 Nathan Karin
Nathan Karin Gizi Wildbaum
Gizi Wildbaum- Department of Immunology and Rappaport Family Institute for Research in the Medical Sciences Rappaport Faculty of Medicine, Technion – Israel Institute of Technology, Haifa, Israel
Chemokines are the key activators of adhesion molecule and also drivers of leukocyte migration to inflammatory sites and are therefore mostly considered as proinflammatory mediators. Many studies, including ours, imply that targeting the function of several key chemokines, but not many others, could effectively suppress inflammatory responses and inflammatory autoimmunity. Along with this, a single chemokine named CXCL10 could be used to induce antitumor immunity, and thereby suppress myeloma. Our working hypothesis is that some chemokines differ from others as aside from being chemoattractants for leukocytes and effective activators of adhesion receptors that possess additional biological properties making them “driver chemokines.” We came up with this notion when studying the interlay between CXCR4 and CXCL12 and between CXCR3 and its three ligands: CXCL9, CXCL10, and CXCL11. The current mini-review focuses on these ligands and their biological properties. First, we elaborate the role of cytokines in directing the polarization of effector and regulatory T cell subset and the plasticity of this process. Then, we extend this notion to chemokines while focusing on CXCL 12 and the CXCR3 ligands. Finally, we elaborate the potential clinical implications of these studies for therapy of autoimmunity, graft-versus-host disease, and cancer.
Introduction
Chemokines are small (~8–14 kDa), secreted proteins, structurally similar to cytokines that regulate cell trafficking through interactions with a subset of seven-transmembrane G protein-coupled receptors (GPCRs) (1–3). Aside from attracting leukocytes to sites of inflammation, chemokines are tightly involved in the activation of adhesion molecules to allow leukocyte extravasation (4–8). This makes them key drivers of inflammation. Studies coming from our laboratory also imply that aside from chemoattraction, some of these chemokines are involved in directing the polarization of CD4+ T cell subsets. This includes the balance between effector T cells subsets (9–11) as well as directing the polarization of effector TH1/Th17 cells into IL-10 producing Tr1-like cells (9–12). The current review focuses on these findings and their biological significance.
Cytokines That Regulate the Balance Between CD4+ T Cells Subsets as Drivers and Regulators of Inflammation
Cytokines are involved in the induction of inflammatory responses by two different, yet complementary, mechanisms: the first includes a direct effect aimed at destructing invading microbes. Two cytokines that posses a major function in this function are tumor necrosis factor alpha (TNF-α) and IL-1β. Consequently, during inflammatory autoimmunity, they are thought to be key mediators of the harmful anti-self distractive response and are, therefore, major targets for therapy of these diseases (13–16). The other mechanism includes directing the functional development (polarization) of CD4+ T cells subsets, and thereby the dynamics of the inflammatory process. The notion that the cytokine milieu at the site of inflammation drives T-cell polarization came from early studies showing that while IL-12 skews the TH1/TH2 balance into IFN-γhigh IL-4low TNFα producing TH1 cells, IL-4 shifts this balance toward IFN-γlow IL-4high TH2 cells, capable of restraining the inflammatory activities of TH1 cells (17–20). Along with this notion, Leonard et al. showed that blocking IL-12 inhibits experimental autoimmune encephalomyelitis (EAE) by shifting the TH1/Th2 balance toward TH2 (21). Another cytokine that has been associated with shifting the TH1/TH2 balance toward TH1 is IL-18 (IGIF) (22). Following this publication, we observed that target neutralization of this cytokine suppresses autoimmunity by interfering in the TH1/TH2 balance toward TH2 (23), and also that targeted expression of its natural inhibitor, IL-18 binding protein (24) at also suppress the disease by the same mechanism (25). A major concern in applying therapies aiming at shifting the TH1/TH2 balanced toward TH2 is that the last are also a subtype of effector T cells that promote IL-4-dependent immunity (26). Thus, shifting anti-self immunity from TH1 to TH2 might result in an unexpected form of self-destructive immunity (27).
In 2005, IL-17-expressing T cells (TH17 cells) were proposed to be a third, independent TH-cell lineage with a role in inflammatory and autoimmune diseases (28). The key cytokines that drive the polarization of these cells vary between rodents and human. In mice, IL-6 together with transforming growth factor-beta (TGF-β) are likely to induce TH17 at early stages of its polarization (together with IL-21) followed by stabilization by IL-23 (29), whereas in human the combination of IL-1 and IL-6, but not TGF-β are key drivers of TH17 polarization (30). More recently, it has been proposed that TH17 cells may also hold anti-inflammatory properties due to potential expression of CD39 and CD73 ectonucleotidases, leading to adenosine release and the subsequent suppression of CD4+ and CD8+ T cell effector functions (31).
The activity of effector T cells is tightly regulated by regulatory T cells that fall into two major subtypes, those expressing the master forkhead box protein 3 (FOXP3) that has a major role in directing their biological properties (32). They suppress the activities of effector T cells and of inflammatory macrophages by various mechanisms, thus maintaining self-tolerance (33–36). Aside from nTregs, FOXP3-positive T cells could be polarized from FOXP3-negative T cells (in vitro) in the presence of transforming growth factor β (TGF-β) (37).
In 1997, Maria Grazia Roncarolo and her coworkers discovered the reciprocal FOXP3-negative IL-10high-producing Tr1 cells (38) that also play a major part in maintenance of self-tolerance (39). These cells could be polarized in vitro by either IL-10 + IL-2 (38) or by the combination of IL-10 + Rapamycin (40) and in human by IL-10 + IFNα (41).
Cytokines and the Plasticity of CD4+ T Cell Subsets
First evidence for potential plasticity in CD4+ T cell subsets have been demonstrated by Anderson et al. in 2007 showing that during chronic cutaneous leishmaniasis TH1 may gain the Tr1-like phenotype and largely produce IL-10 (42). It is not known if these IL10high cells are indeed Tr1 cells, or just IL10high-producing CD4+ T cells, at that time, biomarkers that could distinguish Tr1 cell from other IL10high CD4+ T cells were not yet identified (41, 43). Later IL-27, together with TGFβ, could repolarize TH1 cells into Tr1 (43, 44). As for FOXP3+ Tregs, Chen et al. have shown that coculturing with TGFβ may transform FOXP3−CD4+ T cells into FOXP3+ Tregs, also known as induced Tregs (iTregs) (37). The stability of iTregs in vivo is still questionable.
More recent studies focused on the plasticity between TH17 cells and FOXP3+ Tregs. It appears that expression of Foxp3 by iTreg cells or IL-17 by Th17 cells may not be stable and that there is a great degree of flexibility in their differentiation options as they emerge from an overlapping developmental program (45). Much of the attention has been devoted to exploring the transition from TH17 to iTregs, though a very recent study showed that the inflammatory environment in autoimmune arthritis induces conversion of a subset of Foxp3+ T cells into interleukin-17-producing cells that contribute to disease pathogenesis (46).
These findings should be taken into consideration in designing future therapies aiming at redirecting the polarization of T cell subsets.
The Role of Chemokines in Driving the Functional Development (Polarization) of CD4+ T Cell Subset, are There “Driver” Chemokines?
Chemokines are small (~8–14 kDa), structurally related chemotactic cytokines that regulate cell trafficking through interactions with specific seven-transmembrane, GPCRs (1–3). One of the important features of GPCRs is their ability to transmit diverse signaling cascades upon binding different ligands (47–51). This large family of related molecules is classified on the basis of structural properties, regarding the number and position of conserved cysteine residues, to give two major (CXC and CC) and two minor (C and CX3C) chemokine subfamilies (1–3) (Table 1).
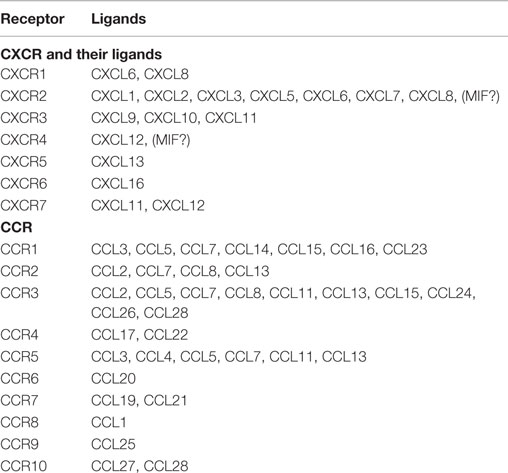
Table 1. Key chemokine receptors and their ligands.
Most of the attention has been drawn to the key role of these chemotactic mediators in promoting lymphocyte migration processes critical for the onset of inflammatory processes with a special interest in inflammatory autoimmune diseases. Reviewing the results of the very many studies in which single chemokines or there receptors were targeted reviles a major paradox; even though most of the 50 known chemokines can direct the migration of the same leukocytes, targeted neutralization of only one chemokine, such as CCL2, CCL3, CCL5, or CXCL10, is sufficient to suppress the entire inflammatory process (10, 52–59). Therefore, the question that begs an answer is why other chemokines that also attract the same type of leukocyte to the autoimmune site do not compensate for the absence of this single chemokine. In addition, it is also not clear why neutralization of as few as eight to 10 of the 50 different chemokines can effectively suppress the attacks in autoimmune inflammatory diseases (10, 52–59). Hence, what are the attributes of this limited number of chemokines that make them so important in the regulation of inflammatory processes?
A partial explanation for this paradigm could be that these chemokines might have other biological actions that are associated with these autoimmune inflammatory diseases. This includes directing the mobilization of various cells types from the bone marrow to the blood, and later their colonization at the inflammatory site, induction of selective migration to specific organs, directing the development cell subtypes (such as CD4+ T cell polarization) or potentiation of innate immune cells. The current review focuses on the role of chemokines on the balance of T cell subsets. CXCL10 is a key driver of TH1 and possibly TH17 polarization and has, therefore, been a major target for neutralization in different autoimmune diseases (9, 10). More recently, we identified two different CXC chemokines that possess anti-inflammatory properties (11, 12).
CXCL12 is an important chemokine that participates in the regulation of tissue homeostasis, immune surveillance, cancer development, and the regulation of inflammatory responses. It is believed that under non-inflammatory conditions, the continuing expression of CXCL12 in tissues that are partially segregated from the immune system, such as the CNS, is important for directing the entry of leukocytes to these sites, as part of immune surveillances (60, 61). We have previously shown that aside from this activity, which in its nature could be proinflammatory, CXCL12 also drives the polarization of CXCR4+ macrophages into the IL-10high M2c-like macrophages (12) that hold anti-inflammatory properties (62) and also of effector CD4+ T cells (CXCR4+) into IL-10high Tr1 cells (12). This may explain why its administration during late stages of EAE leads to rapid remission (12). Based on the above, we thought of generating an Ig-based stabilized protein (CXCL12-Ig) for therapy of various inflammatory autoimmune diseases. Nevertheless, the major involvement of this chemokine in various biological functions, such as homing of stem cells to the bone marrow, homeostasis of neutrophils, angiogenesis, and others precludes it use as a stabilized chemokine for therapy of autoimmune diseases (63).
CXCL11 as a Novel Driver Anti-Inflammatory Chemokine
Is CXCL12 an exception, or are there other chemokine with anti-inflammatory properties?
One of the important features of GPCRs is their ability to transmit diverse signaling cascades upon binding different ligands (47–51). The Nobel prizewinner Robert J. Lefkowitz and his team have previously raised the concept that different ligands binding the same G-coupled receptor may induce diverse signaling cascades resulting in distinct biological activities (50, 64, 65). Even though the mechanistic basis of this feature is not fully understood, its biological and clinical implications are highly significant (50).
We have investigated the interplay between CXCR3 and its three ligands: CXCL9, CXCL10, and CXCL11 on directing the polarization of CD4+ T cells. We observed that while CXCL9 and CXCL10 skew T cell polarization into Th1/Th17 effector cells, CXCL11 drives CD4+ T cell polarization into IL-10-producing Tr-1 (11). We also uncovered the signaling basis of this biased response, and learned that it is GαI independent (11). While CXCL10/CXCR3 interactions drive effector Th1 polarization via STAT1, STAT4, and STAT5 phosphorylation, CXCL11/CXCR3 binding induces an immunotolerizing state that is characterized by IL-10high (Tr1) and IL-4high (Th2) cells and mediated via p70 kinase/mTOR in STAT-3- and STAT-6-dependent pathways (11). CXCL11 binds CXCR3 a higher affinity than CXCL10, suggesting that CXCL11 has potential to mediate and restrain inflammatory autoimmunity (Figure 1). This may explain, in part, why CXCR3-deficient mice develop an extremely severe form of EAE and T1DM (66, 67).
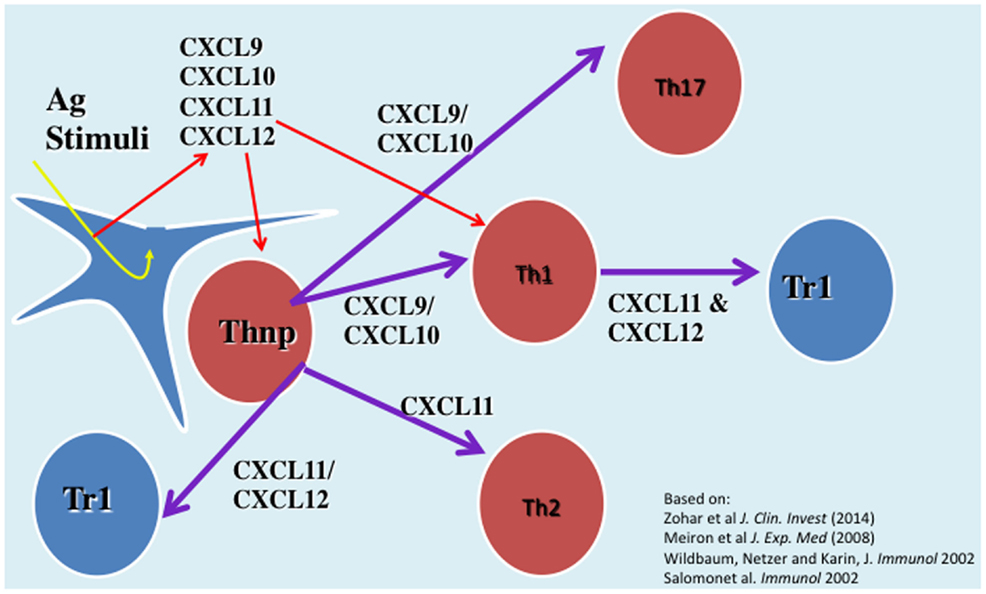
Figure 1. The role of CXC chemokines in driving the polarization and biological function of CD4+ T cell subsets.
Novel Approach for Chemokine-Based Therapy of Inflammatory Autoimmunity GVHD and Cancer Diseases
Thus, far many affords has been spent in studying exploring the therapeutic potential of targeting the interaction between chemokine and their receptors for treating various autoimmune and cancer diseases. This includes antibody-based therapy to single chemokines or their receptors (68, 69), targeted DNA vaccines that that amplify the natural autoantibody titer to chemokines (9, 10, 55, 70), soluble chemokine receptor-based therapy (71, 72), and small molecule-based antagonists to chemokine receptors (73, 74). Some of these studies have been employed in human clinical trials, thus far with very limited successes. It is believed that the major limitation of applying anti-chemokine- or chemokine receptor-based therapies is the redundancy between chemokines and the enhanced in vivo production, once being neutralized (71). The discovery of chemokines with anti-inflammatory properties opens the door for an alterative approach of using stabilized chemokines for therapy of autoimmunity and graft-versus-host disease (GVHD).
Could stabilized chemokines be also used for therapy of cancer diseases? Studies that were initiated in experimental models and recently extended to patients suffering from melanoma showed that blockage of FOXP3+ T cells function by blocking the interaction between immunosuppressive receptor programmed cell death-1 (PD-1) largely expressed on FOXp3+ T cells and its target coreceptor on antigen presenting cells (PDL-1) using anti-PD1 mAb (nivolumab) (75) or anti-PDL-1 mAb (76) suppressed the function of tumor infiltrating Tregs, and thereby enhanced antitumor immunity to suppress tumor development and progression (75, 76). The other successful approach of enhancing antitumor immunity against melanoma included the administration of a mAb (ipilimumab) which blocks cytotoxic T-lymphocyte-associated antigen 4 (CTLA-4) to potentiate an antitumor T-cell response (77). Very recently, combined therapy\ of anti-PD1 (nivolumab) and anti-CTLA-4 (ipilimumab) showed improved efficacy in treating melanoma (78). The observations that CXCL10 enhances effector T cell activities (11) motivated us to explore CXCL10-Ig-based therapy in cancer diseases. Very recently, we showed that indeed administration of CXCL10-Ig in a clinical set-up of myeloma that CXCL10-Ig could be used for immunotherapy of this disease, and that aside from enhancing antitumor immunity, it directly suppresses tumor growth (79). Along with this study, very recently, Barreira da Silva et al. showed that inhibition of DPP4 enzymatic activity enhanced tumor rejection by preserving biologically active CXCL10 and increasing trafficking into the tumor by lymphocytes expressing the counter-receptor CXCR3 (80). We are now exploring combined therapies of CXCL10-Ig with anti-PD1 or anti-CTLA-4 in a melanoma set-up.
Another chemokine that might serve as a target for cancer therapy is CCL1. Its CCR8 receptor is highly expressed on FOXP3+ Tregs and has been associated in their targeted attraction (81, 82). Along with this, Hoelzinger et al. showed that targeting CCL1 might enhance antitumor immunity (83). We are now examining whether its stabilized form (CCL1-Ig) could be used for therapy of inflammatory autoimmunity.
Conclusion
The current review focuses on exploring the involvement of chemokines in directing the polarization and biological function of CD4+ T cells. Thus, far most of the attention has been devoted to exploring the role of cytokines in this property. From a clinically oriented perspective, the findings that chemokines may also polarize Tregs (so far our data shows relevance only for FOXP3-negative Tregs) opens the window of opportunities for using stabilized chemokines for therapy of inflammatory autoimmunity and GVHD, and also for cancer diseases. The basic rational is that the stabilized form of chemokines that induce Tr1-like cells, among them CXCL12 and CXCL11, could be used for therapy of autoimmunity and GVHD, whereas stabilized CXCL10 would be used for cancer therapy.
We find some major differences between CXCL12 and CXCL11 as potential tolerizing chemokines. CXCL12 also renders anti-inflammatory properties in macrophages (12), whereas CXCL11 also polarizes IL-4high Th2 cells (11). We assume that CXCL11 could be a better candidate for being a potential drug since CXCL12 is involved in many biological activities aside from being an immunoregulator, such as neutrophil homeostasis or stem cell homing (63).
Ethics Statement
All experimental work described in the manuscript was approved by the ethical committee of the Technion, according the NIH guideline.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
This study was funded by the Israel Science Foundation (ISF), ICRF, Israel Cancer Association, and the Collek fund of the Technion.
References
1. Luster AD. Chemokines – chemotactic cytokines that mediate inflammation. N Engl J Med (1998) 338:436–45. doi: 10.1056/NEJM199802123380706
2. Zlotnic A, Yoshei O. Chemokines: a new classification system and their role in immunity. Immunity (2000) 12:121–7. doi:10.1016/S1074-7613(00)80165-X
3. Proudfoot AE. Chemokine receptors: multifaceted therapeutic targets. Nat Rev Immunol (2002) 2:106–15. doi:10.1038/nri722
4. Stoolman LM. Adhesion molecules controlling lymphocyte migration. Cell (1989) 56:907–10. doi:10.1016/0092-8674(89)90620-X
5. Elices MJ, Osborn L, Takada Y, Crouse C, Luhowskyj S, Hemler ME, et al. VCAM-1 on activated endothelium interacts with the leukocyte integrin VLA-4 at a site distinct from the VLA-4/fibronectin binding site. Cell (1990) 60:577–84. doi:10.1016/0092-8674(90)90661-W
6. Osborn L. Leukocyte adhesion to endothelium in inflammation. Cell (1990) 62:3–6. doi:10.1016/0092-8674(90)90230-C
7. Springer TA. Adhesion receptors of the immune system. Nature (1990) 346:425–34. doi:10.1038/346425a0
8. Spertini O, Kansas GS, Munro JM, Griffin JD, Tedder TF. Regulation of leukocyte migration by activation of the leukocyte adhesion molecule-1 (LAM-1) selectin. Nature (1991) 349:691–4. doi:10.1038/349691a0
9. Salomon I, Netzer N, Wildbaum G, Schif-Zuck S, Maor G, Karin N. Targeting the function of IFN-gamma-inducible protein 10 suppresses ongoing adjuvant arthritis. J Immunol (2002) 169:2685–93. doi:10.4049/jimmunol.169.5.2685
10. Wildbaum G, Netzer N, Karin N. Plasmid DNA encoding IFN-gamma-inducible protein 10 redirects antigen-specific T cell polarization and suppresses experimental autoimmune encephalomyelitis. J Immunol (2002) 168:5885–92. doi:10.4049/jimmunol.168.11.5885
11. Zohar Y, Wildbaum G, Novak R, Salzman AL, Thelen M, Alon R, et al. CXCL11-dependent induction of FOXP3-negative regulatory T cells suppresses autoimmune encephalomyelitis. J Clin Invest (2014) 124:2009–22. doi:10.1172/JCI71951
12. Meiron M, Zohar Y, Anunu R, Wildbaum G, Karin N. CXCL12 (SDF-1alpha) suppresses ongoing experimental autoimmune encephalomyelitis by selecting antigen-specific regulatory T cells. J Exp Med (2008) 205:2643–55. doi:10.1084/jem.20080730
13. Arend WP, Dayer JM. Inhibition of the production and effects of interleukin-1 and tumor necrosis factor alpha in rheumatoid arthritis. Arthritis Rheum (1995) 38:151–60. doi:10.1002/art.1780380202
14. Feldmann M, Brennan FM, Foxwell BM, Maini RN. The role of TNF alpha and IL-1 in rheumatoid arthritis. Curr Dir Autoimmun (2001) 3:188–99. doi:10.1159/000060522
15. Dayer JM, Krane SM. Anti-TNF-alpha therapy for ankylosing spondylitis – a specific or nonspecific treatment? N Engl J Med (2002) 346:1399–400. doi:10.1056/NEJM200205023461811
16. Ogata H, Hibi T. Cytokine and anti-cytokine therapies for inflammatory bowel disease. Curr Pharm Des (2003) 9:1107–13. doi:10.2174/1381612033455035
17. Mosmann TR, Cherwinski H, Bond MW, Giedlin MA, Coffman RL. Two types of murine helper T cell clone. I. Definition according to profiles of lymphokine activities and secreted proteins. J Immunol (1986) 136:2348–57.
18. Stevens TL, Bossie A, Sanders VM, Fernandez-Botran R, Coffman RL, Mosmann TR, et al. Regulation of antibody isotype secretion by subsets of antigen-specific helper T cells. Nature (1988) 334:255–8. doi:10.1038/334255a0
19. Mosmann TR, Coffman RL. Th1 and Th2 cells: different patterns of lymphokine secretion lead to different functional properties. Annu Rev Immunol (1989) 9:145–73. doi:10.1146/annurev.iy.07.040189.001045
20. Zheng W, Flavell RA. The transcription factor GATA-3 is necessary and sufficient for Th2 cytokine gene expression in CD4 T cells. Cell (1997) 89:587–96. doi:10.1016/S0092-8674(00)80240-8
21. Leonard JP, Waldburger KE, Goldman SJ. Prevention of experimental autoimmune encephalomyelitis by antibodies against interleukin 12. J Exp Med (1995) 181:381–6. doi:10.1084/jem.181.1.381
22. Okamura H, Tsutsi H, Komatsu T, Yutsudo M, Hakura A, Tanimoto T, et al. Cloning of a new cytokine that induces IFN-gamma production by T cells. Nature (1995) 378:88–91. doi:10.1038/378088a0
23. Wildbaum G, Youssef S, Grabie N, Karin N. Neutralizing antibodies to IFN-gamma-inducing factor prevent experimental autoimmune encephalomyelitis. J Immunol (1998) 161:6368–74.
24. Novick D, Kim SH, Fantuzzi G, Reznikov LL, Dinarello CA, Rubinstein M. Interleukin-18 binding protein: a novel modulator of the Th1 cytokine response. Immunity (1999) 10:127–36. doi:10.1016/S1074-7613(00)80013-8
25. Schif-Zuck S, Westermann J, Netzer N, Zohar Y, Meiron M, Wildbaum G, et al. Targeted overexpression of IL-18 binding protein at the central nervous system overrides flexibility in functional polarization of antigen-specific Th2 cells. J Immunol (2005) 174:4307–15. doi:10.4049/jimmunol.174.7.4307
26. Abbas AK, Murphy KM, Sher A. Functional diversity of helper T lymphocytes. Nature (1996) 383:787–93. doi:10.1038/383787a0
27. Pedotti R, Mitchell D, Wedemeyer J, Karpuj M, Chabas D, Hattab EM, et al. An unexpected version of horror autotoxicus: anaphylactic shock to a self-peptide. Nat Immunol (2001) 2:216–22. doi:10.1038/85266
28. Harrington LE, Hatton RD, Mangan PR, Turner H, Murphy TL, Murphy KM, et al. Interleukin 17-producing CD4(+) effector T cells develop via a lineage distinct from the T helper type 1 and 2 lineages. Nat Immunol (2005) 6(11):1123–32. doi:10.1038/ni1254
29. Bettelli E, Korn T, Oukka M, Kuchroo VK. Induction and effector functions of T(H)17 cells. Nature (2008) 453:1051–7. doi:10.1038/nature07036
30. Acosta-Rodriguez EV, Napolitani G, Lanzavecchia A, Sallusto F. Interleukins 1beta and 6 but not transforming growth factor-beta are essential for the differentiation of interleukin 17-producing human T helper cells. Nat Immunol (2007) 8:942–9. doi:10.1038/ni1496
31. Chalmin F, Mignot G, Bruchard M, Chevriaux A, Vegran F, Hichami A, et al. Stat3 and Gfi-1 transcription factors control Th17 cell immunosuppressive activity via the regulation of ectonucleotidase expression. Immunity (2012) 36:362–73. doi:10.1016/j.immuni.2011.12.019
32. Hori S, Nomura T, Sakaguchi S. Control of regulatory T cell development by the transcription factor Foxp3. Science (2003) 299:1057–61. doi:10.1126/science.1079490
33. Sakaguchi S. Naturally arising Foxp3-expressing CD25+CD4+ regulatory T cells in immunological tolerance to self and non-self. Nat Immunol (2005) 6:345–52. doi:10.1038/ni1178
34. Sakaguchi S, Ono M, Setoguchi R, Yagi H, Hori S, Fehervari Z, et al. Foxp3+ CD25+ CD4+ natural regulatory T cells in dominant self-tolerance and autoimmune disease. Immunol Rev (2006) 212:8–27. doi:10.1111/j.0105-2896.2006.00427.x
35. Shevach EM. Mechanisms of foxp3+ T regulatory cell-mediated suppression. Immunity (2009) 30:636–45. doi:10.1016/j.immuni.2009.04.010
36. Wing K, Sakaguchi S. Regulatory T cells exert checks and balances on self tolerance and autoimmunity. Nat Immunol (2009) 11:7–13. doi:10.1038/ni.1818
37. Chen W, Jin W, Hardegen N, Lei KJ, Li L, Marinos N, et al. Conversion of peripheral CD4+CD25- naive T cells to CD4+CD25+ regulatory T cells by TGF-beta induction of transcription factor Foxp3. J Exp Med (2003) 198:1875–86. doi:10.1084/jem.20030152
38. Groux H, O’Garra A, Bigler M, Rouleau M, Antonenko S, De Vries JE, et al. A CD4+ T-cell subset inhibits antigen-specific T-cell responses and prevents colitis. Nature (1997) 389:737–42. doi:10.1038/39614
39. Roncarolo MG, Gregori S, Battaglia M, Bacchetta R, Fleischhauer K, Levings MK. Interleukin-10-secreting type 1 regulatory T cells in rodents and humans. Immunol Rev (2006) 212:28–50. doi:10.1111/j.0105-2896.2006.00420.x
40. Battaglia M, Stabilini A, Draghici E, Gregori S, Mocchetti C, Bonifacio E, et al. Rapamycin and interleukin-10 treatment induces T regulatory type 1 cells that mediate antigen-specific transplantation tolerance. Diabetes (2006) 55:40–9. doi:10.2337/diabetes.55.01.06.db05-0613
41. Gagliani N, Magnani CF, Huber S, Gianolini ME, Pala M, Licona-Limon P, et al. Coexpression of CD49b and LAG-3 identifies human and mouse T regulatory type 1 cells. Nat Med (2013) 19:739–46. doi:10.1038/nm.3179
42. Anderson CF, Oukka M, Kuchroo VJ, Sacks D. CD4(+)CD25(-)Foxp3(-) Th1 cells are the source of IL-10-mediated immune suppression in chronic cutaneous leishmaniasis. J Exp Med (2007) 204:285–97. doi:10.1084/jem.20061886
43. Apetoh L, Quintana FJ, Pot C, Joller N, Xiao S, Kumar D, et al. The aryl hydrocarbon receptor interacts with c-Maf to promote the differentiation of type 1 regulatory T cells induced by IL-27. Nat Immunol (2010) 11:854–61. doi:10.1038/ni.1912
44. Sun J, Dodd H, Moser EK, Sharma R, Braciale TJ. CD4+ T cell help and innate-derived IL-27 induce Blimp-1-dependent IL-10 production by antiviral CTLs. Nat Immunol (2011) 12:327–34. doi:10.1038/ni.1996
45. Zhou L, Chong MM, Littman DR. Plasticity of CD4+ T cell lineage differentiation. Immunity (2009) 30:646–55. doi:10.1016/j.immuni.2009.05.001
46. Joller N, Kuchroo VK. Good guys gone bad: exTreg cells promote autoimmune arthritis. Nat Med (2014) 20:15–7. doi:10.1038/nm.3439
47. Rajagopal S, Kim J, Ahn S, Craig S, Lam CM, Gerard NP, et al. Beta-arrestin- but not G protein-mediated signaling by the “decoy” receptor CXCR7. Proc Natl Acad Sci U S A (2010) 107:628–32. doi:10.1073/pnas.0912852107
48. Blattermann S, Peters L, Ottersbach PA, Bock A, Konya V, Weaver CD, et al. A biased ligand for OXE-R uncouples Galpha and Gbetagamma signaling within a heterotrimer. Nat Chem Biol (2012) 8:631–8. doi:10.1038/nchembio.962
49. Liu JJ, Horst R, Katritch V, Stevens RC, Wuthrich K. Biased signaling pathways in beta2-adrenergic receptor characterized by 19F-NMR. Science (2012) 335:1106–10. doi:10.1126/science.1215802
50. Reiter E, Ahn S, Shukla AK, Lefkowitz RJ. Molecular mechanism of beta-arrestin-biased agonism at seven-transmembrane receptors. Annu Rev Pharmacol Toxicol (2012) 52:179–97. doi:10.1146/annurev.pharmtox.010909.105800
51. Zimmerman B, Beautrait A, Aguila B, Charles R, Escher E, Claing A, et al. Differential beta-arrestin-dependent conformational signaling and cellular responses revealed by angiotensin analogs. Sci Signal (2012) 5:ra33. doi:10.1126/scisignal.2002522
52. Karpus WJ, Lukacs NW, Mcrae BL, Strieter RM, Kunkel SL, Miller SD. An important role for the chemokine macrophage inflammatory protein-1 alpha in the pathogenesis of the T cell-mediated autoimmune disease, experimental autoimmune encephalomyelitis. J Immunol (1995) 155:5003–10.
53. Karpus WJ, Kennedy KJ. MIP-1alpha and MCP-1 differentially regulate acute and relapsing autoimmune encephalomyelitis as well as Th1/Th2 lymphocyte differentiation. J Leukoc Biol (1997) 62:681–7.
54. Karpus WJ, Kennedy KJ, Kunkel SL, Lukacs NW. Monocyte chemotactic protein 1 regulates oral tolerance induction by inhibition of T helper cell 1-related cytokines. J Exp Med (1998) 187:733–41. doi:10.1084/jem.187.5.733
55. Youssef S, Wildbaum G, Maor G, Lanir N, Gour-Lavie A, Grabie N, et al. Long-lasting protective immunity to experimental autoimmune encephalomyelitis following vaccination with naked DNA encoding C-C chemokines. J Immunol (1998) 161:3870–9.
56. Youssef S, Wildbaum G, Karin N. Prevention of experimental autoimmune encephalomyelitis by MIP-1alpha and MCP-1 naked DNA vaccines. J Autoimmun (1999) 13:21–9. doi:10.1006/jaut.1999.0306
57. Izikson L, Klein RS, Charo IF, Weiner HL, Luster AD. Resistance to experimental autoimmune encephalomyelitis in mice lacking the CC chemokine receptor (CCR)2. J Exp Med (2000) 192:1075–80. doi:10.1084/jem.192.7.1075
58. Fife BT, Kennedy KJ, Paniagua MC, Lukacs NW, Kunkel SL, Luster AD, et al. CXCL10 (IFN-gamma-inducible protein-10) control of encephalitogenic CD4+ T cell accumulation in the central nervous system during experimental autoimmune encephalomyelitis. J Immunol (2001) 166:7617–24. doi:10.4049/jimmunol.166.12.7617
59. Fukumoto N, Shimaoka T, Fujimura H, Sakoda S, Tanaka M, Kita T, et al. Critical roles of CXC chemokine ligand 16/scavenger receptor that binds phosphatidylserine and oxidized lipoprotein in the pathogenesis of both acute and adoptive transfer experimental autoimmune encephalomyelitis. J Immunol (2004) 173:1620–7. doi:10.4049/jimmunol.173.3.1620
60. Mccandless EE, Wang Q, Woerner BM, Harper JM, Klein RS. CXCL12 limits inflammation by localizing mononuclear infiltrates to the perivascular space during experimental autoimmune encephalomyelitis. J Immunol (2006) 177:8053–64. doi:10.4049/jimmunol.177.11.8053
61. Cruz-Orengo L, Holman DW, Dorsey D, Zhou L, Zhang P, Wright M, et al. CXCR7 influences leukocyte entry into the CNS parenchyma by controlling abluminal CXCL12 abundance during autoimmunity. J Exp Med (2011) 208:327–39. doi:10.1084/jem.20102010
62. Martinez FO, Sica A, Mantovani A, Locati M. Macrophage activation and polarization. Front Biosci (2008) 13:453–61. doi:10.2741/2692
63. Karin N. The multiple faces of CXCL12 (SDF-1alpha) in the regulation of immunity during health and disease. J Leukoc Biol (2010) 88:463–73. doi:10.1189/jlb.0909602
64. Samama P, Cotecchia S, Costa T, Lefkowitz RJ. A mutation-induced activated state of the beta 2-adrenergic receptor. Extending the ternary complex model. J Biol Chem (1993) 268:4625–36.
65. Luttrell LM, Ferguson SSNG, Daaka Y, Miller WE, Maudsley S, Della Rocca GJ, et al. Arrestin-dependent formation of 2 adrenergic receptor-Src protein kinase complexes. Science (1999) 283:655–61. doi:10.1126/science.283.5402.655
66. Frigerio S, Junt T, Lu B, Gerard C, Zumsteg U, Hollander GA, et al. Beta cells are responsible for CXCR3-mediated T-cell infiltration in insulitis. Nat Med (2002) 8:1414–20. doi:10.1038/nm1202-792
67. Liu L, Huang D, Matsui M, He TT, Hu T, Demartino J, et al. Severe disease, unaltered leukocyte migration, and reduced IFN-gamma production in CXCR3-/- mice with experimental autoimmune encephalomyelitis. J Immunol (2006) 176:4399–409. doi:10.4049/jimmunol.176.7.4399
68. Gazzaniga S, Bravo AI, Guglielmotti A, Van Rooijen N, Maschi F, Vecchi A, et al. Targeting tumor-associated macrophages and inhibition of MCP-1 reduce angiogenesis and tumor growth in a human melanoma xenograft. J Invest Dermatol (2007) 127:2031–41. doi:10.1038/sj.jid.5700827
69. Loberg RD, Ying C, Craig M, Day LL, Sargent E, Neeley C, et al. Targeting CCL2 with systemic delivery of neutralizing antibodies induces prostate cancer tumor regression in vivo. Cancer Res (2007) 67:9417–24. doi:10.1158/0008-5472.CAN-07-1286
70. Youssef S, Maor G, Wildbaum G, Grabie N, Gour-Lavie A, Karin N. C-C chemokine-encoding DNA vaccines enhance breakdown of tolerance to their gene products and treat ongoing adjuvant arthritis. J Clin Invest (2000) 106:361–71. doi:10.1172/JCI9109
71. Izhak L, Wildbaum G, Zohar Y, Anunu R, Klapper L, Elkeles A, et al. A novel recombinant fusion protein encoding a 20-amino acid residue of the third extracellular (E3) domain of CCR2 neutralizes the biological activity of CCL2. J Immunol (2009) 183:732–9. doi:10.4049/jimmunol.0802746
72. Sapir Y, Vitenshtein A, Barsheshet Y, Zohar Y, Wildbaum G, Karin N. A fusion protein encoding the second extracellular domain of CCR5 arrests chemokine-induced cosignaling and effectively suppresses ongoing experimental autoimmune encephalomyelitis. J Immunol (2010) 185:2589–99. doi:10.4049/jimmunol.1000666
73. Burns JM, Summers BC, Wang Y, Melikian A, Berahovich R, Miao Z, et al. A novel chemokine receptor for SDF-1 and I-TAC involved in cell survival, cell adhesion, and tumor development. J Exp Med (2006) 203:2201–13. doi:10.1084/jem.20052144
74. Khan A, Greenman J, Archibald SJ. Small molecule CXCR4 chemokine receptor antagonists: developing drug candidates. Curr Med Chem (2007) 14:2257–77. doi:10.2174/092986707781696618
75. Hamid O, Robert C, Daud A, Hodi FS, Hwu WJ, Kefford R, et al. Safety and tumor responses with lambrolizumab (anti-PD-1) in melanoma. N Engl J Med (2013) 369:134–44. doi:10.1056/NEJMoa1305133
76. Brahmer JR, Tykodi SS, Chow LQ, Hwu WJ, Topalian SL, Hwu P, et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N Engl J Med (2012) 366:2455–65. doi:10.1056/NEJMoa1200694
77. Hodi FS, O’Day SJ, Mcdermott DF, Weber RW, Sosman JA, Haanen JB, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med (2010) 363:711–23. doi:10.1056/NEJMoa1003466
78. Wolchok JD, Kluger H, Callahan MK, Postow MA, Rizvi NA, Lesokhin AM, et al. Nivolumab plus ipilimumab in advanced melanoma. N Engl J Med (2013) 369:122–33. doi:10.1056/NEJMoa1302369
79. Barash U, Zohar Y, Wildbaum G, Beider K, Nagler A, Karin N, et al. Heparanase enhances myeloma progression via CXCL10 down regulation. Leukemia (2014) 28(11):2178–87. doi:10.1038/leu.2014.121
80. Barreira Da Silva R, Laird ME, Yatim N, Fiette L, Ingersoll MA, Albert ML. Dipeptidylpeptidase 4 inhibition enhances lymphocyte trafficking, improving both naturally occurring tumor immunity and immunotherapy. Nat Immunol (2015) 16:850–8. doi:10.1038/ni.3201
81. Iellem A, Mariani M, Lang R, Recalde H, Panina-Bordignon P, Sinigaglia F, et al. Unique chemotactic response profile and specific expression of chemokine receptors CCR4 and CCR8 by CD4(+)CD25(+) regulatory T cells. J Exp Med (2001) 194:847–53. doi:10.1084/jem.194.6.847
82. Coghill JM, Fowler KA, West ML, Fulton LM, Van Deventer H, Mckinnon KP, et al. CC chemokine receptor 8 potentiates donor Treg survival and is critical for the prevention of murine graft-versus-host disease. Blood (2013) 122:825–36. doi:10.1182/blood-2012-06-435735
Keywords: chemokines, T cell subsets, EAE, CXCR3, CXCL11, CXCL10, cancer, immunotherapy
Citation: Karin N and Wildbaum G (2015) The Role of Chemokines in Shaping the Balance Between CD4+ T Cell Subsets and Its Therapeutic Implications in Autoimmune and Cancer Diseases. Front. Immunol. 6:609. doi: 10.3389/fimmu.2015.00609
Received: 31 May 2015; Accepted: 16 November 2015;
Published: 30 November 2015
Edited by:
Wilson Savino, Oswaldo Cruz Foundation, BrazilReviewed by:
Christopher E. Rudd, University of Cambridge, UKAntónio Gil Castro, University of Minho, Portugal
Copyright: © 2015 Karin and Wildbaum. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Nathan Karin, nkarin10@gmail.com