 Carlos I. Falcon-Rodriguez
Carlos I. Falcon-Rodriguez Alvaro R. Osornio-Vargas
Alvaro R. Osornio-Vargas Isabel Sada-Ovalle
Isabel Sada-Ovalle Patricia Segura-Medina
Patricia Segura-Medina- 1Posgrado en Ciencias Biológicas, Universidad Nacional Autónoma de México, Mexico City, Mexico
- 2Departamento de Investigación en Hiperreactividad Bronquial, Instituto Nacional de Enfermedades Respiratorias, Mexico City, Mexico
- 3Department of Pediatrics, University of Alberta, Edmonton, AB, Canada
- 4Laboratorio de Inmunologia Integrativa, Instituto Nacional de Enfermedades Respiratorias, Mexico City, Mexico
Urban air pollution is a serious worldwide problem due to its impact on human health. In the past 60 years, growing evidence established a correlation between exposure to air pollutants and the developing of severe respiratory diseases. Recently particulate matter (PM) is drawing more public attention to various aspects including historical backgrounds, physicochemical characteristics, and its pathological role. Therefore, this review is focused on these aspects. The most famous air pollution disaster happened in London on December 1952; it has been calculated that more than 4,000 deaths occurred during this event. Air pollution is a complex mix of gases and particles. Gaseous pollutants disseminate deeply into the alveoli, allowing its diffusion through the blood–air barrier to several organs. Meanwhile, PM is a mix of solid or liquid particles suspended in the air. PM is deposited at different levels of the respiratory tract, depending on its size: coarse particles (PM10) in upper airways and fine particles (PM2.5) can be accumulated in the lung parenchyma, inducing several respiratory diseases. Additionally to size, the composition of PM has been associated with different toxicological outcomes on clinical and epidemiological, as well as in vivo and in vitro animal and human studies. PM can be constituted by organic, inorganic, and biological compounds. All these compounds are capable of modifying several biological activities, including alterations in cytokine production, coagulation factors balance, pulmonary function, respiratory symptoms, and cardiac function. It can also generate different modifications during its passage through the airways, like inflammatory cells recruitment, with the release of cytokines and reactive oxygen species (ROS). These inflammatory mediators can activate different pathways, such as MAP kinases, NF-κB, and Stat-1, or induce DNA adducts. All these alterations can mediate obstructive or restrictive respiratory diseases like asthma, COPD, pulmonary fibrosis, and even cancer. In 2013, outdoor air pollution was classified as Group 1 by IARC based on all research studies data about air pollution effects. Therefore, it is important to understand how PM composition can generate several pulmonary pathologies.
Introduction
Urban air pollution is a serious problem around the world. Since the Industrial Revolution, the growing use of fuels, electricity demand, and mining activities have been the primary drivers of atmospheric pollution. It was not until after historic high air pollution events that scientists began the study of its impact on health. Three well-documented air pollution episodes occurred in the twentieth century: “The great air pollution disasters” or “The historic pollution episodes.” The first occurred in the Meuse river valley in eastern Belgium. The valley hosted a massive industrial zone with a diverse set of air pollution sources. On December 1930, a combination of low temperature, fog, and low wind speed resulted in the lack of air dispersion, and the consequence was seen as a large accumulation of gaseous and particulate air pollutants in the valley. In 2 days, 6,000 cases of unexpected deaths were observed, mainly impacting the elderly and individuals with preexisting heart and lung diseases (1). The second incident occurred on October 1948, in Donora, PA, USA. The heavily industrialized Monongahela River valley used soft coal as the main fuel. The episode began with persistent cool air and heavy fog and had the sharply irritating pungent odor of sulfur dioxide. While 1 to 2 deaths were expected during the time of the event, an astonishing 18–20 excess in deaths was attributed to the episode (1).
The third episode represented the most severe air pollution disaster and occurred in London during 4 days in December 1952. In the Thames Valley, the meteorological conditions were unusually intense, with cold, stagnant air, dense fog, and a rapid buildup of soot-filled air as a result of a thermal inversion. Approximately 4,000 deaths occurred during that period (2). The cause of death included pneumonia, bronchitis, and heart diseases. Prior to the episode, particle levels averaged a substantial equivalent of 500 μg/m3 of air, and sulfur dioxide levels averaged 0.15 ppm. During the episode, accumulated particles levels arose to 4,500 μg/m3, and sulfur dioxide level reached a substantial 1.3 ppm. The British Smoke Shade methods were used to estimate particles levels based on the dark color of the filter sampler (1). After three events, the scientists started studying the different pathologies or damage caused by air pollution (1). The preexistence of cardiopulmonary disease in individuals aged ≥45 years and during infancy, it was an important condition in 80% of the deaths (1).
In general terms, air pollution is made up of gases and particulate matter (PM). Especially, PM can be transported into the alveoli, depending on its size (3). In recent years, some researchers have reported that air pollution can produce cancer. Lately, outdoor air pollution was classified by the IARC as a group I carcinogen or proven carcinogen for humans (4). However, exposure to PM also produces several other diseases in the respiratory system. Therefore, it is important to establish how the composition of PM can induce several pulmonary pathologies.
Particulate Matter
Particulate matter is defined as solid and/or liquid suspended in the atmosphere, also named aerosol (5). It is generated chiefly through two processes, natural and anthropogenic. The natural process includes phenomena that take place on the earth, such as, sea sprays (6), volcanic eruptions, spontaneous forest fires, and soil erosion (7). The second process involves emissions to the atmosphere, mainly from traffic, other forms of transportation, and industrial sources, such as electricity generation, mining, welding, and building (8). In general, any form of fuel burning, for instance, wood, gas (9), oil-derived diesel, and gasoline (10), generates PM.
Many different types of particles can be found in the atmosphere. If particles are emitted directly to the atmosphere, they will be named primary PM (11, 12), but if they are formed in the atmosphere by gas-to-particle conversion processes, they will be named secondary particles (6). Mineral dust, metals, soot, salt particles, pollen, and spores constitute primary aerosols. On the other hand, secondary aerosols are formed by gases such as sulfates, nitrates, and organic compounds (5). These processes follow three steps that can increase particle size or modify its composition. Nucleation-mode is the first step in new particles generation (13) and depends on gases concentration, humidity, and temperature in the atmosphere (11), and transition of the gaseous phase to liquid or solid phase by condensation or chemical reaction, forming the first nuclei or particles in the atmosphere (5).
The second step is a condensation of hot gases, originating primary aerosols. This event is similar to nucleation (11). The final step in the aerosol formation is coagulation. Whole aerosols formed in previous steps can begin to agglomerate by Brownian motion (14) or turbulence and contact between particles (1). Consequently, particles grow in aerodynamic size (15) forming secondary particles from primary particles (Figure 1).
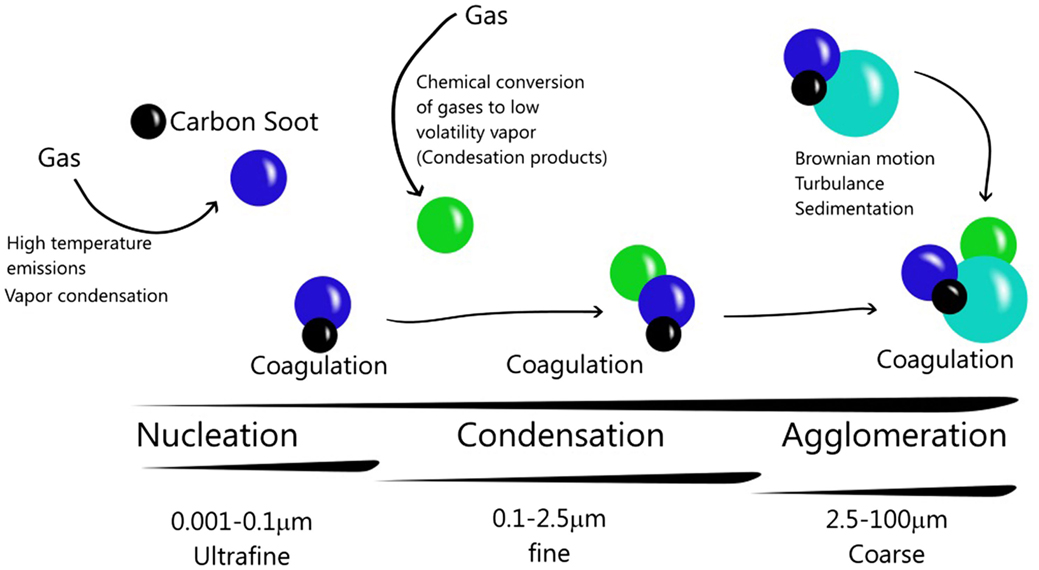
Figure 1. Particulate matter and its atmospheric dynamics. Particles nucleation is generated by gases emission. Condensation can occur by cooling, producing particles. The interaction between primary particles and secondary particles constitute the coagulation. In this way, the particles can increase their size and composition.
Composition and Size
There are different types of PM depending on its source and its composition, e.g., Diesel exhausts particles (DEP), residual oil fly ash (ROFA), and Utah Valley urban air particles (UAP). The first one is produced by diesel combustion and is constituted by transitional metals, such as vanadium and zinc (16), and also by Polycyclic Aromatic Hydrocarbons (PAHs). ROFA is a complex mixture of sulfates and nitrogen compounds, carbon and metals (primarily vanadium) (17). UAP were identified in the United State of America, but became a general denomination for any UAP around the world, due to its high transitional metal levels (18).
Particulate matter is commonly formed by different compounds (Table 1). PM composition can be different among cities, depending on the predominant emission sources. Another important characteristic of particles is the size that depends on the emission sources such as primary aerosol and atmospheric dynamic as a secondary aerosol, already described. The size plays an important role in human airways because it will define the deposition site in the lung. The deposition of aerosol in the human lung occurs through a combination of inertial impaction, gravitational sedimentation, and Brownian diffusion (19). In the atmosphere, different PM sizes can be found, such as the coarse fraction (PM10–2.5) that can penetrate into the upper airways (20) and is deposited through an impaction or sedimentation process (19). The fine fraction (PM2.5) is deposited in the lung, especially in the alveoli (20) through sedimentation and Brownian diffusion processes (19), although it could pass to the systemic circulation (20). PM1 is deposited mainly by Brownian diffusion in the lung (19), but these particles can be translocated from sites in the lung through systemic circulation (21) to the liver, spleen, heart (22), or brain (23). However, they can also arrive to the brain through the olfactory bulb by a trans-synapsis mechanism (23) (Figure 2).

Table 1. The composition of particles.
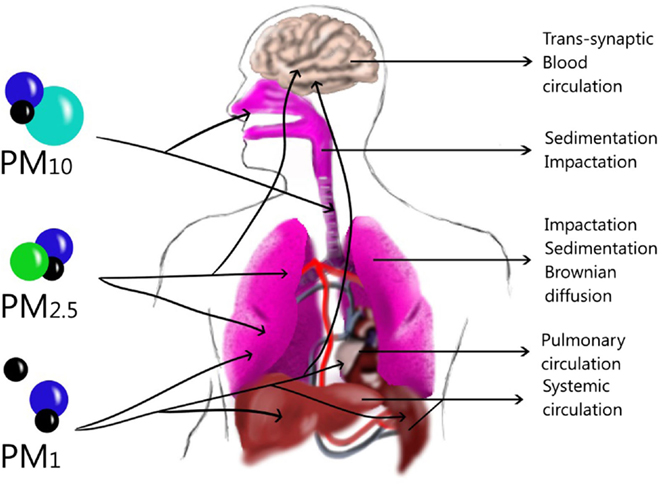
Figure 2. Size and Dynamic of particles in the lung and other tissues. Large particles can be deposited in upper airways through sedimentation or impaction while in the lower airways Brownian diffusion can deposit them in the alveoli. Ultrafine particles can translocate to blood-circulating and be deposited in the liver, spleen or brain, although they might also penetrate through trans-synaptic mechanisms.
Specific Composition and Damage
The complex mix of PM can produce different changes in the tissues, depending on its composition, which includes a water-soluble or a water-insoluble fraction (31). The water-soluble fraction can produce cell signaling, expression of inflammatory mediators, oxidative stress (32) that generates DNA damage via a transition metal-dependent OH formation, implicating an important role of H2O2 (33). In vitro experiments in BEAS-2B have demonstrated that oxidant generation and the concentration of inflammatory cytokine were higher in an exposure with the water-soluble fraction than in an exposure with the insoluble fraction; the production of IL-8 by the former cells increased as well. Likewise, in vivo intratracheal instillation of both water-soluble and insoluble fractions in rats also increased neutrophil incursion and lavage protein concentrations. However, both neutrophil and protein elevation were greater after the exposure to water-soluble fraction (32). Furthermore, water-soluble and insoluble organic aerosols substantially contribute to the oxidative properties of ambient PM (34).
Some researchers have applied statistical methods to demonstrate that certain elements of the particles can produce specific changes. They have used a multivariate technique that analyzes the inter-correlated quantitative dependent variables, called the Principal Component Analysis (PCA) (35). This analysis has demonstrated a strong relationship between IL-6/TNF-α secretion with the presence of Cu and Zn from anthropogenic sources in Mexicali (36). Another study, using BEAS-2B cells exposed to France’s PM from a different season (2008/2009) indicated that inorganic elements and ions were rather related to early oxidative events, whereas PAHs were rather related to later oxidative damage and cytokine secretion such as IL-8 (27). The exposure also produced ROS after PAHs metabolism through the cytochrome P450 activation (37). Some chemical components are preferentially associated with these early oxidative events. Other metals and PAHs have also been associated to oxidative damage and/or cytokine secretion (27).
Respiratory Effects
Particulate matter is easily deposited on bifurcations or angle ramifications of the bronchial tree due to air flow and turbulence, increasing PM interaction with the mucous membrane through an impact process (38). Once deposited on a particular region in the lung, it can penetrate or be absorbed by the mucous layer, generating local damage (39). Many researchers have done in vitro experiments with DEP or carbon black. They have analyzed the effects of particle accumulation in macrophages and their phagocytic capacity (40). PM can produce damage to the whole respiratory apparatus, increasing cellular permeability and reducing the mucocilliary activity by ROS production and cytokine releases. Since 1980, many reports have mentioned that exposure to PM increases cancer and deaths. It is well known that exposure to PM cause pulmonary diseases such as COPD, asthma, and fibrosis (41).
Physiological Alterations
Pulmonary function assessment is the main non-invasive procedure to evaluate respiratory health, identifying ventilation alterations, such as restrictive or obstructive pathologies. For example, FEV1 assesses the volume of forced exhalation in the first second. Another parameter is the forced vital capacity (FVC) that evaluates the amount of air exhaled during the FEV assay. Jie et al. (42) evaluated FVC and FEV on individuals during cooking with coal and non-coal fuels, as well as PM indoor concentration in the kitchen and living room. Despite the absence of outdoor particles, their results showed a significant increase in the relative concentration of PM2.5 in the indoor. The coal smoke was associated with 31.7% decrease in FVC, and 42.0% decrease in FEV1. They conclude that in the kitchen, the fine particles’ relative concentration produces a significant effect on the FVC and FEV1. On the other hand, children exposure to PM10 causes the loss of 23 ml in the FEV1 test for every 1 μg/m3 increment of PM10 in the atmosphere (43). The same finding was observed in women; a reduction of 5.1% in FEV1 per each 7 μg/m3 increment of PM10 (44) was found. In this regard, Forbes et al. (45) mentioned that FEV1 is associated with increased outdoor PM10 concentration. In their analysis, they mentioned that increases in 3 μg/m3 PM10 was associated with a loss of 28 ml in FEV1, mainly in men, elderly people, and ex-smokers.
Also, a Swiss study (SAPALDIA) aimed to evaluate respiratory health in the adult population and the potential association between long-term exposure to air pollution and respiratory health, concluded that PM10 reduced 3.4% the FVC per each increment of 10 μg/m3 of PM10 (46). Even more, patients with a diagnosis of COPD or asthma had a major reduction of the FEV1 when exposed to fine and ultrafine particles (47). Animal models have also been used to evaluate physiological parameters after PM exposures. Barometric plethysmography can also measure respiratory frequency and tidal volume. Exposure to PM increases both parameters (66–103 bpm and 97–190 ml, respectively) in rats (48). Another parameter, Penh (enhanced pause), has been used as well in plethysmographic studies (49). In the murine model, after intranasal instillation of PM1640 (standard reference material), the Penh index increased in a dose-response manner (50). The respiratory physiological changes could be generated by several mechanisms, like the release of molecular mediators that affect the cells, tissue, or systems after the PM exposure.
Immune Response
Perivascular and peribronchiolar inflammation increases after DEP exposure (51). Titanium dioxide nanoparticles (TiO2) rise the number of neutrophils and macrophages recruited in the bronchoalveolar lavage fluid (BALF) (52). In healthy humans, the experimental exposure to DEP (300 μg/m3/h) increased the percentage of inflammatory cells (neutrophils), B and T lymphocytes, and mast cells in the lungs (17, 53). Intratracheal instillation (3.3 mg/kg) of Mexico City particles (PM2.5 and PM10) in a rat model increased the number of inflammatory cells in the lungs (54). This concentration induced lymphocytosis, but higher concentration, such as 5 mg/m3 leads to lymphopenia (20). Some inflammatory proteins, such as IL-1 (55), IL-17 (56), IL-6, IL-8, and TNF-α, increase (57). Exposure to DEP or UAP also increased the same cytokines (58) in in vitro experimental models (59). Also, other cytokines as MIP-1 (2), GM-CSF, IL-1, 2, 4, 5, 10 (60), and IL-13 (17), are affected. Especially, exposure to DEP increases IL-4 (17) and IgE (61). This inflammation process permits diseases development through cytokines activation.
Oxidative Stress
Exposure to fine or ultrafine particles induces ROS-mediated oxidative stress, altering cellular permeability in epithelial cells (62) due to their organic or inorganic content (63). A primary form of ROS is the hydroxyl radical formed by hydrogen peroxide after exposure to PM (64). Also, PM2.5 can produce superoxide leading to the formation of hydrogen peroxide (65). H2O2 is a main free radical in the lung; it can produce cell damage by oxidant stress. Alveolar macrophages and epithelial cells generate oxidants (18). Exposure to Mexico City’s PM changed mRNA expression of several markers of oxidative stress in an in vivo model. Especially, PM10 from the industrial and residential zone induced a significant 3.2-fold and 3.9-fold increase, respectively, as well as the stress-inducible protein HO-1. PM from Industrial area led to a significant 2.5-fold increase in the receptor for oxidized lipoproteins LOX-1 24 h after exposure (54). ROS function as intermediary signaling molecules and can activate the Tyrosine-kinase receptor, MAP kinase, NF-κB, and Stat-1 (66). These signaling pathways activate the transcription and gene expression of molecules related to inflammation, such as TNF-α and IL-1β (67), fibrosis, and apoptosis (18, 66). Oxidative stress can produce damage to DNA inside the lungs, as a consequence, cells die (62). In subjects with or without COPD or lung fibrosis, oxidative stress can regulate the NF-κB transcription, stimulating inflammatory cytokine synthesis (68, 69); exposure to PM can also increase exacerbations and the lung damage.
Asthma
Asthma is a major health issue around the world, which is characterized by airway hyperresponsiveness (70), obstruction (71), and chronic inflammation (72). Another important aspect of this condition is the reversibility of airway obstruction, either spontaneously or following treatment (73). In the asthmatics patients, their pulmonary cells respond to an allergen, producing a Th2 response, including IL-4 and IL-13 (mucous metaplasia), which are required to initiate this response and drive allergen-specific IgE synthesis by B cells; IL-3, which drives basophil development. IL-3, IL-5, and GM-CSF can regulate eosinophil recruitment (74). Also, other interleukins such as IL-17 promote the neutrophilic reaction (75). All the proteins above maintain the asthmatic process in the airways.
Some reports found that the exposure to DEP activates Th2 response and leads to the production of IL-17A (75) and mucous in the bronchiolar epithelium. On the other hand, UAP exposure leads to high level of IgG (animal models) or IgE (humans) production (76) arising the asthma attack (77). Acute exposures to PM can activate the Th2 response, inhibiting INF-γ production (78) and promoting an asthmatic condition. Probably, the chronic exposure enhances Th1 response by activation of IL-12 (79), and in turn trigger to INF-γ (80). Th1 response strongly suppresses the Th2 response (79).
In the asthma animal model, it has been described that exposure to PM increases the asthmatic process, indicating that particles function as an adjuvant in the generation of de novo asthma in mice (76). This characteristic depends on the exposure site because the particles include different components that differ from site to site or city to city (57). PM could be similar to aluminum hydroxide, an indispensable adjuvant in vaccines or the asthma animal model. Both adjuvants, as well as PM, will produce oxidative stress by increasing the production of reactive species oxygen (ROS), IL-1, IL-8, and maturation of B lymphocytes (81). The second mechanism could be related to Th2 response in the lung and also to the production of inflammatory mediators such as IgE or IgG (82).
Chronic Obstructive Pulmonary Disease
Chronic obstructive pulmonary disease (COPD) is a chronic progressive disease that is characterized by an airflow limitation (83), that can be observed in FEV1 or FVC and peak expiratory flow (PEF) (84). This obstruction is not reversible like in asthma. Exacerbation of COPD could be caused by bacteria, viruses (84), cigarette smoking (85), and exposure to indoor and outdoor air pollution (86). It is characterized by chronic inflammation of the airways and lung parenchyma, especially of neutrophils, activated macrophages, and lymphocytes (87). The patients present increased levels of IL-6, TNF-α, and IL-1β (88). A close correlation between high levels of air pollution and COPD clinical manifestation has been described. Patients with a diagnosis of COPD are more susceptible to urban particles, particularly elderly women and patients with severe COPD (89). Chen et al. (90) found a correlation between PM levels (PM2.5 and PM10–2.5) and increased COPD hospitalizations in Vancouver.
In patients with stable COPD showed an increase of proinflammatory mediators such as IL-6, IL-1β, TNF-α, and IL-8, produced by macrophages or epithelial cells (2). IL-6 increased in the systemic circulation in patients during exacerbations, whereas TNF-α and IL-1β was associated with a muscular mass decrease (88). A hypothetical response after PM10 exposure might be that particles produce NF-κB activation, and therefore, increase inflammation and exacerbations in patients with COPD (91). Transcription of TNF-α and IL-1β genes are known to be regulated by NF-κB (67).
Pulmonary Fibrosis
Pulmonary fibrosis is a restrictive disease that presents an irreversible decrement of the vital capacity (63). Furthermore, some cells are implicated such as fibroblast, myofibroblast (92), and macrophage (93), which produce an excess of extracellular matrix components (94) and the pulmonary remodeling like an irreversible distortion of the lung’s architecture (95). In this event, the deposit of collagen fibers is stimulated by TGF-β (96), a potent mediator of fibrogenesis (97). Furthermore, this response is also involved in the production of IL-4 and IL-13 (98). Exposure to ambient particles could lead to pulmonary fibrosis (99), especially the exposure to elements or chemicals such as Al, Si, carbon black, TiO2, silicon oxide, talcum powder, asbestos, and other fibers can cause epithelial damage and rise the levels of IL-2 (100) and IL-8 (66).
PM10 collected in Mexico City can induce increases in the PDGF, a potent mitogen and chemotactic factor for interstitial cells (101), and together with NF-κB, are indispensable in survival factors that inhibits apoptosis and promotes proliferation (102); it also provokes myofibroblast differentiation (66) and production of collagen fibers in the lung (101). Another factor is the proteases activity (20) that increases in airway epithelial A549 cells exposed to PM10 of Mexico city. It was demonstrated an increase in protease activity, especially of MMP-2 and MMP-9 and a decrease in E-cadherin and β-catenin expression (103).
During 5 days, the exposure to TiO2 produced a small increase in procollagen in rat tracheal explants. After 7 days, fine TiO2 significantly increased PDGF-β, TGF-α, or TGF-β levels, compared with animals exposed to ultrafine TiO2; both particle samples produced similar increments of PDGF-A (104). These changes could be associated with oxidative stress, which generates inflammatory infiltrate (99). PM generate ROS, which produces a proinflammatory activity and cytotoxic effects. Proinflammatory effects are mediated by the MAP kinase and NF-κB cascades that are responsible for the expression of cytokines, chemokines, and adhesion molecules (105). In the bleomycin animal model, the exposure to carbon black nanoparticles increased neutrophils and macrophages in the lung. However, lymphocytes or eosinophils do not increase (106), and pulmonary fibrosis worsened in the mouse model (107). Intratracheal instillation of PM increases fibrosis (108) and its fibrogenic mediators such as KC, IL-6, CCL2, and TGF-β1 (109). Metals represents the major source of ROS in PM, which can activate mitogen proteins or nuclear regulating as; nuclear factors such as AP-1 and NFAT (110). However, other elements of PM also produce proteins activation by ROS, the exposures to particles worsen damage in patients with pulmonary fibrosis.
Cancer
Recently, outdoor air pollution was classified as a group I carcinogen by the International Agency for Research on Cancer (4). Particles contain two major components that produce oxidant stress, PAHs, and metals. Both are strong mutagenic and carcinogenesis agents (111), and it has been associated with markers of genetic damage, which may increase the frequency of human cancer (112). Especially the cancers of the trachea, bronchus, or lung represented approximately 7% of total mortality attributable to PM2.5 in 2010 (113). Some studies showed that lung cancer among non-smokers can result by exposure to PM (114), however, the risk of developing lung cancer is higher in smokers (115).
Exposure to UAP or DEP can induce DNA single-strand break, producing a nucleoside, 8 hydroxyguanine, or 8-oxo-7,8-dihydro-2-o-desoxyguanosine (116), that is a predominant product of free radicals, which causes DNA adducts by oxidation (117). Formation of adducts is generated by PAHs metabolism of CYP1A1 and GSTM1 (116). Some reports have mentioned that in vivo exposure to PAHs increases the rate of chromosome aberration, and micronuclei in lymphocytes. In this regard, sister chromatide exchange has been found in lymphocytes of both policemen and drivers in China (112). Furthermore, there is evidence that PAHs can generate deletion (p) of an arm on chromosomes, as well as K-ras and P53 mutations (116), the principal oncogene and tumor suppressor, respectively.
Conclusion
Our ambient plays an important role in the composition and size of the particles; this dynamic process is the main factor to produce specific damage along the airways. In this way, the exposure can increase inflammatory factors and cellular recruitment in the lung, which promotes physiology alterations, resulting in pulmonary diseases as COPD and asthma. Likewise, PM can activate other cellular mediators that produce pulmonary fibrosis. Acute exposure to PM can activate Th2 immune responses; however, chronic exposure changes this profile by activation of Th1, and it triggers pro-fibrotic cytokines as well. The PM components are associated with different damage in accordance with elements such as water-soluble or insoluble fraction. However, all components presents in the PM, form a final complex mixture that will produce or activate inflammatory processes, damage or ROS in the lung. All this changes harm the epithelium, increasing epithelial permeability. In patients with pulmonary diseases, the exposure to PM increases the changes and the lung damage (Figure 3).
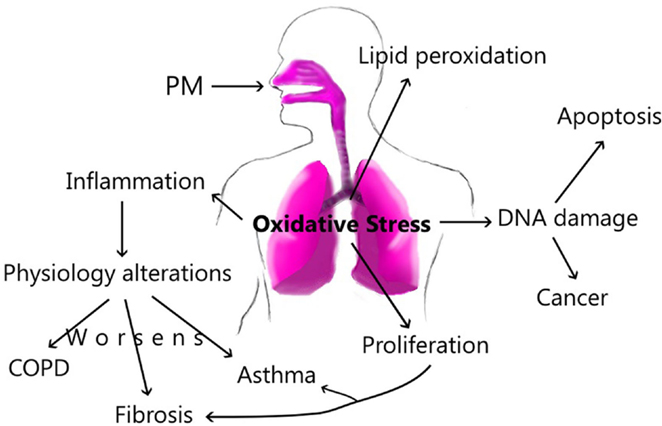
Figure 3. The principal route of damage after PM exposure.
Author Contributions
CF-R and PS-M: responsible for the general design and supervision of the review, writing of the manuscript, and final approval of the version to be published. AO-V and IS-O: writing of the manuscript and final approval of the version to be published.
Conflict of Interest Statement
Authors have no conflict of interests of any kind to declare about this manuscript. The manuscript was produced or written in the absence of any commercial or financial relationships that could create a potential conflict of interest.
The Review Editors Daisuke Kamimura and Hiroki Kimura and the Handling Editor, Masaaki Murakami, declare their shared affiliation, and the Handling Editor states that the review process nevertheless met the standards of a fair and objective review.
References
1. Phalen RF, Phalen RN. Introduction to Air Pollution Science: A Public Health Perspective. Burlington: Jones & Bartlett Learning (2013). p. 89–129.
2. Ling SH, Van Eeden SF. Particulate matter air pollution exposure: role in the development and exacerbation of chronic obstructive pulmonary disease. Int J Chron Obstruct Pulmon Dis (2009) 4:233–43. doi: 10.2147/COPD.S5098
3. Heyder J. Deposition of inhaled particles in the human respiratory tract and consequences for regional targeting in respiratory drug delivery. Proc Am Thorac Soc (2004) 1:315–20. doi:10.1513/pats.200409-046TA
4. IARC. Outdoor Air Pollution a Leading Environmental Cause of Cancer Deaths. Lyon: IARC Press Releases (2013).
5. Curtius J. Nucleation of atmospheric aerosol particles. C R Phys (2006) 7:1027–45. doi:10.1016/j.crhy.2006.10.018
6. Seinfeld JH, Pandis SN. Atmospheric Chemistry and Physics: From Air Pollution to Climate Change. NY: John Wiley & Sons (2012).
7. Vega E, Ruiz H, Escalona S, Cervantes A, Lopez-Veneroni D, Gonzalez-Avalos E, et al. Chemical composition of fine particles in Mexico City during 2003-2004. Atmos Pollut Res (2011) 2:477–83. doi:10.5094/APR.2011.054
8. Pérez-Vidal H, Lunagómez-Rocha M, Acosta-Pérez L. Análisis de partículas suspendidas totales (PST) y partículas fracción respirable (PM10), en Cunduacán. Tabasco: Tabasco Univ Cienc (2010) 26:151–62.
9. Edgerton SA, Bian X, Doran JC, Fast JD, Hubbe JM, Malone EL, et al. Particulate air pollution in Mexico City: a collaborative research project. J Air Waste Manage Assoc (1999) 49:1221–9. doi:10.1080/10473289.1999.10463915
10. Ulrich A, Wichser A, Hess A, Heeb N, Emmenegger L, Czerwinski J, et al. Particle and metal emissions of diesel and gasoline engines – are particle filters appropriate measures? 16th Conference on Combustion Generated Nanoparticles. Zurich: (2012).
11. Raes F, Dingenen RV, Vignati E, Wilson J, Putaud J-P, Seinfeld JH, et al. Formation and cycling of aerosols in the global troposphere. Atmos Environ (2000) 34:4215–40. doi:10.1016/S1352-2310(00)00239-9
12. Whitby KT. The physical characteristics of sulfur aerosols. Atmos Environ (1978) 12:135–59. doi:10.1016/0004-6981(78)90196-8
13. Siingh D, Gautam AS, Kamra AK, Komsaare K. Nucleation events for the formation of charged aerosol particles at a tropical station – preliminary results. Atmos Res (2013) 132–133:239–52. doi:10.1016/j.atmosres.2013.05.024
14. Kim D, Gautam M, Gera D. Parametric studies on the formation of diesel particulate matter via nucleation and coagulation modes. J Aerosol Sci (2002) 33:1609–21. doi:10.1016/S0021-8502(02)00119-2
15. Guo Q, Yang Z, Zhang J. Influence of a combined external field on the agglomeration of inhalable particles from a coal combustion plant. Powder Technol (2012) 227:67–73. doi:10.1016/j.powtec.2011.12.033
16. Osornio-Vargas AR, Bonner JC, Alfaro-Moreno E, Martínez L, García-Cuellar C, Ponce-De-León R, et al. Proinflammatory and cytotoxic effects of Mexico City air pollution particulate matter in vitro are dependent on particle size and composition. Environ Health Perspect (2003) 111:1289–93. doi:10.1289/ehp.5913
17. Huang S-K, Zhang Q, Qiu Z, Chung KF. Mechanistic impact of outdoor air pollution on asthma and allergic diseases. J Thorac Dis (2015) 7:23–33. doi:10.3978/j.issn.2072-1439.2014.12.13
18. Bonner JC. Lung fibrotic responses to particle exposure. Toxicol Pathol (2007) 35:148–53. doi:10.1080/01926230601060009
19. Darquenne C. Aerosol deposition in the human lung in reduced gravity. J Aerosol Med Pulm Drug Deliv (2014) 27:170–7. doi:10.1089/jamp.2013.1079
20. Nemmar A, Holme JA, Rosas I, Schwarze PE, Alfaro-Moreno E. Recent advances in particulate matter and nanoparticle toxicology: a review of the in vivo and in vitro studies. Biomed Res Int (2013) 2013:279371. doi:10.1155/2013/279371
21. Kreyling W, Semmler M, Erbe F, Mayer P, Takenaka S, Schulz H, et al. Translocation of ultrafine insoluble iridium particles from lung epithelium to extrapulmonary organs is size dependent but very low. J Toxicol Environ Health A (2002) 65:1513–30. doi:10.1080/00984100290071649
22. Kreyling WG, Semmler-Behnke M, Moller W. Ultrafine particle-lung interactions: does size matter? J Aerosol Med (2006) 19:74–83. doi:10.1089/jam.2006.19.74
23. Oberdorster G, Sharp Z, Atudorei V, Elder A, Gelein R, Kreyling W, et al. Translocation of inhaled ultrafine particles to the brain. Inhal Toxicol (2004) 16:437–45. doi:10.1080/08958370490439597
24. Steerenberg PA, Van Amelsvoort L, Lovik M, Hetland RB, Alberg T, Halatek T, et al. Relation between sources of particulate air pollution and biological effect parameters in samples from four European cities: an exploratory study. Inhal Toxicol (2006) 18:333–46. doi:10.1080/08958370500515913
25. Schwander S, Okello CD, Freers J, Chow JC, Watson JG, Corry M, et al. Ambient particulate matter air pollution in mpererwe district, kampala, Uganda: a pilot study. J Environ Public Health (2014) 2014:763934. doi:10.1155/2014/763934
26. Garimella S, Deo RN. Neutron activation analysis of atmospheric aerosols from a small Pacific Island Country: a case of Suva, Fiji Islands. Aerosol Air Qual Res (2007) 7:500–17.
27. Dergham M, Lepers C, Verdin A, Cazier F, Billet S, Courcot D, et al. Temporal–spatial variations of the physicochemical characteristics of air pollution particulate matter (PM2.5–0.3) and toxicological effects in human bronchial epithelial cells (BEAS-2B). Environ Res (2015) 137:256–67. doi:10.1016/j.envres.2014.12.015
28. Harrison RM, Yin J. Particulate matter in the atmosphere: which particle properties are important for its effects on health? Sci Total Environ (2000) 249:85–101. doi:10.1016/S0048-9697(99)00513-6
29. Mugica V, Ortiz E, Molina L, De Vizcaya-Ruiz A, Nebot A, Quintana R, et al. PM composition and source reconciliation in Mexico City. Atmos Environ (2009) 43:5068–74. doi:10.1016/j.atmosenv.2009.06.051
30. Calderon-Garciduenas L, Franco-Lira M, Torres-Jardon R, Henriquez-Roldan C, Barragan-Mejia G, Valencia-Salazar G, et al. Pediatric respiratory and systemic effects of chronic air pollution exposure: nose, lung, heart, and brain pathology. Toxicol Pathol (2007) 35:154–62. doi:10.1080/01926230601059985
31. Wang D, Pakbin P, Shafer MM, Antkiewicz D, Schauer JJ, Sioutas C. Macrophage reactive oxygen species activity of water-soluble and water-insoluble fractions of ambient coarse, PM2.5 and ultrafine particulate matter (PM) in Los Angeles. Atmos Environ (2013) 77:301–10. doi:10.1016/j.atmosenv.2013.05.031
32. Ghio AJ. Metals associated with both the water-soluble and insoluble fractions of an ambient air pollution particle catalyze an oxidative stress. Inhal Toxicol (1999) 11:37–49. doi:10.1080/089583799197258
33. Knaapen AM, Shi T, Borm PJ, Schins RP. Soluble metals as well as the insoluble particle fraction are involved in cellular DNA damage induced by particulate matter. Mol Cell Biochem (2002) 234–235:317–26.
34. Verma V, Rico-Martinez R, Kotra N, King L, Liu J, Snell TW, et al. Contribution of water-soluble and insoluble components and their hydrophobic/hydrophilic subfractions to the reactive oxygen species-generating potential of fine ambient aerosols. Environ Sci Technol (2012) 46:11384–92. doi:10.1021/es302484r
35. Abdi H, Williams LJ. Principal Component Analysis. (Vol. 2). NY: Wiley Interdisciplinary Reviews: Computational Statistics (2010). p. 433–59.
36. Osornio-Vargas AR, Serrano J, Rojas-Bracho L, Miranda J, García-Cuellar C, Reyna MA, et al. In vitro biological effects of airborne PM2.5 and PM10 from a semi-desert city on the Mexico–US border. Chemosphere (2011) 83:618–26. doi:10.1016/j.chemosphere.2010.11.073
37. Longhin E, Pezzolato E, Mantecca P, Holme JA, Franzetti A, Camatini M, et al. Season linked responses to fine and quasi-ultrafine Milan PM in cultured cells. Toxicol In Vitro (2013) 27:551–9. doi:10.1016/j.tiv.2012.10.018
38. Shusterman D. The effects of air pollutants and irritants on the upper airway. Proc Am Thorac Soc (2011) 8:101–5. doi:10.1513/pats.201003-027RN
39. Zhang Z, Kleinstreuer C, Kim CS. Effects of curved inlet tubes on air flow and particle deposition in bifurcating lung models. J Biomech (2001) 34:659–69. doi:10.1016/S0021-9290(00)00233-5
40. Mauderly JL. Toxicological and epidemiological evidence for health risks from inhaled engine emissions. Environ Health Perspect (1994) 102:165–71. doi:10.2307/3431948
41. Jones MG, Richeldi L. Air pollution and acute exacerbations of idiopathic pulmonary fibrosis: back to miasma? Eur Respir J (2014) 43:956–9. doi:10.1183/09031936.00204213
42. Jie Y, Houjin H, Xun M, Kebin L, Xuesong Y, Jie X. Relationship between pulmonary function and indoor air pollution from coal combustion among adult residents in an inner-city area of southwest China. Braz J Med Biol Res (2014) 47:982–9. doi:10.1590/1414-431X20144084
43. Mölter A, Agius RM, Vocht F, Lindley A, Gerrard W, Lowe L, et al. Long-term exposure to PM10 and NO2 in association with lung volume and airway resistance in the MAAS birth cohort. Environ Health Perspect (2013) 121:1232–8. doi:10.1289/ehp.1205961
44. Yoshida T, Tuder RM. Pathobiology of cigarette smoke-induced chronic obstructive pulmonary disease. Physiol Rev (2007) 87:1047–82. doi:10.1152/physrev.00048.2006
45. Forbes LJL, Kapetanakis V, Rudnicka AR, Cook DG, Bush T, Stedman JR, et al. Chronic exposure to outdoor air pollution and lung function in adults. Thorax (2009) 64:657–63. doi:10.1136/thx.2008.109389
46. Ackermann-Liebrich U, Leuenberger P, Schwartz J, Schindler C, Monn C, Bolognini G, et al. Lung function and long term exposure to air pollutants in Switzerland. Study on air pollution and lung diseases in adults (SAPALDIA) team. Am J Respir Crit Care Med (1997) 155:122–9. doi:10.1164/ajrccm.155.1.9001300
47. Mccreanor J, Cullinan P, Nieuwenhuijsen MJ, Stewart-Evans J, Malliarou E, Jarup L, et al. Respiratory effects of exposure to diesel traffic in persons with asthma. N Engl J Med (2007) 357:2348–58. doi:10.1056/NEJMoa071535
48. Wichers L, Ledbetter A, Mcgee J, Kellogg R, Rowan W, Nolan J, et al. A method for exposing rodents to resuspended particles using whole-body plethysmography. Part Fibre Toxicol (2006) 3:12. doi:10.1186/1743-8977-3-12
49. Flandre TD, Leroy PL, Desmecht DJM. Effect of somatic growth, strain, and sex on double-chamber plethysmographic respiratory function values in healthy mice. J Appl Physiol (2003) 94:1129–36. doi:10.1152/japplphysiol.00561.2002
50. Archer AJ, Cramton JLH, Pfau JC, Colasurdo G, Holian A. Airway responsiveness after acute exposure to urban particulate matter 1648 in a DO11.10 murine model. Am J Physiol Lung Cell Mol Physiol (2004) 286:L337–43. doi:10.1152/ajplung.00202.2003
51. Song H-M, Jang A-S, Ahn M-H, Takizawa H, Lee S-H, Kwon J-H, et al. Ym1 and Ym2 expression in a mouse model exposed to diesel exhaust particles. Environ Toxicol (2008) 23:110–6. doi:10.1002/tox.20319
52. Bermudez E, Mangum JB, Wong BA, Asgharian B, Hext PM, Warheit DB, et al. Pulmonary responses of mice, rats, and hamsters to subchronic inhalation of ultrafine titanium dioxide particles. Toxicol Sci (2004) 77:347–57. doi:10.1093/toxsci/kfh019
53. Salvi S, Blomberg A, Rudell B, Kelly F, Sandstrom T, Holgate ST, et al. Acute inflammatory responses in the airways and peripheral blood after short-term exposure to diesel exhaust in healthy human volunteers. Am J Respir Crit Care Med (1999) 159:702–9. doi:10.1164/ajrccm.159.3.9709083
54. Snow SJ, De Vizcaya-Ruiz A, Osornio-Vargas A, Thomas RF, Schladweiler MC, Mcgee J, et al. The effect of composition, size, and solubility on acute pulmonary injury in rats following exposure to Mexico City ambient particulate matter samples. J Toxicol Environ Health A (2014) 77:1164–82. doi:10.1080/15287394.2014.917445
55. Steenhof M, Gosens I, Strak M, Godri KJ, Hoek G, Cassee FR, et al. In vitro toxicity of particulate matter (PM) collected at different sites in the Netherlands is associated with PM composition, size fraction and oxidative potential – the RAPTES project. Part Fibre Toxicol (2011) 8:26. doi:10.1186/1743-8977-8-26
56. Van Voorhis M, Knopp S, Julliard W, Fechner JH, Zhang X, Schauer JJ, et al. Exposure to atmospheric particulate matter enhances Th17 polarization through the aryl hydrocarbon receptor. PLoS One (2013) 8:e82545. doi:10.1371/journal.pone.0082545
57. Steerenberg PA, Withagen ET, Van Dalen WJ, Dorma JA, Heisterkamp SH, Van Loveren H, et al. Dose dependency of adjuvant activity of particulate matter from five european sites in three seasons in an ovalbumin–mouse model. Inhal Toxicol (2005) 17:133–45. doi:10.1080/08958370590904490
58. Kafoury RM, Kelley J. Ozone enhances diesel exhaust particles (DEP)-induced interleukin-8 (IL-8) gene expression in human airway epithelial cells through activation of nuclear factors-kappaB (NF-kappaB) and IL-6 (NF-IL6). Int J Environ Res Public Health (2005) 2:403–10. doi:10.3390/ijerph2005030004
59. Manzano-León N, Quintana R, Sánchez B, Serrano J, Vega E, Vázquez-López I, et al. Variation in the composition and in vitro proinflammatory effect of urban particulate matter from different sites. J Biochem Mol Toxicol (2013) 27:87–97. doi:10.1002/jbt.21471
60. Salvi SS, Nordenhall C, Blomberg A, Rudell B, Pourazar J, Kelly FJ, et al. Acute exposure to diesel exhaust increases IL-8 and GRO-alpha production in healthy human airways. Am J Respir Crit Care Med (2000) 161:550–7. doi:10.1164/ajrccm.161.2.9905052
61. Fernvik E, Scharnweber T, Knopp D, Niessner R, Vargaftig BB, Peltre G. Effects of fractions of traffic particulate matter on TH2-cytokines, IgE levels, and bronchial hyperresponsiveness in mice. J Toxicol Environ Health A (2002) 65:1025–45. doi:10.1080/152873902760125200
62. Cachon BF, Firmin S, Verdin A, Ayi-Fanou L, Billet S, Cazier F, et al. Proinflammatory effects and oxidative stress within human bronchial epithelial cells exposed to atmospheric particulate matter (PM2.5 and PM>2.5) collected from Cotonou, Benin. Environ Pollut (2014) 185:340–51. doi:10.1016/j.envpol.2013.10.026
63. Bodlet A, Maury G, Jamart J, Dahlqvist C. Influence of radiological emphysema on lung function test in idiopathic pulmonary fibrosis. Respir Med (2013) 107:1781–8. doi:10.1016/j.rmed.2013.08.039
64. Shi T, Knaapen AM, Begerow J, Birmili W, Borm PJA, Schins RPF. Temporal variation of hydroxyl radical generation and 8-hydroxy-2′-deoxyguanosine formation by coarse and fine particulate matter. Occup Environ Med (2003) 60:315–21. doi:10.1136/oem.60.5.315
65. Dellinger B, Pryor WA, Cueto R, Squadrito GL, Hegde V, Deutsch WA. Role of free radicals in the toxicity of airborne fine particulate matter. Chem Res Toxicol (2001) 14:1371–7. doi:10.1021/tx010050x
66. Byrne JD, Baugh JA. The significance of nanoparticles in particle-induced pulmonary fibrosis. Mcgill J Med (2008) 11:43–50.
67. Lentsch AB, Czermak BJ, Bless NM, Ward PA. NF-kappaB activation during IgG immune complex-induced lung injury: requirements for TNF-alpha and IL-1beta but not complement. Am J Pathol (1998) 152:1327.
68. Lagorio S, Forastiere F, Pistelli R, Iavarone I, Michelozzi P, Fano V, et al. Air pollution and lung function among susceptible adult subjects: a panel study. Environ Health (2006) 5:11. doi:10.1186/1476-069X-5-11
69. García-Diez M, Hoffmann DL, Zilhão J, Heras CDL, Lasheras JA, Montes R, et al. Uranium series dating reveals a long sequence of rock art at Altamira Cave (Santillana del Mar, Cantabria). J Archaeol Sci (2013) 40:4098–106. doi:10.1016/j.jas.2013.05.011
70. Virchow JC, Barnes PJ. Asthma. Semin Respir Crit Care Med (2012) 33:577–8. doi:10.1055/s-0032-1326958
71. Barnes PJ. Immunology of asthma and chronic obstructive pulmonary disease. Nat Rev Immunol (2008) 8:183–92. doi:10.1038/nri2254
72. Keatings VM, Collins PD, Scott DM, Barnes PJ. Differences in interleukin-8 and tumor necrosis factor-alpha in induced sputum from patients with chronic obstructive pulmonary disease or asthma. Am J Respir Crit Care Med (1996) 153:530–4. doi:10.1164/ajrccm.153.2.8564092
73. Nielsen M, Bårnes CB, Ulrik CS. Clinical characteristics of the asthma–COPD overlap syndrome – a systematic review. Int J Chron Obstruct Pulmon Dis (2015) 10:1443–54. doi:10.2147/COPD.S85363
74. Lordan JL, Bucchieri F, Richter A, Konstantinidis A, Holloway JW, Thornber M, et al. Cooperative effects of Th2 cytokines and allergen on normal and asthmatic bronchial epithelial cells. J Immunol (2002) 169:407–14. doi:10.4049/jimmunol.169.1.407
75. Brandt EB, Kovacic MB, Lee GB, Gibson AM, Acciani TH, Le Cras TD, et al. Diesel exhaust particle induction of IL-17A contributes to severe asthma. J Allergy Clin Immunol (2013) 132:1194.e–204.e. doi:10.1016/j.jaci.2013.06.048
76. Ning L, Meiying W, Bramble LA, Schmitz DA, Schauer JJ, Sioutas C, et al. The adjuvant effect of ambient particulate matter is closely reflected by the particulate oxidant potential. Environ Health Perspect (2009) 117:1116–23. doi:10.1289/ehp.0800319
77. Patel MM, Chillrud SN, Deepti KC, Ross JM, Kinney PL. Traffic-related air pollutants and exhaled markers of airway inflammation and oxidative stress in New York City adolescents. Environ Res (2013) 121:71–8. doi:10.1016/j.envres.2012.10.012
78. Jang AS, Choi IS, Takizawa H, Rhim T, Lee JH, Park SW, et al. Additive effect of diesel exhaust particulates and ozone on airway hyperresponsiveness and inflammation in a mouse model of asthma. J Korean Med Sci (2005) 20:759–63. doi:10.3346/jkms.2005.20.5.759
79. Boros DL, Whitfield JR. Enhanced Th1 and dampened Th2 responses synergize to inhibit acute granulomatous and fibrotic responses in murine Schistosomiasis Mansoni. Infect Immun (1999) 67:1187–93.
80. Wang T, Moreno-Vinasco L, Huang Y, Lang G, Linares J, Goonewardena S, et al. Lung responses to ambient particulate matter: genomic analysis and influence on airway hyperresponsiveness. Environ Health Perspect (2008) 116:1500–8. doi:10.1289/ehp.11229
81. Exley C, Siesjö P, Eriksson H. The immunobiology of aluminium adjuvants: how do they really work? Trends Immunol (2010) 31:103–9. doi:10.1016/j.it.2009.12.009
82. Kuroda E, Coban C, Ishii KJ. Particulate adjuvant and innate immunity: past achievements, present findings, and future prospects. Int Rev Immunol (2013) 32:209–20. doi:10.3109/08830185.2013.773326
83. Cazzola M, Macnee W, Martinez FJ, Rabe KF, Franciosi LG, Barnes PJ, et al. Outcomes for COPD pharmacological trials: from lung function to biomarkers. Eur Respir J (2008) 31:416–69. doi:10.1183/09031936.00099306
84. Sapey E, Stockley RA. COPD exacerbations 2: aetiology. Thorax (2006) 61:250–8. doi:10.1136/thx.2005.041822
85. Hodge S, Hodge G, Scicchitano R, Reynolds PN, Holmes M. Alveolar macrophages from subjects with chronic obstructive pulmonary disease are deficient in their ability to phagocytose apoptotic airway epithelial cells. Immunol Cell Biol (2003) 81:289–96. doi:10.1046/j.1440-1711.2003.t01-1-01170.x
86. Mannino DM, Buist AS. Global burden of COPD: risk factors, prevalence, and future trends. Lancet (2007) 370:765–73. doi:10.1016/S0140-6736(07)61380-4
87. Barnes P. Inflammation in COPD. Clin Respir J (2011) 5:1–2. doi:10.1111/j.1752-699X.2011.00265_1.x
88. Barnes P, Celli B. Systemic manifestations and comorbidities of COPD. Eur Respir J (2009) 33:1165–85. doi:10.1183/09031936.00128008
89. Sunyer J, Schwartz J, Tobías A, Macfarlane D, Garcia J, Antó JM. Patients with chronic obstructive pulmonary disease are at increased risk of death associated with urban particle air pollution: a case-crossover analysis. Am J Epidemiol (2000) 151:50–6. doi:10.1093/oxfordjournals.aje.a010121
90. Chen Y, Yang Q, Krewski D, Shi Y, Burnett RT, Mcgrail K. Influence of relatively low level of particulate air pollution on hospitalization for COPD in elderly people. Inhal Toxicol (2004) 16:21–5. doi:10.1080/08958370490258129
91. Macnee W, Donaldson K. Mechanism of lung injury caused by PM10 and ultrafine particles with special reference to COPD. Eur Respir J (2003) 21:47s–51s. doi:10.1183/09031936.03.00403203
92. Phan SH. Genesis of the myofibroblast in lung injury and fibrosis. Proc Am Thorac Soc (2012) 9:148–52. doi:10.1513/pats.201201-011AW
93. Struhar D, Kivity S. Alveolar macrophages in idiopathic pulmonary fibrosis. Am Rev Respir Dis (1985) 131:803–4. doi:10.1164/arrd.1985.131.5.803b
94. Wynn TA. Integrating mechanisms of pulmonary fibrosis. J Exp Med (2011) 208:1339–50. doi:10.1084/jem.20110551
95. Selman M, King TE, Pardo A. Idiopathic pulmonary fibrosis: prevailing and evolving hypotheses about its pathogenesis and implications for therapy. Ann Intern Med (2001) 134:136–51. doi:10.7326/0003-4819-134-2-200101160-00015
96. Tatler AL, Jenkins G. TGF-β activation and lung fibrosis. Proc Am Thorac Soc (2012) 9:130–6. doi:10.1513/pats.201201-003AW
97. Wang Q, Usinger W, Nichols B, Gray J, Xu L, Seeley TW, et al. Cooperative interaction of CTGF and TGF-b in animal models of fibrotic disease. Fibrogenesis Tissue Repair (2011) 4:4. doi:10.1186/1755-1536-4-4
98. Liu T, Jin H, Ullenbruch M, Hu B, Hashimoto N, Moore B, et al. Regulation of found in inflammatory zone 1 expression in bleomycin-induced lung fibrosis: role of IL-4/IL-13 and mediation via STAT-6. J Immunol (2004) 173:3425–31. doi:10.4049/jimmunol.173.5.3425
99. Tanjore H, Blackwell TS, Lawson WE. Emerging evidence for endoplasmic reticulum stress in the pathogenesis of idiopathic pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol (2012) 302:L721–9. doi:10.1152/ajplung.00410.2011
100. Szema AM, Reeder RJ, Harrington AD, Schmidt M, Liu J, Golightly M, et al. Iraq dust is respirable, sharp, and metal-laden and induces lung inflammation with fibrosis in mice via IL-2 upregulation and depletion of regulatory T cells. J Occup Environ Med (2014) 56:243–51. doi:10.1097/JOM.0000000000000119
101. Bonner JC, Rice AB, Lindroos PM, O’Brien PO, Dreher KL, Rosas I, et al. Induction of the lung myofibroblast PDGF receptor system by urban ambient particles from Mexico City. Am J Respir Cell Mol Biol (1998) 19:672–80. doi:10.1165/ajrcmb.19.4.3176
102. Romashkova JA, Makarov SS. NF-[kappa]B is a target of AKT in anti-apoptotic PDGF signalling. Nature (1999) 401:86–90. doi:10.1038/43474
103. Morales-Bárcenas R, Chirino YI, Sánchez-Pérez Y, Osornio-Vargas ÁR, Melendez-Zajgla J, Rosas I, et al. Particulate matter (PM10) induces metalloprotease activity and invasion in airway epithelial cells. Toxicol Lett (2015) 237:167–73. doi:10.1016/j.toxlet.2015.06.001
104. Churg A, Gilks B, Dai J. Induction of fibrogenic mediators by fine and ultrafine titanium dioxide in rat tracheal explants. Am J Physiol (1999) 277:L975–82.
105. Li N, Xia T, Nel AE. The role of oxidative stress in ambient particulate matter-induced lung diseases and its implications in the toxicity of engineered nanoparticles. Free Radic Biol Med (2008) 44:1689–99. doi:10.1016/j.freeradbiomed.2008.01.028
106. Saputra D, Yoon J-H, Park H, Heo Y, Yang H, Lee EJ, et al. Inhalation of carbon black nanoparticles aggravates pulmonary inflammation in mice. Toxicol Res (2014) 30:83–90. doi:10.5487/TR.2014.30.2.083
107. Decologne N, Wettstein G, Kolb M, Margetts P, Garrido C, Camus P, et al. Bleomycin induces pleural and subpleural fibrosis in the presence of carbon particles. Eur Respir J (2010) 35:176–85. doi:10.1183/09031936.00181808
108. Kamata H, Tasaka S, Inoue K-I, Miyamoto K, Nakano Y, Shinoda H, et al. Carbon black nanoparticles enhance bleomycin-induced lung inflammatory and fibrotic changes in mice. Exp Biol Med (2011) 236:315–24. doi:10.1258/ebm.2011.010180
109. Kamata H, Sasatomo T, Ken-Ichiro I, Naoki H, Rina T, Hirohisa T, et al. Carbon black nanoparticle enhances bleomycin-induced inflammatory and fibrotic changes in the lung. A58. Animal Models of Pulmonary Fibrosis. American Thoracic Society (2010). p. A1960–1960.
110. Valko M, Rhodes CJ, Moncol J, Izakovic M, Mazur M. Free radicals, metals and antioxidants in oxidative stress-induced cancer. Chem Biol Interact (2006) 160:1–40. doi:10.1016/j.cbi.2005.12.009
111. De Oliveira Galvão MF, De Melo Cabral T, De André PA, De Fátima Andrade M, De Miranda RM, Saldiva PHN, et al. Cashew nut roasting: chemical characterization of particulate matter and genotocixity analysis. Environ Res (2014) 131:145–52. doi:10.1016/j.envres.2014.03.013
112. Loomis D, Huang W, Chen G. The international agency for research on cancer (IARC) evaluation of the carcinogenicity of outdoor air pollution: focus on China. Chin J Cancer (2014) 33:189. doi:10.5732/cjc.014.10028
113. Hamra GB, Guha N, Cohen A, Laden F, Raaschou-Nielsen O, Samet JM, et al. Outdoor particulate matter exposure and lung cancer: a systematic review and meta-analysis. Environ Health Perspect (2014) 122:906–11. doi:10.1289/ehp/1408092
114. Turner MC, Krewski D, Pope Iii CA, Chen Y, Gapstur SM, Thun MJ. Long-term ambient fine particulate matter air pollution and lung cancer in a large cohort of never-smokers. Am J Respir Crit Care Med (2011) 184:1374–81. doi:10.1164/rccm.201106-1011OC
115. Puett RC, Hart JE, Yanosky JD, Spiegelman D, Wang M, Fisher JA, et al. Particulate matter air pollution exposure, distance to road, and incident lung cancer in the nurses’ health study cohort. Environ Health Perspect (2014) 122(9):926–32. doi:10.1289/ehp.1307490
116. Vineis P, Husgafvel-Pursiainen K. Air pollution and cancer: biomarker studies in human populations. Carcinogenesis (2005) 26:1846–55. doi:10.1093/carcin/bgi216
Keywords: air pollution, particulate matter, COPD, asthma, fibrosis, inflammation
Citation: Falcon-Rodriguez CI, Osornio-Vargas AR, Sada-Ovalle I and Segura-Medina P (2016) Aeroparticles, Composition, and Lung Diseases. Front. Immunol. 7:3. doi: 10.3389/fimmu.2016.00003
Received: 11 September 2015; Accepted: 05 January 2016;
Published: 20 January 2016
Edited by:
Masaaki Murakami, Hokkaido University, JapanCopyright: © 2016 Falcon-Rodriguez, Osornio-Vargas, Sada-Ovalle and Segura-Medina. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Patricia Segura-Medina, cHNlZ3VyYUB1bmFtLm14