 Aline Ignacio
Aline Ignacio
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol., 19 February 2016
Sec. Microbial Immunology
Volume 7 - 2016 | https://doi.org/10.3389/fimmu.2016.00054
This article is part of the Research TopicMicrobial and environmental factors in autoimmune and inflammatory diseasesView all 18 articles
The mammalian gastrointestinal tract harbors a diverse microbial community with which dynamic interactions have been established over millennia of coevolution. Commensal bacteria and their products are sensed by innate receptors expressed in gut epithelia and in gut-associated immune cells, thereby promoting the proper development of mucosal immune system and host homeostasis. Many studies have demonstrated that host–microbiota interactions play a key role during local and systemic immunity. Therefore, this review will focus on how innate sensing of the gut microbiota and their metabolites through inflammasome and toll-like receptors impact the modulation of a distinct set of inflammatory and autoimmune diseases. We believe that a better understanding of the fine-tuning that governs host–microbiota interactions will further improve common prophylactic and therapeutic applications.
The mammalian gastrointestinal (GI) tract harbors more than 500 bacterial species that have protective, metabolic, and trophic roles and are a constant source of stimulation for the immune system (1). Surveillance mechanisms of the innate immune system control the communication of the gut microbiota with the internal environment, preventing their penetration and systemic spread, and maintaining intestinal homeostasis.
The GI innate immune system consists primarily of a physical barrier, which is composed of intestinal epithelial cells (IECs), represented by absorptive enterocytes, mucus-producing goblet cells, hormone-producing enteroendocrine cells, and Paneth cells, which produce antimicrobial peptides and lectins, among other molecules. The selective permeability of the epithelial barrier allows nutrients absorption while contributing to the immune responses by providing microbial products, which can be recognized through pattern recognition receptors (PRRs), including toll-like receptors (TLRs), nucleotide-binding domain leucine-rich repeat-containing receptors (NLRs), RIG-I like receptors (RLRs), C-type lectin family, and AIM2-like receptors (ALRs) (2), triggering different intracellular signaling cascades against pathogen-associated molecular patterns (PAMPs) or damage-associated molecular patterns (DAMPs).
The relationship between the intestinal epithelium and the gut microbiota is not restricted to prevent host invasion by commensals. Bacteria-mediated fermentation of food that cannot be fully digested in the mammalian intestine releases products such as short-chain fatty acids (SCFAs), which are used as fuel by IECs. In turn, the host provides a stable, nutrient-rich ecosystem for commensals to thrive. The microfold (M) cells are specialized epithelial cells of the mucosa-associated lymphoid tissue found in the follicle-associated epithelium (FAE) that mediate the uptake and transepithelial transport of luminal antigens to intraepithelial immune cells, such as macrophages and dendritic cells, which are responsible for presenting antigens to lymphocytes (3). In the intestinal lamina propria, macrophages and dendritic cell-mediated antigen uptake, either directly from the intestinal lumen by extending projections between IECs or indirectly by M cells, results in cytokine production, which drives either inflammatory Th17 or regulatory T cells, and T-cell-dependent and -independent IgA class-switching responses (4–6).
In this review, we focused on the interactions between the gut microbiota and two distinct groups of molecules, the innate sensors, namely, TLRs and the inflammasomes, which are typically composed of the innate sensors NLRs and ALRs, the adaptor protein apoptosis-associated speck-like protein containing a caspase-recruitment domain (ASC), and the proinflammatory caspase-1. Increasing efforts have been pointed out toward the understanding of how innate sensing of commensals promotes homeostasis and immunity. Here, we review the impact of innate sensing of the gut microbiota on inflammatory and autoimmune diseases’ outcomes.
Toll-like receptors are type I transmembrane proteins with an extracellular horseshoe-shaped domain-containing leucine-rich repeats (LRRs) and a cytoplasmic tail containing a toll/IL-1 receptor (TIR) domain (7). They correspond to one of the five main families of PRRs, which recognize a variety of PAMPs and DAMPs.
Toll-like receptors are capable of forming both homodimers and heterodimers to detect different types of ligands. TLR2/TLR1 heterodimers recognize triacylated lipopeptides (8), and it has been recently demonstrated that they can also be activated by α-synuclein, a protein associated with Parkinson’s disease (9). On the other hand, diacylated lipopeptides are recognized by TLR2/TLR6 heterodimers (8).
A recent study has shown that human immunodeficiency virus (HIV-1) structural proteins can activate TLR2- and TLR2-related heterodimers (10). A variety of viral proteins from measles virus (MV), hepatitis C virus (HCV), human cytomegalovirus (hCMV), and herpes simplex (HSV) have also been indicated as TLR2 activators (11–14). Lipoteichoic acid (LTA), zymosan, and peptidoglycan also trigger TLR2 signaling when TLR2/TLR6/CD14, TLR2/Dectin-1, and TLR2/CD14 are assembled, respectively (15–20).
TLR3 recognizes viral double-stranded RNA (dsRNA) (21) and is also capable of recognizing DAMPs released from damaged tissues (22), while TLR7 and TLR8 detect single-stranded RNA (ssRNA) found during viral replication (23–25). TLR4 is the main sensor for lipopolysaccharides (LPS) from Gram-negative bacteria (26) and also detects viral motifs (27). Flagellin is sensed by TLR5 (28), and TLR9 detects unmethylated CpG sequences in DNA molecules of bacterial and viral genomes (29). TLR10 is expressed in humans, but not in mice, making the search for a specific ligand experimentally difficult. It has been recently shown that TLR10 expressed in IECs responds to an unidentified component of Listeria monocytogenes, but it requires the presence of TLR2 for inducing NFκB activation (30). This opens the possibility that TLR10 acts as a coreceptor of TLR2, as it is phylogenetically related to TLR1 and TLR6 (31). Furthermore, other authors have demonstrated anti-inflammatory properties of TLR10 expressed in human peripheral blood mononuclear cells (PBMCs) (32).
TLR11 is expressed in mice and as a non-functional pseudogene in humans and has been related to prevention of Salmonella infection, the detection of uropathogenic Escherichia coli, and the recognition of a profiling-like molecule from Toxoplasma gondii (33–35). TLR12 and TLR13 are not expressed in humans but are expressed in mice and are capable of detecting the profiling-like molecule of T. gondii and 23S rRNA, respectively (36, 37). Besides PAMPs, endogenous ligands activate TLRs in the absence of infection (38).
Toll-like receptor–ligand interactions lead to dimerization of extracellular domains and results in dimerization of their cytoplasmic TIR domains. This structural modification is recognized by TIR domains on the adaptor proteins myeloid differentiation primary response gene 88 (MyD88), MyD88 adapter-like (MAL)/TIR domain-containing adapter protein (TIRAP), TIR domain-containing adapter-inducing interferon-β (TRIF), and TRIF-related adaptor molecule (TRAM), triggering downstream signaling pathways, which culminate in the expression of inflammatory cytokines, several anti-viral and anti-pathogen proteins, leading to the initiation of the adaptive immune response. The Myd88-dependent signaling pathway is common to all TLRs, except TLR3, which is dependent on TRIF. TLR4 uses both MyD88- and TRIF-dependent signaling pathways (7).
The detection of common molecules shared by pathogenic and non-pathogenic bacteria should trigger the same inflammatory response, but paradoxically, the recognition of commensal bacteria by intestinal receptors is somehow regulated to not result in inflammatory responses but to induce tolerance. Moreover, these TLR-mediated signals contribute to the intestinal homeostasis by regulating IECs proliferation and epithelial integrity (39–42). Interestingly, the localization, distribution, and expression of TLRs in the intestinal epithelium seem to be directly related to their role in maintaining homeostasis (43). IECs have a polarized organization, with an apical pole facing the intestinal lumen and a basolateral one that communicates with other IECs and the lamina propria. TLRs are differentially distributed through the polarized organization of IECs and in different quantities along the entire GI tract.
The human esophagus expresses TLR4, low levels of TLR1 and TLR5, and high levels of TLR2 and TLR3 mRNA in epithelial cells (44, 45). In gastric epithelial cells, it is possible to find expression of TLR2, TLR4, and TLR5, as both mRNA and protein (46). In the human small intestine, it is possible to find mRNA expression from the TLRs 1–10 (47). Under homeostatic conditions, enteroendocrine IECs from intestinal crypts express TLR1, TLR2, and TLR4 (48), and IECs from human colon express relatively high levels of mRNA for TLR3 and less for TLR2 and TLR4 (49).
Colonic IECs are hyporesponsive to LPS, and it may be due to epigenetic mechanisms that downregulate MD-2 and TLR4 expression (50). Moreover, TLR3 in murine IECs seems to have an age-dependent expression related to an enhanced response to rotavirus in adult mice (51). On the other hand, the high basolateral expression of TLR5 is directly induced by administration of flagellin to IECs (52). TLR9 apical expression in colonic IECs has been related to the maintenance of the intestinal homeostasis (53).
The FAE presents a differential expression of TLRs in comparison with villous IECs. The murine small intestine expresses TLR2 and TLR9 in both apical and basolateral sides of the FAE, whereas IECs in the villi shows apical expression only. The TLR5 is abundantly expressed in the apical pole of villous IECs and FAE, whereas TLR4 expression is low (54).
It is conceived that TLR-mediated sensing of microbial products has a dual role in promoting the fine-tune balance between proinflammatory and pro-regulatory immune responses. A better understanding of how these molecules are regulated in face of commensals and pathogenic microorganisms will benefit the design of prophylactic and therapeutic approaches.
Bearing in mind the importance of the gut microbiota for the development of the immune system, one might speculate that microbial components could also modulate immunity and trigger inflammatory and autoimmune diseases. Several studies have shown correlations between altered expression of TLRs and inflammatory/autoimmune conditions, and increasing evidence suggests the participation of the gut microbiota during TLRs abnormal signaling and the development of diseases, including type 1 diabetes (T1D), multiple sclerosis (MS), rheumatoid arthritis (RA), and inflammatory bowel diseases (IBDs) (Table 1) (55–58).
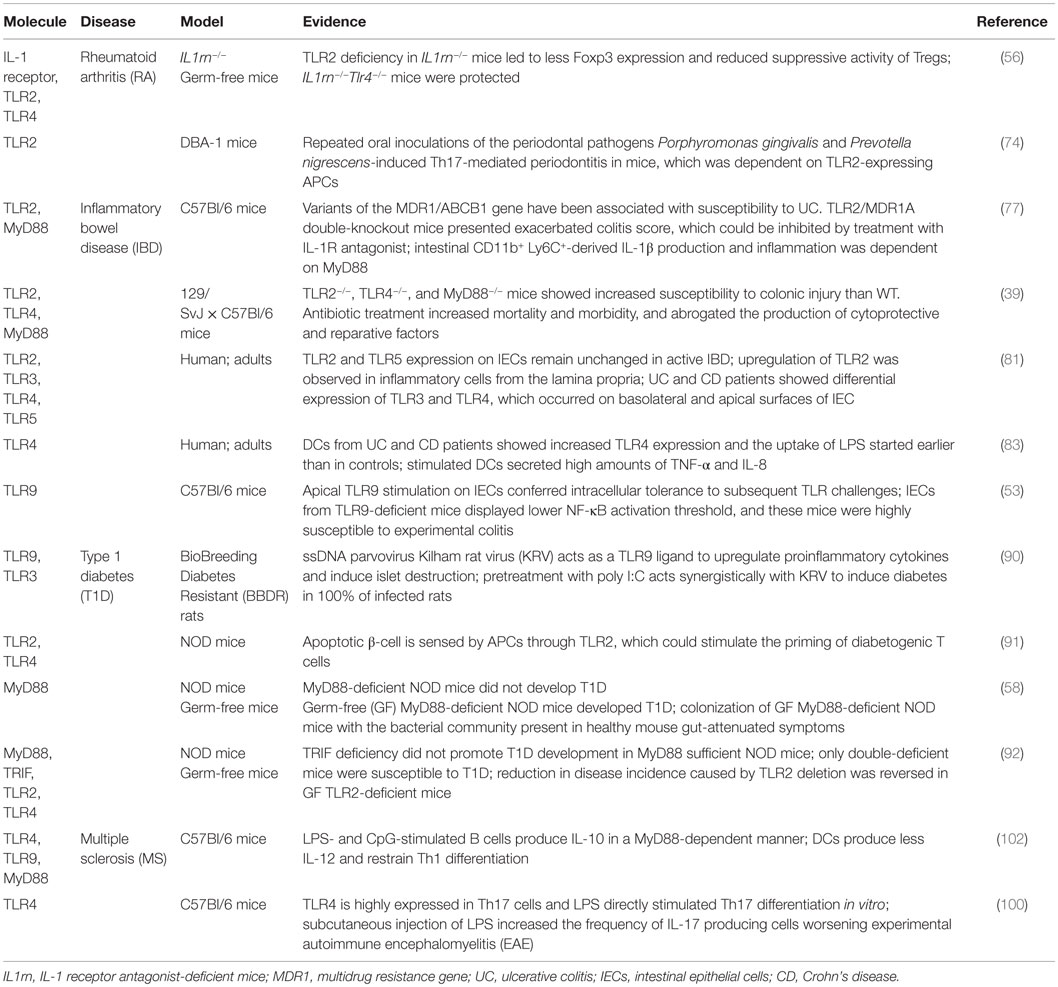
Table 1. Toll-like receptors and related molecules involved in inflammatory and autoimmune diseases.
Rheumatoid arthritis, IBDs, T1D, and MS are classified as multifactorial diseases with genetic and environmental factors contributing to their pathogenesis. Among the environmental factors, the exposition to microbial components has been associated with their development (55, 59–62). The concept called “molecular mimicry” was proposed to explain the role of microbial agents in inflammatory and autoimmune diseases, and according to this concept, cross-reactivity between epitopes from microbes and self-proteins can cause deregulated immune responses, leading to autoantibody production and activation of effectors cells (63).
The major organs exposed to microbial agents and their components are the skin and gut. In the gut, sensing of microbial antigens by innate immune cells and non-immune cells, such as IECs and stromal cells, is mediated by PRRs that recognized PAMPs. It is possible that bacterial components stimulate the mucosal immune cells by penetrating a damaged epithelial barrier or via a paracellular pathway; alternatively, microbial antigens may interact with TLRs at the apical surface of IECs, inducing inflammatory activation of the mucosal immune system. In addition, innate immune cells, such as dendritic cells and macrophages, can also sense PAMPs through TLRs initiating rapid and effective inflammatory responses against microbial invasion (57, 64). As mentioned above, the commensal microbiota is necessary for the constant stimulation of the immune system (1) and TLR-mediated sensing of these microorganisms may play a dual role in disease development as a source of both inflammatory and regulatory signals.
Rheumatoid arthritis is an autoimmune disease characterized by chronic inflammation in which cartilage and bone of the affected subjects are progressively destroyed in multiple joints (55). Several immune and resident cells, such as chondrocytes and fibroblasts, contribute to the development and progression of RA. Release of proinflammatory cytokines, such as tumor-necrosis factor (TNF)-α and interleukin-1 (IL-1), mainly by macrophages, activation of Th17 lymphocytes, and production of autoantibodies are suggested to play an important role in the disease onset (65). Studies showing the presence of bacterial cell wall components in the joints of RA patients accompanied by changes in their gut microbiota support the idea that commensal bacteria may initiate inflammation in genetically susceptible individuals (66, 67).
Abdollahi-Roodsaz et al. (56) using the IL-1 receptor antagonist-deficient (IL1rn−/−) mice that spontaneously develop T-cell-mediated autoimmune arthritis showed that germ-free (GF) IL1rn−/− presented no signs of arthritis during 20 weeks of follow-up, whereas matched non-GF animals started to develop the disease from the age of 5 weeks. The authors also demonstrated that the activation of TLRs was dependent on the microbial status of the mice and that TLR2 deficiency in IL1rn−/− mice led to less Foxp3+ expression and reduced suppressive activity of Tregs, resulting in an enhancement of clinical and histopathological scores of arthritis. By contrast, IL1rn−/−Tlr4−/− mice were protected against severe disease. It has been shown that TLR4 contributes to more severe arthritis by modulating IL-17 production and Th17 cell expansion (56, 68). In fact, gut-residing segmented filamentous bacteria (SFB) have been shown to drive both IL-1β and IL-6 production, and Th17 development, which promotes arthritis (69, 70). Therefore, one may conceive that a delicate equilibrium in TLR-mediated sensing of the gut microbiota is necessary to maintain homeostasis and prevent certain autoimmune diseases.
Although the gut microbiota has been extensively explored as an etiologic factor of RA, less is known about oral commensal microorganisms. In this context, the Gram-negative commensal bacterium Porphyromonas gingivalis has been a major focus of investigation as it provides a direct link between a specific microorganism and an autoimmune disease (71, 72). In fact, individuals with periodontitis present an increased proportion of P. gingivalis and specific antibodies to this bacterium, which has been linked to RA (73). A recent study showed that periodontal disease (PD) induced by P. gingivalis increases the severity of experimental arthritis through Th17 induction. This periodontal pathogen induces IL-17-producing T cells through TLR2 activation on APCs (74), which suggests that the same TLR molecule may play different roles in RA onset, depending on where the molecule is expressed and with which microorganism the interaction occurs. Thus, more studies are still necessary to better determine the role of TLR-mediated sensing of the microbiota and/or pathogens in autoimmune diseases, such as RA. Mice models with organ-specific TLR expression could be used to shed light on how TLRs contribute to disease onset depending on their localization.
Inflammatory bowel diseases have been extensively studied in regarding of the gut microbiota–immune system interactions. Two major forms of IBD have been investigated: the ulcerative colitis (UC) and Crohn’s disease (CD). Distinctive and complex chronic inflammatory processes characterize both disorders (61, 62), and it has been demonstrated that the intestinal microbiota of patients differ from healthy controls, showing an increase of Enterococcus spp. and Bacteroides spp. accompanied by a decrease of Bifidobacterium spp. and Lactobacillus spp. levels (75, 76), suggesting that the gut microbiota may play a pivotal role in intestinal inflammatory diseases.
Increased susceptibility to severe UC has been associated with variants of the multidrug resistance gene (MDR1/ABCB1). Deletion of TLR2 in MDR1A deficiency resulted in fulminant pancolitis, characterized by expansion of CD11b+ myeloid cells, and a shift toward Th1 immune responses in the lamina propria. An unaltered microbiota was required for colitis exacerbation in TLR2/MDR1A double-knockout mice once protection from colitis was observed upon antibiotic treatment (77). Treg-specific deletion of MyD88 has been recently shown to culminate in deficiency of Treg cells, increase in Th17 cells, and exacerbated experimental colitis. An overgrowth of SFB and increased bacterial translocation was reported, and also impaired antimicrobial IgA responses (78), suggesting that the TLR-mediated sensing of the gut microbiota contributes to Treg cell-dependent maintenance of the intestinal homeostasis. On the other hand, it has been shown that gut microbiota-mediated triggering of intestinal epithelial TLRs is not critical for promoting intestinal inflammation once mice lacking TNF receptor-associated factor 6 (TRAF6), but not MyD88/TRIF, were protected from dextran sodium sulfate (DSS)-induced colitis (79). Therefore, it is becoming clear that not only the microbiota is important during TLRs signaling through colitis development but also where these molecules are expressed.
Apart from the alterations in the gut microbiota composition, Cario and Podolsky have studied TLRs expression in CD and UC patients and showed variations in the expression of some receptors. During homeostasis, TLR2 and TLR4 are presented in small amounts on IECs and lamina propria cells to minimize microbiota recognition and to maintain tolerance (80, 81). So, signaling through those PRRs may determine the balance between immunity and tolerance. The authors observed that TLR2 and TLR5 are expressed on IECs from non-IBD subjects and remain unchanged in active IBD. Upregulation of TLR2 was observed in inflammatory cells from the lamina propria of active IBD patients; differential expression of TLR3 was verified in UC patients, which presented a basolateral expression on IECs; both UC and CD patients showed an abundant TLR4 expression on basolateral and apical surfaces of IECs, respectively, and enhanced expression of TLR4 was also present in the lamina propria of IBD individuals (80).
In DCs, PRRs stimulation induces IL-23 releases, which is an important component of antimicrobial defense, but when excessively produced it favors proinflammatory T-cell response and reduces Foxp3+ T cell differentiation. In this way, DCs may act as a key player in the initiation, continuation, and control of IBD (82). Conventional DCs (cDC) from UC and CD patients during remission phase showed an increased TLR4 expression. After stimulation, these cells secreted higher amounts of TNF-α and IL-8, and the uptake of LPS started earlier and was higher than in controls (83). These data suggest that an aberrant TLR4 signaling in cDC of IBD patients may result in an inflammatory phenotype during the acute phase.
Paradoxical effects of TLRs activation may be present in IBD. Bacterial DNA induces strong Th1 immune responses with high production of TNF-α and IL-8, which is also found in experimental and human IBD (84). On the other hand, bacterial DNA stimulation of IECs via apical TLR9 may result in anti-inflammatory effects, with the inhibition of TNF-α and IL-8 secretion, as well as NFκB activation, reducing colitis severity (53). Furthermore, it has been observed in a CD4+ T-cell-dependent Severe combined immunodeficiency (SCID) transfer model of colitis that pretreatment of donor mice with CpG completely abolished colitis development in SCID recipients in a CpG–TLR9-mediated modulation of T-cell function (85).
Another important protective effect promoted by TLR signaling was recently elucidated by Kawashima et al. (86) showing that dsRNA of lactic acid bacteria (LAB), one major commensal bacteria, triggered interferon-β (IFN-β) production through TLR3 activation pathway and protected mice from experimental colitis. On the other hand, pathogenic bacteria induced much less IFN-β and contained less dsRNA than LAB, indicating that dsRNA was not involved in pathogen-induced IFN-β induction. These results point toward TLR3 as a sensor to commensal bacteria and suggest a mechanism by which this endosomal receptor contributes to anti-inflammatory and protective immune responses.
Paradoxical effects of TLRs participation are also observed in T1D, an organ-specific autoimmune disease in which insulin-producing β cells are mainly destroyed by not only Th1 but also Th17 lymphocytes (87). The autoreactive CD4+ T cells that infiltrate the pancreas present a proinflammatory phenotype, characterized by IFN-γ secretion (88), and support cytotoxic T lymphocytes (Tc), which are responsible for the progressive destruction of β cells (55). Although the later steps of T1D development are well known, the initial steps remain unclear.
The role of TLRs–PAMP interactions during T1D onset has been investigated in several animal models. Studies performed in BioBreeding Diabetes Resistant (BBDR) rats infected with the ssDNA parvoviruses Kilham rat virus (KRV) showed that KRV acts as a TLR9 ligand by upregulating proinflammatory cytokines in pancreatic lymph nodes, thus inducing islet destruction (89). Pretreatment with poly I:C, a TLR3 ligand, acts synergistically with KRV to induce diabetes in 100% of infected rats (90). Also, it has been demonstrated that the activation of APCs through TLR2-mediated sensing of β cells death contributes to T1D initiation in non-obese (NOD) mice (91). In contrast with these findings, Wen et al. (58) investigating the effects of MyD88 gene disruption on disease incidence and progression in NOD mice showed that MyD88-deficient NOD mice did not develop T1D, and that the observed protection was dependent on commensal microorganisms once GF MyD88-deficient NOD mice developed disease. The authors also demonstrated that the gut microbiota composition was changed by MyD88 deficiency and colonization of GF MyD88-deficient NOD mice with the bacterial community termed “altered Schedler’s flora” (ASF), normally present in healthy mouse gut, attenuated T1D. Thus, it can be suggested that in the absence of TLRs signaling, some bacterial groups predominate and induce tolerogenic responses. However, the receptors and signaling pathways involved in microbiota-dependent protection against T1D development remain unclear.
Some protective signals against T1D development triggered by the gut microbiota have been revealed through studies with NOD mice lacking MyD88 crossed with mice deficient for other components of the innate immune response. As ASF bacteria colonization has reduced T1D in GF MyD88-deficient NOD mice, it is possible that several signaling pathways activated by TLR agonists could contribute to protection when MyD88 signaling is absent. In fact, stimulation of TLR3 and TLR4 induces the activation of a MyD88-independent, TRIF-dependent signaling pathway (55), which has been pointed out as a negative regulator of immunity. In this context, Burrows and colleagues (92) have demonstrated that TRIF deficiency did not affect T1D development in Myd88 sufficient NOD mice, while double-deficient mice were more susceptible to T1D. Thus, it could be suggested that TLR3- and TLR4-mediated sensing of the gut microbiota participates in the protection against T1D development in MyD88-deficient NOD mice through tolerating mechanisms involving TRIF signaling.
Protective effects from TLR–microbiota interaction have also been demonstrated in MS studies. The MS is an immune-mediated chronic disease in which demyelination and axonal damage are caused by infiltration of myelin-specific autoreactive Th1 and Th17 cells into the central nervous system (CNS) in either relapsing/remitting or progressive condition (93, 94). CNS-derived peptides, such as the melanocortin antagonist agouti-related peptide (AgRP) and neuropeptide Y (NPY), are capable of modulating food intake and physiological processes that control nutrient absorption, which may influence the gut microbiota composition. In turn, gut microbiota modulates brain functions by releasing SCFAs and antigens, such as lipopolysaccharide (LPS), polysaccharide A (PSA), and LTA (95).
Miyake and colleagues (96) have investigated whether the gut microbiota was altered in MS by comparing the gut microbiota of 20 patients with relapsing–remitting (RR) MS with that of 40 healthy subjects. They found differences in the relative abundance of both archaea and butyrate-producing bacteria when comparing MS patients and healthy individuals and a significant reduction of clostridial species in MS patients. Interestingly, none of the clostridial species that were significantly reduced in the gut microbiota of MS patients overlapped with other spore-forming clostridial species capable of inducing colonic regulatory T cells, which have been associated with protection from autoimmunity and allergies (97). However, it has been shown that reconstitution of the gut microbiota from antibiotic-treated mice with Bacteroides fragilis protected against experimental autoimmune encephalomyelitis (EAE) by a mechanism dependent on PSA-induced IL-10-producing Treg cells, which may rely on TLR2 signaling (98). In fact, B. fragilis-derived PSA converts effector CD4+ T cells into IL-10-producing T cells in vitro by a TLR2-dependent mechanism (99). On the other hand, TLR4-mediated sensing of LPS has been shown to stimulate in vitro differentiation of Th17 cells and subcutaneous LPS injection increased the frequency of IL-17-producing cells in inguinal lymph nodes, worsening EAE (100).
Numerous studies have focused on the role of B cells during MS pathogenesis (101, 102). B cell-deficient C57Bl/6 mice suffer an exacerbate EAE form and transfer of IL-10-producing B cells into IL-10-deficient mice protected them from disease (101). In fact, it has been recently shown that microbiota-driven IL-1β and IL-6 production promotes regulatory B cells differentiation (103). Lampropoulou et al. (102) showed that TLR-activated B cells produce IL-10 in a MyD88-dependent manner by stimulating TLR4 and TLR9 with LPS and CpG oligonucleotides, respectively. These B cells limit the capacity of DC to produce IL-12 and restrain Th1 differentiation. Therefore, new studies are fundamental to elucidate the role of different TLRs in the context of EAE modulation, which seems to depend on microbiota-driven inflammatory and regulatory immune cells.
Inflammasomes are multimeric protein complexes typically composed of a sensor protein, the adaptor protein ASC, and the proinflammatory caspase-1, which can be triggered by a variety of stimuli associated with infection and cellular stress (104). Inflammasome activation results in recruitment of ASC, proteolytic cleavage, and activation of caspase-1, which leads to process and release of the proinflammatory cytokines IL-1β and IL-18 (105). The majority of inflammasomes contain a NLR sensor molecule, namely NLRP1 (NOD-, LRR-, and pyrin domain-containing 1), NLRP3, NLRP6, NLRP7, NLRP12, or NLRC4 (NOD-, LRR-, and CARD-containing 4). However, other two inflammasomes have been described containing the pyrin and HIN domain-containing protein (PYHIN) family members absent in melanoma 2 (AIM2) and IFN-γ-inducible protein 16 (IFI16) as sensor molecules (106). A non-canonical activation pathway of inflammatory caspases 4/5 in humans and caspase-11 in mice has also been described and depends on intracellular sensing of lipopolysaccharide (LPS) released upon Gram-negative bacteria escape from vacuoles (107).
Inflammasome signaling has been extensively studied in macrophages in different contexts (104, 108), but little is known about inflammasome expression and function in cells located in the gut. Intestinal CD11b+F4/80+ mononuclear phagocytes that normally reside in the lamina propria were shown to be anergic to ligands for TLRs or commensals but to produce IL-1β upon NLRC4 activation after infection with pathogenic Salmonella or Pseudomonas (109). More recently, Enterobacteriaceae and the pathobiont Proteus mirabilis were shown to induce robust IL-1β production through NLRP3 activation in newly recruited intestinal Ly6Chigh monocytes upon epithelial injury (110). IECs, the first cellular barrier toward the gut lumen, were also shown to express a variety of inflammasome components such as NAIPs 1, 2, 5, and 6 in mice (hNAIP in humans), NLRC4, NLRP1, NLRP6, AIM2, caspase-1, caspase-11 (-4, in humans), ASC, and IL-18. These inflammasome components contribute to intestinal homeostasis by regulating commensals’ ecology, by restricting pathogens, and by restoring epithelial barrier integrity (105). However, further studies are still necessary to shed light on how inflammasomes are regulated in both hematopoietic and non-hematopoietic intestinal cells in face of commensal microorganisms, diet-derived antigens, and pathogens to maintain homeostasis and systemic immunity.
The NLRP6 inflammasome has been shown to regulate intestinal microbiota ecology as metagenomic analysis has revealed pronounced dysbiosis in NLRP6-deficient mice (111, 112). Changes in the biogeographical distribution of microbiota are also observed in NLRP6 deficiency, leading to accumulation of commensal microorganisms in the colonic crypts. Reduced IL-18 levels and increased relative abundance of bacterial phyla Bacteroidetes (Prevotellaceae) and TM7 have been observed in NLRP6-deficient mice, which present spontaneous intestinal hyperplasia, chemokine (C–C motif) ligand 5 (CCL5)-mediated inflammatory cell recruitment, and exacerbation of DSS-induced colitis. Cross-fostering and cohousing experiments were sufficient to transfer dysbiotic microbiota to neonatal and adult wild-type (WT) mice, leading to increased susceptibility to DSS-induced colitis. Although deficiency in other inflammasome components, including ASC, caspase-1, and IL-18, also led to dysbiosis and exacerbated colitis, deficiency in NLRC4, NLRP10, NLRP12, and AIM2 had no impact on the susceptibility of WT mice to colitis upon cohousing (111). These mice were shown to have a distinct configuration of their microbiota when compared to NLRP6-deficient mice, which might explain the different results and suggests that although inflammasomes share many of their effector molecules, different impacts on microbiota ecology can be observed.
It has been recently shown that mice deficient in NLRP6 and the inflammasome components ASC and caspase-1 lack a thick continuous overlaying inner mucus layer in the gut due to abrogated mucus secretion by IECs and are unable to clear enteric pathogens from mucosal surface (113). As mucus has an important role in regulating host–microbial interactions, it is likely that dysbiosis and increased intestinal inflammation observed in NLRP6 mice are consequences of this reduced mucus secretion by epithelial cells. Deficiency in NLRP6 and IL-18 has also been linked to colitis-related colorectal cancer (CRC) development. Enhanced tumorigenesis in these mice was dependent on microbiota-induced CCL5-driven inflammation and local IL-6 production and could be transferred to WT mice upon cohousing (114).
Corticotropin-releasing hormone (CRH)-mediated reduction of intestinal NLRP6 expression in mice exposed to water-avoidance stress (WAS) has been shown to result in altered gut microbiota and acute small intestinal inflammation. These mice presented intestinal erythema, leukocyte infiltration, increased intestinal permeability, and increased mucosal expression of IL-17 and IL-6. Other inflammasome components that had their expression partially inhibited by WAS were ASC, caspase-1, IL-1β, and IL-18, while no significant impact was observed on NLRP3 expression. As observed for colitis and colitis-related CRC, non-stressed mice developed enteritis upon cohousing with WAS-exposed mice and probiotic therapy prior to WAS reduced intestinal inflammation and prevented WAS-mediated dysbiosis (115).
NLRP3-deficient mice have also been shown to be more susceptible to DSS-induced colitis, to have a dysbiotic microbiota, and to present altered colonic β-defensin expression and decreased antimicrobial capacity. Their neutrophils exhibited impaired chemotaxis and enhanced spontaneous apoptosis (116). Modulation of NLRP3 in the gut epithelium by high-fiber feeding has been suggested to contribute to intestinal homeostasis and protection from colitis through maintenance of a healthy microbiota and due to SCFA-mediated sensing by G-protein-coupled receptors GPR43 and GPR109A and IL-18 release (117). However, upon epithelial injury, members of the gut microbiota, such as Enterobacteriaceae, and in particular the pathobiont P. mirabilis induce NLRP3-mediated IL-1β release by Ly6Chigh monocytes, leading to intestinal inflammation (110).
The influence of inflammasome-mediated sensing of the microbiota goes beyond inflammatory processes in the gut. NLRP3, NLRP6, and inflammasome components ASC, caspase-1, and IL-18-deficiency-associated dysbiosis leads to exacerbated hepatic steatosis and inflammation through influx of TLR4 and TLR9 agonists into the portal circulation, which results in enhanced hepatic TNF-α expression and non-alcoholic steatohepatitis (NASH)/non-alcoholic fatty liver disease (NAFLD) progression. Cohousing experiments were sufficient to exacerbate NASH in WT mice, suggesting that intestinal dysbiosis may govern initial steps of systemic autoinflammatory disorders (118).
It has been shown that Pstpip2cmo mice, which are prone to develop osteomyelitis, present dysbiotic intestinal microbiota characterized by Prevotella outgrowth. Interestingly, these mice were protected from inflammatory bone disease and bone erosion when fed with high-fat diet (HFD), which had a marked impact on intestinal Prevotella reduction and significantly reduced pro-IL-1β expression in neutrophils. Antibiotic treatment was also efficient in reducing pro-IL-1β expression in Pstpip2cmo mice, but cohousing experiments and fecal microbiota transplantation from Pstpip2cmo mice failed to cause disease in WT mice. However, fecal microbiota transplantation from low-fat diet (LFD)-fed diseased Pstpip2cmo mice to young LFD-fed Pstpip2cmo mice by oral gavage promoted the expansion of Prevotella, and significantly accelerated the development of osteomyelitis, whereas transplantation of fecal microbiota from HFD-fed Pstpip2cmo mice to young LFD-fed Pstpip2cmo mice limited Prevotella outgrowth and significantly protected mice from developing osteomyelitis. Caspase-1/8-mediated processing of microbiota-induced pro-IL-1β was necessary to promote autoinflammatory disease in these susceptible mice (119).
Extraintestinal inflammatory processes have also been shown to be partially dependent on inflammasome-mediated sensing of the microbiota metabolite acetate. GF, antibiotic-treated, and GPR43-dificient mice are protected from joint inflammation upon injection of monosodium urate monohydrate (MSU) crystals. It was demonstrated that microbiota reconstitution or acetate administration to GF mice restored MSU crystals-induced inflammation and that GPR43 is at least partially necessary to adequate inflammasome assembly and IL-1β production in response to acetate (120). Therefore, one may suggest that inflammasome-mediated sensing of both intestinal microbiota and their products contributes to control local and systemic inflammatory disorders, which should be considered when designing prophylactic and therapeutic applications.
Many studies have focused on the interactions between inflammasomes and TLRs. In fact, it has been shown that the enteric pathogens E. coli- and Citrobacter rodentium-induced TLR4/TRIF-dependent synthesis of caspase-11 and activation of NLRP3 inflammasome in macrophages (121) and that Pseudomonas aeruginosa infection of macrophages induced TLR-4/TRIF-dependent autophagy, which was attenuated by NLRC4/caspase-1-mediated cleavage of TRIF. Prevention of in vivo caspase-1-mediated cleavage of TRIF resulted in enhanced autophagy, reduced IL-1β production, and increased bacterial clearance (122). L. monocytogenes infection of macrophages was shown to induce TLR/IRAK1/IRAK4-dependent activation of NLRP3 (123, 124), which suggests that inflammasome–TLR interactions may have an important role in the bacterial recognition.
It has been also demonstrated that Clathrin-mediated endocytosis followed by TLR8- and TLR7-mediated recognition of HIV and HCV in monocytes and macrophages resulted in NLRP3 activation, which was independent of type I IFN production (125). Moreover, dendritic cells were shown to express higher levels of NLRP3 in the steady-state condition compared to macrophages and to secrete substantial amounts of mature IL-1β upon stimulation with TLR ligands independently of P2X7 signaling (126). On the other hand, chronic TLR stimulation by LPS has been shown to dampen NLRP3 activation through IL-10 induction (127), and TLR2/TLR4 engagement resulted in upregulation of plasminogen activator inhibitor type 2 (PAI-2), a serine protease inhibitor, which culminated in the stabilization of the autophagic protein Beclin 1 to promote autophagy, reduction of mitochondrial reactive oxygen species, NLRP3 protein level, and pro-IL-1β processing (128).
Although increasing evidence suggests that both TLRs and inflammasomes play an important role in promoting host–microbiota communication, studies regarding the interactions of these molecules in the context of the gut microbiota are still lacking and will further improve our knowledge on how commensal microorganisms and the immune system cooperate in the modulation of inflammatory and autoimmune diseases.
The dynamic interactions that have been established between mammalian hosts and commensal microorganisms over millennia of coevolution resulted in the development of a well-structured immune system, which controls pathogenic infections while tolerates a highly diverse microbiota. Innate sensing of the gut microbiota through TLRs and inflammasomes contributes to intestinal homeostasis by stimulating the development/function of both regulatory and inflammatory cells and by promoting proliferation of IECs, epithelial integrity, mucus secretion, and containment of opportunistic infections. It has become clear that disturbances in the fine-tuning that governs host–microbiota interactions may lead to both local and systemic inflammatory and autoimmune diseases. In this review, we bring many examples of the dualistic role played by the gut microbiota in both promoting and regulating inflammation and autoimmunity. We believe that a better understanding of mechanisms involved in these complex interactions between host and commensals will further improve common prophylactic and therapeutic applications.
AI, CM, and RA wrote the manuscript. NC and RA reviewed the manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We gratefully acknowledge funding provided by CNPq and Sao Paulo State Research Funding Agency (FAPESP) (12/02270-2). RA, AI, and CM are recipients of FAPESP fellowships.
1. Tlaskalova-Hogenova H, Stepankova R, Kozakova H, Hudcovic T, Vannucci L, Tuckova L, et al. The role of gut microbiota (commensal bacteria) and the mucosal barrier in the pathogenesis of inflammatory and autoimmune diseases and cancer: contribution of germ-free and gnotobiotic animal models of human diseases. Cell Mol Immunol (2011) 8(2):110–20. doi: 10.1038/cmi.2010.67
2. Mogensen TH. Pathogen recognition and inflammatory signaling in innate immune defenses. Clin Microbiol Rev (2009) 22(2):240–73; Table of Contents. doi:10.1128/CMR.00046-08
3. Mach J, Hshieh T, Hsieh D, Grubbs N, Chervonsky A. Development of intestinal M cells. Immunol Rev (2005) 206:177–89. doi:10.1111/j.0105-2896.2005.00281.x
4. Rescigno M. Intestinal dendritic cells. Adv Immunol (2010) 107:109–38. doi:10.1016/B978-0-12-381300-8.00004-6
5. Farache J, Koren I, Milo I, Gurevich I, Kim KW, Zigmond E, et al. Luminal bacteria recruit CD103+ dendritic cells into the intestinal epithelium to sample bacterial antigens for presentation. Immunity (2013) 38(3):581–95. doi:10.1016/j.immuni.2013.01.009
6. Schulz O, Pabst O. Antigen sampling in the small intestine. Trends Immunol (2013) 34(4):155–61. doi:10.1016/j.it.2012.09.006
7. Botos I, Segal DM, Davies DR. The structural biology of toll-like receptors. Structure (2011) 19(4):447–59. doi:10.1016/j.str.2011.02.004
8. Pålsson-McDermott EM, O’Neill LA. The potential of targeting toll-like receptor 2 in autoimmune and inflammatory diseases. Ir J Med Sci (2007) 176(4):253–60. doi:10.1007/s11845-007-0103-1
9. Daniele SG, Béraud D, Davenport C, Cheng K, Yin H, Maguire-Zeiss KA. Activation of MyD88-dependent TLR1/2 signaling by misfolded α-synuclein, a protein linked to neurodegenerative disorders. Sci Signal (2015) 8(376):ra45. doi:10.1126/scisignal.2005965
10. Henrick BM, Yao XD, Rosenthal KL; INFANT Study Team. HIV-1 structural proteins serve as PAMPs for TLR2 heterodimers significantly increasing infection and innate immune activation. Front Immunol (2015) 6:426. doi:10.3389/fimmu.2015.00426
11. Bieback K, Lien E, Klagge IM, Avota E, Schneider-Schaulies J, Duprex WP, et al. Hemagglutinin protein of wild-type measles virus activates toll-like receptor 2 signaling. J Virol (2002) 76(17):8729–36. doi:10.1128/JVI.76.17.8729-8736.2002
12. Dolganiuc A, Oak S, Kodys K, Golenbock DT, Finberg RW, Kurt-Jones E, et al. Hepatitis C core and nonstructural 3 proteins trigger toll-like receptor 2-mediated pathways and inflammatory activation. Gastroenterology (2004) 127(5):1513–24. doi:10.1053/j.gastro.2004.08.067
13. Boehme KW, Guerrero M, Compton T. Human cytomegalovirus envelope glycoproteins B and H are necessary for TLR2 activation in permissive cells. J Immunol (2006) 177(10):7094–102. doi:10.4049/jimmunol.177.10.7094
14. Leoni V, Gianni T, Salvioli S, Campadelli-Fiume G. Herpes simplex virus glycoproteins gH/gL and gB bind toll-like receptor 2, and soluble gH/gL is sufficient to activate NF-κB. J Virol (2012) 86(12):6555–62. doi:10.1128/JVI.00295-12
15. Cario E. Barrier-protective function of intestinal epithelial toll-like receptor 2. Mucosal Immunol (2008) 1(Suppl 1):S62–6. doi:10.1038/mi.2008.47
16. Bunk S, Sigel S, Metzdorf D, Sharif O, Triantafilou K, Triantafilou M, et al. Internalization and coreceptor expression are critical for TLR2-mediated recognition of lipoteichoic acid in human peripheral blood. J Immunol (2010) 185(6):3708–17. doi:10.4049/jimmunol.0901660
17. Oliveira-Nascimento L, Massari P, Wetzler LM. The role of TLR2 in infection and immunity. Front Immunol (2012) 3:79. doi:10.3389/fimmu.2012.00079
18. Pinke KH, Lima HG, Cunha FQ, Lara VS. Mast cells phagocyte Candida albicans and produce nitric oxide by mechanisms involving TLR2 and Dectin-1. Immunobiology (2016) 221(2):220–7. doi:10.1016/j.imbio.2015.09.004
19. Karumuthil-Melethil S, Sofi MH, Gudi R, Johnson BM, Perez N, Vasu C. TLR2- and Dectin 1-associated innate immune response modulates T-cell response to pancreatic β-cell antigen and prevents type 1 diabetes. Diabetes (2015) 64(4):1341–57. doi:10.2337/db14-1145
20. Royet J, Dziarski R. Peptidoglycan recognition proteins: pleiotropic sensors and effectors of antimicrobial defences. Nat Rev Microbiol (2007) 5(4):264–77. doi:10.1038/nrmicro1620
21. Alexopoulou L, Holt AC, Medzhitov R, Flavell RA. Recognition of double-stranded RNA and activation of NF-kappaB by toll-like receptor 3. Nature (2001) 413(6857):732–8. doi:10.1038/35099560
22. Vontell R, Supramaniam V, Wyatt-Ashmead J, Gressens P, Rutherford M, Hagberg H, et al. Cellular mechanisms of toll-like receptor-3 activation in the thalamus are associated with white matter injury in the developing brain. J Neuropathol Exp Neurol (2015) 74(3):273–85. doi:10.1097/NEN.0000000000000172
23. Shibata T, Ohto U, Nomura S, Kibata K, Motoi Y, Zhang Y, et al. Guanosine and its modified derivatives are endogenous ligands for TLR7. Int Immunol (2015). doi:10.1093/intimm/dxv062
24. Diebold SS, Kaisho T, Hemmi H, Akira S, Reis e Sousa C. Innate antiviral responses by means of TLR7-mediated recognition of single-stranded RNA. Science (2004) 303(5663):1529–31. doi:10.1126/science.1093616
25. Heil F, Hemmi H, Hochrein H, Ampenberger F, Kirschning C, Akira S, et al. Species-specific recognition of single-stranded RNA via toll-like receptor 7 and 8. Science (2004) 303(5663):1526–9. doi:10.1126/science.1093620
26. Chow JC, Young DW, Golenbock DT, Christ WJ, Gusovsky F. Toll-like receptor-4 mediates lipopolysaccharide-induced signal transduction. J Biol Chem (1999) 274(16):10689–92. doi:10.1074/jbc.274.16.10689
27. Kurt-Jones EA, Popova L, Kwinn L, Haynes LM, Jones LP, Tripp RA, et al. Pattern recognition receptors TLR4 and CD14 mediate response to respiratory syncytial virus. Nat Immunol (2000) 1(5):398–401. doi:10.1038/80833
28. Yoon SI, Kurnasov O, Natarajan V, Hong M, Gudkov AV, Osterman AL, et al. Structural basis of TLR5-flagellin recognition and signaling. Science (2012) 335(6070):859–64. doi:10.1126/science.1215584
29. Bauer S, Kirschning CJ, Häcker H, Redecke V, Hausmann S, Akira S, et al. Human TLR9 confers responsiveness to bacterial DNA via species-specific CpG motif recognition. Proc Natl Acad Sci U S A (2001) 98(16):9237–42. doi:10.1073/pnas.161293498
30. Regan T, Nally K, Carmody R, Houston A, Shanahan F, Macsharry J, et al. Identification of TLR10 as a key mediator of the inflammatory response to Listeria monocytogenes in intestinal epithelial cells and macrophages. J Immunol (2013) 191(12):6084–92. doi:10.4049/jimmunol.1203245
31. Guan Y, Ranoa DR, Jiang S, Mutha SK, Li X, Baudry J, et al. Human TLRs 10 and 1 share common mechanisms of innate immune sensing but not signaling. J Immunol (2010) 184(9):5094–103. doi:10.4049/jimmunol.0901888
32. Oosting M, Cheng SC, Bolscher JM, Vestering-Stenger R, Plantinga TS, Verschueren IC, et al. Human TLR10 is an anti-inflammatory pattern-recognition receptor. Proc Natl Acad Sci U S A (2014) 111(42):E4478–84. doi:10.1073/pnas.1410293111
33. Shi Z, Cai Z, Yu J, Zhang T, Zhao S, Smeds E, et al. Toll-like receptor 11 (TLR11) prevents Salmonella penetration into the murine Peyer patches. J Biol Chem (2012) 287(52):43417–23. doi:10.1074/jbc.M112.411009
34. Anders HJ, Patole PS. Toll-like receptors recognize uropathogenic Escherichia coli and trigger inflammation in the urinary tract. Nephrol Dial Transplant (2005) 20(8):1529–32. doi:10.1093/ndt/gfh922
35. Yarovinsky F, Zhang D, Andersen JF, Bannenberg GL, Serhan CN, Hayden MS, et al. TLR11 activation of dendritic cells by a protozoan profilin-like protein. Science (2005) 308(5728):1626–9. doi:10.1126/science.1109893
36. Andrade WA, Souza Mdo C, Ramos-Martinez E, Nagpal K, Dutra MS, Melo MB, et al. Combined action of nucleic acid-sensing toll-like receptors and TLR11/TLR12 heterodimers imparts resistance to Toxoplasma gondii in mice. Cell Host Microbe (2013) 13(1):42–53. doi:10.1016/j.chom.2012.12.003
37. Oldenburg M, Krüger A, Ferstl R, Kaufmann A, Nees G, Sigmund A, et al. TLR13 recognizes bacterial 23S rRNA devoid of erythromycin resistance-forming modification. Science (2012) 337(6098):1111–5. doi:10.1126/science.1220363
38. Yu L, Wang L, Chen S. Endogenous toll-like receptor ligands and their biological significance. J Cell Mol Med (2010) 14(11):2592–603. doi:10.1111/j.1582-4934.2010.01127.x
39. Rakoff-Nahoum S, Paglino J, Eslami-Varzaneh F, Edberg S, Medzhitov R. Recognition of commensal microflora by toll-like receptors is required for intestinal homeostasis. Cell (2004) 118(2):229–41. doi:10.1016/j.cell.2004.07.002
40. Brown SL, Riehl TE, Walker MR, Geske MJ, Doherty JM, Stenson WF, et al. Myd88-dependent positioning of Ptgs2-expressing stromal cells maintains colonic epithelial proliferation during injury. J Clin Invest (2007) 117(1):258–69. doi:10.1172/JCI29159
41. Mukherji A, Kobiita A, Ye T, Chambon P. Homeostasis in intestinal epithelium is orchestrated by the circadian clock and microbiota cues transduced by TLRs. Cell (2013) 153(4):812–27. doi:10.1016/j.cell.2013.04.020
42. Hyun J, Romero L, Riveron R, Flores C, Kanagavelu S, Chung KD, et al. Human intestinal epithelial cells express interleukin-10 through toll-like receptor 4-mediated epithelial-macrophage crosstalk. J Innate Immun (2015) 7(1):87–101. doi:10.1159/000365417
43. Biswas A, Wilmanski J, Forsman H, Hrncir T, Hao L, Tlaskalova-Hogenova H, et al. Negative regulation of toll-like receptor signaling plays an essential role in homeostasis of the intestine. Eur J Immunol (2011) 41(1):182–94. doi:10.1002/eji.201040479
44. Lim DM, Narasimhan S, Michaylira CZ, Wang ML. TLR3-mediated NF-{kappa}B signaling in human esophageal epithelial cells. Am J Physiol Gastrointest Liver Physiol (2009) 297(6):G1172–80. doi:10.1152/ajpgi.00065.2009
45. Verbeek RE, Siersema PD, Ten Kate FJ, Fluiter K, Souza RF, Vleggaar FP, et al. Toll-like receptor 4 activation in Barrett’s esophagus results in a strong increase in COX-2 expression. J Gastroenterol (2014) 49(7):1121–34. doi:10.1007/s00535-013-0862-6
46. Pimentel-Nunes P, Afonso L, Lopes P, Roncon-Albuquerque R, Gonçalves N, Henrique R, et al. Increased expression of toll-like receptors (TLR) 2, 4 and 5 in gastric dysplasia. Pathol Oncol Res (2011) 17(3):677–83. doi:10.1007/s12253-011-9368-9
47. Nishimura M, Naito S. Tissue-specific mRNA expression profiles of human toll-like receptors and related genes. Biol Pharm Bull (2005) 28(5):886–92. doi:10.1248/bpb.28.886
48. Bogunovic M, Davé SH, Tilstra JS, Chang DT, Harpaz N, Xiong H, et al. Enteroendocrine cells express functional toll-like receptors. Am J Physiol Gastrointest Liver Physiol (2007) 292(6):G1770–83. doi:10.1152/ajpgi.00249.2006
49. Furrie E, Macfarlane S, Thomson G, Macfarlane GT, Group MGB, Bank TTT. Toll-like receptors-2, -3 and -4 expression patterns on human colon and their regulation by mucosal-associated bacteria. Immunology (2005) 115(4):565–74. doi:10.1111/j.1365-2567.2005.02200.x
50. Vamadevan AS, Fukata M, Arnold ET, Thomas LS, Hsu D, Abreu MT. Regulation of toll-like receptor 4-associated MD-2 in intestinal epithelial cells: a comprehensive analysis. Innate Immun (2010) 16(2):93–103. doi:10.1177/1753425909339231
51. Pott J, Stockinger S, Torow N, Smoczek A, Lindner C, McInerney G, et al. Age-dependent TLR3 expression of the intestinal epithelium contributes to rotavirus susceptibility. PLoS Pathog (2012) 8(5):e1002670. doi:10.1371/journal.ppat.1002670
52. Eaves-Pyles T, Bu HF, Tan XD, Cong Y, Patel J, Davey RA, et al. Luminal-applied flagellin is internalized by polarized intestinal epithelial cells and elicits immune responses via the TLR5 dependent mechanism. PLoS One (2011) 6(9):e24869. doi:10.1371/journal.pone.0024869
53. Lee J, Mo JH, Katakura K, Alkalay I, Rucker AN, Liu YT, et al. Maintenance of colonic homeostasis by distinctive apical TLR9 signalling in intestinal epithelial cells. Nat Cell Biol (2006) 8(12):1327–36. doi:10.1038/ncb1500
54. Chabot S, Wagner JS, Farrant S, Neutra MR. TLRs regulate the gatekeeping functions of the intestinal follicle-associated epithelium. J Immunol (2006) 176(7):4275–83. doi:10.4049/jimmunol.176.7.4275
55. Gianchecchi E, Fierabracci A. Gene/environment interactions in the pathogenesis of autoimmunity: new insights on the role of toll-like receptors. Autoimmun Rev (2015) 14(11):971–83. doi:10.1016/j.autrev.2015.07.006
56. Abdollahi-Roodsaz S, Joosten LAB, Koenders MI, Devesa I, Roelofs MF, Radstake TRDJ, et al. Stimulation of TLR2 and TLR4 differentially skews the balance of T cells in a mouse model of arthritis. J Clin Invest (2008) 118(1):205–16. doi:10.1172/jci32639
57. Geremia A, Biancheri P, Allan P, Corazza GR, Di Sabatino A. Innate and adaptive immunity in inflammatory bowel disease. Autoimmun Rev (2014) 13(1):3–10. doi:10.1016/j.autrev.2013.06.004
58. Wen L, Ley RE, Volchkov PY, Stranges PB, Avanesyan L, Stonebraker AC, et al. Innate immunity and intestinal microbiota in the development of Type 1 diabetes. Nature (2008) 455(7216):1109–13. doi:10.1038/nature07336
59. Klareskog L, Padyukov L, Ronnelid J, Alfredsson L. Genes, environment and immunity in the development of rheumatoid arthritis. Curr Opin Immunol (2006) 18(6):650–5. doi:10.1016/j.coi.2006.06.004
60. Milo R, Kahana E. Multiple sclerosis: geoepidemiology, genetics and the environment. Autoimmun Rev (2010) 9(5):A387–94. doi:10.1016/j.autrev.2009.11.010
61. Danese S, Fiocchi C. Medical progress ulcerative colitis. N Engl J Med (2011) 365(18):1713–25. doi:10.1056/NEJMra1102942
62. Chen S-J, Liu X-W, Liu J-P, Yang X-Y, Lu F-G. Ulcerative colitis as a polymicrobial infection characterized by sustained broken mucus barrier. World J Gastroenterol (2014) 20(28):9468–75. doi:10.3748/wjg.v20.i28.9468
63. Kuhn KA, Pedraza I, Demoruelle MK. Mucosal immune responses to microbiota in the development of autoimmune disease. Rheum Dis Clin North Am (2014) 40(4):711–25. doi:10.1016/j.rdc.2014.07.013
64. Abreu MT. Toll-like receptor signalling in the intestinal epithelium: how bacterial recognition shapes intestinal function. Nat Rev Immunol (2010) 10(2):131–44. doi:10.1038/nri2707
65. Rogier R, Koenders MI, Abdollahi-Roodsaz S. Toll-like receptor mediated modulation of T cell response by commensal intestinal microbiota as a trigger for autoimmune arthritis. J Immunol Res (2015) 2015:527696. doi:10.1155/2015/527696
66. Vaahtovuo J, Munukka E, Korkeamäki M, Luukkainen R, Toivanen P. Fecal microbiota in early rheumatoid arthritis. J Rheumatol (2008) 35(8):1500–5.
67. Gérard HC, Wang Z, Wang GF, El-Gabalawy H, Goldbach-Mansky R, Li Y, et al. Chromosomal DNA from a variety of bacterial species is present in synovial tissue from patients with various forms of arthritis. Arthritis Rheum (2001) 44(7):1689–97. doi:10.1002/1529-0131(200107)44:7<1689:AID-ART293>3.0.CO;2-K
68. Abdollahi-Roodsaz S, Joosten LA, Roelofs MF, Radstake TR, Matera G, Popa C, et al. Inhibition of toll-like receptor 4 breaks the inflammatory loop in autoimmune destructive arthritis. Arthritis Rheum (2007) 56(9):2957–67. doi:10.1002/art.22848
69. Wu H-J, Ivanov II, Darce J, Hattori K, Shima T, Umesaki Y, et al. Gut-residing segmented filamentous bacteria drive autoimmune arthritis via T helper 17 cells. Immunity (2010) 32(6):815–27. doi:10.1016/j.immuni.2010.06.001
70. Shaw MH, Kamada N, Kim Y-G, Nunez G. Microbiota-induced IL-1 beta, but not IL-6, is critical for the development of steady-state T(H)17 cells in the intestine. J Exp Med (2012) 209(2):251–8. doi:10.1084/jem.20111703
71. Taneja V. Arthritis susceptibility and the gut microbiome. FEBS Lett (2014) 588(22):4244–9. doi:10.1016/j.febslet.2014.05.034
72. Rosenstein ED, Weissmann G, Greenwald RA. Porphyromonas gingivalis, periodontitis and rheumatoid arthritis. Med Hypotheses (2009) 73(3):457–8. doi:10.1016/j.mehy.2009.04.008
73. Scher JU, Ubeda C, Equinda M, Khanin R, Buischi Y, Viale A, et al. Periodontal disease and the oral microbiota in new-onset rheumatoid arthritis. Arthritis Rheum (2012) 64(10):3083–94. doi:10.1002/art.34539
74. de Aquino SG, Abdollahi-Roodsaz S, Koenders MI, van de Loo FA, Pruijn GJ, Marijnissen RJ, et al. Periodontal pathogens directly promote autoimmune experimental arthritis by inducing a TLR2- and IL-1-driven Th17 response. J Immunol (2014) 192(9):4103–11. doi:10.4049/jimmunol.1301970
75. Qin J, Li R, Raes J, Arumugam M, Burgdorf KS, Manichanh C, et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature (2010) 464(7285):59–U70. doi:10.1038/nature08821
76. Willing B, Halfvarson J, Dicksved J, Rosenquist M, Jarnerot G, Engstrand L, et al. Twin studies reveal specific imbalances in the mucosa-associated microbiota of patients with ileal Crohn’s disease. Inflamm Bowel Dis (2009) 15(5):653–60. doi:10.1002/ibd.20783
77. Ey B, Eyking A, Klepak M, Salzman NH, Goethert JR, Ruenzi M, et al. Loss of TLR2 worsens spontaneous colitis in MDR1A deficiency through commensally induced pyroptosis. J Immunol (2013) 190(11):5676–88. doi:10.4049/jimmunol.1201592
78. Wang S, Charbonnier L-M, Rivas MN, Georgiev P, Li N, Gerber G, et al. MyD88 adaptor-dependent microbial sensing by regulatory T cells promotes mucosal tolerance and enforces commensalism. Immunity (2015) 43(2):289–303. doi:10.1016/j.immuni.2015.06.014
79. Vlantis K, Polykratis A, Welz P-S, Van Loo G, Pasparakis M, Wullaert A. TLR-independent anti-inflammatory function of intestinal epithelial TRAF6 signalling prevents DSS-induced colitis in mice. Gut (2015). doi:10.1136/gutjnl-2014-308323
80. Cario E. Toll-like receptors in inflammatory bowel diseases: a decade later. Inflamm Bowel Dis (2010) 16(9):1583–97. doi:10.1002/ibd.21282
81. Cario E, Podolsky DK. Differential alteration in intestinal epithelial cell expression of toll-like receptor 3 (TLR3) and TLR4 in inflammatory bowel disease. Infect Immun (2000) 68(12):7010–7. doi:10.1128/IAI.68.12.7010-7017.2000
82. Bates J, Diehl L. Dendritic cells in IBD pathogenesis: an area of therapeutic opportunity? J Pathol (2014) 232(2):112–20. doi:10.1002/path.4277
83. Baumgart DC, Thomas S, Przesdzing I, Metzke D, Bielecki C, Lehmann SM, et al. Exaggerated inflammatory response of primary human myeloid dendritic cells to lipopolysaccharide in patients with inflammatory bowel disease. Clin Exp Immunol (2009) 157(3):423–36. doi:10.1111/j.1365-2249.2009.03981.x
84. Jiménez-Dalmaroni MJ, Gerswhin ME, Adamopoulos IE. The critical role of toll-like receptors – from microbial recognition to autoimmunity: a comprehensive review. Autoimmun Rev (2015) 15(1):1–8. doi:10.1016/j.autrev.2015.08.009
85. Obermeier F, Strauch UG, Dunger N, Grunwald N, Rath HC, Herfarth H, et al. In vivo CpG DNA/toll-like receptor 9 interaction induces regulatory properties in CD4+CD62L+ T cells which prevent intestinal inflammation in the SCID transfer model of colitis. Gut (2005) 54(10):1428–36. doi:10.1136/gut.2004.046946
86. Kawashima T, Kosaka A, Yan H, Guo Z, Uchiyama R, Fukui R, et al. Double-stranded RNA of intestinal commensal but not pathogenic bacteria triggers production of protective interferon-beta. Immunity (2013) 38(6):1187–97. doi:10.1016/j.immuni.2013.02.024
87. Bluestone JA, Herold K, Eisenbarth G. Genetics, pathogenesis and clinical interventions in type 1 diabetes. Nature (2010) 464(7293):1293–300. doi:10.1038/nature08933
88. Gomez-Tourino I, Arif S, Eichmann M, Peakman M. T cells in type 1 diabetes: instructors, regulators and effectors: a comprehensive review. J Autoimmun (2015) 66:7–16. doi:10.1016/j.jaut.2015.08.012
89. Lien E, Zipris D. The role of toll-like receptor pathways in the mechanism of type 1 diabetes. Curr Mol Med (2009) 9(1):52–68. doi:10.2174/156652409787314453
90. Zipris D, Lien E, Xie JX, Greiner DL, Mordes JP, Rossini AA. TLR activation synergizes with Kilham rat virus infection to induce diabetes in BBDR rats. J Immunol (2005) 174(1):131–42. doi:10.4049/jimmunol.174.1.131
91. Kim HS, Han MS, Chung KW, Kim S, Kim E, Kim MJ, et al. Toll-like receptor 2 senses beta-cell death and contributes to the initiation of autoimmune diabetes. Immunity (2007) 27(2):321–33. doi:10.1016/j.immuni.2007.06.010
92. Burrows MP, Volchkov P, Kobayashi KS, Chervonsky AV. Microbiota regulates type 1 diabetes through toll-like receptors. Proc Natl Acad Sci U S A (2015) 112(32):9973–7. doi:10.1073/pnas.1508740112
93. Ochoa-Repáraz J, Kasper LH. Gut microbiome and the risk factors in central nervous system autoimmunity. FEBS Lett (2014) 588(22):4214–22. doi:10.1016/j.febslet.2014.09.024
94. Gandhi R, Laroni A, Weiner HL. Role of the innate immune system in the pathogenesis of multiple sclerosis. J Neuroimmunol (2010) 221(1–2):7–14. doi:10.1016/j.jneuroim.2009.10.015
95. Wang Y, Kasper LH. The role of microbiome in central nervous system disorders. Brain Behav Immun (2014) 38:1–12. doi:10.1016/j.bbi.2013.12.015
96. Miyake S, Kim S, Suda W, Oshima K, Nakamura M, Matsuoka T, et al. Dysbiosis in the gut microbiota of patients with multiple sclerosis, with a striking depletion of species belonging to clostridia XIVa and IV clusters. PLoS One (2015) 10(9):e0137429. doi:10.1371/journal.pone.0137429
97. Moudgil KD. Interplay among cytokines and T cell subsets in the progression and control of immune-mediated diseases. Cytokine (2015) 74(1):1–4. doi:10.1016/j.cyto.2015.05.006
98. Ochoa-Repáraz J, Mielcarz DW, Ditrio LE, Burroughs AR, Begum-Haque S, Dasgupta S, et al. Central nervous system demyelinating disease protection by the human commensal Bacteroides fragilis depends on polysaccharide A expression. J Immunol (2010) 185(7):4101–8. doi:10.4049/jimmunol.1001443
99. Round JL, Lee SM, Li J, Tran G, Jabri B, Chatila TA, et al. The toll-like receptor 2 pathway establishes colonization by a commensal of the human microbiota. Science (2011) 332(6032):974–7. doi:10.1126/science.1206095
100. Park J-H, Jeong S-Y, Choi A-J, Kim S-J. Lipopolysaccharide directly stimulates Th17 differentiation in vitro modulating phosphorylation of RelB and NF-kappa B1. Immunol Lett (2015) 165(1):10–9. doi:10.1016/j.imlet.2015.03.003
101. Fillatreau S, Sweenie CH, McGeachy MJ, Gray D, Anderton SM. B cells regulate autoimmunity by provision of IL-10. Nat Immunol (2002) 3(10):944–50. doi:10.1038/ni833
102. Lampropoulou V, Hoehlig K, Roch T, Neves P, Calderón Gómez E, Sweenie CH, et al. TLR-activated B cells suppress T cell-mediated autoimmunity. J Immunol (2008) 180(7):4763–73. doi:10.4049/jimmunol.180.7.4763
103. Rosser EC, Oleinika K, Tonon S, Doyle R, Bosma A, Carter NA, et al. Regulatory B cells are induced by gut microbiota-driven interleukin-1 beta and interleukin-6 production. Nat Med (2014) 20(11):1334–9. doi:10.1038/nm.3680
104. Schroder K, Tschopp J. The inflammasomes. Cell (2010) 140(6):821–32. doi:10.1016/j.cell.2010.01.040
105. Sellin ME, Maslowski KM, Maloy KJ, Hardt W-D. Inflammasomes of the intestinal epithelium. Trends Immunol (2015) 36(8):442–50. doi:10.1016/j.it.2015.06.002
106. Hornung V, Latz E. Intracellular DNA recognition. Nat Rev Immunol (2010) 10(2):123–30. doi:10.1038/nri2690
107. Yang J, Zhao Y, Shao F. Non-canonical activation of inflammatory caspases by cytosolic LPS in innate immunity. Curr Opin Immunol (2015) 32:78–83. doi:10.1016/j.coi.2015.01.007
108. Lamkanfi M, Dixit VM. Mechanisms and functions of inflammasomes. Cell (2014) 157(5):1013–22. doi:10.1016/j.cell.2014.04.007
109. Franchi L, Kamada N, Nakamura Y, Burberry A, Kuffa P, Suzuki S, et al. NLRC4-driven production of IL-1 beta discriminates between pathogenic and commensal bacteria and promotes host intestinal defense. Nat Immunol (2012) 13(5):449–U45. doi:10.1038/ni.2263
110. Seo S-U, Kamada N, Munoz-Planillo R, Kim Y-G, Kim D, Koizumi Y, et al. Distinct commensals induce interleukin-1 beta via NLRP3 inflammasome in inflammatory monocytes to promote intestinal inflammation in response to injury. Immunity (2015) 42(4):744–55. doi:10.1016/j.immuni.2015.03.004
111. Elinav E, Strowig T, Kau AL, Henao-Mejia J, Thaiss CA, Booth CJ, et al. NLRP6 inflammasome regulates colonic microbial ecology and risk for colitis. Cell (2011) 145(5):745–57. doi:10.1016/j.cell.2011.04.022
112. Anand PK, Malireddi RKS, Lukens JR, Vogel P, Bertin J, Lamkanfi M, et al. NLRP6 negatively regulates innate immunity and host defence against bacterial pathogens. Nature (2012) 488(7411):389–93. doi:10.1038/nature11250
113. Wlodarska M, Thaiss CA, Nowarski R, Henao-Mejia J, Zhang J-P, Brown EM, et al. NLRP6 inflammasome orchestrates the colonic host-microbial interface by regulating goblet cell mucus secretion. Cell (2014) 156(5):1045–59. doi:10.1016/j.cell.2014.01.026
114. Hu B, Elinav E, Huber S, Strowig T, Hao L, Hafemann A, et al. Microbiota-induced activation of epithelial IL-6 signaling links inflammasome-driven inflammation with transmissible cancer. Proc Natl Acad Sci U S A (2013) 110(24):9862–7. doi:10.1073/pnas.1307575110
115. Sun Y, Zhang M, Chen C-C, Gillilland M III, Sun X, El-Zaatari M, et al. Stress-induced corticotropin-releasing hormone-mediated nlrp6 inflammasome inhibition and transmissible enteritis in mice. Gastroenterology (2013) 144(7):1478–U272. doi:10.1053/j.gastro.2013.02.038
116. Hirota SA, Ng J, Lueng A, Khajah M, Parhar K, Li Y, et al. NLRP3 inflammasome plays a key role in the regulation of intestinal homeostasis. Inflamm Bowel Dis (2011) 17(6):1359–72. doi:10.1002/ibd.21478
117. Macia L, Tan J, Vieira AT, Leach K, Stanley D, Luong S, et al. Metabolite-sensing receptors GPR43 and GPR109A facilitate dietary fibre-induced gut homeostasis through regulation of the inflammasome. Nat Commun (2015) 6:6734. doi:10.1038/ncomms7734
118. Henao-Mejia J, Elinav E, Jin C, Hao L, Mehal WZ, Strowig T, et al. Inflammasome-mediated dysbiosis regulates progression of NAFLD and obesity. Nature (2012) 482(7384):179–U67. doi:10.1038/nature10809
119. Lukens JR, Gurung P, Vogel P, Johnson GR, Carter RA, McGoldrick DJ, et al. Dietary modulation of the microbiome affects autoinflammatory disease. Nature (2014) 516(7530):246–9. doi:10.1038/nature13788
120. Vieira AT, Macia L, Galvao I, Martins FS, Canesso MCC, Amaral FA, et al. A role for gut microbiota and the metabolite-sensing receptor GPR43 in a murine model of gout. Arthritis Rheumatol (2015) 67(6):1646–56. doi:10.1002/art.39107
121. Gurung P, Malireddi RKS, Anand PK, Demon D, Vande Walle L, Liu Z, et al. Toll or interleukin-1 receptor (TIR) domain-containing adaptor inducing interferon-beta (TRIF)-mediated caspase-11 protease production integrates toll-like receptor 4 (TLR4) protein- and Nlrp3 inflammasome-mediated host defense against enteropathogens. J Biol Chem (2012) 287(41):34474–83. doi:10.1074/jbc.M112.401406
122. Jabir MS, Ritchie ND, Li D, Bayes HK, Tourlomousis P, Puleston D, et al. Caspase-1 cleavage of the TLR adaptor TRIF inhibits autophagy and beta-interferon production during Pseudomonas aeruginosa infection. Cell Host Microbe (2014) 15(2):214–27. doi:10.1016/j.chom.2014.01.010
123. Lin K-M, Hu W, Troutman TD, Jennings M, Brewer T, Li X, et al. IRAK-1 bypasses priming and directly links TLRs to rapid NLRP3 inflammasome activation. Proc Natl Acad Sci U S A (2014) 111(2):775–80. doi:10.1073/pnas.1320294111
124. Fernandes-Alnemri T, Kang S, Anderson C, Sagara J, Fitzgerald KA, Alnemri ES. Cutting edge: TLR signaling licenses IRAK1 for rapid activation of the NLRP3 inflammasome. J Immunol (2013) 191(8):3995–9. doi:10.4049/jimmunol.1301681
125. Chattergoon MA, Latanich R, Quinn J, Winter ME, Buckheit RW III, Blankson JN, et al. HIV and HCV activate the inflammasome in monocytes and macrophages via endosomal toll-like receptors without induction of type 1 interferon. PLoS Pathog (2014) 10(5):e1004082. doi:10.1371/journal.ppat.1004082
126. He Y, Franchi L, Nunez G. TLR agonists stimulate Nlrp3-dependent IL-1 beta production independently of the purinergic P2X7 receptor in dendritic cells and in vivo. J Immunol (2013) 190(1):334–9. doi:10.4049/jimmunol.1202737
127. Gurung P, Li B, Malireddi RKS, Lamkanfi M, Geiger TL, Kanneganti T-D. Chronic TLR stimulation controls NLRP3 inflammasome activation through IL-10 mediated regulation of NLRP3 expression and caspase-8 activation. Sci Rep (2015) 5:14488. doi:10.1038/srep14488
Keywords: microbiota, toll-like receptors, inflammasome, autoimmunity, inflammatory diseases
Citation: Ignacio A, Morales CI, Câmara NOS and Almeida RR (2016) Innate Sensing of the Gut Microbiota: Modulation of Inflammatory and Autoimmune Diseases. Front. Immunol. 7:54. doi: 10.3389/fimmu.2016.00054
Received: 03 November 2015; Accepted: 04 February 2016;
Published: 19 February 2016
Edited by:
Ryo Inoue, Kyoto Prefectural University, JapanReviewed by:
Dolores Correa, Instituto Nacional de Pediatria, MexicoCopyright: © 2016 Ignacio, Morales, Câmara and Almeida. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Rafael Ribeiro Almeida, rafaelbio13@usp.br
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.