 Sushmita Sinha
Sushmita Sinha Alexander W. Boyden
Alexander W. Boyden Farah R. Itani
Farah R. Itani Michael P. Crawford
Michael P. Crawford Nitin J. Karandikar
Nitin J. Karandikar
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 10 December 2015
Sec. Multiple Sclerosis and Neuroimmunology
Volume 6 - 2015 | https://doi.org/10.3389/fimmu.2015.00619
This article is part of the Research Topic Lymphocytes in MS and EAE: more than just a CD4+ world View all 17 articles
The vast majority of studies regarding the immune basis of MS (and its animal model, EAE) have largely focused on CD4+ T-cells as mediators and regulators of disease. Interestingly, CD8+ T-cells represent the predominant T-cell population in human MS lesions and are oligoclonally expanded at the site of pathology. However, their role in the autoimmune pathologic process has been both understudied and controversial. Several animal models and MS patient studies support a pathogenic role for CNS-specific CD8+ T-cells, whereas we and others have demonstrated a regulatory role for these cells in disease. In this review, we describe studies that have investigated the role of CD8+ T-cells in MS and EAE, presenting evidence for both pathogenic and regulatory functions. In our studies, we have shown that cytotoxic/suppressor CD8+ T-cells are CNS antigen-specific, MHC class I-restricted, IFNγ- and perforin-dependent, and are able to inhibit disease. The clinical relevance for CD8+ T-cell suppressive function is best described by a lack of their function during MS relapse, and importantly, restoration of their suppressive function during quiescence. Furthermore, CD8+ T-cells with immunosuppressive functions can be therapeutically induced in MS patients by glatiramer acetate (GA) treatment. Unlike CNS-specific CD8+ T-cells, these immunosuppressive GA-induced CD8+ T-cells appear to be HLA-E restricted. These studies have provided greater fundamental insight into the role of autoreactive as well as therapeutically induced CD8+ T-cells in disease amelioration. The clinical implications for these findings are immense and we propose that this natural process can be harnessed toward the development of an effective immunotherapeutic strategy.
Studies addressing the immunobiology of multiple sclerosis (MS) and its animal model experimental autoimmune encephalomyelitis (EAE) have focused on CD4+ T-cells as the main orchestrators of pathogenesis and regulation. CD8+ T-cells are the most abundant T-cells in CNS lesions of MS patients (1) and exhibit oligoclonal expansion (2–4). This indicates an important role for these cells in the target organ. However, the functional nature of these cells during disease and its treatment is unclear and somewhat controversial. There are abundant CNS-specific (5, 6) and therapeutically induced CD8+ T-cell responses in MS patients (5–8). Recent studies suggest that certain MHC class I alleles can be associated with genetic risk or protection in MS (9–11). Functional roles for some of these MHC class I molecules have been tested in the EAE models. 2D1-TCR humanized transgenic mice, expressing MS risk variant HLA-A3 together with TCR that recognizes myelin proteolipid protein (PLP), develope spontaneous EAE in only 4% of mice and mild EAE early on when immunized with PLP peptide. A quarter of these mice went on to develop a severe disease course with 2D1+-TCR+–CD8+ T-cells present in the CNS of these mice, suggesting a pathogenic role for HLA-A3-restricted myelin-specific CD8+ T-cells (12). However, introduction of HLA-A2 alleles in the same model completely abrogates spontaneous and induced EAE, providing evidence for the protective role for HLA-A2-restricted CD8+ T-cells (12). We are only beginning to understand these responses and here attempt to provide an overview of such studies. We will summarize the evidence for both pathogenic and regulatory functions of CD8+ T-cells in MS and EAE. We will provide an overview of the various cellular and molecular interactions that mediate the role of these cells and develop a model for such functions during disease.
Much of the focus regarding the pathogenesis of EAE has revolved primarily around myelin-specific CD4+ T-cells. Adoptive transfer of CD4+ T-cells isolated from myelin antigen-primed animals is sufficient to induce disease. This observation partly facilitated the overall ignorance surrounding CD8+ T-cells and their potential contribution to disease. A pathogenic role first became evident when a CD8+ T-cell-mediated model of EAE was developed using the self-protein myelin basic protein (MBP) (13). In attempts to prime an MHC class I-restricted T-cell response, C3H.Fej, and C3H MBP-deficient shiverer mice were infected with MBP-expressing vaccinia. CD8+ T-cell lines specific for MBP79–87 drove pathogenesis and demyelination when transferred into wildtype (WT) C3H recipients. Mice developed neurological symptoms including ataxia, spasticity, and lost weight when compared to control animals that received vaccinia-specific CD8+ T-cells. Histologically, perivascular cuffs composed primarily of lymphocytes and macrophages were detected in the brain but not in the spinal cord. IFNγ was found to play an important role in mediating MBP-specific CD8+ T-cell-driven disease, as its neutralization reduced severity. The break of peripheral tolerance following viral infection was also shown to induce CD8+ T-cell-mediated CNS autoimmunity (14). In this report, dual TCR-expressing CD8+ T-cells recognizing both viral antigen and MBP triggered disease. Following viral infection, CD8+ T-cells, macrophages, and activated microglia infiltrated both the brain and spinal cord. Clinically, mice lost weight and exhibited symptoms of ataxia, impaired mobility, and tail weakness.
CD8+ T-cell-mediated EAE has also been induced in C57BL/6 (B6) mice through transfer of myelin oligodendrocyte glycoprotein (MOG)-specific CD8+ T-cells (15). MOG-specific CD8+ T-cells isolated from mice immunized with MOG35–55 peptide were encephalitogenic, and transferred severe paralytic disease to B6 mice. One caveat to this study is that cells were nylon wool-enriched, calling purity into question. Disease was transferred using <1e6 MOG35–55 CD8+ T-cells and resulted in more severe EAE compared to active immunization. Transferred cells could be re-isolated 6–8 months later, possibly due to additional IL-2 stimulus. How these cells induced pathology was not investigated.
A separate group identified MOG37–46-specific CD8+ T-cells as autoaggressive effectors (16). In this system, MOG-specific CD8+ T-cells were generated following immunization. Restimulation with antigen and IL-2 readily yielded IFNγ from these cells, but not TGFβ or IL-10. These cells, which were found to be H-2Db-restricted, could induce EAE when transferred into SCID or naïve WT B6 recipients. MOG37–46 elicited the best IFNγ response from MOG35–55-primed lymph node cells, although bound MHC poorly. When used to induce active EAE in B6 mice, MOG37–46 led to similar disease as MOG35–55-immunized mice. Using MOG37–50/H-2Db tetramers, MOG-specific CD8+ T-cells were found to persist within the CNS.
While these studies utilized myelin-components to examine the potential pathogenic role of CD8+ T-cells in EAE, non-myelin antigen-driven systems have been used as well. One report describes CD8+ TCR transgenic mice recognizing glial fibrillary acidic protein (GFAP), an intermediate filament protein expressed in the CNS by astrocytes and in various peripheral tissues (17). BG1 transgenic mice are reactive to the GFAP264–274 peptide presented on H-2Kb, and develop spontaneous inflammatory CNS disease by 6–12 months of age. Interestingly, GFAP-expressing vaccinia induced distinct disease pathology compared to spontaneous disease. Lesion localization and clinical manifestations of disease was dependent upon how CNS-reactive CD8+ T-cells were activated. CD8+ T-cells isolated from brains of WT BG1 mice were poor secretors of IFNγ, IL-17A, and granzyme B, suggesting alternative effector mechanisms.
Efforts to study the role of Src homology 2 domain-containing protein tyrosine phosphatase (SHP-2) in EAE demonstrated that disease could be ameliorated through phosphatase inhibition (18). The competitive inhibitor, NSC-87877 led to reduced demyelination and blocked CD8+ but not CD4+ T-cell migration into the CNS, suggesting a pathogenic role for CD8+ T-cells in this model.
A study of engineered transgenic NOD mice expressing a MOG35–55-reactive TCR (1C6) lends further support for pathogenic CD8+ T-cells in EAE (19). 1C6 mice spontaneously generated MOG-specific CD4+ and CD8+ T-cells that secrete pro-inflammatory cytokines. 1C6 CD8+ T-cells could recognize MOG35–55 in the context of MHC class I and II, and when adoptively transferred into NOD. Scid recipients, induced optic neuritis and mild EAE, while 1C6 CD4+ T-cells induced severe EAE.
CD8+ T-cells’ ability to target CNS components has also been evaluated in several viral models (20–22). LCMV GP33 peptide-specific CD8+ T-cells can induce lesions in cultured murine neurons presenting GP33 in MHC class I. While this report relies on peptide pulsing and artificial upregulation of MHC class I, viral infection-induced upregulation of class I has been demonstrated in Borna disease virus-infected rat neuronal cultures, which could be targeted by antiviral CD8+ T-cells, eventually leading to apoptosis of neurons (23). Although electrical signals were not initially disrupted in this model and longer incubation times were needed for neuronal apoptosis, another study has demonstrated impaired murine neuronal signaling following neuron/CD8+ T-cell interactions along with eventual apoptosis which interestingly occurred independent of perforin/granzymes (24). To this end, IFNγ-production from CNS CD8+ T-cells and subsequent IFNγ signaling in neurons has been shown to be significant for intracranial LCMV disease in mice (25). In another study, OT-I CD8+ T-cells formed immune synapses with MHC class I (H-2Kb)-expressing axons presenting SIINFEKL peptide, and loss of axon integrity was observed. Additionally, axonal injury was dependent upon antigen-specific TCR recognition and granzyme B (26).
Another report also described mice expressing neo-self antigen in oligodendrocytes (ODCs) targeted by transgenic CD8+ T-cells (27). In this model, ovalbumin was expressed exclusively in the cytosol of ODCs and therefore ignored by CD4+ T-cells and B-cells. Following immunization, mild EAE was observed in some ODC–OVA mice. Studies using double transgenic ODC–OVA/OT-I mice demonstrate treatment with D1 mAb (specific for H-2Kb/OVA) prevented the lethal EAE normally observed in these animals (28). Double transgenic mice were also given D1 prophylactically, which in certain instances led to spontaneous disease remission.
CD8+ T-cells have also been shown to indirectly influence CNS autoimmunity. Tc17 cells, coined for their ability to produce IL-17A, were detected in the lymph nodes and CNS of MOG37–50 EAE mice (29). Tc17s differ from conventional CD8+ T-cells regarding granzyme B and IFNγ expression, and thus are impaired in their cytotoxic capacity. In a separate study implementing CD4+ and CD8+ T-cell co-transfer, Tc17 cells were found to help CD4+ Th17 cells accumulate in the CNS and induce EAE (30). Furthermore, their ability to produce IL-17A was required to render CD4+ T-cells encephalitogenic.
While evidence exists to suggest a pathogenic role for CD8+ T-cells in MS and EAE (reviewed in Ref. (31) and discussed above), there is a growing body of evidence supporting the opposite conclusion – CD8+ T-cells play an important regulatory role in the pathogenesis of MS and MS-like disease. Ultimately, CD8+ T-cell subsets likely perform varying effector functions in the context of MS/EAE. However, the seeming discrepancy is in part due to a lack of concrete in vivo evidence demonstrating a cytotoxic effect of CD8+ T-cells in MS lesions. Furthermore, it has been demonstrated that depletion of CD8+ T-cells prior to EAE induction results in exacerbated disease (32). Similar results are seen in mice lacking MHC class I (although a role for NK cells can be argued) (33) and in CD8-deficient mice (32, 34, 35). This is in addition to work from our lab, which clearly demonstrated – in marked contrast to their CD4+ counterparts – neuroantigen-specific CD8+ T-cells failed to adoptively transfer EAE disease to naïve recipient mice (36). We have seen this protective CD8+ T-cells phenotype very robustly in several models of EAE (37).
The notion of a regulatory CD8+ T-cell subset (CD8+ Tregs) in MS is not a new idea. Studies spanning several decades point to the suppressive potential of CD8+ T-cells in MS patients (5–8, 38–41). In lieu of these examples, T-cell-mediated tolerance studies have largely focused on CD4+CD25+Foxp3+ T-cells. Although full appreciation of CD8+ Treg function and significance in MS and EAE is lacking, the last 15 years have seen a steady growth toward this understanding.
CD8+ T-cells’ suppressive ability has been described in many mouse models, including cancer (42), diabetes (43), colitis (44), SLE-like disease (45), Grave’s disease (46), and transplant tolerance (47). Inhibitory CD8+ T-cell subsets involved in autoimmunity in both mice and humans have been exhaustively reviewed in Ref. (48). These regulatory CD8+ T-cells have been extensively studied in T1D where it has been shown that low-avidity autoreactive CD8+ T-cells convert into memory-like autoregulatory cells and blunt diabetes progression (49, 50). However, CD8+ Treg participation in EAE is less-widely studied. Moreover, unlike murine CD4+Foxp3+ Tregs, a universal CD8+ Treg phenotype has yet to be described. For example, in EAE, CD8+CD28− T-cells have been shown to play an inhibitory role (32) while others show CD8+CD122+ T-cells to be protective (51–53). Little is known concerning the induction of these cells in MS-like disease, though the involvement of one subtype versus another surely is influenced by disease setting and may depend on the cell’s antigen specificity/MHC-restriction. Studies of anterior chamber-associated immune deviation (ACAID) represent some of the best efforts to understand antigen-specific CD8+ Tregs, which appear to be Qa-1-restricted (54–56). Several ACAID studies further complicate the CD8+ Treg phenotyping picture (e.g., Foxp3+, CD94+, CD103+, TGFβ-producing, etc.) (56–60). Interestingly, immune deviation can be elicited against myelin antigens (61, 62), pointing to the potential role for Qa-1-restricted CD8+ T-cells in EAE disease. Qa-1-restricted CD8+ T-cells have been described as being important for protection in MBP-driven EAE (63). We have demonstrated that Qa-1-restricted CD8+ T-cells suppress EAE. We have also demonstrated that GA treatment induces CD8+ Treg in mice, and that these CD8+ T-cells are required for GA to be therapeutically effective in ameliorating EAE disease (64).
While little is still known about Qa-1-restricted CD8+ Tregs, even less was understood about CNS-specific CD8+ T-cells until very recently. We observed the surprising result that neuroantigen-specific CD8+ T-cells could suppress EAE induction and even ameliorate established EAE disease (36). To our knowledge, this was the first documentation of neuroantigen-specific CD8+ Tregs in mice. In our recently published and unpublished results, adoptive transfer of both MOG35–55- and PLP178–191-specific CD8+ T-cells can suppress EAE (34, 65). Due to mechanistic studies, we will elaborate upon later that these cells are quite distinct from previously described Qa-1-restricted CD8+ Tregs (37).
Recent work has suggested a role of IL-10-producing CD8+ T-cells in diminishing disease pathology in virus-induced encephalitis models. These IL-10-producing CD8+ T-cells display a more functional profile including increased expression of pro-inflammatory cytokines and chemokines, are immunosuppressive, and their presence in the CNS following Coronavirus infection reduces tissue destruction and morbidity in these mice (66).
Advancement in therapy for MS patients, particularly cellular immunotherapy, necessitates the full understanding of regulatory immune cell interplay. Studies concerning the functional interactions between CD8+ Tregs and other cells in the context of MS and MS-like disease are therefore of paramount interest. The next several sections will provide mechanistic insights into CD8+ T-cell-mediated modulation of other immune cells including CD4+ T-cells and antigen presenting cell (APC) populations.
Qa-1-restricted CD8+ T-cells have been shown to modulate EAE disease through action on CD4+ T-cells. It has been demonstrated in a model of MBP-driven EAE that CD4+ T-cell vaccination protocol-mediated protection against EAE disease is dependent on the presence of Qa-1-restricted CD8+ T-cells that recognize specific TCRVβ molecules on MBP-reactive CD4+ T-cells (63). In this particular example, CD8+ T-cells mediated their control by preferentially suppressing Th1 CD4+ T-cells during EAE. While this report did not directly test cytotoxic killing as a means of suppression, the group had previously established this capability in T-cell vaccination scenarios. Data from another group later confirmed a cytotoxic effect by demonstrating that CD8αα+TCRαβ+ T-cells from lines that recognize TCRVβ8.2+ (MBP-reactive) CD4+ T-cells could protect against EAE disease in recipient mice by the targeted killing of these pathogenic cells via Qa-1-recognition (67).
We have showed that the disease-ameliorating effect of GA-therapy in EAE is dependent upon Qa-1-restricted CD8+ Tregs (64). In this report, we demonstrated that the protective ability of CD8+ T-cells was completely lost or diminished when unable to produce IFNγ or perforin, respectively. These CD8+ T-cells could kill GA-loaded target T-cells and even limited the proliferation of ex vivo neuroantigen-specific CD4+ T-cells (64). Furthermore, the GA-induced Qa-1-restricted CD8+ T-cells in this study were important for generation of CD4+ Tregs (64). These GA-specific CD8+ T-cells have the potential to kill GA-expressing CD4+ T-cells and limit proliferation of neuroantigen-specific and anti-CD3-stimulated CD4+ T-cells (8, 40). We have also demonstrated that GA therapy, whose effects require CD8+ T-cells in mice (64), was able to increase the induction of CD4+CD25+ Tregs from the CD4+CD25− T-cell population in MS patient blood (40).
Distinct from the non-classical HLA-E-like Qa-1-restricted murine CD8+ Tregs, we have also demonstrated the existence of neuroantigen-specific CD8+ Tregs in MS and EAE. Neuroantigen-specific, MHC class Ia-restricted CD8+ T-cells can kill MOG-loaded CD4+ T-cells in mice (34, 36) and mediate their disease-ameliorating effects via the targeting of encephalitogenic CD4+ T-cells during EAE disease (34). We have also demonstrated an ability of neuroantigen-specific CD8+ Tregs to induce anti-inflammatory profiles in CD4+ T-cells during EAE (65). Importantly, we have also shown that neuroantigen-specific CD8+ T-cells are detectable in MS patient blood, and possess capacity to suppress CD4+ T-cell proliferation (5, 68).
The potential for CD8+ T-cells to alter CD4+ T-cell priming through direct effects on DCs is worth investigation. CD8+CD28− T-cells have been implicated as regulators of EAE disease. It has been demonstrated that DCs have reduced costimulatory molecule (CD80, CD86, and CD40) expression after culture with CD8+CD28− T regulatory cells, rendering these DCs as substandard APCs (32). It has been similarly demonstrated that DCs cultured with CD8+CD122+ T-cells had a reduction in CD80/86 and MHC molecules and showed inferior antigen-presentation ability compared to DCs cultured with CD8+CD122− T-cells (52).
While it remains unclear whether Qa-1-restricted CD8+ Tregs have a direct effect on DCs, we have shown that neuroantigen-specific CD8+ Tregs can both kill and suppress antigen presentation of MOG-loaded bulk APCs (contains DCs) (36). Interestingly, we have demonstrated that neuroantigen-specific CD8+ Tregs have little effect on DC surface expression of MHC or costimulatory molecules, but rather shift the inflammatory profiles of CD11c+ DCs from IL-12 to IL-10 (65). Early human MS work from our lab points to the potential of GA therapy-induced CD8+ Treg-mediated killing of APCs, as CD4+ T-cells were only a part of the larger target pool (8).
Another potential mechanism of suppression is CD8+ T-cell-mediated regulation of monocytes or macrophages, which are present in MS lesions and important for pathology in the CNS of EAE mice. Interestingly, GA treatment has been demonstrated to affect monocyte populations in EAE. For example, anti-inflammatory type II monocytes are induced in GA-treated mice, which can shift inflammatory cytokine profiles toward immunosuppressive IL-10, expand Th2 cells, and induce CD4+ Tregs capable of ameliorating EAE (69). We have observed similar results and have further demonstrated that the action of GA on monocytes elicits CD8+ Tregs and actually requires CD8+ T-cells for its ameliorative effects in EAE (64). This GA-induced monocyte-CD8+ T-cell interaction is largely unknown in MS, as is the effect of GA-induced CD8+ T-cell targeting of other macrophage populations. While a direct link to CD8+ T-cells has yet to be confirmed, studies from us and others have shown modulation of monocytes following GA therapy in humans (40, 70, 71). As mentioned in the section above, GA-induced CD8+ T-cell-mediated killing of APC populations like dendritic cells and monocytes/macrophages while unconfirmed, cannot be ruled out, as CD4+ T cells were only a portion of a larger affected target pool (8). Refining these assays for direct detection of killed targets is needed going forward.
Beyond GA-induced CD8+ Tregs, neuroantigen-specific CD8+ Tregs could conceivably modulate monocytes/macrophages in EAE. We have demonstrated that these cells can kill MOG-loaded bulk APCs, which may contain monocytes/macrophages, and can suppress their antigen presentation (36). However, we did not observe a substantial neuroantigen-specific CD8+ Treg effect on monocytes during EAE (65). Furthermore, neuroantigen-specific CD8+ Tregs from MS patients do not appear to specifically target monocytes. More work is needed to understand the potential functional interactions between CD8+ T-cells and monocytes/macrophages during MS and MS-like disease, and may ultimately be a GA treatment-specific phenomenon.
In light of depletion therapy success, more focus is now being given to B-cells and their role in MS. The literature supports both a pathogenic (72–82) and regulatory (76, 83–94) role for B-cells in MS/EAE, and it is intriguing to speculate about the potential immune cell interplay between CD4+ T-cells, B-cells, and CD8+ T-cells therein. There is evidence in the literature to support a B-cell effect on CD8+ T-cells (54, 55, 82, 95–100). Many of these reports point to B-cell antigen presentation to CD8+ T-cells and even a B-cell requirement for CD8+ Treg function in some models. There is also literature supporting a role for Bregs in controlling CD8+ T-cell responses (17, 101–105). Additionally, CD8+ T-cells can be detected in follicles and modulate B-cell biology, such as germinal centers and antibody production (45, 106–111). The significance of these CD8+ T cell and B-cell subset interactions in the context of MS/EAE remains to be seen.
Due to the inherent complexity of studying CD8+ T-cell function in the human brain, only circumstantial evidence exists regarding a pathogenic role for CD8+ T-cells in MS.
CD8+ T-cells are the most abundant T-cells found in the CNS lesions of MS patients, far outnumbering CD4+ T-cells (1). In patients with active disease, CD8+ T-cells were detected in increasing amounts from the center to the edge of the lesions studied (112). The CD8/CD4 ratio is shown to have been as high as 50/1 in the lymphocytic perivascular cuffs at the edge of active plaques (113). CD8+ T-cells displaying activated and memory phenotypes (suggesting previous interaction with local antigens) have also been detected in the CNS and CSF of MS patients (3, 114). CD8+ T-cell clones have also been shown to move throughout the affected CNS and into normal appearing white matter (NAWM) (112). One study demonstrated that there is diffuse infiltration by CD8+ T-cells combined with microglial activation and meningeal inflammation in the NAWM of MS patients (115).
Unfortunately, assigning function to these CD8+ T-cells remains a challenging task, although speculations have been made that CD8+ T-cells present in the CNS lesions of MS patients may be cytotoxic toward CNS cells including glia and axons. CD8+ MHC class I-restricted myelin peptide-specific T-cells have been shown to cause injury to human ODCs in vitro (116). Similarly, an MBP-specific memory phenotype CD8+ T-cell line generated from the peripheral blood of MS patients, in addition to secreting IFNγ and TNFα, was able to lyse COS-MBP/HLA-A2-transfected cells that were presenting endogenous MBP (114).
CD8+ T-cells have also been detected near or attached to ODCs and demyelinated axons in MS patients (117–119). Importantly, MHC class I molecules are present on astrocytes, ODCs, neurons, and endothelial cells (120, 121). Furthermore, MHC class I molecules are upregulated – depending on disease severity – and can be induced by IFNγ (121). CNS blood vessel endothelium as well as several APCs also express MHC class I molecules, which can cross-present exogenous peptides (122). Thus, it is not surprising that CD8+ T-cells have been demonstrated to interact with APCs at CNS plaque margins (119). The potentially detrimental nature of this interaction is supported by a study that showed that the amount of CD8+ T-cells and macrophages present in an MS lesion is proportional to the amount of acute axonal damage present (123).
Effector cytokines from CD8+ T-cells can also enhance their cytotoxic function and activate other immune cells to amplify inflammatory cascades in the CNS. For example, neuroantigen-specific CD8+ T-cells present in the peripheral blood express IFNγ and TNFα in response to their cognate antigen ex vivo (6, 124, 125). IFNγ- and IL-17-producing CD8+ T-cells can be recruited into the CNS when responding to apoptotic T-cell-associated self-epitopes (126). One report demonstrated that CD8+ but not CD4+ T-cells from patients with acute RRMS had increased ability to be recruited in inflamed CNS venules (127). Additionally, CD8+ IL-17-secreting T-cell numbers have been shown to be significantly elevated in acute CNS lesions of MS patients (128). IFNγ- and IL-17-secreting CD8+CD161+ T-cells were also found to be elevated in the peripheral blood of MS patients (129). Higher frequency of CD8+ T-cells expressing cytotoxic molecules like perforin has been shown to be present in MS patients, particularly during a relapse (130).
In light of the present literature, it can be appreciated that CD8+ T-cells in MS and other autoimmune diseases are phenotypically and functionally diverse, and can potentially regulate the pathogenic immune processes. Besides cytolytic molecules like perforin and granzyme, CD8+ T-cells are armed with immunosuppressive cytokines, such as IL-10, that can dampen the inflammatory response.
The evidence for CD8+ T-cell regulatory function in MS has existed for a long time and has been largely ignored by the field. CD8+ T-cells from the peripheral blood of MS patients displaying reduced levels of suppressor function was the first report that suggested a regulatory function for CD8+ T-cells in MS (131). This was followed by another study that demonstrated a similar defect in CD8+ T-cell-mediated suppression in patients with chronic progressive MS (132). Since then, mounting evidence has accumulated in the field of MS disease and others that collectively points toward a regulatory role for CD8+ T-cells in autoimmune diseases (50, 133, 134). More recently, our lab has provided direct evidence for CD8+ T-cell regulatory function in MS and has established clinical correlations with the disease activity (31).
As in the mouse, phenotypic identification of human CD8+ Tregs has been challenging. Human CD8+CD28− T-cells have been shown to possess suppressor activity and are the most extensively studied population of CD8+ Tregs. In MS, they were found to be present at significantly reduced frequency in the blood of RRMS patients as compared to healthy donors (135). Although it is not a marker for CD8+ Tregs, FoxP3-expressing CD8+ T-cells are present in human blood. They possess regulatory activity (136), which is Foxp3-dependent (137), and are associated with autoimmune diseases such as IBD and MS (133, 138). CD8+FoxP3+ cells are present at reduced levels in the CSF of MS patients during acute exacerbation (137). CD8+CXCR3+ T-cells are human counterparts of the well-known regulatory CD8+CD122+ T-cells found in mouse. Human CD8+CXCR3+ T-cells are suppressive in nature and their function is IL-10-dependent (139). Although all of these CD8+ Treg subsets have potent immunosuppressive functions, so far their antigen specificity remains unknown.
Our lab showed for the first time that CNS-specific CD8+ T-cells have potent suppressor activity toward myelin antigen-specific CD4+ T-cells (5). These CNS-specific CD8+ T-cells were reactive to several myelin antigens including MOG, PLP, MBP, MAG and others and are present in the peripheral blood of healthy donors and MS patients (6). Mechanistically, these CNS-specific CD8+ T-cells are MHC class I-restricted, and their suppressive function is IFNγ-and perforin-dependent (5, 68). Our findings lend credence to the hypothesis that CNS-specific CD8+ T-cells in the CNS would function to dampen the inflammatory response by targeting pathogenic CD4+ T-cells and APCs, rather than causing damage themselves. Phenotypically, these cells are CD8+CD27−CD28−CD45RO−CD62L−CD57+ or a terminally differentiated subset of CD8+ T-cells (68).
Similar to Qa1-restricted CD8+ T in murine models, HLA-E-restricted CD8+ T-cells in humans perform a regulatory function and are involved in the maintenance of self-tolerance (140). The nature of NKG2 receptors present on CD8+ T-cells determines the functional outcome of their interaction with Qa1-expressing T-cell targets. For example, NKG2C-expressing CD8+ T-cells suppress Qa1-expressing target T-cells while NKG2A-expressing CD8+ T-cells get suppressed by these targets, and therefore cannot perform regulatory functions. A recent study showed reduced expression of FoxP3 and CD122 in NKG2C-expressing CD8+ T-cells from MS patients compared to healthy controls, suggesting a reduced regulatory potential of these cells in MS patients (41).
Although, there are only a handful of studies that report the phenotypic and functional significance of CD8+ T-cells in MS patients, one prominent feature that emerges from these studies is an underlying defect in the CD8+ Treg component. Of note, this defect is found specifically during MS relapses. Since a relapse represents the active phase of the disease, any significant differences in the phenotype and functions of immune cells between relapse and remission may be directly correlated with the immunopathogenesis of MS. Interestingly, frequency of circulating CD8+FoxP3+ T-cells was found to be significantly lower in the peripheral blood of MS patients during relapse as compared to remission (138). Another study showed that CD8+CD25+CD28− T-cells harbored potent suppressive activity and were lower in MS patients during relapse when compared to healthy controls (141). Importantly, treatment with glucocorticoids leads to a significant increase in the frequency of these CD8+ Tregs in the blood of MS patients. This was an interesting observation, suggesting that recovery from relapse under glucocorticoid treatment might be mediated by the regulatory function of CD8+ T-cells. Furthermore, deficiency in CD8+ Treg function is not limited to the blood, as evidenced by the significantly reduced CD8+ T-cell cloning frequency in the CSF during MS relapse as compared to remission, suggesting loss of CD8+ Tregs in the CSF during relapse (142).
Our own studies show that the terminally differentiated CD8+ T-cell pool, which harbors the CNS-specific CD8+ Tregs, is significantly reduced during MS relapse as compared to remission (68). Furthermore, relapses in MS are associated with significantly lower CNS-specific CD8+ T-cell suppressor ability, while this potential in MS patients during quiescence is similar to healthy donors, suggesting a role with disease activity (5). Of clinical significance, we showed that the CNS-specific CD8+ Treg suppressive function is restored in MS patients during remission and this recovery in CD8+ Treg-mediated suppression correlated with the distance in time from an acute clinical episode. This suggests that the correction of the neuroantigen-specific CD8+ suppressor deficit would correlate with recovery from an acute relapse (5). One caveat to the study is that the quiescence samples could still potentially have pseudo relapses in the CNS in the absence of any clinical signs. Nonetheless, these findings raise the possibility that reduction in CNS-specific CD8+ T-cell suppression might be used as a marker to predict relapses in MS patients.
Although etiology of MS remains unknown, epidemiological studies suggest an association between Epstein–Barr virus (EBV) and MS (143). EBV-reactive CD8+ T-cells are present in the peripheral blood of MS patients (144). By using high throughput sequencing, a recent study demonstrated intrathecal enrichment of EBV-reactive CD8+ T-cells in MS patients (145). However, the function of these CD8+ T-cells in the CNS remains speculative. Interestingly, adoptive immunotherapy with in vitro-expanded autologous EBV-specific CD8+ T-cells in secondary progressive MS had no adverse effects and was associated with clinical improvement and reduced disease activity on MRI (146). This study suggests that the EBV-specific CD8+ T-cells in the CNS of MS patients might be playing a regulatory role by limiting EBV-infected B-cells and antibody production.
The pathogenic function of CD8+ T-cells in MS is believed to be largely derived from its cytotoxic potential toward CNS tissues including glial cells and axons. However, there is a clear lack of evidence in this area in human MS. Interestingly, a recent study demonstrated that CD4+ but not CD8+ T-cells from peripheral blood of MS patients expressed NKG2C and had elevated levels of cytotoxic molecules FasL, granzyme B, and perforin. Intriguingly, these CD4+ T-cells were cytotoxic toward HLA-E-positive human ODCs in vitro (147). This study suggested a novel mechanism for CNS damage in MS which is, in contrast to the widely held view, potentially mediated by CD4+ T-cells.
Although the pathogenic role of CD8+ T-cells in MS remains largely speculative, the studies discussed above strongly suggest that there is now ample evidence for the regulatory role for CD8+ T-cell subsets in the disease process. Lack of their regulatory function specifically during relapses should be probed further, as this could be a major underlying factor leading to relapse in MS.
The majority of drugs used for the long-term management of MS are immunomodulatory in nature. The precise mechanisms by which these drugs act are under constant investigation. We have convincingly demonstrated that CD8+ Tregs not only exist physiologically but can also be induced therapeutically by GA treatment. Both, CD4+ and CD8+ T-cells reactive to GA are present in the peripheral blood of healthy donors and MS patients (7). Although CD4+ T-cell responses are comparable between the two groups, untreated MS patients have reduced GA-induced CD8+ T-cell responses and this deficiency is corrected after GA therapy (7). Functionally, these GA-reactive CD8+ T-cells are HLA-E-restricted and have a strong suppressive potential against CD4+ T-cells (8). Interestingly, GA-reactive CD8+ T-cells obtained from untreated MS patients have reduced suppressor ability and GA therapy restores the CD8+ T-cell suppressive potential in MS patients (8). These were the pioneering findings that linked the regulatory function of CD8+ T-cells with the therapeutic action of the drug. The proof of principle came from our EAE studies discussed above where we showed that GA does not work in the absence of CD8+ T-cells in mice (64), suggesting that CD8+ T-cells are absolutely required for GA action and all the other reported immunomodulatory effects of GA might lie downstream to the induction of CD8+ Tregs by the drug. The idea is also supported by our surprising observation that GA reverses the CD4/CD8 T-cell ratio and increases CD8+ T-cell-mediated suppression as early as 12 h after GA therapy initiation in humans (40). Similar to our findings, a 1-year follow-up study after IFNβ treatment showed expansion of regulatory CD8+ T-cell subsets (CD8+CD25+ and CD8+CD25+CD28−) in the responder cohort (148). Another study found a higher frequency of regulatory CXCR3+CD8+ T-cells 6 months after IFNβ therapy (149). Collectively, these studies suggest that therapeutic induction of CD8+ Tregs might be the underlying factor in other MS therapies as well. Natalizumab treatment results in a decreased CD4+/CD8+ ratio in the CSF and peripheral blood of MS patients (150). Fingolimod therapy is associated with altering the cytokine status of CD8+ T-cells in peripheral blood (151). However, detailed dissection of the role of CD8+ T-cells has not been performed in the setting of these treatments.
The potential roles of CD8+ T-cells in MS is summarized in the model shown in Figure 1, where the various pieces of evidence supporting the potential of CD8+ T-cells for both pathogenic and regulatory roles in MS/EAE disease are depicted. On the pathogenic side of the model, CD8+ T-cells, whose antigenic specificity has yet to be fully elucidated, have been shown to be involved in several disease-driving mechanisms, ranging from cytotoxicity and demyelination to pro-inflammatory cytokine production. This is in addition to hallmark activation behavior in disease lesions, such as oligoclonal expansion and IFNγ production. Interestingly, this fails to rule out the activation of a regulatory population, as indicated in the bottom portion of the model. As illustrated on the regulatory side – to which our lab has made several novel contributions – several lines of evidence exist demonstrating the regulatory mechanisms performed by CD8+ T-cells in the context of MS/EAE, which can either be neuroantigen specific (MHC class 1a-restricted) or GA/Copaxone® specific (HLA-E/Qa-1-restricted). Their protective functions, which seem to depend on IFNγ and perforin production, range from direct cytotoxicity to pathogenic CD4+ T-cells to modulation of pro-inflammatory cytokine profiles to inhibition of APC function. It is still unclear to what extent CD8+ T-cells affect other cell populations such as B-cells, but some evidence demonstrates a suppressive effect on monocytes and macrophages. These all serve to suppress CNS auto-inflammation and protect myelinated axons – effectively limiting EAE disease pathogenesis. The potential role for these CD8+ Tregs in ultimately modulating MS disease is of high interest.
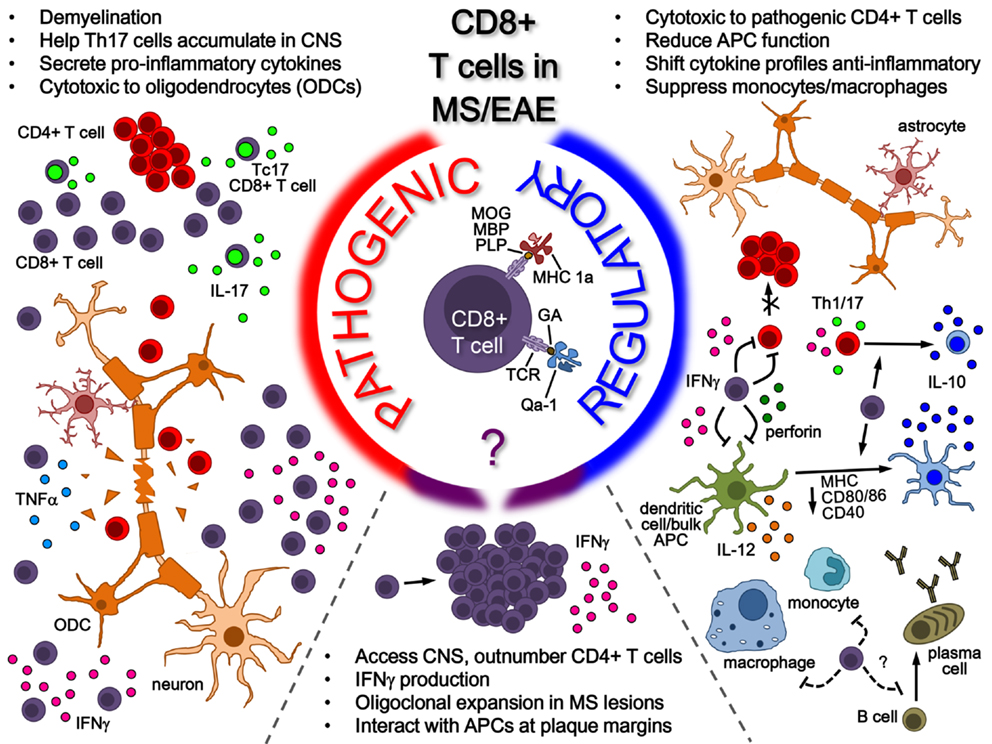
Figure 1. Role of CD8+ T-cells in MS/EAE. While the antigenic specificity of pathogenic CD8+ T-cells remains unknown, their pathogenic function is mainly attributed to pro-inflammatory cytokine secretion (in the peripheral immune system and potentially in the CNS) as well as cytotoxicity toward oligodendrocytes in the CNS. On the other side, several lines of evidence indicate a regulatory role for CD8+ T-cells in both MS and EAE. Neuroantigen-specific autoregulatory T-cells are classically MHC Class I restricted, whereas there are also examples of HLA-E/Qa1-restricted regulatory T-cells that may be naturally occurring or induced through therapy. Mechanisms for CD8+ T-cell-mediated regulation include secretion of cytokines such as IL-10 and IFNγ, cytotoxicity toward pathogenic immune cells and modulation of APC functions, both in the periphery and possibly in the CNS.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
This work was supported, in part, by grant awards (to NJK) from the NIH and National MS Society.
1. Booss J, Esiri MM, Tourtellotte WW, Mason DY. Immunohistological analysis of T lymphocyte subsets in the central nervous system in chronic progressive multiple sclerosis. J Neurol Sci (1983) 62:219–32. doi:10.1016/0022-510X(83)90201-0
2. Babbe H, Roers A, Waisman A, Lassmann H, Goebels N, Hohlfeld R, et al. Clonal expansions of CD8(+) T cells dominate the T cell infiltrate in active multiple sclerosis lesions as shown by micromanipulation and single cell polymerase chain reaction. J Exp Med (2000) 192:393–404. doi:10.1084/jem.192.3.393
3. Jacobsen M, Cepok S, Quak E, Happel M, Gaber R, Ziegler A, et al. Oligoclonal expansion of memory CD8+ T cells in cerebrospinal fluid from multiple sclerosis patients. Brain (2002) 125:538–50. doi:10.1093/brain/awf059
4. Junker A, Ivanidze J, Malotka J, Eiglmeier I, Lassmann H, Wekerle H, et al. Multiple sclerosis: T-cell receptor expression in distinct brain regions. Brain (2007) 130:2789–99. doi:10.1093/brain/awm214
5. Baughman EJ, Mendoza JP, Ortega SB, Ayers CL, Greenberg BM, Frohman EM, et al. Neuroantigen-specific CD8+ regulatory T-cell function is deficient during acute exacerbation of multiple sclerosis. J Autoimmun (2011) 36:115–24. doi:10.1016/j.jaut.2010.12.003
6. Crawford MP, Yan SX, Ortega SB, Mehta RS, Hewitt RE, Price DA, et al. High prevalence of autoreactive neuroantigen-specific CD8+ T cells in multiple sclerosis revealed by novel flow cytometric assay. Blood (2004) 103:4222–31. doi:10.1182/blood-2003-11-4025
7. Karandikar NJ, Crawford MP, Yan X, Ratts RB, Brenchley JM, Ambrozak DR, et al. Glatiramer acetate (Copaxone) therapy induces CD8(+) T cell responses in patients with multiple sclerosis. J Clin Invest (2002) 109:641–9. doi:10.1172/JCI200214380
8. Tennakoon DK, Mehta RS, Ortega SB, Bhoj V, Racke MK, Karandikar NJ. Therapeutic induction of regulatory, cytotoxic CD8+ T cells in multiple sclerosis. J Immunol (2006) 176:7119–29. doi:10.4049/jimmunol.176.11.7119
9. Brynedal B, Duvefelt K, Jonasdottir G, Roos IM, Akesson E, Palmgren J, et al. HLA-A confers an HLA-DRB1 independent influence on the risk of multiple sclerosis. PLoS One (2007) 2:e664. doi:10.1371/journal.pone.0000664
10. Fogdell-Hahn A, Ligers A, Gronning M, Hillert J, Olerup O. Multiple sclerosis: a modifying influence of HLA class I genes in an HLA class II associated autoimmune disease. Tissue Antigens (2000) 55:140–8. doi:10.1034/j.1399-0039.2000.550205.x
11. Harbo HF, Lie BA, Sawcer S, Celius EG, Dai KZ, Oturai A, et al. Genes in the HLA class I region may contribute to the HLA class II-associated genetic susceptibility to multiple sclerosis. Tissue Antigens (2004) 63:237–47. doi:10.1111/j.0001-2815.2004.00173.x
12. Friese MA, Jakobsen KB, Friis L, Etzensperger R, Craner MJ, McMahon RM, et al. Opposing effects of HLA class I molecules in tuning autoreactive CD8+ T cells in multiple sclerosis. Nat Med (2008) 14:1227–35. doi:10.1038/nm.1881
13. Huseby ES, Liggitt D, Brabb T, Schnabel B, Ohlen C, Goverman J. A pathogenic role for myelin-specific CD8(+) T cells in a model for multiple sclerosis. J Exp Med (2001) 194:669–76. doi:10.1084/jem.194.5.669
14. Ji Q, Perchellet A, Goverman JM. Viral infection triggers central nervous system autoimmunity via activation of CD8+ T cells expressing dual TCRs. Nat Immunol (2010) 11:628–34. doi:10.1038/ni.1888
15. Sun D, Whitaker JN, Huang Z, Liu D, Coleclough C, Wekerle H, et al. Myelin antigen-specific CD8+ T cells are encephalitogenic and produce severe disease in C57BL/6 mice. J Immunol (2001) 166:7579–87. doi:10.4049/jimmunol.166.12.7579
16. Ford ML, Evavold BD. Specificity, magnitude, and kinetics of MOG-specific CD8+ T cell responses during experimental autoimmune encephalomyelitis. Eur J Immunol (2005) 35:76–85. doi:10.1002/eji.200425660
17. Sasaki K, Bean A, Shah S, Schutten E, Huseby PG, Peters B, et al. Relapsing-remitting central nervous system autoimmunity mediated by GFAP-specific CD8 T cells. J Immunol (2014) 192:3029–42. doi:10.4049/jimmunol.1302911
18. Luo Q, Sun Y, Gong FY, Liu W, Zheng W, Shen Y, et al. Blocking initial infiltration of pioneer CD8(+) T-cells into the CNS via inhibition of SHP-2 ameliorates experimental autoimmune encephalomyelitis in mice. Br J Pharmacol (2014) 171:1706–21. doi:10.1111/bph.12565
19. Anderson AC, Chandwaskar R, Lee DH, Sullivan JM, Solomon A, Rodriguez-Manzanet R, et al. A transgenic model of central nervous system autoimmunity mediated by CD4+ and CD8+ T and B cells. J Immunol (2012) 188:2084–92. doi:10.4049/jimmunol.1102186
20. Getts MT, Richards MH, Miller SD. A critical role for virus-specific CD8(+) CTLs in protection from Theiler’s virus-induced demyelination in disease-susceptible SJL mice. Virology (2010) 402:102–11. doi:10.1016/j.virol.2010.02.031
21. Haring JS, Pewe LL, Perlman S. Bystander CD8 T cell-mediated demyelination after viral infection of the central nervous system. J Immunol (2002) 169:1550–5. doi:10.4049/jimmunol.169.3.1550
22. Nicholson SM, Dal Canto MC, Miller SD, Melvold RW. Adoptively transferred CD8+ T lymphocytes provide protection against TMEV-induced demyelinating disease in BALB/c mice. J Immunol (1996) 156:1276–83.
23. Medana I, Martinic MA, Wekerle H, Neumann H. Transection of major histocompatibility complex class I-induced neurites by cytotoxic T lymphocytes. Am J Pathol (2001) 159:809–15. doi:10.1016/S0002-9440(10)61755-5
24. Meuth SG, Herrmann AM, Simon OJ, Siffrin V, Melzer N, Bittner S, et al. Cytotoxic CD8+ T cell-neuron interactions: perforin-dependent electrical silencing precedes but is not causally linked to neuronal cell death. J Neurosci (2009) 29:15397–409. doi:10.1523/JNEUROSCI.4339-09.2009
25. Kreutzfeldt M, Bergthaler A, Fernandez M, Bruck W, Steinbach K, Vorm M, et al. Neuroprotective intervention by interferon-gamma blockade prevents CD8+ T cell-mediated dendrite and synapse loss. J Exp Med (2013) 210:2087–103. doi:10.1084/jem.20122143
26. Sauer BM, Schmalstieg WF, Howe CL. Axons are injured by antigen-specific CD8(+) T cells through a MHC class I- and granzyme B-dependent mechanism. Neurobiol Dis (2013) 59:194–205. doi:10.1016/j.nbd.2013.07.010
27. Cao Y, Toben C, Na SY, Stark K, Nitschke L, Peterson A, et al. Induction of experimental autoimmune encephalomyelitis in transgenic mice expressing ovalbumin in oligodendrocytes. Eur J Immunol (2006) 36:207–15. doi:10.1002/eji.200535211
28. Na SY, Eujen H, Gobel K, Meuth SG, Martens K, Wiendl H, et al. Antigen-specific blockade of lethal CD8 T-cell mediated autoimmunity in a mouse model of multiple sclerosis. J Immunol (2009) 182:6569–75. doi:10.4049/jimmunol.0804200
29. Huber M, Heink S, Grothe H, Guralnik A, Reinhard K, Elflein K, et al. A Th17-like developmental process leads to CD8(+) Tc17 cells with reduced cytotoxic activity. Eur J Immunol (2009) 39:1716–25. doi:10.1002/eji.200939412
30. Huber M, Heink S, Pagenstecher A, Reinhard K, Ritter J, Visekruna A, et al. IL-17A secretion by CD8+ T cells supports Th17-mediated autoimmune encephalomyelitis. J Clin Invest (2013) 123:247–60. doi:10.1172/JCI63681
31. Sinha S, Itani FR, Karandikar NJ. Immune regulation of multiple sclerosis by CD8+ T cells. Immunol Res (2014) 59:254–65. doi:10.1007/s12026-014-8529-9
32. Najafian N, Chitnis T, Salama AD, Zhu B, Benou C, Yuan X, et al. Regulatory functions of CD8+CD28- T cells in an autoimmune disease model. J Clin Invest (2003) 112:1037–48. doi:10.1172/JCI17935
33. Linker RA, Rott E, Hofstetter HH, Hanke T, Toyka KV, Gold R. EAE in beta-2 microglobulin-deficient mice: axonal damage is not dependent on MHC-I restricted immune responses. Neurobiol Dis (2005) 19:218–28. doi:10.1016/j.nbd.2004.12.017
34. Ortega SB, Kashi VP, Tyler AF, Cunnusamy K, Mendoza JP, Karandikar NJ. The disease-ameliorating function of autoregulatory CD8 T cells is mediated by targeting of encephalitogenic CD4 T cells in experimental autoimmune encephalomyelitis. J Immunol (2013) 191:117–26. doi:10.4049/jimmunol.1300452
35. Weiss HA, Millward JM, Owens T. CD8+ T cells in inflammatory demyelinating disease. J Neuroimmunol (2007) 191:79–85. doi:10.1016/j.jneuroim.2007.09.011
36. York NR, Mendoza JP, Ortega SB, Benagh A, Tyler AF, Firan M, et al. Immune regulatory CNS-reactive CD8+T cells in experimental autoimmune encephalomyelitis. J Autoimmun (2010) 35:33–44. doi:10.1016/j.jaut.2010.01.003
37. Ortega SB, Kashi VP, Cunnusamy K, Franco J, Karandikar NJ. Autoregulatory CD8 T cells depend on cognate antigen recognition and CD4/CD* myelin determinants. Neurol Neuroimmunol Neuroinflamm (2015) 2:e170. doi:10.1212/NXI.0000000000000170
38. Antel J, Bania M, Noronha A, Neely S. Defective suppressor cell function mediated by T8+ cell lines from patients with progressive multiple sclerosis. J Immunol (1986) 137:3436–9.
39. Bania MB, Antel JP, Reder AT, Nicholas MK, Arnason BG. Suppressor and cytolytic cell function in multiple sclerosis. Effects of cyclosporine A and interleukin 2. J Clin Invest (1986) 78:582–6. doi:10.1172/JCI112612
40. Ayers CL, Mendoza JP, Sinha S, Cunnusamy K, Greenberg BM, Frohman EM, et al. Modulation of immune function occurs within hours of therapy initiation for multiple sclerosis. Clin Immunol (2013) 147:105–19. doi:10.1016/j.clim.2013.02.015
41. Pannemans K, Broux B, Goris A, Dubois B, Broekmans T, Van Wijmeersch B, et al. HLA-E restricted CD8+ T cell subsets are phenotypically altered in multiple sclerosis patients. Mult Scler (2013) 20:790–801. doi:10.1177/1352458513509703
42. Grosso JF, Kelleher CC, Harris TJ, Maris CH, Hipkiss EL, De Marzo A, et al. LAG-3 regulates CD8+ T cell accumulation and effector function in murine self- and tumor-tolerance systems. J Clin Invest (2007) 117:3383–92. doi:10.1172/JCI31184
43. Wang R, Han G, Song L, Wang J, Chen G, Xu R, et al. CD8+ regulatory T cells are responsible for GAD-IgG gene-transferred tolerance induction in NOD mice. Immunology (2009) 126:123–31. doi:10.1111/j.1365-2567.2008.02884.x
44. Endharti AT, Okuno Y, Shi Z, Misawa N, Toyokuni S, Ito M, et al. CD8+CD122+ regulatory T cells (Tregs) and CD4+ Tregs cooperatively prevent and cure CD4+ cell-induced colitis. J Immunol (2011) 186:41–52. doi:10.4049/jimmunol.1000800
45. Kim HJ, Verbinnen B, Tang X, Lu L, Cantor H. Inhibition of follicular T-helper cells by CD8(+) regulatory T cells is essential for self tolerance. Nature (2010) 467:328–32. doi:10.1038/nature09370
46. Saitoh O, Abiru N, Nakahara M, Nagayama Y. CD8+CD122+ T cells, a newly identified regulatory T subset, negatively regulate Graves’ hyperthyroidism in a murine model. Endocrinology (2007) 148:6040–6. doi:10.1210/en.2007-0300
47. Li XL, Menoret S, Bezie S, Caron L, Chabannes D, Hill M, et al. Mechanism and localization of CD8 regulatory T cells in a heart transplant model of tolerance. J Immunol (2010) 185:823–33. doi:10.4049/jimmunol.1000120
48. Suzuki M, Konya C, Goronzy JJ, Weyand CM. Inhibitory CD8+ T cells in autoimmune disease. Hum Immunol (2008) 69:781–9. doi:10.1016/j.humimm.2008.08.283
49. Shameli A, Clemente-Casares X, Wang J, Santamaria P. Development of memory-like autoregulatory CD8+ T cells is CD4+ T cell dependent. J Immunol (2011) 187:2859–66. doi:10.4049/jimmunol.1101117
50. Tsai S, Shameli A, Yamanouchi J, Clemente-Casares X, Wang J, Serra P, et al. Reversal of autoimmunity by boosting memory-like autoregulatory T cells. Immunity (2010) 32:568–80. doi:10.1016/j.immuni.2010.03.015
51. Lee YH, Ishida Y, Rifa’i M, Shi Z, Isobe K, Suzuki H. Essential role of CD8+CD122+ regulatory T cells in the recovery from experimental autoimmune encephalomyelitis. J Immunol (2008) 180:825–32. doi:10.4049/jimmunol.180.2.825
52. Mangalam AK, Luckey D, Giri S, Smart M, Pease LR, Rodriguez M, et al. Two discreet subsets of CD8 T cells modulate PLP(91-110) induced experimental autoimmune encephalomyelitis in HLA-DR3 transgenic mice. J Autoimmun (2012) 38:344–53. doi:10.1016/j.jaut.2012.02.004
53. Yu P, Bamford RN, Waldmann TA. IL-15-dependent CD8+ CD122+ T cells ameliorate experimental autoimmune encephalomyelitis by modulating IL-17 production by CD4+ T cells. Eur J Immunol (2014) 44:3330–41. doi:10.1002/eji.201444675
54. D’Orazio TJ, Mayhew E, Niederkorn JY. Ocular immune privilege promoted by the presentation of peptide on tolerogenic B cells in the spleen. II. Evidence for presentation by Qa-1. J Immunol (2001) 166:26–32.
55. Skelsey ME, Mayhew E, Niederkorn JY. Splenic B cells act as antigen presenting cells for the induction of anterior chamber-associated immune deviation. Invest Ophthalmol Vis Sci (2003) 44:5242–51. doi:10.1167/iovs.03-0768
56. Chattopadhyay S, O’Rourke J, Cone RE. Implication for the CD94/NKG2A-Qa-1 system in the generation and function of ocular-induced splenic CD8+ regulatory T cells. Int Immunol (2008) 20:509–16. doi:10.1093/intimm/dxn008
57. Keino H, Masli S, Sasaki S, Streilein JW, Stein-Streilein J. CD8+ T regulatory cells use a novel genetic program that includes CD103 to suppress Th1 immunity in eye-derived tolerance. Invest Ophthalmol Vis Sci (2006) 47:1533–42. doi:10.1167/iovs.04-1454
58. Jiang L, Yang P, He H, Li B, Lin X, Hou S, et al. Increased expression of Foxp3 in splenic CD8+ T cells from mice with anterior chamber-associated immune deviation. Mol Vis (2007) 13:968–74.
59. He H, Yang P, Jiang L, Zhang J, Zhao C, Chen L, et al. Upregulation of CD94 on CD8+ T cells in anterior chamber-associated immune deviation. BMC Immunol (2008) 9:53. doi:10.1186/1471-2172-9-53
60. Jiang L, He H, Yang P, Lin X, Zhou H, Huang X, et al. Splenic CD8+ T cells secrete TGF-beta1 to exert suppression in mice with anterior chamber-associated immune deviation. Graefes Arch Clin Exp Ophthalmol (2009) 247:87–92. doi:10.1007/s00417-008-0947-8
61. Farooq SM, Ashour HM. In vitro-induced cell-mediated immune deviation to encephalitogenic antigens. Brain Behav Immun (2014) 35:64–9. doi:10.1016/j.bbi.2013.09.016
62. Farooq SM, Elkhatib WF, Ashour HM. The in vivo and in vitro induction of anterior chamber associated immune deviation to myelin antigens in C57BL/6 mice. Brain Behav Immun (2014) 42:118–22. doi:10.1016/j.bbi.2014.06.010
63. Jiang H, Braunstein NS, Yu B, Winchester R, Chess L. CD8+ T cells control the TH phenotype of MBP-reactive CD4+ T cells in EAE mice. Proc Natl Acad Sci U S A (2001) 98:6301–6. doi:10.1073/pnas.101123098
64. Tyler AF, Mendoza JP, Firan M, Karandikar NJ. CD8 T cells are required for glatiramer acetate therapy in autoimmune demyelinating disease. PLoS One (2013) 8:e66772. doi:10.1371/journal.pone.0066772
65. Kashi VP, Ortega SB, Karandikar NJ. Neuroantigen-specific autoregulatory CD8+ T cells inhibit autoimmune demyelination through modulation of dendritic cell function. PLoS One (2014) 9:e105763. doi:10.1371/journal.pone.0105763
66. Trandem K, Zhao J, Fleming E, Perlman S. Highly activated cytotoxic CD8 T cells express protective IL-10 at the peak of coronavirus-induced encephalitis. J Immunol (2011) 186:3642–52. doi:10.4049/jimmunol.1003292
67. Tang X, Maricic I, Purohit N, Bakamjian B, Reed-Loisel LM, Beeston T, et al. Regulation of immunity by a novel population of Qa-1-restricted CD8alphaalpha+TCRalphabeta+ T cells. J Immunol (2006) 177:7645–55. doi:10.4049/jimmunol.177.11.7645
68. Cunnusamy K, Baughman EJ, Franco J, Ortega SB, Sinha S, Chaudhary P, et al. Disease exacerbation of multiple sclerosis is characterized by loss of terminally differentiated autoregulatory CD8+ T cells. Clin Immunol (2014) 152:115–26. doi:10.1016/j.clim.2014.03.005
69. Weber MS, Prod’homme T, Youssef S, Dunn SE, Rundle CD, Lee L, et al. Type II monocytes modulate T cell-mediated central nervous system autoimmune disease. Nat Med (2007) 13:935–43. doi:10.1038/nm1620
70. Kim HJ, Ifergan I, Antel JP, Seguin R, Duddy M, Lapierre Y, et al. Type 2 monocyte and microglia differentiation mediated by glatiramer acetate therapy in patients with multiple sclerosis. J Immunol (2004) 172:7144–53. doi:10.4049/jimmunol.172.11.7144
71. Weber MS, Starck M, Wagenpfeil S, Meinl E, Hohlfeld R, Farina C. Multiple sclerosis: glatiramer acetate inhibits monocyte reactivity in vitro and in vivo. Brain (2004) 127:1370–8. doi:10.1093/brain/awh163
72. Schluesener HJ, Sobel RA, Linington C, Weiner HL. A monoclonal antibody against a myelin oligodendrocyte glycoprotein induces relapses and demyelination in central nervous system autoimmune disease. J Immunol (1987) 139:4016–21.
73. Lyons JA, Ramsbottom MJ, Cross AH. Critical role of antigen-specific antibody in experimental autoimmune encephalomyelitis induced by recombinant myelin oligodendrocyte glycoprotein. Eur J Immunol (2002) 32:1905–13. doi:10.1002/1521-4141(200207)32:7<1905::AID-IMMU1905>3.0.CO;2-L
74. Svensson L, Abdul-Majid KB, Bauer J, Lassmann H, Harris RA, Holmdahl R. A comparative analysis of B cell-mediated myelin oligodendrocyte glycoprotein-experimental autoimmune encephalomyelitis pathogenesis in B cell-deficient mice reveals an effect on demyelination. Eur J Immunol (2002) 32:1939–46. doi:10.1002/1521-4141(200207)32:7<1939::AID-IMMU1939>3.0.CO;2-S
75. Marta CB, Oliver AR, Sweet RA, Pfeiffer SE, Ruddle NH. Pathogenic myelin oligodendrocyte glycoprotein antibodies recognize glycosylated epitopes and perturb oligodendrocyte physiology. Proc Natl Acad Sci USA (2005) 102:13992–7. doi:10.1073/pnas.0504979102
76. Matsushita T, Yanaba K, Bouaziz JD, Fujimoto M, Tedder TF. Regulatory B cells inhibit EAE initiation in mice while other B cells promote disease progression. J Clin Invest (2008) 118:3420–30. doi:10.1172/JCI36030
77. Weber MS, Prod’homme T, Patarroyo JC, Molnarfi N, Karnezis T, Lehmann-Horn K, et al. B-cell activation influences T-cell polarization and outcome of anti-CD20 B-cell depletion in central nervous system autoimmunity. Ann Neurol (2010) 68:369–83. doi:10.1002/ana.22081
78. Weber MS, Hemmer B, Cepok S. The role of antibodies in multiple sclerosis. Biochim Biophys Acta (2011) 1812:239–45. doi:10.1016/j.bbadis.2010.06.009
79. Wu GF, Alvarez E. The immunopathophysiology of multiple sclerosis. Neurol Clin (2011) 29:257–78. doi:10.1016/j.ncl.2010.12.009
80. Barr TA, Shen P, Brown S, Lampropoulou V, Roch T, Lawrie S, et al. B cell depletion therapy ameliorates autoimmune disease through ablation of IL-6-producing B cells. J Exp Med (2012) 209:1001–10. doi:10.1084/jem.20111675
81. Krumbholz M, Derfuss T, Hohlfeld R, Meinl E. B cells and antibodies in multiple sclerosis pathogenesis and therapy. Nat Rev Neurol (2012) 8:613–23. doi:10.1038/nrneurol.2012.203
82. Pierson ER, Stromnes IM, Goverman JM. B cells promote induction of experimental autoimmune encephalomyelitis by facilitating reactivation of T cells in the central nervous system. J Immunol (2014) 192:929–39. doi:10.4049/jimmunol.1302171
83. Wolf SD, Dittel BN, Hardardottir F, Janeway CA Jr. Experimental autoimmune encephalomyelitis induction in genetically B cell-deficient mice. J Exp Med (1996) 184:2271–8. doi:10.1084/jem.184.6.2271
84. Lyons JA, San M, Happ MP, Cross AH. B cells are critical to induction of experimental allergic encephalomyelitis by protein but not by a short encephalitogenic peptide. Eur J Immunol (1999) 29:3432–9. doi:10.1002/(SICI)1521-4141(199911)29:11<3432::AID-IMMU3432>3.0.CO;2-2
85. Fillatreau S, Sweenie CH, McGeachy MJ, Gray D, Anderton SM. B cells regulate autoimmunity by provision of IL-10. Nat Immunol (2002) 3:944–50. doi:10.1038/ni833
86. Fillatreau S, Gray D, Anderton SM. Not always the bad guys: B cells as regulators of autoimmune pathology. Nat Rev Immunol (2008) 8:391–7. doi:10.1038/nri2315
87. Matsushita T, Horikawa M, Iwata Y, Tedder TF. Regulatory B cells (B10 cells) and regulatory T cells have independent roles in controlling experimental autoimmune encephalomyelitis initiation and late-phase immunopathogenesis. J Immunol (2010) 185:2240–52. doi:10.4049/jimmunol.1001307
88. Van Kaer L. Glatiramer acetate for treatment of MS: regulatory B cells join the cast of players. Exp Neurol (2011) 227:19–23. doi:10.1016/j.expneurol.2010.10.009
89. Ray A, Mann MK, Basu S, Dittel BN. A case for regulatory B cells in controlling the severity of autoimmune-mediated inflammation in experimental autoimmune encephalomyelitis and multiple sclerosis. J Neuroimmunol (2011) 230:1–9. doi:10.1016/j.jneuroim.2010.10.037
90. Mauri C, Bosma A. Immune regulatory function of B cells. Annu Rev Immunol (2012) 30:221–41. doi:10.1146/annurev-immunol-020711-074934
91. Yoshizaki A, Miyagaki T, DiLillo DJ, Matsushita T, Horikawa M, Kountikov EI, et al. Regulatory B cells control T-cell autoimmunity through IL-21-dependent cognate interactions. Nature (2012) 491:264–8. doi:10.1038/nature11501
92. Kalampokis I, Yoshizaki A, Tedder TF. IL-10-producing regulatory B cells (B10 cells) in autoimmune disease. Arthritis Res Ther (2013) 15(Suppl 1):S1. doi:10.1186/ar3907
93. Fillatreau S. Cytokine-producing B cells as regulators of pathogenic and protective immune responses. Ann Rheum Dis (2013) 72(Suppl 2):ii80–4. doi:10.1136/annrheumdis-2012-202253
94. Shen P, Roch T, Lampropoulou V, O’Connor RA, Stervbo U, Hilgenberg E, et al. IL-35-producing B cells are critical regulators of immunity during autoimmune and infectious diseases. Nature (2014) 507:366–70. doi:10.1038/nature12979
95. Noble A, Zhao ZS, Cantor H. Suppression of immune responses by CD8 cells. II. Qa-1 on activated B cells stimulates CD8 cell suppression of T helper 2 responses. J Immunol (1998) 160:566–71.
96. Heit A, Huster KM, Schmitz F, Schiemann M, Busch DH, Wagner H. CpG-DNA aided cross-priming by cross-presenting B cells. J Immunol (2004) 172:1501–7. doi:10.4049/jimmunol.172.3.1501
97. Marino E, Tan B, Binge L, Mackay CR, Grey ST. B-cell cross-presentation of autologous antigen precipitates diabetes. Diabetes (2012) 61:2893–905. doi:10.2337/db12-0006
98. Niederkorn JY, Mayhew E. Role of splenic B cells in the immune privilege of the anterior chamber of the eye. Eur J Immunol (1995) 25:2783–7. doi:10.1002/eji.1830251011
99. D’Orazio TJ, Niederkorn JY. Splenic B cells are required for tolerogenic antigen presentation in the induction of anterior chamber-associated immune deviation (ACAID). Immunology (1998) 95:47–55. doi:10.1046/j.1365-2567.1998.00581.x
100. Ashour HM, Niederkorn JY. Peripheral tolerance via the anterior chamber of the eye: role of B cells in MHC class I and II antigen presentation. J Immunol (2006) 176:5950–7. doi:10.4049/jimmunol.176.10.5950
101. Liu Y, Chen Y, Li Z, Han Y, Sun Y, Wang Q, et al. Role of IL-10-producing regulatory B cells in control of cerebral malaria in Plasmodium berghei infected mice. Eur J Immunol (2013) 43:2907–18. doi:10.1002/eji.201343512
102. Mauri C, Menon M. The expanding family of regulatory B cells. Int Immunol (2015) 27:479–86. doi:10.1093/intimm/dxv038
103. Mutnal MB, Hu S, Schachtele SJ, Lokensgard JR. Infiltrating regulatory B cells control neuroinflammation following viral brain infection. J Immunol (2014) 193:6070–80. doi:10.4049/jimmunol.1400654
104. Siewe B, Wallace J, Rygielski S, Stapleton JT, Martin J, Deeks SG, et al. Regulatory B cells inhibit cytotoxic T lymphocyte (CTL) activity and elimination of infected CD4 T cells after in vitro reactivation of HIV latent reservoirs. PLoS One (2014) 9:e92934. doi:10.1371/journal.pone.0092934
105. Siewe B, Stapleton JT, Martinson J, Keshavarzian A, Kazmi N, Demarais PM, et al. Regulatory B cell frequency correlates with markers of HIV disease progression and attenuates anti-HIV CD8(+) T cell function in vitro. J Leukoc Biol (2013) 93:811–8. doi:10.1189/jlb.0912436
106. Kang YM, Zhang X, Wagner UG, Yang H, Beckenbaugh RD, Kurtin PJ, et al. CD8 T cells are required for the formation of ectopic germinal centers in rheumatoid synovitis. J Exp Med (2002) 195:1325–36. doi:10.1084/jem.20011565
107. Shen H, Whitmire JK, Fan X, Shedlock DJ, Kaech SM, Ahmed R. A specific role for B cells in the generation of CD8 T cell memory by recombinant Listeria monocytogenes. J Immunol (2003) 170:1443–51. doi:10.4049/jimmunol.170.3.1443
108. Wei B, Velazquez P, Turovskaya O, Spricher K, Aranda R, Kronenberg M, et al. Mesenteric B cells centrally inhibit CD4+ T cell colitis through interaction with regulatory T cell subsets. Proc Natl Acad Sci USA (2005) 102:2010–5. doi:10.1073/pnas.0409449102
109. Quigley MF, Gonzalez VD, Granath A, Andersson J, Sandberg JK. CXCR5+ CCR7- CD8 T cells are early effector memory cells that infiltrate tonsil B cell follicles. Eur J Immunol (2007) 37:3352–62. doi:10.1002/eji.200636746
110. Deola S, Panelli MC, Maric D, Selleri S, Dmitrieva NI, Voss CY, et al. Helper B cells promote cytotoxic T cell survival and proliferation independently of antigen presentation through CD27/CD70 interactions. J Immunol (2008) 180:1362–72. doi:10.4049/jimmunol.180.3.1362
111. Wei B, McPherson M, Turovskaya O, Velazquez P, Fujiwara D, Brewer S, et al. Integration of B cells and CD8+ T in the protective regulation of systemic epithelial inflammation. Clin Immunol (2008) 127:303–12. doi:10.1016/j.clim.2008.01.001
112. Traugott U, Reinherz EL, Raine CS. Multiple sclerosis. Distribution of T cells, T cell subsets and Ia-positive macrophages in lesions of different ages. J Neuroimmunol (1983) 4:201–21. doi:10.1016/0165-5728(83)90036-X
113. Hauser SL, Bhan AK, Gilles F, Kemp M, Kerr C, Weiner HL. Immunohistochemical analysis of the cellular infiltrate in multiple sclerosis lesions. Ann Neurol (1986) 19:578–87. doi:10.1002/ana.410190610
114. Zang YC, Li S, Rivera VM, Hong J, Robinson RR, Breitbach WT, et al. Increased CD8+ cytotoxic T cell responses to myelin basic protein in multiple sclerosis. J Immunol (2004) 172:5120–7. doi:10.4049/jimmunol.172.8.5120
115. Kutzelnigg A, Lucchinetti CF, Stadelmann C, Bruck W, Rauschka H, Bergmann M, et al. Cortical demyelination and diffuse white matter injury in multiple sclerosis. Brain (2005) 128:2705–12. doi:10.1093/brain/awh641
116. Jurewicz A, Biddison WE, Antel JP. MHC class I-restricted lysis of human oligodendrocytes by myelin basic protein peptide-specific CD8 T lymphocytes. J Immunol (1998) 160:3056–9.
117. Lassmann H, Bruck W, Lucchinetti CF. The immunopathology of multiple sclerosis: an overview. Brain Pathol (2007) 17:210–8. doi:10.1111/j.1750-3639.2007.00064.x
118. Neumann H, Medana IM, Bauer J, Lassmann H. Cytotoxic T lymphocytes in autoimmune and degenerative CNS diseases. Trends Neurosci (2002) 25:313–9. doi:10.1016/S0166-2236(02)02154-9
119. Serafini B, Rosicarelli B, Magliozzi R, Stigliano E, Capello E, Mancardi GL, et al. Dendritic cells in multiple sclerosis lesions: maturation stage, myelin uptake, and interaction with proliferating T cells. J Neuropathol Exp Neurol (2006) 65:124–41. doi:10.1097/01.jnen.0000199572.96472.1c
120. Hoftberger R, Aboul-Enein F, Brueck W, Lucchinetti C, Rodriguez M, Schmidbauer M, et al. Expression of major histocompatibility complex class I molecules on the different cell types in multiple sclerosis lesions. Brain Pathol (2004) 14:43–50. doi:10.1111/j.1750-3639.2004.tb00496.x
121. Massa PT, Ozato K, McFarlin DE. Cell type-specific regulation of major histocompatibility complex (MHC) class I gene expression in astrocytes, oligodendrocytes, and neurons. Glia (1993) 8:201–7. doi:10.1002/glia.440080307
122. Galea I, Bernardes-Silva M, Forse PA, van Rooijen N, Liblau RS, Perry VH. An antigen-specific pathway for CD8 T cells across the blood-brain barrier. J Exp Med (2007) 204:2023–30. doi:10.1084/jem.20070064
123. Bitsch A, Schuchardt J, Bunkowski S, Kuhlmann T, Bruck W. Acute axonal injury in multiple sclerosis. Correlation with demyelination and inflammation. Brain (2000) 123(Pt 6):1174–83. doi:10.1093/brain/123.6.1174
124. Dressel A, Chin JL, Sette A, Gausling R, Hollsberg P, Hafler DA. Autoantigen recognition by human CD8 T cell clones: enhanced agonist response induced by altered peptide ligands. J Immunol (1997) 159:4943–51.
125. Tsuchida T, Parker KC, Turner RV, McFarland HF, Coligan JE, Biddison WE. Autoreactive CD8+ T-cell responses to human myelin protein-derived peptides. Proc Natl Acad Sci U S A (1994) 91:10859–63. doi:10.1073/pnas.91.23.10859
126. Lolli F, Martini H, Citro A, Franceschini D, Portaccio E, Amato MP, et al. Increased CD8+ T cell responses to apoptotic T cell-associated antigens in multiple sclerosis. J Neuroinflammation (2013) 10:94. doi:10.1186/1742-2094-10-94
127. Battistini L, Piccio L, Rossi B, Bach S, Galgani S, Gasperini C, et al. CD8+ T cells from patients with acute multiple sclerosis display selective increase of adhesiveness in brain venules: a critical role for P-selectin glycoprotein ligand-1. Blood (2003) 101:4775–82. doi:10.1182/blood-2002-10-3309
128. Tzartos JS, Friese MA, Craner MJ, Palace J, Newcombe J, Esiri MM, et al. Interleukin-17 production in central nervous system-infiltrating T cells and glial cells is associated with active disease in multiple sclerosis. Am J Pathol (2008) 172:146–55. doi:10.2353/ajpath.2008.070690
129. Annibali V, Ristori G, Angelini DF, Serafini B, Mechelli R, Cannoni S, et al. CD161(high)CD8+ T cells bear pathogenetic potential in multiple sclerosis. Brain (2011) 134:542–54. doi:10.1093/brain/awq354
130. Giovanni F, Domenico P, Alessandro M, Raffaele I, Viviana N, Katia PA, et al. Circulating CD8+CD56-perforin+ T cells are increased in multiple sclerosis patients. J Neuroimmunol (2011) 24(0–241):137–41. doi:10.1016/j.jneuroim.2011.09.002
131. Antel JP, Nicholas MK, Bania MB, Reder AT, Arnason BG, Joseph L. Comparison of T8+ cell-mediated suppressor and cytotoxic functions in multiple sclerosis. J Neuroimmunol (1986) 12:215–24. doi:10.1016/S0165-5728(86)80005-4
132. Balashov KE, Khoury SJ, Hafler DA, Weiner HL. Inhibition of T cell responses by activated human CD8+ T cells is mediated by interferon-gamma and is defective in chronic progressive multiple sclerosis. J Clin Invest (1995) 95:2711–9. doi:10.1172/JCI117973
133. Brimnes J, Allez M, Dotan I, Shao L, Nakazawa A, Mayer L. Defects in CD8+ regulatory T cells in the lamina propria of patients with inflammatory bowel disease. J Immunol (2005) 174:5814–22. doi:10.4049/jimmunol.174.9.5814
134. Cho BA, Sim JH, Park JA, Kim HW, Yoo WH, Lee SH, et al. Characterization of effector memory CD8+ T cells in the synovial fluid of rheumatoid arthritis. J Clin Immunol (2012) 32:709–20. doi:10.1007/s10875-012-9674-3
135. Mikulkova Z, Praksova P, Stourac P, Bednarik J, Strajtova L, Pacasova R, et al. Numerical defects in CD8+CD28- T-suppressor lymphocyte population in patients with type 1 diabetes mellitus and multiple sclerosis. Cell Immunol (2010) 262:75–9. doi:10.1016/j.cellimm.2010.02.002
136. Tsai YG, Yang KD, Niu DM, Chien JW, Lin CY. TLR2 agonists enhance CD8+Foxp3+ regulatory T cells and suppress Th2 immune responses during allergen immunotherapy. J Immunol (2010) 184:7229–37. doi:10.4049/jimmunol.1000083
137. Correale J, Villa A. Role of CD8+ CD25+ Foxp3+ regulatory T cells in multiple sclerosis. Ann Neurol (2010) 67:625–38. doi:10.1002/ana.21944
138. Frisullo G, Nociti V, Iorio R, Plantone D, Patanella AK, Tonali PA, et al. CD8(+)Foxp3(+) T cells in peripheral blood of relapsing-remitting multiple sclerosis patients. Hum Immunol (2010) 71:437–41. doi:10.1016/j.humimm.2010.01.024
139. Shi Z, Okuno Y, Rifa’i M, Endharti AT, Akane K, Isobe K, et al. Human CD8+CXCR3+ T cells have the same function as murine CD8+CD122+ Treg. Eur J Immunol (2009) 39:2106–19. doi:10.1002/eji.200939314
140. Jiang H, Canfield SM, Gallagher MP, Jiang HH, Jiang Y, Zheng Z, et al. HLA-E-restricted regulatory CD8(+) T cells are involved in development and control of human autoimmune type 1 diabetes. J Clin Invest (2010) 120:3641–50. doi:10.1172/JCI43522
141. Aristimuno C, Navarro J, de Andres C, Martinez-Gines L, Gimenez-Roldan S, Fernandez-Cruz E, et al. Expansion of regulatory CD8+ T-lymphocytes and fall of activated CD8+ T-lymphocytes after i.v. methyl-prednisolone for multiple sclerosis relapse. J Neuroimmunol (2008) 204:131–5. doi:10.1016/j.jneuroim.2008.08.009
142. Correale J, Villa A. Isolation and characterization of CD8+ regulatory T cells in multiple sclerosis. J Neuroimmunol (2008) 195:121–34. doi:10.1016/j.jneuroim.2007.12.004
143. Ascherio A, Munger KL. 99th Dahlem conference on infection, inflammation and chronic inflammatory disorders: Epstein–Barr virus and multiple sclerosis: epidemiological evidence. Clin Exp Immunol (2010) 160:120–4. doi:10.1111/j.1365-2249.2010.04121.x
144. Jilek S, Schluep M, Meylan P, Vingerhoets F, Guignard L, Monney A, et al. Strong EBV-specific CD8+ T-cell response in patients with early multiple sclerosis. Brain (2008) 131:1712–21. doi:10.1093/brain/awn108
145. Lossius A, Johansen JN, Vartdal F, Robins H, Jurate Saltyte B, Holmoy T, et al. High-throughput sequencing of TCR repertoires in multiple sclerosis reveals intrathecal enrichment of EBV-reactive CD8+ T cells. Eur J Immunol (2014) 44:3439–52. doi:10.1002/eji.201444662
146. Pender MP, Khanna R. Epstein-Barr virus-specific adoptive immunotherapy: a new horizon for multiple sclerosis treatment? Immunotherapy (2014) 6:659–61. doi:10.2217/imt.14.43
147. Zaguia F, Saikali P, Ludwin S, Newcombe J, Beauseigle D, McCrea E, et al. Cytotoxic NKG2C+ CD4 T cells target oligodendrocytes in multiple sclerosis. J Immunol (2013) 190:2510–8. doi:10.4049/jimmunol.1202725
148. Aristimuno C, de Andres C, Bartolome M, de las Heras V, Martinez-Gines ML, Arroyo R, et al. IFNbeta-1a therapy for multiple sclerosis expands regulatory CD8+ T cells and decreases memory CD8+ subset: a longitudinal 1-year study. Clin Immunol (2010) 134:148–57. doi:10.1016/j.clim.2009.09.008
149. Dhib-Jalbut S, Sumandeep S, Valenzuela R, Ito K, Patel P, Rametta M. Immune response during interferon beta-1b treatment in patients with multiple sclerosis who experienced relapses and those who were relapse-free in the START study. J Neuroimmunol (2013) 254:131–40. doi:10.1016/j.jneuroim.2012.08.012
150. Serpero LD, Filaci G, Parodi A, Battaglia F, Kalli F, Brogi D, et al. Fingolimod modulates peripheral effector and regulatory T cells in MS patients. J Neuroimmune Pharmacol (2013) 8:1106–13. doi:10.1007/s11481-013-9465-5
Keywords: CD8, multiple sclerosis, EAE, T-cells, immune regulation
Citation: Sinha S, Boyden AW, Itani FR, Crawford MP and Karandikar NJ (2015) CD8+ T-Cells as Immune Regulators of Multiple Sclerosis. Front. Immunol. 6:619. doi: 10.3389/fimmu.2015.00619
Received: 13 August 2015; Accepted: 26 November 2015;
Published: 10 December 2015
Edited by:
Nathalie Arbour, Université de Montreal, CanadaReviewed by:
Roland S. Liblau, Université Paul Sabatier – INSERM, FranceCopyright: © 2015 Sinha, Boyden, Itani, Crawford and Karandikar. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Nitin J. Karandikar, bml0aW4ta2FyYW5kaWthckB1aW93YS5lZHU=
†Sushmita Sinha and Alexander W. Boyden have contributed equally to this work.
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.