
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 14 September 2015
Sec. Microbial Immunology
Volume 6 - 2015 | https://doi.org/10.3389/fimmu.2015.00447
This article is part of the Research Topic Bacterial Exotoxins: how bacteria fight the immune system View all 17 articles
 Sylvia Simon1
Sylvia Simon1 Hubert Hilbi1,2*
Hubert Hilbi1,2*
Bacteria trigger host defense and inflammatory processes, such as cytokine production, pyroptosis, and the chemotactic migration of immune cells toward the source of infection. However, a number of pathogens interfere with these immune functions by producing specific so-called “effector” proteins, which are delivered to host cells via dedicated secretion systems. Air-borne Legionella pneumophila bacteria trigger an acute and potential fatal inflammation in the lung termed Legionnaires’ disease. The opportunistic pathogen L. pneumophila is a natural parasite of free-living amoebae, but also replicates in alveolar macrophages and accidentally infects humans. The bacteria employ the intracellular multiplication/defective for organelle trafficking (Icm/Dot) type IV secretion system and as many as 300 different effector proteins to govern host–cell interactions and establish in phagocytes an intracellular replication niche, the Legionella-containing vacuole. Some Icm/Dot-translocated effector proteins target cell-autonomous immunity or cell migration, i.e., they interfere with (i) endocytic, secretory, or retrograde vesicle trafficking pathways, (ii) organelle or cell motility, (iii) the inflammasome and programed cell death, or (iv) the transcription factor NF-κB. Here, we review recent mechanistic insights into the subversion of cellular immune functions by L. pneumophila.
Phagocytic cells of the innate immune system, such as macrophages, neutrophils, and dendritic cells (DC), produce pathogen recognition receptors (PRRs) comprising families of membrane-bound or cytosolic receptors (1–4). The membrane-bound receptors are toll-like receptors (TLRs) and C-type lectin receptors (CLRs), such as the mannose receptors. TLRs trigger cytokine production by signaling through the adaptor molecules MyD88 or TRIF and activate the transcription factor NF-κB or MAP kinase pathways. Cytosolic receptors are Nod-like receptors (NLRs) involved in inflammatory responses and cell death pathways through the activation of multiprotein complexes termed inflammasomes (5), as well as retinoic acid inducible gene-I (RIG-I)-like receptors (RLRs). Collectively, these receptors recognize pathogen-associated molecular patterns (PAMPs) and danger-associated molecular patterns (DAMPs), thus alerting the infected host cell, if material arising from cell infection or cell damage is present. Subsequent cytokine production promotes the clearance of invading microorganisms. The conserved process of cell-autonomous immunity includes not only inflammasome activation and interleukin (IL)-1β production, as well as autophagy, but also NF-κB- and type I interferon (IFN)-dependent cytokine production (6, 7).
Pathogenic bacteria target eukaryotic cells either as adversaries in the case of potentially bactericidal cells of the metazoan immune system and/or as a rich source of nutrients (8, 9). Accordingly, pathogens developed means to counteract the arsenal of the humoral and cellular components of the innate and acquired immune system and to survive and replicate within eukaryotic cells, including phagocytes (10). To this end, a number of pathogens produce specific so-called “effector” proteins, which are delivered via dedicated secretion systems into host cells, where they interfere with immune functions and cell migration (11).
Legionella pneumophila employs the intracellular multiplication/defective for organelle trafficking (Icm/Dot) type IV secretion system (T4SS) and as many as 300 different effector proteins to govern host–cell interactions. The role and molecular mode of action of effectors involved in pathogen vacuole formation has recently been reviewed in detail (12). Here, we will review mechanistic insights into the subversion of cell-autonomous immunity and cell migration by L. pneumophila.
Legionella pneumophila is an opportunistic pathogen and causes a severe pneumonia termed Legionnaires’ disease. The Gram-negative genus Legionella comprises more than 55 species with several serogroups; yet, at least 85% of human infections are caused by L. pneumophila (13). Evolutionary adaptation allows L. pneumophila to persist in a variety of extra- and intracellular niches. The aerobic bacterium can not only replicate in biofilms but also resists degradation by free-living protozoa and replicates within, e.g., Acanthamoeba, Hartmannella, and Tetrahymena species, as well as in Dictyostelium discoideum, although the latter amoeba is likely not a natural host (14, 15). L. pneumophila is ubiquitously found in natural and technical water systems, including cooling towers, whirlpools, and showers.
Upon inhalation of contaminated aerosols, L. pneumophila resists degradation and replicates within alveolar macrophages, which is a precondition for the onset of disease (16). The acquisition of the pathogen from environmental sources is the only infection route; transmission between humans has never been observed. Since L. pneumophila probably mainly evolved as a parasite of free-living protozoa, the human host represents a dead-end for this “accidental” pathogen. Thus, L. pneumophila likely has not been exposed to a rigorous evolutionary selection to avoid recognition by mammalian PRRs, and accordingly, the bacteria trigger the activation of all PRR families (17).
Most humans and mice are able to clear a Legionella infection, and therefore, the development of a suitable small animal model was crucial. Initial studies using guinea pigs exposed to L. pneumophila-containing aerosols revealed a high susceptibility of these animals, which developed an illness reminiscent of typical human Legionnaires’ disease (18). While most inbred mouse strains are resistant to L. pneumophila infection and disease progression, the A/J mouse strain was found to be susceptible and to present with acute pneumonia that resembled human disease (19). These results correspond to in vitro infections of peritoneal mouse macrophages, which indicated that cells from A/J mice were much more permissive for intracellular replication of L. pneumophila than macrophages from other mouse strains, such as C57BL/6 and BALB/c (20, 21). Macrophages from C57BL/6 and BALB/c mice restrict L. pneumophila by the activation of a programed cell death pathway as an ultimate line of defense against the intracellular pathogen (see below). Accordingly, mice lacking components of this pathway fail to restrict L. pneumophila and faithfully mimic Legionella pathology (17, 22).
In A/J mice, L. pneumophila elicits an acute inflammatory reaction, including production of the cytokines tumor necrosis factor (TNF)-α, IFN-γ, IL-12, and IL-18, which restrict pathogen replication (17, 22). IFN-γ is particularly important to inhibit bacterial growth in monocytes and alveolar macrophages, thus contributing to limiting the infection by L. pneumophila (23–25). These inflammatory cytokines recruit and activate polymorphonuclear neutrophil granulocytes (PMNs) (26–28). PMNs are central innate effector cells that not only resolve L. pneumophila infection but – in concert with IFN-γ-producing natural killer (NK) cells – also secrete cytokines, such as IL-18 (29, 30). In a feedback loop, IFN-γ triggers IL-12 production by DC, which activate NK cells. Therefore, DC are also essential to control L. pneumophila infection (31). Interestingly, DC restrict the intracellular growth of L. pneumophila, despite that the pathogen resides in an apparently non-bactericidal compartment derived from the endoplasmic reticulum (ER) (32).
In permissive macrophages as well as in protozoa, L. pneumophila employs a complex and apparently evolutionarily conserved mechanism to establish its replication-permissive membrane-bound niche, the Legionella-containing vacuole (LCV) (Figure 1). LCV formation is governed by the Icm/Dot T4SS, which translocates approximately 300 different effector proteins into the host cell (12, 33–37). Since L. pneumophila–host interactions are defined to a large extent by the Icm/Dot apparatus, the T4SS represents a major virulence factor of L. pneumophila.
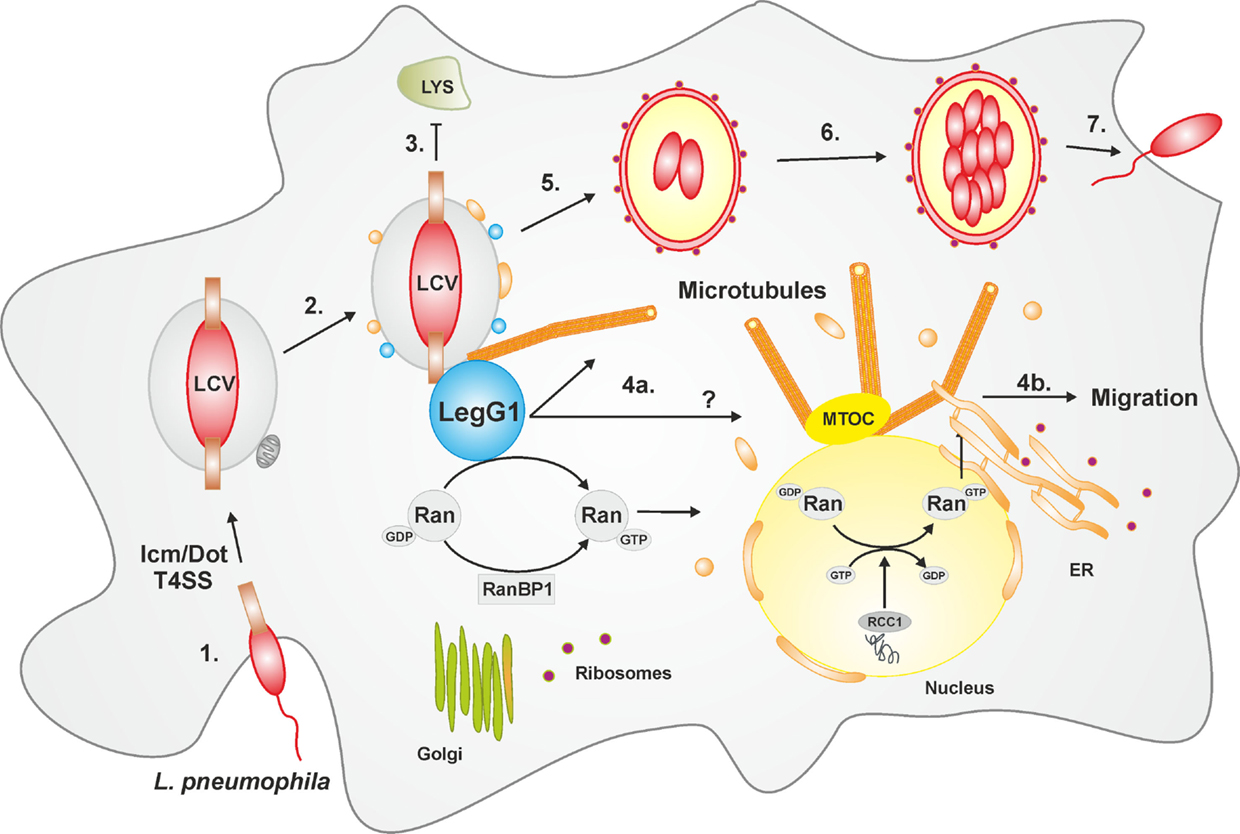
Figure 1. LCV formation and LegG1-dependent modulation of cell migration. The intracellular phases of L. pneumophila can be divided in seven main steps: (1) Adhesion and entry into the host cell via macropinocytosis; (2) Formation of the LCV in an Icm/Dot T4SS-dependent manner and recruitment of vesicles from the ER as well as mitochondria; (3) Evasion from the lysosomal trafficking; (4a) The bacterial effector protein LegG1 activates the GTPase Ran, stabilizes microtubules in the vicinity of the LCV or possibly at a distance (?), and (4b) promotes cell migration. In the nucleus, Ran activation is triggered by the eukaryotic GEF RCC1. (5) The LCV communicates with host vesicle trafficking pathways, and acquires and eventually fuses with the ER; (6) The pathogen compartment turns in a rough-ER-like vacuole, wherein the bacteria replicate; (7) L. pneumophila is released and re-infects new host cells or is transmitted to other environmental niches. Abbreviations: ER, endoplasmic reticulum; LCV, Legionella-containing vacuole; MTOC, microtubule organizing center; Ran, Ras-related nuclear protein; RCC1, regulator of chromosome condensation 1; T4SS, type IV secretion system.
The Icm/Dot T4SS and translocated effectors control every step in the infection process of L. pneumophila (Figure 1), i.e., the uptake (38, 39), inhibition of fusion with lysosomes (40) and acidification (41), subversion of retrograde trafficking (42), as well as interception of secretory vesicle trafficking (43), coalescence with the ER (44–46), and finally, egress from the host cell (47). To interfere with host cell processes, many L. pneumophila effector proteins target pivotal components of eukaryotic membrane dynamics, such as phosphoinositide (PI) lipids and small GTPases (12, 33, 37). The PI lipids PtdIns(4)P and PtdIns(3)P control the secretory and the endosomal pathway, respectively, while the small GTPases Arf1 and Rab1 are pivotal regulators of ER-Golgi secretory trafficking.
Some Icm/Dot substrates subvert PI lipids by using PtdIns(4)P and PtdIns(3)P as membrane anchors, both of which are found on LCVs (33, 48–50). Accordingly, the effectors SidC, SdcA, SidM, LidA, Lpg1101, and Lpg2603 bind PtdIns(4)P (51–57), and the effectors LidA, LptD, RidL, SetA, and LpnE bind PtdIns(3)P (42, 52, 58–60). Two L. pneumophila “CX5R” domain PI phosphatases have been identified: SidF, a PI 3-phosphatase that hydrolyzes PtdIns(3,4)P2 [and also PtdIns(3,4,5)P3] in vitro (61), and SidP, a PI 3-phosphatase that hydrolyzes PtdIns(3)P as well as PtdIns(3,5)P2 in vitro (62). Thus, these PI phosphatases might cause PtdIns(4)P to be formed on and PtdIns(3)P to be removed from LCV membranes. LppA is another Icm/Dot substrate that hydrolyzes polyphosphorylated PIs to mainly produce PtdIns(4)P in vitro (49). However, LppA does not affect the LCV PI pattern in infected cells, but rather functions as a translocated hexakisphosphate inositol phosphatase (phytase), which possibly promotes intracellular replication of L. pneumophila by removing the intracellular micronutrient chelator phytate.
Intracellular multiplication/defective for organelle trafficking T4SS substrates modulate the activity of the small GTPases Arf1 (RalF) (63) or Rab1 (SidM) (55, 64–67) by acting as guanine nucleotide exchange factors (GEFs), GTPase-activating proteins (GAPs; LepB) (68), AMPylase/deAMPylase (SidM, SidD) (69–72), or phosphocholinase/dephosphocholinase (AnkX, Lem3) (73–77). The covalent modifications of Rab1 by AMP or phosphocholine cause a prolonged activation of the GTPase, by preventing its inhibition by the GAP LepB. Similarly, the Icm/Dot substrate LidA binds to activated Rab1 and prevents inactivation by LepB, as well as covalent modifications by SidM or AnkX (64, 78–80). Finally, the PtdIns(4)P-binding effector SidC and its paralog SdcA promote the monoubiquitination of Rab1, in agreement with their function as an E3 ubiquitin ligase containing a catalytic Cys–His–Asp triad (81–83). In summary, after their translocation a number of L. pneumophila Icm/Dot substrates employ PI lipids on the cytoplasmic side of the LCV membrane as membrane anchors. Thus, PI lipids determine at least in part the subcellular localization of the effectors, which show different biochemical activities, including subversion of small GTPases, ubiquitinylation, or binding to and modulation of vesicle trafficking machinery.
Retrograde trafficking in eukaryotes comprises the transport from early or late endosomes through the trans-Golgi network (TGN) to the ER (84). The cation-independent mannose 6-phosphate receptor (CI-MPR) is a well-documented substrate of the retrograde transport pathway. CI-MPR binds mannose present on hydrolases and transports the cargo destined for the endocytic pathway from the TGN to the endosomal system. After releasing the cargo in the lysosomal lumen, the receptor is recycled back through the retrograde pathway to the TGN (85). Proper recycling requires a protein complex termed the retromer, composed of a cargo recognition subcomplex (Vps26, Vps29, and Vps35) and a membrane-deforming subcomplex, which consists of a dimer of sorting nexins (SNXs) (86). The cargo recognition complex is recruited to the membrane by the activated small GTPases Rab5 and Rab7 (87).
Recent findings indicate that L. pneumophila communicates with the retrograde trafficking pathway. The small GTPases Rab5 and Rab7 (88–91), as well as the retromer subunits Vps26, Vps29, and Vps35 (42, 92) localize to the LCV. Interestingly, the Icm/Dot T4SS-translocated substrate RidL functions as a bacterial interactor of the eukaryotic retromer complex (42). RidL, which localizes on LCVs in D. discoideum cells and macrophages, binds to the Vps29 subunit of the cargo recognition subcomplex of the retromer. Subunits of the retromer cargo recognition complex localize to the LCV in an Icm/Dot-dependent but RidL-independent manner. RidL competes with SNX1 and SNX2 in its capacity for membrane binding specifically through PtdIns(3)P, thus possibly triggering the removal of SNXs. By altering the retrograde trafficking cascade, RidL might promote the formation of a non-lysosomal replicative vacuole and intracellular replication of L. pneumophila in protozoan and metazoan phagocytes (42). Earlier work already indicated that the retrograde pathway might control intracellular replication of L. pneumophila. The PtdIns(4,5)P2 5-phosphatase OCRL1 and its D. discoideum homolog Dd5P4 are implicated in retrograde trafficking (93, 94) and localize to LCVs (60). Depletion of OCRL by RNA interference (42) or deletion of Dd5P4 (60) increased intracellular growth of L. pneumophila, indicating that the PI 5-phosphatase OCRL/Dd5P4 indeed restricts pathogen replication.
Autophagy is a major cell-autonomous defense mechanism used by infected cells against intracellular bacteria. The process of macroautophagy is responsible for the degradation of cytoplasmic constituents, such as bacteria or damaged organelles, which are engulfed by autophagosomes and subsequently fuse with lysosomes (95). In the course of microautophagy, the constituents are directly delivered to the lysosomes. Over 30 autophagy-related genes (Atg) have been discovered during the last years (96). An Atg protein essential for autophagy is the microtubule-associated protein light chain 3 (LC3, alias Atg8), which is conjugated to phosphatidylethanolamine and localized on autophagosomal membranes (97). Measurement of LC3/Atg8 during L. pneumophila infection revealed an Icm/Dot-dependent inhibition of autophagy by the bacteria. Intriguingly, the bacterial protein RavZ was identified as an effector required for autophagy inhibition, yet further effectors could be involved, since L. pneumophila lacking ravZ still evaded the autophagy cascade (98). RavZ is a cysteine protease, which irreversibly deconjugates phosphatidylethanolamine from LC3/Atg8, thus reducing its membrane accumulation and activity. While most autophagy factors are not necessary for L. pneumophila replication in D. discoideum (99), amoebae lacking Atg9 do not internalize the pathogen as efficient as wild-type cells, yet allow more effective intracellular growth (100). In summary, these findings suggest that L. pneumophila inhibits retrograde vesicle trafficking as well as autophagy to promote intracellular growth.
The Icm/Dot T4SS is crucially involved in the formation of the LCV, and many L. pneumophila effectors selectively decorate the pathogen vacuole membrane. Intact LCVs can be purified by a straight-forward two-step protocol involving immuno-affinity enrichment using an antibody against the L. pneumophila effector SidC selectively decorating the pathogen vacuole and a secondary antibody coupled to magnetic beads, followed by a conventional density centrifugation step. Proteomics analysis of purified preparations of LCVs from infected D. discoideum amoebae (91) and RAW 264.7 macrophages (90) revealed the presence of 670 and 1150 host proteins, respectively, including 13 small GTPases of the Rab family, as well as the small GTPase Ran and its effector Ran binding protein 1 (RanBP1).
Ran is a member of the Ras superfamily of small GTPases and is fundamental in numerous cellular processes, such as nuclear pore translocation (101), or mitotic spindle assembly and post-mitotic nuclear envelope formation (102, 103). Ran GTPase also plays an important role in cytoplasmic processes involving non-centrosomal microtubules, e.g., endocytic receptor trafficking and retrograde signaling along microtubules in nerve axons (104). Ran can be activated by a nuclear (or in mitotic cells: chromatin bound) Ran GEF termed regulator of chromosome condensation 1 (RCC1) (105). Ran(GTP) is inactivated by the cytoplasmic Ran GTPase-activating protein 1 (RanGAP1) together with RanBP1, which harbors a Ran(GTP)-binding domain (104).
Intriguingly, L. pneumophila produces an Icm/Dot-translocated effector termed LegG1 that harbors eukaryotic-like RCC1 domains (106, 107). The corresponding gene is conserved among the L. pneumophila genomes sequenced to date, including the strains Philadelphia-1, Paris, Lens, Corby, Alcoy, and Lorraine. LegG1 (Lpg1976/PieG) is encoded in the plasticity island of effectors (Pie) gene cluster (108). The C-terminal CAAX tetrapeptide motif of LegG1 is lipidated by the host prenylation machinery (109), thus promoting the membrane localization of the effector to small vesicular structures upon ectopic expression in eukaryotic cells (108).
Recent work revealed that LegG1 is indeed an L. pneumophila virulence factor, which promotes intracellular bacterial replication but is dispensable for uptake (110) (Figure 1). A ΔlegG1 mutant strain is not less cytotoxic than the parental strain, but outcompeted by wild-type L. pneumophila upon co-infection of A. castellanii amoebae. Moreover, Ran, RanBP1, and LegG1 accumulate in an Icm/Dot-dependent manner on the LCV, and the bacterial effector localizes to the cytosolic face of the LCV in L. pneumophila-infected phagocytes. L. pneumophila wild-type but not the ΔlegG1 mutant strain activates Ran on LCVs and in cell lysates; yet, while LegG1 promotes the accumulation of RanBP1 on LCVs, the effector is dispensable for the recruitment of the small GTPase. Several experimental approaches indicated that in infected phagocytes L. pneumophila triggers the polymerization of microtubules in a LegG1-dependent manner (110) (Figure 1). “Microbial microinjection” of LegG1 by Yersinia enterocolitica confirmed the positive effect of LegG1 on microtubule stabilization. Here, Y. enterocolitica harboring a T3SS, but lacking endogenous effectors, translocated LegG1 fused to a type III secretion signal into HeLa cells. Furthermore, while LCVs harboring wild-type L. pneumophila vividly move along microtubules in infected D. discoideum, the pathogen vacuole harboring ΔlegG1 mutant bacteria is stalled (110). In summary, the discovery and characterization of the L. pneumophila Ran activator LegG1 revealed an unexpected role of the small GTPase Ran in the formation of pathogen vacuoles.
Microtubule polarization and dynamics represent pivotal determinants of eukaryotic cell migration (111) (Figure 2). Given the prominent role of Ran and LegG1 on the dynamics of the microtubule cytoskeleton, we investigated the effect of L. pneumophila and LegG1 on host cell motility (112). Studies using D. discoideum amoebae or immune cells, such as RAW 264.7 macrophage-like cells or primary PMN, in different migration assays (under-agarose and Boyden chamber assays), revealed an Icm/Dot-dependent inhibition of migration. Phagocytes infected with wild-type L. pneumophila or Legionella longbeachae showed a substantially reduced migration when compared to cells lacking a functional Icm/Dot T4SS. Uptake and cytotoxicity assays demonstrated that the observed effect is not due to a defect in infection.
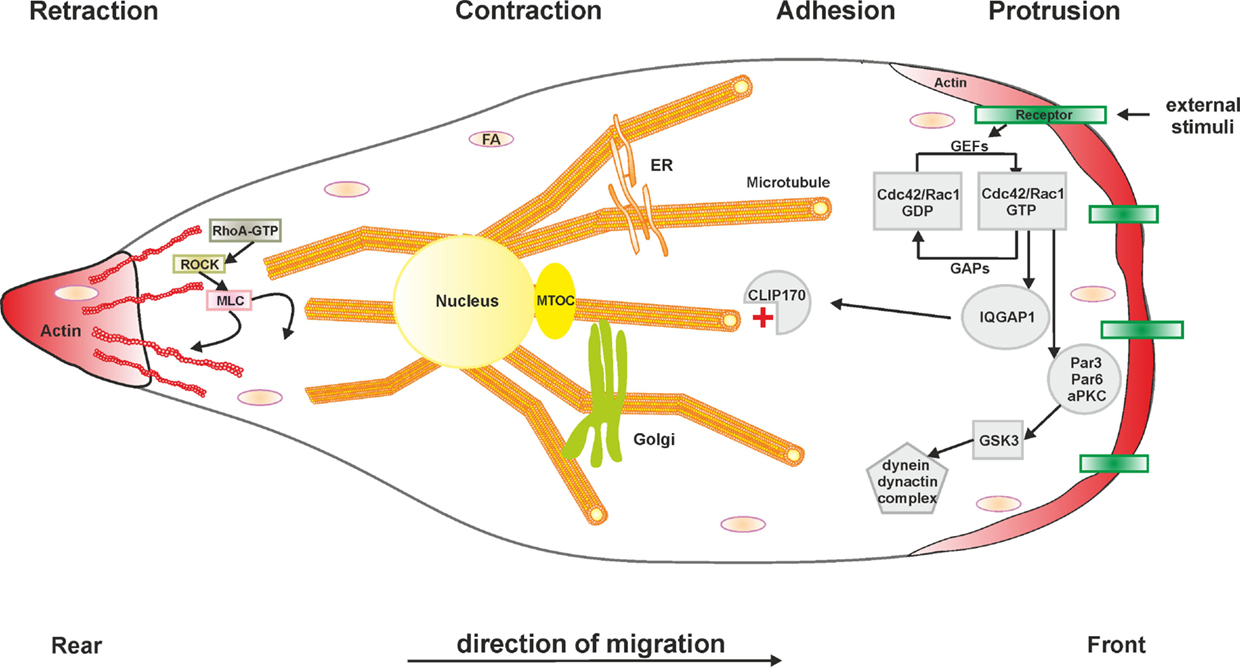
Figure 2. Schematic representation of cell migration. Generally, the cellular migration cycle can be divided into four major steps: (1) protrusion at the leading edge, (2) cell adhesion, (3) contraction to generate the required forces, and (4) retraction at the rear edge. Microtubules and the actin cytoskeleton in concert with Rho GTPases are fundamental components of each phase. Abbreviations: aPKC, atypical protein kinase C; Cdc42, cell division control protein 42; CLIP170, cytoplasmic linker protein 170; FA, focal adhesion; GAP, GTPase-activating protein; GEF, guanine nucleotide exchange factor; GSK3, glycogen synthase kinase 3; IQGAP1, IQ motif-containing GTPase-activating protein 1; MLC, myosin light chain; MTOC, microtubule organizing center; Par3/6, partitioning defective 3/6 homolog; Rac1, Ras-related C3 botulinum toxin substrate 1; RhoA, Ras homolog gene family member A; ROCK, Rho-associated protein kinase. The L. pneumophila effector protein LegG1 promotes cell migration by activating the small GTPase Ran and stabilizing microtubules. Other L. pneumophila factors modulating cell migration are not known.
Based on these findings, the modulation of cell migration by LegG1 was assessed in D. discoideum as well as in RAW 264.7 macrophages or PMN (112) (Figure 1). Interestingly, the ΔlegG1 mutant strain hyper-inhibited the directed migration of phagocytes in the under-agarose assay, even to a larger extent than wild-type L. pneumophila. Overproduction of LegG1 in the ΔlegG1 mutant background re-established the migration range to an extent comparable to cells infected with a ΔicmT mutant strain. Single cell tracking revealed that the forward migration and the velocity of cells infected with wild-type or ΔlegG1 L. pneumophila was impaired. Similarly, upon “microbial microinjection” by Y. enterocolitica, LegG1 was sufficient to stimulate migration of epithelial cells in scratch assays. Moreover, using RNA interference a role of Ran in the LegG1-dependent migration inhibition was demonstrated. Upon depletion of Ran, cells infected with the strain overproducing LegG1 were not able to migrate and to close scratch wounds over time (112). Taken together, the Ran activator LegG1 promotes cell motility by modulating microtubule dynamics and thus antagonizes Icm/Dot-dependent inhibition of cell migration. LegG1 might reverse the otherwise deleterious impact of other L. pneumophila effectors on the host cytoskeleton, and thereby sustain vesicle trafficking and organelle motility required for the establishment and maintenance of LCVs.
Maintenance of LCV integrity and regulation of programed cell death is critical for preserving the intracellular replication niche of L. pneumophila (113). Accordingly, impaired activation of programed cell death turned out to account for the failure of A/J mice to restrict L. pneumophila. The allele conferring sensitivity against L. pneumophila was mapped to the NAIP5 gene (114, 115) within the Lgn1 locus (116). The Naip5 (Birc1e)/Nlrc4 (Ipaf) inflammasome recognizes flagellin and triggers caspase-1 activation, pore formation, and pyroptosis (117–125). Naip5/Nlrc4 inflammasome activation represents a crucial mechanism of L. pneumophila restriction. In agreement with this notion, macrophages as well as DC restrict L. pneumophila replication through a cell death pathway mediated by Naip5, caspase-1, and caspase-3 (126). Yet, neither inflammasomes nor caspases are conserved in amoebae (127).
In addition to PAMPs, such as flagellin, a number of specific Icm/Dot substrates are implicated in the regulation of programed host cell death. The L. pneumophila PI 3-phosphatase SidF inactivates the pro-apoptotic factors BNIP3 and Bcl-rambo by an unknown mechanism, and thereby counteracts cell death induction (128). Furthermore, the effector SdhA plays a role in maintaining the LCV membrane integrity and also contributes to the prevention of cell death (129, 130). Cell death induction in absence of sdhA is suppressed by a secreted bacterial phospholipase A through an unknown mechanism. L. pneumophila lacking sdhA resides in the cytoplasm and triggers caspase-1 activation and IL-1β secretion, as well as macrophage pyroptosis through the DNA-sensing AIM2 inflammasome (131). The Icm/Dot substrate SdhA is also a key suppressor of the type I IFN (IFN-α/β) response to L. pneumophila through nucleic acid-sensing PRRs. Accordingly, RNA from L. pneumophila lacking sdhA triggers the RIG-I-dependent production of type I IFNs (132).
Legionella pneumophila also promotes programed cell death in an Icm/Dot-dependent manner. The phospholipase VipD destabilizes mitochondrial membranes by means of its phospholipase A activity, thus potentially contributing to the release of cytochrome c into the cytosol. This triggers pro-apoptotic caspase-3 activation and subsequently promotes host cell death (133). Analogously, the Icm/Dot-translocated phospholipase PlcC hydrolyzes several lipids, including phosphatidylcholine, phosphatidylglycerol, and phosphatidylinositol, which might destabilize target membranes and cause cell toxicity (134). Stimulation of apoptosis by an intracellular pathogen seems counterintuitive, but might reflect a tight spatial and temporal control of LCV maturation, followed by the release of the bacteria from the pathogen compartment and the host cell at the end of an infection cycle. Thus, the elaborate coordination of anti- and pro-apoptotic factors optimally supports intracellular bacterial proliferation.
Finally, caspases not only regulate cell death during L. pneumophila infection but also control vesicle trafficking pathways and thus contribute to the formation of LCVs. Dependent on the Nlrc4 inflammasome and Naip5, L. pneumophila activates caspase-7 downstream of caspase-1, and consequently, the pathogen is delivered to lysosomes (117, 135). In turn, upon deletion of the caspases, L. pneumophila forms an ER-derived replicative compartment. Similarly (but independently of caspase-1), active caspase-11 restricts the replication of L. pneumophila by promoting the fusion of the pathogen compartment with lysosomes (136).
The transcription factor NF-κB is a master regulator of the mammalian innate immune response and controls the production of anti-apoptotic and pro-survival factors as well as inflammatory mediators (137). Thus, the activation of NF-κB (which is not conserved in amoebae) represents another, more indirect way to prevent host cell death. NF-κB is composed of five Rel family proteins: RelA, RelB, c-Rel, and the precursors p100 and p105, which are processed to their mature forms, p52 and p50. The transcription factor is maintained in its inactive form in the cytoplasm by a family of inhibitors termed IκBs (IκBa, IκBb, IκBe, and Bcl-3).
Bacterial PAMPs, including LPS or flagellin, initiate the NF-κB cascade through TLR2 or TLR5 and the adaptor MyD88. L. pneumophila triggers the production of pro-inflammatory cytokines, such as IL-1β, and the activation of the inflammasome platform through these receptors (138–141). The deletion of single TLRs does not dramatically alter L. pneumophila infection; yet, mice lacking MyD88 fail to produce cytokines, such as NK cell-derived IFN-γ, and are highly susceptible to L. pneumophila infection (142–144, 29). Moreover, mice lacking the IL-1 receptor are impaired for PMN recruitment and bacterial clearance (145). Therefore, IL-1R/MyD88-dependant signaling is critical for host resistance to L. pneumophila infection.
The NF-κB pathway can be activated not only by bacterial PAMPs via a TLR-dependent pathway but also by distinct bacterial effectors (146). The Icm/Dot T4SS and its substrates LnaB and LegK1 strongly induce NF-κB (147–149). While the molecular mechanism of the novel effector LnaB is not understood, LegK1 is a Ser/Thr kinase that phosphorylates the NF-κB inhibitor IκBα, leading to a robust NF-κB activation by triggering the release and nuclear translocation of the transcription factor (150). Since L. pneumophila lacking legK1 is not impaired for intracellular replication, other effectors might also modulate the NF-κB response. Among the five L. pneumophila Icm/Dot substrates that show protein kinase activity in vitro (LegK1–LegK5), LegK2 is a virulence factor, which promotes intracellular replication and efficient recruitment of the ER to LCVs (151).
Translation inhibition by the Icm/Dot substrates Lgt1-3, SidI, and SidL also specifically decreases the production of IκB, even though the effectors are overall cytotoxic (152, 153). To shut down translation, the UDP-glucosyltransferases Lgt1–3 modify the elongation factor eEF1A (154, 155), and SidI inactivates eEF1A/eEF1Bγ by an unknown mechanism (153). Finally, L. pneumophila also modulates host transcription more directly. The Icm/Dot substrate RomA/LegAS4 promotes intracellular replication as a SET domain-containing histone methyltransferase that modifies (immune) gene expression (156, 157). In summary, L. pneumophila modulates protein production and turnover through the activation of NF-κB and by altering the epigenetic pattern, as well as through the inhibition of translation.
The ubiquitous environmental bacterium L. pneumophila triggers an acute and potential fatal pneumonia termed Legionnaires’ disease. The opportunistic pathogen employs the Icm/Dot T4SS and as many as 300 different effector proteins to govern interactions with phagocytes and form an intracellular replication niche, the LCV. Some Icm/Dot-translocated effector proteins interfere with (i) endocytic, secretory, or retrograde vesicle trafficking pathways, (ii) organelle or cell motility, (iii) the inflammasome and programed cell death, or (iv) the transcription factor NF-κB. Future studies will address the molecular mechanisms of action of the many Icm/Dot substrates, which remain uncharacterized to date. The further analysis of L. pneumophila effector proteins will likely continue to provide novel insights into the elaborate pathogen–host interactions between a highly adapted opportunistic pathogen and its phagocytic host cells.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
This work was supported by the Swiss National Science Foundation (SNF; 31003A_153200), the German Research Foundation (DFG; HI 1511/3-1, SPP 1580, SFB 914), and the Bundesministerium für Bildung und Forschung (BMBF; 031A410A; Infect-ERA project EUGENPATH).
1. Chow J, Franz KM, Kagan JC. PRRs are watching you: Localization of innate sensing and signaling regulators. Virology (2015) 479-480:104–9. doi:10.1016/j.virol.2015.02.051
2. Creagh EM, O’Neill LA. TLRs, NLRs and RLRs: a trinity of pathogen sensors that co-operate in innate immunity. Trends Immunol (2006) 27:352–57.
4. Sellge G, Kufer TA. PRR-signaling pathways - Learning from microbial tactics. Semin Immunol (2015) 27:75–84. doi:10.1016/j.smim.2015.03.009
6. de Veer MJ, Holko M, Frevel M, Walker E, Der S, Paranjape JM, et al. Functional classification of interferon-stimulated genes identified using microarrays. J Leukoc Biol (2001) 69:912–20.
7. Schoggins JW, Rice CM. Interferon-stimulated genes and their antiviral effector functions. Curr Opin Virol (2011) 1:519–25. doi:10.1016/j.coviro.2011.10.008
8. Abu Kwaik Y, Bumann D. Microbial quest for food in vivo: ‘nutritional virulence’ as an emerging paradigm. Cell Microbiol (2013) 15:882–90. doi:10.1111/cmi.12138
9. Manske C, Hilbi H. Metabolism of the vacuolar pathogen Legionella and implications for virulence. Front Cell Infect Microbiol (2014) 4:125. doi:10.3389/fcimb.2014.00125
10. Flannagan RS, Cosio G, Grinstein S. Antimicrobial mechanisms of phagocytes and bacterial evasion strategies. Nat Rev Microbiol (2009) 7:355–66. doi:10.1038/nrmicro2128
11. Asrat S, De Jesus DA, Hempstead AD, Ramabhadran V, Isberg RR. Bacterial pathogen manipulation of host membrane trafficking. Annu Rev Cell Dev Biol (2014) 30:79–109. doi:10.1146/annurev-cellbio-100913-013439
12. Finsel I, Hilbi H. Formation of a pathogen vacuole according to Legionella pneumophila: how to kill one bird with many stones. Cell Microbiol (2015) 17:935–50. doi:10.1111/cmi.12450
13. Bangsborg JM. Antigenic and genetic characterization of Legionella proteins: contributions to taxonomy, diagnosis and pathogenesis. APMIS Suppl (1997) 70:1–53.
14. Fields BS. The molecular ecology of legionellae. Trends Microbiol (1996) 4:286–90. doi:10.1016/0966-842X(96)10041-X
15. Steinert M, Heuner K. Dictyostelium as host model for pathogenesis. Cell Microbiol (2005) 7:307–14. doi:10.1111/j.1462-5822.2005.00493.x
16. Hilbi H, Hoffmann C, Harrison CF. Legionella spp. outdoors: colonization, communication and persistence. Environ Microbiol Rep (2011) 3:286–96. doi:10.1111/j.1758-2229.2011.00247.x
17. Massis LM, Zamboni DS. Innate immunity to Legionella pneumophila. Front Microbiol (2011) 2:109. doi:10.3389/fmicb.2011.00109
18. Baskerville A, Fitzgeorge RB, Broster M, Hambleton P, Dennis PJ. Experimental transmission of Legionnaires’ disease by exposure to aerosols of Legionella pneumophila. Lancet (1981) 2:1389–90. doi:10.1016/S0140-6736(81)92803-8
19. Brieland J, Freeman P, Kunkel R, Chrisp C, Hurley M, Fantone J, et al. Replicative Legionella pneumophila lung infection in intratracheally inoculated A/J mice. A murine model of human Legionnaires’ disease. Am J Pathol (1994) 145:1537–46.
20. Yamamoto Y, Klein TW, Newton CA, Widen R, Friedman H. Growth of Legionella pneumophila in thioglycolate-elicited peritoneal macrophages from A/J mice. Infect Immun (1988) 56:370–5.
21. Yoshida S, Goto Y, Mizuguchi Y, Nomoto K, Skamene E. Genetic control of natural resistance in mouse macrophages regulating intracellular Legionella pneumophila multiplication in vitro. Infect Immun (1991) 59:428–32.
22. Newton HJ, Ang DK, Van Driel IR, Hartland EL. Molecular pathogenesis of infections caused by Legionella pneumophila. Clin Microbiol Rev (2010) 23:274–98. doi:10.1128/CMR.00052-09
23. Bhardwaj N, Nash TW, Horwitz MA. Interferon-gamma-activated human monocytes inhibit the intracellular multiplication of Legionella pneumophila. J Immunol (1986) 137:2662–9.
24. Byrd TF, Horwitz MA. Interferon gamma-activated human monocytes downregulate transferrin receptors and inhibit the intracellular multiplication of Legionella pneumophila by limiting the availability of iron. J Clin Invest (1989) 83:1457–65. doi:10.1172/JCI114038
25. Nash TW, Libby DM, Horwitz MA. IFN-gamma-activated human alveolar macrophages inhibit the intracellular multiplication of Legionella pneumophila. J Immunol (1988) 140:3978–81.
26. Blanchard DK, Djeu JY, Klein TW, Friedman H, Stewart WE. Protective effects of tumor necrosis factor in experimental Legionella pneumophila infections of mice via activation of PMN function. J Leukoc Biol (1988) 43:429–35.
27. Blanchard DK, Friedman H, Klein TW, Djeu JY. Induction of interferon-gamma and tumor necrosis factor by Legionella pneumophila: augmentation of human neutrophil bactericidal activity. J Leukoc Biol (1989) 45:538–45.
28. Tateda K, Moore TA, Newstead MW, Tsai WC, Zeng X, Deng JC, et al. Chemokine-dependent neutrophil recruitment in a murine model of Legionella pneumonia: potential role of neutrophils as immunoregulatory cells. Infect Immun (2001) 69:2017–24. doi:10.1128/IAI.69.4.2017-2024.2001
29. Spörri R, Joller N, Albers U, Hilbi H, Oxenius A. MyD88-dependent IFN-gamma production by NK cells is key for control of Legionella pneumophila infection. J Immunol (2006) 176:6162–71. doi:10.4049/jimmunol.176.10.6162
30. Spörri R, Joller N, Hilbi H, Oxenius A. A novel role for neutrophils as critical activators of NK cells. J Immunol (2008) 181:7121–30. doi:10.4049/jimmunol.181.10.7121
31. Ang DK, Oates CV, Schuelein R, Kelly M, Sansom FM, Bourges D, et al. Cutting edge: pulmonary Legionella pneumophila is controlled by plasmacytoid dendritic cells but not type I IFN. J Immunol (2010) 184:5429–33. doi:10.4049/jimmunol.1000128
32. Neild AL, Roy CR. Legionella reveal dendritic cell functions that facilitate selection of antigens for MHC class II presentation. Immunity (2003) 18:813–23. doi:10.1016/S1074-7613(03)00140-7
33. Haneburger I, Hilbi H. Phosphoinositide lipids and the Legionella pathogen vacuole. Curr Top Microbiol Immunol (2013) 376:155–73. doi:10.1007/82_2013_341
34. Hilbi H, Haas A. Secretive bacterial pathogens and the secretory pathway. Traffic (2012) 13:1187–97. doi:10.1111/j.1600-0854.2012.01344.x
35. Hubber A, Roy CR. Modulation of host cell function by Legionella pneumophila type IV effectors. Annu Rev Cell Dev Biol (2010) 26:261–83. doi:10.1146/annurev-cellbio-100109-104034
36. Isberg RR, O’Connor TJ, Heidtman M. The Legionella pneumophila replication vacuole: making a cosy niche inside host cells. Nat Rev Microbiol (2009) 7:13–24. doi:10.1038/nrmicro1967
37. Sherwood RK, Roy CR. A Rab-centric perspective of bacterial pathogen-occupied vacuoles. Cell Host Microbe (2013) 14:256–68. doi:10.1016/j.chom.2013.08.010
38. Hilbi H, Segal G, Shuman HA. Icm/Dot-dependent upregulation of phagocytosis by Legionella pneumophila. Mol Microbiol (2001) 42:603–17. doi:10.1046/j.1365-2958.2001.02645.x
39. Watarai M, Derre I, Kirby J, Growney JD, Dietrich WF, Isberg RR. Legionella pneumophila is internalized by a macropinocytotic uptake pathway controlled by the Dot/Icm system and the mouse lgn1 locus. J Exp Med (2001) 194:1081–96. doi:10.1084/jem.194.8.1081
40. Roy CR, Berger KH, Isberg RR. Legionella pneumophila DotA protein is required for early phagosome trafficking decisions that occur within minutes of bacterial uptake. Mol Microbiol (1998) 28:663–74. doi:10.1046/j.1365-2958.1998.00841.x
41. Xu L, Shen X, Bryan A, Banga S, Swanson MS, Luo ZQ. Inhibition of host vacuolar H+-ATPase activity by a Legionella pneumophila effector. PLoS Pathog (2010) 6:e1000822. doi:10.1371/journal.ppat.1000822
42. Finsel I, Ragaz C, Hoffmann C, Harrison CF, Weber S, Van Rahden VA, et al. The Legionella effector RidL inhibits retrograde trafficking to promote intracellular replication. Cell Host Microbe (2013) 14:38–50. doi:10.1016/j.chom.2013.06.001
43. Kagan JC, Roy CR. Legionella phagosomes intercept vesicular traffic from endoplasmic reticulum exit sites. Nat Cell Biol (2002) 4:945–54. doi:10.1038/ncb883
44. Lu H, Clarke M. Dynamic properties of Legionella-containing phagosomes in Dictyostelium amoebae. Cell Microbiol (2005) 7:995–1007. doi:10.1111/j.1462-5822.2005.00528.x
45. Robinson CG, Roy CR. Attachment and fusion of endoplasmic reticulum with vacuoles containing Legionella pneumophila. Cell Microbiol (2006) 8:793–805. doi:10.1111/j.1462-5822.2005.00666.x
46. Swanson MS, Isberg RR. Association of Legionella pneumophila with the macrophage endoplasmic reticulum. Infect Immun (1995) 63:3609–20.
47. Chen J, De Felipe KS, Clarke M, Lu H, Anderson OR, Segal G, et al. Legionella effectors that promote nonlytic release from protozoa. Science (2004) 303:1358–61. doi:10.1126/science.1094226
48. Hilbi H, Weber S, Finsel I. Anchors for effectors: subversion of phosphoinositide lipids by Legionella. Front Microbiol (2011) 2:91. doi:10.3389/fmicb.2011.00091
49. Weber S, Stirnimann CU, Wieser M, Frey D, Meier R, Engelhardt S, et al. A type IV translocated Legionella cysteine phytase counteracts intracellular growth restriction by phytate. J Biol Chem (2014) 289:34175–88. doi:10.1074/jbc.M114.592568
50. Weber S, Wagner M, Hilbi H. Live-cell imaging of phosphoinositide dynamics and membrane architecture during Legionella infection. MBio (2014) 5:e839–813. doi:10.1128/mBio.00839-13
51. Weber SS, Ragaz C, Reus K, Nyfeler Y, Hilbi H. Legionella pneumophila exploits PI(4)P to anchor secreted effector proteins to the replicative vacuole. PLoS Pathog (2006) 2:e46. doi:10.1371/journal.ppat.0020046
52. Brombacher E, Urwyler S, Ragaz C, Weber SS, Kami K, Overduin M, et al. Rab1 guanine nucleotide exchange factor SidM is a major phosphatidylinositol 4-phosphate-binding effector protein of Legionella pneumophila. J Biol Chem (2009) 284:4846–56. doi:10.1074/jbc.M807505200
53. Del Campo CM, Mishra AK, Wang YH, Roy CR, Janmey PA, Lambright DG. Structural basis for PI(4)P-specific membrane recruitment of the Legionella pneumophila effector DrrA/SidM. Structure (2014) 22:397–408. doi:10.1016/j.str.2013.12.018
54. Dolinsky S, Haneburger I, Cichy A, Hannemann M, Itzen A, Hilbi H. The Legionella longbeachae Icm/Dot substrate SidC selectively binds phosphatidylinositol 4-phosphate with nanomolar affinity and promotes pathogen vacuole-endoplasmic reticulum interactions. Infect Immun (2014) 82:4021–33. doi:10.1128/IAI.01685-14
55. Hubber A, Arasaki K, Nakatsu F, Hardiman C, Lambright D, De Camilli P, et al. The machinery at endoplasmic reticulum-plasma membrane contact sites contributes to spatial regulation of multiple Legionella effector proteins. PLoS Pathog (2014) 10:e1004222. doi:10.1371/journal.ppat.1004222
56. Ragaz C, Pietsch H, Urwyler S, Tiaden A, Weber SS, Hilbi H. The Legionella pneumophila phosphatidylinositol-4 phosphate-binding type IV substrate SidC recruits endoplasmic reticulum vesicles to a replication-permissive vacuole. Cell Microbiol (2008) 10:2416–33. doi:10.1111/j.1462-5822.2008.01219.x
57. Schoebel S, Blankenfeldt W, Goody RS, Itzen A. High-affinity binding of phosphatidylinositol 4-phosphate by Legionella pneumophila DrrA. EMBO Rep (2010) 11:598–604. doi:10.1038/embor.2010.97
58. Harding CR, Mattheis C, Mousnier A, Oates CV, Hartland EL, Frankel G, et al. LtpD is a novel Legionella pneumophila effector that binds phosphatidylinositol 3-phosphate and inositol monophosphatase IMPA1. Infect Immun (2013) 81:4261–70. doi:10.1128/IAI.01054-13
59. Jank T, Bohmer KE, Tzivelekidis T, Schwan C, Belyi Y, Aktories K. Domain organization of Legionella effector SetA. Cell Microbiol (2012) 14:852–68. doi:10.1111/j.1462-5822.2012.01761.x
60. Weber SS, Ragaz C, Hilbi H. The inositol polyphosphate 5-phosphatase OCRL1 restricts intracellular growth of Legionella, localizes to the replicative vacuole and binds to the bacterial effector LpnE. Cell Microbiol (2009) 11:442–60. doi:10.1111/j.1462-5822.2008.01266.x
61. Hsu F, Zhu W, Brennan L, Tao L, Luo ZQ, Mao Y. Structural basis for substrate recognition by a unique Legionella phosphoinositide phosphatase. Proc Natl Acad Sci U S A (2012) 109:13567–72. doi:10.1073/pnas.1207903109
62. Toulabi L, Wu X, Cheng Y, Mao Y. Identification and structural characterization of a Legionella phosphoinositide phosphatase. J Biol Chem (2013) 288:24518–27. doi:10.1074/jbc.M113.474239
63. Nagai H, Kagan JC, Zhu X, Kahn RA, Roy CR. A bacterial guanine nucleotide exchange factor activates ARF on Legionella phagosomes. Science (2002) 295:679–82. doi:10.1126/science.1067025
64. Machner MP, Isberg RR. Targeting of host Rab GTPase function by the intravacuolar pathogen Legionella pneumophila. Dev Cell (2006) 11:47–56. doi:10.1016/j.devcel.2006.05.013
65. Machner MP, Isberg RR. A bifunctional bacterial protein links GDI displacement to Rab1 activation. Science (2007) 318:974–7. doi:10.1126/science.1149121
66. Murata T, Delprato A, Ingmundson A, Toomre DK, Lambright DG, Roy CR. The Legionella pneumophila effector protein DrrA is a Rab1 guanine nucleotide-exchange factor. Nat Cell Biol (2006) 8:971–7. doi:10.1038/ncb1463
67. Schoebel S, Oesterlin LK, Blankenfeldt W, Goody RS, Itzen A. RabGDI displacement by DrrA from Legionella is a consequence of its guanine nucleotide exchange activity. Mol Cell (2009) 36:1060–72. doi:10.1016/j.molcel.2009.11.014
68. Ingmundson A, Delprato A, Lambright DG, Roy CR. Legionella pneumophila proteins that regulate Rab1 membrane cycling. Nature (2007) 450:365–9. doi:10.1038/nature06336
69. Hardiman CA, Roy CR. AMPylation is critical for Rab1 localization to vacuoles containing Legionella pneumophila. MBio (2014) 5:e1035–1013. doi:10.1128/mBio.01035-13
70. Müller MP, Peters H, Blumer J, Blankenfeldt W, Goody RS, Itzen A. The Legionella effector protein DrrA AMPylates the membrane traffic regulator Rab1b. Science (2010) 329:946–9. doi:10.1126/science.1192276
71. Neunuebel MR, Chen Y, Gaspar AH, Backlund PS, Yergey A, Machner MP. De-AMPylation of the small GTPase Rab1 by the pathogen Legionella pneumophila. Science (2011) 333:453–6. doi:10.1126/science.1207193
72. Tan Y, Luo ZQ. Legionella pneumophila SidD is a deAMPylase that modifies Rab1. Nature (2011) 475:506–9. doi:10.1038/nature10307
73. Campanacci V, Mukherjee S, Roy CR, Cherfils J. Structure of the Legionella effector AnkX reveals the mechanism of phosphocholine transfer by the FIC domain. EMBO J (2013) 32:1469–77. doi:10.1038/emboj.2013.82
74. Goody PR, Heller K, Oesterlin LK, Müller MP, Itzen A, Goody RS. Reversible phosphocholination of Rab proteins by Legionella pneumophila effector proteins. EMBO J (2012) 31:1774–84. doi:10.1038/emboj.2012.16
75. Mukherjee S, Liu X, Arasaki K, Mcdonough J, Galan JE, Roy CR. Modulation of Rab GTPase function by a protein phosphocholine transferase. Nature (2011) 477:103–6. doi:10.1038/nature10335
76. Pan X, Lührmann A, Satoh A, Laskowski-Arce MA, Roy CR. Ankyrin repeat proteins comprise a diverse family of bacterial type IV effectors. Science (2008) 320:1651–4. doi:10.1126/science.1158160
77. Tan Y, Arnold RJ, Luo ZQ. Legionella pneumophila regulates the small GTPase Rab1 activity by reversible phosphorylcholination. Proc Natl Acad Sci U S A (2011) 108:21212–7. doi:10.1073/pnas.1114023109
78. Cheng W, Yin K, Lu D, Li B, Zhu D, Chen Y, et al. Structural insights into a unique Legionella pneumophila effector LidA recognizing both GDP and GTP bound Rab1 in their active state. PLoS Pathog (2012) 8:e1002528. doi:10.1371/journal.ppat.1002528
79. Neunuebel MR, Mohammadi S, Jarnik M, Machner MP. Legionella pneumophila LidA affects nucleotide binding and activity of the host GTPase Rab1. J Bacteriol (2012) 194:1389–400. doi:10.1128/JB.06306-11
80. Schoebel S, Cichy AL, Goody RS, Itzen A. Protein LidA from Legionella is a rab GTPase supereffector. Proc Natl Acad Sci U S A (2011) 108:17945–50. doi:10.1073/pnas.1113133108
81. Gazdag EM, Schobel S, Shkumatov AV, Goody RS, Itzen A. The structure of the N-terminal domain of the Legionella protein SidC. J Struct Biol (2014) 186:188–94. doi:10.1016/j.jsb.2014.02.003
82. Horenkamp FA, Mukherjee S, Alix E, Schauder CM, Hubber AM, Roy CR, et al. Legionella pneumophila subversion of host vesicular transport by SidC effector proteins. Traffic (2014) 15:488–99. doi:10.1111/tra.12158
83. Hsu F, Luo X, Qiu J, Teng YB, Jin J, Smolka MB, et al. The Legionella effector SidC defines a unique family of ubiquitin ligases important for bacterial phagosomal remodeling. Proc Natl Acad Sci U S A (2014) 111:10538–43. doi:10.1073/pnas.1402605111
84. Bonifacino JS, Hurley JH. Retromer. Curr Opin Cell Biol (2008) 20:427–36. doi:10.1016/j.ceb.2008.03.009
85. Arighi CN, Hartnell LM, Aguilar RC, Haft CR, Bonifacino JS. Role of the mammalian retromer in sorting of the cation-independent mannose 6-phosphate receptor. J Cell Biol (2004) 165:123–33. doi:10.1083/jcb.200312055
86. Seaman MN. The retromer complex – endosomal protein recycling and beyond. J Cell Sci (2012) 125:4693–702. doi:10.1242/jcs.103440
87. Rojas R, Van Vlijmen T, Mardones GA, Prabhu Y, Rojas AL, Mohammed S, et al. Regulation of retromer recruitment to endosomes by sequential action of Rab5 and Rab7. J Cell Biol (2008) 183:513–26. doi:10.1083/jcb.200804048
88. Clemens DL, Lee BY, Horwitz MA. Deviant expression of Rab5 on phagosomes containing the intracellular pathogens Mycobacterium tuberculosis and Legionella pneumophila is associated with altered phagosomal fate. Infect Immun (2000) 68:2671–84. doi:10.1128/IAI.68.5.2671-2684.2000
89. Clemens DL, Lee BY, Horwitz MA. Mycobacterium tuberculosis and Legionella pneumophila phagosomes exhibit arrested maturation despite acquisition of Rab7. Infect Immun (2000) 68:5154–66. doi:10.1128/IAI.68.9.5154-5166.2000
90. Hoffmann C, Finsel I, Otto A, Pfaffinger G, Rothmeier E, Hecker M, et al. Functional analysis of novel Rab GTPases identified in the proteome of purified Legionella-containing vacuoles from macrophages. Cell Microbiol (2014) 16:1034–52. doi:10.1111/cmi.12256
91. Urwyler S, Nyfeler Y, Ragaz C, Lee H, Mueller LN, Aebersold R, et al. Proteome analysis of Legionella vacuoles purified by magnetic immunoseparation reveals secretory and endosomal GTPases. Traffic (2009) 10:76–87. doi:10.1111/j.1600-0854.2008.00851.x
92. Shevchuk O, Batzilla C, Hägele S, Kusch H, Engelmann S, Hecker M, et al. Proteomic analysis of Legionella-containing phagosomes isolated from Dictyostelium. Int J Med Microbiol (2009) 299:489–508. doi:10.1016/j.ijmm.2009.03.006
93. Choudhury R, Diao A, Zhang F, Eisenberg E, Saint-Pol A, Williams C, et al. Lowe syndrome protein OCRL1 interacts with clathrin and regulates protein trafficking between endosomes and the trans-Golgi network. Mol Biol Cell (2005) 16:3467–79. doi:10.1091/mbc.E05-02-0120
94. van Rahden VA, Brand K, Najm J, Heeren J, Pfeffer SR, Braulke T, et al. The 5-phosphatase OCRL mediates retrograde transport of the mannose 6-phosphate receptor by regulating a Rac1-cofilin signalling module. Hum Mol Genet (2012) 21:5019–38. doi:10.1093/hmg/dds343
95. Glick D, Barth S, Macleod KF. Autophagy: cellular and molecular mechanisms. J Pathol (2010) 221:3–12. doi:10.1002/path.2697
96. Nakatogawa H, Suzuki K, Kamada Y, Ohsumi Y. Dynamics and diversity in autophagy mechanisms: lessons from yeast. Nat Rev Mol Cell Biol (2009) 10:458–67. doi:10.1038/nrm2708
97. Feng Y, Yao Z, Klionsky DJ. How to control self-digestion: transcriptional, post-transcriptional, and post-translational regulation of autophagy. Trends Cell Biol (2015) 25:354–63. doi:10.1016/j.tcb.2015.02.002
98. Choy A, Dancourt J, Mugo B, O’Connor TJ, Isberg RR, Melia TJ, et al. The Legionella effector RavZ inhibits host autophagy through irreversible Atg8 deconjugation. Science (2012) 338:1072–6. doi:10.1126/science.1227026
99. Otto GP, Wu MY, Clarke M, Lu H, Anderson OR, Hilbi H, et al. Macroautophagy is dispensable for intracellular replication of Legionella pneumophila in Dictyostelium discoideum. Mol Microbiol (2004) 51:63–72. doi:10.1046/j.1365-2958.2003.03826.x
100. Tung SM, Unal C, Ley A, Pena C, Tunggal B, Noegel AA, et al. Loss of Dictyostelium ATG9 results in a pleiotropic phenotype affecting growth, development, phagocytosis and clearance and replication of Legionella pneumophila. Cell Microbiol (2010) 12:765–80. doi:10.1111/j.1462-5822.2010.01432.x
101. Stewart M. Molecular mechanism of the nuclear protein import cycle. Nat Rev Mol Cell Biol (2007) 8:195–208. doi:10.1038/nrm2114
102. Clarke PR, Zhang C. Spatial and temporal coordination of mitosis by Ran GTPase. Nat Rev Mol Cell Biol (2008) 9:464–77. doi:10.1038/nrm2410
103. Goodman B, Zheng Y. Mitotic spindle morphogenesis: ran on the microtubule cytoskeleton and beyond. Biochem Soc Trans (2006) 34:716–21. doi:10.1042/BST0340716
104. Yudin D, Fainzilber M. Ran on tracks – cytoplasmic roles for a nuclear regulator. J Cell Sci (2009) 122:587–93. doi:10.1242/jcs.015289
105. Bischoff FR, Ponstingl H. Catalysis of guanine nucleotide exchange on Ran by the mitotic regulator RCC1. Nature (1991) 354:80–2. doi:10.1038/354080a0
106. de Felipe KS, Glover RT, Charpentier X, Anderson OR, Reyes M, Pericone CD, et al. Legionella eukaryotic-like type IV substrates interfere with organelle trafficking. PLoS Pathog (2008) 4:e1000117. doi:10.1371/journal.ppat.1000117
107. de Felipe KS, Pampou S, Jovanovic OS, Pericone CD, Ye SF, Kalachikov S, et al. Evidence for acquisition of Legionella type IV secretion substrates via interdomain horizontal gene transfer. J Bacteriol (2005) 187:7716–26. doi:10.1128/JB.187.22.7716-7726.2005
108. Ninio S, Celli J, Roy CR. A Legionella pneumophila effector protein encoded in a region of genomic plasticity binds to Dot/Icm-modified vacuoles. PLoS Pathog (2009) 5:e1000278. doi:10.1371/journal.ppat.1000278
109. Ivanov SS, Charron G, Hang HC, Roy CR. Lipidation by the host prenyltransferase machinery facilitates membrane localization of Legionella pneumophila effector proteins. J Biol Chem (2010) 285:34686–98. doi:10.1074/jbc.M110.170746
110. Rothmeier E, Pfaffinger G, Hoffmann C, Harrison CF, Grabmayr H, Repnik U, et al. Activation of Ran GTPase by a Legionella effector promotes microtubule polymerization, pathogen vacuole motility and infection. PLoS Pathog (2013) 9:e1003598. doi:10.1371/journal.ppat.1003598
111. Etienne-Manneville S. Microtubules in cell migration. Annu Rev Cell Dev Biol (2013) 29:471–99. doi:10.1146/annurev-cellbio-101011-155711
112. Simon S, Wagner MA, Rothmeier E, Müller-Taubenberger A, Hilbi H. Icm/Dot-dependent inhibition of phagocyte migration by Legionella is antagonized by a translocated Ran GTPase activator. Cell Microbiol (2014) 16:977–92. doi:10.1111/cmi.12258
113. Luo ZQ. Striking a balance: modulation of host cell death pathways by Legionella pneumophila. Front Microbiol (2011) 2:36. doi:10.3389/fmicb.2011.00036
114. Diez E, Lee SH, Gauthier S, Yaraghi Z, Tremblay M, Vidal S, et al. Birc1e is the gene within the Lgn1 locus associated with resistance to Legionella pneumophila. Nat Genet (2003) 33:55–60. doi:10.1038/ng1065
115. Wright EK, Goodart SA, Growney JD, Hadinoto V, Endrizzi MG, Long EM, et al. Naip5 affects host susceptibility to the intracellular pathogen Legionella pneumophila. Curr Biol (2003) 13:27–36. doi:10.1016/S0960-9822(02)01359-3
116. Dietrich WF, Damron DM, Isberg RR, Lander ES, Swanson MS. Lgn1, a gene that determines susceptibility to Legionella pneumophila, maps to mouse chromosome 13. Genomics (1995) 26:443–50. doi:10.1016/0888-7543(95)80161-E
117. Amer A, Franchi L, Kanneganti TD, Body-Malapel M, Ozoren N, Brady G, et al. Regulation of Legionella phagosome maturation and infection through flagellin and host Ipaf. J Biol Chem (2006) 281:35217–23. doi:10.1074/jbc.M604933200
118. Coers J, Vance RE, Fontana MF, Dietrich WF. Restriction of Legionella pneumophila growth in macrophages requires the concerted action of cytokine and Naip5/Ipaf signalling pathways. Cell Microbiol (2007) 9:2344–57. doi:10.1111/j.1462-5822.2007.00963.x
119. Fortier A, De Chastellier C, Balor S, Gros P. Birc1e/Naip5 rapidly antagonizes modulation of phagosome maturation by Legionella pneumophila. Cell Microbiol (2007) 9:910–23. doi:10.1111/j.1462-5822.2006.00839.x
120. Lamkanfi M, Amer A, Kanneganti TD, Munoz-Planillo R, Chen G, Vandenabeele P, et al. The Nod-like receptor family member Naip5/Birc1e restricts Legionella pneumophila growth independently of caspase-1 activation. J Immunol (2007) 178:8022–7. doi:10.4049/jimmunol.178.12.8022
121. Lightfield KL, Persson J, Brubaker SW, Witte CE, Von Moltke J, Dunipace EA, et al. Critical function for Naip5 in inflammasome activation by a conserved carboxy-terminal domain of flagellin. Nat Immunol (2008) 9:1171–8. doi:10.1038/ni.1646
122. Molofsky AB, Byrne BG, Whitfield NN, Madigan CA, Fuse ET, Tateda K, et al. Cytosolic recognition of flagellin by mouse macrophages restricts Legionella pneumophila infection. J Exp Med (2006) 203:1093–104. doi:10.1084/jem.20051659
123. Ren T, Zamboni DS, Roy CR, Dietrich WF, Vance RE. Flagellin-deficient Legionella mutants evade caspase-1- and Naip5-mediated macrophage immunity. PLoS Pathog (2006) 2:e18. doi:10.1371/journal.ppat.0020018
124. Silveira TN, Zamboni DS. Pore formation triggered by Legionella spp. is an Nlrc4 inflammasome-dependent host cell response that precedes pyroptosis. Infect Immun (2010) 78:1403–13. doi:10.1128/IAI.00905-09
125. Zamboni DS, Kobayashi KS, Kohlsdorf T, Ogura Y, Long EM, Vance RE, et al. The Birc1e cytosolic pattern-recognition receptor contributes to the detection and control of Legionella pneumophila infection. Nat Immunol (2006) 7:318–25. doi:10.1038/ni1305
126. Nogueira CV, Lindsten T, Jamieson AM, Case CL, Shin S, Thompson CB, et al. Rapid pathogen-induced apoptosis: a mechanism used by dendritic cells to limit intracellular replication of Legionella pneumophila. PLoS Pathog (2009) 5:e1000478. doi:10.1371/journal.ppat.1000478
127. Hoffmann C, Harrison CF, Hilbi H. The natural alternative: protozoa as cellular models for Legionella infection. Cell Microbiol (2014) 16:15–26. doi:10.1111/cmi.12235
128. Banga S, Gao P, Shen X, Fiscus V, Zong WX, Chen L, et al. Legionella pneumophila inhibits macrophage apoptosis by targeting pro-death members of the Bcl2 protein family. Proc Natl Acad Sci U S A (2007) 104:5121–6. doi:10.1073/pnas.0611030104
129. Creasey EA, Isberg RR. The protein SdhA maintains the integrity of the Legionella-containing vacuole. Proc Natl Acad Sci U S A (2012) 109:3481–6. doi:10.1073/pnas.1121286109
130. Laguna RK, Creasey EA, Li Z, Valtz N, Isberg RRA. Legionella pneumophila-translocated substrate that is required for growth within macrophages and protection from host cell death. Proc Natl Acad Sci U S A (2006) 103:18745–50. doi:10.1073/pnas.0609012103
131. Ge J, Gong YN, Xu Y, Shao F. Preventing bacterial DNA release and absent in melanoma 2 inflammasome activation by a Legionella effector functioning in membrane trafficking. Proc Natl Acad Sci U S A (2012) 109:6193–8. doi:10.1073/pnas.1117490109
132. Monroe KM, Mcwhirter SM, Vance RE. Identification of host cytosolic sensors and bacterial factors regulating the type I interferon response to Legionella pneumophila. PLoS Pathog (2009) 5:e1000665. doi:10.1371/journal.ppat.1000665
133. Zhu W, Hammad LA, Hsu F, Mao Y, Luo ZQ. Induction of caspase 3 activation by multiple Legionella pneumophila Dot/Icm substrates. Cell Microbiol (2013) 15:1783–95. doi:10.1111/cmi.12157
134. Aurass P, Schlegel M, Metwally O, Harding CR, Schroeder GN, Frankel G, et al. The Legionella pneumophila Dot/Icm-secreted effector PlcC/CegC1 together with PlcA and PlcB promotes virulence and belongs to a novel zinc metallophospholipase C family present in bacteria and fungi. J Biol Chem (2013) 288:11080–92. doi:10.1074/jbc.M112.426049
135. Akhter A, Gavrilin MA, Frantz L, Washington S, Ditty C, Limoli D, et al. Caspase-7 activation by the Nlrc4/Ipaf inflammasome restricts Legionella pneumophila infection. PLoS Pathog (2009) 5:e1000361. doi:10.1371/journal.ppat.1000361
136. Akhter A, Caution K, Abu Khweek A, Tazi M, Abdulrahman BA, Abdelaziz DH, et al. Caspase-11 promotes the fusion of phagosomes harboring pathogenic bacteria with lysosomes by modulating actin polymerization. Immunity (2012) 37:35–47. doi:10.1016/j.immuni.2012.05.001
137. Gilmore TD. Introduction to NF-kappaB: players, pathways, perspectives. Oncogene (2006) 25:6680–4. doi:10.1038/sj.onc.1209954
138. Akamine M, Higa F, Arakaki N, Kawakami K, Takeda K, Akira S, et al. Differential roles of toll-like receptors 2 and 4 in in vitro responses of macrophages to Legionella pneumophila. Infect Immun (2005) 73:352–61. doi:10.1128/IAI.73.1.352-361.2005
139. Hawn TR, Berrington WR, Smith IA, Uematsu S, Akira S, Aderem A, et al. Altered inflammatory responses in TLR5-deficient mice infected with Legionella pneumophila. J Immunol (2007) 179:6981–7. doi:10.4049/jimmunol.179.10.6981
140. Hawn TR, Smith KD, Aderem A, Skerrett SJ. Myeloid differentiation primary response gene (88)- and toll-like receptor 2-deficient mice are susceptible to infection with aerosolized Legionella pneumophila. J Infect Dis (2006) 193:1693–702. doi:10.1086/504525
141. Vinzing M, Eitel J, Lippmann J, Hocke AC, Zahlten J, Slevogt H, et al. NAIP and Ipaf control Legionella pneumophila replication in human cells. J Immunol (2008) 180:6808–15. doi:10.4049/jimmunol.180.10.6808
142. Archer KA, Alexopoulou L, Flavell RA, Roy CR. Multiple MyD88-dependent responses contribute to pulmonary clearance of Legionella pneumophila. Cell Microbiol (2009) 11:21–36. doi:10.1111/j.1462-5822.2008.01234.x
143. Archer KA, Roy CR. MyD88-dependent responses involving toll-like receptor 2 are important for protection and clearance of Legionella pneumophila in a mouse model of Legionnaires’ disease. Infect Immun (2006) 74:3325–33. doi:10.1128/IAI.02049-05
144. Neild AL, Shin S, Roy CR. Activated macrophages infected with Legionella inhibit T cells by means of MyD88-dependent production of prostaglandins. J Immunol (2005) 175:8181–90. doi:10.4049/jimmunol.175.12.8181
145. Mascarenhas DP, Pereira MS, Manin GZ, Hori JI, Zamboni DS. Interleukin 1 receptor-driven neutrophil recruitment accounts to MyD88-dependent pulmonary clearance of Legionella pneumophila infection in vivo. J Infect Dis (2015) 211:322–30. doi:10.1093/infdis/jiu430
146. Bartfeld S, Engels C, Bauer B, Aurass P, Flieger A, Brüggemann H, et al. Temporal resolution of two-tracked NF-kappaB activation by Legionella pneumophila. Cell Microbiol (2009) 11:1638–51. doi:10.1111/j.1462-5822.2009.01354.x
147. Abu-Zant A, Jones S, Asare R, Suttles J, Price C, Graham J, et al. Anti-apoptotic signalling by the Dot/Icm secretion system of L. pneumophila. Cell Microbiol (2007) 9:246–64. doi:10.1111/j.1462-5822.2006.00785.x
148. Losick VP, Haenssler E, Moy MY, Isberg RR. LnaB: a Legionella pneumophila activator of NF-kappaB. Cell Microbiol (2010) 12:1083–97. doi:10.1111/j.1462-5822.2010.01452.x
149. Losick VP, Isberg RR. NF-kappaB translocation prevents host cell death after low-dose challenge by Legionella pneumophila. J Exp Med (2006) 203:2177–89. doi:10.1084/jem.20060766
150. Ge J, Xu H, Li T, Zhou Y, Zhang Z, Li S, et al. Legionella type IV effector activates the NF-kappaB pathway by phosphorylating the IkappaB family of inhibitors. Proc Natl Acad Sci U S A (2009) 106:13725–30. doi:10.1073/pnas.0907200106
151. Hervet E, Charpentier X, Vianney A, Lazzaroni JC, Gilbert C, Atlan D, et al. Protein kinase LegK2 is a type IV secretion system effector involved in endoplasmic reticulum recruitment and intracellular replication of Legionella pneumophila. Infect Immun (2011) 79:1936–50. doi:10.1128/IAI.00805-10
152. Fontana MF, Banga S, Barry KC, Shen X, Tan Y, Luo ZQ, et al. Secreted bacterial effectors that inhibit host protein synthesis are critical for induction of the innate immune response to virulent Legionella pneumophila. PLoS Pathog (2011) 7:e1001289. doi:10.1371/journal.ppat.1001289
153. Shen X, Banga S, Liu Y, Xu L, Gao P, Shamovsky I, et al. Targeting eEF1A by a Legionella pneumophila effector leads to inhibition of protein synthesis and induction of host stress response. Cell Microbiol (2009) 11:911–26. doi:10.1111/j.1462-5822.2009.01301.x
154. Belyi Y, Niggeweg R, Opitz B, Vogelsgesang M, Hippenstiel S, Wilm M, et al. Legionella pneumophila glucosyltransferase inhibits host elongation factor 1A. Proc Natl Acad Sci U S A (2006) 103:16953–8. doi:10.1073/pnas.0601562103
155. Belyi Y, Tabakova I, Stahl M, Aktories K. Lgt: a family of cytotoxic glucosyl-transferases produced by Legionella pneumophila. J Bacteriol (2008) 190:3026–35. doi:10.1128/JB.01798-07
156. Li T, Lu Q, Wang G, Xu H, Huang H, Cai T, et al. SET-domain bacterial effectors target heterochromatin protein 1 to activate host rDNA transcription. EMBO Rep (2013) 14:733–40. doi:10.1038/embor.2013.86
Keywords: bacterial pathogenesis, Dictyostelium, inflammasome, Legionella, macrophage, pathogen vacuole, phosphoinositide, small GTPase
Citation: Simon S and Hilbi H (2015) Subversion of cell-autonomous immunity and cell migration by Legionella pneumophila effectors. Front. Immunol. 6:447. doi: 10.3389/fimmu.2015.00447
Received: 30 June 2015; Accepted: 17 August 2015;
Published: 14 September 2015
Edited by:
Katharina F. Kubatzky, University Hospital Heidelberg, GermanyReviewed by:
Dario S. Zamboni, Universidade de São Paulo, BrazilCopyright: © 2015 Simon and Hilbi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hubert Hilbi, Gloriastrasse 30, 8006 Zürich, Switzerland,aGlsYmlAaW1tLnV6aC5jaA==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.