
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 30 September 2016
Sec. Multiple Sclerosis and Neuroimmunology
Volume 7 - 2016 | https://doi.org/10.3389/fimmu.2016.00397
This article is part of the Research Topic Neuroimmune interface in health and diseases View all 15 articles
 Céline Marban1
Céline Marban1 Faezeh Forouzanfar2Amina Ait-Ammar2Faiza Fahmi2Hala El Mekdad2,3Fadoua Daouad2
Faezeh Forouzanfar2Amina Ait-Ammar2Faiza Fahmi2Hala El Mekdad2,3Fadoua Daouad2 Olivier Rohr2,3,4*
Olivier Rohr2,3,4* Christian Schwartz2,3*
Christian Schwartz2,3*
One of the top research priorities of the international AIDS society by the action “Towards an HIV Cure” is the purge or the decrease of the pool of all latently infected cells. This strategy is based on reactivation of latently reservoirs (the shock) followed by an intensifying combination antiretroviral therapy (cART) to kill them (the kill). The central nervous system (CNS) has potential latently infected cells, i.e., perivascular macrophages, microglial cells, and astrocytes that will need to be eliminated. However, the CNS has several characteristics that may preclude the achievement of a cure. In this review, we discuss several limitations to the eradication of brain reservoirs and how we could circumvent these limitations by making it efforts in four directions: (i) designing efficient latency-reversal agents for CNS-cell types, (ii) improving cART by targeting HIV transcription, (iii) improving delivery of HIV drugs in the CNS and in the CNS-cell types, and (iv) developing therapeutic immunization. As a prerequisite to these efforts, we also believe that a better comprehension of molecular mechanisms involved in establishment and persistence of HIV latency in brain reservoirs are essential to design new molecules for strategies aiming to achieve a cure for instance the “shock and kill” strategy.
Combination antiretroviral therapy (cART), introduced in 1996, has radically improved the management of HIV-1 infection and decreased both morbidity and mortality. However, despite initial hopes to cure HIV, treatments were unable to fully eliminate the virus (1–3). Indeed, with very sensitive methods (4–6), a remaining viremia is always noticed in patients on cART. Moreover, HIV RNA returns to a measurable plasma level when cART is disrupted (7, 8). The origin of this persistent viremia is still a matter of debate (9–11). Latent persistence of HIV in long-lived cells, such as the central memory CD4+ T-cells, hematopoietic stem cells, dendritic cells, and cells from the monocyte–macrophages lineage in the form of proviruses have been described (1, 2, 12–19). Moreover, these cells are located in a variety of anatomical sites, including tissues, such as blood, brain, gut-associated lymphoid tissue, bone marrow, and genital tract (20), making it difficult to purge the virus from all the reservoirs.
These latently infected cells are from time to time reactivated and produce HIV particles at low levels, thus explaining the persistence of viremia. An alternative theory, the cryptic ongoing replication states that despite cART, HIV is continuously produced at low levels. The inefficiency of the treatment in cells supporting ongoing replication could be due to poor drug penetration in sanctuaries, such as the brain (21) or by cell-to-cell transfer of the virus (22). In theory, there are critical therapeutic implications for cART as it is expected that during ongoing replication, drug resistance might arise (23–26). The potential mechanisms of HIV persistence have been discussed recently in a review by Hong and Mellors (27).
One of the main debates in the field of HIV reservoir is whether or not the central nervous system (CNS) constitutes a real viral reservoir. Indeed, with its unique features, such as the existence of a blood–brain barrier (BBB) with poor drug penetration, the CNS might be considered as a sanctuary (20) made of specific cell types (28) with reduced immune surveillance. Moreover, the anatomy of the CNS is such that there is poor viral genetic information exchange with the other sites and, thus, might be referred as a compartment (20, 29, 30).
First, we will give our opinion on the existence of viral reservoirs in the CNS referring to excellent recent reviews in this topic. Next, we will discuss the importance to purge these potential viral reservoirs. Indeed, in theory, it is possible to acquire virus resistance to cART if there is an ongoing replication in the brain. Another major concern is the existence of HIV-associated neurocognitive disorders (HAND). In up to 50% of the HIV-infected patients on efficient cART and undetectable virus load (≤50 copies/ml), HAND has been recorded. Several mechanisms are evoked to explain the increase of less severe forms of HAND in which production of some viral proteins occurs during reactivation or cryptic ongoing HIV replication. We will then review the state of art of what is known regarding the molecular mechanisms underlying the establishment and persistence of HIV in the potential reservoirs in the brain and, finally, discuss the profound therapeutic implications of purging reservoirs.
A viral reservoir is an infected cell population that allows persistence of replication-competent virus in patients under cART (20). According to this definition, the only true reservoirs are the resting CD4+ T-cells. Indeed, these cells fulfill all the criteria to be considered as a real reservoir, i.e., presence of integrated virus in long-lived cells, persistence of high levels of virus in a quiescent/latent state in the reservoir and possible reactivation of the virus with the formation of replication-competent particles (31).
There are several evidences that brain cells harbor genome-integrated HIV (28). We know that the virus invades the brain very soon following infection. Virus infection was shown in astrocytes (32), in perivascular macrophages (32), and in microglial cells (33). All three cell types are long-lived cells with perivascular macrophages (34) and astrocytes (35) with a half-life ranging from months and microglial cells with a half-life of years (36). All these cells are infected at high frequency in the brain. Astrocytes, the most abundant cell type in the brain, are infected in up to 19% of the cell population (37). Similar ratio of infected cells has been found among the perivascular macrophages and the microglial cells (33, 38). In addition, several mechanisms, including epigenetic regulation, have been evoked to induce latency in these cells notably in astrocytes and microglial cells (39–42).
Due to ethical and technical problems, it is not possible to evaluate the human brain-infected cells for their capacity to produce replication-competent viruses. However, there are several indirect evidences showing that CNS is a reservoir for HIV. Indeed, HIV DNA has been detected in brain tissues isolated from autopsies of HIV patients whose infection has been controlled by cART (33, 39). Moreover, there is a strong correlation of the amount of HIV DNA found in astrocytes and HIV-associated dementia (HAD) (37). Various animal models have been used to show persistence of HIV infection in the CNS as brain biopsy is not possible. Indeed, several animal models, such as macaque, rats, and humanized BLT mouse, have been used to mimic the condition of HIV-infected patients on cART, which confirmed the presence of viral RNA or viral proteins in the brain (43–45). Specifically, in the macaque model, a mechanism of the establishment of transcriptional HIV latency in the CNS has been suggested (46). They notably showed that interferon beta repressed SIV LTR activity by inducing C/EBPγ expression, a dominant negative isoform of C/EBPβ (47). There are also several evidences supporting continuous CNS perturbation despite an efficient cART (48) with an increase of the prevalence of milder form of HAND. Moreover, in patients under suppressive cART activation of the immune system is still observed in the CNS with some biomarkers, such as neopterin or NFL being detected in the cerebrospinal fluid (CSF) (49). One explanation is the existence of an inflammatory process that might be driven by low-level HIV replication in infected cells (50, 51). Interestingly, neuroimaging data are also in favor of persistent CNS inflammation in patients on cART (52, 53). Finally, development of highly sensitive methods, such as single-copy assay (SCA), has allowed the detection of HIV RNA in the CSF from infected patients on cART or from elite controllers whose HIV RNA level was initially undetectable in the plasma and CSF (54–56). The recent discovery of a CSF viral escape in patients on cART with undetectable plasma HIV RNA but with neurological impairment argue also for the existence of a persistent HIV reservoir in the brain (55–59). In conclusion, there are now several evidences supporting that CNS is a reservoir for HIV even if it is still controversial. Readers will be referred to the following reviews that nourish the debate of whether or not CNS serves as a HIV reservoir (60–63).
The CNS is involved in the control of most functions of the body and mind. The brain operates in a very well controlled microenvironment separated from the other parts of the body by two barriers: the choroid plexus and the BBB. The two barriers, but predominantly the BBB, constitute physical barriers and any perturbation of their integrity will be associated with neurological diseases. There are several other features that make the CNS unique. The CNS has specific immunological features; principally an innate immune response through the perivascular macrophages and the microglial cells. However, the adaptive immune response has also been observed and, thus, contributes to the immune surveillance in the CNS (64, 65). Indeed, leukocytes trafficking to the CSF either by traversing the BBB to the perivascular space or the choroid plexus has been detected (66). More interestingly, in patients having CSF/plasma HIV discordance (patients having higher levels of HIV RNA in CSF than in blood) even at very low levels it was demonstrated that both innate (macrophages and microglial cells) and adaptive (T CD4+ and CD8+ lymphocytes) are involved in CNS injury (67–70). It has been shown that the percentage of a specific set of T CD8+ lymphocytes that expresses interferon γ is higher in the CSF than in blood. Moreover, this higher percentage of T CD8+ cells in CSF versus blood contributes to the occurrence of HAND (67) [reviewed in Ref. (71)]. Within 2 weeks following acute infection by HIV, the virus enters the CNS. There are at least two mechanisms to explain how HIV crosses the BBB, including trafficking of cell free virus and infected cells (72). The well-documented infection of the CNS is accomplished through infected cells and, thus, has been named the “Trojan Horse” mechanism (73). A recent study using natalizumab, an anti-α4 blocking antibody preventing both lymphocytes and monocytes trafficking across the BBB, is in accordance with this mechanism. Indeed, a drastic decrease of SIV DNA in the brain was observed when natalizumab was given to rhesus macaque during acute SIV infection (74). According to this theory, infected monocytes cross the BBB and infect the perivascular macrophages, the microglial cells, and the astrocytes that result in HIV-associated neurological disorders (75). Since the introduction of cART, an important decrease in the incidence of the severe form of HAND has been noticed (76). However, there is an increase of milder form of the infection (up to 50%), which might be largely under diagnosed. Thus, better screening tools to detect HAND are required in the future (77). The reasons for the increase of the prevalence of milder forms of HAND are not fully understood. One explanation might be related to the existence of quiescent/latent viral reservoirs in the CNS that emphasizes the importance of eradicating the reservoirs. Another major concern related to the existence of such quiescent/latent reservoirs in the CNS is that it might be a source of new particles that could replenish the periphery blood. These notions will be discussed in the later chapters.
HIV-associated neurocognitive disorders have been divided into three subgroups according to the Frascati criteria, i.e., asymptomatic neurocognitive impairment (ANI), mild neurocognitive disorder (MND), and HAD (78). These disorders are associated with the entry of HIV into the CNS that occurs almost immediately after systemic infection (79). The more severe form of HAND, i.e., HAD has drastically decreased with the introduction of cART. However, the less severe forms (MND and ANI) have continued with a prevalence ranging from 20% to up to 50%, while keeping in mind that these milder forms are often under diagnosed (80, 81). However, the details of persistence of these less severe forms of HAND in patients on cART are not fully understood. There are at least two hallmarks of HIV infection in the brain, i.e., chronic immune activation and compromised BBB integrity in which the central role for HIV neuropathogenesis is played by the monocytes/macrophages (82–85). Importantly, immune activation still occurs in patients on cART (50, 51). The exact mechanisms of such pathogenesis are not entirely known and rely on two models: a direct and an indirect model (86, 87). In the direct model, infected cells will cause neuronal death through the action of newly synthesized viral proteins, such as Tat, gp120, Vpr, and Nef. The two major viral proteins that lead to neuronal injury are Tat and gp120. Their effects are mediated through their interaction with neuronal cell receptors, such as the NMDA receptor and the chemokine receptors (CCR5 and CXCR4). More details on the mechanisms involved in the neuropathogenesis caused by viral proteins are found in the review (88). In the indirect model, sustained chronic inflammation is induced by secreting perivascular macrophages, microglial cells, and to a lesser extent by astrocytes releasing neurotoxic host factors. Among these secreted products, there are proinflamatory cytokines (TNFα, IL-1β, IL-6, IL-8, and INFα), chemokines (CCL2 and CCL5), and small molecules, such as quinolinic acid and the platelet-activating factor. Moreover, these viral proteins and cellular factors increase the oxidative stress and alter the integrity of the BBB which in turn results in the stimulation of even more infected cells in the brain. Further investigations are needed to decipher the exact mechanisms involved in CNS injury. Interestingly, Tat might be involved in both direct and indirect processes that lead ultimately to neuronal death. Potential roles and functions of Tat in both direct and indirect neurotoxicities have been described elsewhere (89, 90). The importance of Tat is still a matter of debate since there are controversies regarding the amount of Tat present in the CNS cells environment and the amount of Tat used in in vitro experiments. In favor of its importance is the use of Tat transgenic animal model where CNS injury has been observed (91, 92). Therefore, it will be essential to detect Tat in the brain from patients on cART. It is possible that this protein might arise from quiescent/latent reservoirs and, therefore, be responsible for the milder form of HAND. Improvement of cART by targeting the production phase of HIV-1, including transcription appears, therefore, crucial (93). Indeed, current cART is not targeting this step and since the CNS infection occurs almost immediately during acute infection, establishment of infected reservoirs will not be prevented. Moreover, strategies aiming to purge the reservoirs are based on HIV reactivation with the risk that viral proteins, such as Tat will be produced in the brain. HIV-1-mediated neuropathogenesis might also involve a dynamic interaction between astrocytes and peripheral blood mononuclear cells (PBMCs) (94). Indeed, a recent report showed that astrocytes susceptibility to produce HIV infection is enhanced by PBMCs producing interferon γ which in turn inhibit HIV-1 production in PBMCs through the secretion of small glycoprotein, i.e., the Wtns. These later proteins have been shown to be involved in many CNS processes (95), such as synaptic plasticity and neurotransmitter release, which might explain partly HIV-1-mediated neuropathogenesis.
The CNS has two special features making it difficult the achievement of a cure. First of all, the CNS is considered as a sanctuary for HIV by pharmacologic means as it is a site with limited access to antiretroviral drugs (ARV) (96–99). As an outcome, there is a risk to allow the occurrence of virus resistant to the current drugs used in cART. Second, the CNS is also considered as a compartment in which the virus is isolated from other parts of the body (29, 100). Because of poor genetic information exchange with the other sites, neurotropic variants of HIV might be selected, which most likely will not respond to treatment in a similar way than the virus encountered in the CD4+ T-cells, the main target in the body. There are now numerous evidences supporting the fact that the CNS-resident virus has evolved to become macrophage tropic (101). Indeed, sequence analysis of the env gene and of the HIV-1 promoter (LTR) argue for the compartmentalization of HIV variants in the CNS (102–105). Variations in the promoter are important since mechanisms involved in the establishment and persistence of latency in the CNS might differ from the one described in CD4+ T-cells. As mentioned above, this will impact the efficiency of latency-reversing agents (LRA) in strategies aiming to purge the latent/quiescent reservoirs (106, 107). Another major concern regarding the necessity to purge the CNS reservoirs is related to the discovery of CNS viral escape in patients on cART (108). Initial studies have shown occasional cases of virus escapes in the CSF (109, 110). Development of highly sensitive assays has even allowed the detection of CSF HIV RNA, which were not detectable with previous assays (111). Indeed in a report, evaluation of CSF viral escape has been done in a cohort of neurologically asymptomatic patients successfully treated with cART. It was shown that around 10% of these patients had detectable CSF HIV RNA, suggesting that viral escape may be underestimated (112). The recent discovery of a CNS viral escape in a cohort of 14 patients on cART with undetectable plasma HIV RNA but who developed HIV-encephalitis argues for the possibility that CNS is a real reservoir (57). Actually, this study and others raise the question that CNS-specific viral replication can occur in patients on cART from reactivated reservoirs which in theory may have escaped therapy and ultimately lead to drugs resistance (58, 59, 113). Very interestingly a similar drug-privileged site, i.e., the lymphoid tissue has been shown to have low access to drugs (114). The authors notably showed that the virus is continuously produced and might be a source of HIV from which replenishment of blood occurs. However, and contrary to the brain, they do not show that resistance to antiretroviral drugs arises. The authors of this study suggest that this absence of resistance to ARV might be explained by the too low level of drug concentration in lymphoid tissue that is not sufficient to confer competitive advantages to the development of drug-resistant viruses. This study point out to the importance of developing new ways to deliver drugs in all sanctuaries, including brain and lymphoid tissues (115).
Overall, we suggest that it is crucial to eradicate brain reservoirs since ARV-resistant viruses are capable to replenish the systemic circulation from these reservoirs. It will also imply that CSF analysis in patients on cART should be performed more often since it will greatly help assessing the compartmentalization of HIV in the brain and monitoring the efficiency of new treatments (116). Notably, CSF might be used to evaluate HIV drug resistance.
Establishment and persistence of HIV latency occur in brain cells, i.e., perivascular macrophages, microglial cells, and astrocytes. Infection of these cells differs from the infection of blood cells infected, mainly the CD4+ T-cells. Indeed, HIV infection in macrophages is not lytic and these cells are far more resistant to cytopathic effects. Moreover, infected monocyte–macrophage cells are also more resistant to apoptosis, a major obstacle for the eradication of the virus. These cells may harbor latent viruses for months (perivascular macrophages) or for years (microglial cells). Astrocytes are also thought to be infected by HIV-1 despite the lack of the co-receptors CCR5 and CXCR4 probably through the involvement of vesicles (38). However, the infection appears to be non-productive with only early transcripts, such as tat and nef, that are detectable at very low level (117).
Understanding the intimate mechanisms underlying HIV-1 latency in these CNS-specific cells is necessary to develop new and original therapies for viral eradication. The molecular mechanisms underlying these therapies are determined by the cellular specificity of HIV gene transcription and the variability of the LTR found in viruses having evolved in the brain (61, 118). For example, it has been shown in microglial cells that Sp3 and a truncated form of C/EBPβ (NF-IL6) inhibit the basal transcriptional activity of HIV-1 (47). Such a reduced basal and Tat-activated transcriptional activity has also been shown in astrocytes. Transcriptional silencing has been associated with low levels of TAR RNA binding proteins (TRBP) and with mutations of the SP motifs found within the LTR of brain-derived HIV-1. Mutations prevent the transcription factor Sp1 to bind the promoter and, thus, inhibit transcriptional activation (119, 120). However, the main mechanism involved in establishment and persistence of latency involves epigenetic regulation (41, 121, 122). In our laboratory, we showed that the cellular factor COUP-TF interacting protein (CTIP2) is a key factor in the establishment and persistence of HIV latency in microglial cells (123). We notably showed that this protein serves as a platform to anchor several protein complexes having different functions. Indeed, at least two different complexes containing CTIP2 are involved in the establishment and the persistence of HIV-1 latency (Figure 1). Moreover, CTIP2 is also involved in the control of cellular genes of importance for the virus. Among these factors, the cellular cyclin-dependent kinase inhibitor CDKN1A/p21waf has been described to favor HIV-1 gene transcription in the monocyte–macrophage lineage. This effect indirectly favors HIV-1 latency since activation of the p21 gene stimulates viral expression in macrophages (124). Moreover, CTIP2 counteracts HIV-1 Vpr protein that is required for p21 expression (125). We, therefore, suggested that CTIP2 generates a cellular environment disfavoring viral reactivation and, thus, favoring HIV-1 latency.
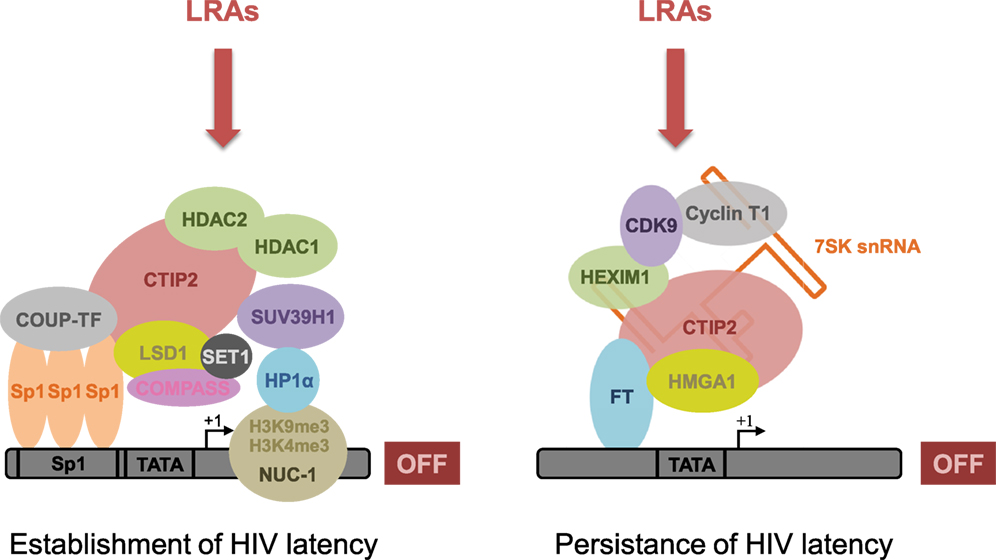
Figure 1. CTIP2 promotes the establishment and persistence of HIV-1 latency through the recruitment of two macromolecular complexes on the HIV-1 promoter in microglial cells. CTIP2 participates in the establishment of HIV-1 latency by recruiting a chromatin-modifying complex at the HIV-1 promoter. This complex consists of two histone deacetylases: HDAC1 and HDAC2 that are responsible for H3K9 deacetylation of Nuc-1, a nucleosome located immediately downstream of the HIV-1 transcriptional start site. The histone methyltransferase SUV39H1 takes also part of the complex and catalyzes the tri-methylation of H3K9 on Nuc-1. Finally, HP1α, a protein associated with heterochromatin, specifically recognizes H3K9me3 and spreads along the HIV-1 promoter, thus creating a domain of heterochromatin refractory to transcription. In parallel, CTIP2 also recruits the histone demethylase complex LSD1/COMPASS/SET1 that, in association with the histone marks H3K9me3 and H3K4me3, contributes to HIV-1 gene silencing and, therefore, the establishment of HIV-1 latency. Besides, by recruiting a transcriptional inhibitory complex at the HIV-1 promoter, CTIP2 is also involved in the prevention of HIV-1 reactivation. This complex is an inactive form of the elongation factor pTEFb and consists of pTEFb, HEXIM1, HMGA1, and the snRNA 7SK. Due to their involvement in HIV-1 establishment and persistence of HIV-1 latency, CTIP2-associated proteins from both complexes constitute new pharmacological targets to reverse HIV-1 latency. Accordingly, new latency-reversing agents (LRAs) are currently being developed or undergoing clinical trials with the aim of reversing HIV-1 latency and depleting HIV-1 reservoirs.
The first CTIP2-associated complex described in our laboratory has been involved in the establishment of HIV-1 latency through the induction of heterochromatin in the vicinity of the viral promoter (Figure 1, left complex). Indeed, we showed that CTIP2 recruits a chromatin-modifying complex through the Sp1 sites of the proximal promoter (42). This complex contains the histone deacetylases HDAC1, HDAC2, and histone methyltransferase SUV39H1 that specifically demethylates lysine 9 of histone H3. This histone modification allows heterochromatin protein 1 (HP1) binding, heterochromatin formation, and hence HIV silencing (42, 126, 127). In a consecutive study, we have shown that CTIP2 interacts physically and functionally with the lysine-specific demethylase (LSD1) to repress HIV-1 transcription and viral expression in a synergistic manner (128). The recruitment of LSD1 at the HIV-1 proximal promoter has been associated with both H3K4me3 and H3K9me3 epigenetic marks, which is linked to the recruitment of hSet1 and WDR5, two members of the hCOMPASS complex, on the HIV-1 promoter (128). Recruitment of CTIP2 on the p21 promoter also induces a heterochromatin environment. Moreover, CTIP2 has been shown to silence p21 gene transcription by creating epigenetic marks of repression, as described above for the HIV-1 promoter (125). Interestingly epigenetic regulation of HIV-1 latency, which was associated with the recruitment of HDACs and SUV39H1 has also been described in astrocytes (40). Finally, in a recent report, investigation of the neuropathology and the molecular alterations associated with CNS latent HIV-1 infections provided evidence that HIV-1 persistence in the brain is associated with high level of CTIP2, HDACs, and HP1 (39).
We also showed that CTIP2 belongs to another complex able to prevent HIV-1 reactivation (Figure 1, right complex) (129). Indeed previous work has shown that CTIP2 represses the late phase, Tat-dependent, of HIV-1 transcription (127). In the absence of the trans-activator factor Tat, an inactive form of the elongation factor pTEFb is found in a multiprotein complex, including 7SK snRNA, CTIP2, and HEXIM1 anchored to viral and cellular gene promoters (129). pTEFb is composed of a regulatory subunit CyclinT1 and a catalytic subunit CDK9, whose kinase activity is involved in the Ser2 phosphorylation of the carboxyl terminal end of the RNA polymerase II and in the phosphorylation of the negative transcriptional elongation factors NELF and DSIF. Following phosphorylation, the RNA pol II processivity significantly increases, which leads to an efficient transcription of genes (130). Interestingly, we have shown that CTIP2 drastically repressed CDK9 kinase activity in this inactive complex, thus, inhibited pTEFb function. Finally, we showed that the cellular protein high mobility group AT-hook 1 (HMGA1), which also belongs to the 7SK snRNA complex recruits the inactive CTIP2/pTEFb complex to the HIV-1 and cellular target promoters (131). As a consequence, protein complexes containing CTIP2 regulate viral and endogenous gene expression, thus favoring HIV-1 persistence. Far more investigations are still needed to decipher the precise molecular mechanisms involved in these processes. We still do not fully understand how the transition from transcription initiation into elongation (which involves pTEFb) is controlled by cellular factors and/or the viral transactivator Tat. We and others hypothesized that the inactive form of the pTEFb complex is part of a 7SK complex that is anchored to the promoter by either CTIP2 (129) or Kap1 (132), thus available for RNApolII elongation through its activation. The transition from the inactive to the active form of the pTEFb complex through the action of Tat is not well understood but may involve a phosphatase (PPM1G/PP2Cγ) that takes apart pTEFb from the 7SK complex (133).
Several considerations already mentioned [emergence of multidrug resistance (24, 113), non-AIDS-related events (134–136) etc.] urge the search of new ways to develop a sterilizing or a functional cure for AIDS (137). The purge of viral reservoirs by the “shock and kill” strategy (138) is a possible approach to achieve such a cure. This strategy aims at purging or at least reducing the size of cellular reservoirs by reactivating HIV transcription (shock) followed by intensive cART therapy and immune activation (kill) (139, 140). As several reports suggested, using LRA alone or in combination have proven the efficiency of this strategy in the reactivation of quiescent/latent HIV from CD4+ T-cells reservoirs (138, 141–145). Several clinical trials have been carried out and some others are in progress or forthcoming (146). This strategy of reactivation needs to work on all potential reservoirs, including brain reservoirs. However, several limitations to the eradication of the brain reservoirs may preclude a cure.
It is essential to decipher the molecular mechanisms underlying HIV persistence in all types of potential reservoirs, since some important differences in those mechanisms have been noticed in all latently infected cell types. For example, LSD1 has been associated with activation of HIV transcription in CD4+ T-cells (147). However, in microglial cells, LSD1 played a role in the establishment of latency (128). LSD1 mediates HIV-1 transcription silencing in microglial by anchoring various factors at the promoter rather than inducing HIV-1 transcription by its own enzymatic activity in CD4+ T-cells. The dual role of LSD1 achieved by different mechanisms in the two main HIV-1 cellular targets points to the complexity of the molecular mechanisms of HIV latency (148). Hence, additional investigations of the epigenetic regulation of HIV latency are needed in order to develop efficient drugs targeting each potential viral reservoir. Furthermore, as mentioned in the previous sections, there are several characteristics of the CNS, which limits a cure by the “shock and kill” strategy:
i. The CNS is a sanctuary with barriers (BBB and choroid plexus) that reduce the access of some of the drugs currently used to the brain (97).
ii. The main cellular targets are astrocytes and CNS-resident macrophages. However, few drugs are able to target the monocyte–macrophages lineage (149) and the effects of cART on HIV replication in astrocytes are unknown or neurotoxic (150).
iii. CNS has long been considered as an immunologically privileged site (151). Therefore, achieving immune activation through cytotoxic T lymphocytes (CTL) activation to eliminate the potential reservoirs may be difficult or even deleterious in the brain.
iv. Another major concern is related to the fact that reactivation of the virus with LRA will lead to the synthesis of neurotoxic viral proteins, such as Tat and the gp120, as there are no drugs currently available targeting HIV transcription. Moreover, reactivation of the virus is often associated with CNS inflammation through macrophage/microglial cell activation (152, 153).
With these limitations evoked in the previous section, it may be difficult to achieve a purge in the CNS. The idea is to eliminate or reduce the pool of latent/quiescent reservoirs with the aim to mimic elite controllers able to control the HIV infection and with very low amount of reservoirs. Introducing cART very early following HIV infection has been proved to be efficient since it limits the size of the latent/quiescent reservoirs (154–156).
In our opinion, achieving a sterilizing cure or a partial functional cure in the brain needs efforts in four directions: (i) designing efficient LRA for CNS-cell types, (ii) improving cART by targeting HIV transcription, (iii) improving delivery of HIV drugs in the CNS and in the CNS-cell types, and (iv) developing therapeutic immunization.
Designing potent LRAs to reactivate HIV-1 transcription from the CNS-cell types is crucial in a “shock and kill” strategy. However, we and others have shown that the molecular mechanisms involved in the establishment and persistence of latency in these cells may differ from the mechanisms involved in the CD4+ T-cells that are currently the main targets for LRAs (106, 107, 137). As a consequence, the outcome in the use of LRAs may differ in CNS-cell types. Several HDAC inhibitors (HDACi) have been tested in the U1 monocyte cell line and in primary cells (astrocytes and macrophages) (106, 107, 157). Preliminary data showed that some LRAs, including panobinostat (158) and JQ1 (159), are relatively non-toxic and efficient to induce HIV reactivation at a therapeutic concentration (106, 107). On the contrary, other LRAs, including disulfiram and vorinostat, which were promising in CD4+ T-cells, were not working at therapeutic concentration in the CNS-cell types (106, 107). Among LRAs, bryostatin-1 is very promising since it can cross the BBB to activate brain Protein Kinase C especially in the two main targets for HIV-1, i.e., microglial cells and astrocytes (142, 160). This PKC activator has already been used in both preclinical trials for Alzheimer disease and in clinical trials to treat cancers [reviewed in Ref. (161)]. Further investigations will be needed to characterize new targets, such as the hCompass complex, recruited on the viral promoter by LSD1 in microglial cells. Preclinical studies in animal models are also needed to test the efficacy of LRAs. Combinations of LRAs have to be tested in vitro and in vivo as well, since they may work in a synergistic manner as described (142, 162). Using combination of LRAs with lower dose may also prevent some drug side effects when used alone at a higher concentration [reviewed in Ref. (163)]. Finally, a recent pilot study has suggested that administration of panobinostat, a potent activator of HIV transcription in CNS-cell types, was not associated with side effect in the brain as assessed by CSF biomarkers, such as neopterin, C reactive protein, and IP-10 (164).
Improving cART by targeting HIV transcription is also crucial since there are currently no drugs targeting this step (93). Moreover, reactivation of HIV leads to the synthesis of neurotoxic viral proteins, such as Tat. We and others discussed in details the importance of targeting this step and readers are referred to these recent reviews (93, 165). Particularly, inhibitors may be developed against the two main targets that control HIV transcription, i.e., the cellular factor NF-KB and the viral transactivator Tat. Since NF-KB also plays a central role in inflammation, new drugs targeting this factor will also prevent or at least reduce chronic inflammation in the brain (166, 167). It is also important to target the viral transactivator Tat since this factor is involved in the regulation of HIV-1 and its secreted form induces neuronal death by direct neurotoxicity. Several molecules, especially natural compounds deserve attention (168, 169). We believe that characterization of new targets associated with the exploitation of new technologies, such as bioengineering, high-throughput screening, computer-aided drug design, and combinatorial chemistry, will considerably improve the discovery of new drugs. Among the molecules that deserve attention, we can mention the dCA, a chemical derivative of corticostatin, a natural steroidal alkaloid isolated from a sponge (170–172). A promising Tat inhibitor has been recently isolated from the plant Tripterygium wilfordii and named triptolide (173). This molecule, which is currently in phase III of a clinical trial, inhibits both HIV replication and transcription by increasing the proteasomal degradation of Tat. Another family of protein that deserves attention is the DING proteins (pDINGs), a family of potential therapeutic agents against HIV-1 (174–178). These molecules discovered in bacteria, plants, and animals have been reported to inhibit HIV transcription. In addition, it has been shown that a phosphorylated form of pDING is a neuroprotective factor and could be used to reduce neuro inflammation due to HIV-1 (88, 179).
Another major limitation to purge brain reservoirs is related to the poor access of the drugs in the CNS due to the presence of barriers, such as the BBB. Moreover, drugs have to target macrophages and astrocytes. Indeed, it has been shown that all drugs, except protease inhibitors, display reduced activity in macrophages compared to CD4+ T-cells (180). We have already mentioned that some LRAs have no effect in the CNS-cell types at a therapeutic concentration. Different mechanisms have been evoked to explain the lower EC50 values of these drugs in macrophages/microglial cells. Drug penetration may be reduced due to the differential expression of efflux transporter and multidrug resistance proteins (181, 182). Several ways are explored to overcome these limitations and discussed in other reviews (183, 184). Improvement of both bioavailability and bio-distribution of drugs used in cART will increase the access of these drugs to the brain. Among the approaches used to improve drug delivery in the brain, there is the development of carriers, such as liposomes, dendrimers, and micelles. A particularly promising approach is based on polymeric nanomedicines that raise hope for eradication of HIV from all potential reservoirs [reviewed in Ref. (185–187)]. Increase in treatment efficacy and tolerance may be expected, hence favoring patient adherence. These later strategies may also increase the distribution of drugs in CNS-cell types, such as astrocytes and macrophages/microglial cells. Indeed, macrophages/microglial cells constitute an important but neglected barrier for HIV eradication, which will need efforts to circumvent (149, 188). Several conventional and new therapeutics against HIV-1 in macrophages, including PI3K/Akt blocking agents, carbohydrate-binding agents, and small interfering RNAs, have been discussed elsewhere and deserve real attention (184, 189).
Immune-based therapeutics should also be considered since the size of the reservoir following treatment with LRAs is not reduced and need immune activation to clear them (190). In particular, CTL activation has been shown to clear HIV-1 from infected CD4+ T-cells (140). Previous studies done in animal models argued in favor of the importance of CTL in the clearance of HIV-1 infected macrophages in the brain (191, 192). A CD8+ T-cell response appears essential in the control of other brain infections, such as toxoplasmosis (193). Dealing with immune activation is not easy and constitutes a challenge for strategies aiming to eradicate HIV-1 reservoirs. These approaches need further investigations and development of adequate animal models to ensure the feasibility of such treatments (194, 195). Another unexplored non-conventional way to clear reactivated latently infected cells is based on the use of neutralizing antibodies against HIV-1 with promising results obtained in humanized mice (143, 196).
Reducing the size of reservoirs is fundamental for HIV+ patients to control their viral replication without any treatment, a situation typical for elite controllers. The purge of HIV reservoirs constitutes, therefore, one of the top research priority of the International AIDS Society (IAS) through the action “Toward an HIV Cure.” We may expect to get a sterilizing cure by eradicating the virus from all the reservoirs but a more realistic view would be a functional cure through the reduction of the pool of cellular reservoirs. A major problem is to reduce/eradicate reservoirs located in the CNS. There are now numerous direct and indirect arguments for the existence of a pool of quiescent/latent reservoirs in the brain even if it has not been demonstrated in human yet. The strategy called “shock and kill” enables reactivation of quiescent/latent reservoirs followed by an intensive cART to clear the reservoirs. Several pilot clinical trials have been done and some are ongoing and upcoming. The results of trials are encouraging but also point to the need of additional interventions, such as immune activation, in order to clear the reservoirs. This immune activation approach is needed to eliminate or reduce brain reservoirs but might be difficult since the CNS has several unique characteristics. Indeed, the CNS is a pharmacological sanctuary and a compartment isolated from the other parts of the body. In addition, latently infected cells in the brain, i.e., astrocytes and macrophages/microglial cells are rather different from the main memory T-cells reservoir. Altogether, intense efforts are needed in several directions, including the design of efficient LRAs for CNS-cell types, improving cART by targeting HIV transcription, improving delivery of HIV drugs in the CNS and in the CNS-cell types and developing therapeutic immunization therapies in order to overcome the above discussed limitations. We believe that we are at a crossroads to achieve a cure for HIV. Indeed, there are several adequate animal models (non-human primate, humanized mice, etc.) to test the efficiency of strategies aiming to purge reservoirs. Identification of new targets and the availability of new technologies will also allow the design of new original drugs. In particular, new natural compounds and their derivatives could help in the design of new class of molecules targeting HIV-1 transcription a step not yet targeted by cART. This is especially crucial in a strategy aiming to reactivate latent CNS-cell types. Finally, hope rises also with the advent of nanotechnologies. Although still in the early stage, nanotechnologies could be used in drug transport to enable drugs to reach both the brain (by crossing barriers such as the BBB) and the CNS-cell types (by crossing cell membranes). Dosage is expected lower and in consequence less toxicity and a better adherence to treatment is awaited.
CM revised the manuscript and made substantial contributions to its final content and design. AA-A, FFo, FFa, FD, and HM revised the manuscript. OR and CS drafted the manuscript. All authors read and approved the final manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
This work was supported by grants from the Agence Nationale de Recherches sur le SIDA (ANRS) to OR, CS, and AA-A, Sidaction, Ligue contre le cancer to OR and CS, from Institut Universitaire de France to OR and from H2020-MSCA-RISE-691119 – EU4HIVCURE to OR.
1. Chun TW, Carruth L, Finzi D, Shen X, DiGiuseppe JA, Taylor H, et al. Quantification of latent tissue reservoirs and total body viral load in HIV-1 infection. Nature (1997) 387(6629):183–8. doi:10.1038/387183a0
2. Chun TW, Stuyver L, Mizell SB, Ehler LA, Mican JA, Baseler M, et al. Presence of an inducible HIV-1 latent reservoir during highly active antiretroviral therapy. Proc Natl Acad Sci U S A (1997) 94(24):13193–7. doi:10.1073/pnas.94.24.13193
3. Zhang L, Ramratnam B, Tenner-Racz K, He Y, Vesanen M, Lewin S, et al. Quantifying residual HIV-1 replication in patients receiving combination antiretroviral therapy. N Engl J Med (1999) 340(21):1605–13. doi:10.1056/NEJM199905273402101
4. Bouchat S, Delacourt N, Kula A, Darcis G, Van Driessche B, Corazza F, et al. Sequential treatment with 5-Aza-2’-deoxycytidine and deacetylase inhibitors reactivates HIV-1. EMBO Mol Med (2016) 8(2):117–38. doi:10.15252/emmm.201505557
5. Di Mascio M, Dornadula G, Zhang H, Sullivan J, Xu Y, Kulkosky J, et al. In a subset of subjects on highly active antiretroviral therapy, human immunodeficiency virus type 1 RNA in plasma decays from 50 to <5 copies per milliliter, with a half-life of 6 months. J Virol (2003) 77(3):2271–5. doi:10.1128/JVI.77.3.2271-2275.2003
6. Dornadula G, Zhang H, VanUitert B, Stern J, Livornese L Jr, Ingerman MJ, et al. Residual HIV-1 RNA in blood plasma of patients taking suppressive highly active antiretroviral therapy. JAMA (1999) 282(17):1627–32. doi:10.1001/jama.282.17.1627
7. Harrigan PR, Whaley M, Montaner JS. Rate of HIV-1 RNA rebound upon stopping antiretroviral therapy. AIDS (1999) 13(8):F59–62. doi:10.1097/00002030-199905280-00001
8. Zhang L, Chung C, Hu BS, He T, Guo Y, Kim AJ, et al. Genetic characterization of rebounding HIV-1 after cessation of highly active antiretroviral therapy. J Clin Invest (2000) 106(7):839–45. doi:10.1172/JCI10565
9. Crowe S, Zhu T, Muller WA. The contribution of monocyte infection and trafficking to viral persistence, and maintenance of the viral reservoir in HIV infection. J Leukoc Biol (2003) 74(5):635–41. doi:10.1189/jlb.0503204
10. Shen L, Siliciano RF. Viral reservoirs, residual viremia, and the potential of highly active antiretroviral therapy to eradicate HIV infection. J Allergy Clin Immunol (2008) 122(1):22–8. doi:10.1016/j.jaci.2008.05.033
11. Maldarelli F. Targeting viral reservoirs: ability of antiretroviral therapy to stop viral replication. Curr Opin HIV AIDS (2010) 6(1):49–56. doi:10.1097/COH.0b013e32834134ea
12. Chun TW, Finzi D, Margolick J, Chadwick K, Schwartz D, Siliciano RF. In vivo fate of HIV-1-infected T cells: quantitative analysis of the transition to stable latency. Nat Med (1995) 1(12):1284–90. doi:10.1038/nm1295-1284
13. Finzi D, Hermankova M, Pierson T, Carruth LM, Buck C, Chaisson RE, et al. Identification of a reservoir for HIV-1 in patients on highly active antiretroviral therapy. Science (1997) 278(5341):1295–300. doi:10.1126/science.278.5341.1295
14. Perelson AS, Essunger P, Cao Y, Vesanen M, Hurley A, Saksela K, et al. Decay characteristics of HIV-1-infected compartments during combination therapy. Nature (1997) 387(6629):188–91. doi:10.1038/387188a0
15. Wong JK, Hezareh M, Gunthard HF, Havlir DV, Ignacio CC, Spina CA, et al. Recovery of replication-competent HIV despite prolonged suppression of plasma viremia. Science (1997) 278(5341):1291–5. doi:10.1126/science.278.5341.1291
16. Bailey JR, Sedaghat AR, Kieffer T, Brennan T, Lee PK, Wind-Rotolo M, et al. Residual human immunodeficiency virus type 1 viremia in some patients on antiretroviral therapy is dominated by a small number of invariant clones rarely found in circulating CD4+ T cells. J Virol (2006) 80(13):6441–57. doi:10.1128/JVI.00591-06
17. Alexaki A, Liu Y, Wigdahl B. Cellular reservoirs of HIV-1 and their role in viral persistence. Curr HIV Res (2008) 6(5):388–400. doi:10.2174/157016208785861195
18. Alexaki A, Wigdahl B. HIV-1 infection of bone marrow hematopoietic progenitor cells and their role in trafficking and viral dissemination. PLoS Pathog (2008) 4(12):e1000215. doi:10.1371/journal.ppat.1000215
19. Coleman CM, Wu L. HIV interactions with monocytes and dendritic cells: viral latency and reservoirs. Retrovirology (2009) 6:51. doi:10.1186/1742-4690-6-51
20. Eisele E, Siliciano RF. Redefining the viral reservoirs that prevent HIV-1 eradication. Immunity (2012) 37(3):377–88. doi:10.1016/j.immuni.2012.08.010
21. Varatharajan L, Thomas SA. The transport of anti-HIV drugs across blood-CNS interfaces: summary of current knowledge and recommendations for further research. Antiviral Res (2009) 82(2):A99–109. doi:10.1016/j.antiviral.2008.12.013
22. Sigal A, Kim JT, Balazs AB, Dekel E, Mayo A, Milo R, et al. Cell-to-cell spread of HIV permits ongoing replication despite antiretroviral therapy. Nature (2011) 477(7362):95–8. doi:10.1038/nature10347
23. Griffiths PD. A perspective on antiviral resistance. J Clin Virol (2009) 46(1):3–8. doi:10.1016/j.jcv.2009.06.017
24. Kozal MJ. Drug-resistant human immunodefiency virus. Clin Microbiol Infect (2009) 15(Suppl 1):69–73. doi:10.1111/j.1469-0691.2008.02687.x
25. Nachega JB, Marconi VC, van Zyl GU, Gardner EM, Preiser W, Hong SY, et al. HIV treatment adherence, drug resistance, virologic failure: evolving concepts. Infect Disord Drug Targets (2011) 11(2):167–74. doi:10.2174/187152611795589663
26. Seligman SJ. Possibility of HIV-1 resistance mutations in cerebrospinal fluid from persons receiving suppressive therapy. J Infect Dis (2011) 204(1):174. doi:10.1093/infdis/jir234 author reply 174–75
27. Hong FF, Mellors JW. Changes in HIV reservoirs during long-term antiretroviral therapy. Curr Opin HIV AIDS (2015) 10(1):43–8. doi:10.1097/COH.0000000000000119
28. Kramer-Hämmerle S, Rothenaigner I, Wolff H, Bell JE, Brack-Werner R. Cells of the central nervous system as targets and reservoirs of the human immunodeficiency virus. Virus Res (2005) 111(2):194–213. doi:10.1016/j.virusres.2005.04.009
29. Nickle DC, Jensen MA, Shriner D, Brodie SJ, Frenkel LM, Mittler JE, et al. Evolutionary indicators of human immunodeficiency virus type 1 reservoirs and compartments. J Virol (2003) 77(9):5540–6. doi:10.1128/JVI.77.9.5540-5546.2003
30. Le Douce V, Herbein G, Rohr O, Schwartz C. Molecular mechanisms of HIV-1 persistence in the monocyte-macrophage lineage. Retrovirology (2010) 7(1):32. doi:10.1186/1742-4690-7-32
31. Blankson JN, Persaud D, Siliciano RF. The challenge of viral reservoirs in HIV-1 infection. Annu Rev Med (2002) 53:557–93. doi:10.1146/annurev.med.53.082901.104024
32. Churchill MJ, Gorry PR, Cowley D, Lal L, Sonza S, Purcell DFJ, et al. Use of laser capture microdissection to detect integrated HIV-1 DNA in macrophages and astrocytes from autopsy brain tissues. J Neurovirol (2006) 12(2):146–52. doi:10.1080/13550280600748946
33. Thompson KA, Cherry CL, Bell JE, McLean CA. Brain cell reservoirs of latent virus in presymptomatic HIV-infected individuals. Am J Pathol (2011) 179(4):1623–9. doi:10.1016/j.ajpath.2011.06.039
34. Koppensteiner H, Brack-Werner R, Schindler M. Macrophages and their relevance in human immunodeficiency virus type I infection. Retrovirology (2012) 9(January):82. doi:10.1186/1742-4690-9-82
35. Sofroniew MV, Vinters HV. Astrocytes: biology and pathology. Acta Neuropathol (2010) 119(1):7–35. doi:10.1007/s00401-009-0619-8
36. Soulet D, Rivest S. Bone-marrow-derived microglia: myth or reality? Curr Opin Pharmacol (2008) 8(4):508–18. doi:10.1016/j.coph.2008.04.002
37. Churchill MJ, Wesselingh SL, Cowley D, Pardo CA, McArthur JC, Brew BJ, et al. Extensive astrocyte infection is prominent in human immunodeficiency virus-associated dementia. Ann Neurol (2009) 66(2):253–8. doi:10.1002/ana.21697
38. Gray LR, Turville SG, Hitchen TL, Wan-Jung C, Ellett AM, Salimi H, et al. HIV-1 entry and trans-infection of astrocytes involves CD81 vesicles. PLoS One (2014) 9(2):e90620. doi:10.1371/journal.pone.0090620
39. Desplats P, Dumaop W, Smith D, Adame A, Everall I, Letendre S, et al. Molecular and pathologic insights from latent HIV-1 infection in the human brain. Neurology (2013) 80(15):1415–23. doi:10.1212/WNL.0b013e31828c2e9e
40. Narasipura SD, Kim S, Al-Harthi L. Epigenetic regulation of HIV-1 latency in astrocytes. J Virol (2014) 88(5):3031–8. doi:10.1128/JVI.03333-13
41. Redel L, Le Douce V, Cherrier T, Marban C, Janossy A, Aunis D, et al. HIV-1 regulation of latency in the monocyte-macrophage lineage and in CD4+ T lymphocytes. J Leukoc Biol (2010) 87(4):575–88. doi:10.1189/jlb.0409264
42. Marban C, Suzanne S, Dequiedt F, de Walque S, Redel L, Van Lint C, et al. Recruitment of chromatin-modifying enzymes by CTIP2 promotes HIV-1 transcriptional silencing. EMBO J (2007) 26(2):412–23. doi:10.1038/sj.emboj.7601516
43. Petry H, Lüke W. Infection of Macaque monkeys with simian immunodeficiency virus: an animal model for neuro-AIDS. Intervirology (1997) 40(2–3):112–21. doi:10.1159/000150538
44. Gorantla S, Gendelman HE, Poluektova LY. Can humanized mice reflect the complex pathobiology of HIV-associated neurocognitive disorders? J Neuroimmune Pharmacol (2012) 7(2):352–62. doi:10.1007/s11481-011-9335-y
45. Vigorito M, Connaghan KP, Chang SL. The HIV-1 transgenic rat model of neuroHIV. Brain Behav Immun (2015) 48(August):336–49. doi:10.1016/j.bbi.2015.02.020
46. Barber SA, Gama L, Dudaronek JM, Voelker T, Tarwater PM, Clements JE. Mechanism for the establishment of transcriptional HIV latency in the brain in a simian immunodeficiency virus-Macaque model. J Infect Dis (2006) 193(7):963–70. doi:10.1086/500983
47. Schwartz C, Catez P, Rohr O, Lecestre D, Aunis D, Schaeffer E. Functional interactions between C/EBP, Sp1, and COUP-TF regulate human immunodeficiency virus type 1 gene transcription in human brain cells. J Virol (2000) 74(1):65–73. doi:10.1128/JVI.74.1.65-73.2000
48. Hellmuth J, Valcour V, Spudich S. CNS reservoirs for HIV: implications for eradication. J Virus Erad (2015) 1(2):67–71.
49. Jessen Krut J, Mellberg T, Price RW, Hagberg L, Fuchs D, Rosengren L, et al. Biomarker evidence of axonal injury in neuroasymptomatic HIV-1 patients. PLoS One (2014) 9(2):e88591. doi:10.1371/journal.pone.0088591
50. Yilmaz A, Price RW, Spudich S, Fuchs D, Hagberg L, Gisslén M. Persistent intrathecal immune activation in HIV-1-infected individuals on antiretroviral therapy. J Acquir Immune Defic Syndr (2008) 47(2):168–73. doi:10.1097/QAI.0b013e31815ace97
51. Edén A, Price RW, Spudich S, Fuchs D, Hagberg L, Gisslén M. Immune activation of the central nervous system is still present after >4 years of effective highly active antiretroviral therapy. J Infect Dis (2007) 196(12):1779–83. doi:10.1086/523648
52. Harezlak J, Cohen R, Gongvatana A, Taylor M, Buchthal S, Schifitto G, et al. Predictors of CNS injury as measured by proton magnetic resonance spectroscopy in the setting of chronic HIV infection and CART. J Neurovirol (2014) 20(3):294–303. doi:10.1007/s13365-014-0246-6
53. Harezlak J, Buchthal S, Taylor M, Schifitto G, Zhong J, Daar E, et al. Persistence of HIV-associated cognitive impairment, inflammation, and neuronal injury in era of highly active antiretroviral treatment. AIDS (2011) 25(5):625–33. doi:10.1097/QAD.0b013e3283427da7
54. Dahl V, Peterson J, Spudich S, Lee E, Shacklett BL, Price RW, et al. Single-copy assay quantification of HIV-1 RNA in paired cerebrospinal fluid and plasma samples from elite controllers. AIDS (2013) 27(7):1145–9. doi:10.1097/QAD.0b013e32835cf235
55. Dahl V, Gisslen M, Hagberg L, Peterson J, Shao W, Spudich S, et al. An example of genetically distinct HIV type 1 variants in cerebrospinal fluid and plasma during suppressive therapy. J Infect Dis (2014) 209(10):1618–22. doi:10.1093/infdis/jit805
56. Dahl V, Peterson J, Fuchs D, Gisslen M, Palmer S, Price RW. Low levels of HIV-1 RNA detected in the cerebrospinal fluid after up to 10 years of suppressive therapy are associated with local immune activation. AIDS (2014) 28(15):2251–8. doi:10.1097/QAD.0000000000000400
57. Lescure F-X, Moulignier A, Savatovsky J, Amiel C, Carcelain G, Molina J-M, et al. CD8 encephalitis in HIV-infected patients receiving cART: a treatable entity. Clin Infect Dis (2013) 57(1):101–8. doi:10.1093/cid/cit175
58. Canestri A, Lescure F-X, Jaureguiberry S, Moulignier A, Amiel C, Marcelin AG, et al. Discordance between cerebral spinal fluid and plasma HIV replication in patients with neurological symptoms who are receiving suppressive antiretroviral therapy. Clin Infect Dis (2010) 50(5):773–8. doi:10.1086/650538
59. Peluso MJ, Ferretti F, Peterson J, Lee E, Fuchs D, Boschini A, et al. Cerebrospinal fluid HIV escape associated with progressive neurologic dysfunction in patients on antiretroviral therapy with well controlled plasma viral load. AIDS (2012) 26(14):1765–74. doi:10.1097/QAD.0b013e328355e6b2
60. Brew BJ, Robertson K, Wright EJ, Churchill M, Crowe SM, Cysique LA, et al. HIV eradication symposium: will the brain be left behind? J Neurovirol (2015) 21(3):322–34. doi:10.1007/s13365-015-0322-6
61. Churchill MJ, Cowley DJ, Wesselingh SL, Gorry PR, Gray LR. HIV-1 transcriptional regulation in the central nervous system and implications for HIV cure research. J Neurovirol (2015) 21(3):290–300. doi:10.1007/s13365-014-0271-5
62. Churchill MJ, Deeks SG, Margolis DM, Siliciano RF, Swanstrom R. HIV reservoirs: what, where and how to target them. Nat Rev Microbiol (2015) 14(1):55–60. doi:10.1038/nrmicro.2015.5
63. Gray LR, Roche M, Flynn JK, Wesselingh SL, Gorry PR, Churchill MJ. Is the central nervous system a reservoir of HIV-1? Curr Opin HIV AIDS (2014) 9(6):552–8. doi:10.1097/COH.0000000000000108
64. Ransohoff RM, Engelhardt B. The anatomical and cellular basis of immune surveillance in the central nervous system. Nat Rev Immunol (2012) 12(9):623–35. doi:10.1038/nri3265
65. Romo-González T, Chavarría A, érez-H JP. Central nervous system: a modified immune surveillance circuit? Brain Behav Immun (2012) 26(6):823–9. doi:10.1016/j.bbi.2012.01.016
66. Ransohoff RM, Kivisäkk P, Kidd G. Three or more routes for leukocyte migration into the central nervous system. Nat Rev Immunol (2003) 3(7):569–81. doi:10.1038/nri1130
67. Schrier RD, Hong S, Crescini M, Ellis R, érez-Santiago JP, Spina C, et al. Cerebrospinal fluid (CSF) CD8+ T-cells that express interferon-gamma contribute to HIV associated neurocognitive disorders (HAND). PLoS One (2015) 10(2):e0116526. doi:10.1371/journal.pone.0116526
68. Nightingale S, Michael BD, Fisher M, Winston A, Nelson M, Taylor S, et al. CSF/plasma HIV-1 RNA discordance even at low levels is associated with up-regulation of host inflammatory mediators in CSF. Cytokine (2016) 83(July):139–46. doi:10.1016/j.cyto.2016.04.004
69. Ho EL, Ronquillo R, Altmeppen H, Spudich SS, Price RW, Sinclair E. Cellular composition of cerebrospinal fluid in HIV-1 infected and uninfected subjects. PLoS One (2013) 8(6):e66188. doi:10.1371/journal.pone.0066188
70. Kowarik MC, Grummel V, Wemlinger S, Buck D, Weber MS, Berthele A, et al. Immune cell subtyping in the cerebrospinal fluid of patients with neurological diseases. J Neurol (2014) 261(1):130–43. doi:10.1007/s00415-013-7145-2
71. Spudich SS. Immune activation in the central nervous system throughout the course of HIV infection. Curr Opin HIV AIDS (2016) 11(2):226–33. doi:10.1097/COH.0000000000000243
72. Hong S, Banks WA. Role of the immune system in HIV-associated neuroinflammation and neurocognitive implications. Brain Behav Immun (2015) 45(March):1–12. doi:10.1016/j.bbi.2014.10.008
73. Williams DW, Veenstra M, Gaskill PJ, Morgello S, Calderon TM, Berman JW. Monocytes mediate HIV neuropathogenesis: mechanisms that contribute to HIV associated neurocognitive disorders. Curr HIV Res (2014) 12(2):85–96. doi:10.2174/1570162X12666140526114526
74. Campbell JH, Ratai E-M, Autissier P, Nolan DJ, Tse S, Miller AD, et al. Anti-α4 antibody treatment blocks virus traffic to the brain and Gut early, and stabilizes CNS injury late in infection. PLoS Pathog (2014) 10(12):e1004533. doi:10.1371/journal.ppat.1004533
75. Elbirt D, Mahlab-Guri K, Bezalel-Rosenberg S, Gill H, Attali M, Asher I. HIV-associated neurocognitive disorders (HAND). Isr Med Assoc J (2015) 17(1):54–9.
76. Tan IL, McArthur JC. HIV-associated neurological disorders: a guide to pharmacotherapy. CNS Drugs (2012) 26(2):123–34. doi:10.2165/11597770-000000000-00000
77. Brouillette M-J, Mayo N, Fellows LK, Lebedeva E, Higgins J, Overton ET, et al. A better screening tool for HIV-associated neurocognitive disorders: is it what clinicians need? AIDS (2015) 29(8):895–902. doi:10.1097/QAD.0000000000000152
78. Antinori A, Arendt G, Becker JT, Brew BJ, Byrd DA, Cherner M, et al. Updated research nosology for HIV-associated neurocognitive disorders. Neurology (2007) 69(18):1789–99. doi:10.1212/01.WNL.0000287431.88658.8b
79. Valcour V, Chalermchai T, Sailasuta N, Marovich M, Lerdlum S, Suttichom D, et al. Central nervous system viral invasion and inflammation during acute HIV infection. J Infect Dis (2012) 206(2):275–82. doi:10.1093/infdis/jis326
80. Zipursky AR, Gogolishvili D, Rueda S, Brunetta J, Carvalhal A, McCombe JA, et al. Evaluation of brief screening tools for neurocognitive impairment in HIV/AIDS: a systematic review of the literature. AIDS (2013) 27(15):2385–401. doi:10.1097/QAD.0b013e328363bf56
81. Valcour V, Paul R, Chiao S, Wendelken LA, Miller B. Screening for cognitive impairment in human immunodeficiency virus. Clin Infect Dis (2011) 53(8):836–42. doi:10.1093/cid/cir524
82. da Fonseca AC, Matias CD, Garcia C, Amaral R, Geraldo LH, Freitas C, et al. The impact of microglial activation on blood-brain barrier in brain diseases. Front Cell Neurosci (2014) 8:362. doi:10.3389/fncel.2014.00362
83. Zhang Y-L, Ouyang Y-B, Liu L-G, Chen D-X. Blood-brain barrier and neuro-AIDS. Eur Rev Med Pharmacol Sci (2015) 19(24):4927–39.
84. Atluri VS, Hidalgo M, Samikkannu T, Venkata Kurapati KR, Jayant RD, Sagar V, et al. Effect of human immunodeficiency virus on blood-brain barrier integrity and function: an update. Front Cell Neurosci (2015) 9(January):212. doi:10.3389/fncel.2015.00212
85. Burdo TH, Lackner A, Williams KC. Monocyte/macrophages and their role in HIV neuropathogenesis. Immunol Rev (2013) 254(1):102–13. doi:10.1111/imr.12068
86. Saylor D, Dickens AM, Sacktor N, Haughey N, Slusher B, Pletnikov M, et al. HIV-associated neurocognitive disorder – pathogenesis and prospects for treatment. Nat Rev Neurol (2016) 12(4):234–48. doi:10.1038/nrneurol.2016.27
87. Zayyad Z, Spudich S. Neuropathogenesis of HIV: from initial neuroinvasion to HIV-associated neurocognitive disorder (HAND). Curr HIV/AIDS Rep (2015) 12(1):16–24. doi:10.1007/s11904-014-0255-3
88. Rao VR, Ruiz AP, Prasad VR. Viral and cellular factors underlying neuropathogenesis in HIV associated neurocognitive disorders (HAND). AIDS Res Ther (2014) 11(January):13. doi:10.1186/1742-6405-11-13
89. Bagashev A, Sawaya BE. Roles and functions of HIV-1 tat protein in the CNS: an overview. Virol J (2013) 10(January):358. doi:10.1186/1743-422X-10-358
90. Li W, Li G, Steiner J, Nath A. Role of tat protein in HIV neuropathogenesis. Neurotox Res (2009) 16(3):205–20. doi:10.1007/s12640-009-9047-8
91. Kim BO, Liu Y, Ruan Y, Xu ZC, Schantz L, He JJ. Neuropathologies in transgenic mice expressing human immunodeficiency virus type 1 tat protein under the regulation of the astrocyte-specific glial fibrillary acidic protein promoter and doxycycline. Am J Pathol (2003) 162(5):1693–707. doi:10.1016/S0002-9440(10)64304-0
92. Chang JR, Mukerjee R, Bagashev A, Del Valle L, Chabrashvili T, Hawkins BJ, et al. HIV-1 tat protein promotes neuronal dysfunction through disruption of microRNAs. J Biol Chem (2011) 286(47):41125–34. doi:10.1074/jbc.M111.268466
93. Le Douce V, Ait-Amar A, Far Forouzan F, Fahmi F, Quiel J, El Mekdad H, et al. Improving combination antiretroviral therapy by targeting HIV-1 gene transcription. Expert Opin Ther Targets (2016) 18:1–14. doi:10.1080/14728222.2016.1198777
94. Richards MH, Narasipura SD, Kim S, Seaton MS, Lutgen V, Al-Harthi L. Dynamic interaction between astrocytes and infiltrating PBMCs in context of NeuroAIDS. Glia (2015) 63(3):441–51. doi:10.1002/glia.22763
95. Angers S, Moon RT. Proximal events in Wnt signal transduction. Nat Rev Mol Cell Biol (2009) 10(7):468–77. doi:10.1038/nrm2717
96. Cory TJ, Schacker TW, Stevenson M, Fletcher CV. Overcoming pharmacologic sanctuaries. Curr Opin HIV AIDS (2013) 8(3):190–5. doi:10.1097/COH.0b013e32835fc68a
97. Eisfeld C, Reichelt D, Evers S, Husstedt I. CSF penetration by antiretroviral drugs. CNS Drugs (2013) 27(1):31–55. doi:10.1007/s40263-012-0018-x
98. Letendre S, Marquie-Beck J, Capparelli E, Best B, Clifford D, Collier AC, et al. Validation of the CNS penetration-effectiveness rank for quantifying antiretroviral penetration into the central nervous system. Arch Neurol (2008) 65(1):65–70. doi:10.1001/archneurol.2007.31
99. Zink MC, Brice AK, Kelly KM, Queen SE, Gama L, Li M, et al. Simian immunodeficiency virus-infected Macaques treated with highly active antiretroviral therapy have reduced central nervous system viral replication and inflammation but persistence of viral DNA. J Infect Dis (2010) 202(1):161–70. doi:10.1086/653213
100. Arrildt KT, Joseph SB, Swanstrom R. The HIV-1 Env protein: a coat of many colors. Curr HIV/AIDS Rep (2012) 9(1):52–63. doi:10.1007/s11904-011-0107-3
101. Joseph SB, Arrildt KT, Sturdevant CB, Swanstrom R. HIV-1 target cells in the CNS. J Neurovirol (2015) 21(3):276–89. doi:10.1007/s13365-014-0287-x
102. Gorry PR, Francella N, Lewin SR, Collman RG. HIV-1 envelope-receptor interactions required for macrophage infection and implications for current HIV-1 cure strategies. J Leukoc Biol (2014) 95(1):71–81. doi:10.1189/jlb.0713368
103. Thomas ER, Dunfee RL, Stanton J, Bogdan D, Taylor J, Kunstman K, et al. Macrophage entry mediated by HIV Envs from brain and lymphoid tissues is determined by the capacity to use low CD4 levels and overall efficiency of fusion. Virology (2007) 360(1):105–19. doi:10.1016/j.virol.2006.09.036
104. Watkins BA, Dorn HH, Kelly WB, Armstrong RC, Potts BJ, Michaels F, et al. Specific tropism of HIV-1 for microglial cells in primary human brain cultures. Science (1990) 249(4968):549–53. doi:10.1126/science.2200125
105. Ait-Khaled M, McLaughlin JE, Johnson MA, Emery VC. Distinct HIV-1 long terminal repeat quasispecies present in nervous tissues compared to that in lung, blood and lymphoid tissues of an AIDS patient. AIDS (1995) 9(7):675–83. doi:10.1097/00002030-199507000-00002
106. Gray LR, Cowley D, Welsh C, Lu HK, Brew BJ, Lewin SR, et al. CNS-specific regulatory elements in brain-derived HIV-1 strains affect responses to latency-reversing agents with implications for cure strategies. Mol Psychiatry (2016) 21(4):574–84. doi:10.1038/mp.2015.111
107. Gray LR, On H, Roberts E, Lu HK, Moso MA, Raison JA, et al. Toxicity and in vitro activity of HIV-1 latency-reversing agents in primary CNS cells. J Neurovirol (2016) 22(4):455–63. doi:10.1007/s13365-015-0413-4
108. Ferretti F, Gisslen M, Cinque P, Price RW. Cerebrospinal fluid HIV escape from antiretroviral therapy. Curr HIV/AIDS Rep (2015) 12(2):280–8. doi:10.1007/s11904-015-0267-7
109. Spudich S, Lollo N, Liegler T, Deeks SG, Price RW. Treatment benefit on cerebrospinal fluid HIV-1 levels in the setting of systemic virological suppression and failure. J Infect Dis (2006) 194(12):1686–96. doi:10.1086/508750
110. Garvey LJ, Everitt A, Winston A, Mackie NE, Benzie A. Detectable cerebrospinal fluid HIV RNA with associated neurological deficits, despite suppression of HIV replication in the plasma compartment. AIDS (2009) 23(11):1443–4. doi:10.1097/QAD.0b013e32832d077c
111. Dahl V, Lee E, Peterson J, Spudich SS, Leppla I, Sinclair E, et al. Raltegravir treatment intensification does not alter cerebrospinal fluid HIV-1 infection or immunoactivation in subjects on suppressive therapy. J Infect Dis (2011) 204(12):1936–45. doi:10.1093/infdis/jir667
112. Edén A, Fuchs D, Hagberg L, Nilsson S, Spudich S, Svennerholm B, et al. HIV-1 viral escape in cerebrospinal fluid of subjects on suppressive antiretroviral treatment. J Infect Dis (2010) 202(12):1819–25. doi:10.1086/657342
113. Nijhuis M, van Maarseveen NM, Boucher CA. Antiviral resistance and impact on viral replication capacity: evolution of viruses under antiviral pressure occurs in three phases. Handb Exp Pharmacol (2009) 189:299–320. doi:10.1007/978-3-540-79086-0_11
114. Lorenzo-Redondo R, Fryer HR, Bedford T, Kim E-Y, Archer J, Kosakovsky Pond SL, et al. Persistent HIV-1 replication maintains the tissue reservoir during therapy. Nature (2016) 530(7588):51–6. doi:10.1038/nature16933
115. Licht A, Alter G, Deeks SG, Fukazawa Y, Lum R, Okoye AA, et al. A drug-free zone – lymph nodes as a safe haven for HIV. Cell Host Microbe (2016) 19(3):275–6. doi:10.1016/j.chom.2016.02.018
116. Almeida SM. Cerebrospinal fluid analysis in the HIV infection and compartmentalization of HIV in the central nervous system. Arq Neuropsiquiatr (2015) 73(7):624–9. doi:10.1590/0004-282X20150071
117. Gorry PR, Chi O, Thorpe J, Bannwarth S, Thompson KA, Gatignol A, et al. Astrocyte infection by HIV-1: mechanisms of restricted virus replication, and role in the pathogenesis of HIV-1-associated dementia. Curr HIV Res (2003) 1(4):463–73. doi:10.2174/1570162033485122
118. Rohr O, Marban C, Aunis D, Schaeffer E. Regulation of HIV-1 gene transcription: from lymphocytes to microglial cells. J Leukoc Biol (2003) 74(5):736–49. doi:10.1189/jlb.0403180
119. Gray LR, Cowley D, Crespan E, Welsh C, Mackenzie C, Wesselingh SL, et al. Reduced basal transcriptional activity of central nervous system-derived HIV type 1 long terminal repeats. AIDS Res Hum Retroviruses (2013) 29(2):365–70. doi:10.1089/AID.2012.0138
120. Ong CL, Thorpe JC, Gorry PR, Bannwarth S, Jaworowski A, Howard JL, et al. Low TRBP levels support an innate human immunodeficiency virus type 1 resistance in astrocytes by enhancing the PKR antiviral response. J Virol (2005) 79(20):12763–72. doi:10.1128/JVI.79.20.12763-12772.2005
121. Van Lint C, Bouchat S, Marcello A. HIV-1 transcription and latency: an update. Retrovirology (2013) 10(January):67. doi:10.1186/1742-4690-10-67
122. Mbonye U, Karn J. Transcriptional control of HIV latency: cellular signaling pathways, epigenetics, happenstance and the hope for a cure. Virology (2014) 454-455:328–39. doi:10.1016/j.virol.2014.02.008
123. Le Douce V, Cherrier T, Riclet R, Rohr O, Schwartz C. The many lives of CTIP2: from AIDS to cancer and cardiac hypertrophy. J Cell Physiol (2014) 229(5):533–7. doi:10.1002/jcp.24490
124. Vazquez N, Greenwell-Wild T, Marinos NJ, Swaim WD, Nares S, Ott DE, et al. Human immunodeficiency virus type 1-induced macrophage gene expression includes the p21 gene, a target for viral regulation. J Virol (2005) 79(7):4479–91. doi:10.1128/JVI.79.7.4479-4491.2005
125. Cherrier T, Suzanne S, Redel L, Calao M, Marban C, Samah B, et al. p21(WAF1) gene promoter is epigenetically silenced by CTIP2 and SUV39H1. Oncogene (2009) 28(38):3380–9. doi:10.1038/onc.2009.193
126. Marban C, Redel L, Suzanne S, Van Lint C, Lecestre D, Chasserot-Golaz S, et al. COUP-TF interacting protein 2 represses the initial phase of HIV-1 gene transcription in human microglial cells. Nucleic Acids Res (2005) 33(7):2318–31.
127. Rohr O, Lecestre D, Chasserot-Golaz S, Marban C, Avram D, Aunis D, et al. Recruitment of tat to heterochromatin protein HP1 via interaction with CTIP2 inhibits human immunodeficiency virus type 1 replication in microglial cells. J Virol (2003) 77(9):5415–27. doi:10.1128/JVI.77.9.5415-5427.2003
128. Le Douce V, Colin L, Redel L, Cherrier T, Herbein G, Aunis D, et al. LSD1 cooperates with CTIP2 to promote HIV-1 transcriptional silencing. Nucleic Acids Res (2012) 40(5):1904–15. doi:10.1093/nar/gkr857
129. Cherrier T, Le Douce V, Eilebrecht S, Riclet R, Marban C, Dequiedt F, et al. CTIP2 is a negative regulator of P-TEFb. Proc Natl Acad Sci U S A (2013) 110(31):12655–60. doi:10.1073/pnas.1220136110
130. Bres V, Yoh SM, Jones KA. The multi-tasking P-TEFb complex. Curr Opin Cell Biol (2008) 20(3):334–40. doi:10.1016/j.ceb.2008.04.008
131. Eilebrecht S, Le Douce V, Riclet R, Targat B, Hallay H, Van Driessche B, et al. HMGA1 recruits CTIP2-repressed P-TEFb to the HIV-1 and cellular target promoters. Nucleic Acids Res (2014) 42(8):4962–71. doi:10.1093/nar/gku168
132. McNamara RP, Reeder JE, McMillan EA, Bacon CW, McCann JL, D’Orso I. KAP1 recruitment of the 7SK snRNP complex to promoters enables transcription elongation by RNA polymerase II. Mol Cell (2016) 61(1):39–53. doi:10.1016/j.molcel.2015.11.004
133. McNamara RP, McCann JL, Gudipaty SA, D’Orso I. Transcription factors mediate the enzymatic disassembly of promoter-bound 7SK snRNP to locally recruit P-TEFb for transcription elongation. Cell Rep (2013) 5(5):1256–68. doi:10.1016/j.celrep.2013.11.003
134. Achhra AC, Amin J, Law MG, Emery S, Gerstoft J, Gordin FM, et al. Immunodeficiency and the risk of serious clinical endpoints in a well studied cohort of treated HIV-infected patients. AIDS (2010) 24(12):1877–86. doi:10.1097/QAD.0b013e32833b1b26
135. Deeks SG, Phillips AN. HIV infection, antiretroviral treatment, ageing, and non-AIDS related morbidity. BMJ (2009) 338:a3172. doi:10.1136/bmj.a3172
136. Ferry T, Raffi F, Collin-Filleul F, Dupon M, Dellamonica P, Waldner A, et al. Uncontrolled viral replication as a risk factor for non-AIDS severe clinical events in HIV-infected patients on long-term antiretroviral therapy: APROCO/COPILOTE (ANRS CO8) cohort study. J Acquir Immune Defic Syndr (2009) 51(4):407–15. doi:10.1097/QAI.0b013e3181acb65f
137. Le Douce V, Janossy A, Hallay H, Ali S, Riclet R, Rohr O, et al. Achieving a cure for HIV infection: do we have reasons to be optimistic? J Antimicrob Chemother (2012) 67(5):1063–74. doi:10.1093/jac/dkr599
138. Kumar A, Darcis G, Van Lint C, Herbein G. Epigenetic control of HIV-1 post integration latency: implications for therapy. Clin Epigenetics (2015) 7(1):103. doi:10.1186/s13148-015-0137-6
139. Deeks SG, Autran B, Berkhout B, Benkirane M, Cairns S, Chomont N, et al. Towards an HIV cure: a global scientific strategy. Nat Rev Immunol (2012) 12(8):607–14. doi:10.1038/nri3262
140. Shan L, Deng K, Shroff NS, Durand CM, Rabi SA, Yang H-C, et al. Stimulation of HIV-1-specific cytolytic T lymphocytes facilitates elimination of latent viral reservoir after virus reactivation. Immunity (2012) 36(3):491–501. doi:10.1016/j.immuni.2012.01.014
141. Bouchat S, Gatot J-S, Kabeya K, Cardona C, Colin L, Herbein G, et al. Histone methyltransferase inhibitors induce HIV-1 recovery in resting CD4(+) T cells from HIV-1-infected HAART-treated patients. AIDS (2012) 26(12):1473–82. doi:10.1097/QAD.0b013e32835535f5
142. Darcis G, Kula A, Bouchat S, Fujinaga K, Corazza F, Ait-Ammar A, et al. An in-depth comparison of latency-reversing agent combinations in various in vitro and ex vivo HIV-1 latency models identified bryostatin-1+JQ1 and ingenol-B+JQ1 to potently reactivate viral gene expression. PLoS Pathog (2015) 11(7):e1005063. doi:10.1371/journal.ppat.1005063
143. Halper-Stromberg A, Lu C-L, Klein F, Horwitz JA, Bournazos S, Nogueira L, et al. Broadly neutralizing antibodies and viral inducers decrease rebound from HIV-1 latent reservoirs in humanized mice. Cell (2014) 158(5):989–99. doi:10.1016/j.cell.2014.07.043
144. Jiang G, Mendes EA, Kaiser P, Wong DP, Tang Y, Cai I, et al. Synergistic reactivation of latent HIV expression by ingenol-3-angelate, PEP005, targeted NF-kB signaling in combination with JQ1 induced P-TEFb activation. PLoS Pathog (2015) 11(7):e1005066. doi:10.1371/journal.ppat.1005066
145. Jiang G, Mendes EA, Kaiser P, Sankaran-Walters S, Tang Y, Weber MG, et al. Reactivation of HIV latency by a newly modified ingenol derivative via protein kinase Cδ-NF-κB signaling. AIDS (2014) 28(11):1555–66. doi:10.1097/QAD.0000000000000289
146. Spivak AM, Planelles V. HIV-1 eradication: early trials (and tribulations). Trends Mol Med (2016) 22(1):10–27. doi:10.1016/j.molmed.2015.11.004
147. Sakane N, Kwon HS, Pagans S, Kaehlcke K, Mizusawa Y, Kamada M, et al. Activation of HIV transcription by the viral tat protein requires a demethylation step mediated by lysine-specific demethylase 1 (LSD1/KDM1). PLoS Pathog (2011) 7(8):e1002184. doi:10.1371/journal.ppat.1002184
148. Yatim A, Benne C, Sobhian B, Laurent-Chabalier S, Deas O, Judde J-G, et al. NOTCH1 nuclear interactome reveals key regulators of its transcriptional activity and oncogenic function. Mol Cell (2012) 48(3):445–58. doi:10.1016/j.molcel.2012.08.022
149. Watters SA, Mlcochova P, Gupta RK. Macrophages: the neglected barrier to eradication. Curr Opin Infect Dis (2013) 26(6):561–6. doi:10.1097/QCO.0000000000000014
150. Vivithanaporn P, Asahchop EL, Acharjee S, Baker GB, Power C. HIV protease inhibitors disrupt astrocytic glutamate transporter function and neurobehavioral performance. AIDS (2016) 30(4):543–52. doi:10.1097/QAD.0000000000000955
151. Muldoon LL, Alvarez JI, Begley DJ, Boado RJ, Del Zoppo GJ, Doolittle ND, et al. Immunologic privilege in the central nervous system and the blood-brain barrier. J Cereb Blood Flow Metab (2013) 33(1):13–21. doi:10.1038/jcbfm.2012.153
152. Nath A, Clements JE. Eradication of HIV from the brain: reasons for pause. AIDS (2011) 25(5):577–80. doi:10.1097/QAD.0b013e3283437d2f
153. Yadav A, Collman RG. CNS inflammation and macrophage/microglial biology associated with HIV-1 infection. J Neuroimmune Pharmacol (2009) 4(4):430–47. doi:10.1007/s11481-009-9174-2
154. Kulpa DA, Chomont N. HIV persistence in the setting of antiretroviral therapy: when, where and how does HIV hide? J Virus Erad (2015) 1(2):59–66.
155. Sáez-Cirión A, Bacchus C, Hocqueloux L, vettand-Fenoel VA, Girault I, Lecuroux C, et al. Post-treatment HIV-1 controllers with a long-term virological remission after the interruption of early initiated antiretroviral therapy ANRS VISCONTI study. PLoS Pathog (2013) 9(3):e1003211. doi:10.1371/journal.ppat.1003211
156. Chun TW, Moir S, Fauci AS. HIV reservoirs as obstacles and opportunities for an HIV cure. Nat Immunol (2015) 16(6):584–9. doi:10.1038/ni.3152
157. Chauhan A. Enigma of HIV-1 latent infection in astrocytes: an in-vitro study using protein kinase C agonist as a latency reversing agent. Microbes Infect (2015) 17(9):651–9. doi:10.1016/j.micinf.2015.05.006
158. Rasmussen TA, Søgaard OS, Brinkmann C, Wightman F, Lewin SR, Melchjorsen J, et al. Comparison of HDAC inhibitors in clinical development: effect on HIV production in latently infected cells and T-cell activation. Human Vaccines & Immunotherapeutics (2013) 9(5):993–1001. doi:10.4161/hv.23800
159. Banerjee C, Archin N, Michaels D, Belkina AC, Denis GV, Bradner J, et al. BET bromodomain inhibition as a novel strategy for reactivation of HIV-1. J Leukoc Biol (2012) 92(6):1147–54. doi:10.1189/jlb.0312165
160. Díaz L, Martínez-Bonet M, Sánchez J, Fernández-Pineda A, Jiménez JL, Muñoz E, et al. Bryostatin activates HIV-1 latent expression in human astrocytes through a PKC and NF-ĸB-dependent mechanism. Sci Rep (2015) 5:12442. doi:10.1038/srep12442
161. Kollár P, Rajchard J, Balounová Z, Pazourek J. Marine natural products: bryostatins in preclinical and clinical studies. Pharm Biol (2014) 52(2):237–42. doi:10.3109/13880209.2013.804100
162. Laird GM, Bullen CK, Rosenbloom DI, Martin AR, Hill AL, Durand CM, et al. Ex vivo analysis identifies effective HIV-1 latency-reversing drug combinations. J Clin Invest (2015) 125(5):1901–12. doi:10.1172/JCI80142
163. Darcis G, Van Driessche B, Van Lint C. Preclinical shock strategies to reactivate latent HIV-1: an update. Curr Opin HIV AIDS (2016) 11(4):388–93. doi:10.1097/COH.0000000000000288
164. Rasmussen TA, Tolstrup M, Jon Møller H, Brinkmann CR, Olesen R, Erikstrup C, et al. Activation of latent human immunodeficiency virus by the histone deacetylase inhibitor panobinostat: a pilot study to assess effects on the central nervous system. Open Forum Infect Dis (2015) 2(1):ofv037. doi:10.1093/ofid/ofv037
165. Mousseau G, Mediouni S, Valente ST. Targeting HIV transcription: the quest for a functional cure. Curr Top Microbiol Immunol (2015) 389(January):121–45. doi:10.1007/82_2015_435
166. Vitiello M, Galdiero M, Finamore E, Galdiero S, Galdiero M. NF-κB as a potential therapeutic target in microbial diseases. Mol Biosyst (2012) 8(4):1108–20. doi:10.1039/c2mb05335g
167. Pateras I, Giaginis C, Tsigris C, Patsouris E, Theocharis S. NF-κB signaling at the crossroads of inflammation and atherogenesis: searching for new therapeutic links. Expert Opin Ther Targets (2014) 18(9):1089–101. doi:10.1517/14728222.2014.938051
168. Kurapati KR, Atluri VS, Samikkannu T, Garcia G, Nair MP. Natural products as anti-HIV agents and role in HIV-associated neurocognitive disorders (HAND): a brief overview. Front Microbiol (2015) 6(January):1444. doi:10.3389/fmicb.2015.01444
169. Cos P, Maes L, Vanden Berghe D, Hermans N, Pieters L, Vlietinck A. Plant substances as anti-HIV agents selected according to their putative mechanism of action. J Nat Prod (2004) 67(2):284–93. doi:10.1021/np034016p
170. Aoki S, Watanabe Y, Sanagawa M, Setiawan A, Kotoku N, Kobayashi M. Cortistatins A, B, C, and D, anti-angiogenic steroidal alkaloids, from the marine sponge corticium simplex. J Am Chem Soc (2006) 128(10):3148–9. doi:10.1021/ja057404h
171. Mousseau G, Clementz MA, Bakeman WN, Nagarsheth N, Cameron M, Shi J, et al. An analog of the natural steroidal alkaloid cortistatin A potently suppresses tat-dependent HIV transcription. Cell Host Microbe (2012) 12(1):97–108. doi:10.1016/j.chom.2012.05.016
172. Mousseau G, Valente ST. Didehydro-cortistatin A: a new player in HIV-therapy? Expert Rev Anti Infect Ther (2016) 14(2):145–8. doi:10.1586/14787210.2016.1122525
173. Wan Z, Chen X. Triptolide inhibits human immunodeficiency virus type 1 replication by promoting proteasomal degradation of tat protein. Retrovirology (2014) 11(January):88. doi:10.1186/s12977-014-0088-6
174. Cherrier T, Elias M, Jeudy A, Gotthard G, Le Douce V, Hallay H, et al. Human-phosphate-binding-protein inhibits HIV-1 gene transcription and replication. Virol J (2011) 8:352. doi:10.1186/1743-422X-8-352
175. Darbinian-Sarkissian N, Darbinyan A, Otte J, Radhakrishnan S, Sawaya BE, Arzumanyan A, et al. p27(SJ), a novel protein in St John’s Wort, that suppresses expression of HIV-1 genome. Gene Ther (2006) 13(4):288–95. doi:10.1038/sj.gt.3302649
176. Lesner A, Shilpi R, Ivanova A, Gawinowicz MA, Lesniak J, Nikolov D, et al. Identification of X-DING-CD4, a new member of human DING protein family that is secreted by HIV-1 resistant CD4(+) T cells and has anti-viral activity. Biochem Biophys Res Commun (2009) 389(2):284–9. doi:10.1016/j.bbrc.2009.08.140
177. Shilpi RY, Sachdeva R, Simm M. Cellular resistance to HIV-1 infection in target cells coincides with a rapid induction of X-DING-CD4 mRNA: indication of the unique host innate response to virus regulated through function of the X-DING-CD4 gene. Innate Immun (2012) 18(4):563–70. doi:10.1177/1753425911426893
178. Suh A, Le Douce V, Rohr O, Schwartz C, Scott K. Pseudomonas DING proteins as human transcriptional regulators and HIV-1 antagonists. Virol J (2013) 10(January):234. doi:10.1186/1743-422X-10-234
179. Darbinian N, Khalili K, Amini S. Neuroprotective activity of pDING in response to HIV-1 tat. J Cell Physiol (2014) 229(2):153–61. doi:10.1002/jcp.24392
180. Gavegnano C, Schinazi RF. Antiretroviral therapy in macrophages: implication for HIV eradication. Antivir Chem Chemother (2009) 20(2):63–78. doi:10.3851/IMP1374
181. Moreau A, Le Vee M, Jouan E, Parmentier Y, Fardel O. Drug transporter expression in human macrophages. Fundam Clin Pharmacol (2011) 25(6):743–52. doi:10.1111/j.1472-8206.2010.00913.x
182. Dallas S, Schlichter L, Bendayan R. Multidrug resistance protein (MRP) 4- and MRP 5-mediated efflux of 9-(2-Phosphonylmethoxyethyl)adenine by microglia. J Pharmacol Exp Ther (2004) 309(3):1221–9. doi:10.1124/jpet.103.063966
183. Giacalone G, Hillaireau H, Fattal E. Improving bioavailability and biodistribution of anti-HIV chemotherapy. Eur J Pharm Sci (2015) 75(July):40–53. doi:10.1016/j.ejps.2015.04.011
184. Ramana LN, Anand AR, Sethuraman S, Krishnan UM. Targeting strategies for delivery of anti-HIV drugs. J Control Release (2014) 192(October):271–83. doi:10.1016/j.jconrel.2014.08.003
185. Lenjisa JL, Woldu MA, Satessa GD. New hope for eradication of HIV from the body: the role of polymeric nanomedicines in HIV/AIDS pharmacotherapy. J Nanobiotechnology (2014) 12(January):9. doi:10.1186/1477-3155-12-9
186. Das MK, Sarma A, Chakraborty T. Nano-ART and NeuroAIDS. Drug Deliv Transl Res (2016) 6(5):452–72. doi:10.1007/s13346-016-0293-z
187. Nair M, Jayant RD, Kaushik A, Sagar V. Getting into the brain: potential of nanotechnology in the management of NeuroAIDS. Adv Drug Deliv Rev (2016) 103(August):202–17. doi:10.1016/j.addr.2016.02.008
188. Abbas W, Tariq M, Iqbal M, Kumar A, Herbein G. Eradication of HIV-1 from the macrophage reservoir: an uncertain goal? Viruses (2015) 7(4):1578–98. doi:10.3390/v7041578
189. Kumar A, Herbein G. The macrophage: a therapeutic target in HIV-1 infection. Mol Cell Ther (2014) 2(January):10. doi:10.1186/2052-8426-2-10
190. Rasmussen TA, Tolstrup M, Søgaard OS. Reversal of latency as part of a cure for HIV-1. Trends Microbiol (2016) 24(2):90–7. doi:10.1016/j.tim.2015.11.003
191. Poluektova LY, Munn DH, Persidsky Y, Gendelman HE. Generation of cytotoxic T cells against virus-infected human brain macrophages in a murine model of HIV-1 encephalitis. J Immunol (2002) 168(8):3941–9.
192. Persidsky Y, Ramirez SH, Haorah J, Kanmogne GD. Blood-brain barrier: structural components and function under physiologic and pathologic conditions. J Neuroimmune Pharmacol (2006) 1(3):223–36. doi:10.1007/s11481-006-9025-3
193. Landrith TA, Harris TH, Wilson EH. Characteristics and critical function of CD8+ T cells in the toxoplasma-infected brain. Semin Immunopathol (2015) 37(3):261–70. doi:10.1007/s00281-015-0487-3
194. Brockman MA, Jones RB, Brumme ZL. Challenges and opportunities for T-Cell-mediated strategies to eliminate HIV reservoirs. Front Immunol (2015) 6(January):506. doi:10.3389/fimmu.2015.00506
195. Ensoli B, Cafaro A, Monini P, Marcotullio S, Ensoli F. Challenges in HIV vaccine research for treatment and prevention. Front Immunol (2014) 5(January):417. doi:10.3389/fimmu.2014.00417
Keywords: brain, reservoirs, latency, cure, cART, HIV transcription
Citation: Marban C, Forouzanfar F, Ait-Ammar A, Fahmi F, El Mekdad H, Daouad F, Rohr O and Schwartz C (2016) Targeting the Brain Reservoirs: Toward an HIV Cure. Front. Immunol. 7:397. doi: 10.3389/fimmu.2016.00397
Received: 01 July 2016; Accepted: 20 September 2016;
Published: 30 September 2016
Edited by:
Ihssane Zouikr, RIKEN Brain Science Institute, JapanReviewed by:
Taisuke Izumi, Frederick National Laboratory for Cancer Research, USACopyright: © 2016 Marban, Forouzanfar, Ait-Ammar, Fahmi, El Mekdad, Daouad, Rohr and Schwartz. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Olivier Rohr, b2xpdmllci5yb2hyQHVuaXN0cmEuZnI=;
Christian Schwartz, c2Nod2FydHouY2hyaXN0aWFuQHVuaXN0cmEuZnI=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.