 Annette Bruchfeld
Annette Bruchfeld Mårten Wendt
Mårten Wendt Edmund J. Miller
Edmund J. Miller- 1Department of Renal Medicine, Clinical Science Intervention and Technology (CLINTEC), Karolinska Institutet, Karolinska University Hospital, Stockholm, Sweden
- 2Feinstein Institute for Medical Research, Manhasset, NY, USA
- 3Hofstra University School of Medicine, Hempstead, NY, USA
Macrophage migration inhibitory factor (MIF) is a proinflammatory cytokine implicated in acute and chronic inflammatory conditions, including sepsis, autoimmune disease, atherogenesis, plaque instability, and pulmonary arterial hypertension. MIF in plasma and urine is significantly elevated in patients with acute kidney injury (AKI) and elevated MIF in serum is associated with markers of oxidative stress, endothelial dysfunction, arterial stiffness, and markers of myocardial damage in chronic kidney disease (CKD). Furthermore, MIF seems to be involved in vascular processes and cardiovascular disease associated with CKD, glomerulonephritis, autosomal dominant polycystic kidney disease, and possibly also in progression to renal failure. Moreover, in active anti-neutrophil cytoplasmatic antibody-associated vasculitis, plasma MIF levels have been shown to be significantly elevated as compared with samples from patients in remission. A significant difference in the genotype frequency of high production MIF -173 G/C genotype has been found in end-stage renal disease, compared to controls. Inhibition of MIF in a diabetic nephropathy model ameliorated blood glucose and albuminuria and in a model of adult polycystic kidney disease cyst growth was delayed. Preclinical studies support a potential therapeutic role for MIF in AKI and in a number of CKDs, whereas these data in human disease are still observational. Future interventional studies are needed to delineate the role of MIF as a treatment target in clinical kidney disease.
Introduction
Macrophage migration inhibitory factor (MIF) was one of the first cytokines that was identified after being isolated from the supernatants of T-lymphocytes, and initially described as a soluble factor with macrophage migration-inhibiting properties (1–3). It has later been shown that MIF is produced by a number of other cells, such as monocytes, macrophages, granulocytes, endocrine cells, epithelial cells, and endothelial cells (4, 5).
Migration inhibitory factor is a pleiotropic upstream proinflammatory integral mediator of the innate immune system, stimulating the release of multiple cytokines, including tumor necrosis factor (TNF)-α, with CD 74 being a binding receptor promoting the recruitment of leukocytes into inflammatory sites in a chemokine-like fashion (6). Three-dimensional X-ray crystallography has revealed that the MIF molecule contains a hydrophobic pocket, which has been identified as the proinflammatory active site of MIF (7) and compounds binding to this region decrease downstream MIF signaling (8, 9).
Migration inhibitory factor has been implicated in the pathogenesis of sepsis, autoimmune diseases, such as rheumatoid arthritis, systemic lupus erythematosus, and cardiovascular disease (CVD) (6, 10–13). In atherosclerosis animal models, aortic inflammation was reduced, and neointimal plaques were stabilized after administration of anti-MIF antibody (14, 15).
Chronic kidney disease (CKD) is a state of chronic inflammation with major implications for morbidity and mortality driven by a significant increased risk for CVD (16, 17). Delineating the role of inflammatory markers in atherosclerotic and inflammatory disease in CKD is therefore of considerable interest. Whether MIF has an important role in this area is not well known. This article therefore aims at reviewing available data on the role of MIF in acute kidney injury (AKI), CKD, diabetic nephropathy, inflammatory kidney disease, and genetic aspects of MIF and kidney disease.
MIF and AKI
Urinary MIF has previously been reported to be increased, and associated with the severity of renal injury, in human glomerulonephritis and has also been suggested as a potential biomarker for acute kidney damage in acute pyelonephritis (18, 19). Similar findings have been demonstrated in kidney transplantation (20). Augmented plasma levels of MIF seem to be an early and predictive event of AKI in septic patients admitted to the ICU (21). In preclinical models, MIF stimulates leukocyte chemotaxis as well as tissue infiltration of leukocytes and induces multiorgan damage affecting both lungs and kidneys (6, 22–24). In a recent paper by Stefaniak et al., it was shown that increased plasma levels of MIF in patients undergoing liver transplantation was significantly more predictive than serum creatinine for AKI and the need for renal replacement therapy postoperatively (25).
MIF and CKD, Implications for Cardiovascular Disease
The prevalence of CKD worldwide is 10–12% and its incidence is even greater in the elderly (26, 27). Systemic low-grade inflammation is associated with loss of renal function, and the uremic phenotype is also linked to premature aging and accelerated atherosclerosis (28, 29). CVD is a major challenge in this patient population in which mortality rates due to CVD are about 10–20 times higher in dialysis patients than those of the general population (30). A number of proinflammatory factors have been investigated, such as C-reactive protein (CRP), interleukin-6 (IL-6), TNF, and high-mobility group box-1 protein (HMGB1), and increased circulating levels have in most cases been shown to be associated with poor outcome (22–34). It has however been suggested that retention by reduced cytokine excretion or degradation in the kidney, not only increased production, may play a role for the elevated cytokine levels (35).
We have previously shown that circulating serum levels of MIF are significantly elevated in CKD stage 3–5 patients (n = 257), compared with controls (n = 53) in a cross-sectional study (36). MIF levels were also associated positively with markers of oxidative stress and endothelial activation, such as 8-hydroxy-2-deoxyguanosine (8-OH-dG) levels and ICAM-1 levels, but not with inflammatory markers, such as CRP, IL-6, and TNF. However, in contrast to most previously described cytokines, we observed no correlation between MIF and glomerular filtration rate (GFR).
Rammos et al. recently showed that plasma MIF levels correlated negatively with endothelial function by flow-mediated dilation of the brachial artery, and positively with arterial stiffness indices using applanation tonometry in patients with end-stage renal disease (ESRD). In a multivariate regression model, MIF was an independent predictor for arterial stiffness. A correlation between high MIF and high-sensitive troponin I also suggested an association with myocardial injury in these patients (37). Taken together, these studies indicate that MIF may play a role in vascular disease associated with CKD, but further studies are needed.
MIF, Pulmonary Arterial Hypertension, and CKD
Pulmonary arterial hypertension (PAH) is characterized by endothelial dysfunction, vasoconstriction, and pulmonary vascular remodeling. Haddad et al. have shown that AKI is relatively common in individuals with PAH and is a strong predictor of early death (38). Furthermore, in the study of Shah et al. (39), the severity of CKD in patients with PAH was directly related to the risk of death.
Recent studies have shown clear links among MIF, PAH, and the concomitant pulmonary vasoconstriction and vascular remodeling (40, 41). The release of MIF and the vascular changes are due, at least in part, to increased oxidative stress (42). Our study in CKD stage 3–5 patients showed that elevated serum MIF concentrations are associated with markers of oxidative stress (plasma 8-OH-dG levels) and endothelial activation (ICAM-1) (36). This further suggests possible links between MIF in CKD and the associated pulmonary vascular changes and cardiac changes.
MIF and Diabetic Kidney Disease
Diabetic nephropathy is one of the leading causes of CKD and dialysis dependency. Albuminuria and impaired GFR predominantly account for the increased mortality observed in type 2 diabetes (43). Accumulating evidence has revealed that immunological and inflammatory mechanisms may play a significant role in the development and progression of diabetic nephropathy, in addition to non-immunological factors (44, 45). Elevated MIF levels have been found in patients with impaired glucose tolerance and type 2 diabetes and have also been associated with coronary events in these patient populations (46–48). MIF protein expression and urinary MIF excretion, the latter preceding the onset of microalbuminuria, have been demonstrated in a diabetic mouse model (49). Glomerular and tubulointerstitial mRNA expressions of the MIF receptor CD74 were shown to be increased in Pima Indians with type 2 diabetes and diabetic nephropathy (50). As shown by Wang et al., treatment of the diabetic db/db mice with the MIF inhibitor ISO-1 significantly decreased blood glucose levels and albuminuria in these mice, suggesting that MIF inhibition may be a potential therapeutic strategy in diabetic nephropathy (51).
MIF and Glomerulonephritis
It has been established that upregulation of renal MIF mRNA expression in the endothelium, glomerular, and tubular epithelial cells is closely related to macrophage accumulation and renal tissue lesions in experimental glomerulonephritis. By contrast, in the normal kidney, MIF mRNA and protein are largely restricted to tubular epithelial cells and some glomerular visceral and parietal epithelial cells (52). By using a neutralizing anti-MIF antibody, it was possible to partially reverse established crescentic glomerulonephritis (53). In human disease, a marked increase in both glomerular and tubular MIF mRNA and protein expression has been demonstrated in proliferative forms of GN, correlating with leukocyte infiltration, histologic damage, and renal function impairment (54). Elevated MIF concentrations have been measured in peripheral blood T cells and monocytes from patients with IgA nephropathy (IgAN), which is the most common form of primary glomerulonephritis and is characterized by IgAN immune complexes in the glomerular mesangium, proliferation of mesangial cells, infiltration of inflammatory cells, and progressive glomerular injury. MIF overproduction was also correlated with the intensity of acute exacerbation in these patients (55, 56). Polymeric IgAN isolated from patients with IgAN was able to induce MIF production in human mesangial cells, and anti-MIF treatment was shown ameliorate kidney injury and reduce glomerular TGF-β 1 expression in an experimental model of IgAN (57, 58).
MIF and Vasculitis
Granulomatosis with polyangiitis (GPA) and microscopic polyangiitis (MPA) are diseases characterized by systemic small vessel necrotizing inflammation, commonly affecting the kidneys and with a close association with the presence of anti-neutrophil cytoplasmatic antibodies (ANCAs), thus known as ANCA-associated vasculitides (AAV). A number of inflammatory cells and proinflammatory mediators have been implicated in the pathogenesis, which is believed to be initiated by priming of neutrophils and monocytes in the circulation by inflammatory stimuli (59). Increased levels of circulating MIF in serum have been found particularly in active AAV patients, but not in patients with other forms of vasculitis, such as giant cell arteritis and polyarteritis nodosa (60). We studied MIF in a prospective study of incident AAV patients at induction treatment, and at follow-up at 3 and 6 months and found elevated plasma levels at baseline. MIF decreased significantly during follow-up when most patients were in remission, but remained still elevated as compared with controls at all time points (61). MIF levels did not correlate with CRP, creatinine, or organ involvement. MIF has intriguingly been shown to be induced from macrophages by low concentrations of glucocorticoids (GC), possibly acting as a counter-regulator for glucocorticoid action (62). However, we found no correlation between MIF levels and GC exposure possibly due to the unphysiologically high GC treatment doses at the time of sampling. Finally, since there is a known association between thyroid disease and AAV in addition to antithyroid drugs being associated with the development of ANCA and vasculitis (63, 64), we investigated the role of thyroid hormone activity and MIF in AAV. The thyroid hormone thyroxine (T4) and its dextrorotatory isomer (dextrothyroxine; d-T4), but not triiodothyronine (T3), bind to the hydrophobic pocket within the MIF molecule and has been shown to be a potent inhibitor of the inflammatory activity of MIF in a dose-dependent manner, which was clearly demonstrated by administration of exogenous d-T4 to mice with severe sepsis (65). Administration of the hormonally inactive d-T4 significantly improved survival, even in mice that had previously undergone thyroidectomy. In patients with severe sepsis, low plasma T4 concentrations were inversely correlated with plasma MIF concentrations (65). In our human study, there was a strong correlation over time between the baseline MIF/T4 ratio and the MIF/T4 ratio at 6 months in AAV patients in remission (61). Both the preclinical and clinical data therefore suggest that blocking the inflammatory active site of MIF may both reduce inflammatory responses and improve the availability of T4.
MIF and Autosomal Dominant Polycystic Kidney Disease
Autosomal dominant polycystic kidney disease (ADPKD) is an inherited autosomal dominant disease characterized by renal cyst formation and is associated with renal interstitial inflammation and fibrosis. The most common is the PKD1 mutation that encodes for polycystin-1 (PC1), which frequently leads to loss of kidney function in mid-life (66). It was recently shown that MIF regulates cyst growth in a murine ADPKD model through several mechanisms and is also accumulated in cyst fluid of human ADPKD kidneys (67). In polycystin-1-deficient mice, recruitment and retention of renal macrophages were dependent on MIF, which promoted cyst expansion. By deleting MIF or by pharmacological inhibition, cyst growth was delayed in murine ADPKD models. Macrophage recruitment was associated with the upregulation of monocyte chemotactic protein 1 (MCP-1) and inflammatory cytokine TNF-α, which further induced MIF affecting renal epithelial cells and cyst development (68). These findings may potentially be very important as the therapeutic options in slowing ADPKD progression until recently have been limited. New drugs, such as tolvaptan, a vasopressin V(2)-receptor antagonist, have recently been approved after demonstrating an effect on the increased rate of total kidney volume and slowing down renal function decline. However, the long-term effect of tolvaptan is unclear and side effects may limit its use (68).
MIF and Genetics
Migration inhibitory factor corresponding gene has known polymorphisms in the -794 CATT(5–8) repeat and the single-nucleotide polymorphism (SNP) -173*G/C and is associated with increased susceptibility and severity of a number of inflammatory and autoimmune conditions (69, 70).
The frequency of high-producer MIF -173 G/C genotype was higher (10.1%) in ESRD than in controls (1.2%), suggesting that it may play a role in progression to renal failure. However, there was no clear association between the MIF genotype and type of kidney disease in ESRD (71). In children with idiopathic nephrotic syndrome, the high-producer MIF -173*C allele was significantly more common than in controls. Furthermore, this allele was more common in steroid-resistance cases and was also associated with significantly higher probability of ESRD compared with G/G homozygous patients within 5 years from onset (72). In a recent study, it was shown that patients with GPA have an increased frequency of high-expression MIF CATT, and higher plasma MIF levels. In a murine model of granulomatous vasculitis, higher MIF expression increased mortality and pulmonary granulomas while injection of anti-MIF mAb protected mice from dying suggesting a role for MIF in the pathogenesis of GPA (73).
Conclusion
We have here described the current evidence of MIF being a mediator in a number of diseases and conditions associated with kidney disease (Figure 1). Both MIF and its receptor CD74 may be potential biomarkers in these disorders and possible targets for pharmacological modulation. However, since MIF is also constitutively expressed it may be problematical to interfere with MIF activity in an interventional setting. Also, while we have discussed the possible detrimental effects of MIF, especially those associated with its inflammatory active site, the molecule can also be protective in certain circumstances. In particular, in addition to the inflammatory site, MIF also has an intrinsic thiol protein oxidoreductase activity. We, and others, have shown that this enzyme activity is protective against oxidative injury induced by ischemia reperfusion injury (74, 75).
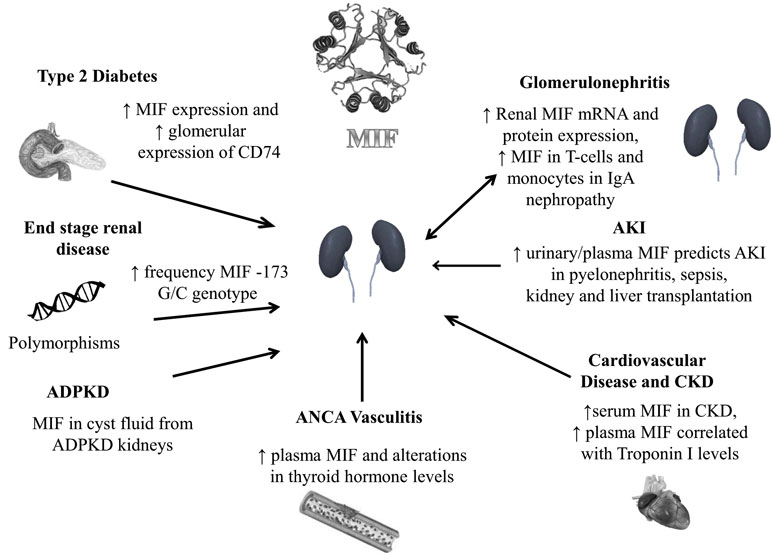
Figure 1. MIF and human kidney disease. Autosomal dominant polycystic kidney disease (ADPKD), acute kidney injury (AKI), chronic kidney disease (CKD).
Recently, Thiele et al. demonstrated that there are two redox-dependent conformational MIF isoforms. Oxidized MIF (oxMIF) is selectively expressed in the plasma and on the cell surface of immune cells of patients with different inflammatory diseases, but not in healthy individuals and is specifically recognized by three monoclonal antibodies (mAbs) directed against MIF. The authors also found a clear correlation between disease severity and the oxMIF/Cr ratio in the urine in patients with acute lupus nephritis, but not in patients with SLE without renal manifestations or in remission. Anti-oxMIF mAbs alleviated disease severity in a rat model of crescentic glomerulonephritis, interestingly with further improvement in synergy with GC (76). However, while there is much evidence, from preclinical studies, for the participatory role of MIF in the pathogenesis if several conditions few clinical trials of anti-MIF agents have been recorded. Anti-MIF mAb therapy is currently in phase I trials both for solid tumors (NCT01765790) and for lupus nephritis (NCT01541670). Whether inhibition of MIF or oxMIF may offer promising therapies in clinical conditions, such as AKI, CKD, diabetic nephropathy, inflammatory kidney diseases, and ADPKD, needs to be elaborated in future interventional studies.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. David JR. Delayed hypersensitivity in vitro: its mediation by cellfree substances formed by lymphoid cell-antigen interaction. Proc Natl Acad Sci U S A (1966) 56:72–7. doi: 10.1073/pnas.56.1.72
2. Bloom BR, Bennett B. Mechanism of a reaction in vitro associated with delayed-type hypersensitivity. Science (1966) 153:80–2. doi:10.1126/science.153.3731.80
3. Bloom BR, Shevach E. Requirement for T cells in the production of migration inhibitory factor. J Exp Med (1975) 142:1306–11. doi:10.1084/jem.142.5.1306
4. Calandra T, Roger T. Macrophage migration inhibitory factor: a regulator of innate immunity. Nat Rev Immunol (2003) 3(10):791–800. doi:10.1038/nri1200
5. Nishihira J, Koyama Y, Mizue Y. Identification of macrophage migration inhibitory factor (MIF) in human vascular endothelial cells and its induction by lipopolysaccharide. Cytokine (1998) 10(3):199–205. doi:10.1006/cyto.1997.0276
6. Bernhagen J, Krohn R, Lue H, Gregory JL, Zernecke A, Koenen RR, et al. MIF is a noncognate ligand of CXC chemokine receptors in inflammatory and atherogenic cell recruitment. Nat Med (2007) 13(5):587–96. doi:10.1038/nm1567
7. Al-Abed Y, Dabideen D, Aljabari B, Valster A, Messmer D, Ochani M, et al. ISO-1 binding to the tautomerase active site of MIF inhibits its pro-inflammatory activity and increases survival in severe sepsis. J Biol Chem (2005) 280:36541–4. doi:10.1074/jbc.C500243200
8. Suzuki M, Sugimoto H, Nakagawa A, Tanaka I, Nishihira J, Sakai M. Crystal structure of the macrophage migration inhibitory factor from rat liver. Nat Struct Biol (1996) 3(3):259–66. doi:10.1038/nsb0396-259
9. Al-Abed Y, VanPatten S. MIF as a disease target: ISO-1 as a proof-of-concept therapeutic. Future Med Chem (2011) 3(1):45–63. doi:10.4155/fmc.10.281
10. Lehmann LE, Novender U, Schroeder S, Pietsch T, von Spiegel T, Putensen C, et al. Plasma levels of macrophage migration inhibitory factor are elevated in patients with severe sepsis. Intensive Care Med (2001) 27(8):1412–5. doi:10.1007/s001340101022
11. Leech M, Metz C, Hall P, Hutchinson P, Gianis K, Smith M, et al. Macrophage migration inhibitory factor in rheumatoid arthritis: evidence of proinflammatory function and regulation by glucocorticoids. Arthritis Rheum (1999) 42(8):1601–8.
12. Foote A, Briganti EM, Kipen Y, Santos L, Leech M, Morand EF. Macrophage migration inhibitory factor in systemic lupus erythematosus. J Rheumatol (2004) 31(2):268–73.
13. Zernecke A, Bernhagen J, Weber C. Macrophage migration inhibitory factor in cardiovascular disease. Circulation (2008) 117(12):1594–602. doi:10.1161/CIRCULATIONAHA.107.729125
14. Burger-Kentischer A, Göbel H, Kleemann R, Zernecke A, Bucala R, Leng L, et al. Reduction of the aortic inflammatory response in spontaneous atherosclerosis by blockade of macrophage migration inhibitory factor (MIF). Atherosclerosis (2006) 184(1):28–38. doi:10.1016/j.atherosclerosis.2005.03.028
15. Schober A, Bernhagen J, Thiele M, Zeiffer U, Knarren S, Roller M, et al. Stabilization of atherosclerotic plaques by blockade of macrophage migration inhibitory factor after vascular injury in apolipoprotein E-deficient mice. Circulation (2004) 109(3):380–5. doi:10.1161/01.CIR.0000109201.72441.09
16. Stenvinkel P, Ketteler M, Johnson RJ, Lindholm B, Pecoits-Filho R, Riella M, et al. IL-10, IL-6, and TNF-alpha: central factors in the altered cytokine network of uremia – the good, the bad, and the ugly. Kidney Int (2005) 67(4):1216–33. doi:10.1111/j.1523-1755.2005.00200.x
17. Go AS, Chertow GM, Fan D, McCulloch CE, Hsu CY. Chronic kidney disease and the risks of death, cardiovascular events, and hospitalization. N Engl J Med (2004) 351(13):1296–305. doi:10.1056/NEJMoa041031
18. Brown FG, Nikolic-Paterson DJ, Hill PA, Isbel NM, Dowling J, Metz CM, et al. Urine macrophage migration inhibitory factor reflects the severity of renal injury in human glomerulonephritis. J Am Soc Nephrol (2002) 13(Suppl 1):S7–13.
19. Hong MY, Tseng CC, Chuang CC, Chen CL, Lin SH, Lin CF. Urinary macrophage migration inhibitory factor serves as a potential biomarker for acute kidney injury in patients with acute pyelonephritis. Mediators Inflamm (2012) 2012:381358. doi:10.1155/2012/381358
20. Brown FG, Nikolic-Paterson DJ, Chadban SJ, Dowling J, Jose M, Metz CN, et al. Urine macrophage migration inhibitory factor concentrations as a diagnostic tool in human renal allograft rejection. Transplantation (2001) 71:1777–83. doi:10.1097/00007890-200106270-00013
21. Payen D, Lukaszewicz AC, Legrand M, Gayat E, Faivre V, Megarbane B, et al. A multicentre study of acute kidney injury in severe sepsis and septic shock: association with inflammatory phenotype and HLA genotype. PLoS One (2012) 7:e35838. doi:10.1371/journal.pone.0035838
22. Takahashi K, Koga K, Linge HM, Zhang Y, Lin X, Metz CN, et al. Macrophage CD74 contributes to MIF-induced pulmonary inflammation. Respir Res (2009) 10:33. doi:10.1186/1465-9921-10-33
23. Lai KN, Leung JC, Metz CN, Lai FM, Bucala R, Lan HY. Role for macrophage migration inhibitory factor in acute respiratory distress syndrome. J Pathol (2003) 199:496–508. doi:10.1002/path.1291
24. Lan HY, Bacher M, Yang N, Mu W, Nikolic-Paterson DJ, Metz C, et al. The pathogenic role of macrophage migration inhibitory factor in immunologically induced kidney disease in the rat. J Exp Med (1997) 185:1455–65. doi:10.1084/jem.185.8.1455
25. Stefaniak J, Schiefer J, Miller EJ, Krenn CG, Baron DM, Faybik P. Macrophage migration inhibitory factor as a potential predictor for requirement of renal replacement therapy after orthotopic liver transplantation. Liver Transpl (2015) 21(5):662–9. doi:10.1002/lt.24103
26. Mills KT, Xu Y, Zhang W, Bundy JD, Chen CS, Kelly TN, et al. A systematic analysis of worldwide population-based data on the global burden of chronic kidney disease in 2010. Kidney Int (2015) 88(5):950–7. doi:10.1038/ki.2015.230
27. Stevens LA, Li S, Wang C, Huang C, Becker BN, Bomback AS, et al. Prevalence of CKD and comorbid illness in elderly patients in the United States: results from the kidney early evaluation program (KEEP). Am J Kidney Dis (2010) 55(3 Suppl 2):S23–33. doi:10.1053/j.ajkd.2009.09.035
28. Stenvinkel P. Chronic kidney disease: a public health priority and harbinger of premature cardiovascular disease. J Intern Med (2010) 268:456–67. doi:10.1111/j.1365-2796.2010.02269.x
29. Kooman JP, Kotanko P, Schols AM, Shiels PG, Stenvinkel P. Chronic kidney disease and premature ageing. Nat Rev Nephrol (2014) 10:732–42. doi:10.1038/nrneph.2014.185
30. Foley RN, Parfrey PS, Sarnak MJ. Clinical epidemiology of cardiovascular disease in chronic renal disease. Am J Kidney Dis (1998) 32(5 Suppl 3):S112–9. doi:10.1053/ajkd.1998.v32.pm9820470
31. Stenvinkel P, Larsson TE. Chronic kidney disease: a clinical model of premature aging. Am J Kidney Dis (2013) 62:339–51. doi:10.1053/j.ajkd.2012.11.051
32. Honda H, Qureshi AR, Heimbürger O, Barany P, Wang K, Pecoits-Filho R, et al. Serum albumin, C-reactive protein, interleukin 6, and fetuin a as predictors of malnutrition, cardiovascular disease, and mortality in patients with ESRD. Am J Kidney Dis (2006) 47:139–48. doi:10.1053/j.ajkd.2005.09.014
33. Meuwese CL, Snaedal S, Halbesma N, Stenvinkel P, Dekker FW, Qureshi AR, et al. Trimestral variations of C-reactive protein, interleukin-6 and tumour necrosis factor-alpha are similarly associated with survival in haemodialysis patients. Nephrol Dial Transplant (2011) 26:1313–8. doi:10.1093/ndt/gfq557
34. Bruchfeld A, Qureshi AR, Lindholm B, Barany P, Yang L, Stenvinkel P. High mobility group box protein-1 correlates with renal function in chronic kidney disease (CKD). Mol Med (2008) 14:109–15. doi:10.2119/2007-00107.Bruchfeld
35. Gupta J, Mitra N, Kanetsky PA, Devaney J, Wing MR, Reilly M, et al. Association between albuminuria, kidney function, and inflammatory biomarker profile in CKD in CRIC. Clin J Am Soc Nephrol (2012) 7:1938–46. doi:10.2215/CJN.03500412
36. Bruchfeld A, Carrero JJ, Qureshi AR, Lindholm B, Barany P, Heimburger O, et al. Elevated serum macrophage migration inhibitory factor (MIF) concentrations in chronic kidney disease (CKD) are associated with markers of oxidative stress and endothelial activation. Mol Med (2009) 15(3–4):70–5. doi:10.2119/molmed.2008.00109
37. Rammos C, Hendgen-Cotta UB, Sobierajski J, Adamczyk S, Hetzel GR, Kleophas W, et al. Macrophage migration inhibitory factor is associated with vascular dysfunction in patients with end-stage renal disease. Int J Cardiol (2013) 168(6):5249–56. doi:10.1016/j.ijcard.2013.08.021
38. Haddad F, Fuh E, Peterson T, Skhiri M, Kudelko KT, De Jesus Perez V, et al. Incidence, correlates, and consequences of acute kidney injury in patients with pulmonary arterial hypertension hospitalized with acute right-side heart failure. J Card Fail (2011) 17(7):533–9. doi:10.1016/j.cardfail.2011.03.003
39. Shah SJ, Thenappan T, Rich S, Tian L, Archer SL, Gomberg-Maitland M. Association of serum creatinine with abnormal hemodynamics and mortality in pulmonary arterial hypertension. Circulation (2008) 117(19):2475–83. doi:10.1161/CIRCULATIONAHA.107.719500
40. Zhang B, Luo Y, Liu ML, Wang J, Xu DQ, Dong MQ, et al. Macrophage migration inhibitory factor contributes to hypoxic pulmonary vasoconstriction in rats. Microvasc Res (2012) 83(2):205–12. doi:10.1016/j.mvr.2011.09.014
42. Ahmed MN, Zhang Y, Codipilly C, Zaghloul N, Patel D, Wolin M, et al. Extracellular superoxide dismutase overexpression can reverse the course of hypoxia-induced pulmonary hypertension. Mol Med (2012) 18(1):38–46. doi:10.2119/molmed.2011.00339
43. Afkarian M, Sachs MC, Kestenbaum B, Hirsch IB, Tuttle KR, Himmelfarb J, et al. Kidney disease and increased mortality risk in type 2 diabetes. J Am Soc Nephrol (2013) 24(2):302–8. doi:10.1681/ASN.2012070718
44. Tuttle KR. Linking metabolism and immunology: diabetic nephropathy is an inflammatory disease. J Am Soc Nephrol (2005) 16(6):1537–8. doi:10.1681/ASN.2005040393
45. Mora C, Navarro JF. Inflammation and diabetic nephropathy. Curr Diab Rep (2006) 6(6):463–8. doi:10.1007/s11892-006-0080-1
46. Herder C, Kolb H, Koenig W, Haastert B, Müller-Scholze S, Rathmann W, et al. Association of systemic concentrations of macrophage migration inhibitory factor with impaired glucose tolerance and type 2 diabetes: results from the cooperative health research in the region of Augsburg, Survey 4 (KORA S4). Diabetes Care (2006) 29(2):368–71. doi:10.2337/diacare.29.02.06.dc05-1474
47. Makino A, Nakamura T, Hirano M, Kitta Y, Sano K, Kobayashi T, et al. High plasma levels of macrophage migration inhibitory factor are associated with adverse long-term outcome in patients with stable coronary artery disease and impaired glucose tolerance or type 2 diabetes mellitus. Atherosclerosis (2010) 213(2):573–8. doi:10.1016/j.atherosclerosis.2010.09.004
48. Schöttker B, Herder C, Rothenbacher D, Roden M, Kolb H, Müller H, et al. Proinflammatory cytokines, adiponectin, and increased risk of primary cardiovascular events in diabetic patients with or without renal dysfunction: results from the ESTHER study. Diabetes Care (2013) 36(6):1703–11. doi:10.2337/dc12-1416
49. Watanabe T, Tomioka NH, Doshi M, Watanabe S, Tsuchiya M, Hosoyamada M. Macrophage migration inhibitory factor is a possible candidate for the induction of microalbuminuria in diabetic db/db mice. Biol Pharm Bull (2013) 36(5):741–7. doi:10.1248/bpb.b12-00741
50. Sanchez-Niño MD, Sanz AB, Ihalmo P, Lassila M, Holthofer H, Mezzano S, et al. The MIF receptor CD74 in diabetic podocyte injury. J Am Soc Nephrol (2009) 20(2):353–62. doi:10.1681/ASN.2008020194
51. Wang Z, Wei M, Wang M, Chen L, Liu H, Ren Y, et al. Inhibition of macrophage migration inhibitory factor reduces diabetic nephropathy in type II diabetes mice. Inflammation (2014) 37(6):2020–9. doi:10.1007/s10753-014-9934-x
52. Lan HY, Mu W, Yang N, Meinhardt A, Nikolic-Paterson DJ, Ng YY, et al. De Novo renal expression of macrophage migration inhibitory factor during the development of rat crescentic glomerulonephritis. Am J Pathol (1996) 149(4):1119–27.
53. Yang N, Nikolic-Paterson DJ, Ng YY, Mu W, Metz C, Bacher M, et al. Reversal of established rat crescentic glomerulonephritis by blockade of macrophage migration inhibitory factor (MIF): potential role of MIF in regulating glucocorticoid production. Mol Med (1998) 4(6):413–24.
54. Lan HY, Yang N, Nikolic-Paterson DJ, Yu XQ, Mu W, Isbel NM, et al. Expression of macrophage migration inhibitory factor in human glomerulonephritis. Kidney Int (2000) 57(2):499–509. doi:10.1046/j.1523-1755.2000.00869.x
55. Berthoux FC, Mohey H, Afiani A. Natural history of primary IgA nephropathy. Semin Nephrol (2008) 28(1):4–9. doi:10.1016/j.semnephrol.2007.10.001
56. Matsumoto K, Kanmatsuse K. Increased production of macrophage migration inhibitory factor by T cells in patients with IgA nephropathy. Am J Nephrol (2001) 21(6):455–64. doi:10.1159/000046649
57. Leung JC, Tang SC, Chan LY, Tsang AW, Lan HY, Lai KN. Polymeric IgA increases the synthesis of macrophage migration inhibitory factor by human mesangial cells in IgA nephropathy. Nephrol Dial Transplant (2003) 18(1):36–45. doi:10.1093/ndt/18.1.36
58. Leung JC, Chan LY, Tsang AW, Liu EW, Lam MF, Tang SC, et al. Anti-macrophage migration inhibitory factor reduces transforming growth factor-beta 1 expression in experimental IgA nephropathy. Nephrol Dial Transplant (2004) 19(8):1976–85. doi:10.1093/ndt/gfh323
59. Jennette JC, Falk RJ. Pathogenesis of antineutrophil cytoplasmic autoantibody-mediated disease. Nat Rev Rheumatol (2014) 10(8):463–73. doi:10.1038/nrrheum.2014.103
60. Becker H, Maaser C, Mickholz E, Dyong A, Domschke W, Gaubitz M. Relationship between serum levels of macrophage migration inhibitory factor and the activity of antineutrophil cytoplasmic antibody-associated vasculitides. Clin Rheumatol (2006) 25(3):368–72. doi:10.1007/s10067-005-0045-9
61. Wendt M, Börjesson O, Avik A, Bratt J, Anderstam B, Qureshi AR, et al. Macrophage migration inhibitory factor (MIF) and thyroid hormone alterations in antineutrophil cytoplasmic antibody (ANCA)-associated vasculitis (AAV). Mol Med (2013) 19:109–14. doi:10.2119/molmed.2012.00352
62. Calandra T, Bernhagen J, Metz CN, Spiegel LA, Bacher M, Donnelly T, et al. MIF as a glucocorticoid-induced modulator of cytokine production. Nature (1995) 377(6544):68–71. doi:10.1038/377068a0
63. Lionaki S, Hogan SL, Falk RJ, Joy MS, Chin H, Jennette CE, et al. Association between thyroid disease and its treatment with ANCA small-vessel vasculitis: a case-control study. Nephrol Dial Transplant (2007) 22(12):3508–15. doi:10.1093/ndt/gfm493
64. Slot MC, Links TP, Stegeman CA, Tervaert JW. Occurrence of antineutrophil cytoplasmic antibodies and associated vasculitis in patients with hyperthyroidism treated with antithyroid drugs: a long-term follow-up study. Arthritis Rheum (2005) 53(1):108–13. doi:10.1002/art.20927
65. Al-Abed Y, Metz CN, Cheng KF, Aljabari B, VanPatten S, Blau S. Thyroxine is a potential endogenous antagonist of macrophage migration inhibitory factor (MIF) activity. Proc Natl Acad Sci U S A (2011) 108(20):8224–7. doi:10.1073/pnas.1017624108
66. Cornec-Le Gall E, Audrézet MP, Chen JM, Hourmant M, Morin MP, Perrichot R, et al. Type of PKD1 mutation influences renal outcome in ADPKD. J Am Soc Nephrol (2013) 24(6):1006–13. doi:10.1681/ASN.2012070650
67. Chen L, Zhou X, Fan LX, Yao Y, Swenson-Fields KI, Gadjeva M, et al. Macrophage migration inhibitory factor promotes cyst growth in polycystic kidney disease. J Clin Invest (2015) 125(6):2399–412. doi:10.1172/JCI80467
68. Torres VE, Chapman AB, Devuyst O, Gansevoort RT, Grantham JJ, Higashihara E, et al. Tolvaptan in patients with autosomal dominant polycystic kidney disease. N Engl J Med (2012) 367(25):2407–18. doi:10.1056/NEJMoa1205511
69. Renner P, Roger T, Calandra T. Macrophage migration inhibitory factor: gene polymorphisms and susceptibility to inflammatory diseases. Clin Infect Dis (2005) 41:513–9. doi:10.1086/432009
70. Gao L, Flores C, Fan-Ma S, Miller EJ, Moitra J, Moreno L, et al. Macrophage migration inhibitory factor in acute lung injury: expression, biomarker, and associations. Transl Res (2007) 150(1):18–29. doi:10.1016/j.trsl.2007.02.007
71. Tripathi G, Borkar M, Akhter A, Sankhwar SN, Sharma RK, Agrawal S. Association of proinflammatory cytokines with end stage renal disease. Cytokine (2010) 50(3):278–83. doi:10.1016/j.cyto.2010.02.019
72. Vivarelli M, D’Urbano LE, Stringini G, Ghiggeri GM, Caridi G, Donn R, et al. Association of the macrophage migration inhibitory factor -173*C allele with childhood nephrotic syndrome. Pediatr Nephrol (2008) 23(5):743–8. doi:10.1007/s00467-007-0729-2
73. Sreih AG, Ezzeddine R, Fan J, Leng L, Carette S, Cuthbertson D, et al. The Role of Macrophage Migration Inhibitory Factor (MIF) and MIF Gene Polymorphisms in the Pathogenesis of Granulomatosis with Polyangiitis. Abstract. Boston: American College of Rheumatology (2014).
74. Koga K, Kenessey A, Powell SR, Sison CP, Miller EJ, Ojamaa K. Macrophage migration inhibitory factor provides cardioprotection during ischemia/reperfusion by reducing oxidative stress. Antioxid Redox Signal (2011) 14(7):1191–202. doi:10.1089/ars.2010.3163
75. Miller EJ, Li J, Leng L, McDonald C, Atsumi T, Bucala R, et al. Macrophage migration inhibitory factor stimulates AMP-activated protein kinase in the ischaemic heart. Nature (2008) 451(7178):578–82. doi:10.1038/nature06504
Keywords: MIF, AKI, CKD, glomerulonephritis, vasculitis, MIF gene polymorphism, diabetic nephropathy, ADPKD
Citation: Bruchfeld A, Wendt M and Miller EJ (2016) Macrophage Migration Inhibitory Factor in Clinical Kidney Disease. Front. Immunol. 7:8. doi: 10.3389/fimmu.2016.00008
Received: 03 August 2015; Accepted: 11 January 2016;
Published: 26 January 2016
Edited by:
Heidi Noels, Institute for Molecular Cardiovascular Research, GermanyReviewed by:
Alberto Ortiz, Instituto de Investigación Sanitaria de la Fundación Jiménez Díaz, SpainXiaogang Li, The University of Kansas Medical Center, USA
Copyright: © 2016 Bruchfeld, Wendt and Miller. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Annette Bruchfeld, YW5uZXR0ZS5icnVjaGZlbGRAa2kuc2U=