 Camilla F. Wenceslau
Camilla F. Wenceslau Cameron G. McCarthy
Cameron G. McCarthy- Department of Physiology, Augusta University, Augusta, GA, USA
The major pathophysiological characteristic of systemic inflammatory response syndrome (SIRS) and sepsis is the loss of control of vascular tone and endothelial barrier dysfunction. These changes are attributed to pro-inflammatory mediators. It has been proposed that in patients and rats without infection, cell components from damaged tissue are the primary instigators of vascular damage. Mitochondria share several characteristics with bacteria, and when fragments of mitochondria are released into the circulation after injury, they are recognized by the innate immune system. N-Formyl peptides are common molecular signatures of bacteria and mitochondria and are known to play a role in the initiation of inflammation by activating the formyl peptide receptor (FPR). We have demonstrated that infusion of mitochondrial N-formyl peptides (F-MIT) leads to sepsis-like symptoms, including vascular leakage. We have also observed that F-MIT, via FPR activation, elicits changes in cytoskeleton-regulating proteins in endothelial cells. Therefore, we hypothesize that these FPR-mediated changes in cytoskeleton can cause endothelial cell contraction and, consequently vascular leakage. Here, we propose that endothelial FPR is a key contributor to impaired barrier function in SIRS and sepsis patients following trauma.
Introduction
Systemic inflammatory response syndrome (SIRS) and sepsis are the principal causes of death in intensive care units, with mortality rates between 30 and 70% (1–3) The diagnosis of sepsis requires confirmation of bacterial growth in blood cultures, as well as the presence of two or more of the following symptoms: hypothermia or hyperthermia, tachycardia, tachypnea, and leukocytopenia or leukocytosis (1, 2). It is noteworthy that only about one-third to one-half of patients meeting these criteria were subsequently confirmed to have sepsis (1–3). Accordingly, all remaining patients are diagnosed with SIRS, a sepsis-like condition. Therefore, diagnosis of SIRS requires the same symptoms, minus the presence of infection. One major pathophysiological characteristic of SIRS and sepsis is the breakdown of the endothelial barrier function results in the loss of fluid into the extravascular space that leads to edema in several tissues (1, 2, 4).
The circulatory responses to sepsis are characterized by an initial hyperdynamic phase, where there is a reduction in systemic vascular resistance due to peripheral vasodilation and unresponsiveness to vasoconstrictor drugs are observed (5). Later, the myocardial dysfunction caused by pro-inflammatory mediators leads to a hypodynamic phase, where cardiac output significantly falls and both the systolic and the diastolic functions of the heart are greatly impaired (5). The endothelium lining of the vascular walls is a major target of sepsis, especially during the initial hyperdynamic phase, and damage of endothelial cells accounts for much of the pathology of septic shock (4, 5). The increase in endothelial permeability and dysfunction has been associated with several factors, including hypoxia, oxidative stress and augmented TNF-α levels (4). Other interleukins (e.g., IL-1, IL-2, and IL-6) are also contributors (6). In line, IL-6 is known to be a clinically suitable biomarker for sepsis, and Waage and coworkers (7) observed high levels of IL-6 and its association with sepsis in patients with meningococcal infection (7). Corroborating these data, Lo and others (8) demonstrated that IL-6 induced redistribution of vascular-endothelial cadherin, which is the major component of endothelial adhesion and barrier function via adherens junctions. Vascular endothelial cadherin-regulated protein complexes that join adjacent endothelial cells and prevent leukocyte emigration and vascular leakage (6, 8). Therefore, disruption of vascular endothelial cadherins function results in trans-endothelial flow of fluid and interstitial edema. Also, it has been observed that other molecules released during acute inflammation such as bradykinin, thrombin, VEGF, and histamine result in endothelial activation and massive increases in glycocalyx expression of endothelial leukocyte adhesion molecule 1, intercellular adhesion molecule 1 (ICAM-1), and vascular cell adhesion molecule 1 (VCAM-1) (9). The increased expression of these proteins leads to leukocyte rolling, adherence, and migration, which initiate the inflammatory damage to endothelium and endo organs (9). Furthermore, it is well known that exacerbated production of nitric oxide by the inducible form of nitric oxide synthase (iNOS) contributes to vascular leakage and hyporeactivity. Nevertheless, pharmacological interventions using NOS inhibitors have not been successful. Currently, there are no therapies for blocking vascular leakage in SIRS and sepsis, given that the molecular mechanisms regulating vascular permeability are not completely understood.
Contraction of Endothelial Cell: A New Paradigm for the Regulation of Vascular Leakage
Although the concept that active contraction of endothelial cells was first suggested by Majno in 1961 (10, 11), currently the intracellular events regulating endothelial contractile activity is still unknown. Just like in other type of cells, the dynamic assembly, disassembly, and reorganization of the actin and myosin cytoskeleton regulate endothelial cells contraction (12). Accordingly, Goeckeler and Wysolmerski (12) reported that thrombin stimulation results in rapid sustained isometric contraction in endothelial cells that increases twofold within 5 min and remains elevated for 60 min. Also, they observed that myosin light chain (MLC) phosphorylation precedes the development of isometric tension (12). Supporting these data, it has been shown that transfection of constitutively active MLCK induces MLC phosphorylation associated with increase in permeability in endothelial cells (13). On the other hand, inhibition of MLC phosphorylation with an MLCK antagonist greatly attenuates the increase in venular permeability in response to soluble inflammatory mediators (14).
More recently, it has been demonstrated that not only the interaction of actin and myosin is necessary for endothelial contraction but also changes in actin polymerization have been associated with the development of isometric force. In many cell types, including endothelial cells, the actin cytoskeleton is a highly dynamic structure that undergoes polymerization and depolymerization based upon cellular demand (15). It is known that actin polymerization occurs in two steps, nucleation and elongation. Nucleation occurs when three actin monomers bind together and provides a site for elongation. Elongation occurs when ATP bound globular (G)-actin binds and grows to form filamentous (F)-actin (15). Reorganization of F-actin, which is a fundamental unit for actin-based cytoskeleton structures, is paramount for endothelial cell contraction and barrier function. Several permeability factors, including angiogenic and inflammatory mediators, trigger signaling pathway in endothelial cell that enhance F-actin polymerization and actomyosin contractility.
It well known that Rho family of p21 small GTP-binding proteins are associated with the direct regulation of actin cytoskeleton (16). RhoA induces actin polymerization at focal adhesions by activating formin-homology protein Dia1, a potent activator of nucleation and elongation of actin filaments, and inhibits actin filament disassembly by inactivation of ADF/cofilin, a family of actin-binding proteins, which disassembles actin filaments. Additionally, RhoA promotes contractility by activating the myosin light-chain kinase through ROCK kinase (17). An interesting study by Gorovoy et al. (18) provide strong evidence that increased RhoA activity results in vascular leakage in mouse lung. In this study, it was observed that increased RhoA activity due to deletion of one its inhibitory proteins, RhoGDI, causes a loss of endothelial junctional integrity and breakdown in endothelial barrier function (18).
Mitogen-activated protein (MAP) kinases are a family of stress activated enzymes that initiate signaling cascades in response to several stimuli, including inflammation and injury. It has previously shown that p38 MAPK kinase leads to reorganization of the actin cytoskeleton to form stress fibers and increase in vascular permeability (19). Furthermore, It was demonstrated in a recently study by using atomic force microscopy and a combination of confocal microscopy methods that thermal injury induces venular hyperpermeability and that serum from burned rats induces endothelium cells actin rearrangement and contraction (20). However, inhibition of the p38 MAPK ameliorates resulting vascular dysfunction by significantly reducing endothelial cells contraction (20).
Despite new information about the pathophysiology of molecular mechanisms regulating vascular permeability, this disruption continues to be treated inappropriately and this contributes to an unacceptably high mortality rate in patients with SIRS and sepsis. Therefore, understanding vascular function and the causes for this disturbance would ultimately provide starting points for therapies designed to treat these devastating diseases.
Mitochondrial N-Formyl Peptides, Formyl Peptide Receptor, and Endothelial Cell Cytoskeleton
It has been proposed that in patients and rats without infection, cell components from damaged tissue can initiate the genesis of SIRS (21–25). These cell components are collectively called damage-associated molecular patterns (DAMPs). DAMPs are endogenous molecules that are released from cells following injury. For evolutionary reasons, mitochondria share several characteristics with bacteria, and when fragments of mitochondria are released into the circulation, they are recognized by the innate immune system. Due to protein translation initiation by formyl-methionine in both bacteria and mitochondria, N-formyl peptides are common molecular signatures of bacteria and mitochondria and are known to play a role in the initiation of inflammation by activating the formyl peptide receptor (FPR) (22, 26, 27). The FPR has been identified as a subfamily of G-protein-coupled receptors (27). FPR-1 and FPR-2 are expressed at high levels on leukocytes and mediate cell chemotaxis (27). However, it has been demonstrated that mitochondrial DAMPs suppress pulmonary immune response via FPR-1 and -2 (28). FPR activation leads to cytoskeletal rearrangements, making N-formyl peptides potent inducers of neutrophil F-actin formation. Stimulation of human neutrophils with the formylated peptide (fMLP, bacteria derived), known to result in a prompt rise of the calcium, also induced a rapid decrease of G-actin content and increase of filamentous actin, F-actin, content (29). A reduction of the fMLP induced calcium transient to about 25 nM, resulted in a less pronounced decrease of G-actin content and increase of F-actin content (30). FPR activation has also been demonstrated to signal through the Rho family protein cell division control protein 42 (Cdc42) to activate Rac- and ARP2/3-dependent pathways leading to actin nucleation (30). Recent evidence suggests that FPR is a membrane mechanosensor that senses the mechanical fluid stress and signals intracellular cascades. Blockage of FPR reduces pseudopod retraction response to fluid stress in neutrophils (31).
It is known though, that a major mechanism by which immune system causes organ damage is due to neutrophil-mediated increases in endothelial cell permeability. Nevertheless, in a recent study from Sun and collaborators (32) demonstrated that mitochondrial DNA isolated from human liver induced dose-dependent rise in endothelial permeability both in the presence and absence of neutrophils (32). In the absence of neutrophils, however, permeability changes were transient and decayed relatively quickly. In the presence of neutrophils, the increases in permeability were sustained and even intensified over time. In addition, Sun et al. demonstrated that non-formylated mitochondrial proteins cause changes in endothelial cell permeability directly. However, bacteria-derived fMLP did not induce any change in endothelial permeability (32).
We have observed that both mitochondrial N-formyl peptides (formylated peptide corresponding to the NH2-terminus of mitochondria NADPH dehydrogenase subunit 6; F-MIT) and fMLP (bacteria derived) induce vascular leakage and exacerbated vasodilatation in resistance arteries, and that a FPR antagonist inhibits these responses (22). F-MIT, but not non-formylated peptides or mitochondrial DNA, induced severe hypotension via FPR activation and histamine release (22). Supporting these data, rats that underwent hemorrhagic shock increased plasma levels of F-MIT associated with lung damage, and antagonism of FPR ameliorated this response.
As described above, FPR activation leads to cytoskeleton rearrangement in immune and non-immune cells. Therefore, because FPR is present in endothelial cells and because we observed that F-MIT induces vascular leakage due to FPR activation (22), we questioned if F-MIT via FPR activation could lead to changes in cytoskeleton-regulating proteins in primary cultures of human aortic endothelial cells (HAEC). To answer this question, we treated HAEC with F-MIT (20 min, 10 μM) in the presence or absence of a cocktail of FPR antagonists (FPR-1 antagonist, cyclosporine H: CsH, 1 μmol/L and FPR-2 antagonist, WRW4, 10 μmol/L). Treatment with F-MIT rapidly increased RhoA/ROCK (Rho: 1.8-fold vs. Veh; ROCK: 1.4-fold vs. Veh, p < 0.05), CDC42 (twofold vs. Veh, p < 0.05), and phospho-MLC Thr/Ser19 (1.5-fold vs. Veh, p < 0.05) in HAEC. These changes were abolished in the presence of FPR antagonist. These data suggest that F-MIT leads to changes in endothelial cell cytoskeleton independent of leukocyte activation. Given that we provided strong evidence FPR induced RhoA and CDC42 expression, and these proteins are linked with endothelial contraction via actin polymerization, we infer that F-MIT elicits endothelial contraction and subsequent vascular leakage not only via actomyosin interaction but also via actin polymerization.
Hypothesis
Our new mechanistic hypothesis is that F-MIT released from trauma/cell damage activate FPR leading to changes in endothelial cell cytoskeleton which subsequently induces endothelial contraction and vascular permeability (Figure 1).
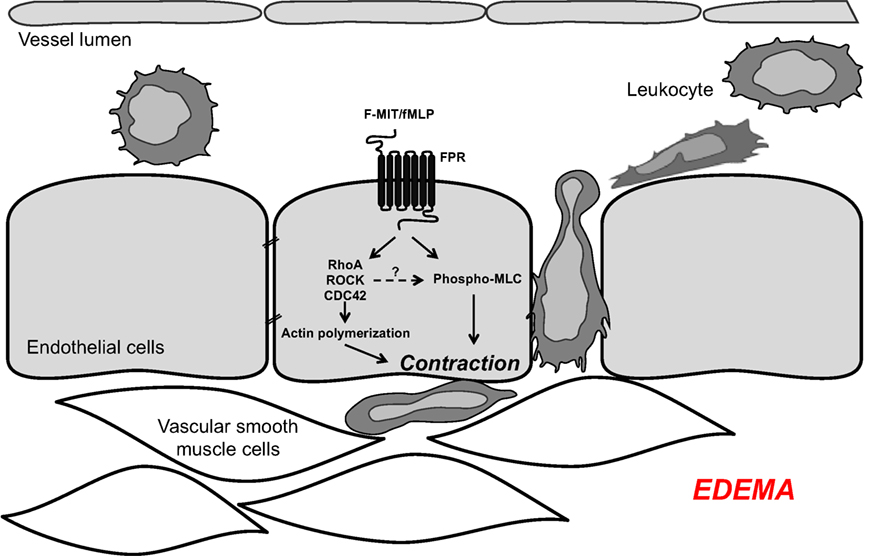
Figure 1. Mitochondrial N-formyl peptides (FMIT) trigger endothelial cell contraction and vascular leakage via formyl peptide receptor (FPR) activation, contributing to the loss of control of vascular tone and edema in trauma-induced SIRS and sepsis.
Evaluation of the Hypothesis
The inability to treat or prevent SIRS and sepsis in trauma patients may be due to our limited understanding of the molecular pathways that govern the disruption of normal vascular control and responsiveness. The proposed hypothesis is significant because it may offer a novel cellular and molecular mechanism underlying the development of arterial dysfunction in SIRS and extend our knowledge about how mitochondria-derived molecules mediate endothelial barrier dysfunction.
In the present study, we have observed that F-MIT, via FPR activation, elicits changes in cytoskeleton-regulating proteins in endothelial cells. Therefore, we infer that this interaction can lead to endothelial contraction, increased vascular leakage, and attenuated barrier function as observed in SIRS patients following trauma. Corroborating this hypothesis, we previously observed that F-MIT induced vascular leakage in conduit and resistance arteries from Wistar rats (22). Based on these findings, it is reasonable to speculate that in conditions of enhanced FPR activation (e.g., tissue damage/trauma), FPR-associated signaling leads increases in endothelial permeability, which subsequently contributes to end organ damage. Therefore, we believe that the FPR may be a target for the treatment of SIRS and sepsis.
Author Contributions
CW analyzed the data in HAEC and wrote, discussed, and reviewed the manuscript. CM performed the experiments in HAEC, discussed, and reviewed the manuscript. RW reviewed the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Dr. Kiyoshi Itagaki for the invitation to write this hypothesis article.
Funding
This work was funded by American Heart Association (#14POST20490292 and #13PRE14080019) and National Institutes of Health.
References
1. Riedemann NC, Guo RF, Ward PA. Novel strategies for the treatment of sepsis. Nat Med (2003) 9:517–24. doi: 10.1038/nm0503-517
2. Rangel-Frausto MS, Pittet D, Costigan M, Hwang T, Davis CS, Wenzel RP. The natural history of the systemic inflammatory response syndrome (SIRS). A prospective study. JAMA (1995) 273(2):117–23. doi:10.1001/jama.273.2.117
3. Brun-Buisson C. The epidemiology of the systemic inflammatory response. Intensive Care Med (2000) 26:S64–74. doi:10.1007/s001340051121
4. Peters K, Unger RE, Brunner J, Kirkpatrick CJ. Molecular basis of endothelial dysfunction in sepsis. Cardiovasc Res (2003) 60:49–57. doi:10.1016/S0008-6363(03)00397-3
5. Kotsovolis G, Kallaras K. The role of endothelium and endogenous vasoactive substances in sepsis. Hippokratia (2010) 14(2):88–93.
6. Krüttgen A, Rose-John S. Interleukin-6 in sepsis and capillary leakage syndrome. J Interferon Cytokine Res (2012) 32(2):60–5. doi:10.1089/jir.2011.0062
7. Waage A, Brandtzaeg P, Halstensen A, Kierulf P, Espevik T. The complex pattern of cytokines in serum from patients with meningococcal septic shock. Association between interleukin 6, interleukin 1, and fatal outcome. J Exp Med (1989) 169(1):333–8. doi:10.1084/jem.169.1.333
8. Lo CW, Chen MW, Hsiao M, Wang S, Chen CA, Hsiao SM, et al. IL-6 trans-signaling in formation and progression of malignant ascites in ovarian cancer. Cancer Res (2011) 71:424–34. doi:10.1158/0008-5472.CAN-10-1496
9. Chelazzi C, Villa G, Mancinelli P, De Gaudio AR, Adembri C. Glycocalyx and sepsis-induced alterations in vascular permeability. Crit Care (2015) 19:26. doi:10.1186/s13054-015-0741-z
10. Majno G, Palade GE. Studies on inflammation. I. The effect of histamine and serotonin on vascular permeability: an electron microscopic study. J Biophys Biochem Cytol (1961) 11:571. doi:10.1083/jcb.11.3.571
11. Majno G, Palade GE, Schoefl G. Studies on inflammation. II. The site of action of histamine and serotonin along the vascular tree: a topographic study. J Biophys Biochem Cytol (1961) 11:607. doi:10.1083/jcb.11.3.571
12. Goeckeler ZM, Wysolmerski RB. Myosin light chain kinase-regulated endothelial cell contraction: the relationship between isometric tension, actin polymerization, and myosin phosphorylation. J Cell Biol (1995) 130(3):613–27. doi:10.1083/jcb.130.3.613
13. Tinsley JH, De Lanerolle P, Wilson E, Ma W, Yuan SY. Myosin light chain kinase transference induces myosin light chain activation and endothelial hyperpermeability. Am J Physiol Cell Physiol (2000) 279(4):C1285–9.
14. Yuan Y, Huang Q, Wu HM. Myosin light chain phosphorylation: modulation of basal and agonist-stimulated venular permeability. Am J Physiol (1997) 272(3 Pt 2):H1437–43.
15. Prasain N, Stevens T. The actin cytoskeleton in endothelial cell phenotypes. Microvasc Res (2009) 77(1):53–63. doi:10.1016/j.mvr.2008.09.012
16. Spiering D, Hodgson L. Dynamics of the Rho-family small GTPases in actin regulation and motility. Cell Adh Migr (2011) 5(2):170–80. doi:10.4161/cam.5.2.14403
17. Saarikangas J, Zhao H, Lappalainen P. Regulation of the actin cytoskeleton-plasma membrane interplay by phosphoinositides. Physiol Rev (2010) 90(1):259–89. doi:10.1152/physrev.00036.2009
18. Gorovoy M, Neamu R, Niu J, Vogel S, Predescu D, Miyoshi J, et al. RhoGDI-1 modulation of the activity of monomeric RhoGTPase RhoA regulates endothelial barrier function in mouse lungs. Circ Res (2007) 101:50–8. doi:10.1161/CIRCRESAHA.106.145847
19. Kayyali US, Pennella CM, Trujillo C, Villa O, Gaestel M, Hassoun PM. Cytoskeletal changes in hypoxic pulmonary endothelial cells are dependent on MAPK-activated protein kinase MK2. J Biol Chem (2002) 277(45):42596–602. doi:10.1074/jbc.M205863200
20. Wang S, Huang Q, Guo J, Guo X, Sun Q, Brunk UT, et al. Local thermal injury induces general endothelial cell contraction through p38 MAP kinase activation. APMIS (2014) 122(9):832–41. doi:10.1111/apm.12226
21. Wenceslau CF, McCarthy CG, Szasz T, Spitler K, Goulopoulou S, Webb RC, et al. Mitochondrial damage-associated molecular patterns and vascular function. Eur Heart J (2014) 35:1172–9. doi:10.1093/eurheartj/ehu047
22. Wenceslau CF, McCarthy CG, Szasz T, Goulopoulou S, Webb RC. Mitochondrial N-formyl peptides induce cardiovascular collapse and sepsis-like syndrome. Am J Physiol Heart Circ Physiol (2015) 308:H768–77. doi:10.1152/ajpheart.00779.2014
23. Zhang Q, Raoof M, Chen Y, Sumi Y, Sursal T, Junger W, et al. Mitochondrial DAMPs cause inflammatory responses to injury. Nature (2010) 464:104–7. doi:10.1038/nature08780
24. Wenceslau CF, McCarthy CG, Goulopoulou S, Szasz T, NeSmith EG, Webb RC. Mitochondrial-derived N-formyl peptides: novel links between trauma, vascular collapse and sepsis. Med Hypotheses (2013) 81(4):532–5. doi:10.1016/j.mehy.2013.06.026
25. Wenceslau CF, Szasz T, McCarthy CG, Baban B, NeSmith E, Webb RC. Mitochondrial N-formyl peptides cause airway contraction and lung neutrophil infiltration via formyl peptide receptor activation. Pulm Pharmacol Ther (2016) 37:49–56. doi:10.1016/j.pupt.2016.02.005
26. Rabiet MJ, Huet E, Boulay F. Human mitochondria-derived N-formylated peptides are novel agonists equally active on FPR and FPRL1, while Listeria monocytogenes-derived peptides preferentially activate FPR. Eur J Immunol (2005) 35:2486–95. doi:10.1002/eji.200526338
27. Le Y, Murphy PM, Wang JM. Formyl peptide receptors revisited. Trends Immunol (2002) 23:541–8. doi:10.1016/S1471-4906(02)02316-5
28. Li H, Itagaki K, Sandler N, Gallo D, Galenkamp A, Kaczmarek E, et al. Mitochondrial damage-associated molecular patterns from fractures suppress pulmonary immune responses via formyl peptide receptors 1 and 2. J Trauma Acute Care Surg (2015) 78(2):272–9. doi:10.1097/TA.0000000000000509
29. Bengtsson T, Stendahl O, Andersson T. The role of the cytosolic free Ca2+ transient for fMet-Leu-Phe induced actin polymerization in human neutrophils. Eur J Cell Biol (1986) 42(2):338–43.
30. Glogauer M, Hartwig J, Stossel T. Two pathways through Cdc42 couple the N-formyl receptor to actin nucleation in permeabilized human neutrophils. J Cell Biol (2000) 150(4):785–96. doi:10.1083/jcb.150.4.785
31. Makino A, Glogauer M, Bokoch GM, Chien S, Schmid-Schönbein GW. Control of neutrophil pseudopods by fluid shear: role of Rho family GTPases. Am J Physiol Cell Physiol (2005) 288(4):C863–71. doi:10.1152/ajpcell.00358.2004
Keywords: formyl peptide receptor, mitochondria N-formyl peptides, endothelial cells, vascular leakage, SIRS
Citation: Wenceslau CF, McCarthy CG and Webb RC (2016) Formyl Peptide Receptor Activation Elicits Endothelial Cell Contraction and Vascular Leakage. Front. Immunol. 7:297. doi: 10.3389/fimmu.2016.00297
Received: 03 June 2016; Accepted: 22 July 2016;
Published: 02 August 2016
Edited by:
Kiyoshi Itagaki, Harvard Medical School, USACopyright: © 2016 Wenceslau, McCarthy and Webb. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Camilla F. Wenceslau, Y2FtaWxsYXdlbmNlc2xhdUBob3RtYWlsLmNvbQ==