 Amanda E. I. Proudfoot
Amanda E. I. Proudfoot Mariagrazia Uguccioni
Mariagrazia Uguccioni
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Immunol. , 19 May 2016
Sec. Inflammation
Volume 7 - 2016 | https://doi.org/10.3389/fimmu.2016.00183
This article is part of the Research Topic Regulation of inflammation, its resolution and therapeutic targeting View all 11 articles
Chemokine biology is mediated by more complex interactions than simple monomolecular ligand–receptor interactions, as chemokines can form higher order quaternary structures, which can also be formed after binding to glycosaminoglycans (GAGs) on endothelial cells, and their receptors are found as dimers and/or oligomers at the cell surface. Due to the complexity of the chemokine binding and signaling system, several mechanisms have been proposed to provide an explanation for the synergy observed between chemokines in leukocyte migration. Pioneering studies on interactions between different chemokines have revealed that they can act as antagonists, or synergize with other chemokines. The synergism can occur at different levels, involving either two chemokine receptors triggered simultaneously or sequentially exposed to their agonists, or the activation of one type of chemokine receptor triggered by chemokine heterocomplexes. In addition to the several chemokines that, by forming a heterocomplex with chemokine receptor agonists, act as enhancers of molecules of the same family, we have recently identified HMGB1, an endogenous damage-associated molecular patterns (DAMPs) molecule, as an enhancer of the activity of CXCL12. It is now evident that synergism between chemokines is crucial at the very early stage of inflammation. In addition, the low-affinity interaction with GAGs has recently been shown to induce cooperativity allowing synergy or inhibition of activity by displacement of other ligands.
Chemokines are key regulators of leukocyte migration and function, playing fundamental roles both in physiological and pathological immune responses, such as inflammatory diseases (1). The chemokine system includes ~50 ligands, which engage a panel of over 20 chemokine receptors in a promiscuous fashion, which are differentially expressed by all leukocytes and many non-hematopoietic cells (2). Proper tissue distribution of distinct leukocyte subsets, under normal and pathological conditions, is guaranteed by the resulting combinatorial diversity in cell responsiveness to chemokines.
To mediate their activity chemokines bind to cell surface receptors which belong to the largest branch of the γ subfamily of rhodopsin-like G protein-coupled receptors (GPCRs) (3), a receptor superfamily which represents the most successful target of small molecule inhibitors for treating diseases affecting different systems in modern pharmacology. All chemokine receptors couple to heterotrimeric Gαi-proteins and accordingly most responses can be fully inhibited by treatment of cells with Bordetella pertussis toxin. Today, a total of 19 signaling receptors have been identified: 7 CXCRs (CXCR1–6 and CXCR8), 10 CCRs (CCR1–10), CX3R3, and CKR1. In addition, there are four “atypical” receptors that use alternative signaling pathways, and act mainly by sequestering and degrading the chemokines present in the microenvironment (4). Thus, the ~50 chemokines outnumber their receptors indicating that a receptor can bind more than one chemokine. In addition, several chemokines can also bind to multiple receptors (2, 5). Novel findings indicate that polysialylation of CCR7, the central chemokine receptor controlling immune cell trafficking to secondary lymphatic organs, is essential for the recognition of the CCR7 ligand CCL21 (6), and that the glycosylation pattern of this receptor shapes receptor signaling (7), suggesting that this further level of control might be shared with other chemokine receptors.
A vast range of in situ experiments, aimed at understanding which chemokines are produced under specific circumstances, has revealed that a variety of chemokines can be concomitantly produced at the target sites of leukocyte trafficking and homing (8–12). This renders the chemokine system a good target for therapy and has promoted the search by pharmaceutical companies for small molecule chemokine antagonists (13–16). While we understand the effects of different chemokines singly, much less is known about the potential consequences of the concomitant expression of multiple chemokines and their interaction with other inflammatory molecules (17, 18).
The suggestion that chemokines might have additional regulatory mechanisms started with the identification of natural chemokine antagonists. Many reports have demonstrated that certain chemokines can also antagonize non-cognate chemokine receptors, by altering agonist-induced signaling and abrogating cellular responses via several mechanisms, including occupancy of the chemokine receptor-binding pocket or signaling through Rac-2 (19–24).
The studies on possible regulatory mechanisms continued when three reports showed that chemokines can synergize to enhance leukocyte functions in response to chemoattractants. The first described a bovine chemokine, regakine 1 that induces enhanced neutrophil migration when combined with CXCL7, CXCL8, and C5a. The receptor or the mechanism of regakine-1-induced synergism is not known. Competition with labeled C5a for binding to neutrophils or receptor-transfected cell lines demonstrated that regakine 1 does not alter receptor recognition. The protein kinase inhibitors 2′ amino 3′ methoxyflavone (PD98059), wortmannin, and staurosporine had no effect on the synergy between C5a and regakine 1 (25). The second study showed that migration of natural IFN-producing cells, a subpopulation of murine and human lymphocytes, to the CXCR3 agonists requires stimulation of CXCR4 by CXCL12. The mechanism by which CXCL12 induces enhanced migration in response to CXCR3 agonists is yet unknown. CXCL12 does not upregulate the expression of CXCR3 and does not increase the affinity of CXCR3 for its agonists (26). The third report (27) showed the same enhanced migration, on human plasmacytoid dendritic cells, in response to CXCR3 agonists induced by stimulation with CXCL12 as observed by Krug et al. (26). These reports undoubtedly indicate, as for the natural antagonist chemokines, that it is necessary to carefully analyze the effects that the concomitant expression of chemokines can have on cell functions and to elucidate the molecular mechanisms governing cell activities at sites of inflammation. Synergism can thus occur at different levels, involving either two chemokine receptors triggered simultaneously or sequentially exposed to their agonists (26–30). We have identified a further mechanism by which chemokines, forming chemokine heteromeric complexes, can activate one type of chemokine receptor (Figure 1A) (31): (i) CXCL13 enhances CCL19 and CCL21 triggering of CCR7 (32); (ii) CXCL10 enhances CCL22 triggering of CCR4 (33); (iii) CCL19 and CCL21 enhance the activity of CCR2 ligands and protect them from degradation (34); and (iv) CXCL9 enhances migration induced by CXCL12 on CXCR4+/CXCR3− malignant B cells (35). Other groups have also shown that the synergism between a chemokine agonist and a non-ligand chemokine can enhance the activity of selective chemokine receptors (36–40).
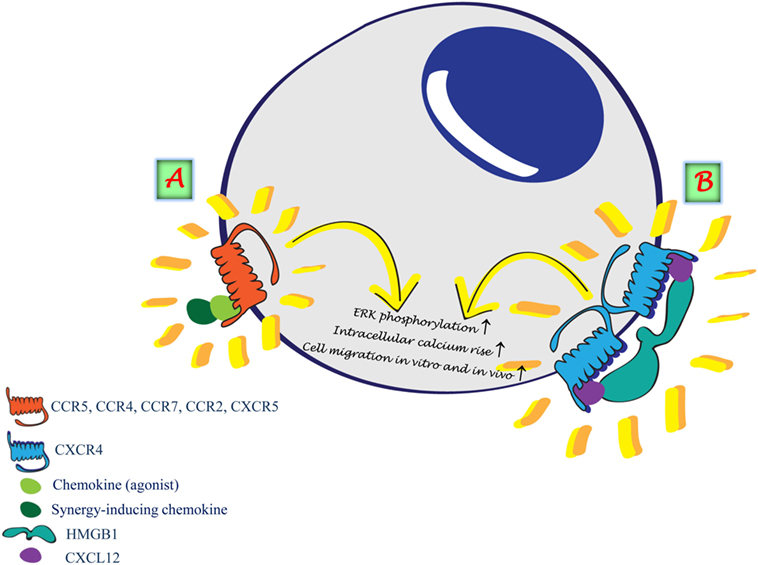
Figure 1. Synergism induced by the formation of heterocomplexes. (A) Heterocomplex formed between two chemokines renders the agonist more potent on the selective receptor. (B) HMGB1 forms a heterocomplex with CXCL12 enhancing CXCL12 potency on CXCR4.
Chemokines have a second important interaction with cell surface expressed glycosaminoglycans (GAGs), which mediates their immobilization on the endothelial surface in order to provide their directional signal (41–43). This interaction was shown to be essential for their ability to recruit cells in vivo by the loss of activity of chemokine variants, which had abrogated GAG-binding capacity (44). Without the interaction with endothelial GAGs, most chemokines would be washed away from the local production site, especially under flow conditions, diluted to a concentration below the threshold required for binding, and distributed uniformly throughout the vasculature such that no localized chemotactic signal is generated for leukocytes to allow directional mobilization. Furthermore, differential binding to GAGs plays an important role in localization. Neutrophil recruitment to the lung is greater in response to chemokines that bind GAGs less strongly. This was demonstrated both by mutants of CXCL8 with abrogated GAG binding as well as comparison of another neutrophil chemoattractant, CXCL1. Although increased recruitment was postulated to be mediated by the stronger GAG binder, lower binding capacity resulted in enhanced recruitment, demonstrating that the tissue microenvironment plays a pivotal role in the spatial formation of chemokine gradients and defining GAGs functions (45, 46).
Recently, binding to cell surface GAGs has identified more subtle roles in chemokine biology, where competitive binding of chemokines to GAGs can either induce cooperative enhancement of activity or inhibition of activity by displacement of certain chemokines. Cooperative enhancement has been demonstrated for both classical receptors as well as atypical or non-signaling receptors such as CCX-CKR/ACKR4 (47). In both cases, competitive displacement of the chemokines from GAGs was shown to be responsible for the effects, using modified chemokines lacking the GAG-binding sequence. The competitive displacement is limited to chemokines which bind GAGs strongly such as CCL11, CXCL12, and CXCL13, compared with low-affinity binders, such as CCL3 and CCL4, being unable to induce this synergy.
A similar phenomenon was observed for CCL18, an interesting chemokine in that is has been shown to be unregulated in many pathological conditions, yet its receptor remained elusive until it was shown recently to activate CCR8 (48). Moreover, CCL18 is always present at considerably higher concentrations in the circulation than most chemokines, and it was shown to displace certain chemokines bound to heparin (49). This property led to the hypothesis that it could prevent the recruitment of leukocytes by these chemokines by removing them from the endothelial surface.
Since chemokine cooperativity via GAG binding would allow chemokines to activate their cognate receptors at lower chemokine concentrations, it is likely that in vivo, this phenomenon would extend the range from which chemokines can induce recruitment of leukocytes (50). GAG binding and/or formation of heterocomplexes can definitively contribute to the fine-tuning modulation of chemokine activities occurring in vivo.
It is well established that many chemokines exist in equilibrium between the monomeric and dimeric state, and even as higher order oligomers (51–53). It is therefore clear that chemokine biology is more complex than simple monomolecular ligand–receptor interactions. It has been shown in vitro that the quaternary structure of chemokines influences the affinity of binding to GAGs (54, 55). Moreover, in vitro studies have suggested that dimerization may also occur after binding to GAGs on endothelial cells (56). In fact, this phenomenon is essential for certain chemokines in vivo since obligate monomers of the proinflammatory chemokines, CCL2, CCL4, and CCL5, are unable to recruit cells when injected into the peritoneal cavity (44).
It is however important to note that alterations in GAG composition can occur in several pathological conditions (57–59). In addition, chemokine receptors can be found as dimers and/or oligomers at the cell surface (60–62). Due to the complexity of chemokine binding and signaling (63), several mechanisms have been proposed to provide an explanation for synergy between chemokines in leukocyte migration. It is now evident that the synergism between chemokines is crucial at the very early stage of inflammation, as in vivo disruption of pro-atherogenic heteromers of CCL5 and CXCL4 resulted in a significant decrease in atherosclerotic lesion formation (38, 64). Moreover, disruption of the heteromers, formed between CCL5 and the α-defensin HNP1, attenuated monocyte and macrophage recruitment in a mouse model of myocardial infarction (65). On the contrary, the study of the role of synergy-inducing chemokines in the tumor microenvironment is at its infancy, as it has been shown in vitro that the distinct co-expression of B and T cell attractant chemokines, present in the tumor microenvironment, control cell trafficking of both tumor-infiltrating lymphocytes and malignant B cells (35).
Under inflammatory conditions, the cross talk between different molecules plays a crucial role in reaching the balance in tissue regeneration. A complete system for the detection, containment, and repair of damage caused to cells in the organism requires warning signals for the cells to respond. These warning signals are called endogenous damage-associated molecular patterns (DAMPs) or alarmins. In addition to the several chemokines that act as enhancers of molecules of the same family, by forming a heterocomplex with chemokine receptor agonists, we have recently identified HMGB1, an alarmin, as an enhancer of the activity of CXCL12 (Figure 1B) (66–68). The heterocomplex HMGB1/CXCL12 can be disrupted with a specific molecule, glycyrrhizin, which inhibits cell influx into the injured tissue. This indicates that a number of components, in addition to the direct activation of the receptor via a selective agonist, can regulate chemokine functions via a direct interaction with chemokines or chemokine receptors. Multiple chemokines within inflamed tissues selectively enhance each other’s migratory functions, depending on their concentrations, proximity, and simultaneous exposure to leukocytes. The mechanisms underlying the involvement of endogenous DAMPs in chronic diseases are still largely unexplored, and the interaction with other molecules might be a possible approach to understand their targets and functions. The interaction between chemokines and inflammatory molecules needs to be taken into account when chemokine cleavage by proteolysis, or chemokine degradation by atypical chemokine receptors, would be beneficial to achieve a resolving microenvironment favorable for resolution of inflammation by abrogating chemokine signals and the recruitment of inflammatory cells (69). The heterocomplex HMGB1/CXCL12 was demonstrated to prevent CXCL12 degradation (70), similarly to the observation that the complex CCL19/CCL7 prevents CCL7 degradation by the atypical receptor ACKR2 (34).
The chemokine system remains a promising biological target for the development of new therapeutic tools for the treatment of immunological disorders. Nevertheless, drug discovery programs have not yet produced successful drugs targeting the chemokine system for the treatment of inflammatory diseases. Most of the competitive chemokine receptor antagonists developed by all major pharma companies have been disappointingly unsuccessful when tested in clinical trials (71), and as a matter of fact, the only two small molecule inhibitors approved by the FDA do not target inflammation. Taking into account GAGs-binding properties, synergy induced by heterocomplexes formed with non-ligand chemokines or inflammatory molecules, and the possibility that the heterocomplexes might induce differential signaling pathways, will certainly help in elaborating the biology involved in this family and will surely contribute to the successful development of inhibitors of the chemokine system as therapeutics.
All authors listed have made substantial, direct, and intellectual contribution to the work and approved it for publication.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Special thanks to Dr. Giorgia Brambilla Pisoni for her work in transforming our data on the synergism into a visual figure.
The authors would like to thank, for the support received for the studies on the modulation of chemokines activities, the Swiss National Science Foundation (3100A0-143718/1 and 141773-RM3 to MU); European Union’s Programs for research, technological development and demonstration under grant agreements INNOCHEM – LSHB-CT-2005-518167 (FP6), DEC-VAC – LSHP-CT-2005-018685 (FP6), ADITEC – 280873 (FP7), and TIMER – 281608 (FP7). Further support was obtained by the San Salvatore Foundation, the Novartis Foundation (to MU), the Helmut Horten Foundation, the Institute for Arthritis Research, and the Gottfried and Julia Bangerter-Rhyner-Foundation.
1. Charo IF, Ransohoff RM. The many roles of chemokines and chemokine receptors in inflammation. N Engl J Med (2006) 354(6):610–21. doi:10.1056/NEJMra052723
2. Bachelerie F, Ben-Baruch A, Burkhardt AM, Combadiere C, Farber JM, Graham GJ, et al. International Union of Pharmacology. LXXXIX. Update on the extended family of chemokine receptors and introducing a new nomenclature for atypical chemokine receptors. Pharmacol Rev (2014) 66(1):1–79. doi:10.1124/pr.113.007724
3. Griffith JW, Sokol CL, Luster AD. Chemokines and chemokine receptors: positioning cells for host defense and immunity. Annu Rev Immunol (2014) 32:659–702. doi:10.1146/annurev-immunol-032713-120145
4. Vacchini A, Locati M, Borroni EM. Overview and potential unifying themes of the atypical chemokine receptor family. J Leukoc Biol (2016). doi:10.1189/jlb.2MR1015-477R
6. Kiermaier E, Moussion C, Veldkamp CT, Gerardy-Schahn R, de Vries I, Williams LG, et al. Polysialylation controls dendritic cell trafficking by regulating chemokine recognition. Science (2016) 351(6269):186–90. doi:10.1126/science.aad0512
7. Hauser MA, Kindinger I, Laufer JM, Spate AK, Bucher D, Vanes SL, et al. Distinct CCR7 glycosylation pattern shapes receptor signaling and endocytosis to modulate chemotactic responses. J Leukoc Biol (2016). doi:10.1189/jlb.2VMA0915-432RR
8. Agace WW, Amara A, Roberts AI, Pablos JL, Thelen S, Uguccioni M, et al. Constitutive expression of stromal derived factor-1 by mucosal epithelia and its role in HIV transmission and propagation. Curr Biol (2000) 10(6):325–8. doi:10.1016/S0960-9822(00)00380-8
9. Manzo A, Paoletti S, Carulli M, Blades MC, Barone F, Yanni G, et al. Systematic microanatomical analysis of CXCL13 and CCL21 in situ production and progressive lymphoid organization in rheumatoid synovitis. Eur J Immunol (2005) 35(5):1347–59. doi:10.1002/eji.200425830
10. Mazzucchelli L, Blaser A, Kappeler A, Scharli P, Laissue JA, Baggiolini M, et al. BCA-1 is highly expressed in Helicobacter pylori-induced mucosa-associated lymphoid tissue and gastric lymphoma. J Clin Invest (1999) 104(10):R49–54. doi:10.1172/JCI7830
11. Smith JR, Braziel RM, Paoletti S, Lipp M, Uguccioni M, Rosenbaum JT. Expression of B-cell-attracting chemokine 1 (CXCL13) by malignant lymphocytes and vascular endothelium in primary central nervous system lymphoma. Blood (2003) 101(3):815–21. doi:10.1182/blood-2002-05-1576
12. Uguccioni M, Gionchetti P, Robbiani DF, Rizzello F, Peruzzo S, Campieri M, et al. Increased expression of IP-10, IL-8, MCP-1, and MCP-3 in ulcerative colitis. Am J Pathol (1999) 155(2):331–6. doi:10.1016/S0002-9440(10)65128-0
13. de Zeeuw D, Bekker P, Henkel E, Hasslacher C, Gouni-Berthold I, Mehling H, et al. The effect of CCR2 inhibitor CCX140-B on residual albuminuria in patients with type 2 diabetes and nephropathy: a randomised trial. Lancet Diabetes Endocrinol (2015) 3(9):687–96. doi:10.1016/S2213-8587(15)00261-2
14. Proudfoot AE, Power CA, Rommel C, Wells TN. Strategies for chemokine antagonists as therapeutics. Semin Immunol (2003) 15(1):57–65. doi:10.1016/S1044-5323(02)00128-8
15. Wells TN, Power CA, Shaw JP, Proudfoot AE. Chemokine blockers – therapeutics in the making? Trends Pharmacol Sci (2006) 27(1):41–7. doi:10.1016/j.tips.2005.11.001
16. Yokoyama W, Kohsaka H, Kaneko K, Walters M, Takayasu A, Fukuda S, et al. Abrogation of CC chemokine receptor 9 ameliorates collagen-induced arthritis of mice. Arthritis Res Ther (2014) 16(5):445. doi:10.1186/s13075-014-0445-9
17. Loos T, Dekeyzer L, Struyf S, Schutyser E, Gijsbers K, Gouwy M, et al. TLR ligands and cytokines induce CXCR3 ligands in endothelial cells: enhanced CXCL9 in autoimmune arthritis. Lab Invest (2006) 86(9):902–16. doi:10.1038/labinvest.3700453
18. Panzer U, Uguccioni M. Prostaglandin E2 modulates the functional responsiveness of human monocytes to chemokines. Eur J Immunol (2004) 34(12):3682–9. doi:10.1002/eji.200425226
19. Fulkerson PC, Zhu H, Williams DA, Zimmermann N, Rothenberg ME. CXCL9 inhibits eosinophil responses by a CCR3- and Rac2-dependent mechanism. Blood (2005) 106(2):436–43. doi:10.1182/blood-2005-02-0489
20. Fulkerson PC, Zimmermann N, Brandt EB, Muntel EE, Doepker MP, Kavanaugh JL, et al. Negative regulation of eosinophil recruitment to the lung by the chemokine monokine induced by IFN-gamma (Mig, CXCL9). Proc Natl Acad Sci U S A (2004) 101(7):1987–92. doi:10.1073/pnas.0308544100
21. Ogilvie P, Bardi G, Clark-Lewis I, Baggiolini M, Uguccioni M. Eotaxin is a natural antagonist for CCR2 and an agonist for CCR5. Blood (2001) 97(7):1920–4. doi:10.1182/blood.V97.7.1920
22. Ogilvie P, Paoletti S, Clark-Lewis I, Uguccioni M. Eotaxin-3 is a natural antagonist for CCR2 and exerts a repulsive effect on human monocytes. Blood (2003) 102(3):789–94. doi:10.1182/blood-2002-09-2773
23. Petkovic V, Moghini C, Paoletti S, Uguccioni M, Gerber B. Eotaxin-3/CCL26 is a natural antagonist for CC chemokine receptors 1 and 5. A human chemokine with a regulatory role. J Biol Chem (2004) 279(22):23357–63. doi:10.1074/jbc.M309283200
24. Petkovic V, Moghini C, Paoletti S, Uguccioni M, Gerber B. I-TAC/CXCL11 is a natural antagonist for CCR5. J Leukoc Biol (2004) 76(3):701–8. doi:10.1189/jlb.1103570
25. Gouwy M, Struyf S, Mahieu F, Put W, Proost P, Van Damme J. The unique property of the CC chemokine regakine-1 to synergize with other plasma-derived inflammatory mediators in neutrophil chemotaxis does not reside in its NH2-terminal structure. Mol Pharmacol (2002) 62(1):173–80. doi:10.1124/mol.62.1.173
26. Krug A, Uppaluri R, Facchetti F, Dorner BG, Sheehan KC, Schreiber RD, et al. IFN-producing cells respond to CXCR3 ligands in the presence of CXCL12 and secrete inflammatory chemokines upon activation. J Immunol (2002) 169(11):6079–83. doi:10.4049/jimmunol.169.11.6079
27. Vanbervliet B, Bendriss-Vermare N, Massacrier C, Homey B, de Bouteiller O, Briere F, et al. The inducible CXCR3 ligands control plasmacytoid dendritic cell responsiveness to the constitutive chemokine stromal cell-derived factor 1 (SDF-1)/CXCL12. J Exp Med (2003) 198(5):823–30. doi:10.1084/jem.20020437
28. Gouwy M, Struyf S, Berghmans N, Vanormelingen C, Schols D, Van Damme J. CXCR4 and CCR5 ligands cooperate in monocyte and lymphocyte migration and in inhibition of dual-tropic (R5/X4) HIV-1 infection. Eur J Immunol (2011) 41(4):963–73. doi:10.1002/eji.201041178
29. Gouwy M, Struyf S, Noppen S, Schutyser E, Springael JY, Parmentier M, et al. Synergy between coproduced CC and CXC chemokines in monocyte chemotaxis through receptor-mediated events. Mol Pharmacol (2008) 74(2):485–95. doi:10.1124/mol.108.045146
30. Mellado M, Rodriguez-Frade JM, Vila-Coro AJ, Fernandez S, Martin de AA, Jones DR, et al. Chemokine receptor homo- or heterodimerization activates distinct signaling pathways. EMBO J (2001) 20(10):2497–507. doi:10.1093/emboj/20.10.2497
31. Cecchinato V, D’Agostino G, Raeli L, Uguccioni M. Chemokine interaction with synergy-inducing molecules: fine tuning modulation of cell trafficking. J Leukoc Biol (2015). doi:10.1189/jlb.1MR1015-457R
32. Paoletti S, Petkovic V, Sebastiani S, Danelon MG, Uguccioni M, Gerber BO. A rich chemokine environment strongly enhances leukocyte migration and activities. Blood (2005) 105(9):3405–12. doi:10.1182/blood-2004-04-1648
33. Sebastiani S, Danelon G, Gerber B, Uguccioni M. CCL22-induced responses are powerfully enhanced by synergy inducing chemokines via CCR4: evidence for the involvement of first beta-strand of chemokine. Eur J Immunol (2005) 35(3):746–56. doi:10.1002/eji.200525800
34. Kuscher K, Danelon G, Paoletti S, Stefano L, Schiraldi M, Petkovic V, et al. Synergy-inducing chemokines enhance CCR2 ligand activities on monocytes. Eur J Immunol (2009) 39(4):1118–28. doi:10.1002/eji.200838906
35. Venetz D, Ponzoni M, Schiraldi M, Ferreri AJ, Bertoni F, Doglioni C, et al. Perivascular expression of CXCL9 and CXCL12 in primary central nervous system lymphoma: T-cell infiltration and positioning of malignant B cells. Int J Cancer (2010) 127(10):2300–12. doi:10.1002/ijc.25236
36. Gijsbers K, Gouwy M, Struyf S, Wuyts A, Proost P, Opdenakker G, et al. GCP-2/CXCL6 synergizes with other endothelial cell-derived chemokines in neutrophil mobilization and is associated with angiogenesis in gastrointestinal tumors. Exp Cell Res (2005) 303(2):331–42. doi:10.1016/j.yexcr.2004.09.027
37. Gouwy M, Schiraldi M, Struyf S, Van Damme J, Uguccioni M. Possible mechanisms involved in chemokine synergy fine tuning the inflammatory response. Immunol Lett (2012) 145(1–2):10–4. doi:10.1016/j.imlet.2012.04.005
38. Koenen RR, von Hundelshausen P, Nesmelova IV, Zernecke A, Liehn EA, Sarabi A, et al. Disrupting functional interactions between platelet chemokines inhibits atherosclerosis in hyperlipidemic mice. Nat Med (2009) 15(1):97–103. doi:10.1038/nm.1898
39. von Hundelshausen P, Koenen RR, Sack M, Mause SF, Adriaens W, Proudfoot AE, et al. Heterophilic interactions of platelet factor 4 and RANTES promote monocyte arrest on endothelium. Blood (2005) 105(3):924–30. doi:10.1182/blood-2004-06-2475
40. Weber C, Koenen RR. Fine-tuning leukocyte responses: towards a chemokine ‘interactome’. Trends Immunol (2006) 27(6):268–73. doi:10.1016/j.it.2006.04.002
41. Hamel DJ, Sielaff I, Proudfoot AE, Handel TM. Chapter 4. Interactions of chemokines with glycosaminoglycans. Methods Enzymol (2009) 461:71–102. doi:10.1016/S0076-6879(09)05404-4
42. Kuschert GSV, Coulin F, Power CA, Proudfoot AEI, Hubbard RE, Hoogewerf AJ, et al. Glycosaminoglycans interact selectively with chemokines and modulate receptor binding and cellular responses. Biochemistry (1999) 38(39):12959–68. doi:10.1021/bi990711d
43. Rot A. Neutrophil attractant/activation protein-1 (interleukin-8) induces in vitro neutrophil migration by haptotactic mechanism. Eur J Immunol (1993) 23:303–6. doi:10.1002/eji.1830230150
44. Proudfoot AE, Handel TM, Johnson Z, Lau EK, LiWang P, Clark-Lewis I, et al. Glycosaminoglycan binding and oligomerization are essential for the in vivo activity of certain chemokines. Proc Natl Acad Sci U S A (2003) 100(4):1885–90. doi:10.1073/pnas.0334864100
45. Gangavarapu P, Rajagopalan L, Kolli D, Guerrero-Plata A, Garofalo RP, Rajarathnam K. The monomer-dimer equilibrium and glycosaminoglycan interactions of chemokine CXCL8 regulate tissue-specific neutrophil recruitment. J Leukoc Biol (2012) 91(2):259–65. doi:10.1189/jlb.0511239
46. Tanino Y, Coombe DR, Gill SE, Kett WC, Kajikawa O, Proudfoot AE, et al. Kinetics of chemokine-glycosaminoglycan interactions control neutrophil migration into the airspaces of the lungs. J Immunol (2010) 184(5):2677–85. doi:10.4049/jimmunol.0903274
47. Verkaar F, van OJ, van der Lee MM, van Lith LH, Watts AO, Rops AL, et al. Chemokine cooperativity is caused by competitive glycosaminoglycan binding. J Immunol (2014) 192(8):3908–14. doi:10.4049/jimmunol.1302159
48. Islam SA, Ling MF, Leung J, Shreffler WG, Luster AD. Identification of human CCR8 as a CCL18 receptor. J Exp Med (2013) 210(10):1889–98. doi:10.1084/jem.20130240
49. Krohn SC, Bonvin P, Proudfoot AE. CCL18 exhibits a regulatory role through inhibition of receptor and glycosaminoglycan binding. PLoS One (2013) 8(8):e72321. doi:10.1371/journal.pone.0072321
50. Adage T, Konya V, Weber C, Strutzmann E, Fuchs T, Zankl C, et al. Targeting glycosaminoglycans in the lung by an engineered CXCL8 as a novel therapeutic approach to lung inflammation. Eur J Pharmacol (2015) 748:83–92. doi:10.1016/j.ejphar.2014.12.019
51. Campanella GS, Grimm J, Manice LA, Colvin RA, Medoff BD, Wojtkiewicz GR, et al. Oligomerization of CXCL10 is necessary for endothelial cell presentation and in vivo activity. J Immunol (2006) 177(10):6991–8. doi:10.4049/jimmunol.177.10.6991
52. Das ST, Rajagopalan L, Guerrero-Plata A, Sai J, Richmond A, Garofalo RP, et al. Monomeric and dimeric CXCL8 are both essential for in vivo neutrophil recruitment. PLoS One (2010) 5(7):e11754. doi:10.1371/journal.pone.0011754
53. Rajarathnam K, Sykes BD, Kay CM, Dewald B, Geiser T, Baggiolini M, et al. Neutrophil activation by monomeric interleukin-8. Science (1994) 264(5155):90–2. doi:10.1126/science.8140420
54. Dyer DP, Salanga CL, Volkman BF, Kawamura T, Handel TM. The dependence of chemokine-glycosaminoglycan interactions on chemokine oligomerization. Glycobiology (2016) 26(3):312–26. doi:10.1093/glycob/cwv100
55. Joseph PR, Mosier PD, Desai UR, Rajarathnam K. Solution NMR characterization of chemokine CXCL8/IL-8 monomer and dimer binding to glycosaminoglycans: structural plasticity mediates differential binding interactions. Biochem J (2015) 472(1):121–33. doi:10.1042/BJ20150059
56. Hoogewerf AJ, Kuschert GSV, Proudfoot AEI, Borlat F, Clark-Lewis I, Power CA, et al. Glycosaminoglycans mediate cell surface oligomerization of chemokines. Biochemistry (1997) 36:13570–8. doi:10.1021/bi971125s
57. Monneau Y, Arenzana-Seisdedos F, Lortat-Jacob H. The sweet spot: how GAGs help chemokines guide migrating cells. J Leukoc Biol (2015). doi:10.1189/jlb.3MR0915-440R
58. Tarbell JM, Cancel LM. The glycocalyx and its significance in human medicine. J Intern Med (2016). doi:10.1111/joim.12465
59. Taylor KR, Gallo RL. Glycosaminoglycans and their proteoglycans: host-associated molecular patterns for initiation and modulation of inflammation. FASEB J (2006) 20(1):9–22. doi:10.1096/fj.05-4682rev
60. Johnson Z, Schwarz M, Power CA, Wells TN, Proudfoot AE. Multi-faceted strategies to combat disease by interference with the chemokine system. Trends Immunol (2005) 26(5):268–74. doi:10.1016/j.it.2005.03.001
61. Springael JY, Urizar E, Parmentier M. Dimerization of chemokine receptors and its functional consequences. Cytokine Growth Factor Rev (2005) 16(6):611–23. doi:10.1016/j.cytogfr.2005.05.005
62. Thelen M, Munoz LM, Rodriguez-Frade JM, Mellado M. Chemokine receptor oligomerization: functional considerations. Curr Opin Pharmacol (2010) 10(1):38–43. doi:10.1016/j.coph.2009.09.004
63. Corbisier J, Gales C, Huszagh A, Parmentier M, Springael JY. Biased signaling at chemokine receptors. J Biol Chem (2015) 290(15):9542–54. doi:10.1074/jbc.M114.596098
64. Back M, Weber C, Lutgens E. Regulation of atherosclerotic plaque inflammation. J Intern Med (2015) 278(5):462–82. doi:10.1111/joim.12367
65. Alard JE, Ortega-Gomez A, Wichapong K, Bongiovanni D, Horckmans M, Megens RT, et al. Recruitment of classical monocytes can be inhibited by disturbing heteromers of neutrophil HNP1 and platelet CCL5. Sci Transl Med (2015) 7(317):317ra196. doi:10.1126/scitranslmed.aad5330
66. Schiraldi M, Raucci A, Munoz LM, Livoti E, Celona B, Venereau E, et al. HMGB1 promotes recruitment of inflammatory cells to damaged tissues by forming a complex with CXCL12 and signaling via CXCR4. J Exp Med (2012) 209(3):551–63. doi:10.1084/jem.20111739
67. Venereau E, Casalgrandi M, Schiraldi M, Antoine DJ, Cattaneo A, De Marchis F, et al. Mutually exclusive redox forms of HMGB1 promote cell recruitment or proinflammatory cytokine release. J Exp Med (2012) 209(9):1519–28. doi:10.1084/jem.20120189
68. Venereau E, Schiraldi M, Uguccioni M, Bianchi ME. HMGB1 and leukocyte migration during trauma and sterile inflammation. Mol Immunol (2013) 55(1):76–82. doi:10.1016/j.molimm.2012.10.037
69. Ortega-Gomez A, Perretti M, Soehnlein O. Resolution of inflammation: an integrated view. EMBO Mol Med (2013) 5(5):661–74. doi:10.1002/emmm.201202382
70. Campana L, Bosurgi L, Bianchi ME, Manfredi AA, Rovere-Querini P. Requirement of HMGB1 for stromal cell-derived factor-1/CXCL12-dependent migration of macrophages and dendritic cells. J Leukoc Biol (2009) 86(3):609–15. doi:10.1189/jlb.0908576
Keywords: chemokines, cell migration, synergy, oligomerization, glycosaminoglycan
Citation: Proudfoot AEI and Uguccioni M (2016) Modulation of Chemokine Responses: Synergy and Cooperativity. Front. Immunol. 7:183. doi: 10.3389/fimmu.2016.00183
Received: 25 February 2016; Accepted: 29 April 2016;
Published: 19 May 2016
Edited by:
Olivier Garraud, Institut National de la Transfusion Sanguine, FranceReviewed by:
Markus Bosmann, Gutenberg University Mainz, GermanyCopyright: © 2016 Proudfoot and Uguccioni. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mariagrazia Uguccioni, bWFyaWFncmF6aWEudWd1Y2Npb25pQGlyYi51c2kuY2g=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.