 Gabriel Arellano
Gabriel Arellano Payton A. Ottum
Payton A. Ottum Lilian I. Reyes
Lilian I. Reyes Paula I. Burgos
Paula I. Burgos Rodrigo Naves
Rodrigo Naves
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Immunol., 29 September 2015
Sec. Immunological Tolerance and Regulation
Volume 6 - 2015 | https://doi.org/10.3389/fimmu.2015.00492
This article is part of the Research TopicSearching for immune tolerance manipulating new molecules and exploiting new concepts on lymphocyte biologyView all 14 articles
The role of interferon (IFN)-γ in multiple sclerosis (MS) and its animal model, experimental autoimmune encephalomyelitis (EAE), has remained as an enigmatic paradox for more than 30 years. Several studies attribute this cytokine a prominent proinflammatory and pathogenic function in these pathologies. However, accumulating evidence shows that IFN-γ also plays a protective role inducing regulatory cell activity and modulating the effector T cell response. Several innate and adaptive immune cells also develop opposite functions strongly associated with the production of IFN-γ in EAE. Even the suppressive activity of different types of regulatory cells is dependent on IFN-γ. Interestingly, recent data supports a stage-specific participation of IFN-γ in EAE providing a plausible explanation for previous conflicting results. In this review, we will summarize and discuss such literature, emphasizing the protective role of IFN-γ on immune cells. These findings are fundamental to understand the complex role of IFN-γ in the pathogenesis of these diseases and can provide basis for potential stage-specific therapy for MS targeting IFN-γ-signaling or IFN-γ-producing immune cells.
Interferon (IFN)-γ is the only type II IFN family member. It is secreted by activated immune cells, mainly T and natural killer (NK) cells, but also B cells, NKT cells, and professional antigen presenting cells (APC). IFN-γ binds to a heterodimeric receptor, IFNGR, expressed ubiquitously on almost all cell types. Given its pleiotropic functions, IFN-γ plays a pivotal role in orchestrating immune system homeostasis (1–4). Historically, IFN-γ production has been considered the hallmark of T helper (Th)1 cells driving inflammation and autoimmunity, such as multiple sclerosis (MS). MS is an inflammatory and demyelinating disorder of the central nervous system (CNS) and is the leading cause of non-traumatic neurological disability in young adults (5). According to the clinical course, MS can be classified in different types: relapsing-remitting disease (RRMS), consisting of acute recurrent attacks followed by a variable degree of recovery, and progressive forms characterized by chronic and irreversible neurological disability (6).
To date, experimental autoimmune encephalomyelitis (EAE) remains as the animal model most widely used to study the immunopathological mechanisms and therapeutic approaches to MS (7, 8). EAE is induced by immunization with myelin-derived antigens in adjuvant or by the adoptive transfer of activated myelin-specific T cells into syngeneic naive hosts. First, an initiation/inductive phase occurs, where innate and adaptive immune cells are antigen stimulated in the periphery. That is followed by the effector phase characterized by an acute immune cell infiltration into the CNS, and a later chronic phase of inflammation and axonal damage (9).
Discrepant results have been reported in relation to the role of IFN-γ in EAE and MS (3, 4, 10). Factors such as dose, site specificity, and timing of action as well as interaction with other cytokines and cells can determine the net effect of IFN-γ (3, 10, Ottum et al., in preparation). Recent evidence supports a, not mutually exclusive, stage-specific role of IFN-γ in EAE providing an explanation to these controversial results and a model whereby this cytokine can both promote and limit the development of these pathologies. In this same Research Topic, we have reviewed the opposing roles of IFN-γ on CNS-resident cells in EAE and MS (Ottum et al., in preparation). Here, we will review the evidence on IFN-γ’s dual role in the cells of the immune system in these same pathologies.
Initially, a positive association between increased levels of IFN-γ and demyelinating lesions in the CNS in MS and EAE attributed this cytokine a pathological role (11–15). In mice, passive immunization of healthy animals with encephalitogenic Th1 lymphocytes producing IFN-γ was sufficiently capable of inducing EAE (16). Besides, mice deficient in T-bet, a transcription factor that drives Th1 differentiation, were protected from developing EAE (17, 18). The proinflammatory effects of IFN-γ were confirmed in a pilot clinical study showing that seven of eighteen RRMS patients treated with IFN-γ exhibited symptom exacerbations (19). Consistently, secondary progressive MS patients (SPMS) treated with antibodies against IFN-γ exhibited slightly reduced clinical symptoms (20).
However, subsequent studies have challenged the notion that IFN-γ is pathogenic, and there is accumulating evidence proposing a protective role for IFN-γ in EAE and MS. Systemic or intraventricular injection of IFN-γ in EAE mice reduced the severity of disease symptoms, morbidity, and mortality (21, 22), and systemic IFN-γ treatment in chronic-relapsing EAE (CREAE) significantly delayed the appearance of relapses (23). Likewise, anti-IFN-γ therapy exacerbated EAE symptoms and made a mice strain resistant to EAE susceptible to developing disease (21–26). These results have been corroborated using animals deficient in the IFN-γ gene, which showed increased incidence of EAE, earlier disease onset and more severe symptoms compared with control mice (27–29). Even more, animals lacking IFNGR developed EAE with higher susceptibility, severity, and lethality (30–32). Passive transfer of encephalitogenic splenic cells from EAE-induced IFNGR-deficient mice into either wild-type (WT) or IFNGR-deficient recipient mice led to the development of EAE, but only WT mice recovered from illness (33). Interestingly, in tumor necrosis factor (TNF)-α receptor-deficient mice, a higher frequency of Th1 cells and enhanced mRNA expression of IFN-γ in the CNS was associated with a milder EAE (34).
Finally, in the marmoset EAE model, administration of human IFN-γ did not aggravate clinical symptoms, and by contrast, there was a trend to delay the appearance of the neurological episodes associated with less inflammation and demyelination during the EAE late phase (35). Regarding MS, induction of endogenous IFNs production in progressive MS patients showed that some patients with improving symptoms had high levels of serum IFN-γ, while clinical worsening was related with low serum IFN-γ levels (36).
The opposing activities that IFN-γ has in MS and the different models of EAE remain unresolved. However, collective evidence has shown that these paradoxical functions likely reflect a disease stage-specific opposing role of IFN-γ in EAE: promoting pathogenesis during the initiation phase but immunosuppression in the effector phase. Delivery of an intrathecal IFN-γ expression system during the initiation phase triggered an earlier disease onset followed by recovery, while overexpression of IFN-γ in the chronic phase resulted in disease amelioration (37). Consistently, intraventricular injection of IFN-γ during the initiation phase in CREAE mice increased the number of relapses (38). More recently, Naves et al. showed that IFNGR-deficient mice exhibited delayed disease onset followed by a more severe chronic phase, compared to WT mice (31). Similar results have been found analyzing mice lacking the IFN-γ gene or injecting an anti-IFN-γ neutralizing antibody during EAE progression (39). Furthermore, the administration of IFN-γ to EAE mice during the inductive period led to disease exacerbation, while such treatment was protective during the effector phase (31). Interestingly, the immunosuppressive activity of IFN-γ required functional type I IFN signaling and signal transducer and activator of transcription (STAT)-1 (31). In this way, stage-specific functions of IFN-γ can reconcile previous conflicting results in EAE and might also explain the mixed outcome reported in RRMS patients treated with IFN-γ (19).
Compelling evidence shows that IFN-γ exerts opposing effects on immune cells during the development of EAE and MS. In addition, several innate and adaptive immune cells play a dual role during the progression of these diseases associated with their IFN-γ production (Figure 1). Below, we will review and discuss this literature, focusing on the less-known protective face of IFN-γ (Table 1).
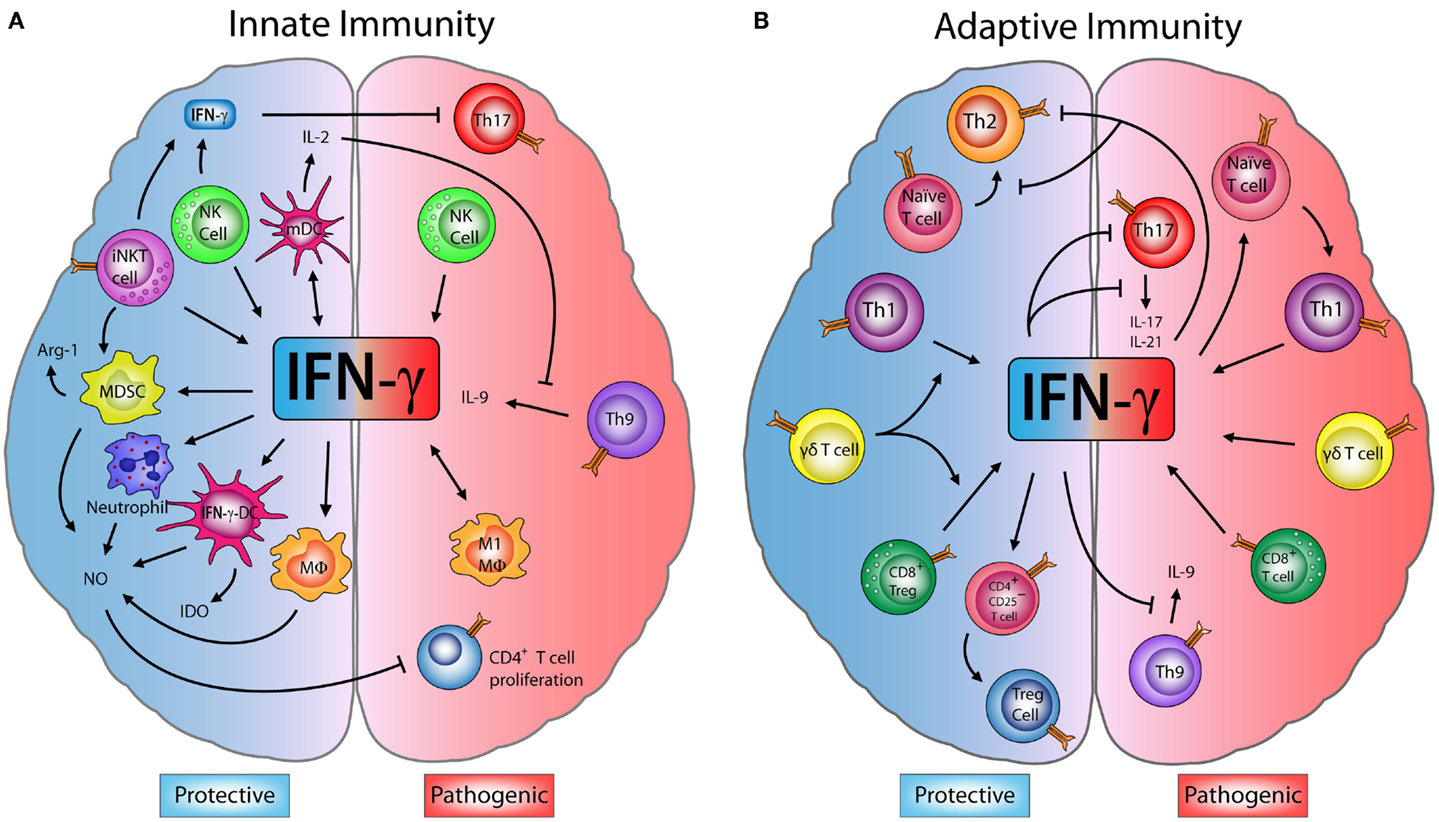
Figure 1. Dual role of IFN-γ in innate and adaptive immune cells in EAE. (A) Innate immunity: M1-macrophages (M1-MΦ) and natural killer (NK) cells produce interferon (IFN)-γ, which has a pathogenic role exacerbating encephalomyelitis autoimmune experimental (EAE) symptoms. However, some studies have shown that IFN-γ produced by NK and invariant NKT (iNKT) cells inhibits effector Th17 cells, decreasing the disease severity. IFN-γ induces the production of nitric oxide (NO) in neutrophils, MΦ, myeloid-derived suppressor cells (MDSCs), and IFN-γ-induced dendritic cells (IFN-γ-DC). NO can directly inhibit the proliferation of CD4+ T cells. IFN-γ also induces the expression of indoleamine 2,3-dioxygenase (IDO) in IFN-γ-DC and arginase-1 (Arg-1) by MDSC, enzymes that can suppress inflammation. Furthermore, IFN-γ induces IL-27 production by mature dendritic cells (mDC) which blocks Th9 differentiation and IL-9 production, controlling disease progression. (B) Adaptive immunity: IFN-γ secreted by Th1 and CD8+ T cells has an inflammatory effect and can drive the onset and progression of EAE. Despite this, IFN-γ is able to block Th9 cells, while Th1-secreted IFN-γ inhibits Th2 and Th17 effector cells. Interestingly, IFN-γ can induce CD4+CD25+ regulatory T cells (Tregs) increasing their FOXP3 expression. Upon transfer, these IFN-γ-induced Tregs limit the severity of EAE. Moreover, IFN-γ production by CD8+ regulatory T cells (CD8+ Tregs) also reduces EAE symptoms. Finally, IFN-γ produced by γδ T cells worsens EAE, but has a regulatory role on the production of IFN-γ by T cells, which is necessary to limit disease.

Table 1. The protective effects of IFN-γ and IFN-γ-producing immune cells in EAE and MS.
IFN-γ controls the infiltration of macrophages and neutrophils into the CNS regulating the course of EAE (74). Animals deficient in IFN-γ or IFNGR generate an atypical disease affecting mainly the brainstem and cerebellum with increased expression of CXCL2, favoring the recruitment of high numbers of CXCR2-mediated neutrophils; while in conventional EAE, IFN-γ leads to increased CCL2 levels guiding macrophage infiltration into the spinal cord mediated by CCR2 (40–42). Macrophages and neutrophils produce high levels of nitric oxide (NO), which has both pathogenic and regulatory functions in neuroinflammation (75). Interestingly, IFN-γ is a primary inducer of NO and mice deficient in inducible nitric oxide synthase (iNOS) develop a severe form of EAE (76, 77). Willenborg et al. showed that peritoneal exudate cells (PEC), characterized by a high presence of macrophages, are able to inhibit the extensive proliferation of splenocytes from IFNGR-deficient mice with EAE by IFN-γ-dependent NO production (33). Neutrophils and myeloid-derived suppressor cells (MDSCs) with high expression of Gr-1 also exhibited potent suppressor activity in EAE, inhibiting T cell proliferation through a mechanism that was absolutely dependent on IFN-γ and NO (43–45). Additionally, IFN-γ along with interleukin (IL)-4, secreted by activated invariant NKT (iNKT) cells, stimulated MDSCs to suppress EAE via iNOS and arginase (arg)-1 expression (51).
NK cells play both a regulatory and pathogenic role in EAE and MS (39, 46, 78–86). Although the underlying mechanisms are poorly understood, several studies suggest that IFN-γ-producing NK cells might be driving this duality in a location and stage-dependent manner (39, 46–49, 87). NK cells have been identified as the main source of IFN-γ production in the initiation stage of EAE, which might be necessary for migration of pathogenic T cells into the CNS (39). Interestingly, early but not late depletion of NK cells significantly delayed the onset of disease (39). IL-18 and IL-21 are two key cytokines involved in NK cell functional maturation (88, 89). Administration of IL-21 before EAE immunization promoted higher IFN-γ production by NK cells and induced a significantly enhanced acute phase with more intense CNS cell infiltration compared to untreated mice (90). However, IL-21 treatment failed to induce augmentation of IFN-γ production and had no effect on disease progression when applied for one week starting a few days before disease onset (90). Similarly, IL-18 injection in WT mice at the time of immunization enhanced disease severity promoting autoreactive Th1 cell development through the induction of IFN-γ by NK cells (87). Moreover, IFN-γ signaling in NK cells was required to restore EAE susceptibility in IL-18 defective mice (87). Taken together, these results suggest that early IFN-γ production by NK cells mainly contributes to the initiation, but not progression, of EAE pathogenesis. By contrast, once NK cells infiltrate the CNS, they assume a protective role suppressing myelin-reactive Th17 cells via modulation of microglia activation. This effect was CNS compartment-restricted and was perforin and IFN-γ-dependent (46). Therefore, signals and/or components generated in the CNS during the effector phase might be inducing protective functions in infiltrating NK cells. Indeed, peptides complexed with the chaperone heat shock protein (Hsp) 70 derived from inflamed brain of EAE mice have been described as promotors of the immunotolerogenic activity of NK cells in EAE. Suppressive effects of Hsp-peptide complex-activated NK cells correlated with high production of IFN-γ and resulted in inhibition of Th17 cells (47–49).
NK cell subtypes and iNKT cells may also have IFN-γ-mediated suppressive activity in MS and EAE. Regarding MS, evidence in two independent cohorts of RRMS patients demonstrated that the classically inhibitory CD3−CD56brightCD16− NK cells from RRMS patients have impaired expansion in response to IL-12, and severely diminished IFN-γ production compared to healthy control NK cells (50). In addition, in vivo activation of iNKT cells at the same time as EAE induction significantly ameliorated disease progression through mechanisms dependent on IFN-γ alone (52) or synergistically with IL-4 and IL-10, resulting in inhibition of the Th17 response (53).
Dendritic cells (DC) are professional APC important to maintain the balance between immunity and tolerance. In EAE, they efficiently present myelin antigens in order to prime and polarize naïve T cells. They also help regulate EAE severity as evidenced by disease exacerbation in DC deficient mice (91, 92). The regulatory effects of DC are partly due to the IFN-γ-induced production of IL-27 that suppressed the differentiation and encephalitogenicity of Th9 cells. It also inhibited the production of IL-9 by both Th9 and Th17 cells. This suppression was partially dependent on STAT-1 and T-bet and was necessary to regulate EAE severity (55). Remarkably, splenic DC exposed to IFN-γ for 48 hours exhibited an immature and tolerogenic phenotype (tol-DC). These tol-DC decreased disease severity in Lewis rats and relapse frequency in SJL/J and B6 mouse models when transferred during the inductive phase (54). EAE amelioration was accompanied by reduced macrophage activation and CD4+ T cell CNS infiltration, compared to control mice. The therapeutic activity was dependent on an antigen-specific IFN-γ pathway, involving increased DC expression of indoleamine 2,3-dioxygenase (IDO), which induced CD4+ T cell apoptosis (54).
CD4+ T (Th) cells proliferate and differentiate into various subtypes in response to antigen stimulation and their microenvironment in order to exert specific effector or regulatory functions (93). Effector CD4+ T (Teff) cell lineages, such as Th1 cells, Th2 cells, Th17 cells, and Th9 cells, and regulatory T cells (Tregs) can be distinguished by the cytokines they produce and the transcription factors essential for their differentiation. These T cells also exhibit functional and phenotypical plasticity expressing cytokines and/or transcription factors of other lineages (94, 95).
Classically, IFN-γ is known for promoting the differentiation of Th1 cells and inhibiting the Th2 immune response which may contribute to neuroinflammation (5, 10, 96, 97). Despite its inflammatory activity, IFN-γ increased apoptosis and inhibited proliferation of CD4+CD44high (activated) T lymphocytes from both the spleen and CNS of EAE mice (57). Notably, it also inhibited in vitro proliferation of T cell receptor (TCR)-activated peripheral blood mononuclear cells (PBMC) from progressive MS patients in a dose-dependent manner (56). Mice depleted of IFN-γ or IFN-γ signaling developed more severe EAE, atypical neurological symptoms, and increased Th17-characteristic inflammation. These data underscore an important anti-inflammatory function of IFN-γ in EAE: the inhibition of pathogenic Th17 cell differentiation and cytokine production (5, 31, 58–62, 96). Besides, it has been shown that IFN-γ has a STAT-1-mediated direct inhibitory effect on pathogenic Th9 cells (55). Interestingly, another study identified a non-pathogenic Th1 cell subset with high IFN-γ expression, capable of restraining EAE development during early stages of disease by suppressing Th17 cells in an IFN-γ-dependent manner (63). The inhibitory mechanism involved the activation of STAT-1 and IL-21 expression via induction of T-bet (60, 62). Despite the ability of IFN-γ to directly and indirectly inhibit Th17 cells, a pathogenic population of Th1 cells has been identified in EAE and MS that also expresses IL-17. This capacity to express both cytokines (IFN-γ and IL-17) may be due to the plasticity of Th17 cells, which can undergo a shift toward the Th1 phenotype (95, 98, 99).
Several studies have shown that γδ T cells are present in the CNS of MS patients and EAE mice (100). Given that activated γδ T cells have the capacity to produce high expression of Th1 and Th17 cytokines, they might contribute to the induction or maintenance of neuroinflammation. However, efforts to determine a role for these cells have given contradictory results. While some studies have found that depletion of γδ T cells resulted in reduced severity of EAE, other reports have described disease aggravation (100). Regarding IFN-γ, evidence suggests that during early EAE, γδ T cells may act either as a main source of this cytokine (101) or regulate IFN-γ expression in other cell types, including CD4+ and CD8+ T cells (64). Indeed, SJL/J mice depleted of γδ T cells showed a significant reduction of IFN-γ expression in the CNS at all stages of EAE (102). Other studies have shown that mice deficient in γδ T cells that are reconstituted with γδ T cells lacking IFN-γ expression developed a significantly delayed and attenuated EAE. This suggested that IFN-γ production by γδ T cells may be central to initial inflammatory events (101). Despite this, Ponomarev et al. proposed that γδ T cells are required to promote CNS-restricted production of sufficient levels of IFN-γ necessary for EAE recovery (64).
It has been reported that IFN-γ is important to the function of Tregs in EAE and MS. Reduced FoxP3 expression and lower frequency and function of Tregs was reported in IFN-γ-deficient mice with EAE, in comparison to EAE-induced WT mice (65). Remarkably, treatment of CD4+CD25− T cells from WT or IFN-γ-deficient mice with IFN-γ alone or with additional TCR stimulation led to their conversion into Tregs expressing CD25 and FOXP3 (65). These IFN-γ-induced Tregs effectively inhibited EAE disease progression when adoptively transferred into IFN-γ-deficient mice. Human CD4+CD25− T cells from healthy volunteers were similarly converted into functionally active Tregs ex vivo upon IFN-γ stimulation (65).
A new subpopulation of Tregs expressing T-bet, CXCR3 and IFN-γ, named Th1-like Tregs has been reported in healthy individuals (103) and have regulatory functions focused on Th1-mediated inflammatory diseases (104–108). Interestingly, these cells were also described in MS and EAE (109, 110). An increased frequency of Th1-like Tregs with reduced suppressive function was reported in untreated RRMS patients compared to healthy controls (110). In this case, addition of IFN-γ neutralizing antibodies recovered their functionality suggesting that IFN-γ might contribute to their reduced immunomodulatory capacity (110).
Several studies have demonstrated that IFN-γ production by CD8+ T cells is a major mediator of EAE induced by cytotoxic T lymphocytes (CTL) (111–114). One of these investigations showed that atypical EAE induced by intrathecal transfer of myelin basic protein (MBP)-specific CD8+ T cells in C3H mice was ameliorated by co-injection with neutralizing antibodies for IFN-γ (112). Other studies have identified subsets of regulatory CD8+ T cells (CD8+ Tregs) that suppress EAE development via IFN-γ-dependent mechanisms. Both therapeutic and prophylactic transfer of myelin oligodendroctye glycoprotein MOG-induced CD8+ T cells into mice with EAE ameliorated disease suppressing the chronic phase, but not affecting the disease onset or acute phase (66, 115). Strikingly, this protective function was lost when IFN-γ-deficient MOG-induced-CD8+ T cells were transferred before EAE induction in WT mice, but was enhanced when IFN-γ production was stimulated in MOG-specific CD8+ T cells before cell transfer (66). Furthermore, in those studies reporting a pathogenic function, myelin-specific CD8+ T cell lines used to passively induce EAE were generated from CD8+ T cells isolated during the inductive phase (111, 112). In contrast, regulatory myelin-specific CD8+ T cells were obtained during the chronic phase of disease (66, 115). Taken together, these results reinforce the notion of a stage-specific IFN-γ-dependent regulation, mediated in this case by CNS-specific regulatory CD8+ T cells.
A naturally occurring CD8+ Tregs subset was identified that expressed latency-associated peptide (LAP) on their cell surface and produced more IFN-γ than their LAP− counterparts. Adoptive transfer of these cells previous to myelin immunization improved EAE recovery mediated by their IFN-γ production (67). A CD8αα+TCRαβ+ T cell subset capable of preventing EAE when stimulated with a TCR-derived peptide before MBP-peptide immunization in H-2u mouse strains has also been described (68, 69, 116). Interestingly, the vaccine failed to prevent EAE development in IFN-γ-deficient mice and resulted in delayed disease onset but worsened disease severity compared to control mice, suggesting an important stage-specific role for IFN-γ signaling in CD8αα+TCRαβ+ T cell-mediated protection (70, 116). A CD8+ Tregs subtype expressing high levels of CD38 ectonucleotidase suppressed Teff cell proliferation in a non-antigen specific, cell-to-cell contact, and IFN-γ-dependent fashion, resulting in ameliorated EAE (71). Finally, IFN-γ-producing CD8+ T cells induced by glatiramer acetate (GA), a therapy for MS, suppressed EAE in mice via an IDO-dependent mechanism, suggesting that the immunomodulatory action of GA is mediated at least in part by IFN-γ production by CD8+ T cells (72). Consistently, GA-specific CD8+ T cells from GA-treated RRMS patients tended to produce more IFN-γ than CD8+ T cells from untreated patients (73). In progressive MS patients, CTL had impaired IL-2 induced IFN-γ production and decreased ability to suppress proliferation of TCR-stimulated autologous lymphocytes (56).
The effect of IFN-γ on B cells in the neuroinflammatory context of MS and EAE is unclear. Bar-Or and colleagues demonstrated that CD19+ B cells isolated from RRMS patients had significantly increased production of lymphotoxin (TNF-β) and TNF-α in response to IFN-γ and insignificant changes in IL-10 production (117). In marmoset EAE, exogenous administration of IFN-γ caused no significant clinical change in disease; however, there was a significant decrease in plasma IgG specific to MOG peptides (35).
Recent studies support the notion that IFN-γ exerts a stage-specific role during EAE development. Strikingly, several innate and adaptive immune cells develop opposite activities during EAE progression, which is related to their production of IFN-γ in a stage-specific manner. Furthermore, the suppressive activity of different types of immune regulatory cells is IFN-γ-dependent. Taken together, these data provide a mechanistic basis explaining the previous controversial results in relation to the role of IFN-γ in EAE and MS. Delineating the varying activities of IFN-γ as well as the role of IFN-γ-producing immune cells during the course of EAE and MS will not only provide insight into the complex role of IFN-γ in these diseases but might also lead to therapies targeting IFN-γ signaling or IFN-γ-producing immune cells. These treatments can be helpful to a selective group of MS patients or during a specific stage of disease.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
This review was supported by DIUSS N° 2012-0004-R (LIR), FONDECYT 1140049 (RN) and 1141211 (PIB), and the CONICYT Doctoral fellowship 21130452 (GA).
1. Borden EC, Sen GC, Uze G, Silverman RH, Ransohoff RM, Foster GR, et al. Interferons at age 50: past, current and future impact on biomedicine. Nat Rev Drug Discov (2007) 6(12):975–90. doi: 10.1038/nrd2422
2. Farrar MA, Schreiber RD. The molecular cell biology of interferon-gamma and its receptor. Annu Rev Immunol (1993) 11:571–611. doi:10.1146/annurev.iy.11.040193.003035
3. Shachar I, Karin N. The dual roles of inflammatory cytokines and chemokines in the regulation of autoimmune diseases and their clinical implications. J Leukoc Biol (2013) 93(1):51–61. doi:10.1189/jlb.0612293
4. Kelchtermans H, Billiau A, Matthys P. How interferon-gamma keeps autoimmune diseases in check. Trends Immunol (2008) 29(10):479–86. doi:10.1016/j.it.2008.07.002
5. Nylander A, Hafler DA. Multiple sclerosis. J Clin Invest (2012) 122(4):1180–8. doi:10.1172/JCI58649
6. Mahad DH, Trapp BD, Lassmann H. Pathological mechanisms in progressive multiple sclerosis. Lancet Neurol (2015) 14(2):183–93. doi:10.1016/S1474-4422(14)70256-X
7. Steinman L, Zamvil SS. How to successfully apply animal studies in experimental allergic encephalomyelitis to research on multiple sclerosis. Ann Neurol (2006) 60(1):12–21. doi:10.1002/ana.20913
8. Baxter AG. The origin and application of experimental autoimmune encephalomyelitis. Nat Rev Immunol (2007) 7(11):904–12. doi:10.1038/nri2190
9. Kurschus FC. T cell mediated pathogenesis in EAE: molecular mechanisms. Biomed J (2015) 38(3):183–93. doi:10.4103/2319-4170.155590
10. Lara Sanvito, Constantinescu CS, Bruno Gran, ‘t Hart Bert A. The multifaceted role of interferon-γ central nervous system autoimmune demyelination. Open Autoimmunity J (2010) 2:151–9.
11. Olsson T. Cytokines in neuroinflammatory disease: role of myelin autoreactive T cell production of interferon-gamma. J Neuroimmunol (1992) 40(2–3):211–8. doi:10.1016/0165-5728(92)90135-8
12. Brosnan CF, Cannella B, Battistini L, Raine CS. Cytokine localization in multiple sclerosis lesions: correlation with adhesion molecule expression and reactive nitrogen species. Neurology (1995) 45(6 Suppl 6):S16–21. doi:10.1212/WNL.45.6_Suppl_6.S16
13. Renno T, Krakowski M, Piccirillo C, Lin JY, Owens T. TNF-alpha expression by resident microglia and infiltrating leukocytes in the central nervous system of mice with experimental allergic encephalomyelitis. Regulation by Th1 cytokines. J Immunol (1995) 154(2):944–53.
14. Renno T, Lin JY, Piccirillo C, Antel J, Owens T. Cytokine production by cells in cerebrospinal fluid during experimental allergic encephalomyelitis in SJL/J mice. J Neuroimmunol (1994) 49(1–2):1–7. doi:10.1016/0165-5728(94)90174-0
15. Merrill JE, Kono DH, Clayton J, Ando DG, Hinton DR, Hofman FM. Inflammatory leukocytes and cytokines in the peptide-induced disease of experimental allergic encephalomyelitis in SJL and B10.PL mice. Proc Natl Acad Sci USA (1992) 89(2):574–8. doi:10.1073/pnas.89.2.574
16. Pettinelli CB, McFarlin DE. Adoptive transfer of experimental allergic encephalomyelitis in SJL/J mice after in vitro activation of lymph node cells by myelin basic protein: requirement for Lyt 1+ 2- T lymphocytes. J Immunol (1981) 127(4):1420–3.
17. Bettelli E, Sullivan B, Szabo SJ, Sobel RA, Glimcher LH, Kuchroo VK. Loss of T-bet, but not STAT1, prevents the development of experimental autoimmune encephalomyelitis. J Exp Med (2004) 200(1):79–87. doi:10.1084/jem.20031819
18. Nath N, Prasad R, Giri S, Singh AK, Singh I. T-bet is essential for the progression of experimental autoimmune encephalomyelitis. Immunology (2006) 118(3):384–91. doi:10.1111/j.1365-2567.2006.02385.x
19. Panitch HS, Hirsch RL, Schindler J, Johnson KP. Treatment of multiple sclerosis with gamma interferon: exacerbations associated with activation of the immune system. Neurology (1987) 37(7):1097–102. doi:10.1212/WNL.37.7.1097
20. Skurkovich S, Boiko A, Beliaeva I, Buglak A, Alekseeva T, Smirnova N, et al. Randomized study of antibodies to IFN-gamma and TNF-alpha in secondary progressive multiple sclerosis. Mult Scler (2001) 7(5):277–84. doi:10.1191/135245801681137960
21. Billiau A, Heremans H, Vandekerckhove F, Dijkmans R, Sobis H, Meulepas E, et al. Enhancement of experimental allergic encephalomyelitis in mice by antibodies against IFN-gamma. J Immunol (1988) 140(5):1506–10.
22. Voorthuis JA, Uitdehaag BM, De Groot CJ, Goede PH, van der Meide PH, Dijkstra CD. Suppression of experimental allergic encephalomyelitis by intraventricular administration of interferon-gamma in Lewis rats. Clin Exp Immunol (1990) 81(2):183–8. doi:10.1111/j.1365-2249.1990.tb03315.x
23. Heremans H, Dillen C, Groenen M, Martens E, Billiau A. Chronic relapsing experimental autoimmune encephalomyelitis (CREAE) in mice: enhancement by monoclonal antibodies against interferon-gamma. Eur J Immunol (1996) 26(10):2393–8. doi:10.1002/eji.1830261019
24. Duong TT, St Louis J, Gilbert JJ, Finkelman FD, Strejan GH. Effect of anti-interferon-gamma and anti-interleukin-2 monoclonal antibody treatment on the development of actively and passively induced experimental allergic encephalomyelitis in the SJL/J mouse. J Neuroimmunol (1992) 36(2–3):105–15. doi:10.1016/0165-5728(92)90042-J
25. Lublin FD, Knobler RL, Kalman B, Goldhaber M, Marini J, Perrault M, et al. Monoclonal anti-gamma interferon antibodies enhance experimental allergic encephalomyelitis. Autoimmunity (1993) 16(4):267–74. doi:10.3109/08916939309014645
26. Duong TT, Finkelman FD, Singh B, Strejan GH. Effect of anti-interferon-gamma monoclonal antibody treatment on the development of experimental allergic encephalomyelitis in resistant mouse strains. J Neuroimmunol (1994) 53(1):101–7. doi:10.1016/0165-5728(94)90069-8
27. Krakowski M, Owens T. Interferon-gamma confers resistance to experimental allergic encephalomyelitis. Eur J Immunol (1996) 26(7):1641–6. doi:10.1002/eji.1830260735
28. Ferber IA, Brocke S, Taylor-Edwards C, Ridgway W, Dinisco C, Steinman L, et al. Mice with a disrupted IFN-gamma gene are susceptible to the induction of experimental autoimmune encephalomyelitis (EAE). J Immunol (1996) 156(1):5–7.
29. Sabatino JJ Jr, Shires J, Altman JD, Ford ML, Evavold BD. Loss of IFN-gamma enables the expansion of autoreactive CD4+ T cells to induce experimental autoimmune encephalomyelitis by a nonencephalitogenic myelin variant antigen. J Immunol (2008) 180(7):4451–7. doi:10.4049/jimmunol.180.7.4451
30. Willenborg DO, Fordham S, Bernard CC, Cowden WB, Ramshaw IA. IFN-gamma plays a critical down-regulatory role in the induction and effector phase of myelin oligodendrocyte glycoprotein-induced autoimmune encephalomyelitis. J Immunol (1996) 157(8):3223–7.
31. Naves R, Singh SP, Cashman KS, Rowse AL, Axtell RC, Steinman L, et al. The interdependent, overlapping, and differential roles of type I and II IFNs in the pathogenesis of experimental autoimmune encephalomyelitis. J Immunol (2013) 191(6):2967–77. doi:10.4049/jimmunol.1300419
32. Xiao BG, Ma CG, Xu LY, Link H, Lu CZ. IL-12/IFN-gamma/NO axis plays critical role in development of Th1-mediated experimental autoimmune encephalomyelitis. Mol Immunol (2008) 45(4):1191–6. doi:10.1016/j.molimm.2007.07.003
33. Willenborg DO, Fordham SA, Staykova MA, Ramshaw IA, Cowden WB. IFN-gamma is critical to the control of murine autoimmune encephalomyelitis and regulates both in the periphery and in the target tissue: a possible role for nitric oxide. J Immunol (1999) 163(10):5278–86.
34. Wheeler RD, Zehntner SP, Kelly LM, Bourbonniere L, Owens T. Elevated interferon gamma expression in the central nervous system of tumour necrosis factor receptor 1-deficient mice with experimental autoimmune encephalomyelitis. Immunology (2006) 118(4):527–38. doi:10.1111/j.1365-2567.2006.02395.x
35. Jagessar SA, Gran B, Heijmans N, Bauer J, Laman JD, ‘t Hart BA, et al. Discrepant effects of human interferon-gamma on clinical and immunological disease parameters in a novel marmoset model for multiple sclerosis. J Neuroimmune Pharmacol (2012) 7(1):253–65. doi:10.1007/s11481-012-9384-x
36. Bever CT Jr, Panitch HS, Levy HB, McFarlin DE, Johnson KP. Gamma-interferon induction in patients with chronic progressive MS. Neurology (1991) 41(7):1124–7. doi:10.1212/WNL.41.7.1124
37. Furlan R, Brambilla E, Ruffini F, Poliani PL, Bergami A, Marconi PC, et al. Intrathecal delivery of IFN-gamma protects C57BL/6 mice from chronic-progressive experimental autoimmune encephalomyelitis by increasing apoptosis of central nervous system-infiltrating lymphocytes. J Immunol (2001) 167(3):1821–9. doi:10.4049/jimmunol.167.3.1821
38. Tanuma N, Shin T, Kogure K, Matsumoto Y. Differential role of TNF-alpha and IFN-gamma in the brain of rats with chronic relapsing autoimmune encephalomyelitis. J Neuroimmunol (1999) 96(1):73–9. doi:10.1016/S0165-5728(99)00018-1
39. Dungan LS, McGuinness NC, Boon L, Lynch MA, Mills KH. Innate IFN-gamma promotes development of experimental autoimmune encephalomyelitis: a role for NK cells and M1 macrophages. Eur J Immunol (2014) 44(10):2903–17. doi:10.1002/eji.201444612
40. Tran EH, Prince EN, Owens T. IFN-gamma shapes immune invasion of the central nervous system via regulation of chemokines. J Immunol (2000) 164(5):2759–68. doi:10.4049/jimmunol.164.5.2759
41. Lees JR, Golumbek PT, Sim J, Dorsey D, Russell JH. Regional CNS responses to IFN-gamma determine lesion localization patterns during EAE pathogenesis. J Exp Med (2008) 205(11):2633–42. doi:10.1084/jem.20080155
42. Stoolman JS, Duncker PC, Huber AK, Segal BM. Site-specific chemokine expression regulates central nervous system inflammation and determines clinical phenotype in autoimmune encephalomyelitis. J Immunol (2014) 193(2):564–70. doi:10.4049/jimmunol.1400825
43. Zehntner SP, Brickman C, Bourbonniere L, Remington L, Caruso M, Owens T. Neutrophils that infiltrate the central nervous system regulate T cell responses. J Immunol (2005) 174(8):5124–31. doi:10.4049/jimmunol.174.8.5124
44. Zhu B, Bando Y, Xiao S, Yang K, Anderson AC, Kuchroo VK, et al. CD11b+Ly-6C(hi) suppressive monocytes in experimental autoimmune encephalomyelitis. J Immunol (2007) 179(8):5228–37. doi:10.4049/jimmunol.179.8.5228
45. Pastor S, Minguela A, Mi W, Ward ES. Autoantigen immunization at different sites reveals a role for anti-inflammatory effects of IFN-gamma in regulating susceptibility to experimental autoimmune encephalomyelitis. J Immunol (2009) 182(9):5268–75. doi:10.4049/jimmunol.0800681
46. Hao J, Liu R, Piao W, Zhou Q, Vollmer TL, Campagnolo DI, et al. Central nervous system (CNS)-resident natural killer cells suppress Th17 responses and CNS autoimmune pathology. J Exp Med (2010) 207(9):1907–21. doi:10.1084/jem.20092749
47. Galazka G, Jurewicz A, Domowicz M, Cannella B, Raine CS, Selmaj K. HINT1 peptide/Hsp70 complex induces NK-cell-dependent immunoregulation in a model of autoimmune demyelination. Eur J Immunol (2014) 44(10):3026–44. doi:10.1002/eji.201444694
48. Galazka G, Jurewicz A, Orlowski W, Stasiolek M, Brosnan CF, Raine CS, et al. EAE tolerance induction with Hsp70-peptide complexes depends on H60 and NKG2D activity. J Immunol (2007) 179(7):4503–12. doi:10.4049/jimmunol.179.7.4503
49. Galazka G, Stasiolek M, Walczak A, Jurewicz A, Zylicz A, Brosnan CF, et al. Brain-derived heat shock protein 70-peptide complexes induce NK cell-dependent tolerance to experimental autoimmune encephalomyelitis. J Immunol (2006) 176(3):1588–99. doi:10.4049/jimmunol.176.3.1588
50. Lunemann A, Tackenberg B, DeAngelis T, da Silva RB, Messmer B, Vanoaica LD, et al. Impaired IFN-gamma production and proliferation of NK cells in multiple sclerosis. Int Immunol (2011) 23(2):139–48. doi:10.1093/intimm/dxq463
51. Parekh VV, Wu L, Olivares-Villagomez D, Wilson KT, Van Kaer L. Activated invariant NKT cells control central nervous system autoimmunity in a mechanism that involves myeloid-derived suppressor cells. J Immunol (2013) 190(5):1948–60. doi:10.4049/jimmunol.1201718
52. Furlan R, Bergami A, Cantarella D, Brambilla E, Taniguchi M, Dellabona P, et al. Activation of invariant NKT cells by alphaGalCer administration protects mice from MOG35-55-induced EAE: critical roles for administration route and IFN-gamma. Eur J Immunol (2003) 33(7):1830–8. doi:10.1002/eji.200323885
53. Mars LT, Araujo L, Kerschen P, Diem S, Bourgeois E, Van LP, et al. Invariant NKT cells inhibit development of the Th17 lineage. Proc Natl Acad Sci USA (2009) 106(15):6238–43. doi:10.1073/pnas.0809317106
54. Xiao BG, Wu XC, Yang JS, Xu LY, Liu X, Huang YM, et al. Therapeutic potential of IFN-gamma-modified dendritic cells in acute and chronic experimental allergic encephalomyelitis. Int Immunol (2004) 16(1):13–22. doi:10.1093/intimm/dxh003
55. Murugaiyan G, Beynon V, Pires Da Cunha A, Joller N, Weiner HL. IFN-gamma limits Th9-mediated autoimmune inflammation through dendritic cell modulation of IL-27. J Immunol (2012) 189(11):5277–83. doi:10.4049/jimmunol.1200808
56. Balashov KE, Khoury SJ, Hafler DA, Weiner HL. Inhibition of T cell responses by activated human CD8+ T cells is mediated by interferon-gamma and is defective in chronic progressive multiple sclerosis. J Clin Invest (1995) 95(6):2711–9. doi:10.1172/JCI117973
57. Chu CQ, Wittmer S, Dalton DK. Failure to suppress the expansion of the activated CD4 T cell population in interferon gamma-deficient mice leads to exacerbation of experimental autoimmune encephalomyelitis. J Exp Med (2000) 192(1):123–8. doi:10.1084/jem.192.1.123
58. Park H, Li Z, Yang XO, Chang SH, Nurieva R, Wang YH, et al. A distinct lineage of CD4 T cells regulates tissue inflammation by producing interleukin 17. Nat Immunol (2005) 6(11):1133–41. doi:10.1038/ni1261
59. Berghmans N, Nuyts A, Uyttenhove C, Van Snick J, Opdenakker G, Heremans H. Interferon-gamma orchestrates the number and function of Th17 cells in experimental autoimmune encephalomyelitis. J Interferon Cytokine Res (2011) 31(7):575–87. doi:10.1089/jir.2010.0137
60. Yeh WI, McWilliams IL, Harrington LE. Autoreactive Tbet-positive CD4 T cells develop independent of classic Th1 cytokine signaling during experimental autoimmune encephalomyelitis. J Immunol (2011) 187(10):4998–5006. doi:10.4049/jimmunol.1100031
61. Mangalam AK, Luo N, Luckey D, Papke L, Hubbard A, Wussow A, et al. Absence of IFN-gamma increases brain pathology in experimental autoimmune encephalomyelitis-susceptible DRB1*0301.DQ8 HLA transgenic mice through secretion of proinflammatory cytokine IL-17 and induction of pathogenic monocytes/microglia into the central nervous system. J Immunol (2014) 193(10):4859–70. doi:10.4049/jimmunol.1302008
62. Yeh WI, McWilliams IL, Harrington LE. IFNgamma inhibits Th17 differentiation and function via Tbet-dependent and Tbet-independent mechanisms. J Neuroimmunol (2014) 267(1–2):20–7. doi:10.1016/j.jneuroim.2013.12.001
63. Wildbaum G, Zohar Y, Karin N. Antigen-specific CD25- Foxp3- IFN-gamma(high) CD4+ T cells restrain the development of experimental allergic encephalomyelitis by suppressing Th17. Am J Pathol (2010) 176(6):2764–75. doi:10.2353/ajpath.2010.090855
64. Ponomarev, Novikova M, Yassai M, Szczepanik M, Gorski J, Dittel BN. Gamma delta T cell regulation of IFN-gamma production by central nervous system-infiltrating encephalitogenic T cells: correlation with recovery from experimental autoimmune encephalomyelitis. J Immunol (2004) 173(3):1587–95. doi:10.4049/jimmunol.173.3.1587
65. Wang Z, Hong J, Sun W, Xu G, Li N, Chen X, et al. Role of IFN-γ in induction of Foxp3 and conversion of CD4+CD25-T cells to CD4+ Tregs. J Clin Invest (2006) 116:2434–41. doi:10.1172/JCI25826
66. Ortega SB, Kashi VP, Tyler AF, Cunnusamy K, Mendoza JP, Karandikar NJ. The disease-ameliorating function of autoregulatory CD8 T cells is mediated by targeting of encephalitogenic CD4 T cells in experimental autoimmune encephalomyelitis. J Immunol (2013) 191(1):117–26. doi:10.4049/jimmunol.1300452
67. Chen ML, Yan BS, Kozoriz D, Weiner HL. Novel CD8+ Treg suppress EAE by TGF-beta- and IFN-gamma-dependent mechanisms. Eur J Immunol (2009) 39(12):3423–35. doi:10.1002/eji.200939441
68. Tang X, Maricic I, Kumar V. Anti-TCR antibody treatment activates a novel population of nonintestinal CD8 alpha alpha+ TCR alpha beta+ regulatory T cells and prevents experimental autoimmune encephalomyelitis. J Immunol (2007) 178(10):6043–50. doi:10.4049/jimmunol.178.10.6043
69. Tang X, Maricic I, Purohit N, Bakamjian B, Reed-Loisel LM, Beeston T, et al. Regulation of immunity by a novel population of Qa-1-restricted CD8alphaalpha+TCRalphabeta+ T cells. J Immunol (2006) 177(11):7645–55. doi:10.4049/jimmunol.177.11.7645
70. Beeston T, Smith TR, Maricic I, Tang X, Kumar V. Involvement of IFN-gamma and perforin, but not Fas/FasL interactions in regulatory T cell-mediated suppression of experimental autoimmune encephalomyelitis. J Neuroimmunol (2010) 229(1–2):91–7. doi:10.1016/j.jneuroim.2010.07.007
71. Bahri R, Bollinger A, Bollinger T, Orinska Z, Bulfone-Paus S. Ectonucleotidase CD38 demarcates regulatory, memory-like CD8+ T cells with IFN-gamma-mediated suppressor activities. PLoS One (2012) 7(9):e45234. doi:10.1371/journal.pone.0045234
72. Tyler AF, Mendoza JP, Firan M, Karandikar NJ. CD8 T cells are required for glatiramer acetate therapy in autoimmune demyelinating disease. PLoS One (2013) 8(6):e66772. doi:10.1371/journal.pone.0066772
73. Karandikar NJ, Crawford MP, Yan X, Ratts RB, Brenchley JM, Ambrozak DR, et al. Glatiramer acetate (Copaxone) therapy induces CD8(+) T cell responses in patients with multiple sclerosis. J Clin Invest (2002) 109(5):641–9. doi:10.1172/JCI14380
74. Lee E, Chanamara S, Pleasure D, Soulika AM. IFN-gamma signaling in the central nervous system controls the course of experimental autoimmune encephalomyelitis independently of the localization and composition of inflammatory foci. J Neuroinflammation (2012) 9:7. doi:10.1186/1742-2094-9-7
75. Willenborg DO, Staykova M, Fordham S, O’Brien N, Linares D. The contribution of nitric oxide and interferon gamma to the regulation of the neuro-inflammation in experimental autoimmune encephalomyelitis. J Neuroimmunol (2007) 191(1–2):16–25. doi:10.1016/j.jneuroim.2007.09.007
76. Fenyk-Melody JE, Garrison AE, Brunnert SR, Weidner JR, Shen F, Shelton BA, et al. Experimental autoimmune encephalomyelitis is exacerbated in mice lacking the NOS2 gene. J Immunol (1998) 160(6):2940–6.
77. Sahrbacher UC, Lechner F, Eugster HP, Frei K, Lassmann H, Fontana A. Mice with an inactivation of the inducible nitric oxide synthase gene are susceptible to experimental autoimmune encephalomyelitis. Eur J Immunol (1998) 28(4):1332–8. doi:10.1002/(SICI)1521-4141(199804)28:04<1332::AID-IMMU1332>3.3.CO;2-7
78. Maghazachi AA. Role of natural killer cells in multiple sclerosis. ISRN Immunol (2012) 2012:14. doi:10.5402/2012/795075
79. Segal BM. The role of natural killer cells in curbing neuroinflammation. J Neuroimmunol (2007) 191(1–2):2–7. doi:10.1016/j.jneuroim.2007.09.006
80. Kaur G, Trowsdale J, Fugger L. Natural killer cells and their receptors in multiple sclerosis. Brain (2013) 136(Pt 9):2657–76. doi:10.1093/brain/aws159
81. Takahashi K, Aranami T, Endoh M, Miyake S, Yamamura T. The regulatory role of natural killer cells in multiple sclerosis. Brain (2004) 127(Pt 9):1917–27. doi:10.1093/brain/awh219
82. Zhang B, Yamamura T, Kondo T, Fujiwara M, Tabira T. Regulation of experimental autoimmune encephalomyelitis by natural killer (NK) cells. J Exp Med (1997) 186(10):1677–87. doi:10.1084/jem.186.10.1677
83. Matsumoto Y, Kohyama K, Aikawa Y, Shin T, Kawazoe Y, Suzuki Y, et al. Role of natural killer cells and TCR gamma delta T cells in acute autoimmune encephalomyelitis. Eur J Immunol (1998) 28(5):1681–8. doi:10.1002/(SICI)1521-4141(199805)28:05<1681::AID-IMMU1681>3.0.CO;2-T
84. Winkler-Pickett R, Young HA, Cherry JM, Diehl J, Wine J, Back T, et al. In vivo regulation of experimental autoimmune encephalomyelitis by NK cells: alteration of primary adaptive responses. J Immunol (2008) 180(7):4495–506. doi:10.4049/jimmunol.180.7.4495
85. Morandi B, Bramanti P, Bonaccorsi I, Montalto E, Oliveri D, Pezzino G, et al. Role of natural killer cells in the pathogenesis and progression of multiple sclerosis. Pharmacol Res (2008) 57(1):1–5. doi:10.1016/j.phrs.2007.11.003
86. Xu W, Fazekas G, Hara H, Tabira T. Mechanism of natural killer (NK) cell regulatory role in experimental autoimmune encephalomyelitis. J Neuroimmunol (2005) 163(1–2):24–30. doi:10.1016/j.jneuroim.2005.02.011
87. Shi FD, Takeda K, Akira S, Sarvetnick N, Ljunggren HG. IL-18 directs autoreactive T cells and promotes autodestruction in the central nervous system via induction of IFN-gamma by NK cells. J Immunol (2000) 165(6):3099–104. doi:10.4049/jimmunol.165.6.3099
88. Brady J, Hayakawa Y, Smyth MJ, Nutt SL. IL-21 induces the functional maturation of murine NK cells. J Immunol (2004) 172(4):2048–58. doi:10.4049/jimmunol.172.4.2048
89. Strengell M, Matikainen S, Siren J, Lehtonen A, Foster D, Julkunen I, et al. IL-21 in synergy with IL-15 or IL-18 enhances IFN-gamma production in human NK and T cells. J Immunol (2003) 170(11):5464–9. doi:10.4049/jimmunol.170.11.5464
90. Vollmer TL, Liu R, Price M, Rhodes S, La Cava A, Shi FD. Differential effects of IL-21 during initiation and progression of autoimmunity against neuroantigen. J Immunol (2005) 174(5):2696–701. doi:10.4049/jimmunol.174.5.2696
91. Xie ZX, Zhang HL, Wu XJ, Zhu J, Ma DH, Jin T. Role of the immunogenic and tolerogenic subsets of dendritic cells in multiple sclerosis. Mediators Inflamm (2015) 2015:513295. doi:10.1155/2015/513295
92. Ganguly D, Haak S, Sisirak V, Reizis B. The role of dendritic cells in autoimmunity. Nat Rev Immunol (2013) 13(8):566–77. doi:10.1038/nri3477
93. Zhu J, Yamane H, Paul WE. Differentiation of effector CD4 T cell populations (*). Annu Rev Immunol (2010) 28:445–89. doi:10.1146/annurev-immunol-030409-101212
94. Barnes MJ, Powrie F. Hybrid Treg cells: steel frames and plastic exteriors. Nat Immunol (2009) 10:563–4. doi:10.1038/ni0609-563
95. Kleinewietfeld M, Hafler DA. The plasticity of human Treg and Th17 cells and its role in autoimmunity. Semin Immunol (2013) 25:305–12. doi:10.1016/j.smim.2013.10.009
96. Legroux L, Arbour N. Multiple sclerosis and T lymphocytes: an entangled story. J Neuroimmune Pharmacol (2015). doi:10.1007/s11481-015-9614-0
97. Kuchroo VK, Anderson AC, Waldner H, Munder M, Bettelli E, Nicholson LB. T cell response in experimental autoimmune encephalomyelitis (EAE): role of self and cross-reactive antigens in shaping, tuning, and regulating the autopathogenic T cell repertoire. Annu Rev Immunol (2002) 20:101–23. doi:10.1146/annurev.immunol.20.081701.141316
98. Kurschus FC, Croxford AL, Heinen AP, Wortge S, Ielo D, Waisman A. Genetic proof for the transient nature of the Th17 phenotype. Eur J Immunol (2010) 40(12):3336–46. doi:10.1002/eji.201040755
99. Hirota K, Duarte JH, Veldhoen M, Hornsby E, Li Y, Cua DJ, et al. Fate mapping of IL-17-producing T cells in inflammatory responses. Nat Immunol (2011) 12(3):255–63. doi:10.1038/ni.1993
100. Paul S, Shilpi, Lal G. Role of gamma-delta (gammadelta) T cells in autoimmunity. J Leukoc Biol (2015) 97(2):259–71. doi:10.1189/jlb.3RU0914-443R
101. Wohler JE, Smith SS, Zinn KR, Bullard DC, Barnum SR. Gammadelta T cells in EAE: early trafficking events and cytokine requirements. Eur J Immunol (2009) 39(6):1516–26. doi:10.1002/eji.200839176
102. Rajan AJ, Klein JD, Brosnan CF. The effect of gammadelta T cell depletion on cytokine gene expression in experimental allergic encephalomyelitis. J Immunol (1998) 160(12):5955–62.
103. Duhen T, Duhen R, Lanzavecchia A, Sallusto F, Campbell DJ. Functionally distinct subsets of human FOXP3+ Treg cells that phenotypically mirror effector Th cells. Blood (2012) 119(19):4430–40. doi:10.1182/blood-2011-11-392324
104. Daniel V, Sadeghi M, Wang H, Opelz G. CD4+CD25+Foxp3+IFN-gamma+ human induced T regulatory cells are induced by interferon-gamma and suppress alloresponses nonspecifically. Hum Immunol (2011) 72(9):699–707. doi:10.1016/j.humimm.2011.05.020
105. Venigalla RK, Guttikonda PJ, Eckstein V, Ho AD, Sertel S, Lorenz HM, et al. Identification of a human Th1-like IFNgamma-secreting Treg subtype deriving from effector T cells. J Autoimmun (2012) 39(4):377–87. doi:10.1016/j.jaut.2012.06.004
106. Koch Ma, Tucker-heard G, Perdue NR, Killebrew JR, Urdahl B, Campbell DJ. T-bet controls regulatory T cell homeostasis and function during type-1 inflammation. Nat Immunol (2009) 10:595–602. doi:10.1038/ni.1731.T-bet
107. Koch Ma, Thomas KN, Perdue NR, Smigiel KS, Srivastava S, Campbell DJ. T-bet+ Treg cells undergo abortive Th1 cell differentiation due to impaired expression of IL-12 receptor β2. Immunity (2012) 37:501–10. doi:10.1016/j.immuni.2012.05.031.T-bet
108. Zheng J, Liu Y, Qin G, Lam KT, Guan J, Xiang Z, et al. Generation of human Th1-like regulatory CD4+ T cells by an intrinsic IFN-gamma- and T-bet-dependent pathway. Eur J Immunol (2011) 41(1):128–39. doi:10.1002/eji.201040724
109. Esposito M, Ruffini F, Bergami A, Garzetti L, Borsellino G, Battistini L, et al. IL-17- and IFN-γ-secreting Foxp3+ T cells infiltrate the target tissue in experimental autoimmunity. J Immunol (2010) 185:7467–73. doi:10.4049/jimmunol.1001519
110. Dominguez-Villar M, Baecher-Allan CM, Hafler Da . Identification of T helper type 1-like, Foxp3+ regulatory T cells in human autoimmune disease. Nature medicine (2011) 17:673–5. doi:10.1038/nm.2389
111. Ford ML, Evavold BD. Specificity, magnitude, and kinetics of MOG-specific CD8+ T cell responses during experimental autoimmune encephalomyelitis. Eur J Immunol (2005) 35(1):76–85. doi:10.1002/eji.200425660
112. Huseby ES, Liggitt D, Brabb T, Schnabel B, Ohlen C, Goverman J. A pathogenic role for myelin-specific CD8(+) T cells in a model for multiple sclerosis. J Exp Med (2001) 194(5):669–76. doi:10.1084/jem.194.5.669
113. Bettini M, Rosenthal K, Evavold BD. Pathogenic MOG-reactive CD8+ T cells require MOG-reactive CD4+ T cells for sustained CNS inflammation during chronic EAE. J Neuroimmunol (2009) 213(1–2):60–8. doi:10.1016/j.jneuroim.2009.05.017
114. Sun D, Whitaker JN, Huang Z, Liu D, Coleclough C, Wekerle H, et al. Myelin antigen-specific CD8+ T cells are encephalitogenic and produce severe disease in C57BL/6 mice. J Immunol (2001) 166(12):7579–87. doi:10.4049/jimmunol.166.12.7579
115. York NR, Mendoza JP, Ortega SB, Benagh A, Tyler AF, Firan M, et al. Immune regulatory CNS-reactive CD8+T cells in experimental autoimmune encephalomyelitis. J Autoimmun (2010) 35(1):33–44. doi:10.1016/j.jaut.2010.01.003
116. Smith TR, Kumar V. Revival of CD8+ Treg-mediated suppression. Trends Immunol (2008) 29(7):337–42. doi:10.1016/j.it.2008.04.002
Keywords: interferon-gamma, experimental autoimmune encephalomyelitis, multiple sclerosis, innate immunity, adaptive immunity, neuroinflammation
Citation: Arellano G, Ottum PA, Reyes LI, Burgos PI and Naves R (2015) Stage-specific role of interferon-gamma in experimental autoimmune encephalomyelitis and multiple sclerosis. Front. Immunol. 6:492. doi: 10.3389/fimmu.2015.00492
Received: 29 July 2015; Accepted: 09 September 2015;
Published: 29 September 2015
Edited by:
Sergio Quezada, University College London Cancer Institute, UKReviewed by:
Ari Waisman, University Medical Center of the Johannes-Gutenberg University of Mainz, GermanyCopyright: © 2015 Arellano, Ottum, Reyes, Burgos and Naves. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Rodrigo Naves, Programa de Inmunología, Facultad de Medicina, Instituto de Ciencias Biomédicas, Universidad de Chile, Independencia 1027, Santiago 8380453, Chile,cm9kcmlnb25hdmVzQG1lZC51Y2hpbGUuY2w=
†Gabriel Arellano and Payton A. Ottum have contributed equally to this work.
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.