 Aleksander Keselman
Aleksander Keselman Nicola Heller
Nicola Heller
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol., 16 November 2015
Sec. Immunological Tolerance and Regulation
Volume 6 - 2015 | https://doi.org/10.3389/fimmu.2015.00568
This article is part of the Research TopicDouble-edge consequence of enhanced immune reactivity in females: Better protection against infection but increased susceptibility to autoimmune diseasesView all 9 articles
Asthma is a chronic airway inflammatory disease that affects ~300 million people worldwide. It is characterized by airway constriction that leads to wheezing, coughing, and shortness of breath. The most common treatments are corticosteroids and β2-adrenergic receptor antagonists, which target inflammation and airway smooth muscle constriction, respectively. The incidence and severity of asthma is greater in women than in men, and women are more prone to develop corticosteroid-resistant or “hard-to-treat” asthma. Puberty, menstruation, pregnancy, menopause, and oral contraceptives are known to contribute to disease outcome in women, suggesting a role for estrogen and other hormones impacting allergic inflammation. Currently, the mechanisms underlying these sex differences are poorly understood, although the effect of sex hormones, such as estrogen, on allergic inflammation is gaining interest. Asthma presents as a heterogeneous disease. In typical Th2-type allergic asthma, interleukin (IL)-4 and IL-13 predominate, driving IgE production and recruitment of eosinophils into the lungs. Chronic Th2-inflammation in the lung results in structural changes and activation of multiple immune cell types, leading to a deterioration of lung function over time. Most immune cells express estrogen receptors (ERα, ERβ, or the membrane-bound G-protein-coupled ER) to varying degrees and can respond to the hormone. Together these receptors have demonstrated the capacity to regulate a spectrum of immune functions, including adhesion, migration, survival, wound healing, and antibody and cytokine production. This review will cover the current understanding of estrogen signaling in allergic inflammation and discuss how this signaling may contribute to sex differences in asthma and allergy.
Allergic disorders are characterized by Th2 responses against innocuous environmental factors that result in eosinophilic inflammation and the production of interleukin (IL)-4, IL-13, and IgE. Genetic, behavioral, sex-specific, and environmental factors contribute to the rising incidence of allergic diseases. Environmental factors that contribute to allergies are extremely broad and include a variety of food, plant, insect, and pollutant products. Allergic sensitization initiates the “atopic march,” and chronic exposure to environmental insults at epithelial surfaces leads to the development of a spectrum of allergic disorders ranging from dermatitis to asthma [reviewed in Ref. (1, 2)]. Allergic diseases are increasingly common in the Western world and to date have no cure. Current treatments broadly include antihistamines, anticongestants, antileukotrienes, bronchodilators, and steroids. Th2 (IL-4, IL-13 and IL-5) cytokine-neutralizing antibodies are currently in phase II and phase III of clinical trials with promising results (3–6). However, hard-to-treat allergies limit the effectiveness of current therapies, especially in the case of asthma.
Asthma is an allergic inflammation of the respiratory tract that affects >300 million people worldwide. It has a prevalence of 1 in 12 adults and 1 in 11 children (7, 8). Respiratory inflammation associated with asthma leads to fibrosis and airway constriction, which presents as wheezing and shortness of breath. The epithelium serves as the interface between allergens and the underlying immune system and thus initiates inflammation by secreting cytokines and chemokines such as IL-5, regulated on activation, normal T cell expressed and secreted (RANTES or CCL5), IL-22, IL-25, thymic stromal lymphopoietin (TSLP), and IL-33 [reviewed in Ref. (9)]. Th2-polarized T cells are abundant in the draining lymph nodes of the lungs, circulating blood, and bronchoalveolar lavage (BAL) of patients with asthma- and allergen-challenged mice. They are central to orchestrating inflammation by secreting cytokines, such as IL-4, IL-5, IL-13, and granulocyte macrophage colony-stimulating factor (GM-CSF). The BAL of asthmatic patients and allergen-challenged mice contains abundant eosinophils and M2-polarized macrophages, which together secrete a variety of inflammation-promoting factors (10–12) [reviewed in Ref. (13)]. These factors include chemokines that recruit more lymphocytes into the airways, prostaglandins that promote bronchoconstriction, and matrix metalloproteinases (MMPs) that promote fibrotic tissue remodeling. Histologically, the airways are infiltrated by leukocytes, and the surrounding smooth muscle is thickened from enhanced collagen deposition. In hard-to-treat cases of steroid-resistant asthma, neutrophils infiltrate the BAL as a result of Th17 responses and IL-8 production.
Asthma incidence and severity exhibit sex differences, and there is mounting clinical and animal model evidence that implicates sex hormones, particularly estrogen, in being key mediators of these differences [reviewed in Ref. (14)]. In the following sections, we will summarize these sex differences in clinical asthma in humans and elaborate on the effects of cycling estrogen levels on asthma severity and incidence in women. Although other sex hormones such as androgens and progesterone influence the immune system and the action of estrogen in cells, we will focus this review on the studies of estrogen on the immune system. Our emphasis here will be the effect of estrogen on innate immune cells, with some discussion of the adaptive immune system. We will describe the estrogen receptors (ERs) and their regulation of gene expression. We will then review recent insights from animal models regarding the impact of estrogen signaling on allergic inflammation, the various cell types involved, and how this may account for the sex difference observed in asthma.
The onset and severity of asthma and allergies typically exhibit sex differences, although the results of clinical surveys differ somewhat depending on geography, likely highlighting environmental, cultural, and dietary factors. Virtually all atopic allergies affect boys more than girls in childhood. Numerous studies that have focused on questionnaire-based and clinician-diagnosed school children with atopic dermatitis have reported a male-dominant incidence that persists from birth until 6–11 years of age (15–17). Conversely, a Danish study reported that while the onset of atopic dermatitis was slightly delayed in girls (2.5 years of age) compared to that in boys (2 years of age), the overall prevalence did not differ by sex (18). The perceived prevalence of peanut allergy in Great Britain exhibits female predominance in general, although after age 15, that trend is reversed (19). A Norwegian study found girls exhibit a higher incidence of food allergies than boys after 18 months of age (20). Allergic rhinoconjunctivitis seems to preferentially affect males beyond the age of 13 (21). The onset of puberty, however, reverses that trend for most allergic disorders, including asthma (22). Women over the age of 25 account for >62% of hospitalizations due to acute asthma attacks and, in 2009, accounted for 64% of asthma-related deaths (23, 24). Roughly 40% of women experience premenstrual exacerbations of asthma and women are more likely to be non-responsive to corticosteroid therapy (25, 26).
Asthma symptoms in women seem to change with various life-stage factors such as menstruation, pregnancy, and menopause (27–30). The incidence of allergy and asthma peaks in women after puberty and strongly subsides with age (31). Roughly 33–52% of asthmatic women report premenstrual worsening of symptoms (32–35). Almost 50% of women hospitalized for asthma symptoms are identified as premenstrual (36). Furthermore, spikes in menstrual hormones correlate with impaired peak expiratory flow rate and thus reduced lung function (37). Lung function is generally worse during the late follicular phase, when estrogen levels are highest (38). Abrupt estrogen spikes are alleviated with oral contraceptive use, which was shown to stabilize asthma symptoms in women (39). Yet, the worsening of symptoms with oral contraceptive use has also been reported (40). Pregnancy causes a large increase in circulating estrogen that starts with ovum fertilization and falls to prepregnancy levels shortly after delivery. Approximately 70% of asthmatic women report alleviation of symptoms during pregnancy (28, 35, 41–43). Symptom severity seems to return to normal within 3 months postpartum. Interestingly, postpartum symptom severity worsens with increasing number of births (44). Estrogen levels drop during menopause and are lower in asthmatic women than in healthy women (45). Menopause typically results in exacerbated asthma symptoms and leads to the development of new asthma in previously unaffected women (46). Hormone replacement therapy (HRT) increases estrogen levels and alleviates symptoms in postmenopausal asthmatic women (45). However, HRT use has also been linked to a 19% increased risk of hospitalization per year of use in menopausal women with asthma (47). Furthermore, rates of new asthma diagnosis have been reported to increase after HRT initiation but diminish with cessation of HRT (48). Because asthma comprises several distinct endotypes, this likely contributes to the seemingly conflicting outcomes of oral contraceptive and HRT use in women.
These survey studies collectively suggest that estrogen levels correlate with symptom severity and likely contribute to sex differences in asthma. In general, women have a greater incidence and severity of asthma than do men (49–51). However, the alleviation of symptoms in women during stages of life that are accompanied by high levels of circulating estrogen suggests that the inflammatory function of estrogen may be dose-specific. This idea is echoed in in vitro and animal studies covered later in this review. To understand how estrogens impact the immune system, we will first give some background on ER biology in the next section.
Estrogen signaling regulates reproductive physiology and gene expression in many tissues and cell types. Not surprisingly, the failure to regulate estrogen signaling is associated with a variety of human diseases, including breast and endometrial cancer, cardiovascular disease, osteoporosis, and Alzheimer’s disease [reviewed in Ref. (52)]. Like all hormones, estrogen readily penetrates the cell membrane. In the cytosol, it encounters ERs, triggers their dimerization, and liberates them from an inactive complex with heat shock protein (HSP)90 (53). The ERs then translocate to the nucleus and engage estrogen response elements (EREs) on target-gene promoters (53). However, this signaling network exhibits several layers of regulatory complexity that result in pleiotropic effects on various tissues and cell types. This diversity in the biological functions of estrogen is achieved through the expression of several ER isoforms that have the capacity to interact with various transcriptional coactivators and corepressors as well as transcription factors to elicit an array of cellular responses. In fact, estrogen is known to elicit non-genomic effects on cells via membrane-bound receptors that crosstalk with an array of cellular signaling networks (54, 55). Furthermore, phosphorylation of the nuclear ERs can mimic ligand binding and thus induce ligand-independent responses (56–58). In the following section, we will discuss the current understanding of the estrogen signaling pathway.
The nuclear ERs exist in two main isoforms termed ERα and ERβ, which are part of a large superfamily of type I nuclear receptors. The nuclear receptor superfamily members exhibit a conserved structure consisting of regions A–F (Figure 1). ERα and ERβ both contain an N-terminal activation function-1 (AF-1) domain within regions A and B, a zinc-finger containing a DNA-binding domain in the centrally located region C, and a C-terminal AF-2 domain within regions E/F that facilitates dimerization, association with Hsp90, and ligand binding via the ligand-binding domain (59). Region D consists of a hinge between the N- and C-terminal halves of the receptor. In the absence of ligand, ERα and ERβ are bound to heat shock proteins that restrict their activity. Ligand binding, however, releases the receptors from this complex and allows their engagement of target gene promoters. Several serine and tyrosine residues on the receptors are also subject to phosphorylation that enhances receptor activity [reviewed in Ref. (60)]. Upon ligation, the DNA-binding activity of the AF-1 domain and the dimerization activity of the AF-2 domain facilitate transcriptional regulation through recruitment of >40 different coactivators, including histone acetyltransferases, ubiquitin ligases, arginine methyltransferases, and transcription factors.

Figure 1. Estrogen receptor isoforms. The domain organization of human estrogen receptor α and β isoforms are illustrated above. The size of the full-length, truncated, and elongated isoforms is indicated.
Estrogen receptor α consists of the full-length 66 kilodalton (kDa) isoform and alternatively spliced truncated 36 and 46 kDa isoforms that result from internal ATG transcription start codons. The 46 kDa isoform lacks the AF-1 domain, and 36 kDa isoform lacks both the AF-1 and AF-2 domains preventing efficient transcriptional activity but permitting heterodimerization with the full-length receptor (61, 62). Human macrophages primarily express the 46 kDa isoform of ERα and to a lesser extent the full-length 66 receptor and the 46 kDa isoform is estrogen inducible (63). Furthermore, the transition of monocytes along the monocyte-macrophage axis is accompanied by an upregulation of the 46 kDa ERα (63). Both isoforms can localize to the membrane. The 5′ flanking region of the 36 kDa isoform contains several putative transcription factor-binding sites, including NF-κB, glucocorticoid receptor (GR), specificity protein (SP)1, and activator protein 1, but is suppressed by full-length 66 kDa ERα (64). ERα-36 is the only ERα isoform expressed in human peripheral blood monocytes and suppresses proinflammatory cytokine production in response to LPS (65). Other truncated isoforms have been identified in a variety of human cancers and rat cells although their expression in healthy human tissue is poorly understood (66, 67). ERβ consists of at least five different truncated isoforms that lack either the AF-1 or AF-2 domain.
The conventional linear pathway of nuclear receptor signaling whereby ligated hormone receptors regulate gene expression at well-defined promoter elements has been revised in recent years. When bound to ligand, ERα and ERβ form either homo- or heterodimers and engage EREs on target gene promoters [reviewed in Ref. (68)]. Intriguingly, although EREs can be functional even as far as 2 kilobases (kb) from their target promoter and exhibit activity either upstream or downstream from the target gene, approximately one-third of estrogen-regulated genes in humans lack EREs altogether (69). Rather, association of ERs with SP-1 mediates indirect ER–DNA binding and utilizes the coactivator-recruiting activity of ERs to drive gene expression independently of EREs. This form of ERE-independent gene induction has been identified for low-density lipoprotein receptor, endothelial nitric oxide synthase (eNOS), c-fos, cyclin D1, and the retinoic acid receptor 1α (70–74). Other transcription factors, such as NF-κB, c-jun, and cyclic adenosine monophosphate (cAMP) response element-binding protein have also been shown to recruit ERs for ERE-independent sites (75). Interestingly, human ERα and ERβ seem to have opposing effects on these promoters, with ERα-enhancing and ERβ-repressing gene expression. This phenomenon may reflect a defective AF-1 domain in human ERβ (76).
The canonical steroid receptor coactivator (SRC) family associated with ERs is termed the SRC/p160 family. The members of this family are referred to by various names in the literature but consist of SRC-1 (also termed NCoA-1), SRC-2 (also termed TIF2, GRIP1, or NCoA-2), and SRC-3 (also termed RAC3, pCIP, ACTR, AIB1, TRAM-1, or NCoA-3). These coactivators contain a nuclear receptor box that consists of an LXXLL motif that fits within the hydrophobic groove formed by the 12 α-helices of the AF-2 domain (77). Unligated or tamoxifen-bound ERs can also interact with nuclear receptor corepressors N-CoR and SMRT, which inhibit the recruitment of SRC/p160 and thus repress transcription at ERE sites (78).
Estrogen receptor α and ERβ contain conserved serine residues that are phosphorylated in response to ligand binding as well as independently of ligand binding. Serines-104, -106, and -118 have been shown to be phosphorylated by cyclin-dependent kinases after estrogen binding (79). These sites reside within the AF-1 domain and enhance ER activity. In addition, ligand-independent activation of the ERs by growth factors and cyclic AMP has been described. In fact, Erk1/2 can directly phosphorylate serines-104 and -118 within the AF-1 domain of ERα and induce dimerization and activation in response to either epidermal growth factor or estrogen signaling (60, 80, 81). Ribosomal S6 kinase and protein kinase B (Akt) have been shown to phosphorylate serine-167 in response to growth factor signaling (82, 83). Protein kinase A activation and cAMP treatment of cells lead to phosphorylation of serine-236, which resides within the DNA-binding domain (84, 85). A tyrosine residue (Tyr-537) within the AF-2 domain is also phosphorylated in ERs and serves as a docking site for the Src homology-2 domain of c-Src (86). The general outcome of these phosphorylation events is enhanced receptor activity. Several groups have compared mutants with alanine substitutions at these serine sites to wild-type receptors using ERE reporter assays and found that the inability to phosphorylate these sites translates to impaired target gene expression (56, 80, 87, 88).
Estrogen receptors not only regulate reproductive and developmental physiology but also participate in hormone-independent signaling by interacting with growth factor and other cell-signaling networks. As described earlier, ER signaling is regulated by several factors, including the relative abundance of receptor isoforms, interactions with coactivators/corepressors, and DNA-remodeling proteins, receptor phosphorylation, and inputs from other signaling networks. This variety in regulation permits profound functional versatility and provides an avenue for hormonal influence over many biological pathways and processes, like allergic inflammation. More research is required to ultimately understand how ER signaling interacts with non-hormonal pathways. With respect to asthma, there is an imperative for greater understanding of the pathophysiology of various endotypes that comprise the disease so that the potential contribution of ER signaling and gene induction can be clarified.
In the last few decades, our understanding of the profound effect of sex hormones on leukocyte biology has expanded dramatically [reviewed in Ref. (89)]. Castration and hormone replacement studies in mice have been instrumental in connecting anecdotal evidence of sex differences in humans to the underlying cellular mechanisms. Cell-specific knockout mice have provided the essential tools to isolate the role of individual ER isoforms and dissect their involvement in the inflammatory process. Studies in selective ER knockout animals have revealed a complex interplay between receptor isoforms, sex hormone levels, cell types, and biological outcomes. The following section will summarize the current understanding of estrogen signaling in cells of the innate immune system that are relevant to the pathogenesis of allergic inflammation in the lung during asthma.
In recent years, the role of macrophages in allergic inflammation has garnered increased attention. In asthma, alveolar macrophages have emerged as important cellular mediators of inflammation and tissue remodeling [reviewed in Ref. (90–92)]. Allergic asthma is considered to be a Th2 disease and is thus accompanied by IL-4/13-polarized M2 alveolar macrophages in the airways. Intratracheal instillation of alternatively activated M2 macrophages prior to ovalbumin (OVA) challenge results in enhanced eosinophil and effector T cell infiltration into the BAL in mice (93). Macrophage depletion studies have revealed a dual role for alveolar macrophages in eliciting and containing allergic lung inflammation in mice (94). Resident alveolar macrophages are inherently immunosuppressive and secrete anti-inflammatory factors, such as transforming growth factor β (TGFβ) and IL-10 (95, 96). In the allergic setting, however, the bronchoalveolar space is infiltrated by inflammatory M2-polarized macrophages that facilitate inflammation by secreting chemokines, such as chemokine c–c motif ligand 2 (CCL2), CCL17, Ym1, and found in inflammatory zone 1 (Fizz1 or Relmα) (97). Depletion of these macrophages in mice results in impaired IL-5 production, eosinophil influx, and airway remodeling (97). In the house, dust mite model of mouse allergic lung inflammation, female mice have more M2-like (Ym1+) cells, whereas males have more M1-like (interferon regulatory factor 5) cells in histological lung cross-sections (98). Likewise, female mice exhibit more eosinophils, effector T cells, and M2-polarized macrophages in the lungs than do males in the OVA model of lung inflammation (93). Furthermore, the production of lipid inflammatory mediators like leukotrienes via 5-lipoxygenase is enhanced in female leukocytes compared to that in male leukocytes, but this difference is due to suppression of the pathway by testosterone (99). This central role of macrophages in allergic lung inflammation has warranted studies addressing the potential influence of sex hormones on macrophage polarization and inflammatory cytokine production.
Bone marrow-derived macrophages (BMMs) have served as a proxy in vitro model for studies addressing the effect of estrogen on macrophage biology. Several groups have measured macrophage cytokine production in response to interferon γ (IFNγ) or IL-4 in the context of either exogenous estrogen treatment or macrophages from ER knockout mice (Figure 2). IL-4-induced M2 polarization is enhanced with the addition of exogenous estrogen at micromolar doses in mouse BMMs (100). Estrogen not only increases arginase 1 (Arg1) activity and Fizz1 expression in IL-4-polarized BMMs but also suppresses inducible NOS (iNOS) expression in IFNγ/lipopolysaccharide (LPS)-treated BMMs. Furthermore, these effects were determined to be mediated through ERα signaling, as they were mimicked with propyl pyrazole triol (PPT), a selective ERα agonist, and not diarylpropionitrile (DPN), a selective ERβ agonist. Likewise, BMMs from LysM-CRE ERαflox/flox mice fail to upregulate Arg1 and Fizz1 in response to IL-4 and exhibit enhanced iNOS responses to IFNγ/LPS. Physiological doses of estrogen (picomolar to nanomolar range) suppress LPS-induced tumor necrosis factor α (TNFα) expression in mouse BMMs (101). BMMs from ovariectomized (OVx) mice exhibit decreased TGFβ1 mRNA, but exogenous treatment with estrogen rescues this phenotype, suggesting that estrogen may contribute to tissue remodeling in allergic inflammation through TGFβ1 induction (102).
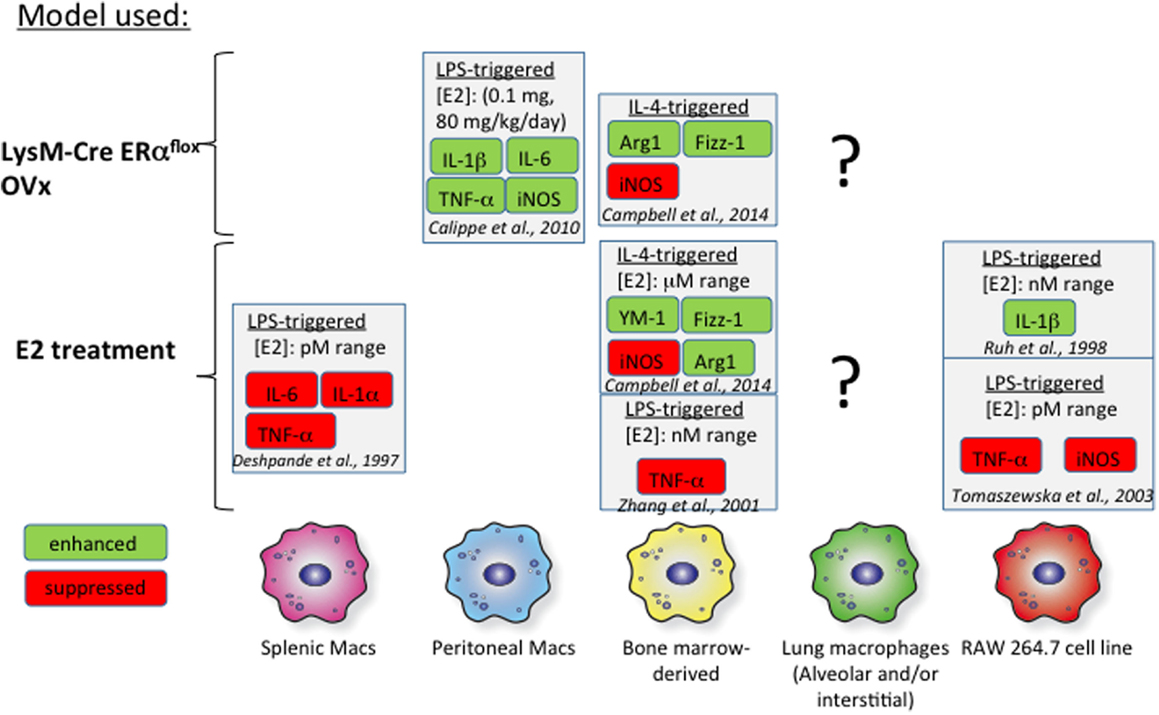
Figure 2. Effects of estrogen in vitro on macrophages. The effects of estrogen on macrophages from various tissues are illustrated above. The findings of several studies are summarized and referenced in the figure. The effects of estrogen on proinflammatory mediators are highly dependent on the tissue of origin. There is a general lack of data focusing on the impact of estrogen signaling on alveolar macrophages, indicated by the question marks.
Studies addressing estrogen signaling in splenic, wound-derived, and peritoneal macrophages have revealed heterogeneity in responses between macrophages obtained from different sources (Figure 2). Signaling through ERα with 1 μM PPT in murine wound-derived macrophages enhances M1 (iNOS and TNFα) and M2 (Arg1 and Ym1) responses following polarization with IFNγ/LPS and IL-4, respectively (100). Conversely, signaling through ERβ with 1 μM DPN has the opposite effect on M1 and M2 wound-derived macrophages. Thioglycollate elicitation of peritoneal macrophages is impaired in OVx mice replenished with estrogen (103). However, these thioglycollate-elicited macrophages in estrogen-replenished OVx mice are proinflammatory in nature and express more IL-1β, IL-6, iNOS, and TNFα than do cells from OVx mice. These effects of estrogen were determined to arise from ERα-mediated inhibition of phosphoinositide 3-kinase. Splenic macrophages treated with lower doses of estrogen (picomolar range) exhibit impaired LPS-induced TNFα production compared to that in untreated cells. These studies highlight a lack of uniformity in study design but also suggest that macrophages exhibit heterogeneity with respect to estrogen signaling. Furthermore, macrophage subsets found from different tissue sources exhibit unique phenotypes, complicating our ability to relate studies that use mouse BMMs or peritoneal and splenic macrophages to alveolar macrophage biology (104, 105). Thus, more in vivo studies are needed that examine the effect of estrogen on alveolar macrophages in the setting of allergic inflammation. A stable mouse or human alveolar macrophage cell line from would also provide a more appropriate in vitro model for understanding lung inflammation.
Dendritic cells (DCs) are essential initiators of the adaptive immune responses that facilitate sensitization to allergens. Estrogen is intricately involved in DC function from differentiation to maturation and proinflammatory cytokine production. GM-CSF-induced DC differentiation from mouse bone marrow precursors is dramatically impaired when conducted using hormone-free serum in the culture medium (106). Adding estrogen (picomolar range) into the culture medium of these cells restores DC differentiation and responsiveness to LPS, as well as their capacity for T cell priming. Bone marrow from ERα−/− mice is defective in generating DCs with GM-CSF, although the addition of 10 nm estrogen partially resolved that, suggesting that other ERs are also important in DC differentiation (106). In fact, GM-CSF-generated bone marrow-derived CD11b+CD11c+ DCs express both ERα and ERβ (107). A CD11c+CD11bint subset that emerges from 100 pM estrogen-enhanced GM-CSF-generated bone marrow-derived DCs expresses langerin and exhibits other features of Langerhans’ cells (107). Thus, estrogen has an enhancing effect on DC differentiation from myeloid progenitors.
The effects of estrogen on DC maturation and cytokine production have been addressed in several in vitro and in vivo models. Upon encountering pathogen-associated molecular patterns (PAMPs), DCs mature by downregulating endocytosis and upregulating the antigen presentation apparatus along with cytokine production. Human plasmacytoid DCs (pDCs) exhibit profound sex differences when stimulated with the toll-like receptors 7/8 ligand R-848 with pDCs obtained from premenopausal women being more responsive by expressing IFNα and TNFα compared to age-matched males (108). Estrogen therapy in menopausal women dramatically increased the cytokine responsiveness of blood pDCs to TLR ligation, and this was recapitulated in an OVx mouse model (108).
Plasmacytoid DCs are known to be potent Th2-priming antigen-presenting cells, and they are more abundant in the blood of patients with asthma compared to healthy individuals (109, 110). IL-4-mediated depletion of pDCs is impaired in patients with asthma compared to healthy controls, despite no measurable difference in IL-4Rα expression (109). Conversely, pDCs also drive tolerance to allergens in OVA-challenged mice, and their depletion exacerbates eosinophil influx and tissue remodeling in the lung (111). Further, the adoptive transfer of pDCs back into depleted mice rescues this exacerbation. The adoptive transfer of pDCs cultured with house-dust mite allergen into mice results in an expansion of FoxP3+ Tregs as well as IL-10 production following allergen challenge (112). Human monocyte-derived DCs treated with ~70 μM estrogen exhibit induction of MCP-1, IL-8, and IL-6 but an unaltered immature phenotype with respect to major histocompatibility complex II (MHCII) and costimulatory molecule expression (113). Splenic DCs, however, respond to estrogen (10 nM – 1 μM) by upregulating CD40 and MHCII (114). At high doses (in the micromolar range), estrogen downregulates endocytosis in splenic DCs and promotes survival as well as IL-6 and IL-10 productions. Likewise, estrogen enhances T-cell proliferation in culture with LPS-activated DCs, suggesting that it promotes the capacity of mature DCs to stimulate T-cell responses. Further, estrogen treatment (1 nM) of mouse bone marrow enhances GM-CSF-induced DC differentiation and LPS/CpG-induced IL-12 production (115). In vivo experiments that focus on the effects of estrogen on DC-mediated T-cell priming are lacking. However, one group found that administration of estrogen to OVx mice enhanced Th1 responses to a subcutaneous OVA challenge and that the response required ERα expression in the hematopoietic compartment (116). The adoptive transfer of estrogen-treated DCs (~73 nM) was shown to alleviate inflammation in experimental allergic encephalomyelitis (EAE) (117). In that model, estrogen administration in Lewis rats impaired the capacity of DCs to prime Th1 and Th2 T-cell response.
Like macrophages, these studies suggest that estrogen has differing effects on DCs, depending on their tissue of origin as well as the species being studied. Bone marrow- and monocyte-derived DCs exhibit enhanced inflammatory cytokine production and toll-like receptor ligand responsiveness with estrogen treatment. In splenic DCs, estrogen promotes the capacity for T-cell priming. In the EAE model, however, DC function seems to be less inflammatory with estrogen treatment (118). More studies are needed to discern the various dose-dependent effects of estrogen on DC subsets from different tissues. Studies in asthma are particularly important because lung DCs have a major role in priming T cells against allergens (119, 120).
Eosinophils are a hallmark cell type of allergic inflammation. These cells are recruited from the blood by IL-5, which is produced in response to epithelial TSLP, IL-25, and IL-33. Eosinophils are recruited as mature cells and elicit antihelminth immunity and inadvertent tissue damage by degranulation. Eosinophil granules contain several different effector molecules. Major basic protein, eosinophil peroxidase, eosinophil cationic protein, and eosinophil-derived neurotoxin are among the granular content of eosinophils. They have both distinct and overlapping effects on target cells. In general, these granular proteins exert toxic effects on target cells by a variety of mechanisms, including pore formation, ribonuclease activity, and the production of reactive oxygen species. Eosinophils are also significant sources of cytokines, such as IL-4 and IL-13, and thus promote allergic inflammation once these cytokines are released.
Estrogen has been shown to regulate eosinophil migration, adhesion, survival, and degranulation. Its influence on eosinophil biology became clear as early as the 1960s, when it was first reported to induce eosinophilic infiltration into the rat uterus as a hallmark of uterine development and tissue remodeling (121–123). Additionally, the number of eosinophils in the uterus was found to follow the estrous cycle, peaking at the estrus phase and dipping at the diestrus phase (124). Uterine eosinophils are virtually absent from OVx animals and late in pregnancy.
Eosinophils are a key cell type involved in the pathogenesis of asthma and allergy and are known to express ERα and membrane-bound G-protein-coupled receptor (GPR-1) in mice and humans (125, 126). The sex differences associated with allergic inflammation, and in particular asthma, are most apparent with the varying degrees of eosinophilia in asthma models in male and female mice. Female mice exhibit greater eosinophilic infiltration into the bronchoalveolar and parenchymal spaces than do male mice, coincident with increased allergen-specific IgE, increased cluster of differentiation 4 (CD4)+ T- and B-cell accumulation, and impaired lung function (127, 128). Treatment of allergen-challenged mice with ERα antagonists, such as tamoxifen and ICI 182,780, dramatically reduces BAL eosinophil numbers and Th2 cytokines, such as IL-5 and IL-13 (125, 129). Furthermore, exogenous estrogen dose dependently induces both IL-5 and IL-13 in mediastinal lymph node cultures from allergic animals. Intriguingly, ovariectomy prior to OVA sensitization impairs eosinophil influx, which is restored with exogenous estrogen (129). However, ovariectomy of mice after sensitization to OVA exhibit enhanced eosinophilia, suggesting that estrogen may on the one hand promote T-cell priming against allergens but on the other control eosinophil infiltration after challenge. Conversely, Dimitropoulou et al. (130) reported a dose-dependent reduction in eosinophil infiltration into the BAL of estrogen-treated, OVA-challenged, OVx mice. These discrepancies may reflect differences in experimental approaches and doses of estrogen replacement used. Estrogen administration triggers eosinophil degranulation in rats in vitro and in vivo (131). Although in a model of acute thioglycollate-induced sterile peritonitis, estrogen plays an inhibitory role in eosinophil influx and survival in mice (132). The administration of estrogen in OVx mice reduces peritoneal eosinophils and IL-5 production in this model, and these are restored in ERα gene knockout mice. Furthermore, administration of the GPR-1 agonist, G-1, to C57BL/6 and BALB/c mice subjected to an OVA-induced allergic inflammation model reduces lung eosinophilia, IL-5, and eotaxin production through induction of IL-10 (133). However, human eosinophils exhibit enhanced eotaxin-induced chemotaxis and survival with G-1 treatment (113). Thus, estrogen impacts eosinophils in different ways depending on the receptors engaged, the species used, the tissue in which the signaling is taking place, and whether the cells are analyzed in vivo or in vitro. Some of the known effects of estrogen on eosinophils that would increase allergic inflammation are summarized in Figure 3.
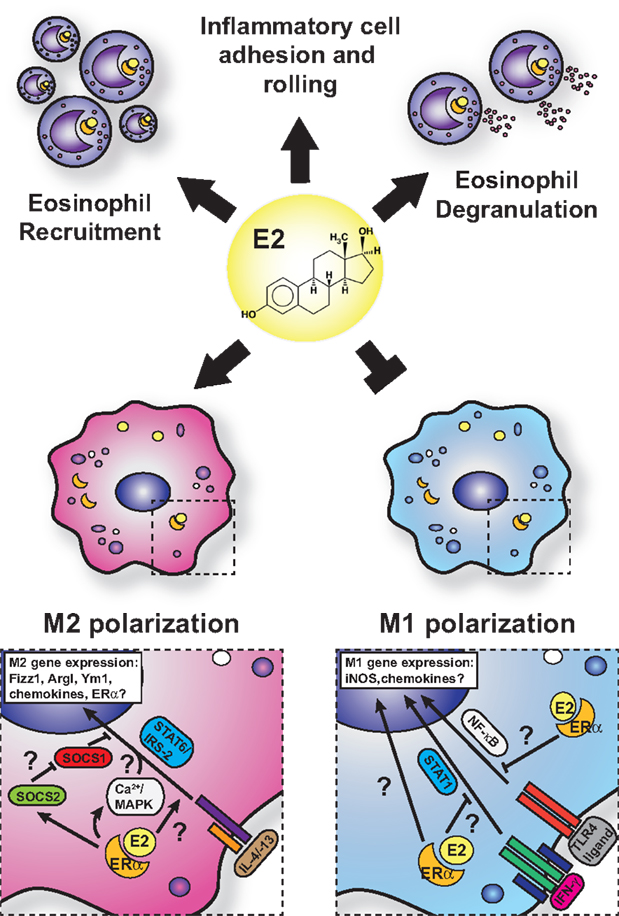
Figure 3. Current model for estrogen signaling in polarization in macrophages in the setting of allergic lung inflammation. The general consensus is that estrogen enhances M2 polarization while suppressing M1-polarization in macrophage. However, mechanistic insights are lacking, and questions remain regarding the interface of ERα and IL-4 signaling.
Mast cells are detectable in the sputum of asthmatic patients, but sex differences with respect to mast cell function have not been adequately explored (134). Nevertheless, their numbers increase and decrease in female reproductive tissues with the estrous cycle, suggesting that estrogen regulates mast cell migration and biology. Mast cells from rat and human cell lines as well as mouse bone marrow-derived mast cells express ERα but not ERβ (135). In rat mammary glands, mast cells accumulate during the estrus and diestrus phases of the estrous cycle (136). After ovariectomy, mast cells decrease dramatically, but their numbers are restored with estrogen replacement. Estrogen induces IgE-dependent and -independent mast cell degranulation in rats (135, 137). These effects are dependent on Ca2+ and thus are non-genomic although the expression of GPR-1 was not evaluated in this study. Mouse models of lung allergen challenge clearly suggest that mast cells orchestrate inflammation, but they do not accumulate in BAL (138). To our knowledge, no studies have investigated the effect of sex on the contribution of mast cells to allergic inflammation in mice.
The airway epithelium provides front-line defense against invading pathogens and environmental insults in the lung by balancing gas exchange with innate immune function. Airway epithelial cells express pattern recognition receptors that enable them to detect and respond to PAMP and danger-associated molecular pattern by secreting cytokines and chemokines to drive inflammation. In asthma, the airway epithelium recruits innate and adaptive immune cells via cytokines, such as IL-25 and IL-33, and chemokines, such as CCL2, CCL17, and CCL20. Furthermore, these cells secrete TGFβ, which promotes tissue remodeling. Human bronchial epithelial cells express both ERα and ERβ and are responsive to estrogen exposure. Treatment of these cells with 10 nM estrogen induces endothelial NOS expression and nitric oxide production that results in bronchodilation (139). Similarly, the same dose of estrogen relaxes human airway smooth muscle and thereby induces bronchodilation (140). Estrogen replacement in OVx mice significantly reduces acetylcholine responsiveness (141). These studies suggest that estrogen is protective against airway constriction and thus may be therapeutic in asthma. However, estrogen treatment of human airway epithelium also induces MUC5B secretion, which may contribute to airway lumen obstruction (142).
Currently, very little is known regarding the effect of estrogen on proinflammatory cytokine production by airway epithelial cells. In patients with cystic fibrosis, estrogen treatment reduces epithelial IL-8 production, which is thought to predispose women to Pseudomonas infection (143). Thus, estrogen may be a double-edged sword with respect to its bronchodilation and inflammatory properties. The outcome on lung function is likely the net result of a balance between increased allergic inflammation and smooth muscle relaxation/increased bronchodilation.
Estrogen signaling has profound effects on adaptive immunity. Consequently, women are generally at greater risk than men for pathology associated with immune responses to infection, autoimmune diseases, and allergies. The underlying cellular basis for these sex differences is currently being evaluated. Estrogen signaling is known to be important for thymic development and antibody production, although pregnancy levels of estrogen induce thymic attrition and suppress proinflammatory responses in mice and humans (144, 145). Thus, physiological estrogen levels can be functionally grouped into several categories that reflect various developmental stages of life. Concentrations of circulating estrogen vary between 40–152 pM and 1–150 nM in menopausal and pregnant women, respectively [reviewed in Ref. (14)]. Research approaches, including ER knockout, overiectomy of animals followed by estrogen replacement, and comparisons of estrogen responses in healthy and diseased lymphocytes, have provided important insights into the cellular basis for sex differences. This section will briefly summarize our current understanding of how estrogen signaling affects T and B lymphocytes and the consequences that may contribute to sex differences in health and disease.
The effects of estrogen on T cells are complex and context and dose specific. As discussed later, estrogen has the capacity to enhance Th1, Th2, and Treg responses. At concentrations found during pregnancy (1–150 nM), estrogen promotes IFNγ production by CD4+ T cells. At those same concentrations, estrogen enhances ex vivo production of IL-10, IFNγ, and TNFα by proteolipid-specific CD4+ T cells from patients with multiple sclerosis compared to that by cells from healthy patients and cells exposed to proteolipid alone (146). Interestingly, higher doses of estrogen seem to inhibit TNFα production but have no effect on IL-4 production. Similarly, replacement of estrogen at pregnancy-level concentrations in OVx mice suppresses T cell-mediated type IV hypersensitivity reaction (147, 148).
Non-obese diabetic (NOD) mice exhibit sex differences in the cytokine profile of islet-infiltrating CD4+ T cells, as females produce more IFNγ and less IL-4 than do males (149). Estrogen replacement in OVx NOD mice, at levels present during pregnancy, enhances this IFNγ production in islet infiltrates. Lambert et al. (150) reported similar enhancement of IFNγ production by ConA-activated splenic CD4+ T cells from C57BL/6 mice stimulated with pregnancy-level concentrations of estrogen. Likewise, subcutaneous OVA sensitization in OVx C57BL/6 mice leads to an estrogen-dependent increase in IFNγ+ CD4+ T cells within the draining lymph nodes, an effect that is abrogated in ERα knockout mice (116). Conversely, estrogen at pregnancy concentrations increased the expression of IL-4 and GATA-3 in a dose-dependent manner by CD3/CD28-cultured splenic T cells from C57BL/6 mice but not in those from ERα knockout mice (151). Concentrations of estrogen measured during pregnancy also promote FoxP3 and CD25 expression on mouse T cells in vivo and in vitro in an ERα-dependent manner (152). Furthermore, ERα expression in T cells is required for estrogen-mediated protection in the mouse model of EAE via the reduction of IFNγ and IL-17 expression by CD4+ T cells (153). Indeed, patients with multiple sclerosis show improvement during pregnancy and this is likely due to elevated estrogen (154). Collectively, these findings suggest that concentrations of estrogen found during pregnancy enhance IFNγ, IL-10, and IL-4 production by CD4+ T cells and thus may protect the fetus against Th17-mediated immunopathology.
In general, estrogen enhances B-cell maturation and antibody production. Cervical mucus IgA levels peak in cycling women prior to ovulation and are increased by oral contraceptives (155). Human blood mononuclear cells cultured with sheep red blood cells (SRBC) likewise produce more anti-SRBC antibodies in response to increasing doses of estrogen, an effect that is ablated with tamoxifen (156). Interestingly, in this model, estrogen was found to interfere with CD8+ T-cell suppression of B-cell maturation. Indeed, estrogen enhances IgG and IgM production in humans, and neutralization of IL-10 impairs this response (157, 158). B cells from patients with systemic lupus erythematosus produce increased anti-dsDNA antibodies in response to estrogen also because of IL-10 production (159). Additionally, high concentrations of estrogen (micromolar range) increase the amount of antibody produced by pokeweed mitogen-stimulated mouse peripheral blood mononuclear cells (145). Importantly, estrogen and estrogenic compounds enhance IgE production by mouse splenocytes and thereby may contribute to allergic inflammation (160).
To date, studies addressing the role of estrogen and other sex hormones in allergy and immunity have been confronted with major experimental limitations, resulting in a lack of consensus over confounding data. In vitro experiments utilizing exogenous treatment of cells with agonists or antagonists to ERs are a good starting point but hardly address how these cells behave in the context of the inflamed lung in vivo. Furthermore, there must be a uniformity regarding the dose, duration, and delivery of estrogen treatment in either in vitro models or estrogen replacement in vivo models. Exceeding normal physiological doses (into the micromolar range) significantly contributes to confounding reports as estrogen exhibits dose-specific properties and has the potential to affect an ever-growing spectrum of cellular pathways. There is also an inherent difficulty in gaining mechanistic insights from either estrogen replacement in OVx mice or from global ER knockout mice since there are systemic consequences to these animal models and allergic responses are complex multicellular processes. Thus, the development of more sophisticated mouse models is imperative to address the impact of estrogen on the various phases of allergic inflammation and on the key specific cell types in a controlled manner. For this purpose, the employment of inducible Cre recombinase under cell-specific promoters to target floxed ER alleles for knockout would be very helpful.
Many questions remain to be addressed regarding the role of estrogen in allergic inflammation. As indicated in Figure 2, the impact of estrogen on alveolar macrophages has been overlooked. Since these cells are important modulators of allergic lung inflammation, more attention should be focused on how estrogen affects their biology in the setting of allergic lung inflammation. The estrogen-mediated effects of changes in macrophage function, tissue remodeling and fibrosis, as well as eosinophil and monocyte recruitment are yet to be explored. Furthermore, the potential recruitment of inflammatory monocytes and the impact of estrogen on their trafficking and polarization have yet to be addressed. The respective contribution of ERα, ERβ, and GPR-1 to the function of innate and adaptive immunities in allergic inflammation needs to be addressed using a targeted and inducible knockout in vivo. Furthermore, the potential role of ERs in steroid resistance has major therapeutic promise. Whether ERs interfere with GR signaling in hard-to-treat asthma that mostly affects women is unknown. Alveolar macrophages may play a role in this pathology, since alveolar macrophages from patients with asthma exhibit relative resistance to dexamethasone compared to healthy individuals (161–164).
Allergic disorders exhibit considerable sex-specific differences in prevalence, severity, age of onset, and clinical outcomes. These differences likely reflect the effects of sex hormones on the immune system, as well as other lifestyle factors. Adult asthma is clinically dominated by women, and the contribution of estrogen to this sex difference is becoming clear. However, the apparent lack of uniformity and consistency between experimental approaches used to address this issue has led to numerous confounding and conflicting results. Furthermore, asthma is a heterogeneous disease, and a one-size-fits-all approach to therapy is no longer considered reasonable. Therefore, it is imperative that the several different endotypes that comprise asthma be fully characterized and suited with appropriate rodent and in vitro models that will enable us to address potential therapeutic avenues. The Th2 paradigm used in animal models needs to be reconsidered, as it addresses only 50% of clinical asthma. To date, the most common treatments for asthma are inhaled glucocorticoids for suppression of inflammatory gene expression and β2-adrenergic receptor agonists for inhibition of bronchoconstriction. Women are exceptionally burdened with hard-to-treat, corticosteroid-resistant, neutrophilic asthma (165–167). The potential interactions between estrogen and glucocorticoid receptors have not been addressed in corticosteroid-resistant asthma despite their sharing coactivator binding partners. Furthermore, the persistent use of glucocorticoids results in significant side effects, such as cataracts and osteoporosis among others (168, 169). Neutralizing antibodies to IL-4Rα and IL-13, such as dupilumab and lebrikizumab, respectively, do show promise in clinical effectiveness but only in select patients with typical Th2-type asthma (170, 171). There is now an apparent shift in thinking that focuses on matching particular treatments to patients presenting with the appropriate biomarkers. This approach, however, requires extensive research with appropriate models. Estrogen signaling affects virtually every cell of the immune system. It is thus an appealing avenue for therapy but must certainly be carried out in a cell-specific and targeted manner that does not interfere with reproductive physiology. To achieve this goal, we must revise animal models to address the role of estrogen in allergic inflammation in a more reductionist manner. Cell-specific ER knockout models are essential for understanding the effect of estrogen signaling on a given cell type without dramatically altering the biology of the animal. The development of promoter-specific inducible CRE models permits such approaches. Such models, coupled with in vitro biochemical studies, may help to identify specific interactions between an ER isoform and a coactivator that facilitate enhanced allergic inflammation in women and can thus be targeted therapeutically.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Aleksander Keselman and Nicola M. Heller are supported by NIH/NHLBI RO1 HL124477.
1. Dharmage SC, Lowe AJ, Matheson MC, Burgess JA, Allen KJ, Abramson MJ. Atopic dermatitis and the atopic march revisited. Allergy (2014) 69:17–27. doi:10.1111/all.12268
2. Bantz SK, Zhu Z, Zheng T. The atopic march: progression from atopic dermatitis to allergic rhinitis and asthma. J Clin Cell Immunol (2014) 5:202.
3. Blanchard C, Mishra A, Saito-Akei H, Monk P, Anderson I, Rothenberg ME. Inhibition of human interleukin-13-induced respiratory and oesophageal inflammation by anti-human-interleukin-13 antibody (CAT-354). Clin Exp Allergy (2005) 35:1096–103. doi:10.1111/j.1365-2222.2005.02299.x
4. Leckie MJ, ten Brinke A, Khan J, Diamant Z, O’Connor BJ, Walls CM, et al. Effects of an interleukin-5 blocking monoclonal antibody on eosinophils, airway hyper-responsiveness, and the late asthmatic response. Lancet (2000) 356:2144–8. doi:10.1016/S0140-6736(00)03496-6
5. Flood-Page P, Swenson C, Faiferman I, Matthews J, Williams M, Brannick L, et al. A study to evaluate safety and efficacy of mepolizumab in patients with moderate persistent asthma. Am J Respir Crit Care Med (2007) 176:1062–71. doi:10.1164/rccm.200701-085OC
6. Haldar P, Brightling CE, Hargadon B, Gupta S, Monteiro W, Sousa A, et al. Mepolizumab and exacerbations of refractory eosinophilic asthma. N Engl J Med (2009) 360:973–84. doi:10.1056/NEJMoa0808991
7. Masoli M, Fabian D, Holt S, Beasley R. The global burden of asthma: executive summary of the GINA Dissemination Committee report. Allergy (2004) 59:469–78. doi:10.1111/j.1398-9995.2004.00526.x
8. Akinbami LJ, Moorman JE, Bailey C, Zahran HS, King M, Johnson CA, et al. Trends in asthma prevalence, health care use, and mortality in the United States, 2001-2010. NCHS Data Brief (2012) 94:1–8.
9. Gandhi VD, Vliagoftis H. Airway epithelium interactions with aeroallergens: role of secreted cytokines and chemokines in innate immunity. Front Immunol (2015) 6:147. doi:10.3389/fimmu.2015.00147
10. Bousquet J, Chanez P, Lacoste JY, Barneon G, Ghavanian N, Enander I, et al. Eosinophilic inflammation in asthma. N Engl J Med (1990) 323:1033–9. doi:10.1056/NEJM199010113231505
11. Robbe P, Draijer C, Borg TR, Luinge M, Timens W I, Wouters M, et al. Distinct macrophage phenotypes in allergic and nonallergic lung inflammation. Am J Physiol Lung Cell Mol Physiol (2015) 308:L358–67. doi:10.1152/ajplung.00341.2014
12. Rossi AG, Haslett C, Hirani N, Greening AP, Rahman I, Metz CN, et al. Human circulating eosinophils secrete macrophage migration inhibitory factor (MIF). Potential role in asthma. J Clin Invest (1998) 101:2869–74. doi:10.1172/JCI1524
13. Liu YC, Zou XB, Chai YF, Yao YM. Macrophage polarization in inflammatory diseases. Int J Biol Sci (2014) 10:520–9. doi:10.7150/ijbs.8879
14. Townsend EA, Miller VM, Prakash YS. Sex differences and sex steroids in lung health and disease. Endocr Rev (2012) 33:1–47. doi:10.1210/er.2010-0031
15. Smidesang I, Saunes M, Storro O, Oien T, Holmen TL, Johnsen R, et al. Atopic dermatitis among 2-year olds; high prevalence, but predominantly mild disease – the PACT study, Norway. Pediatr Dermatol (2008) 25:13–8. doi:10.1111/j.1525-1470.2007.00574.x
16. Peroni DG, Piacentini GL, Bodini A, Boner AL. Preschool asthma in Italy: prevalence, risk factors and health resource utilization. Respir Med (2009) 103:104–8. doi:10.1016/j.rmed.2008.07.016
17. Dirven-Meijer PC, Glazenburg EJ, Mulder PG, Oranje AP. Prevalence of atopic dermatitis in children younger than 4 years in a demarcated area in central Netherlands: the West Veluwe Study Group. Br J Dermatol (2008) 158:846–7. doi:10.1111/j.1365-2133.2007.08407.x
18. Halkjaer LB, Loland L, Buchvald FF, Agner T, Skov L, Strand M, et al. Development of atopic dermatitis during the first 3 years of life: the Copenhagen prospective study on asthma in childhood cohort study in high-risk children. Arch Dermatol (2006) 142:561–6. doi:10.1001/archderm.142.5.561
19. Emmett SE, Angus FJ, Fry JS, Lee PN. Perceived prevalence of peanut allergy in Great Britain and its association with other atopic conditions and with peanut allergy in other household members. Allergy (1999) 54:380–5. doi:10.1034/j.1398-9995.1999.00768.x
20. Lovik M, Namork E, Fæste C, Egaas E. The Norwegian National reporting system and register of severe allergic reactions to food. Nor J Epidemiol (2004) 14:155–60.
21. Shamssain M. Trends in the prevalence and severity of asthma, rhinitis and atopic eczema in 6- to 7- and 13- to 14-yr-old children from the north-east of England. Pediatr Allergy Immunol (2007) 18:149–53. doi:10.1111/j.1399-3038.2006.00498.x
22. Govaere E, Van Gysel D, Verhamme KM, Doli E, De Baets F. The association of allergic symptoms with sensitization to inhalant allergens in childhood. Pediatr Allergy Immunol (2009) 20:448–57. doi:10.1111/j.1399-3038.2008.00805.x
23. Baibergenova A, Thabane L, Akhtar-Danesh N, Levine M, Gafni A, Leeb K. Sex differences in hospital admissions from emergency departments in asthmatic adults: a population-based study. Ann Allergy Asthma Immunol (2006) 96:666–72. doi:10.1016/S1081-1206(10)61063-0
24. Centers for Disease Control and Prevention. N. C. f. H. S. C. W. O.-l. D., Compiled from Compressed Mortality File 1999-2009 Series 20 No. 20. (2012). Available from: http://wonder.cdc.gov/
25. Vrieze A, Postma DS, Kerstjens HA. Perimenstrual asthma: a syndrome without known cause or cure. J Allergy Clin Immunol (2003) 112:271–82. doi:10.1067/mai.2003.1676
27. Rao CK, Moore CG, Bleecker E, Busse WW, Calhoun W, Castro M, et al. Characteristics of perimenstrual asthma and its relation to asthma severity and control: data from the Severe Asthma Research Program. Chest (2013) 143:984–92. doi:10.1378/chest.12-0973
28. Schatz M, Harden K, Forsythe A, Chilingar L, Hoffman C, Sperling W, et al. The course of asthma during pregnancy, post partum, and with successive pregnancies: a prospective analysis. J Allergy Clin Immunol (1988) 81:509–17. doi:10.1016/0091-6749(88)90187-X
29. Troisi RJ, Speizer FE, Willett WC, Trichopoulos D, Rosner B. Menopause, postmenopausal estrogen preparations, and the risk of adult-onset asthma. A prospective cohort study. Am J Respir Crit Care Med (1995) 152:1183–8. doi:10.1164/ajrccm.152.4.7551368
30. Farha S, Asosingh K, Laskowski D, Hammel J, Dweik RA, Wiedemann HP, et al. Effects of the menstrual cycle on lung function variables in women with asthma. Am J Respir Crit Care Med (2009) 180:304–10. doi:10.1164/rccm.200904-0497OC
31. Wolkewitz M, Rothenbacher D, Low M, Stegmaier C, Ziegler H, Radulescu M, et al. Lifetime prevalence of self-reported atopic diseases in a population-based sample of elderly subjects: results of the ESTHER study. Br J Dermatol (2007) 156:693–7. doi:10.1111/j.1365-2133.2006.07659.x
32. Chandler MH, Schuldheisz S, Phillips BA, Muse KN. Premenstrual asthma: the effect of estrogen on symptoms, pulmonary function, and beta 2-receptors. Pharmacotherapy (1997) 17:224–34.
33. Eliasson O, Scherzer HH, DeGraff AC Jr. Morbidity in asthma in relation to the menstrual cycle. J Allergy Clin Immunol (1986) 77:87–94. doi:10.1016/0091-6749(86)90328-3
34. Gibbs CJ, Coutts II, Lock R, Finnegan OC, White RJ. Premenstrual exacerbation of asthma. Thorax (1984) 39:833–6. doi:10.1136/thx.39.11.833
35. Wulfsohn NL, Politzer WM. Bronchial asthma during menses and pregnancy. S Afr Med J (1964) 38:173.
36. Skobeloff EM, Spivey WH, Silverman R, Eskin BA, Harchelroad F, Alessi TV. The effect of the menstrual cycle on asthma presentations in the emergency department. Arch Intern Med (1996) 156:1837–40. doi:10.1001/archinte.156.16.1837
37. Lam SM, Huang SC. Premenstrual asthma: report of a case with hormonal studies. J Microbiol Immunol Infect (1998) 31:197–9.
38. Pauli BD, Reid RL, Munt PW, Wigle RD, Forkert L. Influence of the menstrual cycle on airway function in asthmatic and normal subjects. Am Rev Respir Dis (1989) 140:358–62. doi:10.1164/ajrccm/140.2.358
39. Tan KS, McFarlane LC, Lipworth BJ. Modulation of airway reactivity and peak flow variability in asthmatics receiving the oral contraceptive pill. Am J Respir Crit Care Med (1997) 155:1273–7. doi:10.1164/ajrccm.155.4.9105066
40. Derimanov GS, Oppenheimer J. Exacerbation of premenstrual asthma caused by an oral contraceptive. Ann Allergy Asthma Immunol (1998) 81:243–6. doi:10.1016/S1081-1206(10)62820-7
41. Gluck JC, Gluck P. The effects of pregnancy on asthma: a prospective study. Ann Allergy (1976) 37:164–8.
42. Juniper EF, Daniel EE, Roberts RS, Kline PA, Hargreave FE, Newhouse MT. Improvement in airway responsiveness and asthma severity during pregnancy. A prospective study. Am Rev Respir Dis (1989) 140:924–31. doi:10.1164/ajrccm/140.4.924
43. Sims CD, Chamberlain GV, de Swiet M. Lung function tests in bronchial asthma during and after pregnancy. Br J Obstet Gynaecol (1976) 83:434–7. doi:10.1111/j.1471-0528.1976.tb00860.x
44. Jenkins MA, Dharmage SC, Flander LB, Douglass JA, Ugoni AM, Carlin JB, et al. Parity and decreased use of oral contraceptives as predictors of asthma in young women. Clin Exp Allergy (2006) 36:609–13. doi:10.1111/j.1365-2222.2006.02475.x
45. Kos-Kudla B, Ostrowska Z, Marek B, Ciesielska-Kopacz N, Sieminska L, Kajdaniuk D, et al. Hormone replacement therapy in postmenopausal asthmatic women. J Clin Pharm Ther (2000) 25:461–6. doi:10.1046/j.1365-2710.2000.00310.x
46. Foschino Barbaro MP, Costa VR, Resta O, Prato R, Spanevello A, Palladino GP, et al. Menopausal asthma: a new biological phenotype? Allergy (2010) 65:1306–12. doi:10.1111/j.1398-9995.2009.02314.x
47. Bonnelykke K, Raaschou-Nielsen O, Tjonneland A, Ulrik CS, Bisgaard H, Andersen ZJ. Postmenopausal hormone therapy and asthma-related hospital admission. J Allergy Clin Immunol (2015) 135(813–816):e815. doi:10.1016/j.jaci.2014.11.019
48. Barr RG, Wentowski CC, Grodstein F, Somers SC, Stampfer MJ, Schwartz J, et al. Prospective study of postmenopausal hormone use and newly diagnosed asthma and chronic obstructive pulmonary disease. Arch Intern Med (2004) 164:379–86. doi:10.1001/archinte.164.4.379
49. Wang L, Wang K, Gao X, Paul TK, Cai J, Wang Y. Sex difference in the association between obesity and asthma in U.S. adults: findings from a national study. Respir Med (2015) 109:955–62. doi:10.1016/j.rmed.2015.06.001
50. Yunginger JW, Reed CE, O’Connell EJ, Melton LJ III, O’Fallon WM, Silverstein MD. A community-based study of the epidemiology of asthma. Incidence rates, 1964-1983. Am Rev Respir Dis (1992) 146:888–94. doi:10.1164/ajrccm/146.4.888
51. Almqvist C, Worm M, Leynaert B. Impact of gender on asthma in childhood and adolescence: a GA2LEN review. Allergy (2008) 63:47–57.
52. Jia M, Dahlman-Wright K, Gustafsson JA. Estrogen receptor alpha and beta in health and disease. Best Pract Res Clin Endocrinol Metab (2015) 29:557–68. doi:10.1016/j.beem.2015.04.008
53. Fliss AE, Benzeno S, Rao J, Caplan AJ. Control of estrogen receptor ligand binding by Hsp90. J Steroid Biochem Mol Biol (2000) 72:223–30. doi:10.1016/S0960-0760(00)00037-6
54. Pappas TC, Gametchu B, Watson CS. Membrane estrogen receptors identified by multiple antibody labeling and impeded-ligand binding. FASEB J (1995) 9:404–10.
55. Filardo EJ. Epidermal growth factor receptor (EGFR) transactivation by estrogen via the G-protein-coupled receptor, GPR30: a novel signaling pathway with potential significance for breast cancer. J Steroid Biochem Mol Biol (2002) 80:231–8. doi:10.1016/S0960-0760(01)00190-X
56. Kato S, Endoh H, Masuhiro Y, Kitamoto T, Uchiyama S, Sasaki H, et al. Activation of the estrogen receptor through phosphorylation by mitogen-activated protein kinase. Science (1995) 270:1491–4. doi:10.1126/science.270.5241.1491
57. Ignar-Trowbridge DM, Pimentel M, Parker MG, McLachlan JA, Korach KS. Peptide growth factor cross-talk with the estrogen receptor requires the A/B domain and occurs independently of protein kinase C or estradiol. Endocrinology (1996) 137:1735–44. doi:10.1210/endo.137.5.8612509
58. Demay F, De Monti M, Tiffoche C, Vaillant C, Thieulant ML. Steroid-independent activation of ER by GnRH in gonadotrope pituitary cells. Endocrinology (2001) 142:3340–7. doi:10.1210/endo.142.8.8337
59. Chambraud B, Berry M, Redeuilh G, Chambon P, Baulieu EE. Several regions of human estrogen receptor are involved in the formation of receptor-heat shock protein 90 complexes. J Biol Chem (1990) 265:20686–91.
60. Lannigan DA. Estrogen receptor phosphorylation. Steroids (2003) 68:1–9. doi:10.1016/S0039-128X(02)00110-1
61. Wang Z, Zhang X, Shen P, Loggie BW, Chang Y, Deuel TF. Identification, cloning, and expression of human estrogen receptor-alpha36, a novel variant of human estrogen receptor-alpha66. Biochem Biophys Res Commun (2005) 336:1023–7. doi:10.1016/j.bbrc.2005.08.226
62. Figtree GA, McDonald D, Watkins H, Channon KM. Truncated estrogen receptor alpha 46-kDa isoform in human endothelial cells: relationship to acute activation of nitric oxide synthase. Circulation (2003) 107:120–6. doi:10.1161/01.CIR.0000043805.11780.F5
63. Murphy AJ, Guyre PM, Wira CR, Pioli PA. Estradiol regulates expression of estrogen receptor ERalpha46 in human macrophages. PLoS One (2009) 4:e5539. doi:10.1371/journal.pone.0005539
64. Zou Y, Ding L, Coleman M, Wang Z. Estrogen receptor-alpha (ER-alpha) suppresses expression of its variant ER-alpha 36. FEBS Lett (2009) 583:1368–74. doi:10.1016/j.febslet.2009.03.047
65. Pelekanou V, Kampa M, Kiagiadaki F, Deli A, Theodoropoulos P, Agrogiannis G, et al. Estrogen anti-inflammatory activity on human monocytes is mediated through cross-talk between estrogen receptor ERalpha36 and GPR30/GPER1. J Leukoc Biol (2015). doi:10.1189/jlb.3A0914-430RR
66. Chaidarun SS, Alexander JM. A tumor-specific truncated estrogen receptor splice variant enhances estrogen-stimulated gene expression. Mol Endocrinol (1998) 12:1355–66. doi:10.1210/mend.12.9.0170
67. Shao R, Weijdegard B, Fernandez-Rodriguez J, Egecioglu E, Zhu C, Andersson N, et al. Ciliated epithelial-specific and regional-specific expression and regulation of the estrogen receptor-beta2 in the fallopian tubes of immature rats: a possible mechanism for estrogen-mediated transport process in vivo. Am J Physiol Endocrinol Metab (2007) 293:E147–58. doi:10.1152/ajpendo.00101.2007
68. Marino M, Galluzzo P, Ascenzi P. Estrogen signaling multiple pathways to impact gene transcription. Curr Genomics (2006) 7:497–508. doi:10.2174/138920206779315737
69. Sathya G, Li W, Klinge CM, Anolik JH, Hilf R, Bambara RA. Effects of multiple estrogen responsive elements, their spacing, and location on estrogen response of reporter genes. Mol Endocrinol (1997) 11:1994–2003. doi:10.1210/mend.11.13.0039
70. Li C, Briggs MR, Ahlborn TE, Kraemer FB, Liu J. Requirement of Sp1 and estrogen receptor alpha interaction in 17beta-estradiol-mediated transcriptional activation of the low density lipoprotein receptor gene expression. Endocrinology (2001) 142:1546–53. doi:10.1210/endo.142.4.8096
71. Chambliss KL I, Yuhanna S, Anderson RG, Mendelsohn ME, Shaul PW. ERbeta has nongenomic action in caveolae. Mol Endocrinol (2002) 16:938–46. doi:10.1210/me.16.5.938
72. Duan R, Porter W, Safe S. Estrogen-induced c-fos protooncogene expression in MCF-7 human breast cancer cells: role of estrogen receptor Sp1 complex formation. Endocrinology (1998) 139:1981–90. doi:10.1210/endo.139.4.5870
73. Castro-Rivera E, Samudio I, Safe S. Estrogen regulation of cyclin D1 gene expression in ZR-75 breast cancer cells involves multiple enhancer elements. J Biol Chem (2001) 276:30853–61. doi:10.1074/jbc.M103339200
74. Sun G, Porter W, Safe S. Estrogen-induced retinoic acid receptor alpha 1 gene expression: role of estrogen receptor-Sp1 complex. Mol Endocrinol (1998) 12:882–90. doi:10.1210/me.12.6.882
75. O’Lone R, Frith MC, Karlsson EK, Hansen U. Genomic targets of nuclear estrogen receptors. Mol Endocrinol (2004) 18:1859–75. doi:10.1210/me.2003-0044
76. Paech K, Webb P, Kuiper GG, Nilsson S, Gustafsson J, Kushner PJ, et al. Differential ligand activation of estrogen receptors ERalpha and ERbeta at AP1 sites. Science (1997) 277:1508–10. doi:10.1126/science.277.5331.1508
77. Shiau AK, Barstad D, Loria PM, Cheng L, Kushner PJ, Agard DA, et al. The structural basis of estrogen receptor/coactivator recognition and the antagonism of this interaction by tamoxifen. Cell (1998) 95:927–37. doi:10.1016/S0092-8674(00)81717-1
78. Shang Y, Hu X, DiRenzo J, Lazar MA, Brown M. Cofactor dynamics and sufficiency in estrogen receptor-regulated transcription. Cell (2000) 103:843–52. doi:10.1016/S0092-8674(00)00188-4
79. Joel PB, Traish AM, Lannigan DA. Estradiol and phorbol ester cause phosphorylation of serine 118 in the human estrogen receptor. Mol Endocrinol (1995) 9:1041–52. doi:10.1210/me.9.8.1041
80. Bunone G, Briand PA, Miksicek RJ, Picard D. Activation of the unliganded estrogen receptor by EGF involves the MAP kinase pathway and direct phosphorylation. EMBO J (1996) 15:2174–83.
81. Curtis SW, Washburn T, Sewall C, DiAugustine R, Lindzey J, Couse JF, et al. Physiological coupling of growth factor and steroid receptor signaling pathways: estrogen receptor knockout mice lack estrogen-like response to epidermal growth factor. Proc Natl Acad Sci U S A (1996) 93:12626–30. doi:10.1073/pnas.93.22.12626
82. Frodin M, Gammeltoft S. Role and regulation of 90 kDa ribosomal S6 kinase (RSK) in signal transduction. Mol Cell Endocrinol (1999) 151:65–77. doi:10.1016/S0303-7207(99)00061-1
83. Martin MB, Franke TF, Stoica GE, Chambon P, Katzenellenbogen BS, Stoica BA, et al. A role for Akt in mediating the estrogenic functions of epidermal growth factor and insulin-like growth factor I. Endocrinology (2000) 141:4503–11. doi:10.1210/en.141.12.4503
84. Le Goff P, Montano MM, Schodin DJ, Katzenellenbogen BS. Phosphorylation of the human estrogen receptor. Identification of hormone-regulated sites and examination of their influence on transcriptional activity. J Biol Chem (1994) 269:4458–66.
85. Chen D, Pace PE, Coombes RC, Ali S. Phosphorylation of human estrogen receptor alpha by protein kinase A regulates dimerization. Mol Cell Biol (1999) 19:1002–15.
86. Migliaccio A, Castoria G, Di Domenico M, de Falco A, Bilancio A, Lombardi M, et al. Steroid-induced androgen receptor-oestradiol receptor beta-Src complex triggers prostate cancer cell proliferation. EMBO J (2000) 19:5406–17. doi:10.1093/emboj/19.20.5406
87. Joel PB, Smith J, Sturgill TW, Fisher TL, Blenis J, Lannigan DA. pp90rsk1 regulates estrogen receptor-mediated transcription through phosphorylation of Ser-167. Mol Cell Biol (1998) 18:1978–84. doi:10.1128/MCB.18.4.1978
88. Ali S, Metzger D, Bornert JM, Chambon P. Modulation of transcriptional activation by ligand-dependent phosphorylation of the human oestrogen receptor A/B region. EMBO J (1993) 12:1153–60.
89. Straub RH. The complex role of estrogens in inflammation. Endocr Rev (2007) 28:521–74. doi:10.1210/er.2007-0001
90. Balhara J, Gounni AS. The alveolar macrophages in asthma: a double-edged sword. Mucosal Immunol (2012) 5:605–9. doi:10.1038/mi.2012.74
91. Moreira AP, Hogaboam CM. Macrophages in allergic asthma: fine-tuning their pro- and anti-inflammatory actions for disease resolution. J Interferon Cytokine Res (2011) 31:485–91. doi:10.1089/jir.2011.0027
92. Yang M, Kumar RK, Hansbro PM, Foster PS. Emerging roles of pulmonary macrophages in driving the development of severe asthma. J Leukoc Biol (2012) 91:557–69. doi:10.1189/jlb.0711357
93. Melgert BN, Oriss TB, Qi Z, Dixon-McCarthy B, Geerlings M, Hylkema MN, et al. Macrophages: regulators of sex differences in asthma? Am J Respir Cell Mol Biol (2010) 42:595–603. doi:10.1165/rcmb.2009-0016OC
94. Bang BR, Chun E, Shim EJ, Lee HS, Lee SY, Cho SH, et al. Alveolar macrophages modulate allergic inflammation in a murine model of asthma. Exp Mol Med (2011) 43:275–80. doi:10.3858/emm.2011.43.5.028
95. Khalil N, Whitman C, Zuo L, Danielpour D, Greenberg A. Regulation of alveolar macrophage transforming growth factor-beta secretion by corticosteroids in bleomycin-induced pulmonary inflammation in the rat. J Clin Invest (1993) 92:1812–8. doi:10.1172/JCI116771
96. Katsura Y, Harada N, Harada S, Ishimori A, Makino F, Ito J, et al. Characteristics of alveolar macrophages from murine models of OVA-induced allergic airway inflammation and LPS-induced acute airway inflammation. Exp Lung Res (2015) 41(7):370–82. doi:10.3109/01902148.2015.1044137
97. Lee YG, Jeong JJ, Nyenhuis S, Berdyshev E, Chung S, Ranjan R, et al. Recruited alveolar macrophages, in response to airway epithelial-derived monocyte chemoattractant protein 1/CCl2, regulate airway inflammation and remodeling in allergic asthma. Am J Respir Cell Mol Biol (2015) 52:772–84. doi:10.1165/rcmb.2014-0255OC
98. Draijer C, Robbe P, Boorsma CE, Hylkema MN, Melgert BN. Characterization of macrophage phenotypes in three murine models of house-dust-mite-induced asthma. Mediators Inflamm (2013) 2013:632049. doi:10.1155/2013/632049
99. Pergola C, Dodt G, Rossi A, Neunhoeffer E, Lawrenz B, Northoff H, et al. ERK-mediated regulation of leukotriene biosynthesis by androgens: a molecular basis for gender differences in inflammation and asthma. Proc Natl Acad Sci U S A (2008) 105:19881–6. doi:10.1073/pnas.0809120105
100. Campbell L, Emmerson E, Williams H, Saville CR, Krust A, Chambon P, et al. Estrogen receptor-alpha promotes alternative macrophage activation during cutaneous repair. J Invest Dermatol (2014) 134:2447–57. doi:10.1038/jid.2014.175
101. Zhang X, Wang L, Zhang H, Guo D, Qiao Z, Qiao J. Estrogen inhibits lipopolysaccharide-induced tumor necrosis factor-alpha release from murine macrophages. Methods Find Exp Clin Pharmacol (2001) 23:169–73. doi:10.1358/mf.2001.23.4.634640
102. Gao Y, Qian WP, Dark K, Toraldo G, Lin AS, Guldberg RE, et al. Estrogen prevents bone loss through transforming growth factor beta signaling in T cells. Proc Natl Acad Sci U S A (2004) 101:16618–23. doi:10.1073/pnas.0404888101
103. Calippe B, Douin-Echinard V, Delpy L, Laffargue M, Lelu K, Krust A, et al. 17Beta-estradiol promotes TLR4-triggered proinflammatory mediator production through direct estrogen receptor alpha signaling in macrophages in vivo. J Immunol (2010) 185:1169–76. doi:10.4049/jimmunol.0902383
104. Gautier EL, Chow A, Spanbroek R, Marcelin G, Greter M, Jakubzick C, et al. Systemic analysis of PPARgamma in mouse macrophage populations reveals marked diversity in expression with critical roles in resolution of inflammation and airway immunity. J Immunol (2012) 189:2614–24. doi:10.4049/jimmunol.1200495
105. Zaslona Z, Wilhelm J, Cakarova L, Marsh LM, Seeger W, Lohmeyer J, et al. Transcriptome profiling of primary murine monocytes, lung macrophages and lung dendritic cells reveals a distinct expression of genes involved in cell trafficking. Respir Res (2009) 10:2. doi:10.1186/1465-9921-10-2
106. Paharkova-Vatchkova V, Maldonado R, Kovats S. Estrogen preferentially promotes the differentiation of CD11c+ CD11b(intermediate) dendritic cells from bone marrow precursors. J Immunol (2004) 172:1426–36. doi:10.4049/jimmunol.172.3.1426
107. Mao A, Paharkova-Vatchkova V, Hardy J, Miller MM, Kovats S. Estrogen selectively promotes the differentiation of dendritic cells with characteristics of langerhans cells. J Immunol (2005) 175:5146–51. doi:10.4049/jimmunol.175.8.5146
108. Seillet C, Laffont S, Tremollieres F, Rouquie N, Ribot C, Arnal JF, et al. The TLR-mediated response of plasmacytoid dendritic cells is positively regulated by estradiol in vivo through cell-intrinsic estrogen receptor alpha signaling. Blood (2012) 119:454–64. doi:10.1182/blood-2011-08-371831
109. Uchida Y, Kurasawa K, Nakajima H, Nakagawa N, Tanabe E, Sueishi M, et al. Increase of dendritic cells of type 2 (DC2) by altered response to IL-4 in atopic patients. J Allergy Clin Immunol (2001) 108:1005–11. doi:10.1067/mai.2001.119741
110. Matsuda H, Suda T, Hashizume H, Yokomura K, Asada K, Suzuki K, et al. Alteration of balance between myeloid dendritic cells and plasmacytoid dendritic cells in peripheral blood of patients with asthma. Am J Respir Crit Care Med (2002) 166:1050–4. doi:10.1164/rccm.2110066
111. de Heer HJ, Hammad H, Soullie T, Hijdra D, Vos N, Willart MA, et al. Essential role of lung plasmacytoid dendritic cells in preventing asthmatic reactions to harmless inhaled antigen. J Exp Med (2004) 200:89–98. doi:10.1084/jem.20040035
112. Tsuchida T, Matsuse H, Fukahori S, Kawano T, Tomari S, Fukushima C, et al. Effect of respiratory syncytial virus infection on plasmacytoid dendritic cell regulation of allergic airway inflammation. Int Arch Allergy Immunol (2012) 157:21–30. doi:10.1159/000324676
113. Bengtsson AK, Ryan EJ, Giordano D, Magaletti DM, Clark EA. 17beta-estradiol (E2) modulates cytokine and chemokine expression in human monocyte-derived dendritic cells. Blood (2004) 104:1404–10. doi:10.1182/blood-2003-10-3380
114. Yang L, Hu Y, Hou Y. Effects of 17beta-estradiol on the maturation, nuclear factor kappa B p65 and functions of murine spleen CD11c-positive dendritic cells. Mol Immunol (2006) 43:357–66. doi:10.1016/j.molimm.2005.02.012
115. Siracusa MC, Overstreet MG, Housseau F, Scott AL, Klein SL. 17beta-estradiol alters the activity of conventional and IFN-producing killer dendritic cells. J Immunol (2008) 180:1423–31. doi:10.4049/jimmunol.180.3.1423
116. Maret A, Coudert JD, Garidou L, Foucras G, Gourdy P, Krust A, et al. Estradiol enhances primary antigen-specific CD4 T cell responses and Th1 development in vivo. Essential role of estrogen receptor alpha expression in hematopoietic cells. Eur J Immunol (2003) 33:512–21. doi:10.1002/immu.200310027
117. Pettersson A, Ciumas C, Chirsky V, Link H, Huang YM, Xiao BG. Dendritic cells exposed to estrogen in vitro exhibit therapeutic effects in ongoing experimental allergic encephalomyelitis. J Neuroimmunol (2004) 156:58–65. doi:10.1016/j.jneuroim.2004.07.004
118. Papenfuss TL, Powell ND, McClain MA, Bedarf A, Singh A I, Gienapp E, et al. Estriol generates tolerogenic dendritic cells in vivo that protect against autoimmunity. J Immunol (2011) 186:3346–55. doi:10.4049/jimmunol.1001322
119. Lambrecht BN, Salomon B, Klatzmann D, Pauwels RA. Dendritic cells are required for the development of chronic eosinophilic airway inflammation in response to inhaled antigen in sensitized mice. J Immunol (1998) 160:4090–7.
120. van Rijt LS, Jung S, Kleinjan A, Vos N, Willart M, Duez C, et al. In vivo depletion of lung CD11c+ dendritic cells during allergen challenge abrogates the characteristic features of asthma. J Exp Med (2005) 201:981–91. doi:10.1084/jem.20042311
121. Ross R, Klebanoff SJ. The eosinophilic leukocyte. Fine structure studies of changes in the uterus during the estrous cycle. J Exp Med (1966) 124:653–60. doi:10.1084/jem.124.4.653
122. Lee YH, Howe RS, Sha SJ, Teuscher C, Sheehan DM, Lyttle CR. Estrogen regulation of an eosinophil chemotactic factor in the immature rat uterus. Endocrinology (1989) 125:3022–8. doi:10.1210/endo-125-6-3022
123. Luque EH, Munoz de Toro MM, Ramos JG, Rodriguez HA, Sherwood OD. Role of relaxin and estrogen in the control of eosinophilic invasion and collagen remodeling in rat cervical tissue at term. Biol Reprod (1998) 59:795–800. doi:10.1095/biolreprod59.4.795
124. Rytomaa T. Organ distribution and histochemical properties of eosinophil granulocytes in rat. Acta Pathol Microbiol Scand Suppl (1960) 50(Suppl 140):1–118.
125. Cai Y, Zhou J, Webb DC. Estrogen stimulates Th2 cytokine production and regulates the compartmentalisation of eosinophils during allergen challenge in a mouse model of asthma. Int Arch Allergy Immunol (2012) 158:252–60. doi:10.1159/000331437
126. Tamaki M, Konno Y, Kobayashi Y, Takeda M, Itoga M, Moritoki Y, et al. Expression and functional roles of G-protein-coupled estrogen receptor (GPER) in human eosinophils. Immunol Lett (2014) 160:72–8. doi:10.1016/j.imlet.2014.03.012
127. Melgert BN, Postma DS, Kuipers I, Geerlings M, Luinge MA, van der Strate BW, et al. Female mice are more susceptible to the development of allergic airway inflammation than male mice. Clin Exp Allergy (2005) 35:1496–503. doi:10.1111/j.1365-2222.2005.02362.x
128. Blacquiere MJ, Hylkema MN, Postma DS, Geerlings M, Timens W, Melgert BN. Airway inflammation and remodeling in two mouse models of asthma: comparison of males and females. Int Arch Allergy Immunol (2010) 153:173–81. doi:10.1159/000312635
129. Riffo-Vasquez Y, Ligeiro de Oliveira AP, Page CP, Spina D, Tavares-de-Lima W. Role of sex hormones in allergic inflammation in mice. Clin Exp Allergy (2007) 37:459–70. doi:10.1111/j.1365-2222.2007.02670.x
130. Dimitropoulou C, Drakopanagiotakis F, Chatterjee A, Snead C, Catravas JD. Estrogen replacement therapy prevents airway dysfunction in a murine model of allergen-induced asthma. Lung (2009) 187:116–27. doi:10.1007/s00408-008-9129-z
131. Tchernitchin AN, Barrera J, Arroyo P, Mena MA, Vilches K, Grunert G. Degranulatory action of estradiol on blood eosinophil leukocytes in vivo and in vitro. Agents Actions (1985) 17:60–6. doi:10.1007/BF01966683
132. Douin-Echinard V, Calippe B, Billon-Gales A, Fontaine C, Lenfant F, Tremollieres F, et al. Estradiol administration controls eosinophilia through estrogen receptor-alpha activation during acute peritoneal inflammation. J Leukoc Biol (2011) 90:145–54. doi:10.1189/jlb.0210073
133. Itoga M, Konno Y, Moritoki Y, Saito Y, Ito W, Tamaki M, et al. Correction: G-protein-coupled estrogen receptor agonist suppresses airway inflammation in a mouse model of asthma through IL-10. PLoS One (2015) 10:e0136326. doi:10.1371/journal.pone.0136326
134. Bradley BL, Azzawi M, Jacobson M, Assoufi B, Collins JV, Irani AM, et al. Eosinophils, T-lymphocytes, mast cells, neutrophils, and macrophages in bronchial biopsy specimens from atopic subjects with asthma: comparison with biopsy specimens from atopic subjects without asthma and normal control subjects and relationship to bronchial hyperresponsiveness. J Allergy Clin Immunol (1991) 88:661–74.
135. Zaitsu M, Narita S, Lambert KC, Grady JJ, Estes DM, Curran EM, et al. Estradiol activates mast cells via a non-genomic estrogen receptor-alpha and calcium influx. Mol Immunol (2007) 44:1977–85. doi:10.1016/j.molimm.2006.09.030
136. Jing H, Wang Z, Chen Y. Effect of oestradiol on mast cell number and histamine level in the mammary glands of rat. Anat Histol Embryol (2012) 41:170–6. doi:10.1111/j.1439-0264.2011.01120.x
137. Vliagoftis H, Dimitriadou V, Boucher W, Rozniecki JJ, Correia I, Raam S, et al. Estradiol augments while tamoxifen inhibits rat mast cell secretion. Int Arch Allergy Immunol (1992) 98:398–409. doi:10.1159/000236217
138. Cai Y, Kumar RK, Zhou J, Foster PS, Webb DC. Ym1/2 promotes Th2 cytokine expression by inhibiting 12/15(S)-lipoxygenase: identification of a novel pathway for regulating allergic inflammation. J Immunol (2009) 182:5393–9. doi:10.4049/jimmunol.0803874
139. Townsend EA, Meuchel LW, Thompson MA, Pabelick CM, Prakash YS. Estrogen increases nitric-oxide production in human bronchial epithelium. J Pharmacol Exp Ther (2011) 339:815–24. doi:10.1124/jpet.111.184416
140. Townsend EA, Thompson MA, Pabelick CM, Prakash YS. Rapid effects of estrogen on intracellular Ca2+ regulation in human airway smooth muscle. Am J Physiol Lung Cell Mol Physiol (2010) 298:L521–30. doi:10.1152/ajplung.00287.2009
141. Degano B, Prevost MC, Berger P, Molimard M, Pontier S, Rami J, et al. Estradiol decreases the acetylcholine-elicited airway reactivity in ovariectomized rats through an increase in epithelial acetylcholinesterase activity. Am J Respir Crit Care Med (2001) 164:1849–54. doi:10.1164/ajrccm.164.10.2102009
142. Choi HJ, Chung YS, Kim HJ, Moon UY, Choi YH, Van Seuningen I, et al. Signal pathway of 17beta-estradiol-induced MUC5B expression in human airway epithelial cells. Am J Respir Cell Mol Biol (2009) 40:168–78. doi:10.1165/rcmb.2007-0377OC
143. Chotirmall SH, Greene CM, Oglesby IK, Thomas W, O’Neill SJ, Harvey BJ, et al. 17Beta-estradiol inhibits IL-8 in cystic fibrosis by up-regulating secretory leucoprotease inhibitor. Am J Respir Crit Care Med (2010) 182:62–72. doi:10.1164/rccm.201001-0053OC
144. Erlandsson MC, Ohlsson C, Gustafsson JA, Carlsten H. Role of oestrogen receptors alpha and beta in immune organ development and in oestrogen-mediated effects on thymus. Immunology (2001) 103:17–25. doi:10.1046/j.1365-2567.2001.01212.x
145. Paavonen T, Andersson LC, Adlercreutz H. Sex hormone regulation of in vitro immune response. Estradiol enhances human B cell maturation via inhibition of suppressor T cells in pokeweed mitogen-stimulated cultures. J Exp Med (1981) 154:1935–45. doi:10.1084/jem.154.6.1935
146. Gilmore W, Weiner LP, Correale J. Effect of estradiol on cytokine secretion by proteolipid protein-specific T cell clones isolated from multiple sclerosis patients and normal control subjects. J Immunol (1997) 158:446–51.
147. Carlsten H, Verdrengh M, Taube M. Additive effects of suboptimal doses of estrogen and cortisone on the suppression of T lymphocyte dependent inflammatory responses in mice. Inflamm Res (1996) 45:26–30. doi:10.1007/BF02263501
148. Gregory MS, Duffner LA, Faunce DE, Kovacs EJ. Estrogen mediates the sex difference in post-burn immunosuppression. J Endocrinol (2000) 164:129–38. doi:10.1677/joe.0.1640129
149. Bao M, Yang Y, Jun HS, Yoon JW. Molecular mechanisms for gender differences in susceptibility to T cell-mediated autoimmune diabetes in nonobese diabetic mice. J Immunol (2002) 168:5369–75. doi:10.4049/jimmunol.168.10.5369
150. Karpuzoglu-Sahin E, Zhi-Jun Y, Lengi A, Sriranganathan N, Ansar Ahmed S. Effects of long-term estrogen treatment on IFN-gamma, IL-2 and IL-4 gene expression and protein synthesis in spleen and thymus of normal C57BL/6 mice. Cytokine (2001) 14:208–17. doi:10.1006/cyto.2001.0876
151. Lambert KC, Curran EM, Judy BM, Milligan GN, Lubahn DB, Estes DM. Estrogen receptor alpha (ERalpha) deficiency in macrophages results in increased stimulation of CD4+ T cells while 17beta-estradiol acts through ERalpha to increase IL-4 and GATA-3 expression in CD4+ T cells independent of antigen presentation. J Immunol (2005) 175:5716–23. doi:10.4049/jimmunol.175.9.5716
152. Polanczyk MJ, Carson BD, Subramanian S, Afentoulis M, Vandenbark AA, Ziegler SF, et al. Cutting edge: estrogen drives expansion of the CD4+CD25+ regulatory T cell compartment. J Immunol (2004) 173:2227–30. doi:10.4049/jimmunol.173.4.2227
153. Lelu K, Laffont S, Delpy L, Paulet PE, Perinat T, Tschanz SA, et al. Estrogen receptor alpha signaling in T lymphocytes is required for estradiol-mediated inhibition of Th1 and Th17 cell differentiation and protection against experimental autoimmune encephalomyelitis. J Immunol (2011) 187:2386–93. doi:10.4049/jimmunol.1101578
154. Confavreux C, Hutchinson M, Hours MM, Cortinovis-Tourniaire P, Moreau T. Rate of pregnancy-related relapse in multiple sclerosis. Pregnancy in Multiple Sclerosis Group. N Engl J Med (1998) 339:285–91. doi:10.1056/NEJM199807303390501
155. Franklin RD, Kutteh WH. Characterization of immunoglobulins and cytokines in human cervical mucus: influence of exogenous and endogenous hormones. J Reprod Immunol (1999) 42:93–106. doi:10.1016/S0165-0378(98)00086-2
156. Clerici E, Bergamasco E, Ferrario E, Villa ML. Influence of sex steroids on the antigen-specific primary antibody response in vitro. J Clin Lab Immunol (1991) 34:71–8.
157. Sthoeger ZM, Chiorazzi N, Lahita RG. Regulation of the immune response by sex hormones. I. In vitro effects of estradiol and testosterone on pokeweed mitogen-induced human B cell differentiation. J Immunol (1988) 141:91–8.
158. Kanda N, Tamaki K. Estrogen enhances immunoglobulin production by human PBMCs. J Allergy Clin Immunol (1999) 103:282–8. doi:10.1016/S0091-6749(99)70503-8
159. Kanda N, Tsuchida T, Tamaki K. Estrogen enhancement of anti-double-stranded DNA antibody and immunoglobulin G production in peripheral blood mononuclear cells from patients with systemic lupus erythematosus. Arthritis Rheum (1999) 42:328–37. doi:10.1002/1529-0131(199902)42:2<328::AID-ANR16>3.0.CO;2-#
160. Han D, Denison MS, Tachibana H, Yamada K. Effects of estrogenic compounds on immunoglobulin production by mouse splenocytes. Biol Pharm Bull (2002) 25:1263–7. doi:10.1248/bpb.25.1263
161. Bhavsar P, Hew M, Khorasani N, Torrego A, Barnes PJ, Adcock I, et al. Relative corticosteroid insensitivity of alveolar macrophages in severe asthma compared with non-severe asthma. Thorax (2008) 63:784–90. doi:10.1136/thx.2007.090027
162. Goleva E, Hauk PJ, Hall CF, Liu AH, Riches DW, Martin RJ, et al. Corticosteroid-resistant asthma is associated with classical antimicrobial activation of airway macrophages. J Allergy Clin Immunol (2008) 122(550–559):e553. doi:10.1016/j.jaci.2008.07.007
163. Kraft M, Hamid Q, Chrousos GP, Martin RJ, Leung DY. Decreased steroid responsiveness at night in nocturnal asthma. Is the macrophage responsible? Am J Respir Crit Care Med (2001) 163:1219–25. doi:10.1164/ajrccm.163.5.2002058
164. Simoes DC, Psarra AM, Mauad T, Pantou I, Roussos C, Sekeris CE, et al. Glucocorticoid and estrogen receptors are reduced in mitochondria of lung epithelial cells in asthma. PLoS One (2012) 7:e39183. doi:10.1371/journal.pone.0039183
165. Convery RP, Leitch DN, Bromly C, Ward RJ, Bartlett G, Hendrick DJ. Effect of inhaled fluticasone propionate on airway responsiveness in treatment-naive individuals – a lesser benefit in females. Eur Respir J (2000) 15:19–24. doi:10.1183/09031936.00.15101900
166. Dijkstra A, Vonk JM, Jongepier H, Koppelman GH, Schouten JP, ten Hacken NH, et al. Lung function decline in asthma: association with inhaled corticosteroids, smoking and sex. Thorax (2006) 61:105–10. doi:10.1136/thx.2004.039271
167. Abraham B, Antó JM, Barreiro E, Bel EHD, Bonsignore G, Bousquet J, et al. The ENFUMOSA cross-sectional European multicentre study of the clinical phenotype of chronic severe asthma. European Network for Understanding Mechanisms of Severe Asthma. Eur Respir J (2003) 22:470–7. doi:10.1183/09031936.03.00261903
168. Frenkel B, White W, Tuckermann J. Glucocorticoid-induced osteoporosis. Adv Exp Med Biol (2015) 872:179–215. doi:10.1007/978-1-4939-2895-8_8
169. Schacke H, Docke WD, Asadullah K. Mechanisms involved in the side effects of glucocorticoids. Pharmacol Ther (2002) 96:23–43. doi:10.1016/S0163-7258(02)00297-8
170. Wenzel S, Ford L, Pearlman D, Spector S, Sher L, Skobieranda F, et al. Dupilumab in persistent asthma with elevated eosinophil levels. N Engl J Med (2013) 368:2455–66. doi:10.1056/NEJMoa1304048
Keywords: estrogen, allergy, asthma, inflammation, sex difference, macrophages, IL-4 and IL-13
Citation: Keselman A and Heller N (2015) Estrogen Signaling Modulates Allergic Inflammation and Contributes to Sex Differences in Asthma. Front. Immunol. 6:568. doi: 10.3389/fimmu.2015.00568
Received: 29 August 2015; Accepted: 23 October 2015;
Published: 16 November 2015
Edited by:
Xin M. Luo, Virginia Tech, USAReviewed by:
Sylvie Guerder, Center of Pathophysiology of Toulouse Purpan – INSERM U1043, FranceCopyright: © 2015 Keselman and Heller. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Nicola Heller, bmhlbGxlckBqaG1pLmVkdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.