 Ludovica F. Buttó
Ludovica F. Buttó Monika Schaubeck
Monika Schaubeck Dirk Haller*
Dirk Haller*
- Chair of Nutrition and Immunology, Technische Universität München, Freising-Weihenstephan, Germany
Crohn’s disease (CD) is a systemic chronic inflammatory condition mainly characterized by discontinuous transmural pathology of the gastrointestinal tract and frequent extraintestinal manifestations with intermittent episodes of remission and relapse. Genome-wide association studies identified a number of risk loci that, catalyzed by environmental triggers, result in the loss of tolerance toward commensal bacteria based on dysregulated innate effector functions and antimicrobial defense, leading to exacerbated adaptive immune responses responsible for chronic immune-mediated tissue damage. In this review, we discuss the inter-related role of changes in the intestinal microbiota, epithelial barrier integrity, and immune cell functions on the pathogenesis of CD, describing the current approaches available to investigate the molecular mechanisms underlying the disease. Substantial effort has been dedicated to define disease-associated changes in the intestinal microbiota (dysbiosis) and to link pathobionts to the etiology of inflammatory bowel diseases. A cogent definition of dysbiosis is lacking, as well as an agreement of whether pathobionts or complex shifts in the microbiota trigger inflammation in the host. Among the rarely available animal models, SAMP/Yit and TNFdeltaARE mice are the best known displaying a transmural CD-like phenotype. New hypothesis-driven mouse models, e.g., epithelial-specific Caspase8−/−, ATG16L1−/−, and XBP1−/− mice, validate pathway-focused function of specific CD-associated risk genes highlighting the role of Paneth cells in antimicrobial defense. To study the causal role of bacteria in initiating inflammation in the host, the use of germ-free mouse models is indispensable. Unraveling the interactions of genes, immune cells and microbes constitute a criterion for the development of safe, reliable, and effective treatment options for CD.
Introduction
Crohn’s disease (CD) is one of the two dominant phenotypes of inflammatory bowel diseases (IBD) characterized by chronic and relapsing inflammation of intestinal segments (1). There is overwhelming evidence corroborating the notion that the pathophysiology of CD is under the control of several contributing factors, including heritable traits, environmental cues, abnormalities in intestinal mucosal barrier integrity (2) and function (3), immune regulation (4, 5), and gut microbiota (6). Exaggerated immune responses are presumably directed against normal commensal enteric bacteria in genetically susceptible hosts. Host genetic susceptibility may be related to defective mucosal barrier function and/or bacterial killing, leading to an overexposure to luminal antigens and to inadequate immunoregulation, resulting in abnormal responses and tissue damage (7–11).
Genetic Evidence for Microbe–Host Interaction in Crohn’s Disease
Genome-wide association studies (GWAS) have postulated that the heritability of IBD may arise from polymorphisms in associated genes, each contributing in an additive fashion to the overall disease risk (12–15). The most recent GWAS meta-analysis has ascertained 163 susceptibility loci for IBD, revealing similar genetic predispositions in childhood and adult onsets (15). A number of these genetic risk factors are related to innate immunity, most specifically to microbial recognition and its subsequent elimination. The first susceptibility gene described and associated to increased risk of developing CD in Caucasian populations is the nucleotide-binding oligomerization domain 2 (NOD2, also called CARD15) gene. NOD2 gene encodes a cytoplasmic pathogen-recognition receptor (PRR) that recognizes muramyldipeptide (MDP), a component of both Gram-positive and Gram-negative cell walls, and therefore it is thought to play a role in the clearance of intracellular bacteria (16–20).
Additional CD-susceptibility genes that affect bacterial killing include autophagy-related protein 16-1 (ATG16L1) and immunity-related GTPase M (IRGM) (21–26). ATG16L1 gene encodes for an integral protein of the autophagy pathway. Autophagy is an important host defense mechanism for handling intracellular microorganisms and for the response to cellular stress, and it requires the generation of curved membranes to envelop cytoplasmic organelles and intracellular pathogens (27). A working hypothesis is that variants in components of the autophagy machinery may result in reduced pathogen clearance and intracellular homeostasis. IRGM gene encodes for one of the IRG proteins that orchestrate immunity toward intracellular pathogens including autophagy functions (28). While Irgm-deficient mice do not consistently exhibit spontaneous intestinal inflammation, they are much more susceptible to dextrate sodium sulfate (DSS) colitis compared to the WT counterparts, developing acute epithelial injury, loss of Paneth cell granules, altered antimicrobial peptide (AMP) production, and authophagy function of the epithelium (29).
Further CD-susceptibility loci related to aberrant microbial recognition and handling encompass toll-like receptor 4 (TLR4) (30), leucine-rich repeat kinase-2 (LRRK2) (31), and neutrophil cytosolic factor-4 (NCF4) (25). TLR4 is critical for host defense against Gram-negative bacteria since it recognizes lipopolysaccharide (LPS), and it is expressed on intestinal epithelial cells (IECs) and myeloid cells, such as macrophages (MΦs) and dendritic cells (DCs) (32). Two single nucleotide polymorphisms of human TLR4, D299G, and T399I lead to hyporesponsiveness to LPS by interfering with recruitment of TLR adapter molecules, such as myeloid differentiation primary response 88 (MYD88) and TIR domain-containing adaptor-inducing interferon β (TRIF) (33–35). LRRK2 gene is highly expressed on bone marrow-derived myeloid cells, but it is not expressed in IECs (36). Lrrk2-deficient mice do not develop spontaneous inflammation, but they are more susceptible to DSS experimental colitis compared to WT littermates, suggesting a role for innate immune cells in IBD progression (37). NCF4 encodes the p40-phox subunit of nicotinamide adenine dinucleotide phosphate oxidase that is crucial for reactive oxygen species (ROS) production by phagocytic cells in response to microbial infection. Genetic alterations in this CD-susceptibility gene result in abnormal neutrophil and MΦ recruitment, exacerbated cytokine secretion, impaired ROS production, and reduced bacterial clearance in CD patients (38, 39).
A proposed mechanism for the pathogenesis of CD is an exaggerated T (TH1/TH17) cell response toward luminal microbiota, which results in the breakdown of the mucosal tolerance to enteric bacteria (40). High levels of antibodies against these microbes correlate with disease progression, and TH1 and TH17 cells are massively recruited to mucosal sites and secrete cytokines, such as TNF-α, IFN-γ, IL-17, and IL-22, which participate to the inflammatory state in CD patients (41–45). GWAS indicated that IL-23 receptor gene and five additional genes involved in TH17 differentiation (IL12B, JAK2, STAT3, CCR6, and TNFSF15) are associated with susceptibility to CD (40). Anti-IL-12/IL-23 antibody therapy, which targets both TH1 and TH17 cells, is effective in CD (42, 46). Finally, the exact specificity of these T cells, their pathways of activation, and the mechanisms of their dysfunction in CD are still poorly understood. These aspects have been elegantly discussed by others (47, 48) and will not be examined in the current review.
Compelling evidence regarding the marginal role of heritable traits in triggering the onset and causing flares in CD has been provided by GWAS with monozygotic twin cohorts. Monozygotic twins have an (almost) identical genetic make-up, and they also have presumably similar environmental exposure and dietary habits in early childhood. They also have a similar gut microbial composition when healthy (49). Despite familial aggregation being a risk factor for developing IBD (2, 50), germline variation in CD loci of monozygotic twins only accounts for ~25% of estimated heritability, suggesting that the contribution of genetic factors to the etiology of CD is modest (51, 52). Differential exposure to environmental factors (so-called exposome) might account for the low level of concordance in monozygotic twins. Another working hypothesis is that T and B cell receptors (TCRs and BCRs) are not identical in monozygotic twins and that specific receptor rearrangements predispose to an immune-related sensitivity or autoimmune disease (53). In addition, a potential reason for relatively low concordance rates in monozygotic twins may relate to epigenetic changes. The study by Satsangi’s research team provides compelling evidence for epigenic modifications in several regions of the genome of pediatric and adult CD patients (54, 55).
Sensing of Microbes
Good fences make good neighbors
(Robert Frost, 1914) (56)
The mammalian intestine is inhabited by complex bacterial communities that contribute to the host metabolism and immunity. It remains under investigation how the host maintains homeostasis toward these bacterial populations, discriminating between commensal and pathogenic bacteria-derived signals. A growing body of evidence suggests that IECs contribute to mucosal homeostasis through the integration of microbial signals and interaction with immunocompetent cells (57). Current hypotheses suggest that the breakdown of the intestinal epithelial barrier function leads to increased translocation of food and bacterial antigens, to loss of immune tolerance to commensal bacteria, and to chronic intestinal inflammation, triggering the onset of CD. IECs contribute to tissue homeostasis by maintaining a rigorous spatial separation between microbiota and host, which limits immune activation and promotes induction of tolerance. Specialized IECs may sense the intestinal bacterial community through PRRs, such as TLRs and NOD receptor, and preserve mucosal homeostasis exploiting several strategies to restrict the colonization of resident and pathogenic bacteria and avoid immune overactivation. Processes engaged to this end encompass the secretion of an apical mucus layer by globlet cells (58), the secretion of AMPs by Paneth cells (59, 60), and the transport of secretory immunoglobulin across the epithelial barrier (61, 62). Mucus production has been shown to increase in germ-free (GF) mice exposed to TLR ligands, suggesting that bacteria-derived signals regulate goblet cell functions (58). Paneth cells are specialized cells in the crypts of the small intestine from which they exert a role in crypt homeostasis and maintenance of the intestinal stem-cell niche by secreting AMPs that keeps commensal and pathogenic bacteria at bay and regulate the composition of the gut microflora (63, 64). The expression of AMPs, such as RegIIIγ, is dependent on the recognition of bacterial molecules through Myd88-dependent TLR sensing. Evidence of this mechanism is provided by the proof that epithelial cell-specific Myd88-knockout mice and RegIIIγ-knockout mice loose the segregation between the microbiota and the intestinal surface, leading to a dramatic increase in mucosal-associated bacteria in the terminal ileum (65). Similar loss in spatial organization has been observed in biopsies from CD patients, characterized by lower AMP levels and consequently increased mucolytic bacteria and compromised mucus layer (66–68). Similarly, Muc2-deficient mice develop spontaneous colitis (69).
Small numbers of bacteria that breach epithelial barrier are killed by MΦs or can survive briefly in DCs promoting the induction of IgA by T-dependent and T-independent mechanisms (70). Hence, secretory IgA is selectively induced by DCs loaded with commensal bacteria resulting in local secretion of IgA that limits the penetration and overgrowth of commensal bacteria (71–73). The host may as well produce secretory IgA that is pathogen specific, but it is believed that indigenous bacteria are bound by low-affinity IgA, whereas enteric pathogens are highly coated with IgA (74). Flavell group recently demonstrated that GF mice colonized with highly IgA-coated taxa from IBD patients were much more susceptible to DSS colitis and displayed a bacteria-enriched inner mucus layer compared to GF mice humanized with low IgA-coated bacteria (75). These findings suggest that IgA coating is a defense mechanism that the host engages to mark and distinguish disease-driven bacteria from other members of the intestinal microbiota. The transport of IgA across the intestinal epithelium is modulated by the expression of the polymeric immunoglobulin receptor (pIgR) on the basolateral membrane of IECs. It has been reported that commensal bacteria may activate Myd88- and NF-κB-dependent signaling in IECs and consequently promote the expression of pIgR (62, 76). In addition, microbiota-specific TH17 cells may induce epithelial pIgR expression, therefore promoting intestinal IgA secretion and contributing to intestinal homeostasis. This finding is corroborated by the evidence that IL-17 receptor-deficient mice display reduced IgA levels in the gut that can be restored by TH17 cell transfer, and increased bacterial translocation and colitis scores upon DSS treatment (77). The serum of CD patients is enriched with high levels of IgA autoantibodies (78, 79). Furthermore, CD patients harbor a stronger humoral immune response against intestinal bacteria than healthy subjects, as shown by the detection of elevated IgG-coated bacteria in CD stool samples (80). This finding corroborates the concept of loss of mucosal tolerance for intestinal bacterial communities in CD patients.
Intestinal epithelial cells are currently emerging as key mediators of inflammatory and immune mechanisms in mucosal tissues. The transcription factor NF-κB has been shown to play a crucial role to control epithelial integrity and immune homeostasis toward gut bacteria in gnotobiotic mice and rats (81–83). Functional proof for the role of NF-kB signaling in the epithelium was provided by an elegant study from Nenci et al. (84). Mice-harboring IEC deletion of Nemo, upstream modulator of NF-κB, displayed destruction of intestinal barrier, dramatic translocation of bacteria from the lumen into the mucosa, and developed spontaneous colitis that was Myd88 and TNF receptor-1 dependent (84). Consistent with this protective role of inflammation-related signaling in the epithelium, the absence of TLR-related mechanism conferred increased susceptibility to DSS-induced colitis (85).
Nevertheless, the question remains how much TLR-related microbial sensing confers protection rather than tissue inflammation. It has been proposed that low expression of TLRs at the epithelial level, especially TLR2 and TLR4, is associated with mucosal homeostasis by maintaining a hyporesponsive state toward the presence of commensal bacteria (86–88). Notably, epithelial tolerance to microbial ligands seems to occur immediately after birth in order to promote a stable intestinal host–microbe homeostasis (89). Nevertheless, the high turnover rate of the epithelium may require additional immunosuppressive mediators of the mucosa such as transforming growth factor (TGF)-β and prostaglandin (PG) J2 in order to maintain epithelial cell homeostasis in response to the constant bacterial challenge (82, 83, 90). In the context of infectious or chronic inflammatory conditions, IFN-γ and TNF-α upregulate TLR expression in IECs (91). Consistently, TLR4 is significantly increased on the apical side of IECs throughout the lower gastrointestinal tract of CD patients (86). With respect to the maintenance of mucosal immune homeostasis, it has also been postulated that the expression of individual TLR is limited to specific cell lineages (92), and it is spatially restricted on the apical or basolateral intestinal surface (93, 94). For instance, TLR1, TLR2, and TLR4 seem to be coexpressed on a subpopulation of human and mouse cells located in the intestinal crypt and belonging to the enteroendocrine lineage (92). Analysis of polarized human IECs in vitro indicated that TLR5 is expressed on the basolateral surface and from this location triggers the production of cytokines and chemokines in response to flagellin (93).
Macrophages and DCs sense microbial components of the intestinal lumen, providing a key link between the microbiota and epithelial barrier functions (95, 96). The primary role of MΦs is to phagocytose cellular debris and microbes and stimulate lymphocytes and other immune cells to respond to the antigen through the secretion of cytokines and chemokines. Depending on the requirements of the surrounding tissue environment and of the stimuli encountered, MΦs express different biological functions exhibiting a remarkable plasticity (97). Gastrointestinal mucosal MΦs play a crucial role in maintaining gut homeostasis (98). Considering the huge bacterial load present in the intestinal lumen, it is likely that commensals (and pathogens) breach the epithelium. Mucosal MΦs are strategically located adjacent or in proximity of the epithelia and along the lamina propria. Intestinal MΦs are characterized by low production of proinflammatory cytokines but with an intact phagocytic ability (99). Indeed, this MΦ population helps to maintain a low level of inflammation in the lamina propria (state of physiologic inflammation), in order to prevent inappropriate immune responses to microbes. This is possible because intestinal MΦs express low level of TLR2 and TLR4, the two main receptors involved in sensing bacterial cell wall components (100). However, even though they may express TLR1, TLR3, and TLR5–9 to different extents (101), mucosal MΦs do not release proinflammatory cytokines in response to TLR ligands. They express low levels of TRIF, Myd88, and TRAF6 proteins, leading to an inability to phosphorylate NF-κB p65 and MAPKs (98). Intestinal MΦs retain a highly phagocytic activity, but they do not present antigen in normal intestinal mucosa as they lack constitutive expression of the costimulatory molecules CD40, CD80, and CD86, displaying a tolerance-inducing phenotype (98). In contrast, intestinal MΦs from CD patients express high levels of costimulatory molecules (102–104), increased activation of NF-κB signaling and oxidative burst activity (105, 106), high expression of TLR2 and TLR4 (107–110), and secretion of cytokines, including TNF-α and IL-23 (111).
Dendritic cells located in the lamina propria can penetrate the epithelium without disrupting the barrier function and acquire antigens from food or directly sample gut-associated bacteria and take them to mesenteric lymph nodes, where they are presented to CD4+ T cells. In contrast to DCs loaded with harmful bacteria that reach systemic secondary lymphoid structures (spleen and lymph nodes) and activate systemic immunity, DCs loaded with commensal bacteria migrate to the MLNs but remain confined in the mucosal lymphoid tissue (112). IECs release mediators (e.g., TGF-β, thymic stromal lymphopoietin, and PG E2) that maintain DCs in a quiescent state and promote the induction of regulatory T cells (113). This mechanism allows the host to develop systemic tolerance in response to commensal colonization which appears compromised in CD patients (114). During inflammation, IL-12 and IL-23 produced by DCs restrain regulatory T cells and promote TH1 or TH17 effector cells, respectively (42). The crucial role of IL-23 in promoting chronic mucosal inflammation has been emphasized by evidence suggesting that this cytokine, produced by innate immune cells, may inhibit FoxP3 Treg cell function and consequently positively modulate TH17 differentiation (115).
Ileitis Phenotype in Mouse Models with Deficiency in Crohn’s Disease-Associated Genes
Genetic polymorphisms associated to CD may alter immune responses to commensal bacteria and mucosal barrier function impacting the microbiota composition in the gut. The possible mechanism for genetic regulation of enteric microbiota include altered Paneth cell function but also altered barrier functions and mucus production, defective secretion of IgA, and altered innate and adaptive immune responses.
Paneth cells are specialized IECs located adjacent to the stem cell zone in the base of the crypts in the small intestine. They are critically involved in host defense against enteric pathogens by secreting AMPs and TNF-α (116, 117). Mutations in genes associated with CD, usually highly expressed in Paneth cells, predispose to the development of ileal lesions in humans (118). In line with this, mouse model with deficiency in several CD-associated genes, including Nod2, Atg16l1, and Xbp1, displays Paneth cell defects and susceptibility to intestinal inflammation (119, 120).
Nucleotide-binding oligomerization domain 2 is a well-acknowledged susceptibility gene for CD (16, 18, 121). NOD2 is thought to play a relevant role in maintaining microbial tolerance at the intestinal barrier (16) and to activate innate and adaptive immunity (63). Three common mutations in the C-terminal leucine-rich repeat region of the NOD2 gene, namely G908R, R702W, and the frameshift deletion mutation L1007, and several other rare polymorphisms have been discovered and associated to increased risk of developing CD. How the NOD2 variants increase susceptibility to CD remains debated. For instance, the frameshift variant L1007 has been shown to decrease NF-κB activity in HEK293 cells stimulated with a number of common bacteria (17). This observation contradicts the evidence that CD clinical specimens are characterized by elevated NF-κB activity (122). One possible explanation is that the mutation in NOD2 leads to a defect in the innate immune response allowing intracellular bacteria to escape the first line of host defense, resulting in an enhanced adaptive response. In support of this hypothesis, elevated proinflammatory cytokine production was detected in splenocytes and blood mononuclear cells from Nod2-deficient mice challenged with TLR2 ligands (123, 124). Furthermore, Nod2−/− mice become more susceptible to colitis as a result of enhanced TLR2 responses characterized by increased production of IL-23 and IL-12 (124). However, the role of NOD2 as negative regulator of TLR2 is controversial since the same L1007 frameshift mutation has been shown to protect mice from systemic infection (and inflammation) by Enterococcus faecalis that is a Gram-positive bacterium and therefore sensed mainly by TLR2 (125). Notably, Nod2−/− mice do not spontaneously develop an inflammatory phenotype (63) but exhibit a higher load of commensal bacteria (i.e., Bacteroidaceae) in the terminal ileum and Peyer’s patches and a reduced ability to prevent pathogenic bacteria colonization (126–128), suggesting a genotype-driven selection of a pathobiont-enriched microbiota. Similarly, the NOD2 variant L1007 is associated with higher colonization of the intestinal mucosa by the Bacteroidaceae in humans (129). It is still not clear whether the observed dysbiosis is the cause or a consequence of the disease, but it is tempting to speculate that NOD2 variants are associated with changes in the composition and load of the commensal microbiota in the terminal ileum that may facilitate disease progression and pathology. In line with this speculation, WT mice develop colitis when recolonized with dysbiotic fecal microbiota from Nod2-deficient mice (130). In addition, crypts and Paneth cells from Nod2−/− mice have attenuated antibacterial activity and decreased expression of α-defensin and cryptidin leading to increased susceptibility to Listeria monocytogenes infection in vivo (63, 126). These findings are in agreement with the deficiency in α-defensin production observed in CD patients (3, 131, 132).
In the gut, bacterial growth and division contribute to the remodeling of components of the bacterial cell wall, i.e., pepdidoglycan (PGN), by bacterial autolysins, e.g., muramidases and amidases (133). During this process, soluble PGN fragments can be transferred to the bloodstream and initiate innate immune responses through PRRs, including NOD2 and TLR2 (134, 135). Specific classes of bacteria, such as Actinomyces and Mycobactera, use hydroxylases to covalently modify MDP-generating moieties that activate more potently the NOD2 pathway. This evidence raises the intriguing possibility that bacterial components may account for the differential activation of immune cells resulting in detrimental effects on a host immunity already compromised, such as in CD.
Nucleotide-binding oligomerization domain 2 has been reported to account for stem cell protection and to contribute to stem cells regeneration via responses triggered by MDP recognition and leading to the recruitment of NF-κB at the membrane surface of IECs (136–139). Conversely, NOD2 mutant 3020insC, which is associated with CD, shows an impaired ability to activate NF-κB following MDP stimulation in vitro (138, 140, 141). In the absence of bacterial invasion into the host cytosolic compartment, it has been postulated that MDP can cross plasma membrane and localize into the cytosol via the plasma membrane transporter, PepT1 (142, 143). During chronic inflammation, such as CD, PepT1 expression has been shown to be upregulated in the colon (144, 145). Nevertheless, a recent study argues against the indication of a role for PepT1 in the development of intestinal inflammation, showing that in animal models resembling Crohn-like ileitis (TNFdeltaARE) and colitis (IL-10−/−, IL-10XTLR2−/−, and Rag2−/−) and in human intestinal tissues from IBD patients, severity of inflammation correlated with lowered PepT1 expression levels (146).
Nucleotide-binding oligomerization domain 2 is involved in the cellular protection mechanism called autophagy, by directly interacting with the ATG16L1. Since both of these genes are CD-susceptibility loci, it has been proposed that loss of autophaghy-related function is implicated in the pathogenesis of CD. DCs (147), MΦs (148), and epithelial cells (149) containing ATG16L1 and NOD2 variants show defects in antibacterial autophagy. In DCs, these defects are associated with an impaired ability to process pathogens (such as Salmonella typhimurium) and present exogenous antigens (such as MDP) to CD4+ T cells (147). Murine Atg16l1- and Nod2-knockout MΦs secrete aberrant IL-1β levels in response to LPS (150) and MDP (122), respectively. These findings highlight that CD-susceptibility genes require interaction with environmental (microbial) cues to manifest a disease phenotype. This is further supported by the observation that GF hypomorphic Atg16l1 mice are disease free, whereas colonized mice display aberrant Paneth cells morphology and antimicrobial protein expression, and they are highly susceptible to DSS-induced colitis (119, 150, 151). This phenotype has been recently associated to dysfunctional IECs due to mutations in the autophagy machinery rather than arising from defects in granule formation in Paneth cells (152). In fact, the author exploited IECs-specific Atg16l1-knockout mice to demonstrate that the deletion of the autophagy-related gene is responsible for reduced Paneth cell number, abnormal granule morphology, reduced expression of AMPs, and increased inflammation and systemic translocation of S. typhimurium compared with control mice (152). Similarly, CD patients carrying the ATG16L1 T300A mutation show an autophagy-associated defect in Paneth cells with granule abnormalities (153). Exploiting mice with epithelial cell-specific Myd88 deletion, Hooper group demonstrated that only invasive bacteria, such as S. typhimurium and E. faecalis, can activate autophagy in IECs in a Myd88-dependent fashion (154). In addition, the authors showed that autophagosome formation in vivo is TRIF independent, while in RAW MΦs autophagy was reported to be depended on this adaptor molecule (155). Nod2−/− mice do not harbor defects in autophagosome formation, rather lack of the intracellular PRR is associated to an increased number of bacteria within ileal IECs compared to WT mice, as shown by FISH analysis (154). This finding suggests that bacterial breach of the intestinal epithelium triggers autophagy in IECs in Nod2-independent manner. Mice with an IEC-specific deletion of the essential autophagy gene Atg5 display decreased numbers of autophagosomes but no histological evidence of pathology and an increased number of intracellular S. typhimurium (154). These findings highlight the crucial role of IECs as a selective barrier that contributes to maintaining mucosal homeostasis by finely tuned communications with gut microbiota and the luminal environment, peripheral tissues, and the immune system (156).
Multiple cellular stress responses were observed in IBD, including endoplasmic reticulum (ER) and mitochondrial (mit) unfolded protein responses (UPRs). The accumulation of misfolded proteins in the ER and mit lumen of IECs activates several processes, including inflammation and UPR signaling pathways, and the integrated stress response (157, 158). TLR signaling and autophagy cooperate in bacterial sensing actively interacting with cellular stress responses, i.e., UPR. UPR signaling is mainly driven by inositol requiring, ER-to-nucleus signaling protein 1α–X-box-binding protein-1 (XBP1) pathway. XBP1 gene deletion may lead to increased ER stress as a consequence of a defective UPR in secretory IECs (i.e., goblet and Paneth cells), thereby affecting their function (120). While Xbp1-deficient mice are protected from ileitis in GF housing, colonized Xbp1−/− mice develop spontaneous transmural inflammation, resembling Crohn’s ileitis phenotype in humans (120, 159). In particular, mice with Xbp1-deficient IECs develop spontaneous enteritis and display ER stress with high level of the chaperone grp78 and C/EBP homologous protein (Chop), Paneth cell loss, and reduced globlet cell number and size, compromised response to pathogenic bacteria (i.e., L. monocytogenes), and they are more susceptible to DSS colitis (120). Overexpression of CHOP in IECs aggravates DSS-induced colitis and impairs mucosal wound healing (160). In vitro, XBP1 deficiency induced ER stress that led to a heightened JNK-dependent proinflammatory response of epithelial cells to flagellin and TNF-α (120). The emerging role of ER stress in the progression of CD is supported by the detection of high levels of ER stress markers (i.e., grp78) in ileal and colonic epithelia of CD patients (120, 161, 162). Generation of multiple cellular stress responses, and consequently inflammation, in the intestinal epithelium is multifactorial and includes genetic and environmental factors (163, 164). Paneth cells from patients with quiescent CD and healthy controls carrying the ATG16L1 T300A risk allele are characterized by increased ER stress (153). This finding indicates an interaction between ER stress mechanisms and autophagic pathways. For instance, CD patients may harbor a defect in barrier function which results in overactivation of Paneth cells by bacterial ligands, requiring a higher demand of secretory AMPs, potentially leading to ER stress (165). It is possible to speculate that progression of ER stress in CD patients may be facilitated by the production of ROS induced by the inflammatory cytokine TNF-α (166).
The evidence of barrier dysfunction in CD patients, associated with the inability to achieve mucosal healing or to seal off the damaged epithelium, confers a role of relevance to tight junction proteins as risk factors for disease development (167). Notably, aberrant (high) tight junction-dependent paracellular permeability (168, 169) and exacerbated TNF-α level has been observed in CD patients as well as in their healthy first-degree relatives (170–173). A proposed mechanism is that inflammatory cytokines, i.e., TNF-α, may activate myosin light chain kinase (MLCK) which induces tight junction disruption ultimately leading to cytoskeletal-related barrier defect (174–177). This phenomenon leads to the activation of systemic T cell-mediated immune responses and impaired barrier function (178). A major validation of this molecular mechanism comes from the observation that wild-type mice treated with MLCK inhibitor and mlck-knockout mice are protected from barrier dysfunction (174, 178). In contrast, transgenic mice expressing mlck constitutively (CA-MLCK Tg) displayed increased paracellular permeability within the small intestine and colon, without developing spontaneous disease (179). Intriguingly, transfer of colitogenic T cells into CA-MLCK Tg mice accelerated the onset and severity of colitis (179). This finding suggests that increased gut permeability is insufficient to trigger disease in the absence of other predisposing factors, which most likely include immune dysregulation and altered luminal microbiota.
What is the Contribution of Microbiota to CD?
The microbial composition in the intestinal tract is considered another potential risk factor in individuals with CD (51, 180). There are several lines of evidence for microbial involvement in IBD. For instance, (i) inflammation occurs in regions with higher bacterial density, such as distal ileum and colon, (ii) GF animals do not develop ileitis (181) or colitis (182), (iii) antibiotics have shown some therapeutic efficacy in IBD patients (183), (iv) the severity of the disease correlates with the bacterial density in the intestinal mucosa, and (v) a large number of studies reported an altered bacterial composition in IBD patients compared to healthy individuals, so-called dysbiosis (184).
The Gastro Intestinal Ecosystem – Who is There?
The mammalian gastrointestinal tract is habitat to taxonomically diverse microorganisms in very close proximity to the host. The totality of these microorganisms, so-called “microbiota,” includes bacteria, viruses, archaea, and eukaryotes (yeasts, protozoa), and their genes represent the “intestinal microbiome.” The human and murine intestines are dominated by the bacterial phyla Bacteroidetes and Firmicutes with a minor proportion represented by Proteobacteria, Actinobacteria, Verrucomicrobia, Tenericutes, and Fusobacteria (185, 186). At lower taxonomic levels, the diversity is very high, with ~100–200 bacterial species per individual (187). Notably, great interindividual differences were observed in the overall microbial community, highlighting the limitation in defining the absolute composition of a “healthy” microbiota (188–191). In the last decades, compelling evidence emerged pointing out the pivotal role of the intestinal ecosystem in defining the host immune homeostasis (e.g., by inducing pro- or anti-inflammatory responses) (192). Therefore, the study of shifts in the intestinal ecosystem is of fundamental importance to understand associations and causalities between gut microbes and immunity.
Characteristics of Dysbiotic Ecosystems
Under physiological conditions, the microbiota shows both plasticity and high resilience, i.e., upon short-term perturbations (e.g., change in dietary pattern), the microbial composition adapts to alterations in the intestinal milieu, though soon resembles a predisturbance state (189). The microbial ecosystem can also be changed without pathologic consequences for the host and stabilize within a new “alternative state” (193). Therefore, the microbiota is capable of adapting to exposomal factors including diet, smoking, antibiotics, or intrinsic factors like host genetics. Nevertheless, diet seems capable to overwrite the influence of genetic imprint (194–196). Rapid resilience to perturbations is a key requirement for the intestinal homeostasis and the host’s health in order to maintain a health-associated composition of the ecosystem (eubiosis). However, this resilience is lost in some pathologic conditions, like IBD. In coexistence with genetic susceptibility to inflammatory processes – like in IBD patients – the microbiota is causative for disease development and is therefore considered as “dysbiotic” (197).
For different pathologies, there is a strong evidence for the role of “dysbiosis,” defined as an alteration in the ecosystem associated to pathology (180, 198–200). While for CD and UC, the connection between dysbiosis and pathology seems well established; also other pathologies like obesity, diabetes, cardiovascular disease, and even depression or multiple sclerosis have been recently connected to intestinal dysbiosis (198, 201–204)
However, dysbiosis displays several features, which will be in the present review categorized in (i) reduced bacterial diversity, (ii) expansion of pathobionts, (iii) changes in the microbial composition, i.e., increase or reduction in indicator species, and (iv) change in microbial functional capacity (205). Thereby, the appearance of these characteristics may occur solitarily, successively, or simultaneously.
The most widely discussed attribute of dysbiosis is reduced bacterial diversity. Based on the numbers of bacterial species and their abundance found within one sample, the alpha diversity can be calculated. Many studies associated lowered bacterial diversity to disease, with the rationale of loss in metabolic redundancy (180, 206–209). Several reports have addressed dysbiosis and decreased diversity in IBD patients’ display compared to healthy individuals (51, 180, 200, 206, 210–214). Furthermore, the ability to outcompete pathogens by a low-diverse microbiota is diminished. In patients who underwent frequent antibiotic treatment, the deteriorated intestinal diversity was shown to increase the risk of infection by opportunistic pathogens, such as Clostridium difficile (215). In animal models, pathogens including Salmonella, Citrobacter rodentium, or enterohemorrhagic Escherichia coli fail to colonize in the presence of a diverse, undisturbed microbiot but elicit pathogenic traits if competing strains are missing (216–219). The mechanisms of competitive exclusion may correspond to rivalry for nutrients or virulence modulation of intruding strains (220).
Dysbiosis may also be simplified by linking it to expansion of pathobionts, i.e., single strains of the commensal microbiota that outgrow and cause detrimental effects in the host. The term pathobiont was defined by Chow and Mazmanian as “…symbiont that is able to promote pathology only when specific genetic or environmental conditions are altered in the host” (221). While pathobionts are found only in low abundance in a healthy microbial setting, they overgrow in dysbiosis and cause disease in the susceptible (e.g., immune compromised) host. Hereby, specificity in the combination of microbe and host susceptibility is required. In animal studies, it was shown that Bacteroides vulgatus induced colitis in HLA/B27-ß2m rats, but not in IL-10−/− mice and even prevented colitis in IL-2−/− mice (222, 223). Early studies regarded Mycobacterium avium subspecies paratuberculosis as the responsible pathobiont or even pathogen in IBD. However, this hypothesis could not be verified, as summarized by Packey and Sartor (224). In stool samples and mucosal specimens from IBD patients, an increased abundance of Enterobacteriaceae is repeatedly observed, and among these the E. coli strain LF82 is discussed to be a pathobiont (225–227). The group around Darfeuille-Michaud was the first to describe adherent-invasive E. coli (AIEC), which selectively colonize the ileum of CD patients, suggesting that dysbiosis in IBD may also relate to strain-specific virulence factors (6, 228–231). The authors showed that the AIEC strain LF82 is able to persist within MΦs and epithelial cells and selectively colonizes the ileum of CD patients.
Apart from single pathobiont alterations, dysbiosis is in most cases regarded as shifts in the overall microbial composition, i.e., simultaneous increased or decreased abundance of certain commensals. Due to the new sequencing techniques and improved databases, great progress has been made in characterizing the intestinal microbiome also in larger cohorts. However, most samples were taken from patients who already underwent some sort of treatment, which exerts changes in the ecosystem and thereby impede conclusive interpretation of findings (232–234). A study from Gevers et al. elegantly solved this issue by picturing the treatment naive microbiome in children recently diagnosed for CD, before shifting the intestinal ecology by different forms of pharmaceutical or nutritional intervention. They described an increase in Enterobacteriaceae preceding the onset of CD, an observation made by other studies as well (199, 234, 235). It is worth noting, however, that small inflammatory lesions may result in changes in microbiota composition prior to the diagnosis of disease. Thus, prospective cohorts would be of high value but demanding considering the low incidence of IBD. Among the multitude of studies performed to detect IBD-associated bacterial taxa, little congruence is found between different cohorts. By combining the sequencing data from several studies, Walters et al. showed shifts in the bacterial composition at different taxonomic levels, which were at least partly consistent for several studies and cohorts (227). They showed higher abundance of Actinobacteria and Bacteroides sp. and a loss of Prevotella sp. in CD patients compared to healthy controls. They also described at lower taxonomic level the loss of some indicator species in IBD, like Faecalibacterium prausnitzii. This health-associated species was found in significantly lower levels in the inflamed intestine compared to healthy specimens and exerts positive immune-regulatory effects on the host (236, 237). Therefore, loss of F. prausnitzii is speculated to be an indication for increased IBD risk (211, 236, 237). However, use of just a single indicator species as diagnostic tool may not be sufficient, as IBD-associated strains may be cohort or individual specific. Nevertheless, the novel approach of using the analysis of overall drifts in the intestinal, preferably stool microbiota as a diagnostic tool is a promising new tactic in IBD diagnosis. Walters et al. highlighted some universally valid disease-related shifts, which may be powerful enough to securely diagnose IBD, by combining the sequencing results from several studies (227). By using stool microbiota, this non-invasive approach is even more advantageous.
By great advances in 16S rRNA profiling methods, shifts in the prevailing bacterial members of the intestinal composition can be more easily assessed and discussed. However, most descriptions of intestinal dysbiotic communities fail to take fungal and viral contributions into consideration. Recently Chehoud et al. could show reduced fungal diversity in pediatric CD accompanied by increased Candida taxa (238). Norman et al. showed marked differences in the intestinal virome in CD and UC patients compared to healthy controls (239). The main difference was an increase in Caudovirales bacteriophages which was not secondary to bacterial dysbiosis, but it is more likely to assume that viral dysbiosis contributes to pathology and changes in the bacterial ecosystem due to a “predator–prey” relationship (240–242). However, studies of the role of other microbial taxa in IBD pathogenesis are awaited and will be essential to unravel dysbiotic patterns.
In addition to bacterial changes in composition and diversity, changes in the functional metabolic capacities are characterizing a dysbiotic ecosystem on a functional level. As shown by the human microbiome project, the healthy intestinal ecosystem may be highly different in composition, while its metabolic activity is highly similar (190). An interesting study in IBD patients by Morgan et al. showed a small perturbation of the intestinal composition, though a quite distinct change in microbial function (235). In dysbiotic conditions, microbial pathways for oxidative stress tolerance, immune evasion, metabolite uptake, and carbohydrate as well as amino acid biosynthesis were upregulated. An increase in carbohydrate metabolism and especially changes in the metabolic capacity to utilize fucose were reported in dysbiotic settings in CD patients or animal models of inflammation by others as well (181, 243–245). Metaproteome analysis in a cohort of CD patients also proved a distinct protein signature associated to CD (246). By correlating these functional shifts to the bacterial ecosystem, Bacteroides-derived proteins related to survival in challenging environments (e.g., DnaKs and other chaperones) were found overrepresented. Dysbiosis may also be described by changes in the intestinal microbial function and consecutively produced metabolites (247, 248). This leads to the assumption that dysbiosis may be more precisely characterized by changes in the microbial function rather than composition.
Dietary fibers are often associated to be reducing the IBD risk, as fibers are metabolized to short-chain fatty acids (SCFA) by microbes in the distal gastrointestinal tract (249). Those SCFA were found to hamper the growth of pathogens, increase the intestinal barrier function, and serve as energy source for colonocytes (250–255). Furthermore, they also facilitate the generation and differentiation of regulatory T cells in the gut and thereby are important to maintain a homeostatic environment (252, 254, 256). Machiels et al. have reported that UC is associated with impairment in SCFA production (237). Though the authors observed a reduction in butyrate-producing Roseburia hominis and F. prausnitzii, as well as a reduction in butyrate levels, they could not draw a direct correlation between these findings. This provides evidence that the microbiota may not only be divergent regarding the abundance of community members but also in their metabolic activity.
Furthermore, the definition of dysbiosis should not be a mere one-sided microbial consideration but should take the host into account as well. Palm et al. showed that IBD patients display an altered immune recognition of a dysbiotic microbiota correlating with an increased and divergent IgA coating (75). By using an animal model of chemically induced colitis, they also transferred the disease susceptibility and thereby proved the true causal role of this IgA-coated ecosystem. Consequently, dysbiosis may also be defined as an “alteration of symbiosis” with pathophysiologic consequence (257).
What Causes Dysbiosis?
The sum of environmental triggers, so-called exposome, including diet, early nutrition (breast or formula feeding), mode of delivery, hygienic milieu (contact to disinfectants or animals), stress, drugs, lifestyle, and geography (e.g., air pollution and location) may contribute to shape the intestinal ecosystem (Figure 1) (185, 208, 258–266). Diet has an enormous impact on the intestinal ecology. For instance, diet high in fat shifts the microbiota toward a more dysbiotic pattern associated with increased risk of intestinal inflammation. David et al. effectively showed the plasticity and the stability of the intestinal ecosystem upon short-term perturbations by feeding mice an exclusively animal- or plant-based diet (267). Animal-based diet promotes bile-tolerant microorganisms, such as Alistipes or Bilophila (194, 267). For instance, the increase in Bilophila is of interest considering that Bilophila wadsworthia was previously shown to exert pathobiont behavior in IL-10−/− (268). In another study, a western diet (rich in fat and sugar) induced dysbiosis and favored the increase in AIEC LF82 upon colonization in genetic susceptible CEABAC10 mice (269). Several other studies showed that dramatic shifts in the intestinal ecosystem are not induced by short-term changes in dietary habits or application of single nutrient but require long-term pressure on the ecosystem (189, 270).
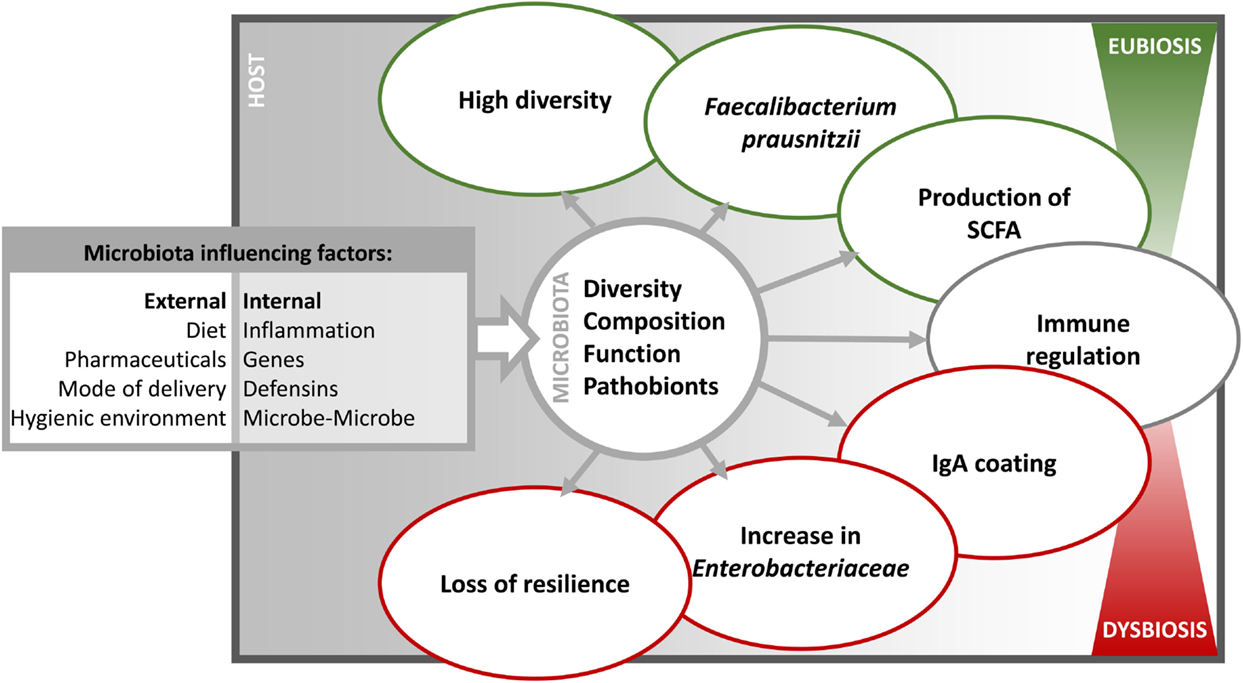
Figure 1. The different influencing factors and characteristics of dysbiosis. The intestinal microbiota is influenced in diversity, composition, function, and prevalence of pathobionts by internal and external factors. Consequently, the microbiota varies in immune relevant characteristics and thereby shifting from a eubiotic to a dysbiotic state.
Beside exposomal factors, also intrinsic factors, i.e., host genetic, shape the intestinal microbiota. Several genes associated with altered immune function, microbial recognition, or antimicrobial defense were shown to influence the intestinal ecosystem. In a meta-analysis, Knights et al. highlighted the significant association between the NOD2 risk allele and increased abundance of Enterobacteriaceae in IBD patients (271). This observation was backed up by cohousing experiments of NOD2-deficient mice which develop a genotype-induced dysbiosis. By transferring this dysbiotic microbiota to WT cage-mates, they became more susceptible to DSS colitis (130). In another study, Rausch et al. showed an association between the intestinal microbiota and the FUT2 (secretor) gene – a physiological trait that regulates gastrointestinal mucosal expression of blood group A and B antigens. Approximately 20% of humans lack the FUT2 gene, which was shown to be a risk factor for CD development (272). Within the group of CD patients, the microbiota from FUT2 carriers differed from non-secretors in composition, diversity, and functionality (245, 273).
In intestinal pathologies, such as IBD, dysbiosis is associated to inflammation. It has been proposed that the shift in bacterial composition is due to the inability of some bacterial taxa to adapt to the inflammatory milieu characterized by increased secretion of AMPs (274). The potential of AMPs to shape the intestinal ecology and their importance in IBD has been shown by others before (275). The inflammatory process may also induce stress or upregulation of virulence-associated genes in the microorganism, as shown in monoassociation studies with the gut commensal strains E. faecalis OG1RF (276) or E. coli NC101 (277, 278). For instance, metalloprotease gelatinase E (GelE) secreted by E. faecalis contributes to the development of chronic intestinal inflammation in mice that are susceptible to intestinal inflammation (IL-10−/− and TNFdeltaARE mice) by impairing epithelial barrier integrity (279). E. faecalis isogenic mutants lacking GelE, enterococcal polysaccharide antigen, or lipoproteins (Lgt) show significantly decreased inflammation in the distal colon of monoassociated IL-10−/− mice and impaired TLR2-mediated activation of DCs (276, 279). E. coli NC101 cells isolated from inflamed IL-10−/− mice displayed upregulated bacterial-stress response genes (e.g., heat shock proteins) compared to isolates from WT mice (277, 278). However, inflammation may also promote growth advantages and virulence of pathogens. For S. typhimurium, it was shown that the host produces tetrathionate under inflammatory circumstances, which consequently promotes the ethanolamine utilization of S. typhimurium and thereby its colonization fitness (280, 281). Also nitrate produced by the host during inflammatory processes is assumed to promote growth of Enterobacteriaceae. Consequently, the often discussed increase of Enterobacteriaceae in IBD may also regard as secondary to inflammatory processes rather than causative (282).
Apart from host-derived factors, also microbe-derived mechanisms exert impact on other commensals. The transfer of single strain or bacterial consortia to GF mice enables to study mechanisms of microbe–microbe interaction. Commensals are capable of inhibiting growth of pathogens by, e.g., competition for nutrients, quorum sensing-mediated colonization repression, as well as expression of virulence factors (220, 283–285). Metabolic interaction (food chain) of commensals and pathobionts are also shaping the intestinal ecosystem, as some organisms depend on the conversion of dietary components or induction of host-derived nutrients by other members of the microbiota (217, 286–288). Also quorum sensing-mediated repression of colonization is regarded as an important mechanism of shaping the intestinal ecosystem, and recent findings from Thompson et al. demonstrate the role of autoinducer-2 in reshaping antibiotic-induced dysbiosis in the gut (284, 289).
The Role of the Microbiota in Animal Models of Intestinal Inflammation
Most dysbiosis-influencing factors cannot be studied under highly controlled and standardized conditions in humans. To discover causative dysbiotic shifts in the microbiota prior to disease onset, large prospective cohort screenings would be necessary, which are hardly feasible due to low incidence rates. Therefore, animal models of intestinal inflammation are gaining more and more importance, as confounders like diet, genetic background, and hygienic environment can be continuously controlled. Genetically driven animal models developing inflammation without any chemical inductor (e.g., DSS or TNBS) are especially appropriate to study the interaction of the intestinal microbiota and pathology, as the chemical compound per se would influence the ecosystem. Ever since the availability of GF and gnotobiotic animal models, great progress has been made to portray microbe–host interaction. Hereby, many IBD mouse models were shown to be free of disease in GF conditions and thereby clearly prove the causality of microbial triggers in IBD development.
The IL-10−/− mouse – incapable of producing the anti-inflammatory cytokine IL-10 – is the best-studied model of colitis. In GF housing, IL-10−/− mice do not develop inflammation, and interestingly the time point of microbial colonization seems to be of importance, as IL-10−/− mice colonized at adult age developed more severe inflammation (182). A more recent model of UC is the Rag2−/− × Tbx21−/− (TRUC) mouse model. Those mice develop UC-like colitis due to the absence of an adaptive immune system. TRUC mice were free of colitis in GF conditions as well (290). Another model protected from inflammation under GF conditions is the TCR alpha-knockout model. These mice are characterized by a defective immunity as well, as αß T cells are absent (291). Also severe combined immunodeficiency (SCID) mice, having an impaired B and T cell functions, are free of colitis in GF-housing conditions (292).
However, all the above-mentioned models display a colitis phenotype. To study mechanisms relevant in CD, which is characterized by ileal involvement, just few animal models are available. The SAMP1/YitFc mouse model is one of the rare models displaying a CD-like inflammation, with still uncertain mechanism of pathology induction (293, 294). Nevertheless, a recent study suggests that inflammation may be mediated by loss of CCL21 signaling and DC migration from the ileal lamina propria to mesenteric lymph nodes (293–297). Another model of CD-like ileitis is the TNFdeltaARE mouse model. These mice have increased TNF levels due to a deletion of the adenosine–uracil-rich element in the TNF transcript (298). The pathology in TNFdeltaARE mice with their transmural ileitis and the Th-1-driven immune response give a very good resemblance to the inflammation in CD patients. In line with this, TNF antibodies, which are a treatment option in CD patients, were shown to hamper inflammation in TNFdeltaARE mice (299). Just recently, the necessity of a microbial trigger for inflammation development was demonstrated, as TNFdeltaARE mice are free of inflammation in GF housing (181). In contrast to SAMP1/YitFc mice, the inflammation in TNFdeltaARE mice is not characterized by the formation of “cobblestone” structures – a thickening of the gut wall with protruding lesions found also in some patients with progressive IBD in humans (300, 301). These divergent three-dimensional structural inflammatory phenotypes again highlight the necessity and basic requirement of different mouse models to study the mechanisms of inflammation development and to be able to characterize the complexity of inflammation in IBD.
Mice with a conditional deletion of caspase 8 (a protease involved in apoptosis regulation) in the IECs (Casp8deltaIEC mice) spontaneously develop chronic inflammation in the terminal ileum due to increased necroptosis (302). Notably, antibiotic-treated Casp8deltaIEC mice were rescued from ileitis, indicating an important role for the intestinal microbiota in the development of the disease (303).
Finally, the development of animal models that mirror human genetic risk factors for IBD or other pathologies may be an important step toward unraveling mechanisms of pathology. The disease-free status of animal models in GF housing is exploited as valuable tool to investigate the development of dysbiosis, its influencing factors, and also mechanisms of microbe–host interaction in gnotobiotic setups.
Dysbiosis and Microbe–Host Interaction in Gnotobiotic Animal Models
Specific mechanisms of disease induction can be studied through the colonization of GF mice with single bacterial strain. Therefore, many colonization studies have been performed for models of intestinal inflammation as well as for other pathologies, including obesity, diabetes, and multiple sclerosis, as recently summarized (304). However, by colonizing immunocompromised GF animals with selected single strain, several obstacles are met. Not all microbes colonize equally, and the induction of inflammation depicts bacteria as well as model-specific traits.
In the IL-10−/− model, several monoassociations with candidate pathobionts failed to induce inflammation, such as Helicobacter hepaticus and E. coli, while other bacteria were effective in developing pathology, such as E. faecalis or B. wadsworthia (279, 305, 306). These findings suggest that an association with only one bacterial strain may not be sufficient to induce the necessary immune maturation required for the establishment of the inflammatory status. Atarashi et al. could show that one strain of Clostridium was not sufficient to induce regulatory immune functions in former GF animals, as this relies on concerted actions of different strains (307, 308). It is tempting to speculate that not all members of the microbiota are equally sufficient to induce pathology but rather requires the interaction with other commensals. An example of this hypothesis encompasses monoassociation of IL-10−/− mice with H. hepaticus or Lactobacillus reuteri, which as single strain did not induce inflammation, while the combination of the two bacteria induced colitis (306). Therefore, current studies tend to use complex microbial settings or defined microbial consortia to ensure sufficient immune maturation in GF animals. Garrett et al. showed that colitis in TRUC mice correlates with increased abundance in Klebsiella pneumonia and Proteus mirabilis. However, the combined colonization of TRUC or Rag2−/− recipient mice with both these two bacterial strains did not induce inflammation. Just a combined colonization of K. pneumonia, P. mirabilis, and a complex microbiota induced inflammation in recipient Rag2−/− mice (290). However, it has to be mentioned that WT cage-mates were also inflamed, suggesting a rather pathogenic than dysbiotic trait of the microbiota. Interestingly, Powell et al. identified Helicobacter typhlonius as a key driver of pathogenesis in TRUC mice (309). The transfer of a complex microbiota from inflamed TRUC mice induced colitis in gnotobiotic TRUC mice as well. However, when the microbiota from antibiotic-treated mice in remission was transferred, the recipients developed only attenuated inflammation (310). In the SCID mouse model, Stepankova et al. showed inflammation development by segmented filamentous bacteria (SFB) only in combination with complex specific pathogen-free (SPF) microbiota (292). SFB are known to be potent inducers of the host’s immune response as summarized elsewhere (292, 311). This also points out the fact that monocolonizations may pose elegant ways to prove causality, whereas complex mechanisms of disease initiation may need more complex interactions. TNFdeltaARE mice colonized with the pathobiont E. coli LF82 did not develop ileitis, whereas the disease was induced upon colonization with microbiota from inflamed donors (181). Those mice showed in SPF housing high variance in ileitis development due to spontaneous shifts in the intestinal microbiota, i.e., inflamed mice displayed microbial patterns distinct from non-inflamed mice. By transferring the microbiota from inflamed TNFdeltaARE to GF mice, the TNFdeltaARE recipients develop inflammation. The WT cage-mates did not develop inflammation, showing that the microbiota was truly dysbiotic and not pathogenic, as susceptibility of the host is needed for inflammation development. By transferring the microbiota from non-inflamed TNFdeltaARE mice, no inflammation was observed in TNFdeltaARE or WT recipients. This clearly pinpoints that the concerted action of a complex microbiota with dysbiotic patterns and the genetic susceptibility are precondition for the CD-like pathology in this model.
The studies mentioned in this review clearly emphasize the role of dysbiosis in intestinal pathology, as well as its multifactorial etiology. Until now, no single microbial trait could be identified as a clear indicator of a dysbiotic state itself. It was rather shown that dysbiosis is characterized by multifaceted variations in community networks and displays specificity for the respective host and inflammatory condition. Several human studies reported dysbiosis before onset of disease (234, 312). In animal models of intestinal inflammation, the causal relationship of a dysbiotic ecosystem in inducing inflammation was effectively shown by transferring the microbiota directly or by cohousing animals and thereby inducing pathology in recipients (130, 181, 290, 310). In these animal models which have different mechanisms of pathology induction, a single microorganism is not capable to induce tissue inflammation but needs the concerted action of a complex microbiota. These observations are further supported by the fact that no single pathobiont has been found that is consistently present in different IBD cohorts. More and more studies show that dysbiosis is characterized by changes in the microbial composition and microbial function rather than mere reduction in diversity or expansion of pathobionts. This may also explain why by now, no single probiotic treatment was found able to restore dysbiosis by administration of just one single probiotic strain, suggesting that one single species may not be powerful enough revert complex dysbiotic shifts.
Rebiosis as Concept for Clinical Therapy
Even though a high number of studies have been performed on the benefits of probiotics in IBD, new meta-analyses show no overwhelming or placebo-superior effect (313, 314). Strain-specific mechanisms were investigated showing the potential of probiotics to enhance intestinal barrier integrity, to counteract proinflammatory cytokines, or to induce an anti-inflammatory immune response (315–317). Newer studies go from single probiotic strain or mixtures with low defined numbers to more complex setups. In C. difficile-induced infection, fecal microbiota transplantation (FMT) seems to be very promising and superior to antibiotic treatment regimens (318–322). Hereby, the complex fecal microbiota of a healthy donor (often a relative or spouse of the patient) is processed, standardized, and subsequently transferred to the patient by either nasogastric route or colonoscopy. Although the bacterial taxa associated with a successful FMT-donor microbiota have not been discovered yet, the subsequent increase in the intestinal microbial diversity of the recipient is in most cases associated with recovery (323). However, due to the novelty of this treatment approach, appropriate cohort numbers and well-controlled trials are to be provided in the future. The generation of biobanks for frozen donor microbiota (e.g., OpenBiome; Microbiome Health Research Institute Inc.) is of utmost importance. A future prospect is FMT capsules for therapeutic treatment of infectious diseases, such as C. difficile infection, which is currently under development (324). Despite the fact that FMT was highly effective in C. difficile infection, its therapeutic implementation in IBD is unclear. A recent study by Moayyedi et al. showed a superior placebo effect of FMT in UC patients, though this effect is donor and time dependent (325). An overall analysis of uncontrolled studies with small patient cohorts shows limited success for FMT as standard therapy in IBD, and the rationale of introducing new antigen pools in a milieu that has been overreacting to microbial stimuli is questionable (326, 327).
The causality of dysbiosis in IBD development has been reported, but clear compositional patterns and functionality are still to be unraveled. These aspects are of importance for basic understanding of which consortia may be effective for therapeutic potential. Finally, the intestinal bacterial community is under the influence of environmental-, host-, and microbial-derived factors, and therefore, microbial composition is not the only trait to be considered in order to choose the suitable donor, but also the recipient genetic background and the exposome should be taken into consideration.
Conclusion and Outlook
Despite a growing body of evidence suggesting a causative role of the intestinal microbiota in CD pathogenesis, the impact of bacteria on disease progression, the development of the various disease phenotypes, the risk to develop therapy refractoriness, or the risk to relapse is completely unclear. An increasing number of sequencing studies in human cohorts show associations of certain bacterial groups and CD development. However, due to the nature of the study or high interindividual variation, they often lack the possibility to depict mechanisms causal for disease initiation. Therefore, animal models of intestinal inflammation are important to show functionality and to depict characteristics of dysbiosis. The possibility to unravel certain mechanisms of interaction in animal models of inflammation is generating important insights. The role of antigenic surface compounds or microbial-derived metabolites is of great interest to elucidate mechanisms of microbiota-induced shifts in intestinal homeostasis. Even though new mouse models were recently generated which display more hypothesis-driven mechanisms of intestinal inflammation, it is evident the paucity of model organisms that accurately reproduce all aspects of multifactorial disease, such as CD.
Questions remain about the reliability of mouse models as tool to draw interpretations on human disease. In some cases, mouse research has led to major advantages in the ability to treat serious conditions. For instance, work on the acute promyelocytic leukemia mouse model resulted in successful treatment for this type of cancer in human patients (328). Knocking out the leptin gene in mice manifested the role this hormone has in regulating appetite and, by extension, preventing obesity (329). In contrast, mice are not always reliable as models for human disease as shown in the case of specific IL-17-deficient mouse strains which generated conflicting results in preclinical models of IBD (330).
Similar questions arise from the use of humanized mice in order to unravel human molecular mechanisms or functions. The humanization process consists in repopulation of mice with human hematopoietic cells or in colonization of the gut of GF mice with human microbiota. The potential of these tools for the study of human immune function and causality of the complex host–microbiota interactions in vivo is apparent. Nevertheless, it should be taken into consideration whether the mouse system is able to support development of human immune cells and whether antigens from human origin enable murine immune system maturation. Additionally, it has been shown before that bacterial strains sharing similar genomic content exert differential ability to stimulate the host response (331, 332), suggesting that molecular and biochemical differences in cell envelope architecture may account for the variation in cytokine profiles. This evidence leads to the hypothesis that some bacterial strains may harbor host specificity, and therefore, isolates specific for the human setup might not be able to induce maturation of the mouse immune system, as previously suggested (333).
Finally, it should be noted that even though GWAS have linked genetic variations to several human conditions, they do not provide information on their functions. Animal models are clearly a fundamental tool to identify gene function, and how mutations in gene associated to the disease alter these functions.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Podolsky DK. The current future understanding of inflammatory bowel disease. Best Pract Res Clin Gastroenterol (2002) 16:933–43. doi:10.1053/bega.2002.0354
2. Buhner S, Buning C, Genschel J, Kling K, Herrmann D, Dignass A, et al. Genetic basis for increased intestinal permeability in families with Crohn’s disease: role of CARD15 3020insC mutation? Gut (2006) 55:342–7. doi:10.1136/gut.2005.065557
3. Wehkamp J, Salzman NH, Porter E, Nuding S, Weichenthal M, Petras RE, et al. Reduced Paneth cell alpha-defensins in ileal Crohn’s disease. Proc Natl Acad Sci U S A (2005) 102:18129–34. doi:10.1073/pnas.0505256102
4. Cobrin GM, Abreu MT. Defects in mucosal immunity leading to Crohn’s disease. Immunol Rev (2005) 206:277–95. doi:10.1111/j.0105-2896.2005.00293.x
5. Xavier RJ, Podolsky DK. Unravelling the pathogenesis of inflammatory bowel disease. Nature (2007) 448:427–34. doi:10.1038/nature06005
6. Darfeuille-Michaud A, Boudeau J, Bulois P, Neut C, Glasser AL, Barnich N, et al. High prevalence of adherent-invasive Escherichia coli associated with ileal mucosa in Crohn’s disease. Gastroenterology (2004) 127:412–21. doi:10.1053/j.gastro.2004.04.061
7. Bouma G, Strober W. The immunological and genetic basis of inflammatory bowel disease. Nat Rev Immunol (2003) 3:521–33. doi:10.1038/nri1132
8. Mowat AM. Anatomical basis of tolerance and immunity to intestinal antigens. Nat Rev Immunol (2003) 3:331–41. doi:10.1038/nri1057
9. Allez M, Mayer L. Regulatory T cells: peace keepers in the gut. Inflamm Bowel Dis (2004) 10:666–76. doi:10.1097/00054725-200409000-00027
10. Sartor RB. Microbial influences in inflammatory bowel diseases. Gastroenterology (2008) 134:577–94. doi:10.1053/j.gastro.2007.11.059
11. Asquith M, Powrie F. An innately dangerous balancing act: intestinal homeostasis, inflammation, and colitis-associated cancer. J Exp Med (2010) 207:1573–7. doi:10.1084/jem.20101330
12. Franke A, McGovern DP, Barrett JC, Wang K, Radford-Smith GL, Ahmad T, et al. Genome-wide meta-analysis increases to 71 the number of confirmed Crohn’s disease susceptibility loci. Nat Genet (2010) 42:1118–25. doi:10.1038/ng.717
13. Anderson CA, Boucher G, Lees CW, Franke A, D’amato M, Taylor KD, et al. Meta-analysis identifies 29 additional ulcerative colitis risk loci, increasing the number of confirmed associations to 47. Nat Genet (2011) 43:246–52. doi:10.1038/ng.764
14. Khor B, Gardet A, Xavier RJ. Genetics and pathogenesis of inflammatory bowel disease. Nature (2011) 474:307–17. doi:10.1038/nature10209
15. Jostins L, Ripke S, Weersma RK, Duerr RH, McGovern DP, Hui KY, et al. Host-microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature (2012) 491:119–24. doi:10.1038/nature11582
16. Hugot JP, Chamaillard M, Zouali H, Lesage S, Cezard JP, Belaiche J, et al. Association of NOD2 leucine-rich repeat variants with susceptibility to Crohn’s disease. Nature (2001) 411:599–603. doi:10.1038/35079107
17. Ogura Y, Bonen DK, Inohara N, Nicolae DL, Chen FF, Ramos R, et al. A frameshift mutation in NOD2 associated with susceptibility to Crohn’s disease. Nature (2001) 411:603–6. doi:10.1038/35079114
18. Cuthbert AP, Fisher SA, Mirza MM, King K, Hampe J, Croucher PJ, et al. The contribution of NOD2 gene mutations to the risk and site of disease in inflammatory bowel disease. Gastroenterology (2002) 122:867–74. doi:10.1053/gast.2002.32415
19. Wells JM, Rossi O, Meijerink M, Van Baarlen P. Epithelial crosstalk at the microbiota-mucosal interface. Proc Natl Acad Sci U S A (2011) 108(Suppl 1):4607–14. doi:10.1073/pnas.1000092107
20. Latz E, Xiao TS, Stutz A. Activation and regulation of the inflammasomes. Nat Rev Immunol (2013) 13:397–411. doi:10.1038/nri3452
21. Baldassano RN, Bradfield JP, Monos DS, Kim CE, Glessner JT, Casalunovo T, et al. Association of the T300A non-synonymous variant of the ATG16L1 gene with susceptibility to paediatric Crohn’s disease. Gut (2007) 56:1171–3. doi:10.1136/gut.2007.122747
22. Hampe J, Franke A, Rosenstiel P, Till A, Teuber M, Huse K, et al. A genome-wide association scan of nonsynonymous SNPs identifies a susceptibility variant for Crohn disease in ATG16L1. Nat Genet (2007) 39:207–11. doi:10.1038/ng1954
23. Parkes M, Barrett JC, Prescott NJ, Tremelling M, Anderson CA, Fisher SA, et al. Sequence variants in the autophagy gene IRGM and multiple other replicating loci contribute to Crohn’s disease susceptibility. Nat Genet (2007) 39:830–2. doi:10.1038/ng2061
24. Prescott NJ, Fisher SA, Franke A, Hampe J, Onnie CM, Soars D, et al. A nonsynonymous SNP in ATG16L1 predisposes to ileal Crohn’s disease and is independent of CARD15 and IBD5. Gastroenterology (2007) 132:1665–71. doi:10.1053/j.gastro.2007.03.034
25. Rioux JD, Xavier RJ, Taylor KD, Silverberg MS, Goyette P, Huett A, et al. Genome-wide association study identifies new susceptibility loci for Crohn disease and implicates autophagy in disease pathogenesis. Nat Genet (2007) 39:596–604. doi:10.1038/ng2032
26. Glas J, Seiderer J, Bues S, Stallhofer J, Fries C, Olszak T, et al. IRGM variants and susceptibility to inflammatory bowel disease in the German population. PLoS One (2013) 8:e54338. doi:10.1371/journal.pone.0054338
27. Deretic V, Saitoh T, Akira S. Autophagy in infection, inflammation and immunity. Nat Rev Immunol (2013) 13:722–37. doi:10.1038/nri3532
28. Singh SB, Davis AS, Taylor GA, Deretic V. Human IRGM induces autophagy to eliminate intracellular mycobacteria. Science (2006) 313:1438–41. doi:10.1126/science.1129577
29. Liu B, Gulati AS, Cantillana V, Henry SC, Schmidt EA, Daniell X, et al. Irgm1-deficient mice exhibit Paneth cell abnormalities and increased susceptibility to acute intestinal inflammation. Am J Physiol Gastrointest Liver Physiol (2013) 305:G573–84. doi:10.1152/ajpgi.00071.2013
30. Meena NK, Verma R, Verma N, Ahuja V, Paul J. TLR4 D299G polymorphism modulates cytokine expression in ulcerative colitis. J Clin Gastroenterol (2013) 47:773–80. doi:10.1097/MCG.0b013e31828a6e93
31. Barrett JC, Hansoul S, Nicolae DL, Cho JH, Duerr RH, Rioux JD, et al. Genome-wide association defines more than 30 distinct susceptibility loci for Crohn’s disease. Nat Genet (2008) 40:955–62. doi:10.1038/ng.175
32. Beutler B. Tlr4: central component of the sole mammalian LPS sensor. Curr Opin Immunol (2000) 12:20–6. doi:10.1016/S0952-7915(99)00046-1
33. Figueroa L, Xiong Y, Song C, Piao W, Vogel SN, Medvedev AE. The Asp299Gly polymorphism alters TLR4 signaling by interfering with recruitment of Myd88 and TRIF. J Immunol (2012) 188:4506–15. doi:10.4049/jimmunol.1200202
34. Ohto U, Yamakawa N, Akashi-Takamura S, Miyake K, Shimizu T. Structural analyses of human toll-like receptor 4 polymorphisms D299G and T399I. J Biol Chem (2012) 287:40611–7. doi:10.1074/jbc.M112.404608
35. Hold GL, Berry S, Saunders KA, Drew J, Mayer C, Brookes H, et al. The TLR4 D299G and T399I SNPs are constitutively active to up-regulate expression of Trif-dependent genes. PLoS One (2014) 9:e111460. doi:10.1371/journal.pone.0111460
36. Gardet A, Benita Y, Li C, Sands BE, Ballester I, Stevens C, et al. LRRK2 is involved in the IFN-gamma response and host response to pathogens. J Immunol (2010) 185:5577–85. doi:10.4049/jimmunol.1000548
37. Liu Z, Lee J, Krummey S, Lu W, Cai H, Lenardo MJ. The kinase LRRK2 is a regulator of the transcription factor NFAT that modulates the severity of inflammatory bowel disease. Nat Immunol (2011) 12:1063–70. doi:10.1038/ni.2113
38. Smith AM, Rahman FZ, Hayee B, Graham SJ, Marks DJ, Sewell GW, et al. Disordered macrophage cytokine secretion underlies impaired acute inflammation and bacterial clearance in Crohn’s disease. J Exp Med (2009) 206:1883–97. doi:10.1084/jem.20091233
39. Somasundaram R, Deuring JJ, Van Der Woude CJ, Peppelenbosch MP, Fuhler GM. Linking risk conferring mutations in NCF4 to functional consequences in Crohn’s disease. Gut (2012) 61:1097. doi:10.1136/gutjnl-2011-301344
40. Duerr RH, Taylor KD, Brant SR, Rioux JD, Silverberg MS, Daly MJ, et al. A genome-wide association study identifies IL23R as an inflammatory bowel disease gene. Science (2006) 314:1461–3. doi:10.1126/science.1135245
41. Fujino S, Andoh A, Bamba S, Ogawa A, Hata K, Araki Y, et al. Increased expression of interleukin 17 in inflammatory bowel disease. Gut (2003) 52:65–70. doi:10.1136/gut.52.1.65
42. Fuss IJ, Becker C, Yang Z, Groden C, Hornung RL, Heller F, et al. Both IL-12p70 and IL-23 are synthesized during active Crohn’s disease and are down-regulated by treatment with anti-IL-12 p40 monoclonal antibody. Inflamm Bowel Dis (2006) 12:9–15. doi:10.1097/01.MIB.0000194183.92671.b6
43. Wolk K, Witte E, Hoffmann U, Doecke WD, Endesfelder S, Asadullah K, et al. IL-22 induces lipopolysaccharide-binding protein in hepatocytes: a potential systemic role of IL-22 in Crohn’s disease. J Immunol (2007) 178:5973–81. doi:10.4049/jimmunol.178.9.5973
44. Dige A, Stoy S, Rasmussen TK, Kelsen J, Hvas CL, Sandahl TD, et al. Increased levels of circulating Th17 cells in quiescent versus active Crohn’s disease. J Crohns Colitis (2013) 7:248–55. doi:10.1016/j.crohns.2012.06.015
45. Chao K, Zhang S, Yao J, He Y, Chen B, Zeng Z, et al. Imbalances of CD4(+) T-cell subgroups in Crohn’s disease and their relationship with disease activity and prognosis. J Gastroenterol Hepatol (2014) 29:1808–14. doi:10.1111/jgh.12592
46. Mannon PJ, Fuss IJ, Mayer L, Elson CO, Sandborn WJ, Present D, et al. Anti-interleukin-12 antibody for active Crohn’s disease. N Engl J Med (2004) 351:2069–79. doi:10.1056/NEJMoa033402
47. Brand S. Crohn’s disease: Th1, Th17 or both? The change of a paradigm: new immunological and genetic insights implicate Th17 cells in the pathogenesis of Crohn’s disease. Gut (2009) 58:1152–67. doi:10.1136/gut.2008.163667
48. Fonseca-Camarillo G, Yamamoto-Furusho JK. Immunoregulatory pathways involved in inflammatory bowel disease. Inflamm Bowel Dis (2015) 21(9):2188–93. doi:10.1097/MIB.0000000000000477
49. Dicksved J, Halfvarson J, Rosenquist M, Jarnerot G, Tysk C, Apajalahti J, et al. Molecular analysis of the gut microbiota of identical twins with Crohn’s disease. ISME J (2008) 2:716–27. doi:10.1038/ismej.2008.37
50. Orholm M, Munkholm P, Langholz E, Nielsen OH, Sorensen TI, Binder V. Familial occurrence of inflammatory bowel disease. N Engl J Med (1991) 324:84–8. doi:10.1056/NEJM199101103240203
51. Willing B, Halfvarson J, Dicksved J, Rosenquist M, Jarnerot G, Engstrand L, et al. Twin studies reveal specific imbalances in the mucosa-associated microbiota of patients with ileal Crohn’s disease. Inflamm Bowel Dis (2009) 15:653–60. doi:10.1002/ibd.20783
52. Halfvarson J. Genetics in twins with Crohn’s disease: less pronounced than previously believed? Inflamm Bowel Dis (2011) 17:6–12. doi:10.1002/ibd.21295
53. Hsu LY, Tan YX, Xiao Z, Malissen M, Weiss A. A hypomorphic allele of ZAP-70 reveals a distinct thymic threshold for autoimmune disease versus autoimmune reactivity. J Exp Med (2009) 206:2527–41. doi:10.1084/jem.20082902
54. Nimmo ER, Prendergast JG, Aldhous MC, Kennedy NA, Henderson P, Drummond HE, et al. Genome-wide methylation profiling in Crohn’s disease identifies altered epigenetic regulation of key host defense mechanisms including the Th17 pathway. Inflamm Bowel Dis (2012) 18:889–99. doi:10.1002/ibd.21912
55. Adams AT, Kennedy NA, Hansen R, Ventham NT, O’leary KR, Drummond HE, et al. Two-stage genome-wide methylation profiling in childhood-onset Crohn’s disease implicates epigenetic alterations at the VMP1/MIR21 and HLA loci. Inflamm Bowel Dis (2014) 20:1784–93. doi:10.1097/MIB.0000000000000179
56. Frost R. Mending wall: a marriage of poetic form and content EDSITEMENT! (2015) National Endowment for the Humanities.
57. Haller D, Bode C, Hammes WP, Pfeifer AM, Schiffrin EJ, Blum S. Non-pathogenic bacteria elicit a differential cytokine response by intestinal epithelial cell/leucocyte co-cultures. Gut (2000) 47:79–87. doi:10.1136/gut.47.1.79
58. Johansson ME, Phillipson M, Petersson J, Velcich A, Holm L, Hansson GC. The inner of the two Muc2 mucin-dependent mucus layers in colon is devoid of bacteria. Proc Natl Acad Sci U S A (2008) 105:15064–9. doi:10.1073/pnas.0803124105
59. Bevins CL, Salzman NH. Paneth cells, antimicrobial peptides and maintenance of intestinal homeostasis. Nat Rev Microbiol (2011) 9:356–68. doi:10.1038/nrmicro2546
60. Gallo RL, Hooper LV. Epithelial antimicrobial defence of the skin and intestine. Nat Rev Immunol (2012) 12:503–16. doi:10.1038/nri3228
61. Johansen FE, Pekna M, Norderhaug IN, Haneberg B, Hietala MA, Krajci P, et al. Absence of epithelial immunoglobulin A transport, with increased mucosal leakiness, in polymeric immunoglobulin receptor/secretory component-deficient mice. J Exp Med (1999) 190:915–22. doi:10.1084/jem.190.7.915
62. Johansen FE, Kaetzel CS. Regulation of the polymeric immunoglobulin receptor and IgA transport: new advances in environmental factors that stimulate pIgR expression and its role in mucosal immunity. Mucosal Immunol (2011) 4:598–602. doi:10.1038/mi.2011.37
63. Kobayashi KS, Chamaillard M, Ogura Y, Henegariu O, Inohara N, Nunez G, et al. Nod2-dependent regulation of innate and adaptive immunity in the intestinal tract. Science (2005) 307:731–4. doi:10.1126/science.1104911
64. Vaishnava S, Behrendt CL, Ismail AS, Eckmann L, Hooper LV. Paneth cells directly sense gut commensals and maintain homeostasis at the intestinal host-microbial interface. Proc Natl Acad Sci U S A (2008) 105:20858–63. doi:10.1073/pnas.0808723105
65. Vaishnava S, Yamamoto M, Severson KM, Ruhn KA, Yu X, Koren O, et al. The antibacterial lectin RegIIIgamma promotes the spatial segregation of microbiota and host in the intestine. Science (2011) 334:255–8. doi:10.1126/science.1209791
66. Buisine MP, Desreumaux P, Debailleul V, Gambiez L, Geboes K, Ectors N, et al. Abnormalities in mucin gene expression in Crohn’s disease. Inflamm Bowel Dis (1999) 5:24–32. doi:10.1002/ibd.3780050105
67. Swidsinski A, Weber J, Loening-Baucke V, Hale LP, Lochs H. Spatial organization and composition of the mucosal flora in patients with inflammatory bowel disease. J Clin Microbiol (2005) 43:3380–9. doi:10.1128/JCM.43.7.3380-3389.2005
68. Fu J, Wei B, Wen T, Johansson ME, Liu X, Bradford E, et al. Loss of intestinal core 1-derived O-glycans causes spontaneous colitis in mice. J Clin Invest (2011) 121:1657–66. doi:10.1172/JCI45538
69. Van Der Sluis M, De Koning BA, De Bruijn AC, Velcich A, Meijerink JP, Van Goudoever JB, et al. Muc2-deficient mice spontaneously develop colitis, indicating that MUC2 is critical for colonic protection. Gastroenterology (2006) 131:117–29. doi:10.1053/j.gastro.2006.04.020
70. Fagarasan S, Kawamoto S, Kanagawa O, Suzuki K. Adaptive immune regulation in the gut: T cell-dependent and T cell-independent IgA synthesis. Annu Rev Immunol (2010) 28:243–73. doi:10.1146/annurev-immunol-030409-101314
71. Fagarasan S, Honjo T. Intestinal IgA synthesis: regulation of front-line body defences. Nat Rev Immunol (2003) 3:63–72. doi:10.1038/nri982
72. Macpherson AJ, Harris NL. Interactions between commensal intestinal bacteria and the immune system. Nat Rev Immunol (2004) 4:478–85. doi:10.1038/nri1373
73. Jung C, Hugot JP, Barreau F. Peyer’s patches: the immune sensors of the intestine. Int J Inflam (2010) 2010:823710. doi:10.4061/2010/823710
74. Slack E, Balmer ML, Fritz JH, Hapfelmeier S. Functional flexibility of intestinal IgA – broadening the fine line. Front Immunol (2012) 3:100. doi:10.3389/fimmu.2012.00100
75. Palm NW, De Zoete MR, Cullen TW, Barry NA, Stefanowski J, Hao L, et al. Immunoglobulin A coating identifies colitogenic bacteria in inflammatory bowel disease. Cell (2014) 158:1000–10. doi:10.1016/j.cell.2014.08.006
76. Bruno ME, Frantz AL, Rogier EW, Johansen FE, Kaetzel CS. Regulation of the polymeric immunoglobulin receptor by the classical and alternative NF-kappaB pathways in intestinal epithelial cells. Mucosal Immunol (2011) 4:468–78. doi:10.1038/mi.2011.8
77. Cao AT, Yao S, Gong B, Elson CO, Cong Y. Th17 cells upregulate polymeric Ig receptor and intestinal IgA and contribute to intestinal homeostasis. J Immunol (2012) 189:4666–73. doi:10.4049/jimmunol.1200955
78. Bolton PM, James SL, Newcombe RG, Whitehead RH, Hughes LE. The immune competence of patients with inflammatory bowel disease. Gut (1974) 15:213–9. doi:10.1136/gut.15.3.213
79. Kazemi-Shirazi L, Gasche CH, Natter S, Gangl A, Smolen J, Spitzauer S, et al. IgA autoreactivity: a feature common to inflammatory bowel and connective tissue diseases. Clin Exp Immunol (2002) 128:102–9. doi:10.1046/j.1365-2249.2002.01804.x
80. Harmsen HJ, Pouwels SD, Funke A, Bos NA, Dijkstra G. Crohn’s disease patients have more IgG-binding fecal bacteria than controls. Clin Vaccine Immunol (2012) 19:515–21. doi:10.1128/CVI.05517-11
81. Haller D, Russo MP, Sartor RB, Jobin C. IKK beta and phosphatidylinositol 3-kinase/Akt participate in non-pathogenic Gram-negative enteric bacteria-induced RelA phosphorylation and NF-kappa B activation in both primary and intestinal epithelial cell lines. J Biol Chem (2002) 277:38168–78.
82. Haller D, Holt L, Kim SC, Schwabe RF, Sartor RB, Jobin C. Transforming growth factor-beta 1 inhibits non-pathogenic Gram negative bacteria-induced NF-kappa B recruitment to the interleukin-6 gene promoter in intestinal epithelial cells through modulation of histone acetylation. J Biol Chem (2003) 278:23851–60. doi:10.1074/jbc.M300075200
83. Ruiz PA, Hoffmann M, Szcesny S, Blaut M, Haller D. Innate mechanisms for Bifidobacterium lactis to activate transient pro-inflammatory host responses in intestinal epithelial cells after the colonization of germ-free rats. Immunology (2005) 115:441–50. doi:10.1111/j.1365-2567.2005.02176.x
84. Nenci A, Becker C, Wullaert A, Gareus R, Van Loo G, Danese S, et al. Epithelial NEMO links innate immunity to chronic intestinal inflammation. Nature (2007) 446:557–61. doi:10.1038/nature05698
85. Rakoff-Nahoum S, Paglino J, Eslami-Varzaneh F, Edberg S, Medzhitov R. Recognition of commensal microflora by toll-like receptors is required for intestinal homeostasis. Cell (2004) 118:229–41. doi:10.1016/j.cell.2004.07.002
86. Cario E, Podolsky DK. Differential alteration in intestinal epithelial cell expression of toll-like receptor 3 (TLR3) and TLR4 in inflammatory bowel disease. Infect Immun (2000) 68:7010–7. doi:10.1128/IAI.68.12.7010-7017.2000
87. Abreu MT, Vora P, Faure E, Thomas LS, Arnold ET, Arditi M. Decreased expression of toll-like receptor-4 and MD-2 correlates with intestinal epithelial cell protection against dysregulated proinflammatory gene expression in response to bacterial lipopolysaccharide. J Immunol (2001) 167:1609–16. doi:10.4049/jimmunol.167.3.1609
88. Otte JM, Cario E, Podolsky DK. Mechanisms of cross hyporesponsiveness to toll-like receptor bacterial ligands in intestinal epithelial cells. Gastroenterology (2004) 126:1054–70. doi:10.1053/j.gastro.2004.01.007
89. Lotz M, Gutle D, Walther S, Menard S, Bogdan C, Hornef MW. Postnatal acquisition of endotoxin tolerance in intestinal epithelial cells. J Exp Med (2006) 203:973–84. doi:10.1084/jem.20050625
90. Ruiz PA, Kim SC, Sartor RB, Haller D. 15-deoxy-delta12,14-prostaglandin J2-mediated ERK signaling inhibits gram-negative bacteria-induced RelA phosphorylation and interleukin-6 gene expression in intestinal epithelial cells through modulation of protein phosphatase 2A activity. J Biol Chem (2004) 279:36103–11. doi:10.1074/jbc.M405032200
91. Abreu MT, Arnold ET, Thomas LS, Gonsky R, Zhou Y, Hu B, et al. TLR4 and MD-2 expression is regulated by immune-mediated signals in human intestinal epithelial cells. J Biol Chem (2002) 277:20431–7. doi:10.1074/jbc.M110333200
92. Bogunovic M, Dave SH, Tilstra JS, Chang DT, Harpaz N, Xiong H, et al. Enteroendocrine cells express functional toll-like receptors. Am J Physiol Gastrointest Liver Physiol (2007) 292:G1770–83. doi:10.1152/ajpgi.00249.2006
93. Gewirtz AT, Navas TA, Lyons S, Godowski PJ, Madara JL. Cutting edge: bacterial flagellin activates basolaterally expressed TLR5 to induce epithelial proinflammatory gene expression. J Immunol (2001) 167:1882–5. doi:10.4049/jimmunol.167.4.1882
94. Lee J, Mo JH, Katakura K, Alkalay I, Rucker AN, Liu YT, et al. Maintenance of colonic homeostasis by distinctive apical TLR9 signalling in intestinal epithelial cells. Nat Cell Biol (2006) 8:1327–36. doi:10.1038/ncb1500
95. Cerovic V, Bain CC, Mowat AM, Milling SW. Intestinal macrophages and dendritic cells: what’s the difference? Trends Immunol (2014) 35:270–7. doi:10.1016/j.it.2014.04.003
96. Rescigno M. Dendritic cell-epithelial cell crosstalk in the gut. Immunol Rev (2014) 260:118–28. doi:10.1111/imr.12181
97. Sica A, Mantovani A. Macrophage plasticity and polarization: in vivo veritas. J Clin Invest (2012) 122:787–95. doi:10.1172/JCI59643
98. Bain CC, Mowat AM. Macrophages in intestinal homeostasis and inflammation. Immunol Rev (2014) 260:102–17. doi:10.1111/imr.12192
99. Smith PD, Smythies LE, Shen R, Greenwell-Wild T, Gliozzi M, Wahl SM. Intestinal macrophages and response to microbial encroachment. Mucosal Immunol (2011) 4:31–42. doi:10.1038/mi.2010.66
100. Kawai T, Akira S. The role of pattern-recognition receptors in innate immunity: update on toll-like receptors. Nat Immunol (2010) 11:373–84. doi:10.1038/ni.1863
101. Abreu MT, Fukata M, Arditi M. TLR signaling in the gut in health and disease. J Immunol (2005) 174:4453–60. doi:10.4049/jimmunol.174.8.4453
102. Rogler G, Andus T, Aschenbrenner E, Vogl D, Falk W, Scholmerich J, et al. Alterations of the phenotype of colonic macrophages in inflammatory bowel disease. Eur J Gastroenterol Hepatol (1997) 9:893–9. doi:10.1097/00042737-199709000-00013
103. Rogler G, Hausmann M, Vogl D, Aschenbrenner E, Andus T, Falk W, et al. Isolation and phenotypic characterization of colonic macrophages. Clin Exp Immunol (1998) 112:205–15. doi:10.1046/j.1365-2249.1998.00557.x
104. Rogler G, Hausmann M, Spottl T, Vogl D, Aschenbrenner E, Andus T, et al. T-cell co-stimulatory molecules are upregulated on intestinal macrophages from inflammatory bowel disease mucosa. Eur J Gastroenterol Hepatol (1999) 11:1105–11. doi:10.1097/00042737-199910000-00006
105. Rogler G, Brand K, Vogl D, Page S, Hofmeister R, Andus T, et al. Nuclear factor kappaB is activated in macrophages and epithelial cells of inflamed intestinal mucosa. Gastroenterology (1998) 115:357–69. doi:10.1016/S0016-5085(98)70202-1
106. Hausmann M, Spottl T, Andus T, Rothe G, Falk W, Scholmerich J, et al. Subtractive screening reveals up-regulation of NADPH oxidase expression in Crohn’s disease intestinal macrophages. Clin Exp Immunol (2001) 125:48–55. doi:10.1046/j.1365-2249.2001.01567.x
107. Hausmann M, Kiessling S, Mestermann S, Webb G, Spottl T, Andus T, et al. Toll-like receptors 2 and 4 are up-regulated during intestinal inflammation. Gastroenterology (2002) 122:1987–2000. doi:10.1053/gast.2002.33662
108. Baumgart DC, Thomas S, Przesdzing I, Metzke D, Bielecki C, Lehmann SM, et al. Exaggerated inflammatory response of primary human myeloid dendritic cells to lipopolysaccharide in patients with inflammatory bowel disease. Clin Exp Immunol (2009) 157:423–36. doi:10.1111/j.1365-2249.2009.03981.x
109. Baumgart DC, Metzke D, Guckelberger O, Pascher A, Grotzinger C, Przesdzing I, et al. Aberrant plasmacytoid dendritic cell distribution and function in patients with Crohn’s disease and ulcerative colitis. Clin Exp Immunol (2011) 166:46–54. doi:10.1111/j.1365-2249.2011.04439.x
110. Brown M, Hughes KR, Moossavi S, Robins A, Mahida YR. Toll-like receptor expression in crypt epithelial cells, putative stem cells and intestinal myofibroblasts isolated from controls and patients with inflammatory bowel disease. Clin Exp Immunol (2014) 178:28–39. doi:10.1111/cei.12381
111. Kamada N, Hisamatsu T, Okamoto S, Chinen H, Kobayashi T, Sato T, et al. Unique CD14 intestinal macrophages contribute to the pathogenesis of Crohn disease via IL-23/IFN-gamma axis. J Clin Invest (2008) 118:2269–80. doi:10.1172/JCI34610
112. Macpherson AJ, Uhr T. Induction of protective IgA by intestinal dendritic cells carrying commensal bacteria. Science (2004) 303:1662–5. doi:10.1126/science.1091334
113. Rescigno M. Intestinal dendritic cells. Adv Immunol (2010) 107:109–38. doi:10.1016/B978-0-12-381300-8.00004-6
114. Adams RJ, Heazlewood SP, Gilshenan KS, O’brien M, McGuckin MA, Florin TH. IgG antibodies against common gut bacteria are more diagnostic for Crohn’s disease than IgG against mannan or flagellin. Am J Gastroenterol (2008) 103:386–96. doi:10.1111/j.1572-0241.2007.01577.x
115. Mus AM, Cornelissen F, Asmawidjaja PS, Van Hamburg JP, Boon L, Hendriks RW, et al. Interleukin-23 promotes Th17 differentiation by inhibiting T-bet and FoxP3 and is required for elevation of interleukin-22, but not interleukin-21, in autoimmune experimental arthritis. Arthritis Rheum (2010) 62:1043–50. doi:10.1002/art.27336
116. Keshav S, Lawson L, Chung LP, Stein M, Perry VH, Gordon S. Tumor necrosis factor mRNA localized to Paneth cells of normal murine intestinal epithelium by in situ hybridization. J Exp Med (1990) 171:327–32. doi:10.1084/jem.171.1.327
117. Ayabe T, Satchell DP, Wilson CL, Parks WC, Selsted ME, Ouellette AJ. Secretion of microbicidal alpha-defensins by intestinal Paneth cells in response to bacteria. Nat Immunol (2000) 1:113–8. doi:10.1038/77783
118. Lala S, Ogura Y, Osborne C, Hor SY, Bromfield A, Davies S, et al. Crohn’s disease and the NOD2 gene: a role for paneth cells. Gastroenterology (2003) 125:47–57. doi:10.1016/S0016-5085(03)00661-9
119. Cadwell K, Liu JY, Brown SL, Miyoshi H, Loh J, Lennerz JK, et al. A key role for autophagy and the autophagy gene Atg16l1 in mouse and human intestinal Paneth cells. Nature (2008) 456:259–63. doi:10.1038/nature07416
120. Kaser A, Lee AH, Franke A, Glickman JN, Zeissig S, Tilg H, et al. XBP1 links ER stress to intestinal inflammation and confers genetic risk for human inflammatory bowel disease. Cell (2008) 134:743–56. doi:10.1016/j.cell.2008.07.021
121. Lesage S, Zouali H, Cezard JP, Colombel JF, Belaiche J, Almer S, et al. CARD15/NOD2 mutational analysis and genotype-phenotype correlation in 612 patients with inflammatory bowel disease. Am J Hum Genet (2002) 70:845–57. doi:10.1086/339432
122. Maeda S, Hsu LC, Liu H, Bankston LA, Iimura M, Kagnoff MF, et al. Nod2 mutation in Crohn’s disease potentiates NF-kappaB activity and IL-1beta processing. Science (2005) 307:734–8. doi:10.1126/science.1103685
123. Netea MG, Kullberg BJ, De Jong DJ, Franke B, Sprong T, Naber TH, et al. NOD2 mediates anti-inflammatory signals induced by TLR2 ligands: implications for Crohn’s disease. Eur J Immunol (2004) 34:2052–9. doi:10.1002/eji.200425229
124. Watanabe T, Kitani A, Strober W. NOD2 regulation of toll-like receptor responses and the pathogenesis of Crohn’s disease. Gut (2005) 54:1515–8. doi:10.1136/gut.2005.071795
125. Kim YG, Shaw MH, Warner N, Park JH, Chen F, Ogura Y, et al. Cutting edge: Crohn’s disease-associated Nod2 mutation limits production of proinflammatory cytokines to protect the host from Enterococcus faecalis-induced lethality. J Immunol (2011) 187:2849–52. doi:10.4049/jimmunol.1001854
126. Petnicki-Ocwieja T, Hrncir T, Liu YJ, Biswas A, Hudcovic T, Tlaskalova-Hogenova H, et al. Nod2 is required for the regulation of commensal microbiota in the intestine. Proc Natl Acad Sci U S A (2009) 106:15813–8. doi:10.1073/pnas.0907722106
127. Rehman A, Sina C, Gavrilova O, Hasler R, Ott S, Baines JF, et al. Nod2 is essential for temporal development of intestinal microbial communities. Gut (2011) 60:1354–62. doi:10.1136/gut.2010.216259
128. Mondot S, Barreau F, Al Nabhani Z, Dussaillant M, Le Roux K, Dore J, et al. Altered gut microbiota composition in immune-impaired Nod2(-/-) mice. Gut (2012) 61:634–5. doi:10.1136/gutjnl-2011-300478
129. Frank DN, Robertson CE, Hamm CM, Kpadeh Z, Zhang T, Chen H, et al. Disease phenotype and genotype are associated with shifts in intestinal-associated microbiota in inflammatory bowel diseases. Inflamm Bowel Dis (2011) 17:179–84. doi:10.1002/ibd.21339
130. Couturier-Maillard A, Secher T, Rehman A, Normand S, De Arcangelis A, Haesler R, et al. NOD2-mediated dysbiosis predisposes mice to transmissible colitis and colorectal cancer. J Clin Invest (2013) 123:700–11. doi:10.1172/JCI62236
131. Fellermann K, Wehkamp J, Herrlinger KR, Stange EF. Crohn’s disease: a defensin deficiency syndrome? Eur J Gastroenterol Hepatol (2003) 15:627–34. doi:10.1097/00042737-200306000-00008
132. Wehkamp J, Harder J, Weichenthal M, Schwab M, Schaffeler E, Schlee M, et al. NOD2 (CARD15) mutations in Crohn’s disease are associated with diminished mucosal alpha-defensin expression. Gut (2004) 53:1658–64. doi:10.1136/gut.2003.032805
133. Strominger JL, Ghuysen JM. Mechanisms of enzymatic bacteriaolysis. Cell walls of bacteri are solubilized by action of either specific carbohydrases or specific peptidases. Science (1967) 156:213–21. doi:10.1126/science.156.3772.213
134. Chamaillard M, Girardin SE, Viala J, Philpott DJ. Nods, Nalps and Naip: intracellular regulators of bacterial-induced inflammation. Cell Microbiol (2003) 5:581–92. doi:10.1046/j.1462-5822.2003.00304.x
135. Cloud-Hansen KA, Peterson SB, Stabb EV, Goldman WE, McFall-Ngai MJ, Handelsman J. Breaching the great wall: peptidoglycan and microbial interactions. Nat Rev Microbiol (2006) 4:710–6. doi:10.1038/nrmicro1486
136. Hisamatsu T, Suzuki M, Reinecker HC, Nadeau WJ, McCormick BA, Podolsky DK. CARD15/NOD2 functions as an antibacterial factor in human intestinal epithelial cells. Gastroenterology (2003) 124:993–1000. doi:10.1053/gast.2003.50153
137. Rosenstiel P, Fantini M, Brautigam K, Kuhbacher T, Waetzig GH, Seegert D, et al. TNF-alpha and IFN-gamma regulate the expression of the NOD2 (CARD15) gene in human intestinal epithelial cells. Gastroenterology (2003) 124:1001–9. doi:10.1016/S0016-5085(03)80546-2
138. Barnich N, Aguirre JE, Reinecker HC, Xavier R, Podolsky DK. Membrane recruitment of NOD2 in intestinal epithelial cells is essential for nuclear factor-{kappa}B activation in muramyl dipeptide recognition. J Cell Biol (2005) 170:21–6. doi:10.1083/jcb.200502153
139. Nigro G, Rossi R, Commere PH, Jay P, Sansonetti PJ. The cytosolic bacterial peptidoglycan sensor Nod2 affords stem cell protection and links microbes to gut epithelial regeneration. Cell Host Microbe (2014) 15:792–8. doi:10.1016/j.chom.2014.05.003
140. Bonen DK, Ogura Y, Nicolae DL, Inohara N, Saab L, Tanabe T, et al. Crohn’s disease-associated NOD2 variants share a signaling defect in response to lipopolysaccharide and peptidoglycan. Gastroenterology (2003) 124:140–6. doi:10.1053/gast.2003.50019
141. Inohara N, Ogura Y, Fontalba A, Gutierrez O, Pons F, Crespo J, et al. Host recognition of bacterial muramyl dipeptide mediated through NOD2. Implications for Crohn’s disease. J Biol Chem (2003) 278:5509–12. doi:10.1074/jbc.C200673200
142. Vavricka SR, Musch MW, Chang JE, Nakagawa Y, Phanvijhitsiri K, Waypa TS, et al. hPepT1 transports muramyl dipeptide, activating NF-kappaB and stimulating IL-8 secretion in human colonic Caco2/bbe cells. Gastroenterology (2004) 127:1401–9. doi:10.1053/j.gastro.2004.07.024
143. Ismair MG, Vavricka SR, Kullak-Ublick GA, Fried M, Mengin-Lecreulx D, Girardin SE. hPepT1 selectively transports muramyl dipeptide but not Nod1-activating muramyl peptides. Can J Physiol Pharmacol (2006) 84:1313–9. doi:10.1139/y06-076
144. Merlin D, Si-Tahar M, Sitaraman SV, Eastburn K, Williams I, Liu X, et al. Colonic epithelial hPepT1 expression occurs in inflammatory bowel disease: transport of bacterial peptides influences expression of MHC class 1 molecules. Gastroenterology (2001) 120:1666–79. doi:10.1016/S0016-5085(01)83470-3
145. Wojtal KA, Eloranta JJ, Hruz P, Gutmann H, Drewe J, Staumann A, et al. Changes in mRNA expression levels of solute carrier transporters in inflammatory bowel disease patients. Drug Metab Dispos (2009) 37:1871–7. doi:10.1124/dmd.109.027367
146. Wuensch T, Ullrich S, Schulz S, Chamaillard M, Schaltenberg N, Rath E, et al. Colonic expression of the peptide transporter PEPT1 is downregulated during intestinal inflammation and is not required for NOD2-dependent immune activation. Inflamm Bowel Dis (2014) 20:671–84. doi:10.1097/01.MIB.0000443336.71488.08
147. Cooney R, Baker J, Brain O, Danis B, Pichulik T, Allan P, et al. NOD2 stimulation induces autophagy in dendritic cells influencing bacterial handling and antigen presentation. Nat Med (2010) 16:90–7. doi:10.1038/nm.2069
148. Travassos LH, Carneiro LA, Ramjeet M, Hussey S, Kim YG, Magalhaes JG, et al. Nod1 and Nod2 direct autophagy by recruiting ATG16L1 to the plasma membrane at the site of bacterial entry. Nat Immunol (2010) 11:55–62. doi:10.1038/ni.1823
149. Homer CR, Richmond AL, Rebert NA, Achkar JP, McDonald C. ATG16L1 and NOD2 interact in an autophagy-dependent antibacterial pathway implicated in Crohn’s disease pathogenesis. Gastroenterology (2010) 139:1630–41, 1641.e1–2. doi:10.1053/j.gastro.2010.07.006
150. Saitoh T, Fujita N, Jang MH, Uematsu S, Yang BG, Satoh T. Loss of the autophagy protein Atg16L1 enhances endotoxin-induced IL-1beta production. Nature (2008) 456:264–8. doi:10.1038/nature07383
151. Cadwell K, Patel KK, Maloney NS, Liu TC, Ng AC, Storer CE, et al. Virus-plus-susceptibility gene interaction determines Crohn’s disease gene Atg16L1 phenotypes in intestine. Cell (2010) 141:1135–45. doi:10.1016/j.cell.2010.05.009
152. Conway KL, Kuballa P, Song JH, Patel KK, Castoreno AB, Yilmaz OH, et al. Atg16l1 is required for autophagy in intestinal epithelial cells and protection of mice from Salmonella infection. Gastroenterology (2013) 145:1347–57. doi:10.1053/j.gastro.2013.08.035
153. Deuring JJ, Fuhler GM, Konstantinov SR, Peppelenbosch MP, Kuipers EJ, De Haar C, et al. Genomic ATG16L1 risk allele-restricted Paneth cell ER stress in quiescent Crohn’s disease. Gut (2014) 63:1081–91. doi:10.1136/gutjnl-2012-303527
154. Benjamin JL, Sumpter R Jr, Levine B, Hooper LV. Intestinal epithelial autophagy is essential for host defense against invasive bacteria. Cell Host Microbe (2013) 13:723–34. doi:10.1016/j.chom.2013.05.004
155. Xu Y, Jagannath C, Liu XD, Sharafkhaneh A, Kolodziejska KE, Eissa NT. Toll-like receptor 4 is a sensor for autophagy associated with innate immunity. Immunity (2007) 27:135–44. doi:10.1016/j.immuni.2007.05.022
156. Haller D. Nutrigenomics and IBD: the intestinal microbiota at the cross-road between inflammation and metabolism. J Clin Gastroenterol (2010) 44(Suppl 1):S6–9. doi:10.1097/MCG.0b013e3181dd8b76
157. Rath E, Berger E, Messlik A, Nunes T, Liu B, Kim SC, et al. Induction of dsRNA-activated protein kinase links mitochondrial unfolded protein response to the pathogenesis of intestinal inflammation. Gut (2012) 61:1269–78. doi:10.1136/gutjnl-2011-300767
158. Rath E, Haller D. Mitochondria at the interface between danger signaling and metabolism: role of unfolded protein responses in chronic inflammation. Inflamm Bowel Dis (2012) 18:1364–77. doi:10.1002/ibd.21944
159. Adolph TE, Tomczak MF, Niederreiter L, Ko HJ, Bock J, Martinez-Naves E, et al. Paneth cells as a site of origin for intestinal inflammation. Nature (2013) 503:272–6. doi:10.1038/nature12599
160. Waldschmitt N, Berger E, Rath E, Sartor RB, Weigmann B, Heikenwalder M, et al. C/EBP homologous protein inhibits tissue repair in response to gut injury and is inversely regulated with chronic inflammation. Mucosal Immunol (2014) 7:1452–66. doi:10.1038/mi.2014.34
161. Heazlewood CK, Cook MC, Eri R, Price GR, Tauro SB, Taupin D, et al. Aberrant mucin assembly in mice causes endoplasmic reticulum stress and spontaneous inflammation resembling ulcerative colitis. PLoS Med (2008) 5:e54. doi:10.1371/journal.pmed.0050054
162. Treton X, Pedruzzi E, Cazals-Hatem D, Grodet A, Panis Y, Groyer A, et al. Altered endoplasmic reticulum stress affects translation in inactive colon tissue from patients with ulcerative colitis. Gastroenterology (2011) 141:1024–35. doi:10.1053/j.gastro.2011.05.033
163. Kaser A, Zeissig S, Blumberg RS. Inflammatory bowel disease. Annu Rev Immunol (2010) 28:573–621. doi:10.1146/annurev-immunol-030409-101225
164. Rath E, Haller D. Inflammation and cellular stress: a mechanistic link between immune-mediated and metabolically driven pathologies. Eur J Nutr (2011) 50:219–33. doi:10.1007/s00394-011-0197-0
165. McGuckin MA, Eri RD, Das I, Lourie R, Florin TH. ER stress and the unfolded protein response in intestinal inflammation. Am J Physiol Gastrointest Liver Physiol (2010) 298:G820–32. doi:10.1152/ajpgi.00063.2010
166. Alzoghaibi MA. Concepts of oxidative stress and antioxidant defense in Crohn’s disease. World J Gastroenterol (2013) 19:6540–7. doi:10.3748/wjg.v19.i39.6540
167. Neurath MF, Travis SP. Mucosal healing in inflammatory bowel diseases: a systematic review. Gut (2012) 61:1619–35. doi:10.1136/gutjnl-2012-302830
168. Wyatt J, Oberhuber G, Pongratz S, Puspok A, Moser G, Novacek G, et al. Increased gastric and intestinal permeability in patients with Crohn’s disease. Am J Gastroenterol (1997) 92:1891–6.
169. Breslin NP, Nash C, Hilsden RJ, Hershfield NB, Price LM, Meddings JB, et al. Intestinal permeability is increased in a proportion of spouses of patients with Crohn’s disease. Am J Gastroenterol (2001) 96:2934–8. doi:10.1111/j.1572-0241.2001.04684.x
170. Mazlam MZ, Hodgson HJ. Peripheral blood monocyte cytokine production and acute phase response in inflammatory bowel disease. Gut (1992) 33:773–8. doi:10.1136/gut.33.6.773
171. Reinecker HC, Steffen M, Witthoeft T, Pflueger I, Schreiber S, Macdermott RP, et al. Enhanced secretion of tumour necrosis factor-alpha, IL-6, and IL-1 beta by isolated lamina propria mononuclear cells from patients with ulcerative colitis and Crohn’s disease. Clin Exp Immunol (1993) 94:174–81. doi:10.1111/j.1365-2249.1993.tb05997.x
172. Dionne S, Hiscott J, D’agata I, Duhaime A, Seidman EG. Quantitative PCR analysis of TNF-alpha and IL-1 beta mRNA levels in pediatric IBD mucosal biopsies. Dig Dis Sci (1997) 42:1557–66. doi:10.1023/A:1018895500721
173. Soderholm JD, Olaison G, Lindberg E, Hannestad U, Vindels A, Tysk C, et al. Different intestinal permeability patterns in relatives and spouses of patients with Crohn’s disease: an inherited defect in mucosal defence? Gut (1999) 44:96–100. doi:10.1136/gut.44.1.96
174. Zolotarevsky Y, Hecht G, Koutsouris A, Gonzalez DE, Quan C, Tom J, et al. A membrane-permeant peptide that inhibits MLC kinase restores barrier function in in vitro models of intestinal disease. Gastroenterology (2002) 123:163–72. doi:10.1053/gast.2002.34235
175. Wang F, Graham WV, Wang Y, Witkowski ED, Schwarz BT, Turner JR. Interferon-gamma and tumor necrosis factor-alpha synergize to induce intestinal epithelial barrier dysfunction by up-regulating myosin light chain kinase expression. Am J Pathol (2005) 166:409–19. doi:10.1016/S0002-9440(10)62264-X
176. Blair SA, Kane SV, Clayburgh DR, Turner JR. Epithelial myosin light chain kinase expression and activity are upregulated in inflammatory bowel disease. Lab Invest (2006) 86:191–201. doi:10.1038/labinvest.3700373
177. Shen L, Su L, Turner JR. Mechanisms and functional implications of intestinal barrier defects. Dig Dis (2009) 27:443–9. doi:10.1159/000233282
178. Clayburgh DR, Barrett TA, Tang Y, Meddings JB, Van Eldik LJ, Watterson DM, et al. Epithelial myosin light chain kinase-dependent barrier dysfunction mediates T cell activation-induced diarrhea in vivo. J Clin Invest (2005) 115:2702–15. doi:10.1172/JCI24970
179. Su L, Shen L, Clayburgh DR, Nalle SC, Sullivan EA, Meddings JB, et al. Targeted epithelial tight junction dysfunction causes immune activation and contributes to development of experimental colitis. Gastroenterology (2009) 136:551–63. doi:10.1053/j.gastro.2008.10.081
180. Manichanh C, Rigottier-Gois L, Bonnaud E, Gloux K, Pelletier E, Frangeul L, et al. Reduced diversity of faecal microbiota in Crohn’s disease revealed by a metagenomic approach. Gut (2006) 55:205–11. doi:10.1136/gut.2005.073817
181. Schaubeck M, Clavel T, Calasan J, Lagkouvardos I, Haange SB, Jehmlich N, et al. Dysbiotic gut microbiota causes transmissible Crohn’s disease-like ileitis independent of failure in antimicrobial defence. Gut (2015). doi:10.1136/gutjnl-2015-309333
182. Sellon RK, Tonkonogy S, Schultz M, Dieleman LA, Grenther W, Balish E, et al. Resident enteric bacteria are necessary for development of spontaneous colitis and immune system activation in interleukin-10-deficient mice. Infect Immun (1998) 66:5224–31.
183. Sokol H. Probiotics and antibiotics in IBD. Dig Dis (2014) 32(Suppl 1):10–7. doi:10.1159/000367820
184. Hunter P. The changing hypothesis of the gut. The intestinal microbiome is increasingly seen as vital to human health. EMBO Rep (2012) 13:498–500. doi:10.1038/embor.2012.68
185. Lozupone CA, Stombaugh JI, Gordon JI, Jansson JK, Knight R. Diversity, stability and resilience of the human gut microbiota. Nature (2012) 489:220–30. doi:10.1038/nature11550
186. Lozupone CA, Stombaugh J, Gonzalez A, Ackermann G, Wendel D, Vazquez-Baeza Y, et al. Meta-analyses of studies of the human microbiota. Genome Res (2013) 23:1704–14. doi:10.1101/gr.151803.112
187. Li J, Jia H, Cai X, Zhong H, Feng Q, Sunagawa S. An integrated catalog of reference genes in the human gut microbiome. Nat Biotechnol (2014) 32:834–41. doi:10.1038/nbt.2942
188. Arumugam M, Raes J, Pelletier E, Le Paslier D, Yamada T, Mende DR, et al. Enterotypes of the human gut microbiome. Nature (2011) 473:174–80. doi:10.1038/nature09944
189. Wu GD, Chen J, Hoffmann C, Bittinger K, Chen YY, Keilbaugh SA. Linking long-term dietary patterns with gut microbial enterotypes. Science (2011) 334:105–8. doi:10.1126/science.1208344
190. Project HM. Structure, function and diversity of the healthy human microbiome. Nature (2012) 486:207–14. doi:10.1038/nature11234
191. Greenblum S, Carr R, Borenstein E. Extensive strain-level copy-number variation across human gut microbiome species. Cell (2015) 160:583–94. doi:10.1016/j.cell.2014.12.038
192. Chu H, Mazmanian SK. Innate immune recognition of the microbiota promotes host-microbial symbiosis. Nat Immunol (2013) 14:668–75. doi:10.1038/ni.2635
193. Faust K, Lahti L, Gonze D, De Vos WM, Raes J. Metagenomics meets time series analysis: unraveling microbial community dynamics. Curr Opin Microbiol (2015) 25:56–66. doi:10.1016/j.mib.2015.04.004
194. Muegge BD, Kuczynski J, Knights D, Clemente JC, Gonzalez A, Fontana L, et al. Diet drives convergence in gut microbiome functions across mammalian phylogeny and within humans. Science (2011) 332:970–4. doi:10.1126/science.1198719
195. Carmody RN, Gerber GK, Luevano JM Jr, Gatti DM, Somes L, Svenson KL, et al. Diet dominates host genotype in shaping the murine gut microbiota. Cell Host Microbe (2015) 17:72–84. doi:10.1016/j.chom.2014.11.010
196. Walter J. Murine gut microbiota-diet trumps genes. Cell Host Microbe (2015) 17:3–5. doi:10.1016/j.chom.2014.12.004
197. Martinez C, Antolin M, Santos J, Torrejon A, Casellas F, Borruel N, et al. Unstable composition of the fecal microbiota in ulcerative colitis during clinical remission. Am J Gastroenterol (2008) 103:643–8. doi:10.1111/j.1572-0241.2007.01592.x
198. Frank DN, St Amand AL, Feldman RA, Boedeker EC, Harpaz N, Pace NR. Molecular-phylogenetic characterization of microbial community imbalances in human inflammatory bowel diseases. Proc Natl Acad Sci U S A (2007) 104:13780–5. doi:10.1073/pnas.0706625104
199. Willing BP, Dicksved J, Halfvarson J, Andersson AF, Lucio M, Zheng Z, et al. A pyrosequencing study in twins shows that gastrointestinal microbial profiles vary with inflammatory bowel disease phenotypes. Gastroenterology (2010) 139:1844–54.e1. doi:10.1053/j.gastro.2010.08.049
200. Lepage P, Hasler R, Spehlmann ME, Rehman A, Zvirbliene A, Begun A, et al. Twin study indicates loss of interaction between microbiota and mucosa of patients with ulcerative colitis. Gastroenterology (2011) 141:227–36. doi:10.1053/j.gastro.2011.04.011
201. Turnbaugh PJ, Hamady M, Yatsunenko T, Cantarel BL, Duncan A, Ley RE, et al. A core gut microbiome in obese and lean twins. Nature (2009) 457:480–4. doi:10.1038/nature07540
202. Berer K, Mues M, Koutrolos M, Rasbi ZA, Boziki M, Johner C, et al. Commensal microbiota and myelin autoantigen cooperate to trigger autoimmune demyelination. Nature (2011) 479:538–41. doi:10.1038/nature10554
203. Karlsson FH, Tremaroli V, Nookaew I, Bergstrom G, Behre CJ, Fagerberg B, et al. Gut metagenome in European women with normal, impaired and diabetic glucose control. Nature (2013) 498:99–103. doi:10.1038/nature12198
204. Koeth RA, Wang Z, Levison BS, Buffa JA, Org E, Sheehy BT, et al. Intestinal microbiota metabolism of L-carnitine, a nutrient in red meat, promotes atherosclerosis. Nat Med (2013) 19:576–85. doi:10.1038/nm.3145
205. Petersen C, Round JL. Defining dysbiosis and its influence on host immunity and disease. Cell Microbiol (2014) 16:1024–33. doi:10.1111/cmi.12308
206. Ott SJ, Musfeldt M, Wenderoth DF, Hampe J, Brant O, Folsch UR, et al. Reduction in diversity of the colonic mucosa associated bacterial microflora in patients with active inflammatory bowel disease. Gut (2004) 53:685–93. doi:10.1136/gut.2003.025403
207. Walker AW, Sanderson JD, Churcher C, Parkes GC, Hudspith BN, Rayment N, et al. High-throughput clone library analysis of the mucosa-associated microbiota reveals dysbiosis and differences between inflamed and non-inflamed regions of the intestine in inflammatory bowel disease. BMC Microbiol (2011) 11:7. doi:10.1186/1471-2180-11-7
208. Hansen R, Russell RK, Reiff C, Louis P, McIntosh F, Berry SH, et al. Microbiota of de-novo pediatric IBD: increased Faecalibacterium prausnitzii and reduced bacterial diversity in Crohn’s but not in ulcerative colitis. Am J Gastroenterol (2012) 107:1913–22. doi:10.1038/ajg.2012.335
209. Sha S, Xu B, Wang X, Zhang Y, Wang H, Kong X, et al. The biodiversity and composition of the dominant fecal microbiota in patients with inflammatory bowel disease. Diagn Microbiol Infect Dis (2013) 75:245–51. doi:10.1016/j.diagmicrobio.2012.11.022
210. Scanlan PD, Shanahan F, O’mahony C, Marchesi JR. Culture-independent analyses of temporal variation of the dominant fecal microbiota and targeted bacterial subgroups in Crohn’s disease. J Clin Microbiol (2006) 44:3980–8. doi:10.1128/JCM.00312-06
211. Sokol H, Pigneur B, Watterlot L, Lakhdari O, Bermudez-Humaran LG, Gratadoux JJ, et al. Faecalibacterium prausnitzii is an anti-inflammatory commensal bacterium identified by gut microbiota analysis of Crohn disease patients. Proc Natl Acad Sci U S A (2008) 105:16731–6. doi:10.1073/pnas.0804812105
212. Sokol H, Seksik P, Furet JP, Firmesse O, Nion-Larmurier I, Beaugerie L, et al. Low counts of Faecalibacterium prausnitzii in colitis microbiota. Inflamm Bowel Dis (2009) 15:1183–9. doi:10.1002/ibd.20903
213. Baumgart DC, Sandborn WJ. Crohn’s disease. Lancet (2012) 380:1590–605. doi:10.1016/S0140-6736(12)60026-9
214. Michail S, Durbin M, Turner D, Griffiths AM, Mack DR, Hyams J, et al. Alterations in the gut microbiome of children with severe ulcerative colitis. Inflamm Bowel Dis (2012) 18:1799–808. doi:10.1002/ibd.22860
215. Buffie CG, Jarchum I, Equinda M, Lipuma L, Gobourne A, Viale A, et al. Profound alterations of intestinal microbiota following a single dose of clindamycin results in sustained susceptibility to Clostridium difficile-induced colitis. Infect Immun (2012) 80:62–73. doi:10.1128/IAI.05496-11
216. Momose Y, Hirayama K, Itoh K. Competition for proline between indigenous Escherichia coli and E. coli O157:H7 in gnotobiotic mice associated with infant intestinal microbiota and its contribution to the colonization resistance against E. coli O157:H7. Antonie Van Leeuwenhoek (2008) 94:165–71. doi:10.1007/s10482-008-9222-6
217. Leatham MP, Banerjee S, Autieri SM, Mercado-Lubo R, Conway T, Cohen PS. Precolonized human commensal Escherichia coli strains serve as a barrier to E. coli O157:H7 growth in the streptomycin-treated mouse intestine. Infect Immun (2009) 77:2876–86. doi:10.1128/IAI.00059-09
218. Endt K, Stecher B, Chaffron S, Slack E, Tchitchek N, Benecke A, et al. The microbiota mediates pathogen clearance from the gut lumen after non-typhoidal Salmonella diarrhea. PLoS Pathog (2010) 6:e1001097. doi:10.1371/journal.ppat.1001097
219. Thevenot J, Etienne-Mesmin L, Denis S, Chalancon S, Alric M, Livrelli V, et al. Enterohemorrhagic Escherichia coli O157:H7 survival in an in vitro model of the human large intestine and interactions with probiotic yeasts and resident microbiota. Appl Environ Microbiol (2013) 79:1058–64. doi:10.1128/AEM.03303-12
220. Kamada N, Kim YG, Sham HP, Vallance BA, Puente JL, Martens EC, et al. Regulated virulence controls the ability of a pathogen to compete with the gut microbiota. Science (2012) 336:1325–9. doi:10.1126/science.1222195
221. Chow J, Mazmanian SK. A pathobiont of the microbiota balances host colonization and intestinal inflammation. Cell Host Microbe (2010) 7:265–76. doi:10.1016/j.chom.2010.03.004
222. Rath HC, Wilson KH, Sartor RB. Differential induction of colitis and gastritis in HLA-B27 transgenic rats selectively colonized with Bacteroides vulgatus or Escherichia coli. Infect Immun (1999) 67:2969–74.
223. Waidmann M, Bechtold O, Frick JS, Lehr HA, Schubert S, Dobrindt U, et al. Bacteroides vulgatus protects against Escherichia coli-induced colitis in gnotobiotic interleukin-2-deficient mice. Gastroenterology (2003) 125:162–77. doi:10.1016/S0016-5085(03)00672-3
224. Packey CD, Sartor RB. Commensal bacteria, traditional and opportunistic pathogens, dysbiosis and bacterial killing in inflammatory bowel diseases. Curr Opin Infect Dis (2009) 22:292–301. doi:10.1097/QCO.0b013e32832a8a5d
225. Baumgart M, Dogan B, Rishniw M, Weitzman G, Bosworth B, Yantiss R, et al. Culture independent analysis of ileal mucosa reveals a selective increase in invasive Escherichia coli of novel phylogeny relative to depletion of Clostridiales in Crohn’s disease involving the ileum. ISME J (2007) 1:403–18. doi:10.1038/ismej.2007.52
226. Lupp C, Robertson ML, Wickham ME, Sekirov I, Champion OL, Gaynor EC, et al. Host-mediated inflammation disrupts the intestinal microbiota and promotes the overgrowth of Enterobacteriaceae. Cell Host Microbe (2007) 2:204. doi:10.1016/j.chom.2007.06.010
227. Walters WA, Xu Z, Knight R. Meta-analyses of human gut microbes associated with obesity and IBD. FEBS Lett (2014) 588:4223–33. doi:10.1016/j.febslet.2014.09.039
228. Boudeau J, Glasser AL, Masseret E, Joly B, Darfeuille-Michaud A. Invasive ability of an Escherichia coli strain isolated from the ileal mucosa of a patient with Crohn’s disease. Infect Immun (1999) 67:4499–509.
229. Carvalho FA, Barnich N, Sivignon A, Darcha C, Chan CH, Stanners CP, et al. Crohn’s disease adherent-invasive Escherichia coli colonize and induce strong gut inflammation in transgenic mice expressing human CEACAM. J Exp Med (2009) 206:2179–89. doi:10.1084/jem.20090741
230. Chassaing B, Koren O, Carvalho FA, Ley RE, Gewirtz AT. AIEC pathobiont instigates chronic colitis in susceptible hosts by altering microbiota composition. Gut (2014) 63:1069–80. doi:10.1136/gutjnl-2013-304909
231. Conte MP, Longhi C, Marazzato M, Conte AL, Aleandri M, Lepanto MS, et al. Adherent-invasive Escherichia coli (AIEC) in pediatric Crohn’s disease patients: phenotypic and genetic pathogenic features. BMC Res Notes (2014) 7:748. doi:10.1186/1756-0500-7-748
232. Cooke EM. Faecal flora of patients with ulcerative colitis during treatment with salicylazosulphapyridine. Gut (1969) 10:565–8. doi:10.1136/gut.10.7.565
233. Andrews CN, Griffiths TA, Kaufman J, Vergnolle N, Surette MG, Rioux KP. Mesalazine (5-aminosalicylic acid) alters faecal bacterial profiles, but not mucosal proteolytic activity in diarrhoea-predominant irritable bowel syndrome. Aliment Pharmacol Ther (2011) 34:374–83. doi:10.1111/j.1365-2036.2011.04732.x
234. Gevers D, Kugathasan S, Denson LA, Vazquez-Baeza Y, Van Treuren W, Ren B, et al. The treatment-naive microbiome in new-onset Crohn’s disease. Cell Host Microbe (2014) 15:382–92. doi:10.1016/j.chom.2014.02.005
235. Morgan XC, Tickle TL, Sokol H, Gevers D, Devaney KL, Ward DV, et al. Dysfunction of the intestinal microbiome in inflammatory bowel disease and treatment. Genome Biol (2012) 13:R79. doi:10.1186/gb-2012-13-9-r79
236. Cao Y, Shen J, Ran ZH. Association between Faecalibacterium prausnitzii reduction and inflammatory bowel disease: a meta-analysis and systematic review of the literature. Gastroenterol Res Pract (2014) 2014:872725. doi:10.1155/2014/872725
237. Machiels K, Joossens M, Sabino J, De Preter V, Arijs I, Eeckhaut V, et al. A decrease of the butyrate-producing species Roseburia hominis and Faecalibacterium prausnitzii defines dysbiosis in patients with ulcerative colitis. Gut (2014) 63:1275–83. doi:10.1136/gutjnl-2013-304833
238. Chehoud C, Albenberg LG, Judge C, Hoffmann C, Grunberg S, Bittinger K, et al. Fungal signature in the gut microbiota of pediatric patients with inflammatory bowel disease. Inflamm Bowel Dis (2015) 21(8):1948–56. doi:10.1097/MIB.0000000000000454
239. Norman JM, Handley SA, Baldridge MT, Droit L, Liu CY, Keller BC. Disease-specific alterations in the enteric virome in inflammatory bowel disease. Cell (2015) 160:447–60. doi:10.1016/j.cell.2015.01.002
240. Duerkop BA, Clements CV, Rollins D, Rodrigues JL, Hooper LV. A composite bacteriophage alters colonization by an intestinal commensal bacterium. Proc Natl Acad Sci U S A (2012) 109:17621–6. doi:10.1073/pnas.1206136109
241. Barr JJ, Auro R, Furlan M, Whiteson KL, Erb ML, Pogliano J, et al. Bacteriophage adhering to mucus provide a non-host-derived immunity. Proc Natl Acad Sci U S A (2013) 110:10771–6. doi:10.1073/pnas.1305923110
242. Reyes A, Wu M, McNulty NP, Rohwer FL, Gordon JI. Gnotobiotic mouse model of phage-bacterial host dynamics in the human gut. Proc Natl Acad Sci U S A (2013) 110:20236–41. doi:10.1073/pnas.1319470110
243. Lin HM, Barnett MP, Roy NC, Joyce NI, Zhu S, Armstrong K, et al. Metabolomic analysis identifies inflammatory and noninflammatory metabolic effects of genetic modification in a mouse model of Crohn’s disease. J Proteome Res (2010) 9:1965–75. doi:10.1021/pr901130s
244. Erickson AR, Cantarel BL, Lamendella R, Darzi Y, Mongodin EF, Pan C, et al. Integrated metagenomics/metaproteomics reveals human host-microbiota signatures of Crohn’s disease. PLoS One (2012) 7:e49138. doi:10.1371/journal.pone.0049138
245. Tong M, McHardy I, Ruegger P, Goudarzi M, Kashyap PC, Haritunians T, et al. Reprograming of gut microbiome energy metabolism by the FUT2 Crohn’s disease risk polymorphism. ISME J (2014) 8:2193–206. doi:10.1038/ismej.2014.64
246. Juste C, Kreil DP, Beauvallet C, Guillot A, Vaca S, Carapito C, et al. Bacterial protein signals are associated with Crohn’s disease. Gut (2014) 63:1566–77. doi:10.1136/gutjnl-2012-303786
247. Marchesi JR, Holmes E, Khan F, Kochhar S, Scanlan P, Shanahan F, et al. Rapid and noninvasive metabonomic characterization of inflammatory bowel disease. J Proteome Res (2007) 6:546–51. doi:10.1021/pr060470d
248. McHardy IH, Goudarzi M, Tong M, Ruegger PM, Schwager E, Weger JR, et al. Integrative analysis of the microbiome and metabolome of the human intestinal mucosal surface reveals exquisite inter-relationships. Microbiome (2013) 1:17. doi:10.1186/2049-2618-1-17
249. Ananthakrishnan AN. Epidemiology and risk factors for IBD. Nat Rev Gastroenterol Hepatol (2015) 12:205–17. doi:10.1038/nrgastro.2015.34
250. Clausen MR, Mortensen PB. Kinetic studies on colonocyte metabolism of short chain fatty acids and glucose in ulcerative colitis. Gut (1995) 37:684–9. doi:10.1136/gut.37.5.684
251. Mariadason JM, Catto-Smith A, Gibson PR. Modulation of distal colonic epithelial barrier function by dietary fibre in normal rats. Gut (1999) 44:394–9. doi:10.1136/gut.44.3.394
252. Maslowski KM, Vieira AT, Ng A, Kranich J, Sierro F, Yu D, et al. Regulation of inflammatory responses by gut microbiota and chemoattractant receptor GPR43. Nature (2009) 461:1282–6. doi:10.1038/nature08530
253. Fukuda S, Toh H, Hase K, Oshima K, Nakanishi Y, Yoshimura K, et al. Bifidobacteria can protect from enteropathogenic infection through production of acetate. Nature (2011) 469:543–7. doi:10.1038/nature09646
254. Smith PM, Howitt MR, Panikov N, Michaud M, Gallini CA, Bohlooly YM, et al. The microbial metabolites, short-chain fatty acids, regulate colonic Treg cell homeostasis. Science (2013) 341:569–73. doi:10.1126/science.1241165
255. Richter JF, Pieper R, Zakrzewski SS, Gunzel D, Schulzke JD, Van Kessel AG. Diets high in fermentable protein and fibre alter tight junction protein composition with minor effects on barrier function in piglet colon. Br J Nutr (2014) 111:1040–9. doi:10.1017/S0007114513003498
256. Arpaia N, Campbell C, Fan X, Dikiy S, Van Der Veeken J, Deroos P, et al. Metabolites produced by commensal bacteria promote peripheral regulatory T-cell generation. Nature (2013) 504:451–5. doi:10.1038/nature12726
257. Dore J, Blottiere H. The influence of diet on the gut microbiota and its consequences for health. Curr Opin Biotechnol (2015) 32:195–9. doi:10.1016/j.copbio.2015.01.002
258. De Filippo C, Cavalieri D, Di Paola M, Ramazzotti M, Poullet JB, Massart S, et al. Impact of diet in shaping gut microbiota revealed by a comparative study in children from Europe and rural Africa. Proc Natl Acad Sci U S A (2010) 107:14691–6. doi:10.1073/pnas.1005963107
259. Dominguez-Bello MG, Costello EK, Contreras M, Magris M, Hidalgo G, Fierer N, et al. Delivery mode shapes the acquisition and structure of the initial microbiota across multiple body habitats in newborns. Proc Natl Acad Sci U S A (2010) 107:11971–5. doi:10.1073/pnas.1002601107
260. Benchimol EI, Fortinsky KJ, Gozdyra P, Van Den Heuvel M, Van Limbergen J, Griffiths AM. Epidemiology of pediatric inflammatory bowel disease: a systematic review of international trends. Inflamm Bowel Dis (2011) 17:423–39. doi:10.1002/ibd.21349
261. Koenig JE, Spor A, Scalfone N, Fricker AD, Stombaugh J, Knight R, et al. Succession of microbial consortia in the developing infant gut microbiome. Proc Natl Acad Sci U S A (2011) 108(Suppl 1):4578–85. doi:10.1073/pnas.1000081107
262. Henderson P, Wilson DC. The rising incidence of paediatric-onset inflammatory bowel disease. Arch Dis Child (2012) 97:585–6. doi:10.1136/archdischild-2012-302018
263. Yatsunenko T, Rey FE, Manary MJ, Trehan I, Dominguez-Bello MG, Contreras M, et al. Human gut microbiome viewed across age and geography. Nature (2012) 486:222–7. doi:10.1038/nature11053
264. Kostic AD, Xavier RJ, Gevers D. The microbiome in inflammatory bowel disease: current status and the future ahead. Gastroenterology (2014) 146:1489–99. doi:10.1053/j.gastro.2014.02.009
265. Musilova S, Rada V, Vlkova E, Bunesova V. Beneficial effects of human milk oligosaccharides on gut microbiota. Benef Microbes (2014) 5:273–83. doi:10.3920/BM2013.0080
266. Backhed F, Roswall J, Peng Y, Feng Q, Jia H, Kovatcheva-Datchary P, et al. Dynamics and stabilization of the human gut microbiome during the first year of life. Cell Host Microbe (2015) 17:690–703. doi:10.1016/j.chom.2015.04.004
267. David LA, Maurice CF, Carmody RN, Gootenberg DB, Button JE, Wolfe BE, et al. Diet rapidly and reproducibly alters the human gut microbiome. Nature (2014) 505:559–63. doi:10.1038/nature12820
268. Devkota S, Wang Y, Musch MW, Leone V, Fehlner-Peach H, Nadimpalli A, et al. Dietary-fat-induced taurocholic acid promotes pathobiont expansion and colitis in Il10-/- mice. Nature (2012) 487:104–8. doi:10.1038/nature11225
269. Martinez-Medina M, Denizot J, Dreux N, Robin F, Billard E, Bonnet R, et al. Western diet induces dysbiosis with increased E. coli in CEABAC10 mice, alters host barrier function favouring AIEC colonisation. Gut (2014) 63:116–24. doi:10.1136/gutjnl-2012-304119
270. Lagkouvardos I, Klaring K, Heinzmann SS, Platz S, Scholz B, Engel KH, et al. Gut metabolites and bacterial community networks during a pilot intervention study with flaxseeds in healthy adult men. Mol Nutr Food Res (2015) 59(8):1614–28. doi:10.1002/mnfr.201500125
271. Knights D, Silverberg MS, Weersma RK, Gevers D, Dijkstra G, Huang H, et al. Complex host genetics influence the microbiome in inflammatory bowel disease. Genome Med (2014) 6:107. doi:10.1186/s13073-014-0107-1
272. McGovern DP, Jones MR, Taylor KD, Marciante K, Yan X, Dubinsky M, et al. Fucosyltransferase 2 (FUT2) non-secretor status is associated with Crohn’s disease. Hum Mol Genet (2010) 19:3468–76. doi:10.1093/hmg/ddq248
273. Rausch P, Rehman A, Kunzel S, Hasler R, Ott SJ, Schreiber S, et al. Colonic mucosa-associated microbiota is influenced by an interaction of Crohn disease and FUT2 (Secretor) genotype. Proc Natl Acad Sci U S A (2011) 108:19030–5. doi:10.1073/pnas.1106408108
274. Cullen TW, Schofield WB, Barry NA, Putnam EE, Rundell EA, Trent MS, et al. Gut microbiota. Antimicrobial peptide resistance mediates resilience of prominent gut commensals during inflammation. Science (2015) 347:170–5. doi:10.1126/science.1260580
275. Salzman NH, Hung K, Haribhai D, Chu H, Karlsson-Sjoberg J, Amir E, et al. Enteric defensins are essential regulators of intestinal microbial ecology. Nat Immunol (2010) 11:76–83. doi:10.1038/ni.1825
276. Ocvirk S, Sava IG, Lengfelder I, Lagkouvardos I, Steck N, Roh JH, et al. Surface-associated lipoproteins link Enterococcus faecalis virulence to colitogenic activity in IL-10-deficient mice independent of their expression levels. PLoS Pathog (2015) 11:e1004911. doi:10.1371/journal.ppat.1004911
277. Patwa LG, Fan TJ, Tchaptchet S, Liu Y, Lussier YA, Sartor RB, et al. Chronic intestinal inflammation induces stress-response genes in commensal Escherichia coli. Gastroenterology (2011) 141:1842–51.e1–10. doi:10.1053/j.gastro.2011.06.064
278. Tchaptchet S, Fan TJ, Goeser L, Schoenborn A, Gulati AS, Sartor RB, et al. Inflammation-induced acid tolerance genes gadAB in luminal commensal Escherichia coli attenuate experimental colitis. Infect Immun (2013) 81:3662–71. doi:10.1128/IAI.00355-13
279. Steck N, Hoffmann M, Sava IG, Kim SC, Hahne H, Tonkonogy SL, et al. Enterococcus faecalis metalloprotease compromises epithelial barrier and contributes to intestinal inflammation. Gastroenterology (2011) 141:959–71. doi:10.1053/j.gastro.2011.05.035
280. Winter SE, Thiennimitr P, Winter MG, Butler BP, Huseby DL, Crawford RW, et al. Gut inflammation provides a respiratory electron acceptor for Salmonella. Nature (2010) 467:426–9. doi:10.1038/nature09415
281. Thiennimitr P, Winter SE, Winter MG, Xavier MN, Tolstikov V, Huseby DL, et al. Intestinal inflammation allows Salmonella to use ethanolamine to compete with the microbiota. Proc Natl Acad Sci U S A (2011) 108:17480–5. doi:10.1073/pnas.1107857108
282. Winter SE, Winter MG, Xavier MN, Thiennimitr P, Poon V, Keestra AM, et al. Host-derived nitrate boosts growth of E. coli in the inflamed gut. Science (2013) 339:708–11. doi:10.1126/science.1232467
283. Reeves AE, Koenigsknecht MJ, Bergin IL, Young VB. Suppression of Clostridium difficile in the gastrointestinal tracts of germfree mice inoculated with a murine isolate from the family Lachnospiraceae. Infect Immun (2012) 80:3786–94. doi:10.1128/IAI.00647-12
284. Hsiao A, Ahmed AM, Subramanian S, Griffin NW, Drewry LL, Petri WA Jr, et al. Members of the human gut microbiota involved in recovery from Vibrio cholerae infection. Nature (2014) 515:423–6. doi:10.1038/nature13738
285. Buffie CG, Bucci V, Stein RR, McKenney PT, Ling L, Gobourne A, et al. Precision microbiome reconstitution restores bile acid mediated resistance to Clostridium difficile. Nature (2015) 517:205–8. doi:10.1038/nature13828
286. Fabich AJ, Jones SA, Chowdhury FZ, Cernosek A, Anderson A, Smalley D, et al. Comparison of carbon nutrition for pathogenic and commensal Escherichia coli strains in the mouse intestine. Infect Immun (2008) 76:1143–52. doi:10.1128/IAI.01386-07
287. Pacheco AR, Curtis MM, Ritchie JM, Munera D, Waldor MK, Moreira CG, et al. Fucose sensing regulates bacterial intestinal colonization. Nature (2012) 492:113–7. doi:10.1038/nature11623
288. Ng KM, Ferreyra JA, Higginbottom SK, Lynch JB, Kashyap PC, Gopinath S, et al. Microbiota-liberated host sugars facilitate post-antibiotic expansion of enteric pathogens. Nature (2013) 502:96–9. doi:10.1038/nature12503
289. Thompson JA, Oliveira RA, Djukovic A, Ubeda C, Xavier KB. Manipulation of the quorum sensing signal AI-2 affects the antibiotic-treated gut microbiota. Cell Rep (2015) 10(11):1861–71. doi:10.1016/j.celrep.2015.02.049
290. Garrett WS, Gallini CA, Yatsunenko T, Michaud M, Dubois A, Delaney ML, et al. Enterobacteriaceae act in concert with the gut microbiota to induce spontaneous and maternally transmitted colitis. Cell Host Microbe (2010) 8:292–300. doi:10.1016/j.chom.2010.08.004
291. Dianda L, Hanby AM, Wright NA, Sebesteny A, Hayday AC, Owen MJ. T cell receptor-alpha beta-deficient mice fail to develop colitis in the absence of a microbial environment. Am J Pathol (1997) 150:91–7.
292. Stepankova R, Powrie F, Kofronova O, Kozakova H, Hudcovic T, Hrncir T, et al. Segmented filamentous bacteria in a defined bacterial cocktail induce intestinal inflammation in SCID mice reconstituted with CD45RBhigh CD4+ T cells. Inflamm Bowel Dis (2007) 13:1202–11. doi:10.1002/ibd.20221
293. Bamias G, Okazawa A, Rivera-Nieves J, Arseneau KO, De La Rue SA, Pizarro TT, et al. Commensal bacteria exacerbate intestinal inflammation but are not essential for the development of murine ileitis. J Immunol (2007) 178:1809–18. doi:10.4049/jimmunol.178.3.1809
294. Pizarro TT, Pastorelli L, Bamias G, Garg RR, Reuter BK, Mercado JR, et al. SAMP1/YitFc mouse strain: a spontaneous model of Crohn’s disease-like ileitis. Inflamm Bowel Dis (2011) 17:2566–84. doi:10.1002/ibd.21638
295. Matsumoto S, Okabe Y, Setoyama H, Takayama K, Ohtsuka J, Funahashi H, et al. Inflammatory bowel disease-like enteritis and caecitis in a senescence accelerated mouse P1/Yit strain. Gut (1998) 43:71–8. doi:10.1136/gut.43.1.71
296. Bamias G, Dahman MI, Arseneau KO, Guanzon M, Gruska D, Pizarro TT, et al. Intestinal-specific TNFalpha overexpression induces Crohn’s-like ileitis in mice. PLoS One (2013) 8:e72594. doi:10.1371/journal.pone.0072594
297. Mikulski Z, Johnson R, Shaked I, Kim G, Nowyhed H, Goodman W, et al. SAMP1/YitFc mice develop ileitis via loss of CCL21 and defects in dendritic cell migration. Gastroenterology (2015) 148:783–93.e5. doi:10.1053/j.gastro.2015.01.027
298. Kontoyiannis D, Pasparakis M, Pizarro TT, Cominelli F, Kollias G. Impaired on/off regulation of TNF biosynthesis in mice lacking TNF AU-rich elements: implications for joint and gut-associated immunopathologies. Immunity (1999) 10:387–98. doi:10.1016/S1074-7613(00)80038-2
299. McNamee EN, Masterson JC, Jedlicka P, Collins CB, Williams IR, Rivera-Nieves J. Ectopic lymphoid tissue alters the chemokine gradient, increases lymphocyte retention and exacerbates murine ileitis. Gut (2013) 62:53–62. doi:10.1136/gutjnl-2011-301272
301. Rodriguez-Palacios A, Kodani T, Kaydo L, Pietropaoli D, Corridoni D, Howell S, et al. Stereomicroscopic 3D-pattern profiling of murine and human intestinal inflammation reveals unique structural phenotypes. Nat Commun (2015) 6:7577. doi:10.1038/ncomms8577
302. Gunther C, Martini E, Wittkopf N, Amann K, Weigmann B, Neumann H, et al. Caspase-8 regulates TNF-alpha-induced epithelial necroptosis and terminal ileitis. Nature (2011) 477:335–9. doi:10.1038/nature10400
303. Gunther C, Buchen B, He GW, Hornef M, Torow N, Neumann H, et al. Caspase-8 controls the gut response to microbial challenges by Tnf-alpha-dependent and independent pathways. Gut (2015) 64:601–10. doi:10.1136/gutjnl-2014-307226
304. Hormannsperger G, Schaubeck M, Haller D. Intestinal microbiota in animal models of inflammatory diseases. ILAR J (2015) 56:179–91. doi:10.1093/ilar/ilv019
305. Kim SC, Tonkonogy SL, Albright CA, Tsang J, Balish EJ, Braun J, et al. Variable phenotypes of enterocolitis in interleukin 10-deficient mice monoassociated with two different commensal bacteria. Gastroenterology (2005) 128:891–906. doi:10.1053/j.gastro.2005.02.009
306. Whary MT, Taylor NS, Feng Y, Ge Z, Muthupalani S, Versalovic J, et al. Lactobacillus reuteri promotes Helicobacter hepaticus-associated typhlocolitis in gnotobiotic B6.129P2-IL-10(tm1Cgn) (IL-10(-/-)) mice. Immunology (2011) 133:165–78. doi:10.1111/j.1365-2567.2011.03423.x
307. Atarashi K, Tanoue T, Shima T, Imaoka A, Kuwahara T, Momose Y, et al. Induction of colonic regulatory T cells by indigenous Clostridium species. Science (2011) 331:337–41. doi:10.1126/science.1198469
308. Atarashi K, Tanoue T, Oshima K, Suda W, Nagano Y, Nishikawa H, et al. Treg induction by a rationally selected mixture of Clostridia strains from the human microbiota. Nature (2013) 500:232–6. doi:10.1038/nature12331
309. Powell N, Walker AW, Stolarczyk E, Canavan JB, Gokmen MR, Marks E, et al. The transcription factor T-bet regulates intestinal inflammation mediated by interleukin-7 receptor+ innate lymphoid cells. Immunity (2012) 37:674–84. doi:10.1016/j.immuni.2012.09.008
310. Rooks MG, Veiga P, Wardwell-Scott LH, Tickle T, Segata N, Michaud M, et al. Gut microbiome composition and function in experimental colitis during active disease and treatment-induced remission. ISME J (2014) 8:1403–17. doi:10.1038/ismej.2014.3
311. Ivanov II, Atarashi K, Manel N, Brodie EL, Shima T, Karaoz U, et al. Induction of intestinal Th17 cells by segmented filamentous bacteria. Cell (2009) 139:485–98. doi:10.1016/j.cell.2009.09.033
312. Olivares M, Neef A, Castillejo G, Palma GD, Varea V, Capilla A, et al. The HLA-DQ2 genotype selects for early intestinal microbiota composition in infants at high risk of developing coeliac disease. Gut (2015) 64:406–17. doi:10.1136/gutjnl-2014-306931
313. Ghouri YA, Richards DM, Rahimi EF, Krill JT, Jelinek KA, Dupont AW. Systematic review of randomized controlled trials of probiotics, prebiotics, and synbiotics in inflammatory bowel disease. Clin Exp Gastroenterol (2014) 7:473–87. doi:10.2147/CEG.S27530
314. Saez-Lara MJ, Gomez-Llorente C, Plaza-Diaz J, Gil A. The role of probiotic lactic acid bacteria and bifidobacteria in the prevention and treatment of inflammatory bowel disease and other related diseases: a systematic review of randomized human clinical trials. Biomed Res Int (2015) 2015:505878. doi:10.1155/2015/505878
315. Macho Fernandez E, Valenti V, Rockel C, Hermann C, Pot B, Boneca IG, et al. Anti-inflammatory capacity of selected lactobacilli in experimental colitis is driven by NOD2-mediated recognition of a specific peptidoglycan-derived muropeptide. Gut (2011) 60:1050–9. doi:10.1136/gut.2010.232918
316. Hummel S, Veltman K, Cichon C, Sonnenborn U, Schmidt MA. Differential targeting of the E-cadherin/beta-catenin complex by gram-positive probiotic lactobacilli improves epithelial barrier function. Appl Environ Microbiol (2012) 78:1140–7. doi:10.1128/AEM.06983-11
317. Von Schillde MA, Hormannsperger G, Weiher M, Alpert CA, Hahne H, Bauerl C, et al. Lactocepin secreted by Lactobacillus exerts anti-inflammatory effects by selectively degrading proinflammatory chemokines. Cell Host Microbe (2012) 11:387–96. doi:10.1016/j.chom.2012.02.006
318. Angelberger S, Reinisch W, Makristathis A, Lichtenberger C, Dejaco C, Papay P, et al. Temporal bacterial community dynamics vary among ulcerative colitis patients after fecal microbiota transplantation. Am J Gastroenterol (2013) 108:1620–30. doi:10.1038/ajg.2013.257
319. Kump PK, Grochenig HP, Lackner S, Trajanoski S, Reicht G, Hoffmann KM, et al. Alteration of intestinal dysbiosis by fecal microbiota transplantation does not induce remission in patients with chronic active ulcerative colitis. Inflamm Bowel Dis (2013) 19:2155–65. doi:10.1097/MIB.0b013e31829ea325
320. Van Nood E, Vrieze A, Nieuwdorp M, Fuentes S, Zoetendal EG, De Vos WM, et al. Duodenal infusion of donor feces for recurrent Clostridium difficile. N Engl J Med (2013) 368:407–15. doi:10.1056/NEJMoa1205037
321. Cui B, Feng Q, Wang H, Wang M, Peng Z, Li P, et al. Fecal microbiota transplantation through mid-gut for refractory Crohn’s disease: safety, feasibility and efficacy trial results. J Gastroenterol Hepatol (2014) 30(1):51–8. doi:10.1111/jgh.12727
322. Cammarota G, Masucci L, Ianiro G, Bibbo S, Dinoi G, Costamagna G, et al. Randomised clinical trial: faecal microbiota transplantation by colonoscopy vs. vancomycin for the treatment of recurrent Clostridium difficile infection. Aliment Pharmacol Ther (2015) 41(9):835–43. doi:10.1111/apt.13144
323. Shahinas D, Silverman M, Sittler T, Chiu C, Kim P, Allen-Vercoe E, et al. Toward an understanding of changes in diversity associated with fecal microbiome transplantation based on 16S rRNA gene deep sequencing. MBio (2012) 3:e338–312. doi:10.1128/mBio.00338-12
324. Youngster I, Russell GH, Pindar C, Ziv-Baran T, Sauk J, Hohmann EL. Oral, capsulized, frozen fecal microbiota transplantation for relapsing Clostridium difficile infection. JAMA (2014) 312:1772–8. doi:10.1001/jama.2014.13875
325. Moayyedi P, Surette MG, Kim PT, Libertucci J, Wolfe M, Onischi C, et al. Fecal microbiota transplantation induces remission in patients with active ulcerative colitis in a randomized, controlled trial. Gastroenterology (2015) 149(1):102–09.e6. doi:10.1053/j.gastro.2015.04.001
326. Ianiro G, Bibbo S, Scaldaferri F, Gasbarrini A, Cammarota G. Fecal microbiota transplantation in inflammatory bowel disease: beyond the excitement. Medicine (Baltimore) (2014) 93:e97. doi:10.1097/MD.0000000000000097
327. Rossen NG, Fuentes S, Van Der Spek MJ, Tijssen JG, Hartman JH, Duflou A, et al. Findings from a randomized controlled trial of fecal transplantation for patients with ulcerative colitis. Gastroenterology (2015) 149:110–18.e4. doi:10.1053/j.gastro.2015.03.045
328. Lallemand-Breitenbach V, Zhu J, Kogan S, Chen Z, De The H. Opinion: how patients have benefited from mouse models of acute promyelocytic leukaemia. Nat Rev Cancer (2005) 5:821–7. doi:10.1038/nrc1719
329. Friedman JM, Mantzoros CS. 20 years of leptin: from the discovery of the leptin gene to leptin in our therapeutic armamentarium. Metabolism (2015) 64:1–4. doi:10.1016/j.metabol.2014.10.023
330. Hueber W, Sands BE, Lewitzky S, Vandemeulebroecke M, Reinisch W, Higgins PD, et al. Secukinumab, a human anti-IL-17A monoclonal antibody, for moderate to severe Crohn’s disease: unexpected results of a randomised, double-blind placebo-controlled trial. Gut (2012) 61:1693–700. doi:10.1136/gutjnl-2011-301668
331. Van Hemert S, Meijerink M, Molenaar D, Bron PA, De Vos P, Kleerebezem M, et al. Identification of Lactobacillus plantarum genes modulating the cytokine response of human peripheral blood mononuclear cells. BMC Microbiol (2010) 10:293. doi:10.1186/1471-2180-10-293
332. Miyauchi E, O’callaghan J, Butto LF, Hurley G, Melgar S, Tanabe S, et al. Mechanism of protection of transepithelial barrier function by Lactobacillus salivarius: strain dependence and attenuation by bacteriocin production. Am J Physiol Gastrointest Liver Physiol (2012) 303:G1029–41. doi:10.1152/ajpgi.00003.2012
Keywords: IEC, dysbiosis, TNF, Paneth cells, barrier dysfunction, microbiota, Crohn’s disease, IBD
Citation: Buttó LF, Schaubeck M and Haller D (2015) Mechanisms of Microbe–Host Interaction in Crohn’s Disease: Dysbiosis vs. Pathobiont Selection. Front. Immunol. 6:555. doi: 10.3389/fimmu.2015.00555
Received: 17 August 2015; Accepted: 16 October 2015;
Published: 19 November 2015
Edited by:
Amélia M. Sarmento, Universidade Fernando Pessoa, PortugalReviewed by:
Wim Van Den Broeck, Ghent University, BelgiumCarl James Yeoman, Montana State University, USA
Copyright: © 2015 Buttó, Schaubeck and Haller. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Dirk Haller, ZGlyay5oYWxsZXJAdHVtLmRl
†Ludovica F. Buttó and Monika Schaubeck have contributed equally to this work.