
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 29 November 2016
Sec. Multiple Sclerosis and Neuroimmunology
Volume 7 - 2016 | https://doi.org/10.3389/fimmu.2016.00544
This article is part of the Research Topic Neuroimmune interface in health and diseases View all 15 articles
 Eric S. Wohleb1,2*
Eric S. Wohleb1,2*
Persistent cognitive and behavioral symptoms that characterize many mental health disorders arise from impaired neuroplasticity in several key corticolimbic brain regions. Recent evidence suggests that reciprocal neuron–microglia interactions shape neuroplasticity during physiological conditions, implicating microglia in the neurobiology of mental health disorders. Neuron–microglia interactions are modulated by several molecular and cellular pathways, and dysregulation of these pathways often have neurobiological consequences, including aberrant neuronal responses and microglia activation. Impaired neuron-microglia interactions are implicated in mental health disorders because rodent stress models lead to concomitant neuronal dystrophy and alterations in microglia morphology and function. In this context, functional changes in microglia may be indicative of an immune state termed parainflammation in which tissue-resident macrophages (i.e., microglia) respond to malfunctioning cells by initiating modest inflammation in an attempt to restore homeostasis. Thus, aberrant neuronal activity and release of damage-associated signals during repeated stress exposure may contribute to functional changes in microglia and resultant parainflammation. Furthermore, accumulating evidence shows that uncoupling neuron–microglia interactions may contribute to altered neuroplasticity and associated anxiety- or depressive-like behaviors. Additional work shows that microglia have varied phenotypes in specific brain regions, which may underlie divergent neuroplasticity observed in corticolimbic structures following stress exposure. These findings indicate that neuron–microglia interactions are critical mediators of the interface between adaptive, homeostatic neuronal function and the neurobiology of mental health disorders.
Mental health disorders, such as anxiety and depression, are a major source of disability leading to significant social and economic burden throughout the world (1–3). While progress has been made in understanding the neurobiology of mental health disorders, it is clear that multiple subtypes exist with varied pathophysiological mechanisms (4). Clinical and preclinical studies show that anxiety- and depressive-like symptoms are linked to altered neuroplasticity in corticolimbic brain regions. In particular, divergent responses are reported; neuronal atrophy and synapse loss in the prefrontal cortex and hippocampus, and neuronal hypertrophy and increased synaptic density in the amygdala and nucleus accumbens (5). Moreover, clinical studies show that many anxiety and depressive symptoms develop or worsen following exposure to psychosocial and environmental stress (6–8). This is pertinent as rodent models of stress can provide insight into the mechanisms that contribute to the neurobiology of anxiety- and depressive-like behaviors. Indeed several stress models, including repeated social defeat and chronic unpredictable stress, recapitulate key neuroplasticity changes that contribute to susceptibility and development of anxiety- and depressive-like behaviors (9–11).
Coinciding with altered neuroplasticity, repeated stress exposure leads to dysregulation of neuroimmune systems that are implicated in mental health disorders (12–15). Microglia are tissue-resident macrophages in the brain that integrate stress-induced neuroimmune signals leading to behavioral consequences (16). Following stress exposure, microglia undergo dynamic alterations in morphology and function within corticolimbic brain regions implicated in anxiety- and depressive-like symptoms (17). In line with these findings, recent studies demonstrate that microglia have an integral role in shaping neuronal responses (i.e., activity) and synaptic elements (i.e., dendrites and dendritic spines) (18), which support adaptive behavior and cognition (19). Thus, stress-associated changes in neuron–microglia interactions may play an integral role in the pathophysiology of mental health disorders. Clinical studies using histological analyses suggest that microglia have altered morphology and function in depressed individuals (20, 21). Moreover, a clinical neuroimaging study showed that individuals experiencing a major depressive episode have enhanced positron emission topography (PET) labeling of the translocator protein (TSPO), a putative marker of neuroinflammation and microglia activation (22). Further studies using microarray gene expression analyses demonstrate increased cytokine and complement pathways in the PFC and hippocampus of postmortem samples obtained from depressed individuals (23, 24). In contrast, clinical reports examining cerebrospinal fluid showed no changes in immune-related markers in depressed patients (25, 26), suggesting that neuroimmune dysregulation may represent a pathophysiological mechanism in a subset of depressed patients. Notably, clinical data showed that individuals with atypical depression had higher levels of inflammatory markers in circulation compared to controls and those with melancholic depression (27). Altogether, clinical studies and preclinical models provide evidence that neuroimmune alterations provoked by stressors may contribute to the neurobiology of mental health disorders in a subset of individuals.
Concomitant neuronal dystrophy and microglia activation implicate reciprocal neuron-microglia interactions in behavioral deficits, and these responses may not directly lead to neuroinflammation per se. As researchers demonstrate the ever-changing form and functions of microglia, it is clear that morphological characteristics may reflect several phenotypes. Thus, microglia appear to display specialized responses that are brain region-dependent and dictated by the stress model and duration. These brain region-specific microglia phenotypes may play a role in divergent neuroplasticity observed in corticolimbic brain regions following stress exposure (28). It is also important to note that stress-induced neuroimmune alterations are modest compared to other pathological situations (29). These neuroimmune processes mediated by microglia do not generally lead to neurotoxicity but may contribute to neuronal dystrophy following stress. In this context, stress-associated functional changes in microglia may be indicative of an immune response similar to parainflammation (30), rather than “neuroinflammation.” This notion of stress-induced parainflammation in the brain will be discussed further in this review. Here, literature are presented showing that neuron–microglia interactions have an integral role in promoting homeostasis in the brain as well as how perturbations in neuron–microglia interactions lead to impaired neuroplasticity. In particular, this review will focus on evidence suggesting that repeated stress exposure leads to dysregulated neuron–microglia interactions and neuroplasticity deficits with implications for mental health and neurological disorders.
The mammalian central nervous system develops circuits of interconnected neurons that underlie complex functions, including cognition and behavior (31–33). Several cell types residing in the brain contribute to the development and maintenance of this neurocircuitry. Important cellular counterpart of neurons are microglia, which are brain-resident macrophages that direct homeostatic functions and mediate immune responses to pathological conditions (13, 34). Microglia are distributed throughout the brain but appear to have varied roles in specific regions and develop unique features based on tissue-specific molecular signals (28, 35, 36). Recent studies reveal that microglia are maintained in the brain through self-renewal (37, 38), suggesting that these long-lived brain-resident macrophages maintain long-term interactions with proximal neurons. Neuron–microglia interactions are mediated by soluble factors as well as contact-dependent mechanisms (39, 40), and reciprocal communication is necessary for adaptive neuroplasticity and behavior. These features place microglia as a critical mediator of neuronal function for better, and for worse (Figure 1).
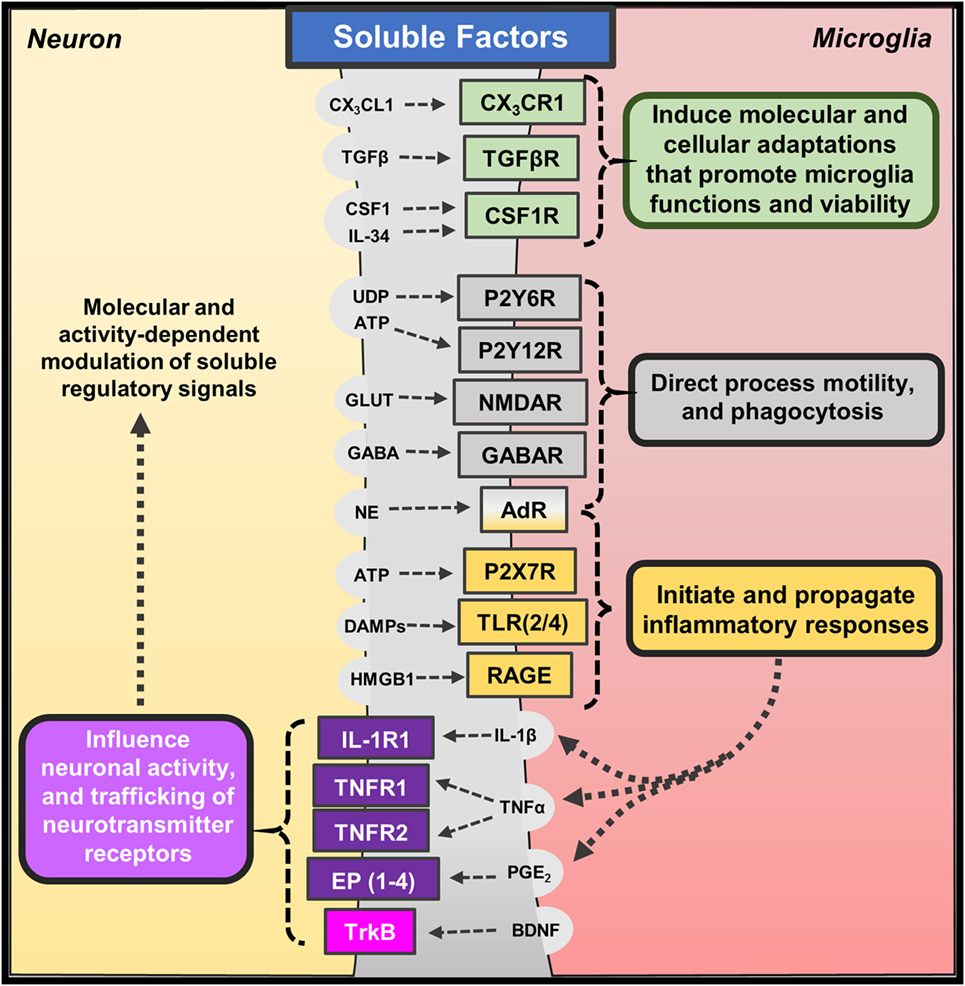
Figure 1. Soluble factors regulating neuron–microglia interactions. Several molecular and cellular pathways mediate neuron–microglia interactions. Neurons release soluble immune-related factors (fractalkine – CX3CL1; transforming growth factor-β – TGFβ; colony-stimulating factor-1 – CSF1; interleukin-34 – IL-34), nucleotides (uridine diphosphate – UDP; adenosine triphosphate – ATP), neurotransmitters (glutamate – GLUT; γ-aminobutyric acid – GABA; norepinephrine – NE), and danger- or damage-associated molecules (DAMPs; high mobility group box 1 – HMGB1) that bind to cognate receptors on microglia. These soluble neuron-derived signals promote many homeostatic functions but can also initiate or propagate neuroinflammation depending on the context. Microglia also release soluble factors, such as cytokines (interleukin-1β – IL-1β; tumor necrosis factor-α – TNFα), prostaglandins (prostaglandin E2 – PGE2), and neurotrophins (brain-derived neurotrophic factor – BDNF), which bind to neuronal receptors. Microglia often release these factors in response to fluctuations in neuron-derived signals; yet, low levels of microglia-derived cytokines are shown to promote homeostatic neuroplasticity through trafficking of neurotransmitter receptors.
Neurons regulate microglia function through soluble factors, including chemokines, cytokines, and neurotransmitters (Figure 1). Neuron-derived fractalkine (CX3CL1) regulates microglia activation through binding to CX3CR1, which is enriched on microglia (41). CX3CL1 is expressed by neurons in two forms (membrane-bound or cleaved soluble proteins) that may transduce different molecular signals (42). The CX3CL1–CX3CR1 pathway is shown to be critical for proper neurodevelopment, and the absence of this pathway significantly impaired brain connectivity leading to social interaction deficits (43–45). In addition, recent work shows that microglia develop a unique phenotype based on soluble molecular cues likely derived from proximal neurons. For instance, transforming growth factor (TGF)-β promotes a unique transcriptional profile that organizes transcriptional pathways within microglia (35, 36). Other studies show that colony-stimulating factor (CSF)-1 and interleukin (IL)-34 signal microglia through CSF1 receptor to regulate their development and viability (46). Indeed, microglia do not develop in mice lacking CSF1 receptor (47), and recent work shows pharmacological blockade of CSF1 receptor caused depletion of microglia in the brain (37). These studies highlight some of the soluble neuron-derived signals that shape microglia morphology and function.
The distinct, surveying phenotype of microglia allows them to rapidly respond to perturbations in their microenvironment along with deviations in neuronal homeostasis and activity (48). These responses are mediated, in part, by neuronal release of nucleotides (i.e., UDP, ATP) (49) and neurotransmitters [i.e., glutamate, GABA, and norepinephrine (NE)] (19). Indeed, acute glutamate uncaging induced microglia chemotaxis and convergence of microglia processes toward sites of increased neuronal activity (50). Other studies show that microglia morphology and process motility is influenced by extracellular glutamate, GABA, and NE (51). The chemotactic properties of ATP appear to be predominantly mediated by microglia expression of P2Y12 receptor. For instance, microglia lacking P2Y12 receptors display similar baseline surveillance of regional microenvironments; however, their motility is impaired during injury responses when nucleotides are released (52). Similar reports suggest that fluctuations in calcium signaling along with ATP signaling through P2Y12 receptors lead to microglia process convergence as well (53). Interestingly, a recent study indicated that microglia expression of P2Y12 receptors is required for proper development of visual cortex and ocular dominance (54). Other purinergic receptors, such as P2Y6, may play a functional role in these responses as UDP signaling through P2Y6 receptors increased microglia-mediated phagocytosis following hippocampal excitotoxicity (55). These findings indicate that nucleotides released following neuronal activity act as attractants for microglia processes, and dysregulated neuroplasticity is observed in the absence of these interactions.
Microglia release soluble factors, including cytokines and prostaglandins, which reciprocally influence and modulate neuronal function (Figure 1). There are significant primary research reports and reviews that describe how cytokines and other immune mediators released by microglia influence neuronal function and neuroplasticity. Indeed, low levels of IL-1β are required for long-term potentiation, while basal levels of TNFα are necessary for proper homeostatic trafficking of AMPA and GABAA receptors, termed synaptic scaling (56, 57) [see also reviews, Ref. (58, 59)]. Based on these studies, it is evident that microglia-mediated cytokine and prostaglandin synthesis can modulate neuronal responses during physiological and pathological conditions. Further studies indicate that microglia-derived cytokines can indirectly affect neurons through gliotransmission mediated by astrocytes (60). For instance, the release of TNFα by activated microglia potentiated astrocyte glutamate release, which can modulate synaptic plasticity and even lead to neurotoxicity (61). In addition, ATP released by microglia is shown to induce glutamate release by astrocytes thereby acutely exciting proximal neurons (62). These indirect signaling pathways may be further augmented during inflammatory conditions. For example, microglia activation and TNFα release increased neuronal hyperactivity and susceptibility to develop seizures (63). Other data show that microglia can support adaptive synaptic plasticity through the release of neurotrophic factors, such as brain-derived neurotrophic factors (BDNF) (64). These findings are not entirely surprising because microglia as the tissue-resident macrophages of the brain mediate pathological processes along with reparative or growth responses (65).
Recent studies also reveal that microglia direct neuronal function through contact-dependent mechanisms, including engulfment of synaptic and dendritic elements (18, 19) (Figure 2). Seminal studies showed that microglia actively phagocytose synapses during neurodevelopment (66). Synapse elimination during neurodevelopment is mediated by complement factor 3 and 1b, which bind synapses with diminished activity, initiating microglia-mediated phagocytosis via complement receptor 3 (CR3; CD11b) (67). Other formative studies showed that microglia perform regular, but brief “synapse sampling” by contacting synapses in the adult brain. Of note, in vivo imaging showed that microglia sampled proximal synapses once per hour and were drawn more frequently to active synapses. Moreover, these studies show that prolonged microglia interactions resulted in synapse loss (68). Other studies show similar activity-dependent microglia-mediated synapse elimination occurs in cortical brain regions (69). In these studies, soluble factors that modulate neuron–microglia interactions may play a prominent role as purinergic signaling along with release of neurotransmitters that rapidly draw microglia processes toward elevated neuronal activity. For instance, glutamate uncaging caused attraction of microglia processes, which subsequently surrounded hyperactive neurons, leading to contact-dependent reductions in neuronal activity (50). While it is clear that microglia can physically remove synapses, it remains to be determined what mechanisms contribute to microglia-mediated reductions in neuronal activity (Figure 2). These findings demonstrate that microglia are directed by soluble neuron-derived cues to initiate contact-dependent regulation of neuronal activity.
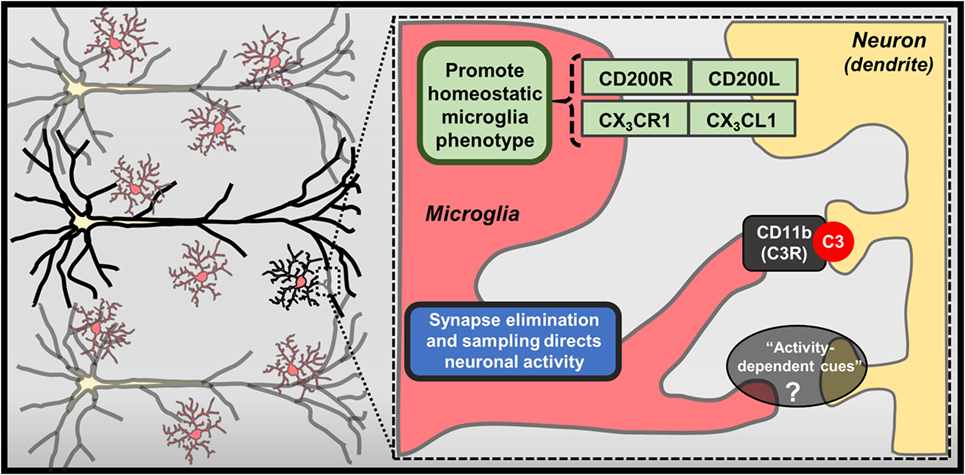
Figure 2. Contact-dependent mechanisms mediating neuron–microglia interactions. Physical interactions between neurons and microglia exist, including membrane-bound CX3CL1 and CD200L, which bind CX3CR1 and CD200R on microglia, respectively. Further recent studies have highlighted specialized mechanisms that contribute to contact-dependent synaptic modulation, such as binding of complement component C3 to synapses and eventual removal of these “tagged” synapses by microglia through CD11b/CR3-mediated phagocytosis. Microglia can also envelop hyperactive neurons and regularly perform “synapse sampling” via activity-dependent mechanisms that are not entirely defined (?).
To further examine the functional role of microglia in various physiological and pathological conditions, several groups have developed methods to deplete microglia (70). Initial studies provide compelling evidence that microglia are necessary for adaptive neuroplasticity and behavior. For instance, mice treated with clodronate liposomes showed robust microglia depletion in the hippocampus, which led to spatial memory decrements as well as reduced sociability. These cognitive and social deficits were recapitulated with widespread microglia depletion using the CSF1 receptor antagonist PLX3397. Of note, cognitive and behavioral consequences of microglia depletion were attenuated following repopulation (71). Other studies revealed that clodronate depletion of microglia resulted in enhanced synapses and excitatory input on hippocampal neurons (72). These neurobiological effects have functional implications as pharmacogenetic microglia depletion caused impairments in the rotarod motor learning task (64). These deficits in motor learning were recapitulated when BDNF expression was selectively deleted from microglia. Despite these findings, other studies indicate that widespread microglia depletion with PLX3397 caused no significant alterations in cognition or behavior (37). The dynamic function of microglia in these depletion studies likely reflects their compartmentalized brain region-specific functions (28, 73, 74). These unique neuron–microglia interactions highlight the complexity of molecular and cellular pathways that regulate neurobiology and behavior.
In the end, reciprocal neuron–microglia interactions are regulated by soluble and contact-dependent pathways. These pathways enable microglia to obtain feedback on neuronal functions and rapidly enact interventions to maintain tissue homeostasis. These neuron–microglia interactions appear to support neuronal homeostasis because perturbations in these pathways often result in neuroplasticity impairments and influence performance in memory-based tasks (Figures 1 and 2). In this context, neuron–microglia interactions may be disrupted during pathological conditions, such as mental health and neurological diseases. Further studies will be reviewed to provide evidence that neuron–microglia interactions may play a critical role in the neurobiology of mental health disorders.
Exposure to psychosocial and environmental stress is shown to cause robust neuronal activation (i.e., cFos, FosB) through the release of glutamate and NE in corticolimbic brain regions, such as the prefrontal cortex, amygdala, and hippocampus (75–78). Further perceived perturbations of homeostasis caused by stress lead to neuroendocrine activation and release of glucocorticoids (GC) into circulation (79). Converging lines of evidence indicate that aberrant neuronal activation coupled with elevated GC levels lead to neuronal dystrophy in corticolimbic brain regions following stress (Figure 3). For instance, repeated stress caused dendritic atrophy and synapse loss on pyramidal neurons in the rat prefrontal cortex (80, 81), and these effects were recapitulated with chronic GC administration (82). Stress-induced atrophy of pyramidal neurons may be related to impaired glutamate receptor (i.e., NMDA, AMPA) expression in the prefrontal cortex, which is dependent on GC receptor activation (83). Recent work also suggests that dysregulation of interneurons may contribute to disrupted microcircuitry in the prefrontal cortex and depressive-like behavior (84, 85). Together, these neurobiological alterations contribute to shifted excitatory–inhibitory tone in the prefrontal cortex, which is proposed to underlie anxiety and depressive symptoms (9, 86, 87) (Figure 3). Further, brain regions that receive PFC projections, including amygdala, hypothalamus, hippocampus, and nucleus accumbens, exhibit disrupted function as well (88, 89).
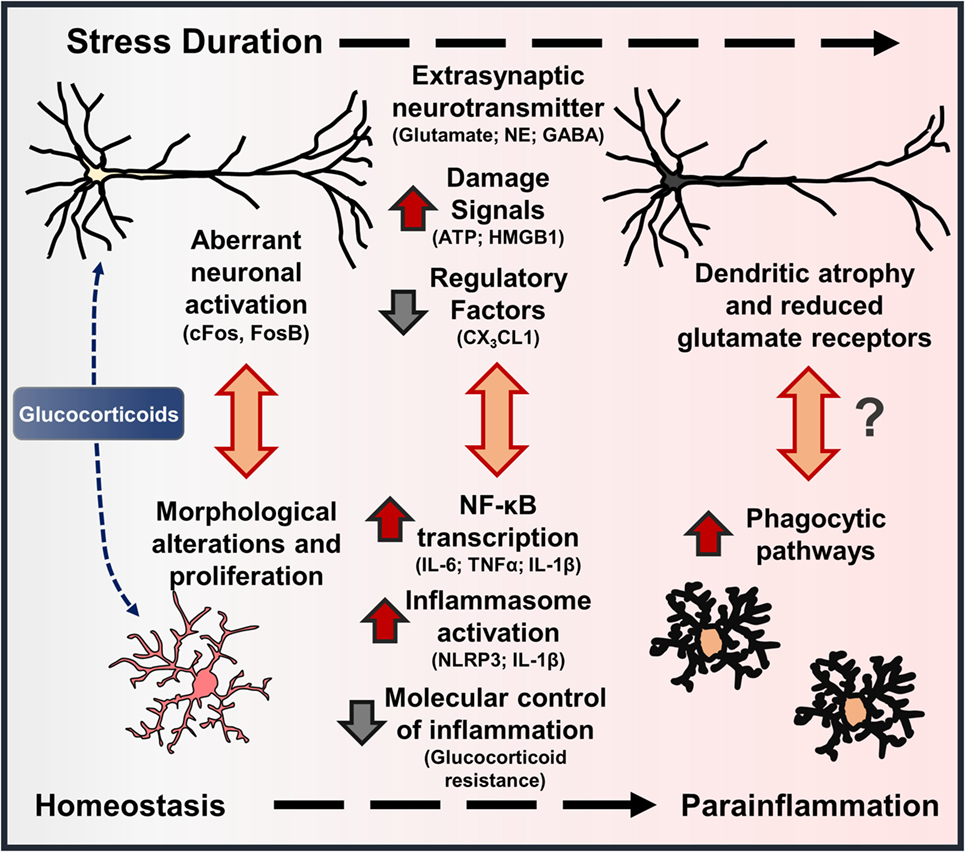
Figure 3. Stress-induced neuronal dystrophy contributes to alterations in microglia function and parainflammation. Models of environmental and psychosocial stress activate characteristic neuroendocrine (i.e., glucocorticoids – GC) and neuroimmune pathways that contribute to neuroplasticity alterations underlying anxiety- and depressive-like behaviors. In this cascade, there are features that show stress-induced “neuroinflammation” resembles parainflammation. As brain-resident macrophages, microglia interact with proximal neurons, and disruptions in neuron–microglia interactions initiate morphological and functional changes in microglia. Repeated stress (i.e., prolonged elevations in glucocorticoids) leads to aberrant neuronal activation, extrasynaptic neurotransmitter levels, and release of neuron-derived damage signals. These signals further promote mild to moderate levels of neuroinflammatory transcription and inflammasome activation in microglia. With persistent or chronic stress, pyramidal neurons undergo dendritic atrophy and reduce glutamate receptors, which may be precipitated by cytokines released by proximal microglia or microglia-mediated synapse elimination. Collectively, these stress-induced neuronal responses and resultant molecular and cellular cascades resemble a state of parainflammation.
Corresponding with altered neuroplasticity in these corticolimbic regions, several reports demonstrate changes in microglia morphology in overlapping brain regions following stress (Figure 3). Indeed, morphological changes in microglia are observed in the prefrontal cortex, amygdala, hypothalamus, hippocampus, and nucleus accumbens following stress exposure (16, 17, 90). Several neuronal and endocrine pathways are shown to cause microglia activation in rodent models of stress. For instance, early work showed that noradrenergic pathways in the brain contribute to elevations in stress-induced pro-inflammatory cytokine expression (91, 92). Further work showed that blockade of β-adrenergic receptors with propranolol prevented microglia activation and pro-inflammatory cytokine gene expression in enriched microglia following repeated social defeat (93). Using inescapable footshock stress, Frank et al. showed that RU486 can prevent stress-induced microglia priming in the hippocampus, suggesting that GCs contribute to these neuroimmune responses as well (94–96). Similar studies using repeated restraint stress showed that pharmacological blockade of NMDA receptors or GC receptors can attenuate stress-associated microglia proliferation, providing evidence that microglia responses are caused by aberrant neuronal activation and neuroendocrine responses (97). In contrast to these findings, recent work by Yirmiya and colleagues showed that chronic stress exposure caused microglia to undergo atrophy in the dentate gyrus of the hippocampus, and these deficits were associated with depressive-like behaviors (98). Further peripheral administration of agents that stimulate microglia function (i.e., LPS, CSF1) reversed microglia atrophy and provided antidepressant effects (98). These compelling findings suggest that brain region-specific signals lead to microglia dysfunction, which can have deleterious effects on neurobiology and behavior as well. Collectively, these studies indicate that dynamic, brain region-specific functional changes in microglia are driven by stress-induced neuroendocrine and neurotransmitter pathways.
Stress-induced microglia activation may also stem from increased release of danger-associated molecular patterns (DAMPs) that bind pattern recognition receptors (PRR) to promote neuroinflammatory signaling through activation of the NLRP3 inflammasome (99). For instance, stress-induced release of the DAMP, high mobility box group (HMGB)-1, caused microglia priming through inflammasome activation and increased pro-inflammatory gene expression (100). In other studies, repeated stress is shown to increase tissue IL-1β levels and depressive-like behaviors; these neuroimmune and behavioral responses are prevented in mice lacking the NLRP3 inflammasome (101, 102). This is consistent with recent work that showed during immobilization stress there is influx of extracellular glutamate and ATP in the hippocampus, followed by elevations in IL-1β and TNFα. Of note, increased release of IL-1β and TNFα is blocked by administration of P2X7 receptor antagonist. In addition, follow-up studies showed that stress-induced depressive-like behaviors were blocked by the P2X7 receptor antagonist and in mice lacking the NLRP3 inflammasome (103). Other reports suggest that increased NLRP3 activation and IL-1β levels in the prefrontal cortex are mediated by microglia following chronic stress, and these responses are reversed with chronic fluoxetine treatment (104). These studies support previous work showing that chronic stress promoted elevations in IL-1β signaling, which contributed to reduced hippocampal neurogenesis and depressive-like behavior (105, 106). Another recent study showed that restraint stress increased TNFα levels in the hippocampus with delayed elevations of several inflammatory mediators in the amygdala, but not in the prefrontal cortex (107). As noted, neuron-derived CX3CL1 acts as integral regulator of microglia function, and deficient signaling can result in neuroplasticity deficits (45). Thus, stress-induced reductions in CX3CL1 may contribute further to microglia activation and anxiety- or depressive-like behaviors (108). These studies provide evidence that fluctuations in neuron-derived signals (i.e., ATP, HMGB1, and CX3CL1) act to regulate microglia activation and subsequent production of pro-inflammatory cytokines with repeated stress exposure, which will influence neuronal function and behavior.
In line with these findings, recent studies show that anxiolytic and antidepressant treatments can block or reverse stress-induced microglia activation. For instance, administration of imipramine following repeated social defeat reduced IL-6 expression in enriched microglia and attenuated depressive-like behaviors following (109). This is consistent with other studies showing selective serotonin reuptake inhibitors produce anti-inflammatory responses in microglia (110). Further work using repeated social defeat demonstrated that pretreatment with benzodiazepines reduced markers of neuroinflammation and diminished anxiety-like behavior (109). Similarly, pretreatment with the non-selective β-adrenergic receptor antagonist propranolol prevented microglia activation and blocked the development of anxiety-like behavior following repeated social defeat (93). These findings raise the question whether these interventions that targeted neurotransmitter systems subsequently attenuated neuroimmune activation or if microglia were the primary effector. In the end, these results suggest that pharmacological treatments may produce anxiolytic or antidepressant effects by simultaneously normalizing neurotransmission and modulating microglia functions. Furthermore, these findings highlight the need to determine cell type-specific pathways that lead to neuronal dystrophy or microglia activation, respectively. As an example, stress-induced microglia activation can be prevented by GC blockade; however, this may be an effect of limiting neuronal dystrophy and diminished release of damage-associated factors or direct effects on microglia. Dissociating the molecular and cellular pathways that contribute to neuronal and microglia adaptations following repeated stress exposure may show novel pharmacological or molecular therapeutic targets.
Based on the overlap of stress-induced neuronal dystrophy and functional changes in microglia, it is evident that stress-induced “neuroinflammation” may reflect an inflammatory state termed parainflammation (30). The notion of parainflammation is related to the condition originally described as “physiological inflammation” by Élie Metchnikoff (111). Indeed, parainflammation is an immune state induced by “stressed or malfunctioning” tissues and is considered to be an intermediate phase between homeostasis and classical inflammation. Moreover, parainflammation appears to be mediated predominantly by tissue-resident macrophages, such as microglia. Thus, microglia activation following stress exposure and reported in depression may represent a state of parainflammation, in part, as a response to neuronal dystrophy. In support of this idea, it is evident that stress-induced microglia production of pro-inflammatory cytokines is modest compared to other pathological conditions (29). Moreover, aberrant neuronal activation and damage signals released during stress exposure appear to provoke microglia activation (99), which contributes to altered neuroplasticity and anxiety- or depressive-like behaviors. Stress duration and intensity likely influence the scale of parainflammation, and these prolonged impairments in neuron–microglia interactions may lead to irreversible neurobiological consequences and mental health disorders (Figure 2).
It is worthwhile to note that parainflammation is not considered an entirely detrimental state because it is provoked by malfunctioning cells, and the objective is to restore tissue homeostasis (112). There is evidence that microglia-mediated activation or “neuroinflammation” in non-pathological conditions may initiate adaptive functions to restore neuronal activity to basal, physiological levels. For instance, repeated peripheral endotoxin challenge caused microglia processes to interfere with inhibitory interneuron synaptic connections on pyramidal neurons in the cortex, which provided neuroprotection through increased neuronal activity (113). In separate studies microglia were shown to play a neuroprotective role during excitotoxic injury, with microglia depletion leading to enhanced susceptibility to neuronal death (114). Consistent with these studies, recent work showed that microglia processes contacted swollen axonal segments during excitotoxic conditions, which normalized the excitability of affected neurons and preserved their viability (115). In these instances, neuron–microglia interactions are critical to maintain homeostasis in the brain but likely lead to undesirable inflammatory consequences. It is critical to point out that in many pathological conditions inflammatory responses are required to limit neuronal death and promote tissue repair processes (65, 116). In this context, stress-induced parainflammation and alterations in microglia function may be aimed to restore neuronal homeostasis. It is possible that these processes are initially protective but may generate pathological consequences with chronic stress exposure. Further studies will need to be performed to establish the dynamic role of microglia in these neurobiological responses.
This is not a novel proposal as recent reviews have proposed that a subset of depressed individuals develop a chronic state of parainflammation that contributes to the pathophysiology underlying their symptoms (4, 117). Others have suggested similar models in which stress exposure causes neuronal microdamage, and neuroinflammatory responses are initiated by microglia to promote repair (118). Depending on the neurocircuitry affected, it is argued that anxiety- or depressive-like symptoms may develop, suggesting that neuroinflammatory pathways may lead to divergent neurobiological changes (118). The neuronal microdamage model is compelling; however, further studies will need to be performed to determine molecular and cellular mediators of these effects. The specific molecular and cellular mediators of stress-induced parainflammation are particularly relevant in the context of potential therapies for mental health disorders. For instance, some common antidepressant drugs, such as selective serotonin reuptake inhibitors, have reported anti-inflammatory effects (119). In other cases, the antidepressant behavioral effects of serotonin reuptake inhibitors were blocked by anti-inflammatory drugs (120). These data demonstrate that dynamic neuron–microglia interactions modulate behavior and suggest that parainflammation may enact microglia-mediated mechanisms that normalize neuronal function. In either case, further studies will need to examine the neuron–microglia interactions that contribute to stress-induced parainflammation, and how interventions can engage these mechanisms to provide therapeutic benefits.
Another important mediator of neuroimmune functions are peripheral immune cells, which propagate immune signals in the brain and influence behavior. Indeed, studies indicate that specific behavioral issues, such as pathological grooming, can be attributed to peripheral hematopoietic immune cells (121). In the context of stress, recent evidence demonstrates a role of peripheral myeloid-derived cells (monocytes and granulocytes) in “neuroinflammation” and anxiety- or depressive-like behavior (14, 90). In these studies, social defeat increased monocyte recruitment to the brain through canonical chemokine pathways (CCL2–CCR2 and CX3CL1–CX3CR1) (108). Moreover, peripheral monocytes may be preferentially recruited to specific brain regions through adhesion molecule expression (122). In the context of neuroinflammation, it is plausible that this is an attempt to restore homeostasis as neuronal stress-associated signals reach upper limits of parainflammation (112). Initial studies show that stress caused peripheral monocytes to infiltrate the brain under defined conditions; however, further work revealed that peripheral monocytes do not significantly contribute to brain-resident microglia populations (123, 124). Further, pharmacological or genetic techniques to limit monocyte trafficking in the brain and neuroinflammation prevent social defeat-induced anxiety-like behaviors (14, 90). Together, these studies revealed that peripheral monocytes reinforce neuroinflammatory processes and highlight a novel neuroimmune axis that promotes mood disturbances (90, 125). Further studies will need to be performed using microglia- or monocyte-specific genetic techniques or depletion to distinguish cell type-specific contributions to reported neuroimmune mechanisms and their influence on neuronal responses.
Microglia are implicated in the neurobiology of several mental health disorders; however, it is unclear how they contribute to divergent neuroplasticity alterations in corticolimbic brain regions. It is plausible that brain region-specific neuron–microglia interactions contribute to divergent neuroplasticity reported. For instance, work by Hinwood and colleagues showed that the putative microglia inhibitor minocycline attenuated alterations in microglia morphology and reduced FosB activation in the medial prefrontal cortex, and prevented working memory deficits following repeated stress exposure (126). These results suggest that microglia activation may modulate neuronal responses and cognitive deficits in a rodent stress model. While there is limited clinical evidence showing microglia modulation of neurons, a recent study showed that elevated C-reactive protein levels in circulation, indicative of low grade peripheral inflammation, is associated with increased glutamate levels in the basal ganglia of depressed patients (127). As noted, microglia may modulate neurobiological and behavioral consequences of psychological stress through the release of pro-inflammatory cytokines (i.e., IL-1β, TNFα, and IL-6) (13, 14, 16). Indeed pro-inflammatory cytokines released by microglia following repeated stress exposure may produce brain region-specific alterations in synaptic plasticity. For instance, TNFα administration on pyramidal neurons derived from the hippocampus increased AMPA receptor trafficking to postsynaptic sites and concomitant reductions in GABAA receptors that caused increased excitability (57, 128). In contrast, TNFα administration on striatal brain slices caused reduced AMPA receptor levels on medium spiny neurons (129). Further TNFα released from microglia led to reduced excitability of medium spiny neurons in the striatum after cocaine administration (130). In these studies, the actions of TNFα are mediated by TNF receptor 1 expressed on neurons, thus varied neurophysiological effects may be dependent on neuron subtype-specific molecular signaling (128). Separate studies showed repeated social defeat-induced microglia-mediated prostaglandin release that attenuated neuronal responses in the ventral tegmental area (VTA). The reduced firing of VTA neurons following repeated social defeat increased social avoidance, indicating that microglia-mediated modulation of this mesolimbic neuronal pathway contributed to the development of depressive-like behavior (131). These findings provide compelling evidence that microglia produce soluble factors eliciting brain region-specific neuronal responses that elicit cognitive and behavioral consequences.
Other studies show that contact-dependent neuron–microglia interactions are critical modulators of neuronal and behavioral responses to stress. In particular, recent work showed that microglia-mediated elimination of synaptic elements may contribute to stress-induced synaptic plasticity deficits in the hippocampus. For instance, 14 days of chronic unpredictable stress increased the presence of dendritic and synaptic elements in the processes of microglia in the CA1 of the hippocampus. The increased presence of neuronal elements in microglia was associated with reduced sucrose preference and impaired long-term potentiation. Moreover, mice lacking CX3CR1 were resilient to CUS, which corresponded with reduced microglia phagocytosis of neuronal elements, attenuated LTP deficits, and normalized sucrose preference (132). It is important to reiterate that mice lacking CX3CR1 have delayed neurodevelopment and display baseline social interaction deficits (43, 44), which may contribute to observed stress resilience. It is unclear if microglia expression of CX3CR1 is necessary for stress-induced activation, but other studies provide evidence that neuronal CX3CL1 activated microglia, which led to the modulation of synaptic strength. For instance, CX3CL1 binding to CX3CR1 on microglia caused increased IL-1β release and downstream molecular mechanisms that altered synaptic plasticity (133). Further it is important to consider that microglia lacking CX3CR1 may be diverted away from homeostatic functions, such as clearance of neural progenitors in the hippocampus, which may exacerbate stress-induced neuropathology (134, 135). These studies demonstrate that microglia can shape neuronal responses to stress through contact-dependent mechanisms, contributing directly to the development of depressive-like behaviors.
The mechanisms that govern microglia-mediated synapse interactions during stress exposure have not been extensively studied; however, as noted microglia are drawn to synapses in an activity-dependent manner. This is pertinent as repeated stress exposure is known to increase neuronal activity in several corticolimbic brain regions, and these neuronal networks are dysregulated in mental health disorders (87, 89). In this context, stress-associated neuronal hyperactivity in specific brain regions may elicit microglia-mediated disruption of synaptic connections with unintended consequences. For instance, microglia disruption of inhibitory synapses on pyramidal neurons may lead to aberrant neuronal activity and extrasynaptic glutamate neurotransmission (136). The potential microglia-mediated exclusion of inhibitory synapses may have consequences on the integrity of interneurons as well. Indeed, recent work indicates that cortical interneurons may be more susceptible to stress-induced dystrophy (85). It is unclear if interneurons are susceptible to stress-induced parainflammation, but deep sequencing techniques in neuron subtype-specific populations may lend insight (137). Further studies will need to expound on these findings to determine how microglia facilitate or disrupt neuroplasticity in stress-responsive brain regions.
In all, microglia have dynamic brain region-specific functions that may contribute to divergent neuroplasticity effects underlying stress-induced anxiety- and depressive-like behavior (5, 13). As the roles of microglia expand, it will be important to characterize brain region-specific phenotypes (28). These studies will undoubtedly reveal unique microglia properties that can be targeted to support adaptive neuronal responses and mental health.
Neurons and microglia utilize bidirectional interactions to shape form and function of both cell types. In models of stress-induced mental health disorders, concomitant alterations in neuroplasticity and microglia function are reported, reflecting disruptions in neuron–microglia interactions. Based on these characteristics stress-induced microglia activation resembles an immune state termed parainflammation, which is aimed to restore neuronal homeostasis. It is possible that stress exposure elicits modest inflammatory responses that divert microglia from their supportive functions, leading to neuroplasticity deficits underlying anxiety- and depressive-like behaviors. Stress-induced microglia dysregulation may also contribute to neurological complications as clinical evidence shows that individuals with prior history of mental health disorders have increased risk for dementia or neurodegenerative disease (138, 139). Thus, impaired neuron–microglia interactions may link psychological stress exposure and mental health disorders with aging and neurodegenerative disease (15, 140). This is plausible as synapse loss is a common pathophysiological feature observed in depressed individuals and early stages of neurodegenerative diseases (141, 142). Further studies should be conducted to determine if impaired neuron–microglia interactions contribute to the link between, psychological stress, mental health, and neurodegenerative disease. In addition, recent work indicates that sex-dependent differences in microglia function exist and these likely have implications for the pathophysiology of mental health and cognitive disorders as well (143–145). In the end, pharmacological or molecular pathways that engage or promote the adaptive and neurotrophic functions of microglia may provide therapeutic benefits for mental health and neurological disorders.
ESW wrote and edited this manuscript.
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author would like to thank Dr. Ronald S. Duman and the Department of Psychiatry at the Yale University School of Medicine for their mentorship and support.
The author was supported through NIH grants (MH045481 and MH093897) to Ronald S. Duman and by departmental support from the University of Cincinnati.
1. Kessler RC, Chiu WT, Demler O, Merikangas KR, Walters EE. Prevalence, severity, and comorbidity of 12-month DSM-IV disorders in the National Comorbidity Survey Replication. Arch Gen Psychiatry (2005) 62(6):617–27. doi:10.1001/archpsyc.62.6.617
2. Kessler RC. Epidemiology of women and depression. J Affect Disord (2003) 74(1):5–13. doi:10.1016/S0165-0327(02)00426-3
3. Murray CJ, Atkinson C, Bhalla K, Birbeck G, Burstein R, Chou D, et al. The state of US health, 1990-2010: burden of diseases, injuries, and risk factors. JAMA (2013) 310(6):591–608. doi:10.1001/jama.2013.13805
4. Gold PW. The organization of the stress system and its dysregulation in depressive illness. Mol Psychiatry (2015) 20(1):32–47. doi:10.1038/mp.2014.163
5. Christoffel DJ, Golden SA, Russo SJ. Structural and synaptic plasticity in stress-related disorders. Rev Neurosci (2011) 22(5):535–49. doi:10.1515/RNS.2011.044
6. Kendler KS, Karkowski LM, Prescott CA. Causal relationship between stressful life events and the onset of major depression. Am J Psychiatry (1999) 156(6):837–41. doi:10.1176/ajp.156.6.837
7. Gilman SE, Trinh NH, Smoller JW, Fava M, Murphy JM, Breslau J. Psychosocial stressors and the prognosis of major depression: a test of Axis IV. Psychol Med (2013) 43(2):303–16. doi:10.1017/S0033291712001080
8. McLaughlin KA, Conron KJ, Koenen KC, Gilman SE. Childhood adversity, adult stressful life events, and risk of past-year psychiatric disorder: a test of the stress sensitization hypothesis in a population-based sample of adults. Psychol Med (2010) 40(10):1647–58. doi:10.1017/S0033291709992121
9. Duman RS, Aghajanian GK, Sanacora G, Krystal JH. Synaptic plasticity and depression: new insights from stress and rapid-acting antidepressants. Nat Med (2016) 22(3):238–49. doi:10.1038/nm.4050
10. Nestler EJ, Hyman SE. Animal models of neuropsychiatric disorders. Nat Neurosci (2010) 13(10):1161–9. doi:10.1038/nn.2647
11. Krishnan V, Nestler EJ. The molecular neurobiology of depression. Nature (2008) 455(7215):894–902. doi:10.1038/nature07455
12. Beumer W, Gibney SM, Drexhage RC, Pont-Lezica L, Doorduin J, Klein HC, et al. The immune theory of psychiatric diseases: a key role for activated microglia and circulating monocytes. J Leukoc Biol (2012) 92(5):959–75. doi:10.1189/jlb.0212100
13. Wohleb ES, Franklin T, Iwata M, Duman RS. Integrating neuroimmune systems in the neurobiology of depression. Nat Rev Neurosci (2016) 17:497–511. doi:10.1038/nrn.2016.69
14. Hodes GE, Kana V, Menard C, Merad M, Russo SJ. Neuroimmune mechanisms of depression. Nat Neurosci (2015) 18(10):1386–93. doi:10.1038/nn.4113
15. Miller AH, Raison CL. The role of inflammation in depression: from evolutionary imperative to modern treatment target. Nat Rev Immunol (2015) 16(1):22–34. doi:10.1038/nri.2015.5
16. Yirmiya R, Rimmerman N, Reshef R. Depression as a microglial disease. Trends Neurosci (2015) 38(10):637–58. doi:10.1016/j.tins.2015.08.001
17. Walker FR, Nilsson M, Jones K. Acute and chronic stress-induced disturbances of microglial plasticity, phenotype and function. Curr Drug Targets (2013) 14(11):1262–76. doi:10.2174/13894501113149990208
18. Tremblay ME, Stevens B, Sierra A, Wake H, Bessis A, Nimmerjahn A. The role of microglia in the healthy brain. J Neurosci (2011) 31(45):16064–9. doi:10.1523/JNEUROSCI.4158-11.2011
19. Wu Y, Dissing-Olesen L, MacVicar BA, Stevens B. Microglia: dynamic mediators of synapse development and plasticity. Trends Immunol (2015) 36(10):605–13. doi:10.1016/j.it.2015.08.008
20. Torres-Platas SG, Cruceanu C, Chen GG, Turecki G, Mechawar N. Evidence for increased microglial priming and macrophage recruitment in the dorsal anterior cingulate white matter of depressed suicides. Brain Behav Immun (2014) 42:50–9. doi:10.1016/j.bbi.2014.05.007
21. Steiner J, Bielau H, Brisch R, Danos P, Ullrich O, Mawrin C, et al. Immunological aspects in the neurobiology of suicide: elevated microglial density in schizophrenia and depression is associated with suicide. J Psychiatr Res (2008) 42(2):151–7. doi:10.1016/j.jpsychires.2006.10.013
22. Setiawan E, Wilson AA, Mizrahi R, Rusjan PM, Miler L, Rajkowska G, et al. Role of translocator protein density, a marker of neuroinflammation, in the brain during major depressive episodes. JAMA Psychiatry (2015) 72(3):268–75. doi:10.1001/jamapsychiatry.2014.2427
23. Shelton RC, Claiborne J, Sidoryk-Wegrzynowicz M, Reddy R, Aschner M, Lewis DA, et al. Altered expression of genes involved in inflammation and apoptosis in frontal cortex in major depression. Mol Psychiatry (2011) 16(7):751–62. doi:10.1038/mp.2010.52
24. Kim S, Hwang Y, Webster MJ, Lee D. Differential activation of immune/inflammatory response-related co-expression modules in the hippocampus across the major psychiatric disorders. Mol Psychiatry (2016) 21(3):376–85. doi:10.1038/mp.2015.79
25. Martinez JM, Garakani A, Yehuda R, Gorman JM. Proinflammatory and “resiliency” proteins in the CSF of patients with major depression. Depress Anxiety (2012) 29(1):32–8. doi:10.1002/da.20876
26. Carpenter LL, Heninger GR, Malison RT, Tyrka AR, Price LH. Cerebrospinal fluid interleukin (IL)-6 in unipolar major depression. J Affect Disord (2004) 79(1–3):285–9. doi:10.1016/S0165-0327(02)00460-3
27. Lamers F, Vogelzangs N, Merikangas KR, de Jonge P, Beekman AT, Penninx BW. Evidence for a differential role of HPA-axis function, inflammation and metabolic syndrome in melancholic versus atypical depression. Mol Psychiatry (2013) 18(6):692–9. doi:10.1038/mp.2012.144
28. Grabert K, Michoel T, Karavolos MH, Clohisey S, Baillie JK, Stevens MP, et al. Microglial brain region-dependent diversity and selective regional sensitivities to aging. Nat Neurosci (2016) 19(3):504–16. doi:10.1038/nn.4222
29. DiSabato D, Quan N, Godbout JP. Neuroinflammation: the devil is in the details. J Neurochem (2016) 139:136–53. doi:10.1111/jnc.13607
30. Medzhitov R. Origin and physiological roles of inflammation. Nature (2008) 454(7203):428–35. doi:10.1038/nature07201
31. Hariri AR, Holmes A. Finding translation in stress research. Nat Neurosci (2015) 18(10):1347–52. doi:10.1038/nn.4111
32. Lerner TN, Ye L, Deisseroth K. Communication in neural circuits: tools, opportunities, and challenges. Cell (2016) 164(6):1136–50. doi:10.1016/j.cell.2016.02.027
33. Deisseroth K. Circuit dynamics of adaptive and maladaptive behaviour. Nature (2014) 505(7483):309–17. doi:10.1038/nature12982
34. Kettenmann H, Kirchhoff F, Verkhratsky A. Microglia: new roles for the synaptic stripper. Neuron (2013) 77(1):10–8. doi:10.1016/j.neuron.2012.12.023
35. Butovsky O, Jedrychowski MP, Moore CS, Cialic R, Lanser AJ, Gabriely G, et al. Identification of a unique TGF-beta-dependent molecular and functional signature in microglia. Nat Neurosci (2014) 17(1):131–43. doi:10.1038/nn.3599
36. Gosselin D, Link VM, Romanoski CE, Fonseca GJ, Eichenfield DZ, Spann NJ, et al. Environment drives selection and function of enhancers controlling tissue-specific macrophage identities. Cell (2014) 159(6):1327–40. doi:10.1016/j.cell.2014.11.023
37. Elmore MR, Najafi AR, Koike MA, Dagher NN, Spangenberg EE, Rice RA, et al. Colony-stimulating factor 1 receptor signaling is necessary for microglia viability, unmasking a microglia progenitor cell in the adult brain. Neuron (2014) 82(2):380–97. doi:10.1016/j.neuron.2014.02.040
38. Bruttger J, Karram K, Wortge S, Regen T, Marini F, Hoppmann N, et al. Genetic cell ablation reveals clusters of local self-renewing microglia in the mammalian central nervous system. Immunity (2015) 43(1):92–106. doi:10.1016/j.immuni.2015.06.012
39. Kierdorf K, Prinz M. Factors regulating microglia activation. Front Cell Neurosci (2013) 7:44. doi:10.3389/fncel.2013.00044
40. Pocock JM, Kettenmann H. Neurotransmitter receptors on microglia. Trends Neurosci (2007) 30(10):527–35. doi:10.1016/j.tins.2007.07.007
41. Cardona AE, Pioro EP, Sasse ME, Kostenko V, Cardona SM, Dijkstra IM, et al. Control of microglial neurotoxicity by the fractalkine receptor. Nat Neurosci (2006) 9(7):917–24. doi:10.1038/nn1715
42. Morganti JM, Nash KR, Grimmig BA, Ranjit S, Small B, Bickford PC, et al. The soluble isoform of CX3CL1 is necessary for neuroprotection in a mouse model of Parkinson’s disease. J Neurosci (2012) 32(42):14592–601. doi:10.1523/JNEUROSCI.0539-12.2012
43. Zhan Y, Paolicelli RC, Sforazzini F, Weinhard L, Bolasco G, Pagani F, et al. Deficient neuron-microglia signaling results in impaired functional brain connectivity and social behavior. Nat Neurosci (2014) 17(3):400–6. doi:10.1038/nn.3641
44. Paolicelli RC, Bolasco G, Pagani F, Maggi L, Scianni M, Panzanelli P, et al. Synaptic pruning by microglia is necessary for normal brain development. Science (2011) 333(6048):1456–8. doi:10.1126/science.1202529
45. Paolicelli RC, Bisht K, Tremblay ME. Fractalkine regulation of microglial physiology and consequences on the brain and behavior. Front Cell Neurosci (2014) 8:129. doi:10.3389/fncel.2014.00129
46. Wang Y, Szretter KJ, Vermi W, Gilfillan S, Rossini C, Cella M, et al. IL-34 is a tissue-restricted ligand of CSF1R required for the development of Langerhans cells and microglia. Nat Immunol (2012) 13(8):753–60. doi:10.1038/ni.2360
47. Ginhoux F, Greter M, Leboeuf M, Nandi S, See P, Gokhan S, et al. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science (2010) 330(6005):841–5. doi:10.1126/science.1194637
48. Nimmerjahn A, Kirchhoff F, Helmchen F. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science (2005) 308(5726):1314–8. doi:10.1126/science.1110647
49. Dissing-Olesen L, LeDue JM, Rungta RL, Hefendehl JK, Choi HB, MacVicar BA. Activation of neuronal NMDA receptors triggers transient ATP-mediated microglial process outgrowth. J Neurosci (2014) 34(32):10511–27. doi:10.1523/JNEUROSCI.0405-14.2014
50. Li Y, Du XF, Liu CS, Wen ZL, Du JL. Reciprocal regulation between resting microglial dynamics and neuronal activity in vivo. Dev Cell (2012) 23(6):1189–202. doi:10.1016/j.devcel.2012.10.027
51. Gyoneva S, Traynelis SF. Norepinephrine modulates the motility of resting and activated microglia via different adrenergic receptors. J Biol Chem (2013) 288(21):15291–302. doi:10.1074/jbc.M113.458901
52. Haynes SE, Hollopeter G, Yang G, Kurpius D, Dailey ME, Gan WB, et al. The P2Y12 receptor regulates microglial activation by extracellular nucleotides. Nat Neurosci (2006) 9(12):1512–9. doi:10.1038/nn1805
53. Eyo UB, Gu N, De S, Dong H, Richardson JR, Wu LJ. Modulation of microglial process convergence toward neuronal dendrites by extracellular calcium. J Neurosci (2015) 35(6):2417–22. doi:10.1523/JNEUROSCI.3279-14.2015
54. Sipe GO, Lowery RL, Tremblay ME, Kelly EA, Lamantia CE, Majewska AK. Microglial P2Y12 is necessary for synaptic plasticity in mouse visual cortex. Nat Commun (2016) 7:10905. doi:10.1038/ncomms10905
55. Koizumi S, Shigemoto-Mogami Y, Nasu-Tada K, Shinozaki Y, Ohsawa K, Tsuda M, et al. UDP acting at P2Y6 receptors is a mediator of microglial phagocytosis. Nature (2007) 446(7139):1091–5. doi:10.1038/nature05704
56. Stellwagen D, Malenka RC. Synaptic scaling mediated by glial TNF-alpha. Nature (2006) 440(7087):1054–9. doi:10.1038/nature04671
57. Stellwagen D, Beattie EC, Seo JY, Malenka RC. Differential regulation of AMPA receptor and GABA receptor trafficking by tumor necrosis factor-alpha. J Neurosci (2005) 25(12):3219–28. doi:10.1523/JNEUROSCI.4486-04.2005
58. Delpech JC, Madore C, Nadjar A, Joffre C, Wohleb ES, Laye S. Microglia in neuronal plasticity: influence of stress. Neuropharmacology (2015) 96(Pt A):19–28. doi:10.1016/j.neuropharm.2014.12.034
59. Yirmiya R, Goshen I. Immune modulation of learning, memory, neural plasticity and neurogenesis. Brain Behav Immun (2011) 25(2):181–213. doi:10.1016/j.bbi.2010.10.015
60. Santello M, Volterra A. TNFalpha in synaptic function: switching gears. Trends Neurosci (2012) 35(10):638–47. doi:10.1016/j.tins.2012.06.001
61. Bezzi P, Domercq M, Brambilla L, Galli R, Schols D, De Clercq E, et al. CXCR4-activated astrocyte glutamate release via TNFalpha: amplification by microglia triggers neurotoxicity. Nat Neurosci (2001) 4(7):702–10. doi:10.1038/89490
62. Pascual O, Ben Achour S, Rostaing P, Triller A, Bessis A. Microglia activation triggers astrocyte-mediated modulation of excitatory neurotransmission. Proc Natl Acad Sci U S A (2012) 109(4):E197–205. doi:10.1073/pnas.1111098109
63. Riazi K, Galic MA, Kuzmiski JB, Ho W, Sharkey KA, Pittman QJ. Microglial activation and TNFalpha production mediate altered CNS excitability following peripheral inflammation. Proc Natl Acad Sci U S A (2008) 105(44):17151–6. doi:10.1073/pnas.0806682105
64. Parkhurst CN, Yang G, Ninan I, Savas JN, Yates JR III, Lafaille JJ, et al. Microglia promote learning-dependent synapse formation through brain-derived neurotrophic factor. Cell (2013) 155(7):1596–609. doi:10.1016/j.cell.2013.11.030
65. Davies LC, Jenkins SJ, Allen JE, Taylor PR. Tissue-resident macrophages. Nat Immunol (2013) 14(10):986–95. doi:10.1038/ni.2705
66. Stevens B, Allen NJ, Vazquez LE, Howell GR, Christopherson KS, Nouri N, et al. The classical complement cascade mediates CNS synapse elimination. Cell (2007) 131(6):1164–78. doi:10.1016/j.cell.2007.10.036
67. Schafer DP, Lehrman EK, Kautzman AG, Koyama R, Mardinly AR, Yamasaki R, et al. Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron (2012) 74(4):691–705. doi:10.1016/j.neuron.2012.03.026
68. Wake H, Moorhouse AJ, Jinno S, Kohsaka S, Nabekura J. Resting microglia directly monitor the functional state of synapses in vivo and determine the fate of ischemic terminals. J Neurosci (2009) 29(13):3974–80. doi:10.1523/JNEUROSCI.4363-08.2009
69. Tremblay ME, Lowery RL, Majewska AK. Microglial interactions with synapses are modulated by visual experience. PLoS Biol (2010) 8(11):e1000527. doi:10.1371/journal.pbio.1000527
70. Waisman A, Ginhoux F, Greter M, Bruttger J. Homeostasis of microglia in the adult brain: review of novel microglia depletion systems. Trends Immunol (2015) 36(10):625–36. doi:10.1016/j.it.2015.08.005
71. Torres L, Danver J, Ji K, Miyauchi JT, Chen D, Anderson ME, et al. Dynamic microglial modulation of spatial learning and social behavior. Brain Behav Immun (2016) 55:6–16. doi:10.1016/j.bbi.2015.09.001
72. Ji K, Akgul G, Wollmuth LP, Tsirka SE. Microglia actively regulate the number of functional synapses. PLoS One (2013) 8(2):e56293. doi:10.1371/journal.pone.0056293
73. Baalman K, Marin MA, Ho TS, Godoy M, Cherian L, Robertson C, et al. Axon initial segment-associated microglia. J Neurosci (2015) 35(5):2283–92. doi:10.1523/JNEUROSCI.3751-14.2015
74. Lawson LJ, Perry VH, Dri P, Gordon S. Heterogeneity in the distribution and morphology of microglia in the normal adult mouse brain. Neuroscience (1990) 39(1):151–70. doi:10.1016/0306-4522(90)90229-W
75. Perrotti LI, Hadeishi Y, Ulery PG, Barrot M, Monteggia L, Duman RS, et al. Induction of deltaFosB in reward-related brain structures after chronic stress. J Neurosci (2004) 24(47):10594–602. doi:10.1523/JNEUROSCI.2542-04.2004
76. Flak JN, Solomon MB, Jankord R, Krause EG, Herman JP. Identification of chronic stress-activated regions reveals a potential recruited circuit in rat brain. Eur J Neurosci (2012) 36(4):2547–55. doi:10.1111/j.1460-9568.2012.08161.x
77. Musazzi L, Treccani G, Popoli M. Functional and structural remodeling of glutamate synapses in prefrontal and frontal cortex induced by behavioral stress. Front Psychiatry (2015) 6:60. doi:10.3389/fpsyt.2015.00060
78. Arnsten AF. Stress signalling pathways that impair prefrontal cortex structure and function. Nat Rev Neurosci (2009) 10(6):410–22. doi:10.1038/nrn2648
79. Ulrich-Lai YM, Herman JP. Neural regulation of endocrine and autonomic stress responses. Nat Rev Neurosci (2009) 10(6):397–409. doi:10.1038/nrn2647
80. Radley JJ, Sisti HM, Hao J, Rocher AB, McCall T, Hof PR, et al. Chronic behavioral stress induces apical dendritic reorganization in pyramidal neurons of the medial prefrontal cortex. Neuroscience (2004) 125(1):1–6. doi:10.1016/j.neuroscience.2004.01.006
81. Radley JJ, Rocher AB, Rodriguez A, Ehlenberger DB, Dammann M, McEwen BS, et al. Repeated stress alters dendritic spine morphology in the rat medial prefrontal cortex. J Comp Neurol (2008) 507(1):1141–50. doi:10.1002/cne.21588
82. Wellman CL. Dendritic reorganization in pyramidal neurons in medial prefrontal cortex after chronic corticosterone administration. J Neurobiol (2001) 49(3):245–53. doi:10.1002/neu.1079
83. Yuen EY, Wei J, Liu W, Zhong P, Li X, Yan Z. Repeated stress causes cognitive impairment by suppressing glutamate receptor expression and function in prefrontal cortex. Neuron (2012) 73(5):962–77. doi:10.1016/j.neuron.2011.12.033
84. Rajkowska G, O’Dwyer G, Teleki Z, Stockmeier CA, Miguel-Hidalgo JJ. GABAergic neurons immunoreactive for calcium binding proteins are reduced in the prefrontal cortex in major depression. Neuropsychopharmacology (2007) 32(2):471–82. doi:10.1038/sj.npp.1301234
85. Lin LC, Sibille E. Somatostatin, neuronal vulnerability and behavioral emotionality. Mol Psychiatry (2015) 20(3):377–87. doi:10.1038/mp.2014.184
86. Ressler KJ, Mayberg HS. Targeting abnormal neural circuits in mood and anxiety disorders: from the laboratory to the clinic. Nat Neurosci (2007) 10(9):1116–24. doi:10.1038/nn1944
87. Price JL, Drevets WC. Neural circuits underlying the pathophysiology of mood disorders. Trends Cogn Sci (2010) 16(1):61–71. doi:10.1016/j.tics.2011.12.011
88. Popoli M, Yan Z, McEwen BS, Sanacora G. The stressed synapse: the impact of stress and glucocorticoids on glutamate transmission. Nat Rev Neurosci (2012) 13(1):22–37. doi:10.1038/nrn3138
89. Ferenczi EA, Zalocusky KA, Liston C, Grosenick L, Warden MR, Amatya D, et al. Prefrontal cortical regulation of brainwide circuit dynamics and reward-related behavior. Science (2016) 351(6268):aac9698. doi:10.1126/science.aac9698
90. Wohleb ES, McKim DB, Sheridan JF, Godbout JP. Monocyte trafficking to the brain with stress and inflammation: a novel axis of immune-to-brain communication that influences mood and behavior. Front Neurosci (2015) 8:447. doi:10.3389/fnins.2014.00447
91. Blandino P Jr, Barnum CJ, Deak T. The involvement of norepinephrine and microglia in hypothalamic and splenic IL-1beta responses to stress. J Neuroimmunol (2006) 173(1–2):87–95. doi:10.1016/j.jneuroim.2005.11.021
92. Johnson JD, Campisi J, Sharkey CM, Kennedy SL, Nickerson M, Greenwood BN, et al. Catecholamines mediate stress-induced increases in peripheral and central inflammatory cytokines. Neuroscience (2005) 135(4):1295–307. doi:10.1016/j.neuroscience.2005.06.090
93. Wohleb ES, Hanke ML, Corona AW, Powell ND, Stiner LM, Bailey MT, et al. β-Adrenergic receptor antagonism prevents anxiety-like behavior and microglial reactivity induced by repeated social defeat. J Neurosci (2011) 31(17):6277–88. doi:10.1523/JNEUROSCI.0450-11.2011
94. Frank MG, Thompson BM, Watkins LR, Maier SF. Glucocorticoids mediate stress-induced priming of microglial pro-inflammatory responses. Brain Behav Immun (2012) 26(2):337–45. doi:10.1016/j.bbi.2011.10.005
95. Frank MG, Watkins LR, Maier SF. Stress- and glucocorticoid-induced priming of neuroinflammatory responses: potential mechanisms of stress-induced vulnerability to drugs of abuse. Brain Behav Immun (2011) 25(Suppl 1):S21–8. doi:10.1016/j.bbi.2011.01.005
96. Frank MG, Baratta MV, Sprunger DB, Watkins LR, Maier SF. Microglia serve as a neuroimmune substrate for stress-induced potentiation of CNS pro-inflammatory cytokine responses. Brain Behav Immun (2007) 21(1):47–59. doi:10.1016/j.bbi.2006.03.005
97. Nair A, Bonneau RH. Stress-induced elevation of glucocorticoids increases microglia proliferation through NMDA receptor activation. J Neuroimmunol (2006) 171(1–2):72–85. doi:10.1016/j.jneuroim.2005.09.012
98. Kreisel T, Frank MG, Licht T, Reshef R, Ben-Menachem-Zidon O, Baratta MV, et al. Dynamic microglial alterations underlie stress-induced depressive-like behavior and suppressed neurogenesis. Mol Psychiatry (2014) 19(6):699–709. doi:10.1038/mp.2013.155
99. Fleshner M, Frank M, Maier SF. Danger signals and inflammasomes: stress-evoked sterile inflammation in mood disorders. Neuropsychopharmacology (2016) 1–47. doi:10.1038/npp.2016.125
100. Weber MD, Frank MG, Tracey KJ, Watkins LR, Maier SF. Stress induces the danger-associated molecular pattern HMGB-1 in the hippocampus of male Sprague Dawley rats: a priming stimulus of microglia and the NLRP3 inflammasome. J Neurosci (2015) 35(1):316–24. doi:10.1523/JNEUROSCI.3561-14.2015
101. Zhang Y, Liu L, Liu YZ, Shen XL, Wu TY, Zhang T, et al. NLRP3 inflammasome mediates chronic mild stress-induced depression in mice via neuroinflammation. Int J Neuropsychopharmacol (2015) 18:1–8. doi:10.1093/ijnp/pyv006
102. Alcocer-Gomez E, de Miguel M, Casas-Barquero N, Nunez-Vasco J, Sanchez-Alcazar JA, Fernandez-Rodriguez A, et al. NLRP3 inflammasome is activated in mononuclear blood cells from patients with major depressive disorder. Brain Behav Immun (2014) 36:111–7. doi:10.1016/j.bbi.2013.10.017
103. Iwata M, Ota KT, Li XY, Sakaue F, Li N, Dutheil S, et al. Psychological stress activates the inflammasome via release of adenosine triphosphate and stimulation of the purinergic type 2X7 receptor. Biol Psychiatry (2016) 80:12–22. doi:10.1016/j.biopsych.2015.11.026
104. Pan Y, Chen XY, Zhang QY, Kong LD. Microglial NLRP3 inflammasome activation mediates IL-1beta-related inflammation in prefrontal cortex of depressive rats. Brain Behav Immun (2014) 41:90–100. doi:10.1016/j.bbi.2014.04.007
105. Koo JW, Russo SJ, Ferguson D, Nestler EJ, Duman RS. Nuclear factor-kappaB is a critical mediator of stress-impaired neurogenesis and depressive behavior. Proc Natl Acad Sci U S A (2010) 107(6):2669–74. doi:10.1073/pnas.0910658107
106. Koo JW, Duman RS. IL-1beta is an essential mediator of the antineurogenic and anhedonic effects of stress. Proc Natl Acad Sci U S A (2008) 105(2):751–6. doi:10.1073/pnas.0708092105
107. Vecchiarelli HA, Gandhi CP, Gray JM, Morena M, Hassan KI, Hill MN. Divergent responses of inflammatory mediators within the amygdala and medial prefrontal cortex to acute psychological stress. Brain Behav Immun (2016) 51:70–91. doi:10.1016/j.bbi.2015.07.026
108. Wohleb ES, Powell ND, Godbout JP, Sheridan JF. Stress-induced recruitment of bone marrow-derived monocytes to the brain promotes anxiety-like behavior. J Neurosci (2013) 33(34):13820–33. doi:10.1523/JNEUROSCI.1671-13.2013
109. Ramirez K, Shea DT, McKim DB, Reader BF, Sheridan JF. Imipramine attenuates neuroinflammatory signaling and reverses stress-induced social avoidance. Brain Behav Immun (2015) 46:212–20. doi:10.1016/j.bbi.2015.01.016
110. Tynan RJ, Weidenhofer J, Hinwood M, Cairns MJ, Day TA, Walker FR. A comparative examination of the anti-inflammatory effects of SSRI and SNRI antidepressants on LPS stimulated microglia. Brain Behav Immun (2012) 26(3):469–79. doi:10.1016/j.bbi.2011.12.011
111. Tauber AI. Metchnikoff and the phagocytosis theory. Nat Rev Mol Cell Biol (2003) 4(11):897–901. doi:10.1038/nrm1244
112. Chovatiya R, Medzhitov R. Stress, inflammation, and defense of homeostasis. Mol Cell (2014) 54(2):281–8. doi:10.1016/j.molcel.2014.03.030
113. Chen Z, Jalabi W, Hu W, Park HJ, Gale JT, Kidd GJ, et al. Microglial displacement of inhibitory synapses provides neuroprotection in the adult brain. Nat Commun (2014) 5:4486. doi:10.1038/ncomms5486
114. Vinet J, Weering HR, Heinrich A, Kalin RE, Wegner A, Brouwer N, et al. Neuroprotective function for ramified microglia in hippocampal excitotoxicity. J Neuroinflammation (2012) 9:27. doi:10.1186/1742-2094-9-27
115. Kato G, Inada H, Wake H, Akiyoshi R, Miyamoto A, Eto K, et al. Microglial contact prevents excess depolarization and rescues neurons from excitotoxicity 1,2,3. eNeuro (2016) 3(3):4–16. doi:10.1523/ENEURO.0004-16.2016
116. Kigerl KA, de Rivero Vaccari JP, Dietrich WD, Popovich PG, Keane RW. Pattern recognition receptors and central nervous system repair. Exp Neurol (2014) 258:5–16. doi:10.1016/j.expneurol.2014.01.001
117. Gold PW, Licinio J, Pavlatou MG. Pathological parainflammation and endoplasmic reticulum stress in depression: potential translational targets through the CNS insulin, klotho and PPAR-gamma systems. Mol Psychiatry (2013) 18(2):154–65. doi:10.1038/mp.2012.167
118. Wager-Smith K, Markou A. Depression: a repair response to stress-induced neuronal microdamage that can grade into a chronic neuroinflammatory condition? Neurosci Biobehav Rev (2011) 35(3):742–64. doi:10.1016/j.neubiorev.2010.09.010
119. Walker FR. A critical review of the mechanism of action for the selective serotonin reuptake inhibitors: do these drugs possess anti-inflammatory properties and how relevant is this in the treatment of depression? Neuropharmacology (2013) 67:304–17. doi:10.1016/j.neuropharm.2012.10.002
120. Warner-Schmidt JL, Vanover KE, Chen EY, Marshall JJ, Greengard P. Antidepressant effects of selective serotonin reuptake inhibitors (SSRIs) are attenuated by antiinflammatory drugs in mice and humans. Proc Natl Acad Sci U S A (2011) 108(22):9262–7. doi:10.1073/pnas.1104836108
121. Chen SK, Tvrdik P, Peden E, Cho S, Wu S, Spangrude G, et al. Hematopoietic origin of pathological grooming in Hoxb8 mutant mice. Cell (2010) 141(5):775–85. doi:10.1016/j.cell.2010.03.055
122. Sawicki CM, McKim DB, Wohleb ES, Jarrett BL, Reader BF, Norden DM, et al. Social defeat promotes a reactive endothelium in a brain region-dependent manner with increased expression of key adhesion molecules, selectins and chemokines associated with the recruitment of myeloid cells to the brain. Neuroscience (2015) 302:151–64. doi:10.1016/j.neuroscience.2014.10.004
123. McKim DB, Niraula A, Tarr AJ, Wohleb ES, Sheridan JF, Godbout JP. Neuroinflammatory dynamics underlie memory impairments after repeated social defeat. J Neurosci (2016) 36(9):2590–604. doi:10.1523/JNEUROSCI.2394-15.2016
124. Wohleb ES, Patterson JM, Sharma V, Quan N, Godbout JP, Sheridan JF. Knockdown of interleukin-1 receptor type-1 on endothelial cells attenuated stress-induced neuroinflammation and prevented anxiety-like behavior. J Neurosci (2014) 34(7):2583–91. doi:10.1523/JNEUROSCI.3723-13.2014
125. Wohleb ES, Delpech JC. Dynamic cross-talk between microglia and peripheral monocytes underlies stress-induced neuroinflammation and behavioral consequences. Prog Neuropsychopharmacol Biol Psychiatry (2016). doi:10.1016/j.pnpbp.2016.04.013
126. Hinwood M, Morandini J, Day TA, Walker FR. Evidence that microglia mediate the neurobiological effects of chronic psychological stress on the medial prefrontal cortex. Cereb Cortex (2012) 22(6):1442–54. doi:10.1093/cercor/bhr229
127. Haroon E, Fleischer CC, Felger JC, Chen X, Woolwine BJ, Patel T, et al. Conceptual convergence: increased inflammation is associated with increased basal ganglia glutamate in patients with major depression. Mol Psychiatry (2016) 21(10):1351–7. doi:10.1038/mp.2015.206
128. Pribiag H, Stellwagen D. TNF-alpha downregulates inhibitory neurotransmission through protein phosphatase 1-dependent trafficking of GABA(A) receptors. J Neurosci (2013) 33(40):15879–93. doi:10.1523/JNEUROSCI.0530-13.2013
129. Lewitus GM, Pribiag H, Duseja R, St-Hilaire M, Stellwagen D. An adaptive role of TNFalpha in the regulation of striatal synapses. J Neurosci (2014) 34(18):6146–55. doi:10.1523/JNEUROSCI.3481-13.2014
130. Lewitus GM, Konefal SC, Greenhalgh AD, Pribiag H, Augereau K, Stellwagen D. Microglial TNF-alpha suppresses cocaine-induced plasticity and behavioral sensitization. Neuron (2016) 90(3):483–91. doi:10.1016/j.neuron.2016.03.030
131. Tanaka K, Furuyashiki T, Kitaoka S, Senzai Y, Imoto Y, Segi-Nishida E, et al. Prostaglandin E2-mediated attenuation of mesocortical dopaminergic pathway is critical for susceptibility to repeated social defeat stress in mice. J Neurosci (2012) 32(12):4319–29. doi:10.1523/JNEUROSCI.5952-11.2012
132. Milior G, Lecours C, Samson L, Bisht K, Poggini S, Pagani F, et al. Fractalkine receptor deficiency impairs microglial and neuronal responsiveness to chronic stress. Brain Behav Immun (2016) 55:114–25. doi:10.1016/j.bbi.2015.07.024
133. Clark AK, Gruber-Schoffnegger D, Drdla-Schutting R, Gerhold KJ, Malcangio M, Sandkuhler J. Selective activation of microglia facilitates synaptic strength. J Neurosci (2015) 35(11):4552–70. doi:10.1523/JNEUROSCI.2061-14.2015
134. Abiega O, Beccari S, Diaz-Aparicio I, Nadjar A, Laye S, Leyrolle Q, et al. Neuronal hyperactivity disturbs ATP microgradients, impairs microglial motility, and reduces phagocytic receptor expression triggering apoptosis/microglial phagocytosis uncoupling. PLoS Biol (2016) 14(5):e1002466. doi:10.1371/journal.pbio.1002466
135. Sierra A, Encinas JM, Deudero JJ, Chancey JH, Enikolopov G, Overstreet-Wadiche LS, et al. Microglia shape adult hippocampal neurogenesis through apoptosis-coupled phagocytosis. Cell Stem Cell (2010) 7(4):483–95. doi:10.1016/j.stem.2010.08.014
136. Hardingham GE, Bading H. Synaptic versus extrasynaptic NMDA receptor signalling: implications for neurodegenerative disorders. Nat Rev Neurosci (2010) 11(10):682–96. doi:10.1038/nrn2911
137. Tasic B, Menon V, Nguyen TN, Kim TK, Jarsky T, Yao Z, et al. Adult mouse cortical cell taxonomy revealed by single cell transcriptomics. Nat Neurosci (2016) 19(2):335–46. doi:10.1038/nn.4216
138. Leonard BE. Inflammation, depression and dementia: are they connected? Neurochem Res (2007) 32(10):1749–56. doi:10.1007/s11064-007-9385-y
139. da Silva J, Goncalves-Pereira M, Xavier M, Mukaetova-Ladinska EB. Affective disorders and risk of developing dementia: systematic review. Br J Psychiatry (2013) 202(3):177–86. doi:10.1192/bjp.bp.111.101931
140. Prinz M, Priller J. Microglia and brain macrophages in the molecular age: from origin to neuropsychiatric disease. Nat Rev Neurosci (2014) 15(5):300–12. doi:10.1038/nrn3722
141. Hong S, Dissing-Olesen L, Stevens B. New insights on the role of microglia in synaptic pruning in health and disease. Curr Opin Neurobiol (2016) 36:128–34. doi:10.1016/j.conb.2015.12.004
142. Hong S, Beja-Glasser VF, Nfonoyim BM, Frouin A, Li S, Ramakrishnan S, et al. Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science (2016) 352(6286):712–6. doi:10.1126/science.aad8373
143. Lenz KM, Nugent BM, Haliyur R, McCarthy MM. Microglia are essential to masculinization of brain and behavior. J Neurosci (2013) 33(7):2761–72. doi:10.1523/JNEUROSCI.1268-12.2013
144. Bollinger JL, Bergeon Burns CM, Wellman CL. Differential effects of stress on microglial cell activation in male and female medial prefrontal cortex. Brain Behav Immun (2016) 52:88–97. doi:10.1016/j.bbi.2015.10.003
Keywords: neuroplasticity, microglia, anxiety, depression, post-traumatic stress disorder, neuroinflammation, parainflammation, neuroimmune
Citation: Wohleb ES (2016) Neuron–Microglia Interactions in Mental Health Disorders: “For Better, and For Worse”. Front. Immunol. 7:544. doi: 10.3389/fimmu.2016.00544
Received: 09 August 2016; Accepted: 16 November 2016;
Published: 29 November 2016
Edited by:
Sanae Hasegawa-Ishii, Pennsylvania State University, USAReviewed by:
Hiroaki Wake, National Institute for Basic Biology, JapanCopyright: © 2016 Wohleb. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Eric S. Wohleb, ZXJpYy53b2hsZWJAdWMuZWR1
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.