 Teresa Krakauer
Teresa Krakauer Kisha Pradhan2
Kisha Pradhan2 Bradley G. Stiles
Bradley G. Stiles
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 02 February 2016
Sec. Microbial Immunology
Volume 7 - 2016 | https://doi.org/10.3389/fimmu.2016.00023
This article is part of the Research Topic Bacterial Exotoxins: how bacteria fight the immune system View all 17 articles
Staphylococcal enterotoxin B (SEB) of Staphylococcus aureus, and related superantigenic toxins produced by myriad microbes, are potent stimulators of the immune system causing a variety of human diseases from transient food poisoning to lethal toxic shock. These protein toxins bind directly to specific Vβ regions of T-cell receptors (TCR) and major histocompatibility complex (MHC) class II on antigen-presenting cells, resulting in hyperactivation of T lymphocytes and monocytes/macrophages. Activated host cells produce excessive amounts of proinflammatory cytokines and chemokines, especially tumor necrosis factor α, interleukin 1 (IL-1), IL-2, interferon γ (IFNγ), and macrophage chemoattractant protein 1 causing clinical symptoms of fever, hypotension, and shock. Because of superantigen-induced T cells skewed toward TH1 helper cells, and the induction of proinflammatory cytokines, superantigens can exacerbate autoimmune diseases. Upon TCR/MHC ligation, pathways induced by superantigens include the mitogen-activated protein kinase cascades and cytokine receptor signaling, resulting in activation of NFκB and the phosphoinositide 3-kinase/mammalian target of rapamycin pathways. Various mouse models exist to study SEB-induced shock including those with potentiating agents, transgenic mice and an “SEB-only” model. However, therapeutics to treat toxic shock remain elusive as host response genes central to pathogenesis of superantigens have only been identified recently. Gene profiling of a murine model for SEB-induced shock reveals novel molecules upregulated in multiple organs not previously associated with SEB-induced responses. The pivotal genes include intracellular DNA/RNA sensors, apoptosis/DNA damage-related molecules, immunoproteasome components, as well as antiviral and IFN-stimulated genes. The host-wide induction of these, and other, antimicrobial defense genes provide evidence that SEB elicits danger signals resulting in multi-organ damage and toxic shock. Ultimately, these discoveries might lead to novel therapeutics for various superantigen-based diseases.
Staphylococcus aureus causes many human and animal diseases, some of them are life-threatening and found throughout the world (1–4). Humans are naturally colonized by S. aureus that often cause no problems and are considered a “harmless” commensal. However, when given an opportunity (i.e., weakened immune system, compromised epidermis, or mucosa, etc.), this bacterium can cause various diseases that become life threatening.
Staphylococcus aureus is a Gram-positive coccus and member of the Micrococcaceae family, which includes non-pathogenic genera found in soil, water, and on skin. Both planktonic- and biofilm-based versions of S. aureus can cause disease in an infected host, subsequently becoming (at times) quite difficult to clear from the body (5, 6). Characteristics of this genus called Staphylococcus (“staphyle” in Greek, meaning grape cluster and “coccus” meaning grain or berry) and species called aureus (“golden” in Latin, as per colony color on agar media), include Gram-positive, non-spore forming, facultative, β-hemolytic, catalase-positive, mannitol fermentation, and salt tolerant (7).
Staphylococcus aureus is known for producing many different protein toxins involved in pathogenesis. Clearly, the bacterium is very good at surviving harsh conditions in/on a host by using a vast array of virulence factors that promote the many diseases caused by infiltrating S. aureus and their toxins. One group of toxins, the staphylococcal enterotoxins (SEs), is the focus of this review. Antibiotic-resistant strains (community-, nursing home-, and hospital-acquired) are a particular problem in health care and its associated economics, requiring effective infection control plans that prevent S. aureus spread locally and globally (8).
Staphylococcus aureus was first described in the early 1880s by a Scotsman, Dr. Alexander Ogston, after microscopy analysis of over a hundred, pus-filled abscesses of human origins (9). S. aureus readily colonizes mammalian epidermis/dermis, mucosa, soft tissues, bone, and medical devices such as catheters. Furthermore, medically relevant strains of the bacterium have become variably resistant to many antibiotics, including methicillin and vancomycin, which negatively impacts psychological and economic aspects of human society (10–13). S. aureus strains that are methicillin resistant (MRSA) are commonly treated first with vancomycin. Both methicillin and vancomycin target bacterial cell-wall synthesis. Other antimicrobials that have been used for fighting S. aureus include (1) linezolid (inhibits protein synthesis at the 50S ribosome); (2) daptomycin (inserts into membranes); (3) fusidic acid (inhibits protein elongation at the ribosome); (4) teicoplanin (like vancomycin, inhibits cell wall synthesis but not used in the United States); and (5) tigecycline (inhibits protein synthesis at the 30S ribosome) (14, 15).
For various reasons, alternative methods for treating antibiotic-resistant strains of S. aureus (i.e., MRSA) are being explored by different groups around the world (16–23). Some of the exotic sources for these novel drugs include snake venom, tree bark/stems/leaves, and carnivorous plants. Risk factors for MRSA colonization include antibiotic exposure, admittance into an intensive care unit, surgery, long-term care residency, and exposure to others carrying MRSA (4, 24). In 2011, the United States Centers for Disease Control and Prevention reported >80,000 MRSA cases that were life threatening, with >11,000 fatalities (14%) (22, 25). This is remarkably a number of deaths greater than that attributed to acquired immunodeficiency syndrome (AIDS) for the same time period. Furthermore, the economic burden of MRSA in the United States is upwards to $13.8 billion/year and represents one of the most costly, acute infectious diseases requiring treatment (10). Invasive forms of MRSA include the following in order of prevalence: bacteremia, pneumonia, cellulitis, osteomyelitis, endocarditis, and septic shock (24). Black males older than 65 years are the most common patients suffering from invasive MRSA; however, the reasons for this are to date unknown (24). MRSA from hospital/health-care settings are perhaps the origins of community strains now seen throughout the human population (4). The latter types of S. aureus are often more susceptible to antibiotics than those from hospital/health-care settings.
From a historical perspective, MRSA was detected within a year after introducing this new beta-lactam antibiotic called methicillin in 1959, further highlighting the rapid adaptability of this microbe (26, 27). Methicillin was meant to overcome the increasingly prevalent, penicillin-resistant strains of S. aureus first detected during the mid-1940s in hospitals and throughout the community. These strains were isolated just 2 years after penicillin’s introduction into clinical practice (27). This pattern of antibiotic resistance is evident with various pathogens, suggesting furthermore the importance of developing truly novel antimicrobials and embracing a paradigm shift for discovering/creating antimicrobials. Perhaps vaccines will play a bigger role in fighting various pathogens, in the future?
Transmission of antibiotic-resistant strains between livestock and humans working with and/or consuming the former is a real problem, poorly understood to date (28–31). Furthermore, aerosol transmission of MRSA from farm animals or human patients in hospital wards, to humans, is also a scary reality (32–34). As just one known example of S. aureus transmission between species, strain CC398 was most commonly associated with asymptomatic colonization of swine, but now causes invasive infections of humans (35). Evidently, farm animals are an important reservoir for disease-causing S. aureus that colonize humans. Humans (~30%) can be asymptomatic carriers of S. aureus strains that possess virulence factor genes encoding antibiotic resistance, toxins, and other proteins that promote some nasty, potentially fatal diseases (1, 3, 36–40) (Table 1). The risk factors are many for children carrying MRSA nasally, and include (1) <4 years of age; (2) being male; (3) family size >4; (4) antibiotic use during previous 3 months; (5) smoking by parents; and (6) sleeping with parents (39). For adults, risk factors that enhance carriage of S. aureus include: (1) being male; (2) having diabetes; (3) use of hormonal contraception by women; (4) close physical contact with others (i.e., sports); and interestingly (5) being a non-smoker (40).

Table 1. Staphylococcus aureu s: some virulence factors and diseases.
Clearly, Homo sapiens are not strangers to hosting S. aureus on, and in, themselves. This bacterium is rather adaptable and employs various virulence factors seemingly important in its survival. Are we, in the near and distant future, able to co-adapt and effectively counter this microbial medical challenge, which is just one of many? We now focus upon one remarkable virulence factor, a family of protein toxins that vary in structure and biological effects upon mammals: the SEs.
From a historical perspective, the SEs (and S. aureus) were first described as “ptomaine” (derived from Greek “ptoma,” meaning corpse) in 1914 by Barber (41), after human consumption of cow’s milk contaminated by staphylococci (from the cow) led to vomiting and diarrhea of unsuspecting visitors. Family members were remarkably resistant to this poisoning effect, perhaps due to repeated exposure and development of toxin-specific immunity. Barber included himself as a “human guinea pig” to prove his point that milk from this one farm, during the hot season in the Philippines, was indeed the medium for enteric illness. His final analysis revealed that short-term storage of the milk at ambient temperature enabled staphylococcal growth and toxin production (41).
Dack et al. reported in 1930 an enteric-acting toxin produced by a “yellow hemolytic Staphylococcus” isolated from sponge cake involved in food poisoning (42). Upon administering culture filtrate of the isolated bacterium grown in the laboratory to a rabbit (intravenous) or humans (n = 3; oral), there was respectively death and uniform nausea/diarrhea. Some of the SEs were first purified by various groups during the 1950s (43–45), further linked to a major cause of food poisoning evident around the world, and found to stimulate specific subsets of T cells leading to shock (1, 46, 47).
Regarding staphylococcal food poisoning and diagnosis, the latter can be done upon suspected food/drink using classic microbiology and agar plates (105 viable S. aureus/gram food), or by direct detection of toxin (47). SE detection in food is most commonly accomplished by employing immunological techniques (i.e., commercially available enzyme/fluorescent immune assays) that are to date limited in detecting relatively few toxin types. However, next generation assays involving mass spectrometry are now being developed for the future pending higher throughput and cost-effectiveness (47).
The SEs are characteristically stable proteins resistant to high heat, low pH, freezing, drying, and proteases. Partly because of their stable nature and powerful effects upon humans at very low doses, the SEs (particularly SEB) have been studied from a biodefense perspective spanning decades of research by various countries (48, 49). SEB is also considered a Category B select agent by various United States federal agencies. SEB, when inhaled, can induce several symptoms within 120 min involving an aching feeling (head and muscles), increased heartbeat, coughing, enteric dysfunction (i.e., nausea, vomiting, and diarrhea), as well as eye irritation (49). Nanogram levels of inhaled SEB are incapacitating while microgram levels can be fatal. SEB adversely affects the immune system, and it is plausible that opportunistic viruses and bacteria can cause further harm to the host exposed to any SE (50–52).
Upon ingestion, the SEs (A–V and counting) are responsible for a prevalent form of food poisoning globally (1, 38, 46, 47, 53). There are five, sequence-based groups of SEs generally recognized to date (1). These single-chain proteins (~25 kDa) are generally composed of two domains containing both alpha helices and beta sheets, separated by a groove, as evidenced by one of the first SEs to be crystallized: SEB (54). Since the mid-2000s, the literature has also adopted the designation of SE-like (SEl) (55). This defines a staphylococcal protein that shares amino acid sequence homology with a previously characterized SE, yet has not to date been tested (or at least reported in the literature) for enteric effects in primates (56). Regarding staphylococcal food poisoning, SEA is the most commonly detected culprit in the United States, followed by SED and SEB. Classic poisoning due to food-based SEs can occur after ingesting processed meats or dairy products contaminated by improper handling (i.e., S. aureus transfer from the skin of a food handler to food) and subsequent storage of food at an elevated temperature conducive to bacterial growth. Depending upon the S. aureus strain, there can be one (or more) SEs released into the tainted food. Following consumption of SEs in food/drink, one may still experience a general malaise 24–72 h later (56). Poisoning by the SEs via many different food types is rarely fatal for healthy individuals, and occurs around the world; however, as is often the case with many diseases, the very young and old represent higher risk groups for severe morbidity and possible death due to SE exposure (57).
Exactly how the SEs cause enteric illness is still remarkably unresolved, but prostaglandins and leukotrienes might mediate the effects (58, 59). Serotonin release in the intestines and vagal nerve stimulation may also be involved in SE-induced emesis (60, 61). The mode of action is seemingly more complex than many other enterotoxins that work directly upon gastric epithelial cells and/or supporting matrix.
The term “superantigen” commonly describes the S. aureus SEs and toxic shock syndrome toxin-1 (TSST-1), while related streptococcal pyrogenic exotoxins (SPEs), also known as superantigens, are produced by Streptococcus pyogenes. Superantigen was first used in late 1980s literature to describe microbial proteins that activate a large population of specific T-cells at very minute (picogram) concentrations (62, 63). Typical “conventional” antigens stimulate far fewer T-cells at higher concentrations. Superantigens and conventional counterparts also differ by: (1) superantigens bind to the exterior of the peptide-binding groove of major histocompatibility complex class II (MHC II); (2) superantigens bind to different MHC II types; (3) superantigenic effects occur without internalization and antigen processing; and (4) T-cell receptor (TCR) recognition of a superantigen: MHC II complex requires the variable region of a TCR β chain (Vβ), not the Vα–Vβ chain used by conventional antigens (62–67). From a virulence perspective, superantigens corrupt host immunity that then enables pathogen survival.
Microbial superantigens are reportedly produced by other bacterial genera (Mycobacterium, Mycoplasma, Yersinia), fungi (Candida), and even viruses (herpes, rabies), thus suggesting a conserved and successful strategy employed throughout Nature (68). Versus superantigens of Staphylococcus and Streptococcus, those from the other aforementioned microbes are poorly characterized to date.
Superantigens interact with both CD4 and CD8 T-cells and the signaling, post-TCR binding, is similar to conventional antigen binding to TCR; however, binding is to specific Vβ regions of TCR unlike conventional antigens. In addition, the interaction of superantigen with MHC class II on antigen-presenting cells (APC) is also different, with superantigens binding outside the peptide-binding groove of MHC class II. Structural and binding studies indicate at least two different binding sites on MHC class II for SE and TSST-1. A common overlapping binding region exists on HLA-DR for these toxins, referred to as the generic MHC class II binding site involving the α chain. An additional binding site with higher affinity can be found within the C-terminus of SEA/SED/SEH, which binds to the HLA-DR β chain in a Zn2+-dependent manner (69–71). The presence of two MHC binding sites allows SEA to cross-link MHC class II on APC and activate monocytes, inducing potent proinflammatory mediators. The binding of staphylococcal superantigens to MHC class II, and bridging to TCR, activates a high percentage of T cells. Two decades of intense investigations focused upon superantigen binding to cellular receptors reveal how superantigens activate cells of the immune system (46, 62, 72–76). Interaction of superantigen with TCR transmits the classical first signal for T-cell activation. The binding of superantigen to costimulatory receptors such as CD28 promotes supramolecular clusters, stabilizes cellular interaction, and optimizes protein kinase signal transduction (77). CD28 co-stimulation enhances mRNA stability of IL-2 and T-cell survival by increased expression of anti-apoptotic Bcl-xl (78). Other cell-surface molecules such as CD2, CD11a/ICAM-1, and ELAM facilitate optimal activation of endothelial and T cells by SEB (79). TCR and costimulatory receptors activate protein tyrosine kinases (PTKs), LCK and ZAP-70, resulting in phospholipase C gamma (PLCγ) activation, the release of intracellular second messengers, and subsequent protein kinase C (PKC) activation (67, 80, 81). Accompanying this T-cell activation is F-actin polymerization and increased intracellular Ca2+. PTK and PKC activation lead to mitogen-activated protein kinase (MAPK), extracellular signal regulated kinase (ERK), and cJun N-terminal kinase (JNK) pathways that activate transcriptional factors NFκB, NF-AT, and AP-1 (67, 81, 82). PKCθ activation leads to CARMA1, Bcl10, and MALT1 (CBM) complexes that induce NFκB transcriptional activation and controls T-cell proliferation (83). Many proinflammatory cytokine genes contain NFκB-binding sites in their promotor region, and are induced by NFκB (84). The NFκB cascade is a major signal transduction pathway for many pattern recognition receptors (PRR), such as toll-like (TLR) and proinflammatory cytokine receptors (85). The cytokines IL-1, TNFα, IFNγ, IL-2, IL-6 and chemokines, specifically MCP-1, are induced by superantigens (74, 75, 86). IL-1 and TNFα also activate fibroblasts, epithelial and endothelial cells to induce NFκB and additional mediators, cell adhesion molecules, and tissue proteases (87). Mediators produced by superantigen-activated cells exert profound damaging effects on the immune and cardiovascular system, culminating in multi-organ dysfunction and lethal shock. PTKs and T-cell cytokines also activate phosphoinositide 3 kinase (PI3K), signaling protein kinase B (Akt), and mammalian target of rapamycin complex 1 (mTORC1) downstream (88). Two signaling molecules, NFκB and mTORC1, are key hubs mediating the major biological responses to SE from TCR, co-stimulator CD28, and cytokine signaling.
T-cell activation via the TCR-CD3 complex subsequently activates membrane proximal PTKs that phosphorylate TCR intracellular components, other cellular substrates, as well as adaptors (67, 80–82). PLCγ cleaves phosphatidylinositol 4,5-bisphosphate, generating second messengers to activate PKC and the proto-oncogene Ras. PTK also activates PI3K, generating several inositol phospholipids and ultimately activating Akt and PKCθ (89). A number of receptors besides TCR, including those for CD28, IL-2 (IL-2R), insulin, growth factor, and the G-protein-coupled receptor (GPCR), also transduce activation signals upon ligand binding via the PI3K pathway. The cytosolic signalsome formed by PKCθ and CBM is located at an immunological synapse formed after T-cell activation by anti-CD3 and -CD28 (89, 90). CBM complexes activate the inhibitor of κB (IκB) kinase complex (IKK) through ubiquitin ligases (91). IKK phosphorylates IκB, releasing NF-κB for nuclear translocation and gene activation. Nuclear NFκB binds DNA coding for proinflammatory cytokines and many other NFκB target genes, activating diverse proinflammatory mediators, as well as pro- and anti-apoptotic molecules.
Toxic shock syndrome toxin-1 induces inositol phospholipid turnover, PKC translocation, and calcium mobilization in human T-cells resembling responses from those of a mitogenic signal (92). SEE uses an alternative LCK-independent pathway by activating PLCβ signaling in T cells (93). This alternative pathway also triggers influx of Ca2+, activates PKC, ERK1/2, and ultimately nuclear translocation of NFκB and NF-AT.
Downstream of PI3K is the serine/threonine kinase Akt, which mediates many diverse biological processes upon specific binding to TCR, growth factors, insulin receptor, tyrosine kinase receptor, and GPCR. Two potent cytokines from superantigen-stimulated T cells, IFNγ and IL-2, also activate the PI3K/Akt/mTORC1 pathway via the transducer Janus kinase 1 (JAK1) after binding IFNγ and IL-2 receptors, respectively (94, 95). Site-specific phosphorylation/dephosphorylation modulates the PI3K/Akt signaling to activate mTORC1. Activation of mTORC1 causes phosphorylation and activation of the ribosomal 40S protein p70S6 kinase (p70S6K) plus eukaryotic initiation factor binding protein 1 (4EBP1) (96–101). Thus mTORC1 controls protein translation essential for G1 to S phase transition (100). An mTORC1-specific inhibitor, rapamycin, blocks SEB-induced T cell proliferation as well as SEB-induced IL-2 and IFNγ in vitro and in vivo (102). Importantly, inhibition of the SEB-induced PI3K/Akt/mTORC1 pathway by rapamycin prevents lethal toxic shock in a mouse model (102).
In vitro and in vivo studies reveal that excessive release of proinflammatory cytokines (IL-1, TNFα, IL-6, and IFNγ) mediates the toxic effects of superantigens (49, 51, 52, 76, 79). Proinflammatory cytokines like IL-1 and TNFα activate the transcriptional factor NFκB in many cell types, thus perpetuating the inflammatory response. IL-1 and TNFα have tissue damaging effects, and together with SEB-induced metalloproteinases (MMP) and the pro-coagulant tissue factor (TF), promote inflammation and coagulation (87, 103). IL-2 from superantigen-activated T cells causes vasodilation, vascular leak, and edema.
Many cell types respond directly to staphylococcal superantigens besides T and APC/monocytes, including B, endothelial, synovial fibroblasts, intestinal epithelial, and mast (58, 79, 104–109). Cross-linking of TCR with MHC class II on B cells by superantigen triggers B cell proliferation and differentiation into plasma cells (104). Stimulation of synovial fibroblasts with superantigens induces chemokine gene expression, raising the possibility that superantigens can trigger chemotactic responses and initiate inflammatory arthritis (105). Human T84 colonic cells increase ion flow after incubation with SEB and PBMC, suggesting that superantigens indirectly affect gut mucosa via the immune system (106). SEB and TSST-1 transcytose intestinal epithelial cell barriers, unlike SEA (107), and TSST-1 can also induce chemotactic cytokines (IL-8 and MIP-3α) from human vaginal epithelial cells (109). S. aureus frequently colonizes vaginal mucosa, producing TSST-1, which penetrates the mucosa through chemokines from epithelial cells (110). Chemokines recruit neutrophils and other immune cells activated furthermore by superantigens. The potent activation of T cells by superantigen interaction of MHC class II and TCR induces T-cell proliferation via cell activation pathways in T cells and APC. The proliferative response as a result of IL-2 induction from superantigen-activated T cells is similar to that induced by mitogens. IL-2 increases vascular permeability, causing edema and multiple organ damage (111). TNFα synergizes with IL-2 to induce pulmonary vascular leak and lymphocyte accumulation (112). The combination of TNFα and IFNγ elaborated by SEB-activated cells disrupt the epithelial barrier, causing edema and vascular leak in gut mucosa (106).
Interleukin 1 interacts with IL-1 receptor 1 (IL-1R1) and an additional accessory protein, triggering downstream signaling molecules like the adaptor myeloid differentiation factor 88 (MyD88), IL-1R- associated protein kinase 1 (IRAK1), and TNF receptor-associated factor 6 (TRAF6) that activate NFκB (113). A set of structurally related receptors, the TLRs, signal with similar intracellular adaptors as those used for IL-1R1, but are not used for superantigen signaling (85, 114). The TLRs are conserved, type-1 transmembrane receptors that recognize pathogen-associated molecular patterns (PAMPs), such as lipoprotein, peptidoglycan, LPS, flagellin, dsRNA, and viral RNA, which stimulate host innate immunity and enhance adaptive immunity (85, 114). SEB reportedly increases cellular expression of TLR2 and TLR4, synergistically promoting lethal shock with LPS (115, 116). Activation of NFκB induces proinflammatory genes as well as pro- and anti-apoptotic genes. An auto-feedback loop exists to downregulate NFκB, which induces IκBα. Other cytosolic PRR sense intracellular damage-associated molecular patterns (DAMPs), triggering inflammasome activation and release of caspases plus IL-1β (117).
TNFα binds to TNF receptor 1 (TNFR1) or TNFR2, activating NFκB and inducing expression of other cytokines, adhesion, as well as co-stimulatory molecules (118, 119). The cytotoxic functions of TNFα are mostly mediated by binding TNFR1 via intracellular death domains that trigger apoptosis through caspase activation. SEB increases expression of CD95 (Fas), a receptor of the TNFR superfamily. Intracellular adaptors TNFR-associated death domain (TRADD), Fas-associated death domain (FADD), and receptor interacting protein kinase (RIP) are used by the TNFR superfamily to activate the caspase 8 cascade, JNK, and NFκB that subsequently elicit apoptosis, cell activation, coagulation, inflammation, and host defense (119). TNFα and IFNγ act synergistically on epithelial cells to increase ion transport, causing cell damage and epithelial leakage (120). The critical role of TNFα in mediating pathological effects of SEB and lethality was recognized early on as anti-TNFα antibodies confer protection from SEB-induced shock in a d-galactosamine (d-gal)-sensitized mouse model (121). Both IL-1 and TNFα enhance the procoagulant activity of TF and activate neutrophils, accounting for tissue damage and organ injury commonly seen in animal models of septic shock and SEB-induced lethal shock.
Type II interferon (IFNγ) binds to IFNγR, which belongs to the family of interferon receptors (IFNR), including the structurally different receptors for type I interferons (IFNα, IFNβ) (122, 123). The signal transducer and activator of transcription 1 (STAT1) is a common signal transducer for both types of IFNR, and phosphorylation of STAT1 by Janus kinases initiates signal transduction. IFNα and IFNβ induce indistinguishable signals, which include anti-proliferative and antiviral activities. Types I and II IFNs have many overlapping activities and stimulate many common interferon-stimulated genes (ISG) (123, 124). However, in addition to host-defense functions, IFNγ also induces immunoproteasomes and expression of MHC class II. IFNγ promotes cell-mediated immunological activities essential for antibacterial defense. The IFNγ-activated JAKs also activate PI3K in a STAT1-independent manner culminating in mTORC1 activation, promoting protein translation (125). IFNγ activates PKC leading to MAPK activation, which is also commonly activated by IL-1, TLR ligands, and TNFα. IFNγ has a critical role in host defense as the induction of immunoproteasome components and antigen-processing peptidases enhance cellular immune responses against pathogens. Both types of IFNs induce apoptosis and many ISGs have antiviral, anti-angiogenic, and immunomodulatory functions. IFNs induce apoptosis by activating death receptors such as Fas (CD95), which then activates the adaptor FADD, leading to subsequent caspase-8 activation. Activation of the caspase 8 cascade causes cell death plus release of cytochrome c and mitochondrial DNA (mtDNA). SEA-induced hepatotoxicity is mediated by FasL and the hepatocellular damage is independent of leukocyte recruitment (126). IFNγ disrupts barrier function and ion transport in superantigen-activated epithelial cells. In vitro, disturbances in epithelial barrier function can be duplicated with IFNγ plus TNFα (106). Recent studies suggest that IFNγ downregulates regulatory T-cells (Treg), accounting for the potent polarizing effects of IFNγ on TH1/TH2 cell differentiation and an inflammatory environment for cell migration and activation (127).
IL-2 binds to IL-2R and activates PI3K and Ras (128). Activation of the PI3K/Akt/mTORC1 pathway and Ras controls proliferation, growth, and differentiation of many cell types. Ras activates the MAPK and ERK cascades, leading to transcriptional activation of AP-1, cJun/Fos, and NFAT. The MAPK cascade induces ER stress and NFκB (129). IL-2 induces vasodilation and increases microvascular permeability by suppressing endothelin-1, causing perivascular edema seen in SEB-induced acute lung injury (130, 131). IL-2-deficient mice are resistant to SEB-induced toxic shock, providing evidence for the critical roles of PI3K/mTORC1 and NFκB (132).
The chemokines IL-8, MCP-1, MIP-1α, and MIP-1β are induced directly by SEB or TSST-1. These chemoattractant chemokines activate leukocytes and influence migration of neutrophils, dendritic cells, and leukocytes (133, 134). Chemokines bind to seven-transmembrane GPCR, induce early Ca2+ flux, activate PLC and signal via the PI3K/mTORC1 pathway (133). Cytokine- and chemokine-activated neutrophils, recruited to sites of tissue injury and inflammation, produce reactive oxygen species (ROS) and activate MMPs contributing to organ dysfunction. MMPs cause tissue degradation and change chemokine interactions with the extracellular matrix (ECM), creating a chemokine gradient affecting cell recruitment (133). Exudates from superantigen-injected air pouches are predominantly neutrophils with some macrophages (135). Both systemic and intranasal administration of SEB cause acute lung injury characterized by increased: (1) expression of adhesion molecules ICAM-1 and VCAM; (2) neutrophil and mononuclear cell infiltrate; (3) endothelial cell injury; and (4) vascular permeability (135–137).
The PI3K/mTORC1 pathway plays a dominant role in superantigen-induced cell proliferation and migration as TCR, CD28, IL-2R, IFNγR, and chemokine receptors all signal through this pathway. IL-1 and TNFα independently activate NFκB via MyD88/TRAF6/IRAK and FADD/TRADD/RIP, respectively (87). TCR and CD28 via PKC also activate NFκB signaling. Other PRR, including surface and cytosolic TLRs, signal via MyD88 to activate NFκB via different adaptors. In addition, TLR3 and TLR4 activate TRIF (toll/IL-1 receptor homology domain-containing adaptor inducing interferon β) signaling, inducing interferon regulatory factors (IRFs) (114). The overlap of PRR signaling by NFκB and IRFs with TCR and co-stimulatory signals by superantigens to activate NFκB and mTORC1 cannot be understated. These pathways activate inflammatory genes, as well as antiviral, anti-apoptotic, and pro-apoptotic molecules. Thus the three initial signals provided by TCR, costimulatory receptors, and cytokines converge on NFκB and mTORC1 to promote host defense against superantigens.
Injection of SEB into mice has been used to study activation-induced apoptosis and T-cell anergy in vivo. This effect may be linked to a rapid (within 1 h) loss of l-selectin on the surface of specific Vβ-bearing T cells, thus resulting in decreased signal transduction (138). Via endocytosis, surface levels of TCR-CD3 decrease ~50% among Vβ-reactive T cells within 30 min after SEB exposure (139). The rapid hyperactivation and proliferation of T cells in mice following an SEB injection is transient, as within 48 h the majority of proliferating T cells is eliminated by activation-induced cell death (140). CD95 mediates elimination of SEB-activated T cells and the residual Vβ-specific cells become anergic, or functionally unresponsive. However, controversy still exists regarding the functional ability and fate of these anergic T cells. After injection of SEB into mice, splenic Vβ8+ T cells are deleted or no longer respond to SEB, and produce less IL-2 and IFNγ. In contrast, others report that these anergic cells synthesize less IL-2 but still secrete IFNγ that can mediate toxic shock following a subsequent dose of SEB (141). An evident paradox is that the anti-inflammatory cytokine IL-10, which protects against SE-induced shock (142), is also produced by SEB-primed T cells. Treg cells likely downregulate superantigen activation in vitro and in vivo.
Immune homeostasis is maintained by Treg cells through a number of mechanisms depending on the “immune environment” (143). Natural and induced Foxp3+ Treg cells control excessive immune activation deleterious to the host. Treg cells also downregulate autoimmune responses, and superantigens can subvert the functional activity of Treg cells in atopic dermatitis (144). Although the immunosuppressive function of Treg cells is well known, the mechanisms of action and involvement of different cell types are still debated. The markers used in identifying different Treg cells are evolving, but the surface receptor cytotoxic T lymphocyte antigen 4 (CTLA4) likely plays a role particularly in superantigen-related counter regulation (145). CTLA4 interacts with costimulatory molecules CD80 and CD86, preventing costimulatory signals in superantigen-activated T cells (146). Systemic inflammatory responses elicited by SEB actually destabilize Treg cells and additional expansion of them in vivo does not protect transgenic mice from SEB-induced toxic shock (147).
The ability of superantigens to cross-link MHC class II and specific TCR Vβ enables these microbial toxins to stimulate the immune system and induce autoimmunity by activating APC and normally quiescent, autoreactive T- and B-cells. Activation results in cytokine and chemokine release, thus mediating a potently acute inflammatory response. Several experimental animal models show that staphylococcal superantigens are arthrogenic (148, 149). TSST-1 exacerbates bacterial cell wall-induced arthritis in rats, possibly linked to accumulating Vβ11+ T cells and IFNγ production within arthritic joints (149). TSST-1 also plays a pivotal role in murine septic arthritis, as the frequency and severity of this disease are increased after intravenous administration of TSST-1-secreting S. aureus (150). SEA or SEB can also induce relapses of experimental autoimmune encephalomyelitis in a murine model for multiple sclerosis (151). How exogenously administered toxin triggers autoimmune processes like arthritis is unknown, but it is likely that proinflammatory cytokines and chemokines produced in response to superantigens facilitate specific recruitment and migration of autoreactive T-cells into synovial tissue and joints. Thus, proinflammatory cytokines and chemokines have acute toxic effects promoting cell activation and recruitment. Following minor tissue injury or inflammation due to superantigen exposure, an increased presence of immune cells might initiate a destructive autoimmune reaction.
Psoriasis and atopic dermatitis also represent autoimmune diseases linked to staphylococcal and streptococcal colonization of skin and subsequent production of superantigens (152). SEB on healthy human skin induces inflammatory reactions, possibly linked to degranulation of cutaneous mast cells (58). T cells from patients with severe atopic dermatitis are apoptotic, which may lead to chronic infections and subsequent worsening of disease (153). Bacterial density on skin can affect relative sensitivity toward these toxins and development of atopic dermatitis.
There are currently no available therapeutics for treating superantigen-induced shock, except for the use of intravenous human immunoglobulin (154, 155). Targeting and neutralizing a superantigen directly is most suitable at early stages of exposure before cell activation and initiation of the proinflammatory cytokine cascade. Therapies targeting superantigens at the receptor level have been extensively reviewed recently (156) and include receptor-blocking peptides derived from toxins, chimeric inhibitors composed of Vβ and MHC class II domains, as well as synthetic blockers of co-stimulatory receptor CD28. However, preventing toxin-receptor interaction is ineffective post-toxin exposure and some inhibitors must be tailored to target individual toxins. Failed sepsis clinical trials of eritoran (anti-endotoxin), a drug that prevents the early steps of receptor interaction, suggest that blocking superantigen–receptor interactions will likely not protect against SEB-induced shock (157).
An important class of therapeutic compounds blocks signal transduction pathways activated by superantigens, and as these events are post-exposure, they are perhaps more amenable to suppressive manipulation. One example is the NFκB cascade containing many upstream activators. In vitro studies indicate numerous genes for cell adhesion molecules, cytokines, chemokines, acute phase proteins, and inducible nitric oxide synthase that are implicated in superantigen-induced lethal shock and contain NFκB binding sites in the promotor/enhancer region (84). Activation of NFĸB leads to the inducible expression of many mediators involved in inflammation and tissue injury seen in SEB-induced lung injury and toxic shock models. Inhibition of NFκB is beneficial, or has no effect, in preventing SEB-induced shock depending upon the mouse model (136, 158, 159); however, NFκB inhibitor must be given early and for a long duration to afford any protection (136).
Pathway inhibitors are used for identifying molecules and signaling pathways crucial for cellular responses to a specific stimulus. The superantigenic properties of SEB make it an “ideal” toxin to study cell activation signals and molecular pathways. An obvious step in testing new therapeutic approaches for SEB-induced shock is finding relevant animal models that mimic important aspects of human disease. Mice are much less susceptible to SEB due to lower affinity of superantigen for mouse MHC class II (51, 62, 160). Potentiating agents such as LPS, viruses, d-gal, and actinomycin D are used together with SEB to sensitize mice (50–52). These synergistic agents alone activate similar host signaling pathways as superantigens, confounding the “pure” host response to SEB. An alternative model utilizes transgenic mice, with either human HLA-DR3 or -DQ8, that lethally respond to SEs without a potentiating agent (161, 162). Another recent, simplified model employs “double-hit,” low dose SEB in C3H/HeJ mice, an LPS-resistant strain (131). Pathological lesions, cytokine response, and time to lethality in this “double-hit” model resemble findings in non-human primates (NHP) and staphylococcal toxic shock syndrome in patients (49). Host signatures arising from “SEB-only” exposure unexpectedly include many IFN-induced genes in multiple organs, not previously linked to SEB pathogenesis (163).
Oligonucleotide microarray analysis in the “double-hit” SEB mouse model reveals induction of danger signals bearing IFN signatures (163). These genes cover five important molecular hubs signaling danger similar to those activated by IFNs and pathogens. The upregulated transcripts present in PBMC, spleen, lung, liver, kidney, and heart include (1) innate mediators; (2) DNA/RNA sensing system; (3) ER stress; (4) metabolic/oxidative stress; and (5) the apoptosis pathway. Proinflammatory cytokines IL-1, IL-6, TNFα, IFNγ, and IFNγ-induced chemokines (CXCL9, CXCL10, CXCL11) are most prominent in SEB-stimulated PBMC, confirming previous observations in vitro and in vivo (51, 86, 135, 161, 162, 164). Activation of other innate host-defense genes include the Fc receptor for phagocytosis, MHC class I and II for increased APC function, as well as cell-surface receptors and adhesion molecules to promote cell recruitment. Z-DNA binding protein 1 (ZBP1), a DNA sensor triggering ISGs via DNA binding (165), is surprisingly upregulated in all tissues from the “double-hit” SEB mouse model (163). T-cell proliferation requires elevated protein translation and increased metabolism, resulting in misfolded proteins and oxidative stress. ER stress response genes such as ubiquitin ligases, immunoproteasome components, proteasome peptidases, and WARS (tryptophanyl-tRNA synthetase) are likely a result of Ca2+ flux, unfolded proteins, and activated PKC following cell activation and increased protein synthesis. Unresolved ER stress activates caspases and apoptosis (166, 167). Enhanced activity of the mitochondrial electron transport chain ultimately leads to oxidative stress, evidenced by activated NADPH oxidase in lung (163). MMP, cathepsins, and other proteases from lysosomes are also seen in this mouse model and in vitro stimulation of PBMC with SEA or SEB (168, 169). Increased ROS and protease levels are major factors in organ injury. Although the “double-hit” mouse model is not perfect, it mimics many in vivo responses of NHP to SEB and there are many similarities of gene expression by mouse and human PBMC incubated with SEB.
The cell death pathway triggered in vitro and in vivo includes genes associated with apoptosis such as FADD, death receptor ligand TRAIL, caspases, CARD, and phospholipid scramblase 1 (PLSCR1). These genes are activated in PBMC and major organs from the “double-hit” SEB model and human PBMC, following SEA or SEB exposure (163, 168, 169). PLSCR1 is implicated in moving phosphatidyl serine to the outside plasma membrane of apoptotic cells. Other danger signals include K+ efflux, particulate ligands such as cholesterol crystals and lysosome destabilization that triggers inflammasome activation via NLRP3 which converts pro-IL-1β to IL-1β through caspase 1 (117). Catabolic enzymes cause destruction of cell matrix, accounting for the liver and lung injury seen in SEB-mediated shock.
Induction of ZBP1 leads to binding of DNA and endosomal TLR9, upregulating antiviral genes via the IRF3 and IRF7 (114, 170). TLR9 activation also promotes NFκB-mediated cytokine gene transcription and inflammasome activation (171). Mitochondria are the most likely source of cytosolic DNA induced by SEB, as ER stress and increased mitochondrial respiration due to T-cell proliferation and activation are known to induce ROS (172). Elevated ROS activates the intrinsic cell death pathway via caspase 9, leading to mitochondrial damage that releases cytochrome c and mtDNA (173). MtDNA acts as a direct inducer of ZBP1. In addition, mtDNA has motifs similar to bacterial DNA (CpG), binds cytosolic TLR9, and promotes activation of NFκB and IRFs. Mitochondrial ROS is also a potent inducer of the inflammasome NLRP3 (117). Both NLRP3 and CARD are identified in microarray studies in vivo and in vitro with superantigen-stimulated PBMC. Cathepsins, another category of “destroyer” molecules identified by microarray in SEB-activated PBMC (169), are indicative of lysosomal rupture which also activates inflammasomes.
Upon comparing the microarray data from an in vivo “SEB-only” mouse model (163), and human PBMC stimulated with SEA (168) or SEB (169), several commonly activated genes importantly induce the pathogenic effects of SEB (Table 2). These genes are activated by IFN and account for the therapeutic effectiveness of rapamycin in preventing SEB-induced shock (102).
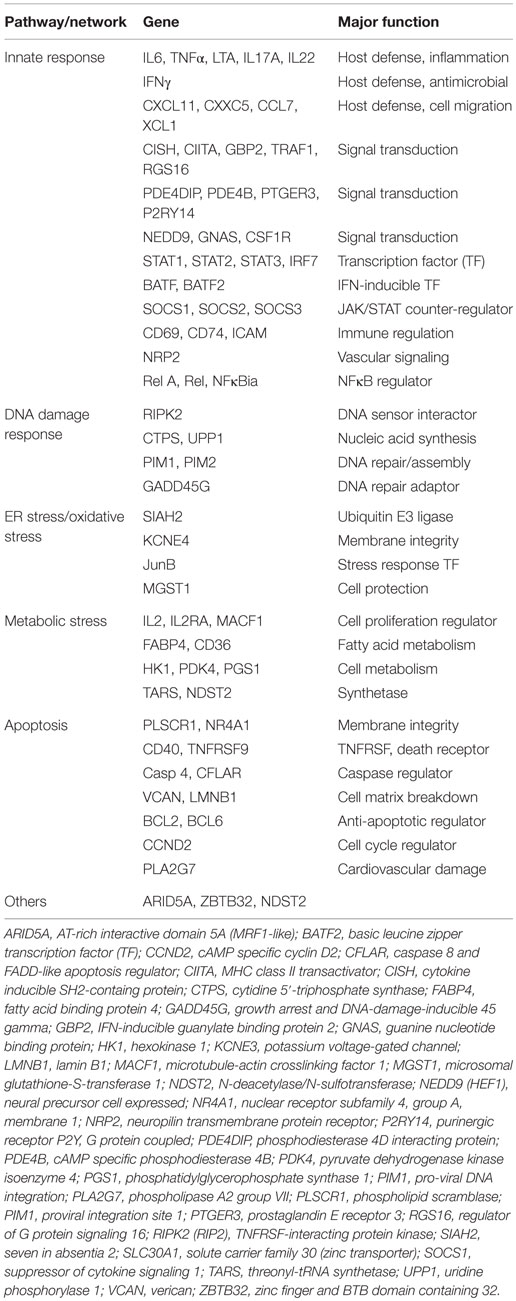
Table 2. Common differentially expressed genes induced by superantigens in vitro and in vivo.
The early induction of three key proinflammatory cytokines, IL-1, TNFα, and IFNγ, work via individual receptor-mediated signaling molecules that, respectively, activate distinct pathways: (1) IL-1R/MyD88/NFκB; (2) TNFαR/FADD/RIP; and (3) IFNγR/JAK/IRF. IL-1, TNFα, and IFNγ have independent and synergistic effects signaling inflammation, caspase activation/cell death, and an antiviral response. IL-1 from inflammasome activation has pleiotropic effects, while TNFα has an established role that initiates cell death through the adaptor FADD, activating caspases 3 and 8. IFNγ triggers innate host defense responses, antiviral genes, apoptotic programs, immunoproteasomes, and has many immunomodulatory functions. Interruption of these concurrent cascades early after SEB exposure is effective in preventing SEB-induced lethal shock.
Decades of drug development against sepsis and septic shock point to the failure of using anti-inflammatory cytokines alone, and early interruption of cytokine release is perhaps a necessary but insufficient target. Drugs also act on multiple targets, some known and unknown, as exemplified by statins that inhibit 3-hydroxy-3-methylglutaryl-coenzyme A with anti-inflammatory effects (174). Statins, used therapeutically to reduce cholesterol, are under consideration for treating various inflammatory diseases (175). Superantigens trigger multiple pathways that cross-regulate each other positively and negatively, thus targeting downstream effectors might be more specific and perhaps interfere less with normal cell function. Knowledge of immunoregulation within activated pathways by SEB enables a better choice of inhibitors.
Apoptosis plays a critical role in sepsis-induced lethality (176). The two pathways leading to apoptosis are operative in SEB-induced lethal shock, as induction of genes for both pathways occurs in the “double-hit” SEB model and other mouse models employing potentiating agents such as d-gal. The death receptor pathway used by the TNFR superfamily with ligands like TNFα and FasL induces cell death following superantigen exposure (121, 126). A second apoptosis pathway, the intrinsic pathway, is dependent on mitochondria and the Bcl2 family of pro- and anti-apoptotic proteins. ROS and loss of mitochondrial transmembrane potential play important roles, as previous studies implicate both in SEB pathogenesis (177). Other cell-surface receptors like CD44 might also contribute to SEB-induced injury, as CD44-knockout mice have elevated liver damage in the d-gal-sensitized SEB model (178). Since d-gal is hepatotoxic, the induction of pro-apoptotic molecules by SEB likely act synergistically with d-gal to promote cell death.
Microarray gene analysis in an “SEB-only” mouse model implicates both extrinsic and intrinsic pathways in SEB-induced apoptosis. Apoptosis plays a role in down-regulating immune responses but simultaneously has devastating effects when apoptotic cells or associated molecules are not removed. Autophagy is a cellular mechanism that removes bacteria, protein aggregates, and damaged organelles to maintain homeostasis (176, 179). A recent study indicates that blocking autophagy augments T-cell activation (180). Degradation and removal of Bcl10, which is part of CBM and a critical component for TCR and costimulatory signaling, is dependent on autophagy (176). IFN induces many genes regulating NFκB and apoptosis. The damage response induced by superantigens likely starts with inflammatory cytokines and apoptotic programs activated by IFNγ and TNFα. DAMPs such as mitochondrial ROS and mtDNA trigger more apoptosis, activate inflammasomes, and induce transcription factors for ISGs. Increasing energy demand and mitochondrial respiratory-chain activity also lead to elevated ROS. Normally, mitochondria damaged by excessive membrane permeability and ROS are removed by a specialized form of autophagy called mitophagy. However, overactivation of PI3K/mTORC1 in superantigen-stimulated cells likely blocks autophagy, resulting in inflammasome activation and accumulation of damaged mitochondria. Rapamycin, a well-known inducer of autophagy, prevents SEB-induced shock by removing damage-inducing molecules and damaged mitochondria. Two other FDA-approved immunosuppressants for organ transplants, cyclosporine A and tacrolimus, do not protect against superantigen-induced shock in NHP and human HLA-DR3 transgenic mice, respectively (181, 182). The calcineurin inhibitor, cyclosporine A, protects d-gal-sensitized mice from SEB-induced shock (121) but does not protect NHP challenged with SEB (181). Tacrolimus suppresses SEB-induced T-cell proliferation in vitro but does not confer protection from toxic shock in transgenic mice (182). Tacrolimus also fails to protect mice from lethal pneumonia induced by superantigen-producing S. aureus (182).
There is good agreement between the genes significantly induced in mouse PBMC from this “double-hit” SEB model and SEA- or SEG-stimulated human PBMC (163, 168). Many genes of the apoptosis-related cell death pathway account for the damage response initiated by SEB. MtDNA is ancestrally related to bacterial DNA (CpG motifs), inducing a “foreign” DNA sensor (ZBP1) or alternatively binding endosomal TLR9 that triggers host defense including type 1 IFN-mediated responses via IRF3 (114). Important clues from animal models, old and new, reveal acute release of proinflammatory cytokines that culminate in damaged organs and lethal shock.
Staphylococcus aureus is a toxin-producing pathogen that causes various diseases found throughout the body. Increasingly, S. aureus becomes more resistant to various therapeutics (i.e., antibiotics) over time, thus our own immune systems must more effectively clear this pathogen. Further knowledge of our immune system will clearly enable us to better thwart S. aureus and other pathogens. Mammals sense invading microbes via conserved PRR for detecting molecular patterns on, or released by, various bacterial, viral, and fungal pathogens. This rapid innate immune response produces proinflammatory mediators, cell activation, and recruitment of inflammatory cells to infection sites. However, some sensors for detecting PAMPs also bind to host DAMPs, confusing the “stranger” versus “danger” signaling. Host response to staphylococcal superantigens typifies the generation of these danger signals as shown in a mouse model of SEB-induced shock. The induction of cell death through apoptotic proteins observed during sepsis or superantigen exposure may provide a common target for therapeutic intervention.
All authors listed have made substantial, direct and intellectual contributions to the work, and approved it for publication.
The views expressed in this publication are those of the authors and do not reflect the official policy or position of the Department of the Army, the Department of Defense, or the U.S. Government.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank DTRA for funding. Brad Stiles acknowledges Wilson College for the continuing support of this, and many other, endeavors.
1. Uchiyama T, Imanishi K, Miyoshi-Akiyama T, Kata H. Staphylococcal superantigens and the diseases they cause. 3rd ed. In: Alouf JE, Popoff MR, editors. The Comprehensive Sourcebook of Bacterial Protein Toxins. London: Academic Press (2006). p. 830–43.
2. Archer NK, Mazaitis MJ, Costerton JW, Leid JG, Powers ME, Shirtliff ME. Staphylococcus aureus biofilms: properties, regulation and roles in human disease. Virulence (2011) 2(5):445–59. doi: 10.4161/viru.2.5.17724
3. Thurlow LR, Joshi GS, Richardson AR. Virulence strategies of the dominant USA300 lineage of community-associated methicillin-resistant Staphylococcus aureus (CA-MRSA). FEMS Immunol Med Microbiol (2012) 65:5–22. doi:10.1111/j.1574-695X.2012.00937.x
4. Chambers HF. The changing epidemiology of Staphylococcus aureus? Emerg Infect Dis (2001) 7:178–82. doi:10.3201/eid0702.010204
5. Abdelhady W, Bayer AS, Seidl K, Nast CC, Kiedrowski MR, Horswill AR, et al. Reduced vancomycin susceptibility in an in vitro catheter-related biofilm model correlates with poor therapeutic outcomes in experimental endocarditis due to methicillin-resistant Staphylococcus aureus. Antimicrob Agents Chemother (2013) 57:1447–54. doi:10.1128/AAC.02073-12
6. Windolf CD, Loegters T, Schoolz M, Windolf J, Flohe S. Lysostaphin-coated titan-implants preventing localized osteitis by Staphylococcus aureus in a mouse model. PLoS One (2014) 9(12):e115940. doi:10.1371/journal.pone.0115940
7. Kloos WE, Smith PB. Staphylococci. In: Lennette EH, Balows A, Hausler WJ, Truant JP, editors. Manual of Clinical Microbiology. Washington, DC: American Society for Microbiology (1980). p. 83–7.
8. David MZ, Medvedev S, Hohmann SF, Ewigman B, Daum RS. Increasing burden of methicillin-resistant Staphylococcus aureus hospitalizations at US academic medical centers, 2003-2008. Infect Control Hosp Epidemiol (2012) 33:782–9. doi:10.1086/666640
10. Lee BY, Singh A, David MZ, Bartsch SM, Slayton RB, Huang SS, et al. The economic burden of community-associated methicillin-resistant Staphylococcus aureus (CA-MRSA). Clin Microbiol Infect (2013) 19:528–36. doi:10.1111/j.1469-0691.2012.03914.x
11. Antonanzas F, Lozano C, Torres C. Economic features of antibiotic resistance: the case of methicillin-resistant Staphylococcus aureus. Pharmacoeconomics (2015) 33(4):285–325. doi:10.1007/s40273-014-0242-y
12. Cornejo-Juarez P, Vilar-Compte D, Perez-Jimenez C, Namendys-Silva SA, Sandoval-Hernandez S, Volkow-Fernandez P. The impact of hospital acquired infections with multidrug-resistant bacteria at an oncological intensive care unit. Int J Infect Dis (2015) 31:31–4. doi:10.1016/j.ijid.2014.12.022
13. Mangen MJ, Bouwknegt M, Friesema IH, Haagsma JA, Kortbeek LM, Tariq L, et al. Cost-of-illness and disease burden of food-related pathogens in the Netherlands, 2011. Int J Food Microbiol (2015) 196:84–93. doi:10.1016/j.ijfoodmicro.2014.11.022
14. Bouza E. New therapeutic choices for infections caused by methicillin-resistant Staphylococcus aureus. Clin Microbiol Infect (2009) 7:44–52. doi:10.1111/j.1469-0691.2009.03091.x
15. Rivera AM, Boucher HW. Current concepts in antimicrobial therapy against select gram-positive organisms: methicillin-resistant Staphylococcus aureus, penicillin-resistant pneumococci, and vancomycin-resistant enterococci. Mayo Clin Proc (2011) 86(12):1230–42. doi:10.4065/mcp.2011.0514
16. Samy RP, Stiles BG, Gopalakrishnakone P, Chow VT. Antimicrobial proteins from snake venoms: direct bacterial damage and activation of innate immunity against Staphylococcus aureus skin infection. Curr Med Chem (2011) 18:5104–13. doi:10.2174/092986711797636108
17. Hiramatsu K, Igarashi M, Morimoto Y, Baba T, Umekita M, Akamatsu Y. Curing bacteria of antibiotic resistance: reverse antibiotics, a novel class of antibiotics in nature. Int J Antimicrob Agents (2012) 39:478–85. doi:10.1016/j.ijantimicag.2012.02.007
18. Arunava K. Antibiotics and bioactive natural products in treatment of methicillin resistant Staphylococcus aureus: a brief review. Pharmacogn Rev (2015) 9:29–34. doi:10.4103/0973-7847.156329
19. Coulibaly K, Zirihi G, Guessennd-Kouadio N, Oussou KR, Dosso M. Antibacterial properties of trunk barks of Terminalia ivorensis, a commercial and medicinal species on methicillin-resistant staphylococci species. Afr Health Sci (2014) 14:753–6. doi:10.4314/ahs.v14i3.35
20. Krychowiak M, Grinholc M, Banasiuk R, Krauze-Baranowska M, Glod D, Kawiak A, et al. Combination of silver nanoparticles and Drosera binata extract as a possible alternative for antibiotic treatment of burn wound infections caused by resistant Staphylococcus aureus. PLoS One (2014) 9:e115727. doi:10.1371/journal.pone.0115727
21. Rose T, Verbeken G, De Vos D, Merabishvilli M, Vaneechoutte M, Lavigne R, et al. Experimental phage therapy of burn wound infection: difficult first steps. Int J Burns Trauma (2014) 4:66–73.
22. Rajamuthiah R, Fuchs BB, Conery AL, Kim W, Jayamani E, Kwon B, et al. Repurposing salicylanilide antihelmintic drugs to combat drug resistant Staphylococcus aureus. PLoS One (2015) 10:e0124595. doi:10.1371/journal.pone.0124595
23. Rempe CS, Burris KP, Woo HL, Goodrich B, Gosnell DK, Tschaplinski TJ, et al. Computational ranking of yerba mate small molecules based on their predicted contribution to antibacterial activity against methicillin-resistant Staphylococcus aureus. PLoS One (2015) 10:e0123925. doi:10.1371/journal.pone.0123925
24. Klevens RM, Morrison MA, Nadle J, Petit S, Gershman K, Ray S, et al. Invasive methicillin-resistant Staphylococcus aureus infections in the United States. JAMA (2007) 298:1763–71. doi:10.1001/jama.298.15.1763
25. CDC. Active Bacterial Core Surveillance (ABCs) Report Emerging Infections Program Network Methicillin-Resistant Staphylococcus aureus (2011). Available from: http://www.cdc.gov/abcs/reports-findings/survreports/mrsa11.pdf
26. Jevons MP. Celbenin-resistant staphylococci. Br Med J (1961) 1:124–5. doi:10.1136/bmj.1.5219.124-a
27. Lowy FD. Antimicrobial resistance: the example of Staphylococcus aureus. J Clin Invest (2003) 111:1265–73. doi:10.1172/JCI18535
28. Attien P, Sina H, Moussaoui W, Zimmermann-Meisse G, Dadie T, Keller D, et al. Mass spectrometry and multiplex antigen assays to assess microbial quality and toxin production of Staphylococcus aureus strains isolated from clinical and food samples. Biomed Res Int (2014) 2014:485620. doi:10.1155/2014/485620
29. Kadariya J, Smith TC, Thapaliya D. Staphylococcus aureus and staphylococcal food-borne disease: an ongoing challenge in public health. Biomed Res Int (2014) 2014:827965. doi:10.1155/2014/827965
30. Nadimpalli M, Rinsky JL, Wing S, Hall D, Stewart J, Larsen J, et al. Persistence of livestock-associated antibiotic-resistant Staphylococcus aureus among industrial hog operation workers in North Carolina over 14 days. Occup Environ Med (2015) 72:90–9. doi:10.1136/oemed-2014-102095
31. Peton V, Le Loir Y. Staphylococcus aureus in veterinary medicine. Infect Genet Evol (2014) 21:602–15. doi:10.1016/j.meegid.2013.08.011
32. Bos ME, Verstappen KM, van Cleef BA, Dohmen W, Dorado-Garcia A, Graveland H, et al. Transmission through air as a possible route of exposure for MRSA. J Expo Sci Environ Epidemiol (2014). doi:10.1038/jes.2014.85
33. Creamer E, Shore AC, Deasy EC, Galvin S, Dolan A, Walley N, et al. Air and surface contamination patterns of methicillin-resistant Staphylococcus aureus on eight acute hospital wards. J Hosp Infect (2014) 86:201–8. doi:10.1016/j.jhin.2013.12.005
34. Patchanee P, Tadee P, Arjkumpa O, Love D, Chanachai K, Alter T, et al. Occurrence and characterization of livestock-associated methicillin-resistant Staphylococcus aureus in pig industries of Northern Thailand. J Vet Sci (2014) 15:529–36. doi:10.4142/jvs.2014.15.4.529
35. Van der Mee-Marquet NL, Corvaglia A, Haenni M, Bertrand X, Franck J-B, Kluytmans J, et al. Emergence of a novel subpopulation of CC398 Staphylococcus aureus infecting animals is a serious hazard for humans. Front Microbiol (2014) 5:00652. doi:10.3389/fmicb.2014.00652
36. Dinges MM, Orwin PM, Schlievert PM. Exotoxins of Staphylococcus aureus. Clin Microbiol Rev (2000) 13:16–34. doi:10.1128/CMR.13.1.16-34.2000
37. Graham PL, Lin SX, Larson EL. A U.S. population-based survey of Staphylococcus aureus colonization. Ann Intern Med (2006) 144:318–25. doi:10.7326/0003-4819-144-5-200603070-00006
38. Tang YW, Stratton CW. Staphylococcus aureus: an old pathogen with new weapons. Clin Lab Med (2010) 30:179–208. doi:10.1016/j.cll.2010.01.005
39. Soltani B, Ardakani AT, Moravveji A, Erami M, Rezaei MH, Moniri R, et al. Risk factors for methicillin resistant Staphylococcus aureus nasal colonization of healthy children. Jundishapur J Microbiol (2014) 7(9):e20025. doi:10.5812/jjm.20025
40. Sollid JUE, Furberg AS, Hanssen AM, Johannessen M. Staphylococcus aureus: determinants of human carriage. Infect Genet Evol (2014) 21:531–41. doi:10.1016/j.meegid.2013.03.020
41. Barber MA. Milk poisoning due to a type of Staphylococcus albus occurring in the udder of a healthy cow. Philippine J Sci (1914) 9:515–9.
42. Dack GM, Cary WE, Woolpert O, Wiggers H. An outbreak of food poisoning proved to be due to a yellow hemolytic staphylococcus. J Prev Med (1930) 4:167–75.
43. Bergdoll MS, Kadavy JL, Surgalla MJ, Dack GM. Partial purification of staphylococcal enterotoxin. Arch Biochem Biophys (1951) 33:259–62. doi:10.1016/0003-9861(51)90104-X
44. Bergdoll MS, Sugiyama H, Dack GM. Staphylococcal enterotoxin. 1. purification. Arch Biochem Biophys (1959) 85:62–9. doi:10.1016/0003-9861(59)90447-3
45. Thatcher FS, Matheson BH, Simon WR. Studies with staphylococcal toxins: III. The application of paper ionophoresis to the resolution of components of toxic concentrates. Can J Microbiol (1955) 1:401–14. doi:10.1139/m55-051
46. Fraser JD, Proft T. The bacterial superantigen and superantigen-like proteins. Immunol Rev (2008) 225:226–43. doi:10.1111/j.1600-065X.2008.00681.x
47. Hennekinne J-A, de Buyser M-L, Dragacci S. Staphylococcus aureus and its food poisoning toxins: characterization and outbreak investigation. FEMS Microbiol Rev (2012) 36:815–36. doi:10.1111/j.1574-6976.2011.00311.x
48. Madsen JM. Toxins as weapons of mass destruction. A comparison and contrast with biological-warfare and chemical-warfare agents. Clin Lab Med (2001) 21:593–605.
49. Ulrich RG, Wilhelmsen CL, Krakauer T. Staphylococcal enterotoxin B and related toxins. In: Zygmund D, editor. Textbook of Military Medicine: Medical Aspects of Biological Warfare. Washington, DC: US Department of Army, Borden Institute (2007). p. 311–22.
50. Schlievert PM. Enhancement of host susceptibility to lethal endotoxin shock by staphylococcal pyrogenic exotoxin type C. Infect Immun (1982) 36:123–8.
51. Stiles BG, Bavari S, Krakauer T, Ulrich RG. Toxicity of staphylococcal enterotoxin potentiated by lipopolysaccharide: major histocompatibility complex class II molecule dependency and cytokine release. Infect Immun (1993) 61:5333–8.
52. Sarawar SR, Blackman MA, Doherty PC. Superantigen shock in mice with an inapparent viral infection. J Infect Dis (1994) 170:1189–94. doi:10.1093/infdis/170.5.1189
53. Loir L, Baron F, Gautier M. Staphylococcus aureus and food poisoning. Genet Mol Res (2003) 2:63–76.
54. Swaminathan S, Furey W, Pletcher J, Sax M. Crystal structure of staphylococcal enterotoxin B, a superantigen. Nature (1992) 359:801–6. doi:10.1038/359801a0
55. Lina G, Bohach GA, Nair SP, Hiramatsu K, Jouvin-Marche E, Mariuzza R. Standard nomenclature for the superantigens expressed by Staphylococcus. J Infect Dis (2004) 189:2334–6. doi:10.1086/420852
56. Bergdoll MS. Monkey feeding test for staphylococcal enterotoxin. Methods Enzymol (1988) 165:324–33. doi:10.1016/S0076-6879(88)65048-8
57. Schelin J, Wallin-Carlquist N, Cohn MT, Lindqvist R, Barker GC, Radstrom P. The formation of Staphylococcus aureus enterotoxin in food environments and advances in risk assessment. Virulence (2011) 2(6):580–92. doi:10.4161/viru.2.6.18122
58. Scheuber PH, Denzliner C, Wilker D, Beck G, Keppler D, Hammer DK. Staphylococcal enterotoxin B as a nonimmunological mast cell stimulus in primates: the role of endogenous cysteinyl leukotrienes. Int Arch Allergy Appl Immunol (1987) 82:289–91. doi:10.1159/000234209
59. Jett M, Brinkley W, Neill R, Gemski P, Hunt R. Staphylococcus aureus enterotoxin B challenge of monkeys: correlation of plasma levels of arachidonic acid cascade products with occurrence of illness. Infect Immun (1999) 58:3494–9.
60. Hu DL, Zhu G, Mori F, Omoe K, Okada M, Wakabayashi K, et al. Staphylococcal enterotoxin induces emesis through increasing serotonin release in intestine and it is downregulated by cannabinoid receptor 1. Cell Microbiol (2007) 9:2267–77. doi:10.1111/j.1462-5822.2007.00957.x
61. Hu DL, Nakane A. Mechanisms of staphylococcal enterotoxin-induced emesis. Eur J Pharmacol (2014) 722:95–107. doi:10.1016/j.ejphar.2013.08.050
62. Choi Y, Kotzin B, Herron L, Callahan J, Marrack P, Kappler J. Interaction of Staphylococcus aureus toxin “superantigens” with human T cells. Proc Natl Acad Sci USA (1989) 86:8941–5. doi:10.1073/pnas.86.22.8941
63. Langley RJ, Fraser JD, Proft T. Bacterial superantigens and superantigen-like toxins. 4th ed. In: Alouf J, Ladant D, Popoff MR, editors. The Comprehensive Sourcebook of Bacterial Protein Toxins. London: Academic Press (2015). p. 911–74.
64. Pontzer CH, Russell JK, Johnson HM. Structural basis for differential binding of staphylococcal enterotoxin A and toxic shock syndrome toxin 1 to class II major histocompatibility molecules. Proc Natl Acad Sci USA (1991) 88:125–8. doi:10.1073/pnas.88.1.125
65. Jablonsky MJ, Subramaniam PS, Johnson HM, Russell JK, Krishna NR. The solution structure of a class II major histocompatibility complex superantigen binding domain. Biochem Biophys Res Commun (1997) 234:660–5. doi:10.1006/bbrc.1997.6692
66. Jardetzky TS, Brown JH, Gorga JC, Stern LJ, Urban RG, Chi YI, et al. Three-dimensional structure of a human class II histocompatibility molecule complexed with superantigen. Nature (1994) 368:711–8. doi:10.1038/368711a0
67. Smith-Garvin JE, Koretzky GA, Jordan MS. T cell activation. Annu Rev Immunol (2009) 27:591–619. doi:10.1146/annurev.immunol.021908.132706
68. Larkin EA, Carman RJ, Krakauer T, Stiles BG. Staphylococcus aureus: the toxic presence of a pathogen extraordinaire. Curr Med Chem (2009) 16:4003–19. doi:10.2174/092986709789352321
69. Chintagumpala MM, Mollick JA, Rich RR. Staphylococcal toxins bind to different sites on HLA-DR. J Immunol (1991) 147:3876–82.
70. Hudson KR, Tiedemann RE, Urban RG, Lowe SC, Strominger JL, Fraser JD. Staphylococcal enterotoxin A has two cooperative binding sites on major histocompatibility complex class II. J Exp Med (1995) 182:711–20. doi:10.1084/jem.182.3.711
71. Mehindate K, Thibodeau J, Dohlsten M, Kalland T, Sékaly RP, Mourad W. Cross-linking MHC class II molecules by staphylococcal enterotoxin A superantigen is a requirement for inflammatory cytokine gene expression. J Exp Med (1995) 182:1573–7. doi:10.1084/jem.182.5.1573
72. Fleischer B, Schrezenmeier H. T cell stimulation by staphylococcal enterotoxins. Clonally variable response and requirement for major histocompatibility complex class II molecules on accessory or target cells. J Exp Med (1988) 167:1697–707. doi:10.1084/jem.167.5.1697
73. Chatila T, Geha RS. Signal transduction by microbial superantigens via MHC class II molecules. Immunol Rev (1993) 131:43–59. doi:10.1111/j.1600-065X.1993.tb01529.x
74. Tiedemann RE, Fraser JD. Cross-linking of MHC class II molecules by staphylococcal enterotoxin A is essential for antigen-presenting cell and T cell activation. J Immunol (1996) 157:3958–66.
75. Carlsson R, Fischer H, Sjogren HO. Binding of staphylococcal enterotoxin A to accessory cells is a requirement for its ability to activate human T cells. J Immunol (1998) 140:2484–8.
76. Anderson MR, Tary-Lehmann M. Staphylococcal enterotoxin-B-induced lethal shock in mice is T-cell-dependent, but disease susceptibility is defined by the non-T-cell compartment. Clin Immunol (2001) 98:85–94. doi:10.1006/clim.2000.4960
77. Cemerski S, Shaw A. Immune synapses in T-cell activation. Curr Opin Immunol (2006) 18:298–304. doi:10.1016/j.coi.2006.03.011
78. Boise LH, Minn AJ, Noel PJ, June CH, Accavitti MA, Lindsten T, et al. CD28 costimulation can promote T cell survival by enhancing the expression of Bcl-xl. Immunity (1995) 3:87–98. doi:10.1016/1074-7613(95)90161-2
79. Krakauer T. Co-stimulatory receptors for the superantigen staphyloccoccal enterotoxin B on human vascular endothelial cells and T cells. J Leukoc Biol (1994) 56:458–63.
80. Weiss A. T lymphocyte activation. 4th ed. In: Paul W, editor. Fundamental Immunology. Philadelphia: Lippincott-Raven (1998). p. 411–47.
81. Van Leeuwen JE, Samelson LE. T cell-antigen receptor signal transduction. Curr Opin Immunol (1999) 11:242–8. doi:10.1016/S0952-7915(99)80040-5
82. Isakov N, Altman A. PKC-theta-mediated signal delivery from the TCR/CD28 surface receptors. Front Immunol (2012) 3:273. doi:10.3389/fimmu.2012.00273
83. Paul S, Schaefer BC. A new look at TCR signaling to NFκB. Trends Immunol (2013) 34:269–81. doi:10.1016/j.it.2013.02.002
84. DiDonato JA, Mercurio F, Karin M. NFκB and the link between inflammation and cancer. Immunol Rev (2012) 246:379–400. doi:10.1111/j.1600-065X.2012.01099.x
85. Takeuchi O, Akira S. Pattern recognition receptors and inflammation. Cell (2010) 140:805–20. doi:10.1016/j.cell.2010.01.022
86. Krakauer T. The induction of CC chemokines in human peripheral blood mononuclear cells by staphylococcal exotoxins and its prevention by pentoxifylline. J Leukoc Biol (1999) 66:158–64.
87. Krakauer T, Vilcek J, Oppenheim JJ. Proinflammatory cytokines: TNF and IL-1 families, chemokines, TGFß and others. 4th ed. In: Paul W, editor. Fundamental Immunology. Philadelphia: Lippincott-Raven (1998). p. 775–811.
88. Deane JA, Fruman DA. Phosphoinositide 3-kinase: diverse roles in immune cell activation. Annu Rev Immunol (2004) 22:563–98. doi:10.1146/annurev.immunol.22.012703.104721
89. Park SG, Schulze-Luehrman J, Hayden MS, Hashimoto N, Ogawa W, Kasuga M, et al. Phosphoinositide-dependent kinase 1 integrates T cell receptor and CD28 co-receptor signaling to effect NFκB induction and T cell activation. Nat Immunol (2009) 10:158–66. doi:10.1038/ni.1687
90. Cartwright NG, Kashyap AK, Schaefer BC. An active kinase domain is required for retention of PKCθ at the immunological synapse. Mol Biol Cell (2011) 22:3491–7. doi:10.1091/mbc.E10-11-0916
91. Clark K, Nanda S, Cohen P. Molecular control of the MEMO family of ubiquitin-binding proteins. Nat Rev Mol Cell Biol (2013) 14:673–85. doi:10.1038/nrm3644
92. Chatila T, Wood N, Parsonnet J, Geha RS. Toxic shock syndrome toxin-1 induces inositol phospholipid turnover, protein kinase C translocation, and calcium mobilization in human T cells. J Immunol (1988) 140:1250–5.
93. Bueno C, Lemke CD, Criado G, Baroja ML, Ferguson SS, Rahman AK, et al. Bacterial superantigens bypass Lck-dependent T cell receptor signaling by activating a Galpha11-dependent, PLC-beta-mediated pathway. Immunity (2006) 25:67–78. doi:10.1016/j.immuni.2006.04.012
94. Ghoreschi K, Laurence A, O’Shea JJ. Janus kinases in immune cell signaling. Immunol Rev (2009) 228:273–87. doi:10.1111/j.1600-065X.2008.00754.x
95. Hulme MA, Wasserfall CH, Atkinson MA, Brusko TM. Central role for interleukin-2 in type 1 diabetes. Diabetes (2012) 61:14–22. doi:10.2337/db11-1213
96. Thomson AW, Turnquist HR, Raimondi G. Immunoregulatory functions of mTOR inhibition. Nat Rev Immunol (2009) 9:324–37. doi:10.1038/nri2546
97. Zoncu R, Sabatini DM, Efeyan A. mTOR: from growth signal integration to cancer, diabetes and ageing. Nat Rev Mol Cell Biol (2011) 12:21–35. doi:10.1038/nrm3025
98. Kim SG, Buel GR, Blenis J. Nutrient regulation of the mTOR complex 1 signaling pathway. Mol Cells (2013) 35:463–73. doi:10.1007/s10059-013-0138-2
99. Laplante M, Sabatini DM. mTOR signaling at a glance. J Cell Sci (2009) 122:3389–94. doi:10.1242/jcs.051011
100. Abraham RT, Wiederrecht OJ. Immunopharmacology of rapamycin. Annu Rev Immunol (1996) 14:483–510. doi:10.1146/annurev.immunol.14.1.483
101. Wullschleger S, Loewith R, Hall MN. TOR signaling in growth and metabolism. Cell (2006) 124:471–84. doi:10.1016/j.cell.2006.01.016
102. Krakauer T, Buckley M, Issaq HJ, Fox SD. Rapamycin protects mice from staphylococcal enterotoxin B-induced toxic shock and blocks cytokine release in vitro and in vivo. Antimicrob Agents Chemother (2010) 54:1125–31. doi:10.1128/AAC.01015-09
103. Mattsson E, Herwald H, Egsten A. Superantigens from Staphylococcus aureus induce procoagulant activity and monocyte tissue factor expression in whole blood and mononuclear cells via IL-1β. J Thromb Haemost (2003) 1:2569–75. doi:10.1111/j.1538-7836.2003.00498.x
104. Stohl W, Elliott JE, Linsley PS. Human T cell-dependent B cell differentiation induced by staphylococcal superantigens. J Immunol (1994) 153:117–27.
105. Mourad W, Mehindate K, Schall T, McColl S. Engagement of MHC class II molecules by superantigen induces inflammatory cytokine gene expression in human rheumatoid fibroblast-like synoviocytes. J Exp Med (1992) 175:613–6. doi:10.1084/jem.175.2.613
106. Lu J, Philpott DJ, Saunders PR, Perdue MH, Yang PC, McKay DM. Epithelial ion transport and barrier abnormalities evoked by superantigen-activated immune cells are inhibited by interleukin-10 but not interleukin-4. J Pharmacol Exp Ther (1998) 287:128–36.
107. Hamad AR, Marrack P, Kappler JW. Transcytosis of staphylococcal superantigen toxins. J Exp Med (1997) 185:1447–54. doi:10.1084/jem.185.8.1447
108. Alber G, Scheuber PH, Reck B, Sailer-Krammer B, Hartmann A, Hammer DK. Role of substance P in immediate-type skin reactions induced by staphylococcal enterotoxin B in unsensitized monkeys. J Allergy Clin Immunol (1989) 84:880–5. doi:10.1016/0091-6749(89)90383-7
109. Peterson M, Ault K, Kremer MJ, Klingelhutz AJ, Davis CC, Squier CA, et al. Innate immune system is activated by stimulation of vaginal epithelial cells with Staphylococcus aureus and toxic shock syndrome toxin 1. Infect Immun (2005) 73:2164–74. doi:10.1128/IAI.73.4.2164-2174.2005
110. Brosnahan AJ, Schlievert PM. Gram positive bacterial superantigen outside-in signaling causes toxic shock syndrome. FEBS J (2011) 278:4649–67. doi:10.1111/j.1742-4658.2011.08151.x
111. Vial T, Descotes J. Immune-mediated side-effects of cytokines in human. Toxicol (1995) 105:31–57. doi:10.1016/0300-483X(95)03124-X
112. Dubinett SM, Huang M, Lichtenstein A, McBride WH, Wang J, Markovitz G, et al. Tumor necrosis factor-alpha plays a central role in interleukin-2 induced pulmonary vascular leak and lymphocyte accumulation. Cell Immunol (1994) 157:170–80. doi:10.1006/cimm.1994.1214
113. Sims JE, Smith DE. The IL-1 family: regulators of immunity. Nat Rev Immunol (2010) 10:89–102. doi:10.1038/nri2691
114. Kawai T, Akira S. Toll-like receptors and their crosstalk with other innate receptors in infection and immunity. Immunity (2011) 34:637–50. doi:10.1016/j.immuni.2011.05.006
115. Hopkins PA, Fraser JD, Pridmore AC, Russell HH, Read RC, Sriskandan S. Superantigen recognition by HLA class II on monocytes up-regulates toll-like receptor 4 and enhances proinflammatory responses to endotoxin. Blood (2005) 105:3655–62. doi:10.1182/blood-2004-07-2523
116. Hopkins PA, Pridmore AC, Ellmerich S, Fraser JD, Russell HH, Read RC, et al. Increased surface toll-like receptor 2 expression in superantigen shock. Crit Care Med (2008) 36:1267–76. doi:10.1097/CCM.0b013e31816a0a78
117. Gross O, Thomas CJ, Guarda G, Tschopp J. The inflammasome: an integrated view. Immunol Rev (2011) 243:136–51. doi:10.1111/j.1600-065X.2011.01046.x
118. Van Herreweghe F, Festjens N, Declercq W, Vandenabeele P. Tumor necrosis factor-mediated cell death: to break or to burst, that’s the question. Cell Mol Life Sci (2010) 67:1567–79. doi:10.1007/s00018-010-0283-0
119. Keystone EC, Ware CF. Tumor necrosis factor and anti-tumor necrosis factor therapies. J Rheumatol (2010) 85:27–39. doi:10.3899/jrheum.091463
120. McKay DM. Bacterial superantigens: provocateurs of gut dysfunction and inflammation? Trends Immunol (2001) 22:497–501. doi:10.1016/S1471-4906(01)02000-2
121. Miethke T, Wahl C, Heeg K, Echtenacher B, Krammer PH, Wagner H. T cell-mediated lethal shock triggered in mice by the superantigen staphylococcal enterotoxin B: critical role of tumor necrosis factor. J Exp Med (1992) 175:91–8. doi:10.1084/jem.175.1.91
122. Dunn GP, Koebel CM, Schreiber RD. Interferons, immunity and cancer immunoediting. Nat Rev Immunol (2006) 6:836–48. doi:10.1038/nri1961
123. Stark GR, Darnell JE. The JAK-STAT pathway at twenty. Immunity (2012) 36:503–14. doi:10.1016/j.immuni.2012.03.013
124. McNab F, Mayer-Barber K, Sher A, Wack A, O’Garra A. Type 1 interferons in infectious disease. Nat Rev Immunol (2015) 15:87–103. doi:10.1038/nri3787
125. Ramana CV, Gil MP, Schreiber RD, Stark GR. Stat-1-dependent and -independent pathways in IFN-dependent signaling. Trends Immunol (2002) 23:96–101. doi:10.1016/S1471-4906(01)02118-4
126. Klintman D, Li X, Sato T, Wang Y, Jeppsson B, Thorlacius H. Staphylococcal enterotoxin A-induced hepatotoxicity is predominantly mediated by Fas ligand (CD95L). Ann Surg (2004) 240(6):1065–72. doi:10.1097/01.sla.0000146159.88918.d2
127. Chang JH, Kim YJ, Han SH, Kang CY. IFN-γ-STAT1 signal regulates the differentiation of inducible Treg: potential role for ROS-mediated apoptosis. Eur J Immunol (2009) 39:1241–51. doi:10.1002/eji.200838913
128. Malek TR, Castro I. Interleukin-2 receptor signaling: at the interface between tolerance and immunity. Immunity (2010) 33:153–65. doi:10.1016/j.immuni.2010.08.004
129. Kyriakis JM, Avruch J. Mammalian MAPK signal transduction pathways activated by stress and inflammation. Physiol Rev (2012) 92:689–737. doi:10.1152/physrev.00028.2011
130. Liu D, Zienkiewicz J, DiGiandomenico A, Hawiger J. Suppression of acute lung inflammation by intracellular peptide delivery of a nuclear import inhibitor. Mol Ther (2009) 17:796–802. doi:10.1038/mt.2009.18
131. Huzella LM, Buckley MJ, Alves DA, Stiles BG, Krakauer T. Central roles for IL-2 and MCP-1 following intranasal exposure to SEB: a new mouse model. Res Vet Sci (2009) 86:241–7. doi:10.1016/j.rvsc.2008.07.020
132. Khan AA, Priya S, Saha B. IL-2 regulates SEB induced toxic shock syndrome in BALB/c mice. PLoS One (2009) 4:e8473. doi:10.1371/journal.pone.0008473
133. Sadik CD, Kim ND, Luster AD. Neutrophils cascading their way to inflammation. Trends Immunol (2011) 32:452–60. doi:10.1016/j.it.2011.06.008
134. Zlotnik A, Yoshie D. The chemokine superfamily revisited. Immunity (2012) 36:705–16. doi:10.1016/j.immuni.2012.05.008
135. Tessier PA, Naccache PH, Diener KR, Gladue RP, Neotem KS, Clark-Lewis I, et al. Induction of acute inflammation in vivo by staphylococcal superantigens. II. Critical role for chemokines, ICAM-1, and TNF-alpha. J Immunol (1998) 161:1204–11.
136. Krakauer T, Buckley M, Huzella LM, Alves D. Critical timing, location and duration of glucocorticoid administration rescues mice from superantigen-induced shock and attenuates lung injury. Int Immunopharmacol (2009) 9:1168–74. doi:10.1016/j.intimp.2009.06.004
137. Neumann B, Engelhardt B, Wagner H, Holzmann B. Induction of acute inflammatory lung injury by staphylococcal enterotoxin B. J Immunol (1997) 158:1862–71.
138. Miethke T, Wahl C, Holzmann B, Heeg K, Wagner H. Bacterial superantigens induce rapid and T cell receptor V beta-selective down-regulation of L-selectin (gp90Mel-14) in vivo. J Immunol (1993) 151:6777–82.
139. Niedergang F, Hemar A, Hewitt CR, Oven MJ, Dautry-Varset A, Alcover A. The Staphylococcus aureus enterotoxin B superantigen induces specific T cell receptor down-regulation by increasing its internalization. J Biol Chem (1995) 270:12839–45. doi:10.1074/jbc.270.21.12839
140. MacDonald HR, Lees RK, Baschieri S, Herrmann T, Lussow AR. Peripheral T-cell reactivity to bacterial superantigens in vivo: the response/anergy paradox. Immunol Rev (1993) 133:105–17. doi:10.1111/j.1600-065X.1993.tb01512.x
141. Florquin S, Amraoul Z, Goldman M. T cells made deficient in interleukin-2 production by exposure to staphylococcal enterotoxin B in vivo are primed for interferon-gamma and interleukin-10 secretion. Eur J Immunol (1995) 25:1148–53. doi:10.1002/eji.1830250503
142. Bean AG, Freiberg RA, Andrade S, Menon S, Zlotnik A. Interleukin 10 protects mice against staphylococcal enterotoxin B-induced lethal shock. Infect Immun (1993) 61:4937–9.
143. Shevach EM. Mechanisms of Fox3+ T regulatory cell-mediated suppression. Immunity (2009) 30:636–45. doi:10.1016/j.immuni.2009.04.010
144. Ou L-S, Goleva E, Hall C, Leung DYM. T regulatory cells in atopic dermatitis and subversion of their activity by superantigens. J Allergy Clin Immunol (2004) 113:756–63. doi:10.1016/j.jaci.2003.12.332
145. Saha B, Harlan DM, Lee KP, June CH, Abe R. Protection against lethal toxic shock by targeted disruption of the CD28 gene. J Exp Med (1996) 183:2675–80. doi:10.1084/jem.183.6.2675
146. Linsley PS, Brady W, Urnes M, Grosmaire LS, Damle NK, Ledbetter JA. CTLA-4 is a second receptor for the B cell activation antigen B7. J Exp Med (1991) 174:561–9. doi:10.1084/jem.174.3.561
147. Tilahun AY, Chowdhary VR, David CS, Rajagopalan G. Systemic inflammatory response elicited by superantigen destabilizes T regulatory cells, rendering them ineffective during toxic shock syndrome. J Immunol (2014) 193:2919–30. doi:10.4049/jimmunol.1400980
148. Schwab JH, Brown RR, Anderle SK, Schlievert PM. Superantigen can reactivate bacterial cell wall-induced arthritis. J Immunol (1993) 150:4151–9.
149. Zhao YX, Abdelnour A, Ljungdahl A, Olsson T, Tarkowski A. Patterns of interferon-gamma mRNA expression in toxic shock syndrome toxin-1 expanded Vβ11+ T lymphocytes. Cell Immunol (1995) 161:28–33. doi:10.1006/cimm.1995.1005
150. Abdelnour AT, Bremell T, Tarkowski A. Toxic shock syndrome toxin 1 contributes to the arthritogenicity of Staphylococcus aureus. J Infect Dis (1994) 170:94–9. doi:10.1093/infdis/170.1.94
151. Brocke S, Gaur A, Piercy C, Gautam A, Gijbels K, Fathman CG, et al. Induction of relapsing paralysis in experimental autoimmune encephalomyelitis by bacterial superantigen. Nature (1993) 365:642–4. doi:10.1038/365642a0
152. Valdimarsson H, Baker BS, Jonsdottir I, Powles A, Fry L. Psoriasis: a T cell-mediated autoimmune disease induced by streptocococcal superantigens? Immunol Today (1995) 16:145–9. doi:10.1016/0167-5699(95)80132-4
153. Yoshino T, Asada H, Sano S, Nakamura T, Itami S, Tamura M, et al. Impaired responses of peripheral blood mononuclear cells to staphylococcal superantigens in patients with severe atopic dermatitis: a role of T cell apoptosis. J Invest Dermatol (2000) 114:281–8. doi:10.1046/j.1523-1747.2000.00878.x
154. Darenberg J, Soderquist B, Normark BH, Norrby-Teglund A. Differences in potency of intravenous polyspecific immunoglobulin G against streptococcal and staphylococcal superantigens: implications for therapy of toxic shock syndrome. Clin Infect Dis (2004) 38:836–42. doi:10.1086/381979
155. Lappin E, Ferguson AJ. Gram-positive toxic shock syndromes. Lancet Infect Dis (2009) 9:281–90. doi:10.1016/S1473-3099(09)70066-0
156. Krakauer T. Update on staphylococcal superantigen-induced signaling pathways and therapeutic interventions. Toxins (2013) 5:1629–54. doi:10.3390/toxins5091629
157. Opal SM, Laterre PF, Francois B, LaRosa SP, Angus DC, Mira JP, et al. Effect of eritoran, an antagonist of MD2-TLR4, on mortality in patients with severe sepsis: the ACCESS randomized trial. JAMA (2013) 309:1154–62. doi:10.1001/jama.2013.2194
158. Krakauer T, Buckley M. Dexamethasone attenuates staphylococcal enterotoxin B-induced hypothermic response and protects mice from superantigen-induced toxic shock. Antimicrob Agents Chemother (2006) 50:391–5. doi:10.1128/AAC.50.1.391-395.2006
159. Tilahun AY, Theuer JE, Patel R, David CS, Rajagopalan G. Detrimental effect of the proteasome inhibitor, bortezomib in bacterial superantigen- and lipopolysaccharide-induced systemic inflammation. Mol Ther (2010) 18:1143–54. doi:10.1038/mt.2010.53
160. Scholl P, Sekaly R, Diez A, Glimcher L, Geha R. Binding of toxic shock syndrome toxin-1 to murine major histocompatibility complex class II molecules. Eur J Immunol (1990) 20:1911–6. doi:10.1002/eji.1830200907
161. Roy CJ, Warfield KL, Welcher BC, Gonzales RF, Larsen T, Hanson J, et al. Human leukocyte antigen-DQ8 transgenic mice: a model to examine the toxicity of aerosolized staphylococcal enterotoxin B. Infect Immun (2005) 73:2452–60. doi:10.1128/IAI.73.4.2452-2460.2005
162. Tilahun AY, Marietta EV, Wu TT, Patel R, David CS, Rajagopalan G. Human leukocyte antigen class II transgenic mouse model unmasks the significant extrahepatic pathology in toxic shock syndrome. Am J Pathol (2011) 178:2760–72. doi:10.1016/j.ajpath.2011.02.033
163. Ferreyra GA, Elinoff JM, Demirkale CY, Starost MF, Buckley M, Munson PJ, et al. Late multiple organ surge in interferon-regulated target genes characterizes staphylococcal enterotoxin B lethality. PLoS One (2014) 9:e88756. doi:10.1371/journal.pone.0088756
164. Krakauer T, Buckley M, Fisher D. Proinflammatory mediators of toxic shock and their correlation to lethality. Mediators Inflamm (2010) 2010:517594. doi:10.1155/2010/517594
165. Stetson DB, Medzhitov R. Recognition of cytosolic DNA activates an IRF3-dependent innate immune response. Immunity (2005) 24:93–103. doi:10.1016/j.immuni.2005.12.003
166. Jimbo A, Fujita E, Kouroku Y, Ohnishi J, Inohara N, Kuida K, et al. ER stress induces caspase-8 activation, stimulating cytochrome c release and caspase-9 activation. Exp Cell Res (2003) 283:156–66. doi:10.1016/S0014-4827(02)00033-2
167. Yoneda T, Imaizumi K, Oono K, Yui D, Gomi F, Katayama T, et al. Activation of caspase-12, an endoplastic reticulum (ER) resident caspase, through tumor necrosis factor receptor-associated factor 2-dependent mechanism in response to the ER stress. J Biol Chem (2001) 276:13935–40. doi:10.1074/jbc.M010677200
168. Dauwalder O, Pachot A, Cazalis MA, Paye M, Faudot C, Badiou C, et al. Early kinetics of the transcriptional response of human leukocytes to staphylococcal superantigenic enterotoxins A and G. Microb Pathog (2009) 47:171–6. doi:10.1016/j.micpath.2009.07.001
169. Mendis C, Das R, Hammamieh R, Royaee A, Yang D, Peel S, et al. Transcriptional response signature of human lymphoid cells to staphylococcal enterotoxin B. Genes Immun (2005) 6:84–94. doi:10.1038/sj.gene.6364160
170. Takaoka A, Wang Z, Choi MK, Yanai H, Negishi H, Ban T, et al. DAI (DLM-1/ZBP1) is a cytosolic DNA sensor and an activator of innate immune response. Nature (2007) 448:501–6. doi:10.1038/nature06013
171. Muruve DA, Pétrilli V, Zaiss AK, White LR, Clark SA, Ross PJ, et al. The inflammasome recognizes cytosolic microbial and host DNA and triggers an innate immune response. Nature (2008) 452:103–7. doi:10.1038/nature06664
172. Zhang Q, Raoof M, Chen Y, Sumi Y, Sursai T, Junger W, et al. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature (2010) 464:104–7. doi:10.1038/nature08780
173. Helmerhorst HJ, Schultz MJ, van der Voort PH, de Jonge E, van Westerloo DJ. Bench-to-bedside review: the effects of hyperoxia during critical illness. Crit Care (2015) 19:284–96. doi:10.1186/s13054-015-0996-4
174. Rosenson RS. Statins in atherosclerosis: lipid-lowering agents with antioxidant capabilities. Atherosclerosis (2004) 173:1–12. doi:10.1016/S0021-9150(03)00239-9
175. Jacobson JR. Statins in endothelial signaling and activation. Antioxid Redox Signal (2009) 11:811–21. doi:10.1089/ARS.2008.2284
176. Hotchkiss RS, Strasser A, McDunn JE, Swanson PE. Cell death in disease: mechanisms and emerging therapeutic concepts. N Engl J Med (2009) 361:1570–83. doi:10.1056/NEJMra0901217
177. Franco-Penteado CF, Desouza I, Teixeira SA, Ribeiro-DaSilva G, De Nucci G, Antunes E. Role of nitric oxide on the increased vascular permeability and neutrophil accumulation induced by staphylococcal enterotoxin B into the mouse paw. Biochem Pharmacol (2001) 15(61):1305–11. doi:10.1016/S0006-2952(01)00573-1
178. McKallip RJ, Fisher M, Gunthert U, Szakai AK, Nagarkatti PS, Nagarkatti M. Role of CD44 and its v7 isoform in staphylococcal enterotoxin B-induced toxic shock: CD44 deficiency on hepatic mononuclear cells leads to reduced activation-induced apoptosis that results in increased liver damage. Infect Immun (2005) 73:50–61. doi:10.1128/IAI.73.1.50-61.2005
179. Martins JD, Liberal J, Silva A, Ferreira I, Neves BM, Cruz MT. Autophagy and inflammasome interplay. DNA Cell Biol (2015) 34:274–81. doi:10.1089/dna.2014.2752
180. Paul S, Schaefer BC. Selective autophagy regulates T cell activation. Autophagy (2012) 8(11):1690–2. doi:10.4161/auto.21581
181. Komisar JL, Weng CF, Oyejide A, Hunt RE, Briscoe C, Tseng J. Cellular and cytokine responses in the circulation and tissue reactions in the lung of rhesus monkeys (Macaca mulatta) pretreated with cyclosporine A and challenged with staphylococcal enterotoxin B. Toxicol Pathol (2001) 29:369–78. doi:10.1080/019262301316905336
Keywords: SEB, superantigens, toxic shock, damage response, therapy, animal models
Citation: Krakauer T, Pradhan K and Stiles BG (2016) Staphylococcal Superantigens Spark Host-Mediated Danger Signals. Front. Immunol. 7:23. doi: 10.3389/fimmu.2016.00023
Received: 01 October 2015; Accepted: 18 January 2016;
Published: 02 February 2016
Edited by:
Katharina F. Kubatzky, University Hospital Heidelberg, GermanyReviewed by:
Howard M. Johnson, University of Florida, USACopyright: © 2016 Krakauer, Pradhan and Stiles. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Teresa Krakauer, dGVyZXNhLmtyYWthdWVyLmNpdkBtYWlsLm1pbA==;
Bradley G. Stiles, YnJhZC5zdGlsZXNAd2lsc29uLmVkdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.