 Nicholas A. Cacalano
Nicholas A. Cacalano
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 11 April 2016
Sec. Cancer Immunity and Immunotherapy
Volume 7 - 2016 | https://doi.org/10.3389/fimmu.2016.00128
This article is part of the Research Topic The two faces of a Killer View all 8 articles
Natural killer (NK) cells, key members of a distinct hematopoietic lineage, innate lymphoid cells, are not only critical effectors that mediate cytotoxicity toward tumor and virally infected cells but also regulate inflammation, antigen presentation, and the adaptive immune response. It has been shown that NK cells can regulate the development and activation of many other components of the immune response, such as dendritic cells, which in turn, modulate the function of NK cells in multiple synergistic feed back loops driven by cell–cell contact, and the secretion of cytokines and chemokines that control effector function and migration of cells to sites of immune activation. The signal transducer and activator of transcription (STAT)-3 is involved in driving almost all of the pathways that control NK cytolytic activity as well as the reciprocal regulatory interactions between NK cells and other components of the immune system. In the context of tumor immunology, NK cells are a first line of defense that eliminates pre-cancerous and transformed cells early in the process of carcinogenesis, through a mechanism of “immune surveillance.” Even after tumors become established, NK cells are critical components of anticancer immunity: dysfunctional NK cells are often found in the peripheral blood of cancer patients, and the lack of NK cells in the tumor microenvironment often correlates to poor prognosis. The pathways and soluble factors activated in tumor-associated NK cells, cancer cells, and regulatory myeloid cells, which determine the outcome of cancer immunity, are all critically regulated by STAT3. Using the tumor microenvironment as a paradigm, we present here an overview of the research that has revealed fundamental mechanisms through which STAT3 regulates all aspects of NK cell biology, including NK development, activation, target cell killing, and fine tuning of the innate and adaptive immune responses.
Natural killer (NK) cells are a critical component of the innate immune response: a first line of defense to protect the host against viral infection and the expansion of cancerous cells. Direct cytolytic activity against infected or transformed cells, on the basis of altered or absent MHC class I molecules (“missing self”), is only one of a number of functions performed by NK cells: they are also critical regulatory cells that interact with and modulate the activity of other components of the immune system, such as dendritic cells (DCs), monocytes/macrophages, endothelial cells, and T lymphocytes. Such regulatory functions are mediated through direct cell–cell interactions as well as via the secretion of immunomodulatory cytokines, which can edit and shape the repertoire of antigen-presenting cells (APCs) and impact the balance of T cell subsets during an adaptive immune response. As a result of this myriad of interactions, NK cells are key regulators of the inflammatory response and have emerged as important members of the innate lymphoid cell (ILC) family, unique lineages of immunomodulatory cells that develop from a distinct compartment within the common lymphoid progenitor population (1, 2).
Evasion of the immune system is one of the classic hallmarks of cancer (3, 4). Tumor cells rapidly evolve to become moving targets by modulating the expression of immunogenic proteins on their surfaces and by producing a host of soluble factors that repress both innate and adaptive immune responses. The critical role played by host defenses in tumor rejection is underscored by studies in both murine disease and gene knockout models of immune function as well as findings in human cancer patients. Specifically, the role of NK cells in early detection (immune surveillance) and elimination of cancerous cells has been demonstrated in many animal models, in which selective deletion of NK cells leads to the spontaneous development of cancer or failure to reject implanted tumor cells (5–8). Likewise, NK cells isolated from human cancer patients often display grossly defective surface marker profile, cytolytic activity, and cytokine production (9–19). Clinically, the critical role of antitumor immunity has been validated by marked advances in cancer therapy, which employ antibodies that target inhibitory immune checkpoints via the CD28–CTLA-4 and PD-1–PD-L1 ligand receptor systems. These novel therapies potentiate antitumor immunity mediated through CD8+ T cells as well as NK cells and have resulted in remarkably effective, durable antitumor immune responses (20–26). Like immune checkpoint inhibitors, therapeutics that target kinases and transcription factors also show great promise as cancer treatments by targeting both the tumor cells as well as components of host immunity. Mechanistically, the molecular basis for NK cell dysfunction in cancer patients is a highly complex phenomenon that integrates both direct effects on the NK cells as well as a range of cell–cell interactions and soluble factors that regulate NK activity. NK cells have become an attractive target for immunotherapy strategies as they are known to mediate direct tumor killing as well as exert a critical “helper” function for adaptive immune responses (27–30).
Unfortunately, therapeutic efforts to potentiate NK-mediated killing of tumor cells have met with little success. Several approaches, involving both in vivo and ex vivo methods to stimulate antitumor NK activity have been disappointing, largely due to (1) molecular evolution of tumors to promote an immunosuppressive microenvironment and (2) the complexity of NK biology and its multiple functions in both innate and adaptive immunity. NK cells are not just tumoricidal lytic machines, and their profound effects on targeting virus-infected cells, cancer stem cells, cytokine and chemokine regulation, and the differentiation of normal and cancerous tissue are only now being fully appreciated.
Specific subpopulations of NK cells, generally distinguished by a CD56bright/CD16− surface profile secrete critical cytokines that can promote the differentiation of normal and transformed tissues, which also impacts antitumor immunity and cancer progression (31). This small sublineage of NK cells can be found in the periphery and represents about 10% of the peripheral NK cells, but they can also be induced following the interaction with sensitive target cells in a process known as “split anergy” (32–38). Thus, NK cells are not only cytotoxic cancer killers but also drive activation and maturation of DCs and T cells, and can induce the differentiation of normal and cancerous stem cells through the secretion of cytokines such as IFN-γ. The finding that cancer stem cells are resistant to chemotherapy and radiation, but are highly sensitive to NK-mediated cytolysis, has suggested that NK cells can be manipulated to target this key population of cells in vivo, either by direct NK-mediated killing or by cytokine-driven differentiation to a population that no longer has self-renewal capacity. A key to reprograming NK cells to effectively kill tumors or induce the differentiation of cancer stem cells, however, critically depends upon our understanding at the molecular level how cancers and the tumor microenvironment repress host anticancer immunity.
Here, we review current advances in the understanding of how the ubiquitously expressed transcription factor signal transducer and activator of transcription (STAT)-3 regulates NK cell biology at multiple levels, including activation of cytolytic function, cytokine-mediated effects, and interactions with other components of the immune system. STAT3 is known to limit all aspects of the NK response, but in this review, we focus on the effects of STAT3 in the context of immune surveillance of cancer and modulation of NK activity in the tumor microenvironment.
Signal transducer and activator of transcription-3 is a downstream effector of multiple cytokines and growth factors that modulate NK cell activity and also drives tumorigenesis and resistance of cancer cells to therapy. It is becoming clear that any efforts to block STAT3 activity in cancer cells are likely to have synergistic and/or deleterious off-target effects on host immune responses mediated through NK cells. It is thus critical to understand the effects of STAT3 signaling on NK activity in order to apply STAT3-targeted therapies in the most effective way.
Signal transducer and activator of transcription-3 is part of a family of cytokine and growth factor-activated transcription factors, which includes STAT1, STAT2, STAT4, STAT5a/b, and STAT6, which are signaling proteins, targets of phosphorylation by Jak and receptor tyrosine kinases (RTKs), as well as transcription factors whose effects on gene expression profoundly impact the immune response, the outcome of viral infections, embryonic development, and cancer biology (39–43). STAT3 is activated by a wide range of cytokines, in particular those that recognize gp130, such as IL-6, IL-11, oncostatin M (OSM), leukemia inhibitory factor (LIF), and ciliary neurotrophic factor (CNTF), as well as cytokines that engage the common gamma (γc) chain, including IL-2, IL-4, IL-7, IL-9, and IL-15. STAT3 is also activated by RTKs, such as the epidermal growth factor receptor (EGFR) and VEGFR, cytoplasmic kinases, such as Src, as well as some cancer therapies, such as ionizing radiation (IR). STAT3 signaling is controlled by several endogenous feedback inhibitors, such as the suppressors of cytokine signaling (SOCS), protein inhibitors of activated STATs (PIAS), and phosphatases such as SHP-2, which dephosphorylates and inactivates cytokine receptor chains (39–43).
In the context of tumor biology, STAT3 is often constitutively activated and phosphorylated in a wide range of epithelial and hematopoietic cancers, largely via paracrine or autocrine stimulation by STAT3-activating cytokines and growth factors. STAT3 drives tumor cell survival and proliferation, epithelial-to-mesenchymal transition (EMT), metastasis, and resistance to chemotherapy and radiation (44–54). In recent years, previously unappreciated critical functions of STAT3 in anticancer immune responses have been revealed in both lymphoid and myeloid lineages. Thus, STAT3-targeted cancer therapies may have a dual function: direct cytostatic or cytotoxic effects on tumor cells and stimulation of cancer destruction and clearance by the immune system. However, the role of STAT3 in anticancer immunity is complex and involves multiple cell lineages, including NK cells. STAT3 targeting in tumors may therefore also have unanticipated, deleterious effects on tumor immunity. Below, we discuss the impact of STAT3 signaling on NK activation and function in the tumor microenvironment.
Several studies have shown that tumor-infiltrating lymphocytes and myeloid cells display constitutive STAT3 phosphorylation, including T cells, DCs, neutrophils, macrophages, and NK cells, which is often driven by the production of cytokines and growth factors by both tumor cells and the infiltrating cells themselves, such as IL-6, IL-10, VEGF, and HGF, among others (55). STAT3 can induce an immunosuppressive tumor microenvironment with both direct and indirect effects on NK cells.
To address the role of STAT3 activation in the hematopoietic compartment, Kortylewski et al. used an inducible system to delete the STAT3 gene in bone marrow-derived cells (55–58). Mice expressing a floxed STAT3 gene in combination with Cre driven from the interferon-inducible Mx1 promoter were treated with poly I:C to induce interferon and activate STAT3 gene deletion in hematopoietic cells. Targeting STAT3 in the bone marrow did not have any effect on NK cell development in vivo; overall numbers and cytotoxic activity toward standard YAC-1 lymphoma targets in the knockout mice were comparable to WT animals. However, following tumor challenge with the B16 melanoma cell line, splenic NK cells from STAT3-targeted mice displayed enhanced cytolytic activity compared to WT tumor-bearing mice. Further, subcutaneous growth of B16 melanoma cells or the MB49 bladder cancer cell line was virtually abolished in STAT3-targeted mice, suggesting that STAT3 exerts a cell-autonomous immunosuppressive effect in hematopoietic cells in the context of an antitumor response.
To exclude the possibility that tumor rejection in STAT3−/− mice was due to off-target effects of STAT3 deletion on non-hematopoietic tissues, tumor growth studies in lethally irradiated WT mice reconstituted with either STAT3+/+ or STAT3−/− bone marrow cells were performed, and it was found that growth of B16 or MB49 tumors was completely blocked in mice reconstituted with STAT3−/− bone marrow cells. Although antibody depletion experiments showed that T cells played a critical role in tumor rejection, NK cells were also involved, as NK depletion using anti-asialo-GM1 antibodies partially abrogated the effects of a STAT3 small molecule inhibitor (CPA-7) on rejection of MB49 tumors.
In order to determine the contribution of STAT3 to NK-specific antitumor responses, Gotthardt et al. generated a targeted STAT3 deletion specifically in the NK compartment, which resulted in increased immunosurveillance in murine melanoma and leukemia models (59). In this study, genetic ablation of STAT3 was accomplished using STAT3fl/fl mice crossed to mice expressing Cre driven from the NKp46 natural cytotoxicity receptor (NCR)-1 promoter, which deleted STAT3 exclusively in NK cells (STAT3fl/fl × Ncr1-iCreTg), sparing other hematopoietic lineages. In these mice, NK cell development, proliferation, and numbers were normal; NK cells at all stages of development are present in numbers similar to WT mice, and expression of key NK developmental and functional markers, such as NK1.1, NKp46, CD27, CD11b, NKG2D, as well as inhibitory receptors, such as LY49A, C, G2, and I, KLRG1 and NKG2A, were identical to NK cells from STAT3+/+ animals as well as STAT3fl/fl–Mx1-Cre mice. Some differences were noted, however, as the numbers of DNAM-1(CD226)+ NK cells were increased in STAT3-deficient mice compared to WT. This finding is intriguing, as DNAM-1 is a cell surface receptor that mediates NK cell cytolytic activity by engaging the CD155 (PVR) and CD112 (Nectin-2) ligands on target cells (60–65). In addition, the levels of two key mediators of NK-dependent cytotoxicity, granzyme B, and perforin were elevated at the protein level in STAT3-deficient NK cells, supporting the idea that STAT3 negatively regulates NK activity. In the B16F10 melanoma model, the number and size of metastatic lung nodules was greatly reduced in mice with STAT3-deficient NK cells, following intravenous injection of tumor cells. This was accompanied by an increase in the percentage of DNAM-1+ circulating NK cells in the STAT3-targeted mice and enhanced in vitro cytotoxicity toward B16F10 tumor targets. B16F10 cells express high levels of the DNAM-1 ligand CD155 and thus enhanced lysis of B16F10 tumor cells by STAT3−/− NK cells was abrogated by treatment with a DNAM-1 blocking antibody. It is of note that the authors found no evidence for enhanced in vitro cytotoxicity of STAT3-deficient NK cells against other targets, such as YAC-1 and Rma cells, which express low levels of CD155, suggesting that blocking STAT3 activity in NK cells may only increase DNAM-1/CD155-dependent killing. STAT3-deficient NK cells were also more efficient at killing leukemic cells in vivo. The size of subcutaneous tumors following injection of v-abl-transformed cells was greatly reduced in mice with the NK-specific STAT3 knockout. Consistent with the data obtained with subcutaneous tumors, it was shown that targeting STAT3 also prolonged the survival of mice injected intravenously with v-abl-transformed cells or infected with a v-abl-encoding retrovirus.
Intrinsically, STAT3 activation in NK cells often appears to have a negative regulatory role, as deletion of STAT3 in the NK compartment generally results in improved antitumor immunity. However, STAT3 activation in tumor cells, as well as in other components of the immune response, can provide feedback regulation of NK cells in an indirect manner as well. The antitumor activity and cytokine profile of NK cells depends on a complex interplay among activating and inhibitory cell surface receptors as well as a cocktail of soluble factors. The integration of multiple signals regulates the migration of NK cells to the tumor microenvironment, NK-mediated recognition of target cells, and downstream signaling that controls lytic and helper functions. STAT3 has been shown to play a role in all of these processes.
Tumor cells often constitutively produce survival or growth factors that enhance tumorigenesis, resistance to therapy, EMT, and metastasis (47, 57, 58, 66). Activated STAT3 in tumor cells impacts anticancer immunity as well, due to its ability to regulate cytokine and chemokine expression by the tumor as well as the expression of growth factors and surface markers on infiltrating immune cells. It has been known for some time that constitutive STAT3 activation in tumor cells can regulate their susceptibility to NK-mediated cytolysis and the infiltration and migration of NK cells within the tumor microenvironment. Constitutive STAT3 phosphorylation in tumor cells is usually driven by the overproduction of key cytokines and growth factors, such as IL-6, IL-10, EGF, HGF, Her2/Neu, and VEGF, or their receptors, as well as aberrantly activated cytoplasmic kinases, such as Src, among others (57). STAT3 activation in tumor cells can regulate their sensitivity to NK-mediated killing by altering the expression of NK-activating ligands on the cell surface. In addition, STAT3-driven cytokine expression in tumors induces the secretion of immunosuppressive cytokines and represses proinflammatory cytokine expression, leading toward a dampening of immune responses in the tumor microenvironment. The relationship between STAT3 activation in tumor cells and components of the innate and adaptive immune responses is complex and multifactorial.
A key proximal event in antitumor immunity is the migration of lymphoid and myeloid cells to the tumor microenvironment. NK cells depend on chemotactic factors secreted by tumor cells, stroma, and other lymphoid and myeloid cells to reach the site of action. While proinflammatory signaling promotes the migration of NK cells to the microenvironment, continued proinflammatory “costimulatory” signals trigger NK-mediated cytokine production and lytic function. Major factors that control NK cell migration to the tumor microenvironment are chemokines and cytokines. NK cells express a range of receptors for soluble factors and thus can respond to a number of chemotactic factors, such as TNF-α, IL-12, RANTES, macrophage chemotactic protein (MCP)2–3, IL-2, MIP1alpha, IL-8, CXCL9, CXCL12, CCL2 (MCP-1), CCL3, CX3CL1 (fractalkine), CXCL10, CXCL12, CCL21 (SLC), and CCL19 (ELC) (67–71). Dysregulated NK migration to the tumor microenvironment has been suggested by studies that found low numbers of NK cells associated with tumors in colorectal cancer (72). Further, experimental models of cancer have shown that gene therapy with CCL2 and CX3CL1/fractalkine can stimulate tumor rejection by increased infiltration and activation of NK cells (73–76).
Burdelya et al. demonstrated that several NK chemotactic factors secreted by tumor cells are regulated by STAT3 in the B16 and K1735 murine melanoma models (77). It was shown that increased STAT3 activity of K1735 subclones correlated to the decreased lymphocyte infiltration of subcutaneous tumors. Treatment of tumor cell lines with a platinum-containing STAT3 inhibitor (CPA-7) or expression of dominant-negative STAT3 (STAT3β) splice variant resulted in increased secretion of chemoattractants, such as RANTES, IFN-γ-inducible protein (IP-10), MCP-1, MIP-2, and T cell activation (TCA)-3. In vivo alteration of the cytokine and chemokine profile induced migration of T cells, NK cells, neutrophils, and macrophages to the tumor microenvironment.
Similarly, Ihara et al. demonstrated similar effects of STAT3 targeting in a carcinogen-driven model of non-small cell lung carcinoma (NSCLC) (78). Using a lung-specific, tetracycline-inducible Cre-lox system to delete STAT3, it was found that the growth of urethane-induced tumors in the lungs of STAT3-targeted mice was significantly reduced relative to tumors generated in WT mice. Cell autonomous and non-immune effects of STAT3 deletion in tumor cells were excluded, as markers of proliferation, angiogenesis, and apoptosis were similar in tumors from WT or STAT3−/− mice. In contrast, numbers of lymphoid and myeloid cells in bronchoalveolar lavage fluid were increased in the STAT3−/− mice, as were the levels of proinflammatory cytokines such as IFN-γ, TNF-α, and IL-6. Microarray analysis of the tumors revealed that targeting STAT3 increased the expression of multiple chemokines, such as CCL2, CCL9, CCL12, CCL17, and CCL21c, and induced downregulation of MHC class I molecules H2-D1 and H2-K1 in the tumor cells. These results indicated that STAT3 inhibition can increase NK cell migration to the tumor microenvironment and enhance tumor cell sensitivity to NK-mediated lysis. We have recently shown that knock down of key proteins in tumor cells and non-transformed healthy cells drives dedifferentiation of tumors and results in the activation of NK cell function (37).
On the other hand, some studies have shown that STAT3 can enhance NK-mediated immunosurveillance, specifically in a model of BCR/Abl-dependent leukemia. Putz et al. measured subcutaneous tumor formation in bone marrow cells transformed by BCR/Ablp185 or v-Ablp160 (79). STAT3 was targeted by interferon treatment of STAT3fl/fl–Mx1-Cre bone marrow cells or by transfecting with a STAT3-specific shRNA lentiviral construct. STAT3-targeted tumors grew larger than WT tumors, with a more invasive phenotype and a higher mitotic index. Cytokine profiling revealed lower levels of proinflammatory mediators IL-6, IFN-γ, IL-17, and CCL5 produced by STAT3-deficient tumors relative to WT controls. Numbers of infiltrating NK cells, as well as CD4+ and CD8+ T cells, were significantly reduced in the tumor microenvironment, with no change in the numbers of Gr1+/CD11b+ MDSCs. STAT3-targeted tumor cells were more resistant to lysis by purified primary NK cells in vitro, and the growth differences between STAT3-targeted and WT tumors in vivo were abrogated in mice lacking NK cells. This study supports previous work implicating NK cells as the major lymphoid population responsible for eradication of BCR/Abl-driven leukemias and suggests that STAT3-targeting strategies may severely impair NK-mediated antitumor immune responses in BCR/Abl leukemias. These conflicting findings indicate that STAT3-targeting strategies in cancer can result in either enhancement or inhibition of anticancer immunity, depending on context and tumor type.
A critical mechanism of NK-mediated recognition of target cells is via the NKG2D NK-activating receptor (80–90). NKG2D is a transmembrane receptor expressed on the surface of NK cells, which physically associates with the DAP10 signaling chain in humans. The fully functional complex is a hexamer, with one NKG2D homodimer physically associated with two Dap10 homodimers. Recognition of NKG2D ligands, which include the MHC class I polypeptide-related chain (MIC)A, MICB, and members of the Rae-1, ULBP, Mult1, and H60 families, induces phosphorylation of a Y–X–X–M motif on the cytoplasmic tail of DAP10, and activates NK degranulation and killing of target cells. NKG2D engagement triggers both PI-3 kinase and MAPK signaling pathways and can override inhibitory signals via MHC class I recognition by killer inhibitory receptors (KIRs). The importance of NKG2D-mediated elimination of tumor cells is underscored by findings in NKG2D gene-targeted mice. In animals lacking NKG2D receptors, NK cell development is normal, but immune surveillance of epithelial and lymphoid cancers is defective (91).
The MICA NKG2D ligand is a stress-induced cell surface molecule that is upregulated in response to genotoxic stimuli, such as chemotherapy or radiation, as well as chromatin disruption and heat shock (89–91). The importance of MICA/NKG2D engagement in the control of cancer has been demonstrated by several studies showing that increased soluble MICA protein in the serum of cancer patients leads to downregulation of NKG2D on NK cells and correlates to poor prognosis (92). STAT3 has been shown to regulate NK lytic activity via transcriptional effects on NK recognition receptors and activating NK ligands on tumor cells.
Bedel et al. demonstrated that pharmacologic or genetic inhibition of STAT3 in a human colorectal cell line (HT29) resulted in increased NK cell degranulation (CD107a expression) and IFN-γ production in an in vitro coculture assay (93). HT29 is highly resistant to NK-mediated lysis, which correlates to high levels of constitutive STAT3 phosphorylation and protein expression relative to NK-sensitive human tumors. Targeting STAT3 in HT29 with siRNA increased IFN-γ production of freshly isolated human NK cells when cocultured with HT29. Pretreatment of cocultures with anti-NKG2D antibody inhibited IFN-γ secretion and NK-mediated cytotoxicity of STAT3-targeted HT29. The effect of anti-NKG2D antibodies was attributed to increased expression of NKG2D ligands MICA and ULBP2 in STAT3-deficient HT29 cells; MICA expression was increased approximately ninefold, following treatment HT29 with a pharmacologic inhibitor of STAT3 (STA21). STAT3 inhibition increased MICA expression in human GBM and breast cancer cell lines as well, and STAT3 was shown to bind directly to a putative STAT-binding site in the MICA promoter. Pharmacologic STAT3 inhibition increased MICA promoter activity, and expression of a constitutively active STAT3 mutant (STAT3C) blocked MICA expression in a human stromal cell line, demonstrating that STAT3 acts as a MICA transcriptional repressor in this system.
Likewise, Fionda et al. demonstrated a molecular link between glycogen synthase kinase (GSK)-3, STAT3, and MICA regulation in multiple myeloma (MM) (94). It was found that pharmacologic inhibition of GSK-3 downregulated MICA expression and increased the sensitivity of human MM cell lines to NK-mediated cytotoxicity. The effect of GSK-3 inhibition was dependent on downregulation of STAT3 phosphorylation and reduced STAT3 binding to the MICA promoter. It was also shown that expression of a constitutively active STAT3 mutant repressed MICA expression in MM cell lines and reversed the effect of GSK-3 inhibitors in MM.
In contrast, some STAT3-activating stimuli can increase NKG2D expression on NK cells in both mice and humans. Zhu et al. have shown that IL-10 and IL-21 can induce high levels of NKG2D, DAP10, and DAP12 expression on the surface of primary human NK cells, and small-molecule STAT3 inhibitors downregulated NKG2D expression on primary human NK cells from healthy donors (95). Likewise, STAT3-deficient NK cells from gene-targeted mice displayed lower NKG2D surface expression than WT controls. The in vivo relevance of STAT3-mediated NKG2D regulation was supported by the finding that NK cells from patients with inactivating STAT3 mutations in conditions, such as hyper IgE syndrome (HIES), displayed lower NKG2D surface expression relative to normal NKs.
In agreement with these studies, IL-21 was shown to augment NKG2D-dependent killing in several murine tumor models (96). Interleukin-21 pretreatment significantly extended survival of mice in a lymphoma model and blocked tumor metastasis in murine models of breast, lung, and prostate cancer. The protective effects of IL-21 were reversed upon treatment of mice with anti-NKG2D antibodies or NK-depleting anti-NK1.1 antibodies. These studies suggest that IL-21-driven STAT3 activation regulates NKG2D expression, although other IL-21-dependent pathways, such as ERK MAP kinase and PI-3 kinase/Akt, are likely to also play a role.
On the other hand, Burgess et al. have shown that IL-21 can inhibit the expression of NKG2D and the adapter/signaling protein DAP10 on IL-2-activated primary human NK cells (97). Interestingly, IL-21 inhibited DAP10 gene promoter activity and reduced surface expression of NKG2D but increased the NK NCR NKp30 and 2B4, suggesting that IL-21 can reprogram NK specificity by editing the recognition receptor repertoire on the cell surface. The different effects of IL-21 in these studies might be explained by the complex interplay between STAT molecules. Interleukin-21 can activate both STAT1 and STAT3, which exert opposing effects in T cells, in particular on the expression of different STAT-dependent genes such as IFN-γ. Thus, it is possible that whether STAT3 has a positive or negative effect on NKG2D or DAP-10 expression depends on the STAT1/STAT3 balance at the time of stimulation (98).
Taken together, STAT3 targeting of tumors may promote NK-mediated cytotoxicity by increasing MICA expression on target cells but can have both positive and negative effects on NK-mediated recognition of tumors by modulating the expression of NK-activating receptors. These conflicting data indicate that the role of STAT3 in the regulation of NK ligands and receptors likely depends on cellular context, the balance between STAT1 and STAT3 activation, method of stimulation, and assay conditions used in the studies.
The programmed cell death (PD)-1 (CD279)/PD-L1 (B7-H1 and CD274) immune checkpoint pathway has been the focus of intense investigation and clinical application in the last several years. Inducibly expressed on activated T cells, PD-1 is considered a marker of lymphoid cell “exhaustion” or chronic stimulation. Engagement of PD-1 on T cells via its ligands PD-L1 or PD-L2 on the surface of lymphoid cells, macrophages, IFN-stimulated epithelial cells, DCs, or tumor cells blocks T cell receptor (TCR) signaling on cytolytic CD8+ cells via the recruitment of the SHP-2 phosphatase and dephosphorylation of the TCR complex (99–106). Tumor cells from various tissues upregulate PD-L1 expression, which inhibits T cell-mediated antitumor immunity. Much is known of PD-1 signaling in T cells, and remarkable, durable antitumor immune responses have been achieved in melanoma and lung cancer using therapeutic antibodies that block PD-1/PD-L1 engagement (107–115). PD-1/PD-L1 blockade, as a monotherapy or in combination with IR and epigenetic targeting, has shown great promise as a novel emerging cancer treatment and is currently being tested clinically for several other solid and hematopoietic cancers (116–119). Although PD-1 is known to be upregulated on NK cells, its role in the regulation of NK-mediated immune surveillance and tumor killing is poorly understood.
At the molecular level, novel high-resolution, high-throughput imaging studies have demonstrated that PD-1 on the surface of NK cells is situated within the immunologic synapse upon engagement with a target cell. This work provides evidence that PD-1 is physically associated with the cytolytic signaling machinery, with access to NK activating receptors and provides evidence that the composition of the NK immunologic synapse can promote PD-1-mediated dephosphorylation of NK recognition structures and blunt lysis of tumor cells (120).
Marzec et al. have demonstrated that NPM/ALK-transformed T cell lymphoma cells escape immune destruction via induction of PD-L1. PD-L1 expression was STAT3-dependent and STAT3 bound directly to the PD-L1 promoter, which has four putative STAT-binding sites (121). Further, Benson et al. have shown that NK cells from MM patients express PD-1 at a much higher level than NK cells from normal donors (122). The functional significance of PD-1 expression was demonstrated by increased NK-mediated lysis of autologous MM cells following treatment with an anti-PD-L1-blocking antibody (CT-011). The authors also suggest that the immunomodulatory effects of the therapeutic lenalidomide can, in part, be explained by its ability to downregulate PD-L1 expression on primary MM cells, leading to increased NK-mediated lysis and synergy with CT-011. Thus, in at least some tumor models, NK-mediated immune surveillance can be inhibited by STAT3-dependent increases in PD-L1 expression. It is also of interest to note that most highly differentiated tumors express PD-L1 and are found to be resistant to NK cell-mediated cytotoxicity (37). Figure 1 and Table 1 summarize the direct effects of STAT3 activation on the expression of NK activating receptors as well as inhibition of cytotoxicity and migration by downregulation of NK ligands and chemotactic factors in tumor cells.
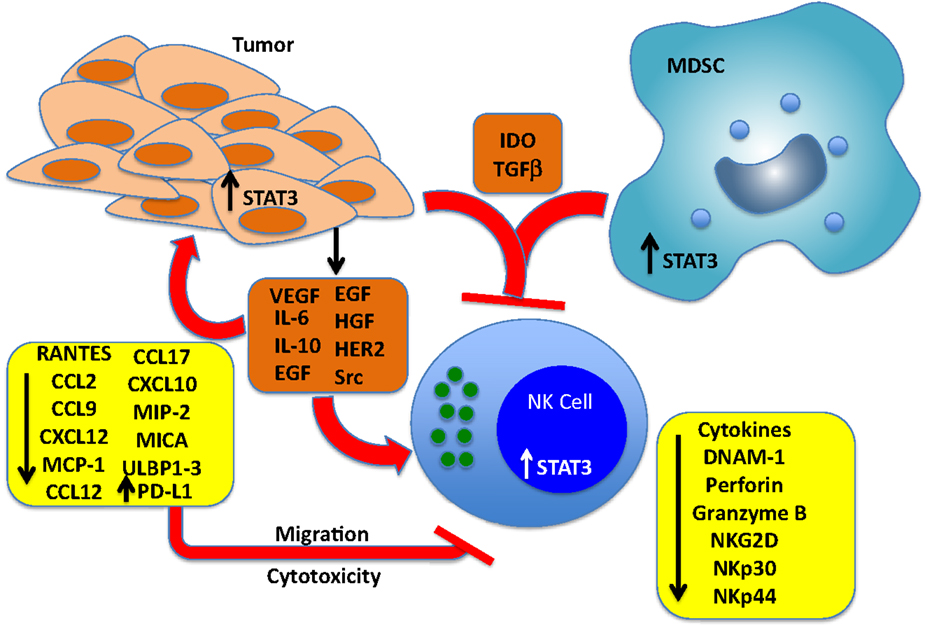
Figure 1. Effects of cancer cell-derived factors that drive STAT3 activation in tumors, NK cells, and myeloid-derived suppressor cells (MDSCs). Tumors produce multiple soluble factors that activate STAT3 in cells of the tumor microenvironment. Among the direct effects of STAT3 activation in NK cells include blunting the expression of key NK activating receptors DNAM-1 NKp30, NKp44, and NKG2D, and inhibition of perforin A and granzyme B expression, which blunts cytotoxic activity. STAT3 activation in tumor cells represses the expression of NK chemotactic factors, which reduces the recruitment of NK cells to the tumor microenvironment, and blunts the expression of NKG2D ligands MICA and ULBP1/2/3, which renders the cells resistant to NK-mediated killing. STAT3 has also been shown to increase the levels of PD-L1, which can engage PD-1 expressed on NK cells and inhibit cytotoxic responses. Finally, IDO and TGF-beta produced by both tumor cells and MDSCs block NK cell development, proliferation, and activation.
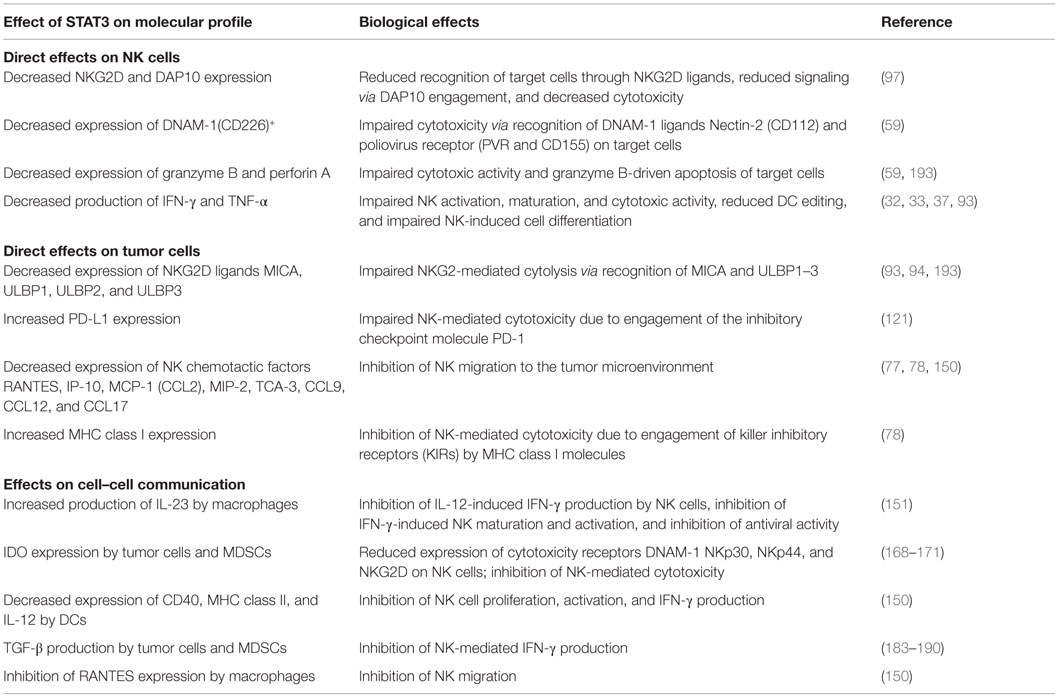
Table 1. Summary of STAT3-mediated regulation of NK function.
Perhaps the most important (and complex) role for STAT3 in NK function is as a regulator of cell–cell communication within the tumor microenvironment. STAT3 controls numerous indirect effects on NKs through cytokine secretion by tumor cells, developmental regulation of DCs, as well as chemokine and cytokine production by DCs, macrophages, and MDSCs, all of which exert feedback controls on NK cell migration, activation, and lytic activity (4, 123–129).
Mutually activating, “helper” interactions between NK cells and DCs have been shown to promote the functional capabilities of both cell types as well as inducing the differentiation of CD4+ and CD8+ T cells, while downregulating immunosuppressive lineages such as Tregs and myeloid-derived suppressor cells (MDSCs) (124, 130–132). Interleukin-18, produced by epithelial cells and macrophages, “primes” NKs responses to DC-derived IL-12 and activates IFN-γ production. Reciprocally, activated NKs produce potent chemoattractants for immature DCs, CCL3, and CCL4, which induce DC migration to the tumor site and drive DC maturation. Interferon gamma and TNF-α produced by activated NK cells can induce polarization of DCs, and NKs also target and kill tolerogenic, immature autologous DCs, increasing the ratio of mature:immature DCs. Thus, NK cells can not only provide helper function to DCs but can also help shape the DC repertoire toward an immunogenic phenotype through a mechanism known as “dendritic cell editing” (28, 125, 133, 134). Inhibition of NK cell function via STAT3 activation can therefore skew the DC lineage toward an immature, immunosuppressive phenotype. Likewise, disruption of DC maturation or function can impair NK cell-mediated killing and cytokine production. On the other end of the spectrum, MDSCs can negatively regulate NK cell function through multiple secreted factors, some of which are, at least in part, STAT3 dependent (Figure 2).
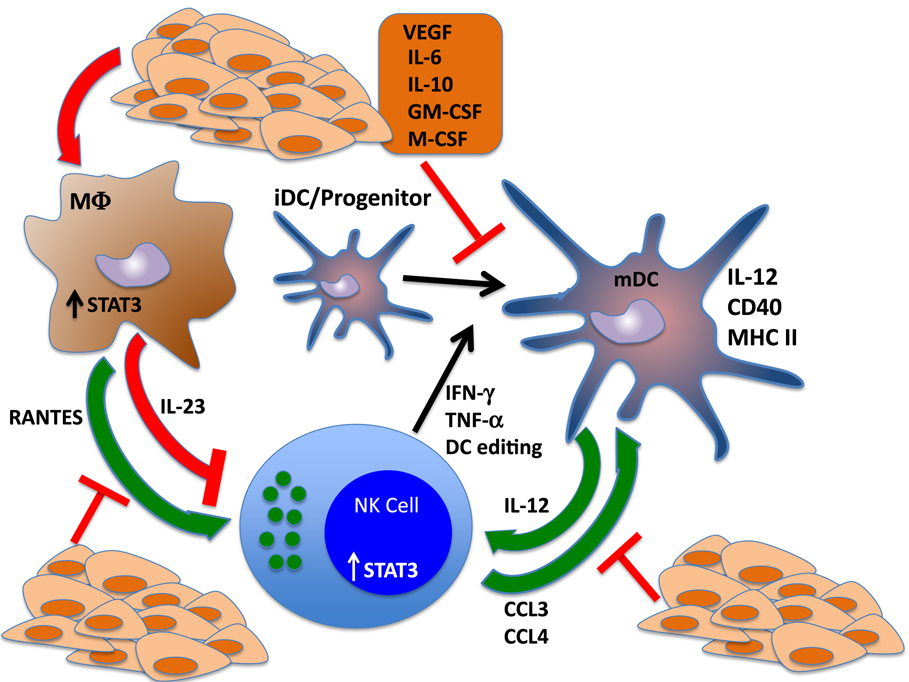
Figure 2. STAT3 regulates reciprocal interactions between NK cells and other components of the immune response. NK cells interact with dendritic cells (DCs) in regulatory loops that amplify the activation and function of each cell type. NK cells produce cytokines that drive the differentiation and polarization of DCs to mature, fully functional antigen-presenting cells. Further, NK cells produce potent chemotactic factors CCL3 and CCL4, which recruit DCs to the tumor microenvironment and can shape the DC repertoire via cytotoxic elimination of tolerogenic, immature DCs. These functions drive the adaptive immune response by potentiating DC-mediated activation of T cells. Reciprocally, DCs drive NK cell maturation through the production of IL-12. Tumor cell production of cytokines that trigger STAT3 in NK cells and DCs send the antitumor immune response into a tailspin by blocking NK-mediated DC editing and polarization, which inhibits IL-12 release and further impairs NK cell activation and function. STAT3 activation of tumor-associated macrophages also inhibits the immune response by inducing the production of the immunosuppressive cytokine IL-23 and blocking the secretion of NK chemotactic factor RANTES.
Several studies have shown that STAT3 activation can disrupt interactions between immunocompetent cells in the tumor microenvironment and induce an immunosuppressive milieu. Blocking STAT3 activation in murine tumor models alters the cytokine profile of tumor cells and tumor-infiltrating hematopoietic cells, and re-establishes proinflammatory, tumoricidal activity.
Early studies in cancer patients and murine tumor models revealed that DCs from the tumor microenvironment were defective in antigen presentation. In particular, it was shown that DCs grown from bone marrow precursors of tumor-bearing mice were efficient APCs, but mature splenic DCs from the same mice were defective, suggesting that tumors produced soluble factors that inhibited DC development (135–141). Likewise, several groups found reduced numbers of peripheral DCs in patients with cancers of the kidney, skin, and colon. DCs from cancer patients and animal models were defective in antigen presentation function and failed to effectively induce the B7-1 and B7-2 costimulatory molecules (142, 143). Cancer patients were also shown to produce increased numbers of LIN−/HLA-DR− immature myeloid cells in the peripheral blood, indicating defective myelopoiesis induced by tumor factors.
Gabrilovich et al. demonstrated that growth factors produced by tumor cells, in particular vascular endothelial growth factor (VEGF), IL-6, IL-10, macrophage colony stimulating factor (M-CSF), and granulocyte–macrophage colony stimulating factor (GM-CSF), induced severe developmental defects in DCs as well as general dysfunction of myelopoiesis (144–147), and later studies showed that these effects can be traced to STAT3 activation in myeloid progenitors. Supernatants from human breast and colon adenocarcinoma cell lines impaired the differentiation of human CD34+ hematopoietic stem cells (HSCs) to functional DCs but did not affect the total number of multipotent stem cells or the antigen presentation function of mature DCs from the periphery, demonstrating a highly specific effect of secreted factors on DC differentiation. The inhibitory effect of the tumor supernatants was reversed by preincubation with anti-VEGF neutralizing antibodies and partially overcome with blocking antibodies specific for M-CSF.
Nefedova et al. subsequently demonstrated that conditioned medium from the murine CT26 colon carcinoma or C3 sarcoma cell lines could trigger Jak2/STAT3 signaling in hematopoietic progenitor cells (HPCs) and resulted in the abnormal accumulation of Gr1+/CD11b+ immature myeloid cells in an in vitro DC differentiation assay (148, 149). Likewise, CD11c+/IAd+ immature DCs, differentiated in the presence of tumor supernatants, displayed much higher STAT3 DNA binding activity compared to control DCs and were impaired in their ability to stimulate T cell proliferation in an allogeneic mixed lymphocyte reaction (MLR). Expression of a dominant-negative STAT3 mutant in HSCs reversed the effects of tumor supernatants. Conversely, expression of a constitutively active STAT3 mutant resulted in the accumulation of Gr-1+/CD11b+ immature myeloid cells that poorly differentiated into DCs in vitro, demonstrating the causative role of STAT3 in defective DC development.
Wang et al. have shown that in three murine cancer models with constitutive STAT3 activation, B16 (melanoma), SCK-1 9 (breast carcinoma), and CT26 (colon carcinoma), expression of a dominant-negative STAT3 variant, STAT3β, or a STAT3-targeting siRNA increased the levels of the proinflammatory cytokines IFNβ, TNF-α, and IL-6, as well as the chemokines RANTES and CXCL10 (IP-10) in the tumor cells, with no effect on IL-4 and IL-10 production (150). Furthermore, conditioned media from STAT3β-expressing B16 tumor cells induced proinflammatory mediators nitric oxide (NO) and RANTES in peritoneal macrophages and TNF-α in neutrophils. STAT3-targeted B16 tumors in vivo also contained higher levels of infiltrating macrophages, neutrophils, and lymphocytes compared to tumors derived from the B16 parental cell line.
Conditioned medium from STAT3-targeted B16 melanoma and CT26 colon carcinoma cells also altered the surface phenotype of DCs, inducing higher expression levels of DC activation and maturation markers IL-12, CD40, and MHC class II molecules. In vivo, STAT3β+ B16 and CT26 tumor cells triggered DC activation and enhanced infiltration of tumor-specific CD8+ T cells. In reciprocal experiments, constitutive STAT3 activation in transformed cells induced the production of secreted factors that inhibited DC maturation, reduced the numbers of CD11c+, MHC class II+ cells, and blocked IL-12 production in an in vitro DC differentiation assay.
The findings from these studies indicate that STAT3 activation is at the apex of a cascade, beginning in tumor cells, which determines the cytokine profile of the cancer and triggers further STAT3 activation in lymphoid and myeloid populations within the tumor microenvironment, promoting immunosuppression and tumor escape. STAT3-driven inhibitory effects on the production of chemokines and inflammatory mediators by tumor cells, inhibition of DC maturation, and the production of IL-12 can block NK activity within the tumor microenvironment (Figure 2).
Signal transducer and activator of transcription-3 signaling has been shown to regulate the cytokine balance and functional interactions between NKs and other hematopoietic cells in the tumor microenvironment. In particular, STAT3 regulates the levels of two key cytokines, IL-12 and the procarcinogenic IL-23, which are related cytokines that share a common ligand chain (IL12p40), as well as a common receptor chain (IL-12Rb1), but exert opposing effects on antitumor immunity (151). Both cytokines are produced primarily by macrophages and DCs, but they differ critically in their signaling properties. Interleukin-12 is a STAT4 activator, while IL-23 is a potent STAT3 inducer. There is also evidence that IL-12 and IL-23 cross inhibit each other during an inflammatory response (152, 153). Interleukin-12 is a critical cytokine that induces the maturation and functional activation of NK cells; it has been shown that NK cell differentiation and effector function are defective in IL-12- and IL-12 receptor-deficient mice, and patients with IL-12/IL-12R mutations are immunodeficient (154–158). In the context of an antitumor immune response, administration of recombinant IL-12 has antitumor effects, and IL-12 knockout mice were found to be more susceptible to ultraviolet light (UV)-induced tumors and skin papillomas (159). Conversely, Langowski et al. have shown that mice with a targeted deletion of the IL-23p19 subunit are completely resistant to TPA-induced skin papillomas, demonstrating a tumor promoting function for IL-23 (160, 161).
A recent study has shown that STAT3 signaling controls the IL-12:IL-23 ratio in the tumor microenvironment (151). When subcutaneously grown B16 melanoma tumors were dissociated and tumor-associated myeloid cells purified, it was found that tumor-infiltrating CD11b+ c-macrophages secreted high levels of IL-23, whereas tumor-associated DCs (CD11b+c+) cells produced very little IL-23. Conversely, neither macrophages nor DCs within the B16 tumor microenvironment secreted significant amounts of IL-12. When STAT3 expression was ablated in the hematopoietic compartment using the STAT3fl/fl–Mx1-Cre system, the levels of macrophage-derived IL-23 was greatly reduced and tumor-associated DCs secreted increased amounts of IL-12. STAT3 was shown to be a direct transcriptional activator of the IL-12p19 gene, and IL-23 in the tumor microenvironment inhibited IL-12 production by tumor-associated DCs. STAT3-dependent reciprocal regulation of IL-23 and IL-12 was demonstrated in melanoma (B16), bladder (MB49), and colon (MB38) cancer models and targeting STAT3 in reduced tumor growth in vivo.
These studies demonstrate that the ability of the immune system to reject a tumor depends critically on a delicate balance of secreted factors and cell–cell interactions. Altering these conditions through aberrant STAT3 activation can establish an immunosuppressive circuit between tumor cells and infiltrating macrophages, DCs and NK cells, driven by STAT3 activation in all three subpopulations. Immunosuppressive reprograming of the tumor microenvironment results in a negative feedback loop, wherein STAT3 activation in NK cells inhibits direct tumor cell cytolysis and DC maturation by inhibiting NK-dependent IFN-γ and TNF production and blocking the release of antigenic proteins by NK-mediated lysis of tumor targets; STAT3 activation in the myeloid compartment blocks the production of IL-12 by DCs and triggers the secretion of IL-23 by tumor-associated macrophages, which further impairs NK cell activation and skews the DC repertoire toward an immunosuppressive phenotype by preventing NK-mediated lysis of immature, tolerogenic DCs. Crosstalk between NK cells, macrophages, and DCs has further immunosuppressive effects on the adaptive immune response by inhibiting the proper activation of CD4+ and CD8+ T cells and promoting the infiltration, development, and activation of MDSCs and CD4+, FoxP3+ regulatory T cells (TRegs). Thus, a STAT3-activating tumor microenvironment, which begins with the production of cytokines and growth factors by tumor cells, triggers a STAT3 cascade in tumor-infiltrating immune cells that has long-range, profound effects on host antitumor immunity that allows the cancer to escape detection and destruction (Figure 2).
Myeloid-derived suppressor cells from both humans and mice produce indoleamine 2,3-dioxygenase (IDO), which negatively regulates the function of both myeloid and lymphoid lineages within the tumor microenvironment. IDO catalyzes the rapid catabolism of tryptophan to kynurenine, which inhibits proliferation and effector function of T and NK cells (162–167). Expression of IDO in the tumor microenvironment inhibits NK function by downregulation of activating NK receptors DNAM-1, NKp30, NKp44, and NKG2D, inhibition of IFN-γ production, increased IL-4 and IL-13 secretion, and induction of NK cell apoptosis (168–171). Strikingly, IDO is also overexpressed by tumor cells, as well as tumor-associated fibroblasts, and mesenchymal stem cells (MSC); regardless of the source, IDO has been shown to profoundly inhibit NK-mediated cytokine production, expression of natural cytolytic receptors (NCRs), and tumor cell cytolysis.
Yu et al. identified an “immature” subset of MDSCs (CD33+CD13+CD14−CD15−), induced from CD33+ progenitors upon coculture with the human breast cancer cell line MDA-MB-231, that produced high levels of IDO (172, 173). In this MDSC subpopulation, STAT3 was required for NFκB-inducing kinase (NIK) phosphorylation and activation of the non-canonical NFκB signaling pathway, which upregulated IDO expression by direct activation of the IDO promoter. A Jak/STAT inhibitor, JSI-124 (cucurbitacin I), blocked STAT3 phosphorylation, NFκB activation, and IDO production by MDSCs. In tumor sections from 30 breast cancer patients, the percentage of CD33+/pSTAT3+ co-expressing cells was 80, suggesting a critical role for this pathway in driving IDO expression in MDSCs associated with primary human breast tumors.
Constitutive, STAT3-dependent IDO expression in drug-resistant tumor cells has been described by Campia et al. (174). Doxorubicin (Dox)-resistant clones of human colon (HT29), lung (A549), and leukemia (K562) cell lines, a murine chemoresistant breast cancer cell line (JC), and human metastatic mesothelioma (HMM) were shown to express high levels of IDO1 and displayed enhanced subcutaneous growth in vivo relative to chemosensitive parental cell lines. Enhanced tumor growth was blocked by treatment of mice with an IDO1 inhibitor, 5-Br-Brassinin. Expression of IDO correlated to enhanced Jak1, STAT1 and STAT3 phosphorylation, and the expression of STAT3 targets that induce IDO expression, such as IL-6, IL-1β, IL-4, IL-13, CD40L, and TNF-α. Interestingly, the expression of PIAS3, an endogenous negative feedback inhibitor of STAT3 signaling, was downregulated in drug-resistant A549 cells relative to the parental cell line. These results suggest that constitutive STAT3 signaling and IDO expression in drug-resistant tumors may be enhanced by the inactivation of STAT3-specific feedback controls such as PIAS3.
Other studies have linked STAT3 and the aryl hydrocarbon receptor (AHR) to IDO expression in human NSCLC and ovarian cancer (175). Litzenberger demonstrated that IDO can drive its own production via an IDO–AHR–IL-6–STAT3 signaling pathway. IDO metabolizes tryptophan to kynenurine, which binds to the AHR, transactivating target genes such as IL-6. Interleukin-6 activates STAT3, which drives IDO transcription, sustaining the cycle.
Likewise, Chen et al. have demonstrated that IDO is induced in human breast fibroblasts (RMF-EG) following co-incubation with COX-2-overexpressing breast cancer cells (176). It was shown that COX-2-dependent production of PGE2 in human breast cancer cell lines induced IDO, and targeting of the PGE2 receptor EP4 abrogated IDO induction. Two STAT-binding GAS elements were identified in the IDO promoter. STAT3 binding to the IDO GAS elements was increased in fibroblasts when cocultured with breast cancer cells, and siRNA targeting of STAT3 abolished IDO induction by a EP4 agonist.
In summary, there are multiple pathways by which STAT3 indirectly modulates the function of NK cells, either by the production of tumor-derived immunosuppressive factors, such as IDO, or by driving the development of immunosuppressive myeloid populations, such as MDSCs, which secrete IDO, transforming growth factor (TGF)-β, or other soluble factors that block NK development, cytolytic function, or cytokine production (Figure 1; Table 1).
Another key regulatory pathway that influences NK cytolytic and helper functions involves TGF-β. It has been well documented that TGF-β is produced by both tumor cells and tumor-infiltrating myeloid cells (177–182). The biological effects of TGF-β in tumorigenesis have been extensively characterized, and it has been shown to both inhibit and promote tumor activity, depending on the context. Tumor cells often escape the growth inhibitory effects of TGF-β by converting its signaling to promote invasive, metastatic behavior, and can drive EMT. TGF-β regulates the immune system on many levels and modulates the function of most lymphoid and myeloid lineages.
In the tumor microenvironment, high levels of TGF-β are attributed to several mechanisms, including overexpression by tumor cells and cancer-associated fibroblasts (CAFs), as well as induced expression in tumor-infiltrating MDSCs and regulatory T cells (Tregs). Several immunosuppressive TGF-β target genes have been identified, including FoxP3, which drives the development of Tregs (181, 182).
Transforming growth factor-β impairs NK function by reducing the expression levels of NK activating receptors NKG2D and NKp30, blocking the production of IFN-γ and chemokines, and inhibiting NK cell degranulation by impairing release of granzyme A and granzyme B (183–190). More indirectly, TGF-β also impairs DC-driven activation of T and NK cells by reducing the expression of IL-12, MHC class II molecules, and the costimulatory molecules CD80 and CD86 (191).
In several studies, disruption of STAT3 signaling in tumor cells was shown to drive down TGF-β expression and potentiate NK-mediated killing of tumor targets. Sui et al. demonstrated a link between STAT3, TGF-β, and tumor surveillance by NK cells in a model of hepatocellular carcinoma (HCC) (192). Treatment of two murine HCC cell lines, Hepa1–6 and H22, with a STAT3-binding site decoy oligonucleotide impaired tumor growth in vivo and prolonged survival of animals with subcutaneous tumors. Numbers of peripheral NK cells (CD3−/DX5+) and cytotoxic function of liver- or spleen-derived lymphocytes against H22 target cells was increased in mice bearing STAT3-targeted H22 tumors relative to NK cells from mice injected with the parental H22 cell line. Peripheral and tumor-associated NK cells from STAT3-targeted tumor-bearing mice also expressed elevated levels of NK activation markers NKG2D, CD69, and Fas ligand (FasL). Critical involvement of NK cells in tumor rejection was demonstrated by showing that STAT3 deletion impaired H22 growth in nude mice, which lack T cells but have functional NK cells. Antibody-mediated depletion experiments in immunocompetent mice showed that the inhibitory effect of STAT3 targeting was greatly reduced when NK cells were ablated with an anti-asialo-GMP antibody, and adoptive transfer of purified CD3−/DX5+ NK cells from tumor-bearing mice inhibited H22 tumor growth and extended survival of recipient mice. The role of TGF-β was demonstrated by antibody blocking experiments in which anti-IL-10 plus anti-TGF-β antibodies could reproduce the effects of STAT3 targeting of H22 cells in in vivo tumor growth experiments.
Likewise, Sun et al. demonstrated a critical link between STAT3 blockade and inhibition of TGFβ production in HCC cell lines, leading to increased sensitivity to NK-mediated lysis (193). Human HCC cells lines Hep2G, H7402, and PLC/PRF/5 were more sensitive to killing by human PBMCs and NK cell lines NKL or NK-92 when transfected with a STAT3-binding decoy oligonucleotide. A major effect of STAT3 targeting in HCC was increased expression of NKG2D ligands ULBP1, ULBP2, and ULBP3, and enhanced NK-mediated killing of STAT3-targeted HCC cells was partially reversed by treatment with an anti-ULBP3 neutralizing antibody.
Further, supernatants from STAT3-targeted HCC cells enhanced the cytolytic activity of NK cell lines against HCC tumor targets, suggesting that the profile of secreted factors by the tumor cells was altered by STAT3 inhibition. Supernatants from STAT3-targeted tumor cells increased the expression of NK activation markers, such as CD69, NKG2D, Fas ligand, granzyme B, perforin, and IFN-γ. Cytokine profiling identified TGF-β, IL-10, and IL-8 as downregulated in all HCC cell lines following STAT3 targeting, while IFN-γ was consistently upregulated. The importance of TGF-β in regulating NK-mediated tumor killing was demonstrated by increased HCC cell lysis following anti-TGF-β treatment. Conversely, increased NK-mediated tumor cell killing conferred by STAT3 decoy oligonucleotide was reversed by the addition of exogenous TGF-β. The immunotherapeutic potential of blocking TGF-β has been suggested by some recent studies in preclinical models. Alvarez et al. showed that a neutralizing anti-TGF-β antibody could reverse NK inhibition within the tumor microenvironment in the murine Lewis lung carcinoma (LLC) model of NSCLC (194).
Tumor cells become addicted to STAT3 signaling via the constitutive expression of STAT3-activating growth factors and cytokines or constitutively active mutations of RTKs or cytoplasmic kinases. However, STAT3 dependence is often further driven by inactivation of endogenous negative feedback mechanisms that normally control STAT3 signaling. Among these mechanisms, the SOCS are a large superfamily of STAT inhibitors, some of which bind directly to activated kinases or receptor chains and blunt cytokine and growth factor-driven STAT activation (195–198). STAT3 dependence in tumors is often accompanied by epigenetic silencing of the SOCS1 or SOCS3 genes via CpG island hypermethylation and histone deacetylation, which greatly enhances the cell’s sensitivity to STAT3 activation, increasing both the magnitude and duration of a STAT3-dependent stimulus. Selective pressure to repress SOCS3 expression in STAT3-addicted tumors has been shown in cancers of the brain, esophagus, prostate, lung, bone marrow, colon, liver, cervix, and head/neck (199–206), which suggests that SOCS3 inhibition may not only promote tumor cell proliferation and metastasis but also drive tumor cell-mediated immunosuppression.
Modulation of SOCS3 in tumor-infiltrating myeloid cells may also contribute to an immunosuppressive environment that represses NK activity. Yu et al. have recently shown that SOCS3-deficient myeloid cells (SOCS3MyeKO) displayed enhanced STAT3 phosphorylation and increased levels of IL-1, IL-6, TNF-α, and iNOS (207). SOCS3MyeKO bone marrow cells preferentially differentiated into MDSC-like cells when cultured in conditioned medium from TRAMP-C1 prostate cancer cells, and TRAMP-C1 tumor growth was accelerated in SOCS3MyeKO compared to WT mice, indicating that SOCS3 can control the extent of MDSC production and may indirectly regulate NK activity in the tumor microenvironment.
In contrast, it has been shown that SOCS3 may drive the generation of immunosuppressive DCs. Mesenchymal stem/stromal cells can induce HSCs to differentiate into CD11b+, Interleukin-10-dependent regulatory DCs (208). This novel differentiation pathway was highly dependent on IL-10-driven chromatin reorganization and activation of the SOCS3 locus and could be reversed by siRNA-mediated SOCS3 targeting. This study suggests that SOCS3 can contribute to an immunosuppressive phenotype by skewing the DC developmental program in hematopoietic progenitors.
Analysis of SOCS3 in NK cells suggests that it may have a distinct, direct role in this lineage as well. Braunschweig et al. demonstrated that SOCS3 can regulate NK cytolytic activity, using the NK-92 human NK cell line (209). Small interfering RNA-mediated targeting of SOCS3 resulted in increased STAT3 phosphorylation and NK-mediated killing of tumor targets, which suggests that SOCS3 may be a negative regulator of NK activity and therapeutic targeting of SOCS3 in NK cells may potentiate killing of tumor targets.
Likewise, Xu et al. demonstrated that the chemokine CCL2 can inhibit NK cytolytic function by inducing SOCS3 and partially blocking perforin expression (210). CCL2 impaired the cytolytic activity of freshly isolated CD56+ human NK cells toward K562 targets, which coincided with SOCS3 induction and partially repressed perforin expression. Transfection of a SOCS3-specific siRNA reversed both the perforin repression and inhibition of cytolytic activity.
Chemokines, such as CCL2, which are normally critical for NK migration and tumor elimination, can in fact become immunosuppressive in a tumor microenvironment lacking key NK-activating “costimulatory” cytokines. Without each component of the process, CCL2 may repress NK activation through a SOCS3-dependent mechanism.
These findings suggest a dual role for SOCS3 in tumor immunity. On the one hand, when expressed in cancer cells and MDSC precursors, SOCS3 can promote antitumor immunity by blocking the effects of STAT3-inducing, immunosuppressive cytokines. On the other hand, in NK cells and DC precursors, SOCS3 may be a key mediator of STAT3-driven immunosuppression. The in vivo relevance of these observations will depend on the generation tissue-specific knockout animals with targeted SOCS3 deletion in DCs and NK cells.
The PIAS is another family of feedback inhibitors of STAT activation that are aberrantly modulated in tumors. It comprised four members: PIAS1, PIAS3, PIASx, and PIASy that can directly bind to STATs and block phosphorylation, DNA binding, and transactivation (211, 212); STAT3 is targeted primarily by the PIAS3 protein. While PIAS proteins can be overexpressed in some cancers (213), PIAS3 expression is repressed in anaplastic lymphoma, glioblastoma, mesothelioma, and NSCLC, which correlates to constitutive STAT3 activation (214–217). Thus, by promoting STAT3 activation, repression of PIAS3 expression in tumor cells can indirectly contribute to NK cell dysfunction by altering the cytokine profile of the cancer. In addition, some studies suggest that inactivation of PIAS3 may be involved in tumor-associated immunosuppression via IDO expression in chemoresistant cancers.
These studies suggest an important link between tumor-mediated immunosuppression and other, early events in tumorigenesis, especially epigenetic silencing of potential tumor suppressor genes. The role of SOCS3 as a negative feedback inhibitor of STAT3 signaling often targets it for CpG island methylation in cells that are addicted to STAT3 signaling and, as an additional selective advantage, may induce or enhance the immunosuppressive potential of STAT3-addicted tumors. Moreover, downregulation of SOCS3 in the myeloid compartment can contribute to increased STAT3 phosphorylation, activation of a MDSC developmental pathway, and the secretion of immunosuppressive cytokines.
The STAT3 transcription factor has been known for many years to drive tumorigenesis, cancer cell survival, proliferation, metastasis, and resistance to anticancer agents. Only relatively recently has its role in the immune system’s response to cancer been fully appreciated. STAT3 regulates antitumor immunity at virtually every level, affecting both lymphoid and myeloid compartments, innate and adaptive immunity, effector cells and APCs, cell migration to the tumor microenvironment, cytolytic and helper function, and the secretion of cytokines and growth factors that modulate immune responses. NK cells are greatly affected by activated STAT3 both intrinsically and extrinsically. Hematopoietic and NK-specific STAT3 genetic ablation have demonstrated that STAT3 is not critical for normal NK development or proliferation. However, STAT3 negatively regulates NK activation and tumor cell killing, as STAT3-deficient NK cells generally exhibit enhanced cytolytic activity and cytokine secretion in vitro and in vivo. The molecular basis of this regulation is not well understood, although it likely involves STAT3-mediated repression of NK activating receptors, such as DNAM-1, as well as cytolytic mediators, such as perforin and granzyme B.
Aside from the direct effects of STAT3 activation in NK cells, constitutive STAT3 phosphorylation in transformed cells can indirectly impair NK function by regulating the expression of ligands for NK activating receptors as well as important immune checkpoint proteins. For example, tumors can escape NK-mediated recognition and lysis through STAT3-dependent downregulation of the NKG2D ligand MICA. This is likely to impair a critical mechanism to eliminate tumor cells that have been treated with DNA damaging agents such as radiation. Likewise, STAT3-driven PD-L1 expression in tumor cells may dampen antitumor immunity by engaging the PD-1 receptor on the surface of NK cells.
Perhaps more importantly, STAT3 activation in tumor cells triggers a cascade of cytokine and growth factor production that reprograms the tumor microenvironment, skewing it toward immunosuppression. Cytokines produced by tumor cells activate STAT3 in NK cells, DCs, T cells, and macrophages that profoundly affect each population intrinsically and via critical crosstalk mechanisms that shut down both innate and adaptive immunity. STAT3 activation in DCs results in impaired IL-12 production, leading to defective NK activation and reduced cytolytic activity toward tumor targets as well as tolerogenic, immature DCs. Reciprocally, poorly activated NK cells fail to produce critical cytokines (IFN-γ and TNF-α) to DCs and T cells, which impairs DC maturation, antigen presentation, and T cell activation. Likewise, STAT3 activation in macrophages alters the balance between IL-12 and IL-23, which enhances carcinogenesis and impairs NK function. This complex web of inhibitory interactions between tumor cells and components of innate and adaptive immunity can send the immune system into a downward spiral in the tumor microenvironment. Finally, STAT3 activation in tumor cells impairs the migration of virtually every component of the immune response to the tumor microenvironment, such that responding cells cannot reach the site of the cancer; targeting STAT3 in tumors alters their cytokine/growth factor profile, resulting in increased migration to microenvironment and potentially enhancing antigen presentation and immune effector functions.
The role of STAT3 in anticancer immunity and therapy becomes even more complex in light of studies suggesting that STAT3 can promote NK-mediated tumor immunity. Some reports have shown that STAT3 can increase the expression of the NK activating receptor NKG2D and promote NK-mediated immunosurveillance in some lymphoma models. Thus, biological targeting of STAT3 in tumor cells may not only have synergistic effects by potentiating the function of NK cells and other components of the immune response but also produce inhibitory off-target effects on NK cells in certain cancers. This raises the possibility that STAT3 inhibitors may work best when combined with one or more of the novel, potent immune checkpoint inhibitors that target PD-1, PD-L1, or CTLA-4, inhibition of immunosuppressive factors, such as IDO, or treatment with chromatin modifiers that release SOCS genes from epigenetic repression in tumor cells. Molecular profiling of tumor tissue, as well as infiltrating lymphoid and myeloid cells, may be of great value to predict the possible immunologic outcome of treating cancer patients with STAT3 inhibitors.
NC researched and wrote the article.
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author’s laboratory is currently funded by the Department of Radiation Oncology at UCLA.
1. Cella M, Miller H, Song C. Beyond NK cells: the expanding universe of innate lymphoid cells. Front Immunol (2014) 5:1–11. doi:10.3389/fimmu.2014.00282
2. Artis D, Spits H. The biology of innate lymphoid cells. Nature (2015) 517:293–301. doi:10.1038/nature14189
3. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell (2011) 144:646–74. doi:10.1016/j.cell.2011.02.013
4. Hanahan D, Coussens LM. Accessories to the crime: functions of cells recruited to the tumor microenvironment. Cancer Cell (2012) 21:309–22. doi:10.1016/j.ccr.2012.02.022
5. Vivier E, Tomasello E, Baratin M, Walzer T, Ugolini S. Functions of natural killer cells. Nat Immunol (2008) 9:503–10. doi:10.1038/ni1582
6. Montaldo E, Vacca P, Moretta L, Mingari MC. Development of human natural killer cells and other innate lymphoid cells. Semin Immunol (2014) 26:107–13. doi:10.1016/j.smim.2014.01.006
7. Moretta L, Montaldo E, Vacca P, Del Zotto G, Moretta F, Merli P, et al. Human natural killer cells: origin, receptors, function, and clinical applications. Int Arch Allergy Immunol (2014) 164:253–64. doi:10.1159/000365632
8. Shifrin N, Raulet DH, Ardolino M. NK cell self tolerance, responsiveness and missing self recognition. Semin Immunol (2014) 26:138–44. doi:10.1016/j.smim.2014.02.007
9. Keating SE, Ní Chorcora C, Dring MM, Stallings RL, O’Meara A, Gardiner CM. Increased frequencies of the killer immunoglobulin-like receptor genes KIR2DL2 and KIR2DS2 are associated with neuroblastoma. Tissue Antigens (2015) 86:172–7. doi:10.1111/tan.12608
10. Rocca YS, Roberti MP, Arriaga JM, Amat M, Bruno L, Pampena MB, et al. Altered phenotype in peripheral blood and tumor-associated NK cells from colorectal cancer patients. Innate Immun (2013) 19:76–85. doi:10.1177/1753425912453187
11. Carrega P, Morandi B, Costa R, Frumento G, Forte G, Altavilla G, et al. Natural killer cells infiltrating human nonsmall-cell lung cancer are enriched in CD56 bright CD16(-) cells and display an impaired capability to kill tumor cells. Cancer (2008) 112:863–75. doi:10.1002/cncr.23239
12. Schleypen JS, Baur N, Kammerer R, Nelson PJ, Rohrmann K, Gröne EF, et al. Cytotoxic markers and frequency predict functional capacity of natural killer cells infiltrating renal cell carcinoma. Clin Cancer Res (2006) 12:718–25. doi:10.1158/1078-0432.CCR-05-0857
13. Platonova S, Cherfils-Vicini J, Damotte D, Crozet L, Vieillard V, Validire P, et al. Profound coordinated alterations of intratumoral NK cell phenotype and function in lung carcinoma. Cancer Res (2011) 71:5412–22. doi:10.1158/0008-5472.CAN-10-4179
14. Koo KC, Shim DH, Yang CM, Lee SB, Kim SM, Shin TY, et al. Reduction of the CD16(-)CD56bright NK cell subset precedes NK cell dysfunction in prostate cancer. PLoS One (2013) 8:e78049. doi:10.1371/journal.pone.0078049
15. Nishimura Y, Kumagai-Takei N, Matsuzaki H, Lee S, Maeda M, Kishimoto T, et al. Functional alteration of natural killer cells and cytotoxic T lymphocytes upon asbestos exposure and in malignant mesothelioma patients. Biomed Res Int (2015) 2015:238431. doi:10.1155/2015/238431
16. Messaoudene M, Périer A, Fregni G, Neves E, Zitvogel L, Cremer I, et al. Characterization of the microenvironment in positive and negative sentinel lymph nodes from melanoma patients. PLoS One (2015) 10:e0133363. doi:10.1371/journal.pone.0133363
17. Bottino C, Dondero A, Bellora F, Moretta L, Locatelli F, Pistoia V, et al. Natural killer cells and neuroblastoma: tumor recognition, escape mechanisms, and possible novel immunotherapeutic approaches. Front Immunol (2014) 5:56. doi:10.3389/fimmu.2014.00056
18. Cai L, Zhang Z, Zhou L, Wang H, Fu J, Zhang S, et al. Functional impairment in circulating and intrahepatic NK cells and relative mechanism in hepatocellular carcinoma patients. Clin Immunol (2008) 2008(129):428–37. doi:10.1016/j.clim.2008.08.012
19. Pietra G, Manzini C, Rivara S, Vitale M, Cantoni C, Petretto A, et al. Melanoma cells inhibit natural killer cell function by modulating the expression of activating receptors and cytolytic activity. Cancer Res (2012) 72:1407–15. doi:10.1158/0008-5472.CAN-11-2544
20. Wölfle SJ, Strebovsky J, Bartz H, Sähr A, Arnold C, Kaiser C, et al. PD-L1 expression on tolerogenic APCs is controlled by STAT-3. Eur J Immunol (2011) 41:413–24. doi:10.1002/eji.201040979
21. Sumpter TL, Thomson AW. The STATus of PD-L1 (B7-H1) on tolerogenic APCs. Eur J Immunol (2011) 41:286–90. doi:10.1002/eji.201041353
22. Azoulay-Alfaguter I, Strazza M, Pedoeem A, Mor A. The coreceptor programmed death 1 inhibits T-cell adhesion by regulating Rap1. J Allergy Clin Immunol (2015) 135:564–7. doi:10.1016/j.jaci.2014.07.055
23. Pedoeem A, Azoulay-Alfaguter I, Strazza M, Silverman GJ, Mor A. Programmed death-1 pathway in cancer and autoimmunity. Clin Immunol (2014) 153:145–52. doi:10.1016/j.clim.2014.04.010
24. Topalian SL, Drake CG, Pardoll DM. Immune checkpoint blockade: a common denominator approach to cancer therapy. Cancer Cell (2015) 27:450–61. doi:10.1016/j.ccell.2015.03.001
25. Brahmer JR, Pardoll DM. Immune checkpoint inhibitors: making immunotherapy a reality for the treatment of lung cancer. Cancer Immunol Res (2013) 1:85–91. doi:10.1158/2326-6066.CIR-13-0078
26. Topalian SL, Drake CG, Pardoll DM. Targeting the PD-1/B7-H1(PD-L1) pathway to activate anti-tumor immunity. Curr Opin Immunol (2012) 24:207–12. doi:10.1016/j.coi.2011.12.009
27. Cantoni C, Grauwet K, Pietra G, Parodi M, Mingari MC, Maria A, et al. Role of NK cells in immunotherapy and virotherapy of solid tumors. Immunotherapy (2015) 7(8):861–82. doi:10.2217/imt.15.53
28. Gras Navarro A, Björklund AT, Chekenya M. Therapeutic potential and challenges of natural killer cells in treatment of solid tumors. Front Immunol (2015) 6:202. doi:10.3389/fimmu.2015.00202
29. Mahmood S, Upreti D, Sow I, Amari A, Nandagopal S, Kung SK. Bidirectional interactions of NK cells and dendritic cells in immunotherapy: current and future perspective. Immunotherapy (2015) 7:301–8.
30. Zheng Y, Dou Y, Duan L, Cong C, Gao A, Lai Q, et al. Using chemo-drugs or irradiation to break immune tolerance and facilitate immunotherapy in solid cancer. Cell Immunol (2015) 294:54–9. doi:10.1016/j.cellimm.2015.02.003
31. Poli A, Michel T, Thérésine M, Andrès E, Hentges F, Zimmer J. CD56bright natural killer (NK) cells: an important NK cell subset. Immunology (2009) 126:458–65. doi:10.1111/j.1365-2567.2008.03027.x
32. Jewett A, Man YG, Tseng HC. Dual functions of natural killer cells in selection and differentiation of stem cells; role in regulation of inflammation and regeneration of tissues. J Cancer (2013) 4:12–24. doi:10.7150/jca.5519
33. Jewett A, Tseng H-C. Tumor induced inactivation of natural killer cell cytotoxic function; implication in growth, expansion and differentiation of cancer stem cells. J Cancer (2011) 2:443–57. doi:10.7150/jca.2.443
34. Jewett A, Head C, Cacalano NA. Emerging mechanisms of immunosuppression in oral cancers. J Dent Res (2006) 85:1061–73. doi:10.1177/154405910608501201
35. Jewett A, Bonavida B. Target-induced inactivation and cell death by apoptosis in a subset of human NK cells. Immunol J (1996) 156:907–15.
36. Jewett A, Bonavida B. Target-induced anergy of natural killer cytotoxic function is restricted to the NK-target conjugate subset. Cell Immunol (1995) 160:91–7. doi:10.1016/0008-8749(95)80013-9
37. Tseng HC, Arasteh A, Paranjpe A, Teruel A, Yang W, Behel A, et al. Increased lysis of stem cells but not their differentiated cells by natural killer cells; de-differentiation or re-programming activates NK cells. PLoS One (2010) 5:e11590. doi:10.1371/journal.pone.0011590
38. Magister Š, Tseng HC, Bui VT, Kos J, Jewett A. Regulation of split anergy in natural killer cells by inhibition of cathepsins C and H and cystatin F. Oncotarget (2015) 6(26):22310–27. doi:10.18632/oncotarget.4208
39. Cacalano NA, Johnston JA. Interleukin-2 signaling and inherited immunodeficiency. Am J Hum Genet (1999) 65:287–93. doi:10.1086/302518
40. Stark GR, Darnell JE Jr. The JAK-STAT pathway at twenty. Immunity (2012) 36(4):503–14. doi:10.1016/j.immuni.2012.03.013
41. Schindler C, Plumlee C. Inteferons pen the JAK-STAT pathway. Semin Cell Dev Biol (2008) 19:311–8. doi:10.1016/j.semcdb.2008.08.010
42. Schindler C, Levy DE, Decker T. JAK-STAT signaling: from interferons to cytokines. J Biol Chem (2007) 282:20059–63. doi:10.1074/jbc.R700016200
43. Villarino AV, Kanno Y, Ferdinand JR, O’Shea JJ. Mechanisms of Jak/STAT signaling in immunity and disease. J Immunol (2015) 194:21–7. doi:10.4049/jimmunol.1401867
44. Cao Q, Li YY, He WF, Zhang ZZ, Zhou Q, Liu X, et al. Interplay between microRNAs and the STAT3 signaling pathway in human cancers. Physiol Genomics (2013) 45:1206–14. doi:10.1152/physiolgenomics.00122.2013
45. Vogel TP, Milner JD, Cooper MA. The Ying and Yang of STAT3 in human disease. J Clin Immunol (2015) 35(7):615–23. doi:10.1007/s10875-015-0187-8
46. Yu H, Lee H, Herrmann A, Buettner R, Jove R. Revisiting STAT3 signalling in cancer: new and unexpected biological functions. Nat Rev Cancer (2014) 14:736–46. doi:10.1038/nrc3818
47. Li N, Grivennikov SI, Karin M. The unholy trinity: inflammation, cytokines, and STAT3 shape the cancer microenvironment. Cancer Cell (2011) 19:429–31. doi:10.1016/j.ccr.2011.03.018
48. Taniguchi K, Karin M. IL-6 and related cytokines as the critical lynchpins between inflammation and cancer. Semin Immunol (2014) 26:54–74. doi:10.1016/j.smim.2014.01.001
49. Aggarwal BB, Kunnumakkara AB, Harikumar KB, Gupta SR, Tharakan ST, Koca C, et al. Signal transducer and activator of transcription-3, inflammation, and cancer: how intimate is the relationship? Ann N Y Acad Sci (2009) 1171:59–76. doi:10.1111/j.1749-6632.2009.04911.x
50. Wendt MK, Balanis N, Carlin CR, Schiemann WP. STAT3 and epithelial-mesenchymal transitions in carcinomas. JAKSTAT (2014) 3:e28975. doi:10.4161/jkst.28975
51. Zhu QC, Gao RY, Wu W, Qin HL. Epithelial-mesenchymal transition and its role in the pathogenesis of colorectal cancer. Asian Pac J Cancer Prev (2013) 14:2689–98. doi:10.7314/APJCP.2013.14.5.2689
52. Duan S, Tsai Y, Keng P, Chen Y, Lee SO, Chen Y. IL-6 signaling contributes to cisplatin resistance in non-small cell lung cancer via the up-regulation of anti-apoptotic and DNA repair associated molecules. Oncotarget (2015) 6:27651–60. doi:10.18632/oncotarget.4753
53. Lau J, Ilkhanizadeh S, Wang S, Miroshnikova YA, Salvatierra NA, Wong RA, et al. STAT3 blockade inhibits radiation-induced malignant progression in glioma. Cancer Res (2015) 75(20):4302–11. doi:10.1158/0008-5472.CAN-14-3331
54. Yu H, Pardoll D, Jove R. STATs in cancer inflammation and immunity: a leading role for STAT3. Nat Rev Cancer (2009) 9:798–809. doi:10.1038/nrc2734
55. Kortylewski M, Kujawski M, Wang T, Wei S, Zhang S, Pilon-Thomas S, et al. Inhibiting Stat3 signaling in the hematopoietic system elicits multicomponent antitumor immunity. Nat Med (2005) 11:1314–21. doi:10.1038/nm1325
56. Kortylewski M, Yu H. Role of Stat3 in suppressing anti-tumor immunity. Curr Opin Immunol (2008) 20:228–33. doi:10.1016/j.coi.2008.03.010
57. Kortylewski M, Yu H. Stat3 as a potential target for cancer immunotherapy. J Immunother (2007) 30:131–9. doi:10.1097/01.cji.0000211327.76266.65
58. Yu H, Kortylewski M, Pardoll D. Crosstalk between cancer and immune cells: role of STAT3 in the tumour microenvironment. Nat Rev Immunol (2007) 7:41–51. doi:10.1038/nri1995
59. Gotthardt D, Putz EM, Straka E, Kudweis P, Biaggio M, Poli V, et al. Loss of STAT3 in murine NK cells enhances NK cell-dependent tumor surveillance. Blood (2014) 124:2370–9. doi:10.1182/blood-2014-03-564450
60. Vacca P, Cantoni C, Prato C, Fulcheri E, Moretta A, Moretta L, et al. Regulatory role of NKp44, NKp46, DNAM-1 and NKG2D receptors in the interaction between NK cells and trophoblast cells. Evidence for divergent functional profiles of decidual versus peripheral NK cells. Int Immunol (2008) 20:1395–405. doi:10.1093/intimm/dxn105
61. Cerboni C, Fionda C, Soriani A, Zingoni A, Doria M, Cippitelli M, et al. The DNA damage response: a common pathway in the regulation of NKG2D and DNAM-1 ligand expression in normal, infected, and cancer cells. Front Immunol (2014) 4:508. doi:10.3389/fimmu.2013.00508
62. de Andrade LF, Smyth MJ, Martinet L. DNAM-1 control of natural killer cells functions through nectin and nectin-like proteins. Immunol Cell Biol (2014) 92:237–44. doi:10.1038/icb.2013.95
63. Fionda C, Soriani A, Zingoni A, Santoni A, Cippitelli M. NKG2D and DNAM-1 ligands: molecular targets for NK cell-mediated immunotherapeutic intervention in multiple myeloma. Biomed Res Int (2015) 2015:178698. doi:10.1155/2015/178698
64. Morisaki T, Onishi H, Katano M. Cancer immunotherapy using NKG2D and DNAM-1 systems. Anticancer Res (2012) 32:2241–7.
65. Elishmereni M, Bachelet I, Levi-Schaffer F. DNAM-1: an amplifier of immune responses as a therapeutic target in various disorders. Curr Opin Investig Drugs (2008) 9:491–6.
66. Kida H, Ihara S, Kumanogoh A. Involvement of STAT3 in immune evasion during lung tumorigenesis. Oncoimmunology (2013) 2(1):e22653. doi:10.4161/onci.22653
67. Gismondi A, Santoni A. Migration of NK cells. In: Badolato R, Sozzani S, editors. Lymphocyte Trafficking in Health and Disease. Basel: Birkhauser Verlag (2006). 95 p.
68. Allavena P, Bianchi G, Zhou D, van Damme J, Jilek P, Sozzani S, et al. Induction of natural killer cell migration by monocyte chemotactic protein-1, -2 and -3. Eur J Immunol (1994) 24:3233–6. doi:10.1002/eji.1830241249
69. Gregiore C, Chasson L, Luci C, Tomasello E, Geissmann F, Vivier E, et al. The trafficking of natural killer cells. Immunol Rev (2007) 220:169–82. doi:10.1111/j.1600-065X.2007.00563.x
70. Nokihara H, Yanagawa H, Nishioka Y, Yano S, Mukaida N, Matsushima K, et al. Natural killer cell-dependent suppression of systemic spread of human lung adenocarcinoma cells by monocyte chemoattractant protein-1 gene transfection in severe combined immunodeficient mice. Cancer Res (2000) 60:7002–7.
71. Mukaida N, Sasaki S, Baba T. Chemokines in cancer development and progression and their potential as targeting molecules for cancer treatment. Mediators Inflamm (2014) 2014:170381. doi:10.1155/2014/170381
72. Halama N, Braun M, Kahlert C, Spille A, Quack C, Rahbari N, et al. Natural killer cells are scarce in colorectal carcinoma tissue despite high levels of chemokines and cytokines. Clin Can Res (2011) 17:678–89. doi:10.1158/1078-0432.CCR-10-2173
73. Tsuchiyama T, Nakamoto Y, Sakai Y, Marukawa Y, Kitahara M, Mukaida N, et al. Prolonged, NK cell-mediated antitumor effects of suicide gene therapy combined with monocyte chemoattractant protein-1 against hepatocellular carcinoma. J Immunol (2007) 178:574–83. doi:10.4049/jimmunol.178.1.574
74. Lavergne E, Combadière B, Bonduelle O, Iga M, Gao JL, Maho M, et al. Fractalkine mediates natural killer-dependent antitumor responses in vivo. Cancer Res (2003) 63:7468–74.
75. Tang L, Hu HD, Hu P, Lan YH, Peng ML, Chen M, et al. Gene therapy with CX3CL1/fractalkine induces antitumor immunity to regress effectively mouse hepatocellular carcinoma. Gene Ther (2007) 14:1226–34. doi:10.1038/sj.gt.3302959
76. Zeng Y, Huebener N, Fest S, Weixler S, Schroeder U, Gaedicke G, et al. Fractalkine (CX3CL1)- and interleukin-2-enriched neuroblastoma microenvironment induces eradication of metastases mediated by T cells and natural killer cells. Cancer Res (2007) 67:2331–8. doi:10.1158/0008-5472.CAN-06-3041
77. Burdelya L, Kujawski M, Niu G, Zhong B, Wang T, Zhang S, et al. Stat3 activity in melanoma cells affects migration of immune effector cells and nitric oxide-mediated antitumor effects. J Immunol (2005) 174:3925–31. doi:10.4049/jimmunol.174.7.3925
78. Ihara S, Kida H, Arase H, Tripathi LP, Chen Y-A, Kimura T, et al. Inhibitory roles of signal transducer and activator of transcription 3 in antitumor immunity during carcinogen-induced lung tumorigenesis. Cancer Res (2012) 72:2990–9. doi:10.1158/0008-5472.CAN-11-4062
79. Putz EM, Hoelzl MA, Baeck J, Bago-Horvath Z, Schuster C, Reichholf B, et al. Loss of STAT3 in lymphoma relaxes NK cell-mediated tumor surveillance. Cancers (Basel) (2014) 6:193–210. doi:10.3390/cancers6010193
80. Chretien AS, Le Roy A, Vey N, Prebet T, Blaise D, Fauriat C, et al. Cancer-induced alterations of NK-mediated target recognition: current and investigational pharmacological strategies aiming at restoring NK-mediated anti-tumor activity. Front Immunol (2014) 5:122. doi:10.3389/fimmu.2014.00122
81. Marcus A, Gowen BG, Thompson TW, Iannello A, Ardolino M, Deng W, et al. Recognition of tumors by the innate immune system and natural killer cells. Adv Immunol (2014) 122:91–128. doi:10.1016/B978-0-12-800267-4.00003-1
82. Baychelier F, Vieillard V. The modulation of the cell-cycle: a sentinel to alert the NK cells of dangers. Front Immunol (2013) 4:325. doi:10.3389/fimmu.2013.00325
83. Lam RA, Chwee JY, Le Bert N, Sauer M, Pogge von Strandmann E, Gasser S. Regulation of self-ligands for activating natural killer cell receptors. Ann Med (2013) 45:384–94. doi:10.3109/07853890.2013.792495
84. Gasser S, Raulet DH. The DNA damage response arouses the immune system. Cancer Res (2006) 66:3959–62. doi:10.1158/0008-5472.CAN-05-4603
85. Lanier LL. NKG2D receptor and its ligands in host defense. Cancer Immunol Res (2015) 3:575–82. doi:10.1158/2326-6066.CIR-15-0098
86. Chen D, Gyllensten U. MICA polymorphism: biology and importance in cancer. Carcinogenesis (2014) 35(12):2633–42. doi:10.1093/carcin/bgu215
87. Goto K, Kato N. MICA SNPs and the NKG2D system in virus-induced HCC. J Gastroenterol (2015) 50:261–72. doi:10.1007/s00535-014-1000-9
88. Champsaur M, Lanier LL. Effect of NKG2D ligand expression on host immune responses. Immunol Rev (2010) 235:267–85. doi:10.1111/j.0105-2896.2010.00893.x
89. Huergo-Zapico L, Acebes-Huerta A, López-Soto A, Villa-Álvarez M, Gonzalez-Rodriguez AP, Gonzalez S. Molecular bases for the regulation of NKG2D ligands in cancer. Front Immunol (2014) 5:106. doi:10.3389/fimmu.2014.00106
90. Spear P, Wu MR, Sentman ML, Sentman CL. NKG2D ligands as therapeutic targets. Cancer Immun (2013) 13:8.
91. Guerra N, Tan YX, Joncker NT, Choy A, Gallardo F, Xiong N, et al. NKG2D-deficient mice are defective in tumor surveillance in models of spontaneous malignancy. Immunity (2008) 28:571–80. doi:10.1016/j.immuni.2008.02.016
92. Arreygue-Garcia NA, Daneri-Navarro A, del Toro-Arreola A, Cid-Arregui A, Gonzalez-Ramella O, Jave-Suarez LF, et al. Augmented serum level of major histocompatibility complex class I-related chain A (MICA) protein and reduced NKG2D expression on NK and T cells in patients with cervical cancer and precursor lesions. BMC Cancer (2008) 8:16. doi:10.1186/1471-2407-8-16
93. Bedel R, Thiery-Vuillemin A, Grandclement C, Balland J, Remy-Martin JP, Kantelip B, et al. Novel role for STAT3 in transcriptional regulation of NK immune cell targeting receptor MICA on cancer cells. Cancer Res (2011) 71:1615–26. doi:10.1158/0008-5472.CAN-09-4540
94. Fionda C, Malgarini G, Soriani A, Zingoni A, Cecere F, Iannitto ML, et al. Inhibition of glycogen synthase kinase-3 increases NKG2D ligand MICA expression and sensitivity to NK cell-mediated cytotoxicity in multiple myeloma cells: role of STAT3. J Immunol (2013) 190:6662–72. doi:10.4049/jimmunol.1201426
95. Zhu S, Phatarpekar PV, Denman CJ, Senyukov VV, Somanchi SS, Nguyen-Jackson HT, et al. Transcription of the activating receptor NKG2D in natural killer cells is regulated by STAT3 tyrosine phosphorylation. Blood (2014) 124:403–11. doi:10.1182/blood-2013-05-499707
96. Takaki R, Hayakawa Y, Nelson A, Sivakumar PV, Hughes S, Smyth MJ, et al. IL-21 enhances tumor rejection through a NKG2D-dependent mechanism. J Immunol (2005) 175(4):2167–73.
97. Burgess SJ, Marusina AI, Pathmanathan I, Borrego F, Coligan JE. IL-21 down-regulates NKG2D/DAP10 expression on human NK and CD81 T cells. J Immunol (2006) 176:1490–7. doi:10.4049/jimmunol.176.3.1490
98. Wan CK, Andraski AB, Spolski R, Li P, Kazemian M, Oh J, et al. Opposing roles of STAT1 and STAT3 in IL-21 function in CD4+ T cells. Proc Natl Acad Sci U S A (2015) 112:9394–9. doi:10.1073/pnas.1511711112
99. Ramsay AG. Immune checkpoint blockade immunotherapy to activate anti-tumour T-cell immunity. Br J Haematol (2013) 162:313–25. doi:10.1111/bjh.12380
100. Sharma P, Allison JP. Immune checkpoint targeting in cancer therapy: toward combination strategies with curative potential. Cell (2015) 161:205–14. doi:10.1016/j.cell.2015.03.030
101. Sharma P, Allison JP. The future of immune checkpoint therapy. Science (2015) 348:56–61. doi:10.1126/science.aaa8172
102. Page DB, Postow MA, Callahan MK, Allison JP, Wolchok JD. Immune modulation in cancer with antibodies. Annu Rev Med (2014) 65:185–202. doi:10.1146/annurev-med-092012-112807
103. Monteiro J. Timeline: checkpoint blockade. Cell (2015) 162:1434. doi:10.1016/j.cell.2015.08.019
104. Littman DR. Releasing the brakes on cancer immunotherapy. Cell (2015) 162:1186–90. doi:10.1016/j.cell.2015.08.038
105. Kyi C, Postow MA. Checkpoint blocking antibodies in cancer immunotherapy. FEBS Lett (2014) 588:368–76. doi:10.1016/j.febslet.2013.10.015
106. Ito A, Kondo S, Tada K, Kitano S. Clinical development of immune checkpoint inhibitors. Biomed Res Int (2015) 2015:605478. doi:10.1155/2015/605478
107. Anagnostou VK, Brahmer JR. Cancer immunotherapy: a future paradigm shift in the treatment of non-small cell lung cancer. Clin Cancer Res (2015) 21:976–84. doi:10.1158/1078-0432.CCR-14-1187
108. Parmiani G, Maccalli C, Maio M. Integrating immune checkpoint blockade with anti-neo/mutated antigens reactivity to increase the clinical outcome of immunotherapy. Vaccines (Basel) (2015) 3:420–8. doi:10.3390/vaccines3020420
109. Kreamer KM. Immune checkpoint blockade: a new paradigm in treating advanced cancer. J Adv Pract Oncol (2014) 5:418–31.
110. Callahan MK, Wolchok JD. Clinical activity, toxicity, biomarkers, and future development of CTLA-4 checkpoint antagonists. Semin Oncol (2015) 42:573–86. doi:10.1053/j.seminoncol.2015.05.008
111. Mashima E, Inoue A, Sakuragi Y, Yamaguchi T, Sasaki N, Hara Y, et al. Nivolumab in the treatment of malignant melanoma: review of the literature. Onco Targets Ther (2015) 8:2045–51. doi:10.2147/OTT.S62102
112. Hamid O, Robert C, Daud A, Hodi FS, Hwu WJ, Kefford R, et al. Safety and tumor responses with lambrolizumab (anti-PD-1) in melanoma. N Engl J Med (2013) 369:134–44. doi:10.1056/NEJMoa1305133
113. Robert C, Schachter J, Long GV, Arance A, Grob JJ, Mortier L, et al. Pembrolizumab versus ipilimumab in advanced melanoma. N Engl J Med (2015) 372:2521–32. doi:10.1056/NEJMoa1503093
114. Homet Moreno B, Ribas A. Anti-programmed cell death protein-1/ligand-1 therapy in different cancers. Br J Cancer (2015) 112:1421–7. doi:10.1038/bjc.2015.124
115. Shin DS, Ribas A. The evolution of checkpoint blockade as a cancer therapy: what’s here, what’s next? Curr Opin Immunol (2015) 33:23–35. doi:10.1016/j.coi.2015.01.006
116. Knaus HA, Kanakry CG, Luznik L, Gojo I. Immunomodulatory drugs II: immune checkpoint agents in acute leukemia. Curr Drug Targets (2015).
117. Cohen J, Sznol M. Therapeutic combinations of immune-modulating antibodies in melanoma and beyond. Semin Oncol (2015) 42:488–94. doi:10.1053/j.seminoncol.2015.02.014
118. Harvey RD. Immunologic and clinical effects of targeting PD-1 in lung cancer. Clin Pharmacol Ther (2014) 96:214–23. doi:10.1038/clpt.2014.74
119. Covre A, Coral S, Di Giacomo AM, Taverna P, Azab M, Maio M. Epigenetics meets immune checkpoints. Semin Oncol (2015) 42:506–13. doi:10.1053/j.seminoncol.2015.02.003
120. Jang JH, Huang Y, Zheng P, Jo MC, Bertolet G, Zhu MX, et al. Imaging of cell-cell communication in a vertical orientation reveals high-resolution structure of immunological synapse and novel PD-1 dynamics. J Immunol (2015) 195:1320–30. doi:10.4049/jimmunol.1403143
121. Marzec M, Zhang Q, Goradia A, Raghunath PN, Liu X, Paessler M, et al. Oncogenic kinase NPM/ALK induces through STAT3 expression of immunosuppressive protein CD274 (PD-L1, B7–H1). Proc Natl Acad Sci U S A (2008) 105:20852–7. doi:10.1073/pnas.0810958105
122. Benson DM Jr, Bakan CE, Mishra A, Hofmeister CC, Efebera Y, Becknell B, et al. The PD-1/PD-L1 axis modulates the natural killer cell versus multiple myeloma effect: a therapeutic target for CT-011, a novel monoclonal anti-PD-1 antibody. Blood (2010) 116:2286–94. doi:10.1182/blood-2010-02-271874
123. Ferlazzo G, Morandi B. Cross-talks between natural killer cells and distinct subsets of dendritic cells. Front Immunol (2014) 5:159. doi:10.3389/fimmu.2014.00159
124. Wong JL, Mailliard RB, Moschos SJ, Edington H, Lotze MT, Kirkwood JM, et al. Helper activity of NK cells during the dendritic cell-mediated induction of melanoma-specific cytotoxic T cells. J Immunother (2011) 34:270–8. doi:10.1097/CJI.0b013e31820b370b
125. Ferlazzo G, Moretta L. Dendritic cell editing by natural killer cells. Crit Rev Oncog (2014) 19:67–75. doi:10.1615/CritRevOncog.2014010827
126. Marvel D, Gabrilovich DI. Myeloid-derived suppressor cells in the tumor micro-environment: expect the unexpected. J Clin Invest (2015) 125:3356–64. doi:10.1172/JCI80005
127. Parker KH, Beury DW, Ostrand-Rosenberg S. Myeloid-derived suppressor cells: critical cells driving immune suppression in the tumor microenvironment. Adv Cancer Res (2015) 128:95–139. doi:10.1016/bs.acr.2015.04.002
128. Talmadge JE, Gabrilovich DI. History of myeloid-derived suppressor cells. Nat Rev Cancer (2013) 13:739–52. doi:10.1038/nrc3581
129. Moretta A, Marcenaro E, Parolini S, Ferlazzo G, Moretta L. NK cells at the interface between innate and adaptive immunity. Cell Death Differ (2008) 15:226–33. doi:10.1038/sj.cdd.4402170
130. Castriconi R, Della Chiesa M, Moretta A. Shaping of adaptive immunity by innate interactions. C R Biol (2004) 327:533–7. doi:10.1016/j.crvi.2003.12.001
131. Pampena MB, Levy EM. Natural killer cells as helper cells in dendritic cell cancer vaccines. Front Immunol (2015) 6:13. doi:10.3389/fimmu.2015.00013
132. Sconocchia G, Eppenberger S, Spagnoli GC, Tornillo L, Droeser R, Caratelli S, et al. NK cells and T cells cooperate during the clinical course of colorectal cancer. Oncoimmunology (2014) 3:e952197. doi:10.4161/21624011.2014.952197
133. Vivier E, Raulet DH, Moretta A, Caligiuri MA, Zitvogel L, Lanier LL, et al. Innate or adaptive immunity? The example of natural killer cells. Science (2011) 331:44–9. doi:10.1126/science.1198687
134. Chaix J, Tessmer MS, Hoebe K, Fuséri N, Ryffel B, Dalod M, et al. Cutting edge: priming of NK cells by IL-18. J Immunol (2008) 181:1627–31. doi:10.4049/jimmunol.181.3.1627
135. Gabrilovich DI, Ciernik IF, Carbone DP. Dendritic cells in antitumor immune responses. I. Defective antigen presentation in tumor-bearing hosts. Cell Immunol (1996) 170:101–10. doi:10.1006/cimm.1996.0139
136. Gabrilovich DI, Nadaf S, Corak J, Berzofsky JA, Carbone DP. Dendritic cells in anti-tumor immune responses. II. Dendritic cells grown from bone marrow precursors, but not mature DC from tumor-bearing mice are effective antigen carriers in the therapy of established tumors. Cell Immunol (1996) 170:111. doi:10.1006/cimm.1996.0140
137. Gabrilovich DI, Corak J I, Ciernik F, Kavanaugh D, Carbone DP. Decreased antigen presentation by dendritic cells in patients with breast cancer. Clin Cancer Res (1997) 3:483.
138. Almand B, Clark JI, Nikitina E, English NR, Knight SC, Carbone DP, et al. Increased production of immature myeloid cells in cancer patients: a mechanism of immunosuppression in cancer. J Immunol (2001) 166:678. doi:10.4049/jimmunol.166.1.678
139. Almand B, Resser J, Lindman B, Nadaf S, Clark J, Kwon E, et al. Clinical significance of defective dendritic cell differentiation in cancer. Clin Cancer Res (2000) 6:1755.
140. Thurnher M, Radmayar C, Ramoner R, Ebner S, Bock G, Klocker H, et al. Human renal-cell carcinoma tissue contains dendritic cells. Int J Cancer (1996) 67:1. doi:10.1002/(SICI)1097-0215(19960927)68:1<1::AID-IJC1>3.0.CO;2-V
141. Nestle FO, Burg G, Fah J, Wrone-Smith T, Nickoloff BJ. Human sunlight-induced basal-cell-carcinoma-associated dendritic cells are deficient in T cell co-stimulatory molecules and are impaired as antigen-presenting cells. Am J Pathol (1997) 150:641.
142. Chaux P, Moutet M, Faivre J, Martin F, Martin M. Inflammatory cells infiltrating human colorectal carcinomas express HLA class II but not B7-1 and B7-2 costimulatory molecules of the T-cell activation. Lab Invest (1996) 74:975.
143. Chaux P, Favre N, Martin M, Martin F. Tumor-infiltrating dendritic cells are defective in their antigen-presenting function and inducible B7 expression in rats. Int J Cancer (1997) 72:619. doi:10.1002/(SICI)1097-0215(19970807)72:4<619::AID-IJC12>3.0.CO;2-6
144. Gabrilovich DI, Chen HL, Girgis KR, Cunningham HT, Meny GM, Nadaf S, et al. Production of vascular endothelial growth factor by human tumors inhibits the functional maturation of dendritic cells. Nat Med (1996) 2:1096–103. doi:10.1038/nm1096-1096
145. Gabrilovich DI, Ishida T, Oyama T, Ran S, Kravtsov V, Nadaf S, et al. Vascular endothelial growth factor inhibits the development of dendritic cells and dramatically affects the differentiation of multiple hematopoietic lineages in vivo. Blood (1998) 92:4150.
146. Gabrilovich DI, Velders MP, Sotomayor EM, Kast WM. Mechanism of immune dysfunction in cancer mediated by immature Gr-1+ myeloid cells. J Immunol (2001) 166:5398–406. doi:10.4049/jimmunol.166.9.5398
147. Gabrilovich D. Mechanisms and functional significance of tumour-induced dendritic-cell defects. Nat Rev Immunol (2004) 4(12):941–52. doi:10.1038/nri1498
148. Nefedova Y, Cheng P, Gilkes D, Blaskovich M, Beg AA, Sebti SM, et al. Activation of dendritic cells via inhibition of Jak2/STAT3 signaling. J Immunol (2005) 175:4338–46. doi:10.4049/jimmunol.175.7.4338
149. Nefedova Y, Huang M, Kusmartsev S, Bhattacharya R, Cheng P, Salup R, et al. Hyperactivation of STAT3 is involved in abnormal differentiation of dendritic cells in cancer. J Immunol (2004) 172:464–74. doi:10.4049/jimmunol.172.1.464
150. Wang T, Niu G, Kortylewski M, Burdelya L, Shain K, Zhang S, et al. Regulation of the innate and adaptive immune responses by Stat-3 signaling in tumor cells. Nat Med (2004) 10:48–54. doi:10.1038/nm976
151. Kortylewski M, Xin H, Kujawski M, Lee H, Liu Y, Harris T, et al. Regulation of the IL-23 and IL-12 balance by Stat3 signaling in the tumor microenvironment. Cancer Cell (2009) 15:114–23. doi:10.1016/j.ccr.2008.12.018
152. Sieve AN, Meeks KD, Lee S, Berg RE. A novel immunoregulatory function for IL-23: inhibition of IL-12 dependent IFN-γ production. Eur J Immunol (2010) 40:2236–47. doi:10.1002/eji.200939759
153. Floss DM, Schröder J, Franke M, Scheller J. Insights into IL-23 biology: from structure to function. Cytokine Growth Factor Rev (2015) 26(5):569–78. doi:10.1016/j.cytogfr.2015.07.005
154. Magram J, Connaughton SE, Warrier RR, Carvajal DM, Wu CY, Ferrante J, et al. IL-12-deficient mice are defective in IFN gamma production and type 1 cytokine responses. Immunity (1996) 4:471–81. doi:10.1016/S1074-7613(00)80413-6
155. Sanal O, Turkkani G, Gumruk F, Yel L, Secmeer G, Tezcan I, et al. A case of interleukin-12 receptor beta-1 deficiency with recurrent leishmaniasis. Pediatr Infect Dis J (2007) 26:366–8. doi:10.1097/01.inf.0000258696.64507.0f
156. Gazzinelli RT, Hieny S, Wynn TA, Wolf S, Sher A. Interleukin 12 is required for the T-lymphocyte-independent induction of interferon gamma by an intracellular parasite and induces resistance in T-cell-deficient hosts. Proc Natl Acad Sci U S A (1993) 90:6115–9. doi:10.1073/pnas.90.13.6115
157. Haraguchi S, Day NK, Nelson RP Jr, Emmanuel P, Duplantier JE, Christodoulou CS, et al. Interleukin 12 deficiency associated with recurrent infections. Proc Natl Acad Sci U S A (1998) 95:13125–9. doi:10.1073/pnas.95.22.13125
158. Palamaro L, Giardino G, Santamaria F, Romano R, Fusco A, Montella S, et al. Interleukin 12 receptor deficiency in a child with recurrent bronchopneumonia and very high IgE levels. Ital J Pediatr (2012) 2012(38):46.
159. Meeran SM, Mantena SK, Meleth S, Elmets CA, Katiyar SK. Interleukin-12-deficient mice are at greater risk of UV radiation-induced skin tumors and malignant transformation of papillomas to carcinomas. Mol Cancer Ther (2006) 5:825–32. doi:10.1158/1535-7163.MCT-06-0003
160. Langowski JL, Kastelein RA, Oft M. Swords into plowshares: IL-23 repurposes tumor immune surveillance. Trends Immunol (2007) 28:207–12. doi:10.1016/j.it.2007.03.006
161. Langowski JL, Zhang X, Wu L, Mattson JD, Chen T, Smith K, et al. IL-23 promotes tumour incidence and growth. Nature (2006) 442:461–5.
162. Sucher R, Kurz K, Weiss G, Margreiter R, Fuchs D, Brandacher G. IDO-mediated tryptophan degradation in the pathogenesis of malignant tumor disease. Int J Tryptophan Res (2010) 3:113–20.
163. Wobser M, Voigt H, Houben R, Eggert AO, Freiwald M, Kaemmerer U, et al. Dendritic cell based antitumor vaccination: impact of functional indoleamine 2,3-dioxygenase expression. Cancer Immunol Immunother (2007) 56:1017–24. doi:10.1007/s00262-006-0256-1
164. Gostner JM, Becker K, Überall F, Fuchs D. The potential of targeting indoleamine 2,3-dioxygenase for cancer treatment. Expert Opin Ther Targets (2015) 19:605–15. doi:10.1517/14728222.2014.995092
165. Pallotta MT, Fallarino F, Matino D, Macchiarulo A, Orabona C. AhR-mediated, non-genomic modulation of IDO1 function. Front Immunol (2014) 5:497. doi:10.3389/fimmu.2014.00497
166. Ball HJ, Jusof FF, Bakmiwewa SM, Hunt NH, Yuasa HJ. Tryptophan-catabolizing enzymes – party of three. Front Immunol (2014) 5:485. doi:10.3389/fimmu.2014.00485
167. Mándi Y, Vécsei L. The kynurenine system and immunoregulation. J Neural Transm (2012) 119:197–209. doi:10.1007/s00702-011-0681-y
168. Ling W, Zhang J, Yuan Z, Ren G, Zhang L, Chen X, et al. Mesenchymal stem cells use IDO to regulate immunity in tumor microenvironment. Cancer Res (2014) 74:1576–87. doi:10.1158/0008-5472.CAN-13-1656
169. Pfeifer S, Schreder M, Bolomsky A, Graffi S, Fuchs D, Sahota SS, et al. Induction of indoleamine-2,3 dioxygenase in bone marrow stromal cells inhibits myeloma cell growth. J Cancer Res Clin Oncol (2012) 138:1821–30. doi:10.1007/s00432-012-1259-2
170. Spaggiari GM, Capobianco A, Abdelrazik H, Becchetti F, Mingari MC, Moretta L. Mesenchymal stem cells inhibit natural killer-cell proliferation, cytotoxicity, and cytokine production: role of indoleamine 2,3-dioxygenase and prostaglandin E2. Blood (2008) 111:1327–33. doi:10.1182/blood-2007-02-074997
171. Peng YP, Zhang JJ, Liang WB, Tu M, Lu ZP, Wei JS, et al. Elevation of MMP-9 and IDO induced by pancreatic cancer cells mediates natural killer cell dysfunction. BMC Cancer (2014) 14:738. doi:10.1186/1471-2407-14-738
172. Yu J, Wang Y, Yan F, Zhang P, Li H, Zhao H, et al. Noncanonical NF-κB activation mediates STAT3-stimulated IDO upregulation in myeloid-derived suppressor cells in breast cancer. J Immunol (2014) 193:2574–86. doi:10.4049/jimmunol.1400833
173. Yu J, Du W, Yan F, Wang Y, Li H, Cao S, et al. Myeloid-derived suppressor cells suppress antitumor immune responses through IDO expression and correlate with lymph node metastasis in patients with breast cancer. J Immunol (2013) 190:3783–97. doi:10.4049/jimmunol.1201449
174. Campia I, Buondonno I, Castella B, Rolando B, Kopecka J, Gazzano E, et al. An autocrine cytokine/JAK/STAT-signaling induces kynurenine synthesis in multidrug resistant human cancer cells. PLoS One (2015) 10:e0126159. doi:10.1371/journal.pone.0126159
175. Litzenburger UM, Opitz CA, Sahm F, Rauschenbach KJ, Trump S, Winter M, et al. Constitutive IDO expression in human cancer is sustained by an autocrine signaling loop involving IL-6, STAT3 and the AHR. Oncotarget (2014) 5:1038–51. doi:10.18632/oncotarget.1637
176. Chen JY, Li CF, Kuo CC, Tsai KK, Hou MF, Hung WC. Cancer/stroma interplay via cyclooxygenase-2 and indoleamine 2,3-dioxygenase promotes breast cancer progression. Breast Cancer Res (2014) 16:410. doi:10.1186/s13058-014-0410-1
177. Yu Y, Xiao CH, Tan LD, Wang QS, Li XQ, Feng YM. Cancer-associated fibroblasts induce epithelial-mesenchymal transition of breast cancer cells through paracrine TGF-β signalling. Br J Cancer (2014) 110:724–32. doi:10.1038/bjc.2013.768
178. Pardali K, Moustakas A. Actions of TGF-beta as tumor suppressor and pro-metastatic factor in human cancer. Biochim Biophys Acta (2007) 1775:21–62.
179. Chaudhury A, Howe PH. The tale of transforming growth factor-beta (TGFbeta) signaling: a soigné enigma. IUBMB Life (2009) 61:929–39. doi:10.1002/iub.239
180. Brunen D, Willems SM, Kellner U, Midgley R, Simon I, Bernards R. TGF β: an emerging player in drug resistance. Cell Cycle (2013) 12:2960–8. doi:10.4161/cc.26034
181. Padua D, Massagué J. Roles of TGFbeta in metastasis. Cell Res (2009) 19:89–102. doi:10.1038/cr.2008.316
182. Massagué J. TGFβ signalling in context. Nat Rev Mol Cell Biol (2012) 13:616–30. doi:10.1038/nrm3434
183. Castriconi R, Dondero A, Bellora F, Moretta L, Castellano A, Locatelli F, et al. Neuroblastoma-derived TGF-β1 modulates the chemokine receptor repertoire of human resting NK cells. J Immunol (2013) 190:5321–8. doi:10.4049/jimmunol.1202693
184. Castriconi R, Cantoni C, Della Chiesa M, Vitale M, Marcenaro E, Conte R, et al. Transforming growth factor beta 1 inhibits expression of NKp30 and NKG2D receptors: consequences for the NK-mediated killing of dendritic cells. Proc Natl Acad Sci U S A (2003) 100:4120–5. doi:10.1073/pnas.0730640100
185. Lee J-C, Lee K-M, Kim D-W, Heo DS. Elevated TGF-h1 secretion and down-modulation of NKG2D underlies impaired NK cytotoxicity in cancer patients. J Immunol (2004) 2004(172):7335–40. doi:10.4049/jimmunol.172.12.7335
186. Rook AH, Kehrl JH, Wakefield LM, Roberts AB, Sporn MB, Burlington DB, et al. Effects of transforming growth factor beta on the functions of natural killer cells: depressed cytolytic activity and blunting of interferon responsiveness. J Immunol (1986) 136:3916–20.
187. Bellone G, Aste-Amezaga M, Trinchieri G, Rodeck U. Regulation of NK cell functions byTGF-h1. J Immunol (1995) 155:1066–73.
188. Park YP, Choi SC, Kiesler P, Gil-Krzewska A, Borrego F, Weck J, et al. Complex regulation of human NKG2D-DAP10 cell surface expression: opposing roles of the γc cytokines and TGF-β1. Blood (2011) 118:3019–27. doi:10.1182/blood-2011-04-346825
189. Song H, Hur DY, Kim KE, Park H, Kim T, Kim CW, et al. IL-2/IL-18 prevent the down-modulation of NKG2D by TGF-beta in NK cells via the c-Jun N-terminal kinase (JNK) pathway. Cell Immunol (2006) 242:39–45. doi:10.1016/j.cellimm.2006.09.002
190. Geissmann F, Revy P, Regnault A, Lepelletier Y, Dy M, Brousse N, et al. TGF-beta 1 prevents the noncognate maturation of human dendritic Langerhans cells. J Immunol (1999) 162:4567–75.
191. Chen L, Qiu M, He W, Huang A, Liu J. Functional study of immature dendritic cells co-transfected with IL-10 and TGF-beta 1 genes in vitro. Mol Biol Rep (2012) 39:6633–9. doi:10.1007/s11033-012-1468-4
192. Sui Q, Zhang J, Sun X, Zhang C, Han Q, Tian Z. NK cells are the crucial antitumor mediators when STAT3-mediated immunosuppression is blocked in hepatocellular carcinoma. J Immunol (2014) 193:2016–23. doi:10.4049/jimmunol.1302389
193. Sun X, Sui Q, Zhang C, Tian Z, Zhang J. Targeting blockage of STAT3 in hepatocellular carcinoma cells augments NK cell functions via reverse hepatocellular carcinoma-induced immune suppression. Mol Cancer Ther (2013) 12:2885–96. doi:10.1158/1535-7163.MCT-12-1087
194. Alvarez M, Bouchlaka MN, Sckisel GD, Sungur CM, Chen M, Murphy WJ. Anti-TGF-β reveals competition between increased antitumor effects using IL-2 with mouse NK and CD8 T cells. J Immunol (2014) 193:1709–16. doi:10.4049/jimmunol.1400034
195. Linossi EM, Babon JJ, Hilton DJ, Nicholson SE. Suppression of cytokine signaling: the SOCS perspective. Cytokine Growth Factor Rev (2013) 24:241–8. doi:10.1016/j.cytogfr.2013.03.005
196. Inagaki-Ohara K, Kondo T, Ito M, Yoshimura A. SOCS, inflammation, and cancer. JAKSTAT (2013) 2:e24053. doi:10.4161/jkst.24053
197. Yoshimura A, Suzuki M, Sakaguchi R, Hanada T, Yasukawa H. SOCS, inflammation, and autoimmunity. Front Immunol (2012) 3:20. doi:10.3389/fimmu.2012.00020
198. Johnston JA. Are SOCS suppressors, regulators, and degraders? J Leukoc Biol (2004) 75:743–8. doi:10.1189/jlb.1003507
199. Wu WY, Kim H, Zhang CL, Meng XL, Wu ZS. Loss of suppressors of cytokine signaling 3 promotes aggressiveness in hepatocellular carcinoma. J Invest Surg (2014) 27:197–204. doi:10.3109/08941939.2013.873098
200. Isomoto H. Epigenetic alterations in cholangiocarcinoma-sustained IL-6/STAT3 signaling in cholangiocarcinoma due to SOCS3 epigenetic silencing. Digestion (2009) 79(Suppl 1):2–8. doi:10.1159/000167859
201. Kim MH, Kim MS, Kim W, Kang MA, Cacalano NA, Kang SB, et al. Suppressor of cytokine signaling (SOCS) genes are silenced by DNA hypermethylation and histone deacetylation and regulate response to radiotherapy in cervical cancer cells. PLoS One (2015) 10:e0123133. doi:10.1371/journal.pone.0123133
202. Boosani CS, Agrawal DK. Methylation and microRNA-mediated epigenetic regulation of SOCS3. Mol Biol Rep (2015) 42:853–72. doi:10.1007/s11033-015-3860-3
203. Lindemann C, Hackmann O, Delic S, Schmidt N, Reifenberger G, Riemenschneider MJ. SOCS3 promoter methylation is mutually exclusive to EGFR amplification in gliomas and promotes glioma cell invasion through STAT3 and FAK activation. Acta Neuropathol (2011) 122:241–51. doi:10.1007/s00401-011-0832-0
204. Weber A, Hengge UR, Bardenheuer W, Tischoff I, Sommerer F, Markwarth A, et al. SOCS-3 is frequently methylated in head and neck squamous cell carcinoma and its precursor lesions and causes growth inhibition. Oncogene (2005) 24:6699–708. doi:10.1038/sj.onc.1208818
205. He B, You L, Xu Z, Mazieres J, Lee AY, Jablons DM. Activity of the suppressor of cytokine signaling-3 promoter in human non-small-cell lung cancer. Clin Lung Cancer (2004) 5:366–70. doi:10.3816/CLC.2004.n.015
206. He B, You L, Uematsu K, Zang K, Xu Z, Lee AY, et al. SOCS-3 is frequently silenced by hypermethylation and suppresses cell growth in human lung cancer. Proc Natl Acad Sci U S A (2003) 100:14133–8. doi:10.1073/pnas.2232790100
207. Yu H, Liu Y, McFarland BC, Deshane JS, Hurst DR, Ponnazhagan S, et al. SOCS3 deficiency in myeloid cells promotes tumor development: involvement of STAT3 activation and myeloid-derived suppressor cells. Cancer Immunol Res (2015) 3:727–40. doi:10.1158/2326-6066.CIR-15-0004
208. Liu X, Qu X, Chen Y, Liao L, Cheng K, Shao C, et al. Mesenchymal stem/stromal cells induce the generation of novel IL-10-dependent regulatory dendritic cells by SOCS3 activation. J Immunol (2012) 189:1182–92. doi:10.4049/jimmunol.1102996
209. Braunschweig A, Poehlmann TG, Busch S, Schleussner E, Markert UR. Signal transducer and activator of transcription 3 (STAT3) and suppressor of cytokine signaling (SOCS3) balance controls cytotoxicity and IL-10 expression in decidual-like natural killer cell line NK-92. Am J Reprod Immunol (2011) 66:329–35. doi:10.1111/j.1600-0897.2011.00989.x
210. Xu X, Wang Q, Deng B, Wang H, Dong Z, Qu X, et al. Monocyte chemoattractant protein-1 secreted by decidual stromal cells inhibits NK cells cytotoxicity by up-regulating expression of SOCS3. PLoS One (2012) 7:e41869. doi:10.1371/journal.pone.0041869
211. Shuai K, Liu B. Regulation of gene-activation pathways by PIAS proteins in the immune system. Nat Rev Immunol (2005) 5:593–605. doi:10.1038/nri1667
212. Levy C, Lee YN, Nechushtan H, Schueler-Furman O, Sonnenblick A, Hacohen S, et al. Identifying a common molecular mechanism for inhibition of MITF and STAT3 by PIAS3. Blood (2006) 107:2839–45. doi:10.1182/blood-2005-08-3325
213. Wang L, Banerjee S. Differential PIAS3 expression in human malignancy. Oncol Rep (2004) 11:1319–24.
214. Brantley EC, Nabors LB, Gillespie GY, Choi YH, Palmer CA, Harrison K, et al. Loss of protein inhibitors of activated STAT-3 expression in glioblastoma multiforme tumors: implications for STAT-3 activation and gene expression. Clin Cancer Res (2008) 14:4694–704. doi:10.1158/1078-0432.CCR-08-0618
215. Kluge A, Dabir S, Vlassenbroeck I, Eisenberg R, Dowlati A. Protein inhibitor of activated STAT3 expression in lung cancer. Mol Oncol (2011) 5:256–64. doi:10.1016/j.molonc.2011.03.004
216. Zhang Q, Raghunath PN, Xue L, Majewski M, Carpentieri DF, Odum N, et al. Multilevel dysregulation of STAT3 activation in anaplastic lymphoma kinase-positive T/null-cell lymphoma. J Immunol (2002) 168:466–74. doi:10.4049/jimmunol.168.1.466
Keywords: signal transducer and activator of transcription, STAT3 transcription factor, natural killer cells, chemokines, cytokines, cancer immunology
Citation: Cacalano NA (2016) Regulation of Natural Killer Cell Function by STAT3. Front. Immunol. 7:128. doi: 10.3389/fimmu.2016.00128
Received: 03 October 2015; Accepted: 21 March 2016;
Published: 11 April 2016
Edited by:
Masoud H. Manjili, Virginia Commonwealth University Massey Cancer Center, USAReviewed by:
Gregory B. Lesinski, The Ohio State University Comprehensive Cancer Center, USACopyright: © 2016 Cacalano. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Nicholas A. Cacalano, bmNhY2FsYW5AdWNsYS5lZHU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.