 Koelina Ganguly
Koelina Ganguly Jeevan Giddaluru
Jeevan Giddaluru Avery August
Avery August Nooruddin Khan
Nooruddin Khan- 1Department of Biotechnology and Bioinformatics, School of Life Sciences, University of Hyderabad, Hyderabad, India
- 2Department of Microbiology and Immunology, College of Veterinary Medicine, Cornell University, New York, NY, USA
Immunological programing of immune cells varies in response to changing environmental signals. This process is facilitated by modifiers that regulate the translational fate of mRNAs encoding various immune mediators, including cytokines and chemokines, which in turn determine the rapid activation, tolerance, and plasticity of the immune system. RNA-binding proteins (RBPs) recruited by the specific sequence elements in mRNA transcripts are one such modifiers. These RBPs form RBP–RNA complexes known as “riboclusters.” These riboclusters serve as RNA sorting machinery, where depending upon the composition of the ribocluster, translation, degradation, or storage of mRNA is controlled. Recent findings suggest that this regulation of mRNA homeostasis is critical for controlling the immune response. Here, we present the current knowledge of the ribocluster-mediated post-transcriptional regulation of immune mediators and highlight recent findings regarding their implications for the pathogenesis of acute or chronic inflammatory diseases.
Introduction
The transcriptome of an immune cell is subjected to various stages of regulation in the nucleus and cytoplasm before being translated into proteins (1–5). In addition to the basic transcriptional and translational regulation, there exists an intermediate post-transcriptional regulatory (PTR) event, which programs the immunological response according to the changing environmental conditions. This PTR includes the various stages of regulation, including splicing, editing, translation, and decay of mRNAs. Gene transcription and nuclear processes leading to the formation of mature mRNAs in the nucleus are followed by nucleocytoplasmic mRNA trafficking and their exposure to a series of regulatory mechanisms in the cytoplasm. The various functions of dynamic ribonucleoprotein (RNP) complexes in coordinating these nucleocytoplasmic regulatory events have been shown to drive a plethora of immunological responses inside the cells (1–5). The first step in the nuclear post-transcriptional regulatory series is splicing, which involves intron removal and exon joining facilitated by spliceosome complex, formed from the “U” class of small nuclear RNPs (snRNPs). In some cases, splicing also includes a more complex event, the alternative splicing that involves the insertion or excision of specific introns, exons, or regulatory domains in the mature mRNA (6). This is mediated by different classes of RNA-binding proteins (RBPs), including heterogeneous nuclear RNPs (hnRNPs) and serine–arginine RBPs (SR RBPs) (7, 8). The second type of nuclear regulation is editing, which involves the alteration of the nucleotide content of RNA and the subsequent protein translation. The most predominant form of RNA editing in mammals is aided by the adenosine deaminases acting on RNA (ADAR) family of double-stranded RNA-binding enzymes that are responsible for deamination of adenine to inosine (5). Once the functional mature mRNA transcripts exit the nucleus for translation in the cytoplasm, they are exposed to different classes of RBPs based on the mRNA’s cis-regulatory sequences. These RBPs aid in the careful orchestration of mRNA decay in stress granules (SG)/processing bodies (P-bodies) or translation in polysomes, which collectively form an intricate post-transcriptional event (9).
Immune cells can sense and process a wide variety of extracellular and intracellular stress signals via cellular stress sensors. Upon activation by cellular stress sensors, the α-subunit of eukaryotic initiation factor 2 (eIF2-α) is phosphorylated, resulting in the attenuation of active translational complex and polysome formation. This results in a pool of translationally stalled uncommitted mRNA transcripts, which then recruit specific RBPs. This recruitment is determined by the cis-acting regulatory elements, such as adenine–uridine-rich elements (AREs) at their 3′-untranslated region (UTR), and forms a RNP complex known as a “ribocluster” (10). The RBPs in the “ribocluster” dictate the location and functionality of mRNAs, determining whether it will be translated or decayed (11). These processes are meticulously coordinated and intricately linked and depend upon the ability of RBPs to interact with the cis-elements, such as AREs at their 3′-UTR of the mRNA transcripts (11, 12). The mRNA transcripts of many immunological mediators, including cytokines and chemokines, have regulatory sequences, such as ARE at 3′-UTR, which allow tight regulation and usage of mRNAs to fine-tune immunological responses as per the cellular requirement (13, 14). The emerging wealth of information regarding RBPs and riboclustering lends credence to its importance in the maintenance of immune homeostasis and programing of the immune response (14). This system has the potential to regulate a wide range of the immune response through the maintenance of equilibrium between synthesis and degradation of the mRNAs that drives immunological reactions, the innate inflammatory responses, immune cell fates, and adaptive host defenses. A better understanding of riboclustering in regulating these integrated pathways might provide leeway toward a development of novel therapeutics. In this review, we present a survey of the current knowledge about riboclustering-mediated post-transcriptional regulation of immune mediators and further highlight recent findings regarding their implications in the pathogenesis of acute or chronic inflammatory diseases.
Allies in Riboclustering
As introduced in the previous section, riboclusters of spliced mature mRNA transcripts and various RBPs control cytokine and chemokine mRNAs for either subsequent protein expression or exosomal decay (15). The assembly of these riboclusters is largely dependent on the 3′-UTRs (16), the binding of trans-factors like RBPs and non-coding RNAs, such as miRNAs. In this section, we discuss RBPs and cis-elements in more detail.
RNA-Binding Proteins
RNA-binding proteins, such as tristetraproline (TTP), T cell-restricted antigen 1 (TIA1), TIA1-related protein (TIAR), ZCCHC11, Regnase-1 (also named Zc3h12a, Mcpip1), and ARE-binding degradation factor 1 (AUF1), bind to specific cis-element sequences of cytokine and chemokine mRNAs to form RNP complexes (10). The various RNA-binding domains of different RBPs (14) that have been described so far include RNA recognition motifs (found in TIA1, TIAR, CUGBP2, AUF1, AUF2, and HuR), zinc finger domain (found in BRF1, BRF2, and TTP), and K homology domain [found in fragile X-related protein 1 (FXR1P) and KH-type splicing regulatory protein (KSRP)] (14). Most of the RBPs are found to translocate between the nucleus and cytoplasm (17), and, therefore, riboclustering is hypothesized to dictate the cytoplasmic localization of the bound transcripts and thereby their fate. Various in vitro studies have shown that multiple RBPs gain entry onto an RNA transcript, suggesting a cooperative (18) or competitive (19) function by the RBPs to modulate the stabilization or destabilization of a common target transcript.
Adenine–Uridine-Rich Elements
A few decades ago, clusters of AREs were identified at the 3′-UTRs of newly cloned cytokine mRNAs transcript and were reported to regulate mRNA metabolism (20). The role of ARE in mRNA regulation was quickly confirmed by the decay of heterologous reporter transcripts fused to ARE sequences derived from the 3′-UTRs of granulocyte–monocyte colony-stimulating factor (GM-CSF) mRNA (21). These AREs can act as decisive cis-acting post-transcriptional gene regulatory elements (10). Table 1 provides an interaction profile of the ARE-bearing immunological mediators and proto-oncogenes with specific RBPs. The basic structural components of these AREs are pentamers, nonamers, or clusters of adenine–uridine-rich repeats. These AREs can modulate the cytokine and chemokine levels in cells either independently or by recruiting different groups of RBPs. Table 2 shows the classification of AREs on the basis of sequence information and destabilization kinetics. The information in the table is collected from the database of human ARE-bearing mRNAs created by Bakheet et al. (http://brp.kfshrc.edu.sa/ARED/) (22). This database also suggests that 8% of the mRNA transcribed from human genome bears AREs (22). Much later, it was found that various pro-inflammatory genes, such as those of cytokines, chemokines, and other pro-inflammatory proteins, undergo ARE-mediated decay (AMD) after being subjected to riboclustering (16). However, in the recent years, it has been pointed out that some of these mRNAs are not subjected to decay but rather are stabilized post-transcriptionally when bound to separate class of RBPs (14).
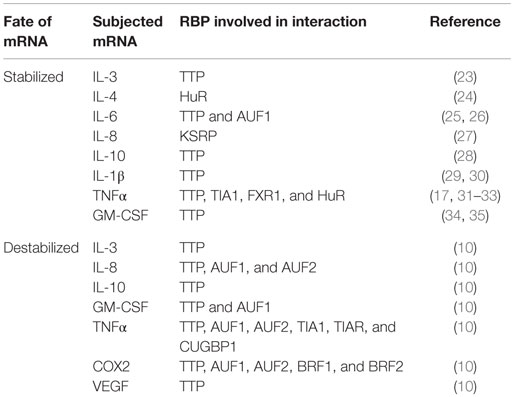
Table 1. Fate of transcripts determined by the interaction with specific RBPs.
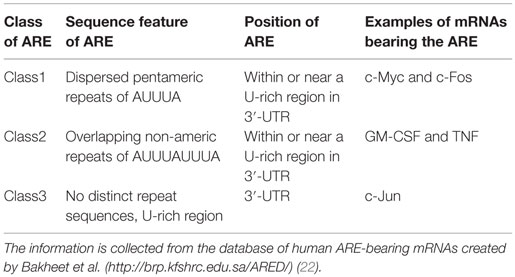
Table 2. Classification of ARE sequences on the basis of sequence features.
Mechanism of Riboclustering
Upstream Regulators Involved in the Formation of Riboclusters
The endoplasmic reticulum (ER) is the site for the post-translational modification, folding, and sorting of proteins destined for the secretory pathway of the cell. Calcium disequilibrium or disruption in post-translational modification leads to accumulation of misfolded or unfolded proteins in ER, resulting in ER stress, which can have negative implications on various cellular functions (36). Diseases, such as diabetes, cancer, and neurodegenerative disorders, are often associated with ER stress (37–41). ER stress triggers the activation of transmembrane signaling proteins, including activating transcription factor 6 (ATF6), inositol-requiring protein-1α (IRE1α), and protein kinase RNA (PKR)-like ER kinase (PERK). This pathway helps to establish ER homeostasis by increasing chaperone expression, sequestering mRNAs from polysomes, and ER-assisted degradation (ERAD) of the misfolded proteins in the cytosol (42). During amino acid depletion, the intracellular sensor general control non-derepressible 2 (GCN2) gets activated by the accumulation of uncharged tRNAs, while HRI senses heme deprivation within the cell. As a part of the antiviral response, PKR gets activated by viral dsRNA, interferons, and growth factors (43). The four vital sensors of stress pathway, namely GCN2, PERK, PKR, and HRI, undergo activation by phosphorylation in response to the above-mentioned stress conditions. Phosphorylated sensor molecules, in turn, phosphorylate eIF2-α (44) and prevent the recruitment of eIF2–GTP–methionyl initiator tRNA onto the 40S ribosomal subunit. This, in turn, leads to the translation initiation failure owing to stalled formation of 43S preinitiation complex and its subsequent binding to eI4F, and other factors required for the assembly of the active polysomes (44). Alternatively, nutrient deprivation or infection activates TSC proteins that negatively regulate the metabolic checkpoint control molecule mammalian target of rapamycin complex 1 (mTORC1). Upon deactivation of mTOR, eIF4E-binding proteins (eIF4E BP1 and BP2) are capable of inhibiting the formation of the preinitiation complex by preventing the interaction of eIF4F complex to the 5′ cap of mRNA (45). Thus, when cells are exposed to oxidative damage, infection, or nutrient deprivation, there is a transient decrease in global protein synthesis as a mode of adaptation to environmental stresses. This results in a pool of translationally stalled mRNAs and disintegrated translation machinery. These uncommitted mRNA transcripts then recruit specific RBPs, determined by the sequence elements in the regulatory domains, to form “riboclusters” (10).
The composition of riboclusters is in constant flux, with some RBPs gaining or losing access during the mRNA’s journey from transcription to translation or decay. The RBPs in the ribocluster dictate the location and functionality of mRNAs as to whether it will be subjected to translation in polysomes, saved in a cache in SGs for further use, or otherwise decayed in P-bodies (11). All of these processes are highly coordinated and intricately linked and depend upon the ability of RBPs to interact with the regulatory sequences or structures present in the UTRs of the mRNA transcripts (11, 12). The presence of such regulatory sequences or elements in the mRNA transcripts encoding many immunological mediators, including cytokines and chemokines (13, 14), allows coordinated regulation and usage to tune immunological responses “on” or “off” as per the cellular requirement.
The Downstream Process
In cells that are exposed to stress, various RBPs, such as TIA1 and TIAR, interact with stalled mRNAs and propel translation preinitiation complexes to discrete cytoplasmic foci of SGs and P-bodies (46). P-bodies bear all the decay enzymes, including 5′–3′ mRNA decay enzyme Xrn1, decapping enzyme, and others (11). The sorted mRNA transcripts destined for translational arrest are delivered from the SGs to the P-bodies either for degradation or temporary silencing. However, the RBP-mediated export of the mRNAs in the SGs from polysomes is reversible in nature (12). Higher concentrations of stabilizing factors, such as HuR, ensure the rescue of the transcripts from SGs and result in translation initiation, while increased recruitment of destabilizing factors, such as TTP, BRF1, BRF2, and others, leads to SG formation and subsequent mRNA decay (13, 47). TIA1 has been found to coprecipitate with KSRP from the TNFα ARE (14), which may explain the temporal sequence of events involved in post-transcriptional regulation of the TNFα mRNAs. After being bound to TIA1/TIAR, TNFα mRNAs are removed from polysomes and are targeted for decay within the P-bodies in association with KSRP (48). Figure 1 shows the current understanding of the mechanism that connects the stress pathway and post-transcriptional regulation of immunological mediators, including cytokines and chemokines.
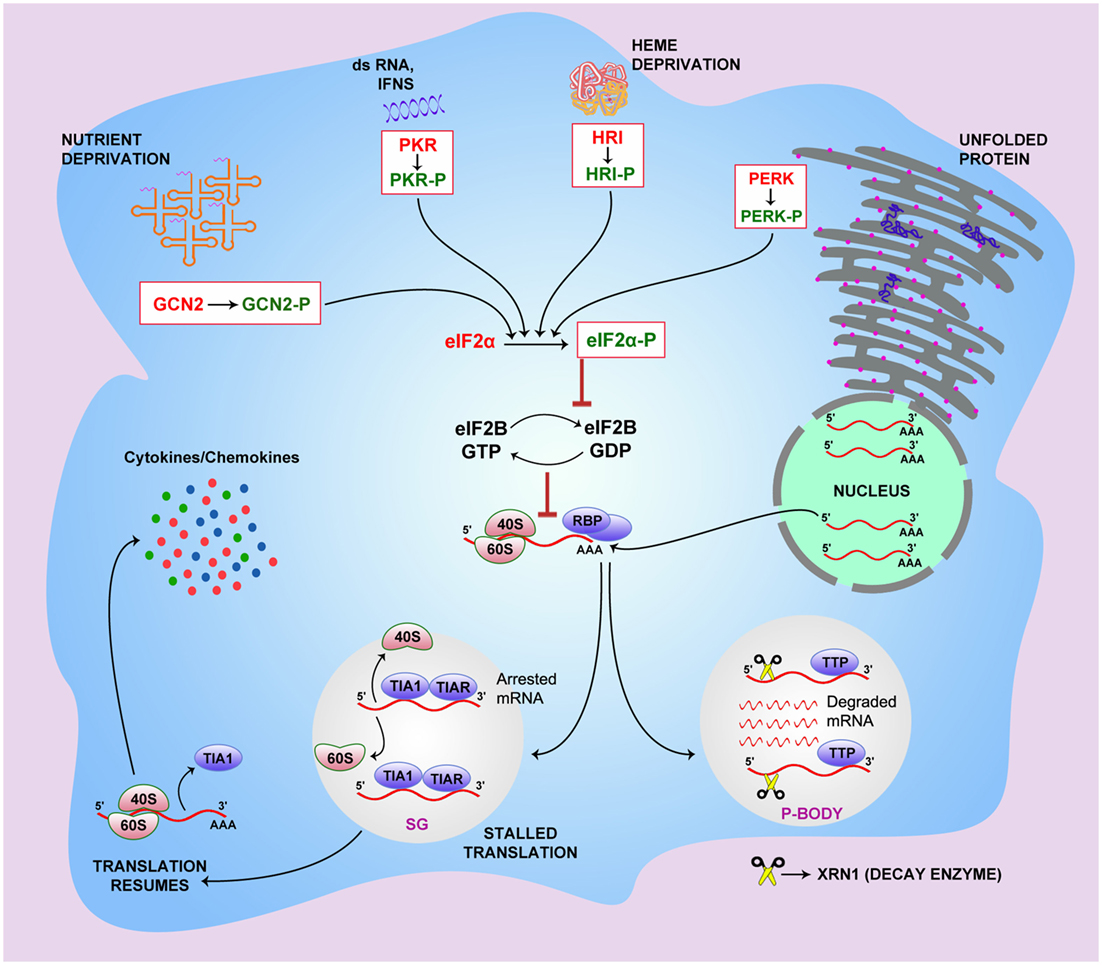
Figure 1. The link between integrated stress pathway and riboclustering. General control non-derepressible 2 (GCN2), PKR-like endoplasmic reticulum kinase (PERK), protein kinase RNA (PKR), and heme-regulated initiation factor 2-alpha kinase (HRI) stress sensors get activated when the cell is subjected to specific stress conditions. PERK senses endoplasmic reticulum stress caused by misfolded or unfolded proteins, GCN2 senses accumulation of uncharged tRNAs under conditions of amino acid unavailability, and the HRI molecule senses heme deprivation within the cell. PKR gets activated by viral dsRNA, interferons, and growth factors. Upon activation, these sensor molecules undergo phosphorylation, which subsequently phosphorylate eukaryotic initiation factor 2-α (eIF2-α), preventing the recruitment of eIF2–GTP–methionyl initiator tRNA onto the 40S ribosomal subunit. This, in turn, leads to the translation initiation failure owing to retarded GTP–GDP exchange and stalled formation of 43S preinitiation complex. In cells exposed to stress, RBPs, including T cell-restricted antigen 1 (TIA1) and TIA1-related protein (TIAR), bind to stalled mRNAs and propel preinitiation complexes of translation to stress granules (SGs) for temporary silencing. The sorted mRNA transcripts, destined for translational arrest, are delivered to the processing bodies (P-bodies) for degradation using decay enzymes like 5′–3′ mRNA decay enzyme Xrn1, decapping enzyme, and others. RBP-mediated export of the mRNAs in the SGs from polysomes is reversible. Higher concentrations of stabilizing factors, such as HuR, ensure rescue of the transcripts from SGs leading to translation initiation, while increased recruitment of destabilizing factors, such as TTP, BRF1, and BRF2, leads to SG formation and subsequent mRNA decay in P-bodies.
Riboclustering in Innate Immunity: Initiation, Perpetuation, and Resolution of Inflammation
The immune system has to respond to invading pathogens by generating a pro-inflammatory response, which eventually has to be suppressed, allowing a return to homeostasis. In response to such changing immune microenvironments, riboclusters change their composition and cytosolic locations in the responding immune cells, and thereby play essential roles in driving the initiation through the resolution phase of inflammation by a prudent balancing of the pro-inflammatory and anti-inflammatory cytokines and chemokines. Pathogen products, such as lipopolysaccharides (LPS), dsRNA, and flagellin, are recognized by pathogen-associated molecular patterns (PAMPs) or PAMP-recognizing receptors (PRRs). Common PRRs, such as toll-like receptors (TLRs) and NOD-like receptors (NLRs) (49–51), are present on/in immune cells, such as antigen-presenting cells (APCs). These receptors subsequently initiate a set of signaling cascades culminating in the activation of different transcription factors, such as nuclear factor kappa B (NF-κB), activator protein-1 (AP-1), interferon regulatory factors (IRFs), and CCAAT/enhancer-binding protein β (C/EBPβ) (52). The activation of different transcription factors is marked by an elevation in mRNA transcripts encoding for pro-inflammatory cytokines, such as IL-1β, IL-6, and TNFα (52).
Activation of these PRRs also triggers riboclustering via the p38-mitogen-activated protein kinase (MAPK) pathway, which phosphorylates TTP at specific serine residues (Ser 52,178) (53). Phosphorylation of TTP reduces its binding to cytokine mRNAs, allowing these mRNAs to be translated. The reversion of TTP from its inactive phosphorylated state to its active (i.e., mRNA-destabilizing) dephosphorylated state is prevented by the recruitment of the 14-3-3 protein, which blocks the interaction of TTP with decay enzymes and phosphatase 2α, thus retaining its phosphorylated state (54). Alternative to the p38-MAPK signaling pathway, several other pathways have been identified in the regulation of cytokine expression, such as TNFα. Initiation factor eIF4G-mediated recruitment of Mnk1 (MAPK signal-integrating kinases) onto the 5′ cap-binding protein eIF4E results in phosphorylation and inactivation of translation silencer hnRNPA1, which regulates TNFα expression. This is manifested in the release of TNFα mRNA from hnRNPA1, followed by cap-dependent translation initiation of TNFα upon triggering of this pathway (55–57).
In an analogous pathway, signals from growth factor activate the PI3-kinase pathway, which then activates protein kinase B/AKT, which in turn phosphorylates the secondary modifiers BRF1 and KSRP. Phosphorylated BRF1 and KSRP phosphorylate TTP, as in the case of the p38-MAPK pathway. Upon phosphorylation, the TTP loses its ability to bind to cytokine mRNA, such as IL-3, thereby resulting in the stabilization and translation of IL-3, a major cytokine in hematopoiesis (58, 59). Figure 2 gives an overview of the signaling pathways involved in this process.
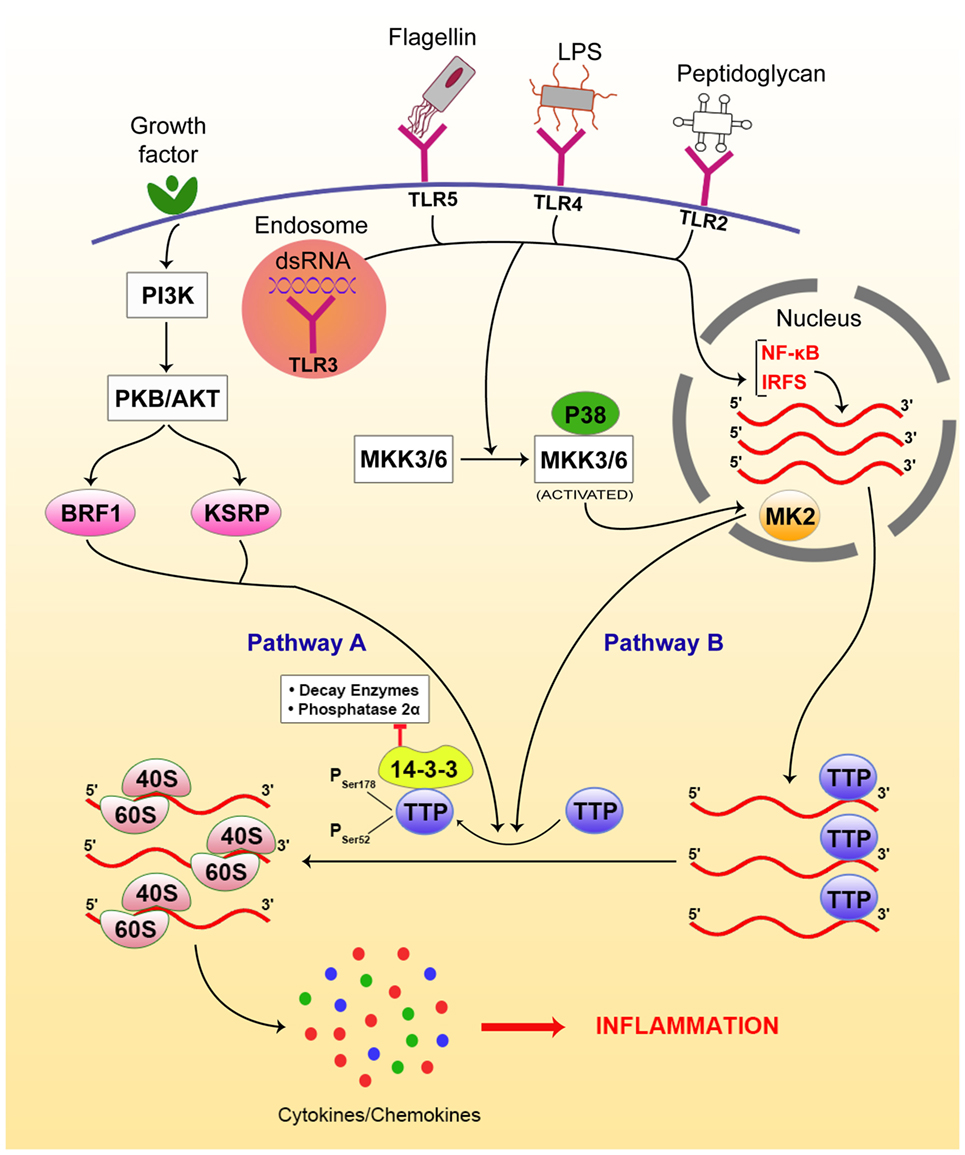
Figure 2. Riboclustering-mediated signaling pathways that regulate innate inflammatory modulators, including cytokines/chemokines. There are two dominant pathways that mediate the process. Toll-like receptors (TLRs) bind to specific ligands, get activated, and induce p38-mitogen-activated protein kinase (MAPK) pathway via nuclear factor (NF)-κB pathway signaling intermediates. On the other hand, the growth factor receptors activate phosphatidylinositol 3-kinase (PI3K), activating protein kinase B (PKB/AKT), which phosphorylates BRF and KSRP. Both pathways contribute to post-transcriptional regulation of cytokine/chemokine mRNAs. Upon phosphorylation by either of the above pathways, TTP gets inactivated and looses access from the 3′-UTRs of the mRNA transcripts, freeing them from SGs and allowing their translation, resulting in inflammation.
These pathways control the cytokines involved in the resolution of inflammation and maintenance of tissue homeostasis mainly through the production of anti-inflammatory mediators, such as IL-4, IL-10, and TGFβ (60). The decay of the pro-inflammatory cytokine mRNAs is another essential determinant of inflammatory inactivation (60) and is promoted by the dephosphorylation of either the destabilization factor, TTP (53, 61), or that of the secondary modifiers BRF1 and KSRP. Once dephosphorylated, they tether to the subjected mRNAs leading to their sequestration to the SGs or P-bodies from the cytosol.
The importance of TTP is illustrated by the fact that TTP knockout mice are highly susceptible to severe inflammatory challenges marked by chronic lung and joint pathology, dermatitis, cachexia, and myeloid hyperplasia (60). However, these conditions can be ameliorated by blocking pro-inflammatory mediators TNFα, CCL3, IL-23, and IL-17 (62–66). Translational silencers TIA1 and TIAR stall formation of translation preinitiation complexes and propel the arrested pro-inflammatory mRNAs from the polysomes to the SGs or P-bodies for degradation (9). Thus, deletion of TIA1 results in aggravation of syndromes in TTP-deficient mice (67). Furthermore, this pathway of mRNA homeostasis can be controlled by the anti-inflammatory cytokines IL-4, IL-10, and TGFβ, which promote TTP expression (68–70).
Besides TTP, dephosphorylation of Regnase-1 protein is also involved in the destabilization of inflammatory cytokine mRNA, such as IL-6. Upon TLR activation, Regnase-1 gets phosphorylated and deactivated in a similar manner to TTP. While in resting condition or anti-inflammatory state, dephosphorylation results in the activation of Regnase-1, allowing it to gain access to the 3′-UTR of IL-6 mRNA and subject the latter to decay (71). Similarly, AUF1 is reported to form dimers and bind to AREs of pro-inflammatory mRNAs (72). Knockout studies show that AUF1-deficit mice are susceptible to acute and chronic inflammatory dermatitis because of high levels of pro-inflammatory cytokines. This clearly indicates that AUF1 is a crucial regulator of the immune response that attenuates the translation of selective pro-inflammatory cytokine transcripts (29, 73).
Other destabilizing factors, such as Roquin proteins 1 and 2, not only target the ARE sequences of the mRNAs but also bind to the constitutive decay elements (CDEs) of TNFα mRNA resulting in its exonucleolytic decay as mediated by TTP in ARE-mediated (AMD) (74). Indeed, knockout experiments suggest that mice lacking Roquin proteins are highly susceptible to TNFα-mediated inflammatory syndromes (75, 76).
Viral ssRNA and dsRNA are detected by the intracellular receptors TLR 7/8 and TLR 3/9, retinoic acid-inducible gene-I (RIG-I), melanoma-differentiation-associated gene 5 (Mda-5), and DHX33 [DEAH (Asp–Glu–Ala–His) Box Polypeptide 33], respectively (49, 77, 78). This interaction results in the production of interferons, which initiate several antiviral processes inside the cell. Interferon activates PKR, which triggers shut down in host translation through blockage of eIF2 (79). It also induces ADAR enzymes to control the translation or decay of the existing mRNAs by ARE-mediated post-transcriptional regulation (5).
Certain RBPs can both repress and de-repress inflammatory mechanisms, depending on the RNP configurations with which they are associated at a certain point of time. For example, the RBP HuR was initially thought to be a stabilizing RBP in the context of ARE-bearing mRNAs. However, mice lacking HuR are more prone to hypersensitive immune disorders due to increased levels of ARE-bearing pro-inflammatory molecules, which support a destabilizing role of HuR (18, 80). However, HuR can play both pro-inflammatory as well as anti-inflammatory roles (80), since HuR and TIA1 cooperatively inhibit translation of pro-inflammatory cytokines, such as IL-1β and TNFα (18). On the other hand, HuR has been found to inhibit AMD by TTP and AUF1, while itself acting synergistically with TIA1 in promoting such decay process (18).
Additionally, other translational blockades are activated inside the cell in cooperation with RBPs that drive the anti-inflammatory processes. When pro-inflammatory modulators reach a critical concentration; they drive autocrine and paracrine regulatory pathways. For instance, IFNγ directs diversion of ribosomal protein L13a onto GAIT (IFNγ-activated inhibitor of translation) elements of mRNA transcripts, such as CCL22, CXCL13, and CCL8, and curbs their translation (81–83). Moreover, the equilibrium between the inflammation and tissue homeostasis is maintained in part by the serine/threonine protein kinase B (Akt)/mTOR pathway (84). For example, during infection with virulent L. pneumophila (85), mTOR-activating kinase Akt gets degraded, and TSC proteins are released, culminating in the inhibition of mTORC1-mediated translation while pro-inflammatory cytokines are readily translated (85). By contrast, during infection with avirulent L. pneumophila, mTOR complex 1 (mTORC1) is activated by loss of TSC proteins, resulting in increased synthesis of immunoregulatory molecules and decay of mRNAs encoding for pro-inflammatory factors (86). The ability of mTOR to regulate translation inhibition and elevated expression of pro-inflammatory cytokines may be due to ARE-BPs. Indeed, metabolic alterations that activate AMP-activated protein kinase can inhibit mTOR-related translation and destabilize RBPs (87, 88). Furthermore, the inactivated mTOR was still able to bind to the destabilizing RBP TIAR, thus leaving the 3′ARE sequences in the mRNA unoccupied (89). This renders the cytokine mRNAs to be ready for translation and could explain their higher concentrations in the absence of active TOR in the cell. Thus, the interactions with different ARE-BPs may be crucial in this mTOR-mediated counterbalance between perpetuation and repression of inflammation.
Riboclustering and Other Post-transcriptional Modifications in Adaptive Immunity
Alongside the innate responses, the adaptive arm of the immune system plays a crucial role in the immune response. RBP-mediated riboclustering and other PTRs have been noted to be critical for maturation, selection, activation, and tolerance of adaptive immune cells. Although its role in B cell biology is not well studied, its role in the genesis and generation of T cell responses has been well-described. Below, we focus on some of these aspects.
T Cell Maturation and Thymic Tolerance
In the thymus, progenitor T cells develop into naive T cells via sequential developmental stages from CD4−/CD8− double negative (DN) T cells to CD4+/CD8+ double positive (DP) T cells, and finally to lineage-specific naive T cells with rearranged T cell receptor (TCR) αβ or γδ receptors. Each of these events is controlled in part by RNP complexes regulated by Notch–Delta and pre-TCR signaling. In developing thymocytes in the thymic cortex, Notch mRNA is controlled by AMD imposed by two TTP variants, ZFP36L1 and ZFP36L2 (90). Before the replacement of Notch receptor by a pre-TCR receptor, ZFP36L1 and ZFP36L2 are subjected to Notch signaling induced phosphorylation (59). During the rearrangements of pre-TCR β receptor mRNAs, the upstream frame shift proteins (Upf1 and 2) are involved in removing the unfolded or misfolded variants (91, 92). Ribosomal protein S6, a major substrate of mTOR, also plays a major role in this process as S6-deficient mice exhibit blocked transition of DN to DP thymocytes due in part of the inability to counteract p53-mediated genotoxic shock resulting from the lack of functional ribosomes (93). The successful transcription of the TCRα gene in DP cells is critical for the development of T cells, allowing expression of the complete receptor for lineage committed naive T cells and allowing them to leave the thymus to participate in the immune response in the periphery. TCRα gene transcription is attenuated by the splicing functions of CUGBP family of RBPs (94). Upon induction by pre-TCR signaling, the CUGBP family protein CUGBP Elav-like family member 2 (CELF2) cause rearrangements in the lymphoid enhancer-binding factor 1 (LEF1) transcription factor mRNA, allowing LEF1 to bind to the enhancer region of the gene encoding TCRα in the DP cells, leading to the development of the final mature TCR αβ complex (94).
T cells with mature TCR also bear CD3 receptor complexes, which transduce TCR signals from antigen-bound major histocompatibility complex (MHC). The strength of TCR–MHC interaction along with co-stimulation from the other accessory surface proteins guides the selection of non-self-reactive CD4+ or CD8+ cells (positive selection), or elimination of self-reactive CD4+ or CD8+ cells (negative selection) in the thymus. These intricately orchestrated selection processes are regulated at multiple stages by RBPs. The CD3ζ chain is the major signaling subunit of the CD3 receptor complex of the TCR. Proper splicing of CD3ζ pre-mRNA is essential for effective TCR signaling, since improperly spliced CD3ζ mRNA lacking either a part of the coding region or a part of their 3′-UTR cannot recruit HuR, and the mRNA becomes prone to degradation. In the absence of functional CD3ζ, the TCRαβ cannot deliver the proper signal required for T cell thymic selection, resulting in the removal of the T cell (95). CD3 chains also interact with ZAP 70, a TCR-associated kinase, which is phosphorylated by Lck, a receptor-proximal Src kinase. Lck is in turn activated by transmembrane tyrosine phosphatase CD45, which dephosphorylates the C-terminal inhibitory tyrosine residue (Tyr505) on Lck (96). CD45 exists in various alternatively spliced isoforms with different functions. The shorter isoform CD45RO is the less active isoform on the surface of immature T cells, while the longer one is the fully functional variant CD45RB and is expressed on the mature CD4 and CD8 T cells. Serine/arginine-rich RBPs, arginine/serine splicing factor (ASF) and SRSF2, and the heterogeneous nuclear RNP hnRNPL are primarily responsible for alternative splicing of the CD45 pre-mRNA (97–99). The knockout experiments have identified that RBP HuR plays crucial role in TCR signaling by differential targeting of the mRNAs with AREs (100). The HuR-deficit cells and hnRNPL-deficient cells show similar phenotypes in the migration of mature T cells from thymic cortex to the medulla and finally into the peripheral circulation (99, 101), suggesting that this pathway also regulates the export of T cell from the thymus to the periphery. Figure 3 gives a brief overview of the role of PTRs in T cell development, maturation, and commitment in the thymus and periphery, respectively.
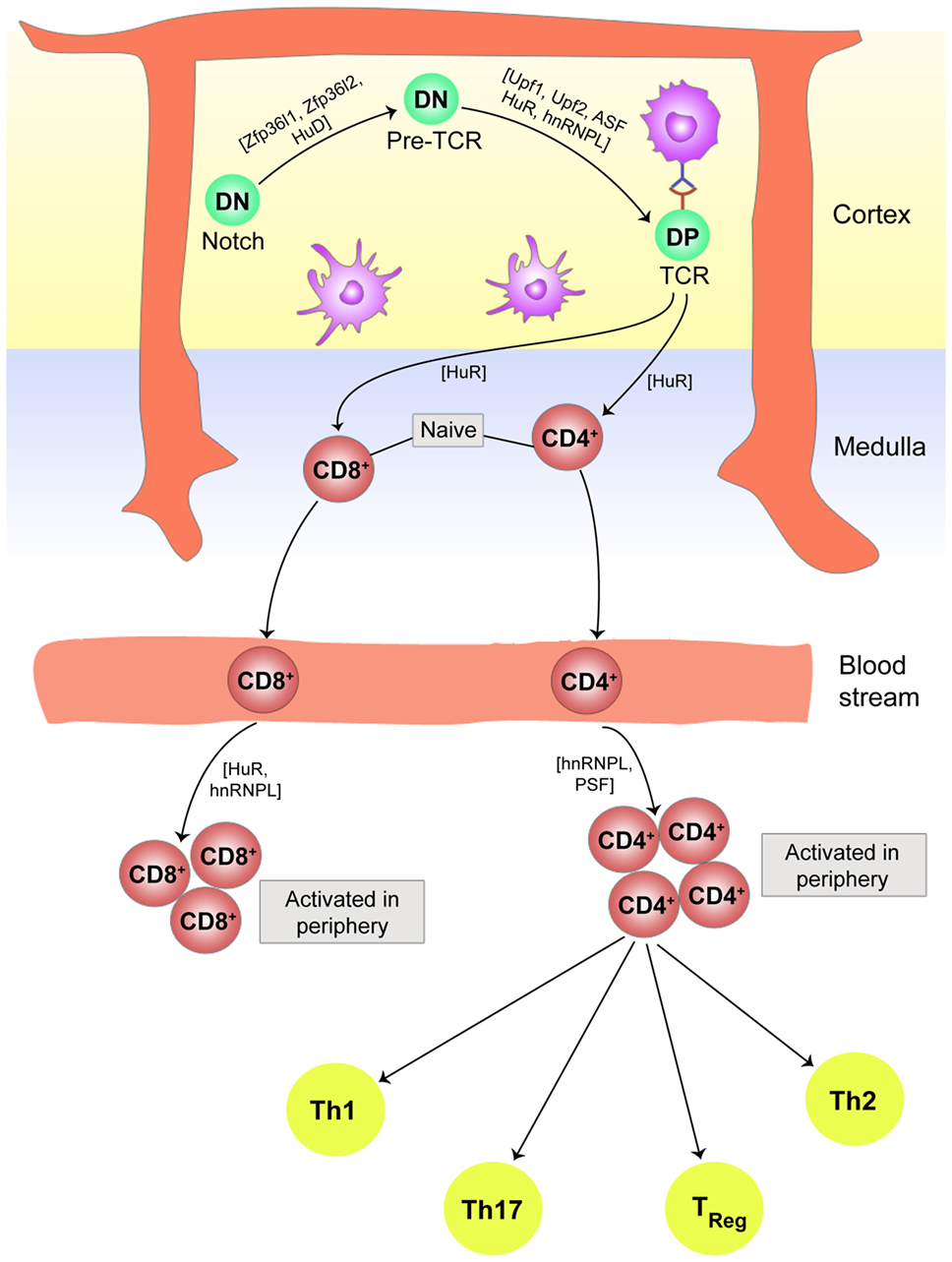
Figure 3. Post-transcriptional regulation plays an essential role in T cell development, maturation, and commitment. In the thymic cortex, double negative (DN) thymocytes experience Notch signaling followed by development of pre-T cell receptors (TCR) and finally end up as double positive (DP) T cells with fully processed TCR. The entire path traversed by a naive T cell to single positive (SP) CD4 or CD8 T cell, to its movement into the peripheral blood stream for activation after antigen, is controlled in part by ribonucleoprotein complexes. In the periphery, activated CD4+ T cells differentiate into subsets of Th17, Th1, Th2, T regulatory cells, and other effector T cells dependent on the cytokine environment in the vicinity. Cytokines and chemokines are in turn modulated by riboclustering, depending upon the nature of the invading pathogen.
T Cell Anergy and Peripheral Tolerance
Once the T cells mature and leave the thymus, they can recognize the antigen in the secondary lymphoid organs. There are typically three sets of signals for successful T cell activation and clonal expansion. The first signal comes from the binding of the TCR to its cognate peptide-bound MHC on the APCs. The second signal is provided by co-stimulation from the CD28–B7 interaction. The third signal is imparted by the cytokines secreted by immune cells in the vicinity. The different isoforms of CD45 are expressed by naive, activated, or memory T cells. Naive T cells express a longer isoform of CD45 (CD45RA) while activated, and memory T cells express the spliced shorter form (CD45RO). This stage-restricted splicing phenomenon of CD45 is aided by hnRNPLL, a paralog of hnRNPL and PSF or polypyrimidine tract-binding protein (PTB)-associated splicing factor (102, 103). A genetic mutation in hnRNPLL results in defective binding to CD45 mRNA, and improper alternate splicing result in decreased number of memory T cells (103).
The co-stimulatory signals from the CD28 superfamily prevent T cell anergy and allow the activated T helper cells to differentiate. The T cell co-stimulatory molecules OX-40 and ICOS are under post-transcriptional regulation by the decay factors Roquin-1, Roquin-2, and Regnase-1 (104). The deletion of these factors either individually or in combination interfere with the T cell co-stimulation (76, 105). Furthermore, the miRNA-mediated silencing of Roquin proteins leads to the activation of CD28, OX-40, and integrin LFA1 in CD4+ T cells (105). This subsequently activates the PIN1 prolyl isomerase, which targets the phosphorylated RBPs to form proline isomers (106). This alteration in the conformation of the RBPs reduces their affinity for the ARE sites of translationally arrested transcripts. A similar level of control exists during T cell activation as a result of CD40 and CD40L interaction, with cytoplasmic stabilization of CD40L pre-mRNA being under the control of nuclear RNPs hnRNPL and nucleolin (107). By contrast, in the absence of CD28 co-stimulation resulting in T cell anergy, mRNAs accumulate, ultimately leading to translational inhibition. This is controlled by the mTORC1 pathway (108).
Cytokine signaling imparts a tertiary function that is required for T cell activation, clonal expansion, and downstream effector functions. The IL-2 functions in autocrine signaling for the T helper cell proliferation. The 3′-UTR of IL-2 mRNA can recruit destabilizing factors TTP, Regnase-1, and the nuclear transcription factor NF90 (109). Moreover, in primed CD4+ T helper cells, the IL-4 mRNA is sequestered in the SGs by the aid of TIA1 until a second exposure to the antigenic signal (110). The HuR can also stabilize the mRNA for other cytokines, such as IL-17, which was recently found to be responsible for autoimmune neuroinflammation (111).
Once the mature T cells are out into the periphery, they undergo another round of scrutiny, distinctively called peripheral tolerance. Self-reactive T cells undergo elimination by apoptosis or programed cell death. This is controlled in part by the cell surface protein Fas/CD95. The Fas/CD95 undergoes alternative splicing, giving rise to two isoforms, a membrane-bound form that facilitates apoptosis and a soluble form that opposes the same. HuR, hnRNP C1, and PTB favor the inclusion of the sixth exon in Fas mRNA, while TIA1, TIAR, and hnRNPA1 block this process (112). The evidence for the regulation of this process by RBPs include that fact that HuR-deficit T cells are resistant to apoptosis (101, 113), and Fas splicing is arrested when U2 snRNPs are cleaved by effector caspases (114). Given the role of Fas/FasL in autoimmune disorders, this suggests a link between PTRs and autoimmune disorders.
Conclusion and Future Prospects
Immune cells exhibit a wide array of temporally and spatially regulated functions in response to a plethora of intracellular and extracellular signals. For example, during inflammation, cytokine mRNAs are induced and gradually subside when the causal agent is removed from the system leading to tissue homeostasis and regeneration. This prompt response of the host cell to the invader and subsequent return to homeostasis suggest the involvement of PTRs, including riboclustering. The mRNA transcripts encoding some of the major effector molecules of this immune response are either kept in cache until they are translated to manifest its effector machinery depending on the needs of the cell or are subjected to alternative splicing, leading to the formation of various isoforms of T or B cell surface molecules. There is strong evidence that riboclustering plays a critical role in this process, and can, therefore, be said to be a key immune-sorting mechanism for these cells. The PTR events are carefully orchestrated for precise coordination of regulatory actions of RBPs and non-coding RNAs, on coding RNA transcripts. Future research should focus on developing approaches to identify these specific RNA targets for individual RBPs, and the cumulative roles of these RNP complexes in response to various signals. To date, various approaches used to understand these functions include overexpression, knockdown, or knockout experiments. However, these approaches come with significant caveats including the fact that a point or domain mutation in RBPs, complete shutdown, or overexpression in the cell may result in stress-related alterations that may be misleading in extrapolating the function of the RBP. Thus, system biological approaches or other experimental approaches promise to reveal more about the crucial regulatory system in the cell. Macromolecular tracer techniques and live cell imaging can also be beneficial in this respect. The future research on riboclustering and elucidation of the molecular pathways involved may usher novel therapeutic approaches and decipher probable drug targets for inflammatory diseases, autoimmune disorders, and cancer.
Author Contributions
KG wrote the paper, JG made the figures, AA critically revised the manuscript, and NK contributed to both writing and critical revision of the paper.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
This work has been financially supported by the Department of Biotechnology, Government of India (BT/PR8624/MED/29/798/2013), the Nano Mission Council, Department of Science and Technology, Government of India [SR/NM/NS-1040/2013(G)], the National Institutes of Health (AI108958), and by the American Society for Microbiology/Indo-US Science and Technology Forum (IUSSTF) and Indo-US Professorship in Microbiology (to AA).
References
1. Bhatt DM, Pandya-Jones A, Tong A-J, Barozzi I, Lissner MM, Natoli G, et al. Transcript dynamics of proinflammatory genes revealed by sequence analysis of subcellular RNA fractions. Cell (2012) 150:279–90. doi:10.1016/j.cell.2012.05.043
2. Bjur E, Larsson O, Yurchenko E, Zheng L, Gandin V, Topisirovic I, et al. Distinct translational control in CD4+ T cell subsets. PLoS Genet (2013) 9:e1003494. doi:10.1371/journal.pgen.1003494
3. Hao S, Baltimore D. The stability of mRNA influences the temporal order of the induction of genes encoding inflammatory molecules. Nat Immunol (2009) 10:281–8. doi:10.1038/ni.1699
4. Hao S, Baltimore D. RNA splicing regulates the temporal order of TNF-induced gene expression. Proc Natl Acad Sci U S A (2013) 110:11934–9. doi:10.1073/pnas.1309990110
5. Wang IX, So E, Devlin JL, Zhao Y, Wu M, Cheung VG. ADAR regulates RNA editing, transcript stability, and gene expression. Cell Rep (2013) 5:849–60. doi:10.1016/j.celrep.2013.10.002
6. Wahl MC, Will CL, Lührmann R. The spliceosome: design principles of a dynamic RNP machine. Cell (2009) 136:701–18. doi:10.1016/j.cell.2009.02.009
7. Kornblihtt AR, Schor IE, Alló M, Dujardin G, Petrillo E, Muñoz MJ. Alternative splicing: a pivotal step between eukaryotic transcription and translation. Nat Rev Mol Cell Biol (2013) 14:153–65. doi:10.1038/nrm3525
8. Cooper TA, Wan L, Dreyfuss G. RNA and disease. Cell (2009) 136:777–93. doi:10.1016/j.cell.2009.02.011
9. Anderson P, Kedersha N. RNA granules: post-transcriptional and epigenetic modulators of gene expression. Nat Rev Mol Cell Biol (2009) 10:430–6. doi:10.1038/nrm2694
10. Palanisamy V, Jakymiw A, Van Tubergen E, D’Silva N, Kirkwood K. Control of cytokine mRNA expression by RNA-binding proteins and microRNAs. J Dent Res (2012) 91:651–8. doi:10.1177/0022034512437372
11. Parker R, Sheth U. P-bodies and the control of mRNA translation and degradation. Mol Cell (2007) 25:635–46. doi:10.1016/j.molcel.2007.02.011
12. Bhattacharyya SN, Habermacher R, Martine U, Closs EI, Filipowicz W. Relief of microRNA-mediated translational repression in human cells subjected to stress. Cell (2006) 125:1111–24. doi:10.1016/j.cell.2006.04.031
13. Stoecklin G, Anderson P. In a tight spot: ARE-mRNAs at processing bodies. Genes Dev (2007) 21:627–31. doi:10.1101/gad.1538807
14. Anderson P. Post-transcriptional control of cytokine production. Nat Immunol (2008) 9:353–9. doi:10.1038/ni1584
15. Chen C-Y, Gherzi R, Ong S-E, Chan EL, Raijmakers R, Pruijn GJ, et al. AU binding proteins recruit the exosome to degrade ARE-containing mRNAs. Cell (2001) 107:451–64. doi:10.1016/S0092-8674(01)00578-5
16. Hamilton TA, Novotny M, Datta S, Mandal P, Hartupee J, Tebo J, et al. Chemokine and chemoattractant receptor expression: post-transcriptional regulation. J Leukoc Biol (2007) 82:213–9. doi:10.1189/jlb.1206754
17. Piecyk M, Wax S, Beck AR, Kedersha N, Gupta M, Maritim B, et al. TIA-1 is a translational silencer that selectively regulates the expression of TNF-α. EMBO J (2000) 19:4154–63. doi:10.1093/emboj/19.15.4154
18. Katsanou V, Papadaki O, Milatos S, Blackshear PJ, Anderson P, Kollias G, et al. HuR as a negative posttranscriptional modulator in inflammation. Mol Cell (2005) 19:777–89. doi:10.1016/j.molcel.2005.08.007
19. Linker K, Pautz A, Fechir M, Hubrich T, Greeve J, Kleinert H. Involvement of KSRP in the post-transcriptional regulation of human iNOS expression – complex interplay of KSRP with TTP and HuR. Nucleic Acids Res (2005) 33:4813–27. doi:10.1093/nar/gki797
20. Caput D, Beutler B, Hartog K, Thayer R, Brown-Shimer S, Cerami A. Identification of a common nucleotide sequence in the 3′-untranslated region of mRNA molecules specifying inflammatory mediators. Proc Natl Acad Sci U S A (1986) 83:1670–4. doi:10.1073/pnas.83.6.1670
21. Shaw G, Kamen R. A conserved AU sequence from the 3′ untranslated region of GM-CSF mRNA mediates selective mRNA degradation. Cell (1986) 46:659–67. doi:10.1016/0092-8674(86)90341-7
22. Bakheet T, Williams BR, Khabar KS. ARED 3.0: the large and diverse AU-rich transcriptome. Nucleic Acids Res (2006) 34:D111–4. doi:10.1093/nar/gkj052
23. Stoecklin G, Ming X-F, Looser R, Moroni C. Somatic mRNA turnover mutants implicate tristetraprolin in the interleukin-3 mRNA degradation pathway. Mol Cell Biol (2000) 20:3753–63. doi:10.1128/MCB.20.11.3753-3763.2000
24. Yarovinsky TO, Butler NS, Monick MM, Hunninghake GW. Early exposure to IL-4 stabilizes IL-4 mRNA in CD4+ T cells via RNA-binding protein HuR. J Immunol (2006) 177:4426–35. doi:10.4049/jimmunol.177.7.4426
25. Neininger A, Kontoyiannis D, Kotlyarov A, Winzen R, Eckert R, Volk H-D, et al. MK2 targets AU-rich elements and regulates biosynthesis of tumor necrosis factor and interleukin-6 independently at different post-transcriptional levels. J Biol Chem (2002) 277:3065–8. doi:10.1074/jbc.C100685200
26. Paschoud S, Dogar AM, Kuntz C, Grisoni-Neupert B, Richman L, Kühn LC. Destabilization of interleukin-6 mRNA requires a putative RNA stem-loop structure, an AU-rich element, and the RNA-binding protein AUF1. Mol Cell Biol (2006) 26:8228–41. doi:10.1128/MCB.01155-06
27. Winzen R, Thakur BK, Dittrich-Breiholz O, Shah M, Redich N, Dhamija S, et al. Functional analysis of KSRP interaction with the AU-rich element of interleukin-8 and identification of inflammatory mRNA targets. Mol Cell Biol (2007) 27:8388–400. doi:10.1128/MCB.01493-07
28. Stoecklin G, Tenenbaum SA, Mayo T, Chittur SV, George AD, Baroni TE, et al. Genome-wide analysis identifies interleukin-10 mRNA as target of tristetraprolin. J Biol Chem (2008) 283:11689–99. doi:10.1074/jbc.M709657200
29. Lu J-Y, Sadri N, Schneider RJ. Endotoxic shock in AUF1 knockout mice mediated by failure to degrade proinflammatory cytokine mRNAs. Genes Dev (2006) 20:3174–84. doi:10.1101/gad.1467606
30. Chen Y-L, Huang Y-L, Lin N-Y, Chen H-C, Chiu W-C, Chang C-J. Differential regulation of ARE-mediated TNFα and IL-1β mRNA stability by lipopolysaccharide in RAW264. 7 cells. Biochem Biophys Res Commun (2006) 346:160–8. doi:10.1016/j.bbrc.2006.05.093
31. Garnon J, Lachance C, Di Marco S, Hel Z, Marion D, Ruiz MC, et al. Fragile X-related protein FXR1P regulates proinflammatory cytokine tumor necrosis factor expression at the post-transcriptional level. J Biol Chem (2005) 280:5750–63. doi:10.1074/jbc.M401988200
32. Carballo E, Lai WS, Blackshear PJ. Feedback inhibition of macrophage tumor necrosis factor-α production by tristetraprolin. Science (1998) 281:1001–5. doi:10.1126/science.281.5379.1001
33. Wang JG, Collinge M, Ramgolam V, Ayalon O, Fan XC, Pardi R, et al. LFA-1-dependent HuR nuclear export and cytokine mRNA stabilization in T cell activation. J Immunol (2006) 176:2105–13. doi:10.4049/jimmunol.176.4.2105
34. Carballo E, Lai WS, Blackshear PJ. Evidence that tristetraprolin is a physiological regulator of granulocyte-macrophage colony-stimulating factor messenger RNA deadenylation and stability. Blood (2000) 95:1891–9.
35. Grosset C, Boniface R, Duchez P, Solanilla A, Cosson B, Ripoche J. In vivo studies of translational repression mediated by the granulocyte-macrophage colony-stimulating factor AU-rich element. J Biol Chem (2004) 279:13354–62. doi:10.1074/jbc.M308003200
36. Douglas PM, Dillin A. Protein homeostasis and aging in neurodegeneration. J Cell Biol (2010) 190:719–29. doi:10.1083/jcb.201005144
37. Schröder M, Kaufman RJ. ER stress and the unfolded protein response. Mutat Res (2005) 569:29–63. doi:10.1016/j.mrfmmm.2004.06.056
38. Marciniak SJ, Ron D. Endoplasmic reticulum stress signaling in disease. Physiol Rev (2006) 86:1133–49. doi:10.1152/physrev.00015.2006
39. Ron D, Walter P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Biol (2007) 8:519–29. doi:10.1038/nrm2199
40. Wek RC, Cavener DR. Translational control and the unfolded protein response. Antioxid Redox Signal (2007) 9:2357–72. doi:10.1089/ars.2007.1764
41. Hotamisligil GS. Endoplasmic reticulum stress and the inflammatory basis of metabolic disease. Cell (2010) 140:900–17. doi:10.1016/j.cell.2010.02.034
42. Sano R, Reed JC. ER stress-induced cell death mechanisms. Biochim Biophys Acta (2013) 1833:3460–70. doi:10.1016/j.bbamcr.2013.06.028
43. Goh KC, deVeer MJ, Williams BR. The protein kinase PKR is required for p38 MAPK activation and the innate immune response to bacterial endotoxin. EMBO J (2000) 19:4292–7. doi:10.1093/emboj/19.16.4292
44. Kedersha N, Stoecklin G, Ayodele M, Yacono P, Lykke-Andersen J, Fritzler MJ, et al. Stress granules and processing bodies are dynamically linked sites of mRNP remodeling. J Cell Biol (2005) 169:871–84. doi:10.1083/jcb.200502088
45. Hay N, Sonenberg N. Upstream and downstream of mTOR. Genes Dev (2004) 18:1926–45. doi:10.1101/gad.1212704
47. Franks TM, Lykke-Andersen J. TTP and BRF proteins nucleate processing body formation to silence mRNAs with AU-rich elements. Genes Dev (2007) 21:719–35. doi:10.1101/gad.1494707
48. Chou C-F, Mulky A, Maitra S, Lin W-J, Gherzi R, Kappes J, et al. Tethering KSRP, a decay-promoting AU-rich element-binding protein, to mRNAs elicits mRNA decay. Mol Cell Biol (2006) 26:3695–706. doi:10.1128/MCB.26.10.3695-3706.2006
49. Takeuchi O, Akira S. Pattern recognition receptors and inflammation. Cell (2010) 140:805–20. doi:10.1016/j.cell.2010.01.022
50. Beutler BA. TLRs and innate immunity. Blood (2009) 113:1399–407. doi:10.1182/blood-2008-07-019307
51. Palm NW, Medzhitov R. Pattern recognition receptors and control of adaptive immunity. Immunol Rev (2009) 227:221–33. doi:10.1111/j.1600-065X.2008.00731.x
52. Mino T, Takeuchi O. Post-transcriptional regulation of cytokine mRNA controls the initiation and resolution of inflammation. Biotechnol Genet Eng Rev (2013) 29:49–60. doi:10.1080/02648725.2013.801236
53. Chrestensen CA, Schroeder MJ, Shabanowitz J, Hunt DF, Pelo JW, Worthington MT, et al. MAPKAP kinase 2 phosphorylates tristetraprolin on in vivo sites including Ser178, a site required for 14-3-3 binding. J Biol Chem (2004) 279:10176–84. doi:10.1074/jbc.M310486200
54. Sun L, Stoecklin G, Van Way S, Hinkovska-Galcheva V, Guo R-F, Anderson P, et al. Tristetraprolin (TTP)-14-3-3 complex formation protects TTP from dephosphorylation by protein phosphatase 2a and stabilizes tumor necrosis factor-α mRNA. J Biol Chem (2007) 282:3766–77. doi:10.1074/jbc.M607347200
55. Pyronnet S, Imataka H, Gingras AC, Fukunaga R, Hunter T, Sonenberg N. Human eukaryotic translation initiation factor 4G (eIF4G) recruits mnk1 to phosphorylate eIF4E. EMBO J (1999) 18:270–9. doi:10.1093/emboj/18.1.270
56. Waskiewicz AJ, Johnson JC, Penn B, Mahalingam M, Kimball SR, Cooper JA. Phosphorylation of the cap-binding protein eukaryotic translation initiation factor 4E by protein kinase Mnk1 in vivo. Mol Cell Biol (1999) 19:1871–80. doi:10.1128/MCB.19.3.1871
57. Buxadé M, Parra JL, Rousseau S, Shpiro N, Marquez R, Morrice N, et al. The Mnks are novel components in the control of TNFα biosynthesis and phosphorylate and regulate hnRNP A1. Immunity (2005) 23:177–89. doi:10.1016/j.immuni.2005.06.009
58. Schmidlin M, Lu M, Leuenberger SA, Stoecklin G, Mallaun M, Gross B, et al. The ARE-dependent mRNA-destabilizing activity of BRF1 is regulated by protein kinase B. EMBO J (2004) 23:4760–9. doi:10.1038/sj.emboj.7600477
59. Benjamin D, Schmidlin M, Min L, Gross B, Moroni C. BRF1 protein turnover and mRNA decay activity are regulated by protein kinase B at the same phosphorylation sites. Mol Cell Biol (2006) 26:9497–507. doi:10.1128/MCB.01099-06
60. Kafasla P, Skliris A, Kontoyiannis DL. Post-transcriptional coordination of immunological responses by RNA-binding proteins. Nat Immunol (2014) 15:492–502. doi:10.1038/ni.2884
61. Stoecklin G, Stubbs T, Kedersha N, Wax S, Rigby WF, Blackwell TK, et al. MK2-induced tristetraprolin: 14-3-3 complexes prevent stress granule association and ARE-mRNA decay. EMBO J (2004) 23:1313–24. doi:10.1038/sj.emboj.7600163
62. Kang J-G, Amar MJ, Remaley AT, Kwon J, Blackshear PJ, Wang PY, et al. Zinc finger protein tristetraprolin interacts with CCL3 mRNA and regulates tissue inflammation. J Immunol (2011) 187:2696–701. doi:10.4049/jimmunol.1101149
63. Kratochvill F, Machacek C, Vogl C, Ebner F, Sedlyarov V, Gruber AR, et al. Tristetraprolin-driven regulatory circuit controls quality and timing of mRNA decay in inflammation. Mol Syst Biol (2011) 7:560. doi:10.1038/msb.2011.93
64. Molle C, Zhang T, de Lendonck LY, Gueydan C, Andrianne M, Sherer F, et al. Tristetraprolin regulation of interleukin 23 mRNA stability prevents a spontaneous inflammatory disease. J Exp Med (2013) 210:1675–84. doi:10.1084/jem.20120707
65. Qiu L-Q, Stumpo DJ, Blackshear PJ. Myeloid-specific tristetraprolin deficiency in mice results in extreme lipopolysaccharide sensitivity in an otherwise minimal phenotype. J Immunol (2012) 188:5150–9. doi:10.4049/jimmunol.1103700
66. Taylor GA, Carballo E, Lee DM, Lai WS, Thompson MJ, Patel DD, et al. A pathogenetic role for TNFα in the syndrome of cachexia, arthritis, and autoimmunity resulting from tristetraprolin (TTP) deficiency. Immunity (1996) 4:445–54. doi:10.1016/S1074-7613(00)80411-2
67. Phillips K, Kedersha N, Shen L, Blackshear PJ, Anderson P. Arthritis suppressor genes TIA-1 and TTP dampen the expression of tumor necrosis factor α, cyclooxygenase 2, and inflammatory arthritis. Proc Natl Acad Sci U S A (2004) 101:2011–6. doi:10.1073/pnas.0400148101
68. Suzuki K, Nakajima H, Ikeda K, Maezawa Y, Suto A, Takatori H, et al. IL-4-Stat6 signaling induces tristetraprolin expression and inhibits TNF-α production in mast cells. J Exp Med (2003) 198:1717–27. doi:10.1084/jem.20031701
69. Schaljo B, Kratochvill F, Gratz N, Sadzak I, Sauer I, Hammer M, et al. Tristetraprolin is required for full anti-inflammatory response of murine macrophages to IL-10. J Immunol (2009) 183:1197–206. doi:10.4049/jimmunol.0803883
70. Blanco FF, Sanduja S, Deane NG, Blackshear PJ, Dixon DA. Transforming growth factor β regulates P-body formation through induction of the mRNA decay factor tristetraprolin. Mol Cell Biol (2014) 34:180–95. doi:10.1128/MCB.01020-13
71. Iwasaki H, Takeuchi O, Teraguchi S, Matsushita K, Uehata T, Kuniyoshi K, et al. The I [kappa] B kinase complex regulates the stability of cytokine-encoding mRNA induced by TLR-IL-1R by controlling degradation of regnase-1. Nat Immunol (2011) 12:1167–75. doi:10.1038/ni.2137
72. White EJ, Brewer G, Wilson GM. Post-transcriptional control of gene expression by AUF1: mechanisms, physiological targets, and regulation. Biochim Biophys Acta (2013) 1829:680–8. doi:10.1016/j.bbagrm.2012.12.002
73. Sadri N, Schneider RJ. Auf1/Hnrnpd-deficient mice develop pruritic inflammatory skin disease. J Invest Dermatol (2009) 129:657–70. doi:10.1038/jid.2008.298
74. Leppek K, Schott J, Reitter S, Poetz F, Hammond MC, Stoecklin G. Roquin promotes constitutive mRNA decay via a conserved class of stem-loop recognition motifs. Cell (2013) 153:869–81. doi:10.1016/j.cell.2013.04.016
75. Vinuesa CG, Cook MC, Angelucci C, Athanasopoulos V, Rui L, Hill KM, et al. A RING-type ubiquitin ligase family member required to repress follicular helper T cells and autoimmunity. Nature (2005) 435:452–8. doi:10.1038/nature03555
76. Pratama A, Ramiscal RR, Silva DG, Das SK, Athanasopoulos V, Fitch J, et al. Roquin-2 shares functions with its paralog Roquin-1 in the repression of mRNAs controlling T follicular helper cells and systemic inflammation. Immunity (2013) 38:669–80. doi:10.1016/j.immuni.2013.01.011
77. Schroder K, Tschopp J. The inflammasomes. Cell (2010) 140:821–32. doi:10.1016/j.cell.2010.01.040
78. Mitoma H, Hanabuchi S, Kim T, Bao M, Zhang Z, Sugimoto N, et al. The DHX33 RNA helicase senses cytosolic RNA and activates the NLRP3 inflammasome. Immunity (2013) 39:123–35. doi:10.1016/j.immuni.2013.07.001
79. Nakayama Y, Plisch EH, Sullivan J, Thomas C, Czuprynski CJ, Williams B, et al. Role of PKR and Type I IFNs in viral control during primary and secondary infection. PLoS Pathog (2010) 6:e1000966. doi:10.1371/journal.ppat.1000966
80. Yiakouvaki A, Dimitriou M, Karakasiliotis I, Eftychi C, Theocharis S, Kontoyiannis DL. Myeloid cell expression of the RNA-binding protein HuR protects mice from pathologic inflammation and colorectal carcinogenesis. J Clin Invest (2012) 122:48. doi:10.1172/JCI45021
81. Mukhopadhyay R, Jia J, Arif A, Ray PS, Fox PL. The GAIT system: a gatekeeper of inflammatory gene expression. Trends Biochem Sci (2009) 34:324–31. doi:10.1016/j.tibs.2009.03.004
82. Arif A, Chatterjee P, Moodt RA, Fox PL. Heterotrimeric GAIT complex drives transcript-selective translation inhibition in murine macrophages. Mol Cell Biol (2012) 32:5046–55. doi:10.1128/MCB.01168-12
83. Poddar D, Basu A, Baldwin WM, Kondratov RV, Barik S, Mazumder B. An extraribosomal function of ribosomal protein L13a in macrophages resolves inflammation. J Immunol (2013) 190:3600–12. doi:10.4049/jimmunol.1201933
84. Weichhart T, Costantino G, Poglitsch M, Rosner M, Zeyda M, Stuhlmeier KM, et al. The TSC-mTOR signaling pathway regulates the innate inflammatory response. Immunity (2008) 29:565–77. doi:10.1016/j.immuni.2008.08.012
85. Ivanov SS, Roy CR. Pathogen signatures activate a ubiquitination pathway that modulates the function of the metabolic checkpoint kinase mTOR. Nat Immunol (2013) 14:1219–28. doi:10.1038/ni.2740
86. Arranz A, Doxaki C, Vergadi E, de la Torre YM, Vaporidi K, Lagoudaki ED, et al. Akt1 and Akt2 protein kinases differentially contribute to macrophage polarization. Proc Natl Acad Sci U S A (2012) 109:9517–22. doi:10.1073/pnas.1119038109
87. Wang W, Fan J, Yang X, Fürer-Galban S, de Silanes IL, von Kobbe C, et al. AMP-activated kinase regulates cytoplasmic HuR. Mol Cell Biol (2002) 22:3425–36. doi:10.1128/MCB.22.10.3425-3436.2002
88. Hahn-Windgassen A, Nogueira V, Chen C-C, Skeen JE, Sonenberg N, Hay N. Akt activates the mammalian target of rapamycin by regulating cellular ATP level and AMPK activity. J Biol Chem (2005) 280:32081–9. doi:10.1074/jbc.M502876200
89. Damgaard CK, Lykke-Andersen J. Translational coregulation of 5′ TOP mRNAs by TIA-1 and TIAR. Genes Dev (2011) 25:2057–68. doi:10.1101/gad.17355911
90. Hodson DJ, Janas ML, Galloway A, Bell SE, Andrews S, Li CM, et al. Deletion of the RNA-binding proteins ZFP36L1 and ZFP36L2 leads to perturbed thymic development and T lymphoblastic leukemia. Nat Immunol (2010) 11:717–24. doi:10.1038/ni.1901
91. Frischmeyer-Guerrerio PA, Montgomery RA, Warren DS, Cooke SK, Lutz J, Sonnenday CJ, et al. Perturbation of thymocyte development in nonsense-mediated decay (NMD)-deficient mice. Proc Natl Acad Sci U S A (2011) 108:10638–43. doi:10.1073/pnas.1019352108
92. Weischenfeldt J, Damgaard I, Bryder D, Theilgaard-Mönch K, Thoren LA, Nielsen FC, et al. NMD is essential for hematopoietic stem and progenitor cells and for eliminating by-products of programmed DNA rearrangements. Genes Dev (2008) 22:1381–96. doi:10.1101/gad.468808
93. Šulić S, Panić L, Barkić M, Merćep M, Uzelac M, Volarević S. Inactivation of S6 ribosomal protein gene in T lymphocytes activates a p53-dependent checkpoint response. Genes Dev (2005) 19:3070–82. doi:10.1101/gad.359305
94. Mallory MJ, Jackson J, Weber B, Chi A, Heyd F, Lynch KW. Signal-and development-dependent alternative splicing of LEF1 in T cells is controlled by CELF2. Mol Cell Biol (2011) 31:2184–95. doi:10.1128/MCB.05170-11
95. Moulton VR, Kyttaris VC, Juang Y-T, Chowdhury B, Tsokos GC. The RNA-stabilizing protein HuR regulates the expression of ζ chain of the human T cell receptor-associated CD3 complex. J Biol Chem (2008) 283:20037–44. doi:10.1074/jbc.M710434200
96. D’Oro U, Ashwell JD. Cutting edge: the CD45 tyrosine phosphatase is an inhibitor of Lck activity in thymocytes. J Immunol (1999) 162:1879–83.
97. Moulton VR, Grammatikos AP, Fitzgerald LM, Tsokos GC. Splicing factor SF2/ASF rescues IL-2 production in T cells from systemic lupus erythematosus patients by activating IL-2 transcription. Proc Natl Acad Sci U S A (2013) 110:1845–50. doi:10.1073/pnas.1214207110
98. Wang H-Y, Xu X, Ding J-H, Bermingham JR, Fu X-D. SC35 plays a role in T cell development and alternative splicing of CD45. Mol Cell (2001) 7:331–42. doi:10.1016/S1097-2765(01)00181-2
99. Gaudreau M-C, Heyd F, Bastien R, Wilhelm B, Möröy T. Alternative splicing controlled by heterogeneous nuclear ribonucleoprotein L regulates development, proliferation, and migration of thymic pre-T cells. J Immunol (2012) 188:5377–88. doi:10.4049/jimmunol.1103142
100. Mukherjee N, Corcoran DL, Nusbaum JD, Reid DW, Georgiev S, Hafner M, et al. Integrative regulatory mapping indicates that the RNA-binding protein HuR couples pre-mRNA processing and mRNA stability. Mol Cell (2011) 43:327–39. doi:10.1016/j.molcel.2011.06.007
101. Papadaki O, Milatos S, Grammenoudi S, Mukherjee N, Keene JD, Kontoyiannis DL. Control of thymic T cell maturation, deletion and egress by the RNA-binding protein HuR. J Immunol (2009) 182:6779–88. doi:10.4049/jimmunol.0900377
102. Heyd F, Lynch KW. Phosphorylation-dependent regulation of PSF by GSK3 controls CD45 alternative splicing. Mol Cell (2010) 40:126–37. doi:10.1016/j.molcel.2010.09.013
103. Wu Z, Jia X, de la Cruz L, Su X-C, Marzolf B, Troisch P, et al. Memory T cell RNA rearrangement programmed by heterogeneous nuclear ribonucleoprotein hnRNPLL. Immunity (2008) 29:863–75. doi:10.1016/j.immuni.2008.11.004
104. Glasmacher E, Hoefig KP, Vogel KU, Rath N, Du L, Wolf C, et al. Roquin binds inducible costimulator mRNA and effectors of mRNA decay to induce microRNA-independent post-transcriptional repression. Nat Immunol (2010) 11:725–33. doi:10.1038/ni.1902
105. Vogel KU, Edelmann SL, Jeltsch KM, Bertossi A, Heger K, Heinz GA, et al. Roquin paralogs 1 and 2 redundantly repress the Icos and Ox40 costimulator mRNAs and control follicular helper T cell differentiation. Immunity (2013) 38:655–68. doi:10.1016/j.immuni.2012.12.004
106. Esnault S, Braun RK, Shen Z-J, Xiang Z, Heninger E, Love RB, et al. Pin1 modulates the type 1 immune response. PLoS One (2007) 2:e226. doi:10.1371/journal.pone.0000226
107. Vavassori S, Covey LR. Post-transcriptional regulation in lymphocytes: the case of CD154. RNA Biol (2009) 6:259–65. doi:10.4161/rna.6.3.8581
108. Zheng Y, Delgoffe GM, Meyer CF, Chan W, Powell JD. Anergic T cells are metabolically anergic. J Immunol (2009) 183:6095–101. doi:10.4049/jimmunol.0803510
109. Li M, Cao W, Liu H, Zhang W, Liu X, Cai Z, et al. MCPIP1 down-regulates IL-2 expression through an ARE-independent pathway. PLoS One (2012) 7:e49841. doi:10.1371/journal.pone.0049841
110. Scheu S, Stetson DB, Reinhardt RL, Leber JH, Mohrs M, Locksley RM. Activation of the integrated stress response during T helper cell differentiation. Nat Immunol (2006) 7:644–51. doi:10.1038/ni1338
111. Chen Z, Lin F, Gao Y, Li Z, Zhang J, Xing Y, et al. FOXP3 and RORγt: transcriptional regulation of Treg and Th17. Int Immunopharmacol (2011) 11:536–42. doi:10.1016/j.intimp.2010.11.008
112. Izquierdo JM, Majós N, Bonnal S, Martínez C, Castelo R, Guigó R, et al. Regulation of Fas alternative splicing by antagonistic effects of TIA-1 and PTB on exon definition. Mol Cell (2005) 19:475–84. doi:10.1016/j.molcel.2005.06.015
113. von Roretz C, Lian XJ, Macri A, Punjani N, Clair E, Drouin O, et al. Apoptotic-induced cleavage shifts HuR from being a promoter of survival to an activator of caspase-mediated apoptosis. Cell Death Differ (2013) 20:154–68. doi:10.1038/cdd.2012.111
Keywords: inflammation, stress granules, polysomes, mRNA stability, T cell maturation, thymic and peripheral tolerance
Citation: Ganguly K, Giddaluru J, August A and Khan N (2016) Post-transcriptional Regulation of Immunological Responses through Riboclustering. Front. Immunol. 7:161. doi: 10.3389/fimmu.2016.00161
Received: 31 January 2016; Accepted: 15 April 2016;
Published: 29 April 2016
Edited by:
Ivan Zanoni, University of Milano-Bicocca, ItalyReviewed by:
Roberto Spreafico, University of California Los Angeles, USAMaura Rossetti, University of California Los Angeles, USA
Copyright: © 2016 Ganguly, Giddaluru, August and Khan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Nooruddin Khan, bm9vciYjeDAwMDQwO3VvaHlkLmFjLmlu