
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Immunol. , 08 September 2015
Sec. Multiple Sclerosis and Neuroimmunology
Volume 6 - 2015 | https://doi.org/10.3389/fimmu.2015.00463
This article is part of the Research Topic The emerging role of monocyte-derived cells in the central nervous system View all 6 articles
 Agnieszka Wlodarczyk1
Agnieszka Wlodarczyk1 Oriane Cédile1
Oriane Cédile1 Kirstine Nolling Jensen1
Kirstine Nolling Jensen1 Agathe Jasson1,2
Agathe Jasson1,2 Jyothi Thyagabhavan Mony1
Jyothi Thyagabhavan Mony1 Reza Khorooshi1
Reza Khorooshi1 Trevor Owens1*
Trevor Owens1*
Inflammation is a series of processes designed for eventual clearance of pathogens and repair of damaged tissue. In the context of autoimmune recognition, inflammatory processes are usually considered to be pathological. This is also true for inflammatory responses in the central nervous system (CNS). However, as in other tissues, neuroinflammation can have beneficial as well as pathological outcomes. The complex role of encephalitogenic T cells in multiple sclerosis and its animal model experimental autoimmune encephalomyelitis (EAE) may derive from heterogeneity of the myeloid cells with which these T cells interact within the CNS. Myeloid cells, including resident microglia and infiltrating bone marrow-derived cells, such as dendritic cells (DC) and monocytes/macrophages [bone marrow-derived macrophages (BMDM)], are highly heterogeneous populations that may be involved in neurotoxicity and also immunoregulation and regenerative processes. Better understanding and characterization of myeloid cell heterogeneity is essential for future development of treatments controlling inflammation and inducing neuroprotection and neuroregeneration in diseased CNS. Here, we describe and compare three populations of myeloid cells: CD11c+ microglia, CD11c− microglia, and CD11c+ blood-derived cells in terms of their pathological versus protective functions in the CNS of mice with EAE. Our data show that CNS-resident microglia include functionally distinct subsets that can be distinguished by their expression of CD11c. These subsets differ in their expression of Arg-1, YM1, iNOS, IL-10, and IGF-1. Moreover, in contrast to BMDM/DC, both subsets of microglia express protective interferon-beta (IFNβ), high levels of colony-stimulating factor-1 receptor, and do not express the Th1-associated transcription factor T-bet. Taken together, our data suggest that CD11c+ microglia, CD11c− microglia, and infiltrating BMDM/DC represent separate and distinct populations and illustrate the heterogeneity of the CNS inflammatory environment.
The contribution of myeloid cells to central nervous system (CNS) function is increasingly appreciated. This is especially relevant in CNS inflammation, where they can act not only as participants in the immune attack but also as regulators of the immune response and promoters of neuronal and synaptic protection and regeneration (1). A key aspect of the inflammatory response within the CNS is the recruitment and reactivation of infiltrating CD4+ T cells and direction of their effector function within the tissue (2). Their role in multiple sclerosis (MS) and its animal model experimental autoimmune encephalomyelitis (EAE) is complex. Although there is a consensus that inhibition or blockade of such responses has therapeutic benefit in MS and EAE [reviewed in Ref. (3)], there are numerous reports of beneficial effect of the activation of such T cells for myelin clearance, remyelination, and repair (4, 5). The cells implicated in interaction with CD4+ T cells within the inflamed CNS are myeloid cells, such as infiltrating dendritic cells (DC) and monocytes/macrophages, and also resident microglia (6–9). These are very heterogeneous populations of immune cells that can dampen inflammation or contribute to pathological events. Better characterization of the heterogeneity of blood-derived as well as CNS-resident myeloid cells and their interaction with T cells is crucial for understanding how to effectively control inflammation and induce neuroprotection and neuroregeneration in diseases like MS.
Microglia are cells of myeloid lineage considered to be CNS macrophages [reviewed in Ref. (10)]. However, they differ in terms of origin: while macrophages originate from fetal liver, microglia emerge from yolk sac and colonize the CNS early during neonatal development (11). Microglia are autonomously maintained through proliferation and in normal circumstances, they are not replaced by bone marrow (BM)-derived cells (12). Based on many phenotypic similarities between microglia and macrophages, the discrimination between these two populations as well as other BM- or blood-derived cells during neuroinflammation is challenging. The difference in the expression level of cell membrane tyrosine phosphatase CD45 can be used to discriminate CD45low microglia from CD45high blood-derived cells by flow cytometry (13, 14). In addition, other common markers used for microglial identification include CD11b, CD11c, Iba1, and CX3CR1 (10, 15–20). However, these can also be expressed by macrophages as well as monocytes and DC. Recent development of a double knock-in red (CCR2)/green (CX3CR1) mouse has allowed the discrimination of CNS-resident microglia from infiltrating leukocytes, microglia being CX3CR1-positive but, in contrast to infiltrating immune cells, CCR2-negative (21).
In this study, we aimed to examine the heterogeneity amongst microglia as well as to compare them to infiltrating blood-derived myeloid cells in EAE. We show that CD11c+ microglia, CD11c− microglia, and CD11c+ blood-derived cells are functionally distinct populations of myeloid cells within the inflamed CNS. Our findings illustrate the heterogeneity of the CNS inflammatory environment, reflecting the complexity of immunological processes that lead to pathology, tissue protection, or immunoregulation.
Female C57BL/6j bom (B6) mice aged 6–8 weeks were obtained from Taconic Europe A/S (Lille Skensved, Denmark) and CX3CR1gfp/gfp were obtained from The Jackson Laboratory (Bar Harbor, ME, USA) and maintained in the Biomedical Laboratory, University of Southern Denmark (Odense). All experiments were approved by the Danish Animal Experiments Inspectorate (approval number 2014-15-0201-00369).
To induce NMO-like lesions, 7–10-week-old female B6 mice received IgG antibodies isolated from NMO patients and human complement by injection in the striatum, as described in Ref. (22). The use of human material was approved by The Committee on Biomedical Research Ethics for the Region of Southern Denmark (ref. no. S-20080142).
In order to induce demyelination, 7–12-week-old female B6 mice were fed with cuprizone for 4 weeks as described in Ref. (23).
Seven to ten weeks old female mice were immunized by injecting subcutaneously 100 μl of an emulsion containing 100 μg of myelin oligodendrocyte glycoprotein (MOG)p35–55 (TAG Copenhagen A/S, Frederiksberg, Denmark) in incomplete freunds adjuvant (DIFCO, Alberstslund, Denmark) supplemented with 400 μg H37Ra Mycobacterium tuberculosis (DIFCO). Bordetella pertussis toxin (300 ng; Sigma-Aldrich, Brøndby, Denmark) in 200 μl of PBS was injected intraperitoneally at day 0 and day 2. Animals were monitored daily from day 5 and scored on a 6-point scale as follows: 0, no symptoms; 0.5, partial loss of tail tonus; 1, complete loss of tail tonus; 2, difficulty to right, 3, paresis in one or both hind legs; 4, paralysis in one or both hind legs; 5, front limb paresis; and 6, moribund. Severe EAE usually developed 14–18 days after immunization and was defined as a score of 3–5.
Mice were anesthetized with 0.2 mg pentobarbital (200 mg/ml; Glostrup Apotek, Glostrup, Denmark) per gram body weight and intracardially perfused with ice-cold PBS. CNS tissue (Brain and SC from EAE model, Ipsi lateral part of the brain from NMO-like disease, and whole brain from Cuprizone-induced demyelination) was collected and a single cell suspension was generated by forcing through a 70 μm cell strainer (BD Biosciences). Mononuclear cells were collected after centrifugation on 37% Percoll (GE Healthcare Bio-sciences AB, Brøndby, Denmark). Spleens were digested with collagenase D (0.5 mg/ml; Roche, Hvidovre-Copenhagen) and DNase I (40 μg/ml, Roche) for 30 min at 37°C. Supernatant was collected and supplemented with 100 mM EDTA for 5 min at 37°C and passed through a 70 μm cell strainer (BD Biosciences, Albertslund, Denmark). Red blood cells were lysed with a 0.83% NH4Cl solution. Splenocytes and CNS mononuclear cells were then incubated with anti-Fc receptor (Clone 2.4G2; 1 μg/ml; BD Pharmingen) and Syrian hamster IgG (50 μg/ml; Jackson Immuno Research Laboratories Inc., Skanderborg, Denmark) in PBS 2% fetal bovine serum (FBS). CNS mononuclear cells were stained with PE-anti-mouse CD45 (Biolegend, Copenhagen, Denmark), PerCP-Cy5.5-anti-mouse CD11b (Biolegend) and biotinylated-anti-mouse CD11c (BD Pharmingen) antibodies in PBS 2% FBS. Splenocytes were stained with PE-anti-mouse CD11c antibodies in PBS 2% FBS and CD11c+ cells were sorted on a FACSAria™ III cell sorter (BD Biosciences, Albertslund, Denmark). After excluding doublets (FSC-H, FSC-W and SSC-H, SSC-W) (Figures 1A–C), CNS cell populations were gated based on isotype control antibodies as CD45dimCD11b+CD11c− (CD11c− microglia), CD45dimCD11b+CD11c+ (CD11c+ microglia), and CD45highCD11c+ (BMDM/DC) (Figures 1D,E) and were sorted on a FACSAria™ III cell sorter (BD Biosciences).
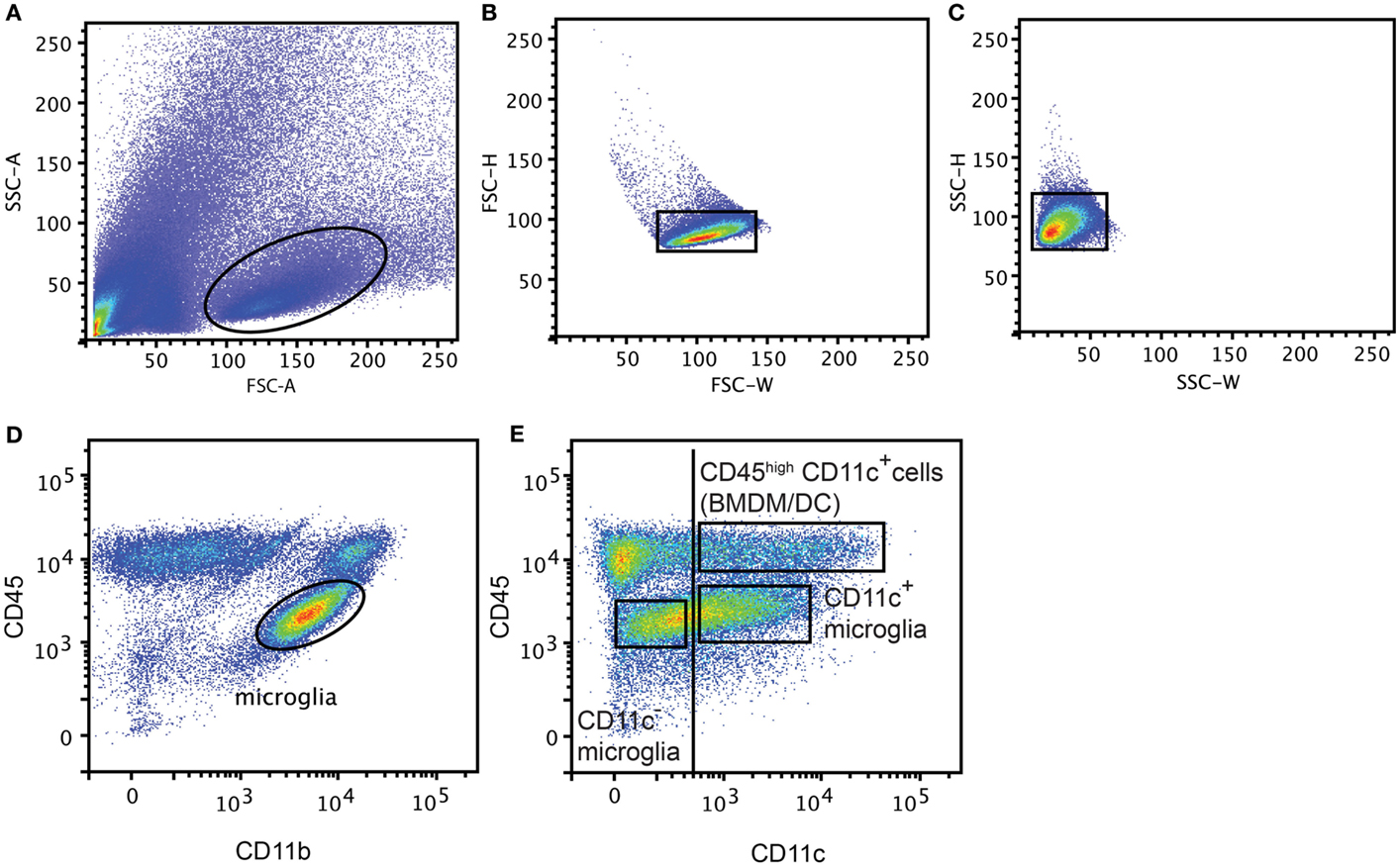
Figure 1. Gating strategy for flow cytometry and FACS-sorting. Representative flow cytometry profiles from individual central nervous system suspensions prepared from mice with severe EAE, showing gating strategy used for flow cytometry and FACS-sorting. First doublets were excluded from live gate (A) based on FSC-H, FSC-W (B) and SSC-H, SSC-W (C), then microglia were gated based on CD45 and CD11b expression (D). CD11c− microglia, CD11c+ microglia, and BMDM/DC population were discriminated based on CD45 and CD11c expression and gated based on isotype control antibodies (vertical line) and sorted (E).
B6 mice received 100 μl of 1 mg/ml BrdU by daily intraperitoneal injection, starting from day 10 after immunization. Mice with severe EAE were sacrificed and analyzed by flow cytometry using BrdU flow kit (BD Pharmingen) according to the manufacturer’s protocol. Data were acquired on an LSRII™ flow cytometer and analyzed using FACSDiva™ 7 software (BD Biosciences).
Sorted splenic CD11c+ cells from individual mice as well as CD11c+ microglia, CD11c− microglia, and CD45highCD11c+ cells from pools of 2–3 mice were placed in RLT buffer (Qiagen, Copenhagen, Denmark). Total RNA was extracted using RNeasy columns as per the manufacturer’s protocol (Qiagen). Reverse transcription was performed with M-MLV reverse transcriptase (Invitrogen) according to the manufacturer’s protocol.
Quantitative real-time PCR (qPCR) was performed with 1 μl cDNA in a 25 μl reaction volume containing Maxima® Probe/ROX qPCR Master mix (Fermentas, St. Leon-rot, Germany), forward and reverse primers (800 nM; from TAG Copenhagen A/S, Frederiksberg, Denmark) and probe (200 nM; Applied Biosystems, Nærum, Denmark and TAG Copenhagen A/S, Frederiksberg, Denmark). Primer and probe sequences were as follows: IFNβ: For: GCGTTCCTGCTGTGCTTCTC; Rev: TTGAAGTCCGCCCTGTAGGT; Probe: CGGAAATGTCAGGAGCT; IRF7: For: CACCCCCATCTTCGACTTCA; Rev: CCAAAACCCAGGTAGATGGTGTA; Probe: CACTTTCTTCCGAGAACT; IFR3: For: CACCCCAAGAAAATCCACTGA; Rev: AGGCGGTCACCTCGAACTC; Probe: TAGCTGAGGAACAATG. TaqMan® PreAmp Master Mix Kits were used for T-bet: Mm01299452; YM1: Mm00657889; ARG1: Mm00475988; iNOS: Mm00440502; CSF1R: Mm01266652. For IGF1, SYBR Green/ROX qPCR Master Mix (2X) Probe/ROX qPCR Master mix (Fermentas, St. Leon-rot, Germany) was used with forward and reverse primers (800 nM) from TAG Copenhagen A/S). Primer sequences were as follows: IGF1: For: CCG AGG GGC TTT TAC TTC AAC AA; Rev: CGG AAG CAA CAC TCA TCC ACA A. PCR reactions were done on an ABI Prism 7300 Sequence Detection System (Applied Biosystems). Results were expressed relative to 18S rRNA (2ΔCT method) as endogenous control (TaqMan® Ribosomal RNA control Reagents kit; Applied Biosystems). cDNA was diluted 1/500 for 18S rRNA analysis.
Fifty-micrometer sections (coronal and sagittal) from 4% PFA-fixed, frozen brains of PBS-perfused B6 mice were cut on a cryostat and stored in de Olmos cryoprotectant solution (24) at −14°C. Sections were washed in PBS and incubated for 30 min with 10% methanol and 10% H2O2 in PBS to block endogenous peroxidase. Alternatively 12 μm spinal cord sections were fixed with 4% PFA. After repeated rinses with 0.2% Triton X100 in PBS (PBST), sections were incubated for 1 h in 3% BSA in PBS to block unspecific binding. Next, sections were incubated overnight at 4°C with corresponding primary antibodies: rabbit anti-Iba1 (Wako, Neuss, Germany), hamster anti-CD11c (25) (kindly provided by Dr. Diego Gomez-Nicola; Centre for Biological Sciences, University of Southampton, SO16 6YD, Southampton, UK), rat anti-mouse Mac-1/CD11b (clone MCA711, AbD Serotec, Kidlington, UK), and rabbit anti-T-bet (H-210, Santa Cruz Biotechnology). Following primary antibody incubation, the sections were washed with PBST and incubated for 1 h with the appropriate secondary antibody: goat anti-Armenian hamster IgG-HRP (Santa Cruz Biotechnology, Heidelberg, Germany), goat anti-rabbit Alexa 488 (Invitrogen, Taastrup, Denmark), goat anti-rat Alexa 488 or biotinylated goat anti-rabbit (Abcam, Cambridge, UK) and PE-streptavidin (Biolegend). To visualize CD11c following secondary antibody incubation, the sections were washed with PBS and incubated for 4 min with TSA™ Plus Cy3 System (PerkinElmer, Skovlunde, Denmark). After immunofluorescent labeling, the sections were counterstained with DAPI and mounted with Fluorescence Mounting Medium (DAKO, Glostrup, Denmark). The sections were visualized on an Olympus FV1000MPE Confocal microscope and analyzed by FV10-ASW 4.06 software (Olympus, Denmark).
All experiments were repeated at least three times and data are presented as means ± SEM. Statistical significance was assessed using the two-tailed Mann–Whitney U-test (GraphPad Prism 4). P-values <0.05 were considered significant.
CD11b and CD11c form integrin heterodimers with CD18 and function as complement receptors. Both can be expressed by microglia [reviewed in Ref. (10)]. However, CD11c expression is usually attributed to activation of these cells. In humans, CD11c was shown to be constitutively expressed at a low level in microglia and upregulated in Alzheimer’s disease (AD) (26). CD11c+ Iba1+ cells were found in developing mouse brain as early as E16, they were also evident during postnatal neurodevelopment P2 (27), while in adult brain they were hardly visible (20, 27). We have confirmed prominence of CD11c-positive cells in the developing brain, most of them being CCR2− CX3CR1+ Iba1+ microglia (unpublished data). In adult mice, only very few CD11c+ cells are present in the steady state, averaging 2–3% of total microglia (Figure 2A). However, their proportions increase dramatically during active neuroinflammation, such as in EAE (19), cuprizone-induced demyelination (23), and in a mouse model for neuromyelitis optica (Figure 2A), and also in an animal model for AD (28). Due to many phenotypic overlaps, bone marrow-derived macrophages (BMDM)/DC and microglia within the inflamed CNS are difficult to distinguish from each other by histology. Nevertheless, we assessed the localization of CD11c+ cells within the inflamed spinal cord. We identified three different populations of myeloid cells within the lesion: Iba1+ CD11c+ and Iba1+CD11c− cells, that are likely to include microglia/BMDM, as well as CD11c+ Iba1− cells, most likely DC. Beside differential expression of these markers, they show different morphology, ranging from round, as well as dendritic-like CD11c+ Iba1− cells to amoeboid double-positive and Iba1-single positive cells. All of them were uniformly distributed within the lesion, with obvious possibility for interaction with T cells and with each other (Figure 2B). These data emphasize the high morphological and phenotypic heterogeneity of myeloid cell populations in neuroinflammation.
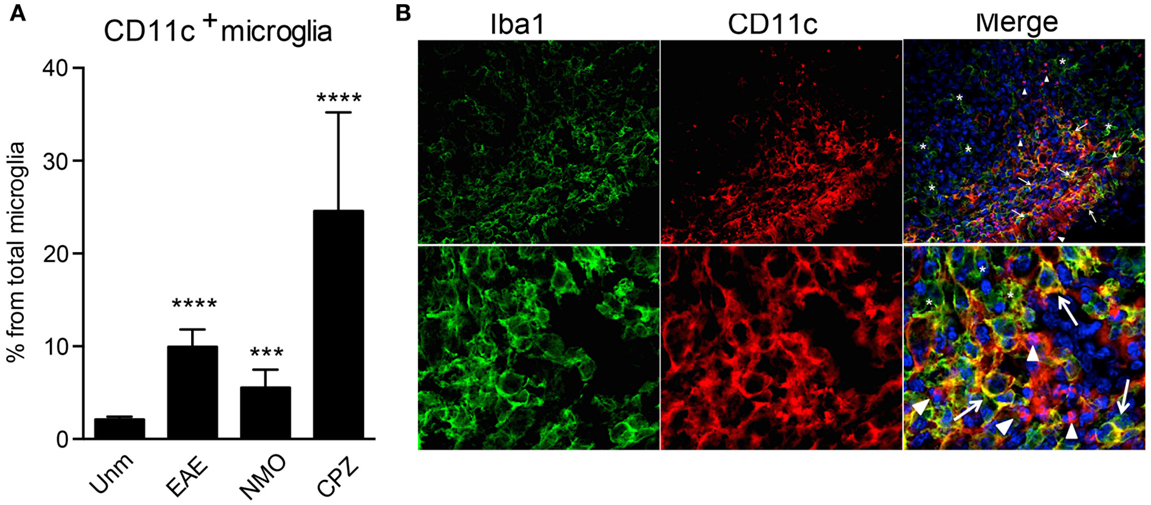
Figure 2. CD11c+ microglia emerge in response to neuroinflammation. (A) Flow cytometry analysis shows a significant increase of CD11c+ microglia, presented as a percentage of total microglia, in EAE, NMO-like disease, and cuprizone-induced demyelination. Data are presented as means ± SEM of three individual experiments (n ≥ 6), **P < 0.01 ***P < 0.005, ****P < 0.001. (B) Representative confocal microscopic analysis of spinal cord from mice with severe EAE from three individual experiments. Arrowheads point to CD11c (red) single positive cells, asterisks point to Iba1 (green) single positive cells, and arrows point to cells co-expressing CD11c marker with Iba1.
Fractalkine receptor CX3CR1 and its ligand CX3CL1 are expressed within the CNS by microglia and neurons, respectively (29). Their interaction plays an important role in neurodevelopment and has also been implicated in neuroinflammation. Disruption of CX3CR1–CX3CL1 axis leads to microglial activation and hypersensitivity and may result in neurotoxicity [(18) and reviewed in Ref. (30)]. For instance, mice deficient in CX3CR1 have been reported to develop more severe EAE symptoms than WT animals (31). Here, we show that mice lacking CX3CR1 are more susceptible to EAE, with a significantly earlier onset and higher incidence of the disease (Figure 3). This data support conclusions from BM chimeras (31) that CX3CR1 regulates myeloid cell and possibly microglial responses, which in this case would act to dampen neuroinflammation, as also evidenced in CX3CR1-deficient mice (18).
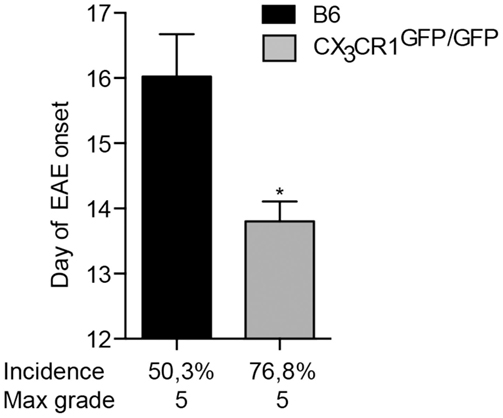
Figure 3. CX3CR1-deficient mice are more susceptible to EAE. CX3CR1gfp/gfp and B6 mice were immunized with (MOG)p35–55 and monitored daily for development of EAE. The data shows significantly earlier onset, higher incidence of EAE in CX3CR1-deficient mice than in B6 mice. Data are presented as means ± SEM of at least four individual experiments (n ≥ 4), *P < 0.05.
Microglial phenotypic heterogeneity may also reflect functional status (1, 32), as has been described for macrophages [reviewed in Ref. (33)]. Classically activated macrophages secrete inflammatory cytokines and mediators involved in host defense, such as tumor necrosis factor (TNFα), IL-12, or nitric oxide (NO) produced by inducible NO synthase (iNOS). By contrast, alternatively activated macrophages that are involved in the regulation of the immune response to limit inflammation and promote tissue repair express arginase-1 (ARG1) or chitinase-like proteins, such as YM1. Such cells also secrete growth factors, such as insulin-like growth factor 1 (IGF-1) or anti-inflammatory cytokine IL-10 (33). We have addressed functional correlates to CD11c phenotypic distinction for microglia sorted from the CNS of B6 and CX3CR1GFP/GFP mice with EAE, by analysis of mRNA levels of ARG1, YM1, iNOS, IL-10, and IGF1. All of them showed clear-cut preferential expression by one or another microglial subset (Figure 4). There was no difference in TNFα expression between these microglial populations (not shown). These results, therefore, point to functional distinctions between CD11c+ and CD11c− microglia. Moreover, CX3CR1 deficiency did not influence M1/M2-associated gene expression in microglia except for YM1, which was upregulated in CX3CR1-deficient microglia (Figure 4). These data suggest that the susceptibility to EAE in CX3CR1-deficient mice is not dependent on microglial M1/M2 phenotype.
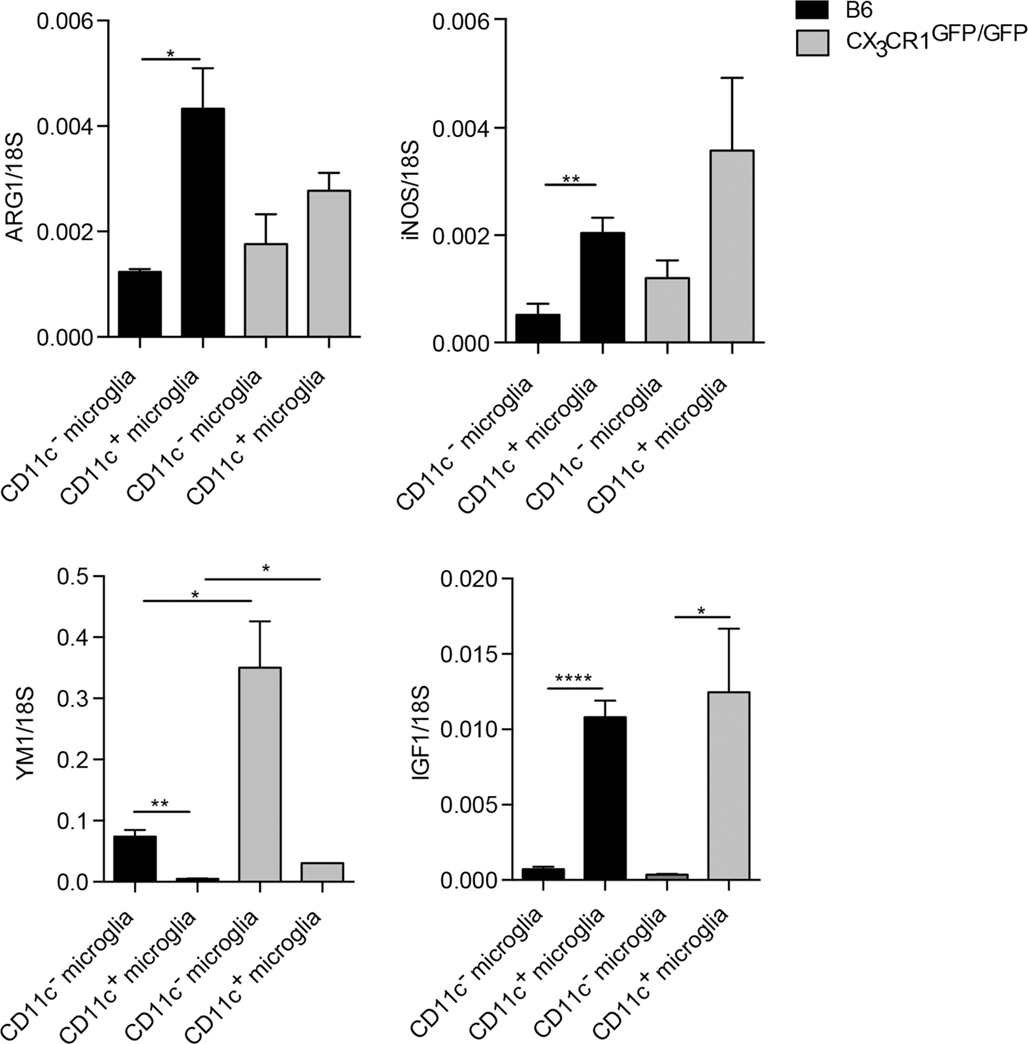
Figure 4. Function-related gene expression by CD11c− and CD11c+ microglia in CX3CR1gfp/gfp and B6 mice in EAE. Expression of IL-10, ARG1, YM1, iNOS, and IGF-1 in fluorescence-activated cell sorted CD11c+ microglia and CD11c− microglia from the central nervous system from mice with severe EAE were analyzed by quantitative real-time PCR. Data are presented as means ± SEM of three individual experiments (n ≥ 5, where n represents a pool of 2–3 individual mice); *P < 0.05; **P < 0.01.
We were further interested in differences between microglia and blood-derived myeloid cells; thus, we compared mRNA levels of ARG1, YM1, iNOS, IL-10, and IGF1 between microglia populations and CD45highCD11c+ cells (BMDM/DC). Here, we show that BMDM/DC express significantly higher levels of ARG1, iNOS, and IL-10 than both populations of microglia, they also expressed significantly more transcripts of YM1 than CD11c+ microglia, but less than CD11c− microglia (Figure 5). Interestingly, while CD11c+ microglia showed high expression of IGF1 transcripts, neither CD11c− microglia nor infiltrating CD45high cells expressed significant levels of this growth factor (Figure 5). Association of IGF1 expression with CD11c+ microglia has also been reported in glatiramer acetate-treated transgenic mice with an AD-like phenotype and has been suggested as a mechanistic basis for the ability of CD11c+ microglia to promote neurogenesis (34).
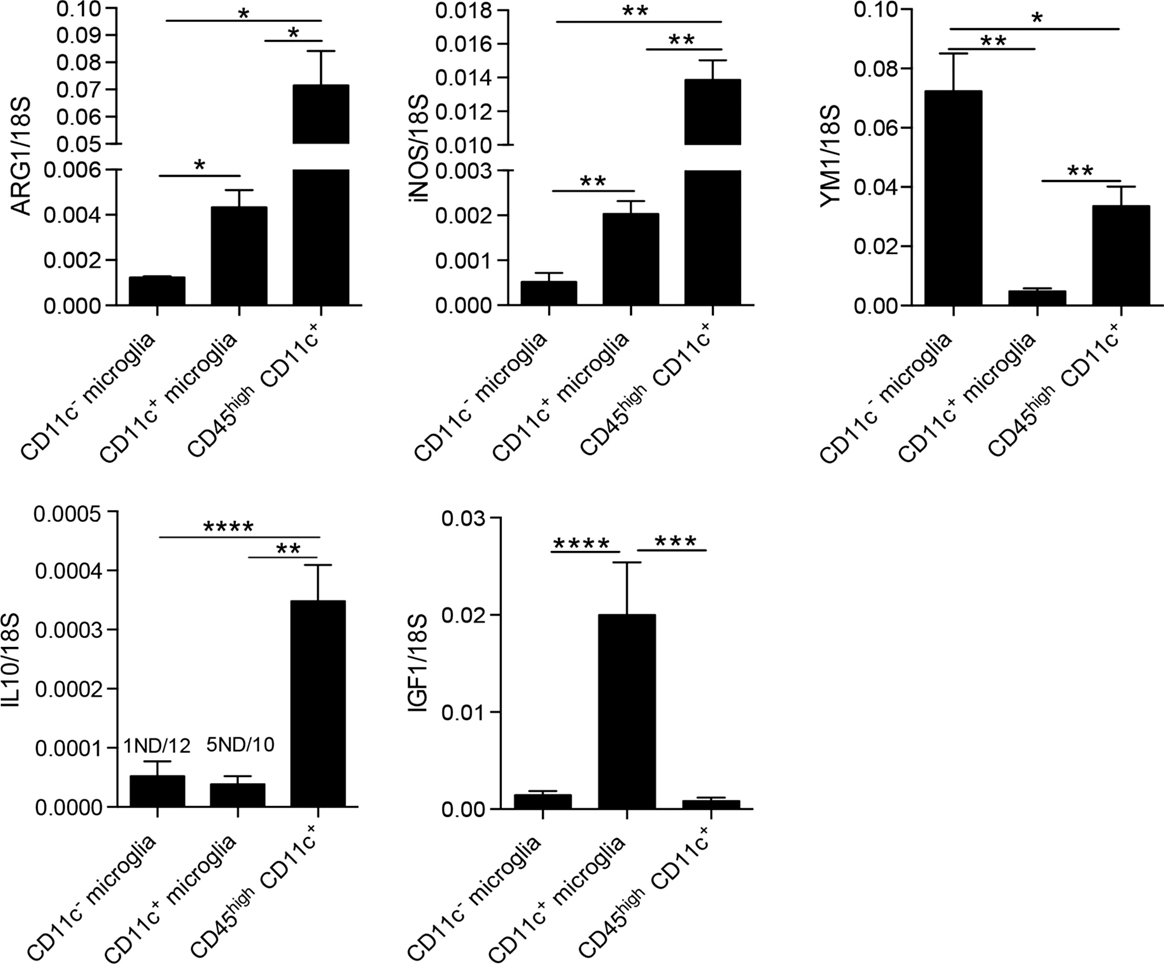
Figure 5. Comparison of expression of M1/M2-associated genes in myeloid cells in CNS in EAE. Expression of ARG1, YM1, iNOS, IL-10, and IGF-1 in fluorescence-activated cell sorted CD11c+ microglia and CD11c− microglia and CD45highCD11c+ infiltrating cells (BMDM/DC) from the central nervous system from mice with severe EAE were analyzed by quantitative real-time PCR. Data are presented as means ± SEM of three individual experiments (n ≥ 5, where n represents a pool of 2–3 individual mice); *P < 0.05; **P < 0.01.
Since neurogenesis may be considered as a part of the anti-inflammatory or protective spectrum, we asked whether these cells additionally selectively expressed other regulatory cytokines. It was recently published that IFNβ produced by a subpopulation of microglia at the peak of EAE facilitated clearance of myelin debris, thus, contributing to possible explanations for amelioration of EAE as well as MS by IFNβ (35). Moreover, we have shown that experimental induction of microglial-derived IFNβ was protective in EAE (36). We, therefore, asked whether CD11c+ microglia expressed IFNβ in EAE. Contrary to our expectations, there was no difference in expression of IFNβ mRNA by CD11c+ and CD11c− microglia isolated from the CNS of mice with EAE (Figure 6A). On this basis, there was, therefore, no evidence for a selective regulatory role for CD11c+ microglia. By contrast, infiltrating BMDM/DC expressed very low levels of IFNβ mRNA comparable to unmanipulated splenic CD11c+ cells, consistent with lack of a regulatory role for these cells, distinct from CNS-resident microglia. However, all of these brain myeloid populations may respond to type I IFN, as evidenced by equivalent expression of IRF7 and IRF3 (Figures 6B,C). Interestingly, IRF3 was upregulated in BMDM/DC compared to unmanipulated splenic CD11c+ cells (Figure 6C), suggesting ongoing active response within the CNS. These data further suggest a beneficial role for microglia in demyelinating diseases as well as emphasize functional distinctions between them and infiltrating BMDM/DC. Peripherally administered IFNβ is used as a first-line therapy for MS (37). IFNβ, constitutively expressed by microglial cells, is increased in EAE and play a protective role in the CNS (35, 36). Whether this protection might involve IFNβ-driven DC response may be speculated but would be consistent with the expression of the IFNβ response gene IRF7 by DC, which we have shown.
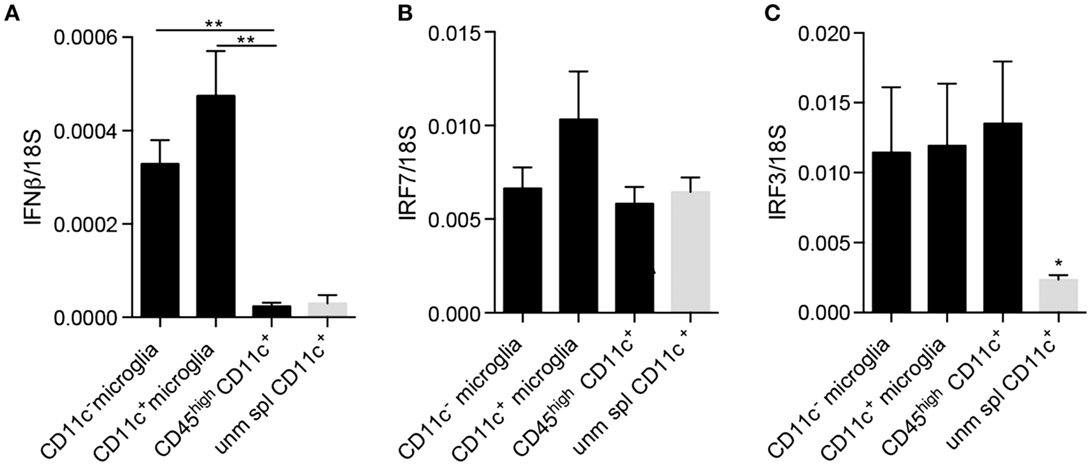
Figure 6. IFNβ and IFN regulatory factors expression by microglia and infiltrating BMDM/DC in EAE. Expression of IFNβ (A), IRF7 (B), and IRF3 (C) by sorted myeloid cells (CD11c+ microglia, CD11c− microglia and CD45highCD11c+ BMDM/DC) from the CNS of mice with severe EAE as well as unmanipulated splenic CD11c+ cells was analyzed by RT-qPCR. Data are presented as means ± SEM of three individual experiments (n ≥ 5, where n represents a pool of 2–3 individual mice) **P < 0.01.
Our findings support observations made by Yamasaki et al. who used a combination of differential reporter gene expression and morphological correlates in EAE to infer that whereas BMDM were implicated in myelin stripping at nodes of Ranvier, microglia were not intimately associated with the demyelinating axon. Their location, relative morphology, and gene expression profile pointed to microglial involvement in phagocytosis and clearance of debris (38). Together with our findings, now based on consensus phenotypic distinction between microglia and BMDM, microglia can be seen to play a beneficial role in CNS inflammation, as has been discussed elsewhere (39).
Microglia have been shown to be a self-renewing population within the CNS. Their proliferation in response to neurodegeneration and their maintenance in adult mice were reported to be dependent on macrophage colony-stimulating factor receptor (CSF1R) signaling (25, 40). CSF1R signaling in response to its ligands, CSF1 and IL-34, induces microglial proliferation (40), suggesting tonic self-renewal as a component of normal homeostasis. Given the emergence of CD11c+ microglia in neuroinflammation, we asked whether CSF1R signaling might play a selective role in expansion of these cells. We used BrdU incorporation to assess microglial proliferation. Both CD11c+ and CD11c− populations of microglia proliferated equivalently during EAE (Figures 7A,B), arguing against proliferation per se as basis for relative expansion of CD11c+ cells. Consistent with this, there was no significant difference in expression of CSF1R by CD11c+ and CD11c− microglia (Figure 7C). Strikingly, infiltrating BMDM/DC (CD45highCD11c+ cells) expressed very low levels of CSF1R (Figure 7C). This again emphasizes that differences between subsets of CNS-resident microglia are minor when both are compared to BMDM/DC.
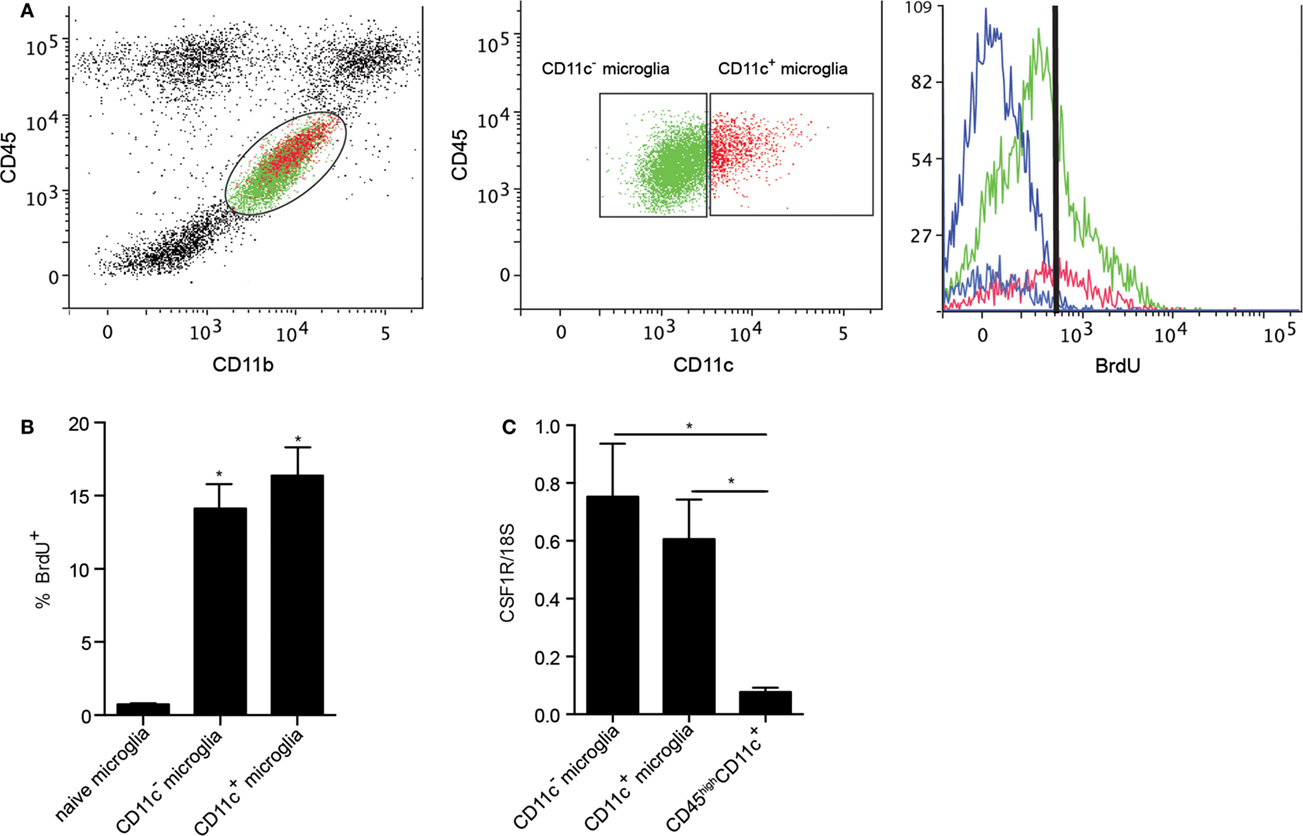
Figure 7. Proliferation of microglial and BMDM/DC populations during EAE. Representative flow cytometry profiles of six individual central nervous system suspensions prepared from mice with severe EAE showing BrdU incorporation by CD11c− microglia (green) and CD11c+ microglia (red) based on negative control (blue) (A). Flow cytometry analysis shows a significant increase in percentage of proliferating (incorporating BrdU) microglia during EAE compared to naive controls (B). Expression of CSF1R in fluorescence-activated cell sorted myeloid cells (CD11c+ microglia, CD11c− microglia, and CD45highCD11c+) from the central nervous system was analyzed by quantitative real-time PCR (C). Data are presented as means ± SEM of three individual experiments (n ≥ 5, where n represents a pool of 2–3 individual mice) *P < 0.05.
We have shown that CD11c+ microglia in EAE express MHC-II and co-stimulatory molecules CD80 and CD86, and that these cells have potent ability to induce proliferation of antigen-primed CD4+ T cells (19). This has led to speculation that they may reflect a more DC-like subset of microglia, as was previously suggested, largely on the basis of morphology and CD11c expression (41). However, they differ from DC in their relative expression of Th1- and Th17-inducing cytokines and in their quantitative ability to induce such T cell responses, CD11c+ microglia being, noticeably, less effective (19). We have now further explored possible overlap between CD11c+ microglia and DC by comparison of their expression of the transcription factor T-bet. T-bet was first isolated as a novel Th1-specific T box transcription factor that controls the expression of the hallmark Th1 cytokine, IFNγ (42). T-bet is also expressed by other IFNγ-producing cells including CD8+ T cells and NK cells, and has been shown to be expressed by IgG2a+ memory B cells, where it plays a role in IFNγ-mediated class switching as well as controlling expression of the chemokine receptor CXCR3 [reviewed in Ref. (43)]. T-bet is classically associated with the Th1 CD4+ T cell subset but also controls IL23R expression by Th17 (44). It has been implicated in regulation of effector responses by CD8+ T cells (45, 46) and in control of Th-1 expression of granulocyte–macrophage colony-stimulating factor (GM-CSF or CSF2) (47), which has been proposed to be the link between pathogenic T cells and infiltrating myeloid cells (48). T-bet was reported to be required for Th1 and Th17 encephalitogenicity (49), although this has been contradicted by later studies (50). Earlier studies had shown that T-bet was also expressed by DC and regulated DC functions (45, 46), and although reported to play a role in IFNγ production by DC, that is unlikely to be a major activity of this cell type. Expression of T-bet by DC was critical for Th1 induction and for the generation of T cell memory (43). T-bet was later shown to play a role in DC control of mast cell precursor migration (51). We compared levels of T-bet mRNA in microglia and BMDM/DC isolated from the CNS of mice with EAE. Whereas CD45highCD11c+ BMDM/DC robustly expressed high levels of T-bet mRNA, it was undetectable in most microglial samples, regardless of CD11c expression (Figure 8A). The expression of T-bet is, therefore, a selective property of infiltrating cells, likely to be specific for DC based on previous reports, and does not occur in CNS-resident microglia.
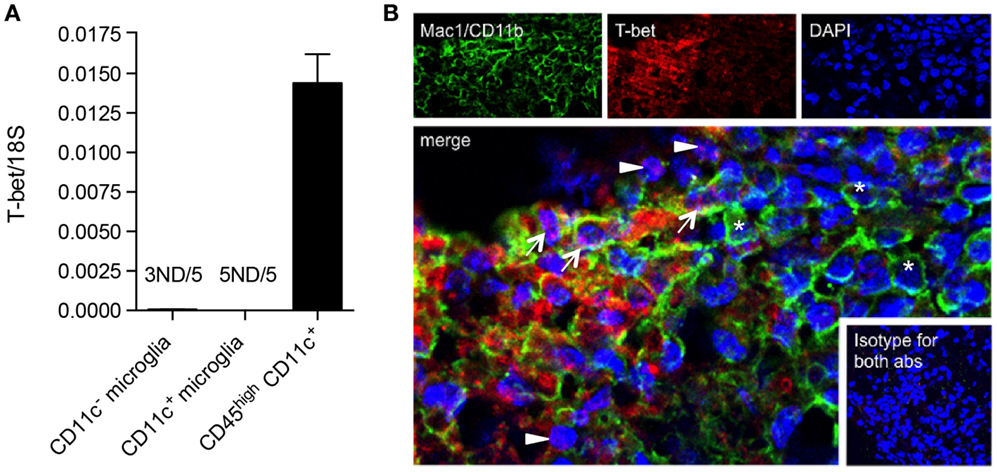
Figure 8. T-bet is expressed by infiltrating BMDM/DC but not by microglia in EAE. (A) Expression of T-bet in sorted myeloid cells (CD11c+ microglia, CD11c− microglia, and CD45highCD11c+) from the central nervous system from mice with severe EAE was analyzed by quantitative real-time PCR. Data are presented as means ± SEM of three individual experiments (n ≥ 5, where n represents a pool of 2–3 individual mice). ND, not detected. (B) Representative confocal microscopic analysis of spinal cord from mice with severe EAE for four individual experiments. Arrowheads point to T-bet (red) single positive cells, asterisks point to Mac1/CD11b (green) single positive cells, and arrows point to cells co-expressing T-bet marker with Mac1/CD11b.
Glimcher and colleagues showed that DC express T-bet (45). Microglial expression had not previously been reported and our findings, somewhat surprisingly given the many overlaps in myeloid cell phenotype and their involvement in Th1 induction and chemokine responses, indicate that, in fact, these brain macrophages do not express significant levels of T-bet. Thus, it was of interest that such a high proportion of T-bet-expressing cells in EAE infiltrates should turn out to be DC rather than T cells.
We were interested whether T-bet+ DC co-localized with microglial cells within EAE lesions. Using immunohistochemistry and immunofluorescence protocols, we showed that although some CD4+ cells co-stained with T-bet, as expected, the majority of T-bet+ cells did not express CD4 (data not shown), but stained for Mac-1/CD11b (Figure 8B). These represent the sorted CD45high BMDM/DC, which we showed to be the only myeloid cell source of T-bet mRNA (Figure 8A). EAE lesions are, therefore, highly heterogeneous, containing regulatory microglia with differential ability to present antigen to and induce cytokine responses from infiltrating T cells as well as infiltrating BMDM/DC that show no regulatory phenotype, many of which express T-bet.
All together, these data suggest that T-bet may be useful as a potential marker (along with myeloid markers, such as CD11b) for infiltrating DC in neuroinflammation, given that these populations of myeloid cells are morphologically indistinguishable.
We, thus, show that CNS-resident microglia that are maintained at least in part by proliferation include functionally distinct subsets that can be distinguished by expression of CD11c. These subsets differ in their expression of ARG1, YM1, iNOS, IL-10, and IGF1. These markers have been used as a basis for a M1/M2 phenotype classification for macrophages and were extended to describe microglia. Although both subpopulations of microglia clearly differed from each other, they did not show a pure M1 or M2 phenotype. This further supports the detailed transcriptional analysis of LPS-activated macrophages, which has revealed limits to such a binary distinction (52) and is in line with other studies where microglia have been shown to express M1 and M2-related genes simultaneously (53, 54). It is, thus, probably more useful to define phenotypic/functional correlates on a case-by-case basis as, for instance, in Ref. (55). The issue of M1/M2 microglial phenotype and its limitations have been thoroughly discussed in Ref. (39).
We further show that despite these phenotypic and functional distinctions, microglia differ from blood-derived or BMDM/DC in that they express the regulatory cytokine IFNβ, to which BMDM/DC may respond but do not express, and that BMDM/DC express the Th1-associated transcription factor T-bet, whereas microglia do not. The fact that DC and microglia co-localize within inflammatory lesions in EAE emphasizes that the microenvironment in which autoreactive T cells are activated is complex and includes both regulatory and pro-pathologic myeloid cells that themselves may exert effector functions in the inflamed CNS.
Over two decades of research on CD4+ T cell subsets based on their cytokine production profile has led to major increases in understanding of the inflammatory process. Myeloid cells in neuroinflammation also show heterogeneity and may need to be equivalently thoroughly studied in order to better describe their functions and their interaction with T cell subsets and neural cells in the CNS.
AW and TO designed the work. AW, KJ, AJ, JM, RK, and OC performed the experiments. AW, OC, and TO analyzed the data and wrote the paper. All authors read and approved the final manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
This work has been supported by grants to TO from Lundbeckfonden (2012-12307, 2014-717), NovoNordiskFonden (NNF13OC0006131), Scleroseforeningen (R399-A27689), and Region of Southern Denmark Health Research.
We thank Dina Dræby and Pia Nyborg Nielsen for expert technical assistance, Inger Andersen and Lars Vitved for help with cell sorting, Zsolt Illes and Justyna Okarmus for collaboration on cuprizone experiments. We are grateful to Nasrin Asgari (Department Neurology, Vejle Hospital, Denmark) for providing us with NMO-IgG and to Diego Gomez-Nicola (University of Southampton, UK) for providing anti-CD11c antibody. The bioimaging experiments reported in this paper were performed at DaMBIC, a bioimaging research core facility, at the University of Southern Denmark. DaMBIC was established by an equipment grant from the Danish Agency for Science Technology and Innovation and by internal funding from the University of Southern Denmark.
1. Gertig U, Hanisch UK. Microglial diversity by responses and responders. Front Cell Neurosci (2014) 8:101. doi:10.3389/fncel.2014.00101
2. Goverman J. Autoimmune T cell responses in the central nervous system. Nat Rev Immunol (2009) 9:393–407. doi:10.1038/nri2550
3. Chen SJ, Wang YL, Fan HC, Lo WT, Wang CC, Sytwu HK. Current status of the immunomodulation and immunomediated therapeutic strategies for multiple sclerosis. Clin Dev Immunol (2012) 2012:970789. doi:10.1155/2012/970789
4. Schwartz M, Moalem G, Leibowitz-Amit R, Cohen IR. Innate and adaptive immune responses can be beneficial for CNS repair. Trends Neurosci (1999) 22:295–9. doi:10.1016/S0166-2236(99)01405-8
5. Hvilsted Nielsen H, Toft-Hansen H, Lambertsen KL, Owens T, Finsen B. Stimulation of adult oligodendrogenesis by myelin-specific T cells. Am J Pathol (2011) 179:2028–41. doi:10.1016/j.ajpath.2011.06.006
6. Aloisi F, Ria F, Columba-Cabezas S, Hess H, Penna G, Adorini L. Relative efficiency of microglia, astrocytes, dendritic cells and B cells in naive CD4+ T cell priming and Th1/Th2 cell restimulation. Eur J Immunol (1999) 29:2705–14. doi:10.1002/(SICI)1521-4141(199909)29:09<2705::AID-IMMU2705>3.0.CO;2-1
7. Chabot S, Williams G, Hamilton M, Sutherland G, Yong VW. Mechanisms of IL-10 production in human microglia-T cell interaction. J Immunol (1999) 162:6819–28.
8. Matyszak MK, Denis-Donini S, Citterio S, Longhi R, Granucci F, Ricciardi-Castagnoli P. Microglia induce myelin basic protein-specific T cell anergy or T cell activation, according to their state of activation. Eur J Immunol (1999) 29:3063–76. doi:10.1002/(SICI)1521-4141(199910)29:10<3063::AID-IMMU3063>3.0.CO;2-G
9. Becher B, Blain M, Antel JP. CD40 engagement stimulates IL-12 p70 production by human microglial cells: basis for Th1 polarization in the CNS. J Neuroimmunol (2000) 102:44–50. doi:10.1016/S0165-5728(99)00152-6
10. Hanisch UK, Kettenmann H. Microglia: active sensor and versatile effector cells in the normal and pathologic brain. Nat Neurosci (2007) 10:1387–94. doi:10.1038/nn1997
11. Ginhoux F, Greter M, Leboeuf M, Nandi S, See P, Gokhan S, et al. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science (2010) 330:841–5. doi:10.1126/science.1194637
12. Ajami B, Bennett JL, Krieger C, Tetzlaff W, Rossi FM. Local self-renewal can sustain CNS microglia maintenance and function throughout adult life. Nat Neurosci (2007) 10:1538–43. doi:10.1038/nn2014
13. Sedgwick JD, Schwender S, Imrich H, Dorries R, Butcher GW, Ter Meulen V. Isolation and direct characterization of resident microglial cells from the normal and inflamed central nervous system. Proc Natl Acad Sci U S A (1991) 88:7438–42. doi:10.1073/pnas.88.16.7438
14. Ford AL, Goodsall AL, Hickey WF, Sedgwick JD. Normal adult ramified microglia separated from other central nervous system macrophages by flow cytometric sorting. Phenotypic differences defined and direct ex vivo antigen presentation to myelin basic protein-reactive CD4+ T cells compared. J Immunol (1995) 154:4309–21.
15. Perry VH, Hume DA, Gordon S. Immunohistochemical localization of macrophages and microglia in the adult and developing mouse brain. Neuroscience (1985) 15:313–26. doi:10.1016/0306-4522(85)90215-5
16. Giulian D, Baker TJ. Characterization of ameboid microglia isolated from developing mammalian brain. J Neurosci (1986) 6:2163–78.
17. Ito D, Imai Y, Ohsawa K, Nakajima K, Fukuuchi Y, Kohsaka S. Microglia-specific localisation of a novel calcium binding protein, Iba1. Brain Res Mol Brain Res (1998) 57:1–9. doi:10.1016/S0169-328X(98)00040-0
18. Cardona AE, Pioro EP, Sasse ME, Kostenko V, Cardona SM, Dijkstra IM, et al. Control of microglial neurotoxicity by the fractalkine receptor. Nat Neurosci (2006) 9:917–24. doi:10.1038/nn1715
19. Wlodarczyk A, Lobner M, Cedile O, Owens T. Comparison of microglia and infiltrating CD11c(+) cells as antigen presenting cells for T cell proliferation and cytokine response. J Neuroinflammation (2014) 11:57. doi:10.1186/1742-2094-11-57
20. Immig K, Gericke M, Menzel F, Merz F, Krueger M, Schiefenhovel F, et al. CD11c-positive cells from brain, spleen, lung, and liver exhibit site-specific immune phenotypes and plastically adapt to new environments. Glia (2015) 63:611–25. doi:10.1002/glia.22771
21. Mizutani M, Pino PA, Saederup N, Charo IF, Ransohoff RM, Cardona AE. The fractalkine receptor but not CCR2 is present on microglia from embryonic development throughout adulthood. J Immunol (2012) 188:29–36. doi:10.4049/jimmunol.1100421
22. Khorooshi R, Wlodarczyk A, Asgari N, Owens T. Neuromyelitis optica-like pathology is dependent on type I interferon response. Exp Neurol (2013) 247:744–7. doi:10.1016/j.expneurol.2013.02.005
23. Remington LT, Babcock AA, Zehntner SP, Owens T. Microglial recruitment, activation, and proliferation in response to primary demyelination. Am J Pathol (2007) 170:1713–24. doi:10.2353/ajpath.2007.060783
24. Khorooshi R, Owens T. Detection and cellular localization of phospho-STAT2 in the central nervous system by immunohistochemical staining. Methods Mol Biol (2013) 967:179–88. doi:10.1007/978-1-62703-242-1_13
25. Gomez-Nicola D, Fransen NL, Suzzi S, Perry VH. Regulation of microglial proliferation during chronic neurodegeneration. J Neurosci (2013) 33:2481–93. doi:10.1523/JNEUROSCI.4440-12.2013
26. Akiyama H, McGeer PL. Brain microglia constitutively express beta-2 integrins. J Neuroimmunol (1990) 30:81–93. doi:10.1016/0165-5728(90)90055-R
27. Bulloch K, Miller MM, Gal-Toth J, Milner TA, Gottfried-Blackmore A, Waters EM, et al. CD11c/EYFP transgene illuminates a discrete network of dendritic cells within the embryonic, neonatal, adult, and injured mouse brain. J Comp Neurol (2008) 508:687–710. doi:10.1002/cne.21668
28. Butovsky O, Kunis G, Koronyo-Hamaoui M, Schwartz M. Selective ablation of bone marrow-derived dendritic cells increases amyloid plaques in a mouse Alzheimer’s disease model. Eur J Neurosci (2007) 26:413–6. doi:10.1111/j.1460-9568.2007.05652.x
29. Nishiyori A, Minami M, Ohtani Y, Takami S, Yamamoto J, Kawaguchi N, et al. Localization of fractalkine and CX3CR1 mRNAs in rat brain: does fractalkine play a role in signaling from neuron to microglia? FEBS Lett (1998) 429:167–72. doi:10.1016/S0014-5793(98)00583-3
30. Wolf Y, Yona S, Kim KW, Jung S. Microglia, seen from the CX3CR1 angle. Front Cell Neurosci (2013) 7:26. doi:10.3389/fncel.2013.00026
31. Garcia JA, Pino PA, Mizutani M, Cardona SM, Charo IF, Ransohoff RM, et al. Regulation of adaptive immunity by the fractalkine receptor during autoimmune inflammation. J Immunol (2013) 191:1063–72. doi:10.4049/jimmunol.1300040
32. Hanisch UK. Functional diversity of microglia – how heterogeneous are they to begin with? Front Cell Neurosci (2013) 7:65. doi:10.3389/fncel.2013.00065
33. Mosser DM, Edwards JP. Exploring the full spectrum of macrophage activation. Nat Rev Immunol (2008) 8:958–69. doi:10.1038/nri2448
34. Butovsky O, Koronyo-Hamaoui M, Kunis G, Ophir E, Landa G, Cohen H, et al. Glatiramer acetate fights against Alzheimer’s disease by inducing dendritic-like microglia expressing insulin-like growth factor 1. Proc Natl Acad Sci U S A (2006) 103:11784–9. doi:10.1073/pnas.0604681103
35. Kocur M, Schneider R, Pulm AK, Bauer J, Kropp S, Gliem M, et al. IFNbeta secreted by microglia mediates clearance of myelin debris in CNS autoimmunity. Acta Neuropathol Commun (2015) 3:20. doi:10.1186/s40478-015-0192-4
36. Khorooshi R, Morch MT, Holm TH, Berg CT, Dieu RT, Draeby D, et al. Induction of endogenous type I interferon within the central nervous system plays a protective role in experimental autoimmune encephalomyelitis. Acta Neuropathol (2015) 130:107–18. doi:10.1007/s00401-015-1418-z
37. Compston A, Coles A. Multiple sclerosis. Lancet (2008) 372:1502–17. doi:10.1016/S0140-6736(08)61620-7
38. Yamasaki R, Lu H, Butovsky O, Ohno N, Rietsch AM, Cialic R, et al. Differential roles of microglia and monocytes in the inflamed central nervous system. J Exp Med (2014) 211:1533–49. doi:10.1084/jem.20132477
39. Biber K, Owens T, Boddeke E. What is microglia neurotoxicity (Not)? Glia (2014) 62:841–54. doi:10.1002/glia.22654
40. Elmore MR, Najafi AR, Koike MA, Dagher NN, Spangenberg EE, Rice RA, et al. Colony-stimulating factor 1 receptor signaling is necessary for microglia viability, unmasking a microglia progenitor cell in the adult brain. Neuron (2014) 82:380–97. doi:10.1016/j.neuron.2014.02.040
41. Fischer HG, Reichmann G. Brain dendritic cells and macrophages/microglia in central nervous system inflammation. J Immunol (2001) 166:2717–26. doi:10.4049/jimmunol.166.4.2717
42. Szabo SJ, Kim ST, Costa GL, Zhang X, Fathman CG, Glimcher LH. A novel transcription factor, T-bet, directs Th1 lineage commitment. Cell (2000) 100:655–69. doi:10.1016/S0092-8674(00)80702-3
43. Lazarevic V, Glimcher LH, Lord GM. T-bet: a bridge between innate and adaptive immunity. Nat Rev Immunol (2013) 13:777–89. doi:10.1038/nri3536
44. Gocke AR, Cravens PD, Ben LH, Hussain RZ, Northrop SC, Racke MK, et al. T-bet regulates the fate of Th1 and Th17 lymphocytes in autoimmunity. J Immunol (2007) 178:1341–8. doi:10.4049/jimmunol.178.3.1341
45. Lugo-Villarino G, Maldonado-Lopez R, Possemato R, Penaranda C, Glimcher LH. T-bet is required for optimal production of IFN-gamma and antigen-specific T cell activation by dendritic cells. Proc Natl Acad Sci U S A (2003) 100:7749–54. doi:10.1073/pnas.1332767100
46. Sullivan BM, Juedes A, Szabo SJ, Von Herrath M, Glimcher LH. Antigen-driven effector CD8 T cell function regulated by T-bet. Proc Natl Acad Sci U S A (2003) 100:15818–23. doi:10.1073/pnas.2636938100
47. Cravens PD, Hussain RZ, Zacharias TE, Ben LH, Herndon E, Vinnakota R, et al. Lymph node-derived donor encephalitogenic CD4+ T cells in C57BL/6 mice adoptive transfer experimental autoimmune encephalomyelitis highly express GM-CSF and T-bet. J Neuroinflammation (2011) 8:73. doi:10.1186/1742-2094-8-73
48. Codarri L, Greter M, Becher B. Communication between pathogenic T cells and myeloid cells in neuroinflammatory disease. Trends Immunol (2013) 34:114–9. doi:10.1016/j.it.2012.09.007
49. Yang Y, Weiner J, Liu Y, Smith AJ, Huss DJ, Winger R, et al. T-bet is essential for encephalitogenicity of both Th1 and Th17 cells. J Exp Med (2009) 206:1549–64. doi:10.1084/jem.20082584
50. Spath S, Becher B. T-bet or not T-bet: taking the last bow on the autoimmunity stage. Eur J Immunol (2013) 43:2810–3. doi:10.1002/eji.201344109
51. Alcaide P, Jones TG, Lord GM, Glimcher LH, Hallgren J, Arinobu Y, et al. Dendritic cell expression of the transcription factor T-bet regulates mast cell progenitor homing to mucosal tissue. J Exp Med (2007) 204:431–9. doi:10.1084/jem.20060626
52. Reynier F, De Vos AF, Hoogerwerf JJ, Bresser P, Van Der Zee JS, Paye M, et al. Gene expression profiles in alveolar macrophages induced by lipopolysaccharide in humans. Mol Med (2012) 18:1303–11. doi:10.2119/molmed.2012.00230
53. Mantovani A, Sica A, Locati M. Macrophage polarization comes of age. Immunity (2005) 23:344–6. doi:10.1016/j.immuni.2005.10.001
54. Ajmone-Cat MA, Mancini M, De Simone R, Cilli P, Minghetti L. Microglial polarization and plasticity: evidence from organotypic hippocampal slice cultures. Glia (2013) 61:1698–711. doi:10.1002/glia.22550
Keywords: microglia, EAE, CD11c, myeloid cells, CNS, M1/M2
Citation: Wlodarczyk A, Cédile O, Jensen KN, Jasson A, Mony JT, Khorooshi R and Owens T (2015) Pathologic and protective roles for microglial subsets and bone marrow- and blood-derived myeloid cells in central nervous system inflammation. Front. Immunol. 6:463. doi: 10.3389/fimmu.2015.00463
Received: 03 July 2015; Accepted: 25 August 2015;
Published: 08 September 2015
Edited by:
Attila Szabo, University of Debrecen, HungaryReviewed by:
Jennifer Gommerman, University of Toronto, CanadaCopyright: © 2015 Wlodarczyk, Cédile, Jensen, Jasson, Mony, Khorooshi and Owens. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Trevor Owens, Department of Neurobiology Research, Institute for Molecular Medicine, University of Southern Denmark, J.B Winslowsvej 25, Odense DK-5000, Denmark,dG93ZW5zQGhlYWx0aC5zZHUuZGs=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.