 Francesca Di Rosa
Francesca Di Rosa Thomas Gebhardt
Thomas Gebhardt
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 16 February 2016
Sec. Immunological Memory
Volume 7 - 2016 | https://doi.org/10.3389/fimmu.2016.00051
This article is part of the Research Topic Bone marrow T cells at the center stage in immunological memory View all 11 articles
Changes in T cell trafficking accompany the naive to memory T cell antigen-driven differentiation, which remains an incompletely defined developmental step. Upon priming, each naive T cell encounters essential signals – i.e., antigen, co-stimuli and cytokines – in a secondary lymphoid organ; nevertheless, its daughter effector and memory T cells recirculate and receive further signals during their migration through various lymphoid and non-lymphoid organs. These additional signals from tissue microenvironments have an impact on immune response features, including T cell effector function, expansion and contraction, memory differentiation, long-term maintenance, and recruitment upon antigenic rechallenge into local and/or systemic responses. The critical role of T cell trafficking in providing efficient T cell memory has long been a focus of interest. It is now well recognized that naive and memory T cells have different migratory pathways, and that memory T cells are heterogeneous with respect to their trafficking. We and others have observed that, long time after priming, memory T cells are preferentially found in certain niches such as the bone marrow (BM) or at the skin/mucosal site of pathogen entry, even in the absence of residual antigen. The different underlying mechanisms and peculiarities of resulting immunity are currently under study. In this review, we summarize key findings on BM and tissue-resident memory (TRM) T cells and revisit some issues in memory T cell maintenance within such niches. Moreover, we discuss BM seeding by memory T cells in the context of migration patterns and protective functions of either recirculating or TRM T cells.
When a pathogen attacks either skin or mucosa, primary immune responses are initiated in the draining lymph nodes (LN) and in some cases in the spleen. In these secondary lymphoid organs, mature antigen-presenting dendritic cells (DC) prime T cells to undergo huge clonal expansion and differentiation into short-lived effector and long-lived memory T cells. Both types of antigen-experienced T cell migrate far from the original priming site, displaying increased capacity to migrate to inflamed tissues as well as to the bone marrow (BM) as compared with naive T cells. Activation-induced changes in T cell membrane expression of chemokine receptors, integrins, and other adhesion molecules underlie this homing behavior. At the end of effective responses, when there is little or no residual antigen left, effector T cells die, leaving behind a small population of long-lived memory T cells, ready to provide protection in case of challenge with the same antigen. Memory T cells can be found all over the body, with a peculiar enrichment either in the BM or at the port of pathogen entry.
We will examine the evidence showing that memory T cells home to the BM and persist over time within this organ, being in constant exchange with blood T cell pool, whereas memory T cells residing at the port of pathogen entry (skin, etc.) are sessile. Moreover, we will review recent data and speculations on the niches wherein either BM or tissue-resident memory (TRM) T cells are maintained over time. We will discuss how BM memory T cells contribute to systemic memory, while TRM cells participate in local protection.
The BM consists of islets of hematopoietic BM interspersed with fatty areas, all contained within spongy bone and inside central cavities of long bones. It has long been known that in healthy individuals BM contains mature T cells, which can mediate graft-versus-host disease in T-replete BM transplantation settings. T cells represent about 3–8% of total nucleated BM cells, and have a typically reduced CD4/CD8 T cell ratio, as compared with blood (1, 2). BM T cells include also regulatory CD4 T cells (3). No lymphatic drainage is present, thus BM exchanges with the rest of the body occur only through blood circulation.
Upon T cell priming induced via different routes, T cell contraction is less pronounced in the BM than in the spleen and other organs, and is followed by long-lasting persistence of BM antigen-specific memory T cells (4–9). The BM also contains a high proportion of memory-phenotype T cells, i.e., a heterogeneous subset defined by the expression of activation/memory markers, which increases with aging and includes memory T cells specific for previously encountered antigens (10–12). BM memory T cells contain both central memory (TCM) and effector memory (TEM) T cells, two subsets of recirculating memory T cells identified in blood, having respectively high or low expression of the LN homing receptor CCR7 and distinct homing potential (13–16). Thus, the BM is often described as a “reservoir” for long-lived memory T cells (14, 15, 17).
Pivotal experiments in sheep showed that T cells labeled in situ in the BM migrated out of the organ and reached the spleen and other secondary lymphoid organs (18), suggesting that the BM represents a temporary stopping point for recirculating memory T cells (2). In agreement with this notion, parabiosis experiments showed that about 2 weeks after surgery leading to anastomoses of blood vessels between two CD45-congenic mice, comparable numbers of CD45.1+ and CD45.2+ antigen-specific memory CD8 T cells were found in the BM of each parabiotic mouse (19). Furthermore, intra-vital dynamic imaging studies demonstrated that naive and memory CD8 T cells injected either into the carotid artery or intravenously entered the BM parenchyma of mouse skull and constantly crawled in it (14, 20). Competition among “rival” memory T cells for lodging into the BM was suggested by adoptive transfer experiments showing that memory-phenotype T cells entered BM more easily into young than in thymectomized old mice, where an existing memory T cell pool precluded their free access (11). Such competition with host T cells was lacking when BM T cell recipients were RAG1-deficient mice (21). Thus, it appears that most BM T cells are motile recirculating cells. Some authors argued that the majority if not all of the BM memory T cells are non-migratory cells that permanently inhabit the BM; however, this speculation was based on cell phenotype, activation state, and gene expression analysis (22, 23) and did not take into account the in vivo data, including those obtained by in situ labeling, parabiosis, intra-vital dynamic imaging, and adoptive transfer (11, 14, 18–20). Nevertheless, the possibility that, similarly to thymus, LN, and spleen (24, 25), the BM also contains a few TRM cells cannot be excluded. For example, parabiosis experiments demonstrated that 3–5% of the antigen-specific memory T cells present in spleen and LN reside permanently in specific locations, i.e., the spleen marginal zone and red pulp and the LN sinuses (25).
In respect to the molecular players of memory T cell homing into the BM, memory CD8 T cells slow down and roll in BM microvessels via L-, P-, and E-selectin-mediated interactions (14). The BM tropism of memory T cells is supported by their high expression of the integrin VLA-4 (α4β1) and strong response to the BM chemokine CXCL12 (11, 14, 26). Conversely, only a few BM CD8 T cells express cutaneous lymphocyte antigen (CLA) and CCR9, involved in T cell homing to skin and gut, respectively (27). CD4 T cells lodge into the BM via molecular mechanisms at least partially similar to those of CD8 T cells. Expression of β1-integrin by CD4 T cells is required for their retention in the BM (28). In addition, CD4 T cell homing to BM is greatly reduced by anti-α2-integrin antibodies (21), suggesting a pivotal role for α2-integrin-mediated interactions, e.g., between the T cell integrin VLA-2 (α2β1) and type I collagen, which is highly abundant in bone. Both CD4 and CD8 T cell localization in the BM was compromised when mice lacked the adhesion molecule VCAM-1 (29).
Molecular regulation of T cell egress from the BM involves Sphingosine-1-phosphate (S1P) interaction with its receptor S1P1 (30). S1P levels in the BM are lower than in plasma, so that CD4 and CD8 T cells responding to S1P concentration gradient are normally recruited into the blood, unless S1P1 is pharmacologically inhibited by FTY720 (30). In agreement with the inhibition exerted by CD69 on S1P1 membrane expression and function (31), it was observed that CD69 ko memory CD4 T cells accumulated in lower numbers in the BM as compared with their WT counterparts (32). However, CD69 deficiency did not cause CD4 T cell increase in blood, implying a more complex scenario (32). Since CD69+ cells but not CD69− cells were associated with laminin+ stromal cells in the BM, it was proposed that CD69 could mediate retention of memory CD4 T cells within the BM (32). Taken together, these results suggest that CD69 regulates local T cell retention in the BM by a variety of mechanisms.
Certain infectious agents and cytokines can modulate BM T cell exchange with blood, for instance in Human Immunodeficiency Virus (HIV)-infected individuals, CD4 T cells migrate to BM more rapidly than in healthy controls (33). Moreover, after Hematopoietic Stem Cell (HSC)-mobilization procedures, some T cell subsets and especially regulatory T cells increase in the blood, suggesting that they are mobilized from the BM (34, 35). Conversely, high levels of type I IFN induced by acute viral infections block T cell egress from lymphoid organs by a CD69-dependent mechanism (31). Further studies are required to define the rules governing changes in BM T cell recruitment into the blood upon peripheral and systemic immune responses (30, 36).
Memory T cells specific for previously encountered antigens are commonly found in the BM of healthy subjects as well as of individuals affected by infectious, immune-mediated, and neoplastic diseases. For example, human BM from tetanus toxoid-vaccinated individuals contains tetanus-specific CD4 T cells (22, 37), which can be transferred with BM grafts (37). Pathogen-specific CD4 and CD8 T cells were present not only in the BM of immune individuals after resolution of acute infections (22) but also in the BM of subjects infected by persistent viruses, including Cytomegalovirus (CMV), Epstein–Barr Virus (EBV), and Human Hepatitis C Virus (HCV) (16, 27, 38, 39). BM from neoplastic patients contained tumor antigen-specific T cells, even without intentional immunization (40, 41). Moreover, auto-reactive T cells were found in the BM from individuals affected by autoimmune diseases (42).
In some of these cases, antigens were expressed in the BM, e.g., in EBV-infected subjects (27) or in idiopathic thrombocytopenia purpura patients (42). Though undetected, tumor antigen-positive micro-metastatic cells could be present in patients with solid tumors (41, 43). However, the presence of antigen in the BM was not a reasonable possibility in other cases, for example long time after vaccination against tetanus (22, 37). In rodents, antigen-primed CD4 and CD8 T cell migration and retention into the BM were observed after peripheral immunization procedures that were unlikely to result in the presence of antigen in the BM (7, 44, 45). Hence, it appears that long-lived antigen-specific memory T cells lodge in the BM during immune responses to a variety of antigens, either localized in the BM or not (2).
Furthermore, mouse BM can be a major site of naive CD4 and CD8 T cell priming under conditions of disrupted lymphocyte trafficking in splenectomized mice (46). Blood-borne antigens can reach the BM and induce CD4 and CD8 T cell priming via BM-resident APC (20, 47). Moreover, circulating DC and even neutrophils can transport antigens to BM (48, 49). Altogether, these results show that, under certain circumstances, BM can function as a secondary lymphoid organ.
T cell contacts with other cells in BM niches and molecular interactions influencing T cell activation, proliferation, or survival in BM niches appear heterogeneous. Microscopy studies of BM sections documented that most mouse memory CD4 and CD8 T cells contacted IL-7+ VCAM-1+ stromal cells (21, 23, 50), whereas some were in proximity to either F4/80+ or CD11c+ cells (21, 51). Clusters of memory T cells and IL-15-producing cells were observed in human BM sections (52) while a population of 4-1BBL-expressing radioresistant stromal cells supporting memory CD8 T cell survival was described in mouse BM (51). In addition to 4-1BBL, other TNF family members have been involved in T cell survival within BM niches, e.g., GITRL (53–57).
Elegant cellular barcoding experiments in mice clearly demonstrated that after pathogen infection via a systemic route, pathogen-specific CD8 T cells from the BM and those from blood, spleen, and LN all derive from the same set of precursors (58). BM memory T cells exhibit some phenotypic differences when compared with corresponding cells from lymphoid periphery and blood (7, 8, 22, 56, 59, 60). For example, a high proportion of BM memory CD4 and CD8 T cells express CD69 (22, 23, 32). Moreover, memory CD8 T cells from both mouse and human BM have a lower membrane expression of CD127, i.e., the IL-7Rα chain (8, 22, 59, 61), with the exception of antigen-specific CD8 T cells from lymphocytic choriomeningitis virus (LCMV)-infected mice (23). The TNF-R family member GITR is selectively upregulated by a fraction of mouse BM memory CD8 T cells (56). Downregulation of CD127 and upregulation of GITR were both observed in BM but not in spleen samples from WT mice, while they were lost in IL-15 ko mice, suggesting that they are indirect evidence of IL-15 stimulation in BM (56, 60). In agreement with these observations, adoptively transferred splenic CD8 T cells converted to BM-phenotype after entry into the BM (56, 60). Moreover, phospho-STAT-5 and phospho-p38 MAPK were increased in freshly isolated BM CD8 T cells as compared with corresponding spleen cells, possibly reflecting molecular events in the BM, e.g., signaling by IL-15 and TNF family members (59).
No major differences were found by gene-expression analysis of memory T cell paired samples obtained from either BM or blood/spleen, for example mouse BM and spleen CD127+ memory CD8 T cells (23) and human BM and blood CD69− memory CD4 T cells (22). This may not be surprising, considering T cell recirculation and the shared ancestry between memory T cells present in BM and in other lymphoid organs (58). After in vitro stimulation, memory CD8 T cells freshly isolated from BM strikingly changed their transcriptional profile (23). Of note, BM and spleen memory CD8 T cells had a roughly similar global transcription profile following 2 days of in vitro stimulation with anti-CD3+ anti-CD28 beads (23), suggesting that the two types of cell shared a common set of genes poised to be expressed after activation (62, 63).
Taken together, these findings support the view that most BM memory T cells cannot be identified as a distinguished subset. They also suggest that BM memory T cells integrate various signals received in the organ, so that their activation state is different from that of recirculating memory T cells from other sources. However, after either egress from the BM or experimental isolation, memory T cells only transiently retain some traits of the stimulation received in the organ (59).
Although the majority of BM memory T cells are quiescent, as demonstrated by staining with the cell cycle marker Ki67 (22, 23), a small percentage of them divides under steady state (8, 9, 60). Indeed, memory T cells have a higher rate of local proliferation in the BM than in spleen and LN, as demonstrated by a variety of experimental approaches (8, 9, 57, 59, 60, 64). For example, by Bromodeoxyuridine, carboxyfluorescein diacetate succinimidyl ester (CFSE), or DNA content assays, BM antigen-specific memory CD8 T cells contained a higher percentage of dividing cells than corresponding cells in spleen, LN, liver, and blood (8, 9). An increased proliferation in the BM was also observed in the case of naive CD8 T cells (59), although as expected their turnover was much lower than that of memory CD8 T cells (65). Estimates of T cell numbers strengthened the view that the BM gives a major contribution to long-term memory T cell maintenance, as well as to the homeostatic regulation of both memory and naive CD8 T cell numbers (2, 8, 9, 66, 67).
CD8 T cell proliferation in the BM is supported by local stimuli, including a dominant role for IL-15, as demonstrated by in vivo experiments with IL-15 ko and IL-15Rα ko mice (60). When tested in vitro, purified BM CD8 T cells did not exhibit an enhanced proliferative response to IL-7, IL-15, or IL-21 as compared with spleen CD8 T cells (59). Adoptively transferred CD8 T cells originally obtained from either BM or spleen similarly expanded in vivo after host injection with Poly I:C, a synthetic dsRNA analog inducing type I IFN and IL-15 (59), as well as upon secondary antigen challenge (68). Taken together, these data support the notion that BM T cells do not exhibit an intrinsic higher proliferative capability, but rather they are constantly stimulated in the BM (59).
Lack of memory T cell lodging into the BM can have a strong impact on memory T cell responses. For example, in CD69 ko mice and in mice treated with anti-α2 integrin antibodies, CD4 T cells were greatly reduced in the BM and help for B cell response was defective (21, 32). Memory CD8 T cells lacking the transcription factor eomesodermin did not populate the BM niche and had impaired long-term persistence and secondary expansion (69). In contrast, β1 integrin-deficient CD4 T cells were not retained in the BM niche but proliferated normally in response to antigenic peptide plus LPS injection (28). Likewise, in the absence of CXCR4, anti-LCMV memory CD8 T cells were defective in migration into the BM, but displayed normal, or even greater, expansion upon secondary challenge with pathogen (70). Further studies are required to clarify these discrepancies, possibly due to pleiotropic effects of key molecule deficiency, and/or differences in the measured aspect of memory response, e.g., T cell self-renewal in the memory phase, T cell expansion upon secondary response, or anti-pathogen protection, after different types of rechallenge.
Bone marrow T cells exert potent antigen-specific effector function, as demonstrated by either adoptive transfers in immunodeficient mice (5) or in vitro studies (7). T cells from BM of patients with different types of solid and hematological cancers were able to kill autologous tumor cells (40, 71, 72). Adoptive transfer of BM T cells from breast cancer patients in immunodeficient mice induced regression of xenotransplanted autologous tumors, while blood T cells were not as effective (41). Clinical trials with autologous BM T cells have been initiated in various tumors (73), and the first trial in multiple myeloma patients looks very promising (74).
Moreover, it has been proposed that BM-based T cell proliferation is one of the species-specific advantages allowing Simian Immunodeficiency Virus (SIV)-infected Sooty Mangabeys (SM) but not Rhesus Macaques (RM) to maintain normal CD4 T cell counts (75). Notably, SIV infection does not cause immunodeficiency in SM, whereas it progresses to AIDS in RM, similarly to HIV infection in humans (75).
T cell-derived cytokines can modulate hematopoiesis, implying that BM T cells can contribute to shaping hematopoiesis during both acute and chronic infections (76). For example, IFN-γ released by T cells can either induce IL-6 production by BM stromal cells, in turn augmenting myelopoiesis (77), or also act directly on HSC (78–80).
Bone marrow can play a detrimental role in some T-cell mediated diseases by maintaining pathogenic T cells. In mouse models, pathogenic autoreactive T cells were found in the BM in organ-specific autoimmune diseases, e.g., type I diabetes (81) and chronic uveitis (82). Similarly, in mice with inflammatory colitis, pathogenic CD4 T cells were found in the BM (83). Interestingly, maintenance of pathogenic CD4 T cells required IL-7 in the BM, but not in the colon (84). Thus, it was proposed that, in the disease remission phase, colitogenic CD4 T cells persisted in the BM (83, 84).
Moreover, T cell effector function in the BM can stimulate pathological bone resorption, by activating osteoclasts. It is well established that CD4 T cells recruited in joints and periodontal tissue of patients affected by rheumatoid arthritis and periodontitis, respectively, stimulate osteoclastogenesis by producing IL-17 and RANK-L (85–88). Recently, a subset of osteoclastogenic Th17 TNF-α producing cells has been identified in PBMC from patients with Crohn’s disease, and it has been proposed that these cells can migrate to the BM and mediate bone loss, in agreement with mouse models (89–91). Notably, in a mouse model of breast cancer, pro-osteoclastogenic BM T cells favored the establishment of skeletal metastases by inducing osteolytic lesions (92).
Finally, T cells regulate physiological processes occurring in the BM, i.e., normal hematopoiesis and bone tissue homeostasis. Surprisingly, the maintenance of normal bone mass and bone mineral density in physiological conditions is promoted by T cells, which stimulate the production of the RANK-L decoy receptor osteoprotegerin by B cells, through CD40L/CD40 interaction (93). A cross-talk between T cells and hematopoietic precursors occurs in the BM in normal healthy conditions (94, 95). For example, it has been shown that BM T cells sustain normal granulopoiesis (94), while regulatory T cells inhibit excessive T cell-production of the granulopoiesis-promoting cytokines GM-CSF, TNF, and IL-6, thus allowing for sufficient B lymphopoiesis (95). Regulatory T cells in the BM are required for HSC engraftment upon transplantation (96, 97), and likewise might protect normal HSC and their niches from destructive immune responses (97). Taken together, these results suggest that BM T cells are engaged in a complex interplay with other cells in the local environment, contributing to maintain bone and BM integrity and function.
In addition to the BM and secondary lymphoid organs, the body’s surfaces such as the linings of the skin, gut, and reproductive tract also harbor large numbers of CD4 and CD8 T cells (6, 98–100). Most of these peripheral T cells are antigen-experienced memory cells and are generally believed to provide specific immunity against renewed infection with previously encountered pathogens. Given their location in close proximity to the external environment, it appears likely that some of these memory T cells also recognize commensal microbiota, and such T cell–microbiota interactions have been proposed to fine-tune peripheral immunity (101, 102).
While it is clear that T cells recirculate between peripheral tissues and the blood via the lymphatic system (103–105), there is recent evidence for a non-recirculating population of memory T cells that remain localized to peripheral tissues and never return to the blood (106, 107). Such TRM cells are best characterized for the CD8 subset and have been described in a large number of peripheral organs, including skin, gut, brain, salivary glands, lungs, female reproductive tract, and others (106, 107). Furthermore, non-recirculating memory T cells also exist in lymphoid organs such as LN and thymus (24, 25).
Various experimental strategies, such as organ transplantation, sex-mismatched adoptive T cell transfer, and parabiosis in mice have unequivocally demonstrated that TRM cells can persist in peripheral tissues in disconnection from the pool of recirculating T cells in the blood (108–111). In line with this, CD8 TRM cells are often found in specialized microanatomical compartments such as the epithelial layers of skin and gut where they are sequestered from direct lymphatic drainage by the underlying epithelial basement membrane.
These epithelial CD8 TRM cells commonly express surface receptors such as CD103 (the αE integrin subunit), CD69, and the integrin VLA-1 (α1β1), which are variously involved in T cell retention and persistence (107, 112). For instance, genetic deficiency in CD103 expression results in defective generation of CD8 TRM cells in gut mucosa (113, 114), abolished accumulation of CD8 T cells in islet grafts (115), as well as a gradual loss of TRM cells from skin and lung mucosa (116, 117). Similarly, genetic or functional ablation of VLA-1 causes a dramatic decline in memory CD8 T cell numbers in lung after pulmonary virus infection (118). While these adhesion molecules are likely to mediate the tethering of TRM cells to their microenvironment, integrin binding and ligation may also support T cell survival and functional activity (119, 120). By contrast, the surface molecule CD69 can act to block the functional activity of the tissue exit receptor, S1P1, and thereby inhibit tissue egress of effector T cells with the potential to undergo local differentiation into TRM cells (31, 121). In addition to this posttranslational regulation of S1P1 function, the progressive transcriptional downregulation of S1P1 expression appears to be a critical checkpoint in the TRM maturation pathway in a variety of tissues (117, 122). The high level surface expression of CD69 by fully developed TRM cells further echoes the absence of S1P1 expression, rather than recent T cell receptor activation, as both molecules physically interact with each other, resulting in mutual inhibition of surface expression (123).
In addition to epithelial CD103+CD8+ TRM cells, phenotypically and anatomically distinct CD103− CD8+ TRM cells, including those that form clusters at sites of intestinal infection with Yersinia pseudotuberculosis, have also been described more recently (100, 124). Likewise, presumably non-recirculating or long-term retained memory T cells have been identified among CD4+ memory T cells in a variety of tissues, including skin, as well as lung, gut, and vaginal mucosa (125–129). Some of these also express markers initially described for CD8+ TRM cells, such as CD69 and CD103, although the role of these molecules in tissue persistence of CD4+ TRM cells remains largely unknown. It is therefore likely that TRM cells exist among both CD4+ and CD8+ memory T cells, and future studies will have to compare the molecular and functional relationship between those different types of TRM cells. Nevertheless, given that the molecular pathways underpinning the maturation and persistence of TRM cells have so far most extensively been studied for CD103+ CD8+ TRM cells, we will focus our attention on this subset for the remainder of this article.
The highest frequencies of CD8+CD103+ TRM cells are found in previously infected or inflamed tissue (108, 110, 130), where TRM cells develop from infiltrating effector T cells during lesion resolution (114, 117). Accordingly, peripheral infection can be regarded as the principle inducer of TRM cells in barrier organs such as skin and mucosa. In addition, systemic infections that generate a large pool of memory precursor T cells disseminating throughout the body can give rise to TRM populations in a variety of barrier tissues and internal organs (6, 111, 113, 131). Likewise, repeated immunization has been shown to progressively build up considerable numbers of TRM cells even in non-infected areas of skin (110).
Tissue-resident memory precursor cells are found within the KLRG1− effector population that also contains the precursors of circulating memory CD8 T cells (114, 117, 132). Of note, TRM precursor effector cells rapidly cease to migrate through peripheral tissues after pathogen clearance (111, 130), meaning that the pool of TRM cells is established early after infection and remains stable with minimal external input thereafter.
The generation of TRM cells in infected skin is regulated by a complex interplay of processes such as peripheral infiltration, tissue exit, local positioning, and survival, which ultimately results in the maturation of a long-lived population of TRM cells in the epidermal layer. Indeed, epithelial infiltration is critical for TRM formation, at least in skin, since effector T cells that are prevented from epidermal entry by pertussis toxin treatment fail to generate long-lived CD103+ memory cells (117). This compartment-specific generation of T cell memory in skin likely reflects the anatomical restriction of essential differentiation and survival factors such as TGF-β and IL-15 that are expressed at high levels in the epithelial layer. Further supporting this assumption, CD8 T cells fail to acquire a CD103+ TRM phenotype and are lost from skin over time in absence of TGF-β and IL-15 signaling (117).
In addition to these roles of TGF-β and IL-15 in TRM differentiation and survival in skin, the same or other essential cytokines and local factors may be involved in the establishment of TRM populations in an organ- or location-specific manner (113, 114, 116). Local antigen recognition for instance is strictly required for the establishment of long-lived TRM populations in organs such as brain, sensory ganglia, and lungs (120, 130, 133, 134). By contrast, in many other peripheral tissues and internal organs, including skin, gut, kidneys, and salivary glands, local antigen recognition appears to be dispensable for TRM development (113, 130). Nevertheless, it is conceivable that local antigen stimulation enhances TRM accumulation, possibly through the downregulation of tissue exit receptors such as CCR7 or through direct effects on local proliferation and survival of TRM precursor cells.
Importantly, local memory differentiation by tissue infiltrating effector T cells is accompanied by the progressive acquisition of a specific transcriptional program that distinguishes TRM cells from their TEM and TCM counterparts in the circulation (117, 122). Interestingly, the majority of these TRM-specific transcripts appear to be imprinted by the organ of residency. Skin CD8 TRM cells for instance express CCR8 whereas those in gut mucosa express CCR9 (130), and both chemokine receptors have been implicated in organ-specific homing to the respective anatomical locations. In addition, approximately a quarter of the TRM-specific transcripts are commonly up or downregulated in different CD103+ TRM cell populations from skin, gut, and lungs when compared to recirculating memory cells. This TRM-specific core transcriptional profile across a variety of organs comprises genes encoding for migration and adhesion molecules (e.g., Itga, Itga1, and S1pr1), transcription factors (e.g., Eomes, Hobit, and Litaf), immunoregulatory molecules (e.g., Cd244, Ctla-4, and Icos), and various enzymes (e.g., Inapp4b, Cmah, and Qpct) and likely echoes a common molecular program underpinning the formation, persistence, and function of CD103+ CD8 TRM cells in peripheral tissues (130).
Future work will have to define how stably this transcriptional profile is imprinted in TRM cells, and whether TRM cells can adopt alternative transcriptional and functional programs on renewed antigenic challenge or experimental release from the tissue context. Relevant to this, it will be important to test whether the same epigenetic modifications that regulate chromatin accessibility and gene transcriptional programs in recirculating memory T cells (62, 63) also operate in TRM cells. Likewise, the extent to which the CD8 TRM transcriptional signatures and their putative epigenetic regulators overlap with those in CD103− and/or CD4 TRM cells is currently unknown and needs to be tested. Regardless, there is accumulating evidence that a considerable proportion of T cells in peripheral non-lymphoid tissues adopt a phenotype and transcriptional profile that clearly distinguishes them from recirculating memory T cells.
Owing to their strategic location at body surfaces, TRM cells can immediately sense invading pathogens and initiate a rapid local immune response (112). In skin, for instance, TRM cells use dendritic protrusions to probe their environment, a behavior that reflects their active contribution to skin immune surveillance (109, 135). Following activation by their pathogen-specific T cell receptors, TRM cells produce a number of proinflammatory mediators, including the cytokines IFN-γ, TNF-α, and IL-2 (112, 136–139). These mediators drive the local activation and recruitment of innate and adaptive immune cells and also act on stromal and parenchymal cells to confer a resistance to pathogen dissemination (112, 136, 138, 139). In addition, TRM cell may directly lyse infected target cells, although the contribution of cytotoxicity in TRM-mediated protection remains to be demonstrated.
Tissue-resident memory cells have been shown to provide an exceptional level of immediate protection from renewed infection with a broad variety of viral and bacterial pathogens (108–110, 114, 134, 135, 138, 140). Furthermore, TRM cells generated by immunization strategies combining T cell priming with local treatment with inflammatory adjuvants provide a level of protection from de novo infection in skin and mucosa that is far superior to what can be achieved by circulating memory cells (130). Likewise, local immunizations with human papilloma virus vectors or treatment with T cell-attracting chemokines have been shown to generate protective TRM cells in vaginal mucosa and the female reproductive tract (141, 142).
Since these experiments confirm that TRM cells do not rely on antigen for long-term persistence, unlike other approaches used to generate T-cell immunity in peripheral tissues (143), TRM cells are now regarded as promising mediators of long-lived peripheral immunity for future vaccines (144). Importantly in this respect, a series of landmark papers have demonstrated the existence of Herpes Simplex Virus (HSV)-specific CD8+ memory T cells resident at the dermal-epidermal junction in human genital skin (145–147). Remarkably, these resident T cells share transcriptional commonalities with HSV-specific TRM cells in mice (148) and are involved in preventing genital lesions upon asymptomatic HSV-2 shedding in skin (147). Furthermore, CD8+ memory T cells with a CD103+CD69+VLA-1+ TRM phenotype also exist in other human tissues (129, 149, 150). Such observations strongly argue that TRM cells are critical mediators of peripheral immunity in both mice and humans, and it is has been speculated that one of their principle functions is in dealing with recurrent or persistent infections in defined anatomical niches such as epithelial or neuronal compartments (151).
While in such cases, TRM cells exert highly beneficial protective functions, pathogenic TRM responses may also cause tissue pathology. Supporting this notion, accumulation and aberrant activation of TRM cells have been described in localized and recurrent human diseases such as skin autoimmunity and transplant rejection (152). Early studies using xenotransplantation of pre-psoriatic human skin onto mice, for instance, have demonstrated that graft-derived human TRM cells, in absence of circulating memory T cells, are sufficient to drive development of psoriasis lesions through localized production of inflammatory cytokines (153, 154). Similarly, IFN-γ producing epidermal TRM cells can initiate skin lesions upon ingestion of drugs that cause recurrent fixed drug eruptions (155), and CD8+ T cells with a TRM phenotype have been described in alopecia and vitiligo lesions (156, 157). Another impressive demonstration of TRM-mediated pathology in humans stems from a recent study that defined donor-derived TRM cells as the major drivers of localized rejection responses after face transplantation (158). Finally, CD103+CD8+ T cells make up a considerable proportion of tumor-infiltrating lymphocytes in carcinomas and brain tumors, and their peri-tumoural accumulation is associated with improved clinical outcomes (159–162).
In summary, an accumulating body of literature implies a critical role of protective as well as pathogenic TRM responses in infectious and inflammatory diseases in humans. While so far this has most intensively been studied in easily accessible organs such as skin, it is tempting to speculate that TRM cells act as central mediators of localized diseases also in lungs, gut, and many other internal organs. Future studies will have to further clarify the role of protective and pathogenic TRM cell responses in humans and will have to explore avenues for their prophylactic or therapeutic manipulation in experimental and clinical settings.
The postulated division of labor between recirculating memory T cells and TRM cells offers a novel view of both memory maintenance and response to antigenic rechallenge that integrates and broadens the previous perspective based on TCM/TEM paradigm (13).
Long-lived persistence of memory T cells is achieved in the steady state by different mechanisms. Recirculating memory T cells rely on a finely tuned equilibrium between quiescence and homeostatic proliferation, which is mostly achieved within the BM niches wherein these cells temporarily stop. In contrast, TRM cells live permanently as sessile non-migratory cells within skin/mucosal niches, wherein they survive in a quiescent state (Figure 1A).
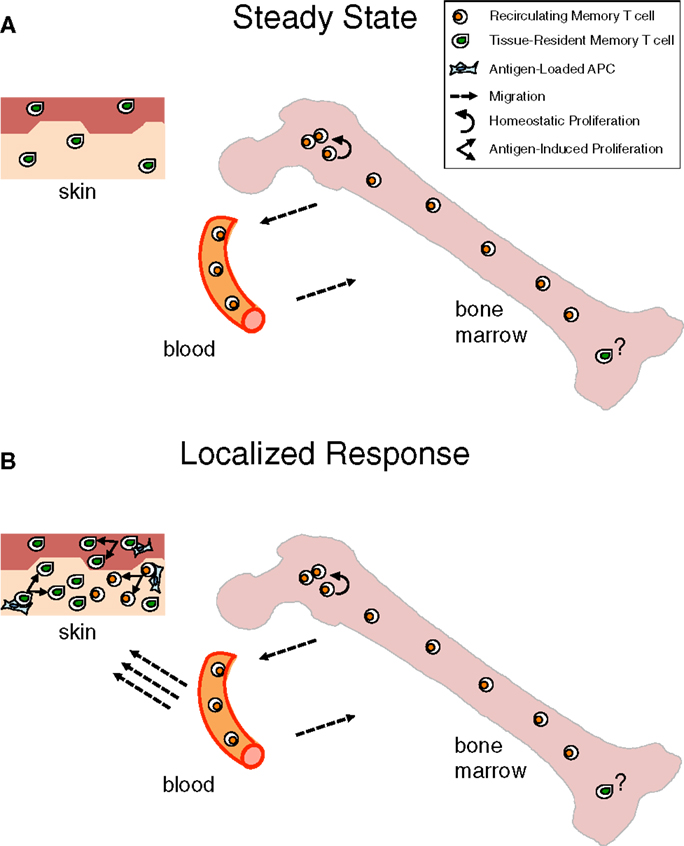
Figure 1. Contribution of bone marrow memory T cells and tissue-resident memory T cells to long-lasting immunity. (A) Under steady state, Bone Marrow (BM) memory T cells are in constant exchange with the recirculating memory T cell pool, while tissue resident memory T (TRM) cells are fixed within peripheral organs, e.g., skin, in a quiescent state. Although the majority of BM memory T cells are quiescent, a minor proportion of them self-renew within BM niches, mostly in response to cytokines, e.g., IL-15. This homeostatic proliferation counterbalances losses due to cell death and contributes to maintain constant numbers of recirculating memory T cells over time (8, 9). The putative possibility that a few TRM cells exist in the BM is depicted. (B) When infection is localized in peripheral organs, TRM cells represent a first line of defense that is reinforced by the arrival of recirculating memory T cells, acting as a second line of protection. Antigen presented by APC in the peripheral tissue leads to immediate antigen-specific expansion and effector response of the TRM and, with some delay, of the newly recruited memory T cells (163, 164).
It appears that recirculating memory T cells and TRM cells provide respectively systemic immunity and immediate protection at the port of pathogen entry. Yet, the two types of memory T cells act in concert for tissue protection as recirculating memory T cells are recruited to skin or mucosal sites of secondary challenge, resulting in both more efficient local effector response and boosting of systemic memory (Figure 1B). Naturally, recirculating memory T cells arrive with some delay in the tissue, while TRM cells are already there for highly efficient immediate protection.
Taking into consideration that TRM cells have been identified not only at the epithelial barriers of the body but also in lymphoid and/or internal organs, e.g., LN and brain (106, 107), it is likely that each organ harbors some TRM cells and that BM is not an exception to this rule (Figure 1). The peculiarity of the BM would be that under steady state most memory T cells in it belong to the recirculating pool. Some heterogeneity might exist in the kinetics of recirculating memory T cell transit into the BM, with some cells just quickly passing through and others stopping over during their journey, thus inhabiting for some time the BM niches wherein they receive signals for survival/homeostatic proliferation (Figure 2).
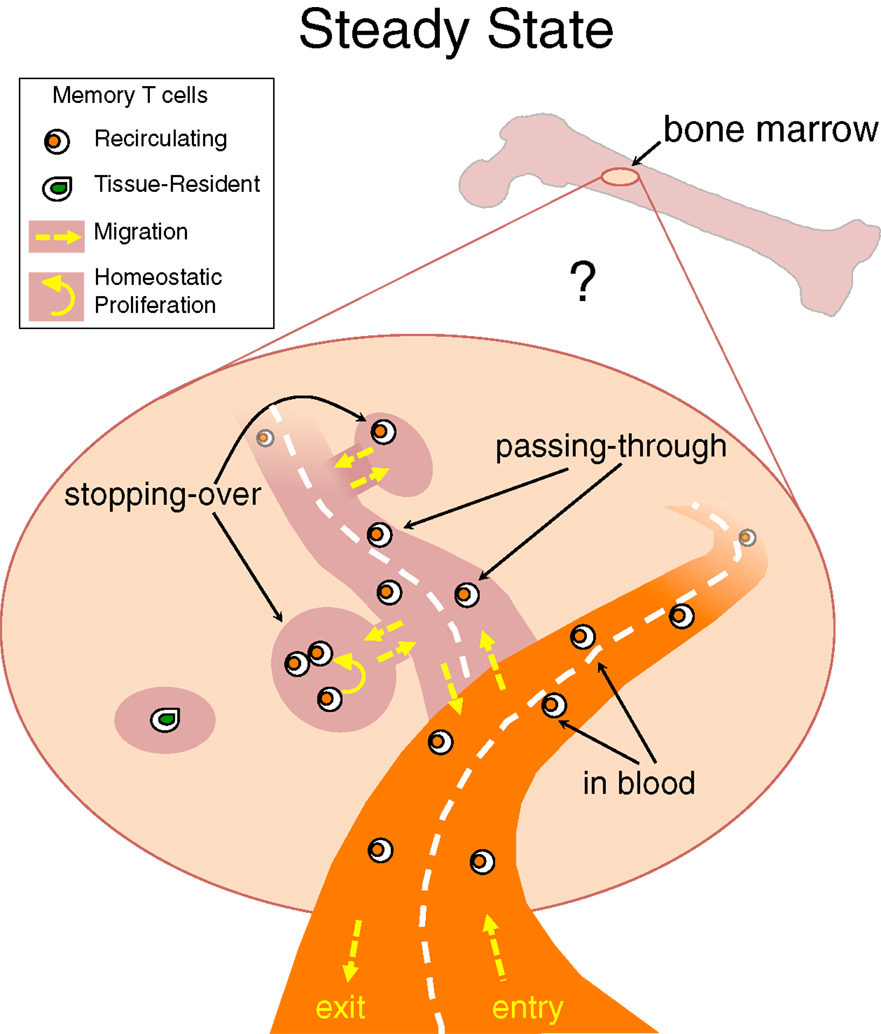
Figure 2. Stopping-over, passing-through and tissue-resident memory T cells in bone marrow. Under steady state, memory T cells migrate into the BM and then circulate back to the blood, with poorly defined kinetics. It is possible that some recirculating memory T cells quickly transit through the BM parenchyma while others stop over for some time within BM niches. A few memory T cells might stay permanently in BM niches and never return to the blood, representing the equivalent of tissue-resident memory (TRM) T cells identified in other organs.
Despite their different migration pathways, positioning and role in immunity, both TRM and BM T cells participate in a local network of cellular and molecular interactions in the organ where they are located, influencing normal tissue homeostasis and organ function. In the case of BM T cells, it has been shown that they normally regulate hematopoiesis, as well as bone metabolism. In respect with TRM cells located in barrier organs, it is conceivable that they protect host’s health and sometimes contribute to disease in several manners, for example they might shape the gut microbiota composition, with a possible indirect impact on metabolic syndrome, obesity-related disorders, inflammatory bowel disease, and colorectal cancer (101, 165). Some of the unsolved questions on BM memory T cells and TRM cells are listed in Box 1.
Box 1. Unsolved questions on bone marrow (BM) memory T cells and tissue-resident memory (TRM) T cells.
Tissue-specific signals for differentiation: After naive T cell priming, what are the distinct signals regulating differentiation of either recirculating memory T cells or TRM cells? Do recirculating memory T cells integrate signals for differentiation they receive over time in diverse environments (e.g., within BM, spleen, and LN)? Do TRM cells lodged in diverse tissues receive in each location a specific combination of signals for differentiation?
Tissue-specific signals for survival/homeostatic proliferation: What are the external signals regulating the long-lasting maintenance of recirculating memory T cells within BM niches and/or TRM cells within tissue niches? What is the role of external signals (including antigen, IL-15, TGF-β, etc.) in the quiescence/self-renewal of BM memory T cells and/or in the survival in a quiescent state of TRM cells?
Heterogeneity: If TRM cells receive a specific combination of signals for differentiation and maintenance in each organ or location, and if recirculating memory T cells integrate diverse signals over time, are TRM cells, taken as a whole, expected to be more heterogeneous than recirculating memory T cells? Is it conceivable that several different subsets of location-specific TRM cells do exist?
Intracellular networks orchestrating differentiation: What are the intracellular molecular pathways regulating differentiation and stable genetic imprinting or plasticity of TRM and recirculating memory T cells? How do key intracellular molecules regulate epigenetic marking, gene transcription, protein translation, metabolic state of TRM and of recirculating memory T cells?
Regulation of absolute cell numbers: Is homeostatic maintenance of memory T cell numbers independently regulated for TRM cells and for recirculating memory T cells (including those in BM, spleen, and LN)?
CD4 and CD8 T cells: Do TRM cells resemble recirculating memory T cells in respect to the shared and/or specific protective functions of the CD4 and the CD8 T cell subsets? Are differentiation and maintenance of the CD4 TRM similar to those of the CD8 TRM?
Aging: Are there changes occurring with aging in the distribution of tasks between TRM and recirculating memory T cells?
Normal tissue homeostasis and organ function: Do BM memory T cells and TRM cells play a role in maintaining normal tissue homeostasis and organ function?
FR conceptualized the article, prepared the box and the figures, and wrote the section on BM T cells; TG wrote the section on TRM cells; FR and TG worked together on the remaining sections.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank A. Hayday, P. Matzinger, R. Pabst, and A. Santoni for discussion, and P. Matzinger and A. Natalini for help with the figures.
FD is supported by CTN01_00177_962865 (Medintech) grant from Ministero dell’Università e delle Ricerca (MIUR). TG is supported by grant and fellowship funding from the National Health and Medical Research Council Australia, the University of Melbourne, and the Silvia and Charles Viertel Charitable Foundation.
1. Westermann J, Pabst R. Distribution of lymphocyte subsets and natural killer cells in the human body. Clin Investig (1992) 70:539–44. doi:10.1007/BF00184787
2. Di Rosa F, Pabst R. The bone marrow: a nest for migratory memory T cells. Trends Immunol (2005) 26:360–6. doi:10.1016/j.it.2005.04.011
3. Zou L, Barnett B, Safah H, Larussa VF, Evdemon-Hogan M, Mottram P, et al. Bone marrow is a reservoir for CD4+CD25+ regulatory T cells that traffic through CXCL12/CXCR4 signals. Cancer Res (2004) 64:8451–5. doi:10.1158/0008-5472.CAN-04-1987
4. Benner R, Meima F, van der Meulen GM. Antibody formation in mouse bone marrow. II. Evidence for a memory-dependent phenomenon. Cell Immunol (1974) 13:95–106. doi:10.1016/0008-8749(74)90230-5
5. Slifka MK, Whitmire JK, Ahmed R. Bone marrow contains virus-specific cytotoxic T lymphocytes. Blood (1997) 90:2103–8.
6. Masopust D, Vezys V, Marzo AL, Lefrancois L. Preferential localization of effector memory cells in nonlymphoid tissue. Science (2001) 291:2413–7. doi:10.1126/science.1058867
7. Di Rosa F, Santoni A. Bone marrow CD8 T cells are in a different activation state than those in lymphoid periphery. Eur J Immunol (2002) 32:1873–80. doi:10.1002/1521-4141(200207)32:7<1873::AID-IMMU1873>3.0.CO;2-P
8. Parretta E, Cassese G, Barba P, Santoni A, Guardiola J, Di Rosa F. CD8 cell division maintaining cytotoxic memory occurs predominantly in the bone marrow. J Immunol (2005) 174:7654–64. doi:10.4049/jimmunol.174.12.7654
9. Becker TC, Coley SM, Wherry EJ, Ahmed R. Bone marrow is a preferred site for homeostatic proliferation of memory CD8 T cells. J Immunol (2005) 174:1269–73. doi:10.4049/jimmunol.174.3.1269
10. Price PW, Cerny J. Characterization of CD4+ T cells in mouse bone marrow. I. Increased activated/memory phenotype and altered TCR Vbeta repertoire. Eur J Immunol (1999) 29:1051–6. doi:10.1002/(SICI)1521-4141(199903)29:03<1051::AID-IMMU1051>3.0.CO;2-Y
11. Di Rosa F, Santoni A. Memory T-cell competition for bone marrow seeding. Immunology (2003) 108:296–304. doi:10.1046/j.1365-2567.2003.01593.x
12. Herndler-Brandstetter D, Landgraf K, Tzankov A, Jenewein B, Brunauer R, Laschober GT, et al. The impact of aging on memory T cell phenotype and function in the human bone marrow. J Leukoc Biol (2012) 91:197–205. doi:10.1189/jlb.0611299
13. Sallusto F, Lenig D, Forster R, Lipp M, Lanzavecchia A. Two subsets of memory T lymphocytes with distinct homing potentials and effector functions. Nature (1999) 401:708–12. doi:10.1038/44385
14. Mazo IB, Honczarenko M, Leung H, Cavanagh LL, Bonasio R, Weninger W, et al. Bone marrow is a major reservoir and site of recruitment for central memory CD8+ T cells. Immunity (2005) 22:259–70. doi:10.1016/j.immuni.2005.01.008
15. Zhang X, Dong H, Lin W, Voss S, Hinkley L, Westergren M, et al. Human bone marrow: a reservoir for “enhanced effector memory” CD8+ T cells with potent recall function. J Immunol (2006) 177:6730–7. doi:10.4049/jimmunol.177.10.6730
16. Letsch A, Knoedler M, Na IK, Kern F, Asemissen AM, Keilholz U, et al. CMV-specific central memory T cells reside in bone marrow. Eur J Immunol (2007) 37:3063–8. doi:10.1002/eji.200636930
17. Akatsuka Y, Torikai H, Inamoto Y, Tsujimura K, Morishima Y, Kodera Y, et al. Bone marrow may be a reservoir of long-lived memory T cells specific for minor histocompatibility antigen. Br J Haematol (2006) 135:413–4. doi:10.1111/j.1365-2141.2006.06313.x
18. Pabst R, Miyasaka M, Dudler L. Numbers and phenotype of lymphocytes emigrating from sheep bone marrow after in situ labelling with fluorescein isothiocyanate. Immunology (1986) 59:217–22.
19. Klonowski KD, Williams KJ, Marzo AL, Blair DA, Lingenheld EG, Lefrancois L. Dynamics of blood-borne CD8 memory T cell migration in vivo. Immunity (2004) 20:551–62. doi:10.1016/S1074-7613(04)00103-7
20. Milo I, Sapoznikov A, Kalchenko V, Tal O, Krauthgamer R, van Rooijen N, et al. Dynamic imaging reveals promiscuous crosspresentation of blood-borne antigens to naive CD8+ T cells in the bone marrow. Blood (2013) 122:193–208. doi:10.1182/blood-2012-01-401265
21. Tokoyoda K, Zehentmeier S, Hegazy AN, Albrecht I, Grun JR, Lohning M, et al. Professional memory CD4+ T lymphocytes preferentially reside and rest in the bone marrow. Immunity (2009) 30:721–30. doi:10.1016/j.immuni.2009.03.015
22. Okhrimenko A, Grun JR, Westendorf K, Fang Z, Reinke S, von Roth P, et al. Human memory T cells from the bone marrow are resting and maintain long-lasting systemic memory. Proc Natl Acad Sci U S A (2014) 111:9229–34. doi:10.1073/pnas.1318731111
23. Sercan Alp O, Durlanik S, Schulz D, McGrath M, Grun JR, Bardua M, et al. Memory CD8(+) T cells colocalize with IL-7(+) stromal cells in bone marrow and rest in terms of proliferation and transcription. Eur J Immunol (2015) 45:975–87. doi:10.1002/eji.201445295
24. Hofmann M, Oschowitzer A, Kurzhals SR, Kruger CC, Pircher H. Thymus-resident memory CD8+ T cells mediate local immunity. Eur J Immunol (2013) 43:2295–304. doi:10.1002/eji.201343519
25. Schenkel JM, Fraser KA, Masopust D. Cutting edge: resident memory CD8 T cells occupy frontline niches in secondary lymphoid organs. J Immunol (2014) 192:2961–4. doi:10.4049/jimmunol.1400003
26. Cinamon G, Shinder V, Alon R. Shear forces promote lymphocyte migration across vascular endothelium bearing apical chemokines. Nat Immunol (2001) 2:515–22. doi:10.1038/88710
27. Palendira U, Chinn R, Raza W, Piper K, Pratt G, Machado L, et al. Selective accumulation of virus-specific CD8+ T cells with unique homing phenotype within the human bone marrow. Blood (2008) 112:3293–302. doi:10.1182/blood-2008-02-138040
28. DeNucci CC, Shimizu Y. Beta1 integrin is critical for the maintenance of antigen-specific CD4 T cells in the bone marrow but not long-term immunological memory. J Immunol (2011) 186:4019–26. doi:10.4049/jimmunol.1003566
29. Koni PA, Joshi SK, Temann UA, Olson D, Burkly L, Flavell RA. Conditional vascular cell adhesion molecule 1 deletion in mice: impaired lymphocyte migration to bone marrow. J Exp Med (2001) 193:741–54. doi:10.1084/jem.193.6.741
30. Maeda Y, Seki N, Sato N, Sugahara K, Chiba K. Sphingosine 1-phosphate receptor type 1 regulates egress of mature T cells from mouse bone marrow. Int Immunol (2010) 22:515–25. doi:10.1093/intimm/dxq036
31. Shiow LR, Rosen DB, Brdickova N, Xu Y, An J, Lanier LL, et al. CD69 acts downstream of interferon-alpha/beta to inhibit S1P1 and lymphocyte egress from lymphoid organs. Nature (2006) 440:540–4. doi:10.1038/nature04606
32. Shinoda K, Tokoyoda K, Hanazawa A, Hayashizaki K, Zehentmeier S, Hosokawa H, et al. Type II membrane protein CD69 regulates the formation of resting T-helper memory. Proc Natl Acad Sci U S A (2012) 109:7409–14. doi:10.1073/pnas.1118539109
33. Chen JJ, Huang JC, Shirtliff M, Briscoe E, Ali S, Cesani F, et al. CD4 lymphocytes in the blood of HIV(+) individuals migrate rapidly to lymph nodes and bone marrow: support for homing theory of CD4 cell depletion. J Leukoc Biol (2002) 72:271–8.
34. Gurman G, Dilek I, Arslan O, Arat M, Beksac M, Ilhan O, et al. The effect of G-CSF on lymphocyte subsets and CD34+ cells in allogeneic stem cell transplantation. Transfus Apher Sci (2001) 24:23–8. doi:10.1016/S0955-3886(00)00123-5
35. Condomines M, Quittet P, Lu ZY, Nadal L, Latry P, Lopez E, et al. Functional regulatory T cells are collected in stem cell autografts by mobilization with high-dose cyclophosphamide and granulocyte colony-stimulating factor. J Immunol (2006) 176:6631–9. doi:10.4049/jimmunol.176.11.6631
36. Lopez CB, Hermesh T. Systemic responses during local viral infections: type I IFNs sound the alarm. Curr Opin Immunol (2011) 23:495–9. doi:10.1016/j.coi.2011.06.003
37. Vavassori M, Maccario R, Moretta A, Comoli P, Wack A, Locatelli F, et al. Restricted TCR repertoire and long-term persistence of donor-derived antigen-experienced CD4+ T cells in allogeneic bone marrow transplantation recipients. J Immunol (1996) 157:5739–47.
38. Racanelli V, Frassanito MA, Leone P, Brunetti C, Ruggieri S, Dammacco F. Bone marrow of persistently hepatitis C virus-infected individuals accumulates memory CD8+ T cells specific for current and historical viral antigens: a study in patients with benign hematological disorders. J Immunol (2007) 179:5387–98. doi:10.4049/jimmunol.179.8.5387
39. Guerreiro M, Na IK, Letsch A, Haase D, Bauer S, Meisel C, et al. Human peripheral blood and bone marrow Epstein-Barr virus-specific T-cell repertoire in latent infection reveals distinct memory T-cell subsets. Eur J Immunol (2010) 40:1566–76. doi:10.1002/eji.200940000
40. Jahn B, Bergmann L, Weidmann E, Brieger J, Fenchel K, Schwulera U, et al. Bone marrow-derived T-cell clones obtained from untreated acute myelocytic leukemia exhibit blast directed autologous cytotoxicity. Leuk Res (1995) 19:73–82. doi:10.1016/0145-2126(94)00119-U
41. Feuerer M, Beckhove P, Bai L, Solomayer EF, Bastert G, Diel IJ, et al. Therapy of human tumors in NOD/SCID mice with patient-derived reactivated memory T cells from bone marrow. Nat Med (2001) 7:452–8. doi:10.1038/86523
42. Olsson B, Andersson PO, Jernas M, Jacobsson S, Carlsson B, Carlsson LM, et al. T-cell-mediated cytotoxicity toward platelets in chronic idiopathic thrombocytopenic purpura. Nat Med (2003) 9:1123–4. doi:10.1038/nm921
43. Letsch A, Keilholz U, Assfalg G, Mailander V, Thiel E, Scheibenbogen C. Bone marrow contains melanoma-reactive CD8+ effector T cells and, compared with peripheral blood, enriched numbers of melanoma-reactive CD8+ memory T cells. Cancer Res (2003) 63:5582–6.
44. Marshall DR, Turner SJ, Belz GT, Wingo S, Andreansky S, Sangster MY, et al. Measuring the diaspora for virus-specific CD8+ T cells. Proc Natl Acad Sci U S A (2001) 98:6313–8. doi:10.1073/pnas.101132698
45. Isogai S, Miyata S, Taha R, Yoshizawa Y, Martin JG, Hamid Q. CD4+ T cells migrate from airway to bone marrow after antigen inhalation in rats. J Allergy Clin Immunol (2004) 113:455–61. doi:10.1016/j.jaci.2003.11.035
46. Tripp RA, Topham DJ, Watson SR, Doherty PC. Bone marrow can function as a lymphoid organ during a primary immune response under conditions of disrupted lymphocyte trafficking. J Immunol (1997) 158:3716–20.
47. Feuerer M, Beckhove P, Garbi N, Mahnke Y, Limmer A, Hommel M, et al. Bone marrow as a priming site for T-cell responses to blood-borne antigen. Nat Med (2003) 9:1151–7. doi:10.1038/nm914
48. Cavanagh LL, Bonasio R, Mazo IB, Halin C, Cheng G, van der Velden AW, et al. Activation of bone marrow-resident memory T cells by circulating, antigen-bearing dendritic cells. Nat Immunol (2005) 6:1029–37. doi:10.1038/ni1249
49. Duffy D, Perrin H, Abadie V, Benhabiles N, Boissonnas A, Liard C, et al. Neutrophils transport antigen from the dermis to the bone marrow, initiating a source of memory CD8+ T cells. Immunity (2012) 37:917–29. doi:10.1016/j.immuni.2012.07.015
50. Tokoyoda K, Hauser AE, Nakayama T, Radbruch A. Organization of immunological memory by bone marrow stroma. Nat Rev Immunol (2010) 10:193–200. doi:10.1038/nri2727
51. Lin GH, Edele F, Mbanwi AN, Wortzman ME, Snell LM, Vidric M, et al. Contribution of 4-1BBL on radioresistant cells in providing survival signals through 4-1BB expressed on CD8(+) memory T cells in the bone marrow. Eur J Immunol (2012) 42:2861–74. doi:10.1002/eji.201242503
52. Herndler-Brandstetter D, Landgraf K, Jenewein B, Tzankov A, Brunauer R, Brunner S, et al. Human bone marrow hosts polyfunctional memory CD4+ and CD8+ T cells with close contact to IL-15-producing cells. J Immunol (2011) 186:6965–71. doi:10.4049/jimmunol.1100243
53. Pulle G, Vidric M, Watts TH. IL-15-dependent induction of 4-1BB promotes antigen-independent CD8 memory T cell survival. J Immunol (2006) 176:2739–48. doi:10.4049/jimmunol.176.5.2739
54. Sabbagh L, Snell LM, Watts TH. TNF family ligands define niches for T cell memory. Trends Immunol (2007) 28:333–9. doi:10.1016/j.it.2007.06.001
55. Allam A, Conze DB, Giardino Torchia ML, Munitic I, Yagita H, Sowell RT, et al. The CD8+ memory T-cell state of readiness is actively maintained and reversible. Blood (2009) 114:2121–30. doi:10.1182/blood-2009-05-220087
56. Snell LM, Lin GH, Watts TH. IL-15-dependent upregulation of GITR on CD8 memory phenotype T cells in the bone marrow relative to spleen and lymph node suggests the bone marrow as a site of superior bioavailability of IL-15. J Immunol (2012) 188:5915–23. doi:10.4049/jimmunol.1103270
57. Lin GH, Snell LM, Wortzman ME, Clouthier DL, Watts TH. GITR-dependent regulation of 4-1BB expression: implications for T cell memory and anti-4-1BB-induced pathology. J Immunol (2013) 190:4627–39. doi:10.4049/jimmunol.1201854
58. Gerlach C, van Heijst JW, Swart E, Sie D, Armstrong N, Kerkhoven RM, et al. One naive T cell, multiple fates in CD8+ T cell differentiation. J Exp Med (2010) 207:1235–46. doi:10.1084/jem.20091175
59. Cassese G, Parretta E, Pisapia L, Santoni A, Guardiola J, Di Rosa F. Bone marrow CD8 cells down-modulate membrane IL-7R{alpha} expression and exhibit increased STAT-5 and p38 MAPK phosphorylation in the organ environment. Blood (2007) 110:1960–9. doi:10.1182/blood-2006-09-045807
60. Quinci AC, Vitale S, Parretta E, Soriani A, Iannitto ML, Cippitelli M, et al. IL-15 inhibits IL-7Ralpha expression by memory-phenotype CD8(+) T cells in the bone marrow. Eur J Immunol (2012) 42:1129–39. doi:10.1002/eji.201142019
61. Kudernatsch RF, Letsch A, Guerreiro M, Lobel M, Bauer S, Volk HD, et al. Human bone marrow contains a subset of quiescent early memory CD8(+) T cells characterized by high CD127 expression and efflux capacity. Eur J Immunol (2014) 44:3532–42. doi:10.1002/eji.201344180
62. Weng NP, Araki Y, Subedi K. The molecular basis of the memory T cell response: differential gene expression and its epigenetic regulation. Nat Rev Immunol (2012) 12:306–15. doi:10.1038/nri3173
63. Youngblood B, Hale JS, Ahmed R. Memory CD8 T cell transcriptional plasticity. F1000Prime Rep (2015) 7:38. doi:10.12703/P7-38
64. Nemoto Y, Kanai T, Makita S, Okamoto R, Totsuka T, Takeda K, et al. Bone marrow retaining colitogenic CD4+ T cells may be a pathogenic reservoir for chronic colitis. Gastroenterology (2007) 132:176–89. doi:10.1053/j.gastro.2006.10.035
65. Tough DF, Sprent J. Turnover of naive- and memory-phenotype T cells. J Exp Med (1994) 179:1127–35. doi:10.1084/jem.179.4.1127
66. Parretta E, Cassese G, Santoni A, Guardiola J, Vecchio A, Di Rosa F. Kinetics of in vivo proliferation and death of memory and naive CD8 T cells: parameter estimation based on 5-bromo-2’-deoxyuridine incorporation in spleen, lymph nodes, and bone marrow. J Immunol (2008) 180:7230–9. doi:10.4049/jimmunol.180.11.7230
67. Di Rosa F. T-lymphocyte interaction with stromal, bone and hematopoietic cells in the bone marrow. Immunol Cell Biol (2009) 87:20–9. doi:10.1038/icb.2008.84
68. Graef P, Buchholz VR, Stemberger C, Flossdorf M, Henkel L, Schiemann M, et al. Serial transfer of single-cell-derived immunocompetence reveals stemness of CD8(+) central memory T cells. Immunity (2014) 41:116–26. doi:10.1016/j.immuni.2014.05.018
69. Banerjee A, Gordon SM, Intlekofer AM, Paley MA, Mooney EC, Lindsten T, et al. Cutting edge: the transcription factor eomesodermin enables CD8+ T cells to compete for the memory cell niche. J Immunol (2010) 185:4988–92. doi:10.4049/jimmunol.1002042
70. Chaix J, Nish SA, Lin WH, Rothman NJ, Ding L, Wherry EJ, et al. Cutting edge: CXCR4 is critical for CD8+ memory T cell homeostatic self-renewal but not rechallenge self-renewal. J Immunol (2014) 193:1013–6. doi:10.4049/jimmunol.1400488
71. Beckhove P, Feuerer M, Dolenc M, Schuetz F, Choi C, Sommerfeldt N, et al. Specifically activated memory T cell subsets from cancer patients recognize and reject xenotransplanted autologous tumors. J Clin Invest (2004) 114:67–76. doi:10.1172/JCI200420278
72. Noonan K, Matsui W, Serafini P, Carbley R, Tan G, Khalili J, et al. Activated marrow-infiltrating lymphocytes effectively target plasma cells and their clonogenic precursors. Cancer Res (2005) 65:2026–34. doi:10.1158/0008-5472.CAN-04-3337
73. Domschke C, Ge Y, Bernhardt I, Schott S, Keim S, Juenger S, et al. Long-term survival after adoptive bone marrow T cell therapy of advanced metastasized breast cancer: follow-up analysis of a clinical pilot trial. Cancer Immunol Immunother (2013) 62:1053–60. doi:10.1007/s00262-013-1414-x
74. Noonan KA, Huff CA, Davis J, Lemas MV, Fiorino S, Bitzan J, et al. Adoptive transfer of activated marrow-infiltrating lymphocytes induces measurable antitumor immunity in the bone marrow in multiple myeloma. Sci Transl Med (2015) 7:288ra78. doi:10.1126/scitranslmed.aaa7014
75. Paiardini M, Cervasi B, Engram JC, Gordon SN, Klatt NR, Muthukumar A, et al. Bone marrow-based homeostatic proliferation of mature T cells in nonhuman primates: implications for AIDS pathogenesis. Blood (2009) 113:612–21. doi:10.1182/blood-2008-06-159442
76. Takizawa H, Boettcher S, Manz MG. Demand-adapted regulation of early hematopoiesis in infection and inflammation. Blood (2012) 119:2991–3002. doi:10.1182/blood-2011-12-380113
77. Schurch CM, Riether C, Ochsenbein AF. Cytotoxic CD8+ T cells stimulate hematopoietic progenitors by promoting cytokine release from bone marrow mesenchymal stromal cells. Cell Stem Cell (2014) 14:460–72. doi:10.1016/j.stem.2014.01.002
78. Baldridge MT, King KY, Boles NC, Weksberg DC, Goodell MA. Quiescent haematopoietic stem cells are activated by IFN-gamma in response to chronic infection. Nature (2010) 465:793–7. doi:10.1038/nature09135
79. de Bruin AM, Demirel O, Hooibrink B, Brandts CH, Nolte MA. Interferon-gamma impairs proliferation of hematopoietic stem cells in mice. Blood (2013) 121:3578–85. doi:10.1182/blood-2012-05-432906
80. de Bruin AM, Voermans C, Nolte MA. Impact of interferon-gamma on hematopoiesis. Blood (2014) 124(16):2479–86. doi:10.1182/blood-2014-04-568451
81. Li R, Perez N, Karumuthil-Melethil S, Vasu C. Bone marrow is a preferential homing site for autoreactive T-cells in type 1 diabetes. Diabetes (2007) 56:2251–9. doi:10.2337/db07-0502
82. Oh HM, Yu CR, Lee Y, Chan CC, Maminishkis A, Egwuagu CE. Autoreactive memory CD4+ T lymphocytes that mediate chronic uveitis reside in the bone marrow through STAT3-dependent mechanisms. J Immunol (2011) 187:3338–46. doi:10.4049/jimmunol.1004019
83. Nemoto Y, Kanai T, Kameyama K, Shinohara T, Sakamoto N, Totsuka T, et al. Long-lived colitogenic CD4+ memory T cells residing outside the intestine participate in the perpetuation of chronic colitis. J Immunol (2009) 183:5059–68. doi:10.4049/jimmunol.0803684
84. Nemoto Y, Kanai T, Takahara M, Oshima S, Nakamura T, Okamoto R, et al. Bone marrow-mesenchymal stem cells are a major source of interleukin-7 and sustain colitis by forming the niche for colitogenic CD4 memory T cells. Gut (2013) 62:1142–52. doi:10.1136/gutjnl-2012-302029
85. Kong YY, Feige U, Sarosi I, Bolon B, Tafuri A, Morony S, et al. Activated T cells regulate bone loss and joint destruction in adjuvant arthritis through osteoprotegerin ligand. Nature (1999) 402:304–9. doi:10.1038/46303
86. Kotake S, Udagawa N, Takahashi N, Matsuzaki K, Itoh K, Ishiyama S, et al. IL-17 in synovial fluids from patients with rheumatoid arthritis is a potent stimulator of osteoclastogenesis. J Clin Invest (1999) 103:1345–52. doi:10.1172/JCI5703
87. Teng YT, Nguyen H, Gao X, Kong YY, Gorczynski RM, Singh B, et al. Functional human T-cell immunity and osteoprotegerin ligand control alveolar bone destruction in periodontal infection. J Clin Invest (2000) 106:R59–67. doi:10.1172/JCI10763
88. Sato K, Suematsu A, Okamoto K, Yamaguchi A, Morishita Y, Kadono Y, et al. Th17 functions as an osteoclastogenic helper T cell subset that links T cell activation and bone destruction. J Exp Med (2006) 203:2673–82. doi:10.1084/jem.20061775
89. Ciucci T, Ibanez L, Boucoiran A, Birgy-Barelli E, Pene J, Abou-Ezzi G, et al. Bone marrow Th17 TNFalpha cells induce osteoclast differentiation, and link bone destruction to IBD. Gut (2015) 64:1072–81. doi:10.1136/gutjnl-2014-306947
90. van Hogezand RA, Hamdy NA. Skeletal morbidity in inflammatory bowel disease. Scand J Gastroenterol (2006) 243:59–64. doi:10.1080/00365520600664276
91. Redlich K, Smolen JS. Inflammatory bone loss: pathogenesis and therapeutic intervention. Nat Rev Drug Discov (2012) 11:234–50. doi:10.1038/nrd3669
92. Monteiro AC, Leal AC, Goncalves-Silva T, Mercadante AC, Kestelman F, Chaves SB, et al. T cells induce pre-metastatic osteolytic disease and help bone metastases establishment in a mouse model of metastatic breast cancer. PLoS One (2013) 8:e68171. doi:10.1371/journal.pone.0068171
93. Li Y, Toraldo G, Li A, Yang X, Zhang H, Qian WP, et al. B cells and T cells are critical for the preservation of bone homeostasis and attainment of peak bone mass in vivo. Blood (2007) 109:3839–48. doi:10.1182/blood-2006-07-037994
94. Monteiro JP, Benjamin A, Costa ES, Barcinski MA, Bonomo A. Normal hematopoiesis is maintained by activated bone marrow CD4+ T cells. Blood (2005) 105:1484–91. doi:10.1182/blood-2004-07-2856
95. Kim S, Park K, Choi J, Jang E, Paik DJ, Seong RH, et al. Foxp3+ regulatory T cells ensure B lymphopoiesis by inhibiting the granulopoietic activity of effector T cells in mouse bone marrow. Eur J Immunol (2015) 45:167–79. doi:10.1002/eji.201444532
96. Muller AM, Poyser J, Kupper NJ, Burnett C, Ko RM, Kohrt HE, et al. Donor hematopoiesis in mice following total lymphoid irradiation requires host T-regulatory cells for durable engraftment. Blood (2014) 123:2882–92. doi:10.1182/blood-2013-10-530212
97. Fujisaki J, Wu J, Carlson AL, Silberstein L, Putheti P, Larocca R, et al. In vivo imaging of Treg cells providing immune privilege to the haematopoietic stem-cell niche. Nature (2011) 474:216–9. doi:10.1038/nature10160
98. Clark RA, Chong B, Mirchandani N, Brinster NK, Yamanaka K, Dowgiert RK, et al. The vast majority of CLA+ T cells are resident in normal skin. J Immunol (2006) 176:4431–9. doi:10.4049/jimmunol.176.7.4431
99. Reinhardt RL, Khoruts A, Merica R, Zell T, Jenkins MK. Visualizing the generation of memory CD4 T cells in the whole body. Nature (2001) 410:101–5. doi:10.1038/35065111
100. Steinert EM, Schenkel JM, Fraser KA, Beura LK, Manlove LS, Igyarto BZ, et al. Quantifying memory CD8 T cells reveals regionalization of immunosurveillance. Cell (2015) 161:737–49. doi:10.1016/j.cell.2015.03.031
101. Hand TW, Dos Santos LM, Bouladoux N, Molloy MJ, Pagan AJ, Pepper M, et al. Acute gastrointestinal infection induces long-lived microbiota-specific T cell responses. Science (2012) 337:1553–6. doi:10.1126/science.1220961
102. Naik S, Bouladoux N, Linehan JL, Han SJ, Harrison OJ, Wilhelm C, et al. Commensal-dendritic-cell interaction specifies a unique protective skin immune signature. Nature (2015) 520:104–8. doi:10.1038/nature14052
103. Bromley SK, Yan S, Tomura M, Kanagawa O, Luster AD. Recirculating memory T cells are a unique subset of CD4+ T cells with a distinct phenotype and migratory pattern. J Immunol (2013) 190:970–6. doi:10.4049/jimmunol.1202805
104. Mackay CR, Marston WL, Dudler L. Naive and memory T cells show distinct pathways of lymphocyte recirculation. J Exp Med (1990) 171:801–17. doi:10.1084/jem.171.3.801
105. Yawalkar N, Hunger RE, Pichler WJ, Braathen LR, Brand CU. Human afferent lymph from normal skin contains an increased number of mainly memory/effector CD4(+) T cells expressing activation, adhesion and co-stimulatory molecules. Eur J Immunol (2000) 30:491–7. doi:10.1002/1521-4141(200002)30:2<491::AID-IMMU491>3.0.CO;2-H
106. Gebhardt T, Mueller SN, Heath WR, Carbone FR. Peripheral tissue surveillance and residency by memory T cells. Trends Immunol (2013) 34:27–32. doi:10.1016/j.it.2012.08.008
107. Schenkel JM, Masopust D. Tissue-resident memory T cells. Immunity (2014) 41:886–97. doi:10.1016/j.immuni.2014.12.007
108. Gebhardt T, Wakim LM, Eidsmo L, Reading PC, Heath WR, Carbone FR. Memory T cells in nonlymphoid tissue that provide enhanced local immunity during infection with herpes simplex virus. Nat Immunol (2009) 10:524–30. doi:10.1038/ni.1718
109. Gebhardt T, Whitney PG, Zaid A, Mackay LK, Brooks AG, Heath WR, et al. Different patterns of peripheral migration by memory CD4+ and CD8+ T cells. Nature (2011) 477:216–9. doi:10.1038/nature10339
110. Jiang X, Clark RA, Liu L, Wagers AJ, Fuhlbrigge RC, Kupper TS. Skin infection generates non-migratory memory CD8+ T(RM) cells providing global skin immunity. Nature (2012) 483:227–31. doi:10.1038/nature10851
111. Masopust D, Choo D, Vezys V, Wherry EJ, Duraiswamy J, Akondy R, et al. Dynamic T cell migration program provides resident memory within intestinal epithelium. J Exp Med (2010) 207:553–64. doi:10.1084/jem.20090858
112. Gebhardt T, Mackay LK. Local immunity by tissue-resident CD8+ memory T cells. Front Immunol (2012) 3:340. doi:10.3389/fimmu.2012.00340
113. Casey KA, Fraser KA, Schenkel JM, Moran A, Abt MC, Beura LK, et al. Antigen-independent differentiation and maintenance of effector-like resident memory T cells in tissues. J Immunol (2012) 188:4866–75. doi:10.4049/jimmunol.1200402
114. Sheridan BS, Pham QM, Lee YT, Cauley LS, Puddington L, Lefrancois L. Oral infection drives a distinct population of intestinal resident memory CD8(+) T cells with enhanced protective function. Immunity (2014) 40:747–57. doi:10.1016/j.immuni.2014.03.007
115. Feng Y, Wang D, Yuan R, Parker CM, Farber DL, Hadley GA. CD103 expression is required for destruction of pancreatic islet allografts by CD8(+) T cells. J Exp Med (2002) 196:877–86. doi:10.1084/jem.20020178
116. Lee YT, Suarez-Ramirez JE, Wu T, Redman JM, Bouchard K, Hadley GA, et al. Environmental and antigen receptor-derived signals support sustained surveillance of the lungs by pathogen-specific cytotoxic T lymphocytes. J Virol (2011) 85:4085–94. doi:10.1128/JVI.02493-10
117. Mackay LK, Rahimpour A, Ma JZ, Collins N, Stock AT, Hafon ML, et al. The developmental pathway for CD103(+)CD8+ tissue-resident memory T cells of skin. Nat Immunol (2013) 14:1294–301. doi:10.1038/ni.2744
118. Ray SJ, Franki SN, Pierce RH, Dimitrova S, Koteliansky V, Sprague AG, et al. The collagen binding alpha1beta1 integrin VLA-1 regulates CD8 T cell-mediated immune protection against heterologous influenza infection. Immunity (2004) 20:167–79. doi:10.1016/S1074-7613(04)00021-4
119. Le Floc’h A, Jalil A, Vergnon I, Le Maux Chansac B, Lazar V, Bismuth G, et al. Alpha E beta 7 integrin interaction with E-cadherin promotes antitumor CTL activity by triggering lytic granule polarization and exocytosis. J Exp Med (2007) 204:559–70. doi:10.1084/jem.20061524
120. Wakim LM, Woodward-Davis A, Bevan MJ. Memory T cells persisting within the brain after local infection show functional adaptations to their tissue of residence. Proc Natl Acad Sci U S A (2010) 107:17872–9. doi:10.1073/pnas.1010201107
121. Mackay LK, Braun A, Macleod BL, Collins N, Tebartz C, Bedoui S, et al. Cutting edge: CD69 interference with sphingosine-1-phosphate receptor function regulates peripheral T cell retention. J Immunol (2015) 195(5):2059–63. doi:10.4049/jimmunol.1402256
122. Skon CN, Lee JY, Anderson KG, Masopust D, Hogquist KA, Jameson SC. Transcriptional downregulation of S1pr1 is required for the establishment of resident memory CD8+ T cells. Nat Immunol (2013) 14:1285–93. doi:10.1038/ni.2745
123. Bankovich AJ, Shiow LR, Cyster JG. CD69 suppresses sphingosine 1-phosophate receptor-1 (S1P1) function through interaction with membrane helix 4. J Biol Chem (2010) 285:22328–37. doi:10.1074/jbc.M110.123299
124. Bergsbaken T, Bevan MJ. Proinflammatory microenvironments within the intestine regulate the differentiation of tissue-resident CD8(+) T cells responding to infection. Nat Immunol (2015) 16:406–14. doi:10.1038/ni.3108
125. Purwar R, Campbell J, Murphy G, Richards WG, Clark RA, Kupper TS. Resident memory T cells (T(RM)) are abundant in human lung: diversity, function, and antigen specificity. PLoS One (2011) 6:e16245. doi:10.1371/journal.pone.0016245
126. Teijaro JR, Turner D, Pham Q, Wherry EJ, Lefrancois L, Farber DL. Cutting edge: tissue-retentive lung memory CD4 T cells mediate optimal protection to respiratory virus infection. J Immunol (2011) 187:5510–4. doi:10.4049/jimmunol.1102243
127. Iijima N, Iwasaki A. T cell memory. A local macrophage chemokine network sustains protective tissue-resident memory CD4 T cells. Science (2014) 346:93–8. doi:10.1126/science.1257530
128. Watanabe R, Gehad A, Yang C, Scott LL, Teague JE, Schlapbach C, et al. Human skin is protected by four functionally and phenotypically discrete populations of resident and recirculating memory T cells. Sci Transl Med (2015) 7:279ra39. doi:10.1126/scitranslmed.3010302
129. Sathaliyawala T, Kubota M, Yudanin N, Turner D, Camp P, Thome JJ, et al. Distribution and compartmentalization of human circulating and tissue-resident memory T cell subsets. Immunity (2013) 38:187–97. doi:10.1016/j.immuni.2012.09.020
130. Mackay LK, Stock AT, Ma JZ, Jones CM, Kent SJ, Mueller SN, et al. Long-lived epithelial immunity by tissue-resident memory T (TRM) cells in the absence of persisting local antigen presentation. Proc Natl Acad Sci U S A (2012) 109:7037–42. doi:10.1073/pnas.1202288109
131. Masopust D, Vezys V, Wherry EJ, Barber DL, Ahmed R. Cutting edge: gut microenvironment promotes differentiation of a unique memory CD8 T cell population. J Immunol (2006) 176:2079–83. doi:10.4049/jimmunol.176.4.2079
132. Joshi NS, Cui W, Chandele A, Lee HK, Urso DR, Hagman J, et al. Inflammation directs memory precursor and short-lived effector CD8(+) T cell fates via the graded expression of T-bet transcription factor. Immunity (2007) 27:281–95. doi:10.1016/j.immuni.2007.07.010
133. Wakim LM, Smith J, Caminschi I, Lahoud MH, Villadangos JA. Antibody-targeted vaccination to lung dendritic cells generates tissue-resident memory CD8 T cells that are highly protective against influenza virus infection. Mucosal Immunol (2015) 8:1060–71. doi:10.1038/mi.2014.133
134. Wu T, Hu Y, Lee YT, Bouchard KR, Benechet A, Khanna K, et al. Lung-resident memory CD8 T cells (TRM) are indispensable for optimal cross-protection against pulmonary virus infection. J Leukoc Biol (2014) 95:215–24. doi:10.1189/jlb.0313180
135. Ariotti S, Beltman JB, Chodaczek G, Hoekstra ME, van Beek AE, Gomez-Eerland R, et al. Tissue-resident memory CD8+ T cells continuously patrol skin epithelia to quickly recognize local antigen. Proc Natl Acad Sci U S A (2012) 109:19739–44. doi:10.1073/pnas.1208927109
136. Ariotti S, Hogenbirk MA, Dijkgraaf FE, Visser LL, Hoekstra ME, Song JY, et al. T cell memory. Skin-resident memory CD8(+) T cells trigger a state of tissue-wide pathogen alert. Science (2014) 346:101–5. doi:10.1126/science.1254803
137. McMaster SR, Wilson JJ, Wang H, Kohlmeier JE. Airway-resident memory CD8 T cells provide antigen-specific protection against respiratory virus challenge through rapid IFN-gamma production. J Immunol (2015) 195:203–9. doi:10.4049/jimmunol.1402975
138. Schenkel JM, Fraser KA, Beura LK, Pauken KE, Vezys V, Masopust D. T cell memory. Resident memory CD8 T cells trigger protective innate and adaptive immune responses. Science (2014) 346:98–101. doi:10.1126/science.1254536
139. Schenkel JM, Fraser KA, Vezys V, Masopust D. Sensing and alarm function of resident memory CD8(+) T cells. Nat Immunol (2013) 14:509–13. doi:10.1038/ni.2568
140. Hofmann M, Pircher H. E-cadherin promotes accumulation of a unique memory CD8 T-cell population in murine salivary glands. Proc Natl Acad Sci U S A (2011) 108:16741–6. doi:10.1073/pnas.1107200108
141. Cuburu N, Graham BS, Buck CB, Kines RC, Pang YY, Day PM, et al. Intravaginal immunization with HPV vectors induces tissue-resident CD8+ T cell responses. J Clin Invest (2012) 122:4606–20. doi:10.1172/JCI63287
142. Shin H, Iwasaki A. A vaccine strategy that protects against genital herpes by establishing local memory T cells. Nature (2012) 491:463–7. doi:10.1038/nature11522
143. Hansen SG, Vieville C, Whizin N, Coyne-Johnson L, Siess DC, Drummond DD, et al. Effector memory T cell responses are associated with protection of rhesus monkeys from mucosal simian immunodeficiency virus challenge. Nat Med (2009) 15:293–9. doi:10.1038/nm.1935
144. Kupper TS. Old and new: recent innovations in vaccine biology and skin T cells. J Invest Dermatol (2012) 132:829–34. doi:10.1038/jid.2011.400
145. Zhu J, Hladik F, Woodward A, Klock A, Peng T, Johnston C, et al. Persistence of HIV-1 receptor-positive cells after HSV-2 reactivation is a potential mechanism for increased HIV-1 acquisition. Nat Med (2009) 15:886–92. doi:10.1038/nm.2006
146. Zhu J, Koelle DM, Cao J, Vazquez J, Huang ML, Hladik F, et al. Virus-specific CD8+ T cells accumulate near sensory nerve endings in genital skin during subclinical HSV-2 reactivation. J Exp Med (2007) 204:595–603. doi:10.1084/jem.20061792
147. Zhu J, Peng T, Johnston C, Phasouk K, Kask AS, Klock A, et al. Immune surveillance by CD8alphaalpha+ skin-resident T cells in human herpes virus infection. Nature (2013) 497:494–7. doi:10.1038/nature12110
148. Peng T, Zhu J, Phasouk K, Koelle DM, Wald A, Corey L. An effector phenotype of CD8+ T cells at the junction epithelium during clinical quiescence of herpes simplex virus 2 infection. J Virol (2012) 86:10587–96. doi:10.1128/JVI.01237-12
149. Piet B, de Bree GJ, Smids-Dierdorp BS, van der Loos CM, Remmerswaal EB, von der Thusen JH, et al. CD8(+) T cells with an intraepithelial phenotype upregulate cytotoxic function upon influenza infection in human lung. J Clin Invest (2011) 121:2254–63. doi:10.1172/JCI44675
150. Woodberry T, Suscovich TJ, Henry LM, August M, Waring MT, Kaur A, et al. Alpha E beta 7 (CD103) expression identifies a highly active, tonsil-resident effector-memory CTL population. J Immunol (2005) 175:4355–62. doi:10.4049/jimmunol.175.7.4355
151. Bevan MJ. Memory T cells as an occupying force. Eur J Immunol (2011) 41:1192–5. doi:10.1002/eji.201041377
152. Clark RA. Resident memory T cells in human health and disease. Sci Transl Med (2015) 7:269rv1. doi:10.1126/scitranslmed.3010641
153. Boyman O, Hefti HP, Conrad C, Nickoloff BJ, Suter M, Nestle FO. Spontaneous development of psoriasis in a new animal model shows an essential role for resident T cells and tumor necrosis factor-alpha. J Exp Med (2004) 199:731–6. doi:10.1084/jem.20031482
154. Conrad C, Boyman O, Tonel G, Tun-Kyi A, Laggner U, de Fougerolles A, et al. Alpha1beta1 integrin is crucial for accumulation of epidermal T cells and the development of psoriasis. Nat Med (2007) 13:836–42. doi:10.1038/nm1605
155. Mizukawa Y, Yamazaki Y, Teraki Y, Hayakawa J, Hayakawa K, Nuriya H, et al. Direct evidence for interferon-gamma production by effector-memory-type intraepidermal T cells residing at an effector site of immunopathology in fixed drug eruption. Am J Pathol (2002) 161:1337–47. doi:10.1016/S0002-9440(10)64410-0
156. Harris JE, Harris TH, Weninger W, Wherry EJ, Hunter CA, Turka LA. A mouse model of vitiligo with focused epidermal depigmentation requires IFN-gamma for autoreactive CD8(+) T-cell accumulation in the skin. J Invest Dermatol (2012) 132:1869–76. doi:10.1038/jid.2011.463
157. Xing L, Dai Z, Jabbari A, Cerise JE, Higgins CA, Gong W, et al. Alopecia areata is driven by cytotoxic T lymphocytes and is reversed by JAK inhibition. Nat Med (2014) 20:1043–9. doi:10.1038/nm.3645
158. Lian CG, Bueno EM, Granter SR, Laga AC, Saavedra AP, Lin WM, et al. Biomarker evaluation of face transplant rejection: association of donor T cells with target cell injury. Mod Pathol (2014) 27:788–99. doi:10.1038/modpathol.2013.249
159. Cresswell J, Robertson H, Neal DE, Griffiths TR, Kirby JA. Distribution of lymphocytes of the alpha(E)beta(7) phenotype and E-cadherin in normal human urothelium and bladder carcinomas. Clin Exp Immunol (2001) 126:397–402. doi:10.1046/j.1365-2249.2001.01652.x
160. Djenidi F, Adam J, Goubar A, Durgeau A, Meurice G, de Montpreville V, et al. CD8+CD103+ tumor-infiltrating lymphocytes are tumor-specific tissue-resident memory T cells and a prognostic factor for survival in lung cancer patients. J Immunol (2015) 194:3475–86. doi:10.4049/jimmunol.1402711
161. Webb JR, Milne K, Nelson BH. Location, location, location: CD103 demarcates intraepithelial, prognostically favorable CD8 tumor-infiltrating lymphocytes in ovarian cancer. Oncoimmunology (2014) 3:e27668. doi:10.4161/onci.27668
162. Webb JR, Milne K, Watson P, Deleeuw RJ, Nelson BH. Tumor-infiltrating lymphocytes expressing the tissue resident memory marker CD103 are associated with increased survival in high-grade serous ovarian cancer. Clin Cancer Res (2014) 20:434–44. doi:10.1158/1078-0432.CCR-13-1877
163. Wakim LM, Gebhardt T, Heath WR, Carbone FR. Cutting edge: local recall responses by memory T cells newly recruited to peripheral nonlymphoid tissues. J Immunol (2008) 181:5837–41. doi:10.4049/jimmunol.181.9.5837
164. Wakim LM, Waithman J, van Rooijen N, Heath WR, Carbone FR. Dendritic cell-induced memory T cell activation in nonlymphoid tissues. Science (2008) 319:198–202. doi:10.1126/science.1151869
Keywords: memory T cells, migration, recirculation, bone marrow, tissue-resident T cells, CD8 T cells
Citation: Di Rosa F and Gebhardt T (2016) Bone Marrow T Cells and the Integrated Functions of Recirculating and Tissue-Resident Memory T Cells. Front. Immunol. 7:51. doi: 10.3389/fimmu.2016.00051
Received: 09 December 2015; Accepted: 01 February 2016;
Published: 16 February 2016
Edited by:
Peter Katsikis, Erasmus University Medical Center, NetherlandsReviewed by:
David Hildeman, Cincinnati Children’s Hospital, USACopyright: © 2016 Di Rosa and Gebhardt. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Francesca Di Rosa, ZnJhbmNlc2NhLmRpcm9zYUB1bmlyb21hMS5pdA==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.