 Borja Sánchez
Borja Sánchez Arancha Hevia1
Arancha Hevia1
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 20 November 2015
Sec. Immunological Tolerance and Regulation
Volume 6 - 2015 | https://doi.org/10.3389/fimmu.2015.00594
This article is part of the Research Topic Double-edge consequence of enhanced immune reactivity in females: Better protection against infection but increased susceptibility to autoimmune diseases View all 9 articles
Autoimmune diseases, such as systemic lupus erythematosus (SLE), are caused by a complex interaction of environmental-, genetic-, and sex-related factors. Although SLE has traditionally been considered independent from the microbiota, recent work published during the last 5 years suggests a strong connection between SLE and the composition of our gut commensals as one of the main environmental factors linked to this disease. Preliminary data have evidenced that (i) interaction of certain microbial-derived molecules with specific cell receptors and (ii) the influence of certain commensal microorganisms over specific immune cell subsets plays an important role in the pathogenesis of SLE and SLE-like diseases. In addition, epigenetic changes driven by certain microbial groups have been recently proposed as an additional link between gut microbiota and SLE. As immune responses elicited against commensal bacteria are deeply dependent on the composition of the latter, and as microbial populations can be modified by dietary interventions, identifying the precise gut microorganisms responsible for worsening the SLE symptoms is of crucial importance for this and other SLE-related diseases, including antiphospholipid syndrome or lupus nephritis. In this minireview, the current knowledge on the relationships between microbes and SLE and SLE-related diseases is compiled and discussed.
The term human microbiota describes the ensemble of microorganisms inhabiting our body, which account for 10 times more cells than our own cells, with the colon being the place in the body populated by the largest number of microbes. Alterations of gut microbial populations (also termed intestinal dysbiosis) have been associated with a large number of autoimmune and chronic inflammatory diseases during the last 10 years, for instance, rheumatoid arthritis, type 1 diabetes, inflammatory bowel disease, and recently systemic lupus erythematosus (SLE) (1, 2).
Our microbiota and immune system have co-evolved in a symbiotic way; while bacteria helps in digestion, presence of certain commensals triggers different signals that drive proper maturation of our immune system. In turn, the colon provides the right environment to host these populations. Microbiota composition can be quickly modified by diet or by the use of certain living bacteria, known as probiotics. Intestinal microbiota influences the development of autoimmune diseases, and so this influence might be modulated through specific diets or with the administration of specific probiotic strains (3). Scientific evidence suggests that every type of microorganism might have a specific mechanism of action over the host immune system, balancing anti- and pro-inflammatory responses given some host factors, such as the genetic background, sex, and the relative abundance of a given microorganism in the whole microbiota (4).
In the last years, some studies have identified different microbial molecules, notably located at the surface level, able to drive those immunological effects, among which extracellular polysaccharides and proteins, peptides, and teichoic acids are noteworthy (5). The aim of the present minireview is to compile and discuss current evidence of the potential influence of gut microbes on SLE pathogeny and other related autoimmune diseases.
Systemic lupus erythematosus is a complex disease with remarkable heterogeneity in its clinical features, which includes malar rash, photosensitivity, and renal disorders among others. About half of the patients develop renal symptoms within the first year, and in fact kidney-specific molecules are the main target for immunosuppressive treatments. From the immunological point of view, SLE is characterized by aberrant responses at different levels, including uncontrolled T-cell differentiation and activation, abnormal polyclonal B-cell activation/proliferation, and autoantibody (IgGs) production linked to immune complex formation (6).
The key immunological characteristic of SLE is B-cell hyperactivity, which results in the production of a wide set of autoantibodies recognizing more than 100 different ligands, such as nuclear DNA or histones, ribonucleoproteins, or cardiolipin (7). Binding of these autoantibodies to self-antigens induces the formation of immune complexes, whose accumulation triggers molecular inflammation and leads to severe damages due to immunological clearance. For instance, injuries at the glomerular level are responsible for lupus nephritis (LN), the severity of the symptoms correlating with the autoantibody titers and, therefore, with the amount of immune complexes formed (8).
The SLE diagnosis cannot be done only on a serological basis, in spite of the abundant array of autoantibodies generated during its onset and development. SLE is diagnosed when 4 out of 11 specific criteria are fulfilled, the presence of elevated autoantibody titers representing only two of them (9). As B-cell hyperactivity underlies this auto-antigen generation, this cell type is target for several treatments, including monoclonal antibodies binding to specific B-cell-surface antigens (CD19, CD20, CD22) to trigger apoptosis/lysis, or peptides/proteins blocking binding of B-cell activating factors to B-cell receptors (BLyS, APRIL), thus, aborting maturation and proliferation (10). Remarkably, genetic susceptibility of the B-cell receptor gene is a factor favoring induction of B-cell hyperactivity by external stimuli and, therefore, conditioning an individual to autoimmunity (11).
In general, total IgG-class antibody titers in plasma were shown to be doubled in a SLE cohort regarding the controls (12). As it has been shown that specific IgGs raised against the gut microbiota displayed lower titers during active SLE episodes compared to inactive periods, it is conceivable to hypothesize that these specific antibodies are sequestered in immune complexes, contributing to the pathology of the disease, and opening the door to the involvement of certain bacteria at least in SLE relapses (12).
The term “autoimmune disease” denotes a disorder where the immune system recognizes self-molecules as foreign, a process in which microbes may have a direct or indirect influence. Direct mechanisms involve growing and temporal persistence of the bacterium within the gut, as well as the secretion or release of different molecular mediators. On the contrary, indirect mechanisms involve the production of epigenetic changes in human cells driven by the presence of the bacterium. For these reasons, the relationship and interactions among commensal microbes, immune system, and epigenetics are an emerging field of great interest in autoimmune disease research (13).
It should be highlighted that host sex conditions the populations inhabiting our gut, which may condition the severity of the symptoms, as in SLE (14). In turn, the configuration of host immunity at both innate and adaptive levels may condition relative abundances within those gut commensal populations, establishing a complicated feedback between relative microorganism abundances, host sex and the way to shape the type of immune responses against self and foreign antigens (15).
Commensal microbiota composition has a deep effect in terms of immunomodulation, whatever the immunological context of the host. This statement is very well illustrated by a single bacterial species, the segmented filamentous bacteria (SFB), and might be applicable to every bacterial population. SFB have the ability to expand the set of Th17 cells, which are involved in many human autoimmune diseases (16). The Th17 response is pathogenic in certain animal models of autoimmune diseases such as rheumatoid arthritis or certain IBD types, but protective in others such as type 1 diabetes (17–19), so the specific effect of a single bacterium can be either positive or negative, depending on the immunological status of the host. Moreover, SFB effect varies depending on the host sex, and so Th17 stimulation confers protection in females but not in males in certain murine models of autoimmune diseases (18, 19).
Among the different autoimmune diseases, SLE has been considered independent from the microbiota, with few scientific evidences dating back to the 1970s and 1980s. If we examine carefully the scientific literature, we can find some evidences, suggesting a link between the presence of certain bacteria and SLE. For instance, cell wall deficient forms (CWD) of Propionibacterium acnes, Corynebacterium sp., Staphylococcus epidermis, and Streptococcus sp. were isolated in cutaneous lesions of patients affected by SLE and other SLE-related disorders such as cutaneous lupus erythematosus (LE) (20). Perhaps the strongest evidence linking a bacterial antigen and the production of self-antibodies in SLE-related murine models is lipopolysaccharide (LPS) (21). Injection of LPS in mice induced production of anti-ds DNA antibodies, which were associated with an increased formation of immune complexes in kidneys and an exacerbation of the LN symptoms, including glomerular dysfunction and chronic kidney dysfunction (22). Regarding LPS and the generation of autoantibodies in murine SLE models, two studies dating from the mid-1970s are available (23, 24). The situation has drastically changed during the last 5 years with the publication of new data in animal models and in other related autoimmune diseases. In contrast to other autoimmune diseases, we have less scientific evidences of direct relationship between presence of commensal microorganisms and SLE pathogenesis. One of the first evidences of this potential association was the isolation of Helicobacter pylori in the 67% of kidney biopsies from a cohort of the SLE-related disease LN (25). Apart from this isolated case, most of the microbial evidence in human SLE derives from animal models and in vitro experiments (26). These evidences of the involvement of gut microbiota on SLE and SLE-related autoimmune diseases are represented in Figure 1.
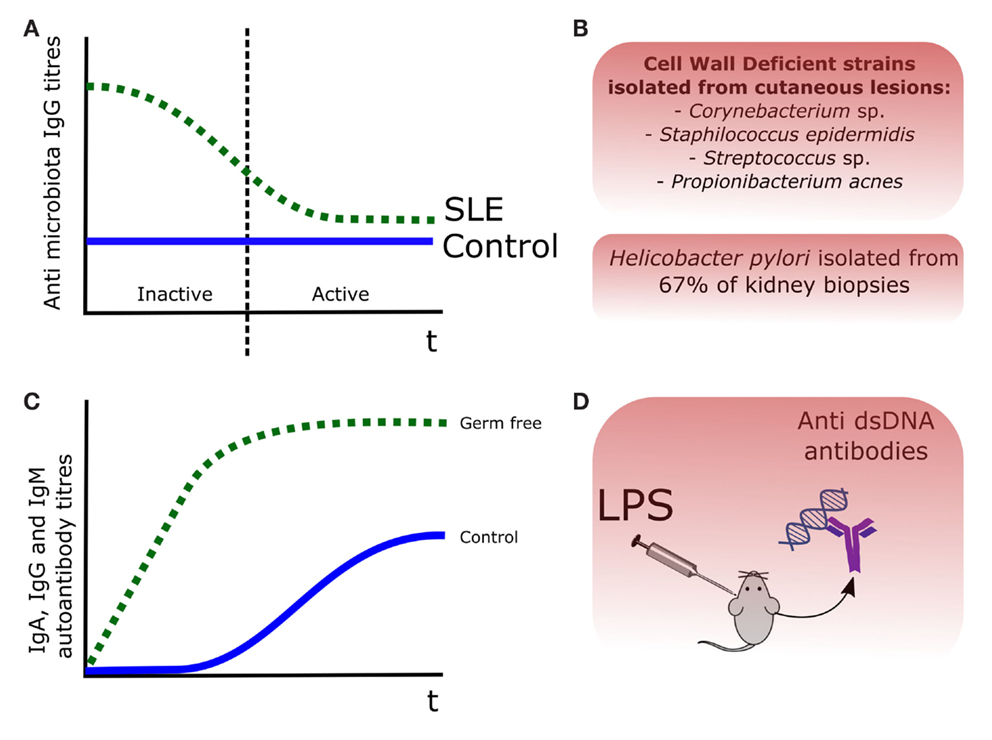
Figure 1. Evidences of the role of the commensal microbiota in SLE pathogenesis. (A) Antibody titers were increased in SLE patients, being those recognizing the gut microbiota decreased during the disease episodes, probably after binding to certain bacterial members (12). (B) Cell wall deficient forms (CWD) of commensal bacteria were isolated from skin lesions in LE patients (20), and Helicobacter pylori was isolated in the 67% of kidney biopsies from a LN cohort (25). (C) In the pristane animal model of SLE, production of autoantibodies was lower and delayed, in germ-free versus control mice (27). (D) Injection of LPS in mice induced production of anti-dsDNA antibodies (22).
The potential importance of gut bacteria in SLE may be deduced from the pristane-induced animal model (27). Pristane, or 2,6,10,14-tetramethylpentadecane, is a saturated alkane present in shark liver oil able to induce an autoimmune disease similar to SLE in mice (28). Pristane administration was associated with different degrees of hypergammaglobulinemia in conventional housed and microbiota-free mice; more concisely, presence of gut microbiota was associated with lower IgM but higher IgA and IgG titers. Interestingly, production of autoantibodies was markedly lower and delayed in microbiota-free mice with respect to control mice, suggesting the existence of a microbiota-triggered stimulus in this murine SLE model (27).
Some microbial structures have the ability to activate autoreactive T cells in certain host genetic backgrounds prone to autoimmunity. For instance, both bacterial-DNA and different cell-wall components, induced the production of anti-double-stranded DNA (dsDNA) autoantibodies in mice (29). Molecular mimicry has been hypothesized as the mechanism connecting the infection of Burkholderia sp. and the exacerbation of SLE symptoms (30). Molecular mimicry occurs when an antibody raised against a given antigen is able to recognize another antigen from a different molecule, for instance, two different amino acid epitopes coming from two proteins (31). In the framework of autoimmune diseases, antibodies developed against bacterial antigens during infection are supposed to recognize self-antigens, inducing formation of immune complexes that contribute, for instance, to renal damages. On the contrary, certain bacterial molecules may activate autoreactive T cells.
There are several examples suggesting an involvement of molecular mimicry in SLE-related diseases (Table 1). Antibodies developed against dsDNA were shown to bind to the sequence ARVLWRATH from cytochrome B 561 and to sequence RAGTDEGFG from one of the transcription regulators from Burkholderia sp. (30). Another example has been documented in an autoimmune disease-denominated glomerulonephritis. In this case, the amino acid residues 72–80 of the bacterial adhesin FimH and the human lysosomal-associated membrane protein (hLAMP-2) are recognized by autoantibodies (32). FimH is a fimbria subunit that is present in Gram-negative enteropathogens, among which species from the genus Escherichia, Klebsiella, and Proteus. Infections by Gram-negative bacteria can be positioned just before glomerulonephritis onset, as well as before detection of autoantibodies raised against hLAMP-2 (33), suggesting a potential role of a bacterial infection with the generation of autoantibodies and with the onset of the disease. Molecular mimicry may also be the mechanism underlying the activation of the T-cell population reactive against certain SLE antigens such as Ro60, a protein binding to small, non-coding RNAs termed “Y RNAs” (34). Although authors claimed that multiple commensal microbial peptides are able to activate T cells reactive against Ro60, no further information on the amino acid sequences is available.
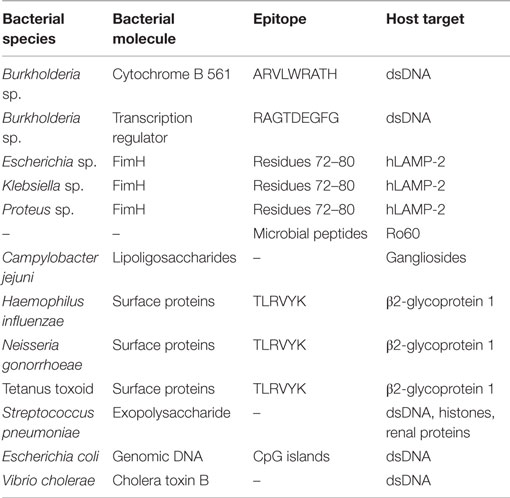
Table 1. Molecular mimicry between molecules of bacterial origin and host structures.
Molecular mimicry involving bacterial molecules is also involved in the generation of autoantibodies in other autoimmune diseases, such as antiphospholipid syndrome (APS) (16). In this disease, certain surface molecules such as lipoligosaccharides from Campylobacter jejuni are believed to mimic gangliosides from the human nerves, generating and maintaining the production of autoantibodies. Notably, certain bacterial sequences mimic the self-targets of aPLs autoantibodies, such as TLRVYK sequence from β2-glycoprotein 1, which shows homology to surface proteins from Haemophilus influenzae, Neisseria gonorrhoeae, or the tetanus toxoid (16). Several antibodies developed against pneumococcal antigens showed cross-reactivity against self-antigens, notably dsDNA (35). Eight out of those antibodies were also able to recognize bacterial exopolysaccharide, DNA, and histones, as well as glomerular structures such as some renal proteins (36). Additional evidence of molecular mimicry underlying bacterial infections are the antibodies raised against CpG DNA of Escherichia coli and the cholera toxin B of Vibrio cholerae, which promoted production of DNA autoantibodies in mice models (37).
Superantigens are a class of toxins produced by many bacteria and viruses, which have the capacity to massively activate immune cells, by binding simultaneously to major histocompatibility complex class II proteins on antigen-presenting cells, and to the specific T-cell receptors on activated T cells (38). Superantigens are involved, among other pathological processes such as bacterial toxic shocks, in inducing autoimmunity by activating self-reactive T cells. Different bacterial molecules act as antigens and are involved in the trigger and progression of many autoimmune diseases. For instance, relapses of granulomatosis, an autoimmune disorder affecting kidneys, is directly correlated with a previous infection of superantigen producing Staphylococcus aureus strains in the upper respiratory tract (39). In this process, the adhesion properties of the staphylococcal acid phosphatase have a pivotal role through its binding to endothelial cells (40).
It should be mentioned that although molecular mimicry and superantigen involvement are well-explained in animal models, further research is needed in order to identify specific interactions between microbiota and immune system responsible for the generation and maintenance of autoreactive T cells in the human host, as well as for the production of autoantibodies.
During the last years, high-throughput technologies have made it possible to deeply characterize the composition of the microbial populations inhabiting our gut, and to associate certain composition patterns, or the absence/presence of certain species with different disorders. One of the first in vivo studies, suggesting a relationship between alterations in the gut microbiota and a female-biased autoimmune disease, was published 4 years ago using a model of type 1 diabetes, the non-obese diabetic (NOD) mice (it should be noted that in humans the type 1 diabetes is not sex-biased) (18, 19). In this work, gut colonization by the Th17 inducing SFB was associated with the absence of development of type 1 diabetes. Moreover, knock out lines of the NOD mice lacking the myeloid differentiation primary response gene (MyD88), which is an adaptor molecule downstream the signaling pathway of certain toll-like receptors (TLRs), resulted in changes on the microbiota composition and conferred a protective effect on type 1 diabetes (41). These results strongly suggested a role for the microbiota composition in the context of female-biased autoimmune diseases, such as SLE or the APS.
In the moment where this manuscript was drafted, two metagenomic studies concerning SLE were available, one in the murine MRL/lpr model and the other in humans (1, 2, 42). From a microbial point of view, the situation is slightly different in humans when compared to mice. MRL/lpr mouse females showed higher numbers of the families Lachnospiraceae and Bacteroidetes, and lower numbers of Bifidobacteriaceae and Erysipelotrichaceae (42). Overall, the human situation was summarized by decreases on the Firmicutes/Bacteroidetes ratio in the samples corresponding to SLE patients (1, 2). Decreases in this ratio have also been observed in human type-2 diabetes and in Crohn’s disease when compared to control populations (43, 44).
Being more precise, pronounced increases in the Bacteroidetes/Prevotellaceae groups, and decreases of the Lachnospiraceae/Ruminococcaceae members were observed in the human SLE cohort (1, 2). Some of the species belonging to the Lachnospiraceae family comprise butyrate producers, such as Roseburia sp. or Butyrivibrio sp. Butyrate production in the human gut is relevant since it promotes differentiation of Tregs, a T-cell type able to suppress any kind of pro-inflammatory response (45).
Whereas the same observations were reported for Bacteroidetes populations in mice, Lachnospiraceae and Ruminococcaceae families showed opposite behaviors. This might be a direct consequence of the mutation carried by MRL/lpr mice (Faslpr), which makes them deficient in Fas-mediated signaling. This genetic deficiency makes T cells refractory to butyrate-induced apoptosis, and this in turn may have a consequence in the composition of the intestinal microbiota (46). Decreases in members of the family Lachnospiraceae have been proposed as a parameter for monitoring disease activity in inflammatory bowel disease, and it remains to be validated whether this can also be applied to SLE (47). For this purpose, further metagenomic studies, involving larger cohorts, will be needed.
One of the factors that has delayed the study of the microbial influence on SLE pathogenesis was the absence of correlation between symptoms and breeding conditions in murine models, as this strongly affect the microbial load of the animals (4). Some scientific studies performed in the last years suggested an important role for two receptors of the innate immune system, TLR7 and TLR9, in SLE pathogenesis; the involvement of TLR4 being also under investigation (48). TLRs recognize specific microorganism ligands, and are one of the first lines of defense against external threats. Among the 10 TLRs identified so far in humans (12 in mice), TLR7 recognizes single-stranded viral RNA (ssRNA), whereas TLR9 recognizes both bacterial/viral dsDNA. The main TLR4 ligand is LPS, but it can also recognize different proteins such as viral glycoproteins, heat-shock proteins, and fibronectin, among others.
Expression of both TLR7/9 genes is increased in SLE patients when compared to healthy controls and this expression level correlates positively with levels of typical serological pro-inflammatory markers, such as IL-6, IFN-gamma, and TNF-alpha (49). Self-RNA and self-DNA are main targets for autoantibody generation in autoimmune diseases, and the complex nucleic acid – antibody seems to contribute, through an altered recognition by TLR7 and TLR9, to the aberrant immune responses observed in SLE (50). This includes activation and expansion of autoreactive B and T cells (51); in fact, triggering of autoreactive memory B cells by bacterial/viral DNA through the action of TLR9 has been proposed to be implicated in SLE relapses (52).
Results from animal models have shown that alterations of the innate immune system through the action of specific receptors, such as TLRs, correlate with SLE symptoms. Duplication of TLR7 gene in a SLE mouse model (Yaa mice), has been shown to be directly related to the disease by an excessive auto-RNA signaling through this receptor (53). In fact, the number of TLR7 molecules is directly related to the risk of the development of SLE in mice, supporting the role of the innate immune system in the onset of this disease. Duplication or overexpression of the tlr7 gene resulted in increased titters of RNA-specific autoantibodies, whereas deficiencies in the TLR9 signalization pathway involved increased production of anti-DNA/anti-chromatin antibodies (54).
An important part of the microbial populations inhabiting our body are virus and bacteriophages, which are referred collectively as the human virome. The first evidence suggesting that viruses were involved in the triggering and development of SLE was the higher IFN-α levels found in the blood of a SLE cohort (55). Infection by Epstein–Barr virus (EBV) has been correlated in LN through a higher production of anti-Sm antibodies, which are autoantibodies directed against seven proteins of different ribonucleoproteins (56). Certain experimental studies indicated a connection between an abnormal immune response to EBV and SLE; more concisely, it has been observed that SLE patients developed specific anti-EBV IgG antibodies that were absent in healthy controls, and vice versa (57). From an epidemic point of view, EBV infection is associated with a higher risk of developing SLE and other autoimmune diseases, such as multiple sclerosis (58). As EBV infects B cells (and other immune cells) inducing the production of pro-inflammatory cytokines, this fact and an aberrant immune response to this virus could be linked to SLE. Further research will elucidate the precise role of viruses in SLE.
From a nutrition point of view, most of the work in SLE has been traditionally focused on the immunomodulatory effect of single dietary components with a demonstrated role on the immune system. Although very little is known about the impact of diet on microbiota in SLE subjects, there is some scientific evidence supporting the hypothesis that some of the dietary impact on SLE pathology could be achieved through modulation of the gut microbiota (4). Currently, there is not enough information to estimate if a dietary intervention could work on preventing SLE relapses. However, evidence from other diseases with an associated dysbiosis showed the impact of several dietary components in balancing certain microorganism populations, which may be applied to SLE.
It has been reported that a diet restricted in carbohydrates or fat, balanced the gut microbiota increasing the proportion of Bacteroidetes in obese subjects with high Firmicutes/Bacteroidetes ratios (compared to lean controls) (59). In this sense, Western diets, characterized by their richness in animal proteins and saturated fats, have also been correlated with a greater abundance of Bacteroidetes (60). On the contrary, diets supplemented with whole grains produced an increase in Firmicutes/Bacteroidetes ratio (61), and the administration of a low-fat/high-fiber diet has been correlated positively with Firmicutes (62). In consonance with these findings, lower levels of Bacteroidetes were observed in African children with a long-term consumption of diets rich in whole grains, dietary fibers, and vegetables supporting the health benefits of whole grains by means of microbiota modulation (63).
To the best of our knowledge, there is no intervention study in the literature to test the effects of the amount and type of fats included in diet on SLE microbiota. A descriptive work from our research group did not support the association between fats or saturate fatty acids at moderate amounts in a well-balanced diet and fecal microbiota in a SLE cohort (64). It is noteworthy that some of the effects ascribed to fat intake are dose- and food-dependent, i.e., they show different effects depending on the food where they are found. For instance, presence of orange juice in a high-fat meal decreased the impact of fats on oxidative stress (65). For this reason, some authors have suggested studying the interactions between the different components of a meal, rather to focus on the effects of a single ingredient (66).
Recently, we have reported on the association between the intake of fruits, such as oranges and apples, and decreases in specific microorganism populations in SLE patients (64). It is important to consider that these fruits are natural dietary sources of polyphenols and fibers with a high potential to modulate microbiota. Specific phenolic compounds like dihydrochalcones from apples, along with dietary fiber, are degraded by bifidobacteria promoting its growth (64, 67, 68). These kinds of changes could be of special interest for SLE patients given the immunomodulatory effect attributed to some strains of the genus Bifidobacterium (69). For instance Bifidobacterium bifidum LMG13195 strain promoted the expansion of Treg cells, a T-cell population favoring mucosal homeostasis (70, 71). Other works showed positive associations between dietary flavones and the levels of Blautia sp., a member of the Clostridium cluster XIVa. As this microorganism is also involved in the expansion of Treg cells, promoting its growth through the diet may be interesting in order to maintain/restore immune homeostasis (72).
Finally, it is very interesting to mention the effect of vitamin A in the diet. A dietary intervention with vitamin A in animal models has returned improvements of the SLE pathogenesis, notably in the reduction of both proteinuria and glomerulonephritis, and has been shown to globally reduce the LN symptoms (73, 74). Another dietary intervention study showed that the intake of retinoic acid, which is the active form of vitamin A (retinol), restored the lactobacilli levels that were decreased during SLE development in mice (42). This is not the only known effect of vitamin A on our gut commensals, as the growth of SFB is influenced by the levels of this vitamin, and as stated before this bacterial group appears to play crucial roles in the development of several SLE-like diseases through the expansion of the Th17 cell subset (4).
In this review, we have shown that gut bacteria composition may have a role in SLE as deduced from SLE-related autoimmune diseases, female-biased or not. Potential mechanisms involved are summarized in Figure 2. With the incoming of novel metagenomic and metatranscriptomic studies focused on SLE microbiota, we will undoubtedly describe the precise role of the different microbial population on SLE and other autoimmune diseases. Altered microbial colonization in the gut (such as abnormal Firmicutes:Bacteroidetes ratio) might contribute to a misbalanced immune response responsible for the triggering or the relapses of the disease. In this sense, it might be hypothesized that excessive Bacteroides signaling through our innate immune system might have a role in SLE pathogenesis, although this is a speculative statement that deserves further investigation.
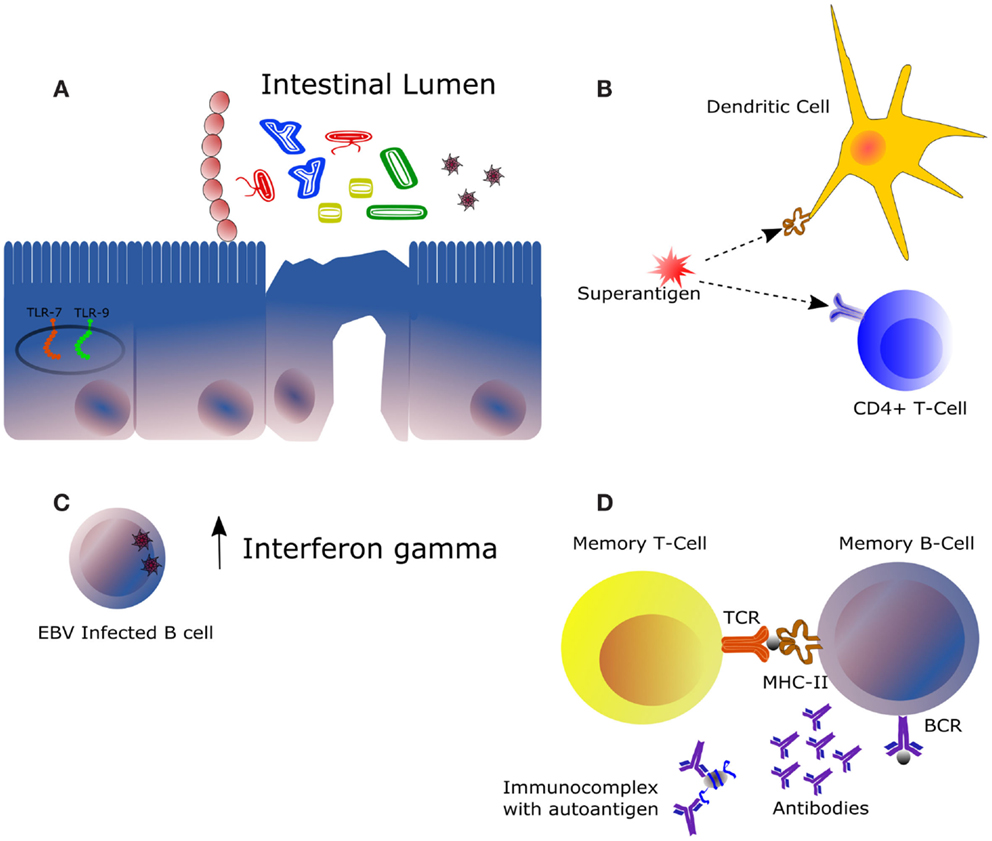
Figure 2. Main mechanisms of potential influence of the intestinal microbiota in the framework of autoimmune diseases. (A) The immunomodulatory effect of the intestinal microbiota is highly dependent on its composition and on the immunological status of the host. Some bacteria trigger downstream effector responses, such as Th1, Th17, or Th2, whereas other acts by expanding the subset of regulatory T cells. Expression of certain receptors recognizing bacterial and viral DNA, mainly TLR7 and TLR9, is increased in the context of SLE. (B) Superantigens are molecules activating both innate (represented by a dendritic cell) and adaptative immunity (represented by a T-cell clone), being this the mechanism of action of some pathogens notably at the level of the upper respiratory tract. (C) Infection of the Epstein–Barr virus in B cells could be responsible for the increased levels of interferon gamma observed in the serum of certain SLE patients. IFNγ is produced mainly by natural killer and natural killer T cells during innate immune response and by CD4+ Th1 and CD8+ cytotoxic T lymphocyte cells during adaptative immune response. (D) Autoreactive B and T cells may be activated by antigens from bacterial origin leading to production of auto antibodies. This similarity between certain bacteria antigens, such as peptides (represented by the gray sphere) and autoantigens at the molecular level, is denominated molecular mimicry. Clearance of immune complexes is directly related to damages at the kidney level. In a context of autoimmunity, increased production of autoantibodies involves higher formation of such complexes.
Scientific results evidenced how some surface-associate proteins and DNA/RNA from both bacteria and viruses are wrongly recognized by the immune system. This molecular mimicry lead autoreactive immune cells to attack self-structures in response to bacterial exposure. Therefore, future research will elucidate whether SLE patients also develop antibodies against the commensal microbiota, and if molecules derived from are able to activate autoreactive T cells through molecular mimicry (1, 2).
Dietary interventions will undoubtedly be of great importance to generate new designs of functional foods focused to correct dysbiosis observed in SLE and other autoimmune diseases. For instance, administration of specific commensals, used as probiotics, might be designed to specifically treat SLE. Use of microorganisms can be dual, as they are able to both displace undesired populations and modulate host immune responses. In this sense, the usefulness of experimental treatments such as fecal microbiota transplantation separated and normalized from the rest of feces components through density gradients, or application of immunomodulatory peptides obtained from our commensal microbiota might be important in the future.
BS and AM conceived the manuscript. BS, AH, and SG drafted the manuscript and all the authors revised the final version prior submission.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
This research was funded by Grants AGL2013-44039-R and AGL2010-14952 from the Spanish “Plan Estatal de I + D + I”. BS and AH were recipients of a Ramón y Cajal postdoctoral contract and a FPI grant, respectively, from the Spanish Ministry of Economy and Competitiveness.
1. Hevia A, Lopez P, Suarez A, Jacquot C, Urdaci MC, Margolles A, et al. Association of levels of antibodies from patients with inflammatory bowel disease with extracellular proteins of food and probiotic bacteria. Biomed Res Int (2014) 2014:1–8. doi: 10.1155/2014/351204
2. Hevia A, Milani C, López P, Cuervo A, Arboleya S, Duranti S, et al. Intestinal dysbiosis associated with systemic lupus erythematosus. MBio (2014) 5:e1548–1514. doi:10.1128/mBio.01548-14
3. Hill C, Guarner F, Reid G, Gibson GR, Merenstein DJ, Pot B, et al. The International Scientific Association for Probiotics and Prebiotics consensus statement on the scope and appropriate use of the term probiotic. Nat Rev Gastroenterol Hepatol (2014) 11:506–14. doi:10.1038/nrgastro.2014.66
4. Vieira SM, Pagovich OE, Kriegel MA. Diet, microbiota and autoimmune diseases. Lupus (2014) 23:518–26. doi:10.1177/0961203313501401
5. Ruiz L, Hevia A, Bernardo D, Margolles A, Sanchez B. Extracellular molecular effectors mediating probiotic attributes. FEMS Microbiol Lett (2014) 359:1–11. doi:10.1111/1574-6968.12576
6. Mok CC, Lau CS. Pathogenesis of systemic lupus erythematosus. J Clin Pathol (2003) 56:481–90. doi:10.1136/jcp.56.7.481
7. Sherer Y, Gorstein A, Fritzler MJ, Shoenfeld Y. Autoantibody explosion in systemic lupus erythematosus: more than 100 different antibodies found in SLE patients. Semin Arthritis Rheum (2004) 34:501–37. doi:10.1016/j.semarthrit.2004.07.002
8. Santiago-Raber ML, Borel P, Uematsu S, Akira S, Izuil S. Role of TLR7 and TLR9 in a murine model of SLE. Swiss Med Wkly (2008) 138:15s–15s. doi:10.1186/ar2771
9. Hochberg MC. Updating the American college of rheumatology revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum (1997) 40:1725–1725. doi:10.1002/art.1780400928
10. Cambridge G, Leandro MJ, Isenberg DA, Teodorescu M, Ehrenstein MR, Edwards JCW. B cell depletion therapy in systemic lupus erythematosus: effect on autoantibody and anti-microbial antibody profiles. Arthritis Rheum (2004) 50:S227–8. doi:10.1002/art.22211
11. Cambier JC. Autoimmunity risk alleles: hotspots in B cell regulatory signaling pathways. J Clin Investig (2013) 123:1928–31. doi:10.1172/JCI69289
12. Apperloorenkema HZ, Bootsma H, Mulder BI, Kallenberg CGB, Vanderwaaij D. Host-microflora interaction in systemic lupus-erythematosus (Sle) – circulating antibodies to the indigenous bacteria of the intestinal-tract. Epidemiol Infect (1995) 114:133–41. doi:10.1017/S0950268800051980
13. Edwards CJ, Costenbader KH. Epigenetics and the microbiome: developing areas in the understanding of the aetiology of lupus. Lupus (2014) 23:505–6. doi:10.1177/0961203314531636
14. Markle JG, Fish EN. SeXX matters in immunity. Trends Immunol (2014) 35:97–104. doi:10.1016/j.it.2013.10.006
15. Zhang HS, Sparks JB, Karyala SV, Settlage R, Luo XM. Host adaptive immunity alters gut microbiota. ISME J (2015) 9:770–81. doi:10.1038/ismej.2014.165
16. Ruff WE, Vieira SM, Kriegel MA. The role of the gut microbiota in the pathogenesis of antiphospholipid syndrome. Curr Rheumatol Rep (2015) 17:472. doi:10.1007/s11926-014-0472-1
17. Wu HJ, Ivanov II, Darce J, Hattori K, Shima T, Umesaki Y, et al. Gut-residing segmented filamentous bacteria drive autoimmune arthritis via T helper 17 cells. Immunity (2010) 32:815–27. doi:10.1016/j.immuni.2010.06.001
18. Kriegel MA, Sefik E, Hill JA, Wu HJ, Benoist C, Mathis D. Sex-specific effects of segmented filamentous bacteria in the autoimmune-prone NOD mouse strain-segregation with diabetes protection in females but not males. Arthritis Rheum (2011) 63:S918–918. doi:10.1002/art.33310
19. Kriegel MA, Sefik E, Hill JA, Wu HJ, Benoist C, Mathis D. Naturally transmitted segmented filamentous bacteria segregate with diabetes protection in nonobese diabetic mice. Proc Natl Acad Sci U S A (2011) 108:11548–53. doi:10.1073/pnas.1108924108
20. Cantwell AR, Kelso DW, Jones JE. Histologic observations of coccoid forms suggestive of cell-wall deficient bacteria in cutaneous and systemic lupus-erythematosus. Int J Dermatol (1982) 21:526–37. doi:10.1111/j.1365-4362.1982.tb01198.x
21. Gilkeson GS, Grudier JP, Karounos DG, Pisetsky DS. Induction of anti-double stranded DNA antibodies in normal mice by immunization with bacterial-DNA. J Immunol (1989) 142:1482–6.
22. Granholm NA, Cavallo T. Long-lasting effects of bacterial lipopolysaccharide promote progression of lupus nephritis in Nzb/W mice. Lupus (1994) 3:507–14. doi:10.1177/096120339400300614
23. Fournie GJ, Lambert PH, Miescher PA. Release of DNA in circulating blood and induction of anti-DNA antibodies after injection of bacterial lipopolysaccharides. J Exp Med (1974) 140:1189–206. doi:10.1084/jem.140.5.1189
24. Izui S, Lambert PH, Fournie GJ, Turler H, Miescher PA. Features of systemic lupus-erythematosus in mice injected with bacterial lipopolysaccharides – identification of circulating DNA and renal localization of DNA-anti-DNA complexes. J Exp Med (1977) 145:1115–30. doi:10.1084/jem.145.5.1115
25. Li Q, Lin X, Wu Z, He L, Wang W, Cao Q, et al. Immuno-histochemistry analysis of Helicobacter pylori antigen in renal biopsy specimens from patients with glomerulonephritis. Saudi J Kidney Dis Transpl (2013) 24:751–8. doi:10.4103/1319-2442.113871
26. Kronbichler A, Kerschbaum J, Mayer G. The influence and role of microbial factors in autoimmune kidney diseases: a systematic review. J Immunol Res (2015) 2015:858027. doi:10.1155/2015/858027
27. Hamilton KJ, Satoh M, Swartz J, Richards HB, Reeves WH. Influence of microbial stimulation on hypergammaglobulinemia and autoantibody production in pristane-induced lupus. Clin Immunol Immunopathol (1998) 86:271–9. doi:10.1006/clin.1997.4481
28. Perry D, Sang A, Yin YM, Zheng YY, Morel L. Murine models of systemic lupus erythematosus. J Biomed Biotechnol (2011) 2011:1–19. doi:10.1155/2011/271694
30. Zhang W, Reichlin M. A possible link between infection with burkholderia bacteria and systemic lupus erythematosus based on epitope mimicry. Clin Dev Immunol (2008) 2008:1–7. doi:10.1155/2008/683489
32. Kain R, Exner M, Brandes R, Ziebermayr R, Cunningham D, Alderson CA, et al. Molecular mimicry in pauci-immune focal necrotizing glomerulonephritis. Nat Med (2008) 14:1088–96. doi:10.1038/nm.1874
33. Roth AJ, Brown MC, Smith RN, Badhwar AK, Parente O, Chung Hc, et al. Anti-LAMP-2 antibodies are not prevalent in patients with antineutrophil cytoplasmic autoantibody glomerulonephritis. J Am Soc Nephrol (2012) 23:545–55. doi:10.1681/ASN.2011030273
34. Szymula A, Szczerba B, Bagavant H, Fu SM, Deshmukh U. A case for microbial involvement in the activation of lupus-antigen reactive T cells. J Immunol (2012) 188:171–39.
35. Kowal C, Weinstein A, Diamond B. Molecular mimicry between bacterial and self antigen in a patient with systemic lupus erythematosus. Eur J Immunol (1999) 29:1901–11. doi:10.1002/(SICI)1521-4141(199906)29:06<1901::AID-IMMU1901>3.0.CO;2-L
36. Chowdhry IA, Kowal C, Hardin J, Zhou Z, Diamond B. Autoantibodies that bind glomeruli: cross-reactivity with bacterial antigen. Arthritis Rheum (2005) 52:2403–10. doi:10.1002/art.21143
37. Deng GM, Tsokos GC. Cholera toxin B accelerates disease progression in lupus-prone mice by promoting lipid raft aggregation. J Immunol (2008) 181:4019–26. doi:10.4049/jimmunol.181.6.4019
38. Kearney DE, Wang W, Redmond HP, Wang JH. Bacterial superantigens enhance the in vitro proinflammatory response and in vivo lethality of the TLR2 agonist bacterial lipoprotein. J Immunol (2011) 187:5363–9. doi:10.4049/jimmunol.1003747
39. Popa ER, Stegeman CA, Abdulahad WH, van der Meer B, Arends J, Manson WM, et al. Staphylococcal toxic-shock-syndrome-toxin-1 as a risk factor for disease relapse in Wegener’s granulomatosis. Rheumatology (2007) 46:1029–33. doi:10.1093/rheumatology/kem022
40. Brons RH, Bakker HI, Van Wijk RT, Van Dijk NW, Kobold ACM, Limburg PC, et al. Staphylococcal acid phosphatase binds to endothelial cells via charge interaction; a pathogenic role in Wegener’s granulomatosis? Clin Exp Immunol (2000) 119:566–73. doi:10.1046/j.1365-2249.2000.01172.x
41. Wen L, Ley RE, Volchkov PY, Stranges PB, Avanesyan L, Stonebraker AC, et al. Innate immunity and intestinal microbiota in the development of type 1 diabetes. Nature (2008) 455:U1109–1110. doi:10.1038/nature07336
42. Zhang HS, Liao XF, Sparks JB, Luo XM. Dynamics of gut microbiota in autoimmune lupus. Appl Environ Microbiol (2014) 80:7551–60. doi:10.1128/AEM.02676-14
43. Larsen N, Vogensen FK, van den Berg FW, Nielsen DS, Andreasen AS, Pedersen BK, et al. Gut microbiota in human adults with type 2 diabetes differs from non-diabetic adults. PLoS One (2010) 5:e9085. doi:10.1371/journal.pone.0009085
44. Man SM, Kaakoush NO, Mitchell HM. The role of bacteria and pattern-recognition receptors in Crohn’s disease. Nat Rev Gastroenterol Hepatol (2011) 8:152–68. doi:10.1038/nrgastro.2011.3
45. Singh N, Gurav A, Sivaprakasam S, Brady E, Padia R, Shi H, et al. Activation of Gpr109a, receptor for niacin and the commensal metabolite butyrate, suppresses colonic inflammation and carcinogenesis. Immunity (2014) 40:128–39. doi:10.1016/j.immuni.2013.12.007
46. Zimmerman MA, Singh N, Martin PM, Thangaraju M, Ganapathy V, Waller JL, et al. Butyrate suppresses colonic inflammation through HDAC1-dependent Fas upregulation and Fas-mediated apoptosis of T cells. Am J Physiol Gastrointest Liver Physiol (2012) 302:G1405–15. doi:10.1152/ajpgi.00543.2011
47. Berry D, Reinisch W. Intestinal microbiota: a source of novel biomarkers in inflammatory bowel diseases? Best Pract Res Clin Gastroenterol (2013) 27:47–58. doi:10.1016/j.bpg.2013.03.005
48. Richez C, Blanco P, Rifkin I, Moreau JF, Schaeverbeke T. Role for toll-like receptors in autoimmune disease: the example of systemic lupus erythematosus. Joint Bone Spine (2011) 78:124–30. doi:10.1016/j.jbspin.2010.09.005
49. Lyn-Cook BD, Xie C, Oates J, Treadwell E, Word B, Hammons G, et al. Increased expression of toll-like receptors (TLRs) 7 and 9 and other cytokines in systemic lupus erythematosus (SLE) patients: ethnic differences and potential new targets for therapeutic drugs. Mol Immunol (2014) 61:38–43. doi:10.1016/j.molimm.2014.05.001
50. Celhar T, Magalhes R, Fairhurst AM. TLR7 and TLR9 in SLE: when sensing self goes wrong. Immunol Res (2012) 53:58–77. doi:10.1007/s12026-012-8270-1
51. Kessel A, Peri R, Haj T, Snir A, Slobodin G, Sabo E, et al. IVIg attenuates TLR-9 activation in B cells from SLE patients. J Clin Immunol (2011) 31:30–8. doi:10.1007/s10875-010-9469-3
52. Capolunghi F, Rosado MM, Cascioli S, Girolami E, Bordasco S, Vivarelli M, et al. Pharmacological inhibition of TLR9 activation blocks autoantibody production in human B cells from SLE patients. Rheumatology (2010) 49:2281–9. doi:10.1093/rheumatology/keq226
53. Pisitkun P, Deane JA, Difilippantonio MJ, Tarasenko T, Satterthwaite AB, Bolland S. Genetic modifiers of SLE and Btk-dependent Anti-RNA B cells in Yaa mice due to TLR7 gene duplication. Arthritis Rheum (2006) 54:S776–776.
54. Santiago-Raber ML, Baudino L, Izui S. Emerging roles of TLR7 and TLR9 in murine SLE. J Autoimmun (2009) 33:231–8. doi:10.1016/j.jaut.2009.10.001
55. Lichtman EI, Helfgott SM, Kriegel MA. Emerging therapies for systemic lupus erythematosus – focus on targeting interferon-alpha. Clin Immunol (2012) 143:210–21. doi:10.1016/j.clim.2012.03.005
56. Yu XX, Yao CW, Tao JL, Yang C, Luo MN, Li SM, et al. The expression of renal Epstein-Barr virus markers in patients with lupus nephritis. Exp Ther Med (2014) 7:1135–40. doi:10.3892/etm.2014.1578
57. Fattal I, Shental N, Molad Y, Gabrielli A, Pokroy-Shapira E, Oren S, et al. Epstein-Barr virus antibodies mark systemic lupus erythematosus and scleroderma patients negative for anti-DNA. Immunology (2014) 141:276–85. doi:10.1111/imm.12200
58. Casiraghi C, Horwitz MS. Epstein-Barr virus and autoimmunity: the role of a latent viral infection in multiple sclerosis and systemic lupus erythematosus pathogenesis. Future Virol (2013) 8:173–82. doi:10.2217/fvl.12.136
59. Ley RE, Turnbaugh PJ, Klein S, Gordon JI. Microbial ecology – human gut microbes associated with obesity. Nature (2006) 444:1022–3. doi:10.1038/4441022a
60. Yatsunenko T, Rey FE, Manary MJ, Trehan I, Dominguez-Bello MG, Contreras M, et al. Human gut microbiome viewed across age and geography. Nature (2012) 486:222–7. doi:10.1038/nature11053
61. Martínez I, Lattimer JM, Hubach KL, Case JA, Yang J, Weber CG, et al. Gut microbiome composition is linked to whole grain-induced immunological improvements. ISME J (2013) 7:269–80. doi:10.1038/ismej.2012.104
62. Wu GD, Chen J, Hoffmann C, Bittinger K, Chen YY, Keilbaugh SA, et al. Linking long-term dietary patterns with gut microbial enterotypes. Science (2011) 334:105–8. doi:10.1126/science.1208344
63. De Filippo C, Cavalieri D, Di Paola M, Ramazzotti M, Poullet JB, Massart S, et al. Impact of diet in shaping gut microbiota revealed by a comparative study in children from Europe and rural Africa. Proc Natl Acad Sci U S A (2010) 107:14691–6. doi:10.1073/pnas.1005963107
64. Cuervo A, Hevia A, Lopez P, Suarez A, Sanchez B, Margolles A, et al. Association of polyphenols from oranges and apples with specific intestinal microorganisms in systemic lupus erythematosus patients. Nutrients (2015) 7:1301–17. doi:10.3390/nu7021301
65. Ghanim H, Sia CL, Upadhyay M, Korzeniewski K, Viswanathan P, Abuaysheh S, et al. Orange juice neutralizes the proinflammatory effect of a high-fat, high-carbohydrate meal and prevents endotoxin increase and toll-like receptor expression. Am J Clin Nutr (2010) 91:940–9. doi:10.3945/ajcn.2009.28584
66. Moreira APB, Texeira TFS, Ferreira AB, Peluzio MDG, Alfenas RDG. Influence of a high-fat diet on gut microbiota, intestinal permeability and metabolic endotoxaemia. Br J Nutr (2012) 108:801–9. doi:10.1017/S0007114512001213
67. Sembries S, Dongowski G, Jacobasch G, Mehrlander K, Will F, Dietrich H. Effects of dietary fibre-rich juice colloids from apple pomace extraction juices on intestinal fermentation products and microbiota in rats. Br J Nutr (2003) 90:607–15. doi:10.1079/BJN2003925
68. Sembries S, Dongowski G, Mehrlander K, Will F, Dietrich H. Physiological effects of extraction juices from apple, grape, and red beet pomaces in rats. J Agric Food Chem (2006) 54:10269–80. doi:10.1021/jf0618168
69. Konieczna P, Akdis CA, Quigley EM, Shanahan F, O’Mahony L. Portrait of an immunoregulatory Bifidobacterium. Gut Microbes (2012) 3:261–6. doi:10.4161/gmic.20358
70. Lopez P, Gonzalez-Rodriguez I, Gueimonde M, Margolles A, Suarez A. Immune response to Bifidobacterium bifidum strains support Treg/Th17 plasticity. PLoS One (2011) 6:e24776. doi:10.1371/journal.pone.0024776
71. Lopez P, Gonzalez-Rodriguez I, Sanchez B, Ruas-Madiedo P, Suarez A, Margolles A, et al. Interaction of Bifidobacterium bifidum LMG13195 with HT29 cells influences regulatory-T-cell-associated chemokine receptor expression. Appl Environ Microbiol (2012) 78:2850–7. doi:10.1128/AEM.07581-11
72. Atarashi K, Tanoue T, Shima T, Imaoka A, Kuwahara T, Momose Y, et al. Induction of colonic regulatory T cells by indigenous Clostridium species. Science (2011) 331:337–41. doi:10.1126/science.1198469
73. de Lema GP, Lucio-Cazana FJ, Molina A, Luckow B, Schmid H, de Wit C, et al. Retinoic acid treatment protects MRL/lpr lupus mice from the development of glomerular disease. Kidney Int (2004) 66:1018–28. doi:10.1111/j.1523-1755.2004.00850.x
Keywords: intestinal microbiota, systemic lupus erythematosus, molecular mimicry, toll-like receptors, dietary intervention
Citation: Sánchez B, Hevia A, González S and Margolles A (2015) Interaction of Intestinal Microorganisms with the Human Host in the Framework of Autoimmune Diseases. Front. Immunol. 6:594. doi: 10.3389/fimmu.2015.00594
Received: 30 September 2015; Accepted: 05 November 2015;
Published: 20 November 2015
Edited by:
Xin M. Luo, Virginia Tech, USAReviewed by:
Muriel Moser, Université Libre de Bruxelles, BelgiumCopyright: © 2015 Sánchez, Hevia, González and Margolles. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Borja Sánchez, Ym9yamEuc2FuY2hlekBjc2ljLmVz
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.