 Sakshi Malik
Sakshi Malik Amit Awasthi
Amit Awasthi
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 29 January 2016
Sec. Multiple Sclerosis and Neuroimmunology
Volume 7 - 2016 | https://doi.org/10.3389/fimmu.2016.00014
This article is part of the Research Topic Lymphocytes in MS and EAE: more than just a CD4+ world View all 17 articles
γδ (gamma–delta) T cells, a small population of unconventional T cells, have been found in central nervous system lesions of multiple sclerosis (MS) patients, but their function in disease activity is not clearly understood. Previous studies in experimental autoimmune encephalomyelitis (EAE) were inconsistent in identifying their specific roles in suppressing or promoting disease pathogenesis. Emerging advancements in the biology of γδ T cells especially in the context of their being the major initial producers of IL-17, suggested their crucial role in pathogenesis of EAE. In addition, γδ T cells express high levels of IL-23R and IL-1R, which further enhance their effector functions in the pathogenesis of EAE. Nonetheless, activated heterogeneous γδ T cells display functional dichotomy, which is crucial in determining the outcomes of tissue inflammation in EAE. In this review, we discussed recent advances in understanding the biology of γδ T cells in tissue inflammation as well as their roles in suppressing or promoting the development of EAE.
γδ (gamma–delta) T cells comprise a small fraction (~1–5%) of the total blood lymphocytes of mice and humans and are more commonly localized in mucosal tissue and skin where they constitute a major population (up to 50%) of lymphocytes (1). The identification of an unusually rearranged γ chain of the T cell receptor (TCR) gene led to the discovery of γδ T cells (2, 3). After the identification of γδ T cells as a new subset of T cells, it became clear that these cell types, unlike their αβ (alpha-beta) T cell counterparts, possess features of both innate and adaptive immune cells (4, 5). Moreover, γδ T cells have also been recognized as non-conventional innate-like cells as they share several features of innate immune cells, such as surface expression of Toll-like receptors (TLRs) (6). In addition, γδ T cells acquire preactivated phenotypes of effector and memory T cells during their early development (6).
The antigen recognition, activation, and effector functions of γδ T cells are different than those of their αβ T cell counterparts. Unlike αβ+ T cells, γδ T cells can be activated with or without their cognate TCR ligands and appear to induce an early burst of inflammatory cytokine that initiates effective and progressive αβ T cell responses in tissue inflammation during experimental autoimmune encephalomyelitis (EAE) (7–10). These unusual unique features of γδ T cells make them an early effector T cells during an immune response in inflamed tissue.
αβ+ CD4+ T cells are crucial for inducing tissue inflammation in EAE. It has been convincingly elucidated that IL-17-producing Th17 cells are the major driver in inducing pathogenesis of EAE. Ablation of Th17 cells or absence of IL-17 significantly reduces the severity of inflammation in EAE (11). Similarly, the absence of Th17 cell-associated genes, such as Rorc, a master transcription factor for Th17 cells development, and IL-23R also attenuate inflammation in EAE (12, 13). Interestingly, γδ T cells express higher level of IL-23R on their surface, which raised an interesting possibility that IL-23-responsive γδ T cells may contribute to the severity of tissue inflammation in EAE (8). Furthermore, GWAS studies suggested a genetic association of IL-23R with MS (14).
Although the bona fide antigens were identified for γδ T cells, still not much is known about their antigenic repertoire and restrictions (15). In addition to their antigens, γδ T cells can be activated by TLRs to induce various inflammatory cytokines, such as IFN-γ, IL-4, IL-17, IL-21, and IL-22 (6, 16).
Unlike αβ+ T cells, antigen recognition by the TCR of γδ T cells does not require antigen processing and presentation by MHC molecules (17, 18). Moreover, deficiencies of MHC class II and β2 microglobulin do not affect the development of γδ T cells and their repertoire remain intact, which suggest that the generation of γδ T cells is apparently independent of both class I and II molecules (19, 20). Interestingly, non-classical MHC class Ib molecules T10 and T22 are described as the natural ligands for murine γδ T cells (21, 22). Similarly, human class I-like molecules MICA and MICB were also suggested as natural antigens for human γδ T cells (21, 23–25). Interestingly, alterations in the expression of these ligands are induced by infection or tissue inflammation or stress, which can provide early danger-signal to initiate the activation of γδ T cells even in the absence of αβ+ T cells activation (15, 16).
The functions of γδ T cells in different pathophysiological conditions are driven by their tissue-specific distributions and tropism. At steady state, γδ T cells are predominantly localized in epithelial surfaces of liver, skin, and mucosal surfaces of digestive, respiratory, and reproductive organs (15, 16). Moreover, the distribution of γδ T cells to the above mentioned epithelial and mucosal surfaces is often driven by their specific expression of invariant or closely related γδ TCRs; for example, Vγ6Vδ1 TCR-expressing γδ T cells mostly accumulate in the lung, peritoneum, and reproductive organs, while Vγ5Vδ1-bearing γδ T cells predominantly reside in the epithelial surface of the skin (16). In addition to their tissue localization, cellular distribution, pathophysiological conditions, and inflammatory signals also determine the activation and phenotypic plasticity of γδ T cells.
Upon activation, γδ T cells can produce the effector cytokines of Th1, Th2, and Th17 cells, such as IFN-γ, IL-4, and IL-17, respectively, therefore contribute to specific effector function in Th1, Th2, and Th17 cell-associated tissue inflammation (26). Interestingly, IL-23 stimulation of γδ T cells rapidly induces IL-17 production (6, 13, 27) to initiate tissue inflammation and enhance CD4+ αβ Th17 cells responses during EAE (7). It is apparent that γδ T cells play critical role in the induction and pathogenesis of EAE (15). Nonetheless, the regulatory role of γδ T cells is also suggested in EAE.
The functions of γδ T cells are not only critically required for elimination of intra- and extracellular pathogens and tissue surveillance in cancer but are also associated with multiple organ-specific autoimmunity, such as type 1 diabetes, arthritis, inflammatory bowel disease (IBD), and MS (16).
There are multiple subtypes of γδ T cells that are involved in the pathogenesis of EAE and can be identified based on the usage of their variable regions for both γ and δ genes (28, 29). Unlike the mucosal surfaces and the skin, which usually harbor higher frequency of γδ T cells, a smaller frequency of γδ T cells can be found within the central nervous system (CNS) in steady state of untreated naive mice (30, 31). Although the role of γδ T cells in the CNS at steady state is not precisely understood, it might be possible that their presence within the CNS could be required for carrying out immune surveillance function. Nonetheless, the frequency of γδ T cells profoundly increases within the CNS in EAE; and moreover, their distribution within the CNS can be classified based on their TCR usage during different phases of EAE (28). At the initial phase of EAE, CNS-infiltrating γδ T cells show a limited repertoire, including Vδ1, Vδ4, Vδ5, Vγ1–3, and Vγ6, while almost all the Vγ and Vδ transcripts can be found in the brain at the chronic or later phase of the disease (28). Although lymph nodes of EAE mice contained most of the Vγ transcripts during all phases of disease, a limited repertoire of γδ T cells was also observed within the CNS at the initial phase of the disease. Though Vγ6 (also known as DV7s6) expressing γδ T cells are predominantly located in mucosa, but they can also be found within the CNS at the initial phase of EAE. However, the precise antigen specificity of CNS-localized Vγ6 γδ T cells is not clearly understood in EAE. Since γδ T cells do not appear to recognize myelin basic protein (MBP) as antigen; therefore, Vγ6 T cells might be recruited to the CNS in EAE in response to the heat shock protein (HSP), which is expressed on stressed autologous cells (32). In addition, another possibility is that Vγ6 γδ T cells could recognize self-antigens that mimic bacterial peptide in the CNS during inflammation as this subset of γδ T cells is known to recognize microbial antigens (bacterial peptide) at mucosal surfaces (15, 33). Furthermore, γδ T cells are suggested to be functionally dichotomous on the basis of their TCR usage in EAE; Vγ1 subset preferentially regulates while Vγ4 subset further enhances tissue inflammation in EAE (34). Further analysis revealed that the Vγ1 subset is predominantly prevalent in spleen in all phases of EAE, and in fact, about 35–50% of total splenic γδ T cells are found to be Vγ1 in EAE (34). However, a small percentage of Vγ1 γδ T cells are also found in the CNS during EAE. Emerging literature suggested that Vγ1 γδ T cells act as regulatory cells and were shown to suppress tissue inflammation during the acute phase of EAE by enhancing the functions of Foxp3+ regulatory T (Treg) cells. Moreover, it is proposed that Vγ1 subset of γδ T cells highly express CCL4, which can bind to CCR5 on Treg cells and promote their suppressive functions in EAE (34). Consistent with their regulatory role in EAE, CNS-sorted Vγ1 γδ T cells from EAE mice do not express high amounts of IL-17A, IL-17F, IL-23R, and GM-CSF, which further reinforce their regulatory function in EAE (34). Thus, it is suggested that Vγ1 subset might shift the balance away from Th17 cells while promoting the proliferation and suppressive functions of Treg cells during EAE.
Yet, another subset of γδ T cells, Vγ4 predominates in the CNS during EAE. These cells typically responds to self-antigens by producing pro inflammatory cytokine, such as IL-17, which in turn can directly act on stromal cells and induce migration of lymphocytes across blood brain barrier in EAE (8, 27). Interestingly, the IL-17-producing Vγ4 γδ T cells also expressed other Th17 cell-associated molecules, such as Rorc, IL-22, IL-1R, and IL-1β (6, 34), which further suggested to contribute to inflammation and exacerbation of EAE (34). In addition to EAE, IL-17-producing Vγ4 γδ T cells are shown to promote collagen-induced arthritis (CIA), as antibody-mediated depletion of Vγ4 γδ T cells resulted in attenuated tissue inflammation in CIA (35). It is proposed that adjuvant rather antigen expands IL-17-producing Vγ4 γδ T cells in CIA.
The ability of γδ T cells to produce IL-17 innately in response to IL-23 in EAE could be attributed to Vγ4 subset of γδ T cells as they highly express IL-23R on their surface (6, 7, 27). Nonetheless, it is not clearly understood whether natural ligand or antigen of Vγ4 γδ T cells can induce strong IL-17 response in EAE. In addition to Vγ4 γδ T cells, Vγ6 γδ T cells, which primarily resides under the skin also express IL-23R on their surface, and therefore might be contributing to IL-17-mediated inflammation in the CNS of EAE mice (27).
Interestingly, in addition to IL-23R and Vγ4, the differential expression of CD27 can also identify γδT17 cells (IL-17-producing γδ T cells). CD27+ γδ T cells produce IFN-γ while CD27− γδ T cells secrete IL-17 suggested that the surface expression CD27 can differentially mark IL-17- and IFN-γ-producing γδ T cells (36).
Furthermore, structural and functional heterogeneity of γδ T cells in EAE can be further contributed by different mice strain. Olive et al. have reported amplification of Vγ5 transcript in C57Bl/6 mice during EAE while this transcript was not detected in the CNS of SJL/J mice, suggesting that the infiltrating population of γδ T cells in CNS during disease can be varied on the basis of mouse strains (28).
After the identification of Th17 cells as a separate lineage of helper T (Th) cells, it became clear that they, together, with Th1 cells, play a crucial role in EAE (37, 38). Before the identification of Th17 cells, IFN-γ-producing Th1 cells were thought to be the primary effector cell type involved in the disease induction of EAE, which has puzzled immunologist for a very long time as both IFN-γ- and IFN-γR-deficient animals had exacerbated tissue inflammation in EAE (39). In addition, the deficiencies of IL-12p35 (IL-12) and IL-12Rβ2 (IL-12 receptor), which are critically required for the development of Th1 cells, also enhanced the development of EAE (37). Taken together, it is clearly suggested that Th1 cells are not the primary effector T cell subsets involved in development of EAE. In fact, Th1 cell-associated molecules, such as IFN-γ, IL-12, and IL-12R, negatively regulate disease and tissue inflammation in EAE (11). Nonetheless, Th1 cells also critical for the development of EAE, as Th1 cells were found in the CNS in active EAE. In fact, a sizable population of IFN-γ and IL-17 double positive CD4+ T cells was found within the CNS at the peak of EAE (40).
Seminal studies demonstrated that TGF-β1 and IL-6 are required for the differentiation of Th17 cells (41–43). IL-6 strongly induces IL-21 in Th17 cells, which creates feed forward loop to further amplify the generation of Th17 cells (44–46). The role of Th17 cells and IL-17 was further demonstrated by using IL-17-deficient mice, as Il-17−/− animals develop attenuated EAE with delayed onset. Moreover, the adoptive transfer of Il-17−/− CD4+ T cells is inefficient in transferring EAE, suggesting that IL-17 is crucial for tissue inflammation and disease pathogenesis (47).
Similar to Th17 cells, IL-6 and TGF-β are also crucial for the generation of γδT17 cells (8, 48). Tgfb−/− and Smad3−/− mice harbor reduced precursor frequency of γδT17 cells in thymus (48). On the other hand, Il6−/− mice have shown reduced frequency of peripheral γδT17 cells (8). Taken together, similar to Th17 cells differentiation, TGF-β and IL-6 are crucial for the generation of γδT17 cells.
Importantly, the precise role of γδ T cells was demonstrated in EAE using Tcrd−/− mice (15, 49). Mice lacking TCR delta chain gene develop less severe EAE with reduced infiltration of αβ+ T cells in their CNS (49). Similarly, depletion of γδ T cells by anti-GL3 antibody before the onset or at chronic phase of EAE reduces the severity and clinical signs of EAE (50). Moreover, antibody-mediated depletion of γδ T cells regulates the influx of proinflammatory cytokines, such as IL-1, IL-6, TNF-α, lymphotoxin, and IFN-γ, further suggesting an essential role of γδ T cells in contributing to the pathogenesis of EAE (50). Furthermore, it is demonstrated that the depletion of γδ T cells from MBP-reactive lymph node cells transferred attenuated EAE with reduced T cells proliferation and IL-12 secretion (51). Moreover, replenishing γδ T cells population not only enhanced the severity of EAE but also restored the IL-12 production and T cells proliferation (51).
In addition, a detailed systematic analysis of γδ T cells was performed to understand their distribution in different phases of EAE (52). Interestingly, an increased frequency of γδ T cells (up to 12% of total CD3+ T cells) was found in the CNS during the acute phase while the percentage of γδ T cells decreased (from 12 to 5% of total CD3+ T cells) during the recovery phase of EAE (52). Since the frequency of myelin-specific Foxp3+ Treg cells increases during recovery phase of EAE, it is possible that the increased number of Foxp3+ Tregs contributes in controlling the expansion of γδ T cells population during recovery phase of EAE (8, 53). Interestingly, the contraction of γδ T cells population was restricted only to the CNS, as their percentages in spleen remained low (~2% of total CD3+ T cells) during all phases of EAE. This implies that γδ T cells selectively accumulate in the target tissue during tissue inflammation to enhance severity of inflammation in EAE (52).
Although αβ+ CD4+ T cells are suggested to be the primary source of IL-17 in infection and autoimmune inflammation, γδT cells can be a potent source of IL-17, and in some cases, even more dominant than Th17 cells (6, 33, 54). In fact, in the model of Fas-ligand-induced inflammation in which injecting FasL-expressing tumor cells into peritoneum of mice induces enhanced production of IL-17 from non-conventional T cells (55). Interestingly, the majority of these IL-17-producing cells were γδ T cells as compared to αβ Th17 cells in this particular model (55). Similarly, γδ T cells isolated from Mycobacterium-infected lung and spleen produce massive amounts of IL-17 as compared to αβ Th17 cells (56). Furthermore, in other model of infection, such as Escherichia coli, Bacillus subtilis, and experimental sepsis, γδ T cells, rather than αβ+ Th17 cells, are the primary source of IL-17 (33, 57). Hence, in certain conditions, γδ T cells appear to have an inherent ability to rapidly produce substantial amounts of IL-17 without being primed.
Although, initial studies identified that IL-17-producing γδ T cells are essential for clearing infections, the role of γδ T cells are also suggested for inducing autoimmune inflammation and propagation of autoimmune diseases, including EAE (15).
In addition to Th17 cells, Th1 cells were also implicated in the development of EAE (58). In fact, many studies suggested that myelin-specific Th1 cells adoptively transfer EAE (58). Interestingly, the initiation of EAE development by adoptively transferred myelin-specific Th1 cells resulted in recruitment of IL-17-producing host cells (IL-17hc) to the CNS (59). Further cellular characterization revealed that γδ T cells comprising almost 60% of the total IL-17hc (59). Moreover, in the absence of IL-17hc, myelin-specific Th1 cells transferred less severe EAE, suggesting the requirement of host production of IL-17, largely by γδ T cells, in the development of EAE (59).
Progression and development of tissue inflammation in EAE are primarily mediated by infiltrating mononuclear cells, which produce proinflammatory cytokines. Among other CNS-infiltrating cells, γδ T cells predominantly and rapidly produce proinflammatory cytokines to further enhance tissue inflammation in EAE. Like conventional αβ T cells, γδ T cells also expand in secondary lymphoid organs upon immunization with MOG/CFA. Once migrated to the CNS in β2 integrin-independent manner, these γδ T cells further expand and accumulate shortly before the peak of EAE and produce IFN-γ, TNF-α, and IL-17 to further enhance disease progression (60, 61).
Unlike αβ Th17 cells, which require primary (TCR), secondary (costimulation) and cytokine signals (TGF-β1 + IL-6) to produce IL-17, γδ T cells can produce IL-17 with cytokine signals (IL-23 and IL-1β) alone in the absence of primary and secondary signals (7). This peculiar feature of γδ T cells make them superior IL-17 producers by capturing the initial burst of proinflammatory cytokines produced by dendritic cells (DCs) and macrophages in response to TLR and NLR activation in EAE. The ability of γδ T cells to generate an initial burst of IL-17 in the absence of activation of αβ T cells is critical for initiating CNS inflammation, as Tcrd−/− mice develop less severe EAE with reduced production of IL-17 (7, 49). Moreover, αβ T cells from Tcrd−/− mice produce lower amounts of IL-17 as compared to αβ T cells from wild-type mice (8), which clearly suggested that the presence of γδ T cells is essentially required for optimal production of IL-17 by αβ T cells. Interestingly, Il1r−/− mice are substantially more resistant to EAE development (62); however, reconstituting IL-1R-sufficient γδ T cells into Il1r−/− mice prior to MOG immunization enhances progression of EAE, suggesting that IL-1β–IL-1R interaction on γδ T cells is essential for promoting tissue inflammation in EAE (7). Furthermore, stimulation of γδ T cells with IL-1β together with IL-23 synergistically enhanced IL-17 production in the absence of TCR stimulation (7). In addition to IL-17, other Th17 cell-associated cytokines, such as IL-17F, IL-21, and IL-22, were also produced by γδ T cells upon their activation with IL-1β and IL-23 (Figure 1). Consistently, culture supernatant of IL-1β− and IL-23-stimulated γδ T cells further enhanced IL-17 production from αβ+ CD4+ T cells (7, 8). Neutralization of IL-21 and IL-17 reduced IL-17 induction from αβ+ CD4+ T cells induced by culture supernatant of IL-1β- and IL-23-stimulated γδ T cells (7, 8). In fact, it is suggested that the combination of IL-1β- and IL-23-stimulated γδ T cells provides early burst of IL-21, which not only enhances production of IL-17 by the γδ T cells but it can also amplify the generation of Th17 cells (8, 11, 45) (Figure 1).
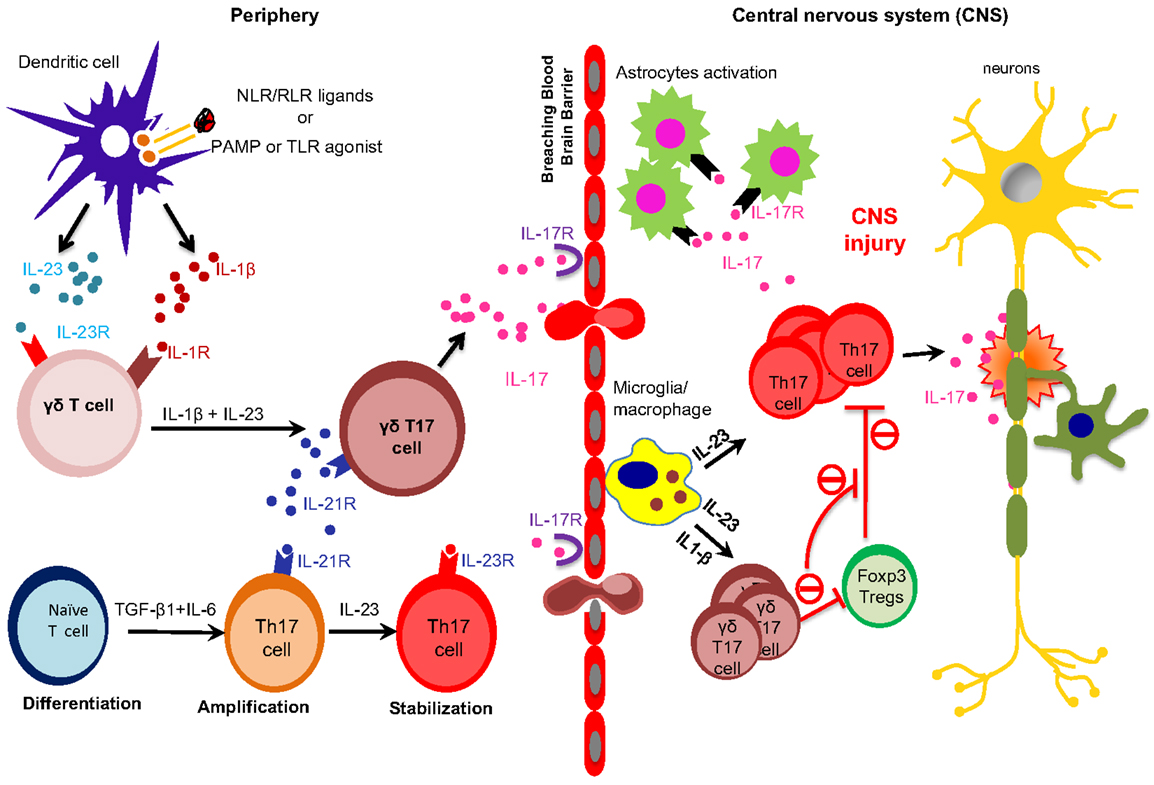
Figure 1. Peripherally primed γδ T cells execute their effector functions in the CNS in EAE. TLRs and NLRs activated dendritic cells (DCs) and macrophages produce proinflammatory cytokines, such as IL-6, IL-23 and IL-1β. IL-23 and IL-1β are sensed by IL-23R-expressing γδ T cells, which in turn produce early burst of IL-17 during early phase of EAE. On the other hand, IL-6 together with TGF-β induce the differentiation of Th17 cells. γδT17 cells produce IL-21, which further amplify their own generation and also amplify the generation of Th17 cells. Differentiated γδT17 and αβ Th17 cells breach the blood brain barrier to execute their effector functions within the CNS during EAE. Activated microglia/macrophages produce IL-23 within the CNS to promote the generation of γδT17 and Th17 cells. Inflammatory γδT17 cells promote CNS injury in EAE by enhancing the effector functions of Th17 cells and restraining the suppressive functions of Tregs cells.
In addition to IL-21, another common γ chain family cytokine, IL-2 also play a role in generation of γδT17 cells. IL-2, which is known to suppress Th17 cells (63), promotes γδT17 cells generation, as Il2−/− and Cd25−/− mice selectively reduced the frequency of γδT17 cells (64). Interestingly, the new subset of IL-15-producing γδ T cells (γδT15) was recently identified in EAE (65). γδT15 cells suggested to enhance tissue inflammation in EAE by enhancing the functions of CD44hi memory T and Th17 cells (65). However, whether these γδT15 cells express other inflammatory cytokines, such as IL-17 and GM-CSF, are not clear. In summary, various cytokines signals are required for the generation of γδT17 cells; and interestingly, some of these cytokines can directly activate γδ T cells without the requirement of TCR activation. Taken together, the initial burst of proinflammatory cytokines produce by γδ T cells is crucial for induction of EAE.
IL-18, an IL-1 family cytokine, also known as IFN-γ-inducing factor. It has been shown that IL-18 further enhances the development of IL-12-induced Th1 cells. Moreover, Th1 cells sensitized with IL-18 enhance their disease promoting effector functions in EAE by activating IFN-γ-producing NK cells (66). The function of IL-18 in EAE was described using IL-18R1-deficient animals. Il18r1−/− mice were completely resistant to development of EAE, suggesting the role of IL-18R in inducing encephalitogenic T cells in disease (67). Moreover, the engagement of IL-18Rα on antigen-presenting cells is essential for generation of pathogenic Th17 cells during EAE (67). In fact, caspase-1-processed cytokines IL-1β and IL-18 predominantly promote innate production of IL-17 from γδ T cells in EAE (9). Immunization with CFA, which contains heat-killed cell wall of Mycobacterium tuberculosis, activates caspase-1 via NLRP3 inflammasome to induce active forms of IL-1β and IL-18 from DCs. Inhibition of caspase-1 by its specific inhibitor suppresses EAE development and IL-17 production from γδ T cells (9). Similar to IL-23R, γδ T cells also express IL-18R constitutively on their surface even in the steady state (Figure 2). On the contrary, the expression of IL-18R on CD4+ T cells is induced in inflammatory conditions during EAE, suggesting that γδ T cells, and not CD4+ T cells, respond first to the IL-18 in order to induce IL-17 production. It has been shown that the combination of IL-18 together with IL-23 rapidly induced innate production of IL-17 from γδ T cells in the absence of TCR stimulation (Figure 2). This initial burst of IL-17 from γδ T cells may be required for initiation of EAE and the development of pathogenic Th17 cells. It is, however, unclear whether coexpression of IL-23R and IL-18R on γδ T cells make them more pathogenic in initiating EAE.
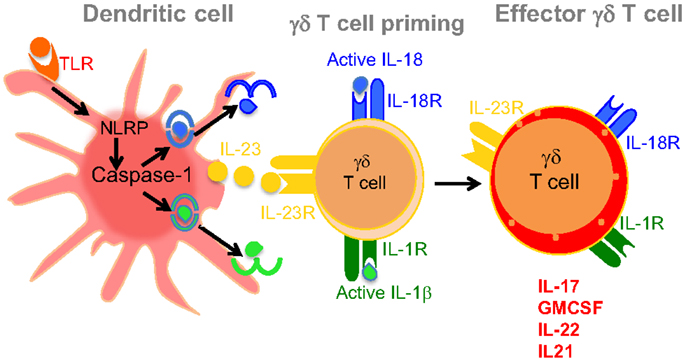
Figure 2. Dendritic cell-derived IL-23, IL-1β, and IL-18 mediates induction of effector γδT17 cells. Ligation of TLRs on the surface of DCs induces caspase-1 activation in inflammasome-dependent manner. Activated caspase-1 cleaves pro IL-1β and IL-18 into their active forms as shown in the figure. Activated DCs also produce IL-23, which together with IL-1β or IL-18 promote the induction of proinflammatory cytokines, such as IL-17, GM-CSF, IL-21, and IL-22. These effector γδ T cells initiate disease induction and help αβ+ CD4+ T cells to induce EAE.
In addition to IL-17 and IFN-γ, GM-CSF is also essentially required for the development of EAE. GM-CSF-deficient mice are resistant to the development of EAE with reduced infiltration of effector T cells into the CNS (68). Rostami et al. reported that the neutralization of GM-CSF-attenuated tissue inflammation in EAE (69). Taken together, it is clearly suggested that GM-CSF is required for the induction of encephalitogenic T cells in EAE. In fact, both Th1 and Th17 cells were shown to produce GM-CSF, which can further enhance the encephalitogenicity of these effector T cells in mediating the development of EAE. Moreover, it is proposed that GM-CSF is critical for the induction of pathogenic Th17 cells in EAE. Although both IL-12 and IL-23 can induce the production of GM-CSF by the effector T cells, it is clearly demonstrated that IL-23, but not IL-12, signaling is critically required for GM-SCF production in EAE (70–72). Similarly, exposure of IL-23 enhances the pathogenic functions of Th17 cells mediated by GM-CSF in EAE. In addition to CD4+ T cells, macrophages, and NK cells, γδ T cells produce high amounts of GM-CSF, which contributes to neuroinflammation of CNS in EAE (68). In fact, γδ T cells are the major innate source of GM-CSF in the CNS during EAE development (10). Combination of IL-23 together with IL-1β promotes GM-CSF production from γδ T cells in the absence of TCR stimulation (10). Moreover, the production of GM-CSF induced by IL-23 and IL-1β was compromised in Il1r−/− γδ T cells. In fact, production of GM-CSF by CNS-infiltrating γδ T cells is abolished in Il1r−/− mice, suggesting that IL-1 signaling is crucial for generation of GM-SCF-producing γδ T cells in EAE (Figure 2) (10). Caspase-1, which is required for active IL-1β production, is also critical in inducing GM-CSF from γδ T cells, as caspase1−/− γδ T cells are defective in GM-CSF production. In fact, caspase1−/− and Il1b−/− mice share a similar EAE phenotype, suggesting a specific role of caspase-1 and downstream IL-1β in regulating the induction of GM-CSF during EAE (10). It is suggested that GM-CSF contribute to the development of EAE by enhancing the functions of CNS-resident myeloid cells, including microglial cells (70). Although IL-1 signaling is required for the generation of GM-CSF-producing γδ T cells within the CNS during EAE, it is not identified which subtype of γδ T cells predominantly produce GM-CSF during disease. Moreover, IL-1- and IL-23-mediated inductions of GM-CSF in γδ T cells are dependent on MyD88 signaling, as γδ T cells from MyD88-deficient mice severely reduced GM-CSF production (10). Since MyD88 is a major downstream signaling component of TLR signaling pathway, it might be possible that ligation of TLRs on γδ T cells can also induce IL-17 production (57). To precisely understand the role TLRs in generating γδT17 cells, Dong et al. used IL-17-RFP⋅KI mice to understand the cellular source of IL-17 in EAE in response to TLR4 ligation. Using a faithful IL-17 reporter system, Dong et al. clearly demonstrated that the expression of TLR4 is high on IL-17+ as compared to IL-17-γδ T cells (31). In addition to IL-17 expression, IL-23-stimulation strongly enhanced the expression of TLR4 on γδ T cells. Moreover, the combination of IL-23 together with LPS further enhanced the secretion of IL-17 from γδ T cells (31). In addition to induced IL-17 production, TLR4 signaling also enhanced the survival of γδ T cells, which can further contribute in enhancing tissue inflammation in EAE. Taken together, TLRs especially TLR4 plays an essential role in inducing the development of IL-17-producing γδ T cells and their survival.
Regulatory T cells are critical for maintaining immune homeostasis of the host as loss of these cells either by naturally occurring mutation or cellular ablation leads to overwhelming activation of effector T cell-mediated multiple organ failure of the host (73–75). The critical functions of Treg cells were described in various models of autoimmune diseases, including EAE (53, 76). Using MOG tetramer and Foxp3-GFP⋅KI mice, it has been demonstrated that myelin–antigen-specific Treg cells are primed and expanded during the priming phase of EAE (53). Similar to effector T cells, these myelin-specific Treg cells can effectively migrate to the CNS (53). Although their frequency within the CNS is lower during the peak of EAE, strikingly, the population of Treg cells outnumber the population of effector T cells within the CNS at recovery phase of EAE (53). These CNS-accumulated Treg cells produce both IL-10 and TGF-β, which help in resolving the inflammation at the recovery phase of EAE. Interestingly, both Tr1 and Treg cells were shown to produce IL-10 in the CNS during the recovery phase of EAE (53, 77). In spite of their presence in the CNS at the peak of EAE, Treg cells failed to suppress proliferation and effector functions of CNS-accumulated effector T cells (53). Interestingly, cytokine analysis of CNS-accumulated effector CD4+ T cells revealed a strikingly higher production of proinflammatory cytokines, such as IL-6, TNF-α, and IL-21, which can be accounted for the failure of suppressive functions of Treg cells in EAE (53, 78). Interestingly, the higher frequency of γδ T cells together with αβ effector T cells found to be accumulated within the CNS (7, 8, 53, 76). In general, γδ T cells have high expression of IL-23R, in fact, all the γδ T cells present in the CNS at the peak of EAE exclusively expressed IL-23R (8, 13). Moreover, the frequency of IL-23R+ γδ T cells contracts while the frequency of Tregs cells increases during the recovery phase of EAE (8, 53). This raised an interesting possibility that the presence of γδ T cells within the CNS might promote the functions of inflammatory αβ+ T cells while hampering the suppressive functions of Treg cells in EAE (Figure 1). In fact, Tcrd−/− mice mount-attenuated effector αβ T cells response in EAE, supporting the fact that the presence of γδ T cells are essential for effective CD4+ T cells effector functions in EAE (49). Interestingly, Korn et al. suggested a mechanism by which γδ T cells enhanced the effector functions of CD4+ T cells during inflammation (8). Activation of γδ T cells with IL-23 produced soluble factors, which make αβ+ T cells refractory to Treg cell-mediated suppression, as cellular supernatant of IL-23-activated γδ T cells inhibited the suppressive functions of Treg cell (7, 8). It has been demonstrated that Treg cells can lose their suppressive functions in the presence of inflammatory environment. In fact, IL-6 makes αβ effector T cells refractory to the suppressive activity of Tregs cells (8, 53, 78). In addition, IL-6 has also been shown to inhibit TGF-β-induced de novo conversion of conventional T cells into Treg cells (41, 42). Similarly, Kuchroo et al. has demonstrated that IL-21, in addition to IL-6, can also suppress TGF-β-mediated de novo conversion of conventional T cells into Treg cells. Interestingly, IL-23R-stimulated γδ T cells not only block the conversion of conventional T cells into Treg cells but also make αβ+ effector T cells refractory to Treg cells suppression in vivo (8). This clearly indicates that the presence of γδ T cells at the site of tissue inflammation within the CNS indirectly promote the effector functions of αβ+ T cells by restraining their de novo conversion into Treg cells and inhibiting the suppressive functions of Treg cells in EAE (7, 8) (Figure 1). Similarly, the role of IL-23 in restraining the suppressive functions of Treg is well described in intestinal inflammation, as the frequency of inducible Foxp3+ Treg (iTreg) cells increases in the absence of IL-23 (79). However, it is not clear whether appearance of increased frequency of iTreg cells in the absence of IL-23–IL-23R signaling in the intestinal inflammation is due to loss of IL-23R+ γδ T cells functions, which are known to suppress the conversion of conventional T cells into Treg cells (8). Moreover, the importance of γδ T cells in mediating the inhibition of suppressive functions of Treg cells was further elucidated in Tcrd−/− mice, as these mice develop attenuated EAE with reduced production of IL-17 due to increased frequency of Treg cells (7, 49). Strikingly, anti-CD25 antibody-mediated depletion of Treg cells in Tcrd−/− mice enhanced the development of EAE with increased production of IL-17 (8). Altogether, it suggests that γδ T cells are crucial cellular component in promoting inflammation in EAE by restraining the regulatory functions of Treg cells and promoting the functions inflammatory αβ T cells (Figure 2).
While some models of EAE suggest that γδ T cells are pathogenic, others suggest that they modulate disease; thus, their precise role in pathogenesis is unclear. Both disease-promoting and disease-preventing functions of γδ T cells were documented in EAE. Deficiency of γδ T cells on B10⋅PL background develop a chronic EAE as compared to the development of monophasic acute EAE in the control mice (30). It has been further shown that γδ T cells regulate chronic inflammation by Fas–FasL-mediated killing of CNS-infiltrating inflammatory T cells (30). These studies clearly suggested the protective role of γδ T cells in EAE development.
Although recent literature on γδ T cells in context of IL-17 production implicated the pathogenic role of these cell types in EAE, a number of studies have ascribed the protective role of γδ T cells in EAE (30, 80, 81). A number of factors, such as using different mice strains in combination with either depleting antibodies or genetic manipulation of γδ T cells, might be contributing to these conflicting observations. Treatment of mice with UC7-13D5 anti-γδ antibody accelerates the onset of EAE (80). Similar results were obtained with the usage of UC7-13D5 antibody in other models of autoimmunity. It is partially identified that different subtypes of γδ T cells such as Vγ1 produce regulatory or Vγ4 and Vγ6 produce inflammatory cytokines (Table 1); therefore, it is possible that the treatment of UC7-13D5 antibody may alter this ratio and activate different subtypes of γδ T cell populations by cross-linking their TCR at different phases of EAE, which results in different outcome of disease. Nonetheless, it was not clearly understood whether anti-pan γδ T cells antibody depletes or activates γδ T cells by cross-linking their TCR in EAE (80). Using Tcrd-GFP knock-in mice, it has been clearly demonstrated that treatment with anti-pan γδ T cell antibodies activates, rather than depletes, γδ T cells and therefore exacerbating EAE (34). In addition, Tcrd−/− mice develop chronic inflammation in some mouse model of EAE (82). Tcrd−/− mice are devoid of δ TCR, which allow γδ T cells not to be activated by their TCR stimulation; however, the number of γδ T cells in these mice remains unchanged. This indicates that TCR-independent activation of γδ T cells can still occur in Tcrd−/− mice. We have discussed those different subsets of γδ T cells play opposite roles in EAE development. An interesting dichotomy has been established among Vγ1 and Vγ4 subsets of γδ T cells in EAE, which further provide a logical explanation for previously published contradictory results. Specific antibody-mediated activation of Vγ4 γδ T cells promote the development of EAE associated with enhanced production of IL-17 (34). On the other hand, specific antibody-mediated activation of Vγ1 γδ T cells suppressed EAE development (34). Interestingly, it has recently shown that γδ T cells can be activated with proinflammatory cytokines without the requirement of their TCR signals. To further identify the pathogenic or protective role of γδ T cells in EAE, a detailed study, including the involvement of various subtypes of γδ T cells, is required with more definitive tools. Nonetheless, accumulated literature in other autoimmunity has suggested that γδ T cells might play a pathogenic role in EAE. We have summarized the chief findings of γδ T cells in EAE in Table 2.

Table 1. Major γδ T cells subset in mouse.
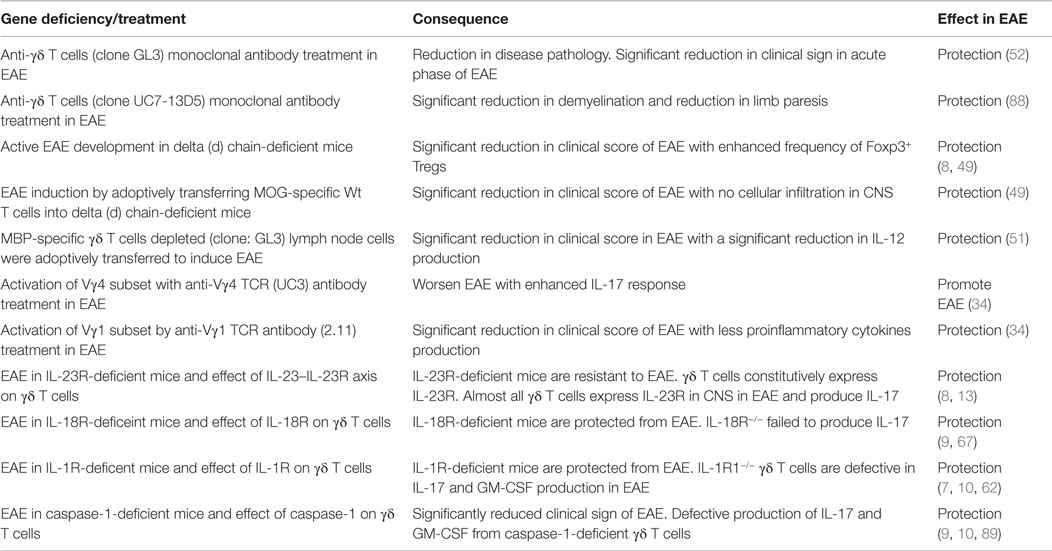
Table 2. Chief findings of γδ T cells in EAE.
Multiple sclerosis is demyelinating disease of CNS, which is caused by inflammatory T cells. In addition to αβ+ CD4+ T cells, γδ T cells were also clearly implicated in the disease pathogenesis in MS. It is shown that γδ T cells are accumulated in the MS plaques (90, 91). A restricted repertoire of γδ T cells was identified in MS lesions. CNS-restricted γδ T cells abundantly express variable gene segments Vδ1 and Vδ2. Furthermore, Vγ9+ γδ T cells circulate abundantly in the blood of MS patients and can be used as an indicator of disease activity (92). With the emerging literature on γδ T cells in EAE, it is indicated the involvement of γδ T cells in the pathogenesis of disease (see Table 1). Mouse data in EAE clearly indicated that IL-17-producing γδ T cells are crucial for disease induction and tissue inflammation in EAE (8, 10). Moreover, the role of IL-23, IL-1, IL-18, and caspase-1 is clearly indicated in enhancing IL-17- and GM-CSF-producing γδ T cells in EAE. Recent advancements in understanding the biology of Th17- and IL-17-producing γδ T cell and their implication in autoimmune diseases, including MS, could suggest new therapeutic targets for MS by targeting Th17- and IL-17-producing γδ T cells populations.
A number of studies have demonstrated a potential role of γδ T cells in the induction and maintenance of demyelinating CNS inflammation. γδ T cells are multifaceted cells, which are equipped with variety of functions to potentially influence all levels of inflammation by recognizing diverse array of antigens, rapid production of inflammatory mediators, and influencing the differentiation of their αβ counterparts. Equipped with functions of both innate and adaptive immune cells, γδ T cells can provide consequential functions in EAE development. Opposing roles of different subtypes of γδ T cells have been described in different mouse strains in EAE. Moreover, the identification of IL-17-producing inflammatory γδ T cells suggested their pathogenic role in EAE. In fact, many of the key questions in autoimmune inflammation, including EAE, were resolved by the discovery of IL-17-secreting Th17 cells. Moreover, clarification on the indispensible role of IL-23–IL-23R axis in Th17 cells also urged researchers to identify the role of IL-23–IL-23R signaling in γδ T cells as they have high expression of IL-23R receptor and therefore are responsive to IL-23 even in steady state – a characteristic which naive αβ T cells lack. This revisits the importance of IL-23 in the settings of EAE since it can influence the generation of two pathogenic subsets Th17 cells and γδT17 cells both of which contributes IL-17 to large extent. Synergistic action of IL-23, IL-1β, and IL-21 induces inflammatory IL-17-producing-γδ T cells, which not only enhance the generation and functions of αβ+ Th17 cells but also obstructs the suppressive functions of Treg cells in EAE. Recently, significant progress has been made in understanding the pathogenic role of γδ T cells in tissue inflammation. Yet more substantial evidences are required on different subtypes of γδ T cells for defining their opposing roles in tissue inflammation and explaining the confounding findings on their pathogenic or protective role in EAE.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
This work was supported by the grants from Department of Biotechnology (DBT), Government of India. AA is recipient of DBT-Wellcome Trust intermediate fellowship (IA/I/12/1/500524) and Innovative Young Biotechnologist Award (BT/05/IYBA/2011) from DBT. SM is a senior research fellow of the Council of Scientific and Industrial Research (CSIR). MW is a post-doctoral fellow supported by DBT-Wellcome Trust. We thank Dr. Kasia Karwacz (Brigham and Women’s Hospital) for her critical and thoughtful comments on the manuscript.
1. Carding SR, Egan PJ. Gammadelta T cells: functional plasticity and heterogeneity. Nat Rev Immunol (2002) 2:336–45. doi:10.1038/nri797
2. Saito H, Kranz DM, Takagaki Y, Hayday AC, Eisen HN, Tonegawa S. Complete primary structure of a heterodimeric T-cell receptor deduced from cDNA sequences. Nature (1984) 309:757–62. doi:10.1038/309757a0
3. Brenner MB, McLean J, Dialynas DP, Strominger JL, Smith JA, Owen FL, et al. Identification of a putative second T-cell receptor. Nature (1986) 322:145–9. doi:10.1038/322145a0
4. Born WK, Jin N, Aydintug MK, Wands JM, French JD, Roark CL, et al. Gammadelta T lymphocytes-selectable cells within the innate system? J Clin Immunol (2007) 27:133–44. doi:10.1007/s10875-007-9077-z
5. O’Brien RL, Roark CL, Jin N, Aydintug MK, French JD, Chain JL, et al. Gammadelta T-cell receptors: functional correlations. Immunol Rev (2007) 215:77–88. doi:10.1111/j.1600-065X.2006.00477.x
6. Martin B, Hirota K, Cua DJ, Stockinger B, Veldhoen M. Interleukin-17-producing gammadelta T cells selectively expand in response to pathogen products and environmental signals. Immunity (2009) 31:321–30. doi:10.1016/j.immuni.2009.06.020
7. Sutton CE, Lalor SJ, Sweeney CM, Brereton CF, Lavelle EC, Mills KH. Interleukin-1 and IL-23 induce innate IL-17 production from gammadelta T cells, amplifying Th17 responses and autoimmunity. Immunity (2009) 31:331–41. doi:10.1016/j.immuni.2009.08.001
8. Petermann F, Rothhammer V, Claussen MC, Haas JD, Blanco LR, Heink S, et al. Gammadelta T cells enhance autoimmunity by restraining regulatory T cell responses via an interleukin-23-dependent mechanism. Immunity (2010) 33:351–63. doi:10.1016/j.immuni.2010.08.013
9. Lalor SJ, Dungan LS, Sutton CE, Basdeo SA, Fletcher JM, Mills KH. Caspase-1-processed cytokines IL-1beta and IL-18 promote IL-17 production by gammadelta and CD4 T cells that mediate autoimmunity. J Immunol (2011) 186:5738–48. doi:10.4049/jimmunol.1003597
10. Lukens JR, Barr MJ, Chaplin DD, Chi H, Kanneganti TD. Inflammasome-derived IL-1beta regulates the production of GM-CSF by CD4(+) T cells and gammadelta T cells. J Immunol (2012) 188:3107–15. doi:10.4049/jimmunol.1103308
11. Korn T, Bettelli E, Oukka M, Kuchroo VK. IL-17 and Th17 cells. Annu Rev Immunol (2009) 27:485–517. doi:10.1146/annurev.immunol.021908.132710
12. Ivanov II, McKenzie BS, Zhou L, Tadokoro CE, Lepelley A, Lafaille JJ, et al. The orphan nuclear receptor RORgammat directs the differentiation program of proinflammatory IL-17+ T helper cells. Cell (2006) 126:1121–33. doi:10.1016/j.cell.2006.07.035
13. Awasthi A, Riol-Blanco L, Jager A, Korn T, Pot C, Galileos G, et al. Cutting edge: IL-23 receptor gfp reporter mice reveal distinct populations of IL-17-producing cells. J Immunol (2009) 182:5904–8. doi:10.4049/jimmunol.0900732
14. Nunez C, Dema B, Cenit MC, Polanco I, Maluenda C, Arroyo R, et al. IL23R: a susceptibility locus for celiac disease and multiple sclerosis? Genes Immun (2008) 9:289–93. doi:10.1038/gene.2008.16
15. Blink SE, Miller SD. The contribution of gammadelta T cells to the pathogenesis of EAE and MS. Curr Mol Med (2009) 9:15–22. doi:10.2174/156652409787314516
16. Paul S, Shilpi S, Lal G. Role of gamma-delta (gammadelta) T cells in autoimmunity. J Leukoc Biol (2015) 97:259–71. doi:10.1189/jlb.3RU0914-443R
17. Chien YH, Konigshofer Y. Antigen recognition by gammadelta T cells. Immunol Rev (2007) 215:46–58. doi:10.1111/j.1600-065X.2006.00470.x
18. Konigshofer Y, Chien YH. Gammadelta T cells – innate immune lymphocytes? Curr Opin Immunol (2006) 18:527–33. doi:10.1016/j.coi.2006.07.008
19. Correa I, Bix M, Liao NS, Zijlstra M, Jaenisch R, Raulet D. Most gamma delta T cells develop normally in beta 2-microglobulin-deficient mice. Proc Natl Acad Sci U S A (1992) 89:653–7. doi:10.1073/pnas.89.2.653
20. Bigby M, Markowitz JS, Bleicher PA, Grusby MJ, Simha S, Siebrecht M, et al. Most gamma delta T cells develop normally in the absence of MHC class II molecules. J Immunol (1993) 151:4465–75.
21. Crowley MP, Reich Z, Mavaddat N, Altman JD, Chien Y. The recognition of the nonclassical major histocompatibility complex (MHC) class I molecule, T10, by the gammadelta T cell, G8. J Exp Med (1997) 185:1223–30. doi:10.1084/jem.185.7.1223
22. Crowley MP, Fahrer AM, Baumgarth N, Hampl J, Gutgemann I, Teyton L, et al. A population of murine gammadelta T cells that recognize an inducible MHC class Ib molecule. Science (2000) 287:314–6. doi:10.1126/science.287.5451.314
23. Wu J, Groh V, Spies T. T cell antigen receptor engagement and specificity in the recognition of stress-inducible MHC class I-related chains by human epithelial gamma delta T cells. J Immunol (2002) 169:1236–40. doi:10.4049/jimmunol.169.3.1236
24. Haas W, Pereira P, Tonegawa S. Gamma/delta cells. Annu Rev Immunol (1993) 11:637–85. doi:10.1146/annurev.iy.11.040193.003225
25. Bauer S, Groh V, Wu J, Steinle A, Phillips JH, Lanier LL, et al. Activation of NK cells and T cells by NKG2D, a receptor for stress-inducible MICA. Science (1999) 285:727–9. doi:10.1126/science.285.5428.727
26. Ferrick DA, Schrenzel MD, Mulvania T, Hsieh B, Ferlin WG, Lepper H. Differential production of interferon-gamma and interleukin-4 in response to Th1- and Th2-stimulating pathogens by gamma delta T cells in vivo. Nature (1995) 373:255–7. doi:10.1038/373255a0
27. Riol-Blanco L, Lazarevic V, Awasthi A, Mitsdoerffer M, Wilson BS, Croxford A, et al. IL-23 receptor regulates unconventional IL-17-producing T cells that control bacterial infections. J Immunol (2010) 184:1710–20. doi:10.4049/jimmunol.0902796
28. Olive C. Gamma delta T cell receptor variable region usage during the development of experimental allergic encephalomyelitis. J Neuroimmunol (1995) 62:1–7. doi:10.1016/0165-5728(95)00081-C
29. Olive C. T cell receptor usage in autoimmune disease. Immunol Cell Biol (1995) 73:297–307. doi:10.1038/icb.1995.46
30. Ponomarev ED, Novikova M, Yassai M, Szczepanik M, Gorski J, Dittel BN. Gamma delta T cell regulation of IFN-gamma production by central nervous system-infiltrating encephalitogenic T cells: correlation with recovery from experimental autoimmune encephalomyelitis. J Immunol (2004) 173:1587–95. doi:10.4049/jimmunol.173.3.1587
31. Reynolds JM, Martinez GJ, Chung Y, Dong C. Toll-like receptor 4 signaling in T cells promotes autoimmune inflammation. Proc Natl Acad Sci U S A (2012) 109:13064–9. doi:10.1073/pnas.1120585109
32. Hirsh MI, Junger WG. Roles of heat shock proteins and gamma delta T cells in inflammation. Am J Respir Cell Mol Biol (2008) 39:509–13. doi:10.1165/rcmb.2008-0090TR
33. Roark CL, Simonian PL, Fontenot AP, Born WK, O’Brien RL. Gammadelta T cells: an important source of IL-17. Curr Opin Immunol (2008) 20:353–7. doi:10.1016/j.coi.2008.03.006
34. Blink SE, Caldis MW, Goings GE, Harp CT, Malissen B, Prinz I, et al. Gammadelta T cell subsets play opposing roles in regulating experimental autoimmune encephalomyelitis. Cell Immunol (2014) 290:39–51. doi:10.1016/j.cellimm.2014.04.013
35. Roark CL, French JD, Taylor MA, Bendele AM, Born WK, O’Brien RL. Exacerbation of collagen-induced arthritis by oligoclonal, IL-17-producing gamma delta T cells. J Immunol (2007) 179:5576–83. doi:10.4049/jimmunol.179.8.5576
36. Ribot JC, deBarros A, Pang DJ, Neves JF, Peperzak V, Roberts SJ, et al. CD27 is a thymic determinant of the balance between interferon-gamma- and interleukin 17-producing gammadelta T cell subsets. Nat Immunol (2009) 10:427–36. doi:10.1038/ni.1717
37. Cua DJ, Sherlock J, Chen Y, Murphy CA, Joyce B, Seymour B, et al. Interleukin-23 rather than interleukin-12 is the critical cytokine for autoimmune inflammation of the brain. Nature (2003) 421:744–8. doi:10.1038/nature01355
38. McGeachy MJ, Cua DJ. Th17 cell differentiation: the long and winding road. Immunity (2008) 28:445–53. doi:10.1016/j.immuni.2008.03.001
39. Ferber IA, Brocke S, Taylor-Edwards C, Ridgway W, Dinisco C, Steinman L, et al. Mice with a disrupted IFN-gamma gene are susceptible to the induction of experimental autoimmune encephalomyelitis (EAE). J Immunol (1996) 156:5–7.
40. Duhen R, Glatigny S, Arbelaez CA, Blair TC, Oukka M, Bettelli E. Cutting edge: the pathogenicity of IFN-gamma-producing Th17 cells is independent of T-bet. J Immunol (2013) 190:4478–82. doi:10.4049/jimmunol.1203172
41. Bettelli E, Carrier Y, Gao W, Korn T, Strom TB, Oukka M, et al. Reciprocal developmental pathways for the generation of pathogenic effector Th17 and regulatory T cells. Nature (2006) 441:235–8. doi:10.1038/nature04753
42. Veldhoen M, Hocking RJ, Atkins CJ, Locksley RM, Stockinger B. TGFbeta in the context of an inflammatory cytokine milieu supports de novo differentiation of IL-17-producing T cells. Immunity (2006) 24:179–89. doi:10.1016/j.immuni.2006.01.001
43. Mangan PR, Harrington LE, O’Quinn DB, Helms WS, Bullard DC, Elson CO, et al. Transforming growth factor-beta induces development of the T(H)17 lineage. Nature (2006) 441:231–4. doi:10.1038/nature04754
44. Zhou L, Ivanov II, Spolski R, Min R, Shenderov K, Egawa T, et al. IL-6 programs T(H)-17 cell differentiation by promoting sequential engagement of the IL-21 and IL-23 pathways. Nat Immunol (2007) 8:967–74. doi:10.1038/ni1488
45. Korn T, Bettelli E, Gao W, Awasthi A, Jager A, Strom TB, et al. IL-21 initiates an alternative pathway to induce proinflammatory T(H)17 cells. Nature (2007) 448:484–7. doi:10.1038/nature05970
46. Nurieva R, Yang XO, Martinez G, Zhang Y, Panopoulos AD, Ma L, et al. Essential autocrine regulation by IL-21 in the generation of inflammatory T cells. Nature (2007) 448:480–3. doi:10.1038/nature05969
47. Komiyama Y, Nakae S, Matsuki T, Nambu A, Ishigame H, Kakuta S, et al. IL-17 plays an important role in the development of experimental autoimmune encephalomyelitis. J Immunol (2006) 177:566–73. doi:10.4049/jimmunol.177.1.566
48. Do JS, Fink PJ, Li L, Spolski R, Robinson J, Leonard WJ, et al. Cutting edge: spontaneous development of IL-17-producing gamma delta T cells in the thymus occurs via a TGF-beta 1-dependent mechanism. J Immunol (2010) 184:1675–9. doi:10.4049/jimmunol.0903539
49. Spahn TW, Issazadah S, Salvin AJ, Weiner HL. Decreased severity of myelin oligodendrocyte glycoprotein peptide 33 – 35-induced experimental autoimmune encephalomyelitis in mice with a disrupted TCR delta chain gene. Eur J Immunol (1999) 29:4060–71. doi:10.1002/(SICI)1521-4141(199912)29:12<4060::AID-IMMU4060>3.0.CO;2-S
50. Rajan AJ, Klein JD, Brosnan CF. The effect of gammadelta T cell depletion on cytokine gene expression in experimental allergic encephalomyelitis. J Immunol (1998) 160:5955–62.
51. Odyniec A, Szczepanik M, Mycko MP, Stasiolek M, Raine CS, Selmaj KW. Gammadelta T cells enhance the expression of experimental autoimmune encephalomyelitis by promoting antigen presentation and IL-12 production. J Immunol (2004) 173:682–94. doi:10.4049/jimmunol.173.1.682
52. Rajan AJ, Gao YL, Raine CS, Brosnan CF. A pathogenic role for gamma delta T cells in relapsing-remitting experimental allergic encephalomyelitis in the SJL mouse. J Immunol (1996) 157:941–9.
53. Korn T, Reddy J, Gao W, Bettelli E, Awasthi A, Petersen TR, et al. Myelin-specific regulatory T cells accumulate in the CNS but fail to control autoimmune inflammation. Nat Med (2007) 13:423–31. doi:10.1038/nm1564
54. Lockhart E, Green AM, Flynn JL. IL-17 production is dominated by gammadelta T cells rather than CD4 T cells during Mycobacterium tuberculosis infection. J Immunol (2006) 177:4662–9. doi:10.4049/jimmunol.177.7.4662
55. Umemura M, Kawabe T, Shudo K, Kidoya H, Fukui M, Asano M, et al. Involvement of IL-17 in Fas ligand-induced inflammation. Int Immunol (2004) 16:1099–108. doi:10.1093/intimm/dxh111
56. Umemura M, Yahagi A, Hamada S, Begum MD, Watanabe H, Kawakami K, et al. IL-17-mediated regulation of innate and acquired immune response against pulmonary Mycobacterium bovis bacille Calmette-Guerin infection. J Immunol (2007) 178:3786–96. doi:10.4049/jimmunol.178.6.3786
57. Shibata K, Yamada H, Hara H, Kishihara K, Yoshikai Y. Resident Vdelta1+ gammadelta T cells control early infiltration of neutrophils after Escherichia coli infection via IL-17 production. J Immunol (2007) 178:4466–72. doi:10.4049/jimmunol.178.7.4466
58. Kuchroo VK, Anderson AC, Waldner H, Munder M, Bettelli E, Nicholson LB. T cell response in experimental autoimmune encephalomyelitis (EAE): role of self and cross-reactive antigens in shaping, tuning, and regulating the autopathogenic T cell repertoire. Annu Rev Immunol (2002) 20:101–23. doi:10.1146/annurev.immunol.20.081701.141316
59. Lees JR, Iwakura Y, Russell JH. Host T cells are the main producers of IL-17 within the central nervous system during initiation of experimental autoimmune encephalomyelitis induced by adoptive transfer of Th1 cell lines. J Immunol (2008) 180:8066–72. doi:10.4049/jimmunol.180.12.8066
60. Smith SS, Barnum SR. Differential expression of beta 2-integrins and cytokine production between gammadelta and alphabeta T cells in experimental autoimmune encephalomyelitis. J Leukoc Biol (2008) 83:71–9. doi:10.1189/jlb.0407263
61. Wohler JE, Smith SS, Zinn KR, Bullard DC, Barnum SR. Gammadelta T cells in EAE: early trafficking events and cytokine requirements. Eur J Immunol (2009) 39:1516–26. doi:10.1002/eji.200839176
62. Sutton C, Brereton C, Keogh B, Mills KH, Lavelle EC. A crucial role for interleukin (IL)-1 in the induction of IL-17-producing T cells that mediate autoimmune encephalomyelitis. J Exp Med (2006) 203:1685–91. doi:10.1084/jem.20060285
63. Laurence A, Tato CM, Davidson TS, Kanno Y, Chen Z, Yao Z, et al. Interleukin-2 signaling via STAT5 constrains T helper 17 cell generation. Immunity (2007) 26:371–81. doi:10.1016/j.immuni.2007.02.009
64. Shibata K, Yamada H, Nakamura R, Sun X, Itsumi M, Yoshikai Y. Identification of CD25+ gamma delta T cells as fetal thymus-derived naturally occurring IL-17 producers. J Immunol (2008) 181:5940–7. doi:10.4049/jimmunol.181.9.5940
65. Wang X, Wei Y, Liu X, Xing C, Han G, Chen G, et al. IL-15-secreting gammadeltaT cells induce memory T cells in experimental allergic encephalomyelitis (EAE) mice. Mol Immunol (2015) 66:402–8. doi:10.1016/j.molimm.2015.04.021
66. Shi FD, Takeda K, Akira S, Sarvetnick N, Ljunggren HG. IL-18 directs autoreactive T cells and promotes autodestruction in the central nervous system via induction of IFN-gamma by NK cells. J Immunol (2000) 165:3099–104. doi:10.4049/jimmunol.165.6.3099
67. Gutcher I, Urich E, Wolter K, Prinz M, Becher B. Interleukin 18-independent engagement of interleukin 18 receptor-alpha is required for autoimmune inflammation. Nat Immunol (2006) 7:946–53. doi:10.1038/ni1377
68. Shi Y, Liu CH, Roberts AI, Das J, Xu G, Ren G, et al. Granulocyte-macrophage colony-stimulating factor (GM-CSF) and T-cell responses: what we do and don’t know. Cell Res (2006) 16:126–33. doi:10.1038/sj.cr.7310017
69. El-Behi M, Ciric B, Dai H, Yan Y, Cullimore M, Safavi F, et al. The encephalitogenicity of T(H)17 cells is dependent on IL-1- and IL-23-induced production of the cytokine GM-CSF. Nat Immunol (2011) 12:568–75. doi:10.1038/ni.2031
70. Ponomarev ED, Shriver LP, Maresz K, Pedras-Vasconcelos J, Verthelyi D, Dittel BN. GM-CSF production by autoreactive T cells is required for the activation of microglial cells and the onset of experimental autoimmune encephalomyelitis. J Immunol (2007) 178:39–48. doi:10.4049/jimmunol.178.1.39
71. Codarri L, Gyulveszi G, Tosevski V, Hesske L, Fontana A, Magnenat L, et al. RORgammat drives production of the cytokine GM-CSF in helper T cells, which is essential for the effector phase of autoimmune neuroinflammation. Nat Immunol (2011) 12:560–7. doi:10.1038/ni.2027
72. Croxford AL, Lanzinger M, Hartmann FJ, Schreiner B, Mair F, Pelczar P, et al. The cytokine GM-CSF drives the inflammatory signature of CCR2 monocytes and licenses autoimmunity. Immunity (2015) 43:502–14. doi:10.1016/j.immuni.2015.08.010
73. Rudensky AY. Regulatory T cells and Foxp3. Immunol Rev (2011) 241:260–8. doi:10.1111/j.1600-065X.2011.01018.x
74. Ramsdell F, Ziegler SF. FOXP3 and scurfy: how it all began. Nat Rev Immunol (2014) 14:343–9. doi:10.1038/nri3650
75. Kim JM, Rasmussen JP, Rudensky AY. Regulatory T cells prevent catastrophic autoimmunity throughout the lifespan of mice. Nat Immunol (2007) 8:191–7. doi:10.1038/ni1428
76. Korn T, Mitsdoerffer M, Croxford AL, Awasthi A, Dardalhon VA, Galileos G, et al. IL-6 controls Th17 immunity in vivo by inhibiting the conversion of conventional T cells into Foxp3+ regulatory T cells. Proc Natl Acad Sci U S A (2008) 105:18460–5. doi:10.1073/pnas.0809850105
77. Awasthi A, Carrier Y, Peron JP, Bettelli E, Kamanaka M, Flavell RA, et al. A dominant function for interleukin 27 in generating interleukin 10-producing anti-inflammatory T cells. Nat Immunol (2007) 8:1380–9. doi:10.1038/ni1541
78. Pasare C, Medzhitov R. Toll pathway-dependent blockade of CD4+CD25+ T cell-mediated suppression by dendritic cells. Science (2003) 299:1033–6. doi:10.1126/science.1078231
79. Izcue A, Hue S, Buonocore S, Arancibia-Carcamo CV, Ahern PP, Iwakura Y, et al. Interleukin-23 restrains regulatory T cell activity to drive T cell-dependent colitis. Immunity (2008) 28:559–70. doi:10.1016/j.immuni.2008.02.019
80. Kobayashi Y, Kawai K, Ito K, Honda H, Sobue G, Yoshikai Y. Aggravation of murine experimental allergic encephalomyelitis by administration of T-cell receptor gammadelta-specific antibody. J Neuroimmunol (1997) 73:169–74. doi:10.1016/S0165-5728(96)00187-7
81. Ponomarev ED, Dittel BN. Gamma delta T cells regulate the extent and duration of inflammation in the central nervous system by a Fas ligand-dependent mechanism. J Immunol (2005) 174:4678–87. doi:10.4049/jimmunol.174.8.4678
82. Koenecke C, Chennupati V, Schmitz S, Malissen B, Forster R, Prinz I. In vivo application of mAb directed against the gammadelta TCR does not deplete but generates “invisible” gammadelta T cells. Eur J Immunol (2009) 39:372–9. doi:10.1002/eji.200838741
83. Welte T, Lamb J, Anderson JF, Born WK, O’Brien RL, Wang T. Role of two distinct gammadelta T cell subsets during West Nile virus infection. FEMS Immunol Med Microbiol (2008) 53:275–83. doi:10.1111/j.1574-695X.2008.00430.x
84. Girardi M, Oppenheim DE, Steele CR, Lewis JM, Glusac E, Filler R, et al. Regulation of cutaneous malignancy by gammadelta T cells. Science (2001) 294:605–9. doi:10.1126/science.1063916
85. Jameson J, Havran WL. Skin gammadelta T-cell functions in homeostasis and wound healing. Immunol Rev (2007) 215:114–22. doi:10.1111/j.1600-065X.2006.00483.x
86. Chen Y, Chou K, Fuchs E, Havran WL, Boismenu R. Protection of the intestinal mucosa by intraepithelial gamma delta T cells. Proc Natl Acad Sci U S A (2002) 99:14338–43. doi:10.1073/pnas.212290499
87. Tsuchiya T, Fukuda S, Hamada H, Nakamura A, Kohama Y, Ishikawa H, et al. Role of gamma delta T cells in the inflammatory response of experimental colitis mice. J Immunol (2003) 171:5507–13. doi:10.4049/jimmunol.171.10.5507
88. Dandekar AA, Perlman S. Virus-induced demyelination in nude mice is mediated by gamma delta T cells. Am J Pathol (2002) 161:1255–63. doi:10.1016/S0002-9440(10)64402-1
89. Furlan R, Martino G, Galbiati F, Poliani PL, Smiroldo S, Bergami A, et al. Caspase-1 regulates the inflammatory process leading to autoimmune demyelination. J Immunol (1999) 163:2403–9.
90. Wucherpfennig KW, Newcombe J, Li H, Keddy C, Cuzner ML, Hafler DA. Gamma delta T-cell receptor repertoire in acute multiple sclerosis lesions. Proc Natl Acad Sci U S A (1992) 89:4588–92. doi:10.1073/pnas.89.10.4588
91. Hvas J, Oksenberg JR, Fernando R, Steinman L, Bernard CC. Gamma delta T cell receptor repertoire in brain lesions of patients with multiple sclerosis. J Neuroimmunol (1993) 46:225–34. doi:10.1016/0165-5728(93)90253-U
Keywords: gamma–delta T cells, Th17 cells, cytokines, inflammation, autoimmunity
Citation: Malik S, Want MY and Awasthi A (2016) The Emerging Roles of Gamma–Delta T Cells in Tissue Inflammation in Experimental Autoimmune Encephalomyelitis. Front. Immunol. 7:14. doi: 10.3389/fimmu.2016.00014
Received: 22 July 2015; Accepted: 12 January 2016;
Published: 29 January 2016
Edited by:
Manu Rangachari, Centre hospitalier universitaire de Québec, CanadaReviewed by:
Girdhari Lal, National Centre for Cell Science, IndiaCopyright: © 2016 Malik, Want and Awasthi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Amit Awasthi, YWF3YXN0aGlAdGhzdGkucmVzLmlu
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.