 Tim D. Hewitson
Tim D. Hewitson Stephen G. Holt1,2
Stephen G. Holt1,2 Edward R. Smith
Edward R. Smith
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 17 September 2015
Sec. Cytokines and Soluble Mediators in Immunity
Volume 6 - 2015 | https://doi.org/10.3389/fimmu.2015.00465
This article is part of the Research Topic The CXCR4 ligand/receptor family and the DPP4 protease in high-risk cardiovascular patients View all 14 articles
The close association between cardiovascular pathology and renal dysfunction is well documented and significant. Patients with conventional risk factors for cardiovascular disease like diabetes and hypertension also suffer renal dysfunction. This is unsurprising if the kidney is simply regarded as a “modified blood vessel” and thus, traditional risk factors will affect both systems. Consistent with this, it is relatively easy to comprehend how patients with either sudden or gradual cardiac and or vascular compromise have changes in both renal hemodynamic and regulatory systems. However, patients with pure or primary renal dysfunction also have metabolic changes (e.g., oxidant stress, inflammation, nitric oxide, or endocrine changes) that affect the cardiovascular system. Thus, cardiovascular and renal systems are intimately, bidirectionally and inextricably linked. Whilst we understand several of these links, some of the mechanisms for these connections remain incompletely explained. Animal models of cardiovascular and renal disease allow us to explore such mechanisms, and more importantly, potential therapeutic strategies. In this article, we review various experimental models used, and examine critically how representative they are of the human condition.
Patients with conventional risk factors for cardiovascular disease (CVD), like diabetes and hypertension, also suffer renal dysfunction. This is unsurprising if the kidney is simply regarded as a “modified blood vessel” with traditional risk factors likely to affect both systems and coexist (1). However, although a problem in one organ system affects the other, the prime mover in this loop may be occult. Animal models have helped us tease apart these associations but have consistently shown that impairment of one organ has detrimental effects on the other at functional, biochemical, and molecular level [reviewed in Ref. (2–5)]. First described by El-Atat et al. (6), these clinical interactions are now collectively known as the cardiorenal syndromes (CRS), which for many years were considered a single diagnostic group, and thus, may have limited or confounded many early mechanistic studies.
Given its importance, and the wide spectrum of primary disease and clinical presentation, a number of classification systems have been proposed. Ronco et al. have classified CRS on time frame (acute, chronic or secondary) and which organ is involved first (heart or kidney) recognizing five different types of the CRS or renocardiac syndromes (RCS) (7, 8). Other approaches are based on pathophysiological links (9–11) (Figure 1), arguing that effects are bidirectional (11), and that temporal differences in organ involvement are artificial (9).
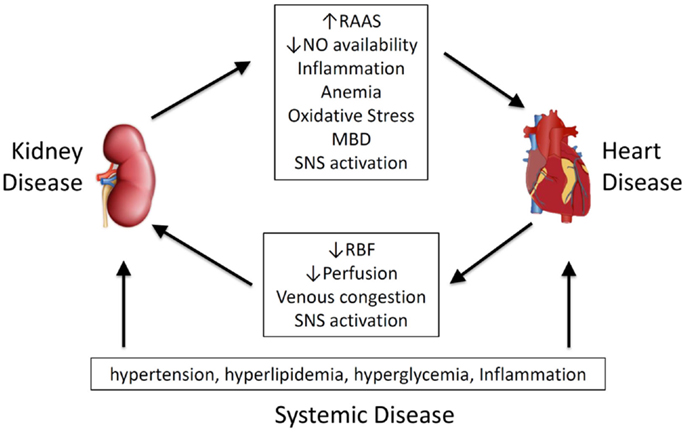
Figure 1. Pathophysiological mediators of cardiorenal syndrome and renocardiac syndrome. MBD, mineral bone disorder; NO, nitric oxide; RAAS, renal–angiotensin–aldosterone system; RBF, renal blood flow; SNS, sympathetic nervous system.
As the natural history is sometimes slow and consequently difficult to explore in clinical trials, adequate experimental modeling of the clinical scenario is crucial to examining mechanisms and potential therapeutic strategies. In this review, we use the Ronco classification (7) to discuss animal approximations of the CRS.
Neurohumoral mechanisms have evolved to maintain a relatively constant blood volume and organ perfusion under continuously changing conditions (11). In the context of a failing pump (heart), vasopressor systems, like the sympathetic nervous system (SNS) and renin–angiotensin–aldosterone system (RAAS), are activated to maintain the hemodynamic balance (12). Vasoconstriction of the efferent artery helps maintain glomerular filtration rate (GFR) in low-output states, but the increased vascular resistance may reduce overall renal perfusion and cause intra-renal hemodynamic changes. This can occur acutely in the setting of abrupt hypotension [e.g., due to myocardial infarction (MI)] or chronically, which over time causes tubular hypoxia and apoptosis leading to a loss of nephron mass and function.
In animal models, MI, produced by ligation of the left anterior descending coronary artery has detrimental effects on the renal function over time. GFR significantly decreases after MI within 4 weeks, and deteriorates further at 16 weeks. Histological analysis reveals greater renal interstitial fibrosis and downstream transforming growth factor-β1 (TGFβ1) signaling (smad2 phosphorylation) at all time-points (13).
In rodents, beta-adrenergic stimulation through multiple isoproterenol injections results in left ventricular (LV) fibrosis (14). It remains unclear if this interference with the SNS has renal consequences, consistent with CRS.
Hemodynamic changes are a well-recognized driving force in the pathophysiology of CRS. A reduction in cardiac output (forward failure) reduces renal blood flow leading to changes in distal tubular chloride and other solute content, resulting in increased renin release from the macula densa and activation of the RAAS (15). Surgical restriction of the carotid artery leads to aortic regurgitation (16) with both cardiac hypertrophy and albuminuria from 5 months (17).
Higher central venous pressures due to congestion may increase both interstitial pressure and efferent pressure (through a decreased afferent-efferent gradient) (5). In these circumstances, elevated venous pressures (backward failure) may reduce renal blood flow and consequently urine output more than a reduction in arterial pressure, ultimately leading to hypoxia and activation of the RAAS. Elevated renal venous pressure alone reduces renal arterial flow and GFR in the pig, with increases in plasma renin activity (18). While maintaining cardiac homeostasis initially, long-term activation of the RAAS eventually leads to progression and myocardial remodeling through fibrosis (3).
As chronic kidney disease (CKD) progresses, the SNS is stimulated as a result of renal ischemia, activation of the RAAS and suppression of nitric oxide (NO) synthesis. This results in hypertension, left ventricular hypertrophy (LVH), and progressive LV dilatation (12). Early cardiac hypertrophy, and subsequent fibrosis due to fibroblast activation (19, 20), are final common contributors to organ dysfunction irrespective of the nature of the initial injury.
Sub-total nephrectomy, often termed 5/6 nephrectomy, is probably the most established method of modeling progressive renal failure seen with loss of renal mass. Although commonly performed in the rat, similar rabbit and mouse models exist but appear less reliable. Rather than mimicking a renal disease per se, sub-total nephrectomy parallels the consequences of reducing functional nephron number. The predominant pathological abnormalities within the remaining kidney are glomerulosclerosis and tubulointerstitial fibrosis (21). It is important to appreciate that two quite different models are encompassed by the expression “5/6 nephrectomy”, namely an ablation model widely used for the study of CRS and a less common ligation model. In the former, one kidney is removed along with ~50% of the contralateral kidney by polar excision 1–2 weeks later, with the rate of renal decline very closely related to the amount of tissue excised (21). In contrast to human chronic kidney disease (CKD), although LVH is a consistent feature (22) and resembles that seen in early human CKD (23), severe hypertension is not usually a feature of this model. Nevertheless early LV diastolic dysfunction is seen with commensurate increases in heart weight (corrected for body weight), and myocyte cross sectional area (24, 25). Other documented pathologies include increased myocardial artery wall thickness, capillary density (26) and interstitial fibrosis (24). Similar changes are seen in mice, but tend to be more strain dependent (4).
While traditional cardiovascular risk factors are highly prevalent in patients with CKD, results of clinical trials focusing on controling such factors have been largely disappointing, and thus, non-traditional risk factors associated with CKD are being increasingly explored.
Anemia. Anemia is a feature of most CKD and related to erythropoietin deficiency (27), yet it is unclear whether it is a mediator of CRS or simply a marker of disease progression (11). There is some experimental evidence that correction of anemia preserves both renal and cardiovascular function. Cardiomyopathy occurs after IV injection of anthracycline antibiotics, such as adriamycin (28, 29), with renal fibrosis and pronounced loss of renal function accompanying LV enlargement (29). In the doxorubicin model administration of darbepoetin can increase Hb to control levels and significantly attenuates the renal dysfunction, renal interstitial fibrosis and LV weight (29). Likewise, erythropoietin improves cardiac function post-MI, an effect seemingly related to the promotion of neovascularization (30). Effects in a combined cardiac-renal model have not been examined.
Inflammation. Progressive renal impairment is characterized by a chronic inflammatory state with elevated tissue and circulating concentrations of cytokines including interleukin-6 (IL-6) and tumor necrosis factor-α (TNFα). The origin of inflammation in renal disease is multifactorial, and involves reduced clearance of pro-inflammatory cytokines, reactive oxygen species and effects of comorbid conditions, such as diabetes (12). Inflammation is also a risk factor for MI and death in uremic patients (31) and a marker of severity and progression of heart failure (32). Experimentally, increased inflammatory cytokine mRNA expression is seen in the heart after renal ischemia-reperfusion injury (33). Dietary phosphate overload directly induces systemic increases in serum and tissue TNFα in a model of adenine-induced chronic renal failure (34). Conversely, renal macrophage infiltration and inflammatory cytokine expression is significantly increased from as early as 3 days post-MI (35). Activation of inflammasome pathways have been seen in both acute and CKD, with infiltrating macrophages specifically implicated (36). It is interesting to note that statins may reduce effects of inflammation in both the kidney and heart and may be protective (37).
Nitric oxide. The nitric oxide synthase (NOS) inhibitor asymmetrical dimethyl arginine (ADMA) accumulates in renal failure (38) and contributes to a reduction in NO systemically and intra-renally. NOS inhibition with N-nitro-l-arginine administration (l-NAME) depletes NO and both exacerbate renal dysfunction, and induce permanent cardiac dysfunction in rats with sub-total renal nephrectomy (39). Further evidence for the role of NO comes from those studies showing that chronic administration of l-NAME on its own also induces heart and kidney damage similar to that found in CRS (40). The model is characterized by a progressive increase in BP over 10 weeks with severe proteinuria, glomerulosclerosis, and tubulointerstitial fibrosis, and elevation in serum creatinine (40). Further, SNS activation is a recognized feature of the l-NAME model. Proteinuria and cardiac hypertrophy induced by chronic l-NAME treatment is abrogated by bilateral renal sympathetic denervation, but not hydralazine, even when blood pressure and NO depletion are equivalent (40).
Oxidative stress. Reactive oxygen species, produced as a result of redox reactions in various cells, have been recognized as key chemical mediators causing cellular damage and organ dysfunction in both CVD and CKD. There is growing evidence that oxidative stress is one of the central mediators of CRS. Increased oxidative stress is seen in patients with cardiac (41) and renal failure (42) and the reduced availability of NO impairs vasodilatation, and reduces renal perfusion (43).
It is now widely hypothesized that interactions between the RAAS system, the SNS and inflammation, may all potentiate CRS through excessive oxidative stress pathways (44). Consistent with this, experimental studies have identified several dysregulated pathways in heart failure and in CKD that lead to increased oxidative stress. Dahl salt-sensitive rats show increased LV NADPH oxidase activity, which is normalized by Angiotensin II blockade (45). In rats with sub-total nephrectomy, mitochondrial respiration in the heart is dysregulated, with cardiomyocytes isolated from uremic animals more susceptible to oxidant induced cell death than their normal counterparts (46).
Protein-bound toxins. Uremic toxins, such as indoxyl sulfate (IS), appear to accelerate the progression of CKD via profibrotic and oxidative pathways. Oral administration of IS in sub-totally nephrectomized rats induced renal tubular injury, renal interstitial fibrosis and glomerular sclerosis, leading to functional impairment (increased serum creatinine and blood urea nitrogen). These changes are associated with increased renal expression of profibrotic genes, such as TGFβ1, tissue inhibitor of metalloproteinases-1, and pro-collagen α1(I). Glomerulosclerosis and renal impairment has also been demonstrated in sub-totally nephrectomized rats receiving indole (47). The presence of IS, not indole, in the urine of indole-loaded animals confirms the protein metabolite hypothesis of IS production (47). IS has effects on cardiac myocytes and can cause cardiac hypertrophy and fibrosis (48, 49).
Neurohormonal disturbance. SNS over-activation is observed early in CKD, stemming from renal ischemia, raised angiotensin II levels, and suppression of NO amongst other causes. It is deleterious causing hypertension, LVH and eventually ventricular dysfunction and dilatation (12). Renal denervation in small animals can be achieved both surgically and chemically. These techniques have been widely used to study SNS activation and the relationship to hypertension in various experimental models of renal disease including sub-total nephrectomy (50) and deoxycorticosterone acetate (DOCA)-salt hypertension (51). Sympathectomy directly prevents onset and progression of albuminuria after chronic cardiac volume overload caused by aortic regurgitation (17).
Mineral bone disorder. The burden of excess CVD in patients with CKD is partly attributed to systemic disturbances in mineral metabolism and changes in bone histomorphometry. This is frequently accompanied by soft tissue calcification (52) especially within the arterial wall, where it is associated with significant mortality and morbidity (53). Patients with CKD also have a preponderance of medial arterial calcification (MAC), as well as greater calcification of intimal lesions. Pathophysiologically, aortic MAC is linked to alterations in vessel compliance, which exposes the heart to changes in vascular compliance and resistance changes in systolic pressure leading to LVH and myocardial fibrosis (54). Impaired aortic recoil results in lower diastolic pressures and a widened pulse pressure and reduced perfusion of coronary arteries, leading to sub-endocardial ischemia (55). Loss of vessel compliance may also impact on renal autoregulation (56). Although not a consistent finding in man (57), rodent models of renal failure demonstrate a convincing relationship between vascular stiffness and calcification scores (58, 59).
Three rodent animal models are commonly employed to study mineral bone disorder (MBD) and its cardiovascular sequelae in the context of CKD: 1) phosphate/vitamin D loading post 5/6 nephrectomy (60); 2) adenine-induced renal failure (61); and 3) the mouse electrocautery model of CKD (62) in strains with a genetic predisposition to vascular calcification (e.g., LDLR−/− or apoE−/−). While these models consistently generate biochemical changes, calcification phenotypes are inconsistent due to differences in diet, study duration and the genetic background of the animals. Indeed, some inbred rodent strains (e.g., C57BL/6 mice and Sprague-Dawley rats) are surprisingly calcification resistant. Without concurrent calcitriol administration and or high phosphate feeding (>1% diet), vascular calcification after 5/6 nephrectomy is only apparent after 24 weeks (63). The mouse electrocautery model shows variable CKD, and mild hyperphosphataemia, even with high phosphate feeding. Interestingly, induction of CKD in transgenic atherosclerotic-prone animals only imparts a modest increase in aortic calcium relative to non-uremic littermates. Induction of milder renal impairment (equivalent to CKD Stage 2) by less intensive cautery of one kidney followed by contralateral nephrectomy (64) provides compelling evidence of the development of MBD in early CKD. The generation of uremic mice with adenine-enriched diets has gained considerable interest; here, animals develop advanced CKD, hyperphosphataemia (even without dietary phosphate loading), severe hyperparathyroidism despite normocalcaemia, and MAC within 4 weeks when on a 0.75% adenine diet (65, 66).
Crucially however, the MBD animal models discussed, thus far are generated by acute injury and rely on the consequent development of CKD. To date, few models of spontaneous CKD with well-characterized MBD and vascular calcification have been described. One such model, heterozygous Han:SPRD (Cy/−) rats (a model of autosomal dominant polycystic kidney disease) develop slowly progressive CKD, hyperphosphataemia, hyperparathyroidism and bone abnormalities but the vascular calcification generated is not progressive and only present in a subset of animals even after 38 weeks (67). Other models of spontaneous CKD (e.g., the Col4a3 null mouse model of human autosomal-recessive Alport syndrome), despite showing convincing biochemical evidence of CKD-MBD, do not exhibit a consistent vascular calcification phenotype (68).
A plethora of in vitro and in vivo studies have evoked the now widely accepted view that vascular calcification is a highly regulated and principally cell-mediated phenomenon that recapitulates many features of physiological ossification (69). There is strong evidence of osteochondrocytic differentiation of vascular cells in the calcified intimal plaques of high-fat fed LDLR−/− and ApoE−/− mice (70, 71), as well as in the arterial media of adenine and mineral stressed 5/6 nephrectomy models (72, 73). However, while de-differentiation of VSMC to a synthetic phenotype is found in some knockout models deficient in calcification inhibitors (e.g., matrix Gla protein (MGP)−/− mice) (74), it is not a consistent finding (e.g., fetuin-A-deficient mice) (75). Moreover, mutations in the regulators of mineralization can manifest themselves quite differently in rodents and man. For instance, MGP−/− mice have massive MAC. In humans, however, inactivating mutations in MGP (Keutel syndrome) exhibit infrequent arterial calcification (76). Conversely, inactivating mutations in Ennp1 encoding the pyrophosphate synthesizing ectoenzyme nucleotide pryophosphatase/phosphodiesterase results in the devastating syndrome, Generalized Arterial Calcification of Infancy (77). Ablation of homologous gene, Npps, in mice however, results in ossification of the spinal ligaments and peri-articular calcification but with relatively minor arterial involvement and only a modestly shortened lifespan (78).
Finally it is worth noting that despite considerable enthusiasm for the role of VSMC phenotype switching as a major mechanism for vascular calcification, the evidence for this phenomenon in human CKD is currently limited to a subset of patients on hemodialysis (79, 80). Indeed, even in this setting, evidence of such changes appear conspicuously absent at some vascular sites exclusively affected by medial calcification (81). An explanation for this variable disease penetrance is not currently forthcoming, although it should also be stressed that analyzes to date have been on small-to-medium sized muscular arteries that are generally free from intimal/atherosclerotic disease involvement and which may, therefore, not be representative of changes occurring in some larger elastic vessels (e.g., aorta). This may in part also explain the apparent disparity with findings in some animal models of uremia, where studies have mainly centered on changes in VSMC phenotype in the aorta and where phenotypic switching would appear to occur relatively early in disease progression (64).
In many cases cardiac and renal pathologies are common to a system-wide perturbation. Relevant models include metabolic syndromes, such as hypertension, diabetes, obesity, liver disease, myeloma, lupus, and other autoimmune disease. Self-evidently these models align closely with the traditional risk factors seen in CVD, and reflect both chronic and acute causes.
A number of experimental models of hypertension exist, including amongst others, inbred models of inherited primary hypertension [spontaneously hypertensive rat (SHR), Milan hypertensive rat, dahl salt-sensitive rat, and transgenic models over expressing renin (mRen2)]. While these consistently display cardiac pathologies, only some have parallel changes in renal function (82), with high pre-glomerular resistance relatively protective in selectively bred SHRs (83). Whilst inherited hypertension is a rare but recognized cause of hypertension in humans (82), in animal models correction of hypertension per se is not sufficient to prevent progression of experimental CRS (84).
Several models of diabetes have shown simultaneous cardiac and renal dysfunction including accelerated models, developed in an attempt to more closely mimic the human condition. The transgenic (mRen-2) rat overexpresses the murine renin gene (85) with elevated angiotensin II activity. Streptozotocin (STZ) induced beta-cell destruction in the mRen-2 rat is an established model of diabetes and its complications. The major advantage being that these rodents develop functional and structural pathology closely mimicking that seen in advanced human diabetic nephropathy (85) and cardiomyopathy (86). In a similar manner, SHR made diabetic with STZ replicate the confounding hypertension seen in diabetic nephropathy (87). However, despite a similar rise in blood pressure, unlike the diabetic mRen2 rat, these animals do not usually progress to renal failure.
STZ administration in the atherosclerotic (ApoE−/−) mouse model accelerates both diabetic renal pathology and atherosclerosis, a major risk factor for MI ischemia (88).
Obese Zucker (OZ) rats (readily available commercially, and extensively studied) are characterized by mild glucose intolerance and peripheral insulin resistance similar to that found in humans with Type 2 diabetes. These abnormalities precede the development of albuminuria and glomerular injury and animals show a parallel deterioration in cardiac output and renal function (89). Ultrastructural studies have shown cardiomyopathy in both the OZ rat, and in mice with a similar mutation (db/db mouse) (90). A comprehensive analysis in the db/db mouse has shown that albuminuria/glomerulopathy and cardiac contractile dysfunction appear after 2–4 months of hyperglycemia (91).
Both acute cholestatic and chronic fibrotic liver disease cause renal and cardiac dysfunction. Bile duct ligation causes acute and chronic renal (92) and cardiac dysfunction (93). Administration of carbon tetrachloride (CCl4) is a model of acute (or if repeated, chronic) hepatic failure causing widespread disruption to cardiac and renal function, although CCl4 generates oxidant injury directly in other organs (94). Other relevant experimental models include toxin induced murine models, as well as some murine models of autoimmune hepatitis and primary biliary cirrhosis (95).
The classic murine model of lupus (as a paradigm for other autoimmune disease) includes genetically predisposed crosses (New Zealand Black crosses) and toxin (pristane) induced models (96). Nevertheless, many of the murine models fail to fully replicate the multisystem manifestations of human lupus, and whilst most replicate lupus nephritis, other organs like skin and arthritis are inconsistently affected, although the NZB murine model does seem to develop pericardial, epicardial and myocardial inflammation (97). Because the murine model is not altogether representative of the human condition, other models including canine and porcine models have been developed (98, 99).
A less common experimental scenario looks at the effect of overlaying renal impairment on cardiac disease, and the reverse. For example sub-total nephrectomy accelerates pathological cardiac remodeling post-MI when performed 4 weeks after infarction with worse ejection fraction in those animals with renal impairment (100). Similarly when insults are reversed e.g., performing a sub-total nephrectomy a week before MI, LV damage is worse and associated with worse creatinine clearance (101) and renal blood flow, and more proteinuria and glomerulosclerosis. Sub-total nephrectomy followed by MI once renal injury is firmly established leads to more pronounced damage in both organs (102).
In sub-totally nephrectomized rats, temporary ligation of the descending branch of the left coronary artery after 3 weeks resulted in a larger area of cardiac necrosis (devoid of mitochondrial oxidation) than sham ligated paired controls. Infarcts after coronary artery ligation (CAL) were larger in animals with even modest renal impairment (84). A major disadvantage of these models is that experimental mortality is high. Although the effect is mild, reducing renal mass by uninephrectomy is also often used as a means of accelerating renal disease, but this is not in itself sufficient to produce a cardiac phenotype (103).
No animal model in isolation reproduces the complexity of different CRS (Table 1). Nevertheless, animal models have provided valuable insights into the pathogenesis of CRS in all its forms (5). Like their counterpart clinical trials, animal studies have highlighted the mechanistic importance of non-traditional risk factors. To this end the RAAS system, SNS, indirect and direct effects of the uremic toxins, anemia, inflammation, neurohormonal factors and disturbances in mineral handling and bone turnover are all implicated causatively. However, although the small animal models frequently employed in these studies are readily amenable to further genetic manipulation and intervention, induction of injury is generally acute and unphysiological, and these models often fail to faithfully recapitulate the pathological features of human CRS.
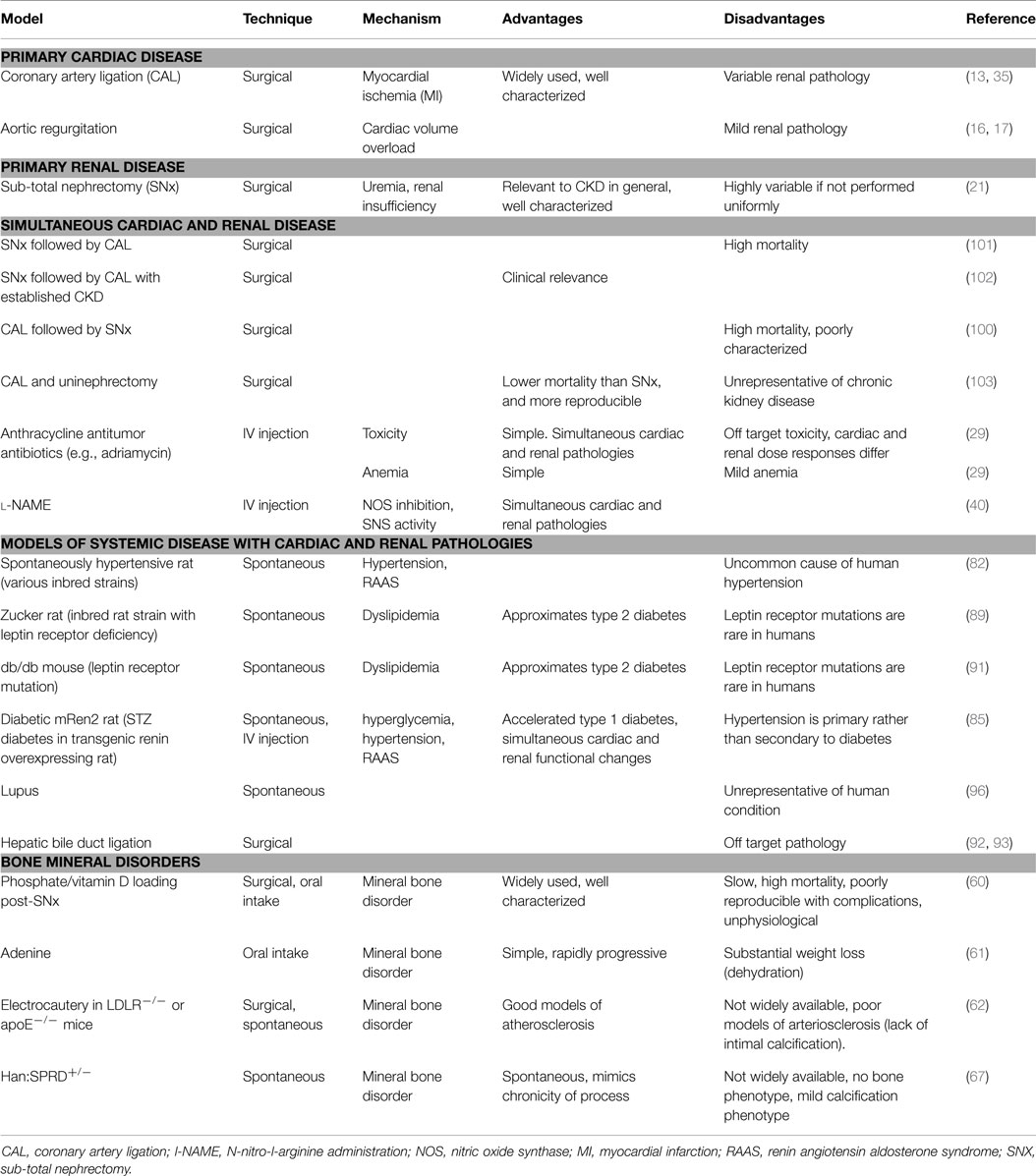
Table 1. Animal models of cardiorenal and renocardiac syndromes, and relative advantages and disadvantages of each.
In this article, we have discussed a range of models that can be used to mimic the mechanisms of human renal and cardiac disease, and examined how representative they are of the human condition. While not perfect, careful and ethical use of animal models offers the opportunity to examine the complex interactions seen in CRS in an accelerated time frame.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The authors are supported by the National Health and Medical Research Council of Australia (APP1078694) and RMH Research Funding Program (grant-in aid).
1. Tomlinson LA, Holt SG. Is the kidney just a modified blood vessel? Unravelling the direction of causality between cardiovascular and renal disease. Atherosclerosis (2011) 216(2):275–6. doi: 10.1016/j.atherosclerosis.2011.02.049
2. Liu S, Lekawanvijit S, Kompa AR, Wang BH, Kelly DJ, Krum H. Cardiorenal syndrome: pathophysiology, preclinical models, management and potential role of uraemic toxins. Clin Exp Pharmacol Physiol (2012) 39(8):692–700. doi:10.1111/j.1440-1681.2011.05632.x
3. Szymanski MK, de Boer RA, Navis GJ, van Gilst WH, Hillege HL. Animal models of cardiorenal syndrome: a review. Heart Fail Rev (2012) 17(3):411–20. doi:10.1007/s10741-011-9279-6
4. Bongartz LG, Braam B, Gaillard CA, Cramer MJ, Goldschmeding R, Verhaar MC, et al. Target organ cross talk in cardiorenal syndrome: animal models. Am J Physiol Renal Physiol (2012) 303(9):F1253–63. doi:10.1152/ajprenal.00392.2012
5. Tsuruya K, Eriguchi M. Cardiorenal syndrome in chronic kidney disease. Curr Opin Nephrol Hypertens (2015) 24(2):154–62. doi:10.1097/MNH.0000000000000099
6. El-Atat FA, Stas SN, McFarlane SI, Sowers JR. The relationship between hyperinsulinemia, hypertension and progressive renal disease. J Am Soc Nephrol (2004) 15(11):2816–27. doi:10.1097/01.ASN.0000133698.80390.37
7. Ronco C, Haapio M, House AA, Anavekar N, Bellomo R. Cardiorenal syndrome. J Am Coll Cardiol (2008) 52(19):1527–39. doi:10.1016/j.jacc.2008.07.051
8. McCullough PA, Haapio M, Mankad S, Zamperetti N, Massie B, Bellomo R, et al. Prevention of cardio-renal syndromes: workgroup statements from the 7th ADQI consensus conference. Nephrol Dial Transplant (2010) 25(6):1777–84. doi:10.1093/ndt/gfq180
9. Hatamizadeh P, Fonarow GC, Budoff MJ, Darabian S, Kovesdy CP, Kalantar-Zadeh K. Cardiorenal syndrome: pathophysiology and potential targets for clinical management. Nat Rev Nephrol (2013) 9(2):99–111. doi:10.1038/nrneph.2012.279
10. Braam B, Joles JA, Danishwar AH, Gaillard CA. Cardiorenal syndrome – current understanding and future perspectives. Nat Rev Nephrol (2014) 10(1):48–55. doi:10.1038/nrneph.2013.250
11. Bock JS, Gottlieb SS. Cardiorenal syndrome: new perspectives. Circulation (2010) 121(23):2592–600. doi:10.1161/CIRCULATIONAHA.109.886473
12. Kumar S, Bogle R, Banerjee D. Why do young people with chronic kidney disease die early? World J Nephrol (2014) 3(4):143–55. doi:10.5527/wjn.v3.i4.143
13. Lekawanvijit S, Kompa AR, Zhang Y, Wang BH, Kelly DJ, Krum H. Myocardial infarction impairs renal function, induces renal interstitial fibrosis, and increases renal KIM-1 expression: implications for cardiorenal syndrome. Am J Physiol Heart Circ Physiol (2012) 302(9):H1884–93. doi:10.1152/ajpheart.00967.2011
14. Samuel CS, Bodaragama H, Chew JY, Widdop RE, Royce SG, Hewitson TD. Serelaxin is a more efficacious antifibrotic than enalapril in an experimental model of heart disease. Hypertension (2014) 64(2):315–22. doi:10.1161/HYPERTENSIONAHA.114.03594
15. Peti-Peterdi J, Harris RC. Macula densa sensing and signaling mechanisms of renin release. J Am Soc Nephrol (2010) 21(7):1093–6. doi:10.1681/ASN.2009070759
16. Noma T, Nishiyama A, Mizushige K, Murakami K, Tsuji T, Kohno M, et al. Possible role of uncoupling protein in regulation of myocardial energy metabolism in aortic regurgitation model rats. FASEB J (2001) 15(7):1206–8.
17. Rafiq K, Noma T, Fujisawa Y, Ishihara Y, Arai Y, Nabi AH, et al. Renal sympathetic denervation suppresses de novo podocyte injury and albuminuria in rats with aortic regurgitation. Circulation (2012) 125(11):1402–13. doi:10.1161/CIRCULATIONAHA.111.064097
18. Doty JM, Saggi BH, Sugerman HJ, Blocher CR, Pin R, Fakhry I, et al. Effect of increased renal venous pressure on renal function. J Trauma (1999) 47(6):1000–3. doi:10.1097/00005373-199907000-00076
19. Hewitson TD. Fibrosis in the kidney: is a problem shared a problem halved? Fibrogenesis Tissue Repair (2012) 5(Suppl 1):S14. doi:10.1186/1755-1536-5-S1-S14
20. Bursac N. Cardiac fibroblasts in pressure overload hypertrophy: the enemy within? J Clin Invest (2014) 124(7):2850–3. doi:10.1172/JCI76628
21. Hewitson TD, Ono T, Becker GJ. Small animal models of kidney disease: a review. Methods Mol Biol (2009) 466:41–57. doi:10.1007/978-1-59745-352-3_4
22. Rambausek M, Ritz E, Mall G, Mehls O, Katus H. Myocardial hypertrophy in rats with renal insufficiency. Kidney Int (1985) 28(5):775–82. doi:10.1038/ki.1985.197
23. Stefanski A, Schmidt KG, Waldherr R, Ritz E. Early increase in blood pressure and diastolic left ventricular malfunction in patients with glomerulonephritis. Kidney Int (1996) 50(4):1321–6. doi:10.1038/ki.1996.444
24. Lekawanvijit S, Kompa AR, Manabe M, Wang BH, Langham RG, Nishijima F, et al. Chronic kidney disease-induced cardiac fibrosis is ameliorated by reducing circulating levels of a non-dialysable uremic toxin, indoxyl sulfate. PLoS One (2012) 7(7):e41281. doi:10.1371/journal.pone.0041281
25. Suzuki H, Schaefer L, Ling H, Schaefer RM, Dammrich J, Teschner M, et al. Prevention of cardiac hypertrophy in experimental chronic renal failure by long-term ACE inhibitor administration: potential role of lysosomal proteinases. Am J Nephrol (1995) 15(2):129–36. doi:10.1159/000168817
26. Amann K, Wiest G, Zimmer G, Gretz N, Ritz E, Mall G. Reduced capillary density in the myocardium of uremic rats – a stereological study. Kidney Int (1992) 42(5):1079–85. doi:10.1038/ki.1992.390
27. Fort J. Chronic renal failure: a cardiovascular risk factor. Kidney Int Suppl (2005) 99:S25–9. doi:10.1111/j.1523-1755.2005.09906.x
28. Mettler FP, Young DM, Ward JM. Adriamycin-induced cardiotoxicity (cardiomyopathy and congestive heart failure) in rats. Cancer Res (1977) 37(8 Pt 1):2705–13.
29. Noiri E, Nagano N, Negishi K, Doi K, Miyata S, Abe M, et al. Efficacy of darbepoetin in doxorubicin-induced cardiorenal injury in rats. Nephron Exp Nephrol (2006) 104(1):e6–14. doi:10.1159/000093258
30. van der Meer P, Lipsic E, Henning RH, Boddeus K, van der Velden J, Voors AA, et al. Erythropoietin induces neovascularization and improves cardiac function in rats with heart failure after myocardial infarction. J Am Coll Cardiol (2005) 46(1):125–33. doi:10.1016/j.jacc.2005.03.044
31. Zebrack JS, Anderson JL, Beddhu S, Horne BD, Bair TL, Cheung A, et al. Do associations with C-reactive protein and extent of coronary artery disease account for the increased cardiovascular risk of renal insufficiency? J Am Coll Cardiol (2003) 42(1):57–63. doi:10.1016/S0735-1097(03)00564-3
32. Levine B, Kalman J, Mayer L, Fillit HM, Packer M. Elevated circulating levels of tumor necrosis factor in severe chronic heart failure. N Engl J Med (1990) 323(4):236–41. doi:10.1056/NEJM199007263230405
33. Kelly KJ. Distant effects of experimental renal ischemia/reperfusion injury. J Am Soc Nephrol (2003) 14(6):1549–58. doi:10.1097/01.ASN.0000064946.94590.46
34. Yamada S, Tokumoto M, Tatsumoto N, Taniguchi M, Noguchi H, Nakano T, et al. Phosphate overload directly induces systemic inflammation and malnutrition as well as vascular calcification in uremia. Am J Physiol Renal Physiol (2014) 306(12):F1418–28. doi:10.1152/ajprenal.00633.2013
35. Cho E, Kim M, Ko YS, Lee HY, Song M, Kim MG, et al. Role of inflammation in the pathogenesis of cardiorenal syndrome in a rat myocardial infarction model. Nephrol Dial Transplant (2013) 28(11):2766–78. doi:10.1093/ndt/gft376
36. Chang A, Ko K, Clark MR. The emerging role of the inflammasome in kidney diseases. Curr Opin Nephrol Hypertens (2014) 23(3):204–10. doi:10.1097/01.mnh.0000444814.49755.90
37. Fiore MC, Jimenez PM, Cremonezzi D, Juncos LI, Garcia NH. Statins reverse renal inflammation and endothelial dysfunction induced by chronic high salt intake. Am J Physiol Renal Physiol (2011) 301(2):F263–70. doi:10.1152/ajprenal.00109.2010
38. MacAllister RJ, Rambausek MH, Vallance P, Williams D, Hoffmann KH, Ritz E. Concentration of dimethyl-L-arginine in the plasma of patients with end-stage renal failure. Nephrol Dial Transplant (1996) 11(12):2449–52. doi:10.1093/oxfordjournals.ndt.a027213
39. Bongartz LG, Braam B, Verhaar MC, Cramer MJ, Goldschmeding R, Gaillard CA, et al. Transient nitric oxide reduction induces permanent cardiac systolic dysfunction and worsens kidney damage in rats with chronic kidney disease. Am J Physiol Regul Integr Comp Physiol (2010) 298(3):R815–23. doi:10.1152/ajpregu.00727.2009
40. Eriguchi M, Tsuruya K, Haruyama N, Yamada S, Tanaka S, Suehiro T, et al. Renal denervation has blood pressure-independent protective effects on kidney and heart in a rat model of chronic kidney disease. Kidney Int (2015) 87(1):116–27. doi:10.1038/ki.2014.220
41. Heymes C, Bendall JK, Ratajczak P, Cave AC, Samuel JL, Hasenfuss G, et al. Increased myocardial NADPH oxidase activity in human heart failure. J Am Coll Cardiol (2003) 41(12):2164–71. doi:10.1016/S0735-1097(03)00471-6
42. Vaziri ND, Dicus M, Ho ND, Boroujerdi-Rad L, Sindhu RK. Oxidative stress and dysregulation of superoxide dismutase and NADPH oxidase in renal insufficiency. Kidney Int (2003) 63(1):179–85. doi:10.1046/j.1523-1755.2003.00702.x
43. Kielstein JT, Bode-Boger SM, Klein G, Graf S, Haller H, Fliser D. Endogenous nitric oxide synthase inhibitors and renal perfusion in patients with heart failure. Eur J Clin Invest (2003) 33(5):370–5. doi:10.1046/j.1365-2362.2003.01149.x
44. Onuigbo MA. RAAS inhibition and cardiorenal syndrome. Curr Hypertens Rev (2014) 10(2):107–11. doi:10.2174/1573402111666141231144228
45. Guo P, Nishiyama A, Rahman M, Nagai Y, Noma T, Namba T, et al. Contribution of reactive oxygen species to the pathogenesis of left ventricular failure in Dahl salt-sensitive hypertensive rats: effects of angiotensin II blockade. J Hypertens (2006) 24(6):1097–104. doi:10.1097/01.hjh.0000226200.73065.5d
46. Taylor D, Bhandari S, Seymour AM. Mitochondrial dysfunction in uremic cardiomyopathy. Am J Physiol Renal Physiol (2015) 308(6):F579–87. doi:10.1152/ajprenal.00442.2014
47. Lekawanvijit S, Kompa AR, Wang BH, Kelly DJ, Krum H. Cardiorenal syndrome: the emerging role of protein-bound uremic toxins. Circ Res (2012) 111(11):1470–83. doi:10.1161/CIRCRESAHA.112.278457
48. Yang K, Xu X, Nie L, Xiao T, Guan X, He T, et al. Indoxyl sulfate induces oxidative stress and hypertrophy in cardiomyocytes by inhibiting the AMPK/UCP2 signaling pathway. Toxicol Lett (2015) 234(2):110–9. doi:10.1016/j.toxlet.2015.01.021
49. Yisireyili M, Shimizu H, Saito S, Enomoto A, Nishijima F, Niwa T. Indoxyl sulfate promotes cardiac fibrosis with enhanced oxidative stress in hypertensive rats. Life Sci (2013) 92(24–26):1180–5. doi:10.1016/j.lfs.2013.05.008
50. Campese VM, Kogosov E, Koss M. Renal afferent denervation prevents the progression of renal disease in the renal ablation model of chronic renal failure in the rat. Am J Kidney Dis (1995) 26(5):861–5. doi:10.1016/0272-6386(95)90456-5
51. Jacob F, Clark LA, Guzman PA, Osborn JW. Role of renal nerves in development of hypertension in DOCA-salt model in rats: a telemetric approach. Am J Physiol Heart Circ Physiol (2005) 289(4):H1519–29. doi:10.1152/ajpheart.00206.2005
52. KDIGO CKD-MBD workgroup. KDIGO clinical practice guideline for the diagnosis, evaluation, prevention, and treatment of chronic kidney disease-mineral and bone disorder (CKD-MBD). Kidney Int Suppl (2009) 113:S1–130. doi:10.1038/ki.2009.188
53. Goodman WG, Goldin J, Kuizon BD, Yoon C, Gales B, Sider D, et al. Coronary-artery calcification in young adults with end-stage renal disease who are undergoing dialysis. N Engl J Med (2000) 342(20):1478–83. doi:10.1056/NEJM200005183422003
54. London GM, Marchais SJ, Guerin AP, Metivier F, Pannier B. Cardiac hypertrophy and arterial alterations in end-stage renal disease: hemodynamic factors. Kidney Int Suppl (1993) 41:S42–9.
55. Briet M, Boutouyrie P, Laurent S, London GM. Arterial stiffness and pulse pressure in CKD and ESRD. Kidney Int (2012) 82(4):388–400. doi:10.1038/ki.2012.131
56. Mitchell GF. Effects of central arterial aging on the structure and function of the peripheral vasculature: implications for end-organ damage. J Appl Physiol (1985) (2008) 105(5):1652–60. doi:10.1152/japplphysiol.90549.2008
57. Sigrist MK, Taal MW, Bungay P, McIntyre CW. Progressive vascular calcification over 2 years is associated with arterial stiffening and increased mortality in patients with stages 4 and 5 chronic kidney disease. Clin J Am Soc Nephrol (2007) 2(6):1241–8. doi:10.2215/CJN.02190507
58. Ng K, Hildreth CM, Phillips JK, Avolio AP. Aortic stiffness is associated with vascular calcification and remodeling in a chronic kidney disease rat model. Am J Physiol Renal Physiol (2011) 300(6):F1431–6. doi:10.1152/ajprenal.00079.2011
59. Sutliff RL, Walp ER, El-Ali AM, Elkhatib S, Lomashvili KA, O’Neill WC. Effect of medial calcification on vascular function in uremia. Am J Physiol Renal Physiol (2011) 301(1):F78–83. doi:10.1152/ajprenal.00533.2010
60. Ejerblad S, Eriksson I, Johansson H. Uraemic arterial disease. An experimental study with special reference to the effect of parathyroidectomy. Scand J Urol Nephrol (1979) 13(2):161–9. doi:10.3109/00365597909181172
61. Katsumata K, Kusano K, Hirata M, Tsunemi K, Nagano N, Burke SK, et al. Sevelamer hydrochloride prevents ectopic calcification and renal osteodystrophy in chronic renal failure rats. Kidney Int (2003) 64(2):441–50. doi:10.1046/j.1523-1755.2003.00126.x
62. Gagnon RF, Duguid WP. A reproducible model for chronic renal failure in the mouse. Urol Res (1983) 11(1):11–4. doi:10.1007/BF00272702
63. Shobeiri N, Adams MA, Holden RM. Vascular calcification in animal models of CKD: a review. Am J Nephrol (2010) 31(6):471–81. doi:10.1159/000299794
64. Fang Y, Ginsberg C, Sugatani T, Monier-Faugere MC, Malluche H, Hruska KA. Early chronic kidney disease-mineral bone disorder stimulates vascular calcification. Kidney Int (2014) 85(1):142–50. doi:10.1038/ki.2013.271
65. Tamagaki K, Yuan Q, Ohkawa H, Imazeki I, Moriguchi Y, Imai N, et al. Severe hyperparathyroidism with bone abnormalities and metastatic calcification in rats with adenine-induced uraemia. Nephrol Dial Transplant (2006) 21(3):651–9. doi:10.1093/ndt/gfi273
66. Ferrari GO, Ferreira JC, Cavallari RT, Neves KR, dos Reis LM, Dominguez WV, et al. Mineral bone disorder in chronic kidney disease: head-to-head comparison of the 5/6 nephrectomy and adenine models. BMC Nephrol (2014) 15:69. doi:10.1186/1471-2369-15-69
67. Moe SM, Chen NX, Seifert MF, Sinders RM, Duan D, Chen X, et al. A rat model of chronic kidney disease-mineral bone disorder. Kidney Int (2009) 75(2):176–84. doi:10.1038/ki.2008.456
68. Stubbs JR, He N, Idiculla A, Gillihan R, Liu S, David V, et al. Longitudinal evaluation of FGF23 changes and mineral metabolism abnormalities in a mouse model of chronic kidney disease. J Bone Miner Res (2012) 27(1):38–46. doi:10.1002/jbmr.516
69. Sage AP, Tintut Y, Demer LL. Regulatory mechanisms in vascular calcification. Nat Rev Cardiol (2010) 7(9):528–36. doi:10.1038/nrcardio.2010.115
70. Al-Aly Z, Shao JS, Lai CF, Huang E, Cai J, Behrmann A, et al. Aortic Msx2-Wnt calcification cascade is regulated by TNF-alpha-dependent signals in diabetic Ldlr-/- mice. Arterioscler Thromb Vasc Biol (2007) 27(12):2589–96. doi:10.1161/ATVBAHA.107.153668
71. Bobryshev YV. Transdifferentiation of smooth muscle cells into chondrocytes in atherosclerotic arteries in situ: implications for diffuse intimal calcification. J Pathol (2005) 205(5):641–50. doi:10.1002/path.1743
72. Neven E, Dauwe S, De Broe ME, D’Haese PC, Persy V. Endochondral bone formation is involved in media calcification in rats and in men. Kidney Int (2007) 72(5):574–81. doi:10.1038/sj.ki.5002353
73. Davies MR, Lund RJ, Hruska KA. BMP-7 is an efficacious treatment of vascular calcification in a murine model of atherosclerosis and chronic renal failure. J Am Soc Nephrol (2003) 14(6):1559–67. doi:10.1097/01.ASN.0000068404.57780.DD
74. Speer MY, Yang HY, Brabb T, Leaf E, Look A, Lin WL, et al. Smooth muscle cells give rise to osteochondrogenic precursors and chondrocytes in calcifying arteries. Circ Res (2009) 104(6):733–41. doi:10.1161/CIRCRESAHA.108.183053
75. Jahnen-Dechent W, Heiss A, Schafer C, Ketteler M. Fetuin-A regulation of calcified matrix metabolism. Circ Res (2011) 108(12):1494–509. doi:10.1161/CIRCRESAHA.110.234260
76. Keutel J, Jorgensen G, Gabriel P. [A new autosomal-recessive hereditary syndrome. Multiple peripheral pulmonary stenosis, brachytelephalangia, inner-ear deafness, ossification or calcification of cartilages]. Dtsch Med Wochenschr (1971) 96(43):1676–1681assim. doi:10.1055/s-0028-1110200
77. Rutsch F, Ruf N, Vaingankar S, Toliat MR, Suk A, Hohne W, et al. Mutations in ENPP1 are associated with ‘idiopathic’ infantile arterial calcification. Nat Genet (2003) 34(4):379–81. doi:10.1038/ng1221
78. Sakamoto M, Hosoda Y, Kojimahara K, Yamazaki T, Yoshimura Y. Arthritis and ankylosis in twy mice with hereditary multiple osteochondral lesions: with special reference to calcium deposition. Pathol Int (1994) 44(6):420–7. doi:10.1111/j.1440-1827.1994.tb01705.x
79. Shroff RC, McNair R, Skepper JN, Figg N, Schurgers LJ, Deanfield J, et al. Chronic mineral dysregulation promotes vascular smooth muscle cell adaptation and extracellular matrix calcification. J Am Soc Nephrol (2010) 21(1):103–12. doi:10.1681/ASN.2009060640
80. Yoshida H, Yokoyama K, Yaginuma T, Ohkido I, Yamamoto H, Utsunomiya Y, et al. Difference in coronary artery intima and media calcification in autopsied patients with chronic kidney disease. Clin Nephrol (2011) 75(1):1–7.
81. O’Neill WC, Adams AL. Breast arterial calcification in chronic kidney disease: absence of smooth muscle apoptosis and osteogenic transdifferentiation. Kidney Int (2013) 85(3):668–76. doi:10.1038/ki.2013.351
82. Pinto YM, Paul M, Ganten D. Lessons from rat models of hypertension: from Goldblatt to genetic engineering. Cardiovasc Res (1998) 39(1):77–88. doi:10.1016/S0008-6363(98)00077-7
83. Ooshima A, Yamori Y, Okamoto K. Cardiovascular lesions in the selectively-bred group of spontaneously hypertensive rats with severe hypertension. Jpn Circ J (1972) 36(8):797–812. doi:10.1253/jcj.36.797
84. Dikow R, Kihm LP, Zeier M, Kapitza J, Tornig J, Amann K, et al. Increased infarct size in uremic rats: reduced ischemia tolerance? J Am Soc Nephrol (2004) 15(6):1530–6. doi:10.1097/01.ASN.0000130154.42061.C6
85. Kelly DJ, Wilkinson-Berka JL, Allen TJ, Cooper ME, Skinner SL. A new model of diabetic nephropathy with progressive renal impairment in the transgenic (mRen-2)27 rat (TGR). Kidney Int (1998) 54(2):343–52. doi:10.1046/j.1523-1755.1998.00019.x
86. Samuel CS, Hewitson TD, Zhang Y, Kelly DJ. Relaxin ameliorates fibrosis in experimental diabetic cardiomyopathy. Endocrinology (2008) 149(7):3286–93. doi:10.1210/en.2008-0250
87. Lian M, Hewitson TD, Wigg B, Samuel CS, Chow F, Becker GJ. Long-term mineralocorticoid receptor blockade ameliorates progression of experimental diabetic renal disease. Nephrol Dial Transplant (2012) 27:906–12. doi:10.1093/ndt/gfr495
88. Forbes JM, Yee LT, Thallas V, Lassila M, Candido R, Jandeleit-Dahm KA, et al. Advanced glycation end product interventions reduce diabetes-accelerated atherosclerosis. Diabetes (2004) 53(7):1813–23. doi:10.2337/diabetes.53.7.1813
89. Joshi D, Gupta R, Dubey A, Shiwalkar A, Pathak P, Gupta RC, et al. TRC4186, a novel AGE-breaker, improves diabetic cardiomyopathy and nephropathy in Ob-ZSF1 model of type 2 diabetes. J Cardiovasc Pharmacol (2009) 54(1):72–81. doi:10.1097/FJC.0b013e3181ac3a34
90. Hayden MR, Sowers JR. Childhood-adolescent obesity in the cardiorenal syndrome: lessons from animal models. Cardiorenal Med (2011) 1(2):75–86. doi:10.1159/000327022
91. Sen S, Chen S, Feng B, Iglarz M, Chakrabarti S. Renal, retinal and cardiac changes in type 2 diabetes are attenuated by macitentan, a dual endothelin receptor antagonist. Life Sci (2012) 91(13–14):658–68. doi:10.1016/j.lfs.2012.03.032
92. Bomzon A, Holt S, Moore K. Bile acids, oxidative stress, and renal function in biliary obstruction. Semin Nephrol (1997) 17(6):549–62.
93. Tajuddin M, Tariq M, Bilgrami NL, Kumar S. Biochemical and pathological changes in the heart following bile duct ligation. Adv Myocardiol (1980) 2:209–12.
94. Suzuki K, Nakagawa K, Yamamoto T, Miyazawa T, Kimura F, Kamei M, et al. Carbon tetrachloride-induced hepatic and renal damages in rat: inhibitory effects of cacao polyphenol. Biosci Biotechnol Biochem (2015) 79(10):1669–75. doi:10.1080/09168451.2015.1039481
95. Liu Y, Meyer C, Xu C, Weng H, Hellerbrand C, ten Dijke P, et al. Animal models of chronic liver diseases. Am J Physiol Gastrointest Liver Physiol (2013) 304(5):G449–68. doi:10.1152/ajpgi.00199.2012
96. Perry D, Sang A, Yin Y, Zheng YY, Morel L. Murine models of systemic lupus erythematosus. J Biomed Biotechnol (2011) 2011:271694. doi:10.1155/2011/271694
97. Pansky B, Freimer EH. Spontaneous carditis in the NZB mouse and its hybrids. Arthritis Rheum (1974) 17(4):403–8. doi:10.1002/art.1780170410
98. Huston DP, Steinberg AD. Animal models of human systemic lupus erythematosus. Yale J Biol Med (1979) 52(3):289–305.
99. Sharabi A, Dayan M, Zinger H, Mozes E. A new model of induced experimental systemic lupus erythematosus (SLE) in pigs and its amelioration by treatment with a tolerogenic peptide. J Clin Immunol (2010) 30(1):34–44. doi:10.1007/s10875-009-9326-4
100. Liu S, Kompa AR, Kumfu S, Nishijima F, Kelly DJ, Krum H, et al. Subtotal nephrectomy accelerates pathological cardiac remodeling post-myocardial infarction: implications for cardiorenal syndrome. Int J Cardiol (2013) 168(3):1866–80. doi:10.1016/j.ijcard.2012.12.065
101. Windt WA, Henning RH, Kluppel AC, Xu Y, de Zeeuw D, van Dokkum RP. Myocardial infarction does not further impair renal damage in 5/6 nephrectomized rats. Nephrol Dial Transplant (2008) 23(10):3103–10. doi:10.1093/ndt/gfn233
102. Bongartz LG, Joles JA, Verhaar MC, Cramer MJ, Goldschmeding R, Tilburgs C, et al. Subtotal nephrectomy plus coronary ligation leads to more pronounced damage in both organs than either nephrectomy or coronary ligation. Am J Physiol Heart Circ Physiol (2012) 302(3):H845–54. doi:10.1152/ajpheart.00261.2011
Keywords: animal models, cardiorenal syndrome, heart, kidney, review
Citation: Hewitson TD, Holt SG and Smith ER (2015) Animal models to study links between cardiovascular disease and renal failure and their relevance to human pathology. Front. Immunol. 6:465. doi: 10.3389/fimmu.2015.00465
Received: 26 June 2015; Accepted: 26 August 2015;
Published: 17 September 2015
Edited by:
Heidi Noels, Institute for Molecular Cardiovascular Research, GermanyReviewed by:
Jaap Joles, University Medical Center Utrecht, NetherlandsCopyright: © 2015 Hewitson, Holt and Smith. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Tim D. Hewitson, Department of Nephrology, Royal Melbourne Hospital (RMH), Grattan Street, Parkville, Melbourne, VIC 3050, Australia,dGltLmhld2l0c29uQG1oLm9yZy5hdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.