increased interstitial fluid and lymphatic flow has a number of effects on the tumor microenvironment, contributing to peritumoral lymphangiogenesis, altering the extracellular matrix and fibroblast differentiation, and promoting the development of lymphoid-like features (178, 181). These lymphoid-like stromal features such as CCL21 expression, required for the homing of naïve T cells, are important components of the tertiary lymphoid structures seen in tumors, which, as discussed above, can show both positive and negative associations with antitumor immunity. Lymphatic flow can also induce the upregulation of transforming growth factor beta (TGFβ) by fibroblasts, leading to myofibroblast differentiation, contraction, and matrix stiffening (182). TGFβ also dampens the innate immune response through effects on the maturation of DCs, natural killer (NK) cells, T cells, neutrophils, and macrophages and supports the differentiation and induction of regulatory T cells (183). TGFβ has been suggested as a link between the mechanics of interstitial fluid pressure, lymphatic flow, and the development of an immunosuppressive tumor microenvironment (181).
Role of LECs in Immune Suppression and Tolerance
Peripheral tolerance is the process by which self-reactive T cells that escape thymic selection are deleted or rendered anergic. Lymphatic flow and the delivery of lymph fluid to the lymph node are required for the induction of new peripheral tolerance (184, 185). Hence, the increased lymphatic flow seen draining tumors may play a critical role in the development of a permissive immune microenvironment. Induction of peripheral tolerance in the draining lymph node is a multistep process involving the transport of antigens and APCs to the lymph node, antigen presentation in the lymph node, and activation of inhibitory pathways including deletion of reactive T cells, anergy, and Treg induction. LECs, both in peripheral tissues and in the lymph node, and lymph node stromal cells have important roles in the induction of tolerance, which is summarized in Figure 4.
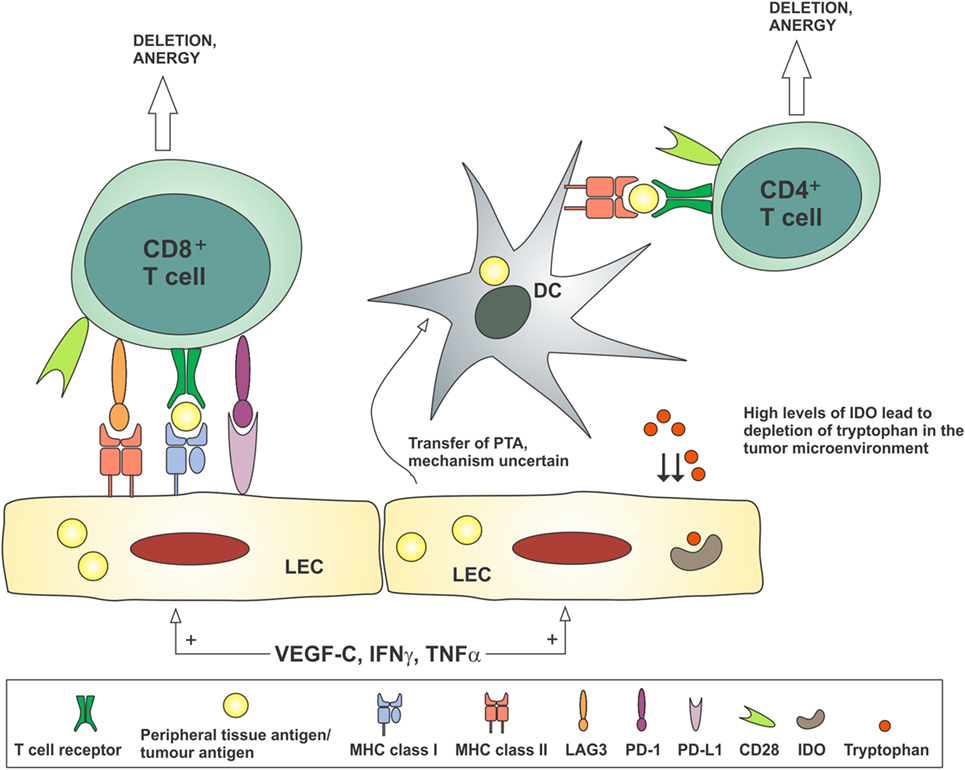
Figure 4. Lymphatic endothelial cells may contribute to the development of tolerance to tumor antigens by antigen presentation to CD8+ T cells in the absence of costimulatory molecules such as CD80/CD86, or in the presence of co-inhibitory molecules such as PD-L1 and LAG3. Peripheral tissue antigens or tumor antigens may be transferred from LECs to dendritic cells, which present these antigens to CD4+ T cells in the absence of costimulatory molecules, thereby inducing anergy. Stimulation of LECs by VEGF-C and inflammatory cytokines TNFα and IFNγ can reduce CD86 expression on dendritic cells and produce IDO, which depletes tryptophan from the microenvironment, thereby preventing the activation of T cells. DC, dendritic cell; IDO, indoleamine 2,3-dioxygenase; IFNγ, interferon-gamma; LEC, lymphatic endothelial cell; PTA, peripheral tissue antigen; TNFα, tumor necrosis factor-alpha; VEGF-C, vascular endothelial growth factor-C.
The development of peripheral tolerance depends on the delivery of soluble antigens and tissue-resident APCs to the draining lymph node. Migration of tissue DCs into initial lymphatics is dependent on CCR7 expression by activated DCs and CCL21 expression on LECs (178). Antigens are carried in the interstitial fluid through the button junctions of the initial lymphatics. Once at the draining lymph node, DCs are guided to the paracortical T cell zone by CCL21 and CCL19. Small antigens are directed into the lymph node via intricate conduits, then taken up and processed by lymph node-resident DCs, while larger antigens are captured and processed by sinus macrophages (186, 187).
Stromal cells within the lymph node, including LECs and fibroblast reticular cells (FRCs), play important structural and physiological roles in the functions of the node. LECs and FRCs express MHC class I molecules as do nearly all nucleated cells (188). However, LECs and FRCs participate in the process of peripheral immune tolerance through ectopic expression of tissue-specific antigens on MHC class I, for example, antigens usually restricted to melanocytes, intestinal epithelium or pancreas, and presentation of these antigens to CD8+ T cells (188, 189). These antigens are not scavenged from the lymph fluid but directly expressed in both an autoimmune regulator (Aire)-dependent manner, as is seen in central tolerance in the thymus, and also in an Aire-independent manner (188). The costimulatory molecules CD40, CD80, and CD86 are not expressed on LECs and FRCs; however, the inhibitory molecule PD-L1 is expressed at high levels (190). Hence, presentation of antigens by LECs and FRCs can result in deletional tolerance of the reactive CD8+ T cells. In addition to this presentation of self-antigens, LECs activated by VEGF-C have also been shown to scavenge and cross-present tumor antigens, leading to the apoptosis of tumor-specific CD8+ T cells (181). MHC class II, expressed by professional APCs including DCs and B cells, is also expressed at low levels by lymph node LECs but not tissue LECs. LECs do not appear to present endogenous antigen on MHC class II molecules but instead act as a reservoir for transfer of antigen to DCs for effective presentation to CD4+ T cells (191). In addition, MHC class II may be a ligand for the co-inhibitory molecule LAG3, resulting in induction of CD8+ T cell tolerance through synergy with PD-1/PD-L1 signaling (191).
Lymphatic endothelial cells and FRCs also prevent the expansion of the activated T cell pool in lymph nodes by expression of NOS 2 and production of nitric oxide (192). LECs stimulated by inflammatory cytokines TNFα and IFNγ can also suppress the ability of DCs to activate and induce T cell proliferation by reducing the expression of the costimulatory molecule CD86 (193) and activating production of IDO (194), an enzyme of the innate immune system that depletes tryptophan, an amino acid essential for the activation of T cells. These features of lymph node stromal cells contribute to ongoing suppression of any immune reactions to self-antigens and may contribute to suppression of responses to tumor antigens.
The contribution of lymphatic flow to tumor immune evasion is supported by the evidence that a permissive environment is created in tumor-draining lymph nodes, the so-called “metastatic niche” [reviewed elsewhere (195)]. The presence of tumor cells in the sentinel lymph node, that is, the first lymph node draining the region of the tumor, is associated with disease progression and often changes clinical management. It is now well established that the sentinel node undergoes changes in stromal and immune cell composition, even before the arrival of tumor cells (196). Lymphangiogenesis and lymphatic remodeling in the lymph node, driven by VEGF-A, VEGF-C, and VEGF-D, are important components of the pre-metastatic niche (197–199). HEVs, which normally support extravasation of naïve lymphocytes into the lymph node parenchyme, are also remodeled, becoming dilated and losing their typical “high” morphology and other molecular characteristics important for lymphocyte trafficking (199, 200). VEGF-D can suppress the proliferation of typical versus remodeled HEVs in the draining lymph node (199). In addition, the recruitment of naïve lymphocytes to the lymph node is impaired in tumor-draining nodes through loss of expression of CCL21 in HEVs, whereas recruitment of inflammatory cell subsets is enhanced in larger venules (201). While tumor-secreted factors such as VEGFs can act directly on LECs and HEVs in lymph nodes, HEV morphology and function are known to be dependent on lymphatic drainage, particularly the trafficking of DCs (202). Therefore, it is likely that lymphatic flow, HEV function, and immune cell composition in tumor-draining lymph nodes are strongly interrelated. The composition and function of immune cells is known to be altered in tumor-draining lymph nodes, with a lower percentage of effector T cells, loss or immaturity of DCs, and higher numbers of Tregs (196). In addition, effector T cells in tumor-draining lymph nodes may be functionally tolerant (203). In a mouse melanoma model, tumor cells implanted into lymph nodes unrelated to the primary tumor were rejected by a specific CD8+ T cell response (204). However, tumor cells introduced into the tumor-draining lymph nodes were able to successfully implant following anergy of the reactive T cells due to MHC class I presentation of tumor antigens (204).
The relationship between tumor lymphangiogenesis, lymphatic remodeling, and the immune response is not yet fully elucidated with some apparently contradictory reports in the literature. Lymphatic vessel density at the invasive margins of tumors has been shown to correlate with metastasis and reduced overall survival in many tumor types, including melanoma, breast cancer, colorectal cancer, and lung cancer [reviewed elsewhere (179)]. Expression of lymphangiogenic factors and their receptors can also be prognostic and predictive of metastatic disease in these tumors. Interactions between VEGF-D and VEGFR-3 can promote the early events of lymphatic metastasis, as demonstrated in a VEGF-D-driven mouse tumor model (205). The proximity of tumor cells expressing VEGF-D to small lymphatic vessels can also be an important determinant of metastasis (206). For the reasons outlined above, increased lymphatic vessel density and lymphatic flow is thought to increase peripheral tolerance and enhance the immunosuppressive microenvironment of both the tumor site and the draining lymph node. Surprisingly, a recent study of human colorectal cancers found that lymphatic vessel density at the invasive margin correlated with the cytotoxic T cell density and inversely correlated with the risk of metastasis (207). Recent analysis of The Cancer Genome Atlas data of human metastatic melanoma samples has shown a correlation between levels of lymphatic gene expression and expression of genes associated with immune infiltration (208). In a mouse model of melanoma, it was found that mice lacking dermal lymphatics showed a lower immune cell infiltrate than mice with intact lymphatic drainage, but that adoptive T cell transfer was more effective in the absence of lymphatic vessels (208). This finding was hypothesized to be due to the lack of Tregs and suppressive macrophages in the tumor microenvironment, allowing the transferred T cells to exert their cytotoxic effects (208). Further investigation of the contribution of lymphatic vessels to the immune infiltrate in tumors and the development of an immunosuppressive environment is needed.
Role of Blood Vascular Endothelial Cells in Immune Suppression and Tolerance
Blood vessel endothelial cells (BECs) also function as semi-professional APCs and can modulate the T cell response. BECs constitutively express both MHC class I and MHC class II molecules and upregulate these in response to inflammatory signals (78). They possess antigen-processing machinery and have been shown to take up and present antigens in vivo and in vitro (209). Critical costimulatory molecules CD80 and CD86 are not expressed on cultured human endothelial cells, rendering them unable to stimulate naïve CD8+ T cells (210). However, limited activation of memory CD8+ T cells that have less stringent costimulatory requirements has been observed (210). Co-inhibitory molecules including PD-L1 and PD-L2 can be expressed by endothelial cells (209, 211). Expression of these immune checkpoint molecules is upregulated by TNFα and can inhibit CD8+ T cell activation (211). Huang et al. demonstrated that endothelial cells derived from B cell lymphomas can express the co-inhibitory molecule TIM-3, which correlated with increased growth and dissemination of lymphoma in a mouse model (212). Expression of the immunosuppressive enzyme IDO has also been demonstrated in endothelial cells in renal cell carcinoma (213).
B7-H3 and B7-H4 are members of the B7 family of immune regulatory molecules, which includes PD-L1 (B7-H1) and PD-L2 (B7-DC) (214). Both molecules are thought to function as co-inhibitory signals limiting T cell activation (215, 216). Expression of B7-H3 on tumor cells and the endothelium of tumor-associated vasculature has been described in ovarian, endometrial, and cervical carcinomas and correlated with higher grade and poor prognosis (217–219). Interestingly, in cervical carcinomas, endothelial B7-H3 expression inversely correlated with CD8+ T cell infiltration (219), whereas there was no correlation in endometrial carcinomas (218). Expression of B7-H3 and B7-H4 has also been demonstrated on tumor vasculature in renal cell carcinomas and is associated with poor prognosis (220, 221). Correlation with TILs has not been reported in this setting. Clearly the endothelial lining of tumor blood vessels has immunomodulatory capabilities, but it remains to be demonstrated conclusively in vivo that tumor endothelial cells take up and present tumor-specific antigens and contribute to the immunosuppressive tumor microenvironment.
Implications for Treatment Strategies
Current clinical therapeutic approaches targeting the tumor vasculature include neutralizing antibodies to VEGF-A (bevacizumab), neutralizing antibodies to VEGFR-2 (ramucirumab), ligand traps (aflibercept), and multi-target tyrosine kinase inhibitors such as sunitinib and sorafenib, which target a range of receptor tyrosine kinases including the VEGF receptors, PDGF receptors, Flt3, and c-kit (222, 223). The ligand trap aflibercept is a recombinant protein containing regions of the extracellular domain of VEGFR-1 and VEGFR-2 fused to the Fc portion of IgG and functions to prevent the binding of VEGF-A, VEGF-B, and PlGF to VEGF receptors, on the cell surface (96). In addition, tyrosine kinase inhibitors targeting the epidermal growth factor receptor (EGFR), now widely used in the treatment of EGFR-mutant lung adenocarcinoma, have also been shown to decrease production of VEGF-A, reduce tumor hypoxia, and possibly have a direct effect on tumor endothelial cells (224, 225). Bevacizumab is the most commonly used and well-studied agent, approved for use in combination with conventional chemotherapy in colorectal, lung, renal cell, and ovarian cancer [reviewed elsewhere (226)]. The mechanism of action of these antiangiogenic therapies is not yet fully understood. Rather than purely starving the tumor of nutrients, these antiangiogenic therapies are also thought to exert their effect by physical normalization of the tumor vasculature and alleviation of hypoxia (147). VEGF-A inhibitors have been shown to reduce the size and tortuosity of tumor vessels, enhance vessel maturation, recruit pericytes, and normalize the basement membrane (149). This results in improved oxygenation and drug delivery to tumors, in part through the ability of normalized vessels to sustain a pressure gradient (151). Vascular normalization has been difficult to demonstrate clinically, as effects may be transient, variable in response to different doses, and occur in only a proportion of tumors. However, studies using advanced magnetic resonance imaging techniques have demonstrated that antiangiogenic therapy can improve tumor perfusion in the clinical setting (227). In a study of cytotoxic chemotherapy combined with VEGF receptor inhibition for the treatment of glioblastoma, patients in whom this improved perfusion was demonstrated had an improved overall survival (227). This finding suggests that vascular normalization can indeed improve access of chemotherapeutic agents to tumors and therefore may also improve the delivery of immunotherapies and the trafficking of immune effector cells. Blocking the VEGF signaling pathway may also act to reduce immunosuppression in the tumor environment.
As outlined in previous sections, the tumor vasculature and the immune microenvironment are intricately linked, with the blood and lymphatic vessels both regulating access of immune cells to the tumor and showing direct immunosuppressive actions through angiogenic factors and endothelial cells. The combination of antiangiogenic therapy and immunotherapy has been explored in a variety of pre-clinical models (Table 1) and forms the basis for a number of current clinical trials (Table 2). Much of the pre-clinical evidence relates to adoptive cell transfer and vaccination strategies, in combination with a wide variety of antiangiogenic therapies including VEGF-A blockade (97, 98, 111), VEGFR-2 blockade (100, 101), ligand traps (99, 112), receptor tyrosine kinase inhibitors (106, 107, 114, 115), irradiation (166), and angiostatic peptides (102, 103, 105, 113). For example, Shrimali et al. demonstrated enhanced tumor infiltration, decreased tumor size, and improved survival when adoptive T cell transfer was combined with treatment with an anti-mouse VEGF-A antibody in a mouse model of melanoma (97). Results from these pre-clinical models suggest that vascular normalization can improve lymphocyte infiltration into tumors and combining antiangiogenic therapy and CAR T cell transfer in solid tumors may be worthy of further investigation in clinical trials.
In the clinical setting, interactions between immune checkpoint inhibitors and the tumor vasculature are beginning to be described. Ipilimumab, an anti-CTLA-4 antibody, shows durable responses in up to 30% of patients with metastatic melanoma (2) and can result in an immune-mediated lymphocytic vasculopathy with resultant vessel obstruction and tumor necrosis (228). In a cohort of patients with advanced melanoma, pre-treatment serum levels of VEGF-A correlated with poor overall survival and poor response to immune checkpoint therapy with ipilimumab (229). Initial promising results have been reported in phase I clinical trials combining ipilimumab and the anti-VEGF-A antibody bevacizumab in advanced melanoma and glioblastoma (109, 117). This combination appears safe and well tolerated (109, 117) and warrants further investigation and comparison to current treatment regimens. Tumor endothelial cells isolated from melanoma patients treated with this combination of ipilimumab and bevacizumab showed variable upregulation of adhesion molecules E-selectin, ICAM-1, and VCAM-1, with resulting enhancement of T cell infiltration into the tumor (109, 230). Changes in levels of circulating chemokines, cytokines, and growth factors were seen following treatment, including increased levels of chemoattractant IP-10 (CXCL10) and decreased levels of VEGF-A (230). Endothelial anergy induced by VEGF-A could be demonstrated in these samples and reversed by the addition of bevacizumab (230). A recent report describes results from a phase I study combining bevacizumab and the anti-PD-L1 antibody atezolizumab in the treatment of advanced renal cell carcinoma (110). Before the addition of atezolizumab, bevacizumab treatment increased the Th1 gene expression signature, which is associated with CD8 T+ cells, NK cells, and Th1 chemokines (110). There was a pronounced increase in intratumoral T cells following combination therapy, suggested to be related to an increase in expression of both CX3CL1 (fractalkine) and its receptor (110). Although not a primary endpoint of this small single-arm study, clinical activity was higher with combination therapy than that has been previously reported with either bevacizumab or atezolizumab alone (110). Each drug may potentiate the effects of the other, controlling tumor angiogenesis and counteracting the immunosuppressive microenvironment. These studies provide important clinical and laboratory data to support further investigation of the use of antiangiogenic agents to enhance immunotherapy.
Following the description of the role of lymphangiogenesis, lymphatic remodeling, and lymphangiogenic factors in promoting tumor metastasis, targeting this signaling axis has been suggested as an adjunct to conventional cancer treatments (231). Analogous to the targeting of angiogenesis through anti-VEGF-A antibody bevacizumab, monoclonal antibodies to VEGF-C (232), VEGF-D (233, 234), and VEGFR-3 (235) have been developed and are being evaluated in both pre-clinical models and clinical trials. Ligand traps that contain components of VEGFR-2 (236) and VEGFR-3 (237) have also been developed, which are designed to block the binding of VEGF-C and VEGF-D to cell surface receptors. Multi-target receptor tyrosine kinase inhibitors such as sunitinib and sorafenib, described above, can also block signaling through VEGFR-3 on LECs (238). As detailed in previous sections, LECs and lymphangiogenic factors can also influence the host immune response to cancer. Consideration should be given to the potential to enhance immunotherapy by targeting lymphangiogenesis through monoclonal antibodies or ligand traps. Blocking the immunomodulatory functions of VEGF-C and VEGF-D and decreasing lymphangiogenesis to reduce the tolerance-promoting effects of LECs may be effective ways to improve immunotherapy approaches such as checkpoint inhibitors or adoptive cell transfer. Pre-clinical evaluation of these combinations will help to delineate the contribution of the lymphatic vasculature to evasion of the host immune response and explore the potential benefit of targeting this component of the microenvironment.
Conclusion
Physiological processes such as the growth and remodeling of blood and lymphatic vessels and the immune response to foreign antigens are altered in the tumor microenvironment, and these alterations contribute to the establishment and progression of cancer. Significant interactions between endothelial cells and immune cells alter the extent and composition of the immune infiltrate in tumors, through both molecular and mechanical means. In addition, lymphangiogenesis and LECs have important roles in the development of tolerance to peripheral tissue antigens, including tumor antigens. The contribution of blood and lymphatic vessels to the modification of the antitumor host immune response in human cancer remains to be fully described. It is not known whether aspects of the tumor vasculature are different in tumors that respond to immunotherapy and those that do not, and if features such as hypoxia, production of angiogenic factors, or lymphatic vessel density may serve as predictive biomarkers. Immunotherapy and antiangiogenic therapy both target aspects of the tumor microenvironment rather than specifically targeting the tumor cells themselves. As such, combination approaches may be required to obtain the full benefit of these therapies. Further investigation of antiangiogenic and antilymphangiogenic therapy as a potential adjunct to immunotherapy may see improvement in the access of CAR T cell therapy to solid tumors and expand the benefits of immune checkpoint inhibition to non-inflamed tumors.
Author Contributions
Conceived the topic and outlined the paper: SF, SH, SS, MA, and RF. Wrote and revised the paper: SH, SF, SS, MA, RF, and BS. Final approval of the version to be published: SH, SF, SS, MA, RF, and BS.
Conflict of Interest Statement
MA and SS are shareholders of Opthea Ltd., which has a commercial interest in antiangiogenesis and anti-lymphangiogenesis in cancer, and Ark Therapeutics (acquired by Premier Veterinary Group PLC), which has an interest in the application of growth factors in vascular disease. The other authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Special thanks to David Byrne for assistance with photography and immunohistochemical staining. We apologize to authors whose work could not be quoted due to space limitations.
Funding
The work is supported by a Program Grant (1053535) from the National Health and Medical Research Council of Australia. SS is supported by Research Fellowship (1060498) from the National Health and Medical Research Council of Australia.
References
1. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell (2011) 144(5):646–74. doi:10.1016/j.cell.2011.02.013
2. Hodi FS, O’Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med (2010) 363(8):711–23. doi:10.1056/NEJMoa1003466
3. Robert C, Long GV, Brady B, Dutriaux C, Maio M, Mortier L, et al. Nivolumab in previously untreated melanoma without BRAF mutation. N Engl J Med (2015) 372(4):320–30. doi:10.1056/NEJMoa1412082
4. Rosenberg JE, Hoffman-Censits J, Powles T, Van der Heijden ME, Balar AV, Necchi A, et al. Atezolizumab in patients with locally advanced and metastatic urothelial carcinoma who have progressed following treatment with platinum-based chemotherapy: a single-arm, multicentre, phase 2 trial. Lancet (2016) 387:1909–20. doi:10.1016/S0140-6736(16)00561-4
5. Ansell SM, Lesokhin AM, Borrello I, Halwani A, Scott EC, Gutierrez M, et al. PD-1 blockade with nivolumab in relapsed or refractory Hodgkin’s lymphoma. N Engl J Med (2015) 372(4):311–9. doi:10.1056/NEJMoa1411087
6. Borghaei H, Paz-Ares L, Horn L, Spigel DR, Steins M, Ready NE, et al. Nivolumab versus docetaxel in advanced nonsquamous non-small-cell lung cancer. N Engl J Med (2015) 373(17):1627–39. doi:10.1056/NEJMoa1507643
7. Brahmer J, Reckamp KL, Baas P, Crino L, Eberhardt WE, Poddubskaya E, et al. Nivolumab versus docetaxel in advanced squamous-cell non-small-cell lung cancer. N Engl J Med (2015) 373(2):123–35. doi:10.1056/NEJMoa1504627
8. Garon EB, Rizvi NA, Hui R, Leighl N, Balmanoukian AS, Eder JP, et al. Pembrolizumab for the treatment of non-small-cell lung cancer. N Engl J Med (2015) 372(21):2018–28. doi:10.1056/NEJMoa1501824
9. Nghiem PT, Bhatia S, Lipson EJ, Kudchadkar RR, Miller NJ, Annamalai L, et al. PD-1 blockade with pembrolizumab in advanced Merkel-cell carcinoma. N Engl J Med (2016) 374(26):2542–52. doi:10.1056/NEJMoa1603702
10. Seiwert TY, Burtness B, Mehra R, Weiss J, Berger R, Eder JP, et al. Safety and clinical activity of pembrolizumab for treatment of recurrent or metastatic squamous cell carcinoma of the head and neck (KEYNOTE-012): an open-label, multicentre, phase 1b trial. Lancet Oncol (2016) 17(7):956–65. doi:10.1016/s1470-2045(16)30066-3
11. Maude SL, Teachey DT, Porter DL, Grupp SA. CD19-targeted chimeric antigen receptor T-cell therapy for acute lymphoblastic leukemia. Blood (2015) 125(26):4017–23. doi:10.1182/blood-201412-580068
12. Topalian SL, Taube JM, Anders RA, Pardoll DM. Mechanism-driven biomarkers to guide immune checkpoint blockade in cancer therapy. Nat Rev Cancer (2016) 16(5):275–87. doi:10.1038/nrc.2016.36
13. Newick K, Moon E, Albelda SM. Chimeric antigen receptor T-cell therapy for solid tumors. Mol Ther Oncolytics (2016) 3:16006. doi:10.1038/mto.2016.6
14. Quigley DA, Kristensen V. Predicting prognosis and therapeutic response from interactions between lymphocytes and tumor cells. Mol Oncol (2015) 9(10):2054–62. doi:10.1016/j.molonc.2015.10.003
15. Teng MW, Galon J, Fridman WH, Smyth MJ. From mice to humans: developments in cancer immunoediting. J Clin Invest (2015) 125(9):3338–46. doi:10.1172/JCI80004
16. Gajewski TF, Schreiber H, Fu YX. Innate and adaptive immune cells in the tumor microenvironment. Nat Immunol (2013) 14(10):1014–22. doi:10.1038/ni.2703
17. Coulie PG, Van den Eynde BJ, van der Bruggen P, Boon T. Tumour antigens recognized by T lymphocytes: at the core of cancer immunotherapy. Nat Rev Cancer (2014) 14(2):135–46. doi:10.1038/nrc3670
18. Lee PP, Yee C, Savage PA, Fong L, Brockstedt D, Weber JS, et al. Characterisation of circulating T cells specific for tumor-associated antigens in melanoma patients. Nat Med (1999) 5(6):677–85. doi:10.1038/9525
19. Gros A, Parkhurst MR, Tran E, Pasetto A, Robbins PF, Ilyas S, et al. Prospective identification of neoantigen-specific lymphocytes in the peripheral blood of melanoma patients. Nat Med (2016) 22(4):433–8. doi:10.1038/nm.4051
20. Fridman WH, Pages F, Sautes-Fridman C, Galon J. The immune contexture in human tumours: impact on clinical outcome. Nat Rev Cancer (2012) 12(4):298–306. doi:10.1038/nrc3245
21. Galon J, Mlecnik B, Bindea G, Angell HK, Berger A, Lagorce C, et al. Towards the introduction of the ‘Immunoscore’ in the classification of malignant tumours. J Pathol (2014) 232(2):199–209. doi:10.1002/path.4287
22. Savas P, Salgado R, Denkert C, Sotiriou C, Darcy PK, Smyth MJ, et al. Clinical relevance of host immunity in breast cancer: from TILs to the clinic. Nat Rev Clin Oncol (2016) 13(4):228–41. doi:10.1038/nrclinonc.2015.215
23. Gajewski TF, Woo SR, Zha Y, Spaapen R, Zheng Y, Corrales L, et al. Cancer immunotherapy strategies based on overcoming barriers within the tumor microenvironment. Curr Opin Immunol (2013) 25(2):268–76. doi:10.1016/j.coi.2013.02.009
24. Teng MW, Ngiow SF, Ribas A, Smyth MJ. Classifying cancers based on T-cell infiltration and PD-L1. Cancer Res (2015) 75(11):2139–45. doi:10.1158/0008-5472.CAN-15-0255
25. Bailey P, Chang DK, Nones K, Johns AL, Patch AM, Gingras MC, et al. Genomic analyses identify molecular subtypes of pancreatic cancer. Nature (2016) 531(7592):47–52. doi:10.1038/nature16965
26. Guinney J, Dienstmann R, Wang X, de Reynies A, Schlicker A, Soneson C, et al. The consensus molecular subtypes of colorectal cancer. Nat Med (2015) 21(11):1350–6. doi:10.1038/nm.3967
27. Cancer Genome Atlas Network. Genomic classification of cutaneous melanoma. Cell (2015) 161(7):1681–96. doi:10.1016/j.cell.2015.05.044
28. Salgado R, Denkert C, Demaria S, Sirtaine N, Klauschen F, Pruneri G, et al. The evaluation of tumor-infiltrating lymphocytes (TILs) in breast cancer: recommendations by an International TILs working group 2014. Ann Oncol (2015) 26(2):259–71. doi:10.1093/annonc/mdu450
29. Azimi F, Scolyer RA, Rumcheva P, Moncrieff M, Murali R, McCarthy SW, et al. Tumor-infiltrating lymphocyte grade is an independent predictor of sentinel lymph node status and survival in patients with cutaneous melanoma. J Clin Oncol (2012) 30(21):2678–83. doi:10.1200/JCO.2011.37.8539
30. Le DT, Uram JN, Wang H, Bartlett BR, Kemberling H, Eyring AD, et al. PD-1 blockade in tumors with mismatch-repair deficiency. N Engl J Med (2015) 372(26):2509–20. doi:10.1056/NEJMoa1500596
31. Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SA, Behjati S, Biankin AV, et al. Signatures of mutational processes in human cancer. Nature (2013) 500(7463):415–21. doi:10.1038/nature12477
32. Snyder A, Makarov V, Merghoub T, Yuan J, Zaretsky JM, Desrichard A, et al. Genetic basis for clinical response to CTLA-4 blockade in melanoma. N Engl J Med (2014) 371(23):2189–99. doi:10.1056/NEJMoa1406498
33. Rivzi NA, Hellmann MD, Snyder A, Kvistborg P, Makarov V, Havel JJ, et al. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science (2015) 348(6230):124–8. doi:10.1126/science.aaa1348
34. Remark R, Becker C, Gomez JE, Damotte D, Dieu-Nosjean MC, Sautes-Fridman C, et al. The non-small cell lung cancer immune contexture. A major determinant of tumor characteristics and patient outcome. Am J Respir Crit Care Med (2015) 191(4):377–90. doi:10.1164/rccm.201409-1671PP
35. Slaney CY, Kershaw MH, Darcy PK. Trafficking of T cells into tumors. Cancer Res (2014) 74(24):7168–74. doi:10.1158/0008-5472.CAN-14-2458
36. Masopust D, Schenkel JM. The integration of T cell migration, differentiation and function. Nat Rev Immunol (2013) 13(5):309–20. doi:10.1038/nri3442
37. Mulligan AM, Raitman I, Feeley L, Pinnaduwage D, Nguyen LT, O’Malley FP, et al. Tumoral lymphocytic infiltration and expression of the chemokine CXCL10 in breast cancers from the Ontario familial breast cancer registry. Clin Cancer Res (2013) 19(2):336–46. doi:10.1158/1078-0432.CCR-11-3314
38. Harlin H, Meng Y, Peterson AC, Zha Y, Tretiakova M, Slingluff C, et al. Chemokine expression in melanoma metastases associated with CD8+ T-cell recruitment. Cancer Res (2009) 69(7):3077–85. doi:10.1158/0008-5472.CAN-08-2281
39. Molon B, Ugel S, Del Pozzo F, Soldani C, Zilio S, Avella D, et al. Chemokine nitration prevents intratumoral infiltration of antigen-specific T cells. J Exp Med (2011) 208(10):1949–62. doi:10.1084/jem.20101956
40. Joyce JA, Fearon DT. T cell exclusion, immune privilege, and the tumor microenvironment. Science (2015) 348(6230):74–80. doi:10.1126/science.aaa6204
41. Noman MZ, Hasmim M, Messai Y, Terry S, Kieda C, Janji B, et al. Hypoxia: a key player in antitumor immune response. A review in the theme: cellular responses to hypoxia. Am J Physiol Cell Physiol (2015) 309(9):C569–79. doi:10.1152/ajpcell.00207.2015
42. Molon B, Cali B, Viola A. T cells and cancer: how metabolism shapes immunity. Front Immunol (2016) 7:20. doi:10.3389/fimmu.2016.00020
43. Munn DH, Mellor AL. Indoleamine 2,3 dioxygenase and metabolic control of immune responses. Trends Immunol (2013) 34(3):137–43. doi:10.1016/j.it.2012.10.001
44. Nguyen NT, Kimura A, Nakahama T, Chinen I, Masuda K, Nohara K, et al. Aryl hydrocarbon receptor negatively regulates dendritic cell immunogenicity via a kynurenine-dependent mechanism. Proc Natl Acad Sci U S A (2010) 107(46):19961–6. doi:10.1073/pnas.1014465107
45. Munn DH, Sharma MD, Baban B, Harding HP, Zhang Y, Ron D, et al. GCN2 kinase in T cells mediates proliferative arrest and anergy induction in response to indoleamine 2,3-dioxygenase. Immunity (2005) 22(5):633–42. doi:10.1016/j.immuni.2005.03.013
46. Fallarino F, Grohmann U, You S, McGrath BC, Cavener DR, Vacca C, et al. The combined effects of tryptophan starvation and tryptophan catabolites down-regulate T cell receptor-chain and induce a regulatory phenotype in naive T cells. J Immunol (2006) 176(11):6752–61. doi:10.4049/jimmunol.176.11.6752
47. Sharma MD, Baban B, Chandler P, Hou DY, Singh N, Yagita H, et al. Plasmacytoid dendritic cells from mouse tumor-draining lymph nodes directly activate mature Tregs via indoleamine 2,3-dioxygenase. J Clin Invest (2007) 117(9):2570–82. doi:10.1172/JCI31911
48. Holmgaard RB, Zamarin D, Munn DH, Wolchok JD, Allison JP. Indoleamine 2,3-dioxygenase is a critical resistance mechanism in antitumor T cell immunotherapy targeting CTLA-4. J Exp Med (2013) 210(7):1389–402. doi:10.1084/jem.20130066
49. Moon YW, Hajjar J, Hwu P, Naing A. Targeting the indoleamine 2,3-dioxygenase pathway in cancer. J Immunother Cancer (2015) 3:51. doi:10.1186/s40425-015-0094-9
50. Bronte V, Zanovello P. Regulation of immune responses by l-arginine metabolism. Nat Rev Immunol (2005) 5(8):641–54. doi:10.1038/nri1668
51. Rodriguez PC, Quiceno DG, Zabaleta J, Ortiz B, Zea AH, Piazuelo MB, et al. Arginase I production in the tumor microenvironment by mature myeloid cells inhibits T-cell receptor expression and antigen-specific T-cell responses. Cancer Res (2004) 64:5839–49. doi:10.1158/0008-5472.CAN-04-0465
52. Kostourou V, Cartwright JE, Johnstone AP, Boult JK, Cullis ER, Whitley G, et al. The role of tumour-derived iNOS in tumour progression and angiogenesis. Br J Cancer (2011) 104(1):83–90. doi:10.1038/sj.bjc.6606034
53. Rodriguez PC, Ernstoff MS, Hernandez C, Atkins M, Zabaleta J, Sierra R, et al. Arginase I-producing myeloid-derived suppressor cells in renal cell carcinoma are a subpopulation of activated granulocytes. Cancer Res (2009) 69(4):1553–60. doi:10.1158/0008-5472.CAN-08-1921
54. Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer (2012) 12(4):252–64. doi:10.1038/nrc3239
55. Walker LS, Abbas AK. The enemy within: keeping self-reactive T cells at bay in the periphery. Nat Rev Immunol (2002) 2(1):11–9. doi:10.1038/nri701
56. Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med (2012) 366(26):2443–54. doi:10.1056/NEJMoa1200690
57. Brahmer JR, Tykodi SS, Chow LQ, Hwu WJ, Topalian SL, Hwu P, et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N Engl J Med (2012) 366(26):2455–65. doi:10.1056/NEJMoa1200694
58. Tang H, Wang Y, Chlewicki LK, Zhang Y, Guo J, Liang W, et al. Facilitating T cell infiltration in tumor microenvironment overcomes resistance to PD-L1 blockade. Cancer Cell (2016) 29(3):285–96. doi:10.1016/j.ccell.2016.02.004
59. Kershaw MH, Westwood JA, Darcy PK. Gene-engineered T cells for cancer therapy. Nat Rev Cancer (2013) 13(8):525–41. doi:10.1038/nrc3565
60. Mukai S, Kjaergaard J, Shu S, Plautz GE. Infiltration of tumors by systemically transferred tumor-reactive T lymphocytes is required for antitumor efficacy. Cancer Res (1999) 59(20):5245–9.
61. Craddock JA, Lu A, Bear A, Pule M, Brenner MK, Rooney CM, et al. Enhanced tumor trafficking of GD2 chimeric antigen receptor T cells by expression of the chemokine receptor CCR2b. J Immunother (2010) 33(8):780–8. doi:10.1097/CJI.0b013e3181ee6675
62. Adusumilli PS, Cherkassky L, Villena-Vargas J, Colovos C, Servais E, Plotkin J, et al. Regional delivery of mesothelin-targeted CAR T cell therapy generates potent and long-lasting CD4-dependent tumor immunity. Sci Transl Med (2014) 6(261):261ra151. doi:10.1126/scitranslmed.3010162
63. Beavis PA, Slaney CY, Kershaw MH, Gyorki D, Neeson PJ, Darcy PK. Reprogramming the tumor microenvironment to enhance adoptive cellular therapy. Semin Immunol (2016) 28(1):64–72. doi:10.1016/j.smim.2015.11.003
64. Ganss R, Hanahan D. Tumor microenvironment can restrict the effectiveness of activated antitumour lymphocytes. Cancer Res (1998) 58:4673–81.
65. Pober JS, Tellides G. Participation of blood vessel cells in human adaptive immune responses. Trends Immunol (2012) 33(1):49–57. doi:10.1016/j.it.2011.09.006
66. Ley K, Laudanna C, Cybulsky MI, Nourshargh S. Getting to the site of inflammation: the leukocyte adhesion cascade updated. Nat Rev Immunol (2007) 7(9):678–89. doi:10.1038/nri2156
67. Lawrence MB, Kansas GS, Kunkel EJ, Ley K. Threshold levels of fluid shear promote leukocyte adhesion through selectins (CD62L, P, E). J Cell Biol (1997) 136:717–27. doi:10.1083/jcb.136.3.717
68. Rothlein R, Dustin ML, Marlin SD, Springer TA. A human intercellular adhesion molecule (ICAM-1) distinct from LFA-1. J Immunol (1986) 137:1270–4.
69. Osborn L, Hession C, Tizard R, Vassallo C, Luhowskyj S, Chi-Rosso G, et al. Direct expression cloning of vascular cell adhesion molecule 1, a cytokine-induced endothelial protein that binds to lymphocytes. Cell (1989) 59:1203–11. doi:10.1016/0092-8674(89)90775-7
70. Dustin ML, Rothlein R, Bhan AK, Dinarello CA, Springer TA. Induction by IL-1 and interferon-γ: tissue distribution, biochemistry, and function of a natural adherence molecule (ICAM-1). J Immunol (1986) 186:5024–33.
71. Elices MJ, Osborn L, Takada Y, Crouse C, Luhowskyj S, Hemler ME, et al. VCAM-1 on activated endothelium interacts with the leukocyte integrin VLA-4 at a site distinct from the VLA-4/fibronectin binding site. Cell (1990) 60:577–84. doi:10.1016/0092-8674(90)90661-W
72. Springer TA. Traffic signals on endothelium for lymphocyte recirculation and leukocyte emigration. Annu Rev Physiol (1995) 57:827–72. doi:10.1146/annurev.ph.57.030195.004143
73. Muller WA. Mechanisms of leukocyte transendothelial migration. Annu Rev Pathol (2011) 6:323–44. doi:10.1146/annurev-pathol-011110-130224
74. Clark RA, Huang SJ, Murphy GF, Mollet IG, Hijnen D, Muthukuru M, et al. Human squamous cell carcinomas evade the immune response by down-regulation of vascular E-selectin and recruitment of regulatory T cells. J Exp Med (2008) 205(10):2221–34. doi:10.1084/jem.20071190
75. Afanasiev OK, Nagase K, Simonson W, Vandeven N, Blom A, Koelle DM, et al. Vascular E-selectin expression correlates with CD8 lymphocyte infiltration and improved outcome in Merkel cell carcinoma. J Invest Dermatol (2013) 133(8):2065–73. doi:10.1038/jid.2013.36
76. Bouma-ter Steege JC, Baeten CI, Thijssen VL, Satijn SA, Verhoeven IC, Hillen HF, et al. Angiogenic profile of breast carcinoma determines leukocyte infiltration. Clin Cancer Res (2004) 10:7171–8. doi:10.1158/1078-0432.CCR-04-0742
77. Springer TA. Adhesion receptors of the immune system. Nature (1990) 34:425–34. doi:10.1038/346425a0
78. Choi J, Enis DR, Koh KP, Shiao SL, Pober JS. T lymphocyte-endothelial cell interactions. Annu Rev Immunol (2004) 22:683–709. doi:10.1146/annurev.immunol.22.012703.104639
79. Pober JS, Gimbrone MA, Lapierre LA, Mendrick DL, Fiers W, Rothlein R, et al. Overlapping patterns of activation of human endothelial cells by interleukin 1, tumor necrosis factor, and immune interferon. J Immunol (1986) 137:1893–6.
80. Crusz SM, Balkwill FR. Inflammation and cancer: advances and new agents. Nat Rev Clin Oncol (2015) 12(10):584–96. doi:10.1038/nrclinonc.2015.105
81. Piali L, Fichtel A, Terpe H, Imhof BA, Gisler RH. Endothelial vascular cell adhesion molecule1 expression is suppressed by melanoma and carcinoma. J Exp Med (1995) 181:811–6. doi:10.1084/jem.181.2.811
82. Weishaupt C, Munoz KN, Buzney E, Kupper TS, Fuhlbrigge RC. T-cell distribution and adhesion receptor expression in metastatic melanoma. Clin Cancer Res (2007) 13(9):2549–56. doi:10.1158/1078-0432.CCR-06-2450
83. Griffioen AW, Damen CA, Blijham GH, Groenewegen G. Tumor angiogenesis is accompanied by a decreased inflammatory response of tumor-associated endothelium. Blood (1996) 88(2):667–73.
84. Griffioen AW, Damen CA, Martinotti S, Blijham GH, Groenewegen G. Endothelial intercellular adhesion molecule 1 is suppressed in malignancies: the role of angiogenic factors. Cancer Res (1996) 56:1111–7.
85. Dirkx AE, Oude Egbrink MG, Kuijpers MJE, van der Niet ST, Heijnen VVT, Bouma-ter Steege JC, et al. Tumor angiogenesis modulates leukocyte-vessel wall interactions in vivo by reducing endothelial adhesion molecule expression. Cancer Res (2003) 63:2322–9.
86. Horsman MR, Vaupel P. Pathophysiological basis for the formation of the tumor microenvironment. Front Oncol (2016) 6:66. doi:10.3389/fonc.2016.00066
87. Leung DW, Cachianes G, Kuang WJ, Goeddel DV, Ferrara N. Vascular endothelial growth factor is a secreted angiogenic mitogen. Science (1989) 246:1306–9. doi:10.1126/science.2479986
88. Keck PJ, Hauser SD, Krivi G, Sanzo K, Warren T, Feder J, et al. Vascular permeability factor, an endothelial cell mitogen related to PDGF. Science (1989) 246:1309–12. doi:10.1126/science.2479987
89. Zhang H, Issekutz AC. Down modulation of monocyte transendothelial migration and endothelial adhesion molecule expression by fibroblast growth factor. Am J Pathol (2002) 160(6):2219–30. doi:10.1016/S0002-9440(10)61169-8
90. Griffioen AW, Relou IAM, Gallardo Torres HI, Damen CA, Martinotti S, de Graaf JC, et al. The angiogenic factor bFGF impairs leukocyte adhesion and rolling under flow conditions. Angiogenesis (1998) 2:235–43. doi:10.1023/A:1009289303145
91. Maksan S, Araib PM, Ryschich E, Gebhard MM, Schmidt J. Immune escape mechanism: defective resting and stimulated leukocyte-endothelium interaction in hepatocellular carcinoma of the rat. Dig Dis Sci (2004) 49(5):859–65. doi:10.1023/B:DDAS.0000030100.05979.b7
92. Tromp SC, Oude Egbrink MG, Dings RP, van Velzen S, Slaaf DW, Hillen HFP, et al. Tumor angiogenesis factors reduce leukocyte adhesion in vivo. Int Immunol (2000) 12(5):671–6. doi:10.1093/intimm/12.5.671
93. Flati V, Pastore LI, Griffioen AW, Satijn SA, Toniato E, D’Alimonte I, et al. Endothelial cell anergy is mediated by bFGF through the sustained activation of p38-MAPK and NFkB inhibition. Int J Immunopathol Pharmacol (2006) 19(4):761–73.
94. Dirkx AE, oude Egbrink MG, Castermans K, van der Schaft DW, Thijssen VL, Dings RP, et al. Anti-angiogenesis therapy can overcome endothelial cell anergy and promote leukocyte-endothelium interactions and infiltration in tumors. FASEB J (2006) 20(6):621–30. doi:10.1096/fj.05-4493com
95. Griffioen AW, Damen CA, Mayo KH, Barendsz-Jansen A, Martinotti S, Blijham GH, et al. Angiogenesis inhibitors overcome tumor induced endothelial cell anergy. Int J Cancer (1999) 80:315–9. doi:10.1002/(SICI)1097-0215(19990118)80:2<315::AID-IJC23>3.0.CO;2-L
96. Limaverde-Sousa G, Sternberg C, Ferreira CG. Antiangiogenesis beyond VEGF inhibition: a journey from antiangiogenic single-target to broad-spectrum agents. Cancer Treat Rev (2014) 40(4):548–57. doi:10.1016/j.ctrv.2013.11.009
97. Shrimali RK, Yu Z, Theoret MR, Chinnasamy D, Restifo NP, Rosenberg SA. Antiangiogenic agents can increase lymphocyte infiltration into tumor and enhance the effectiveness of adoptive immunotherapy of cancer. Cancer Res (2010) 70(15):6171–80. doi:10.1158/0008-5472.CAN-10-0153
98. Tao L, Huang G, Shi S, Chen L. Bevacizumab improves the antitumor efficacy of adoptive cytokine-induced killer cells therapy in non-small cell lung cancer models. Med Oncol (2014) 31(1):777. doi:10.1007/s12032-013-0777-3
99. Li B, Lalani AS, Harding TC, Luan B, Koprivnikar K, Huan Tu G, et al. Vascular endothelial growth factor blockade reduces intratumoral regulatory T cells and enhances the efficacy of a GM-CSF-secreting cancer immunotherapy. Clin Cancer Res (2006) 12(22):6808–16. doi:10.1158/1078-0432.CCR-06-1558
100. Manning EA, Ullman JG, Leatherman JM, Asquith JM, Hansen TR, Armstrong TD, et al. A vascular endothelial growth factor receptor-2 inhibitor enhances antitumor immunity through an immune-based mechanism. Clin Cancer Res (2007) 13(13):3951–9. doi:10.1158/1078-0432.CCR-07-0374
101. Huang Y, Yuan J, Righi E, Kamoun WS, Ancukiewicz M, Nezivar J, et al. Vascular normalizing doses of antiangiogenic treatment reprogram the immunosuppressive tumor microenvironment and enhance immunotherapy. Proc Natl Acad Sci U S A (2012) 109(43):17561–6. doi:10.1073/pnas.1215397109
102. Huang KW, Wu HL, Lin HL, Liang PC, Chen PJ, Chen SH, et al. Combining antiangiogenic therapy with immunotherapy exerts better therapeutical effects on large tumors in a woodchuck hepatoma model. Proc Natl Acad Sci U S A (2010) 107(33):14769–74. doi:10.1073/pnas.1009534107
103. Chan SF, Wang HT, Huang KW, Torng PL, Lee HI, Hwang LH. Anti-angiogenic therapy renders large tumors vulnerable to immunotherapy via reducing immunosuppression in the tumor microenvironment. Cancer Lett (2012) 320(1):23–30. doi:10.1016/j.canlet.2012.01.024
104. Shi S, Wang R, Chen Y, Song H, Chen L, Huang G. Combining antiangiogenic therapy with adoptive cell immunotherapy exerts better antitumor effects in non-small cell lung cancer models. PLoS One (2013) 8(6):e65757. doi:10.1371/journal.pone.0065757
105. Dings RP, Vang KB, Castermans K, Popescu F, Zhang Y, Oude Egbrink MG, et al. Enhancement of T-cell-mediated antitumor response: angiostatic adjuvant to immunotherapy against cancer. Clin Cancer Res (2011) 17(10):3134–45. doi:10.1158/1078-0432.CCR-10-2443
106. Huang X, Wong MK, Yi H, Watkins S, Laird AD, Wolf SF, et al. Combined therapy of local and metastatic 4T1 breast tumor in mice using SU6668, an inhibitor of angiogenic receptor tyrosine kinases, and the immunostimulator B7.2-IgG fusion protein. Cancer Res (2002) 62:5727–35.
107. Farsaci B, Donahue RN, Coplin MA, Grenga I, Lepone LM, Molinolo AA, et al. Immune consequences of decreasing tumor vasculature with antiangiogenic tyrosine kinase inhibitors in combination with therapeutic vaccines. Cancer Immunol Res (2014) 2(11):1090–102. doi:10.1158/2326-6066.CIR-14-0076
108. Johansson A, Hamzah J, Payne CJ, Ganss R. Tumor-targeted TNFalpha stabilizes tumor vessels and enhances active immunotherapy. Proc Natl Acad Sci U S A (2012) 109(20):7841–6. doi:10.1073/pnas.1118296109
109. Hodi FS, Lawrence D, Lezcano C, Wu X, Zhou J, Sasada T, et al. Bevacizumab plus ipilimumab in patients with metastatic melanoma. Cancer Immunol Res (2014) 2(7):632–42. doi:10.1158/2326-6066.CIR-14-0053
110. Wallin JJ, Bendell JC, Funke R, Sznol M, Korski K, Jones S, et al. Atezolizumab in combination with bevacizumab enhances antigen-specific T-cell migration in metastatic renal cell carcinoma. Nat Commun (2016) 7:12624. doi:10.1038/ncomms12624
111. Gabrilovich DI, Ishida T, Nadaf S, Ohm JE, Carbone DP. Antibodies to vascular endothelial growth factor enhance the efficacy of cancer immunotherapy by improving endogenous dendritic cell function. Clin Cancer Res (1999) 5:2963–70.
112. Renner DN, Malo CS, Jin F, Parney IF, Pavelko KD, Johnson AJ. Improved treatment efficacy of antiangiogenic therapy when combined with picornavirus vaccination in the GL261 glioma model. Neurotherapeutics (2016) 13(1):226–36. doi:10.1007/s13311-015-0407-1
113. Shi S, Chen L, Huang G. Antiangiogenic therapy improves the antitumor effect of adoptive cell immunotherapy by normalizing tumor vasculature. Med Oncol (2013) 30(4):698. doi:10.1007/s12032-013-0698-1
114. Ozao-Choy J, Ma G, Kao J, Wang GX, Meseck M, Sung M, et al. The novel role of tyrosine kinase inhibitor in the reversal of immune suppression and modulation of tumor microenvironment for immune-based cancer therapies. Cancer Res (2009) 69(6):2514–22. doi:10.1158/0008-5472.CAN-08-4709
115. Yu N, Fu S, Xu Z, Liu Y, Hao J, Zhang A, et al. Synergistic antitumor responses by combined GITR activation and sunitinib in metastatic renal cell carcinoma. Int J Cancer (2016) 138(2):451–62. doi:10.1002/ijc.29713
116. Grenga I, Kwilas AR, Donahue RN, Farsaci B, Hodge JW. Inhibition of the angiopoietin/Tie2 axis induces immunogenic modulation, which sensitizes human tumor cells to immune attack. J Immunother Cancer (2015) 3:52. doi:10.1186/s40425-015-0096-7
117. Carter T, Shaw H, Cohn-Brown D, Chester K, Mulholland P. Ipilimumab and bevacizumab in glioblastoma. Clin Oncol (R Coll Radiol) (2016) 28(10):622–6. doi:10.1016/j.clon.2016.04.042
118. Achen MG, Jeltsch M, Kukk E, Makinen T, Vitali A, Wilks AF, et al. Vascular endothelial growth factor D (VEGF-D) is a ligand for the tyrosine kinases VEGF receptor 2 (Flk1) and VEGF receptor 3 (Flt4). Proc Natl Acad Sci U S A (1998) 95:548–53. doi:10.1073/pnas.95.2.548
119. Joukov V, Pajusola K, Kaipainen A, Chilov D, Lahtinen I, Kukk E, et al. A novel vascular endothelial growth factor, VEGF-C, is a ligand for the Flt4 (VEGFR-3) and KDR (VEGFR-2) receptor tyrosine kinases. EMBO J (1996) 15:290–8.
120. Stacker SA, Caesar C, Baldwin ME, Thornton GE, Williams RA, Prevo R, et al. VEGF-D promotes the metastatic spread of tumor cells via the lymphatics. Nat Med (2001) 7:186–91. doi:10.1038/84635
121. Skobe M, Hawighorst T, Jackson DG, Prevo R, Janes L, Velasco P, et al. Induction of tumor lymphangiogenesis by VEGF-C promotes breast cancer metastasis. Nat Med (2001) 7:192–8. doi:10.1038/84643
122. Mandriota SJ, Jussila L, Jeltsch M, Compagni A, Baetens D, Prevo R, et al. Vascular endothelial growth factor-C-mediated lymphangiogenesis promotes tumour metastasis. EMBO J (2001) 20:672–82. doi:10.1093/emboj/20.4.672
123. Achen MG, Williams RA, Minekus MP, Thornton GE, Stenvers K, Rogers PAW, et al. Localisation of vascular endothelial growth factor-D in malignant melanoma suggests a role in tumor angiogenesis. J Pathol (2001) 193:147–54. doi:10.1002/1096-9896(2000)
124. Achen MG, McColl BK, Stacker SA. Focus on lymphangiogenesis in tumor metastasis. Cancer Cell (2005) 7(2):121–7. doi:10.1016/j.ccr.2005.01.017
125. Salven P, Lymboussaki A, Heikkila P, Jaaskela-Saari H, Enholm B, Aase K, et al. Vascular endothelial growth factors VEGF-B and VEGF-C are expressed in human tumors. Am J Pathol (1998) 153:103–8. doi:10.1016/S0002-9440(10)65550-2
126. Griffioen AW. Anti-angiogenesis: making the tumor vulnerable to the immune system. Cancer Immunol Immunother (2008) 57(10):1553–8. doi:10.1007/s00262-008-0524-3
127. Hansen TF, Christensen R, Andersen RF, Sorensen FB, Johnsson A, Jakobsen A. MicroRNA-126 and epidermal growth factor-like domain 7-an angiogenic couple of importance in metastatic colorectal cancer. Results from the Nordic ACT trial. Br J Cancer (2013) 109(5):1243–51. doi:10.1038/bjc.2013.448
128. Delfortrie S, Pinte S, Mattot V, Samson C, Villain G, Caetano B, et al. Egfl7 promotes tumor escape from immunity by repressing endothelial cell activation. Cancer Res (2011) 71(23):7176–86. doi:10.1158/0008-5472.CAN-11-1301
129. Rosano L, Spinella F, Bagnato A. Endothelin 1 in cancer: biological implications and therapeutic opportunities. Nat Rev Cancer (2013) 13(9):637–51. doi:10.1038/nrc3546
130. Wulfing P, Kersting C, Tio J, Fischer RJ, Wulfing C, Poremba C, et al. Endothelin-1, endothelin-A- and endothelin-B-receptor expression is correlated with vascular endothelial growth factor expression and angiogenesis in breast cancer. Clin Cancer Res (2004) 10:2393–400. doi:10.1158/1078-0432.CCR-03-0115
131. Buckanovich RJ, Facciabene A, Kim S, Benencia F, Sasaroli D, Balint K, et al. Endothelin B receptor mediates the endothelial barrier to T cell homing to tumors and disables immune therapy. Nat Med (2008) 14(1):28–36. doi:10.1038/nm1699
132. Nakashima S, Sugita Y, Miyoshi H, Arakawa F, Muta H, Ishibashi Y, et al. Endothelin B receptor expression in malignant gliomas: the perivascular immune escape mechanism of gliomas. J Neurooncol (2016) 127(1):23–32. doi:10.1007/s11060-015-2017-5
133. Sugita Y, Terasaki M, Nakashima S, Ohshima K, Morioka M, Abe H. Perivascular microenvironment in primary central nervous system lymphomas: the role of chemokines and the endothelin B receptor. Brain Tumor Pathol (2015) 32(1):41–8. doi:10.1007/s10014-014-0206-0
134. Tanaka T, Sho M, Takayama T, Wakatsuki K, Matsumoto S, Migita K, et al. Endothelin B receptor expression correlates with tumour angiogenesis and prognosis in oesophageal squamous cell carcinoma. Br J Cancer (2014) 110(4):1027–33. doi:10.1038/bjc.2013.784
135. Karikoski M, Marttila-Ichihara F, Elima K, Rantakari P, Hollmen M, Kelkka T, et al. Clever-1/stabilin-1 controls cancer growth and metastasis. Clin Cancer Res (2014) 20(24):6452–64. doi:10.1158/1078-0432.CCR-14-1236
136. Motz GT, Santoro SP, Wang LP, Garrabrant T, Lastra RR, Hagemann IS, et al. Tumor endothelium FasL establishes a selective immune barrier promoting tolerance in tumors. Nat Med (2014) 20(6):607–15. doi:10.1038/nm.3541
137. Kzhyshkowska J, Gratchev A, Goerdt S. Stabilin-1, a homeostatic scavenger receptor with multiple functions. J Cell Mol Med (2006) 10(3):635–49. doi:10.2755/jcmm010.003.08
138. Karikoski M, Irjala H, Maksimow M, Miiluniemi M, Granfors K, Hernesniemi S, et al. Clever-1/Stabilin-1 regulates lymphocyte migration within lymphatics and leukocyte entrance to sites of inflammation. Eur J Immunol (2009) 39(12):3477–87. doi:10.1002/eji.200939896
139. Irjala H, Alanen K, Grenman R, Heikkila P, Joensuu H, Jalkanen S. Mannose receptor (MR) and common lymphatic endothelial and vascular endothelial receptor (CLEVER)-1 direct the binding of cancer cells to the lymph vessel endothelium. Cancer Res (2003) 63:4671–6.
140. Irjala H, Elima K, Johansson E, Merinen M, Kontula K, Alanen K, et al. The same endothelial receptor controls lymphocyte traffic both in vascular and lymphatic vessels. Eur J Immunol (2003) 33:815–24. doi:10.1002/eji.200323859
141. Palazon A, Aragones J, Morales-Kastresana A, de Landazuri MO, Melero I. Molecular pathways: hypoxia response in immune cells fighting or promoting cancer. Clin Cancer Res (2012) 18(5):1207–13. doi:10.1158/1078-0432.CCR-11-1591
142. Facciabene A, Peng X, Hagemann IS, Balint K, Barchetti A, Wang LP, et al. Tumour hypoxia promotes tolerance and angiogenesis via CCL28 and T(reg) cells. Nature (2011) 475(7355):226–30. doi:10.1038/nature10169
143. Yan M, Jene N, Byrne D, Millar EKA, O’Toole S, McNeil CM, et al. Recruitment of regulatory T cells is correlated with hypoxia-induced CXCR4 expression, and is associated with poor prognosis in basal-like breast cancers. Breast Cancer Res (2011) 13:R47. doi:10.1186/bcr2869
144. Noman MZ, Desantis G, Janji B, Hasmim M, Karray S, Dessen P, et al. PD-L1 is a novel direct target of HIF-1alpha, and its blockade under hypoxia enhanced MDSC-mediated T cell activation. J Exp Med (2014) 211(5):781–90. doi:10.1084/jem.20131916
145. Voron T, Colussi O, Marcheteau E, Pernot S, Nizard M, Pointet AL, et al. VEGF-A modulates expression of inhibitory checkpoints on CD8+ T cells in tumors. J Exp Med (2015) 212(2):139–48. doi:10.1084/jem.20140559
146. Bellone M, Calcinotto A. Ways to enhance lymphocyte trafficking into tumors and fitness of tumor infiltrating lymphocytes. Front Oncol (2013) 3:231. doi:10.3389/fonc.2013.00231
147. Jain RK. Normalization of the tumour vasculature: an emerging concept in antiangiogenic therapy. Science (2005) 307:58–62. doi:10.1126/science.1104819
148. Farnsworth RH, Lackmann M, Achen MG, Stacker SA. Vascular remodeling in cancer. Oncogene (2014) 33(27):3496–505. doi:10.1038/onc.2013.304
149. Carmeliet P, Jain RK. Principles and mechanisms of vessel normalization for cancer and other angiogenic diseases. Nat Rev Drug Discov (2011) 10(6):417–27. doi:10.1038/nrd3455
150. Jain RK. Normalizing tumor microenvironment to treat cancer: bench to bedside to biomarkers. J Clin Oncol (2013) 31(17):2205–18. doi:10.1200/JCO.2012.46.3653
151. Tong RT, Boucher Y, Kozin SV, Winkler F, Hicklin DJ, Jain RK. Vascular normalisation by vascular endothelial growth factor receptor 2 blockade induces a pressure gradient across the vasculature and improves drug penetration in tumors. Cancer Res (2004) 64:3731–6. doi:10.1158/0008-5472.CAN-04-0074
152. Hahn C, Schwartz MA. Mechanotransduction in vascular physiology and atherogenesis. Nat Rev Mol Cell Biol (2009) 10(1):53–62. doi:10.1038/nrm2596
153. Wragg JW, Durant S, McGettrick HM, Sample KM, Egginton S, Bicknell R. Shear stress regulated gene expression and angiogenesis in vascular endothelium. Microcirculation (2014) 21(4):290–300. doi:10.1111/micc.12119
154. Buchanan CF, Verbridge SS, Vlachos PP, Rylander MN. Flow shear stress regulates endothelial barrier function and expression of angiogenic factors in a 3D microfluidic tumor vascular model. Cell Adh Migr (2014) 8(5):517–24. doi:10.4161/19336918.2014.970001
155. Chiu JJ, Lee PL, Chen CN, Lee CI, Chang SF, Chen LJ, et al. Shear stress increases ICAM-1 and decreases VCAM-1 and E-selectin expressions induced by tumor necrosis factor-[alpha] in endothelial cells. Arterioscler Thromb Vasc Biol (2004) 24(1):73–9. doi:10.1161/01.ATV.0000106321.63667.24
156. Raza A, Franklin MJ, Dudek AZ. Pericytes and vessel maturation during tumor angiogenesis and metastasis. Am J Hematol (2010) 85(8):593–8. doi:10.1002/ajh.21745
157. Jain RK, Booth MF. What brings pericytes to tumor vessels? J Clin Invest (2003) 112(8):1134–6. doi:10.1172/JCI20087
158. Bergers G, Song S, Meyer-Morse N, Bergsland E, Hanahan D. Benefits of targeting both pericytes and endothelial cells in the tumor vasculature with kinase inhibitors. J Clin Invest (2003) 111(9):1287–95. doi:10.1172/JCI17929
159. Ruan J, Luo M, Wang C, Fan L, Yang SN, Cardenas M, et al. Imatinib disrupts lymphoma angiogenesis by targeting vascular pericytes. Blood (2013) 121(26):5192–202. doi:10.1182/blood-2013-03-490763
160. Meng M, Zaorsky NG, Deng L, Wang HH, Chao J, Zhao L, et al. Pericytes: a double-edged sword in cancer therapy. Future Oncol (2015) 11(1):169–79. doi:10.2217/fon.14.123
161. Augustin HG, Koh GY, Thurston G, Alitalo K. Control of vascular morphogenesis and homeostasis through the angiopoietin-Tie system. Nat Rev Mol Cell Biol (2009) 10(3):165–77. doi:10.1038/nrm2639
162. Keskin D, Kim J, Cooke VG, Wu CC, Sugimoto H, Gu C, et al. Targeting vascular pericytes in hypoxic tumors increases lung metastasis via angiopoietin-2. Cell Rep (2015) 10(7):1066–81. doi:10.1016/j.celrep.2015.01.035
163. Coleman RL, Broaddus RR, Bodurka DC, Wolf JK, Burke TW, Kavanagh JJ, et al. Phase II trial of imatinib mesylate in patients with recurrent platinum- and taxane-resistant epithelial ovarian and primary peritoneal cancers. Gynecol Oncol (2006) 101(1):126–31. doi:10.1016/j.ygyno.2005.09.041
164. Raymond E, Brandes AA, Dittrich C, Fumoleau P, Coudert B, Clement PM, et al. Phase II study of imatinib in patients with recurrent gliomas of various histologies: a European organisation for research and treatment of cancer brain tumor group study. J Clin Oncol (2008) 26(28):4659–65. doi:10.1200/JCO.2008.16.9235
165. Hong J, Tobin NP, Rundqvist H, Li T, Lavergne M, Garcia-Ibanez Y, et al. Role of tumor pericytes in the recruitment of myeloid-derived suppressor cells. J Natl Cancer Inst (2015) 107(10):djv209. doi:10.1093/jnci/djv209
166. Hamzah J, Jugold M, Kiessling F, Rigby P, Manzur M, Marti HH, et al. Vascular normalization in Rgs5-deficient tumours promotes immune destruction. Nature (2008) 453(7193):410–4. doi:10.1038/nature06868
167. Manzur M, Hamzah J, Ganss R. Modulation of G protein signaling normalizes tumor vessels. Cancer Res (2009) 69(2):396–9. doi:10.1158/0008-5472.CAN-08-2842
168. Manzur M, Hamzah J, Ganss R. Modulation of the “blood-tumor” barrier improves immunotherapy. Cell Cycle (2008) 7(16):2452–5. doi:10.4161/cc.7.16.6451
169. Jain RK, Martin JD, Stylianopoulos T. The role of mechanical forces in tumor growth and therapy. Annu Rev Biomed Eng (2014) 16:321–46. doi:10.1146/annurev-bioeng-071813-105259
170. Senger DR, Perruzzi CA, Feder J, Dvorak HF. A highly conserved vascular permeability factor secreted by a variety of human and rodent tumor cell lines. Cancer Res (1986) 46:5629–32.
171. Yeo K-T, Wang HH, Nagy JA, Sioussat TM, Ledbetter SR, Hoogewerf AJ, et al. Vascular permeability factor (vascular endothelial growth factor) in guinea pig and human tumor and inflammatory effusions. Cancer Res (1993) 53:2912–8.
172. Zhang L, Conejo-Garcia JR, Katsaros D, Gimotty PA, Massobrio M, Regnani G, et al. Intratumoural T cells, recurrence and survival in epithelial ovarian cancer. N Engl J Med (2003) 348:203–13. doi:10.1056/NEJMoa020177
173. Ager A, May MJ. Understanding high endothelial venules: lessons for cancer immunology. Oncoimmunology (2015) 4(6):e1008791. doi:10.1080/2162402X.2015.1008791
174. Martinet L, Garrido I, Filleron T, Le Guellec S, Bellard E, Fournie JJ, et al. Human solid tumors contain high endothelial venules: association with T- and B-lymphocyte infiltration and favorable prognosis in breast cancer. Cancer Res (2011) 71(17):5678–87. doi:10.1158/0008-5472.CAN-11-0431
175. Dieu-Nosjean MC, Goc J, Giraldo NA, Sautes-Fridman C, Fridman WH. Tertiary lymphoid structures in cancer and beyond. Trends Immunol (2014) 35(11):571–80. doi:10.1016/j.it.2014.09.006
176. Thompson ED, Enriquez HL, Fu YX, Engelhard VH. Tumor masses support naive T cell infiltration, activation and differentiation into effectors. J Exp Med (2010) 207:1791–804. doi:10.1084/jem.20092454
177. Peske JD, Thompson ED, Gemta L, Baylis RA, Fu YX, Engelhard VH. Effector lymphocyte-induced lymph node-like vasculature enables naive T-cell entry into tumours and enhanced anti-tumour immunity. Nat Commun (2015) 6:7114. doi:10.1038/ncomms8114
178. Shields JD, Kourtis IC, Tomei AA, Roberts JM, Swartz MA. Induction of lymphoidlike stroma and immune escape by tumours that express the chemokine CCL21. Science (2010) 328:749–52. doi:10.1126/science.1185837
179. Stacker SA, Williams SP, Karnezis T, Shayan R, Fox SB, Achen MG. Lymphangiogenesis and lymphatic vessel remodelling in cancer. Nat Rev Cancer (2014) 14(3):159–72. doi:10.1038/nrc3677
180. Heldin CH, Rubin K, Pietras K, Ostman A. High interstitial fluid pressure – an obstacle in cancer therapy. Nat Rev Cancer (2004) 4(10):806–13. doi:10.1038/nrc1456
181. Swartz MA, Lund AW. Lymphatic and interstitial flow in the tumour microenvironment: linking mechanobiology with immunity. Nat Rev Cancer (2012) 12(3):210–9. doi:10.1038/nrc3186
182. Ng CP, Hinz B, Swartz MA. Insterstitial fluid flow induces myofibroblast differentiation and collagen alignment in vitro. J Cell Sci (2005) 118:4731–9. doi:10.1242/jcs
183. Flavell RA, Sanjabi S, Wrzesinski SH, Licona-Limon P. The polarization of immune cells in the tumour environment by TGFbeta. Nat Rev Immunol (2010) 10(8):554–67. doi:10.1038/nri2808
184. Lund AW, Duraes FV, Hirosue S, Raghavan VR, Nembrini C, Thomas SN, et al. VEGF-C promotes immune tolerance in B16 melanomas and cross-presentation of tumor antigen by lymph node lymphatics. Cell Rep (2012) 1(3):191–9. doi:10.1016/j.celrep.2012.01.005
185. Friedlaender MH, Baer H. Immunologic tolerance: role of the regional lymph node. Science (1972) 176:312–4. doi:10.1126/science.176.4032.312
186. Card CM, Yu SS, Swartz MA. Emerging roles of lymphatic endothelium in regulating adaptive immunity. J Clin Invest (2014) 124(3):943–52. doi:10.1172/JCI73316
187. Liao S, von der Weid PY. Lymphatic system: an active pathway for immune protection. Semin Cell Dev Biol (2015) 38:83–9. doi:10.1016/j.semcdb.2014.11.012
188. Cohen JN, Guidi CJ, Tewalt EF, Qiao H, Rouhani SJ, Ruddell A, et al. Lymph node-resident lymphatic endothelial cells mediate peripheral tolerance via Aire-independent direct antigen presentation. J Exp Med (2010) 207(4):681–8. doi:10.1084/jem.20092465
189. Fletcher AL, Lukacs-Kornek V, Reynoso ED, Pinner SE, Bellemare-Pelletier A, Curry MS, et al. Lymph node fibroblastic reticular cells directly present peripheral tissue antigen under steady-state and inflammatory conditions. J Exp Med (2010) 207(4):689–97. doi:10.1084/jem.20092642
190. Tewalt EF, Cohen JN, Rouhani SJ, Guidi CJ, Qiao H, Fahl SP, et al. Lymphatic endothelial cells induce tolerance via PD-L1 and lack of costimulation leading to high-level PD-1 expression on CD8 T cells. Blood (2012) 120(24):4772–82. doi:10.1182/blood-2012-04427013
191. Rouhani SJ, Eccles JD, Riccardi P, Peske JD, Tewalt EF, Cohen JN, et al. Roles of lymphatic endothelial cells expressing peripheral tissue antigens in CD4 T-cell tolerance induction. Nat Commun (2015) 6:6771. doi:10.1038/ncomms7771
192. Lukacs-Kornek V, Malhotra D, Fletcher AL, Acton SE, Elpek KG, Tayalia P, et al. Regulated release of nitric oxide by nonhematopoietic stroma controls expansion of the activated T cell pool in lymph nodes. Nat Immunol (2011) 12(11):1096–104. doi:10.1038/ni.2112
193. Podgrabinska S, Kamalu O, Mayer L, Shimaoka M, Snoeck H, Randolph GJ, et al. Inflamed lymphatic endothelium suppresses dendritic cell maturation and function via Mac-1/ICAM-1-dependent mechanism. J Immunol (2009) 183(3):1767–79. doi:10.4049/jimmunol.0802167
194. Norder M, Gutierrez MG, Zicari S, Cervi E, Caruso A, Guzman CA. Lymph node-derived lymphatic endothelial cells express functional costimulatory molecules and impair dendritic cell-induced allogenic T-cell proliferation. FASEB J (2012) 26(7):2835–46. doi:10.1096/fj.12-205278
195. Chin AR, Wang SE. Cancer tills the premetastatic field: mechanistic basis and clinical implications. Clin Cancer Res (2016) 22(15):1–9. doi:10.1158/1078-0432.CCR-16-0028
196. Pereira ER, Jones D, Jung K, Padera TP. The lymph node microenvironment and its role in the progression of metastatic cancer. Semin Cell Dev Biol (2015) 38:98–105. doi:10.1016/j.semcdb.2015.01.008
197. Hirakawa S, Kodama S, Kunstfeld R, Kajiya K, Brown LF, Detmar M. VEGF-A induces tumor and sentinel lymph node lymphangiogenesis and promotes lymphatic metastasis. J Exp Med (2005) 201(7):1089–99. doi:10.1084/jem.20041896
198. Hirakawa S, Brown LF, Kodama S, Paavonen K, Alitalo K, Detmar M. VEGF-C-induced lymphangiogenesis in sentinel lymph nodes promotes tumor metastasis to distant sites. Blood (2007) 109:1010–7. doi:10.1182/blood-200605-021758
199. Farnsworth RH, Karnezis T, Shayan R, Matsumoto M, Nowell CJ, Achen MG, et al. A role for bone morphogenetic protein-4 in lymph node vascular remodeling and primary tumor growth. Cancer Res (2011) 71(20):6547–57. doi:10.1158/0008-5472.CAN-11-0200
200. Qian CN, Berghuis B, Tsarfaty G, Bruch M, Kort EJ, Ditlev J, et al. Preparing the “soil”: the primary tumor induces vasculature reorganization in the sentinel lymph node before the arrival of metastatic cancer cells. Cancer Res (2006) 66(21):10365–76. doi:10.1158/0008-5472.CAN-06-2977
201. Carriere V, Colisson R, Jiguet-Jiglaire C, Bellard E, Bouche G, Al Saati T, et al. Cancer cells regulate lymphocyte recruitment and leukocyte-endothelium interactions in the tumor-draining lymph node. Cancer Res (2005) 65(24):11639–48. doi:10.1158/0008-5472.CAN-05-1190
202. Girard JP, Moussion C, Forster R. HEVs, lymphatics and homeostatic immune cell trafficking in lymph nodes. Nat Rev Immunol (2012) 12(11):762–73. doi:10.1038/nri3298
203. Zippelius A, Batard P, Rubio-Godoy V, Bioley G, Lienard D, Lejeune F, et al. Effector function of human tumor-specific CD8 T cells in melanoma lesions: a state of local functional tolerance. Cancer Res (2004) 64:2865–73. doi:10.1158/0008-5472.CAN-03-3066
204. Preynat-Seauve O, Contassot E, Schuler P, Piguet V, French LE, Huard B. Extralymphatic tumors prepare draining lymph nodes to invasion via a T-cell cross-tolerance process. Cancer Res (2007) 67(10):5009–16. doi:10.1158/0008-5472.CAN-06-4494
205. Matsumoto M, Roufail S, Inder R, Caesar C, Karnezis T, Shayan R, et al. Signaling for lymphangiogenesis via VEGFR-3 is required for the early events of metastasis. Clin Exp Metastasis (2013) 30(6):819–32. doi:10.1007/s10585-013-9581-x
206. Shayan R, Inder R, Karnezis T, Caesar C, Paavonen K, Ashton MW, et al. Tumor location and nature of lymphatic vessels are key determinants of cancer metastasis. Clin Exp Metastasis (2013) 30(3):345–56. doi:10.1007/s10585-012-9541-x
207. Mlecnik B, Bindea G, Kirilovsky A, Angell HK, Obenauf AC, Tosolini M, et al. The tumor microenvironment and Immunoscore are critical determinants of dissemination to distal metastasis. Sci Transl Med (2016) 8(327):327ra26. doi:10.1126/scitranslmed.aad6352
208. Lund AW, Wagner M, Fankhauser M, Steinskog ES, Broggi MA, Spranger S, et al. Lymphatic vessels regulate immune microenvironments in human and murine melanoma. J Clin Invest (2016) 126(9):3389–402. doi:10.1172/JCI79434
209. Carman CV, Martinelli R. T lymphocyte-endothelial interactions: emerging understanding of trafficking and antigen-specific immunity. Front Immunol (2015) 6:603. doi:10.3389/fimmu.2015.00603
210. Epperson DE, Pober JS. Antigen-presenting function of human endothelial cells. Direct activation of resting CD8 T cells. J Immunol (1994) 153:5402–12.
211. Rodig N, Ryan T, Allen JA, Pang H, Grabie N, Chernova T, et al. Endothelial expression of PD-L1 and PD-L2 down-regulates CD8+ T cell activation and cytolysis. Eur J Immunol (2003) 33(11):3117–26. doi:10.1002/eji.200324270
212. Huang X, Bai X, Cao Y, Wu J, Huang M, Tang D, et al. Lymphoma endothelium preferentially expresses Tim-3 and facilitates the progression of lymphoma by mediating immune evasion. J Exp Med (2010) 207(3):505–20. doi:10.1084/jem.20090397
213. Riesenberg R, Weiler C, Spring O, Eder M, Buchner A, Popp T, et al. Expression of indoleamine 2,3-dioxygenase in tumor endothelial cells correlates with long-term survival of patients with renal cell carcinoma. Clin Cancer Res (2007) 13(23):6993–7002. doi:10.1158/1078-0432.CCR-07-0942
214. Zang X, Allison JP. The B7 family and cancer therapy: costimulation and coinhibition. Clin Cancer Res (2007) 13(18 Pt 1):5271–9. doi:10.1158/1078-0432.CCR-07-1030
215. Sica GL, Choi I, Zhu G, Tamada K, Wang S, Tamura H, et al. B7-H4, a molecule of the B7 family, negatively regulates T cell immunity. Immunity (2003) 18:849–61. doi:10.1016/S1074-7613(03)00152-3
216. Suh WK, Gajewska BU, Okada H, Gronski MA, Bertram EM, Dawicki W, et al. The B7 family member B7-H3 preferentially down-regulates T helper type 1-mediated immune responses. Nat Immunol (2003) 4(9):899–906. doi:10.1038/ni967
217. Zang X, Sullivan PS, Soslow RA, Waitz R, Reuter VE, Wilton A, et al. Tumor associated endothelial expression of B7-H3 predicts survival in ovarian carcinomas. Mod Pathol (2010) 23(8):1104–12. doi:10.1038/modpathol.2010.95
218. Brunner A, Hinterholzer S, Riss P, Heinze G, Brustmann H. Immunoexpression of B7-H3 in endometrial cancer: relation to tumor T-cell infiltration and prognosis. Gynecol Oncol (2012) 124(1):105–11. doi:10.1016/j.ygyno.2011.09.012
219. Brustmann H, Igaz M, Eder C, Brunner A. Epithelial and tumor-associated endothelial expression of B7-H3 in cervical carcinoma: relation with CD8+ intraepithelial lymphocytes, FIGO stage, and phosphohistone H3 (PHH3) reactivity. Int J Gynecol Pathol (2015) 34(2):187–95. doi:10.1097/PGP.0000000000000116
220. Crispen PL, Sheinin Y, Roth TJ, Lohse CM, Kuntz SM, Frigola X, et al. Tumor cell and tumor vasculature expression of B7-H3 predict survival in clear cell renal cell carcinoma. Clin Cancer Res (2008) 14(16):5150–7. doi:10.1158/1078-0432.CCR-08-0536
221. Krambeck AE, Thompson RH, Dong H, Lohse CM, Park ES, Kuntz SM, et al. B7-H4 expression in renal cell carcinoma and tumor vasculature: associations with cancer progression and survival. Proc Natl Acad Sci U S A (2006) 103(27):10391–6. doi:10.1073/pnas.0600937103
222. He YC, Halford MM, Achen MG, Stacker SA. Exploring the role of endothelium in the tumour response to anti-angiogenic therapy. Biochem Soc Trans (2014) 42(6):1569–75. doi:10.1042/BST20140173
223. Fontanella C, Ongaro E, Bolzonello S, Guardascione M, Fasola G, Aprile G. Clinical advances in the development of novel VEGFR2 inhibitors. Ann Transl Med (2014) 2(12):123. doi:10.3978/j.issn.2305-5839.2014.08.14
224. Solomon B, Binns D, Roselt P, Weibe LI, McArthur GA, Cullinane C, et al. Modulation of intratumoral hypoxia by the epidermal growth factor receptor inhibitor gefitinib detected using small animal PET imaging. Mol Cancer Ther (2005) 4(9):1417–22. doi:10.1158/1535-7163.MCT-05-0066
225. Solomon B, Hagekyriakou J, Trivett MK, Stacker SA, McArthur GA, Cullinane C. EGFR blockade with ZD1839 (“Iressa”) potentiates the antitumor effects of single and multiple fractions of ionizing radiation in human A431 squamous cell carcinoma. Int J Radiat Oncol Biol Phys (2003) 55(3):713–23. doi:10.1016/S0360-3016(02)04357-2
226. Jayson GC, Kerbel R, Ellis LM, Harris AL. Antiangiogenic therapy in oncology: current status and future directions. Lancet (2016) 388:518–29. doi:10.1016/S0140-6736(15)01088-0
227. Batchelor TT, Gerstner ER, Emblem KE, Duda DG, Kalpathy-Cramer J, Snuderl M, et al. Improved tumor oxygenation and survival in glioblastoma patients who show increased blood perfusion after cediranib and chemoradiation. Proc Natl Acad Sci U S A (2013) 110(47):19059–64. doi:10.1073/pnas.1318022110
228. Hodi FS, Mihm MC, Soiffer RJ, Haluska FG, Butler M, Seiden MV, et al. Biologic activity of cytotoxic T lymphocyte-associated antigen 4 antibody blockade in previously vaccinated metastatic melanoma and ovarian carcinoma patients. Proc Natl Acad Sci U S A (2003) 100(8):4712–7. doi:10.1073/pnas.0830997100
229. Yuan J, Zhou J, Dong Z, Tandon S, Kuk D, Panageas KS, et al. Pretreatment serum VEGF is associated with clinical response and overall survival in advanced melanoma patients treated with ipilimumab. Cancer Immunol Res (2014) 2(2):127–32. doi:10.1158/2326-6066.CIR-13-0163
230. Wu X, Giobbie-Hurder A, Liao X, Lawrence D, McDermott D, Zhou J, et al. VEGF neutralization plus CTLA-4 blockade alters soluble and cellular factors associated with enhancing lymphocyte infiltration and humoral recognition in melanoma. Cancer Immunol Res (2016) 4(10):858–68. doi:10.1158/2326-6066.CIR-16-0084
231. Achen MG, Mann GB, Stacker SA. Targeting lymphangiogenesis to prevent tumour metastasis. Br J Cancer (2006) 94(10):1355–60. doi:10.1038/sj.bjc.6603120
232. Rinderknecht M, Villa A, Ballmer-Hofer K, Neri D, Detmar M. Phage-derived fully human monoclonal antibody fragments to human vascular endothelial growth factor-C block its interaction with VEGF receptor-2 and 3. PLoS One (2010) 5(8):e11941. doi:10.1371/journal.pone.0011941
233. Achen MG, Roufail S, Domagala T, Catimel B, Nice EC, Geleick DM, et al. Monoclonal antibodies to vascular endothelial growth factor-D block its interactions with both VEGF receptor-2 and VEGF-receptor 3. Eur J Biochem (2000) 267:2505–15. doi:10.1046/j.1432-1327.2000.01257.x
234. Davydova N, Roufail S, Streltsov VA, Stacker SA, Achen MG. The VD1 neutralizing antibody to vascular endothelial growth factor-D: binding epitope and relationship to receptor binding. J Mol Biol (2011) 407(4):581–93. doi:10.1016/j.jmb.2011.02.009
235. Persaud K, Tille J, Liu M, Zhu Z, Jimenez X, Pereira DS, et al. Involvement of the VEGF receptor 3 in tubular morphogenesis demonstrated with a human anti-human VEGR-3 monoclonal antibody that antagonises receptor activation by VEGF-C. J Cell Sci (2004) 117:2745–56. doi:10.1242/jcs
236. Albuquerque RJ, Hayashi T, Cho WG, Kleinman ME, Dridi S, Takeda A, et al. Alternatively spliced vascular endothelial growth factor receptor-2 is an essential endogenous inhibitor of lymphatic vessel growth. Nat Med (2009) 15(9):1023–30. doi:10.1038/nm.2018
237. Makinen T, Jussila L, Veikkola T, Karpanen T, Kettunen MI, Pulkkanen KJ, et al. Inhibition of lymphangiogenesis with resulting lymphedema in transgenic mice expressing soluble VEGF receptor-3. Nat Med (2001) 7:199–205. doi:10.1038/84651
238. Kodera Y, Katanasaka Y, Kitamura Y, Tsuda H, Nishio K, Tamura T, et al. Sunitinib inhibits lymphatic endothelial cell functions and lymph node metastasis in a breast cancer model through inhibition of vascular endothelial growth factor receptor 3. Breast Cancer Res (2011) 13:R66. doi:10.1186/bcr2903
Keywords: endothelial cells, lymphatic endothelial cells, angiogenesis inhibitors, tumor immune evasion, immunotherapy
Citation: Hendry SA, Farnsworth RH, Solomon B, Achen MG, Stacker SA and Fox SB (2016) The Role of the Tumor Vasculature in the Host Immune Response: Implications for Therapeutic Strategies Targeting the Tumor Microenvironment. Front. Immunol. 7:621. doi: 10.3389/fimmu.2016.00621
Received: 17 October 2016; Accepted: 07 December 2016;
Published: 20 December 2016
Edited by:
Silvia Della Bella, University of Milan, ItalyReviewed by:
Samuel Bertin, University of California San Diego, USATimothy Padera, Massachusetts General Hospital, USA
Copyright: © 2016 Hendry, Farnsworth, Solomon, Achen, Stacker and Fox. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Stephen B. Fox, c3RlcGhlbi5mb3hAcGV0ZXJtYWMub3Jn