 Katayoun Rezvani
Katayoun Rezvani Rayne H. Rouce
Rayne H. Rouce
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 17 November 2015
Sec. NK and Innate Lymphoid Cell Biology
Volume 6 - 2015 | https://doi.org/10.3389/fimmu.2015.00578
This article is part of the Research Topic NK cell-based cancer immunotherapy View all 21 articles
Natural killer (NK) cells are essential components of the innate immune system and play a critical role in host immunity against cancer. Recent progress in our understanding of NK cell immunobiology has paved the way for novel NK cell-based therapeutic strategies for the treatment of cancer. In this review, we will focus on recent advances in the field of NK cell immunotherapy, including augmentation of antibody-dependent cellular cytotoxicity, manipulation of receptor-mediated activation, and adoptive immunotherapy with ex vivo-expanded, chimeric antigen receptor (CAR)-engineered, or engager-modified NK cells. In contrast to T lymphocytes, donor NK cells do not attack non-hematopoietic tissues, suggesting that an NK-mediated antitumor effect can be achieved in the absence of graft-vs.-host disease. Despite reports of clinical efficacy, a number of factors limit the application of NK cell immunotherapy for the treatment of cancer, such as the failure of infused NK cells to expand and persist in vivo. Therefore, efforts to enhance the therapeutic benefit of NK cell-based immunotherapy by developing strategies to manipulate the NK cell product, host factors, and tumor targets are the subject of intense research. In the preclinical setting, genetic engineering of NK cells to express CARs to redirect their antitumor specificity has shown significant promise. Given the short lifespan and potent cytolytic function of mature NK cells, they are attractive candidate effector cells to express CARs for adoptive immunotherapies. Another innovative approach to redirect NK cytotoxicity towards tumor cells is to create either bispecific or trispecific antibodies, thus augmenting cytotoxicity against tumor-associated antigens. These are exciting times for the study of NK cells; with recent advances in the field of NK cell biology and translational research, it is likely that NK cell immunotherapy will move to the forefront of cancer immunotherapy over the next few years.
Natural killer (NK) cell-mediated cytotoxicity contributes to the innate immune response against various malignancies, including leukemia (1, 2). The antitumor effect of NK cells is a subject of intense investigation in the field of cancer immunotherapy. In this review, we will focus on recent advances in NK cell immunotherapy, including augmentation of antibody-dependent cytotoxicity, manipulation of receptor-mediated activation, and adoptive immunotherapy with ex vivo-expanded, chimeric antigen receptor (CAR)-engineered, or engager-modified NK cells.
Natural killer cells are characterized by the lack of CD3/TCR molecules and by the expression of CD16 and CD56 surface antigens. Around 90% of circulating NK cells are CD56dim, characterized by their distinct ability to mediate cytotoxicity in response to target cell stimulation (3, 4). This subset includes the alloreactive NK cells that play a central role in targeting leukemia cells in the setting of allogeneic hematopoietic stem cell transplant (HSCT) (5). The remaining NK cells, predominantly housed in lymphoid organs, are CD56bright, and although less mature (“unlicensed”) (3, 6, 7), they have a greater capability to secrete and respond to cytokines (8, 9). CD56bright and CD56dim NK cells are also distinguished by their differential expression of FcγRIII (CD16), an integral determinant of NK-mediated antibody-dependent cellular cytotoxicity (ADCC), with CD56dim NK cells expressing high levels of the receptor, while CD56bright NK cells are CD16 dim or negative (6). In contrast to T and B lymphocytes, NK cells do not express rearranged, antigen-specific receptors; rather, NK effector function is dictated by the integration of signals received through germ-line-encoded receptors that can recognize ligands on their cellular targets. Functionally, NK cell receptors are classified as activating or inhibitory. NK cell function, including cytotoxicity and cytokine release, is governed by a balance between signals received from inhibitory receptors, notably the killer Ig-like receptors (KIRs) and the heterodimeric C-type lectin receptor (NKG2A), and activating receptors, in particular the natural cytotoxicity receptors (NCRs) NKp46, NKp30, NKp44, and the C-type lectin-like activating immunoreceptor NKG2D (9).
The inhibitory KIRs (iKIRs) with known HLA ligands include KIR2DL2 and KIR2DL3, which recognize the HLA-C group 1-related alleles characterized by an asparagine residue at position 80 of the α-1 helix (HLA-CAsn80); KIR2DL1, which recognizes the HLA-C group 2-related alleles characterized by a lysine residue at position 80 (HLA-CLys80); and KIR3DL1, which recognizes the HLA-Bw4 alleles (9, 10). NK cells also express several activating receptors that are potentially specific for self-molecules. KIR2DS1 has been shown to interact with group 2 HLA-C molecules (HLA-C2), while KIR2DS2 was recently shown to recognize HLA-A*11 (10, 11). Hence, these receptors require mechanisms to prevent inadvertent activation against normal tissues, processes referred to as “tolerance to self.” Engagement of iKIR receptors by HLA class I leads to signals that block NK-cell triggering during effector responses. These receptors explain the “missing self” hypothesis, which postulates that NK cells survey tissues for normal levels of the ubiquitously expressed MHC class I molecules (12, 13). Upon cellular transformation or viral infection, surface MHC class I expression on the cell surface is often reduced or lost to evade recognition by antitumor T cells. When a mature NK cell encounters transformed cells lacking MHC class I, their inhibitory receptors are not engaged, and the unsuppressed activating signals, in turn, can trigger cytokine secretion and targeted attack of the virus-infected or transformed cell (13, 14). In parallel, cellular stress and DNA damage (occurring in cells during viral or malignant transformation) results in upregulation of “stress ligands” that can be recognized by activating NK receptors. Thus, human tumor cells that have lost self-MHC class I expression or bear “altered-self” stress-inducible proteins are ideal targets for NK recognition and killing (14–16). NK cells directly kill tumor cells through several mechanisms, including release of cytoplasmic granules containing perforin and granzyme (16–18), expression of tumor necrosis factor (TNF) family members, such as FasL or TNF-related apoptosis-inducing ligand (TRAIL), which induce tumor cell apoptosis by interacting with their respective receptors Fas and TRAIL receptor (TRAILR) (16–19) as well as ADCC (9).
Increasing understanding of NK cell biology and their interaction with other cells of the immune system has led to several novel immunotherapeutic approaches as discussed in this review. NK cells produce cytokines that can exert regulatory control of downstream adaptive immune responses by influencing the magnitude of T cell responses, specifically T helper-1 (TH1) function (20). NK cell function, in turn, is regulated by cytokines, such as IL-2, IL-15, IL-12, and IL-18 (21), as well as by interactions with other cell types, such as dendritic cells, macrophages, and mesenchymal stromal cells (10, 22, 23). IL-15 has emerged as a pivotal cytokine required for NK cell development and maintenance. Whereas mice deficient in IL-2 (historically the cytokine of choice to expand and activate NK cells) have normal NK cells, IL-15-deficient mice lack NK cells (24).
Several cytokines are also known to inhibit NK cell activation and function, thus playing a crucial role in tumor escape from NK immune surveillance. Recently, considerable attention has been paid to the inhibitory effects of transforming growth factor-beta (TGF-β) and IL-10 on NK cell cytotoxicity (12, 25, 26). Several groups have shown that secretion of TGF-β by tumor cells results in downregulation of activating receptors, such as NKp30 and NKG2D, with resultant NK dysfunction (25, 26). Similarly, IL-10 production by acute myeloid leukemia (AML) blasts induces upregulation of NKG2A with significant impairment in NK function (3).
The CD56dim subset of NK cells expresses the Fcγ receptor CD16, through which NK cells mount ADCC, providing opportunities for its modulation to augment NK effector function (27, 28). In fact, a number of clinically approved therapeutic antibodies targeting tumor-associated antigens (such as rituximab or cetuximab) function at least partially through triggering NK cell-mediated ADCC. Several studies using mouse tumor models have established that efficient antibody–Fc receptor (FcR) interactions are essential for the efficacy of monoclonal antibody (mAb) therapy, a mainstay of cancer therapy (28, 29). Based on this premise, Romain et al. successfully engineered the Fc region of the IgG mAb, HuM195 targeting the AML leukemia antigen CD33, by introducing the triple mutation S293D/A330L/I332E (DLE). Using timelapse imaging microscopy in nanowell grids (TIMING, a method of analyzing kinetics of thousands of NK cells and mAb-coated targets), they demonstrated that the DLE-HuM195 antibody increased both the quality and quantity of NK cell-mediated ADCC by recruiting NK cells to participate in cytotoxicity via CD16-mediated signaling. NK cells encountering DLE-HuM195-coated targets induced rapid target cell apoptosis by promoting conjugation to multiple target cells (leading to increased “serial killing” of targets), thus inducing apoptosis in twice the number of targets as the wild-type mAb (27).
Additional approaches under investigation to enhance NK cell-mediated ADCC include antibody engineering and therapeutic combination of antibodies predicted to have synergistic activity. For example, mogamulizumab (an anti-CCR4 mAb recently approved in Japan) is defucosylated to increase binding by FcγRIIIA and thereby enhances ADCC. Mogamulizumab successfully induced ADCC activity against CCR4-positive cell lines and inhibited the growth of EBV-positive NK-cell lymphomas in a murine xenograft model (30). These findings suggest that mogamulizumab may be a therapeutic option against EBV-associated T and NK-lymphoproliferative diseases (30). Obinutuzumab (GA101) is a novel type II glycoengineered mAb against CD20 with increased FcγRIII binding and ADCC activity. In contrast to rituximab, GA101 induces activation of NK cells irrespective of their inhibitory KIR expression, and its activity is not negatively affected by KIR/HLA interactions (31). These data show that modification of the Fc fragment to enhance NK-mediated ADCC can be an effective strategy to augment the efficacy of therapeutic mAbs (31).
Although enhanced NK-mediated ADCC occurs in the presence of certain mAbs, in the case of non-engineered mAbs (such as rituximab), this NK-mediated cytotoxicity is typically still under the jurisdiction of KIR-mediated inhibition. However, ADCC responses can be potentiated in vitro in the presence of antibodies that block NK cell inhibitory receptor interaction with MHC class I ligands (32). These include the use of anti-KIR Abs to block the interaction of iKIRs with their cognate HLA class I ligands. To exploit this pathway pharmacologically, a fully humanized anti-KIR mAb 1-7F9 (IPH2101) (33) with the ability to block KIR2DL1/L2/L3 and KIR2DS1/S2 was generated. In vitro, anti-KIR mAbs can augment NK cell-mediated lysis of HLA-C-expressing tumor cells, including autologous AML blasts and autologous CD138+ multiple myeloma (MM) cells (34). Additionally, in a dose-escalation phase 1 clinical trial in elderly patients with AML, 1-7F9 mAb was reported to be safe and could block KIRs for prolonged periods (35). A recombinant version of this mAb with a stabilized hinge (lirilumab) was recently developed. Lirilumab is a fully humanized IgG4 anti-KIR2DL1, -L2, -L3, -S1, and -S2 mAb. The iKIRs targeted by lirilumab collectively recognize virtually all HLA-C alleles, and the blockade of the three KIR2DLs allows targeting of every patient without the need for prior HLA or KIR typing (33, 34). Furthermore, the combination of an anti-KIR mAb with the immunomodulatory drug lenalidomide was shown to potentiate ADCC and is being tested in a phase 1 clinical trial in patients with MM [NCT01217203 (35)]. A potential concern is related to how inhibitory KIR blockade may impact on the ability of NK cells to discriminate self, healthy cells from abnormal virally infected or cancerous cells. Preliminary in vitro data suggest that Ab blockade of iKIRs will preferentially augment the ADCC response, without increasing cytotoxicity against self healthy cells (32). It is reassuring that in the IPH2101 phase 1 studies, no alterations in the expression of major inhibitory or activating NK receptors or frequencies of circulating peripheral lymphocytes were reported, indicating that the Ab does not induce clinically significant targeting of normal cells by NK cells (35). Lin et al. recently reported on the application of an agonistic NK cell-targeted mAb to augment ADCC (36). Following FcR triggering during ADCC, expression of the activation marker CD137 is increased. Agonistic antibodies targeting CD137 have been reported to augment NK-cell function, including degranulation, secretion of IFN-γ, and antitumor cytotoxicity in in vitro and in vivo preclinical models of tumor (36–39). The combination of the agonistic anti-CD137 antibody with rituximab is currently being evaluated in a phase 1 trial in patients with lymphoma [NCT01307267 (35–37)].
Other factors, such as specific CD16 polymorphisms and NKG2D engagement, can also influence ADCC, with certain polymorphisms (such as FcγRIIIa-V158F polymorphism) resulting in a stronger IgG binding (40). These findings are clinically relevant, as supported by the observation that patients with non-Hodgkin lymphoma (NHL) with the FcγRIIIa-V158F polymorphism experienced improved clinical response to rituximab (41, 42). In summary, several antibody combinations designed to boost ADCC have shown promising results in preclinical and early clinical trials, thus warranting further study of this strategy to enhance NK cell activity against tumor cells.
The early studies of adoptive NK cell therapy focused on enhancing the antitumor activity of endogenous NK cells (43). Initial trials of adoptive NK therapy in the autologous setting involved using CD56 beads to select NK cells from a leukapheresis product and subsequently infusing the bead-selected autologous NK cells into patients (43, 44). Infusions were followed by administration of systemic cytokines (most commonly IL-2) to provide additional in vivo stimulation and support their expansion. This strategy met with limited success due to a combination of factors (44). Although cytokine stimulation promoted NK cell activation and resulted in greater cytotoxicity against malignant targets in vitro, only limited in vivo antitumor activity was observed (43–45). Similar findings were observed when autologous NK cells and systemic IL-2 were given as consolidation treatment to patients with lymphoma who underwent autologous BMT (46). The poor clinical outcomes observed with adoptive transfer of ex vivo activated autologous NK cells followed by systemic IL-2 were attributed to three factors: (1) development of severe life-threatening side effects, such as vascular leak syndrome as a result of IL-2 therapy; (2) IL-2-induced expansion of regulatory T cells known to directly inhibit NK cell function and induce activation-induced cell death (47–49); and (3) lack of antitumor effect related to the inhibition of autologous NK cells by self-HLA molecules. Strategies to overcome this autologous “checkpoint,” thus redirecting autologous NK cells to target and kill leukemic blasts are the subject of intense investigation (33–35). These include the use of anti-KIR Abs (such as the aforementioned lirilumab) to block the interaction of inhibitory receptors on the surface of NK cells with their cognate HLA class I ligand.
An alternative strategy is to use allogeneic instead of autologous NK cells, thus taking advantage of the inherent alloreactivity afforded by the “missing self” concept (13). Over the past decade, adoptive transfer of ex vivo-activated or -expanded allogeneic NK cells has emerged as a promising immunotherapeutic strategy for cancer (24, 50–52). Allogeneic NK cells are less likely to be subject to the inhibitory response resulting from NK cell recognition of self-MHC molecules as seen with autologous NK cells. A number of studies have shown that infusion of haploidentical NK cells to exploit KIR/HLA alloreactivity is safe and can mediate impressive clinical activity in some patients with AML (50–52). In fact, algorithms have been developed to ensure selection of stem cell donors with the greatest potential for NK cell alloreactivity for allogeneic HSCT (50).
Promising results in the HSCT setting suggest that the application of this strategy in the non-transplant setting may be a plausible option. Miller et al. were among the first to show that adoptive transfer of ex vivo-expanded haploidentical NK cells after lymphodepleting chemotherapy is safe, and can result in expansion of NK cells in vivo without inducing graft-vs.-host disease (GVHD) (50). In a phase I dose-escalation trial, 43 patients with either hematologic malignancies (poor prognosis AML or Hodgkin lymphoma) or solid tumor (metastatic melanoma or renal cell carcinoma) received up to 2 × 107cells/kg of haploidentical NK cells following either low intensity [low-dose cyclophosphamide (Cy) and methylprednisolone or fludarabine (Flu)] or high intensity regimens (Hi-Cy/Flu). All patients received subcutaneous IL-2 after NK cell infusion. Whereas adoptively infused NK cells persisted only transiently following low intensity regimens, AML patients who received the more intense Hi-Cy/Flu regimen had a marked rise in endogenous IL-15 associated with expansion of donor NK cells and induction of complete remission (CR) in five of 19 very high-risk patients. The superior NK expansion observed after high-dose compared to low-dose chemotherapy was attributed to a combination of factors including prevention of host T cell-mediated rejection and higher levels of cytokines, such as IL-15. These findings provided the first evidence that haploidentical NK cells are safe and can persist and expand in vivo, supporting the proof of concept that NK cells may be applied for the treatment of selected malignancies either alone or as an adjunct to HSCT (50).
Another pivotal pilot study, the NKAML trial (Pilot Study of Haploidentical NK Transplantation for AML), reported that infusion of KIR-HLA-mismatched donor NK cells can reduce the risk of relapse in childhood AML (51). Ten pediatric patients with favorable or intermediate risk AML in first CR were enrolled following completion of 4–5 cycles of chemotherapy. All patients received a low-dose conditioning regimen consisting of Cy/Flu prior to infusion of NK cells (median, 29 × 106/kg NK cells) from a haploidentical donor, followed by six doses of IL-2. NK infusions were well tolerated with limited non-hematologic toxicity. All patients had transient engraftment of NK cells for a median of 10 days (range 2–189 days) with significant expansion of KIR-mismatched NK cells. With a median follow-up of 964 days, all patients remained in remission, suggesting that donor-recipient HLA-mismatched NK cells may reduce the risk of relapse in childhood AML (51).
Other strategies currently under investigation include the infusion of KIR-ligand-mismatched haploidentical NK cells as part of the pre-HSCT conditioning regimen (NCT00402558), and NK cell infusion to prevent relapse or as therapy for minimal residual disease in patients after haploidentical HSCT (NCT01386619).
Natural killer cell-based immunotherapies are also a promising therapeutic option for solid tumors. A number of studies have shown that the presence of intratumoral NK cells correlates with delayed tumor progression and improved outcomes (53–55). However, the successful application of NK cell-based therapies in the solid tumor setting poses a special challenge. In addition to the immune evasion strategies common to hematologic malignancies, such as secretion of immunosuppressive cytokines and downregulation of activating ligands (55–57), additional challenges specific to solid tumors exist; NK cells must not only traffic to sites of disease, but also penetrate the tumor capsule in order to exert their effector function. Furthermore, tumor targets must be inherently susceptible to NK-mediated cytotoxicity (58). Several groups have focused on strategies to alter the tumor microenvironment by targeting myeloid-derived suppressor cells or regulatory T cells (Treg) rather than the tumors themselves (58, 59). In fact, the prospect of combining NK cell-based immunotherapy with approaches to target the immunosuppressive tumor microenvironment or immune checkpoints, such as KIR blockade, is especially relevant to the treatment of solid tumors (55, 58). Several early phase clinical trials have demonstrated the feasibility of adoptive therapy with autologous or allogeneic ex vivo activated/expanded NK cells in patients with refractory solid malignancies [NCT01875601 (60)]; however, outside of the post-HSCT setting (namely in neuroblastoma), limited data on the clinical efficacy of NK cells in eradicating solid tumors exist. Currently, several trials are actively recruiting patients with refractory solid tumors for adoptive NK therapy (including NCT01807468, NCT02130869, and NCT0210089).
Recent approaches to adoptive NK therapy focused on infusion of NK cells that have undergone a process of ex vivo cytokine activation and expansion (61). A number of cytokines (IL-2, IL-12, IL-15, IL-18, IL-21, and type I IFNs) have been studied to activate and expand NK cells ex vivo (62–65). The most extensively studied cytokine is IL-2 (62, 63). This is not surprising, considering IL-2 was the only cytokine available in clinical grade until recently. Nevertheless, NK cells expanded in the presence of IL-12, IL-15, and IL-18, either alone or in combination, have shown remarkable activity against tumor targets in experimental models and offer an attractive strategy for clinical expansion of NK cells (64, 65). IL-15, in particular, is appealing as it does not stimulate Tregs (65). IL-15 has been tested in preclinical models with promising results; however, very high doses were necessary to observe any meaningful in vivo antitumor effects, and toxicity of systemic cytokine administration and cytokine-induced NK-cell apoptosis remained major issues (65). Recently, Miller et al. compared the persistence and in vivo efficacy of adoptively infused freshly activated NK cells (FA-NK) and ex vivo-expanded NK cells (Ex-NK) in a xenotransplantation model. They showed that in vivo NK cell persistence is cytokine dependent, with IL-15 being superior to IL-2. They also reported that cryopreservation of FA-NK or Ex-NK was detrimental to NK cell function, and that culture conditions influence homing, persistence, and expansion of NK cells in vivo (66).
Although the results from the abovementioned trials proved that transient persistence of adoptively transferred NK cells obtained via apheresis is feasible and safe, the requirement of a willing, available donor precludes the widespread applicability of this approach. Hence, more recent efforts have focused on optimizing methods for ex vivo expansion of NK cells from peripheral blood mononuclear cells (PBMCs) collected by a simple blood draw, with a goal of producing large quantities of purified, functionally active NK cells for clinical use. These expansion strategies include the use of “feeder cells,” such as monocytes in the form of irradiated PBMCs, EBV-transformed lymphoblastoid cell lines (EBV-LCLs) or gene-modified, irradiated K562 cells expressing membrane-bound IL-15 or IL-21 and 41BB ligand for costimulation (61, 66–69) in gas-permeable large-scale expansion flasks. These techniques have dramatically increased the yield and activation status of NK cells, potentially overcoming the need for leukapheresis. Because the feeder cells used in these manufacture methods are lethally irradiated prior to use in culture (leaving the remaining feeder cells to be lysed by the expanding NK cells), the risk of infusing viable feeder cells is negligible. However, a number of safeguards have also been incorporated that include monitoring the growth rate of feeder cells and testing for the presence of viable feeder cells at the end of the culture period. Clinical products are, therefore, only released if no viable gene-modified K562 cells or transformed LCLs are present, with strict cutoff values for contaminating B cells and monocytes at the end of the culture period as well (67).
Although these expansion methods can produce large numbers of functionally active NK cells, concomitant expansion of competing cells with immunosuppressive properties, such as Tregs remains problematic. Early studies reported that NK cell infusions from haploidentical donors are able to induce remissions in some patients with AML, but not others (50–52). Several groups, therefore, set out to explore factors that may contribute to the failure of NK expansion in vivo. Bachanova explored the effect of competition between Tregs and NK cells in 57 patients with refractory AML who received lymphodepleting chemotherapy followed by NK cell infusion and IL-2 administration [NCT00274846 and NCT01106950 (70)]. Fifteen patients also received the IL-2-diphtheria toxin fusion protein (IL2DT) to deplete Tregs prior to NK cell infusion. IL2DT treatment was associated with increased donor NK cell persistence and improved CR and disease-free survival at 6 months (33 vs. 5% in patients not receiving IL2DT; P < 0.01). In the IL2DT cohort, NK cell expansion correlated with higher post-chemotherapy serum IL-15 levels (P = 0.002) and effective peripheral blood (PB) Treg depletion (<5%) at day 7 (P < 0.01). This study shed light on the importance of optimizing the cytokine milieu to facilitate the in vivo expansion of adoptively transferred NK cells and identifying ways to abrogate the immunosuppressive elements, such as regulatory T cells.
Although these data are encouraging, adoptive transfer of NK cells under good manufacturing practices (GMP) requires significant infrastructure and specialized processing equipment, thus limiting the availability and scalability of these NK cell therapies to a few specialized institutions (61). Nonetheless, the feasibility of centralized processing and safe delivery of ex vivo-manufactured NK cells for infusion at remote clinics have been demonstrated, suggesting that the practice might become more widespread as procedures are optimized (71). For example, in order to improve access to ex vivo activated NK cells and ease the burden associated with producing cellular products at individual treatment centers, the National Heart, Lung, and Blood Institute (NHLBI, Bethesda, MD, USA) sponsored the Production Assistance for Cellular Therapies (PACT) program. Using this approach, activated NK cells have been sent to other centers for infusion into patients (72, 73).
Since the initial reports of successful adoptive transfer of NK cells (50–52), many groups continue to perform extensive preclinical exploration of the ideal manufacturing strategy for ex vivo activation and expansion of NK cells. Several expansion methods optimized in the preclinical setting have been successfully scaled up for the clinic (61, 67–70). In addition to the six clinical trials of adoptive NK cell therapy for leukemia that have reported their data (48–52, 70), there are currently 12 active clinical trials enrolling patients with hematologic malignancies for NK cell adoptive therapy, a number that is steadily rising.
Although the majority of clinical studies of NK cell immunotherapy have used PB NK cells, several alternative sources of NK cells exist. These include bone marrow, human embryonic stem cells (hESCs), induced pluripotent stem cells [iPCSs (74, 75)], and umbilical cord blood (CB). While the generation of NK cells from hESCs or iPCS has been largely experimental to date, clinical-grade generation and expansion of NK cells from CB-derived CD34+ cells has been successfully achieved (76).
Umbilical CB as a source for NK cells lends additional clinical advantages. CB contains a high percentage of NK cells (77, 78) and serves as an immediate “off-the-shelf” source of NK cells, with less stringent requirements for HLA matching, and lower risk of causing GVHD following infusion due to the naivety of the cord T cell repertoire (77, 78). Although no direct comparison of PB- and CB-derived NKs has been performed in the clinical setting, in vitro studies have identified a number of differences between CB and PB NK cells. CB NK cells form weaker conjugates with target cells due to the lower membrane expression of adhesion molecules on their surface (79, 80). CB NK cells also express higher levels of lectin-like inhibitory receptors (CD94/NKG2A) and lower levels of KIRs, indicating an immature phenotype (81). CB NK cells are similarly sensitive to cytokines for in vivo expansion and persistence (82). However, it appears that the requirements for in vitro expansion of CB NK cells may be different to those required for PB NKs. CB NKs are less responsive to IL-2 stimulation, which may be related to the lower expression of IL-2Rα and reduced activation of the STAT5 signaling pathway as compared with PB NK cells (83). The combination of IL-15 and IL-18, however, can induce significant proliferation and cytokine production by CB NK cells, while the killing capacity of CB NK cells is significantly enhanced after stimulation with IL-15 (83). As with PB-derived NK cells, T-cell contamination is a concern, but can be ameliorated by CD3 depletion. T-cell contamination should be limited to <1–5 × 105/kg (61) to minimize the risk of GVHD. In addition, CD56+ selection reduces B-cell contamination to <1%, which minimizes passenger B lymphocyte-mediated complications, such as EBV-related post-transplant lymphoproliferative disorder (PTLD) and acute hemolytic anemia.
More recently, efforts have focused on optimizing the large-scale expansion of purified CB-derived NK cells. Shah et al. were the first to describe a strategy for expanding NK cells from cryopreserved CB units in which they employed K562-based artificial antigen-presenting cells (aAPCs) expressing membrane-bound IL-21 (clone 9.mbIL21) (77, 84). The clone 9.mbIL21 cell line is GMP-grade and expresses membrane-bound IL-21, 4-1BB ligand, CD64 (FcγRI), and CD86. After only 14 days of culture in a gas-permeable culture system, mean-fold expansion of CB-NK cells was 1848-fold from fresh and 2389-fold from cryopreserved CB with >95% purity for CD56+CD3− NK cells. aAPC-expanded CB-NK cells displayed a phenotype similar to that of expanded PB-NK cells and maintained strong expression of the transcription factors eomesodermin and T-bet. Furthermore, CB-NK cells formed functional immune synapses and efficiently killed various MM targets in vitro. Finally, aAPC-expanded CB-NK cells showed significant in vivo activity against MM in a xenogenic mouse model. These findings highlight a clinically applicable strategy for the generation of highly functional CB-NK cells using an aAPC platform, which can be potentially extended to other hematologic malignancies and solid tumors (77). A number of phase I/II clinical trials are underway to test the feasibility and efficacy of CB-NK cell adoptive therapy in patients with hematologic malignancies (NCT01619761, NCT01729091 NCT02280525, NCT01914263, and NCT00412360) (summarized in Table 1).
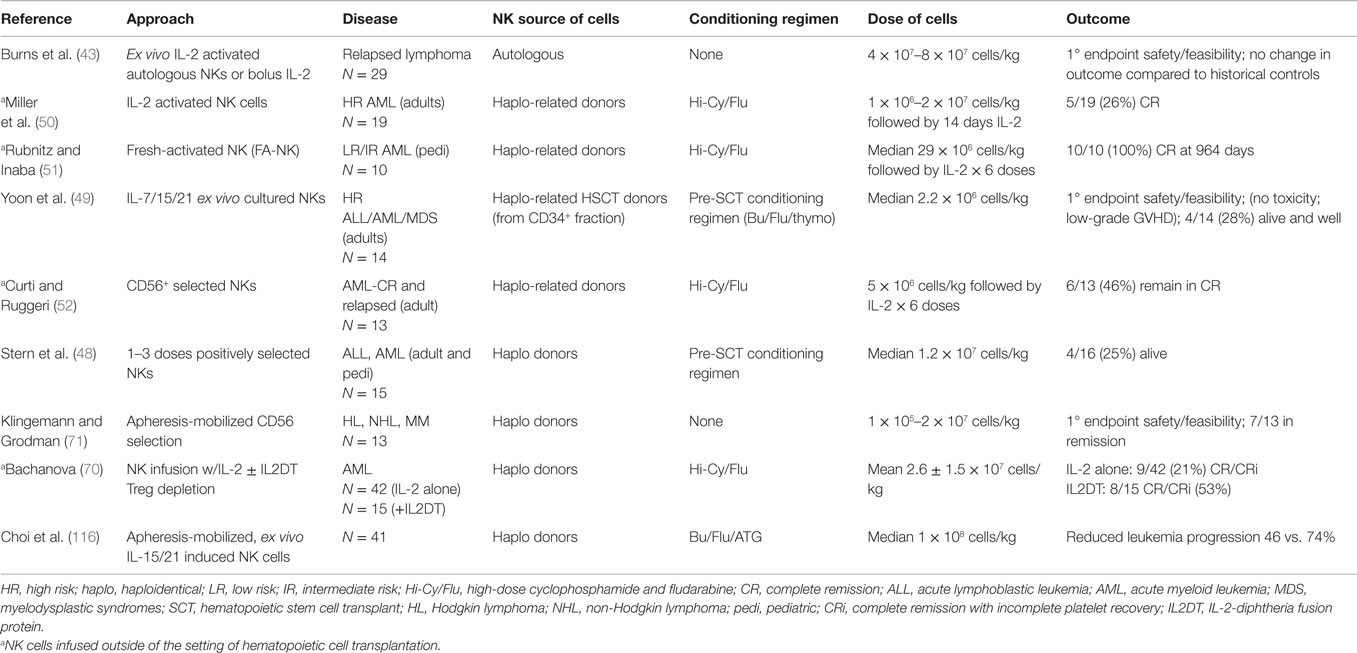
Table 1. Published results of NK adoptive immunotherapy trials in hematologic malignancies.
The adoptive transfer of NK cell lines has several theoretical advantages over the use of patient- or donor-derived NK cells. These are primarily related to the lack of expression of iKIRs, presumed lack of immunogenicity, ease of expansion and availability as an “off-the-shelf” product (85). Several human NK cell lines, such as NK-92 and KHYG-1, have been documented to exert antitumor activity in both preclinical and clinical settings (86–88). NK-92, the most extensively characterized NK-cell line, was established in 1994 from the PB of a male Caucasian patient with NHL. NK-92 cells are IL-2-dependent, harbor a CD2+CD56+CD57+ phenotype and exert potent in vitro cytotoxicity (86). Infusion of up to 1010 cells/m2 NK-92 cells into patients with advanced lung cancer and other advanced malignancies was well tolerated and the cells persisted for a minimum of 48 h with encouraging clinical responses (86, 88–91). However, potential limitations of using NK cell lines, such as NK-92 cells, include the requirement for irradiation to reduce the risk of engrafting cells with potential in vivo tumorigenicity, and the need for pre-infusion conditioning to avoid host rejection. Furthermore, infusion of allogeneic NK cell lines may induce T and B cell alloimmune responses, limiting their in vivo persistence and precluding multiple infusions. A number of studies are testing NK-92 cells (Neukoplast®) in patients with solid tumors, such as Merkel cell cancer and renal cell carcinoma, as well as in hematological malignancies (85).
While results from clinical studies of NK cell adoptive therapy are encouraging (48–52, 70), significant gaps remain in our understanding of the optimal conditions for NK cell infusion. Based on the pioneering work from Rosenberg et al. demonstrating the importance of lymphodepletion to support the expansion of tumor-infiltrating T cells (92) and given its emergence as a key determinant of efficacy with CAR therapy, several groups are actively investigating the ideal preparative regimen to promote the expansion and persistence of adoptively infused NK cells (53, 69, 70, 75). Available data support the use of high-dose Cy/Flu regimen as the frontrunner, considering it is reasonably well tolerated and shown to support the in vivo expansion of NK cells (51, 70). IL-15 is an ideal candidate cytokine for the expansion of NK cells in vivo, especially since it does not promote expansion of regulatory T cells (66), which have been shown to suppress NK cell effector function in IL-2-based trials (69, 70). In a recent phase 1 study in patients with metastatic melanoma or renal cell carcinoma, rhIL-15 was shown to activate NK cells, monocytes, γδ, and CD8 T cells (93). However, as an intravenous bolus dose, rhIL-15 proved too difficult to administer because of significant clinical toxicities (93). Based on these promising data, alternative dosing strategies are being investigated, including continuous intravenous infusions. To this effect, systemic IL-15 along with infusion of donor NK cells are currently being tested in a phase I clinical trial for AML (NCT01385423).
Chimeric antigen receptors have been used extensively to redirect the specificity of T cells against leukemia (94). Recently, use of an anti-CD19-BB-ζ receptor transduced into autologous or allogeneic T cells produced dramatic clinical responses in patients with acute lymphoblastic leukemia (95, 96); however, infusions of activated T cells from an allogeneic source are likely to increase the risk of GVHD. T cell-depleted allogeneic NK cells, by contrast, should not cause GVHD, as predicted by observations in murine models, as well as in patients with leukemia and solid malignancies treated with haploidentical NK cells (50–52). Given their shorter lifespan and potent cytolytic function, mature NK cells provide attractive candidate effector cells to express CARs and, provide an excellent source of off-the-shelf cellular therapy for patients with cancer.
The feasibility of genetically engineering NK cells to express CARs has been shown in the preclinical setting (97, 98). Primary human NK cells, as well as NK-92 cells, have been successfully engineered to express CARs against a number of targets including CD19, CD20, CD244, and HER2 (97). CAR-transduced NK cells mediate efficient in vitro and in vivo killing of tumor targets (97, 98) although to date, no clinical data of CAR NK cell therapy have been reported. Shimasaki et al. recently tested the expression of a receptor containing CD3ζ and 4-1BB signaling molecules (anti-CD19-BB-ζ) in human NK cells after mRNA electroporation using a clinical-grade electroporator. The authors reported adequate transfection efficiency 24 h after electroporation, with median anti-CD19-BB-ζ expression of 40.3% in freshly purified and 61.3% in expanded NK cells. NK cells expressing anti-CD19-BB-ζ secreted IFN-γ in response to CD19-positive target cells. Interestingly, the levels of CAR expression in NK cells after mRNA transfection were comparable to those achieved by retroviral transduction. A large-scale protocol was developed to transfect expanded NK cells, achieving excellent receptor expression and considerable cytotoxicity of CAR-transduced NK cells in xenograft models of B-cell leukemia (99). Another interesting strategy is the development of CAR-modified NK cells that target NKG2D ligands on the surface of tumor cells, rendering NK more cytotoxic against a variety of hematologic and solid malignancies (100). NK cells have also been successfully engineered to target antigens on a variety of solid tumors. For example, an NK-CAR targeting the ganglioside GD2 (present on neuroblastoma cells) has been tested in preclinical studies (101, 102). GD2 is also expressed on breast cancer stem cells, thus raising the potential for its widespread use as a target for immunotherapy (103). Additional antigens targeted by NK CARs include HER2 (overexpressed in a number of solid tumors), CD138, and CS1 (overexpressed in MM) (104, 105).
Although these data support the use of CAR engineering to redirect the specificity of NK cells to augment their cytotoxicity, a number of challenges remain. These include the relative difficulty in expressing exogenous genes in primary human NK cells and the need to expand NK cells in culture to achieve adequate numbers for clinical studies of immunotherapy. To counteract this difficulty, some groups have expressed CARs in the human NK-like cell line NK-92, in an attempt to engineer a uniformly cytolytic effector cell population (106). As previously mentioned, NK-92 cells can be easily expanded in culture and their safety has been shown in phase I clinical trials in human subjects. Thus, CAR-expressing NK-92 cells may offer a practical source of cells for NK cell-based immunotherapeutic trials. In order to prevent the risk of engrafting cells with potential in vivo tumorigenicity, however, NK-92 cells must be irradiated prior to infusion, which may in turn significantly impact their in vivo persistence and long-term antitumor efficacy. Although limited in vivo persistence could prove beneficial once the alloreactive NK cells have eradicated the tumors, a number of studies of adoptive therapy with NK cells and CAR-modified T cells have reported the importance of cell persistence in inducing long-term antitumor response (50, 95, 96).
As with CAR-modified T cell therapy, a number of variables can affect the activation, antitumor efficacy, and persistence of CAR-NK cells. Second and third generation CAR constructs incorporating additional costimulatory domains (e.g., CD28, OX-40, or 4-1BB) have been shown to enhance both in vitro and in vivo activation, and the persistence of CAR T cells. Further studies exploring the optimal vector, construct and transduction method are necessary to identify the “perfect NK CAR.”
When considering the use of CAR-modified effector cells, one must take into account their safety profile. Many of the same concerns raised with CAR-modified T cells may be relevant to CAR-NK cells. These include on-target/off-tumor effects, GVHD, cytokine release syndrome, tumor lysis syndrome, and toxicity to normal tissues due to limited selectivity of the target antigen (107–109). Thus, the necessity of equipping CAR-modified NK cells with a “safety switch” or suicide gene is an important question to explore. While mature allogeneic CAR-engineered NK cells are expected to be short lived, data on the persistence of more immature NK cells, such as those derived from CB, are lacking. Interestingly, a recent study reported that IL15/4-1BBL-activated NK cells infused early after T-depleted allogeneic stem cell transplantation in patients not receiving immunosuppressive prophylaxis could contribute to acute GVHD (110). To this effect, the insertion of a suicide safety switch system, as employed with CAR-modified T cells (111, 112), can provide an efficient means for depletion of these cells if needed. Inducible suicide systems have safely and effectively eradicated GVHD in patients receiving adoptively transferred T cells without causing deleterious effects (112). However, these systems have not been extensively studied in NK cells, and in the absence of clinical data on the in vivo persistence of CAR-modified NK cells, the necessity of a suicide switch in this setting remains unknown.
Despite the growing wealth of preclinical experience with CAR-engineered NK cells, to date, only two clinical studies (both targeting CD19+ malignancies using a retroviral transduced anti-CD19-BB-ζ NK-CAR) have obtained regulatory approval: one is a recently completed pediatric study at St. Jude Children’s Research Hospital, where haploidentical NK cells modified with anti-CD19-BB-ζ CAR were infused into patients with B-ALL (ClinicalTrials.gov.NCT00995137) and the other is an ongoing study at the National University Hospital in Singapore (ClinicalTrials.gov.NCT01974479) using IL-2-activated haploidentical CAR-modified NK cells in pediatric and adult patients with refractory B-ALL (99). The results of these studies have not been reported to date.
An innovative immunoglobulin-based strategy to redirect NK cytotoxicity towards tumor cells is to create either bispecific or trispecific antibodies (BiKE, TriKE) (113). BiKEs are constructed by joining a single-chain Fv against CD16 and a single-chain Fv against a tumor-associated antigen (BiKE), or two tumor-associated antigens (TriKE). Gleason et al. showed that bispecific (bscFv) CD16/CD19 and trispecific (tscFv) CD16/CD19/CD22 engagers directly trigger NK cell activation through CD16, significantly increasing NK cell cytolytic activity and cytokine production against various CD19-expressing B cell lines. The same group also developed and tested a CD16 × 33 BiKE in refractory AML and demonstrated that the potent killing by NK cells could overcome the inhibitory effect of KIR signaling (113, 114).
Notably, activated NK cells lose CD16 (FcRγIII) and CD62L through a metalloprotease called ADAM17, which is expressed on NK cells, which may in turn impact on the efficacy of Fc-mediated cytotoxicity (115). Romee et al. recently showed that selective inhibition of ADAM17 enhances CD16-mediated NK cell function by preserving CD16 on the NK cell surface, thus enhancing ADCC (115). Additionally, Fc-induced production of cytokines by NK cells exposed to rituximab-coated B cell targets can be further enhanced by ADAM17 inhibition. These findings support a role for targeting ADAM17 to prevent CD16 shedding and to improve the efficacy of therapeutic mAbs. The same group subsequently discovered that ADAM17 inhibition enhances CD16 × 33 BiKE responses against primary AML targets (114).
Recent advances in the understanding of NK cell immunobiology have paved the way for novel and innovative anti-cancer therapies. Here, we have discussed a representation of these novel immunotherapeutic strategies to potentiate NK cell function and enhance antitumor activity including ADCC-inducing mAbs, ex vivo activated or genetically modified NK cells and bi- or trispecific engagers (Figure 1).
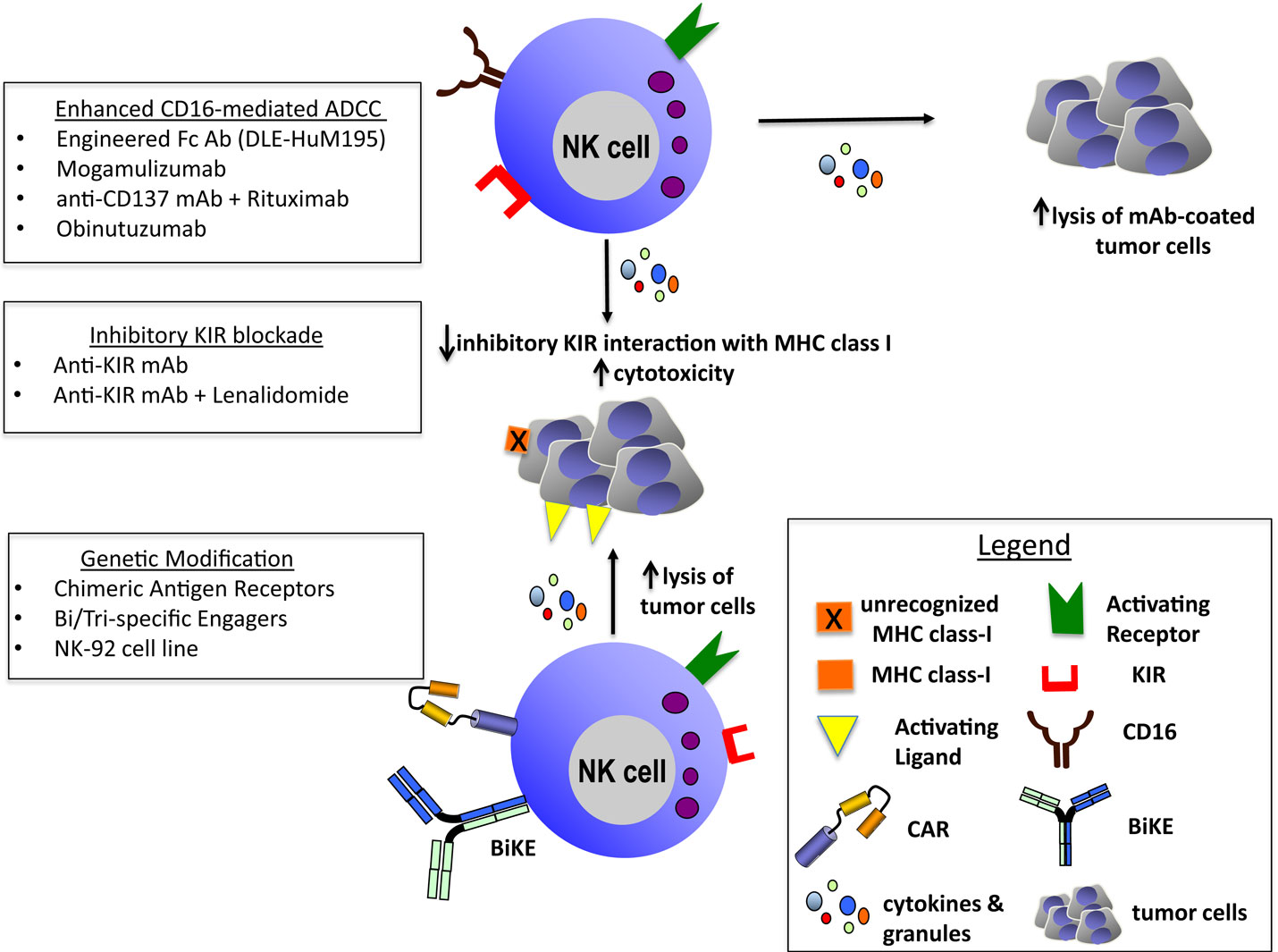
Figure 1. Enhanced CD16-mediated ADCC: engineered Fc Ab (DLE-HuM195) (27), mogamulizumab (30), anti-CD137 mAb + rituximab (36–39), and obinutuzumab (31). Inhibitory KIR blockade: anti-KIRmAb (33–35) and anti-KIR mAb + lenalidomide (35). Genetic modification: chimeric antigen receptors (97–105), bi/trispecific engagers (113, 114), and NK-92 cell line (86–91).
Although experience has shown that adoptive immunotherapy with allogeneic NK cells may be more efficacious than with autologous NK cells, to date, their long-term antitumor benefits have been modest (3). Expansion and persistence of NK cells following infusion appear to be the main determinants of clinical response (50–52, 70), thus underscoring the importance of identifying ways to enhance their persistence and antitumor activity. It is likely that the combination of high-dose lymphodepleting chemotherapy with additional modifications (such as Treg depletion, in vivo administration of cytokines, such as IL-15 or enhancement of CD16-mediated antigen targeting) may maximize NK persistence and efficacy.
In addition, the possibility of third-party “off-the-shelf” products with partially HLA-matched NK cells from CB, third-party donors, or NK cell lines allow the advantage of unlimited sources of cells to improve the practicality of cell therapy. With increasing focus on genetically modifying NK cells to redirect their specificity or engager-modified NK cells, it is likely that NK cells will move to the forefront of cancer therapy over the next few years.
RR and KR wrote and edited the manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The authors’ research efforts are funded in part by the Lymphoma Research Foundation (RHR), Lymphoma SPORE (RHR), MDACC Leukemia SPORE Grant CA (KR), and SINF (KR).
1. Lotzova E, Savary CA, Herberman RB. Inhibition of clonogenic growth of fresh leukemia cells by unstimulated and IL-2 stimulated NK cells of normal donors. Leuk Res (1987) 11(12):1059–66. doi:10.1016/0145-2126(87)90158-5
2. Lowdell MW, Craston R, Samuel D, Wood ME, O’Neill E, Saha V, et al. Evidence that continued remission in patients treated for acute leukaemia is dependent upon autologous natural killer cells. Br J Haematol (2002) 117(4):821–7. doi:10.1046/j.1365-2141.2002.03495.x
3. Davies J, Stringaris K, Barrett AJ, Rezvani K. Opportunities and limitations of NK cells as adoptive therapy for malignant disease. Cytotherapy (2014) 16(11):1453–66. doi:10.1016/j.jcyt.2014.03.009
4. Lanier LL, Le AM, Civin CI, Loken MR, Phillips JH. The relationship of CD16 (Leu-11) and Leu-19 (NKH-1) antigen expression on human peripheral blood NK cells and cytotoxic T lymphocytes. J Immunol (1986) 136(12):4480–6.
5. Locatelli F, Moretta F, Brescia L, Merli P. Natural killer cells in the treatment of high-risk acute leukaemia. Semin Immunol (2014) 26(2):173–9. doi:10.1016/j.smim.2014.02.004
6. Cooper MA, Fehniger TA, Turner SC, Chen KS, Ghaheri BA, Ghayur T, et al. Human natural killer cells: a unique innate immunoregulatory role for the CD56(bright) subset. Blood (2001) 97(10):3146–51. doi:10.1182/blood.V97.10.3146
7. Campbell JJ, Qin S, Unutmaz D, Soler D, Murphy KE, Hodge MR, et al. Unique subpopulations of CD56+ NK and NK-T peripheral blood lymphocytes identified by chemokine receptor expression repertoire. J Immunol (2001) 166(11):6477–82. doi:10.4049/jimmunol.166.11.6477
8. De Maria A, Bozzano F, Cantoni C, Moretta L. Revisiting human natural killer cell subset function revealed cytolytic CD56(dim)CD16+ NK cells as rapid producers of abundant IFN-gamma on activation. Proc Natl Acad Sci U S A (2011) 108:728–32. doi:10.1073/pnas.1012356108
9. Farag S, Caligiuri M. Human natural killer cell development and biology. Blood Rev (2006) 20(3):123–37. doi:10.1016/j.blre.2005.10.001
10. Moretta L, Pietra G, Montaldo E, Vacca P, Pende D, Falco M, et al. Human NK cells: from surface receptors to the therapy of leukemias and solid tumors. Front Immunol (2014) 5:87. doi:10.3389/fimmu.2014.00087
11. Liu J, Xiao Z. Activating killer immunoglobulin-like receptor 2DSs2 binds to HLA A*11. Proc Natl Acad Sci U S A (2014) 111(7):2662–7. doi:10.1073/pnas.1322052111
12. Fauriat C, Long EO, Ljunggren HG, Bryceson YT. Regulation of human NK-cell cytokine and chemokine production by target cell recognition. Blood (2010) 115:2167–76. doi:10.1182/blood-2009-08-238469
14. Campbell K, Hasegawa J. Natural killer cell biology: an update and future directions. J Allergy Clin Immuol. (2013) 32(3):536–44. doi:10.1016/j.jaci.2013.07.006
15. Caligiuri MA. Human natural killer cells. Blood (2008) 112:461–9. doi:10.1182/blood-2007-09-077438
16. Bradley M, Zeytun A, Rafi-Janajreh A, Nagarkatti PS, Nagarkatti M. Role of spontaneous and interleukin-2-induced natural killer cell activity in the cytotoxicity and rejection of Fas1 and Fas- tumor cells. Blood (1998) 92:4248–55.
17. Screpanti V, Wallin RP, Ljunggren HG, Grandien A. A central role for death receptor-mediated apoptosis in the rejection of tumors by NK cells. J Immunol (2001) 167:2068–73. doi:10.4049/jimmunol.167.4.2068
18. Kayagaki N, Yamaguchi N, Nakayama M, Takeda K, Akiba H, Tsutsui H, et al. Expression and function of TNF-related apoptosis-inducing ligand on murine activated NK cells. J Immunol (1999) 163:1906–13.
19. Joncker NT, Shifrin N, Delebecque F, Raulet DH. Mature natural killer cells reset their responsiveness when exposed to an altered MHC environment. J Exp Med (2010) 207(10):2065–72. doi:10.1084/jem.20100570
20. Martin-Fontecha A, Thomsen L, Brett S, Gerard C, Lipp M, Lanzavecchia A, et al. Induced recruitment of NK cells to lymph nodes provides IFN-γ for TH1 priming. Nat Immunol (2004) 5:1260–5. doi:10.1038/ni1138
21. Agaugue S, Marcenaro E. Human natural killer cells exposed to IL-2, IL-12, IL-18 or IL-4 differently modulate priming of naïve T cells by monocyte-derived dendritic cells. Blood (2008) 112(5):1776–83. doi:10.1182/blood-2008-02-135871
22. Ferlazzo G, Tsang ML, Moretta L, Melioli G, Steinman RM, Munz C. Human dendritic cells activate resting natural killer (NK) cells and are recognized via the NKp30 receptor by activated NK cells. J Exp Med (2002) 195(3):343–511. doi:10.1084/jem.20011149
23. Bellora F, Castriconi R, Dondero A, Reggiardo G, Moretta L, Mantovani A, et al. The interaction of human natural killer cells with either unpolarized or polarized macrophages results in different functional outcomes. Proc Natl Acad Sci U S A (2010) 107(50):21641–59. doi:10.1073/pnas.1007654108
24. Murphy WJ, Parham P, Miller JS. NK cells – from bench to clinic. Biol Blood Marrow Transplant (2012) 18(1 Suppl):S2–7. doi:10.1016/j.bbmt.2011.10.033
25. Castriconi R, Dondero A, Bellora F, Moretta L, Castellano A, Locatelli F, et al. Neuroblastoma-derived TGF-beta1 modulates the chemokine receptor repertoire of human resting NK cells. J Immunol. (2013) 190:5321–8. doi:10.4049/jimmunol.1202693
26. Lee HM, Kim KS, Kim J. A comparative study of the effects of inhibitory cytokines on human natural killer cells and the mechanistic features of transforming growth factor-beta. Cell Immunol (2014) 290:52–61. doi:10.1016/j.cellimm.2014.05.001
27. Romain G, Senyukov V, Rey-Villamizar N, Merouane A, Kelton W, Liadi I, et al. Antibody Fc-engineering improves frequency and promotes kinetic boosting of serial killing mediated by NK cells. Blood (2014) 124(22):3241–9. doi:10.1182/blood-2014-04-569061
28. Clynes RA, Towers TL, Presta LG, Ravetch JV. Inhibitory Fc receptors modulate in vivo cytotoxicity against tumor targets. Nat Med (2000) 6(4):443–6. doi:10.1038/74704
29. de Haij S, Jansen JH, Boross P, Beurskens FJ, Bakema JE, Bos DL, et al. In vivo cytotoxicity of type I CD20 antibodies critically depends on Fc receptor ITAM signaling. Cancer Res (2010) 70(8):3209–17. doi:10.1158/0008-5472.CAN-09-4109
30. Kanazawa T, Hiramatsu Y, Iwata S, Siddiquey M, Sato Y, Suzuki M, et al. Anti CCR-4 monoclonal antibody mogamulizumab for the treatment of EBV-associated T-and NK-cell lymphoproliferative diseases. Clin Cancer Res (2014) 20(19):5075–84. doi:10.1158/1078-0432.CCR-14-0580
31. Terszowski G, Klein CKIR. /HLA interactions negatively affect rituximab-but not GA101 (obinutuzumab)-induced antibody-dependent cellular cytotoxicity. J Immunol (2014) 192(12):5618–24. doi:10.4049/jimmunol.1400288
32. Binyamin L, Alpaugh RK, Hughes TL, Lutz CT, Campbell KS, Weiner LM. Blocking NK cell inhibitory self-recognition promotes antibody-dependent cellular cytotoxicity in a model of anti-lymphoma therapy. J Immunol (2008) 180(9):6392–401. doi:10.4049/jimmunol.180.9.6392
33. Kohrt H, Thielens A. Anti-KIR antibody enhancement of anti-lymphoma activity of natural killer cells as monotherapy and in combination with anti-CD20 antibodies. Blood (2014) 123(5):678–86. doi:10.1182/blood-2013-08-519199
34. Romagne F, Andre P, Spee P, Zahn S, Anfossi N, Gauthier L, et al. Preclinical characterization of I-7F9, a novel human anti-KIR receptor therapeutic antibody that augments natural killer-mediated killing of tumor cells. Blood (2009) 114(13):2667–77. doi:10.1182/blood-2009-02-206532
35. Vey N, Bourhis JH, Boissel N, Bordessoule D, Prebet T, Charbonnier A, et al. A phase 1 trial of the anti-inhibitory KIR mAb IPH2101 for AML in complete remission. Blood (2012) 120(22):4317–23. doi:10.1182/blood-2012-06-437558
36. Lin W, Voskens C, Zhang X, Schindler DG, Wood A, Burch E, et al. Fc-dependent expression of CD137 on human NK cells: insights into “agonistic” effects of anti-CD137 monoclonal antibodies. Blood (2008) 112(3):699–707. doi:10.1182/blood-2007-11-122465
37. Benson DM Jr, Bakan CE, Zhang S, Collins SM, Liang J, Srivastava S, et al. IPH2101, a novel anti-inhibitory KIR antibody, and lenalidomide combine to enhance the natural killer cell versus multiple myeloma effect. Blood (2011) 118(24):6387–91. doi:10.1182/blood-2011-06-360255
38. Kohrt HE, Houot R, Goldstein MJ, Weiskopf K, Alizadeh AA, Brody J, et al. CD137 stimulation enhances the antilymphoma activity of anti-CD20 antibodies. Blood (2011) 117(8):2423–32. doi:10.1182/blood-2010-08-301945 Epub 2011/01/05.,
39. Wilcox RA, Tamada K, Strome SE, Chen L. Signaling through NK cell-associated CD137 promotes both helper function for CD8+ cytolytic T cells and responsiveness to IL-2 but not cytolytic activity. J Immunol (2002) 169(8):4230–6. doi:10.4049/jimmunol.169.8.4230
40. Seidel UJ, Schlegel P, Lang P. Natural killer cell mediated antibody-dependent cellular cytotoxicity in tumor immunotherapy with therapeutic antibodies. Front Immunol (2013) 4:76. doi:10.3389/fimmu.2013.00076
41. Cartron G, Dacheux L. Therapeutic activity of humanized anti-CD20 monoclonal antibody and polymorphism in IgG Fc receptor FcgammaRIIIa gene. Blood (2002) 99(3):754–8. doi:10.1182/blood.V99.3.754
42. Weng WK, Levy R. Two immunoglobulin G fragment C receptor polymorphisms independently predict response to rituximab in patients with follicular lymphoma. J Clin Oncol (2012) 21(21):3940–7. doi:10.1200/JCO.2003.05.013
43. Burns LJ, Weisdorf DJ, DeFor TE, Vesole DH, Repka TL, Blazar BR, et al. IL-2-based immunotherapy after autologous transplantation for lymphoma and breast cancer induces immune activation and cytokine release: a phase I/II trial. Bone Marrow Transplant (2003) 32:177–86. doi:10.1038/sj.bmt.1704086
44. Rosenberg SA, Lotze MT, Muul LM, Leitman S, Chang AE, Ettinghausen SE, et al. Observations on the systemic administration of autologous lymphokine-activated killer cells and recombinant interleukin-2 to patients with metastatic cancer. N Engl J Med (1985) 313:1485–92. doi:10.1056/NEJM198512053132327
45. Parkhurst MR, Riley JP, Dudley ME, Rosenberg SA. Adoptive transfer of autologous natural killer cells leads to high levels of circulating natural killer cells but does not mediate tumor regression. Clin Cancer Res (2011) 17:6287–97. doi:10.1158/1078-0432.CCR-11-1347
46. Benyunes MC, Higuchi C. Immunotherapy with interleukin 2 with or without lymphokine-activated killer cells after autologous bone marrow transplantation for malignant lymphoma: a feasibility trial. Bone Marrow Transplant (1995) 16(2):283–8.
47. Cheng M, Chen Y, Xiao W, Sun R, Tian Z. NK cell-based immunotherapy for malignant diseases. Cell Mol Immunology (2013) 10(3):230–52. doi:10.1038/cmi.2013.10
48. Stern M, Passweg JR, Meyer-Monard S, Esser R, Tonn T, Soerensen J, et al. Pre-emptive immunotherapy with purified natural killer cells after haploidentical SCT: a prospective phase II study in two centers. Bone Marrow Transplant (2013) 48:433–8. doi:10.1038/bmt.2012.162
49. Yoon SR, Lee YS, Yang SH, Ahn KH, Lee JH, Lee JH, et al. Generation of donor natural killer cells from CD34+progenitor cells and subsequent infusion after HLA-mismatched allogeneic hematopoietic cell transplantation: a feasibility study. Bone Marrow Transplant (2010) 45(6):1038–46. doi:10.1038/bmt.2009.304
50. Miller JS, Soignier Y. Successful adoptive transfer and in vivo expansion of human haploidentical NK cells in patients with cancer. Blood (2005) 105(8):3051–7. doi:10.1182/blood-2004-07-2974
51. Rubnitz JE, Inaba H. NKAML: a pilot study to determine the safety and feasibility of haploidentical natural killer cell transplantation in childhood acute myeloid leukemia. J Clin Oncol (2010) 28(6):955–9. doi:10.1200/JCO.2009.24.4590
52. Curti A, Ruggeri L. Succesful transfer of alloreactive haploidentical KIR ligand-mismatched natural killer cells after infusion in elderly high risk acute myeloid leukemia patients. Blood (2011) 118(12):3273–9. doi:10.1182/blood-2011-01-329508
53. Rusakiewicz S, Semeraro M, Sarabi M, Desbois M, Locher C, Mendez R, et al. Immune infiltrates are prognostic factors in localized gastrointestinal stromal tumors. Cancer Res (2013) 73(12):3499–510. doi:10.1158/0008-5472.CAN-13-0371
54. Mamessier E, Sylvain A, Thibult ML, Houvenaeghel G, Jacquemier J, Castellano R, et al. Human breast cancer cells enhance self tolerance by promoting evasion from NK cell antitumor immunity. J. Clin. Invest (2011) 121:3609–22. doi:10.1172/JCI45816
55. Navarro A, Bjorklund A, Chekenya M. Therapeutic potential and challenges of natural killer cells in treatment of solid tumors. Front Immunol (2015) 29(6):202. doi:10.3389/fimmu.2015.00202
56. Stringaris K, Sekine T, Khoder A, Alsuliman A, Razzaghi B, Sargeant R, et al. Leukemia-induced phenotypic and functional defects in natural killer cells predict failure to achieve remission in acute myeloid leukemia. Haematologica (2014) 99(5):836–47. doi:10.3324/haematol.2013.087536
57. Rouce R, Shaim H, Sekine T, et al. The TGF-β/SMAD pathway is an important mechanism for NK cell immune evasion in childhood B acute lymphoblastic leukemia. Leukemia (in press).
58. Murray S, Lundqvist A. Targeting the tumor microenvironment to improve natural killer cell-based immunotherapies: on being in the right place at the right time, with resilience. Hum Vaccin Immunother (2015) 24:0. doi:10.1080/21645515.2015.1096458
59. Nausch N, Galani I, Schlecker E, Cerwenka A. Mononuclear myeloid-derived “suppressor” cells express RAE-1 and activate natural killer cells. Blood (2008) 112(10):4080–9. doi:10.1182/blood-2008-03-143776
60. Geller MA, Cooley S, Judson PL, Ghebre R, Carson LF, Argenta PA, et al. A phase II study of allogeneic natural killer cell therapy to treat patients with recurrent ovarian and breast cancer. Cytotherapy (2011) 13(1):98–107. doi:10.3109/14653249.2010.515582
61. Koepsell SA, Miller JS. Natural killer cells: a review of manufacturing and clinical utility. Transfusion (2013) 53(2):404–10. doi:10.1111/j.1537-2995.2012.03724.x
62. Lang P, Pfeiffer M, Teltschik HM, Schlegel P, Feuchtinger T, Ebinger M, et al. Natural killer cell activity influences outcome after T cell depleted stem cell transplantation from matched unrelated and haploidentical donors. Best Pract Res Clin Haematol (2011) 24:403–11. doi:10.1016/j.beha.2011.04.009
63. Pfeiffer M, Schumm M, Feuchtinger T, Dietz K, Handgretinger R, Lang P. Intensity of HLA class I expression and KIR-mismatch determine NK-cell mediated lysis of leukaemic blasts from children with acute lymphatic leukaemia. Br J Haematol (2007) 138:97–100. doi:10.1111/j.1365-2141.2007.06631.x
64. Leong W, Chase JM, Romee R, Schneider SE, Sullivan RP, Cooper MA, et al. Pre-activation with IL-12, IL-15, and IL-18 induces CD25 and a functional high affinity IL-2 receptor on human cytokine-induced memory-like NK cells. Biol Blood Marrow Transplant (2014) 20(4):463–73. doi:10.1016/j.bbmt.2014.01.006
65. Ni J, Miller M, Stojanovic A, Garbi N, Cerwenka A. Sustained effector function of IL-12/15/18-preactivated NK cells against established tumors. J Exp Med (2012) 209:2351–65. doi:10.1084/jem.20120944
66. Miller J, Rooney C, Curtsinger J, McElmurry R, McCullar V, Verneris MR, et al. Expansion and homing of adoptively transferred human natural killer cells in immunodeficient mice varies with product preparation and in vivo cytokine administration: implications for clinical therapy. Biol Blood Marrow Transplant (2014) 20(8):1252–7. doi:10.1016/j.bbmt.2014.05.004
67. Fujisaki H, Kakuda H, Shimasaki N, Imai C, Ma J, Lockey T, et al. Expansion of highly cytotoxic human natural killer cells for cancer cell therapy. Cancer Res (2009) 69:4010–7. doi:10.1158/0008-5472.CAN-08-3712
68. Berg M, Lundqvist A, McCoy P Jr, Samsel L, Fan Y, Tawab A, et al. Clinical-grade ex vivo-expanded human natural killer cells up-regulate activating receptors and death receptor ligands and have enhanced cytolytic activity against tumor cells. Cytotherapy (2009) 11:341–55. doi:10.1080/14653240902807034
69. Lundqvist A, Yokoyama H, Smith A, Berg M, Childs R. Bortezomib treatment and regulatory T-cell depletion enhance the antitumor effects of adoptively infused NK cells. Blood (2009) 113:6120–7. doi:10.1182/blood-2008-11-190421
70. Bachanova C. Clearance of acute myeloid leukemia by haploidentical natural killer cells is improved using IL-2 diphtheria toxin fusion protein. Blood (2014) 123(25):3855–63. doi:10.1182/blood-2013-10-532531
71. Klingemann H, Grodman C. Autologous stem cell transplant recipients tolerate haploidentical related-donor natural killer cell-enriched infusions. Transfusion (2013) 53:412–8. doi:10.1111/j.1537-2995.2012.03764.x
72. Reed W, Noga SJ, Gee AP, Rooney CM, Wagner JE, McCullough J, et al. Production assistance for cellular therapies (PACT): four-year experience from the United States National Heart, Lung, and Blood Institute (NHLBI) contract research program in cell and tissue therapies. Transfusion (2009) 49:786–96. doi:10.1111/j.1537-2995.2008.02027.x
73. Koepsell SA, Kadidlo DM, Fautsch S, McCullough J, Klingemann H, Wagner JE, et al. Successful “in-flight” activation of natural killer cells during long-distance shipping. Transfusion (2013) 53:398–403. doi:10.1111/j.1537-2995.2012.03695.x
74. Woll P, Grzywacz B, Tian X, Marcus RK, Knorr DA, Verneris MR, et al. Human embryonic stem cells differentiate into a homogenous population of natural killer cells with potent in vivo antitumor activity. Blood (2009) 113(24):6094–101. doi:10.1182/blood-2008-06-165225
75. Chouaib S, Pittari G, Nanbakhsh A, ElAyoubi H, Amsellem S, Bourhis JH, et al. Improving the outcome of leukemia by natural killer cell-based immunotherapeutic strategies. Front Immunol (2014) 5:95. doi:10.3389/fimmu.2014.00095
76. Spanholtz J, Preijers F, Tordoir M, Trilsbeek C, Paardekooper J, de Witte T, et al. Clinical-grade generation of active NK cells from cord blood hematopoietic progenitor cells for immunotherapy using a closed-system culture process. PLoS One (2011) 6:e20740. doi:10.1371/journal.pone.0020740
77. Shah N, Martin-Antonio B, Yang H, Ku S, Lee DA, Cooper LJ, et al. Antigen presenting cell-mediated expansion of human umbilical cord blood yields log-scale expansion of natural killer cells with anti-myeloma activity. PLoS One (2013) 8(10):e76781. doi:10.1371/journal.pone.0076781
78. Shaim H, Yvon E. Cord blood: a promising source of allogeneic natural killer cells for immunotherapy. Cytotherapy (2015) 17(1):1–2. doi:10.1016/j.jcyt.2014.12.001
79. Dalle JH, Menezes J, Wagner E, Blagdon M, Champagne J, Champagne MA, et al. Characterization of cord blood natural killer cells: implications for transplantation and neonatal infections. Pediatr Res (2005):57.
80. Tomchuck S, Leung W, Dallas M. Enhanced cytotoxic function of natural killer and CD3+CD56+ cells in cord blood after culture. Biol Blood Marrow Transplant (2015) 21(1):39–49. doi:10.1016/j.bbmt.2014.10.014
81. Della C, Falco M, Podesta M, Locatelli F, Moretta L, Frassoni F, et al. Phenotypic and functional heterogeneity of human NK cells developing after umbilical cord blood transplantation: a role for human cytomegalovirus. Blood (2012) 119(2):399–410. doi:10.1182/blood-2011-08-372003
82. Xing R. Cord blood natural killer cells exhibit impaired lytic immunological synapse formation that is reversed with IL-2 ex vivo expansion. J Immunother (2010) 33(7):684–96. doi:10.1097/CJI.0b013e3181e475e9
83. Alnabhan R, Madrigal A, Saudemont A. Differential activation of cord blood and peripheral blood NK cells by cytokines. Cytotherapy (2015) 17(1):73–85. doi:10.1016/j.jcyt.2014.08.003
84. Denman CJ, Senyukov VV, Somanchi SS, Phatarpekar PV, Kopp LM, Johnson JL, et al. Membrane-bound IL-21 promotes sustained ex vivo proliferation of human natural killer cells. PLoS One (2012) 7:e30264. doi:10.1371/journal.pone.0030264
85. Pittari G, Filippini P, Gentilcore G, Grivel JC, Rutella S. Revving up natural killer cells and cytokine-induced killer cells against hematological malignancies. Front Immunol (2015) 6:230. doi:10.3389/fimmu.2015.00230
86. Klingemann HG, Wong E, Maki GA. cytotoxic NK-cell line (NK-92) for ex vivo purging of leukemia from blood. Biol Blood Marrow Transplant (1996) 2:68–75.
87. Yagita M, Huang C, Umehara H, Matsuo Y, Tabata R, Miyake M, et al. A novel natural killer cell line (KHYG-1) from a patient with aggressive natural killer cell leukemia carrying a p53 point mutation. Leukemia (2000) 14(5):922–30. doi:10.1038/sj.leu.2401769
88. Tonn T, Schwabe D, Klingemann H, Becker S, Esser R, Koehl U, et al. Treatment of patients with advanced cancer with the natural killer cell line NK-92. Cytotherapy (2013) 15(12):1563–70. doi:10.1016/j.jcyt.2013.06.017
89. Swift B, Williams B. Natural killer cell lines preferentially kill clonogenic multiple myeloma cells and decrease myeloma engraftment in a bioluminescent xenograft mouse model. Haematologica (2012) 97(7):1020–8. doi:10.3324/haematol.2011.054254
90. Yan Y, Steinherz P, Klingemann HG, Dennig D, Childs BH, McGuirk J, et al. Antileukemia activity of a natural killer cell line against human leukemias. Clin Cancer Res (1998) 4:2859–68.
91. Tam YK, Miyagawa B, Ho VC, Klingemann HG. Immunotherapy of malignant melanoma in a SCID mouse model using the highly cytotoxic natural killer cell line NK-92. J Hematother (1999) 8:281–90. doi:10.1089/106161299320316
92. Dudley ME, Wunderlich JR, Yang JC, Sherry RM, Topalian SL, Restifo NP, et al. Adoptive cell transfer therapy following non-myeloablative but lymphodepleting chemotherapy for the treatment of patients with refractory metastatic melanoma. Journal of Clinical Oncology (2005) 23:2346–57. doi:10.1200/JCO.2005.00.240
93. Conlon K, Lugli E, Welles H. Redistribution, hyperproliferation, activation of natural killer cells and CD8 T cells, and cytokine production during first-in-human clinical trial of recombinant human interleukin-15 in patients with cancer. J Clin Oncol (2015) 33(1):74–82. doi:10.1200/JCO.2014.57.3329
94. Pegram H, Smith E, Rafiq S, Brentgens R. CAR therapy for hematological cancers: can success seen in the treatment of B-cell acute lymphoblastic leukemia be applied to other hematological malignancies? Immunotherapy (2015) 7(5):545–61. doi:10.2217/imt.15.6
95. Grupp S, Kalos M, Barrett D, Aplenc R, Porter D, Rheingold S, et al. Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. N Engl J Med (2013) 368(16):1509–18. doi:10.1056/NEJMoa1215134
96. Lee DW, Kochenderfer JN, Stetler-Stevenson M, Cui YK, Delbrook C, Feldman SA, et al. T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: a phase 1 dose-escalation trial. Lancet (2015) 385(9967):517–28. doi:10.1016/S0140-6736(14)61403-3
97. Glienke W, Esser R, Priesner C, Suerth J, Schambach A, Wels WS, et al. Advantages and applications of CAR-expressing NK cells. Front Pharmacol (2015) 6:21. doi:10.3389/fphar.2015.00021
98. Tettamanti S, Marin V, Pizzitola I, Magnani CF, Giordano Attianese GM, Cribioli E, et al. Targeting of acute myeloid leukaemia by cytokine-induced killer cells redirected with a novel CD123-specific chimeric antigen receptor. Br J Haematol (2013) 161:389–401. doi:10.1111/bjh.12282
99. Shimasaki N, Fujisaki H, Cho D, Masselli M, Lockey T, Eldridge P, et al. A clinically adaptable method to enhance the cytotoxicity of natural killer cells against B-cell malignancies. Cytotherapy (2012) 14(7):830–40. doi:10.3109/14653249.2012.671519
100. Chang YH, Connolly J, Shimasaki N, Mimura K, Kono K, Campana D. A chimeric receptor with NKG2D specificity enhances natural killer cell activation and killing of tumor cells. Cancer Res (2013) 73(6):1777–86. doi:10.1158/0008-5472.CAN-12-3558
101. Altvater B, Landmeier S, Pscherer S, Temme J, Schweer K, Kailayangiri S, et al. 2B4(CD244) signaling by recombinant antigen-specific chimeric receptors costimulates natural killer cell activation to leukemia and neuroblastoma cells. Clin. Cancer Res (2009) 15(15):4857–66. doi:10.1158/1078-0432.CCR-08-2810
102. Esser R, Muller T, Stefes D, Kloess S, Seidel D, Gillies S, et al. NK cells engineered to express a GD2-specific antigen receptor display built-in ADCC-like activity against tumour cells of neuroectodermal origin. J. Cell Mol. Med (2012) 16(3):569–81. doi:10.1111/j.1582-4934.2011.01343.x
103. Battula VL, Shi Y, Evans KW, Wang RY, Spaeth EL, Jacamo RO, et al. Ganglioside GD2 identifies breast cancer stem cells and promotes tumorigenesis. J Clin Invest (2012) 122(6):2066–78. doi:10.1172/JCI59735
104. Jiang H, Zhang W, Shang P, Zhang H, Fu W, Ye F, et al. Transfection of chimeric anti-CD138 gene enhances natural killer cell activation and killing of multiple myeloma cells. Mol.Oncol (2014) 8(2):297–310. doi:10.1016/j.molonc.2013.12.001
105. Chu J, Deng Y, Benson DM, He S, Hughes T, Zhang J, et al. CS1-specific chimeric antigen receptor (CAR)-engineered natural killer cells enhance in vitro and in vivo antitumor activity against human multiple myeloma. Leukemia (2014) 28(4):917–27. doi:10.1038/leu.2013.279
106. Boissel L, Betancur-Boissel M, Lu W, Krause DS, Van Etten RA, Wels WS, et al. Retargeting NK-92 cells by means of CD19- and CD20-specific chimeric antigen receptors compares favorably with antibody-dependent cellular cytotoxicity. OncoImmunology (2013) 2(10):e26527. doi:10.4161/onci.26527
107. Ferrara JL, Levine JE, Reddy P, Holler E. Graft-versus-host disease. Lancet (2009) 373(9674):1550–61. doi:10.1016/S0140-6736(09)60237-3
108. Stauss H, Morris E. Immunotherapy with gene-modified T cells: limiting side effects provides new challenges. Gene Ther (2013) 20(11):1029–32. doi:10.1038/gt.2013.34
109. Maus M, Grupp S, Porter D, June C. Antibody-modified T cells: CARs take the front seat for hematologic malignancies. Blood (2014) 123(17):2625–35. doi:10.1182/blood-2013-11-492231
110. Shah N, Baird K, Delbrook C, Fleisher T, Kohler M, Rampertaap S, et al. Acute GVHD in patients receiving IL-15/4-1BBL activated NK cells following T cell depleted stem cell transplantation. Blood (2015) 125(5):784–92. doi:10.1182/blood-2014-07-592881
111. Jones B, Lamb L, Goldman F, DiStasi A. Improving the safety of cell therapy products by suicide gene transfer. Front Pharmacol (2014) 5:254. doi:10.3389/fphar.2014.00254
112. Zhou X, Dotti G, Krance R, Martinez C, Naik S, Kamble R, et al. Inducible caspase-9 suicide gene controls adverse effects from alloreplete T cells after haploidentical stem cell transplantation. Blood (2015) 125(26):4103–13. doi:10.1182/blood-2015-02-628354
113. Gleason R. CD16xCD33 bispecific killer cell engager (BiKE) activates NK cells against primary MDS and MDSC CD33+ targets. Blood (2014) 123(19):3016–26. doi:10.1182/blood-2013-10-533398
114. Wiernik A, Foley B, Zhang B, Verneris MR, Warlick E, Gleason MK, et al. Targeting natural killer cells to AML in vitro with a CD16x33 engager. Clin Cancer Res (2013) 19(14):3844–55. doi:10.1158/1078-0432.CCR-13-0505
115. Romee R, Foley B, Lenvik T, Wang Y, Zhang B, Ankarlo D, et al. NK cell CD16 surface expression is regulated by a disintegrin and metalloprotease-17 (ADAM17). Blood (2013) 121(18):3599–608. doi:10.1182/blood-2012-04-425397
Keywords: natural killer cells, adoptive immunotherapy, CAR NK cells, ADCC, anti-KIR antibody, NK-92, transplantation
Citation: Rezvani K and Rouce RH (2015) The Application of Natural Killer Cell Immunotherapy for the Treatment of Cancer. Front. Immunol. 6:578. doi: 10.3389/fimmu.2015.00578
Received: 24 September 2015; Accepted: 29 October 2015;
Published: 17 November 2015
Edited by:
Rafael Solana, University of Cordoba, SpainReviewed by:
Kamalakannan Rajasekaran, Blood Research Institute, USACopyright: © 2015 Rezvani and Rouce. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Katayoun Rezvani, a3JlenZhbmlAbWRhbmRlcnNvbi5vcmc=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.