 Hlumani Ndlovu
Hlumani Ndlovu Mohlopheni J. Marakalala
Mohlopheni J. Marakalala
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 24 October 2016
Sec. Vaccines and Molecular Therapeutics
Volume 7 - 2016 | https://doi.org/10.3389/fimmu.2016.00434
This article is part of the Research Topic Targeting host immunity for treatment of tuberculosis View all 8 articles
Tuberculosis (TB) remains a leading global health problem that is aggravated by emergence of drug-resistant strains, which account for increasing number of treatment-refractory cases. Thus, eradication of this disease will strongly require better therapeutic strategies. Identification of host factors promoting disease progression may accelerate discovery of adjunct host-directed therapies (HDTs) that will boost current treatment protocols. HDTs focus on potentiating key components of host anti-mycobacterial effector mechanisms, and limiting inflammation and pathological damage in the lung. Granulomas represent a pathological hallmark of TB. They are comprised of impressive arrangement of immune cells that serve to contain the invading pathogen. However, granulomas can also undergo changes, developing caseums and cavities that facilitate bacterial spread and disease progression. Here, we review current concepts on the role of granulomas in pathogenesis and protective immunity against TB, drawing from recent clinical studies in humans and animal models. We also discuss therapeutic potential of inflammatory pathways that drive granuloma progression, with a focus on new and existing drugs that will likely improve TB treatment outcomes.
One-third of the world population is latently infected with Mycobacterium tuberculosis (Mtb), the causative agent of tuberculosis (TB). About 5–10% of the latently infected population develops active TB and manifest clinical signs of the disease (1, 2). Active TB is estimated to cause 1.5 million deaths annually, making it one of the most deadly communicable diseases worldwide (3). Efforts to control and eradicate TB are hampered by coinfection with the human immunodeficiency virus (HIV) and emergence of drug-resistant bacteria. In 2014, 480,000 new cases of multidrug-resistant (MDR) TB were reported, and an estimated 9.7% of the MDR-TB patients have extensively drug-resistant (XDR) TB, characterized by resistance to second-line drugs and poor treatment outcomes (3). Therefore, there is an urgent need to develop new therapeutic drugs, an effective vaccine, and reliable diagnostic tests for latent disease in order to achieve the health targets of the newly adopted sustainable development goals (SDGs) aimed at ending the TB epidemic by 2030 (3).
The current standard first-line treatment regimen consisting of isoniazid, rifampicin, ethambutol, and pyrazinamide requires long duration of treatment (at least 6 months for first-line TB therapy and 18–20 months for MDR-TB therapy) to achieve sterilization of infection. Moreover, the toxic side effects of drugs may lead to non-compliance to treatment, thus, opening a window for development of drug resistance. Hence, novel treatment strategies are required to shorten the duration of treatment and improve treatment outcomes particularly in drug-resistant patients. The use of adjunctive host-directed therapies (HDTs) that seek to limit lung pathological damage and boost the host protective armory against Mtb infection is an attractive avenue requiring more research exploration (4–6). In this review, we discuss the role of the granuloma in the progression of TB disease and review HDTs that target pathways implicated in severe inflammation during TB disease.
A prominent pathological feature of TB is the formation of granulomas, driven by both bacterial and host factors in the lungs of infected patients (Figure 1) (6, 7). A granuloma is an organized and compact immunological structure rich with immune cells, such as macrophages, monocytes, dendritic cells (DCs), neutrophils, epithelioid cells, foamy macrophages, and multi-nucleated giant cells (8, 9). This initial structure is surrounded by a layer of lymphocytes giving it an organized solid structure (8, 9).
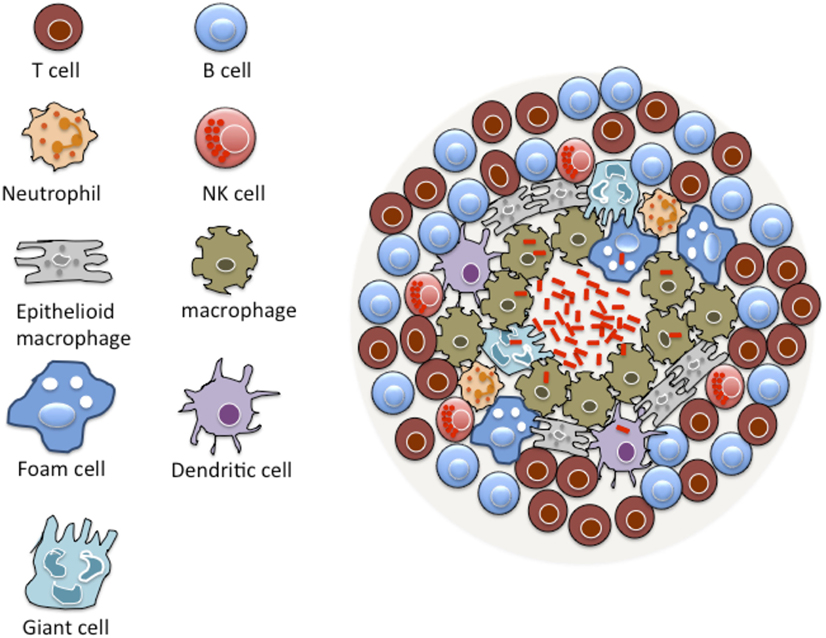
Figure 1. Basic structure of TB granuloma. A granuloma is a compact immunological structure rich with macrophages at the center. Macrophages can undergo specialized transformation differentiating into other cell types, such as epithelioid cells, multi-nucleated giant cells, and foamy macrophages that are accumulated with lipid droplets. A lymphocytic cuff that is largely comprised of B and T cells characterizes the periphery of the granuloma. Many other cell types are known to constitute a granuloma, including neutrophils, dendritic cells, natural killer (NK) cells, and fibroblasts.
Early events that lead to granuloma formation were elucidated through a study by Davis and Ramakrishnan that utilized a transparent embryonic zebrafish model of Mycobacterium marinum (M.m) expressing fluorescent proteins (10). The study showed that bacteria freely replicate within macrophages and that mycobacteria direct recruitment and motility of uninfected macrophages into the surrounding areas of infected cells using the ESX-1/RD1 virulence locus (10). Moreover, mycobacterial virulence protein ESAT-6 induces the secretion of host matrix metalloproteinase-9 (MMP-9) by epithelial cells to drive the recruitment of new macrophages into the granuloma (11, 12). Studies conducted in humans and animal models have confirmed the crucial role of MMP-9 in pathogenesis of TB (7, 13–15). Infected macrophages departing the primary granuloma are involved in establishing secondary granulomas, thus, promoting early dissemination of infection (10). Mycobacterial cell wall glycolipid, trehalose 6,6′-dimycolate (TDM), is also known to trigger immunogenic responses that lead to granuloma formation in mice. The TDM-driven granulomatous response is initiated and maintained by pro-inflammatory cytokines (TNF-α and IL-6) and complement C5 and can be down modulated by lactoferrin (16, 17).
Macrophages within the granuloma develop into specialized cell types, such as epithelioid macrophages, foamy macrophages, and multi-nucleated giant cells that form after fusion of plasma membranes of multiple macrophages (18). Mycobacterial lipids, such as oxygenated mycolic acid, have been shown to trigger the differentiation of human monocyte-derived macrophages (MDM) into foamy macrophages in vitro (19). Murine macrophages infected with bacillus Calmette-Guérin (BCG) differentiate into foamy macrophages through a signaling process that is mediated by toll-like receptor 2 (TLR2) (20). Studies conducted in humans and murine models have associated foamy macrophages with necrotic regions of the granuloma (19). A recent study by Berg et al. showed that snapc1b zebrafish mutants, which were identified in a genetic forward screen by Tobin et al., were hypersusceptible to M.m infection due to early breakdown of granulomas that resulted in release of mycobacteria to a growth permissive extracellular milieu (21, 22). In addition, macrophages from snapc1b mutants displayed vacuolated morphology and had reduced speed of homeostatic migration due to accumulation of undigested debris in the lysosomes (22). This indicates that the migratory capacity and lysosomal functionality of macrophages is crucial for control of bacterial proliferation.
Dendritic cells are among the early arrivals at the site of infection where they partake in sampling of antigen by engulfing bacteria, bacterial products, and infected dying cells, leaving the site of infection and migrating via the lymphatic system to the draining lymph nodes where they possibly trigger acquired immunity (23). A study by Harding et al. showed that inflammatory DCs infected with BCG associated with bacteria-specific T cells, resulting in the formation of new multi-foci lesions, demonstrating their role in granuloma reformation (24). Moreover, Mtb can modulate DC function by impairing antigen presentation and migration to the draining lymph nodes, thereby delaying the development of the adaptive immune response required to arrest bacterial proliferation (23, 25).
Diverse myeloid cell subsets contribute to the granuloma and drive unique pathological outcomes during TB disease. Studies conducted in murine and non-human primate models showed that polymorphonuclear neutrophils (PMNs) are recruited early to the lung during virulent Mtb infection (26–29). PMNs either confer host protection by driving T-cell priming and granuloma formation or promote disease severity depending on the genetic background of experimental mice (30–34). PMNs have been implicated in lung damage during TB disease in studies conducted in mice (35) and humans (36). Kinetics studies in the lungs of C57BL/6 and 129S2 mice detected an accumulation of myeloid-derived suppressor cells (MDSCs) that drive inflammatory responses to Mtb and restrict selected T-cell responses (37, 38). MDSCs have also been identified in pleural effusions and blood of TB patients (39, 40).
The final step in granuloma formation is the arrival of T and B cells that form the lymphocytic cuff at the periphery of the granuloma giving it a solid intact structure (41–45). The arrival of Mtb-specific T cells in the lungs coincides with the arrest of bacterial proliferation 14–21 days after initiation of infection by producing TNF and IFN-γ that enhance macrophage microbicidal activity (1, 46–51). Mice deficient of CD4+ T cells are susceptible to Mtb infection and have decreased cellular infiltration to the lungs (52). Other T cell subsets involved in immune response to Mtb infections in mice and humans include γδ T cells, CD8 T cells and, CD1-restricted CD4/CD8 double negative T cells (53, 54). Development of an optimal Th1 and Th17 immune responses is critical for long-term control of Mtb infection (55, 56). However, an exuberant immune response may be detrimental to the host due to excessive tissue inflammation; hence, a more balanced Th1/Th2/Th17 immune response is more desired to drive host protection, while limiting tissue pathology.
As TB disease progresses, the granuloma may undergo complex remodeling events that result in the formation of a caseum, a cheese-like cellular structure containing necrotic material at the center of the granuloma that is rich in lipids and reportedly hypoxic. Analysis of human TB granulomas by genome-wide microarray found genes associated with lipid sequestration and metabolism to be highly expressed in caseous granulomas (57). Moreover, immunohistological studies revealed that cells surrounding the caseum expressed high levels of proteins involved in lipid metabolism, such as adipophilin, acyl-CoA synthetase long-chain family member 1, and saposin C (57). Destruction of the caseum results in liquefaction and eventually to granuloma cavitation, which permits the release of infectious Mtb into the airways where they are aerosolized in cough droplets and can facilitate bacterial transmission to new hosts (57, 58). The events that lead to granuloma collapse and spread of bacteria are depicted in Figure 2. Histological studies conducted in non-human primates (NHP) have elucidated the complexities of granuloma dissociation in relation to disease progression. A study by Lin et al. found that a discordance exists between the extent of granuloma destruction and the clinical state of disease in NHPs (59). Moreover, a subsequent study by Lin et al. showed that each individual granuloma within the same host behaves independently of others, suggesting that infected individuals have a heteregenous mixture of granulomas with varying immune profile and competence to control the bacteria (60). Hence, the fate of a few granulomas within the host seems to drive the majority of clinical morbidities associated with the disease (57, 60, 61).
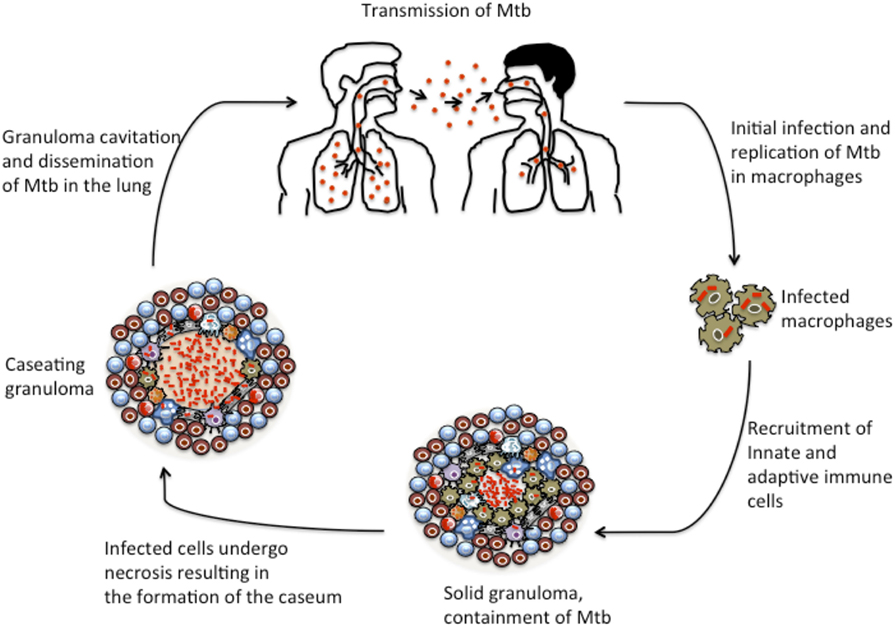
Figure 2. Granuloma development during TB progression. The initial stages of TB infection involve inhalation of M. tuberculosis bacilli into the lung and phagocytosis by resident alveolar macrophages. Mtb elicits local inflammatory response resulting in the recruitment of monocytes and macrophages and other innate immune cells to the site of infection. The macrophages further differentiate into other specialized cells, such as epithelioid macrophages, foamy macrophages, and multi-nucleated giant cells. Upon induction of adaptive immunity, the granuloma gets a peripheral lymphocytic cuff mainly characterized by B and T cells. This is the balanced solid state in which many granulomas persist and restrict the bacilli at their center. However, as the disease progresses, increased necrotic breakdown of granuloma cells leads to the accumulation of caseum, which may result in cavitation of granulomas. Ultimately, as the granuloma cavitates and collapses into the lung, Mtb bacilli are released into the airway.
Dissociation of the granuloma is driven by both host and mycobacterial factors that induce different cell-death modalities in infected cells. Infected granuloma macrophages can die either by apoptosis or necrosis. Apoptotic cell death seems to favor the host by keeping the bacteria encased within dead macrophages that can be readily phagocytosed by newly arriving uninfected macrophages. In contrast, necrotic cell death, which is characterized by the loss of cell membrane integrity, results in leakage of the bacteria into the growth permissive extracellular environment where the bacteria thrive by adopting a characteristic cording phenotype, making it difficult for the bacteria to be engulfed by new macrophages (62, 63). Host-derived TNF-α has been implicated in causing macrophage necrosis during Mtb infection (64, 65). Excessive production of TNF-α drives necrosis of infected macrophages through a programmed pathway called necroptosis that involves the activation of receptor activating protein 1 (RIP1) and RIP3 kinases via the production of reactive oxygen species (ROS) by the mitochondria (64, 65). TNF-α-induced macrophage necrosis is modulated by mitochondrial cyclophilin D via the formation of the mitochondrial permeability transition pore complex and the production of ceramide by the acid sphingomyelinase (65). Inhibition of cyclophilin D with alisporivir and inactivation of acid sphingomyelinase with an anti-depressant drug desipramine restored resistance to M.m infection in zebrafish (65).
Studies conducted in mice and zebrafish have shown that mycobacterial virulence factors can modulate macrophage necrosis by generating specific eicosanoids from arachidonic acid (AA) (21, 66). Virulent Mtb H37Rv strain induces the production of lipoxin A4 (LXA4) by 5- and 15-lipoxygenases to promote necrosis, while H37Ra mutants induced the synthesis of pro-apoptotic prostaglandin E2 (PGE2) by cyclooxygenase (COX) 1 and 2 and prostaglandin synthases (66–68). Mice lacking 5-lipoxygenase (ALOX5−/−) are more resistant to infection with Mtb H37Rv strain, indicating that LXA4 is detrimental to the host (21). The contribution of eicosanoid metabolites in susceptibility to mycobacterial infection was confirmed in a genetic screening study that aimed to identify mutant zebrafish that were hypersusceptible to M.m infection (21). The study identified leukotriene A4 hydrolase (LTA4H), an enzyme involved in the production of leukotriene B4 (LTB4) as a key determinant of host susceptibility to M.m infection (21). LTA4H mediates host susceptibility through two mechanisms that deregulate TNF-α activity: excess LTA4H causes overproduction of LTB4 that drives exuberant production of TNF, while low or absence of LTA4H induces the production of anti-inflammatory LXA4 that suppresses TNF activity leading to uncontrolled bacterial proliferation and macrophage necrosis (21, 64). Similar genetic variants encoding for low or high LTA4H expression have been identified in humans, and individuals who are homozygotes for low or high expression variants of LTA4H get severe TB meningitis with high mortality rate, while heterozygotes with intermediate LTA4H expression get protection (21, 64). Translation of these observations had huge implications for therapy as demonstrated in a Vietnamese cohort of TB meningitis in which adjunctive therapy with broadly anti-inflammatory glucocorticoids was beneficial to a group of patients with high LTA4H expression, while deleterious to patients with low LTA4H expression (21, 64).
A recent study by Marakalala et al. characterized protein and lipid signatures associated with granuloma progression by analyzing different granuloma types (intact, caseous, and cavitary granuloma) from tissue obtained from patients who underwent surgery due to severe lung disease (69). This study utilized laser-capture microdissection, mass spectrometry, and confocal microscopy to generate a molecular map of different regions of each granuloma type. The study identified over 3000 proteins that were differentially expressed between different regions of granuloma types (69). The study found more proteomic diversity within rather than between different granuloma types. Importantly, the proteomic profiles of caseous and cavitary centers were composed largely of pro-inflammatory pathways, including proteins that release pro-inflammatory eicosanoids from AA metabolism (69). In contrast, cellular regions found at the periphery of the granulomas displayed a more anti-inflammatory signature characterized by the abundance of pathways involved in normal cellular biogenesis and some enzymes involved in the synthesis of prostanoids (69). Together, these data suggest that spatial organization of inflammatory responses within the granuloma may determine the pathologic response to TB.
Initially, the granuloma was thought to be a solely host protective structure that “walls off” infecting bacteria (70, 71). However, we argue that the granuloma may represent a standoff between two warring camps: the immune response trying to kill the invading bacteria and the bacteria seeking to defend itself and establish a niche where it can thrive. Earlier histological studies conducted on tuberculous lungs obtained from autopsies in the pre-chemotherapy era chronicled a series of events associated with granuloma destruction (72, 73). Areas of the granuloma that had undergone caseation were correlated with increased bacterial load, while areas with intact granulomas had reduced bacterial numbers, thus, suggesting that the integrity of the granuloma is critical for control of bacterial proliferation (72, 73). This was further corroborated by findings in a zebrafish model, where it was shown that the onset of granuloma caseation is linked to uncontrolled bacterial proliferation (74). Moreover, mice deficient in TNF (48, 49, 75–79), IFN-γ (80), IL-12 (81), signal transducer and activator of transcription 4 (STAT4) (82), and myeloid differentiation primary-response protein 88 (MYD88) (83–85) had poorly formed granulomas and were highly susceptible to Mtb infection, thus, giving more credibility to the host-protective nature of granulomas. Although gene deficient mice are hypersusceptible to Mtb infection, the mechanism for their susceptibility may not depend on granulomatous responses but be driven by the impaired ability of infected macrophages to kill the bacteria (48).
Findings generated over the previous decade suggest that Mtb orchestrates the early events of granuloma formation for its own benefit, has adapted to life in the granuloma, and has developed an arsenal of strategies to evade host-offensive mechanisms. Recent technological advances have enabled us to chronicle the early events of granuloma formation in the context of innate immunity using dynamic imaging tools in a zebrafish model infected with M.m (10, 86). Mycobacteria coordinate granuloma formation by driving the recruitment and chemotactic motility of uninfected macrophages to the granuloma by deploying the ESX-1 secretory system encoded by the RD1 virulence locus (10). The recruitment of uninfected macrophages to the granuloma is further enhanced by the secretion of MMP-9 by epithelial cells activated by the mycobacterial virulence factor ESAT-6 (11, 12). MMP-9 has been identified as a biomarker for severe TB meningitis and is associated with increased mortality in human patients (13, 15). Interestingly, ESAT-6 has also been shown to induce apoptosis of infected cells in vitro (74, 87–89). The bacteria seem to be able to persist within the hostile environment of the granuloma as indicated by the propensity of newly infected macrophages to migrate to existing granulomas than avoid them in both zebrafish and mice with mature granulomas (90, 91). Thus, the ability of the bacteria to coordinate both recruitment and death of macrophages suggests that the bacteria are actively attempting to create an environment ideal for their dissemination and proliferation.
Mycobacteria have adapted to survive in the hostile environment by entering a state of non-replicating persistence characterized by slow growth and metabolic shift that reduces their susceptibility to environmental pressure (92). Moreover, the bacteria have developed strategies to survive the hypoxic environment encountered in caseous granulomas by inducing complex transcriptional networks like DosR regulon (93, 94) and angiogenesis to improve the transport of oxygen to the hypoxic granuloma (95). Suppression of granuloma associated angiogenesis by administration of VEGFA inhibitor reduced bacterial burdens and acted synergistically with first-line drugs to kill the bacteria (95). A study by Zhang et al. identified a set of genes (the “counteractome”) required by Mtb to survive the immunological stress mediated by CD4+ T cells in mice (96). CD4+ T cells starve Mtb of exogenous tryptophan, and deletion of trpE gene required for bacterial tryptophan biosynthesis rendered Mtb susceptible to the immunological stress (96). Interestingly, a combination of IFN-γ administration and inhibition of TrpE by a small molecule 6-FABA resulted in synergistic killing of Mtb in vitro, suggesting that inhibitors of the Mtb “counteractome” can be explored as drug candidates that could potentiate anti-mycobacterial immunity (96).
One of the major challenges associated with current TB treatment protocols is the lengthy duration of therapy that causes poor compliance of patients, resulting in increasing cases of MDR- and XDR-TB and high mortalities among the treatment-refractory patients (6). In order to achieve the WHO goal to eradicate TB by 2050, we need new treatment strategies that will boost currently available drugs, shorten the duration of treatment, and ameliorate inflammation and tissue damage. One strategy that is showing greater promise of assisting in the fight against TB is the development of HDTs that could be used as adjunctive therapy in combination with the currently available standard treatment regimens. HDTs are aimed at augmenting the host’s immunological defense mechanisms and reducing excessive tissue pathology, culminating in improved clinical outcomes reflected in reduced duration of treatment and reduced morbidity and mortality (6). A wide range of HDTs are currently at varying stages of development and clinical testing, including repurposed drugs for diabetes, epilepsy, hypercholesterolemia, asthma, cancer, and arthritis (6).
The focus of this section is to discuss recent advances in the development of HDTs targeting inflammatory pathways associated with granuloma dissociation for which proof-of-principle has been achieved in animal models or clinical settings (Table 1). To have the full view of progress, made to date to develop and test HDTs, readers are referred to other extensive reviews (2, 5, 6, 97–99).
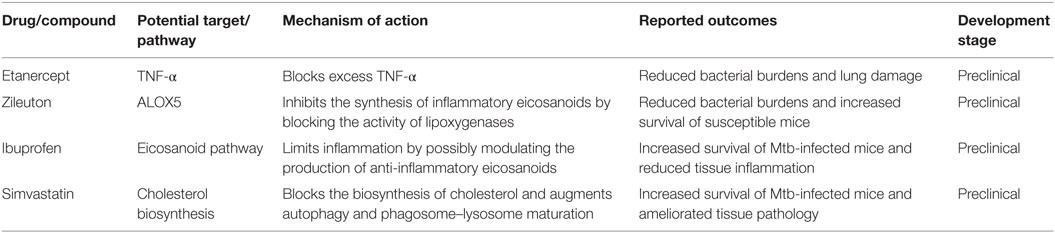
Table 1. List of drugs inhibiting the inflammatory pathways implicated in granuloma dissociation with proven efficacy in animal models [adapted from Ref. (2, 6)].
Although TNF is required to induce macrophage-killing activities (48, 49, 77), excessive- or low-TNF production is detrimental to the host causing augmented tissue pathology (21, 64, 65). Inhibition of TNF-α activity has been successfully used to treat inflammatory diseases, like rheumatoid arthritis (RA), Crohn’s disease, and psoriasis (5). Hence, blocking the activity of TNF has been explored as a potential host-directed therapy for TB. Treatment of Kramnik mice with a TNF-α inhibitor etanercept in conjunction with standard TB treatment reduced pulmonary bacterial burden and ameliorated lung pathology (100). However, the relapse of some of the mice treated with etanercept adjunctive therapy is of concern, albeit the relapse being lower compared with mice that received the standard treatment alone (100). The safety risks of using TNF-α inhibitors have also been demonstrated in human studies where patients receiving TNF-α inhibitors for chronic inflammatory disease like RA and Crohn’s disease can develop reactivation of TB disease (101, 102). Moreover, the presence of polymorphisms in LTA4H that skew TNF production is another confounding factor that needs to be addressed before considering the use of TNF-α inhibitors as adjunctive therapy in humans. Therefore, more studies are still required to improve the safety profile of TNF inhibitors.
As discussed above, pro-inflammatory eicosanoids have been implicated in disease progression by driving necrotic cell death and contribute to increased mortality of mice infected with Mtb. Recently, eicosanoids have been associated with the type I IFN response and contribute to the outcomes of both Mtb and influenza infections (103, 104). Studies conducted in mice and humans have demonstrated a deleterious role for type I IFNs to the host during Mtb infection (105–112). IFN-α/β have been shown to block the production of host protective cytokines IL-1α and IL-1β both in vitro using human and murine cells and in vivo using mice (113–115). Interestingly, IL-1α and IL-1β negatively regulate type I IFNs in murine and human macrophages and inhibit pro-bacterial effects downstream of IFN-β (103). IL-1-driven PGE2 production counter-regulates the function of type I IFN and is beneficial to the host as demonstrated in mice double deficient for IL-1R/IFNAR1 (103). This counter-regulatory mechanism has been shown to be at play in both human and murine cells infected with Mtb (103). The direct administration of PGE2 or blocking 5-lipoxygenase with zileuton rescued the highly susceptible IL-1-knockout mice from Mtb infection and reduced bacterial burdens (103). Therefore, manipulating the balance of eicosanoid by administering PGE2 or zileuton might offer an attractive adjunctive therapy for TB that could be used to improve the disease outcome (103). Interestingly, zileuton has already been approved for clinical use in asthma (103). The role of eicosanoids in driving granuloma dissociation and the potential benefits of targeting specific proteins as candidates for HDTs is summarized in Figure 3.

Figure 3. Targeting inflammatory proteins as host-directed therapies (HDTs) for TB to ameliorate tissue inflammation. Arachidonic acid (AA) is metabolized by 5-lipoxygenase (5-LO) to produce leukotriene A4 (LTA4), a substrate in the subsequent step that is catalyzed by leukotriene A4 hydrolase (LTA4H) to produce the highly inflammatory leukotriene B4 (LTB4). High levels of LTB4 drive excessive TNF-α production by macrophages that may trigger necrotic cell death. Necrotic cell death leads to granuloma dissociation resulting in the formation of caseums and eventually cavities after the necrotic center collapses into the airways. Type I IFNs block the production of host protective IL-1α and IL-1β in vitro and in vivo resulting in reduced production of PGE2. Targeting specific inflammatory proteins with chemical inhibitors or antibodies could be a possible HDT aimed at limiting tissue inflammation and improve treatment outcomes. The proof-of-principle for the interventions depicted in the schematic has been established in animal models.
Inhibition of inflammatory eicosanoids with non-steroidal anti-inflammatory drugs (NSAIDs), such as ibuprofen, has been used as monotherapy for TB in mice with great success (116, 117). Treatment of C3HeB/FeJ (Kramnik) mice with ibuprofen increased survival of Mtb-infected mice by limiting severe tissue pathology and reducing bacterial load compared with untreated control mice (116). This study established a proof-of-principle for the use of NSAIDs as a host-directed adjunctive therapy for TB that can be used in combination with standard chemotherapeutic drugs.
The role of lipids in the dissociation of the granuloma has been demonstrated in a study that utilized genome-wide screening to determine the levels of genes involved in lipid metabolism in human TB lung tissues (57). This study found that genes involved in lipid metabolism were highly expressed in caseous granulomas, and this was further proven by immunohistological staining that confirmed the abundance of proteins involved in lipid metabolism in caseous regions of the granuloma (57). Mtb can utilize host cholesterol to maintain persistent chronic infection. Moreover, lipid bodies present within foamy macrophages have been shown to play a role in reactivation of latent TB disease (118, 119). Therefore, lipids have been targeted as possible host-directed therapy for TB disease using statins; competitive inhibitors of HMG-CoA reductase, an enzyme involved in the biosynthesis of cholesterol (120). Statins have been used clinically to treat patients with coronary disorders and hypercholesterolemia (121) and have been shown to possess immunomodulatory and anti-inflammatory properties (122, 123).
In vitro studies conducted using peripheral blood mononuclear cells (PBMCs) and MDM isolated from patients with familial hypercholesterolemia who underwent statin therapy were found to be resistant to Mtb infection compared with cells from healthy controls (124). In addition, treatment of mice with simvastatin prior to Mtb infection increased survival due to decreased lung bacterial load and improved pathological outcomes (124). The host-protective mechanism was shown to be mediated by the ability of statins to promote phagosomal maturation and autophagy (124). Statins have also been shown to enhance the bactericidal activity of first-line drugs in vitro using infected macrophages (125) and in vivo using a mouse model of chronic TB infection (126). A recent study by Dutta et al. showed that using simvastatin as an adjunctive therapy in combination with the first-line drug rifampicin shortened the duration of treatment in mice (127). Therefore, statins appear to be attractive candidates for host-directed therapy for TB. However, more work is still required to assess the safety, dosage, and efficacy of statins in humans infected with Mtb.
Tuberculosis still remains a huge public health problem with high mortality rate in infected individuals. Moreover, drug resistance has emerged as an enormous problem exerting more strain on health services. This has necessitated an urgent need to develop new drugs that not only target the pathogen but also helps to boost the host’s natural ability to fight the infection. The site that represents a standoff between the host and the pathogen as they seek to establish dominance is the granuloma; a key immunological structure composed of innate and adaptive cells of the immune system. Initially, the granuloma was thought to provide host protection by “walling off” the bacteria; however, evidence emerging from recent studies indicates that the granuloma may be manipulated by the mycobacteria to achieve its dominance in the host (70, 71, 128). Studies have demonstrated that virulence factors secreted by Mtb via the ESX-1 secretory system drive the recruitment of macrophages to the granuloma and influence the cell-death modality of infected macrophages by favoring necrosis mediated by the RIP1/RIP3 necroptosome (64, 65). Moreover, ESAT-6 has been shown to induce the secretion of MMP-9 by epithelial cells to further drive the recruitment of macrophages to the granuloma (11, 13–15).
The granuloma can undergo complex remodeling events driven by both bacterial and host factors resulting in some structural changes that coincide with TB disease progression. Studies have been conducted to understand the mechanism driving granuloma dissociation during TB disease (57, 69). Excessive TNF-α production has been implicated in the destruction of the granuloma in zebrafish (64, 65). Caseating regions of the granuloma have an abundance of proteins associated with lipid metabolism (57). Moreover, pro-inflammatory proteins, such as LTA4H, were highly expressed in caseous regions of the granuloma compared with cellular regions and associated with TNF-α expression (69). Hence, factors that drive granuloma dissociation have been explored as potential HDTs that could be used to improve treatment outcomes in conjunction with currently available standard treatment regimen. More studies are still required to further expand our knowledge of the molecular factors that sway the double-edged nature of the granuloma in favor of the host or the pathogen.
Blocking TNF-α with etanercept in the presence of first-line drugs proved beneficial to host by reducing bacterial load and limiting tissue pathology (100). Moreover, manipulating the balance of pro-inflammatory eicosanoids by directly administering PGE2 or using zileuton to block 5-lipoxygenase increased the survival of highly susceptible mice infected with Mtb (103). Finally, treatment of Mtb-infected mice with statins in the presence or absence of first-line TB drugs increased survival and reduced the duration of TB therapy (124, 127). These studies have established a proof-of-principle for the use of drugs that inhibit the pro-inflammatory pathways involved in granuloma dissociation in murine models. However, further studies are required to determine the safety, dosage, and efficacy of these drugs for use in humans. Challenges encountered with the administration of glucocorticoid steroids in patients with TB meningitis suggests that caution must be exercised in the roll-out of HDTs for TB and that perhaps personalized medicine might offer a better treatment approach.
Although not discussed in this review, a number of drugs currently used clinically to treat diabetes (115), cancer (95, 129), and hypertension (130) have shown some promise in anti-TB defense and can be repurposed for use as adjunctive therapy for the disease (2, 6). These will require more testing to ensure their efficacy and safety to use as adjunctive therapy for TB at the currently approved clinical dosages. To bolster our arsenal of adjunctive HDTs for TB, we need to amplify our efforts to identify potential new targets and improve our understanding of the complex processes that lead to disease progression at both molecular and systemic levels.
Both authors planned the manuscript content, analyzed the literature, and wrote the manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The authors acknowledge funding from the South African Medical Research Council (SAMRC) self-initiated research grant (MM) and the University of Cape Town (HN and MM). We also thank our colleagues at University of Cape Town and Harvard T.H Chan School of Public Health for stimulating discussions.
1. Philips JA, Ernst JD. Tuberculosis pathogenesis and immunity. Annu Rev Pathol (2012) 7:353–84. doi:10.1146/annurev-pathol-011811-132458
2. Rayasam GV, Balganesh TS. Exploring the potential of adjunct therapy in tuberculosis. Trends Pharmacol Sci (2015) 36:506–13. doi:10.1016/j.tips.2015.05.005
3. World Health Organisation. World Health Organisation Tuberculosis Fact Sheet No. 104. Geneva: WHO (2016). Available from: http://www.who.int/mediacentre/factsheets/fs104/en/
4. Worthington RJ, Melander C. Combination approaches to combat multidrug-resistant bacteria. Trends Biotechnol (2013) 31:177–84. doi:10.1016/j.tibtech.2012.12.006
5. Wallis RS, Maeurer M, Mwaba P, Chakaya J, Rustomjee R, Migliori GB, et al. Tuberculosis – advances in development of new drugs, treatment regimens, host-directed therapies, and biomarkers. Lancet Infect Dis (2016) 16:e34–46. doi:10.1016/S1473-3099(16)00070-0
6. Zumla A, Rao M, Wallis RS, Kaufmann SHE, Rustomjee R, Mwaba P, et al. Host-directed therapies for infectious diseases: current status, recent progress, and future prospects. Lancet Infect Dis (2016) 16:e47–63. doi:10.1016/S1473-3099(16)00078-5
7. Taylor JL, Hattle JM, Dreitz SA, Troudt JM, Izzo LS, Basaraba RJ, et al. Role for matrix metalloproteinase 9 in granuloma formation during pulmonary Mycobacterium tuberculosis infection. Infect Immun (2006) 74:6135–44. doi:10.1128/IAI.02048-05
9. Cosma CL, Sherman DR, Ramakrishnan L. The secret lives of the pathogenic mycobacteria. Annu Rev Microbiol (2003) 57:641–76. doi:10.1146/annurev.micro.57.030502.091033
10. Davis JM, Ramakrishnan L. The role of the granuloma in expansion and dissemination of early tuberculous infection. Cell (2009) 136:37–49. doi:10.1016/j.cell.2008.11.014
11. Volkman HE, Pozos TC, Zheng J, Davis JM, Rawls JF, Ramakrishnan L. Tuberculous granuloma induction via interaction of a bacterial secreted protein with host epithelium. Science (2010) 327:466–9. doi:10.1126/science.1179663
12. Van den Steen PE, Dubois B, Nelissen I, Rudd PM, Dwek RA, Opdenakker G. Biochemistry and molecular biology of gelatinase B or matrix metalloproteinase-9 (MMP-9). Crit Rev Biochem Mol Biol (2002) 37:375–536. doi:10.1080/10409230290771546
13. Price NM, Farrar J, Chau TTH, Mai NTH, Hien TT, Friedland JS. Identification of a matrix-degrading phenotype in human tuberculosis in vitro and in vivo. J Immunol (2001) 166:4223–30. doi:10.4049/jimmunol.166.6.4223
14. Sheen P, O’Kane CM, Chaudhary K, Tovar M, Santillan C, Sosa J, et al. High MMP-9 activity characterises pleural tuberculosis correlating with granuloma formation. Eur Respir J (2009) 33:134–41. doi:10.1183/09031936.00127807
15. Elkington PT, Green JA, Emerson JE, Lopez-Pascua LD, Boyle JJ, O’Kane CM, et al. Synergistic up-regulation of epithelial cell matrix metalloproteinase-9 secretion in tuberculosis. Am J Respir Cell Mol Biol (2007) 37:431–7. doi:10.1165/rcmb.2007-0011OC
16. Welsh KJ, Abbott AN, Hwang S-A, Indrigo J, Armitige LY, Blackburn MR, et al. A role for tumour necrosis factor-alpha, complement C5 and interleukin-6 in the initiation and development of the mycobacterial cord factor trehalose 6,6’-dimycolate induced granulomatous response. Microbiology (2008) 154:1813–24. doi:10.1099/mic.0.2008/016923-0
17. Welsh KJ, Hwang S-A, Hunter RL, Kruzel ML, Actor JK. Lactoferrin modulation of mycobacterial cord factor trehalose 6-6’-dimycolate induced granulomatous response. Transl Res (2010) 156:207–15. doi:10.1016/j.trsl.2010.06.001
18. Puissegur M-P, Lay G, Gilleron M, Botella L, Nigou J, Marrakchi H, et al. Mycobacterial lipomannan induces granuloma macrophage fusion via a TLR2-dependent, ADAM9- and 1 integrin-mediated pathway. J Immunol (2007) 178:3161–9. doi:10.4049/jimmunol.178.5.3161
19. Peyron P, Vaubourgeix J, Poquet Y, Levillain F, Botanch C, Bardou F, et al. Foamy macrophages from tuberculous patients’ granulomas constitute a nutrient-rich reservoir for M. tuberculosis persistence. PLoS Pathog (2008) 4:e1000204. doi:10.1371/journal.ppat.1000204
20. D’Avila H, Melo RCN, Parreira GG, Werneck-Barroso E, Castro-Faria-Neto HC, Bozza PT. Mycobacterium bovis bacillus Calmette-Guerin induces TLR2-mediated formation of lipid bodies: intracellular domains for eicosanoid synthesis in vivo. J Immunol (2006) 176:3087–97. doi:10.4049/jimmunol.176.5.3087
21. Tobin DM, Vary JC, Ray JP, Walsh GS, Dunstan SJ, Bang ND, et al. The lta4h locus modulates susceptibility to mycobacterial infection in zebrafish and humans. Cell (2010) 140:717–30. doi:10.1016/j.cell.2010.02.013
22. Berg RD, Levitte S, O’Sullivan MP, O’Leary SM, Cambier CJ, Cameron J, et al. Lysosomal disorders drive susceptibility to tuberculosis by compromising macrophage migration. Cell (2016) 165:139–52. doi:10.1016/j.cell.2016.02.034
23. Wolf AJ, Linas B, Trevejo-Nunez GJ, Kincaid E, Tamura T, Takatsu K, et al. Mycobacterium tuberculosis infects dendritic cells with high frequency and impairs their function in vivo. J Immunol (2007) 179:2509–19. doi:10.4049/jimmunol.179.4.2509
24. Harding JS, Rayasam A, Schreiber HA, Fabry Z, Sandor M. Mycobacterium-infected dendritic cells disseminate granulomatous inflammation. Sci Rep (2015) 5:15248. doi:10.1038/srep15248
25. Wolf AJ, Desvignes L, Linas B, Banaiee N, Tamura T, Takatsu K, et al. Initiation of the adaptive immune response to Mycobacterium tuberculosis depends on antigen production in the local lymph node, not the lungs. J Exp Med (2008) 205:105–15. doi:10.1084/jem.20071367
26. Dorhoi A, Iannaccone M, Maertzdorf J, Nouailles G, Weiner J, Kaufmann SHE. Reverse translation in tuberculosis: neutrophils provide clues for understanding development of active disease. Front Immunol (2014) 5:36. doi:10.3389/fimmu.2014.00036
27. Repasy T, Lee J, Marino S, Martinez N, Kirschner DE, Hendricks G, et al. Intracellular bacillary burden reflects a burst size for Mycobacterium tuberculosis in vivo. PLoS Pathog (2013) 9:e1003190. doi:10.1371/journal.ppat.1003190
28. Gopal R, Monin L, Torres D, Slight S, Mehra S, McKenna KC, et al. S100A8/A9 proteins mediate neutrophilic inflammation and lung pathology during tuberculosis. Am J Respir Crit Care Med (2013) 188:1137–46. doi:10.1164/rccm.201304-0803OC
29. Nouailles G, Dorhoi A, Koch M, Zerrahn J, Weiner J, Faé KC, et al. CXCL5-secreting pulmonary epithelial cells drive destructive neutrophilic inflammation in tuberculosis. J Clin Invest (2014) 124:1268–82. doi:10.1172/JCI72030
30. Blomgran R, Ernst JD. Lung neutrophils facilitate activation of naive antigen-specific CD4+ T cells during Mycobacterium tuberculosis infection. J Immunol (2011) 186:7110–9. doi:10.4049/jimmunol.1100001
31. Kang DD, Lin Y, Moreno J-R, Randall TD, Khader SA. Profiling early lung immune responses in the mouse model of tuberculosis. PLoS One (2011) 6:e16161. doi:10.1371/journal.pone.0016161
32. Seiler P, Aichele P, Bandermann S, Hauser AE, Lu B, Gerard NP, et al. Early granuloma formation after aerosol Mycobacterium tuberculosis infection is regulated by neutrophils via CXCR3-signaling chemokines. Eur J Immunol (2003) 33:2676–86. doi:10.1002/eji.200323956
33. Keller C, Hoffmann R, Lang R, Brandau S, Hermann C, Ehlers S. Genetically determined susceptibility to tuberculosis in mice causally involves accelerated and enhanced recruitment of granulocytes. Infect Immun (2006) 74:4295–309. doi:10.1128/IAI.00057-06
34. Eruslanov EB, Lyadova IV, Kondratieva TK, Majorov KB, Scheglov IV, Orlova MO, et al. Neutrophil responses to Mycobacterium tuberculosis infection in genetically susceptible and resistant mice. Infect Immun (2005) 73:1744–53. doi:10.1128/IAI.73.3.1744-1753.2005
35. Ong CWM, Elkington PT, Friedland JS. Tuberculosis, pulmonary cavitation, and matrix metalloproteinases. Am J Respir Crit Care Med (2014) 190:9–18. doi:10.1164/rccm.201311-2106PP
36. Ong CWM, Elkington PT, Brilha S, Ugarte-Gil C, Tome-Esteban MT, Tezera LB, et al. Neutrophil-derived MMP-8 drives AMPK-dependent matrix destruction in human pulmonary tuberculosis. PLoS Pathog (2015) 11:e1004917. doi:10.1371/journal.ppat.1004917
37. Knaul JK, Jörg S, Oberbeck-Mueller D, Heinemann E, Scheuermann L, Brinkmann V, et al. Lung-residing myeloid-derived suppressors display dual functionality in murine pulmonary tuberculosis. Am J Respir Crit Care Med (2014) 190:1053–66. doi:10.1164/rccm.201405-0828OC
38. Obregón-Henao A, Henao-Tamayo M, Orme IM, Ordway DJ. Gr1(int)CD11b+ myeloid-derived suppressor cells in Mycobacterium tuberculosis infection. PLoS One (2013) 8:e80669. doi:10.1371/journal.pone.0080669
39. du Plessis N, Loebenberg L, Kriel M, von Groote-Bidlingmaier F, Ribechini E, Loxton AG, et al. Increased frequency of myeloid-derived suppressor cells during active tuberculosis and after recent Mycobacterium tuberculosis infection suppresses T-cell function. Am J Respir Crit Care Med (2013) 188:724–32. doi:10.1164/rccm.201302-0249OC
40. Yang B, Wang X, Jiang J, Zhai F, Cheng X. Identification of CD244-expressing myeloid-derived suppressor cells in patients with active tuberculosis. Immunol Lett (2014) 158:66–72. doi:10.1016/j.imlet.2013.12.003
41. Ulrichs T, Kosmiadi GA, Trusov V, Jörg S, Pradl L, Titukhina M, et al. Human tuberculous granulomas induce peripheral lymphoid follicle-like structures to orchestrate local host defence in the lung. J Pathol (2004) 204:217–28. doi:10.1002/path.1628
42. Ehlers S, Schaible UE. The granuloma in tuberculosis: dynamics of a host-pathogen collusion. Front Immunol (2012) 3:411. doi:10.3389/fimmu.2012.00411
43. Guirado E, Schlesinger LS. Modeling the Mycobacterium tuberculosis granuloma – the critical battlefield in host immunity and disease. Front Immunol (2013) 4:98. doi:10.3389/fimmu.2013.00098
44. Mattila JT, Ojo OO, Kepka-Lenhart D, Marino S, Kim JH, Eum SY, et al. Microenvironments in tuberculous granulomas are delineated by distinct populations of macrophage subsets and expression of nitric oxide synthase and arginase isoforms. J Immunol (2013) 191:773–84. doi:10.4049/jimmunol.1300113
45. Ramakrishnan L. Revisiting the role of the granuloma in tuberculosis. Nat Rev Immunol (2012) 12:352–66. doi:10.1038/nri3211
46. Sasindran SJ, Torrelles JB. Mycobacterium tuberculosis infection and inflammation: what is beneficial for the host and for the bacterium? Front Microbiol (2011) 2:2. doi:10.3389/fmicb.2011.00002
47. Cooper AM. Cell-mediated immune responses in tuberculosis. Annu Rev Immunol (2009) 27:393–422. doi:10.1146/annurev.immunol.021908.132703
48. Clay H, Volkman HE, Ramakrishnan L. Tumor necrosis factor signaling mediates resistance to mycobacteria by inhibiting bacterial growth and macrophage death. Immunity (2008) 29:283–94. doi:10.1016/j.immuni.2008.06.011
49. Flynn JL, Goldstein MM, Chan J, Triebold KJ, Pfeffer K, Lowenstein CJ, et al. Tumor necrosis factor-α is required in the protective immune response against Mycobacterium tuberculosis in mice. Immunity (1995) 2:561–72. doi:10.1016/1074-7613(95)90001-2
50. Flesch IEA, Kaufmann SHE. Role of cytokines in tuberculosis. Immunobiology (1993) 189:316–39. doi:10.1016/S0171-2985(11)80364-5
51. Russell DG, Barry CE, Flynn JL. Tuberculosis: what we don’t know can, and does, hurt us. Science (2010) 328:852–6. doi:10.1126/science.1184784
52. Saunders BM, Frank AA, Orme IM, Cooper AM. CD4 is required for the development of a protective granulomatous response to pulmonary tuberculosis. Cell Immunol (2002) 216:65–72. doi:10.1016/S0008-8749(02)00510-5
53. D’Souza CD, Cooper AM, Frank AA, Ehlers S, Turner J, Bendelac A, et al. Novel nonclassic β 2-microglobulin-restricted mechanism influencing early lymphocyte accumulation and subsequent resistance to tuberculosis in the lung. Am J Respir Cell Mol Biol (2000) 23:188–93. doi:10.1165/ajrcmb.23.2.4063
54. Mogues T, Goodrich ME, Ryan L, LaCourse R, North RJ. The relative importance of T cell subsets in immunity and immunopathology of airborne Mycobacterium tuberculosis infection in mice. J Exp Med (2001) 193:271–80. doi:10.1084/jem.193.3.271
55. Nandi B, Behar SM. Regulation of neutrophils by interferon-γ limits lung inflammation during tuberculosis infection. J Exp Med (2011) 208:2251–62. doi:10.1084/jem.20110919
56. Desvignes L, Ernst JD, Banaiee N, Kincaid EZ, Buchwald U, Jacobs WR, et al. Interferon-γ-responsive nonhematopoietic cells regulate the immune response to Mycobacterium tuberculosis. Immunity (2009) 31:974–85. doi:10.1016/j.immuni.2009.10.007
57. Kim MJ, Wainwright HC, Locketz M, Bekker LG, Walther GB, Dittrich C, et al. Caseation of human tuberculosis granulomas correlates with elevated host lipid metabolism. EMBO Mol Med (2010) 2:258–74. doi:10.1002/emmm.201000079
58. Subbian S, Tsenova L, Kim M-J, Wainwright HC, Visser A, Bandyopadhyay N, et al. Lesion-specific immune response in granulomas of patients with pulmonary tuberculosis: a pilot study. PLoS One (2015) 10:e0132249. doi:10.1371/journal.pone.0132249
59. Lin PL, Rodgers M, Smith L, Bigbee M, Myers A, Bigbee C, et al. Quantitative comparison of active and latent tuberculosis in the cynomolgus macaque model. Infect Immun (2009) 77:4631–42. doi:10.1128/IAI.00592-09
60. Lin PL, Ford CB, Coleman MT, Myers AJ, Gawande R, Ioerger T, et al. Sterilization of granulomas is common in active and latent tuberculosis despite within-host variability in bacterial killing. Nat Med (2014) 20:75–9. doi:10.1038/nm.3412
61. Gideon HP, Phuah J, Myers AJ, Bryson BD, Rodgers MA, Coleman MT, et al. Variability in tuberculosis granuloma T cell responses exists, but a balance of pro- and anti-inflammatory cytokines is associated with sterilization. PLoS Pathog (2015) 11:e1004603. doi:10.1371/journal.ppat.1004603
62. Cambier CJ, Falkow S, Ramakrishnan L. Host evasion and exploitation schemes of Mycobacterium tuberculosis. Cell (2014) 159:1497–509. doi:10.1016/j.cell.2014.11.024
63. Bernut A, Herrmann J-L, Kissa K, Dubremetz J-F, Gaillard J-L, Lutfalla G, et al. Mycobacterium abscessus cording prevents phagocytosis and promotes abscess formation. Proc Natl Acad Sci U S A (2014) 111:E943–52. doi:10.1073/pnas.1321390111
64. Tobin DM, Roca FJ, Oh SF, McFarland R, Vickery TW, Ray JP, et al. Host genotype-specific therapies can optimize the inflammatory response to mycobacterial infections. Cell (2012) 148:434–46. doi:10.1016/j.cell.2011.12.023
65. Roca FJ, Ramakrishnan L. TNF dually mediates resistance and susceptibility to mycobacteria via mitochondrial reactive oxygen species. Cell (2013) 153:521–34. doi:10.1016/j.cell.2013.03.022
66. Chen M, Divangahi M, Gan H, Shin DSJ, Hong S, Lee DM, et al. Lipid mediators in innate immunity against tuberculosis: opposing roles of PGE2 and LXA4 in the induction of macrophage death. J Exp Med (2008) 205:2791–801. doi:10.1084/jem.20080767
67. Divangahi M, Behar SM, Remold H. Dying to live: how the death modality of the infected macrophage affects immunity to tuberculosis. Adv Exp Med Biol (2013) 783:103–20. doi:10.1007/978-1-4614-6111-1_6
68. Behar SM, Divangahi M, Remold HG. Evasion of innate immunity by Mycobacterium tuberculosis: is death an exit strategy? Nat Rev Microbiol (2010) 8:668–74. doi:10.1038/nrmicro2387
69. Marakalala MJ, Raju RM, Sharma K, Zhang YJ, Eugenin EA, Prideaux B, et al. Inflammatory signaling in human tuberculosis granulomas is spatially organized. Nat Med (2016) 22:531–8. doi:10.1038/nm.4073
70. Saunders BM, Cooper AM. Restraining mycobacteria: role of granulomas in mycobacterial infections. Immunol Cell Biol (2000) 78:334–41. doi:10.1046/j.1440-1711.2000.00933.x
71. Chao MC, Rubin EJ. Letting sleeping dos lie: does dormancy play a role in tuberculosis? Annu Rev Microbiol (2010) 64:293–311. doi:10.1146/annurev.micro.112408.134043
72. Canetti G. The Tubercle Bacillus in the Pulmonary Lesion of Man: Histobacteriology and Its Bearing on the Therapy of Pulmonary Tuberculosis. New York: Springer (1955).
74. Swaim LE, Connolly LE, Volkman HE, Humbert O, Born DE, Ramakrishnan L. Mycobacterium marinum infection of adult zebrafish causes caseating granulomatous tuberculosis and is moderated by adaptive immunity. Infect Immun (2006) 74:6108–17. doi:10.1128/IAI.00887-06
75. Kindler V, Sappino A-P, Grau GE, Piguet P-F, Vassalli P. The inducing role of tumor necrosis factor in the development of bactericidal granulomas during BCG infection. Cell (1989) 56:731–40. doi:10.1016/0092-8674(89)90676-4
76. Roach DR, Bean AGD, Demangel C, France MP, Briscoe H, Britton WJ. TNF regulates chemokine induction essential for cell recruitment, granuloma formation, and clearance of mycobacterial infection. J Immunol (2002) 168:4620–7. doi:10.4049/jimmunol.168.9.4620
77. Bean AG, Roach DR, Briscoe H, France MP, Korner H, Sedgwick JD, et al. Structural deficiencies in granuloma formation in TNF gene-targeted mice underlie the heightened susceptibility to aerosol Mycobacterium tuberculosis infection, which is not compensated for by lymphotoxin. J Immunol (1999) 162:3504–11.
78. Algood HMS, Lin PL, Flynn JL. Tumor necrosis factor and chemokine interactions in the formation and maintenance of granulomas in tuberculosis. Clin Infect Dis (2005) 41:S189–93. doi:10.1086/429994
79. Chakravarty SD, Zhu G, Tsai MC, Mohan VP, Marino S, Kirschner DE, et al. Tumor necrosis factor blockade in chronic murine tuberculosis enhances granulomatous inflammation and disorganizes granulomas in the lungs. Infect Immun (2008) 76:916–26. doi:10.1128/IAI.01011-07
80. Cooper AM, Dalton DK, Stewart TA, Griffin JP, Russell DG, Orme IM. Disseminated tuberculosis in interferon gamma gene-disrupted mice. J Exp Med (1993) 178:2243–7. doi:10.1084/jem.178.6.2243
81. Cooper AM, Magram J, Ferrante J, Orme IM. Interleukin 12 (IL-12) is crucial to the development of protective immunity in mice intravenously infected with Mycobacterium tuberculosis. J Exp Med (1997) 186:39–45. doi:10.1084/jem.186.1.39
82. Sugawara I, Yamada H, Mizuno S. Relative importance of STAT4 in murine tuberculosis. J Med Microbiol (2003) 52:29–34. doi:10.1099/jmm.0.05026-0
83. Scanga CA, Bafica A, Feng CG, Cheever AW, Hieny S, Sher A. MyD88-deficient mice display a profound loss in resistance to Mycobacterium tuberculosis associated with partially impaired Th1 cytokine and nitric oxide synthase 2 expression. Infect Immun (2004) 72:2400–4. doi:10.1128/IAI.72.4.2400-2404.2004
84. Fremond CM, Yeremeev V, Nicolle DM, Jacobs M, Quesniaux VF, Ryffel B. Fatal Mycobacterium tuberculosis infection despite adaptive immune response in the absence of MyD88. J Clin Invest (2004) 114:1790–9. doi:10.1172/JCI21027
85. Fremond CM, Togbe D, Doz E, Rose S, Vasseur V, Maillet I, et al. IL-1 receptor-mediated signal is an essential component of MyD88-dependent innate response to Mycobacterium tuberculosis infection. J Immunol (2007) 179:1178–89. doi:10.4049/jimmunol.179.2.1178
86. Davis JM, Clay H, Lewis JL, Ghori N, Herbomel P, Ramakrishnan L. Real-time visualization of mycobacterium-macrophage interactions leading to initiation of granuloma formation in zebrafish embryos. Immunity (2002) 17:693–702. doi:10.1016/S1074-7613(02)00475-2
87. Choi H-H, Shin D-M, Kang G, Kim K-H, Park JB, Hur GM, et al. Endoplasmic reticulum stress response is involved in Mycobacterium tuberculosis protein ESAT-6-mediated apoptosis. FEBS Lett (2010) 584:2445–54. doi:10.1016/j.febslet.2010.04.050
88. Derrick SC, Morris SL. The ESAT6 protein of Mycobacterium tuberculosis induces apoptosis of macrophages by activating caspase expression. Cell Microbiol (2007) 9:1547–55. doi:10.1111/j.1462-5822.2007.00892.x
89. Mishra BB, Moura-Alves P, Sonawane A, Hacohen N, Griffiths G, Moita LF, et al. Mycobacterium tuberculosis protein ESAT-6 is a potent activator of the NLRP3/ASC inflammasome. Cell Microbiol (2010) 12:1046–63. doi:10.1111/j.1462-5822.2010.01450.x
90. Cosma CL, Humbert O, Sherman DR, Ramakrishnan L. Trafficking of superinfecting mycobacterium organisms into established granulomas occurs in mammals and is independent of the Erp and ESX-1 mycobacterial virulence loci. J Infect Dis (2008) 198:1851–5. doi:10.1086/593175
91. Cosma CL, Humbert O, Ramakrishnan L. Superinfecting mycobacteria home to established tuberculous granulomas. Nat Immunol (2004) 5:828–35. doi:10.1038/ni1091
92. Wayne LG, Hayes LG. An in vitro model for sequential study of shiftdown of Mycobacterium tuberculosis through two stages of non-replicating persistence. Infect Immun (1996) 64:2062–9.
93. Boon C, Dick T. Mycobacterium bovis BCG response regulator essential for hypoxic dormancy. J Bacteriol (2002) 184:6760–7. doi:10.1128/JB.184.24.6760-6767.2002
94. Leistikow RL, Morton RA, Bartek IL, Frimpong I, Wagner K, Voskuil MI. The Mycobacterium tuberculosis DosR regulon assists in metabolic homeostasis and enables rapid recovery from nonrespiring dormancy. J Bacteriol (2010) 192:1662–70. doi:10.1128/JB.00926-09
95. Oehlers SH, Cronan MR, Scott NR, Thomas MI, Okuda KS, Walton EM, et al. Interception of host angiogenic signalling limits mycobacterial growth. Nature (2014) 517:612–5. doi:10.1038/nature13967
96. Zhang YJ, Reddy MC, Ioerger TR, Rothchild AC, Dartois V, Schuster BM, et al. Tryptophan biosynthesis protects mycobacteria from CD4 T-cell-mediated killing. Cell (2013) 155:1296–308. doi:10.1016/j.cell.2013.10.045
97. Dorhoi A, Kaufmann SHE. Perspectives on host adaptation in response to Mycobacterium tuberculosis: modulation of inflammation. Semin Immunol (2014) 26:533–42. doi:10.1016/j.smim.2014.10.002
98. Zumla A, Rao M, Parida SK, Keshavjee S, Cassell G, Wallis R, et al. Inflammation and tuberculosis: host-directed therapies. J Intern Med (2015) 277:373–87. doi:10.1111/joim.12256
99. Uhlin M, Andersson J, Zumla A, Maeurer M. Adjunct immunotherapies for tuberculosis. J Infect Dis (2012) 205:S325–34. doi:10.1093/infdis/jis197
100. Skerry C, Harper J, Klunk M, Bishai WR, Jain SK. Adjunctive TNF inhibition with standard treatment enhances bacterial clearance in a murine model of necrotic TB granulomas. PLoS One (2012) 7:e39680. doi:10.1371/journal.pone.0039680
101. Wallis RS, Broder MS, Wong JY, Hanson ME, Beenhouwer DO. Granulomatous infectious diseases associated with tumor necrosis factor antagonists. Clin Infect Dis (2004) 38:1261–5. doi:10.1086/383317
102. Keane J, Gershon S, Wise RP, Mirabile-Levens E, Kasznica J, Schwieterman WD, et al. Tuberculosis associated with infliximab, a tumor necrosis factor α-neutralizing agent. N Engl J Med (2001) 345:1098–104. doi:10.1056/NEJMoa011110
103. Mayer-Barber KD, Andrade BB, Oland SD, Amaral EP, Barber DL, Gonzales J, et al. Host-directed therapy of tuberculosis based on interleukin-1 and type I interferon crosstalk. Nature (2014) 511:99–103. doi:10.1038/nature13489
104. Coulombe F, Jaworska J, Verway M, Tzelepis F, Massoud A, Gillard J, et al. Targeted prostaglandin E2 inhibition enhances antiviral immunity through induction of type I interferon and apoptosis in macrophages. Immunity (2014) 40:554–68. doi:10.1016/j.immuni.2014.02.013
105. Manca C, Tsenova L, Freeman S, Barczak AK, Tovey M, Murray PJ, et al. Hypervirulent M. tuberculosis W/Beijing strains upregulate type I IFNs and increase expression of negative regulators of the Jak-Stat pathway. J Interferon Cytokine Res (2005) 25:694–701. doi:10.1089/jir.2005.25.694
106. Ordway D, Henao-Tamayo M, Harton M, Palanisamy G, Troudt J, Shanley C, et al. The hypervirulent Mycobacterium tuberculosis strain HN878 induces a potent TH1 response followed by rapid down-regulation. J Immunol (2007) 179:522–31. doi:10.4049/jimmunol.179.1.522
107. Stanley SA, Johndrow JE, Manzanillo P, Cox JS. The type I IFN response to infection with Mycobacterium tuberculosis requires ESX-1-mediated secretion and contributes to pathogenesis. J Immunol (2007) 178:3143–52. doi:10.4049/jimmunol.178.5.3143
108. Manca C, Tsenova L, Bergtold A, Freeman S, Tovey M, Musser JM, et al. Virulence of a Mycobacterium tuberculosis clinical isolate in mice is determined by failure to induce Th1 type immunity and is associated with induction of IFN-alpha/beta. Proc Natl Acad Sci U S A (2001) 98:5752–7. doi:10.1073/pnas.091096998
109. Berry MPR, Graham CM, McNab FW, Xu Z, Bloch SAA, Oni T, et al. An interferon-inducible neutrophil-driven blood transcriptional signature in human tuberculosis. Nature (2010) 466:973–7. doi:10.1038/nature09247
110. Cliff JM, Lee J-S, Constantinou N, Cho J-E, Clark TG, Ronacher K, et al. Distinct phases of blood gene expression pattern through tuberculosis treatment reflect modulation of the humoral immune response. J Infect Dis (2013) 207:18–29. doi:10.1093/infdis/jis499
111. Maertzdorf J, Repsilber D, Parida SK, Stanley K, Roberts T, Black G, et al. Human gene expression profiles of susceptibility and resistance in tuberculosis. Genes Immun (2011) 12:15–22. doi:10.1038/gene.2010.51
112. Ottenhoff THM, Dass RH, Yang N, Zhang MM, Wong HEE, Sahiratmadja E, et al. Genome-wide expression profiling identifies type 1 interferon response pathways in active tuberculosis. PLoS One (2012) 7:e45839. doi:10.1371/journal.pone.0045839
113. de Paus RA, van Wengen A, Schmidt I, Visser M, Verdegaal EME, van Dissel JT, et al. Inhibition of the type I immune responses of human monocytes by IFN-α and IFN-β. Cytokine (2013) 61:645–55. doi:10.1016/j.cyto.2012.12.005
114. Novikov A, Cardone M, Thompson R, Shenderov K, Kirschman KD, Mayer-Barber KD, et al. Mycobacterium tuberculosis triggers host type I IFN signaling to regulate IL-1 production in human macrophages. J Immunol (2011) 187:2540–7. doi:10.4049/jimmunol.1100926
115. Singhal A, Jie L, Kumar P, Hong GS, Leow MK-S, Paleja B, et al. Metformin as adjunct antituberculosis therapy. Sci Transl Med (2014) 6:263ra159. doi:10.1126/scitranslmed.3009885
116. Vilaplana C, Marzo E, Tapia G, Diaz J, Garcia V, Cardona P-J. Ibuprofen therapy resulted in significantly decreased tissue bacillary loads and increased survival in a new murine experimental model of active tuberculosis. J Infect Dis (2013) 208:199–202. doi:10.1093/infdis/jit152
117. Ivanyi J, Zumla A. Nonsteroidal antiinflammatory drugs for adjunctive tuberculosis treatment. J Infect Dis (2013) 208:185–8. doi:10.1093/infdis/jit153
118. Griffin JE, Pandey AK, Gilmore SA, Mizrahi V, Mckinney JD, Bertozzi CR, et al. Cholesterol catabolism by Mycobacterium tuberculosis requires transcriptional and metabolic adaptations. Chem Biol (2012) 19:218–27. doi:10.1016/j.chembiol.2011.12.016
119. Pandey AK, Sassetti CM. Mycobacterial persistence requires the utilization of host cholesterol. Proc Natl Acad Sci U S A (2008) 105:4376–80. doi:10.1073/pnas.0711159105
120. Endo A. The discovery and development of HMG-CoA reductase inhibitors. J Lipid Res (1992) 33:1569–82.
121. Ray KK, Seshasai SRK, Erqou S, Sever P, Jukema JW, Ford I, et al. Statins and all-cause mortality in high-risk primary prevention. Arch Intern Med (2010) 170:1024. doi:10.1001/archinternmed.2010.182
122. Liao JK, Laufs U. Pleitropic effects of statins. Annu Rev Pharmacol Toxicol (2005) 45:89–118. doi:10.1146/annurev.pharmtox.45.120403.095748
123. Mach F, Kwak B, Mulhaupt F, Myit S. Statins as a newly recognized type of immunomodulator. Nat Med (2000) 6:1399–402. doi:10.1038/82219
124. Parihar SP, Guler R, Khutlang R, Lang DM, Hurdayal R, Mhlanga MM, et al. Statin therapy reduces the Mycobacterium tuberculosis burden in human macrophages and in mice by enhancing autophagy and phagosome maturation. J Infect Dis (2014) 209:754–63. doi:10.1093/infdis/jit550
125. Lobato LS, Rosa PS, Ferreira JdS, Neumann AdS, da Silva MG, do Nascimento DC, et al. Statins increase rifampin mycobactericidal effect. Antimicrob Agents Chemother (2014) 58:5766–74. doi:10.1128/AAC.01826-13
126. Skerry C, Pinn ML, Bruiners N, Pine R, Gennaro ML, Karakousis PC. Simvastatin increases the in vivo activity of the first-line tuberculosis regimen. J Antimicrob Chemother (2014) 69:2453–7. doi:10.1093/jac/dku166
127. Dutta NK, Bruiners N, Pinn ML, Zimmerman MD, Prideaux B, Dartois V, et al. Statin adjunctive therapy shortens the duration of TB treatment in mice. J Antimicrob Chemother (2016) 71:1570–7. doi:10.1093/jac/dkw014
128. Rittershaus ESC, Baek S-H, Sassetti CM. The normalcy of dormancy: common themes in microbial quiescence. Cell Host Microbe (2013) 13:643–51. doi:10.1016/j.chom.2013.05.012
129. Stanley SA, Barczak AK, Silvis MR, Luo SS, Sogi K, Vokes M, et al. Identification of host-targeted small molecules that restrict intracellular Mycobacterium tuberculosis growth. PLoS Pathog (2014) 10:e1003946. doi:10.1371/journal.ppat.1003946
Keywords: tuberculosis, Mycobacterium tuberculosis, granuloma, inflammation, host-directed therapy, drug-resistant TB, MDR-TB, tissue pathology
Citation: Ndlovu H and Marakalala MJ (2016) Granulomas and Inflammation: Host-Directed Therapies for Tuberculosis. Front. Immunol. 7:434. doi: 10.3389/fimmu.2016.00434
Received: 01 September 2016; Accepted: 04 October 2016;
Published: 24 October 2016
Edited by:
Jeffrey K. Actor, UTHealth Science Center, USAReviewed by:
Ying Kong, University of Tennessee Health Science Center, USACopyright: © 2016 Ndlovu and Marakalala. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mohlopheni J. Marakalala, bWoubWFyYWthbGFsYUB1Y3QuYWMuemE=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.