 Payton A. Ottum
Payton A. Ottum Gabriel Arellano
Gabriel Arellano Lilian I. Reyes
Lilian I. Reyes Mirentxu Iruretagoyena
Mirentxu Iruretagoyena Rodrigo Naves
Rodrigo Naves
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Immunol., 30 October 2015
Sec. Immunological Tolerance and Regulation
Volume 6 - 2015 | https://doi.org/10.3389/fimmu.2015.00539
This article is part of the Research TopicSearching for immune tolerance manipulating new molecules and exploiting new concepts on lymphocyte biologyView all 14 articles
Multiple sclerosis (MS) is the principal cause of autoimmune neuroinflammation in humans, and its animal model, experimental autoimmune encephalomyelitis (EAE), is widely used to gain insight about their immunopathological mechanisms for and the development of novel therapies for MS. Most studies on the role of interferon (IFN)-γ in the pathogenesis and progression of EAE have focused on peripheral immune cells, while its action on central nervous system (CNS)-resident cells has been less explored. In addition to the well-known proinflammatory and damaging effects of IFN-γ in the CNS, evidence has also endowed this cytokine both a protective and regulatory role in autoimmune neuroinflammation. Recent investigations performed in this research field have exposed the complex role of IFN-γ in the CNS uncovering unexpected mechanisms of action that underlie these opposing activities on different CNS-resident cell types. The mechanisms behind these two-faced effects of IFN-γ depend on dose, disease phase, and cell development stage. Here, we will review and discuss the dual role of IFN-γ on CNS-resident cells in EAE highlighting its protective functions and the mechanisms proposed.
Multiple sclerosis (MS) and its animal model, experimental autoimmune encephalomyelitis (EAE), are chronic autoimmune diseases of the central nervous system (CNS) characterized by inflammatory infiltrates, demyelination and neurological damage (1, 2). MS and EAE were initially considered to be mediated by interferon (IFN)-γ-expressing T helper (Th) 1 cells (3, 4). Currently, it is most widely accepted that several innate and adaptive immune cell types and immunomodulatory molecules contribute to the disease development and progression (5). Moreover, several studies have challenged the notion that IFN-γ is only pathogenic, and accumulative evidence attributes it a protective role in EAE and MS (6–8). In this same journal research topic, we have reviewed recent data supporting a stage-specific participation of IFN-γ in MS and EAE providing a plausible explanation for previous conflicting results and a model whereby this cytokine can both promote and limit the development of these pathologies (8). However, the majority of these studies have focused on the roles of IFN-γ in immune cells, while its activity in CNS-resident cells remains less explored. In this review, we will begin by discussing evidence that reports opposite roles of IFN-γ in the CNS during EAE development. Then, we will review both the inflammatory and protective effects of IFN-γ on glial cells and neurons in EAE.
Early studies demonstrated that IFN-γ can activate CNS-resident cells and induce expression of major histocompatibility complex (MHC) molecules (3, 9). Direct injection of IFN-γ into the rat CNS induced inflammation and cellular infiltration similar to that observed in EAE (10–13) and potentiated the demyelination process (13). Furthermore, demyelination occurred in transgenic mice expressing IFN-γ under the control of the myelin basic protein (MBP) promoter (14–16). On the contrary, animals with EAE that were injected with IFN-γ systemically or directly into the CNS showed amelioration of clinical symptoms (17–20). Therefore, despite early data reporting an inflammatory effect of IFN-γ in the CNS during EAE development, accumulating evidence has also demonstrated a neuroprotective activity for IFN-γ in this disease.
Classical EAE is characterized by an ascending progressive paralysis dominated by inflammatory lesions in the white matter of the spinal cord and limited brain inflammation (2, 21). However, the absence of either IFN-γ or its receptor (IFNGR) leads to the development of atypical EAE symptoms such as head tilting, ataxia, dystonia, spasticity, and axial-rotation suggestive of brain-associated damage (19, 22–24). This atypical EAE is associated with greater encephalitogenicity of Th1 and Th17 cells and enhanced demyelination in the brainstem and cerebellum (19, 24). Even more, some studies have shown that IFN-γ may exert opposite effects in the brain and spinal cord determining the regional localization of lesions and inflammation in EAE. According to this evidence, IFN-γ induces inflammation in the spinal cord but protection in the brain (21, 25, 26). The mechanisms underlying these differential effects of IFN-γ may involve the regulation of the expression of specific chemokines in the brain versus spinal cord that restrain encephalitogenic T cell brain infiltration (19, 24, 25).
IFN-γ may also exert differential effects on the blood–brain barrier (BBB) and blood-spinal cord barrier (BSCB), resulting in opposite effects on their function and integrity. Supporting this hypothesis, atypical EAE symptoms were significantly ameliorated in mice that only expressed IFNGR in endothelial cells (EC-IFNGR mice). In these EC-IFNGR mice, inflammatory immune cell infiltration and demyelination were significantly inhibited in the brain but not in the spinal cord. In contrast to IFNGR-deficient mice, the functional integrity of the BBB was preserved in EC-IFNGR mice. This could be due to enhanced IFN-γ-induced claudin-5 expression that resulted in increased paracellular tightness of brain endothelial cell cultures (27). Therefore, these results indicate that endothelial expression of IFNGR is necessary for maintaining BBB function and preventing atypical EAE and brain inflammation. Further analysis must be performed in order to establish differential effects of IFN-γ on the BSCB.
There are two groups of glial cells in the CNS: the macroglia, including astrocytes, oligodendrocytes, and ependymal cells, and the microglia. Several studies support the notion that glial cells not only provide functional support to neurons nor are they only a target of autoimmune injury, but are also active players in the development and progression of MS and EAE. Indeed, studies using an adoptive transfer EAE model have suggested that prevention of atypical EAE by IFN-γ is dependent on IFN-γ signaling not only in encephalitogenic T cells but also in glial cells (19, 25). Below, we will review the opposite effects of IFN-γ on glial cells and neurons, highlighting its less known protective functions and the mechanisms whereby IFN-γ is able to exert neuroprotection (Figure 1).
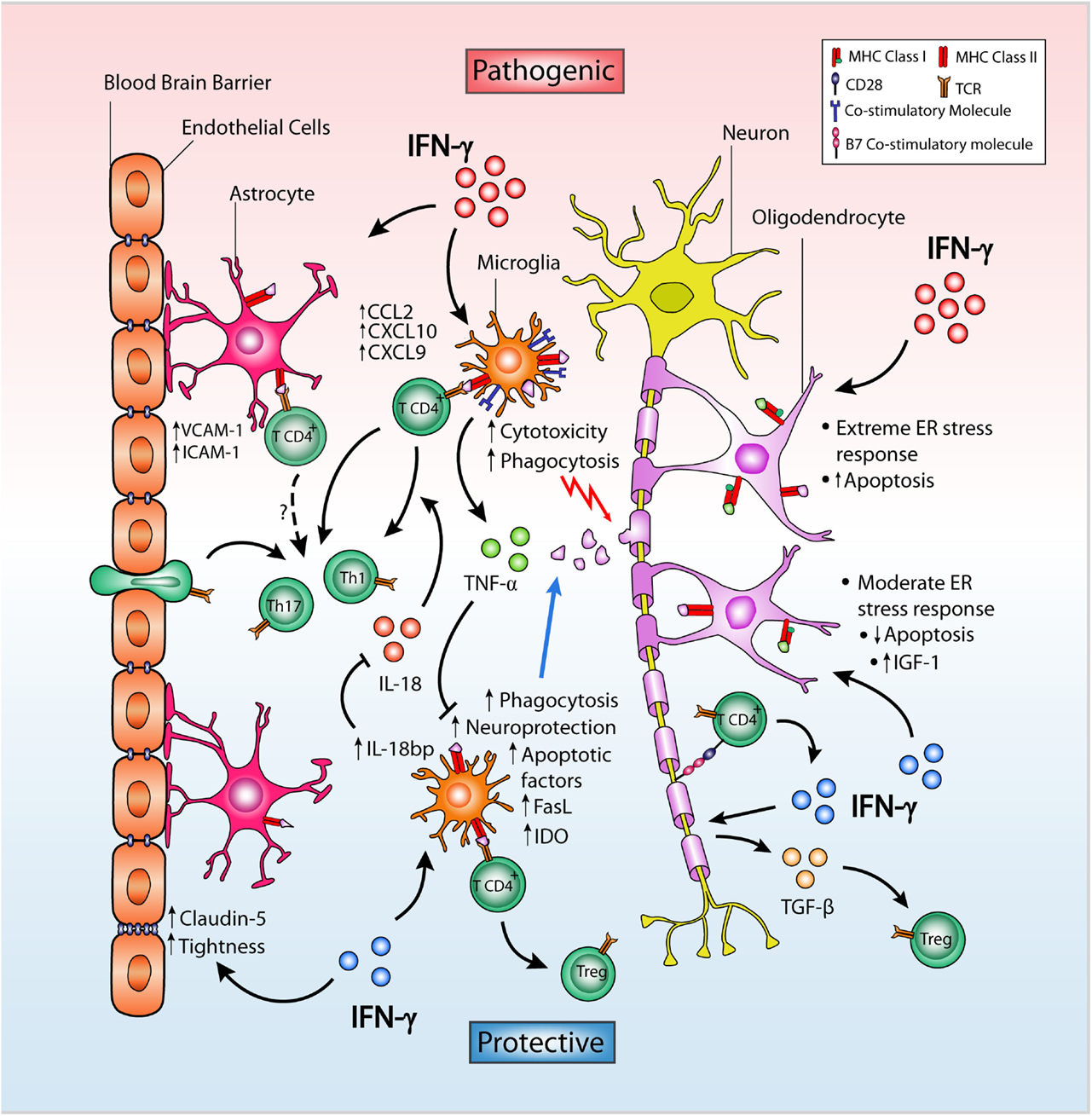
Figure 1. Dual role of IFN-γ on CNS-resident cells in EAE. IFN-γ in the CNS may facilitate helper T cell infiltration and neuroinflammation by inducing expression of VCAM-1, ICAM-1, and the chemokines CCL2, CXCL9, and CXCL10 in astrocytes located in close proximity to the BBB. Despite this, IFN-γ promotes BBB integrity by enhancing the expression and membrane distribution of claudin-5, in turn increasing tightness and ameliorating EAE severity. In microglia, IFN-γ differentially causes pathogenic cytotoxicity or neuroprotection and apoptosis depending on dose. Low-dose IFN-γ can induce tolerogenic microglia, while high doses result in increased expression of MHC class II protein and co-stimulatory molecules, leading to encephalitogenic T cell activation. TNF-α expression is upregulated by IFN-γ in microglia and inhibits their neuroprotective properties. In contrast, IFN-γ upregulates microglial IDO expression limiting T cell proliferation and inducing Fas and FasL expression favoring increased T cell apoptosis. IFN-γ also induces IL-18bp expression in microglia which inhibits the proinflammatory actions of IL-18 inhibiting EAE. In neurons, IFN-γ can induce B7 co-stimulatory molecules and TGF-β expression, promoting a Treg cell population that suppresses EAE. In oligodendrocytes, the effect of IFN-γ depends on both dose and the level of ER stress. By inducing expression of MHC molecules and other immune proteins, IFN-γ overloads the stress response in actively myelinating oligodendrocytes causing cell death, whereas a moderate stress response induced by low dose of IFN-γ on steady-state oligodendrocytes decreases apoptosis and increases IGF-1 levels in the CNS promoting remyelination of post-neuroinflammatory damage.
Oligodendrocytes are CNS-resident myelin-producing cells and thus necessary for remyelination. Loss of oligodendrocytes, demyelination, and axonal damage are hallmarks of MS and EAE. Several in vitro studies have shown detrimental effects of IFN-γ on oligodendrocyte survival (28). Consistently, overexpression of IFN-γ in the CNS results in oligodendrocyte apoptosis and inhibition of myelination during development and after a demye-linating insult (15, 16, 29). However, when exposed to low levels of IFN-γ, oligodendrocytes were protected against oxidative stress and showed enhanced proteasome activity, two important processes preventing the accumulation of oxidized proteins, mitochondrial dysfunction and apoptosis (30). Furthermore, transgenic mice expressing low levels of IFN-γ were protected against chemically induced demyelination with cuprizone, and these animals did not show signs of oligodendroglial death, astrogliosis, or microgliosis compared to wild-type mice exposed to the toxin (31). Interestingly, elevated levels of insulin-like growth factor (IGF)-1 were detected in the CNS of these transgenic mice, which might contribute to the observed protective effects of IFN-γ, as IGF-1 has been demonstrated to inhibit oligodendrocyte apoptosis and promote myelination (31). Therefore, these results indicate that IFN-γ dose is critical in determining the survival of oligodendrocytes.
Those studies performed with transgenic mice have the limitation that overexpression of IFN-γ was induced during myelin formation whereas EAE and MS occur when myelination has been established in adulthood. In order to overcome this pitfall, the same authors developed an elegant in vivo experimental model that allowed temporally regulated delivery of IFN-γ to the CNS through a tetracycline-controllable system. IFN-γ delivery in the CNS that was first detected in the acute phase of EAE ameliorated disease severity and prevented oligodendrocyte loss, demyelination, and axonal damage. By contrast, delivery of this cytokine during the EAE remitting phase delayed disease recovery and inhibited remyelination (32). Interestingly, the effects of this cytokine in oligodendrocytes were mediated by the activation of protein kinase RNA-like endoplasmic reticulum kinase (PERK) and the phosphorylation of the α-subunit of eukaryotic translation initiation factor 2 (eIF2a), factors associated with the endoplasmic reticulum (ER) stress response (29, 32). The authors explained these apparently contradictory results proposing a dual role of IFN-γ on oligodendrocyte survival depending on their developmental stage and cellular stress (33, 34). According to this model, during the active process of myelination such as CNS development or remyelination (i.e., EAE remission), IFN-γ causes an overloaded ER stress response by increasing expression of MHC molecules and other inflammatory proteins in oligodendrocytes with already elevated ER stress, resulting in apoptotic program activation. Conversely, mature oligodendrocytes in adult mice produce significantly lower levels of membrane to maintain myelin homeostasis (i.e., acute EAE). In this case, IFN-γ would induce a moderate and protective ER stress response leading to oligodendrocyte survival and remyelination (33–35). Further investigations have determined that protection of oligodendrocytes in EAE may be mediated through Janus kinase (JAK) and the transcription factor signal transducer and activator of transcription (STAT)-1 signaling, as overexpression of the suppressor of cytokine signal (SOCS)-1, a competitive inhibitor of the IFN-γ-induced JAK/STAT-1 signaling pathway resulted in early EAE onset, enhanced inflammation, and oligodendrocyte apoptosis (30). Instead, the signal transduction pathway activating the transcription factor interferon regulatory factor (IRF)-1 would play a proinflammatory role in oligodendrocytes during EAE progression (36).
Microglia comprise 10–20% of all glial cells and are considered the immune sentinels of the CNS (37). They develop a broad and versatile range of functions involved in inflammation, immunomodulation, and promotion of neural repair that can be mediated in part by IFN-γ (38, 39). IFN-γ potentiates the phagocytic activity of microglia (40) and induces the expression of MHC class II and co-stimulatory molecules. This enables them to function as antigen-presenting cells (APCs) for infiltrating myelin-specific T cells leading to inflammation and demyelination (41–45).
During neuroinflammation, different subtypes of microglia can be distinguished according to their activation status. M1 microglia are primarily associated with an inflammatory phenotype and EAE initiation, while M2 microglia play an anti-inflammatory role and participate in tissue repair and remodeling associated with EAE recovery (5, 46). Despite being a potent activator of microglia and their polarization toward M1, IFN-γ can, at least in part, regulate the dual activity of microglia, modulating both pathogenic and regenerative processes (40, 47–49). How can IFN-γ lead these opposing roles?.
Evidence suggests that IFN-γ dose constitutes a fine-tune mechanism of regulation determining the balance between inflammatory and anti-inflammatory microglia. IFN-γ along with neuroantigen can determine an effector or regulatory helper T cell response modulating the activation state of microglia in a dose-dependent manner (48). Myelin oligodendrocyte glycoprotein (MOG)-specific CD4+ T cells co-cultured with microglia that were pre-activated with a high dose of IFN-γ and MOG peptide were primarily differentiated toward CD4+CD25+FoxP3− effector T cells. Instead, microglia primed with a low dose of both IFN-γ and neuroantigen induced the expansion of stable CD4+CD25+FoxP3+ regulatory T cells (Tregs) capable of suppressing EAE after adoptive transfer (48). Furthermore, low concentrations of IFN-γ enabled microglia to perform neuroprotective functions such as clearance of glutamate, neuronal survival, neurogenesis, and, to a lesser extent, oligodendrogenesis in response to CNS insult (50–52). Remarkably, EAE disease onset was significantly delayed in mice that were stereotaxically injected with microglia activated in vitro by a low dose of IFN-γ into the cerebral ventricles 7 days after immunization (53). By contrast, high concentrations of IFN-γ rendered microglia cytotoxic and impaired their neuroprotective activities (50–53). Interestingly, the dose-dependent paradoxical effects of IFN-γ may be mediated by microglial production of tumor necrosis factor (TNF)-α. Neutralization of TNF-α, whose expression is upregulated in IFN-γ-activated microglia, boosted both the neurogenesis and oligodendrogenesis induced by microglia activated with low-dose IFN-γ, whereas the addition of TNF-α to similar cell cultures prevented these cell renewal processes (52). Thus, low levels of IFN-γ induce beneficial effects on microglia, which are counteracted by the microglial production of TNF-α in response to increasing levels of IFN-γ.
On the other hand, IFN-γ can induce several self-limiting negative feedback mechanisms to restrain the magnitude and duration of its proinflammatory effects on microglia and to provide CNS protection. For example, IFN-γ induces mRNA expression of SOCS-1 which antagonizes IFN-γ-induced STAT-1 activation (54). Indeed, overexpression of SOCS-1 inhibited IFN-γ-induced expression of MHC class II and CD40 in microglia by inhibiting STAT-1-mediated expression of the class II transactivator (CIITA) transcription factor (54, 55). Furthermore, IFN-γ induced microglial cell death through the upregulation of apoptotic proteins, especially bcl-2-associated X protein (Bax), in EAE (56). This process was only observed in the advanced stage of EAE, suggesting a disease stage-specific role of IFN-γ on microglia in EAE, and likely MS. In fact, the activation and subsequent death of microglia induced by IFN-γ was proposed as a possible mechanism underlying MS relapse and remission (56).
Another important regulatory mechanism of IFN-γ is the suppression of T cell functions by localized catabolism of the aminoacid tryptophan, which is essential for cell growth and functioning. Indoleamine-2,3-dioxygenase (IDO), whose expression in microglia is upregulated by the IFN-γ-induced STAT-1 and phosphatidylinositol 3-kinase (PI3K) signaling pathways (57), catabolizes tryptophan to kynurenine, and has been shown to play a protective role in EAE (58). Microglial IDO expression, induced by IFN-γ, reduced extracellular tryptophan and increased kynurenine which suppressed the proliferation of myelin-specific T cells and inhibited production of proinflammatory Th1 cytokines (59). In addition, tryptophan deprivation made T cells more susceptible to apoptosis through Fas-Fas ligand (FasL)-mediated signaling (60), whose expression can be induced in microglia by IFN-γ (61), enabling them with an additional protective mechanism by facilitating apoptosis of myelin-specific T cells.
Additionally, IFN-γ establishes a regulatory feedback limiting the inflammatory activity of interleukin (IL)-18, a member of the IL-1 family produced by microglia that synergizes with IL-12 to induce Th1 polarization and IFN-γ production. IL-18 is upregulated during EAE (62) and its neutralization inhibited EAE (63). In turn, IFN-γ induced the expression of IL-18 binding protein (IL-18bp), an endogenous inhibitor of IL-18, in CNS-resident microglia and infiltrating macrophages during EAE (64). Interestingly, augmented IL-18bp expression in the CNS mediated by an adenoviral vector, blocked the induction of Th17 cells, but not Th1 cells, in the CNS and significantly reduced the incidence and severity of EAE (64). Therefore, these results suggest that IFN-γ-dependent IL-18bp production in microglia might regulate the balance between the Th1 and Th17 responses during autoimmune neuroinflammation.
Astrocytes are the most abundant glial cell population and are essential for brain homeostasis and neuronal function. They play a major role in maintaining both the structure and functional integrity of the BBB, develop metabolic functions, and seclude damaged areas in the CNS. Astrocytes also exhibit a variety of immune functions whereby they may both stimulate and restrain neuroinflammation (65–68). Although the capability of astrocytes to function as APCs in vivo is still controversial, some investigators have identified astrocytes expressing MHC class II molecules (69), co-stimulatory B7 molecules (70), and intercellular adhesion molecule (ICAM)-1 (71) at the edges of active MS lesions. The expression of these antigen-presenting molecules in astrocytes is upregulated by IFN-γ both in vivo and in vitro (12). In line with these results, astrocytes exposed to IFN-γ in vitro can induce proliferation of myelin-specific T cells and Th1 differentiation (66, 68). Importantly, CD4+ T cells activated by astrocytes treated with IFN-γ and pulsed with myelin protein were able to induce EAE by adoptive transfer (72). In addition, IFN-γ upregulated expression of ICAM-1 and vascular cell adhesion molecule (VCAM)-1 on primary astrocytes (72, 73), which might have a direct impact on the extravasation of T cells into the CNS considering the close apposition of astrocyte foot processes to the microvascular endothelium of the BBB.
IFN-γ signaling in astrocytes also plays an important role in triggering expression of a wide range of chemokines involved in the recruitment of inflammatory cells to the CNS in EAE and MS. At the edges of demyelinating MS lesions, astrocytes expressed CCL2, CXCL10, and their respective receptors CCR2 and CXCR3 (74, 75). In EAE, CCL2 and CXCL10 were localized predominantly in astrocytes surrounding inflammatory lesions (76–78) and their expression was abolished in IFNGR-deficient mice (76). In vitro studies have confirmed that both human and murine astrocytes are induced by IFN-γ to produce CCL2, CXCL9, and CXCL10 (79–81).
Selective silencing of IFN-γ signaling in astrocytes using a lentiviral vector significantly ameliorated both actively and passively induced EAE (82). Surprisingly, purified astrocytes from spinal cord of these animals not only exhibited reduced expression of IFN-γ-inducible chemokines such as CXCL9, CXCL10, and CXCL11 but also inhibited expression of chemokines induced by IL-17 signaling such as CXCL1, CXCL2, and CCL20 (82). These results were confirmed in vitro by analyzing astrocytes deficient in IFNGR and suggest that IFN-γ signaling is the common pathway in both Th1 and Th17 cell-mediated EAE (82). In contrast to results depicted in astrocytes, the authors of this study found that selective blocking of IFN-γ signaling in microglia resulted in more severe EAE progression that was associated with significantly enhanced CNS inflammatory infiltration. In addition, IFN-γ signaling restricted microglia proliferation (82). Together, these results suggest that IFN-γ would exert opposing roles on two different CNS-resident cell types in EAE: inflammatory on astrocytes and protective on microglia.
However, another study using transgenic mice expressing a signaling deficient dominant-negative IFNGR1 specifically on astrocytes reached opposite conclusions when analyzing EAE progression (83). Compared to wild-type mice, these animals did not show alteration in the incidence, disease onset, or initiation of clinical symptoms. Instead, during the transition from acute to chronic disease, the clinical score and mortality were significantly increased, suggesting that IFN-γ signaling in astrocytes provides disease stage-specific protection (83). Different methodological strategies used to ablate IFN-γ signaling might explain the discrepant results found between these two studies (82, 83). A posterior study demonstrated that persistent CNS inflammation and progressive disability observed in these mice lacking IFN-γ signaling in astrocytes was associated with elevated expression of IL-6 and sustained proliferation and activation of microglia (84). Taken together, these results indicate that IFN-γ signaling plays an important role in controlling the proliferation of microglia during EAE either directly on microglia (82) or indirectly through astrocytes, (84) and highlight the crucial role of IFN-γ in the signaling network between astrocytes and microglia during EAE pathogenesis.
There is discrepant evidence regarding the relationship between neurons and IFN-γ, mainly described in the non-inflammatory brain, with both protective and detrimental effects (3, 9, 85, 86). In EAE, it has been observed that treatment with nerve growth factor (NGF), a well-known neural factor involved in neuron survival, decreased disease severity, and increased IFNGR expression in spinal cord neurons (87). Even more, neurons were induced to express transforming growth factor (TGF)-β and B7 molecules in response to IFN-γ produced by encephalitogenic T cells. Remarkably, encephalitogenic T cell–neuron interaction induced higher neuron survival and promoted the conversion of encephalitogenic CD4+ T cells to functional CD4+CD25+TGF-β+CTLA-4+ Tregs. The adoptive transfer of these converted Tregs suppressed EAE. Strikingly, the acquisition of this regulatory phenotype occurred in the CNS and was mediated by the production of IFN-γ and TNF-α by T cells (88).
Despite several studies having reported an inflammatory effect of IFN-γ in the CNS during EAE development, accumulating data have also demonstrated that IFN-γ possesses neuroprotective activity in this disease. The recent evidence summarized in this review underscores the duality of IFN-γ on CNS-resident cells in EAE and provides different mechanisms whereby this cytokine exerts these opposing activities. In low doses, IFN-γ induces protection in both microglia and oligodendrocytes, while in high doses it induces disease worsening effects in both glial cell types. In astrocytes, the evidence reveals a primarily disease-promoting role for IFN-γ signaling, although it may be disease stage-specific. Interestingly, IFN-γ also exerts opposing roles in oligodendrocytes depending on the cell maturation status and cellular stress response. Importantly, these findings implicate that defects or fluctuations related to the expression of IFN-γ, its receptor, and/or its signaling pathway may underlie the immunopathogenesis of MS as well as other demyelinating inflammatory diseases. Therefore, delineating the precise role of IFN-γ in the CNS and on CNS-resident cells might provide the basis for fine-tuning the development of CNS-targeted selective therapy.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
This review was supported by FONDECYT 1140049 (RN) and 1141211 (MI), DIUSS N° 2012-0004-R (LR), and the CONICYT Doctoral fellowship 21130452 (GA).
1. Nylander A, Hafler DA. Multiple sclerosis. J Clin Invest (2012) 122(4):1180–8. doi:10.1172/JCI58649
2. Simmons SB, Pierson ER, Lee SY, Goverman JM. Modeling the heterogeneity of multiple sclerosis in animals. Trends Immunol (2013) 34(8):410–22. doi:10.1016/j.it.2013.04.006
3. Olsson T. Cytokines in neuroinflammatory disease: role of myelin autoreactive T cell production of interferon-gamma. J Neuroimmunol (1992) 40(2–3):211–8. doi:10.1016/0165-5728(92)90135-8
4. Fletcher JM, Lalor SJ, Sweeney CM, Tubridy N, Mills KH. T cells in multiple sclerosis and experimental autoimmune encephalomyelitis. Clin Exp Immunol (2010) 162(1):1–11. doi:10.1111/j.1365-2249.2010.04143.x
5. Duffy SS, Lees JG, Moalem-Taylor G. The contribution of immune and glial cell types in experimental autoimmune encephalomyelitis and multiple sclerosis. Mult Scler Int (2014) 2014:285245. doi:10.1155/2014/285245
6. Sanvito L, Constantinescu CS, Gran B, ‘t Hart BA. The multifaceted role of interferon. Open Autoimmun J (2010) 2:151–9. doi:10.2174/1876894601002040151
7. Kelchtermans H, Billiau A, Matthys P. How interferon-gamma keeps autoimmune diseases in check. Trends Immunol (2008) 29(10):479–86. doi:10.1016/j.it.2008.07.002
8. Arellano G, Ottum P, Reyes L, Burgos P, Naves R. Stage-specific role of interferon-gamma in experimental autoimmune encephalomyelitis and multiple sclerosis. Front Immunol (2015) 6:492. doi:10.3389/fimmu.2015.00492
9. Popko B, Corbin JG, Baerwald KD, Dupree J, Garcia AM. The effects of interferon-gamma on the central nervous system. Mol Neurobiol (1997) 14(1–2):19–35. doi:10.1007/BF02740619
10. Simmons RD, Willenborg DO. Direct injection of cytokines into the spinal cord causes autoimmune encephalomyelitis-like inflammation. J Neurol Sci (1990) 100(1–2):37–42. doi:10.1016/0022-510X(90)90010-K
11. Sethna MP, Lampson LA. Immune modulation within the brain: recruitment of inflammatory cells and increased major histocompatibility antigen expression following intracerebral injection of interferon-gamma. J Neuroimmunol (1991) 34(2–3):121–32. doi:10.1016/0165-5728(91)90121-M
12. Vass K, Lassmann H. Intrathecal application of interferon gamma. Progressive appearance of MHC antigens within the rat nervous system. Am J Pathol (1990) 137(4):789–800.
13. Vass K, Heininger K, Schafer B, Linington C, Lassmann H. Interferon-gamma potentiates antibody-mediated demyelination in vivo. Ann Neurol (1992) 32(2):198–206. doi:10.1002/ana.410320212
14. Renno T, Taupin V, Bourbonniere L, Verge G, Tran E, De Simone R, et al. Interferon-gamma in progression to chronic demyelination and neurological deficit following acute EAE. Mol Cell Neurosci (1998) 12(6):376–89. doi:10.1006/mcne.1998.0725
15. Corbin JG, Kelly D, Rath EM, Baerwald KD, Suzuki K, Popko B. Targeted CNS expression of interferon-gamma in transgenic mice leads to hypomyelination, reactive gliosis, and abnormal cerebellar development. Mol Cell Neurosci (1996) 7(5):354–70. doi:10.1006/mcne.1996.0026
16. Horwitz MS, Evans CF, McGavern DB, Rodriguez M, Oldstone MB. Primary demyelination in transgenic mice expressing interferon-gamma. Nat Med (1997) 3(9):1037–41. doi:10.1038/nm0997-1037
17. Voorthuis JA, Uitdehaag BM, De Groot CJ, Goede PH, van der Meide PH, Dijkstra CD. Suppression of experimental allergic encephalomyelitis by intraventricular administration of interferon-gamma in Lewis rats. Clin Exp Immunol (1990) 81(2):183–8. doi:10.1111/j.1365-2249.1990.tb03315.x
18. Heremans H, Dillen C, Groenen M, Martens E, Billiau A. Chronic relapsing experimental autoimmune encephalomyelitis (CREAE) in mice: enhancement by monoclonal antibodies against interferon-gamma. Eur J Immunol (1996) 26(10):2393–8. doi:10.1002/eji.1830261019
19. Naves R, Singh SP, Cashman KS, Rowse AL, Axtell RC, Steinman L, et al. The interdependent, overlapping, and differential roles of type I and II IFNs in the pathogenesis of experimental autoimmune encephalomyelitis. J Immunol (2013) 191(6):2967–77. doi:10.4049/jimmunol.1300419
20. Billiau A, Heremans H, Vandekerckhove F, Dijkmans R, Sobis H, Meulepas E, et al. Enhancement of experimental allergic encephalomyelitis in mice by antibodies against IFN-gamma. J Immunol (1988) 140(5):1506–10.
21. Pierson E, Simmons SB, Castelli L, Goverman JM. Mechanisms regulating regional localization of inflammation during CNS autoimmunity. Immunol Rev (2012) 248(1):205–15. doi:10.1111/j.1600-065X.2012.01126.x
22. Abromson-Leeman S, Bronson R, Luo Y, Berman M, Leeman R, Leeman J, et al. T-cell properties determine disease site, clinical presentation, and cellular pathology of experimental autoimmune encephalomyelitis. Am J Pathol (2004) 165(5):1519–33. doi:10.1016/S0002-9440(10)63410-4
23. Wensky AK, Furtado GC, Marcondes MC, Chen S, Manfra D, Lira SA, et al. IFN-gamma determines distinct clinical outcomes in autoimmune encephalomyelitis. J Immunol (2005) 174(3):1416–23. doi:10.4049/jimmunol.174.3.1416
24. Mangalam AK, Luo N, Luckey D, Papke L, Hubbard A, Wussow A, et al. Absence of IFN-gamma increases brain pathology in experimental autoimmune encephalomyelitis-susceptible DRB1*0301.DQ8 HLA transgenic mice through secretion of proinflammatory cytokine IL-17 and induction of pathogenic monocytes/microglia into the central nervous system. J Immunol (2014) 193(10):4859–70. doi:10.4049/jimmunol.1302008
25. Lees JR, Golumbek PT, Sim J, Dorsey D, Russell JH. Regional CNS responses to IFN-gamma determine lesion localization patterns during EAE pathogenesis. J Exp Med (2008) 205(11):2633–42. doi:10.1084/jem.20080155
26. Stromnes IM, Cerretti LM, Liggitt D, Harris RA, Goverman JM. Differential regulation of central nervous system autoimmunity by T(H)1 and T(H)17 cells. Nat Med (2008) 14(3):337–42. doi:10.1038/nm1715
27. Ni C, Wang C, Zhang J, Qu L, Liu X, Lu Y, et al. Interferon-gamma safeguards blood-brain barrier during experimental autoimmune encephalomyelitis. Am J Pathol (2014) 184(12):3308–20. doi:10.1016/j.ajpath.2014.08.019
28. Pouly S, Becher B, Blain M, Antel JP. Interferon-gamma modulates human oligodendrocyte susceptibility to Fas-mediated apoptosis. J Neuropathol Exp Neurol (2000) 59(4):280–6.
29. Lin W, Harding HP, Ron D, Popko B. Endoplasmic reticulum stress modulates the response of myelinating oligodendrocytes to the immune cytokine interferon-gamma. J Cell Biol (2005) 169(4):603–12. doi:10.1083/jcb.200502086
30. Balabanov R, Strand K, Goswami R, McMahon E, Begolka W, Miller SD, et al. Interferon-gamma-oligodendrocyte interactions in the regulation of experimental autoimmune encephalomyelitis. J Neurosci (2007) 27(8):2013–24. doi:10.1523/JNEUROSCI.4689-06.2007
31. Gao X, Gillig TA, Ye P, D’Ercole AJ, Matsushima GK, Popko B. Interferon-gamma protects against cuprizone-induced demyelination. Mol Cell Neurosci (2000) 16(4):338–49. doi:10.1006/mcne.2000.0883
32. Lin W, Bailey SL, Ho H, Harding HP, Ron D, Miller SD, et al. The integrated stress response prevents demyelination by protecting oligodendrocytes against immune-mediated damage. J Clin Invest (2007) 117(2):448–56. doi:10.1172/JCI29571
33. Lin W, Kemper A, Dupree JL, Harding HP, Ron D, Popko B. Interferon-gamma inhibits central nervous system remyelination through a process modulated by endoplasmic reticulum stress. Brain (2006) 129(Pt 5):1306–18. doi:10.1093/brain/awl044
34. Lin W, Popko B. Endoplasmic reticulum stress in disorders of myelinating cells. Nat Neurosci (2009) 12(4):379–85. doi:10.1038/nn.2273
35. Lees JR, Cross AH. A little stress is good: IFN-gamma, demyelination, and multiple sclerosis. J Clin Invest (2007) 117(2):297–9. doi:10.1172/JCI31254
36. Ren Z, Wang Y, Tao D, Liebenson D, Liggett T, Goswami R, et al. Overexpression of the dominant-negative form of interferon regulatory factor 1 in oligodendrocytes protects against experimental autoimmune encephalomyelitis. J Neurosci (2011) 31(23):8329–41. doi:10.1523/JNEUROSCI.1028-11.2011
37. Schwartz M, Butovsky O, Bruck W, Hanisch UK. Microglial phenotype: is the commitment reversible? Trends Neurosci (2006) 29(2):68–74. doi:10.1016/j.tins.2005.12.005
38. Benveniste EN. Role of macrophages/microglia in multiple sclerosis and experimental allergic encephalomyelitis. J Mol Med (1997) 75(3):165–73. doi:10.1007/s001090050101
40. Merson TD, Binder MD, Kilpatrick TJ. Role of cytokines as mediators and regulators of microglial activity in inflammatory demyelination of the CNS. Neuromolecular Med (2010) 12(2):99–132. doi:10.1007/s12017-010-8112-z
41. Wong GH, Bartlett PF, Clark-Lewis I, Battye F, Schrader JW. Inducible expression of H-2 and Ia antigens on brain cells. Nature (1984) 310(5979):688–91. doi:10.1038/310688a0
42. Suzumura A, Mezitis SG, Gonatas NK, Silberberg DH. MHC antigen expression on bulk isolated macrophage-microglia from newborn mouse brain: induction of Ia antigen expression by gamma-interferon. J Neuroimmunol (1987) 15(3):263–78. doi:10.1016/0165-5728(87)90121-4
43. Menendez Iglesias B, Cerase J, Ceracchini C, Levi G, Aloisi F. Analysis of B7-1 and B7-2 costimulatory ligands in cultured mouse microglia: upregulation by interferon-gamma and lipopolysaccharide and downregulation by interleukin-10, prostaglandin E2 and cyclic AMP-elevating agents. J Neuroimmunol (1997) 72(1):83–93. doi:10.1016/S0165-5728(96)00155-5
44. Murphy AC, Lalor SJ, Lynch MA, Mills KH. Infiltration of Th1 and Th17 cells and activation of microglia in the CNS during the course of experimental autoimmune encephalomyelitis. Brain Behav Immun (2010) 24(4):641–51. doi:10.1016/j.bbi.2010.01.014
45. Matsumoto Y, Ohmori K, Fujiwara M. Immune regulation by brain cells in the central nervous system: microglia but not astrocytes present myelin basic protein to encephalitogenic T cells under in vivo-mimicking conditions. Immunology (1992) 76(2):209–16.
46. Goldmann T, Prinz M. Role of microglia in CNS autoimmunity. Clin Dev Immunol (2013) 2013:208093. doi:10.1155/2013/208093
47. Bruck W, Porada P, Poser S, Rieckmann P, Hanefeld F, Kretzschmar HA, et al. Monocyte/macrophage differentiation in early multiple sclerosis lesions. Ann Neurol (1995) 38(5):788–96. doi:10.1002/ana.410380514
48. Ebner F, Brandt C, Thiele P, Richter D, Schliesser U, Siffrin V, et al. Microglial activation milieu controls regulatory T cell responses. J Immunol (2013) 191(11):5594–602. doi:10.4049/jimmunol.1203331
49. Prineas JW, Graham JS. Multiple sclerosis: capping of surface immunoglobulin G on macrophages engaged in myelin breakdown. Ann Neurol (1981) 10(2):149–58. doi:10.1002/ana.410100205
50. Shaked I, Tchoresh D, Gersner R, Meiri G, Mordechai S, Xiao X, et al. Protective autoimmunity: interferon-gamma enables microglia to remove glutamate without evoking inflammatory mediators. J Neurochem (2005) 92(5):997–1009. doi:10.1111/j.1471-4159.2004.02954.x
51. Butovsky O, Talpalar AE, Ben-Yaakov K, Schwartz M. Activation of microglia by aggregated beta-amyloid or lipopolysaccharide impairs MHC-II expression and renders them cytotoxic whereas IFN-gamma and IL-4 render them protective. Mol Cell Neurosci (2005) 29(3):381–93. doi:10.1016/j.mcn.2005.03.005
52. Butovsky O, Ziv Y, Schwartz A, Landa G, Talpalar AE, Pluchino S, et al. Microglia activated by IL-4 or IFN-gamma differentially induce neurogenesis and oligodendrogenesis from adult stem/progenitor cells. Mol Cell Neurosci (2006) 31(1):149–60. doi:10.1016/j.mcn.2005.10.006
53. Butovsky O, Landa G, Kunis G, Ziv Y, Avidan H, Greenberg N, et al. Induction and blockage of oligodendrogenesis by differently activated microglia in an animal model of multiple sclerosis. J Clin Invest (2006) 116(4):905–15. doi:10.1172/JCI26836
54. Wesemann DR, Dong Y, O’Keefe GM, Nguyen VT, Benveniste EN. Suppressor of cytokine signaling 1 inhibits cytokine induction of CD40 expression in macrophages. J Immunol (2002) 169(5):2354–60. doi:10.4049/jimmunol.169.5.2354
55. O’Keefe GM, Nguyen VT, Ping Tang LL, Benveniste EN. IFN-gamma regulation of class II transactivator promoter IV in macrophages and microglia: involvement of the suppressors of cytokine signaling-1 protein. J Immunol (2001) 166(4):2260–9. doi:10.4049/jimmunol.166.4.2260
56. Takeuchi H, Wang J, Kawanokuchi J, Mitsuma N, Mizuno T, Suzumura A. Interferon-gamma induces microglial-activation-induced cell death: a hypothetical mechanism of relapse and remission in multiple sclerosis. Neurobiol Dis (2006) 22(1):33–9. doi:10.1016/j.nbd.2005.09.014
57. Yadav MC, Burudi EM, Alirezaei M, Flynn CC, Watry DD, Lanigan CM, et al. IFN-gamma-induced IDO and WRS expression in microglia is differentially regulated by IL-4. Glia (2007) 55(13):1385–96. doi:10.1002/glia.20544
58. Kwidzinski E, Bunse J, Aktas O, Richter D, Mutlu L, Zipp F, et al. Indolamine 2,3-dioxygenase is expressed in the CNS and down-regulates autoimmune inflammation. FASEB J (2005) 19(10):1347–9. doi:10.1096/fj.04-3228fje
59. Platten M, Ho PP, Youssef S, Fontoura P, Garren H, Hur EM, et al. Treatment of autoimmune neuroinflammation with a synthetic tryptophan metabolite. Science (2005) 310(5749):850–5. doi:10.1126/science.1117634
60. Lee GK, Park HJ, Macleod M, Chandler P, Munn DH, Mellor AL. Tryptophan deprivation sensitizes activated T cells to apoptosis prior to cell division. Immunology (2002) 107(4):452–60. doi:10.1046/j.1365-2567.2002.01526.x
61. Badie B, Schartner J, Vorpahl J, Preston K. Interferon-gamma induces apoptosis and augments the expression of Fas and Fas ligand by microglia in vitro. Exp Neurol (2000) 162(2):290–6. doi:10.1006/exnr.1999.7345
62. Jander S, Stoll G. Differential induction of interleukin-12, interleukin-18, and interleukin-1beta converting enzyme mRNA in experimental autoimmune encephalomyelitis of the Lewis rat. J Neuroimmunol (1998) 91(1–2):93–9. doi:10.1016/S0165-5728(98)00162-3
63. Wildbaum G, Youssef S, Grabie N, Karin N. Neutralizing antibodies to IFN-gamma-inducing factor prevent experimental autoimmune encephalomyelitis. J Immunol (1998) 161(11):6368–74.
64. Millward JM, Lobner M, Wheeler RD, Owens T. Inflammation in the central nervous system and Th17 responses are inhibited by IFN-gamma-Induced IL-18 binding protein. J Immunol (2010) 185(4):2458–66. doi:10.4049/jimmunol.0902153
65. Miljkovic D, Timotijevic G, Mostarica Stojkovic M. Astrocytes in the tempest of multiple sclerosis. FEBS Lett (2011) 585(23):3781–8. doi:10.1016/j.febslet.2011.03.047
66. Chastain EM, Duncan DS, Rodgers JM, Miller SD. The role of antigen presenting cells in multiple sclerosis. Biochim Biophys Acta (2011) 1812(2):265–74. doi:10.1016/j.bbadis.2010.07.008
67. Nair A, Frederick TJ, Miller SD. Astrocytes in multiple sclerosis: a product of their environment. Cell Mol Life Sci (2008) 65(17):2702–20. doi:10.1007/s00018-008-8059-5
68. Dong Y, Benveniste EN. Immune function of astrocytes. Glia (2001) 36(2):180–90. doi:10.1002/glia.1107
69. Zeinstra E, Wilczak N, Streefland C, De Keyser J. Astrocytes in chronic active multiple sclerosis plaques express MHC class II molecules. Neuroreport (2000) 11(1):89–91. doi:10.1097/00001756-200001170-00018
70. Zeinstra E, Wilczak N, De Keyser J. Reactive astrocytes in chronic active lesions of multiple sclerosis express co-stimulatory molecules B7-1 and B7-2. J Neuroimmunol (2003) 135(1–2):166–71. doi:10.1016/S0165-5728(02)00462-9
71. Sobel RA, Mitchell ME, Fondren G. Intercellular adhesion molecule-1 (ICAM-1) in cellular immune reactions in the human central nervous system. Am J Pathol (1990) 136(6):1309–16.
72. Tan L, Gordon KB, Mueller JP, Matis LA, Miller SD. Presentation of proteolipid protein epitopes and B7-1-dependent activation of encephalitogenic T cells by IFN-gamma-activated SJL/J astrocytes. J Immunol (1998) 160(9):4271–9.
73. Rosenman SJ, Shrikant P, Dubb L, Benveniste EN, Ransohoff RM. Cytokine-induced expression of vascular cell adhesion molecule-1 (VCAM-1) by astrocytes and astrocytoma cell lines. J Immunol (1995) 154(4):1888–99.
74. Van Der Voorn P, Tekstra J, Beelen RH, Tensen CP, Van Der Valk P, De Groot CJ. Expression of MCP-1 by reactive astrocytes in demyelinating multiple sclerosis lesions. Am J Pathol (1999) 154(1):45–51. doi:10.1016/S0002-9440(10)65249-2
75. Tanuma N, Shin T, Kogure K, Matsumoto Y. Differential role of TNF-alpha and IFN-gamma in the brain of rats with chronic relapsing autoimmune encephalomyelitis. J Neuroimmunol (1999) 96(1):73–9. doi:10.1016/S0165-5728(99)00018-1
76. Carter SL, Muller M, Manders PM, Campbell IL. Induction of the genes for Cxcl9 and Cxcl10 is dependent on IFN-gamma but shows differential cellular expression in experimental autoimmune encephalomyelitis and by astrocytes and microglia in vitro. Glia (2007) 55(16):1728–39. doi:10.1002/glia.20587
77. Glabinski AR, Tani M, Strieter RM, Tuohy VK, Ransohoff RM. Synchronous synthesis of alpha- and beta-chemokines by cells of diverse lineage in the central nervous system of mice with relapses of chronic experimental autoimmune encephalomyelitis. Am J Pathol (1997) 150(2):617–30.
78. Ransohoff RM, Hamilton TA, Tani M, Stoler MH, Shick HE, Major JA, et al. Astrocyte expression of mRNA encoding cytokines IP-10 and JE/MCP-1 in experimental autoimmune encephalomyelitis. FASEB J (1993) 7(6):592–600.
79. Salmaggi A, Gelati M, Dufour A, Corsini E, Pagano S, Baccalini R, et al. Expression and modulation of IFN-gamma-inducible chemokines (IP-10, Mig, and I-TAC) in human brain endothelium and astrocytes: possible relevance for the immune invasion of the central nervous system and the pathogenesis of multiple sclerosis. J Interferon Cytokine Res (2002) 22(6):631–40. doi:10.1089/10799900260100114
80. Hayashi M, Luo Y, Laning J, Strieter RM, Dorf ME. Production and function of monocyte chemoattractant protein-1 and other beta-chemokines in murine glial cells. J Neuroimmunol (1995) 60(1–2):143–50. doi:10.1016/0165-5728(95)00064-9
81. Zhou ZH, Chaturvedi P, Han YL, Aras S, Li YS, Kolattukudy PE, et al. IFN-gamma induction of the human monocyte chemoattractant protein (hMCP)-1 gene in astrocytoma cells: functional interaction between an IFN-gamma-activated site and a GC-rich element. J Immunol (1998) 160(8):3908–16.
82. Ding X, Yan Y, Li X, Li K, Ciric B, Yang J, et al. Silencing IFN-gamma binding/signaling in astrocytes versus microglia leads to opposite effects on central nervous system autoimmunity. J Immunol (2015) 194(9):4251–64. doi:10.4049/jimmunol.1303321
83. Hindinger C, Bergmann CC, Hinton DR, Phares TW, Parra GI, Hussain S, et al. IFN-gamma signaling to astrocytes protects from autoimmune mediated neurological disability. PLoS One (2012) 7(7):e42088. doi:10.1371/journal.pone.0042088
84. Savarin C, Hinton DR, Valentin-Torres A, Chen Z, Trapp BD, Bergmann CC, et al. Astrocyte response to IFN-gamma limits IL-6-mediated microglia activation and progressive autoimmune encephalomyelitis. J Neuroinflammation (2015) 12(1):79. doi:10.1186/s12974-015-0293-9
85. Gonzalez-Perez O, Jauregui-Huerta F, Galvez-Contreras AY. Immune system modulates the function of adult neural stem cells. Curr Immunol Rev (2010) 6(3):167–73. doi:10.2174/157339510791823772
86. Hammarberg H, Lidman O, Lundberg C, Eltayeb SY, Gielen AW, Muhallab S, et al. Neuroprotection by encephalomyelitis: rescue of mechanically injured neurons and neurotrophin production by CNS-infiltrating T and natural killer cells. J Neurosci (2000) 20(14):5283–91.
87. Stampachiacchiere B, Aloe L. Differential modulatory effect of NGF on MHC class I and class II expression in spinal cord cells of EAE rats. J Neuroimmunol (2005) 169(1–2):20–30. doi:10.1016/j.jneuroim.2005.07.022
Keywords: interferon-gamma, experimental autoimmune encephalomyelitis, multiple sclerosis, glial cells, neurons, central nervous system, neuroinflammation
Citation: Ottum PA, Arellano G, Reyes LI, Iruretagoyena M and Naves R (2015) Opposing roles of interferon-gamma on cells of the central nervous system in autoimmune neuroinflammation. Front. Immunol. 6:539. doi: 10.3389/fimmu.2015.00539
Received: 21 August 2015; Accepted: 08 October 2015;
Published: 30 October 2015
Edited by:
Sergio Quezada, University College London Cancer Institute, UKReviewed by:
Ari Waisman, University Medical Center of the Johannes-Gutenberg University of Mainz, GermanyCopyright: © 2015 Ottum, Arellano, Reyes, Iruretagoyena and Naves. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Rodrigo Naves, cm9kcmlnb25hdmVzQG1lZC51Y2hpbGUuY2w=
†Payton A. Ottum and Gabriel Arellano have contributed equally to this work.
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.