 Nele Claes
Nele Claes Judith Fraussen
Judith Fraussen Piet Stinissen1
Piet Stinissen1 Raymond Hupperts
Raymond Hupperts Veerle Somers
Veerle Somers
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 21 December 2015
Sec. Multiple Sclerosis and Neuroimmunology
Volume 6 - 2015 | https://doi.org/10.3389/fimmu.2015.00642
This article is part of the Research Topic Lymphocytes in MS and EAE: more than just a CD4+ world View all 17 articles
Multiple sclerosis (MS) is a severe disease of the central nervous system (CNS) characterized by autoimmune inflammation and neurodegeneration. Historically, damage to the CNS was thought to be mediated predominantly by activated pro-inflammatory T cells. B cell involvement in the pathogenesis of MS was solely attributed to autoantibody production. The first clues for the involvement of antibody-independent B cell functions in MS pathology came from positive results in clinical trials of the B cell-depleting treatment rituximab in patients with relapsing-remitting (RR) MS. The survival of antibody-secreting plasma cells and decrease in T cell numbers indicated the importance of other B cell functions in MS such as antigen presentation, costimulation, and cytokine production. Rituximab provided us with an example of how clinical trials can lead to new research opportunities concerning B cell biology. Moreover, analysis of the antibody-independent B cell functions in MS has gained interest since these trials. Limited information is present on the effects of current immunomodulatory therapies on B cell functions, although effects of both first-line (interferon, glatiramer acetate, dimethyl fumarate, and teriflunomide), second-line (fingolimod, natalizumab), and even third-line (monoclonal antibody therapies) treatments on B cell subtype distribution, expression of functional surface markers, and secretion of different cytokines by B cells have been studied to some extent. In this review, we summarize the effects of different MS-related treatments on B cell functions that have been described up to now in order to find new research opportunities and contribute to the understanding of the pathogenesis of MS.
Multiple sclerosis (MS) is a chronic inflammatory disease of the central nervous system (CNS), characterized by demyelination in white and gray matter regions, axonal degeneration, and gliosis (1). MS is the most common chronic neurological disease in young adults affecting more women than men (three to one) with an incidence of 7/100,000 and a prevalence of 120/100,000 in Northern Europe (1). The diagnosis of MS is mostly preceded by a clinically isolated syndrome (CIS), which is the first clinical manifestation of a demyelinating disease that has not met the criteria of MS yet (2). Different clinical subtypes of MS are described. About 80% of the patients present with relapsing-remitting (RR) MS, which is characterized by disease exacerbations with periods of functional improvement (3). Over time, about 60% of the RRMS patients develop secondary progressive (SP) MS (4). About 10–20% of MS patients show progressive accumulation of disability from onset, referred to as primary progressive (PP) MS (2). According to the revised definitions of MS, above mentioned MS subtypes can present themselves in an inactive and active form (2). The underlying process of disease progression is not completely understood (5). Most MS therapies are primarily designed as treatment for RRMS patients, where there is marked inflammation.
Current data support the conceptual idea of MS as a complex heterogeneous disease caused by interactions between the environment, genetic susceptibility, and a dysbalanced immune system (6–8). Traditionally, T cells were considered as critical immune components required for the induction of MS pathogenesis. Recently, compelling evidence is present highlighting B cells as central components of the disease as well (9, 10). Autoreactive T cells are activated in the periphery most likely via molecular mimicry or bystander activation and home through a disrupted blood–brain barrier (BBB) to the CNS, where they are reactivated by antigen-presenting cells. This triggers the production of different mediators, such as chemokines and cytokines, by T cells, microglia, and other cells of the CNS. This will in turn initiate the recruitment of other inflammatory cells, including B cells and macrophages. B cells have the ability to cross the BBB and undergo stimulation, antigen-driven affinity maturation, and clonal expansion (11). The inflammatory reaction of T, B, and other immune cells leads to demyelinated lesions throughout the CNS (3).
As B cell involvement in MS has become more evident in recent years, more data have been collected concerning the effects of B cells in MS pathogenesis. Proof of B cell involvement in MS is described thoroughly further on in the review. Both B cell subtype distribution and B cell effector functions are important contributors to the disease. These processes are first described in more detail in order to fully understand how these processes are affected in MS patients and modulated by different MS treatments.
B cell development starts in the bone marrow where a hematopoietic stem cell evolves into an immature CD19+ B cell (Figure 1) (12). Transitional B cells (CD19+CD38++CD24++ or CD19+CD27−IgD+CD38+) enter the circulation and mature into naive B cells (CD19+IgD+CD27−). Upon antigen recognition, naive B cells proliferate into short-lived plasma blasts (CD19+CD138++ or CD19+CD27+CD38++) or plasma cells (CD38+CD138+) that produce low-affinity antibodies for a few days or further mature into memory B cells (CD19+CD27+) in a germinal center (GC) reaction. A proportion of memory B cells remains non-class-switched memory cells (CD19+IgD+CD27+), while others lose their immunoglobulin (Ig)D expression following isotype switching (CD19+IgD−CD27+). This classically results in the surface expression of IgG, IgA, or IgE, although a small proportion of memory B cells preserve IgM surface expression, namely IgM only memory B cells (CD19+IgD−CD27+IgM++) (13–17). A proportion of the memory B cells further matures into plasma blasts and long-lived plasma cells.
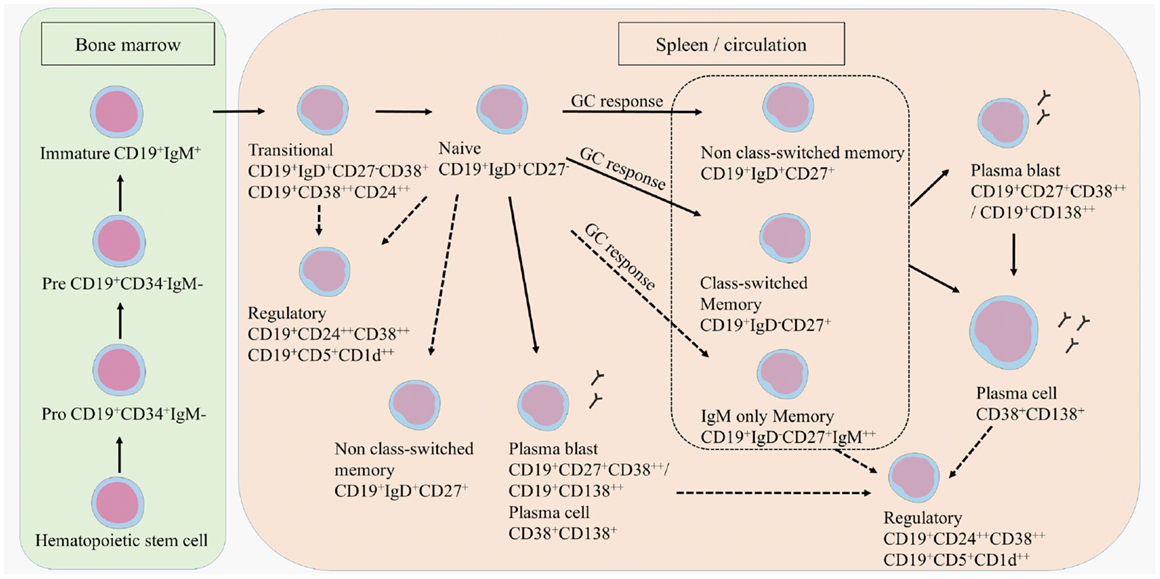
Figure 1. B cell development. B cells develop in the bone marrow and enter the circulation as transitional B cells. B cells remain naive until they encounter an antigen after which they differentiate into plasma blasts, short-lived plasma cells, or further mature into class-switched or non-class-switched memory B cells in a GC response. However, non-class-switched memory B cells can also be formed independent of a GC. A proportion of the memory B cells further develops into plasma blasts and/or plasma cells. Regulatory B cells are characterized within the transitional, naive, memory, and plasma blast or plasma cell population. Potential developmental routes are indicated with the dotted lines.
T cell subtypes important for providing help in the GC reactions are follicular helper T cells (TFH), follicular regulatory T cells (TFR), but also Th17 cells that can all induce or regulate GC formation and isotype switching (18–21). Regulatory B cells (Bregs) have been identified more recently by their function in immune regulation via the production of IL-10 (22, 23). Bregs could be enriched from transitional B cells, CD27+ memory B cells and plasma cells. Surface markers to characterize Bregs are still not clearly defined, although in humans CD24, CD38, CD5, and CD1d are mostly used (24–26).
Compositional changes of B cell subtypes in the peripheral blood (PB) are evidenced, shifting the balance toward more pro-inflammatory responses and less regulation. It is thought that memory B cells, plasma blasts and plasma cells preferentially cross the disrupted BBB and migrate into the CNS of MS patients, where they dominate the B cell pool and exert different effector functions (11, 27–35). During MS relapses, the percentage of PB memory B cells is increased (36). As TFH and TFR cells contribute to a normal GC response wherein potential autoantibodies are eliminated, the altered TFH and TFR function observed in MS patients can result in an inadequate GC response and the production of autoantibodies in the PB (18, 19).
In contrast to an increased percentage of memory B cells in PB, the proportion of Bregs was decreased in MS patients, while unchanged compared to healthy donors in other studies (37–40). Breg function was shown to be preserved as no differences were observed between MS patients and healthy donors in the ability of Bregs to inhibit proliferation of CD4+CD25− T responder cells (40).
B cells exert multiple effector functions, which are relevant to the pathogenesis and therapy of MS (9). First, B cells differentiate into antibody-secreting plasma blasts and plasma cells and produce antigen-specific antibodies (Figure 2). IgG from MS patients caused demyelination and axonal damage in a complement-dependent manner when using both in vivo and in vitro models (41, 42). Plasmapheresis and immunoadsorption in order to remove antibodies and complement factors already showed promising results as treatment for MS patients with steroid-resistant relapses (43, 44). In MS, different antibody targets have been described, including myelin basic protein (MBP), myelin oligodendrocyte glycoprotein (MOG), neurofilament, sperm-associated antigen 16 (SPAG16), coronin-1a, heat shock proteins, and other components of the CNS, emphasizing the diversity and complexity of the antibody response (45–54). An extensive review on different antibody targets is found in Ref. (45).
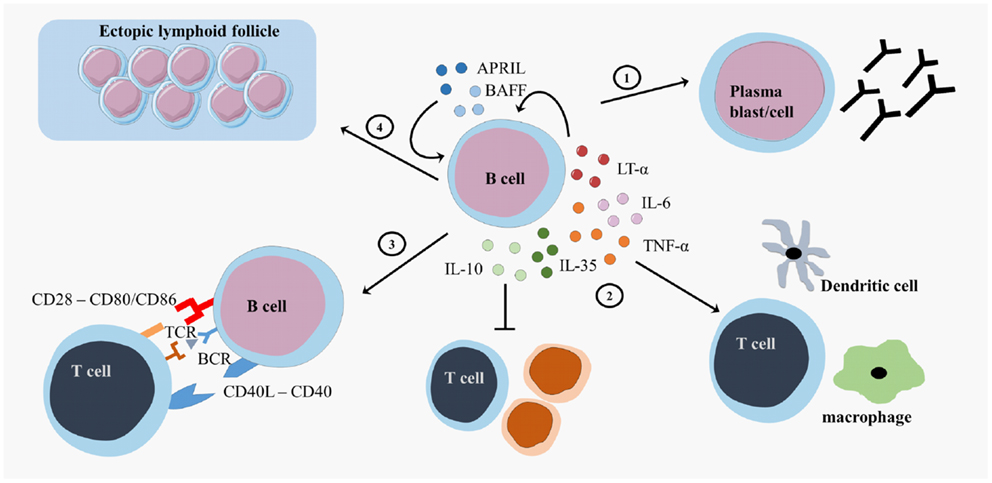
Figure 2. B cell effector functions. B cells exert different effector functions. B cells evolve into plasma blasts or plasma cells and produce antibodies (1). B cells produce different pro-inflammatory cytokines (lymphotoxin (LT)-α, tumor necrosis factor (TNF)-α, interleukin (IL)-6 or regulatory cytokines (IL-10, IL-35)) that influence other immune cells (2). B cells present antigens to T cells and provide costimulatory signals in order to induce appropriate T cell responses (3). B cells form ectopic lymphoid follicles that support the inflammatory responses (4). CD, cluster of differentiation; CD40L, CD40 ligand; APRIL, a proliferation-inducing ligand; BAFF, B cell activating factor; TCR, T cell receptor; BCR, B cell receptor.
Second, B cells form GC-like structures, ectopic lymphoid follicles, outside of secondary lymphoid organs at sites of inflammation (Figure 2). These follicles harbor a local source of class-switched Igs that contribute to the immune response and are detected as oligoclonal bands (OCB) in the cerebrospinal fluid (CSF) of MS patients (55–57). These OCB in the CSF of MS patients were one of the first findings for B cell involvement in MS (58, 59). Intrathecal B cells are the local source for these OCB in the CSF, contributing to inflammation, and the destruction of the myelin sheet in the CNS (60). B cells migrate to the CNS using surface markers such as C–X–C motif receptor (CXCR)3, CXCR5, and CC chemokine receptor (CCR)5. The CNS has a fostering environment in which the production of CXCL10 and CXCL13 attracts B cells (61). In the meninges of MS patients, these migrated B cells form ectopic GC structures (57).
Third, B cells serve as highly effective and selective antigen-presenting cells leading to optimal antigen-specific T cell expansion, memory formation, and cytokine production (Figure 2) (62–64). After antigen binding by the B cell receptor (BCR), the antigen is internalized, processed, and expressed on the surface of the B cells as a complex with major histocompatibility complex (MHC)-I or II molecules. Additional to antigen-presentation molecules, costimulatory molecules, such as CD80, CD86, and CD40, are expressed on B cells and contribute to optimal T cell activation (65). Myelin reactive peripheral B cells can induce CD4+ T cell responses in a proportion of MS patients (66). Additionally, B cell expression of the costimulatory molecules CD80 and CD86 is higher in MS patients than healthy controls (67, 68).
Finally, B cells support or regulate effector immune functions via the secretion of different cytokines (Figure 2). B cell activation factor (BAFF) and A Proliferation-Inducing Ligand (APRIL) are important survival factors for B cells and plasma cells, thereby maintaining the B cell pool (69). BAFF expression is upregulated in active and inactive MS lesions (70, 71). Maintaining BAFF expression within certain limits in order to balance pro-inflammatory and regulatory B cell subtypes can be an important feature for MS therapies. B cells support pro-inflammatory functions through secretion of tumor necrosis factor alpha (TNF-α), interleukin (IL)-6, and lymphotoxin alpha (LT-α) and exert regulatory functions via the production of IL-10 and IL-35 (22, 23, 72–75). In healthy individuals, transitional B cells perform regulatory functions by producing IL-10, thereby suppressing antigen-mediated T cell activity (26). Within the CD27+ memory B cell and plasma cell population, IL-10 and IL-35 producing Bregs can be enriched, showing that more mature B cells can also have regulatory functions next to antibody production and T cell activation (23, 25, 76–78). B cells from MS patients showed an increased production of IL-6, an increased LT-α/IL-10 ratio and increased LT-α and TNF-α production after stimulation in vitro (70). In addition, B cells from untreated MS patients secreted more pro-inflammatory IL-6 and less regulatory IL-10 than those from healthy controls (37, 79, 80).
Additional proof of B cell involvement in MS came from analysis of BCR sequences and genetic and animal studies. Analysis of Ig heavy chain variable sequences (VH) of intrathecal B cells from MS patients showed a restricted usage of Ig VH gene segments, pointing to a chronic antigen-driven B cell response in MS patients (81–83). Genetic studies in MS identified susceptibility genes that show a strong association with B cell function, such as HLA-DRB1*1501, HLA-DRB5*0101, and HLA-DQB1*0602 (84). Also observations from clinical trials of the B cell-depleting anti-CD20 monoclonal antibody rituximab indicated the importance of antibody-independent B cell functions in the pathogenesis of MS. These clinical studies showed an unchanged level of total Ig and a decrease in CSF T cell numbers, providing additional proof that B cells highly interact with T cells in MS (85–87).
Other information about the involvement of B cells in the pathogenesis of MS is available from experimental autoimmune encephalomyelitis (EAE), the animal model of MS. The role of B cells in EAE has long-time been neglected as B cells are not essential contributors to EAE models based on peptide immunization. More recent studies using recombinant MOG protein immunization have highlighted the role of B cells in EAE induction and pathology (88). The dual role of B cells in EAE was indicated by the use of anti-CD20 treatment, as disease exacerbation was evident when depleting Bregs before EAE induction while disease severity decreased when depleting memory B cells after EAE induction (22, 89, 90). B cells were essential for the generation of optimal pathogenic CD4+ T cell responses and differentiation of MOG specific T-helper (Th)1 and Th17 cells (91). In B cell deficient mice, EAE induction by adoptive transfer of activated T cells was reduced and reactivation of infiltrated T cells was impaired (92). Further, B cell-specific MHC class II-deficient mice were resistant to EAE induction and exhibited diminished Th1 and Th17 responses (93). Hence, B cells can promote EAE induction by acting as antigen-presenting cells. Moreover, B cell antigen presentation was proven to be crucial for maximal disease in EAE, further emphasizing the importance of B cells in MS pathogenesis (94).
Recently, a direct link between peripheral and intrathecal B cells was demonstrated. Clonally expanded autoreactive B cells with signs of affinity maturation were, next to the CSF, found in the PB of MS patients (82, 95). Further, expanded B cell clones were found both in the PB/draining cervical lymph nodes and the CSF, indicating a complex crosstalk between the periphery and the CNS in MS pathogenesis (27, 96). Exchange of B cells between the CSF and the PB may suggest that B cells carry antigen from the CNS to peripheral secondary lymphoid organs (11). Primed T cells then migrate to the CNS where residing B cells may further promote T cell activation. These data underline the importance of using therapeutics based on the inhibition of B cell transmigration into the CNS or that induce peripheral B cell depletion (11, 27, 96, 97). Additionally, autoreactive B cells can be removed from the B cell pool via both a central and a peripheral checkpoint. It seems that especially the peripheral tolerance checkpoint is defective, as shown by the equal proportion of polyreactive and anti-nuclear transitional B cells in MS patients and healthy donors (normal central B cell tolerance) and the increased proportion of mature naive B cells from MS patients reactive toward peripheral and CNS self antigens (defective peripheral B cell tolerance) (98). This defect is probably due to impaired Treg function that leads to the accumulation of autoreactive B cells (99). All these observations strengthen the idea that PB B cells contribute to the pathogenic B cell pool present in the CNS of MS patients and are involved in MS pathogenesis both by antibody-dependent and -independent B cell functions. Thus, investigating PB B cells and the effects of treatment on peripheral B cell functions may contribute to our understanding of the pathogenesis of MS (80, 85–87).
This review summarizes how current MS treatments influence B cell functions. At the moment, numerous FDA approved MS treatments or drugs in clinical trials can be subdivided in first-, second- and third-line therapies (Tables 1–3). Generally established first-line therapies include interferon-beta (IFN-β) and glatiramer acetate (GA), while fingolimod and natalizumab are considered to be second-line treatments. The recently approved oral drugs teriflunomide and dimethyl fumarate (DMF) are oral treatments used as first-line treatment for MS (100–105). Second- and third-line antibody treatments are rituximab, alemtuzumab, ocrelizumab, ofatumumab, and antibodies that target BAFF and APRIL. Modulating B cell functions is an important tool for treating MS patients, although information on the effects of therapy on B cell functions is limited. Investigating the effects of treatment on B cell functions is of potential relevance to the efficacy of such treatments and it will help to increase our insight into the involvement of PB B cells in MS pathogenesis.
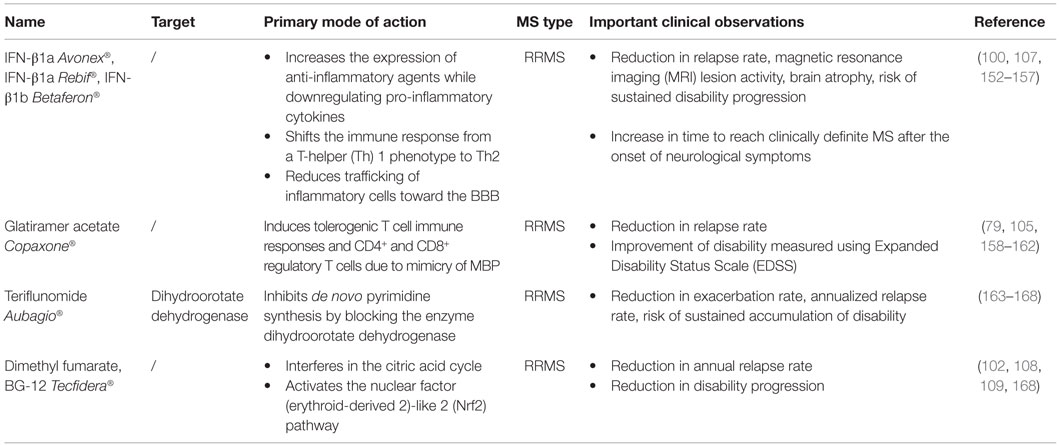
Table 1. Overview of first-line MS treatments.

Table 2. Overview of second-line MS treatments.
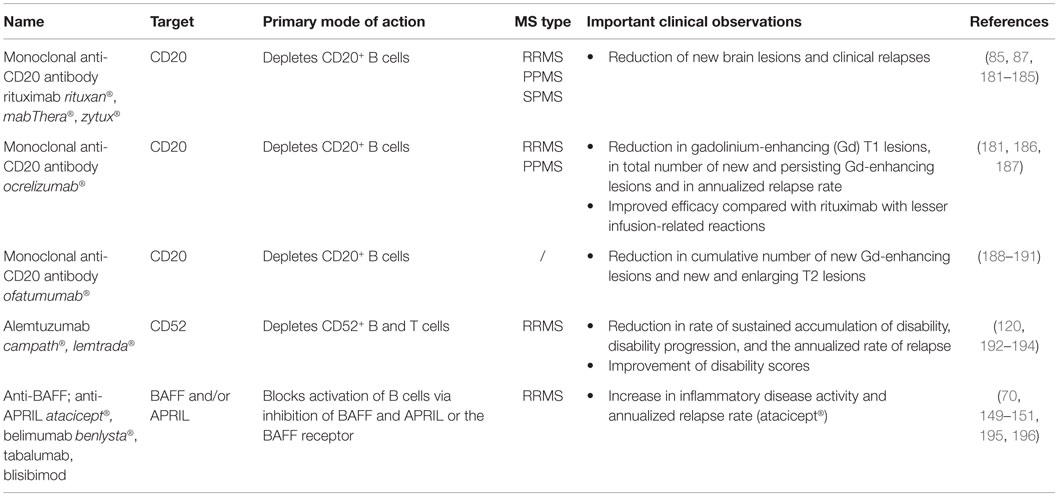
Table 3. Overview of third-line MS treatments.
Total B cell numbers and percentages in the PB were changed during treatment, both in cross-sectional and longitudinal studies, with an increase in the frequency of CD19+ B cells in IFN-β-treated MS patients and a decrease in GA- and DMF-treated MS patients (Figure 3) (79, 106–110). Different studies indicated that the percentage of B cells was increased in the PB and decreased in the CSF of natalizumab-treated MS patients, due to the inhibition of lymphocyte migration into the CNS (106, 111–118). This increase in PB B cells was observed up to 30 months after start of the treatment (112). Opposite effects were observed in fingolimod-treated MS patients where total B cell numbers in the PB were diminished because of the lymphocyte entrapment within secondary lymphoid organs. No changes were observed in CSF B cell numbers under fingolimod treatment (118, 119). In a study with 69 RRMS patients treated with rituximab, a decrease of 95% in the percentage of CD20+ B cells was evidenced from 2 weeks after treatment until 24 weeks (87). By week 48, B cells returned to 31% of baseline values. Alemtuzumab treatment caused a general depletion of both T and B cells in the PB of treated patients (120).
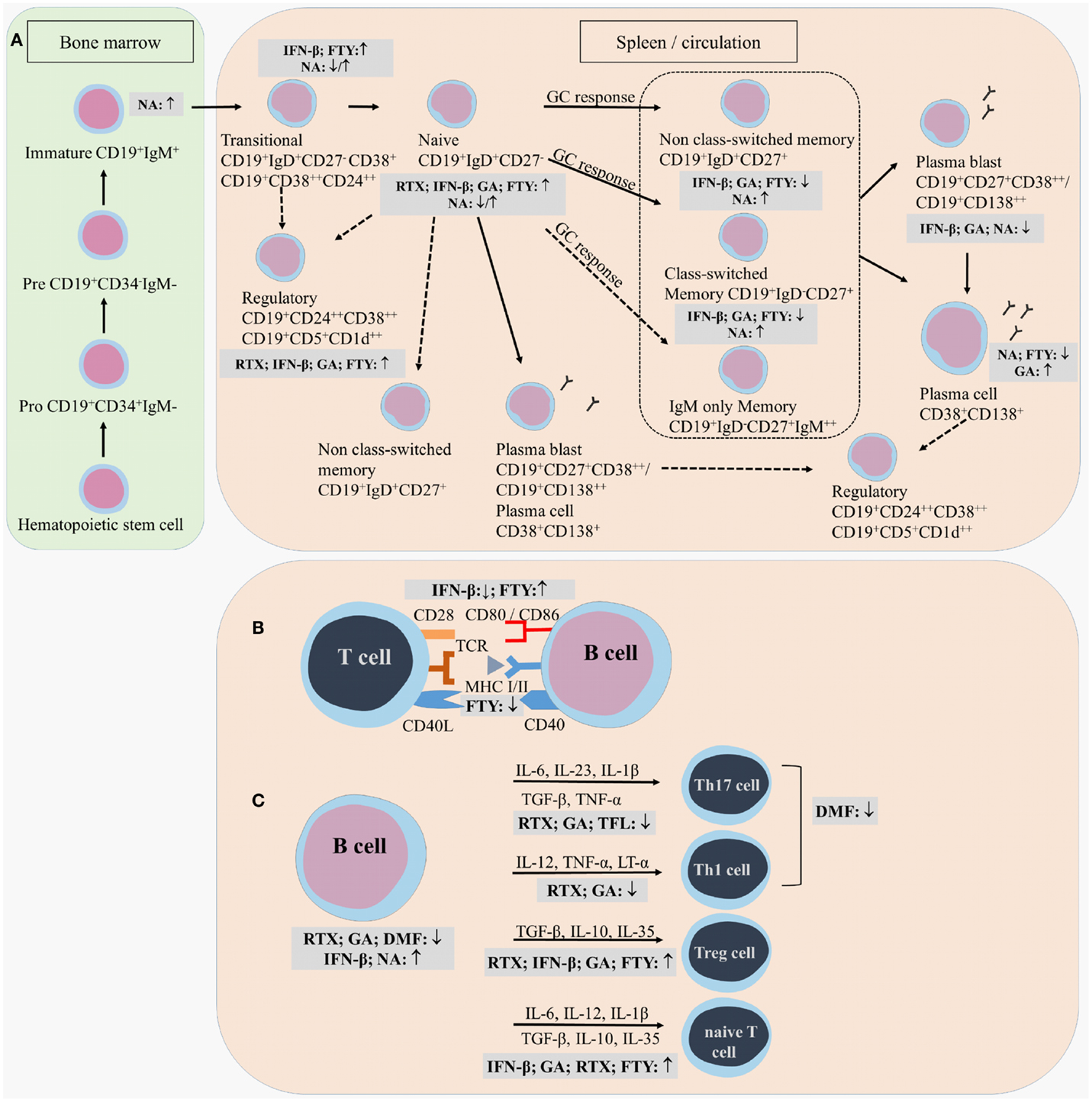
Figure 3. Effects of immunomodulatory therapy on B cell subtype distribution and function. B cell development in the bone marrow and periphery (A), antigen presentation and costimulatory molecules expressed on the B cell surface (B) and B cell cytokine production (C) are shown together with the effects of treatment on the different B cell subtypes and functions. CD, cluster of differentiation; IFN-β, interferon-β; FTY, fingolimod; GA, glatiramer acetate; NA, natalizumab; DMF, dimethyl fumarate; TFL, teriflunomide; RTX, rituximab; IL, interleukin; TGF, transforming growth factor; TNF, tumor necrosis factor; Th, T helper.
Different effects on B cell subtype distribution were demonstrated using different MS treatments. An increased frequency of immature and transitional B cells was generally evidenced under different treatments, including IFN-β, natalizumab, fingolimod, and during repopulation following rituximab or alemtuzumab treatment (106, 107, 111, 113, 120–124). These reports all point toward an increased output of B cells from the bone marrow under immunomodulatory treatment. In this regard, an increased release of lymphoid committed progenitor cells was shown during natalizumab therapy in MS (112). However, in a cross-sectional study with 8 natalizumab-treated RRMS patients, a significant decrease in the percentage of transitional B cells was evidenced (106). Also in fingolimod treated MS patients, the output of newly produced B cells or immature B cells from the bone marrow was decreased (121).
Therapeutic effects on frequencies of Bregs have only been described in relation to the use of fingolimod, rituximab and alemtuzumab therapy. In 48 fingolimod treated MS patients, a proportional increase of Bregs was recently described compared to 74 untreated MS patients and 70 healthy controls (125). During repopulation after B cell depletion by rituximab or alemtuzumab, naive B cells with an increased expression of CD38 and CD5, which are described as Bregs, were predominantly present, both in MS and other autoimmune diseases (31, 122, 126).
Peripheral blood naive B cells were increased in IFN-β, GA, natalizumab, and fingolimod-treated MS patients in comparison with treatment-naive MS patients in different cross-sectional and longitudinal studies (79, 106, 115, 119, 127). This indicates that the B cell population shifts toward a less disease promoting B cell pool after different MS treatments. For GA and natalizumab, this could not be reproduced in other studies where a decreased frequency of naive B cells was observed or no change in B cell subtype distribution at all (106, 107, 127). However, no information was available about the treatment duration, which makes it difficult to compare the studies.
Also contributing to a less disease promoting B cell phenotype is the significant decrease in the frequency of non-class-switched, class-switched memory B cells, and plasma blasts in both cross-sectional and longitudinal studies of IFN-β-, GA-, and fingolimod-treated MS patients, even when using different B cell classifications (36, 79, 106, 107, 119, 125, 127–129). Although a decrease in the proportion of plasma blasts was observed in natalizumab-treated MS patients, a higher percentage of memory and marginal zone B cells was reported (112, 114, 115, 127, 130). This memory B cell increase is probably due to the reduced retention of memory B cells in the spleen (112). In the CSF, natalizumab treatment particularly depleted CD5+ B cells and plasma blasts (131).
Data on B cell subtype distribution are missing for DMF and teriflunomide-treated MS patients. In vitro studies have shown that teriflunomide induces cell cycle arrest in B cells without inducing apoptotic cell death (101, 132, 133). Moreover, the effects of different third-line treatments on B cell subtype distribution is poorly investigated in MS as not all treatments are FDA approved and clinical trials are ongoing. To our knowledge, no data are available on the repopulation of B cells after discontinuation of the B cell-depleting therapies ocrelizumab and ofatumumab. Further research is warranted to increase the understanding of the exact mechanism of action and to investigate restoration of the immune balance following depletion therapies.
From this overview, we can conclude that immunomodulatory treatment of MS patients induces a shift in the distribution of B cell subtypes toward a more regulatory or anti-inflammatory phenotype. This is of high clinical importance as a disturbed balance between the different B cell subtypes is observed in MS. For different MS treatments, the effects on B cell subtype distribution have already been investigated to some extent, still conflicting data are present. This is probably due to variation in measurement time points and B cell characterization strategies. Furthermore, as each treatment requires a different time to reach a steady state of immunological parameters and treatment efficiency, it is difficult to compare study results. Therefore, it is essential to use a longitudinal design of the study and take into account the pharmacodynamical properties of the treatment, since some treatment effects could get lost when only measuring in a cross-sectional manner. B cell subtype analysis can also be highly relevant in the search for new markers for progressive multifocal leukoencephalopathy (PML) in natalizumab-treated patients, as B cells were described as potential carriers of the John Cunningham (JC) virus into the CNS (134). Other research is focused on finding risk factors for the development of PML during natalizumab treatment (112, 130, 135, 136).
Here, we present the available data on the effect of immunomodulatory treatment on antibody-dependent and -independent B cell functions. These include antibody production, antigen presentation, costimulation, migration, and cytokine production (Figure 3).
Glatiramer acetate treatment did not change serum levels of total IgG and IgM in MS patients, but in vitro levels of IgG and IgM antibodies were increased after stimulation of PB B cells from these patients (79). Natalizumab-treated MS patients showed lower levels of IgM in both serum and CSF and lower anti-neurofilament light antibodies in the serum than non-natalizumab-treated MS patients (116, 137). Longitudinal data of 24 MS patients confirmed these results with a decrease in neurofilament light antibody levels, a decline in total IgG levels in the PB and CSF, and a decline in total IgM in the PB (116, 137). Further, the IgG index, which reflects intrathecal IgG production, was decreased during natalizumab treatment, resulting in the disappearance of OCB in some of the treated MS patients (138). Whether a decline in the anti-neurofilament light antibodies is a consequence of a decrease in total antibody levels is not stated. Additionally, vaccination studies in fingolimod-treated healthy volunteers have demonstrated a mild to moderate decrease in IgG and IgM antibody levels toward some antigens, suggesting that fingolimod could reduce autoantibody production in MS as well (139). Teriflunomide, in contrast, did not influence immune responses toward influenza vaccines, indicating that the protective immune responses are preserved in these patients (140).
The anti-BAFF antibody atacicept® did not show beneficial results in clinical trials for MS and even led to worsening of the disease. More patients with optic neuritis who received atacicept® progressed to clinical definite MS (141). The efficacy of this therapy was proven in a clinical trial for RA wherein circulating IgG and IgA rheumatoid factor (RF) and total IgM, IgA, and IgG levels were reduced (142, 143). These observations indicate that, although MS and RA are both autoimmune diseases in which B cells are involved, different effector mechanisms of B cells are involved in both diseases. Since atacicept® affects antibody-producing plasma cells and clinical efficacy of atacicept® is shown in RA, one can speculate that in RA pathogenesis autoantibody production is more important than in MS pathogenesis. This underlines the multifactorial functions of B cells in autoimmunity.
Most information on effects of treatment on B cell antigen presentation, costimulation, and migration is available for IFN-β. Ex vivo analysis of PB B cells from 15 IFN-β-treated MS patients showed a decreased percentage of CD80, CD86, and CCR5 positive total and CD27− naive B cells compared to untreated MS patients (36). This pointed toward a less migratory and costimulatory phenotype of these B cells in the PB under treatment, which was confirmed in vitro (36, 144, 145). Furthermore, the increase in CD80 positive cells during relapses in MS patients was shown to be counteracted by IFN-β treatment (68). Since CD80 expression is associated with a Th1 phenotype and CD86 expression is associated with a Th2 response, these findings could indicate a shift from Th1 to Th2 in IFN-β treated MS patients (146). Within the CD27+ memory B cell compartment, the percentage of CD86 positive B cells was increased while the percentage of CXCR3 positive cells was decreased in the IFN-β group compared to healthy controls, indicating that memory B cells were less able to migrate to the CNS (36). IFN-β pretreated B cells were less able to induce proliferation of anti-CD3 and anti-CD28 stimulated CD4+ T cells than untreated B cells, further proving the immunomodulatory capacity of IFN-β therapy (144).
In a longitudinal study, B cell expression of the adhesion marker intracellular adhesion molecule (ICAM)-3 was reduced during GA treatment, indicating a potential role for GA in controlling the migration of B cells toward the CNS (147). Other longitudinal data showed a decrease in B cell expression of the antigen-presenting molecule human leukocyte antigen (HLA)-DR/DP/DQ and an increase in CD80 and CD86 costimulatory molecules on PB B cells in fingolimod treated MS patients (119). In contrast, a decreased expression of CD80 and stable CD86 expression was evidenced on B cells from fingolimod treated MS patients when compared to untreated MS patients in another study (128).
No data are present, to our knowledge, concerning the effects of DMF, teriflunomide, natalizumab and the CD20-depleting antibodies like rituximab, ocrelizumab, and ofatumumab on B cell surface expression of antigen presentation, costimulation, and migration markers. Natalizumab treatment could indirectly have an effect on these B cell functions due to the observed B cell subtype redistribution and general immune modulation. Because DMF and teriflunomide are recent FDA approved drugs, more research is warranted to investigate the effects of these treatments on B cell functions. Still, it can be concluded that different MS therapies can influence the interaction of B cells with T cells or other immune cells. As a consequence, inflammatory responses that are detrimental for the CNS are tempered, which is reflected in the clinical outcome of the treated MS patients.
In a cross-sectional study of IFN-β treated RRMS patients, increased serum levels of BAFF were observed compared to healthy controls, untreated, and GA-treated RRMS patients (107, 148). Twelve months after discontinuation of alemtuzumab treatment, increased serum BAFF levels were also observed (122). The BAFF-depleting antibody atacicept® exacerbated MS, which could be due to the decreased functionality of Bregs, as BAFF and APRIL signaling is highly implicated in the survival of Bregs. Still, the exact reason for the observed increased disease activity needs to be elucidated (149–151).
In terms of changes in cytokine production, IFN-β treatment caused induction of IL-10 production by B cells in vitro (144). Although GA did not directly modulate B cell proliferation or cytokine secretion in vitro (9), ex vivo analysis showed an increased secretion of IL-10 by B cells of 22 RRMS patients treated with GA (79). Intracellular flow cytometric analysis of B cells isolated from GA treated MS patients showed no increased frequency of IL-10 positive B cells in the PB of MS patients, indicating that GA does not influence the number of cytokine producing cells but rather the secretion of the cytokines (79). Further, a decreased capacity to secrete LT-α and IL-6 was indicated after B cell stimulation via CD40 and CD40L interaction or via Toll-like receptor triggering (79). An elevated IL-10 production was also evidenced for PB B cells from fingolimod-treated MS patients and repopulated B cells after rituximab treatment (31, 80, 125, 128). In fingolimod treated MS patients, the increased IL-10 production was accompanied by a decreased TNF-α production, while B cells following rituximab treatment secreted less pro-inflammatory cytokines IL-6, LT-α, and TNF-α (31, 80, 125, 128). Limited data is present of the effects of DMF and teriflunomide on the immune function in MS patients. In psoriasis patients, it was shown that DMF altered the immune and T cell cytokine profile (102, 110). Teriflunomide limits the secretion of pro-inflammatory molecules by immune cells, including IL-6 and IL-8 (101).
Thus, similar effects have been observed for all studied treatments on the cytokine production by B cells, correcting the imbalance between regulatory and disease promoting B cell functions in MS. We have to keep in mind that since different B cell subtypes produce different cytokines, by changing B cell subtype distribution, cytokine balances are changed as a secondary effect of the treatment. Data are missing on the effects of treatment on cytokine production by B cells for some FDA approved treatments such as natalizumab and for some treatments in clinical trials such as anti-CD20 monoclonal antibodies. It can be speculated that a potential mode of action by which these treatments contribute to the improvement of MS pathogenesis can be by influencing B cell cytokine production from a pro-inflammatory phenotype toward a more regulatory phenotype, still this needs to be further investigated.
It is eminent that B cells are major players in MS pathogenesis and contribute to the disease via both antibody-dependent and -independent mechanisms. B cells are essential for antigen presentation and costimulation of T cells, for the production of cytokines and to produce antibodies that will target components of the CNS. Thus, focusing on effects of treatment on these cells will help in our understanding of MS pathogenesis. Although initially not designed for that purpose, many MS modifying treatments influence both antibody-dependent and -independent B cell functions. Research on effects of therapy on B cell phenotype and function has demonstrated a shift from pro-inflammatory B cell functions toward more anti-inflammatory and regulatory functions. Still, each treatment influences this balance in its own manner. IFN-β, natalizumab, fingolimod, BAFF and APRIL targeting monoclonal antibodies, rituximab and alemtuzumab, induce compositional changes of the B cells, resulting in a less disease promoting distribution of B cells in both the PB and CSF of MS patients. GA, DMF, and teriflunomide work primarily via modulating B cell cytokine production. Still, all these effector mechanisms of B cells are interconnected and cannot be separated from each other. Investigating the mechanism of action of different treatments in different autoimmune diseases leads to new insights into that specific disease. For example, atacicept® has different clinical effects in RA and MS, indicating different roles of B cells in these diseases. More research is needed since inconsistencies between studies are present due to differences in B cell subtype definition and time point of measurement. Consensus in B cell subtype characterization will have added value in future research. Further, researchers should take into account pharmacodynamics of the compounds in order to decide on the specific time point for measuring B cell characteristics. Analysis of treatment effects on B cell subtype distribution and function can alternatively lead to prognostic knowledge for determining therapy efficiency. Finally, research should focus on finding specific therapies for the treatment of SPMS although initial efforts have been made. Further analysis of B cell functions in MS pathogenesis and the effects of treatment on these functions is hereby important in order to increase insight into the role of B cells in the disease process. This could lead to the development of novel and more specific therapies.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Compston A, Coles A. Multiple sclerosis. Lancet (2008) 372(9648):1502–17. doi:10.1016/S0140-6736(08)61620-7
2. Lublin FD, Reingold SC, Cohen JA, Cutter GR, Sørensen PS, Thompson AJ, et al. Defining the clinical course of multiple sclerosis: the 2013 revisions. Neurology (2014) 83(3):278–86. doi:10.1212/WNL.0000000000000560
3. Noseworthy JH, Lucchinetti C, Rodriguez M, and Weinshenker BG. Multiple sclerosis. N Engl J Med (2000) 343(13):938–52. doi:10.1056/NEJM200009283431307
4. Weiner HL. A shift from adaptive to innate immunity: a potential mechanism of disease progression in multiple sclerosis. J Neurol (2008) 255(Suppl 1):3–11. doi:10.1007/s00415-008-1002-8
5. Fox RJ, Thompson A, Baker D, Baneke P, Brown D, Browne P, et al. Setting a research agenda for progressive multiple sclerosis: the International Collaborative on Progressive MS. Mult Scler (2012) 18(11):1534–40. doi:10.1177/1352458512458169
6. Correale J, Ysrraelit MC, Gaitan MI. Immunomodulatory effects of vitamin D in multiple sclerosis. Brain (2009) 132(Pt 5):1146–60. doi:10.1093/brain/awp033
7. Goris A, Pauwels I, Gustavsen MW, van Son B, Hilven K, Bos SD, et al. Genetic variants are major determinants of CSF antibody levels in multiple sclerosis. Brain (2015) 138(Pt 3):632–43. doi:10.1093/brain/awu405
8. Mallucci G, Peruzzotti-Jametti L, Bernstock JD, Pluchino S. The role of immune cells, glia and neurons in white and gray matter pathology in multiple sclerosis. Prog Neurobiol (2015) 127-128C:1–22. doi:10.1016/j.pneurobio.2015.02.003
9. Ireland SJ, Blazek M, Harp CT, Greenberg B, Frohman EM, Davis LS, et al. Antibody-independent B cell effector functions in relapsing remitting multiple sclerosis: clues to increased inflammatory and reduced regulatory B cell capacity. Autoimmunity (2012) 45(5):400–14. doi:10.3109/08916934.2012.665529
10. Broux B, Stinissen P, Hellings N. Which immune cells matter? The immunopathogenesis of multiple sclerosis. Crit Rev Immunol (2013) 33(4):283–306. doi:10.1615/CritRevImmunol.2013007453
11. von Büdingen HC, Kuo TC, Sirota M, van Belle CJ, Apeltsin L, Glanville J, et al. B cell exchange across the blood-brain barrier in multiple sclerosis. J Clin Invest (2012) 122(12):4533–43. doi:10.1172/JCI63842
12. Pieper K, Grimbacher B, Eibel H. B-cell biology and development. J Allergy Clin Immunol (2013) 131(4):959–71. doi:10.1016/j.jaci.2013.01.046
13. Gray D. Immunological memory: a function of antigen persistence. Trends Microbiol (1993) 1(2):39–41. doi:10.1016/0966-842X(93)90026-N
14. Gray D. Immunological memory. Annu Rev Immunol (1993) 11:49–77. doi:10.1146/annurev.iy.11.040193.000405
15. Banchereau J, Rousset F. Human B lymphocytes: phenotype, proliferation, and differentiation. Adv Immunol (1992) 52:125–262. doi:10.1016/S0065-2776(08)60876-7
16. Klein U, Kuppers R, Rajewsky K. Evidence for a large compartment of IgM-expressing memory B cells in humans. Blood (1997) 89(4):1288–98.
17. Klein U, Rajewsky K, Kuppers R. Human immunoglobulin (Ig)M+IgD+ peripheral blood B cells expressing the CD27 cell surface antigen carry somatically mutated variable region genes: CD27 as a general marker for somatically mutated (memory) B cells. J Exp Med (1998) 188(9):1679–89. doi:10.1084/jem.188.9.1679
18. Dhaeze T, Peelen E, Hombrouck A, Peeters L, Van Wijmeersch B, Lemkens N, et al. Circulating follicular regulatory T cells are defective in multiple sclerosis. J Immunol (2015) 195(3):832–40. doi:10.4049/jimmunol.1500759
19. Dhaeze T, Stinissen P, Liston A, Hellings N. Humoral autoimmunity: a failure of regulatory T cells? Autoimmun Rev (2015) 14(8):735–41. doi:10.1016/j.autrev.2015.04.006
20. King C, Tangye SG, Mackay CR. T follicular helper (TFH) cells in normal and dysregulated immune responses. Annu Rev Immunol (2008) 26:741–66. doi:10.1146/annurev.immunol.26.021607.090344
21. Mitsdoerffer M, Lee Y, Jäger A, Kim HJ, Korn T, Kolls JK, et al. Proinflammatory T helper type 17 cells are effective B-cell helpers. Proc Natl Acad Sci U S A (2010) 107(32):14292–7. doi:10.1073/pnas.1009234107
22. Fillatreau S, Sweenie CH, McGeachy MJ, Gray D, Anderton SM. B cells regulate autoimmunity by provision of IL-10. Nat Immunol (2002) 3(10):944–50. doi:10.1038/ni833
23. Shen P, Roch T, Lampropoulou V, O’Connor RA, Stervbo U, Hilgenberg E, et al. IL-35-producing B cells are critical regulators of immunity during autoimmune and infectious diseases. Nature (2014) 507(7492):366–70. doi:10.1038/nature12979
24. Bouaziz JD, Yanaba K, Tedder TF. Regulatory B cells as inhibitors of immune responses and inflammation. Immunol Rev (2008) 224:201–14. doi:10.1111/j.1600-065X.2008.00661.x
25. Yanaba K, Bouaziz JD, Haas KM, Poe JC, Fujimoto M, Tedder TF. A regulatory B cell subset with a unique CD1dhiCD5+ phenotype controls T cell-dependent inflammatory responses. Immunity (2008) 28(5):639–50. doi:10.1016/j.immuni.2008.03.017
26. Blair PA, Noreña LY, Flores-Borja F, Rawlings DJ, Isenberg DA, Ehrenstein MR, et al. CD19(+)CD24(hi)CD38(hi) B cells exhibit regulatory capacity in healthy individuals but are functionally impaired in systemic lupus erythematosus patients. Immunity (2010) 32(1):129–40. doi:10.1016/j.immuni.2009.11.009
27. Palanichamy A, Apeltsin L, Kuo TC, Sirota M, Wang S, Pitts SJ, et al. Immunoglobulin class-switched B cells form an active immune axis between CNS and periphery in multiple sclerosis. Sci Transl Med (2014) 6(248):248ra106. doi:10.1126/scitranslmed.3008930
28. von Büdingen HC, Harrer MD, Kuenzle S, Meier M, Goebels N. Clonally expanded plasma cells in the cerebrospinal fluid of MS patients produce myelin-specific antibodies. Eur J Immunol (2008) 38(7):2014–23. doi:10.1002/eji.200737784
29. Cepok S, Rosche B, Grummel V, Vogel F, Zhou D, Sayn J, et al. Short-lived plasma blasts are the main B cell effector subset during the course of multiple sclerosis. Brain (2005) 128(Pt 7):1667–76. doi:10.1093/brain/awh486
30. Harp C, Lee J, Lambracht-Washington D, Cameron E, Olsen G, Frohman E, et al. Cerebrospinal fluid B cells from multiple sclerosis patients are subject to normal germinal center selection. J Neuroimmunol (2007) 183(1–2):189–99. doi:10.1016/j.jneuroim.2006.10.020
31. Duddy M, Niino M, Adatia F, Hebert S, Freedman M, Atkins H, et al. Distinct effector cytokine profiles of memory and naive human B cell subsets and implication in multiple sclerosis. J Immunol (2007) 178(10):6092–9. doi:10.4049/jimmunol.178.10.6092
32. Good KL, Avery DT, Tangye SG. Resting human memory B cells are intrinsically programmed for enhanced survival and responsiveness to diverse stimuli compared to naive B cells. J Immunol (2009) 182(2):890–901. doi:10.4049/jimmunol.182.2.890
33. Pascual V, Liu YJ, Magalski A, de Bouteiller O, Banchereau J, Capra JD. Analysis of somatic mutation in five B cell subsets of human tonsil. J Exp Med (1994) 180(1):329–39. doi:10.1084/jem.180.1.329
34. Qin Y, Duquette P, Zhang Y, Talbot P, Poole R, Antel J. Clonal expansion and somatic hypermutation of V(H) genes of B cells from cerebrospinal fluid in multiple sclerosis. J Clin Invest (1998) 102(5):1045–50. doi:10.1172/JCI3568
35. Tangye SG, Avery DT, Deenick EK, Hodgkin PD. Intrinsic differences in the proliferation of naive and memory human B cells as a mechanism for enhanced secondary immune responses. J Immunol (2003) 170(2):686–94. doi:10.4049/jimmunol.170.2.686
36. Niino M, Hirotani M, Miyazaki Y, Sasaki H. Memory and naive B-cell subsets in patients with multiple sclerosis. Neurosci Lett (2009) 464(1):74–8. doi:10.1016/j.neulet.2009.08.010
37. Knippenberg S, Peelen E, Smolders J, Thewissen M, Menheere P, Cohen Tervaert JW, et al. Reduction in IL-10 producing B cells (Breg) in multiple sclerosis is accompanied by a reduced naive/memory Breg ratio during a relapse but not in remission. J Neuroimmunol (2011) 239(1–2):80–6. doi:10.1016/j.jneuroim.2011.08.019
38. Quan C, Yu H, Qiao J, Xiao B, Zhao G, Wu Z, et al. Impaired regulatory function and enhanced intrathecal activation of B cells in neuromyelitis optica: distinct from multiple sclerosis. Mult Scler (2013) 19(3):289–98. doi:10.1177/1352458512454771
39. Habib J, Deng J, Lava N, Tyor W, Galipeau J. Blood B cell and regulatory subset content in multiple sclerosis patients. J Mult Scler (Foster City) (2015) 2(2):1000139.
40. Michel L, Chesneau M, Manceau P, Genty A, Garcia A, Salou M, et al. Unaltered regulatory B-cell frequency and function in patients with multiple sclerosis. Clin Immunol (2014) 155(2):198–208. doi:10.1016/j.clim.2014.09.011
41. Piddlesden SJ, Lassmann H, Zimprich F, Morgan BP, Linington C. The demyelinating potential of antibodies to myelin oligodendrocyte glycoprotein is related to their ability to fix complement. Am J Pathol (1993) 143(2):555–64.
42. Elliott C, Lindner M, Arthur A, Brennan K, Jarius S, Hussey J, et al. Functional identification of pathogenic autoantibody responses in patients with multiple sclerosis. Brain (2012) 135(6):1819–33. doi:10.1093/brain/aws105
43. Heigl F, Hettich R, Arendt R, Durner J, Koehler J, Mauch E. Immunoadsorption in steroid-refractory multiple sclerosis: clinical experience in 60 patients. Atheroscler Suppl (2013) 14(1):167–73. doi:10.1016/j.atherosclerosissup.2012.10.025
44. Keegan M, König F, McClelland R, Brück W, Morales Y, Bitsch A, et al. Relation between humoral pathological changes in multiple sclerosis and response to therapeutic plasma exchange. Lancet (2005) 366(9485):579–82. doi:10.1016/S0140-6736(05)67102-4
45. Fraussen J, Claes N, de Bock L, Somers V. Targets of the humoral autoimmune response in multiple sclerosis. Autoimmun Rev (2014) 13(11):1126–37. doi:10.1016/j.autrev.2014.07.002
46. de Bock L, Somers K, Fraussen J, Hendriks JJ, van Horssen J, Rouwette M, et al. Sperm-associated antigen 16 is a novel target of the humoral autoimmune response in multiple sclerosis. J Immunol (2014) 193(5):2147–56. doi:10.4049/jimmunol.1401166
47. Rouwette M, Noben JP, Van Horssen J, Van Wijmeersch B, Hupperts R, Jongen PJ, et al. Identification of coronin-1a as a novel antibody target for clinically isolated syndrome and multiple sclerosis. J Neurochem (2013) 126(4):483–92. doi:10.1111/jnc.12335
48. Rouwette M, Somers K, Govarts C, De Deyn PP, Hupperts R, Van Wijmeersch B, et al. Novel cerebrospinal fluid and serum autoantibody targets for clinically isolated syndrome. J Neurochem (2012) 123(4):568–77. doi:10.1111/jnc.12005
49. Angelucci F, Mirabella M, Frisullo G, Caggiula M, Tonali PA, Batocchi AP. Serum levels of anti-myelin antibodies in relapsing-remitting multiple sclerosis patients during different phases of disease activity and immunomodulatory therapy. Dis Markers (2005) 21(2):49–55. doi:10.1155/2005/826817
50. Bartos A, Fialová L, Soukupová J, Kukal J, Malbohan I, Pitha J. Antibodies against light neurofilaments in multiple sclerosis patients. Acta Neurol Scand (2007) 116(2):100–7. doi:10.1111/j.1600-0404.2006.00794.x
51. Berger T, Rubner P, Schautzer F, Egg R, Ulmer H, Mayringer I, et al. Antimyelin antibodies as a predictor of clinically definite multiple sclerosis after a first demyelinating event. N Engl J Med (2003) 349(2):139–45. doi:10.1056/NEJMoa022328
52. Cid C, Regidor I, Alcazar A. Anti-heat shock protein 90beta antibodies are detected in patients with multiple sclerosis during remission. J Neuroimmunol (2007) 184(1–2):223–6. doi:10.1016/j.jneuroim.2006.11.001
53. Hedegaard CJ, Chen N, Sellebjerg F, Sørensen PS, Leslie RG, Bendtzen K, et al. Autoantibodies to myelin basic protein (MBP) in healthy individuals and in patients with multiple sclerosis: a role in regulating cytokine responses to MBP. Immunology (2009) 128(1 Suppl):e451–61. doi:10.1111/j.1365-2567.2008.02999.x
54. Lalive PH, Menge T, Delarasse C, Della Gaspera B, Pham-Dinh D, Villoslada P, et al. Antibodies to native myelin oligodendrocyte glycoprotein are serologic markers of early inflammation in multiple sclerosis. Proc Natl Acad Sci U S A (2006) 103(7):2280–5. doi:10.1073/pnas.0510672103
55. Grogan JL, Ouyang W. A role for Th17 cells in the regulation of tertiary lymphoid follicles. Eur J Immunol (2012) 42(9):2255–62. doi:10.1002/eji.201242656
56. Humby F, Bombardieri M, Manzo A, Kelly S, Blades MC, Kirkham B, et al. Ectopic lymphoid structures support ongoing production of class-switched autoantibodies in rheumatoid synovium. PLoS Med (2009) 6(1):e1. doi:10.1371/journal.pmed.0060001
57. Serafini B, Rosicarelli B, Magliozzi R, Stigliano E, Aloisi F. Detection of ectopic B-cell follicles with germinal centers in the meninges of patients with secondary progressive multiple sclerosis. Brain Pathol (2004) 14(2):164–74. doi:10.1111/j.1750-3639.2004.tb00049.x
58. Cole SR, Beck RW, Moke PS, Kaufman DI, Tourtellotte WW. The predictive value of CSF oligoclonal banding for MS 5 years after optic neuritis. Optic Neuritis Study Group. Neurology (1998) 51(3):885–7.
59. Kabat EA, Glusman M, Knaub V. Quantitative estimation of the albumin and gamma globulin in normal and pathologic cerebrospinal fluid by immunochemical methods. Am J Med (1948) 4(5):653–62. doi:10.1016/S0002-9343(48)90389-1
60. Obermeier B, Lovato L, Mentele R, Brück W, Forne I, Imhof A, et al. Related B cell clones that populate the CSF and CNS of patients with multiple sclerosis produce CSF immunoglobulin. J Neuroimmunol (2011) 233(1–2):245–8. doi:10.1016/j.jneuroim.2011.01.010
61. Krumbholz M, Theil D, Cepok S, Hemmer B, Kivisäkk P, Ransohoff RM, et al. Chemokines in multiple sclerosis: CXCL12 and CXCL13 up-regulation is differentially linked to CNS immune cell recruitment. Brain (2006) 129(Pt 1):200–11. doi:10.1093/brain/awh680
62. Crawford A, Macleod M, Schumacher T, Corlett L, Gray D. Primary T cell expansion and differentiation in vivo requires antigen presentation by B cells. J Immunol (2006) 176(6):3498–506. doi:10.4049/jimmunol.176.6.3498
63. Linton PJ, Harbertson J, Bradley LM. A critical role for B cells in the development of memory CD4 cells. J Immunol (2000) 165(10):5558–65. doi:10.4049/jimmunol.165.10.5558
64. Batista FD, Harwood NE. The who, how and where of antigen presentation to B cells. Nat Rev Immunol (2009) 9(1):15–27. doi:10.1038/nri2454
65. Linton PJ, Bautista B, Biederman E, Bradley ES, Harbertson J, Kondrack RM, et al. Costimulation via OX40L expressed by B cells is sufficient to determine the extent of primary CD4 cell expansion and Th2 cytokine secretion in vivo. J Exp Med (2003) 197(7):875–83. doi:10.1084/jem.20021290
66. Harp CT, Ireland S, Davis LS, Remington G, Cassidy B, Cravens PD, et al. Memory B cells from a subset of treatment-naive relapsing-remitting multiple sclerosis patients elicit CD4(+) T-cell proliferation and IFN-gamma production in response to myelin basic protein and myelin oligodendrocyte glycoprotein. Eur J Immunol (2010) 40(10):2942–56. doi:10.1002/eji.201040516
67. Aung LL, Balashov KE. Decreased dicer expression is linked to increased expression of co-stimulatory molecule CD80 on B cells in multiple sclerosis. Mult Scler (2015) 21(9):1131–8. doi:10.1177/1352458514560923
68. Genc K, Dona DL, Reder AT. Increased CD80(+) B cells in active multiple sclerosis and reversal by interferon beta-1b therapy. J Clin Invest (1997) 99(11):2664–71. doi:10.1172/JCI119455
69. Lesley R, Xu Y, Kalled SL, Hess DM, Schwab SR, Shu HB, et al. Reduced competitiveness of autoantigen-engaged B cells due to increased dependence on BAFF. Immunity (2004) 20(4):441–53. doi:10.1016/S1074-7613(04)00079-2
70. Krumbholz M, Meinl E. B cells in MS and NMO: pathogenesis and therapy. Semin Immunopathol (2014) 36(3):339–50. doi:10.1007/s00281-014-0424-x
71. Krumbholz M, Theil D, Derfuss T, Rosenwald A, Schrader F, Monoranu CM, et al. BAFF is produced by astrocytes and up-regulated in multiple sclerosis lesions and primary central nervous system lymphoma. J Exp Med (2005) 201(2):195–200. doi:10.1084/jem.20041674
72. Bar-Or A, Fawaz L, Fan B, Darlington PJ, Rieger A, Ghorayeb C, et al. Abnormal B-cell cytokine responses a trigger of T-cell-mediated disease in MS? Ann Neurol (2010) 67(4):452–61. doi:10.1002/ana.21939
73. Carter NA, Rosser EC, Mauri C. Interleukin-10 produced by B cells is crucial for the suppression of Th17/Th1 responses, induction of T regulatory type 1 cells and reduction of collagen-induced arthritis. Arthritis Res Ther (2012) 14(1):R32. doi:10.1186/ar3736
74. Yoshizaki A, Miyagaki T, DiLillo DJ, Matsushita T, Horikawa M, Kountikov EI, et al. Regulatory B cells control T-cell autoimmunity through IL-21-dependent cognate interactions. Nature (2012) 491(7423):264–8. doi:10.1038/nature11501
75. Meinl E, Krumbholz M, Hohlfeld R. B lineage cells in the inflammatory central nervous system environment: migration, maintenance, local antibody production, and therapeutic modulation. Ann Neurol (2006) 59(6):880–92. doi:10.1002/ana.20890
76. Iwata Y, Matsushita T, Horikawa M, Dilillo DJ, Yanaba K, Venturi GM, et al. Characterization of a rare IL-10-competent B-cell subset in humans that parallels mouse regulatory B10 cells. Blood (2011) 117(2):530–41. doi:10.1182/blood-2010-07-294249
77. Kessel A, Haj T, Peri R, Snir A, Melamed D, Sabo E, et al. Human CD19(+)CD25(high) B regulatory cells suppress proliferation of CD4(+) T cells and enhance Foxp3 and CTLA-4 expression in T-regulatory cells. Autoimmun Rev (2012) 11(9):670–7. doi:10.1016/j.autrev.2011.11.018
78. Fillatreau S. Regulatory plasma cells. Curr Opin Pharmacol (2015) 23:1–5. doi:10.1016/j.coph.2015.04.006
79. Ireland SJ, Guzman AA, O’Brien DE, Hughes S, Greenberg B, Flores A, et al. The effect of glatiramer acetate therapy on functional properties of B cells from patients with relapsing-remitting multiple sclerosis. JAMA Neurol (2014) 71(11):1421–8. doi:10.1001/jamaneurol.2014.1472
80. Barr TA, Shen P, Brown S, Lampropoulou V, Roch T, Lawrie S, et al. B cell depletion therapy ameliorates autoimmune disease through ablation of IL-6-producing B cells. J Exp Med (2012) 209(5):1001–10. doi:10.1084/jem.20111675
81. Owens GP, Ritchie AM, Burgoon MP, Williamson RA, Corboy JR, Gilden DH. Single-cell repertoire analysis demonstrates that clonal expansion is a prominent feature of the B cell response in multiple sclerosis cerebrospinal fluid. J Immunol (2003) 171(5):2725–33. doi:10.4049/jimmunol.171.5.2725
82. Qin Y, Duquette P, Zhang Y, Olek M, Da RR, Richardson J, et al. Intrathecal B-cell clonal expansion, an early sign of humoral immunity, in the cerebrospinal fluid of patients with clinically isolated syndrome suggestive of multiple sclerosis. Lab Invest (2003) 83(7):1081–8. doi:10.1097/01.LAB.0000077008.24259.0D
83. Monson NL, Brezinschek HP, Brezinschek RI, Mobley A, Vaughan GK, Frohman EM, et al. Receptor revision and atypical mutational characteristics in clonally expanded B cells from the cerebrospinal fluid of recently diagnosed multiple sclerosis patients. J Neuroimmunol (2005) 158(1–2):170–81. doi:10.1016/j.jneuroim.2004.04.022
84. International Multiple Sclerosis Genetics Consortium, Wellcome Trust Case Control Consortium 2, Sawcer S, Hellenthal G, Pirinen M, Spencer CC, et al. Genetic risk and a primary role for cell-mediated immune mechanisms in multiple sclerosis. Nature (2011) 476(7359):214–9. doi:10.1038/nature10251
85. Bar-Or A, Calabresi PA, Arnold D, Markowitz C, Shafer S, Kasper LH, et al. Rituximab in relapsing-remitting multiple sclerosis: a 72-week, open-label, phase I trial. Ann Neurol (2008) 63(3):395–400. doi:10.1002/ana.21363
86. Cross AH, Stark JL, Lauber J, Ramsbottom MJ, Lyons JA. Rituximab reduces B cells and T cells in cerebrospinal fluid of multiple sclerosis patients. J Neuroimmunol (2006) 180(1–2):63–70. doi:10.1016/j.jneuroim.2006.06.029
87. Hauser SL, Waubant E, Arnold DL, Vollmer T, Antel J, Fox RJ, et al. B-cell depletion with rituximab in relapsing-remitting multiple sclerosis. N Engl J Med (2008) 358(7):676–88. doi:10.1056/NEJMoa0706383
88. Lyons JA, San M, Happ MP, Cross AH. B cells are critical to induction of experimental allergic encephalomyelitis by protein but not by a short encephalitogenic peptide. Eur J Immunol (1999) 29(11):3432–9. doi:10.1002/(SICI)1521-4141(199911)29:11<3432::AID-IMMU3432>3.0.CO;2-2
89. Wolf SD, Dittel BN, Hardardottir F, Janeway CA Jr. Experimental autoimmune encephalomyelitis induction in genetically B cell-deficient mice. J Exp Med (1996) 184(6):2271–8. doi:10.1084/jem.184.6.2271
90. Matsushita T, Yanaba K, Bouaziz JD, Fujimoto M, Tedder TF. Regulatory B cells inhibit EAE initiation in mice while other B cells promote disease progression. J Clin Invest (2008) 118(10):3420–30. doi:10.1172/JCI36030
91. Weber MS, Prod’homme T, Patarroyo JC, Molnarfi N, Karnezis T, Lehmann-Horn K, et al. B-cell activation influences T-cell polarization and outcome of anti-CD20 B-cell depletion in central nervous system autoimmunity. Ann Neurol (2010) 68(3):369–83. doi:10.1002/ana.22081
92. Pierson ER, Stromnes IM, Goverman JM. B cells promote induction of experimental autoimmune encephalomyelitis by facilitating reactivation of T cells in the central nervous system. J Immunol (2014) 192(3):929–39. doi:10.4049/jimmunol.1302171
93. Molnarfi N, Schulze-Topphoff U, Weber MS, Patarroyo JC, Prod’homme T, Varrin-Doyer M, et al. MHC class II-dependent B cell APC function is required for induction of CNS autoimmunity independent of myelin-specific antibodies. J Exp Med (2013) 210(13):2921–37. doi:10.1084/jem.20130699
94. Parker Harp CR, Archambault AS, Sim J, Ferris ST, Mikesell RJ, Koni PA, et al. B cell antigen presentation is sufficient to drive neuroinflammation in an animal model of multiple sclerosis. J Immunol (2015) 194(11):5077–84. doi:10.4049/jimmunol.1402236
95. Fraussen J, Vrolix K, Claes N, Martinez-Martinez P, Losen M, Hupperts R, et al. Autoantigen induced clonal expansion in immortalized B cells from the peripheral blood of multiple sclerosis patients. J Neuroimmunol (2013) 261(1–2):98–107. doi:10.1016/j.jneuroim.2013.05.002
96. Stern JN, Yaari G, Vander Heiden JA, Church G, Donahue WF, Hintzen RQ, et al. B cells populating the multiple sclerosis brain mature in the draining cervical lymph nodes. Sci Transl Med (2014) 6(248):248ra107. doi:10.1126/scitranslmed.3008879
97. Bankoti J, Apeltsin L, Hauser SL, Allen S, Albertolle ME, Witkowska HE, et al. In multiple sclerosis, oligoclonal bands connect to peripheral B-cell responses. Ann Neurol (2014) 75(2):266–76. doi:10.1002/ana.24088
98. Kinnunen T, Chamberlain N, Morbach H, Cantaert T, Lynch M, Preston-Hurlburt P, et al. Specific peripheral B cell tolerance defects in patients with multiple sclerosis. J Clin Invest (2013) 123(6):2737–41. doi:10.1172/JCI68775
99. Kinnunen T, Chamberlain N, Morbach H, Choi J, Kim S, Craft J, et al. Accumulation of peripheral autoreactive B cells in the absence of functional human regulatory T cells. Blood (2013) 121(9):1595–603. doi:10.1182/blood-2012-09-457465
100. The IFNB Multiple Sclerosis Study Group. Interferon beta-1b is effective in relapsing-remitting multiple sclerosis. I. Clinical results of a multicenter, randomized, double-blind, placebo-controlled trial. Neurology (1993) 43(4):655–61. doi:10.1212/WNL.43.4.655
101. Bar-Or A. Teriflunomide (Aubagio(R)) for the treatment of multiple sclerosis. Exp Neurol (2014) 262(Pt A):57–65. doi:10.1016/j.expneurol.2014.06.005
102. Bomprezzi R. Dimethyl fumarate in the treatment of relapsing-remitting multiple sclerosis: an overview. Ther Adv Neurol Disord (2015) 8(1):20–30. doi:10.1177/1756285614564152
103. Brinkmann V. FTY720 (fingolimod) in Multiple Sclerosis: therapeutic effects in the immune and the central nervous system. Br J Pharmacol (2009) 158(5):1173–82. doi:10.1111/j.1476-5381.2009.00451.x
104. Derfuss T, Kuhle J, Lindberg R, Kappos L. Natalizumab therapy for multiple sclerosis. Semin Neurol (2013) 33(1):26–36. doi:10.1055/s-0033-1343793
105. Johnson KP, Brooks BR, Cohen JA, Ford CC, Goldstein J, Lisak RP, et al. Copolymer 1 reduces relapse rate and improves disability in relapsing-remitting multiple sclerosis: results of a phase III multicenter, double-blind placebo-controlled trial. The Copolymer 1 Multiple Sclerosis Study Group. Neurology (1995) 45(7):1268–76. doi:10.1212/WNL.45.7.1268
106. Haas J, Bekeredjian-Ding I, Milkova M, Balint B, Schwarz A, Korporal M, et al. B cells undergo unique compartmentalized redistribution in multiple sclerosis. J Autoimmun (2011) 37(4):289–99. doi:10.1016/j.jaut.2011.08.003
107. Schubert RD, Hu Y, Kumar G, Szeto S, Abraham P, Winderl J, et al. IFN-beta treatment Requires B cells for efficacy in neuroautoimmunity. J Immunol (2015) 194(5):2110–6. doi:10.4049/jimmunol.1402029
108. Fox RJ, Miller DH, Phillips JT, Hutchinson M, Havrdova E, Kita M, et al. Placebo-controlled phase 3 study of oral BG-12 or glatiramer in multiple sclerosis. N Engl J Med (2012) 367(12):1087–97. doi:10.1056/NEJMoa1206328
109. Gold R, Kappos L, Arnold DL, Bar-Or A, Giovannoni G, Selmaj K, et al. Placebo-controlled phase 3 study of oral BG-12 for relapsing multiple sclerosis. N Engl J Med (2012) 367(12):1098–107. doi:10.1056/NEJMoa1114287
110. Litjens NH, Nibbering PH, Barrois AJ, Zomerdijk TP, Van Den Oudenrijn AC, Noz KC, et al. Beneficial effects of fumarate therapy in psoriasis vulgaris patients coincide with downregulation of type 1 cytokines. Br J Dermatol (2003) 148(3):444–51. doi:10.1046/j.1365-2133.2003.05153.x
111. Krumbholz M, Meinl I, Kümpfel T, Hohlfeld R, Meinl E. Natalizumab disproportionately increases circulating pre-B and B cells in multiple sclerosis. Neurology (2008) 71(17):1350–4. doi:10.1212/01.wnl.0000327671.91357.96
112. Planas R, Jelčić I, Schippling S, Martin R, Sospedra M. Natalizumab treatment perturbs memory- and marginal zone-like B-cell homing in secondary lymphoid organs in multiple sclerosis. Eur J Immunol (2012) 42(3):790–8. doi:10.1002/eji.201142108
113. Lesesve JF, Debouverie M, Decarvalho Bittencourt M, Béné MC. CD49d blockade by natalizumab therapy in patients with multiple sclerosis increases immature B-lymphocytes. Bone Marrow Transplant (2011) 46(11):1489–91. doi:10.1038/bmt.2010.328
114. Putzki N, Baranwal MK, Tettenborn B, Limmroth V, Kreuzfelder E. Effects of natalizumab on circulating B cells, T regulatory cells and natural killer cells. Eur Neurol (2010) 63(5):311–7. doi:10.1159/000302687
115. Skarica M, Eckstein C, Whartenby KA, Calabresi PA. Novel mechanisms of immune modulation of natalizumab in multiple sclerosis patients. J Neuroimmunol (2011) 235(1–2):70–6. doi:10.1016/j.jneuroim.2011.02.010
116. Warnke C, Stettner M, Lehmensiek V, Dehmel T, Mausberg AK, von Geldern G, et al. Natalizumab exerts a suppressive effect on surrogates of B cell function in blood and CSF. Mult Scler (2015) 21(8):1036–44. doi:10.1177/1352458514556296
117. Harrer A, Pilz G, Einhaeupl M, Oppermann K, Hitzl W, Wipfler P, et al. Lymphocyte subsets show different response patterns to in vivo bound natalizumab – a flow cytometric study on patients with multiple sclerosis. PLoS One (2012) 7(2):e31784. doi:10.1371/journal.pone.0031784
118. Kowarik MC, Pellkofer HL, Cepok S, Korn T, Kümpfel T, Buck D, et al. Differential effects of fingolimod (FTY720) on immune cells in the CSF and blood of patients with MS. Neurology (2011) 76(14):1214–21. doi:10.1212/WNL.0b013e3182143564
119. Claes N, Dhaeze T, Fraussen J, Broux B, Van Wijmeersch B, Stinissen P, et al. Compositional changes of B and T cell subtypes during fingolimod treatment in multiple sclerosis patients: a 12-month follow-up study. PLoS One (2014) 9(10):e111115. doi:10.1371/journal.pone.0111115
120. Havrdova E, Horakova D, Kovarova I. Alemtuzumab in the treatment of multiple sclerosis: key clinical trial results and considerations for use. Ther Adv Neurol Disord (2015) 8(1):31–45. doi:10.1177/1756285614563522
121. Chiarini M, Sottini A, Bertoli D, Serana F, Caimi L, Rasia S, et al. Newly produced T and B lymphocytes and T-cell receptor repertoire diversity are reduced in peripheral blood of fingolimod-treated multiple sclerosis patients. Mult Scler (2015) 21(6):726–34. doi:10.1177/1352458514551456
122. Heidt S, Hester J, Shankar S, Friend PJ, Wood KJ. B cell repopulation after alemtuzumab induction-transient increase in transitional B cells and long-term dominance of naive B cells. Am J Transplant (2012) 12(7):1784–92. doi:10.1111/j.1600-6143.2012.04012.x
123. Kessel A, Rosner I, Toubi E. Rituximab: beyond simple B cell depletion. Clin Rev Allergy Immunol (2008) 34(1):74–9. doi:10.1007/s12016-008-8074-1
124. Thompson SA, Jones JL, Cox AL, Compston DA, Coles AJ. B-cell reconstitution and BAFF after alemtuzumab (Campath-1H) treatment of multiple sclerosis. J Clin Immunol (2010) 30(1):99–105. doi:10.1007/s10875-009-9327-3
125. Grützke B, Hucke S, Gross CC, Herold MV, Posevitz-Fejfar A, Wildemann BT, et al. Fingolimod treatment promotes regulatory phenotype and function of B cells. Ann Clin Transl Neurol (2015) 2(2):119–30. doi:10.1002/acn3.155
126. Pers JO, Daridon C, Bendaoud B, Devauchelle V, Berthou C, Saraux A, et al. B-cell depletion and repopulation in autoimmune diseases. Clin Rev Allergy Immunol (2008) 34(1):50–5. doi:10.1007/s12016-007-8015-4
127. Rovituso D, Heller S, Schroeter M, Kleinschnitz C, Kuerten S. B1 cells are unaffected by immune modulatory treatment in remitting-relapsing multiple sclerosis patients. J Neuroimmunol (2014) 272(1–2):86–90. doi:10.1016/j.jneuroim.2014.04.008
128. Miyazaki Y, Niino M, Fukazawa T, Takahashi E, Nonaka T, Amino I, et al. Suppressed pro-inflammatory properties of circulating B cells in patients with multiple sclerosis treated with fingolimod, based on altered proportions of B-cell subpopulations. Clin Immunol (2014) 151(2):127–35. doi:10.1016/j.clim.2014.02.001
129. Nakamura M, Matsuoka T, Chihara N, Miyake S, Sato W, Araki M, et al. Differential effects of fingolimod on B-cell populations in multiple sclerosis. Mult Scler (2014) 20(10):1371–80. doi:10.1177/1352458514523496
130. Sottini A, Capra R, Zanotti C, Chiarini M, Serana F, Ricotta D, et al. Pre-existing T- and B-cell defects in one progressive multifocal leukoencephalopathy patient. PLoS One (2012) 7(4):e34493. doi:10.1371/journal.pone.0034493
131. Villar LM, García-Sánchez MI, Costa-Frossard L, Espiño M, Roldán E, Páramo D, et al. Immunological markers of optimal response to natalizumab in multiple sclerosis. Arch Neurol (2012) 69(2):191–7. doi:10.1001/archneurol.2011.971
132. Li L, Liu J, Delohery T, Zhang D, Arendt C, Jones C. The effects of teriflunomide on lymphocyte subpopulations in human peripheral blood mononuclear cells in vitro. J Neuroimmunol (2013) 265(1–2):82–90. doi:10.1016/j.jneuroim.2013.10.003
133. Ringshausen I, Oelsner M, Bogner C, Peschel C, Decker T. The immunomodulatory drug Leflunomide inhibits cell cycle progression of B-CLL cells. Leukemia (2008) 22(3):635–8. doi:10.1038/sj.leu.2404922
134. Chapagain ML, Nerurkar VR. Human polyomavirus JC (JCV) infection of human B lymphocytes: a possible mechanism for JCV transmigration across the blood-brain barrier. J Infect Dis (2010) 202(2):184–91. doi:10.1086/653823
135. Warnke C, von Geldern G, Markwerth P, Dehmel T, Hoepner R, Gold R, et al. Cerebrospinal fluid JC virus antibody index for diagnosis of natalizumab-associated progressive multifocal leukoencephalopathy. Ann Neurol (2014) 76(6):792–801. doi:10.1002/ana.24153
136. Serana F, Chiarini M, Sottini A, Bertoli D, Giustini V, Tessitore MV, et al. Immunological biomarkers identifying natalizumab-treated multiple sclerosis patients at risk of progressive multifocal leukoencephalopathy. J Neuroimmunol (2014) 277(1–2):6–12. doi:10.1016/j.jneuroim.2014.10.011
137. Amor S, van der Star BJ, Bosca I, Raffel J, Gnanapavan S, Watchorn J, et al. Neurofilament light antibodies in serum reflect response to natalizumab treatment in multiple sclerosis. Mult Scler (2014) 20(10):1355–62. doi:10.1177/1352458514521887
138. Harrer A, Tumani H, Niendorf S, Lauda F, Geis C, Weishaupt A, et al. Cerebrospinal fluid parameters of B cell-related activity in patients with active disease during natalizumab therapy. Mult Scler (2013) 19(9):1209–12. doi:10.1177/1352458512463483
139. Boulton C, Meiser K, David OJ, Schmouder R. Pharmacodynamic effects of steady-state fingolimod on antibody response in healthy volunteers: a 4-week, randomized, placebo-controlled, parallel-group, multiple-dose study. J Clin Pharmacol (2012) 52(12):1879–90. doi:10.1177/0091270011427908
140. Bar-Or A, Freedman MS, Kremenchutzky M, Menguy-Vacheron F, Bauer D, Jodl S, et al. Teriflunomide effect on immune response to influenza vaccine in patients with multiple sclerosis. Neurology (2013) 81(6):552–8. doi:10.1212/WNL.0b013e31829e6fbf
141. Sergott RC, Bennett JL, Rieckmann P, Montalban X, Mikol D, Freudensprung U, et al. ATON: results from a phase II randomized trial of the B-cell-targeting agent atacicept in patients with optic neuritis. J Neurol Sci (2015) 351(1–2):174–8. doi:10.1016/j.jns.2015.02.019
142. Genovese MC, Kinnman N, de La Bourdonnaye G, Pena Rossi C, Tak PP. Atacicept in patients with rheumatoid arthritis and an inadequate response to tumor necrosis factor antagonist therapy: results of a phase II, randomized, placebo-controlled, dose-finding trial. Arthritis Rheum (2011) 63(7):1793–803. doi:10.1002/art.30373
143. Tak PP, Thurlings RM, Rossier C, Nestorov I, Dimic A, Mircetic V, et al. Atacicept in patients with rheumatoid arthritis: results of a multicenter, phase Ib, double-blind, placebo-controlled, dose-escalating, single- and repeated-dose study. Arthritis Rheum (2008) 58(1):61–72. doi:10.1002/art.23178
144. Huang H, Ito K, Dangond F, Dhib-Jalbut S. Effect of interferon beta-1a on B7.1 and B7.2 B-cell expression and its impact on T-cell proliferation. J Neuroimmunol (2013) 258(1–2):27–31. doi:10.1016/j.jneuroim.2013.02.010
145. Ramgolam VS, Sha Y, Marcus KL, Choudhary N, Troiani L, Chopra M, et al. B cells as a therapeutic target for IFN-beta in relapsing-remitting multiple sclerosis. J Immunol (2011) 186(7):4518–26. doi:10.4049/jimmunol.1000271
146. Kuchroo VK, Das MP, Brown JA, Ranger AM, Zamvil SS, Sobel RA, et al. B7-1 and B7-2 costimulatory molecules activate differentially the Th1/Th2 developmental pathways: application to autoimmune disease therapy. Cell (1995) 80(5):707–18. doi:10.1016/0092-8674(95)90349-6
147. Sellner J, Koczi W, Harrer A, Oppermann K, Obregon-Castrillo E, Pilz G, et al. Glatiramer acetate attenuates the pro-migratory profile of adhesion molecules on various immune cell subsets in multiple sclerosis. Clin Exp Immunol (2013) 173(3):381–9. doi:10.1111/cei.12125
148. Krumbholz M, Faber H, Steinmeyer F, Hoffmann LA, Kümpfel T, Pellkofer H, et al. Interferon-beta increases BAFF levels in multiple sclerosis: implications for B cell autoimmunity. Brain (2008) 131(Pt 6):1455–63. doi:10.1093/brain/awn077
149. Yang M, Sun L, Wang S, Ko KH, Xu H, Zheng BJ, et al. Novel function of B cell-activating factor in the induction of IL-10-producing regulatory B cells. J Immunol (2010) 184(7):3321–5. doi:10.4049/jimmunol.0902551
150. Hartung HP, Kieseier BC. Atacicept: targeting B cells in multiple sclerosis. Ther Adv Neurol Disord (2010) 3(4):205–16. doi:10.1177/1756285610371146
151. Kappos L, Hartung HP, Freedman MS, Boyko A, Radü EW, Mikol DD, et al. Atacicept in multiple sclerosis (ATAMS): a randomised, placebo-controlled, double-blind, phase 2 trial. Lancet Neurol (2014) 13(4):353–63. doi:10.1016/S1474-4422(14)70028-6
152. Jacobs LD, Cookfair DL, Rudick RA, Herndon RM, Richert JR, Salazar AM, et al. A phase III trial of intramuscular recombinant interferon beta as treatment for exacerbating-remitting multiple sclerosis: design and conduct of study and baseline characteristics of patients. Multiple Sclerosis Collaborative Research Group (MSCRG). Mult Scler (1995) 1(2):118–35.
153. Paty DW, Li DK. Interferon beta-1b is effective in relapsing-remitting multiple sclerosis. II. MRI analysis results of a multicenter, randomized, double-blind, placebo-controlled trial. UBC MS/MRI Study Group and the IFNB Multiple Sclerosis Study Group. Neurology (1993) 43(4):662–7. doi:10.1212/WNL.43.4.662
154. Panitch H, Goodin D, Francis G, Chang P, Coyle P, O’Connor P, et al. Benefits of high-dose, high-frequency interferon beta-1a in relapsing-remitting multiple sclerosis are sustained to 16 months: final comparative results of the EVIDENCE trial. J Neurol Sci (2005) 239(1):67–74. doi:10.1016/j.jns.2005.08.003
155. Panitch H, Goodin DS, Francis G, Chang P, Coyle PK, O’Connor P, et al. Randomized, comparative study of interferon beta-1a treatment regimens in MS: the EVIDENCE trial. Neurology (2002) 59(10):1496–506. doi:10.1212/01.WNL.0000034080.43681.DA
156. Schwid SR, Thorpe J, Sharief M, Sandberg-Wollheim M, Rammohan K, Wendt J, et al. Enhanced benefit of increasing interferon beta-1a dose and frequency in relapsing multiple sclerosis: the EVIDENCE Study. Arch Neurol (2005) 62(5):785–92. doi:10.1001/archneur.62.5.785
157. Dhib-Jalbut S, Marks S. Interferon-beta mechanisms of action in multiple sclerosis. Neurology (2010) 74(Suppl 1):S17–24. doi:10.1212/WNL.0b013e3181c97d99
158. Mikol DD, Barkhof F, Chang P, Coyle PK, Jeffery DR, Schwid SR, et al. Comparison of subcutaneous interferon beta-1a with glatiramer acetate in patients with relapsing multiple sclerosis (the REbif vs glatiramer acetate in relapsing MS disease [REGARD] study): a multicentre, randomised, parallel, open-label trial. Lancet Neurol (2008) 7(10):903–14. doi:10.1016/S1474-4422(08)70200-X
159. Duda PW, Schmied MC, Cook SL, Krieger JI, Hafler DA. Glatiramer acetate (copaxone) induces degenerate, Th2-polarized immune responses in patients with multiple sclerosis. J Clin Invest (2000) 105(7):967–76. doi:10.1172/JCI8970
160. Hong J, Li N, Zhang X, Zheng B, Zhang JZ. Induction of CD4+CD25+ regulatory T cells by copolymer-I through activation of transcription factor Foxp3. Proc Natl Acad Sci U S A (2005) 102(18):6449–54. doi:10.1073/pnas.0502187102
161. Karandikar NJ, Crawford MP, Yan X, Ratts RB, Brenchley JM, Ambrozak DR, et al. Glatiramer acetate (copaxone) therapy induces CD8(+) T cell responses in patients with multiple sclerosis. J Clin Invest (2002) 109(5):641–9. doi:10.1172/JCI200214380
162. Vieira PL, Heystek HC, Wormmeester J, Wierenga EA, Kapsenberg ML. Glatiramer acetate (copolymer-1, copaxone) promotes Th2 cell development and increased IL-10 production through modulation of dendritic cells. J Immunol (2003) 170(9):4483–8. doi:10.4049/jimmunol.170.9.4483
163. Confavreux C, O’Connor P, Comi G, Freedman MS, Miller AE, Olsson TP, et al. Oral teriflunomide for patients with relapsing multiple sclerosis (TOWER): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Neurol (2014) 13(3):247–56. doi:10.1016/S1474-4422(13)70308-9
164. O’Connor P, Wolinsky JS, Confavreux C, Comi G, Kappos L, Olsson TP, et al. Randomized trial of oral teriflunomide for relapsing multiple sclerosis. N Engl J Med (2011) 365(14):1293–303. doi:10.1056/NEJMoa1014656
165. Vermersch P, Czlonkowska A, Grimaldi LM, Confavreux C, Comi G, Kappos L, et al. Teriflunomide versus subcutaneous interferon beta-1a in patients with relapsing multiple sclerosis: a randomised, controlled phase 3 trial. Mult Scler (2014) 20(6):705–16. doi:10.1177/1352458513507821
166. Freedman MS. Teriflunomide in relapsing multiple sclerosis: therapeutic utility. Ther Adv Chronic Dis (2013) 4(5):192–205. doi:10.1177/2040622313492810
167. Bar-Or A, Pachner A, Menguy-Vacheron F, Kaplan J, Wiendl H. Teriflunomide and its mechanism of action in multiple sclerosis. Drugs (2014) 74(6):659–74. doi:10.1007/s40265-014-0212-x
168. Carrithers MD. Update on disease-modifying treatments for multiple sclerosis. Clin Ther (2014) 36(12):1938–45. doi:10.1016/j.clinthera.2014.08.006
169. Rudick R, Polman C, Clifford D, Miller D, Steinman L. Natalizumab: bench to bedside and beyond. JAMA Neurol (2013) 70(2):172–82. doi:10.1001/jamaneurol.2013.598
170. Polman CH, O’Connor PW, Havrdova E, Hutchinson M, Kappos L, Miller DH, et al. A randomized, placebo-controlled trial of natalizumab for relapsing multiple sclerosis. N Engl J Med (2006) 354(9):899–910. doi:10.1056/NEJMoa044397
171. Rudick RA, Stuart WH, Calabresi PA, Confavreux C, Galetta SL, Radue EW, et al. Natalizumab plus interferon beta-1a for relapsing multiple sclerosis. N Engl J Med (2006) 354(9):911–23. doi:10.1056/NEJMoa044396
172. Cohen JA, Barkhof F, Comi G, Hartung HP, Khatri BO, Montalban X, et al. Oral fingolimod or intramuscular interferon for relapsing multiple sclerosis. N Engl J Med (2010) 362(5):402–15. doi:10.1056/NEJMoa0907839
173. Comi G, O’Connor P, Montalban X, Antel J, Radue EW, Karlsson G, et al. Phase II study of oral fingolimod (FTY720) in multiple sclerosis: 3-year results. Mult Scler (2010) 16(2):197–207. doi:10.1177/1352458509357065
174. Kappos L, Radue EW, O’Connor P, Polman C, Hohlfeld R, Calabresi P, et al. A placebo-controlled trial of oral fingolimod in relapsing multiple sclerosis. N Engl J Med (2010) 362(5):387–401. doi:10.1056/NEJMoa0909494
175. O’Connor P, Comi G, Montalban X, Antel J, Radue EW, de Vera A, et al. Oral fingolimod (FTY720) in multiple sclerosis: two-year results of a phase II extension study. Neurology (2009) 72(1):73–9. doi:10.1212/01.wnl.0000338569.32367.3d
176. Brinkmann V, Billich A, Baumruker T, Heining P, Schmouder R, Francis G, et al. Fingolimod (FTY720): discovery and development of an oral drug to treat multiple sclerosis. Nat Rev Drug Discov (2010) 9(11):883–97. doi:10.1038/nrd3248
177. Fox EJ, Rhoades RW. New treatments and treatment goals for patients with relapsing-remitting multiple sclerosis. Curr Opin Neurol (2012) 25(Suppl):S11–9. doi:10.1097/01.wco.0000413320.94715.e9
178. Mehling M, Johnson TA, Antel J, Kappos L, Bar-Or A. Clinical immunology of the sphingosine 1-phosphate receptor modulator fingolimod (FTY720) in multiple sclerosis. Neurology (2011) 76(8 Suppl 3):S20–7. doi:10.1212/WNL.0b013e31820db341
179. Pelletier D, Hafler DA. Fingolimod for multiple sclerosis. N Engl J Med (2012) 366(4):339–47. doi:10.1056/NEJMct1101691
180. von Wenckstern H, Zimmermann K, Kleuser B. The role of the lysophospholipid sphingosine 1-phosphate in immune cell biology. Arch Immunol Ther Exp (Warsz) (2006) 54(4):239–51. doi:10.1007/s00005-006-0028-9
181. Barun B, Bar-Or A. Treatment of multiple sclerosis with anti-CD20 antibodies. Clin Immunol (2012) 142(1):31–7. doi:10.1016/j.clim.2011.04.005
182. Gürcan HM, Keskin DB, Stern JN, Nitzberg MA, Shekhani H, Ahmed AR. A review of the current use of rituximab in autoimmune diseases. Int Immunopharmacol (2009) 9(1):10–25. doi:10.1016/j.intimp.2008.10.004
183. Lehmann-Horn K, Kinzel S, Feldmann L, Radelfahr F, Hemmer B, Traffehn S, et al. Intrathecal anti-CD20 efficiently depletes meningeal B cells in CNS autoimmunity. Ann Clin Transl Neurol (2014) 1(7):490–6. doi:10.1002/acn3.71
184. Monson NL, Cravens PD, Frohman EM, Hawker K, Racke MK. Effect of rituximab on the peripheral blood and cerebrospinal fluid B cells in patients with primary progressive multiple sclerosis. Arch Neurol (2005) 62(2):258–64. doi:10.1001/archneur.62.2.258
185. Pescovitz MD. Rituximab, an anti-cd20 monoclonal antibody: history and mechanism of action. Am J Transplant (2006) 6(5 Pt 1):859–66. doi:10.1111/j.1600-6143.2006.01288.x
186. Kappos L, Li D, Calabresi PA, O’Connor P, Bar-Or A, Barkhof F, et al. Ocrelizumab in relapsing-remitting multiple sclerosis: a phase 2, randomised, placebo-controlled, multicentre trial. Lancet (2011) 378(9805):1779–87. doi:10.1016/S0140-6736(11)61649-8
187. Kausar F, Mustafa K, Sweis G, Sawaqed R, Alawneh K, Salloum R, et al. Ocrelizumab: a step forward in the evolution of B-cell therapy. Expert Opin Biol Ther (2009) 9(7):889–95. doi:10.1517/14712590903018837
188. Sorensen PS, Lisby S, Grove R, Derosier F, Shackelford S, Havrdova E, et al. Safety and efficacy of ofatumumab in relapsing-remitting multiple sclerosis: a phase 2 study. Neurology (2014) 82(7):573–81. doi:10.1212/WNL.0000000000000125
189. Osterborg A. Ofatumumab, a human anti-CD20 monoclonal antibody. Expert Opin Biol Ther (2010) 10(3):439–49. doi:10.1517/14712590903586239
190. Østergaard M, Baslund B, Rigby W, Rojkovich B, Jorgensen C, Dawes PT, et al. Ofatumumab, a human anti-CD20 monoclonal antibody, for treatment of rheumatoid arthritis with an inadequate response to one or more disease-modifying antirheumatic drugs: results of a randomized, double-blind, placebo-controlled, phase I/II study. Arthritis Rheum (2010) 62(8):2227–38. doi:10.1002/art.27524
191. Reagan JL, Castillo JJ. Ofatumumab as front-line therapy in untreated chronic lymphocytic leukemia. Future Oncol (2014) 10(7):1147–55. doi:10.2217/fon.14.73
192. Cohen JA, Coles AJ, Arnold DL, Confavreux C, Fox EJ, Hartung HP, et al. Alemtuzumab versus interferon beta 1a as first-line treatment for patients with relapsing-remitting multiple sclerosis: a randomised controlled phase 3 trial. Lancet (2012) 380(9856):1819–28. doi:10.1016/S0140-6736(12)61769-3
193. Coles AJ, Fox E, Vladic A, Gazda SK, Brinar V, Selmaj KW, et al. Alemtuzumab more effective than interferon beta-1a at 5-year follow-up of CAMMS223 clinical trial. Neurology (2012) 78(14):1069–78. doi:10.1212/WNL.0b013e31824e8ee7
194. Coles AJ, Twyman CL, Arnold DL, Cohen JA, Confavreux C, Fox EJ, et al. Alemtuzumab for patients with relapsing multiple sclerosis after disease-modifying therapy: a randomised controlled phase 3 trial. Lancet (2012) 380(9856):1829–39. doi:10.1016/S0140-6736(12)61768-1
195. Benson MJ, Dillon SR, Castigli E, Geha RS, Xu S, Lam KP, et al. Cutting edge: the dependence of plasma cells and independence of memory B cells on BAFF and APRIL. J Immunol (2008) 180(6):3655–9. doi:10.4049/jimmunol.180.6.3655
Keywords: multiple sclerosis, B cell subtypes, therapy, antibodies, cytokines, costimulation, antigen presentation
Citation: Claes N, Fraussen J, Stinissen P, Hupperts R and Somers V (2015) B Cells Are Multifunctional Players in Multiple Sclerosis Pathogenesis: Insights from Therapeutic Interventions. Front. Immunol. 6:642. doi: 10.3389/fimmu.2015.00642
Received: 30 June 2015; Accepted: 07 December 2015;
Published: 21 December 2015
Edited by:
Manu Rangachari, CHU de Québec, CanadaReviewed by:
Patrick Burkett, Brigham and Women’s Hospital, USACopyright: © 2015 Claes, Fraussen, Stinissen, Hupperts and Somers. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Somers Veerle, dmVlcmxlLnNvbWVyc0B1aGFzc2VsdC5iZQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.