 Aimé Cézaire Adiko1,2
Aimé Cézaire Adiko1,2 Joel Babdor3,4,5
Joel Babdor3,4,5 Enric Gutiérrez-Martínez6
Enric Gutiérrez-Martínez6 Pierre Guermonprez6
Pierre Guermonprez6 Loredana Saveanu1,2*
Loredana Saveanu1,2*
- 1INSERM U1149, Faculté Bichat Medical School, ELR8252 CNRS, Center for Research on Inflammation, Paris, France
- 2Université Paris Diderot, Sorbonne Paris Cité, Paris, France
- 3INSERM UMR 1163, Laboratory of Cellular and Molecular Mechanisms of Hematological Disorders and Therapeutic Implications, Paris, France
- 4Université Paris Descartes, Sorbonne Paris Cité, Paris, France
- 5Imagine Institute, Paris, France
- 6Laboratory of Phagocyte Immunobiology, Peter Gorer Department of Immunobiology, King’s College London, London, UK
Cross-presentation, in which exogenous antigens are presented via MHC I complexes, is involved both in the generation of anti-infectious and anti-tumoral cytotoxic CD8+ T cells and in the maintenance of immune tolerance. While cross-presentation was described almost four decades ago and while it is now established that some dendritic cell (DC) subsets are better than others in processing and cross-presenting internalized antigens, the involved molecular mechanisms remain only partially understood. Some of the least explored molecular mechanisms in cross-presentation concern the origin of cross-presenting MHC I molecules and the cellular compartments where antigenic peptide loading occurs. This review focuses on MHC I molecules and their intracellular trafficking. We discuss the source of cross-presenting MHC I in DCs as well as the role of the endocytic pathway in their recycling from the cell surface. Next, we describe the importance of the TAP peptide transporter for delivering peptides to MHC I during cross-presentation. Finally, we highlight the impact of innate immunity mechanisms on specific antigen cross-presentation mechanisms in which TLR activation modulates MHC I trafficking and TAP localization.
A Short History of Major Histocompatibility Complex (MHC) Identification
The MHC is a key component of the immune system that allows T lymphocytes to specifically detect foreign antigens in the context of a self-identification system. The discovery of MHC genes, as genetic loci controlling the immune response, originated from organ transplantation experiments and was performed between 1940 and 1970 by George D. Snell, Jean Dausset, and Baruj Bernacerraf, rewarded by the Nobel Prize in 1980 for these discoveries (1). In 1975, while studying the immune response of mice to viruses, Doherty and Zinkernagel demonstrated the relevance of T cell restriction by MHC. They showed that T cells were able to recognize viral peptides only if these peptides are bound to MHC and highlighted for the first time the phenomenon of “MHC restriction” (2). These major discoveries have also been rewarded, in 1996, by a Nobel Prize. The principle of parallel recognition of both self and non-self molecules was the basis for our current understanding of the specificity of the cellular immune response. In parallel, the study of antibody response in inbred congenic animal models led to the discovery of MHC II and established the distinction between MHC I and MHC II [for a comprehensive review on MHC II discovery, see Ref. (3)].
Further research on MHC antigen presentation established that MHC I binds antigenic peptides derived from endogenous antigens and present them to CD8+ T cells, while MHC II binds antigenic peptides from exogenous antigens and present them to CD4+ T cells. This dichotomy is not absolute and exceptions exist at several levels. The most recent, and also one of the most surprising exceptions, is the discovery of an efficient CD8+ T cytotoxic response that is MHC II restricted, in the case of monkey vaccination with a cytomegalovirus vector expressing antigens from the simian immunodeficiency virus (4).
While the concept that CD8+ T cells are exclusively MHC I restricted has been re-evaluated only recently, the idea that MHC I present exclusively endogenous antigens has been reconsidered nearly 40 years ago, when Bevan discovered the phenomenon of CD8+ T cells cross-priming (5). He showed that minor histocompatibility antigens from transplanted cells can elicit a cytotoxic T cell response restricted by host MHC I molecules, which revealed that the source of antigenic peptides presented by MHC I might also be an exogenous antigen, and not only an antigen synthesized inside the presenting cell.
Contribution of Cross-Presentation to Immunity and Self-Tolerance
The presentation of exogenous antigens by MHC I has been named cross-presentation. Following the discovery of CD8+ T cell cross-priming in transplant rejection (5), additional studies demonstrated that cross-presentation also occurs in the case of several cell-associated antigens, such as self-antigens (6), viral antigens (7), or antigens derived from tumoral cells (8). In addition to these typical cell-associated antigens, an important source of cross-presented material is the proteins synthesized by intracellular pathogens, such as Listeria (9), Plasmodium (10), or Leishmania (11). In these infections, the CD8+ T cells response contributes to protective immunity [reviewed in Ref. (12)].
Early during the study of cross-presentation, it was discovered that this process can lead not only to CD8+ T cell activation (T cell cross-priming) but also to T cell tolerance by either T cell anergy or T cell deletion [Cross-tolerance (13)]. How exactly cross-presentation influences the outcome of T cell response is a topic of particular interest that was extensively studied in the last two decade (14). Early after the discovery of tolerogenic abilities of cross-presentation, it has been supposed that there are several types of antigen cross-presenting cells, some which induce predominantly T cell activation and others that promote T cell tolerance. Alternatively, it has been proposed that the same antigen cross-presenting cell can be switched from a tolerogenic to an immunogenic status according to the nature of the engulfed antigenic material. As a result of significant efforts of several laboratories in the field, it became evident that in dendritic cells (DCs), it is the second scenario that reflects the plasticity of the cross-presentation process to orient the final outcome of the immune response toward cross-priming or cross-tolerance.
Overview of Dendritic Cell Populations
Dendritic cells are by far the most effective cross-presenting cells in both homeostatic and inflammatory conditions (15). As widely demonstrated by studies done both in humans and mice, DCs are a very heterogeneous cellular population (16–18). In a homeostatic situation and without mentioning the skin and mucosal DCs, the murine DC system in lymphoid organs is composed of conventional DCs (CD8+ and CD8−) and plasmacytoïd DCs (pDCs). In humans, DCs found in the blood, spleen, and lymph nodes are classified as pDCs, BDCA1+ DCs, and BDCA3+ DCs. Based on gene-expression profiles, their function, and morphology, BDCA3+ DCs are considered to be the equivalent of murine CD8+ DCs and BDCA1+, the equivalent of murine CD8− DCs (19–22). Conventional DCs require for their differentiation the cytokine FMS-like tyrosine kinase 3 ligand (FLT3L) and additional sub-type specific transcription factors, describing differentiation pathways. A unified ontogeny-based nomenclature of mouse and human DCs has been recently proposed for conventional DCs, which are grouped in three main sub-types: classical type 1 DCs (cDC1), classical type 2 DCs (cDC2), and pDCs (23). The cDC1 sub-type is dependent on the transcription factor Baft3 and includes murine CD8+ DCs, murine CD103+ DCs, and human BDCA3+ DCs. The cDC2 sub-type requires the transcription factor IRF4 and includes murine CD4+ DCs and human BDCA1+ DCs, while the differentiation of pDCs requires E2-2 transcription factor.
If in the murine DCs system, there is a degree of specialization of DC subsets for cross-presentation, with splenic CD8+ DCs and tissue resident CD103+ DCs being recognized as the most efficient cross-presenting subsets (16), in humans, all DC subsets display similar levels of ability for cross-presentation ex vivo (24). In addition to the DC populations found in homeostatic conditions, in inflammatory conditions, in both humans and mice, the monocytes can differentiate in several “DC-like” cellular populations known as monocyte-derived DCs. Considering their ontogeny, which is different from that of conventional DCs, the new name “monocyte-derived cells” has been recently proposed for highly heterogeneous cellular populations that arise from monocytes in inflammatory conditions (23). Although sub-types of monocyte-derived cells are able to capture antigens in vivo and to cross present them via MHC I in vitro (25), their role in vivo for the priming of CD8+ T cells remains to be elucidated. The study of these subclasses will become possible as soon as the transcription factors required for their differentiation will be identified, to allow targeted depletion of specific monocyte-derived cells.
Some of DC subsets induce cross-tolerance in steady-state conditions (26–28) and cross-priming when the antigenic material contains a “danger signal” that will stimulate one of the pattern recognition receptors (PRRs) of DCs, such as toll-like-receptors (TLRs), NOD-like receptors (NLRs), or RIG-I-Like Receptors (RLRs) (29). Thus, phagocytosis of apoptotic cells, which lack “danger” signals and do not activate the PRRs, triggers an anti-inflammatory response (30) and cross-tolerance. In contrast, phagocytosis of pathogens, which activates the PRRs, triggers an inflammatory response and cross-priming. Signaling by PRRs thus leads to profound modifications of DCs, which become able to provide signals that are mandatory for cross-priming. These signals include recognition of peptide MHC complex (pMHC I) by the T cell receptor (TCR), followed by the interaction of DC’s co-stimulatory molecules CD80, CD86 with CD28 on T cells and polarized secretion of pro-inflammatory cytokines (IL-12) toward the immunological synapse (31).
In conclusion, a critical feature of DCs is their ability to coordinate antigen presentation, including MHC I cross-presentation, with the physiological context, like the presence of inflammatory cytokines or danger signals from pathogens. Among the complex cell-intrinsic mechanisms that allow this coordination, the regulation of MHC I trafficking during cross-presentation is a critical factor that will be discussed in detail in this review.
Accessory Cells Involved in MHC I Cross-Presentation
In addition to DCs, several cell types were shown to be able to perform cross-presentation in particular situations. These include macrophages (32, 33), liver sinusoidal endothelial cells (LSECs) (34), endothelial cells (35), γδT cells (36), mast cells (37), and B cells (38). Although unusual, these events of cross-presentation might be relevant for CD8+ T cell priming against tumor antigens (33) or poorly vascularized transplanted organs (35). These particular antigen cross-presentation events are usually prompted by inflammatory conditions; with the exception of LSECs-mediated cross-presentation that occurs under homeostatic conditions and leads to CD8+ T cell tolerance (39). Interestingly, even in the presence of inflammatory triggers, such as TLR4 ligands, the antigen cross-presentation by LSECs remains tolerogenic (40), which indicates that cellular biology of antigen processing and cross-presentation in LSECs should be different from how it occurs in DCs.
Molecular Players Involved in MHC I Assembly
MHC I is a heterodimeric complex containing a glycosylated transmembrane protein called heavy chain (HC), a small soluble protein, the beta-2 microglobulin (β2m) and a peptide ligand that is 8–10 aminoacids in length. In all nucleated cells of the body that can present endogenous antigens via MHC I, newly synthesized heavy chains (HC) are translocated in the endoplasmic reticulum (ER), where the HC assembly with β2m and peptide binding occurs. Like many other cells, DCs are also capable to present antigens that are expressed in their cytosol after endogenous protein synthesis from viral or endogenous mRNAs. During endogenous MHC I presentation, the antigenic peptides are produced in the cytosol by the concerted action of several cytosolic proteases, among which the proteasome contributes a major fraction (41). The ABC peptide transporter TAP transports these antigenic peptides from the cytosol in the ER, where they can bind the newly synthesized HC. MHC I assembly in the ER is a complex process in which a series of dedicated chaperons assist the correct folding of HC. An overview of molecular events leading to MHC I assembly and peptide loading in the ER, comprehensively reviewed in Ref. (42–44) is depicted in Figure 1.
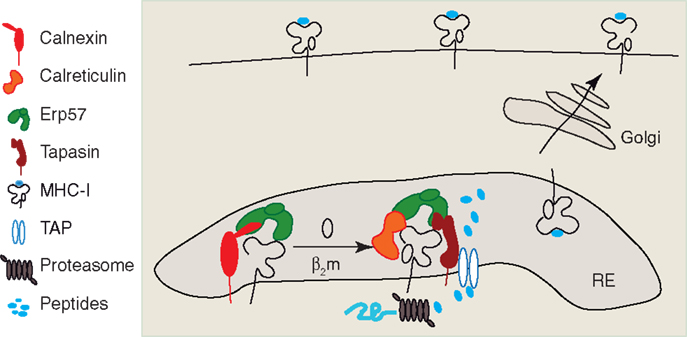
Figure 1. Intracellular assembly of MHC I peptide complex. The newly synthetized heavy chain of MHC I interacts initially with Calnexin and associated thiol oxidoreductase ERp57, and is retained in the ER, where the HC associates with β2m. The HC–β2m complex interacts with the peptide-loading complex (PLC) formed by Calreticulin, ERp57, Tapasin, and the peptide transporter TAP. Antigenic peptide precursors from cytosol are transported by TAP in the ER lumen, where they are processed by ER trimming aminopeptidases (ERAPs). Once the optimal epitope binds the HC–β2m complex, the MHC I is released from the PLC, is packaged into COP-II vesicles, and undergoes the anterograde transport on the secretory pathway. MHC I lacks obvious signal motifs for anterograde transport and the molecular events leading to MHC I packaging into COP-II vesicles remain unknown.
After the binding of a high affinity peptide, MHC I complex may be exported outside ERGIC to reach the cell membrane. Both peptide-loaded and peptide free human MHC I can be visualized by immunoblot and fluorescence microscopy using conformational antibodies (e.g., W6.32 that recognizes peptide-loaded MHC I and HC-10 that detects free HC). It was thus possible to visualize peptide-free HC outside the ER, in the ERGIC, and cis-Golgi. Besides peptide-receptive MHC I (45), other components of MHC I peptide-loading complex, such as functional TAP (46) and calreticulin or tapasin (47), have been detected in ERGIC, suggesting that peptide loading might occur outside ER, in the early secretory pathway. The molecular events involved in the exit of both pMHC and free HC of MHC I from the ER toward the Golgi stacks, remain unknown. The cytosolic tail of HC does not contain any previously described ER–Golgi trafficking signals (48), which suggests that MHC I may bind to cargo receptors, the identity of which remains elusive. Such a cargo was proposed to be Bap31, a transmembrane protein that cycles between ER and Golgi and interacts with both human and murine MHC I (49). However, knockdown of Bap31 does not lead to a decrease in MHC I expression at cell surface (50). Therefore, it has been suggested that additional and redundant mechanisms facilitate MHC I exit from the ER (51).
Pathways of Cell-Surface MHC I Recycling in Non-Professional Antigen-Presenting Cells
Depending on the cell type, between 50 and 180% of plasma membrane surface is constitutively recycled by endocytosis (52). Membrane internalization by clathrin-mediated endocytosis is the best-characterized mechanism of endocytosis [for a review on clathrin-mediated endocytosis, the adaptors involved and the sorting signals recognized by the clathrin adaptors, see Ref. (53)]. Less well characterized, but also very important are the routes of clathrin-independent endocytosis, such as caveolae-mediated uptake, macropinocytosis, and constitutive clathrin-independent uptake [reviewed in Ref. (54, 55)]. These internalization routes permit endocytosis and recycling of plasma membrane proteins lacking any sorting signal for clathrin-mediated endocytosis, including the MHC I. Both fully conformed and empty MHC I are internalized via a clathrin- and dynamin-independent pathway. This pathway is ubiquitous, has been extensively studied in HeLa cells (56, 57), and requires free cholesterol and an active Arf6 GTP-binding protein.
The study of several mutant forms of Arf6 demonstrated that Arf6 is involved in both phosphoinositide kinase PI(4)P5K and phospholipase D activation (58–60). An Arf6-dependent formation of PIP2, the product of PI(4)P5K, might be required for the budding of tubular transport carriers directed to the plasma membrane (61), whereas the products of PLD activation could participate in a later fission step of such tubules in an analogous way to their proposed roles in the fission of COP-I transport carriers (62–64).
The majority of studies on Arf6-dependent endocytosis of MHC I analyzed the fully conformed MHC I recognized specifically by the conformational antibody W6.32. Recently, Zagorac et al. have investigated in parallel the endocytosis of fully conformed MHC I and empty MHC I (visualized by the non-conformational antibody HC-10), but without analyzing their colocalization with Arf6 (65). In this study, several alleles of fully conformed and empty MHC I were found in the same population of early endosomes. This suggests either that both forms of MHC I are internalized by the same route, or that early after their internalization they converge toward the sorting endosomes (SE) independently of how the initial internalization step at the plasma membrane took place. Considering that empty MHC I and conformational MHC I seemed to localize to distinct membrane domains at the cell surface, and that empty MHC I is internalized five to eight times faster than conformational MHC I (66), it is possible that the initial internalization step is different for empty and fully conformed MHC I.
The dichotomy between empty and conformational class I trafficking is obvious in later steps of endocytosis. In these steps, empty MHC I colocalizes with EGF receptor in late endosomes and is directed to lysosomes. In contrast, fully conformed MHC I remains longer in the early endocytic pathway, its localization overlaps with early endosomal antigen 1 and transferrin receptor (65), and reaches endosomal tubular structures similar to the Eps 15 homology domain protein 1 (EDH-1) recycling endosomes that require Arf6 activity (67). As a concluding remark of these studies, one might say that empty MHC I are rapidly internalized and at the level of SE are directed toward the lysosomes, while a part of conformational MHC I recycle back to plasma membrane by a tubular compartment, which requires Arf6, EDH-1 (67), and Rab22 and Rab11 (68, 69) [reviewed in Ref. (51)].
A Potential Role of Ubiquitylation in MHC I Endocytosis
Viral immune evasion mechanisms often depend on viral proteins that interfere with MHC I trafficking. Well-known examples are MHC I retention in the ER by the US3 protein of HCMV (70), cytosolic MHC I degradation by the US11 protein of HCMV (71), MHC I endocytosis promoted by the Nef-1 protein of human immunodeficiency virus (HIV) (72) or by the K3 and K5 proteins of Kaposi sarcoma-associated herpes virus (73). The last example suggests that a potentially important event in MHC I recycling could be the ubiquitylation of lysine 63 of its HC cytosolic tail by the K3 and K5 ubiquitin ligases. Ubiquitin provides a signal for protein incorporation into intralumenal vesicles of multivesicular bodies (MVBs), a subpopulation of late endosomes that fuse with lysosomes (74). Thus, ubiquitylated MHC I is directed to lysosomes where it is degraded (75), allowing virus-infected cells to escape recognition by cytotoxic T cells. Beside the involvement in immune evasion mechanisms, regulation of MHC I by ubiquitin ligases might be a physiological phenomenon, since an endogenous ubiquitin ligase family similar to K3 and K5 proteins was discovered (76). This includes 11 mammalian membrane-associated RING-CH (MARCH) proteins that have been shown to increase the trafficking to late endosomes of several proteins internalized via clathrin-independent endocytosis (77). Among them, MARCH IV and MARCH IX over-expression leads to ubiquitylation and lysosomal degradation of MHC I (78), while MARCH I controls trafficking of MHC II during DCs maturation (79). The pattern of expression of MARCH IV and IX proteins in DC subpopulations, their transcriptional regulation, and their impact on both fully conformed and empty MHC I trafficking in DCs remain to be investigated and are of a great interest for the cellular biology of cross-presentation.
Early Studies of MHC I Trafficking in DCs
Almost everything that we know about endocytosis and recycling of MHC was learnt through the study of non-professional antigen-presenting cells, leading to the current overview of MHC I endocytosis depicted in Figure 2. However, a few studies on MHC I trafficking have been done in DCs, especially in monocytes-derived cells and DC-like cell lines. Early studies on murine DC-like cell line D1 (80) and human monocyte-derived DCs (81), showed that DCs activation by pathogens or TLR ligands increases MHC I biosynthesis and its stability at cell surface. Later studies discovered that MHC I intracellular distribution changes during DCs maturation. Thus, Delamarre et al., when analyzing MHC I localization in murine bone marrow-derived DCs (BM-DCs), demonstrated that in immature BM-DCs MHC I is intracellular, colocalizing with the Golgi marker GM130. Upon BM-DCs maturation by LPS and cluster disruption, the expression of MHC I at cell surface is up-regulated, being sevenfold higher than in immature BM-DCs (82). Like in BM-DCs, in human monocyte-derived DCs and in a DC-like cell line (KG-1), MHC I trafficking is regulated during DCs maturation (83). In immature human DCs, cell-surface MHC I has a short half-life at cell surface and about 60% of MHC I is sequestered in the proximal Golgi apparatus, colocalizing with GM130, γ-adaptin, and GRASP22. Upon DC activation by LPS, MHC I is relocated at cell surface, where it has an increased half-life, in correlation with down regulation of its endocytosis (83). The molecular adaptors that retain MHC I in Golgi remain still unknown, even if a possible role was commented for CD99, which has been reported to be required for MHC I anterograde transport from the ER in B cells (84). Different from monocyte-derived DCs, in immature human Langerhans cells (LCs), MHC I is stored in intracellular vesicles, partially overlapping with HLA-DM. LPS-induced maturation of LCs only modestly increases MHC I biosynthesis, but induces a rapid mobilization of the intracellular pool of MHC I to cell surface (85).
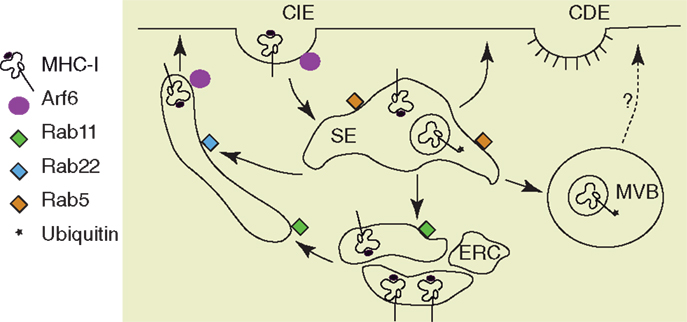
Figure 2. Pathways of MHC I recycling. Mammalian cells have two major pathways of endocytosis, clathrin-dependent endocytosis (CDE) and clathrin-independent endocytosis (CIE). MHC I molecules are internalized via CIE and arrive in sorting endosomes (SE). Depending on their conformation and their ubiquitination status, the MHC I are either recycled or directed to multivesicular bodies (MVB). The recycling of MHC I occurs via the endocytic recycling compartment (ERC) described by the small GTPase Rab11 or via tubular structures dependent on Rab22 and Arf6 (68, 69). Among these MHC I recycling pathways that were described in HeLa cells, only the MHC I from Rab11+ ERC was recently explored for their function in cross-presentation after antigen phagocytosis (118).
These initial studies on MHC I localization in human monocyte-derived DCs and LC indicate that the intracellular localization and trafficking regulation vary from one DC type to another. A careful and systematic examination of MHC I localization in several DC subpopulations has not been done yet. Nevertheless, this will be required in the future, to be able to estimate to what extent the molecular mechanisms regulating MHC I trafficking in monocyte-derived DCs, which will be discussed in subsequent sections of this review, operate in other DC subsets.
Pathways of Antigen Processing during Cross-Presentation
Early studies on molecular mechanisms allowing exogenous antigens presentation on MHC I established that two major pathways of antigen processing exist during cross-presentation: one that is TAP and proteasome-dependent and another that is TAP and proteasome-independent. In the proteasome-independent cross-presentation, known as vacuolar cross-presentation, acidic lysosomal proteases, among which the Catepsin S seems to have a major role (86), generate the MHC I ligands in the endocytic pathway. Therefore, in proteasome-independent cross-presentation, MHC I loading occurs in endocytic vesicles. The vacuolar route of MHC I cross-presentation is considered to be less effective than proteasome-dependent cross-presentation (87). Nevertheless, from the data available today it is problematic to get definitive conclusions on the relative importance of each cross-presentation pathway because proteasome inhibition has pleiotropic cellular effects while TAP deficiency also alters MHC I trafficking.
Contrary to vacuolar cross-presentation in which the antigen processing occurs in the endocytic vesicles, TAP and proteasome-dependent cross-presentation involves transport of exogenous antigen from the endocytic pathway to the cytosol. The unique ability of DCs to export the antigenic material from the endocytic vesicles to the cytosol (88) is probably the key feature that allows them to excel in cross-presentation of exogenous antigens. Antigenic material shuttled into cytosol is digested by the proteasome, which is the main cytosolic broad substrate specificity protease. The proteasome generates precursors of antigenic peptides that are longer than 8–10 amino acids, which is the optimal peptide length for an MHC I ligand (89). Thus, the ligand precursors generated by the proteasome have rarely the optimal length and require further processing by accessory peptidases, either in the cytosol or in the ER [for a review, see Ref. (90)].
Aminoterminal Trimming of Antigen Precursors and MHC I Loading Compartments during Cross-Presentation
The enzymes that perform the final step of antigen processing for MHC I antigen presentation belong to the same subfamily (IRAP subfamily) of M1 metallo-peptidases and are encoded by genes located in the same region of human chromosome 5. In humans, this family includes two ER localized enzymes, ER trimming aminopeptidases (ERAP)1 and ERAP2 (91), and the endosomal enzyme insulin-responsive aminopeptidase (IRAP) (92). Rodents have only two antigen trimming aminopeptidases, IRAP in endosomes, and the equivalent of human ERAP1, ERAAP in the ER (93). ERAPs are soluble, intraluminal proteins, while IRAP is a type II transmembrane protein that bears an intracytosolic domain of 110 amino acids. This cytosolic domain contains all the information required for IRAP trafficking, as demonstrated by extensive study of the enzyme in insulin-responsive tissues (94, 95), where a major cargo of IRAP vesicles is the insulin-regulated glucose transporter Glut4 (96). In DCs, IRAP is found in intracellular vesicles that contain also MHC I molecules, the small GTPase Rab14, and the Q-SNARE Syntaxin 6. These vesicles are recruited to early phagosomes and endosomes containing the internalized antigen and facilitate cross-presentation ability of DCs by antigen trimming function of IRAP (92, 97). Interestingly, IRAP not only colocalizes with internalized MHC I molecules, but also co-immunoprecipitates with MHC I. Why the endosomal trimming aminopeptidase interacts with MHC I is still unknown. The close proximity of IRAP and MHC I molecules might facilitate the loading of MHC I in endosomes and phagosomes, an environment that might be less favorable than the ER for class I loading. Alternatively, IRAP, which bears two di-leucine motifs in its cytosolic domain might interact with clathrin adaptors and direct the vesicular trafficking of MHC I molecules. To understand the functional role of IRAP interaction with MHC I, these hypotheses need further investigation.
The discovery and detailed characterization of the localization of trimming peptidases and their roles in cross-presentation allowed us to speculate on the cellular compartments in which MHC I is loaded with peptides (Figure 3). In the proteasome-dependent cross-presentation pathways, these compartments are the perinuclear ER, the phagosomes, and the specialized endosomes, while in the vacuolar pathway of cross-presentation the MHC I loading occurs in late endosomes or lysosomes.
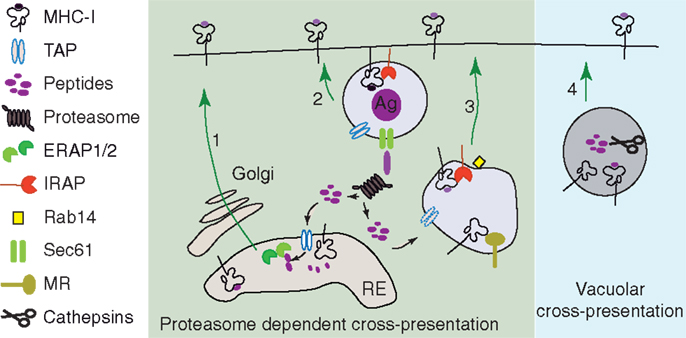
Figure 3. MHC I loading compartments during cross-presentation. (1) The antigen precursors produced in the cytosol of DCs can join the classical MHC I presentation pathway and can be transported by TAP in ER. In ER, the final trimming of antigen precursors is performed by the ER aminopeptidases (91, 93, 121, 122) and the resulting peptides bind to newly synthetized empty MHC I (92, 123, 124). (2 and 3) Proteasome digestion products generated in the cytosol of DCs can be retro-transported in phagosomes (111, 112, 114) or specialized endosomes (117, 125). The final step of antigen processing is probably done by IRAP which is the main trimming aminopeptidase recruited to the phagosome and colocalizing with mannose receptor endosomes (92). (4) Alternatively, in proteasome-independent cross-presentation (vacuolar pathway of cross-presentation), the exogenous antigen is entirely processed in the endocytic pathway by endolysosomal hydrolases (86).
The Intracellular Origin of Cross-Presenting MHC I
Although, theoretically, MHC I loading with exogenous peptides during cross-presentation could take place in the perinuclear ER, several lines of evidence accumulated in the last decades that in, DCs, the main components of antigen processing machinery have access to antigen containing endosomes and phagosomes. This enhanced access is likely to contribute to a high efficiency of the cross-presentation phenomenon, as summarized in the Figure 3. The existence of peptide-receptive MHC I molecules is the primordial ingredient for cross-presentation. Thus, to understand the molecular mechanisms of cross-presentation, the origin of MHC I molecules that are loaded with exogenous peptides in the endocytic pathway is a central question investigated by several recent reports.
In the proteasome-independent vacuolar pathway of cross-presentation, initial studies clearly indicated that the MHC I that are loaded with antigenic peptides are recycling MHC I (98–101), even if the molecular mechanisms of MHC I endocytosis and recycling have not been investigated systematically in that work. However, the most recent exploration of MHC I intracellular localization during cross-presentation gave new insights into MHC I vesicular trafficking. Thus, in a model of viral infections as well as soluble ovalbumin cross-presentation, Lizée et al. demonstrated that a conserved tyrosine in the cytosolic tail of MHC I is required for MHC I endocytosis and its targeting to lysosomal vesicles (102). The lysosomal-targeted class I molecules were probably loaded with antigenic peptides generated in the endocytic pathway, although the proteasome dependence of the cross-presentation models used in these studies (102, 103) has not been investigated. The molecular mechanism by which the tyrosine residue in the cytosolic tail of MHC I favors its endocytosis and lysosomal targeting remains enigmatic and the cytosolic adaptor that recognizes this MHC I trafficking motif remains to be identified.
Additional evidence that the cytosolic tail of MHC I contains a cryptic sorting signal around the previously mentioned tyrosine residue comes from the study of molecular mechanisms by which the HIV down-regulates the cell-surface MHC I level (104–106). In the presence of HIV Nef protein, the cryptic signal YXXXAXXD of MHC I binds the AP-1 clathrin adaptor (107) and this trimolecular complex directs MHC I to endosomes and lysosomes. Interestingly, while in the presence of Nef protein, the cross-presentation by MHC I containing the tyrosine signal (HLA-A2, HLA-B) is compromised, both the clathrin adaptor AP-1 and the tyrosine motif of MHC I are absolutely required for cross-presentation of soluble ovalbumin (108). If these data clearly indicate a role for AP-1 in targeting MHC I from trans Golgi to intracellular vesicles prior to presentation at the cell surface, the nature of these intracellular vesicles nor the mechanism of AP-1 binding to MHC I in the absence of Nef is known. The example of HIV Nef protein that reorients MHC I trafficking at trans Golgi level via AP-1 clathrin adaptor might indicate the existence of an endogenous protein that can substitute for Nef protein and ensure AP-1 binding to MHC I. Such a candidate is the class II invariant chain Ii, which binds AP-1 clathrin adaptor (109) and which has been shown to be required for MHC I sorting to endosomes and lysosomes (110).
The majority of data supporting the role of MHC I cytosolic tail in MHC I trafficking and cross-presentation were obtained in cross-presentation settings that often used soluble antigens and were possibly proteasome-independent. To what extent these findings apply to phagosome-mediated and receptor-targeted cross-presentation, which are usually proteasome-dependent, remains to be elucidated.
Contrary to vacuolar cross-presentation, in the proteasome-dependent cross-presentation, the source of MHC I molecules that are loaded with exogenous peptides has been considered for a long time to be the MHC I from the ER. This assumption is also compatible with the fusion of ER with phagosomal membranes, which provide to phagosomes several components of MHC I antigenic presenting machinery, including MHC I molecules (111–113). It is likely that ER-phagosome fusion results in a mixed population of MHC I molecules, since both Endo H sensitive (ER form) and Endo H resistant (surface form) MHC I can be detected in phagosomes [(114) and our unpublished results]. Several recent data allow us to reconsider this original hypothesis and seem to indicate that the source of MHC I involved in proteasome dependent cross-presentation is not the newly synthetized class I from the ER. Thus, we have showed that preincubation of TAP-deficient DCs at 26°C increases the cell-surface level of MHC I and fully restores the cross-presentation of phagocytized antigens, in a model of proteasome-dependent cross-presentation (115). These experiments suggest that MHC I molecules from the late steps of secretory pathways or from the cell surface are essential in proteasome-dependent cross-presentation of phagosomal antigens. Interestingly, in the same study, the recovery of cell-surface MHC I by low temperature incubation of TAP-deficient DCs did not restore the cross-presentation of antigen targeted ovalbumin, indicating either that the source of loadable MHC I are different between phagocytized and receptor-targeted antigens, or that the phagosomes, but not the endosomes, are equipped with an alternative mechanism for peptide import, such as Sec61 (114).
Further proof in favor of the involvement of the recycling MHC I in proteasome-dependent cross-presentation was provided by a recent study performed in the DC-like cell line DC2.4, in which the cross-presentation of E. Coli expressing ovalbumin was investigated after siRNA-mediated inactivation of 57 Rab proteins (116). Several Rabs were required for optimal cross-presentation, such as Rab3b, 3c, 5b, 8b, 10, 33a, 34, and 35. Among them, Rab3 seems to be required for recycling of cell-surface MHC I, as demonstrated using ZsGreen β2m as a tracker. Additional strong evidence against the implication of ER localized MHC I molecules in cross-presentation of phagosomal antigens result from a very recent study on Rab11 role in MHC I trafficking during cross-presentation that will be presented in the next paragraphs.
Control of MHC I Antigen Cross-Presentation by TLR Signaling
The well-known ability of cross-presentation to induce both T cell priming and T cell tolerance is associated to activation of PRRs (29), among which the most potent and the best studied are the TLRs. The increase in co-stimulatory molecules and pro-inflammatory cytokine production by DCs enhance the potential of TLR-activated DCs to cross-prime the naïve CD8+ T cells. In addition to these effects of TLR activation, it has been recently demonstrated that the intracellular trafficking of two essential components of MHC I antigen-presentation machinery, TAP, and MHC I is regulated by TLR signaling (Figure 4). In a model of mannose receptor (MR) targeted cross-presentation, Burgdorf et al. demonstrated that TAP is enriched in MR endosomes upon DCs stimulation by a TLR4 ligand, the LPS (117). In a model of cross-presentation upon phagocytosis of yeast cells expressing ovalbumin, we detected a moderate increase in the amount of TAP recruited on phagosomes containing yeast cells when compared with the phagosomes containing latex beads (92). More recently, it has been shown that Sec22b-mediated fusion of ERGIC derived vesicles to the phagosome even in the absence of TLR ligands on the phagosomal cargo (113), suggesting that the recruitment of TAP to phagosomal membrane is less dependent on TLR stimulation than the recruitment of MHC I.
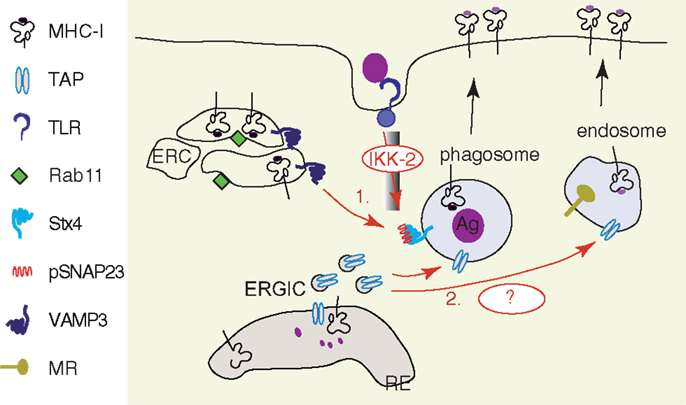
Figure 4. TLRs activation facilitates the recruitment of MHC I and TAP to antigen containing vesicles. (1) Following TLR activation, the downstream signaling cascade involving MyD88 drives phosphorylation of SNAP23 by IkB-Kinase (IKK2). Phosphorylated SNAP23 facilitates MHC I recruitment from Rab11+ ERC to phagosomal membrane, probably by stabilization of SNARE complexes (118). (2) Sec22b-mediated ER–phagosome fusion provides TAP to the phagosomal membrane in the absence of TLR activation (113). A further increase in TAP recruitment to antigen containing vesicles, such as MR+ endosomes (117) and yeast containing phagosomes (92), has been observed after TLR stimulation. The underlying molecular mechanisms remain to be investigated.
The supply of MHC I molecules to the phagosomes is strongly enhanced by TLR stimulation (118). Nair-Gupta and colleagues demonstrated that upon TLR4 stimulation by LPS, the downstream signaling cascade induces the IKK2-dependent phosphorylation of synaptosome-associated protein of 23 kDa (SNAP 23) on the phagosomes containing the TLR ligand. The phosphorylated SNAP23/Syntaxin 4 (STX4) complex recruits the t-SNARE VAMP3 and this molecular complex mediates the membrane fusion between endosomes containing MHC I and phagosomes. The VAMP3 endosomal compartment that contains MHC I is described by the small GTPase Rab11a and is identified as the endosomal recycling compartment (ERC). How MHC I molecules are stored in DCs in the VAMP3+ Rab11+ ERC and if they are directly targeted from the transGolgi network to ERC or if they are internalized from the cell membrane is still unknown. The importance of tyrosine in the cryptic sorting motif of the cytosolic tail of MHC I and the role of AP-1 clathrin adaptor in MHC I targeting to VAMP3+ Rab11a+ ERC remain to be investigated as well. Finally, since SNAP 23 has the ability to interact not only with STX4 and VAMP3, but also with several others SNAREs (STX2, 6, 11, VAMP2, 8), the activation of SNAP 23 by TLR signaling might trigger important changes in vesicular trafficking in DCs. Thus, this regulation of membrane fusion machinery by TLR signaling might be an important checkpoint for antigen presentation and immunogenic capacities of DCs.
Concluding Remarks
Cross-presentation of exogenous antigens via MHC I is a complex phenomenon, which is largely based on the high plasticity of the endocytic system in DCs. The main ingredient for successful cross-presentation is the presence of peptide loadable MHC I in the cellular compartment where the antigenic peptide is produced. While the pathways of intracellular trafficking of MHC I have been fairly well studied in model cell lines, such as HeLa cells, the study of MHC I trafficking in DCs is still in a pioneer stage and the few cellular biology studies focused on mouse BM-DCs. However, we think that the time has come to develop this topic since it could fully benefit from the comprehensive characterization of DC subpopulations and their equivalence between humans and rodents (23). In combination with the availability of high-resolution microscopy and excellent conformation-specific antibodies against human MHC I (119, 120), the characterization of MHC I trafficking in human DC subpopulations became possible.
Considering the well-known ability of signaling through PRRs to increase the immunogenic abilities of DCs, including cross-presentation (29) it is of outstanding interest to determine how TLR signaling regulates MHC I trafficking in DC subpopulations. The recent work of Nair-Gupta et al. has provided some insights on MHC I recruitment from Rab11+ ERC to antigen-containing phagosomes in mouse BM-DCs upon TLR2 and TLR4 activation via the MyD88 signaling pathway (118). To what degree these interesting findings apply to steady-state DC subpopulations or to accessory cross-presenting cells? Are Rab11+ ERCs a TLR-regulated reservoir of MHC I in all cells capable of cross-presentation? Is the TRIF-dependent signaling pathway able to regulate MHC I trafficking? Answering these questions in the near future would pave the way to a mechanistic comprehension of MHC I cross-presentation regulation through modulation of intracellular MHC I trafficking.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Raju TN. The nobel chronicles. 1980: George Davis Snell (1903-96); Jean Baptiste Dausset (b 1916); Baruj Benacerraf (b 1920). Lancet (1999) 354:1738. doi: 10.1016/S0140-6736(05)76734-9
2. Doherty PC, Zinkernagel RM. Enhanced immunological surveillance in mice heterozygous at the H-2 gene complex. Nature (1975) 256:50–2. doi:10.1038/256050a0
3. McDevitt HO. Discovering the role of the major histocompatibility complex in the immune response. Annu Rev Immunol (2000) 18:1–17. doi:10.1146/annurev.immunol.18.1.1
4. Hansen SG, Sacha JB, Hughes CM, Ford JC, Burwitz BJ, Scholz I, et al. Cytomegalovirus vectors violate CD8+ T cell epitope recognition paradigms. Science (2013) 340:1237874. doi:10.1126/science.1237874
5. Bevan MJ. Cross-priming for a secondary cytotoxic response to minor H antigens with H-2 congenic cells which do not cross-react in the cytotoxic assay. J Exp Med (1976) 143:1283–8. doi:10.1084/jem.143.5.1283
6. Kurts C, Heath WR, Carbone FR, Allison J, Miller JF, Kosaka H. Constitutive class I-restricted exogenous presentation of self antigens in vivo. J Exp Med (1996) 184:923–30. doi:10.1084/jem.184.3.923
7. Sigal LJ, Crotty S, Andino R, Rock KL. Cytotoxic T-cell immunity to virus-infected non-haematopoietic cells requires presentation of exogenous antigen. Nature (1999) 398:77–80. doi:10.1038/18038
8. Huang AY, Golumbek P, Ahmadzadeh M, Jaffee E, Pardoll D, Levitsky H. Role of bone marrow-derived cells in presenting MHC class I-restricted tumor antigens. Science (1994) 264:961–5. doi:10.1126/science.7513904
9. Tvinnereim AR, Harty JT. CD8(+) T-cell priming against a nonsecreted Listeria monocytogenes antigen is independent of the antimicrobial activities of gamma interferon. Infect Immun (2000) 68:2196–204. doi:10.1128/IAI.68.4.2196-2204.2000
10. Jung S, Unutmaz D, Wong P, Sano G, De Los Santos K, Sparwasser T, et al. In vivo depletion of CD11c+ dendritic cells abrogates priming of CD8+ T cells by exogenous cell-associated antigens. Immunity (2002) 17:211–20. doi:10.1016/S1074-7613(02)00365-5
11. Ashok D, Schuster S, Ronet C, Rosa M, Mack V, Lavanchy C, et al. Cross-presenting dendritic cells are required for control of Leishmania major infection. Eur J Immunol (2014) 44:1422–32. doi:10.1002/eji.201344242
12. Blanchard N, Shastri N. Topological journey of parasite-derived antigens for presentation by MHC class I molecules. Trends Immunol (2010) 31:414–21. doi:10.1016/j.it.2010.08.004
13. Kurts C, Kosaka H, Carbone FR, Miller JF, Heath WR. Class I-restricted cross-presentation of exogenous self-antigens leads to deletion of autoreactive CD8(+) T cells. J Exp Med (1997) 186:239–45. doi:10.1084/jem.186.2.239
14. Heath WR, Carbone FR. Cross-presentation, dendritic cells, tolerance and immunity. Annu Rev Immunol (2001) 19:47–64. doi:10.1146/annurev.immunol.19.1.47
15. Guermonprez P, Valladeau J, Zitvogel L, Thery C, Amigorena S. Antigen presentation and T cell stimulation by dendritic cells. Annu Rev Immunol (2002) 20:621–67. doi:10.1146/annurev.immunol.20.100301.064828
16. Liu K, Nussenzweig MC. Origin and development of dendritic cells. Immunol Rev (2010) 234:45–54. doi:10.1111/j.0105-2896.2009.00879.x
17. Merad M, Sathe P, Helft J, Miller J, Mortha A. The dendritic cell lineage: ontogeny and function of dendritic cells and their subsets in the steady state and the inflamed setting. Annu Rev Immunol (2013) 31:563–604. doi:10.1146/annurev-immunol-020711-074950
18. Murphy KM. Transcriptional control of dendritic cell development. Adv Immunol (2013) 120:239–67. doi:10.1016/B978-0-12-417028-5.00009-0
19. Bachem A, Guttler S, Hartung E, Ebstein F, Schaefer M, Tannert A, et al. Superior antigen cross-presentation and XCR1 expression define human CD11c+CD141+ cells as homologues of mouse CD8+ dendritic cells. J Exp Med (2010) 207:1273–81. doi:10.1084/jem.20100348
20. Crozat K, Guiton R, Contreras V, Feuillet V, Dutertre CA, Ventre E, et al. The XC chemokine receptor 1 is a conserved selective marker of mammalian cells homologous to mouse CD8alpha+ dendritic cells. J Exp Med (2010) 207:1283–92. doi:10.1084/jem.20100223
21. Jongbloed SL, Kassianos AJ, Mcdonald KJ, Clark GJ, Ju X, Angel CE, et al. Human CD141+ (BDCA-3)+ dendritic cells (DCs) represent a unique myeloid DC subset that cross-presents necrotic cell antigens. J Exp Med (2010) 207:1247–60. doi:10.1084/jem.20092140
22. Poulin LF, Salio M, Griessinger E, Anjos-Afonso F, Craciun L, Chen JL, et al. Characterization of human DNGR-1+ BDCA3+ leukocytes as putative equivalents of mouse CD8alpha+ dendritic cells. J Exp Med (2010) 207:1261–71. doi:10.1084/jem.20092618
23. Guilliams M, Ginhoux F, Jakubzick C, Naik SH, Onai N, Schraml BU, et al. Dendritic cells, monocytes and macrophages: a unified nomenclature based on ontogeny. Nat Rev Immunol (2014) 14:571–8. doi:10.1038/nri3712
24. Segura E, Durand M, Amigorena S. Similar antigen cross-presentation capacity and phagocytic functions in all freshly isolated human lymphoid organ-resident dendritic cells. J Exp Med (2013) 210:1035–47. doi:10.1084/jem.20121103
25. Cheong C, Matos I, Choi JH, Dandamudi DB, Shrestha E, Longhi MP, et al. Microbial stimulation fully differentiates monocytes to DC-SIGN/CD209(+) dendritic cells for immune T cell areas. Cell (2010) 143:416–29. doi:10.1016/j.cell.2010.09.039
26. Hawiger D, Inaba K, Dorsett Y, Guo M, Mahnke K, Rivera M, et al. Dendritic cells induce peripheral T cell unresponsiveness under steady state conditions in vivo. J Exp Med (2001) 194:769–79. doi:10.1084/jem.194.6.769
27. Bonifaz L, Bonnyay D, Mahnke K, Rivera M, Nussenzweig MC, Steinman RM. Efficient targeting of protein antigen to the dendritic cell receptor DEC-205 in the steady state leads to antigen presentation on major histocompatibility complex class I products and peripheral CD8+ T cell tolerance. J Exp Med (2002) 196:1627–38. doi:10.1084/jem.20021598
28. Steinman RM, Hawiger D, Nussenzweig MC. Tolerogenic dendritic cells. Annu Rev Immunol (2003) 21:685–711. doi:10.1146/annurev.immunol.21.120601.141040
29. Palm NW, Medzhitov R. Pattern recognition receptors and control of adaptive immunity. Immunol Rev (2009) 227:221–33. doi:10.1111/j.1600-065X.2008.00731.x
30. Henson PM, Hume DA. Apoptotic cell removal in development and tissue homeostasis. Trends Immunol (2006) 27:244–50. doi:10.1016/j.it.2006.03.005
31. Lanzavecchia A, Sallusto F. Regulation of T cell immunity by dendritic cells. Cell (2001) 106:263–6. doi:10.1016/S0092-8674(01)00455-X
32. Kovacsovics-Bankowski M, Clark K, Benacerraf B, Rock KL. Efficient major histocompatibility complex class I presentation of exogenous antigen upon phagocytosis by macrophages. Proc Natl Acad Sci U S A (1993) 90:4942–6. doi:10.1073/pnas.90.11.4942
33. Asano K, Nabeyama A, Miyake Y, Qiu CH, Kurita A, Tomura M, et al. CD169-positive macrophages dominate antitumor immunity by crosspresenting dead cell-associated antigens. Immunity (2011) 34:85–95. doi:10.1016/j.immuni.2010.12.011
34. Limmer A, Ohl J, Kurts C, Ljunggren HG, Reiss Y, Groettrup M, et al. Efficient presentation of exogenous antigen by liver endothelial cells to CD8+ T cells results in antigen-specific T-cell tolerance. Nat Med (2000) 6:1348–54. doi:10.1038/82161
35. Valujskikh A, Lantz O, Celli S, Matzinger P, Heeger PS. Cross-primed CD8(+) T cells mediate graft rejection via a distinct effector pathway. Nat Immunol (2002) 3:844–51. doi:10.1038/ni831
36. Brandes M, Willimann K, Bioley G, Levy N, Eberl M, Luo M, et al. Cross-presenting human gammadelta T cells induce robust CD8+ alphabeta T cell responses. Proc Natl Acad Sci U S A (2009) 106:2307–12. doi:10.1073/pnas.0810059106
37. Stelekati E, Bahri R, D’Orlando O, Orinska Z, Mittrucker HW, Langenhaun R, et al. Mast cell-mediated antigen presentation regulates CD8+ T cell effector functions. Immunity (2009) 31:665–76. doi:10.1016/j.immuni.2009.08.022
38. Kamphorst AO, Guermonprez P, Dudziak D, Nussenzweig MC. Route of antigen uptake differentially impacts presentation by dendritic cells and activated monocytes. J Immunol (2010) 185:3426–35. doi:10.4049/jimmunol.1001205
39. Knolle PA, Uhrig A, Hegenbarth S, Loser E, Schmitt E, Gerken G, et al. IL-10 down-regulates T cell activation by antigen-presenting liver sinusoidal endothelial cells through decreased antigen uptake via the mannose receptor and lowered surface expression of accessory molecules. Clin Exp Immunol (1998) 114:427–33. doi:10.1046/j.1365-2249.1998.00713.x
40. Crispe IN. The liver as a lymphoid organ. Annu Rev Immunol (2009) 27:147–63. doi:10.1146/annurev.immunol.021908.132629
41. van Endert P. Providing ligands for MHC class I molecules. Cell Mol Life Sci (2011) 68:1467–9. doi:10.1007/s00018-011-0654-1
42. Peaper DR, Cresswell P. Regulation of MHC class I assembly and peptide binding. Annu Rev Cell Dev Biol (2008) 24:343–68. doi:10.1146/annurev.cellbio.24.110707.175347
43. Saunders PM, van Endert P. Running the gauntlet: from peptide generation to antigen presentation by MHC class I. Tissue Antigens (2011) 78:161–70. doi:10.1111/j.1399-0039.2011.01735.x
44. Hulpke S, Tampe R. The MHC I loading complex: a multitasking machinery in adaptive immunity. Trends Biochem Sci (2013) 38:412–20. doi:10.1016/j.tibs.2013.06.003
45. Garstka M, Borchert B, Al-Balushi M, Praveen PV, Kuhl N, Majoul I, et al. Peptide-receptive major histocompatibility complex class I molecules cycle between endoplasmic reticulum and cis-Golgi in wild-type lymphocytes. J Biol Chem (2007) 282:30680–90. doi:10.1074/jbc.M701721200
46. Ghanem E, Fritzsche S, Al-Balushi M, Hashem J, Ghuneim L, Thomer L, et al. The transporter associated with antigen processing (TAP) is active in a post-ER compartment. J Cell Sci (2010) 123:4271–9. doi:10.1242/jcs.060632
47. Paulsson KM, Jevon M, Wang JW, Li S, Wang P. The double lysine motif of tapasin is a retrieval signal for retention of unstable MHC class I molecules in the endoplasmic reticulum. J Immunol (2006) 176:7482–8. doi:10.4049/jimmunol.176.12.7482
48. Dancourt J, Barlowe C. Protein sorting receptors in the early secretory pathway. Annu Rev Biochem (2010) 79:777–802. doi:10.1146/annurev-biochem-061608-091319
49. Ladasky JJ, Boyle S, Seth M, Li H, Pentcheva T, Abe F, et al. Bap31 enhances the endoplasmic reticulum export and quality control of human class I MHC molecules. J Immunol (2006) 177:6172–81. doi:10.4049/jimmunol.177.9.6172
50. Paquet ME, Cohen-Doyle M, Shore GC, Williams DB. Bap29/31 influences the intracellular traffic of MHC class I molecules. J Immunol (2004) 172:7548–55. doi:10.4049/jimmunol.172.12.7548
51. Donaldson JG, Williams DB. Intracellular assembly and trafficking of MHC class I molecules. Traffic (2009) 10:1745–52. doi:10.1111/j.1600-0854.2009.00979.x
52. Steinman RM, Mellman IS, Muller WA, Cohn ZA. Endocytosis and the recycling of plasma membrane. J Cell Biol (1983) 96:1–27. doi:10.1083/jcb.96.1.1
53. Traub LM, Bonifacino JS. Cargo recognition in clathrin-mediated endocytosis. Cold Spring Harb Perspect Biol (2013) 5:a016790. doi:10.1101/cshperspect.a016790
54. Nichols BJ, Lippincott-Schwartz J. Endocytosis without clathrin coats. Trends Cell Biol (2001) 11:406–12. doi:10.1016/S0962-8924(01)02107-9
55. Mayor S, Parton RG, Donaldson JG. Clathrin-independent pathways of endocytosis. Cold Spring Harb Perspect Biol (2014) 6:a016758. doi:10.1101/cshperspect.a016758
56. Radhakrishna H, Donaldson JG. ADP-ribosylation factor 6 regulates a novel plasma membrane recycling pathway. J Cell Biol (1997) 139:49–61. doi:10.1083/jcb.139.1.49
57. Naslavsky N, Weigert R, Donaldson JG. Characterization of a nonclathrin endocytic pathway: membrane cargo and lipid requirements. Mol Biol Cell (2004) 15:3542–52. doi:10.1091/mbc.E04-02-0151
58. Peters PJ, Hsu VW, Ooi CE, Finazzi D, Teal SB, Oorschot V, et al. Overexpression of wild-type and mutant ARF1 and ARF6: distinct perturbations of nonoverlapping membrane compartments. J Cell Biol (1995) 128:1003–17. doi:10.1083/jcb.128.6.1003
59. Honda A, Nogami M, Yokozeki T, Yamazaki M, Nakamura H, Watanabe H, et al. Phosphatidylinositol 4-phosphate 5-kinase alpha is a downstream effector of the small G protein ARF6 in membrane ruffle formation. Cell (1999) 99:521–32. doi:10.1016/S0092-8674(00)81540-8
60. Jovanovic OA, Brown FD, Donaldson JG. An effector domain mutant of Arf6 implicates phospholipase D in endosomal membrane recycling. Mol Biol Cell (2006) 17:327–35. doi:10.1091/mbc.E05-06-0523
61. Shinozaki-Narikawa N, Kodama T, Shibasaki Y. Cooperation of phosphoinositides and BAR domain proteins in endosomal tubulation. Traffic (2006) 7:1539–50. doi:10.1111/j.1600-0854.2006.00480.x
62. Fernandez-Ulibarri I, Vilella M, Lazaro-Dieguez F, Sarri E, Martinez SE, Jimenez N, et al. Diacylglycerol is required for the formation of COPI vesicles in the Golgi-to-ER transport pathway. Mol Biol Cell (2007) 18:3250–63. doi:10.1091/mbc.E07-04-0334
63. Yang JS, Gad H, Lee SY, Mironov A, Zhang L, Beznoussenko GV, et al. A role for phosphatidic acid in COPI vesicle fission yields insights into Golgi maintenance. Nat Cell Biol (2008) 10:1146–53. doi:10.1038/ncb1774
64. Gutierrez-Martinez E, Fernandez-Ulibarri I, Lazaro-Dieguez F, Johannes L, Pyne S, Sarri E, et al. Lipid phosphate phosphatase 3 participates in transport carrier formation and protein trafficking in the early secretory pathway. J Cell Sci (2013) 126:2641–55. doi:10.1242/jcs.117705
65. Zagorac GB, Mahmutefendic H, Tomas MI, Kucic N, Le Bouteiller P, Lucin P. Early endosomal rerouting of major histocompatibility class I conformers. J Cell Physiol (2012) 227:2953–64. doi:10.1002/jcp.23042
66. Mahmutefendic H, Zagorac GB, Tomas MI, Groettrup M, Momburg F, Lucin P. Endosomal trafficking of open major histocompatibility class I conformers – implications for presentation of endocytosed antigens. Mol Immunol (2013) 55:149–52. doi:10.1016/j.molimm.2012.10.008
67. Caplan S, Naslavsky N, Hartnell LM, Lodge R, Polishchuk RS, Donaldson JG, et al. A tubular EHD1-containing compartment involved in the recycling of major histocompatibility complex class I molecules to the plasma membrane. EMBO J (2002) 21:2557–67. doi:10.1093/emboj/21.11.2557
68. Weigert R, Yeung AC, Li J, Donaldson JG. Rab22a regulates the recycling of membrane proteins internalized independently of clathrin. Mol Biol Cell (2004) 15:3758–70. doi:10.1091/mbc.E04-04-0342
69. Barral DC, Cavallari M, Mccormick PJ, Garg S, Magee AI, Bonifacino JS, et al. CD1a and MHC class I follow a similar endocytic recycling pathway. Traffic (2008) 9:1446–57. doi:10.1111/j.1600-0854.2008.00781.x
70. Noriega VM, Hesse J, Gardner TJ, Besold K, Plachter B, Tortorella D. Human cytomegalovirus US3 modulates destruction of MHC class I molecules. Mol Immunol (2012) 51:245–53. doi:10.1016/j.molimm.2012.03.024
71. Wiertz EJ, Jones TR, Sun L, Bogyo M, Geuze HJ, Ploegh HL. The human cytomegalovirus US11 gene product dislocates MHC class I heavy chains from the endoplasmic reticulum to the cytosol. Cell (1996) 84:769–79. doi:10.1016/S0092-8674(00)81054-5
72. Schwartz O, Marechal V, Le Gall S, Lemonnier F, Heard JM. Endocytosis of major histocompatibility complex class I molecules is induced by the HIV-1 Nef protein. Nat Med (1996) 2:338–42. doi:10.1038/nm0396-338
73. Coscoy L, Ganem D. Kaposi’s sarcoma-associated herpesvirus encodes two proteins that block cell surface display of MHC class I chains by enhancing their endocytosis. Proc Natl Acad Sci U S A (2000) 97:8051–6. doi:10.1073/pnas.140129797
74. Williams RL, Urbe S. The emerging shape of the ESCRT machinery. Nat Rev Mol Cell Biol (2007) 8:355–68. doi:10.1038/nrm2162
75. Duncan LM, Piper S, Dodd RB, Saville MK, Sanderson CM, Luzio JP, et al. Lysine-63-linked ubiquitination is required for endolysosomal degradation of class I molecules. EMBO J (2006) 25:1635–45. doi:10.1038/sj.emboj.7601056
76. Nathan JA, Lehner PJ. The trafficking and regulation of membrane receptors by the RING-CH ubiquitin E3 ligases. Exp Cell Res (2009) 315:1593–600. doi:10.1016/j.yexcr.2008.10.026
77. Eyster CA, Cole NB, Petersen S, Viswanathan K, Fruh K, Donaldson JG. MARCH ubiquitin ligases alter the itinerary of clathrin-independent cargo from recycling to degradation. Mol Biol Cell (2011) 22:3218–30. doi:10.1091/mbc.E10-11-0874
78. Bartee E, Mansouri M, Hovey Nerenberg BT, Gouveia K, Fruh K. Downregulation of major histocompatibility complex class I by human ubiquitin ligases related to viral immune evasion proteins. J Virol (2004) 78:1109–20. doi:10.1128/JVI.78.3.1109-1120.2004
79. De Gassart A, Camosseto V, Thibodeau J, Ceppi M, Catalan N, Pierre P, et al. MHC class II stabilization at the surface of human dendritic cells is the result of maturation-dependent MARCH I down-regulation. Proc Natl Acad Sci U S A (2008) 105:3491–6. doi:10.1073/pnas.0708874105
80. Rescigno M, Citterio S, Thery C, Rittig M, Medaglini D, Pozzi G, et al. Bacteria-induced neo-biosynthesis, stabilization, and surface expression of functional class I molecules in mouse dendritic cells. Proc Natl Acad Sci U S A (1998) 95:5229–34. doi:10.1073/pnas.95.9.5229
81. Cella M, Salio M, Sakakibara Y, Langen H, Julkunen I, Lanzavecchia A. Maturation, activation, and protection of dendritic cells induced by double-stranded RNA. J Exp Med (1999) 189:821–9. doi:10.1084/jem.189.5.821
82. Delamarre L, Holcombe H, Mellman I. Presentation of exogenous antigens on major histocompatibility complex (MHC) class I and MHC class II molecules is differentially regulated during dendritic cell maturation. J Exp Med (2003) 198:111–22. doi:10.1084/jem.20021542
83. Ackerman AL, Cresswell P. Regulation of MHC class I transport in human dendritic cells and the dendritic-like cell line KG-1. J Immunol (2003) 170:4178–88. doi:10.4049/jimmunol.170.8.4178
84. Sohn HW, Shin YK, Lee IS, Bae YM, Suh YH, Kim MK, et al. CD99 regulates the transport of MHC class I molecules from the Golgi complex to the cell surface. J Immunol (2001) 166:787–94. doi:10.4049/jimmunol.166.2.787
85. MacAry PA, Lindsay M, Scott MA, Craig JI, Luzio JP, Lehner PJ. Mobilization of MHC class I molecules from late endosomes to the cell surface following activation of CD34-derived human Langerhans cells. Proc Natl Acad Sci U S A (2001) 98:3982–7. doi:10.1073/pnas.071477498
86. Shen L, Sigal LJ, Boes M, Rock KL. Important role of cathepsin S in generating peptides for TAP-independent MHC class I crosspresentation in vivo. Immunity (2004) 21:155–65. doi:10.1016/j.immuni.2004.07.004
87. Sigal LJ, Rock KL. Bone marrow-derived antigen-presenting cells are required for the generation of cytotoxic T lymphocyte responses to viruses and use transporter associated with antigen presentation (TAP)-dependent and -independent pathways of antigen presentation. J Exp Med (2000) 192:1143–50. doi:10.1084/jem.192.8.1143
88. Rodriguez A, Regnault A, Kleijmeer M, Ricciardi-Castagnoli P, Amigorena S. Selective transport of internalized antigens to the cytosol for MHC class I presentation in dendritic cells. Nat Cell Biol (1999) 1:362–8. doi:10.1038/14058
89. Rock KL, Goldberg AL. Degradation of cell proteins and the generation of MHC class I-presented peptides. Annu Rev Immunol (1999) 17:739–79. doi:10.1146/annurev.immunol.17.1.739
90. Weimershaus M, Evnouchidou I, Saveanu L, Van Endert P. Peptidases trimming MHC class I ligands. Curr Opin Immunol (2013) 25:90–6. doi:10.1016/j.coi.2012.10.001
91. Saveanu L, Carroll O, Lindo V, Del Val M, Lopez D, Lepelletier Y, et al. Concerted peptide trimming by human ERAP1 and ERAP2 aminopeptidase complexes in the endoplasmic reticulum. Nat Immunol (2005) 6:689–97. doi:10.1038/ni1208
92. Saveanu L, Carroll O, Weimershaus M, Guermonprez P, Firat E, Lindo V, et al. IRAP identifies an endosomal compartment required for MHC class I cross-presentation. Science (2009) 325:213–7. doi:10.1126/science.1172845
93. Serwold T, Gonzalez F, Kim J, Jacob R, Shastri N. ERAAP customizes peptides for MHC class I molecules in the endoplasmic reticulum. Nature (2002) 419:480–3. doi:10.1038/nature01074
94. Subtil A, Lampson MA, Keller SR, Mcgraw TE. Characterization of the insulin-regulated endocytic recycling mechanism in 3T3-L1 adipocytes using a novel reporter molecule. J Biol Chem (2000) 275:4787–95. doi:10.1074/jbc.275.7.4787
95. Watson RT, Hou JC, Pessin JE. Recycling of IRAP from the plasma membrane back to the insulin-responsive compartment requires the Q-SNARE syntaxin 6 but not the GGA clathrin adaptors. J Cell Sci (2008) 121:1243–51. doi:10.1242/jcs.017517
96. Saveanu L, van Endert P. The role of insulin-regulated aminopeptidase in MHC class I antigen presentation. Front Immunol (2012) 3:57. doi:10.3389/fimmu.2012.00057
97. Weimershaus M, Maschalidi S, Sepulveda F, Manoury B, Van Endert P, Saveanu L. Conventional dendritic cells require IRAP-Rab14 endosomes for efficient cross-presentation. J Immunol (2012) 188:1840–6. doi:10.4049/jimmunol.1101504
98. Reid PA, Watts C. Cycling of cell-surface MHC glycoproteins through primaquine-sensitive intracellular compartments. Nature (1990) 346:655–7. doi:10.1038/346655a0
99. Pfeifer JD, Wick MJ, Roberts RL, Findlay K, Normark SJ, Harding CV. Phagocytic processing of bacterial antigens for class I MHC presentation to T cells. Nature (1993) 361:359–62. doi:10.1038/361359a0
100. Chiu I, Davis DM, Strominger JL. Trafficking of spontaneously endocytosed MHC proteins. Proc Natl Acad Sci U S A (1999) 96:13944–9. doi:10.1073/pnas.96.24.13944
101. Rock KL, Shen L. Cross-presentation: underlying mechanisms and role in immune surveillance. Immunol Rev (2005) 207:166–83. doi:10.1111/j.0105-2896.2005.00301.x
102. Lizee G, Basha G, Tiong J, Julien JP, Tian M, Biron KE, et al. Control of dendritic cell cross-presentation by the major histocompatibility complex class I cytoplasmic domain. Nat Immunol (2003) 4:1065–73. doi:10.1038/ni989
103. Basha G, Lizee G, Reinicke AT, Seipp RP, Omilusik KD, Jefferies WA. MHC class I endosomal and lysosomal trafficking coincides with exogenous antigen loading in dendritic cells. PLoS One (2008) 3:e3247. doi:10.1371/journal.pone.0003247
104. Le Gall S, Erdtmann L, Benichou S, Berlioz-Torrent C, Liu L, Benarous R, et al. Nef interacts with the mu subunit of clathrin adaptor complexes and reveals a cryptic sorting signal in MHC I molecules. Immunity (1998) 8:483–95. doi:10.1016/S1074-7613(00)80553-1
105. Roeth JF, Williams M, Kasper MR, Filzen TM, Collins KL. HIV-1 Nef disrupts MHC-I trafficking by recruiting AP-1 to the MHC-I cytoplasmic tail. J Cell Biol (2004) 167:903–13. doi:10.1083/jcb.200407031
106. Lubben NB, Sahlender DA, Motley AM, Lehner PJ, Benaroch P, Robinson MS. HIV-1 Nef-induced down-regulation of MHC class I requires AP-1 and clathrin but not PACS-1 and is impeded by AP-2. Mol Biol Cell (2007) 18:3351–65. doi:10.1091/mbc.E07-03-0218
107. Jia X, Singh R, Homann S, Yang H, Guatelli J, Xiong Y. Structural basis of evasion of cellular adaptive immunity by HIV-1 Nef. Nat Struct Mol Biol (2012) 19:701–6. doi:10.1038/nsmb.2328
108. Kulpa DA, Del Cid N, Peterson KA, Collins KL. Adaptor protein 1 promotes cross-presentation through the same tyrosine signal in major histocompatibility complex class I as that targeted by HIV-1. J Virol (2013) 87:8085–98. doi:10.1128/JVI.00701-13
109. Hofmann MW, Honing S, Rodionov D, Dobberstein B, Von Figura K, Bakke O. The leucine-based sorting motifs in the cytoplasmic domain of the invariant chain are recognized by the clathrin adaptors AP1 and AP2 and their medium chains. J Biol Chem (1999) 274:36153–8. doi:10.1074/jbc.274.51.36153
110. Basha G, Omilusik K, Chavez-Steenbock A, Reinicke AT, Lack N, Choi KB, et al. A CD74-dependent MHC class I endolysosomal cross-presentation pathway. Nat Immunol (2012) 13:237–45. doi:10.1038/ni.2225
111. Guermonprez P, Saveanu L, Kleijmeer M, Davoust J, Van Endert P, Amigorena S. ER-phagosome fusion defines an MHC class I cross-presentation compartment in dendritic cells. Nature (2003) 425:397–402. doi:10.1038/nature01911
112. Houde M, Bertholet S, Gagnon E, Brunet S, Goyette G, Laplante A, et al. Phagosomes are competent organelles for antigen cross-presentation. Nature (2003) 425:402–6. doi:10.1038/nature01912
113. Cebrian I, Visentin G, Blanchard N, Jouve M, Bobard A, Moita C, et al. Sec22b regulates phagosomal maturation and antigen crosspresentation by dendritic cells. Cell (2011) 147:1355–68. doi:10.1016/j.cell.2011.11.021
114. Ackerman AL, Giodini A, Cresswell P. A role for the endoplasmic reticulum protein retrotranslocation machinery during crosspresentation by dendritic cells. Immunity (2006) 25:607–17. doi:10.1016/j.immuni.2006.08.017
115. Merzougui N, Kratzer R, Saveanu L, Van Endert P. A proteasome-dependent, TAP-independent pathway for cross-presentation of phagocytosed antigen. EMBO Rep (2011) 12:1257–64. doi:10.1038/embor.2011.203
116. Zou L, Zhou J, Zhang J, Li J, Liu N, Chai L, et al. The GTPase Rab3b/3c-positive recycling vesicles are involved in cross-presentation in dendritic cells. Proc Natl Acad Sci U S A (2009) 106:15801–6. doi:10.1073/pnas.0905684106
117. Burgdorf S, Scholz C, Kautz A, Tampe R, Kurts C. Spatial and mechanistic separation of cross-presentation and endogenous antigen presentation. Nat Immunol (2008) 9:558–66. doi:10.1038/ni.1601
118. Nair-Gupta P, Baccarini A, Tung N, Seyffer F, Florey O, Huang Y, et al. TLR signals induce phagosomal MHC-I delivery from the endosomal recycling compartment to allow cross-presentation. Cell (2014) 158:506–21. doi:10.1016/j.cell.2014.04.054
119. Parham P, Barnstable CJ, Bodmer WF. Use of a monoclonal antibody (W6/32) in structural studies of HLA-A,B,C, antigens. J Immunol (1979) 123:342–9.
120. Stam NJ, Spits H, Ploegh HL. Monoclonal antibodies raised against denatured HLA-B locus heavy chains permit biochemical characterization of certain HLA-C locus products. J Immunol (1986) 137:2299–306.
121. Saric T, Chang SC, Hattori A, York IA, Markant S, Rock KL, et al. An IFN-gamma-induced aminopeptidase in the ER, ERAP1, trims precursors to MHC class I-presented peptides. Nat Immunol (2002) 3:1169–76. doi:10.1038/ni859
122. York IA, Chang SC, Saric T, Keys JA, Favreau JM, Goldberg AL, et al. The ER aminopeptidase ERAP1 enhances or limits antigen presentation by trimming epitopes to 8-9 residues. Nat Immunol (2002) 3:1177–84. doi:10.1038/ni860
123. Yan J, Parekh VV, Mendez-Fernandez Y, Olivares-Villagomez D, Dragovic S, Hill T, et al. In vivo role of ER-associated peptidase activity in tailoring peptides for presentation by MHC class Ia and class Ib molecules. J Exp Med (2006) 203:647–59. doi:10.1084/jem.20052271
124. Firat E, Huai J, Saveanu L, Gaedicke S, Aichele P, Eichmann K, et al. Analysis of direct and cross-presentation of antigens in TPPII knockout mice. J Immunol (2007) 179:8137–45. doi:10.4049/jimmunol.179.12.8137
Keywords: MHC class I molecules, cross-presentation, endosomal recycling compartment, tubular endosomes, dendritic cells
Citation: Adiko AC, Babdor J, Gutiérrez-Martínez E, Guermonprez P and Saveanu L (2015) Intracellular transport routes for MHC I and their relevance for antigen cross-presentation. Front. Immunol. 6:335. doi: 10.3389/fimmu.2015.00335
Received: 01 May 2015; Accepted: 15 June 2015;
Published: 02 July 2015
Edited by:
Monique Gannagé, University of Geneva, SwitzerlandReviewed by:
Nicolas Blanchard, Center for Pathophysiology of Toulouse-Purpan, FranceSven Burgdorf, Rheinische Friedrich-Willhelms-University, Germany
Copyright: © 2015 Adiko, Babdor, Gutiérrez-Martínez, Guermonprez and Saveanu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Loredana Saveanu, INSERM U1149, Faculté Bichat Medical School, ELR8252 CNRS, Center for Research on Inflammation, 16 Henri Huchard, 75018, Paris, France,bG9yZWRhbmEuc2F2ZWFudUBpbnNlcm0uZnI=