 Christine Riedhammer
Christine Riedhammer Robert Weissert
Robert Weissert
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 17 June 2015
Sec. Multiple Sclerosis and Neuroimmunology
Volume 6 - 2015 | https://doi.org/10.3389/fimmu.2015.00322
Antigen presentation is in the center of the immune system, both in host defense against pathogens, but also when the system is unbalanced and autoimmune diseases like multiple sclerosis (MS) develop. It is not just by chance that a major histocompatibility complex gene is the major genetic susceptibility locus in MS; a feature that MS shares with other autoimmune diseases. The exact etiology of the disease, however, has not been fully understood yet. T cells are regarded as the major players in the disease, but most probably a complex interplay of altered central and peripheral tolerance mechanisms, T-cell and B-cell functions, characteristics of putative autoantigens, and a possible interference of environmental factors like microorganisms are at work. In this review, new data on all these different aspects of antigen presentation and their role in MS will be discussed, probable autoantigens will be summarized, and comparisons to other autoimmune diseases will be drawn.
Multiple sclerosis (MS) is an inflammatory disease of the central nervous system (CNS), causing a loss of myelin sheaths and a degeneration of axons preferably in the white matter, but also in the cortex (1).
Despite immense exploratory efforts, the pathogenesis and target antigen(s) of MS have so far not or only partially been identified. This problem is also present in other autoimmune disease like rheumatoid arthritis (RA) or type 1 diabetes mellitus (T1D). On the other hand, research has revealed the targets of other diseases like myasthenia gravis (MG) or neuromyelitis optica (NMO), but their exact pathogeneses by the interplay of different immune cells still remain unclear.
Multiple sclerosis is widely thought to be induced and perpetuated by the immune system. T cells are believed to have a central role in the pathogenesis of the disease. It is assumed that defects of central and peripheral tolerance permit an existence of self-reactive T cells, which are activated by antigen-presenting cells (APCs).
As different autoimmune diseases share the feature that risk is conferred by genes that are encoded within the major histocompatibility complex (MHC) locus, antigen presentation generally seems to be of great importance in autoimmune diseases. This review will point out important mechanisms of antigen presentation, autoantigens as well as the contribution of different types of immune cells in autoimmune diseases. The focus will be on MS, but other autoimmune diseases like RA, T1D, MG, NMO, and autoimmune encephalitides (AE) will provide comparisons concerning the genetic background, the ambiguity of the identified or suspected target antigens, and the underlying pathogenic mechanisms.
Peptide fragments that are presented on MHC class I or MHC class II molecules are first processed within the cell before they reach the cell membrane on the respective MHC molecule (2). The processing machineries for MHC class I and MHC class II molecules differ. Peptides derived from the cytosol and the nucleus are presented on MHC class I molecules. For their processing, the proteasome is of paramount importance. Inhibition of proteasomal function can block antigen presentation (3). Interferon (IFN)-γ can induce the immunoproteasome, which leads to the processing of a different set of peptides compared to the constitutive proteasome (4). Apart from the proteasome, also the endoplasmic reticulum (ER) is important for the presentation of peptides on MHC class I molecules. Specialized aminopeptidases are involved in trimming the peptides to a length of 8–10 amino acids (5, 6). Peptides are subsequently loaded on MHC class I molecules. In this process, tapasin and the transporter associated with antigen processing (TAP) are involved, which are expressed in the ER (7). The binding groove of MHC class I molecules is closed, limiting the length of the presented peptides to 8–10 amino acids, even though examples of longer peptides presented on MHC class I molecules exist (8).
Major histocompatibility complex class II molecules bind antigens that are derived from extracellular proteins. These are taken up by endosomes and subsequently processed. The pH in the endosomes is very important for the control of protease activity (9). The binding grooves of MHC class II molecules in the endosomes are normally covered by the invariant chain (10). A specific part of the invariant chain (the CLIP) covers the groove and can be removed by human leukocyte antigen (HLA)-DM (11). Subsequently, peptides that are present in the endosomes can bind in the groove and be presented to T cells. The binding groove of MHC class II molecules is open and presented peptides are generally 14–18 amino acids in length. Under certain conditions, however, also shorter or longer peptides can be presented. While MHC class I molecules are present on all nucleated cells, MHC class II molecules are only present on specialized APCs like dendritic cells (DCs), B cells, and macrophages. In the CNS, microglia are considered as professional APCs (12). Also, astrocytes can present antigen on MHC class II molecules under certain conditions (13). Since the uptake of antigen differs between different types of professional APCs, also the presented set of peptides can differ. In addition, also the intracellular composition of molecules differs regarding the antigen processing machinery (14).
There is cross-presentation of antigens in the MHC class I and MHC class II pathway (15, 16). Different pathways allow this cross-presentation. Phagosomes that take up antigen from the extracellular space can have ER-associated molecules. Also certain immunopeptidases contribute to cross-presentation (17). Cross-presented antigens from extracellular origin are mainly presented on MHC class I molecules of DCs (18).
In regard to autoimmune diseases, the exact sequence of events leading to the presentation of certain sets of autoantigen-derived peptides on APCs in humans is still incompletely deciphered. Potentially, all molecules involved in antigen processing and binding of autoantigens to MHC molecules might influence the emergence of autoimmune disease. Even though the role of the different steps in antigen processing in autoimmune diseases has not been fully established yet, much will be learned over the next years and be of potential importance for the design of new therapies.
The emergence of an autoimmune disease apparently requires the existence of self-reactive cells which escape central and peripheral tolerance mechanisms and become activated in some way. Self-reactive T cells have been shown to be present both in persons with and without autoimmune disease (19–26). How self-tolerance is disrupted in autoimmune disease, however, remains to be ascertained. The understanding of how tolerance mechanisms work is also the basis for a comprehension of potential deficiencies.
The challenge of intrathymic central tolerance induction includes tolerizing T cells reactive with antigens which are not expressed ubiquitously, but only found in particular organs (tissue-restricted antigens, TRAs). The intrathymic expression of TRAs can induce tolerance and prevent development of autoimmune disease (27). This is achieved by negative selection of T cells in the thymic medulla, where T cells exhibiting a high affinity/avidity interaction between their T-cell receptor (TCR) and MHC–self-peptide complexes presented by medullary APCs become apoptotic. Medullary thymic epithelial cells (mTECs) and DCs cooperate to achieve this (28). The thymic transcription factor autoimmune regulator (AIRE) plays a fundamental role in central tolerance induction, as it controls the transcription of genes coding for TRAs (29), and is predisposed, when not working properly, to favor the development of autoimmune disease. Certain single nucleotide polymorphisms (SNPs) in the AIRE gene have been suggested to play a role in autoimmune diseases like RA or MG (30–32). Interestingly, mTECs make use of macroautophagy for presenting antigens also on MHC class II molecules, which enables them to tolerize CD4+ T cells (33). Important candidate autoantigens of MS and other autoimmune diseases seem to be present in the human thymus (34–36). Consequently, a lack of presentation of important self-antigens in the thymus cannot fully account for the existence of autoreactive T cells.
Apart from thymocytes recognizing a peptide that is only rarely presented in the thymus, also thymocytes expressing a TCR whose affinity/avidity for a peptide–MHC complex is too low to eliminate the thymocytes during negative selection are thought to be able to elude central tolerance mechanisms (37).
Post-translational modifications of self-antigens in the periphery can also change the affinity/avidity of the interaction between a TCR and a self-peptide–MHC complex. Thus, the affinity/avidity between a TCR and a self-peptide–MHC complex might be low enough to avoid central tolerance induction, but high enough to lead to an activation of autoreactive T cells in the periphery after a post-translational modification of the antigen. For example, thiopalmitoylation occurs frequently with proteolipid protein (PLP), a putative autoantigen in MS, and it was found that post-translationally modified PLP is more encephalitogenic than the unmodified form (38). Also citrullinated proteins were found more frequently in MS lesions than in control brain tissue (39).
Another interesting aspect is the structural nature of the interaction between a TCR of a potentially self-reactive T cell and its respective peptide–MHC complex in the thymus. An analysis of the crystal structure of a TCR and a self-peptide–MHC complex in an MS patient showed that the TCR bound in a tilted way to the peptide–MHC complex (40). Such structural deviations might result in a low affinity/avidity of the interaction between TCR and self-peptide–MHC complex, so that the T cell can escape from central tolerance. In the periphery, however, the quantity of self-antigen is higher and the affinity/avidity might then be high enough to contribute to the pathogenesis of autoimmune disease. It was shown that autoreactive T-cell clones both in MS and T1D exhibited a lower recruitment of self-peptide–MHC complexes than T cells from the same patients specific for viral peptides. Nevertheless, the autoreactive T-cell clones proliferated vigorously in response to self-antigen, indicating that TCR signaling is still functional in these cells (41). Again, these facts might support an escape of negative selection and a possible activation of these cells in the periphery.
When self-reactive T cells escape central tolerance mechanisms, peripheral tolerance mechanisms are required, which include ignorance, i.e., the physical separation of autoreactive cells and their target, the deletion of autoreactive cells, their inhibition, e.g., by a lack of costimulatory factors and their suppression by regulatory T cells (42).
An important factor for the inhibition of T cells is the function of the cytotoxic T-lymphocyte-associated protein 4 (CTLA-4 = CD152). Its role in MS has been investigated in many studies. A recent meta-analysis of genetic polymorphisms of the CTLA-4 gene did not reveal any associations with MS (43). However, there are several studies pointing toward a significance of costimulatory mechanisms for MS. Myelin-reactive T cells from MS patients were observed to be less sensitive to a lack of costimulation than T cells from controls (44–46). Also, another study found increased T cells insensitive to costimulation in MS patients, which predominantly produced Th1 cytokines (47). In active MS, increased CD80+ cells were found (48).
An important mechanism for the deletion or “activation-induced cell death” of autoreactive cells is the interaction of Fas and Fas ligand, which can induce apoptosis in autoreactive T cells. A higher resistance of T cells to apoptosis in MS patients has been reported in several studies which found decreased expression of Fas and a lower rate of apoptosis induced by stimulation with mitogen in T cells from MS patients (49, 50). New data indicate that Th1 cells seem to be more susceptible for Fas-mediated apoptosis than Th17 cells, both in MS patients and healthy controls (51).
Regulatory T cells are probably the most important contributors to the maintenance of peripheral tolerance. They are mainly defined by the expression of the transcription factor Foxp3 (52). Foxp3+ regulatory T cells are able to suppress proliferation of CD4+CD25− T cells (52, 53). It was found that interactions of thymocytes with MHC class II molecules expressed by mTECs were important for the induction of regulatory T cells (54). The possible role of different regulatory cell types in MS is discussed below in the respective sections.
The CNS as the target organ of the autoimmune response in MS is a special one. It is only a short time ago that it was recognized that the CNS is physiologically supervised by immune cells. Thereby, memory T cells are thought to play an important role (55).
It is believed that myelin-specific T cells have a crucial role in the pathogenesis of MS (56). Naïve myelin-specific T cells, which cannot access areas outside the secondary lymphoid tissues (57), are thought to be activated in the periphery, gain access to the CNS, and then be reactivated by CNS-resident cells presenting self-antigen (37). The CNS is extraordinarily shielded from the periphery by the existence of the brain–blood barrier (BBB) and the brain–cerebrospinal fluid (CSF) barrier. For activated T cells, there are three routes to enter the CNS, i.e., by crossing the BBB or the blood–CSF barrier or by migrating through the carotid arteries into the subarachnoid space (58). The most important pathogenic mechanisms in MS are outlined in Figure 1.
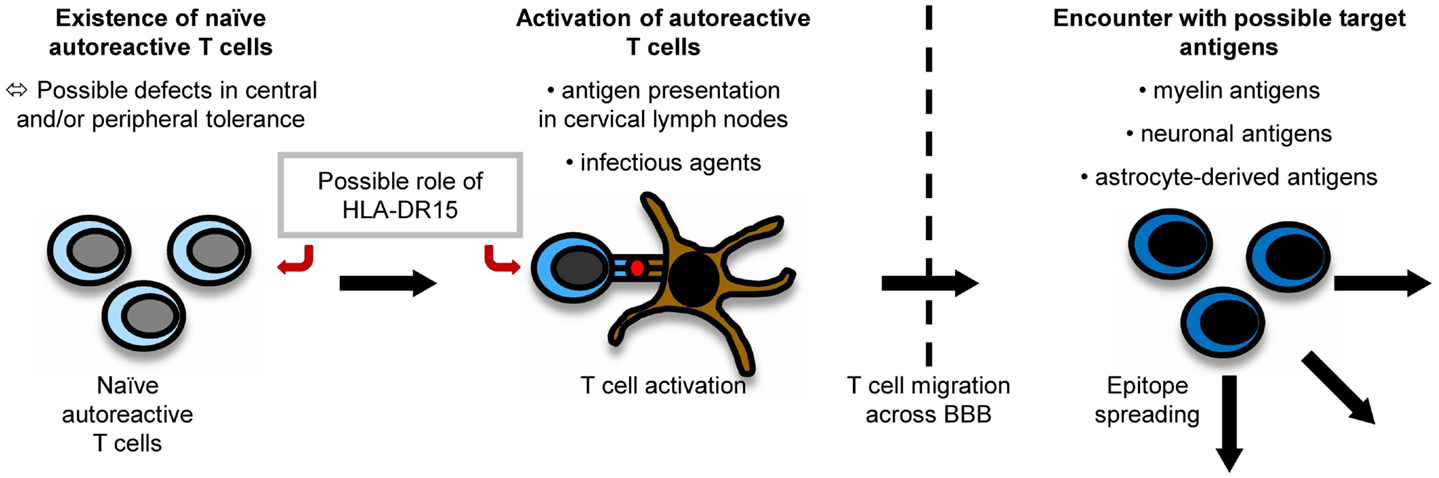
Figure 1. Pathogenesis of MS. The exact pathogenesis of MS is not clear yet. However, it can be imagined that naïve autoreactive T cells exist because of imperfections in central and peripheral tolerance mechanisms. They might then become activated by antigen presentation in cervical lymph nodes or effects of molecular mimicry. Antigen presentation on disease-associated HLA-DR15-molecules might influence both the emergence and activation of autoreactive T cells. When autoreactive T cells reach the CNS by crossing the BBB, they encounter their target antigen and start an inflammatory cascade, in which other antigens become unveiled, triggering the process of epitope spreading.
How T cells specific for CNS antigen are activated in the periphery remains unclear. However, their activation could result from a trigger by an infectious agent or from the presentation of myelin antigens in the cervical lymph nodes (37). Concerning the latter mechanism, CSF is drained via the nasal lymphatic system to the cervical lymph nodes. In experimental autoimmune encephalomyelitis (EAE) induced in marmosets, a relevant animal model of MS, myelin components were detected to be increased in APCs in cervical lymph nodes (59). A study using fine-needle biopsy material of cervical lymph nodes of MS patients and controls also detected more macrophages containing myelin basic protein (MBP) and PLP in MS patients than in controls (60). Therefore, APCs presenting myelin fragments in the cervical lymph nodes might contribute to an activation of naïve self-reactive T cells.
Apart from this concept, also infectious agents are considered to play an important role in triggering autoimmune diseases. In this context, a cross-talk between the innate and adaptive immune system might be relevant: infectious agents can activate APCs via Toll-like receptors (TLR). APCs can then contribute to the activation of self-reactive T cells (61).
Structural similarities between antigens of infectious agents and myelin proteins (molecular mimicry) can induce an activation of naïve autoreactive T cells which recognize peptides derived from infectious agents and self-antigens. Novel data indicate that cross-reactivities do not necessarily require nearly identical amino acid sequences. They also occur when some important motifs are conserved which make sure the overall structures of TCR–peptide–MHC interaction are similar (62). This suggests that cross-reactivities may happen frequently. There has been intensive research on possible infectious triggers of MS: it has been shown that T-cell clones specific for an immunodominant epitope of MBP, namely MBP85–99, also recognize viral peptides, e.g., of Epstein Barr virus (EBV), influenza A virus, herpes simplex virus, human papilloma virus (63), or human herpesvirus-6 (64). Concerning EBV, for example, MS patients seem to exhibit higher antibody titers against certain antigenic components of the virus than controls even before the onset of MS (65). In EAE experiments, it could also be shown that a peptide from H. influenzae mimicking a PLP-peptide can actually induce CNS disease (66).
In the context of molecular mimicry, also self-mimicry has been observed. Transgenic myelin oligodendrocyte glycoprotein (MOG)-deficient mice expressing a MOG-specific TCR develop EAE due to a cross-reactivity between a MOG epitope and neurofilament NF-M (67). Such cross-reactivities could play a role in the induction of axonal damage also in human MS.
Independently from cross-reactivities, infectious agents can lead to a disruption of tolerance to self-antigens by bystander activation. For example, demyelination can be induced when certain immunodeficient (RAG2−/− transgenic) mice are infected with mouse hepatitis virus (MHV), even though the CD8+ T cells they possess are neither specific for MHV nor for CNS antigen, when their T cells are activated by the antigen they recognize (68).
Recently, besides molecular mimicry and bystander activation, another interesting mechanism has been proposed: myelin-specific CD8+ T cells expressing a dual TCR specific for both MBP and viral antigens have been discovered. The activation of such T cells during viral infection might also induce autoimmune reactions (69).
Besides infectious agents, commensal microbiota could be of importance in the pathogenesis of the disease. EAE in mice expressing a transgenic TCR for MOG was found to depend on the presence of the commensal gut flora (70).
During the course of an autoimmune disease, otherwise physiological immunological mechanisms like epitope spreading set in, which contribute to the perpetuation and diversification of the ongoing immune response. Epitope spreading means the expansion of the immune response to epitopes that are different from the initially targeted ones. This process is physiological and helpful in the fight against pathogens, but it also seems to play an important role in the emergence of autoimmune responses.
In EAE, it could be shown that the immune response is first focused on a certain epitope and then spreads to other epitopes during the chronification of the disease (71, 72). Apart from intramolecular epitope spreading (e.g., within different MBP epitopes), also intermolecular epitope spreading, e.g., from MOG to MBP, has been observed in different EAE models (71, 73, 74). In different animal models of MS, it could also be shown that epitope spreading can begin in the CNS (75). Interestingly, also in an animal model using the CNS-resident virus Theiler’s murine encephalomyelitis virus for disease induction, T-cell reactivities against certain myelin epitopes emerged during the course of the disease, which were not due to molecular mimicry (76). Epitope spreading was reported to be associated with clinical relapses in animal models, as T cells reactive with epitopes the immune response had spread to could induce disease in other animals (74).
Both intramolecular (24, 25, 77–79) and intermolecular (80) epitope spreading has been observed in MS patients as well. However, it remains to be proven that this process also plays a pathogenic role in the disease, as some studies could not detect any associations with clinical exacerbations (77, 78).
Epitope spreading is also involved in other autoimmune diseases, complicating the search for the initial target antigens of the autoimmune response and complicating also the development of potent therapies which should ideally operate in all or many patients. Further understanding of this process will be crucial for designing efficient therapies.
CD4+ T cells are widely considered major players in the pathogenesis of MS.
This is in part due to the fact that most of the genetic susceptibility for MS is associated with certain MHC class II alleles (81). CD4+ T cells have also been detected in MS lesions (82). Evidence also comes from a humanized mouse model: transgenic mice expressing the MS-associated DR2-molecule (DRA*0101/DRB1*1501), an MBP-specific TCR derived from MS patients and human CD4 develop disease with symptoms very similar to those in MS and more severe symptoms than mice lacking CD4 expression (83). It is not clear yet, which CD4+ T helper cell subset (Th cell) exerts the most important influence on the disease. The subsets which are discussed most are Th1 cells and Th17 cells. Figure 2 gives an overview of different T-cell subsets, their signature cytokines, and important transcription factors.
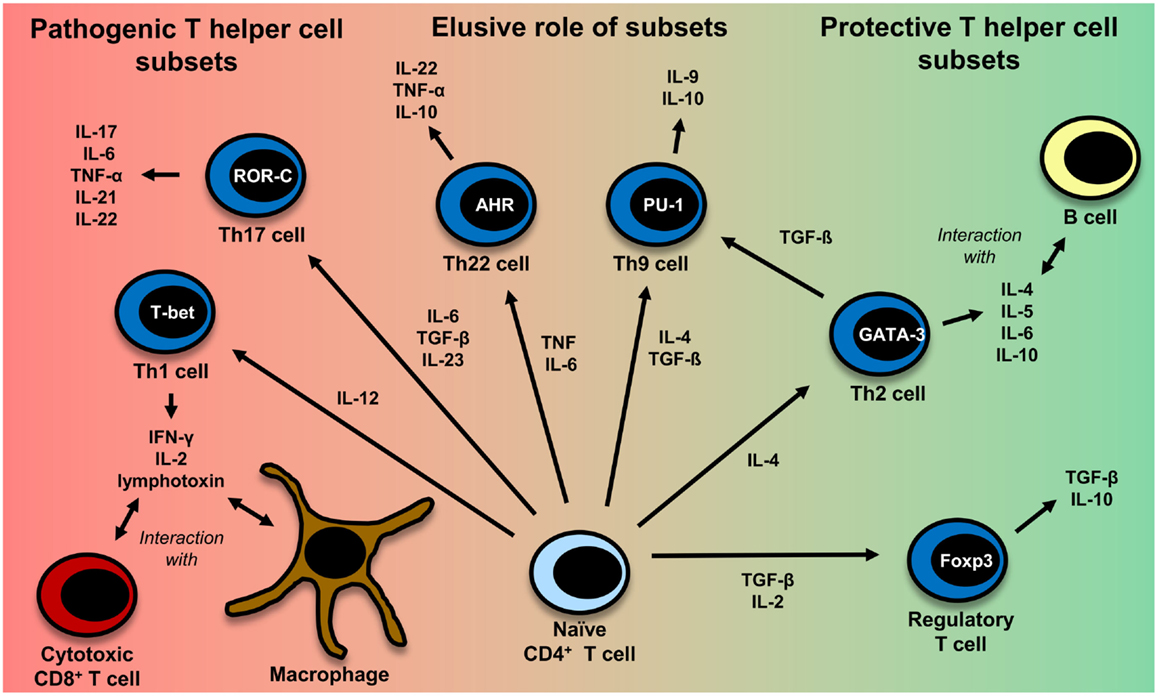
Figure 2. CD4+ T-cell differentiation. Naïve CD4+ T cells can differentiate into different T helper cell subsets. In MS, Th1 and Th17 cells are regarded to be disease-promoting, whereas regulatory T cells and Th2 cells seem to exert a protective effect. The exact role of Th9 and Th22 cells remains to be further clarified. Th1 cells, which predominantly produce TNF (95), IFN-γ, IL-2 (332), and lymphotoxin-α (333), are induced by IL-12 (95). Their master regulator is the transcription factor (TF) T-bet (334). They mainly interact with cytotoxic CD8+ T cells and macrophages (335). Th2 cells, which mainly interact with B cells (335), are induced by IL-4 and secrete IL-4, IL-5, IL-6, and IL-10 (101, 333, 335). GATA-3 is the most important TF of Th2 cells (336). IL-6, TGF-β (93), and IL-23 (95) are important cytokines for the induction of Th17 cells producing IL-17, IL-6, TNFα (95), IL-21 (94), IL-22 (337). RORγt is the master regulator of this T-cell subset (338). Interestingly, TGF-β is also necessary for the induction of regulatory T cells, whereas IL-6 must be absent in this case (93, 117). The master regulator for the induction of regulatory T cells is the TF Foxp3 (52, 339). Regulatory T cells then produce TGF-β and IL-10 (117). Recently also Th9 cells and Th22 have been identified to be a discrete T-cell subset. The induction of Th9 cells is promoted by IL-4 and TGF-β (340). Th9 cells produce IL-9 and to a lesser extent IL-10 (340, 341). Under the influence of TGF-β, Th2 cells can switch their phenotype and become Th9 cells (342). Master regulators for Th9 and Th22 cells have not been identified so far. Important factors for the induction of Th9 cells are PU.1, IRF, BATF, different STAT factors, and TGF-β-induced SMADS (341, 343). Priming of IL-22 producing Th22 cells is promoted by IL-6 and TNF (103). The transcription factor aryl hydrocarbon regulator (AHR) seems to exert an important influence on IL-22 production (344).
Th1 cells have been and are seen as causative agents in MS, since MS-resembling animal models are driven by this T helper cell subset. There is also evidence that this T-cell subset might play a major role in human MS.
Administration of an altered peptide ligand (APL) based on an immunodominant epitope of MBP led to cross-recognition of MBP and the APL, resulting in exacerbations in some patients. The phenotype of the reactive T cells was skewed toward Th1 (84). Also other studies showed (partly by determination of chemokine receptor expression) that Th1 cells are especially increased in MS patients suffering a relapse compared to healthy controls or patients in remission (85–87). In PLP-specific T-cell lines isolated from MS patients during acute attacks, Th1 cytokines were found to be dominant (88) and also MBP-specific T-cell lines in MS patients produced significantly more IFN-γ than T-cell lines from controls (89). A correlation between IFN-γ-secretion (after stimulation with phytohemagglutinin) and contrast-enhancing lesions in MR was also detected (90). Higher levels of myelin-specific high-avidity CD4+ T cells were found in MS patients than in healthy controls, which exhibited a Th1 phenotype and higher IFN-γ production in MS patients (91). Recently, EAE experiments showed that Th1 cells first infiltrate the CNS and are suppressed by regulatory T cells, which stops progression of the disease, whereas Th17 cells infiltrate the CNS later during the disease course (92).
In the last years, interleukin (IL)-17 producing Th17 cells have moved to the focus of research. Important factors for the induction, differentiation, and maintenance of the Th17 cell repertoire are IL-6, transforming growth factor (TGF)-β (93), IL-21 (94), and IL-23 (95). IL-23-deficient mice have been shown to be unable to develop EAE (95). Interestingly, the development of regulatory T cells also requires TGF-β in the absence of IL-6 (93). IL-17 transcripts have been shown increased especially in chronic silent MS lesions (82) and IL-17 positive cells were also found increased in MS lesions (96). In MS patients, more Th17 cells have been detected than in healthy controls and NMO patients (97). A study investigated T-cell subsets in patients with an aggressive disease course who received high-intensity chemotherapy followed by autologous stem cell transplantation, which induced remission in all patients. Interestingly, Th17 cell numbers were still decreased 1 year after transplantation, whereas Th1 cells had reached normal levels, which might speak for an association between the decreased Th17 cells and the remission (98). Higher IL-17 production by myelin-reactive T cells has been reported in MS patients than in controls (99).
Recently, novel T-cell subsets have been detected whose role in MS has already begun to be investigated: Th9 cells reside mainly in the skin, where IL-9 might also play a role in the production of other cytokines like IFN-γ, IL-13, or IL-17 by T cells in the skin (100). Besides Th1 and Th17 cells, also Th9 cells could be shown to induce a milder form of EAE (101).
Similar to Th9 cells, Th22 cells were identified as a distinct T-cell subset to be present in the skin a few years ago (102, 103). Increased fractions of (myelin-reactive) Th22 cells have been reported in MS patients (104).
In MS patients, there does not seem to be a reduction of regulatory T cells in number (105–110), but there is some evidence that regulatory T cells might suppress autoreactive T cells less efficiently compared with healthy controls: regulatory T cells from MS patients seem to be less able to suppress effector T cells (106–108, 110–112) and have a lower cloning frequency than regulatory T cells from healthy controls (106). However, it must be stated that some of these studies assumed the CD4+CD25hi phenotype as characteristic for regulatory T cells. However, after Foxp3 had been discovered, it became clear that by far not all regulatory T cells express CD25 at high levels (53). New regulatory T cells either mature in the thymus or stem from an expansion of regulatory T cells in the periphery. The latter mechanism physiologically gains importance with increasing age. In MS patients, there seems to be a selective lack of regulatory T cells released from the thymus, to which the observed functional impairment might be ascribed. The difference in thymic release of regulatory T cells between MS patients and healthy controls vanishes with increasing age of patients and controls (113–115). This might also partly explain why a functional impairment of regulatory T cells was detected in (younger) MS patients with relapsing-remitting disease (RR-MS) but not in (older) patients with secondary-progressive MS (SP-MS) (109). Interestingly, Th17 cells can only be suppressed by a special subset of regulatory T cells expressing CD39, which were found reduced in number and less functional in RR-MS patients (but not in SP-MS patients) (116).
In EAE experiments, different observations concerning the function of regulatory cells during relapses were made: a study observed that regulatory T cells succeeded in suppressing naïve splenic autoreactive T cells, but not effector T cells from the CNS (in a cytokine milieu containing IL-6 and TNF) at the peak of disease (117). In another EAE study, however, regulatory T cells were found fully functional also during the peak of disease and were shown to alleviate disease by a putative suppression of Th1 cells (92). Similarly, human T cells showed an enhanced expression of genes responsible for suppression of IFN-γ (118). These findings might indicate that during acute attacks the immune system starts a counterregulation, which might lead to remission. Interestingly, regulatory T cells were found increased in the CSF of MS patients compared to controls regardless of an active or inactive disease state (108). In a study investigating autoimmune responses after bone marrow transplantation in rats at the peak of EAE, an induction of regulatory T cells was shown, which are probable to make an important contribution to the achieved remission observed (119).
Recently, also CD8+ T cells have drawn more attention. They are more frequently encountered in MS lesions than CD4+ T cells (120). CD8+ T-cell clones detected in brain lesions were shown to be still present in blood and CSF after several years (121). Another fact supporting the pathogenic role of CD8+ T cells is that MHC class II molecules interacting with CD4+ T cells are mainly expressed on professional APCs. However, MHC class I molecules are expressed on all nucleated cells, including oligodendrocytes or astrocytes and CD8+ T cells can interact with peptides bound to the MHC-I complex. In this context, CD8+ T cells were shown to be able to cause axonal damage (122). Increased reactivities of MBP-specific CD8+ T cells were shown to be present in MS patients compared with healthy controls. The CD8+ T cells identified by this study were predominantly memory T cells (CD45RO+) (123). An increased proportion of memory CD8+ T cells was also reported in the CSF of MS patients (124, 125). Elevated levels of granzyme, which is released by cytotoxic T cells, were found in the CSF of patients during relapse (126). So far, many attempts have failed to demonstrate stable disease-relevant CD8+ T-cell expansions to specific CNS-derived antigens in blood from MS patients. This is in contrast to some interesting studies in EAE, in which the relevance of myelin-specific CD8+ T cells has been shown. Importantly, CD8+ T cells specific for MBP can mediate a form of EAE in which a predominance of lesions is seen in the brain rather than in the spinal cord and less inflammation can be observed than in classical EAE (127).
On the other hand, also regulatory autoantigen-specific CD8+ T-cell populations exist, which are able to destroy pathogenic CD4+ T cells or mediate a suppression of their proliferation by interacting with DCs. Interestingly, these suppressive capabilities were found to be reduced during relapses (128–130).
Even though T cells are widely believed to play the central role in the pathogenesis of MS, there is also evidence supporting a pathogenic role of B cells. The importance of a possible role of B cells in MS is reflected in a study with rituximab, a B-cell depleting anti-CD20 monoclonal antibody (mAb), which was effective in reducing both lesion load measured by MRI and clinical relapses (131). The role of B cells in MS is schematically depicted in Figure 3.
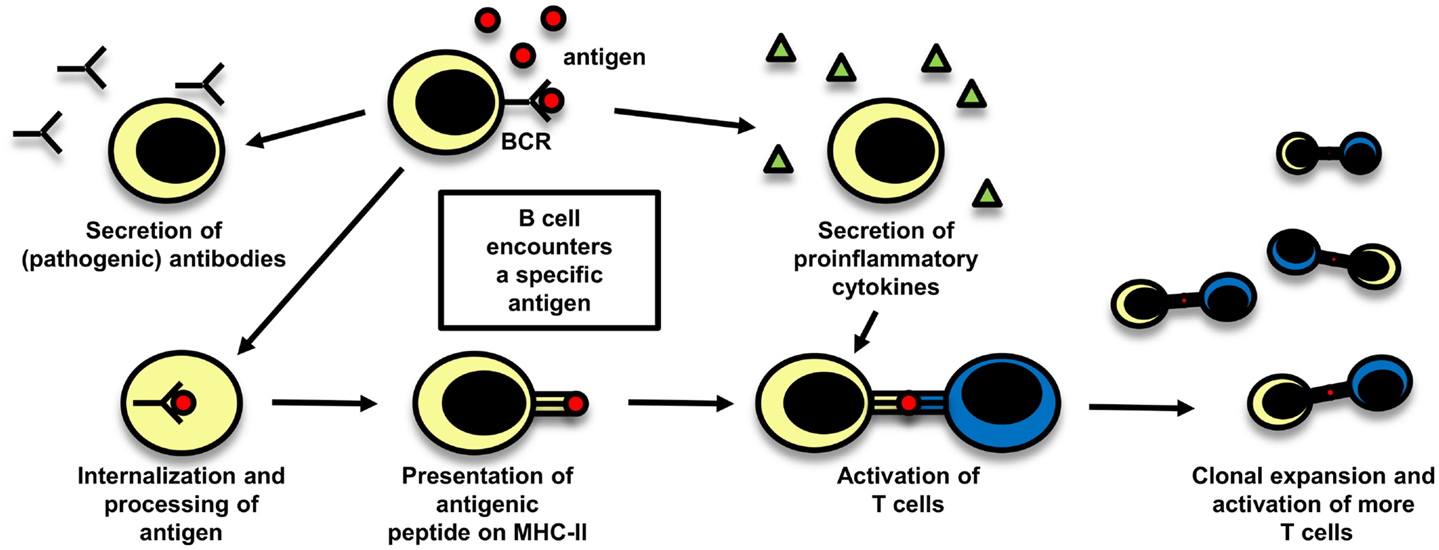
Figure 3. The role of B cells in MS. B cells can play a role in autoimmune disease by different mechanisms which are depicted here: they can secrete potentially pathogenic antibodies, but also their function in cellular immune mechanisms are of importance, either by secreting proinflammatory cytokines and thereby influencing other immune cells or by acting as antigen-presenting cells. After B cells have encountered their specific antigen, they process it, present it on MHC and can become activated. As they are able to expand clonally, they are then capable to activate a big number of T cells.
Most MS patients show IgG oligoclonal bands in their CSF, indicating a local antibody production in the CNS (132). However, the targets of these locally produced antibodies have not yet been fully revealed. MOG-specific autoantibodies have been discovered in acute lesions of MS patients (133, 134).
Apart from their ability to secrete antibodies, the cytokines secreted by B cells can also influence T-cell proliferation in MS patients (135). A further important aspect in their role in cellular immunity is that they can serve as APCs (136). Their ability to expand clonally allows them to activate many T cells. In an EAE model depending on B and T cells, the functions of B cells as APCs were shown to be necessary for the induction of EAE instead of their ability to secrete antibodies (137). Memory B cells from RR-MS patients were shown to induce an increased response of CD4+ T cells to myelin antigens compared to memory B cells from healthy controls, also pointing toward a role of B cells as APCs (138). Clonally expanded B cells have been detected even in the CNS of recently diagnosed MS patients (139). Another interesting aspect concerning B cells as professional APCs is that the B-cell receptors (BCR) consisting of membrane-bound antibodies mostly recognize conformational epitopes of a protein, whereas T cells recognize linear peptides, processed before by APCs (140). The epitopes of an antigen presented by B cells might therefore be different from those presented by thymic epithelial and DCs, including the possibility that T cells specific for epitopes presented by B cells might escape central tolerance induction more easily.
Defects in peripheral (but not central) B-cell tolerance have been reported in MS patients, whereas, interestingly, defects in both peripheral and central B-cell tolerance have been detected in RA and T1D patients (141).
There are also B cells which can adopt regulatory functions, e.g., IL-10-secreting B cells, which can inhibit cytokine secretion (142). The induction of these IL-10-secreting B cells has been found to depend on the presence of IL-21 (which can be secreted, e.g., by Th17 cells) and CD40-dependent cognate interactions with T cells, whereas it can be inhibited by the presence of IFN-γ and TGF-β (143). It is not fully clear yet if the regulatory function of B cells might be defective in MS. Data on a possible reduction of IL-10-secreting B cells in MS patients are contradictory, which is probably also due to different study designs (144–146). Interestingly, parasite infection with helminths in MS patients can induce these IL-10-secreting regulatory B cells (147).
Apart from B cells, also other types of APCs have been shown to be of importance for antigen presentation in MS. Most work regarding the role of APCs in inflammation of the CNS has been conducted in rodent models. Unfortunately, the knowledge about APCs in human MS is still incomplete and not all gathered knowledge can be translated from rodents to humans. DCs are very important for linking innate and adaptive immune responses. Different types of DCs have been described in humans, namely the blood DCs plasmacytoid (p)DC (BDCA2), CD141+ DC (BDCA3), and CD1c+ (BDCA1), as well as the skin DCs, the Langerhans cells, and CD14+ DCs and inflammatory DCs in non-lymphoid tissue (148). All types are capable of presenting antigen, but differ in their expression pattern of molecules, migratory activity, cytokine secretion, and capacity for antigen presentation (149). For example, human CD141+ DC (BDCA3) express mainly TLR3, human CD1c + DC (BDCA1) mainly TLR2 and pDC (BDCA2) mainly TLR7 and TLR9 (150). This leads to different capabilities to react to certain pathogens in the environment. TLR stimulation itself results in DC maturation (151). DCs are required for the priming of naïve T cells (152). Also the capability to cross-present antigen differs between different types of DCs (153). Even though DCs are obviously very important for the immunopathogenesis of MS, so far only limited data are available (154). Also macrophages have been shown to be capable to present antigen in MS (155). In rodent models, perivascular phagocytes in the CNS which present CNS antigens to T cells are of paramount importance in lesion development (156). Such cells can also be found in human MS (157). Within the CNS, microglia can serve as professional APCs (158). A change in the activation pattern of microglia is already seen very early in MS, which could potentially also speak for an increased capacity of antigen presentation (159, 160). Also astrocytes can express MHC class II molecules under certain conditions and present antigens in rodents (13, 161). In humans, so far the role of astrocytes in antigen presentation in CNS inflammation is still incompletely defined.
Multiple sclerosis is not a classical hereditary disease. It is rather defined as a complex genetic disease. This is supported by twin studies, in which for monozygous twins the concordance rate is around 30%, and by findings in siblings of affected individuals who carry a risk of about 2% (162). Most probably, a combination of genetic and also environmental risk factors like smoking (163) contributes to its pathogenesis.
More than 100 risk loci have been identified so far in genome-wide association studies (164), among which the variations within the HLA complex play the most important role. The most significant gene codes for the MHC class II, DR (81). The haplotype HLA-DR2 confers the largest part of the genetic risk for MS in Caucasians. In this MS-associated haplotype, two HLA-DRβ-chains (HLA-DRB1*1501 and HLA-DRB5*0101) pair with the α-chain HLA-DRA*0101, resulting in two heterodimers DR2a (HLA-DRA*0101, HLA-DRB5*0101) and DR2b (HLA-DRA*0101, HLA-DRB1*1501) expressed on the surface of APCs. In this haplotype, three alleles are in linkage disequilibrium and therefore mostly inherited together: HLA-DRB1*1501, HLA-DRB5*0101, and HLA-DQB1*0602 (165). Among these three alleles, HLA-DRB1*1501 confers the greatest genetic risk for MS. Other alleles with increased risk are HLA-DRB1*0301, HLA-DQB1*0201, HLA-DRB1*1303 (81). An association of the HLA-DR15 haplotype in MS with younger age at disease onset was reported, but no correlations with the clinical course, prognosis, or disease type could be detected (166). In contrast, some HLA-A alleles can be attributed some protective effect, most notably the allele HLA-A*0201, but also HLA-A*6801, HLA-A*0205, and HLA-A*0206 have recently been identified as protective (81). The expression of HLA-A*0201 in a humanized transgenic mouse model also protected animals from developing disease, possibly by influencing negative selection processes in the thymus (167).
Apart from the variations in the MHC-locus, 110 other risk-conferring genes have been identified up to now in genome-wide association studies (164). Interestingly, a magnitude of them are genes related to the immune system, which corroborates the hypothesis that MS is an autoimmune disease. Only some important genes can be discussed here. Genes regulating IL-2 signaling, which plays a pivotal role in T-cell activation, have been proposed to be associated with MS, e.g., the IFL-3 gene which regulates IL-2 expression by T cells, but also associations with the IL-2 receptor have been described (81, 164). Concerning the cytokine pathway, also genes coding for IL-7 and its receptor have been linked with a higher risk for MS. Less suppressive effects of regulatory T cells in MS patients was observed in the presence of cells expressing IL-7Rα (110), providing a possible link between the associated gene and a possible function in the disease. Also associations with genes coding for molecules involved in the costimulatory pathway like CD40 or CD80 have been detected. The possible role of defects in this mechanism has been discussed above. Several genes coding for signal transduction molecules (like STAT-3) have been described as well, which are highly relevant for immune cell function. There are also genes related to environmental risk factors such as vitamin D (e.g., CYP27B1) (81).
Table 1 shows some of the susceptibility loci of MS, RA, T1D, and MG in comparison. Even though this list is by far not exhaustive, some interesting aspects can be comprehended: associations with MHC molecules are found not only in MS, but also in RA, T1D, MG, and other autoimmune diseases (168–171). There are also genes coding for molecules involved in similar costimulatory pathways like CD86 or CTLA-4, which contribute to the strength of the peptide–MHC interaction, or for cytokine pathways like IL2RA or TNF in the respective diseases (81, 168). This shows that there may be similar genetic traits leading to the development of different autoimmune diseases.
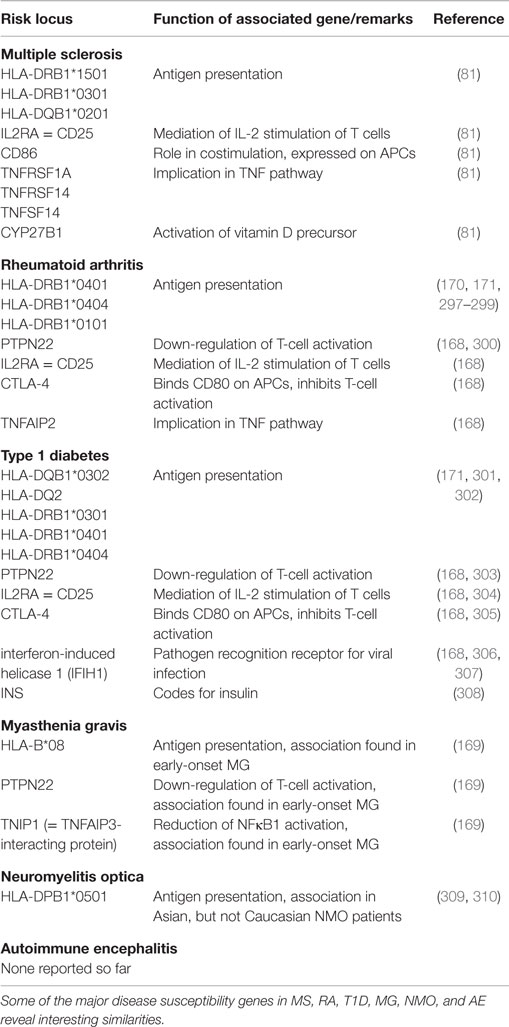
Table 1. Susceptibility genes in different autoimmune diseases.
Genetic studies are able to reveal potential associations of certain genes with autoimmune diseases, but subsequent functional studies are necessary to determine the functional relevance of the identified genes. The functional relevance of most of the genes associated with different autoimmune diseases still remains to be clarified.
The relatively strong association of genes coding for MHC molecules with different autoimmune diseases suggests that the respective genetic variants might affect antigen presentation in a way that the emergence of an autoimmune disease is facilitated. Structural analyses of the MHC loci in different autoimmune diseases revealed certain characteristics of the binding groove, potentially leading to a preferential presentation of certain peptides (171). In general, MHC class II molecules are involved both in negative selection in the thymus by presentation of self-peptides and in antigen presentation in the target tissue. However, the exact mechanisms how risk is conferred by the MHC molecules still remain sketchy. An interesting new aspect was recently observed: in HLA-DR15+ individuals, increased autologous T-cell proliferation was found. In addition, the authors discovered that the HLA-DR15 risk allele in MS is presented on itself, suggesting a possible role of this haplotype as an autoantigen and as a contributor to autologous proliferation (172). Interestingly, also in RA, DRB1*0401-β-chain-peptides were found to be presented by themselves (173).
A future further understanding of the risk-conferring mechanisms of the HLA genes will provide important new insights into the pathogenesis of the respective autoimmune diseases.
Principally, possible candidate autoantigens in MS include myelin antigens, neuronal antigens, and astrocyte-derived antigens. So far, most research has been done on myelin proteins, but there is evidence that also other antigens could serve as possible autoantigens. In the following, some of the antigens derived from the myelin sheath are discussed in detail, which drew most attention of research in the past. Even though these are the most promising candidate autoantigens in MS, it will become clear that the data are not always consistent.
Myelin basic protein is the second most abundant myelin protein after PLP. It is the only myelin component that can be found in both central and peripheral myelin. Thus, a peripheral activation of MBP-specific T cells seems more viable than of T cells reactive with other myelin components, from which they are normally separated.
It has been shown that there are MBP-reactive T cells in MS patients and healthy controls (19, 20). Several studies found significantly more IFN-γ-secreting T cells in response to different MBP–peptides in MS patients than in controls, suggesting a quantitative increase of T cells exhibiting an activated phenotype (89, 174–176). Significantly more MBP-specific T-cell lines were found in the CSF of MS patients than of controls (23).
The central region of MBP84–102/MBP83–99, which can bind to several HLA-DRB molecules, was identified as immunodominant in several studies (177, 178). However, results varied with respect to differences in reactivities to MBP84–102 between MS patients and healthy controls. While some studies found increased reactivity to MBP84–102 in MS patients (20) or an enhanced response to MBP83–99 in MS patients during relapse (87), others could not discover any discrepancies between the two cohorts (179–181). Krogsgaard et al. showed by staining of CNS tissue of HLA-DR2-positive patients with a mAb specific for the HLA-DR2:MBP83–99 complex that this MBP epitope is locally presented in the CNS of MS patients, supporting a possible role as an autoantigen (182). Other epitopes described as immunodominant are MBP13–32 (177) and MBP144–163 (177)/MBP143–168 (23)/MBP151–170 (87). Interestingly, for high-affinity T cells, MBP83–99 was not immunodominant. Instead, MBP13–32, MBP111–129, and MBP146–170 were immunodominant in these T cells. Higher reactivities to these peptides were seen in MS patients (91).
After promising results in animal models, APLs based on the immunodominant epitope MBP83–99 were designed and administered in different phase II clinical trials. In one study, the APL successfully slightly reduced enhancing lesions at a certain dosage and skewed the T-cell response toward a Th2 phenotype, so that the trial had to be stopped because of the occurrence of hypersensitivity reactions (183). However, in another study, the APL induced cross-recognition of MBP83–99 and the APL by T cells in some patients, leading to exacerbation (84). One of the patients included in the latter study suffered two relapses under APL-treatment. Interestingly, before the first relapse, the number of T cells specific for MBP83–99 increased fivefold to decrease again after remission. In the second relapse, T cells specific for PLP190–209 were found expanded (80). This finding shows that myelin reactivity can correlate with disease progression and – besides other pieces of evidence – it also supports the notion of an immune pathogenesis of MS. In other studies, it could also be shown that T-cell reactivities against myelin components can correlate with disease progression, measured by using a disability score (174) or by assessment in MRI (176).
As MBP is an important candidate autoantigen in MS, its relationship with the disease-associated haplotype DR15 is of particular interest. Both HLA-DRB1*1501 and HLA-DRB5*0101 contained in this haplotype can serve as restriction elements for MBP-reactive T cells (177). The immunodominant peptide MBP85–105, for example, seems to be able to be presented by both DRB1*1501 and DRB5*0101 (184, 185) and the complex DR2:MBP85–99 could be detected in tissue lesions (182).
A recent study showed that MBP is degraded by the 26S proteasome without being ubiquitinated beforehand (186). Whether this discovery can be put in context with the pathogenesis of the disease remains to be resolved, but this shows very well how different aspects of antigen presentation might be involved in the emergence of autoimmune disease.
Proteolipid protein is the most abundant myelin protein. There are two isoforms: full-length PLP, which is nearly exclusively expressed in the CNS, and DM20, a splice variant of PLP missing a loop of 25 amino acids, which is expressed in various peripheral organs like the thymus and lymph nodes (35). Only DM20 plays a role in negative selection in the thymus (27). The PLP loop contains the epitope PLP139–151, which seems predisposed to be a target of autoreactive T cells which can escape central tolerance induction in the thymus. However, enhanced reactivities to this epitope could only be detected in some studies (91), but not in others (181, 187).
T-cell responses to PLP seem to be heterogenous (188). Different epitopes have been identified as immunodominant in different studies (24, 180, 181, 187, 189, 190).
Some studies found higher proliferative responses (191) or higher precursor frequencies (23) of PLP-specific T-cell lines in MS patients. Higher reactivities to some PLP epitopes were detected in some studies in MS patients compared to controls (180, 189). However, other studies could not detect any differences in reactivities against their epitopes detected as immunodominant between patients and controls (188, 190).
Myelin oligodendrocyte glycoprotein is a minor component of the myelin sheath. It could not be detected in the human thymus and might therefore evade presentation in central tolerance induction, supporting its possible role as an autoantigen (35). Many different studies have so far assessed T- and B-cell responses to this antigen.
Some studies have detected higher numbers of IFN-γ-secreting cells (192, 193), higher numbers of T cells reactive with DRB1*0401/MOG97–109-tetramer (194), higher proliferative responses of peripheral blood lymphocytes (PBL) (195), or higher proliferative responses of T cells to certain MOG epitopes (196) in MS patients than in healthy controls.
Interestingly, there are also data that MOG could play a role in humoral autoimmunity in MS. MOG-specific autoantibodies have been discovered in acute lesions of MS patients (133, 134). Some studies also found higher levels of MOG-specific antibodies or higher antibody responses to certain MOG epitopes in the serum of MS patients than in control sera (197–202) or in certain clinical subgroups of MS patients (203). However, other studies did not observe any differences between the sera of MS patients and healthy controls or patients with other neurological diseases, or did not detect any MOG-antibodies at all (134, 204, 205). The antibodies were shown to be actually demyelinating in an EAE experiment (200).
Data about a possible association of anti-MOG-antibodies (and anti-MBP-antibodies) with a progression from CIS to definite MS remain controversial (202, 206–210), rendering the use of these antibodies as biomarkers difficult.
There are interindividual differences concerning the specific MOG epitopes causing an antibody response (198, 211) and T-cell reactivities (212), which might also depend on the individual HLA type (213).
Important new data also indicate that MOG can interact with DC-specific intercellular adhesion molecule 3-grabbing non-integrin (DC-SIGN), which can induce IL-10 secretion and suppress T-cell proliferation. This interaction depends on the correct glycosylation state of MOG, which can be altered during inflammation (214). These findings could indicate that altered glycosylation of MOG can potentially result in disruption of tolerance and induction of anti-MOG-specific T- and B-cell responses.
Besides the above described antigens MBP, MOG, and PLP, myelin-associated antigen (MAG), myelin-associated oligodendrocyte basic protein (MOBP), and 2′,3′-cyclic-nucleotide 3′-phosphodiesterase (CNPase) have been shown to evoke T- or/and B-cell responses in patients with MS. Apart from these myelin components, other antigens like S100β protein or transaldolase H are discussed as autoantigens in MS (56). While a-B-crystallin was first thought to be an autoantigen in MS (215), more recent data indicate that it serves as a chaperone and does not fulfill the criteria of an autoantigen (216). In another study, 300 peptides presented on MHC class I and MHC class II molecules in the CNS of MS patients were eluted and identified. Among them, widely investigated proteins like MBP were found, but also non-myelin proteins could be detected (217). Table 2 summarizes the autoantigens discussed in MS. For a comparison with other autoimmune diseases, Tables 3 and 4 summarize some important candidate autoantigens in MG, NMO, T1D, RA, and AE.

Table 2. Autoantigens in MS.
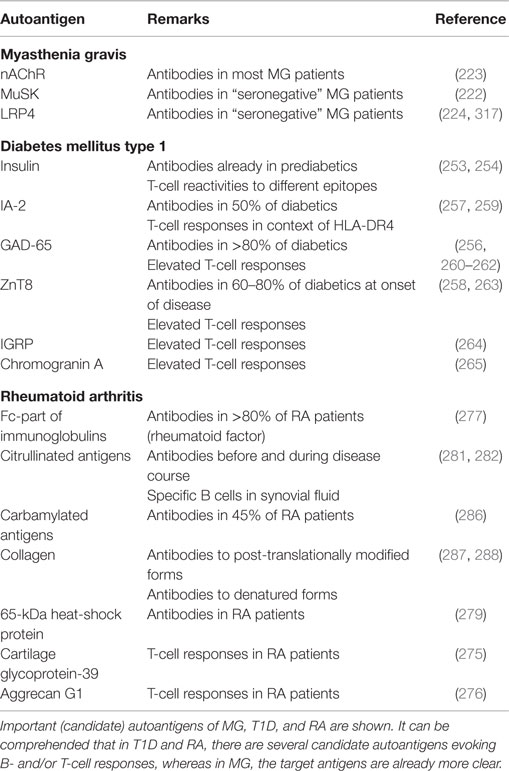
Table 3. Autoantigens in other (peripheral) autoimmune diseases.
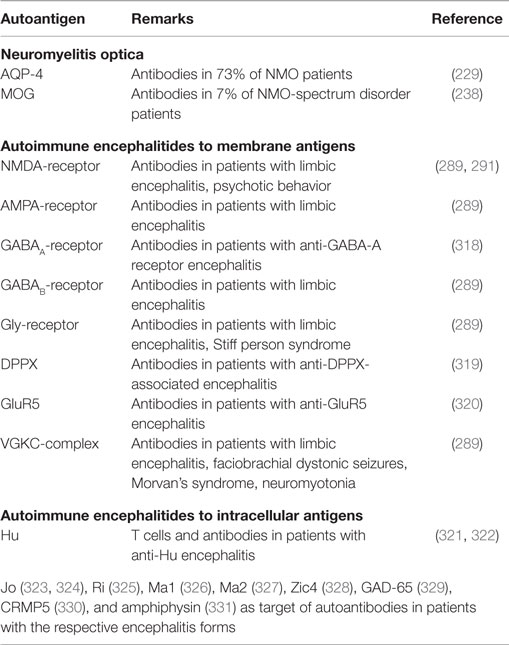
Table 4. Autoantigens in NMO and autoimmune encephalitides as examples of other CNS autoimmune diseases.
Myasthenia gravis is a neurological disease characterized by muscle weakness which worsens by exertion and improves by rest (218).
Some decades ago, antibodies directed against nicotine acetylcholine receptors (nAChR) were detected in most MG patients (219, 220), which could also be shown to be pathogenic by a transfer of sera to mice (221). A considerable part of the 10–20% of MG patients who had long been assessed as seronegative for nAChR-autoantibodies could be shown to possess autoantibodies against the receptor tyrosine kinase MuSK (222) or still possess low-affinity antibodies against the nAChR by employing a more sensitive assay (223). More recently, also low-density lipoprotein receptor-related protein (Lrp4) was identified to be a target of autoantibodies in a small proportion of seronegative MG patients (224). These findings point toward B cells playing the central role in the pathogenesis of MG. But also nAChR-specific T lymphocytes have been discovered (22, 225), consistent with necessary T cell help and activation for an autoantibody production by B cells. Regulatory T cells isolated from thymi of MG patients showed a reduced suppressive capability (226).
Even though B cells and their antibodies seem to play a crucial role for the disease, its exact pathogenesis could not be revealed yet. Complement activation by autoantibodies and crosslinking nAChR leading to their degradation were suggested to be underlying pathogenic mechanisms of the nAChR-autoantibodies (218). But still, the emergence of an autoimmune disease always requires an interplay of different immune cell types and T cells are probable to play an important role. Like in MS, as discussed above, it could also be shown that B cells can serve as APCs in MG (227).
Compared to MS, the autoimmune origin of the disease is evidenced very clearly and the nAChR is well characterized as the target structure of the autoimmune response. But still, the exact etiology of the disease remains to be clarified.
Neuromyelitis optica is a demyelinating disease of the CNS characterized by optic neuritis and longitudinally extensive transverse myelitis (228). Originally, the disease was not differentiated from MS, but important differences between NMO and MS will be pointed out in the following. Autoantibodies directed against aquaporin-4 (AQP-4)-channels were discovered in 73% of sera of NMO patients but not in sera of MS patients (229, 230). Anti-AQP-4 antibodies also seem to have some predictive value concerning the clinical course of the disease (231). AQP-4-channels are expressed on astrocytic foot processes at the BBB (230, 232). Pathological findings indicate a complete loss of AQP-4 in NMO lesions, whereas in MS, AQP-4 staining was enhanced in the periplaque white matter of active lesions, and absent in inactive lesions (232). There is evidence that these antibodies might also be pathogenic. They were found to have cytotoxic effects on astrocytes in the presence of complement (233, 234), increase natural killer cell-mediated cytotoxicity (235, 236), complement-mediated involvement of granulocytes (235, 236), and the permeability of the BBB (235). In animal studies, a more severe disease course of EAE displaying features of NMO was observed (236, 237).
There are also NMO patients who are seronegative for AQP-4-antibodies. In 7% of patients with NMO-spectrum disorders (NMOSD), i.e., patients presenting either with isolated transverse myelitis or optic neuritis, MOG-antibodies were discovered (238). Also AQP-4-antibody-negative patients with classical signs of NMO have been described to possess antibodies to MOG (239). However, MOG-antibodies seem to be less specific for NMO. MOG-antibody-positive patients with NMOSD also seem to show a milder clinical course than AQP-4-positive patients (238). Data in animal models of NMO indicate that the T-cell response is not mandatory for the development of NMO if AQP-4 antibodies are present in serum or CSF. The access of the antibodies to the CNS and their binding to astrocytes appears to be most important (240, 241). Consistent with an important role of a loss of B-cell tolerance, regulatory B cells were found decreased in NMO and secreted less IL-10 (97). Nevertheless, T cells seem to be mandatory for the initial emergence of plasma cells which secrete AQP-4-specific antibodies (242, 243).
So even though NMO can sometimes clinically resemble MS, the mechanisms of their pathogeneses seem to differ greatly in regard to the autoimmune target and the emerging immune response.
Type 1 diabetes is an autoimmune disease in which the β-cells in the Langerhans islands of the pancreas are destroyed, resulting in a lack of insulin. It is thought that T cells play a more decisive role in β-cell destruction than B cells, but B cells were shown to be relevant by producing autoantibodies against islet cell antigens already in prediabetic phases and by presenting antigen to CD4+ and CD8+ T cells (244). Concerning CD4+ T cells, Th1 and Th17 cells seem to be the most significant T helper cell subsets for T1D. The animal model of T1D is mainly driven by Th1 cells, but there is also evidence of a role of Th1 cells in human T1D (245). More IL-17-secreting T cells were detected in recent-onset T1D and IL-17 seems to promote the inflammatory response to β-cells (246, 247). Th17 cells were also found increased in pancreatic-draining lymph nodes (248). Insulin-specific CD8+ T cells could be associated with an autoimmune destruction of transplanted Langerhans islands (249). CD8+ T cells were also more frequent in pancreatic-draining lymph nodes than in those of controls (248). Similar to MS, defects in the suppressive capabilities of regulatory T cells have been detected (250). This was also shown for T cells residing in pancreatic-draining lymph nodes (248). On the other hand, also a higher resistance of effector T cells to suppression by regulatory T cells has been reported (251). Possible defects in central and peripheral B-cell tolerance were also reported (141). Epitope spreading also seems to be an important mechanism in the autoimmune response in T1D, intermolecular epitope spreading was observed even before the clinical onset of the disease (252).
Different antigens are discussed as autoantigens in T1D. Among them, insulin seems to play a major role: in some diabetics, antibodies to insulin can be discovered before insulin treatment (253). Most T1D patients were shown to possess CD8+ T cells reactive with proinsulin peptides (254).
Islet cell autoantibodies (ICA) were detected to be more frequent in T1D patients and were also suggested to have predictive value (255). Many T1D patients also exhibit antibodies against the enzyme glutamic acid decarboxylase (GAD)-65, which is expressed in neurons and in pancreatic β-cells (256). Subsequently, also autoantibodies against tyrosine phosphatase-like islet-antigen 2 (IA-2) (257) and against the zinc transporter ZnT8 (258) were detected in diabetics.
In addition to insulin, T-cell reactivities against several other antigens were identified: not only autoantibodies, but also T-cell responses against IA-2 and GAD-65 were detected, especially in the context of HLA-DR4 (259–262). Also ZnT8 seems to evoke elevated T-cell responses in diabetics (263). Human PBMCs of patients with recent-onset diabetes also showed elevated IFN-γ secretion when stimulated with islet-specific glucose-6-phosphatase catalytic subunit-related protein (IGRP), supporting a possible role as an autoantigen (264). Chromogranin A was also discovered to be a relevant T-cell antigen in human T1D (265).
On the whole, there are several features MS and T1D have in common. Their animal models seem to be mainly driven by autoreactive Th1 cells, but both animal and human data indicate also an important role of Th17 cells and an impaired function of regulatory populations. Their major genetic risk loci code for MHC molecules and several T- and B-cell antigens have been suggested as putative target structures. Interestingly, T1D has also been reported to display cyclic processes in the ongoing β-cell destruction, characterized by damage by autoreactive T cells, epitope spreading, β-cell proliferation, and suppressive activities by regulatory T cells (266). This could resemble a relapsing-remitting course as found in most MS patients.
In RA, it is not fully resolved whether T cells or B cells play the more important role in its pathogenesis, for there are important aspects supporting a relevance of both cell types. T- and B-cell clones have been detected in the joints of RA patients, partly even the same clones were found in different affected joints (267, 268). Like in MS and T1D, a pathogenic role of Th1 and Th17 cells has been suggested. Th1 cells were encountered much more frequently than Th17 cells in the joint of RA patients with established disease (269). On the other hand, increased numbers of Th17 cells and higher IL-17 levels were found in peripheral blood and synovial fluid of patients with untreated early RA (270). Part of their pathogenicity might be due to the effect of IL-17, which among other cytokines leads to an increased production of TNF-α (271), which plays a pivotal role in the pathogenesis of RA. Th17 memory cells can also induce secretion of proinflammatory cytokines by synovial fibroblasts (272). Regulatory T cells in RA have been found less able to suppress the production of proinflammatory cytokines like TNFα in acute phases of the disease (273). B cells do not only secrete potentially pathogenic antibodies, but also their function in cellular immunity plays a role in RA, as T cells were shown to be activated by B cells in synovitis (267). Defects in both peripheral and central B-cell tolerance were observed (141). The efficacy of a treatment with rituximab confirms a pathogenic role of B cells in the disease (274).
Like in MS and T1D, several different antigens are discussed as autoantigens in RA; some of the possible targets in RA will be discussed here.
Examples for putative T-cell targets in RA include the human cartilage glycoprotein-39 (275) and aggrecan G1 (276). Some autoantibodies target the Fc-part of immunoglobulins (277). These antibodies are also referred to as the “rheumatoid factor.” The presence of the rheumatoid factor is associated with a more severe disease course (278). Elevated autoantibodies against 65-kDa heat-shock protein have also been detected in RA patients compared to controls (279). An interesting aspect of the autoimmune response in RA is that it also seems to be directed against post-translational modifications of antigens like citrullination or carbamylation (280). Antibodies against different citrullinated antigens are found significantly more often in RA patients compared to controls and are elevated already before the onset of disease (281). A considerable proportion of B cells found in synovial fluid have been observed to secrete antibodies directed against citrullinated antigens (282). Epitope spreading of the antibody response to different citrullinated peptides has been observed even before the clinical onset of RA (283). Data from animal models indicate that antibodies directed against citrullinated antigens are pathogenic and worsen arthritis (284). Human data indicate that anti-cyclic citrullinated antibodies can lead to complement activation (285). Independently from APCA, antibodies directed against carbamylated antigens have been discovered in about 45% of RA patients (286). In early stages of RA, autoantibodies against post-translationally modified collagen have recently been detected in over 90% or RA patients (287). During the course of the disease, also higher antibody titers to denaturated collagen were found in RA patients than in controls (288).
Like T1D and MS, also RA shows an HLA-association, an important role of Th1 and Th17 cells and possible defects in regulatory mechanisms. In contrast to MS and T1D, autoimmune responses to post-translational modifications have been studied more extensively in RA.
In the last years, several antibody-mediated AE were discovered with immune responses to membrane-bound proteins, which respond well to immunotherapies like plasmapheresis. Symptoms range from psychiatric symptoms, such as personality and behavioral change, agitation and paranoia, to epilepsy, amnesia, and many others. Antibodies against different neuronal antigens were found in different disorders, with most autoantigens directed against certain ion channel proteins or receptors involved in brain signaling. For example, antibodies against N-methyl-d-aspartate (NMDA) receptors, alpha-amino-3-hydroxy-5-methyl-4-isoxazoleprionic acid (AMPA)-receptors, gamma-aminobutyric acid (GABA) type A or B receptors, glycine receptors (GlyR), dipeptidyl-peptidase-like-protein 6 (DPPX), metabotrophic glutamate receptor 5 (GluR5), or voltage-gated potassium channels were detected in patients with different forms of autoimmune encephalitis (289).
As an example, only anti-NMDA-receptor encephalitis is briefly discussed here, as there is evidence of a direct pathogenic relevance of the autoantibodies by leading to receptor internalization (290). The occurring autoantibodies were detected to be directed against heteromers of the NMDA-receptor containing the subunits NR2B or NR2A (291). A pathological study barely found markers of cell cytotoxicity, but large deposits of IgG, pointing toward a functionally primary causative role of antibodies produced by B cells instead of cellular immunity (292). Anti-NMDA-receptor encephalitis as well as other types of autoimmune encephalitis can be of paraneoplastic origin. Associations of anti-NMDA-receptor encephalitis with ovarian teratoma have been described (291). The role of T cells in these encephalitis forms is not well established yet. Possibly they are instrumental for disease initiation by helping B cells to secrete pathogenic antibodies.
There are many intracellularly located target antigens that can cause autoimmune encephalitis. Many of these are induced by paraneoplastic mechanisms, but there are also pure autoimmune conditions. Most probably, T cells and especially CD8+ T cells are of paramount importance by leading to tissue damage (293, 294). For diagnoses, antibodies against such antigens are pivotal. Antigens that have been described as target antigens and to which also antibodies are found in patients are Hu, Yo, Ri, Ma1, Ma2, Zic4, GAD-65, CRMP5, and amphiphysin (Table 4).
Principally, possible autoantigens can be determined by testing the immune reaction against antigens that can be assumed to be targeted in an autoimmune disease because of certain characteristics of the disease (e.g., myelin antigens in the demyelinating disease MS). As described above, there has already been much research investigating the role of myelin antigens in MS.
Another way to determine possible autoantigens would be first to identify possible candidate autoantigens of the disease, e.g., by elution of peptides presented on MHC molecules of APCs in affected individuals or by screening sera of patients for autoantibodies, and afterwards test the actual functional role of the findings. The approach to elute peptides presented on APCs in autoimmunity has been successfully applied in research on different autoimmune diseases like RA or T1D (173, 259, 260, 295), but also in MS (217). The eluted peptides from the CNS of MS patients might have a high potential as autoantigens, since antigen presentation in the healthy CNS is low. In active MS lesions, however, MHC molecules have been observed to be upregulated (82).
In all autoimmune diseases, the importance of the role of peptide–MHC–TCR interactions cannot be underestimated. Antigen presentation is in the center of every autoimmune disease, influencing both tolerance induction in the thymus and self-antigen recognition and immune cell activation in the periphery. This is underlined by the fact that the genetic risk factors of many autoimmune diseases are conferred by genes involved in antigen presentation. The onset of several physiological immune mechanisms like epitope spreading obscures the search for the primary underlying target of the ongoing immune attack. This underlines the great importance of studying immune reactions in recently diagnosed patients or even before diagnosis for eventually being able to determine the primary event in MS and other diseases of a putative autoimmune origin. On the other hand, for more efficient therapy development, the processes which set in later during the course of an autoimmune disease must be taken into account as well. In the discussed autoimmune diseases, the emergence of neo-antigens by tissue destruction and epitope spreading play a role. To give only one example, this might be a reason for the failure of therapeutic trials with peptides based only on MBP in MS treatment. Recently, evidence was adduced that the native conformation of an antigen is of importance for inducing an immune response (296). This is of great relevance for studies investigating possible autoantigens, since antigens are not always employed in their native conformation; which further complicates the search for the principal autoantigen(s).
It is striking that many susceptibility genes conferring risk for different autoimmune diseases code for HLA genes, further emphasizing the extreme relevance of antigen presentation for autoimmune diseases. A comparison of MS with other autoimmune diseases is interesting also in other respects: the exact pathogenesis has not been identified yet in either autoimmune disease. The determination of the (primary) autoantigens is a crucial step in understanding the pathogenesis of a disease. But even when target antigens have been identified like in MG or NMO, the pathogenic mechanism can still remain unclear. Neither has it been resolved which cell types play the dominant role in the induction of an autoimmune disease. The concept of MS being a “T-cell-mediated” autoimmune disease and MG being a “B-cell-mediated” autoimmune disease must be reevaluated. Data indicate that autoimmune diseases emerge from a complex interplay of many different factors of the immune system.
Comparisons of MS with RA or T1D show that there are more diseases whose target antigen(s) have not been fully identified. Until an unequivocal target of the immune attack has been identified in MS and other diseases, there will also remain the question if the causative factors might even be heterogenic.
At all events, a detailed understanding of the etiology of the discussed autoimmune diseases is an enormous challenge, but will remain a prerequisite for the development of new, specific, and more efficient treatment options.
CR and RW outlined the subject of the review, searched for, analyzed, and interpreted the literature and wrote the manuscript. CR and RW agree to be accountable for all aspects of the work.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Lassmann H, Brück W, Lucchinetti CF. The immunopathology of multiple sclerosis: an overview. Brain Pathol (2007) 17:210–8. doi:10.1111/j.1750-3639.2007.00064.x
2. Blum JS, Wearsch PA, Cresswell P. Pathways of antigen processing. Annu Rev Immunol (2013) 31:443–73. doi:10.1146/annurev-immunol-032712-095910
3. Rock KL, Gramm C, Rothstein L, Clark K, Stein R, Dick L, et al. Inhibitors of the proteasome block the degradation of most cell proteins and the generation of peptides presented on MHC class I molecules. Cell (1994) 78:761–71. doi:10.1016/S0092-8674(94)90462-6
4. Gaczynska M, Rock KL, Spies T, Goldberg AL. Peptidase activities of proteasomes are differentially regulated by the major histocompatibility complex-encoded genes for LMP2 and LMP7. Proc Natl Acad Sci U S A (1994) 91:9213–7. doi:10.1073/pnas.91.20.9213
5. Saric T, Chang S, Hattori A, York IA, Markant S, Rock KL, et al. An IFN-gamma-induced aminopeptidase in the ER, ERAP1, trims precursors to MHC class I-presented peptides. Nat Immunol (2002) 3:1169–76. doi:10.1038/ni859
6. York IA, Chang S, Saric T, Keys JA, Favreau JM, Goldberg AL, et al. The ER aminopeptidase ERAP1 enhances or limits antigen presentation by trimming epitopes to 8-9 residues. Nat Immunol (2002) 3:1177–84. doi:10.1038/ni860
7. Sadasivan B, Lehner PJ, Ortmann B, Spies T, Cresswell P. Roles for calreticulin and a novel glycoprotein, tapasin, in the interaction of MHC class I molecules with TAP. Immunity (1996) 5:103–14. doi:10.1016/S1074-7613(00)80487-2
8. Momburg F, Roelse J, Howard JC, Butcher GW, Hämmerling GJ, Neefjes JJ. Selectivity of MHC-encoded peptide transporters from human, mouse and rat. Nature (1994) 367:648–51. doi:10.1038/367648a0
9. Watts C. The exogenous pathway for antigen presentation on major histocompatibility complex class II and CD1 molecules. Nat Immunol (2004) 5:685–92. doi:10.1038/ni1088
10. Riberdy JM, Newcomb JR, Surman MJ, Barbosa JA, Cresswell P. HLA-DR molecules from an antigen-processing mutant cell line are associated with invariant chain peptides. Nature (1992) 360:474–7. doi:10.1038/360474a0
11. Denzin LK, Cresswell P. HLA-DM induces CLIP dissociation from MHC class II alpha beta dimers and facilitates peptide loading. Cell (1995) 82:155–65. doi:10.1016/0092-8674(95)90061-6
12. Ford AL, Foulcher E, Lemckert FA, Sedgwick JD. Microglia induce CD4 T lymphocyte final effector function and death. J Exp Med (1996) 184:1737–45. doi:10.1084/jem.184.5.1737
13. Fontana A, Fierz W, Wekerle H. Astrocytes present myelin basic protein to encephalitogenic T-cell lines. Nature (1984) 307:273–6. doi:10.1038/307273a0
14. Gresser O, Weber E, Hellwig A, Riese S, Régnier-Vigouroux A. Immuno-competent astrocytes and microglia display major differences in the processing of the invariant chain and in the expression of active cathepsin L and cathepsin S. Eur J Immunol (2001) 31:1813–24. doi:10.1002/1521-4141(200106)31:6<1813::AID-IMMU1813>3.0.CO;2-8
15. Ackerman AL, Giodini A, Cresswell P. A role for the endoplasmic reticulum protein retrotranslocation machinery during crosspresentation by dendritic cells. Immunity (2006) 25:607–17. doi:10.1016/j.immuni.2006.08.017
16. Malnati MS, Marti M, LaVaute T, Jaraquemada D, Biddison W, DeMars R, et al. Processing pathways for presentation of cytosolic antigen to MHC class II-restricted T cells. Nature (1992) 357:702–4. doi:10.1038/357702a0
17. Saveanu L, Carroll O, Weimershaus M, Guermonprez P, Firat E, Lindo V, et al. IRAP identifies an endosomal compartment required for MHC class I cross-presentation. Science (2009) 325:213–7. doi:10.1126/science.1172845
18. Segura E, Albiston AL, Wicks IP, Chai SY, Villadangos JA. Different cross-presentation pathways in steady-state and inflammatory dendritic cells. Proc Natl Acad Sci U S A (2009) 106:20377–81. doi:10.1073/pnas.0910295106
19. Martin R, Jaraquemada D, Flerlage M, Richert J, Whitaker J, Long EO, et al. Fine specificity and HLA restriction of myelin basic protein-specific cytotoxic T cell lines from multiple sclerosis patients and healthy individuals. J Immunol (1990) 145:540–8.
20. Ota K, Matsui M, Milford EL, Mackin GA, Weiner HL, Hafler DA. T-cell recognition of an immunodominant myelin basic protein epitope in multiple sclerosis. Nature (1990) 346:183–7. doi:10.1038/346183a0
21. Burns JB, Bartholomew BD, Lobo ST. Isolation of CD45RO+, memory T cells recognizing proteolipid protein from neurologically normal subjects. Cell Immunol (2001) 212:44–50. doi:10.1006/cimm.2001.1842
22. Melms A, Malcherek G, Gern U, Wiethölter H, Müller CA, Schoepfer R, et al. T cells from normal and myasthenic individuals recognize the human acetylcholine receptor: heterogeneity of antigenic sites on the alpha-subunit. Ann Neurol (1992) 31:311–8. doi:10.1002/ana.410310314
23. Zhang J, Markovic-Plese S, Lacet B, Raus J, Weiner HL, Hafler DA. Increased frequency of interleukin 2-responsive T cells specific for myelin basic protein and proteolipid protein in peripheral blood and cerebrospinal fluid of patients with multiple sclerosis. J Exp Med (1994) 179:973–84. doi:10.1084/jem.179.3.973
24. Correale J, McMillan M, McCarthy K, Le T, Weiner LP. Isolation and characterization of autoreactive proteolipid protein-peptide specific T-cell clones from multiple sclerosis patients. Neurology (1995) 45:1370–8. doi:10.1212/WNL.45.7.1370
25. Goebels N, Hofstetter H, Schmidt S, Brunner C, Wekerle H, Hohlfeld R. Repertoire dynamics of autoreactive T cells in multiple sclerosis patients and healthy subjects: epitope spreading versus clonal persistence. Brain (2000) 123:508–18. doi:10.1093/brain/123.3.508
26. Tsuchida T, Parker KC, Turner RV, McFarland HF, Coligan JE, Biddison WE. Autoreactive CD8+ T-cell responses to human myelin protein-derived peptides. Proc Natl Acad Sci U S A (1994) 91:10859–63. doi:10.1073/pnas.91.23.10859
27. Klein L, Klugmann M, Nave KA, Tuohy VK, Kyewski B. Shaping of the autoreactive T-cell repertoire by a splice variant of self protein expressed in thymic epithelial cells. Nat Med (2000) 6:56–61. doi:10.1038/71540
28. Klein L, Hinterberger M, Wirnsberger G, Kyewski B. Antigen presentation in the thymus for positive selection and central tolerance induction. Nat Rev Immunol (2009) 9:833–44. doi:10.1038/nri2669
29. Anderson MS, Venanzi ES, Klein L, Chen Z, Berzins SP, Turley SJ, et al. Projection of an immunological self shadow within the thymus by the aire protein. Science (2002) 298:1395–401. doi:10.1126/science.1075958
30. García-Lozano J, Torres-Agrela B, Montes-Cano M, Ortiz-Fernández L, Conde-Jaldón M, Teruel M, et al. Association of the AIRE gene with susceptibility to rheumatoid arthritis in a European population: a case control study. Arthritis Res Ther (2013) 15:R11. doi:10.1186/ar4141
31. Shao S, Li X, Cen H, Yin Z. Association of AIRE polymorphisms with genetic susceptibility to rheumatoid arthritis in a Chinese population. Inflammation (2014) 37:495–9. doi:10.1007/s10753-013-9763-3
32. Giraud M, Taubert R, Vandiedonck C, Ke X, Lévi-Strauss M, Pagani F, et al. An IRF8-binding promoter variant and AIRE control CHRNA1 promiscuous expression in thymus. Nature (2007) 448:934–7. doi:10.1038/nature06066
33. Wu C, Aichinger M, Nedjic J, Klein L. Thymic epithelial cells use macroautophagy to turn their inside out for CD4 T cell tolerance. Autophagy (2013) 9:931–2. doi:10.4161/auto.24374
34. Adamopoulou E, Tenzer S, Hillen N, Klug P, Rota IA, Tietz S, et al. Exploring the MHC-peptide matrix of central tolerance in the human thymus. Nat Commun (2013) 4:2039. doi:10.1038/ncomms3039
35. Bruno R, Sabater L, Sospedra M, Ferrer-Francesch X, Escudero D, Martínez-Cáceres E, et al. Multiple sclerosis candidate autoantigens except myelin oligodendrocyte glycoprotein are transcribed in human thymus. Eur J Immunol (2002) 32:2737–47. doi:10.1002/1521-4141(2002010)32:10<2737::AID-IMMU2737>3.0.CO;2-0
36. Pribyl TM, Campagnoni CW, Kampf K, Kashima T, Handley VW, McMahon J, et al. The human myelin basic protein gene is included within a 179-kilobase transcription unit: expression in the immune and central nervous systems. Proc Natl Acad Sci U S A (1993) 90:10695–9. doi:10.1073/pnas.90.22.10695
37. Goverman J. Autoimmune T cell responses in the central nervous system. Nat Rev Immunol (2009) 9:393–407. doi:10.1038/nri2550
38. Greer JM, Denis B, Sobel RA, Trifilieff E. Thiopalmitoylation of myelin proteolipid protein epitopes enhances immunogenicity and encephalitogenicity. J Immunol (2001) 166:6907–13. doi:10.4049/jimmunol.166.11.6907
39. Bradford CM, Ramos I, Cross AK, Haddock G, McQuaid S, Nicholas AP, et al. Localisation of citrullinated proteins in normal appearing white matter and lesions in the central nervous system in multiple sclerosis. J Neuroimmunol (2014) 273:85–95. doi:10.1016/j.jneuroim.2014.05.007
40. Sethi DK, Schubert DA, Anders A, Heroux A, Bonsor DA, Thomas CP, et al. A highly tilted binding mode by a self-reactive T cell receptor results in altered engagement of peptide and MHC. J Exp Med (2011) 208:91–102. doi:10.1084/jem.20100725
41. Schubert DA, Gordo S, Sabatino JJ, Vardhana S, Gagnon E, Sethi DK, et al. Self-reactive human CD4 T cell clones form unusual immunological synapses. J Exp Med (2012) 209:335–52. doi:10.1084/jem.20111485
42. Kamradt T, Mitchison NA. Tolerance and autoimmunity. N Engl J Med (2001) 344:655–64. doi:10.1056/NEJM200103013440907
43. Liu J, Zhang H. CTLA-4 gene and the susceptibility of multiple sclerosis: an updated meta-analysis study including 12,916 cases and 15,455 controls. J Neurogenet (2014) 28:153–63. doi:10.3109/01677063.2014.880703
44. Lovett-Racke AE, Trotter JL, Lauber J, Perrin PJ, June CH, Racke MK. Decreased dependence of myelin basic protein-reactive T cells on CD28-mediated costimulation in multiple sclerosis patients. A marker of activated/memory T cells. J Clin Invest (1998) 101:725–30. doi:10.1172/JCI1528
45. Scholz C, Patton KT, Anderson DE, Freeman GJ, Hafler DA. Expansion of autoreactive T cells in multiple sclerosis is independent of exogenous B7 costimulation. J Immunol (1998) 160:1532–8.
46. Oliveira EM, Bar-Or A, Waliszewska AI, Cai G, Anderson DE, Krieger JI, et al. CTLA-4 dysregulation in the activation of myelin basic protein reactive T cells may distinguish patients with multiple sclerosis from healthy controls. J Autoimmun (2003) 20:71–81. doi:10.1016/S0896-8411(02)00106-3
47. Markovic-Plese S, Cortese I, Wandinger KP, McFarland HF, Martin R. CD4+CD28- costimulation-independent T cells in multiple sclerosis. J Clin Invest (2001) 108:1185–94. doi:10.1172/JCI12516
48. Genç K, Dona DL, Reder AT. Increased CD80(+) B cells in active multiple sclerosis and reversal by interferon beta-1b therapy. J Clin Invest (1997) 99:2664–71. doi:10.1172/JCI119455
49. Moreno M, Negrotto L, Río J, Moubarak R, Martín I, Bustamante MF, et al. Activation-induced cell death in T lymphocytes from multiple sclerosis patients. J Neuroimmunol (2014) 272:51–5. doi:10.1016/j.jneuroim.2014.04.007
50. Macchi B, Matteucci C, Nocentini U, Caltagirone C, Mastino A. Impaired apoptosis in mitogen-stimulated lymphocytes of patients with multiple sclerosis. Neuroreport (1999) 10:399–402. doi:10.1097/00001756-199902050-00034
51. Cencioni MT, Santini S, Ruocco G, Borsellino G, De Bardi M, Grasso MG, et al. FAS-ligand regulates differential activation-induced cell death of human T-helper 1 and 17 cells in healthy donors and multiple sclerosis patients. Cell Death Dis (2015) 6:e1741. doi:10.1038/cddis.2015.100
52. Fontenot JD, Gavin MA, Rudensky AY. Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nat Immunol (2003) 4:330–6. doi:10.1038/ni904
53. Fontenot JD, Rasmussen JP, Williams LM, Dooley JL, Farr AG, Rudensky AY. Regulatory T cell lineage specification by the forkhead transcription factor Foxp3. Immunity (2005) 22:329–41. doi:10.1016/j.immuni.2005.01.016
54. Aschenbrenner K, D’Cruz LM, Vollmann EH, Hinterberger M, Emmerich J, Swee LK, et al. Selection of Foxp3+ regulatory T cells specific for self antigen expressed and presented by Aire+ medullary thymic epithelial cells. Nat Immunol (2007) 8:351–8. doi:10.1038/ni1444
55. Kivisäkk P, Mahad DJ, Callahan MK, Trebst C, Tucky B, Wei T, et al. Human cerebrospinal fluid central memory CD4+ T cells: evidence for trafficking through choroid plexus and meninges via P-selectin. Proc Natl Acad Sci U S A (2003) 100:8389–94. doi:10.1073/pnas.1433000100
56. Sospedra M, Martin R. Immunology of multiple sclerosis. Annu Rev Immunol (2005) 23:683–747. doi:10.1146/annurev.immunol.23.021704.115707
57. Butcher EC, Picker LJ. Lymphocyte homing and homeostasis. Science (1996) 272:60–6. doi:10.1126/science.272.5258.60
58. Ransohoff RM, Kivisäkk P, Kidd G. Three or more routes for leukocyte migration into the central nervous system. Nat Rev Immunol (2003) 3:569–81. doi:10.1038/nri1130
59. de Vos AF, van Meurs M, Brok HP, Boven LA, Hintzen RQ, van der Valk P, et al. Transfer of central nervous system autoantigens and presentation in secondary lymphoid organs. J Immunol (2002) 169:5415–23. doi:10.4049/jimmunol.169.10.5415
60. Fabriek BO, Zwemmer JN, Teunissen CE, Dijkstra CD, Polman CH, Laman JD, et al. In vivo detection of myelin proteins in cervical lymph nodes of MS patients using ultrasound-guided fine-needle aspiration cytology. J Neuroimmunol (2005) 161:190–4. doi:10.1016/j.jneuroim.2004.12.018
61. Waldner H, Collins M, Kuchroo VK. Activation of antigen-presenting cells by microbial products breaks self tolerance and induces autoimmune disease. J Clin Invest (2004) 113:990–7. doi:10.1172/JCI19388
62. Birnbaum ME, Mendoza JL, Sethi DK, Dong S, Glanville J, Dobbins J, et al. Deconstructing the peptide-MHC specificity of T cell recognition. Cell (2014) 157:1073–87. doi:10.1016/j.cell.2014.03.047
63. Wucherpfennig KW, Strominger JL. Molecular mimicry in T cell-mediated autoimmunity: viral peptides activate human T cell clones specific for myelin basic protein. Cell (1995) 80:695–705. doi:10.1016/0092-8674(95)90348-8
64. Tejada-Simon MV, Zang YC, Hong J, Rivera VM, Zhang JZ. Cross-reactivity with myelin basic protein and human herpesvirus-6 in multiple sclerosis. Ann Neurol (2003) 53:189–97. doi:10.1002/ana.10425
65. Ascherio A, Munger KL, Lennette ET, Spiegelman D, Hernán MA, Olek MJ, et al. Epstein-Barr virus antibodies and risk of multiple sclerosis: a prospective study. JAMA (2001) 286:3083–8. doi:10.1001/jama.286.24.3083
66. Croxford JL, Olson JK, Anger HA, Miller SD. Initiation and exacerbation of autoimmune demyelination of the central nervous system via virus-induced molecular mimicry: implications for the pathogenesis of multiple sclerosis. J Virol (2005) 79:8581–90. doi:10.1128/JVI.79.13.8581-8590.2005
67. Krishnamoorthy G, Saxena A, Mars LT, Domingues HS, Mentele R, Ben-Nun A, et al. Myelin-specific T cells also recognize neuronal autoantigen in a transgenic mouse model of multiple sclerosis. Nat Med (2009) 15:626–32. doi:10.1038/nm.1975
68. Haring JS, Pewe LL, Perlman S. Bystander CD8 T cell-mediated demyelination after viral infection of the central nervous system. J Immunol (2002) 169:1550–5. doi:10.4049/jimmunol.169.3.1550
69. Ji Q, Perchellet A, Goverman JM. Viral infection triggers central nervous system autoimmunity via activation of CD8+ T cells expressing dual TCRs. Nat Immunol (2010) 11:628–34. doi:10.1038/ni.1888
70. Berer K, Mues M, Koutrolos M, Rasbi ZA, Boziki M, Johner C, et al. Commensal microbiota and myelin autoantigen cooperate to trigger autoimmune demyelination. Nature (2011) 479:538–41. doi:10.1038/nature10554
71. Klehmet J, Shive C, Guardia-Wolff R, Petersen I, Spack EG, Boehm BO, et al. T cell epitope spreading to myelin oligodendrocyte glycoprotein in HLA-DR4 transgenic mice during experimental autoimmune encephalomyelitis. Clin Immunol (2004) 111:53–60. doi:10.1016/j.clim.2003.12.012
72. Lehmann PV, Forsthuber T, Miller A, Sercarz EE. Spreading of T-cell autoimmunity to cryptic determinants of an autoantigen. Nature (1992) 358:155–7. doi:10.1038/358155a0
73. McFarland HI, Lobito AA, Johnson MM, Nyswaner JT, Frank JA, Palardy GR, et al. Determinant spreading associated with demyelination in a nonhuman primate model of multiple sclerosis. J Immunol (1999) 162:2384–90.
74. Vanderlugt CL, Neville KL, Nikcevich KM, Eagar TN, Bluestone JA, Miller SD. Pathologic role and temporal appearance of newly emerging autoepitopes in relapsing experimental autoimmune encephalomyelitis. J Immunol (2000) 164:670–8. doi:10.4049/jimmunol.164.2.670
75. McMahon EJ, Bailey SL, Castenada CV, Waldner H, Miller SD. Epitope spreading initiates in the CNS in two mouse models of multiple sclerosis. Nat Med (2005) 11:335–9. doi:10.1038/nm1202
76. Miller SD, Vanderlugt CL, Begolka WS, Pao W, Yauch RL, Neville KL, et al. Persistent infection with Theiler’s virus leads to CNS autoimmunity via epitope spreading. Nat Med (1997) 3:1133–6. doi:10.1038/nm1097-1133
77. Ristori G, Giubilei F, Giunti D, Perna A, Gasperini C, Buttinelli C, et al. Myelin basic protein intramolecular spreading without disease progression in a patient with multiple sclerosis. J Neuroimmunol (2000) 110:240–3. doi:10.1016/S0165-5728(00)00342-8
78. Uccelli A, Ristori G, Giunti D, Seri M, Montesperelli C, Caroli F, et al. Dynamics of the reactivity to MBP in multiple sclerosis. J Neurovirol (2000) 6(Suppl 2):S52–6.
79. Tuohy VK, Yu M, Weinstock-Guttman B, Kinkel RP. Diversity and plasticity of self recognition during the development of multiple sclerosis. J Clin Invest (1997) 99:1682–90. doi:10.1172/JCI119331
80. Muraro PA, Wandinger K, Bielekova B, Gran B, Marques A, Utz U, et al. Molecular tracking of antigen-specific T cell clones in neurological immune-mediated disorders. Brain (2003) 126:20–31. doi:10.1093/brain/awg021
81. Sawcer S, Hellenthal G, Pirinen M, Spencer CC, Patsopoulos NA, Moutsianas L, et al. Genetic risk and a primary role for cell-mediated immune mechanisms in multiple sclerosis. Nature (2011) 476:214–9. doi:10.1038/nature10251
82. Lock C, Hermans G, Pedotti R, Brendolan A, Schadt E, Garren H, et al. Gene-microarray analysis of multiple sclerosis lesions yields new targets validated in autoimmune encephalomyelitis. Nat Med (2002) 8:500–8. doi:10.1038/nm0502-500
83. Madsen LS, Andersson EC, Jansson L, Krogsgaard M, Andersen CB, Engberg J, et al. A humanized model for multiple sclerosis using HLA-DR2 and a human T-cell receptor. Nat Genet (1999) 23:343–7. doi:10.1038/15525
84. Bielekova B, Goodwin B, Richert N, Cortese I, Kondo T, Afshar G, et al. Encephalitogenic potential of the myelin basic protein peptide (amino acids 83-99) in multiple sclerosis: results of a phase II clinical trial with an altered peptide ligand. Nat Med (2000) 6:1167–75. doi:10.1038/80516
85. Nakajima H, Fukuda K, Doi Y, Sugino M, Kimura F, Hanafusa T, et al. Expression of Th1/Th2-related chemokine receptors on peripheral T cells and correlation with clinical disease activity in patients with multiple sclerosis. Eur Neurol (2004) 52:162–8. doi:10.1159/000081856
86. Shimizu Y, Ota K, Kubo S, Kabasawa C, Kobayashi M, Ohashi T, et al. Association of Th1/Th2-related chemokine receptors in peripheral T cells with disease activity in patients with multiple sclerosis and neuromyelitis optica. Eur Neurol (2011) 66:91–7. doi:10.1159/000329576
87. Tejada-Simon MV, Zang YC, Yang D, Hong J, Li S, Singh RA, et al. Aberrant T cell responses to myelin antigens during clinical exacerbation in patients with multiple sclerosis. Int Immunol (2000) 12:1641–50. doi:10.1093/intimm/12.12.1641
88. Correale J, Gilmore W, McMillan M, Li S, McCarthy K, Le T, et al. Patterns of cytokine secretion by autoreactive proteolipid protein-specific T cell clones during the course of multiple sclerosis. J Immunol (1995) 154:2959–68.
89. Tejada-Simon MV, Hong J, Rivera VM, Zhang JZ. Reactivity pattern and cytokine profile of T cells primed by myelin peptides in multiple sclerosis and healthy individuals. Eur J Immunol (2001) 31:907–17. doi:10.1002/1521-4141(200103)31:3<907::AID-IMMU907>3.0.CO;2-1
90. Calabresi PA, Fields NS, Farnon EC, Frank JA, Bash CN, Kawanashi T, et al. ELI-spot of Th-1 cytokine secreting PBMC’s in multiple sclerosis: correlation with MRI lesions. J Neuroimmunol (1998) 85:212–9. doi:10.1016/S0165-5728(98)00008-3
91. Bielekova B, Sung M, Kadom N, Simon R, McFarland H, Martin R. Expansion and functional relevance of high-avidity myelin-specific CD4+ T cells in multiple sclerosis. J Immunol (2004) 172:3893–904. doi:10.4049/jimmunol.172.6.3893
92. Lowther DE, Chong DL, Ascough S, Ettorre A, Ingram RJ, Boyton RJ, et al. Th1 not Th17 cells drive spontaneous MS-like disease despite a functional regulatory T cell response. Acta Neuropathol (2013) 126:501–15. doi:10.1007/s00401-013-1159-9
93. Bettelli E, Carrier Y, Gao W, Korn T, Strom TB, Oukka M, et al. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature (2006) 441:235–8. doi:10.1038/nature04753
94. Korn T, Bettelli E, Gao W, Awasthi A, Jäger A, Strom TB, et al. IL-21 initiates an alternative pathway to induce proinflammatory TH17 cells. Nature (2007) 448:484–7. doi:10.1038/nature05970
95. Langrish CL, Chen Y, Blumenschein WM, Mattson J, Basham B, Sedgwick JD, et al. IL-23 drives a pathogenic T cell population that induces autoimmune inflammation. J Exp Med (2005) 201:233–40. doi:10.1084/jem.20041257
96. Kebir H, Kreymborg K, Ifergan I, Dodelet-Devillers A, Cayrol R, Bernard M, et al. Human TH17 lymphocytes promote blood-brain barrier disruption and central nervous system inflammation. Nat Med (2007) 13:1173–5. doi:10.1038/nm1651
97. Quan C, Yu H, Qiao J, Xiao B, Zhao G, Wu Z, et al. Impaired regulatory function and enhanced intrathecal activation of B cells in neuromyelitis optica: distinct from multiple sclerosis. Mult Scler (2013) 19:289–98. doi:10.1177/1352458512454771
98. Darlington PJ, Touil T, Doucet J, Gaucher D, Zeidan J, Gauchat D, et al. Diminished Th17 (not Th1) responses underlie multiple sclerosis disease abrogation after hematopoietic stem cell transplantation. Ann Neurol (2013) 73:341–54. doi:10.1002/ana.23784
99. Venken K, Hellings N, Hensen K, Rummens J, Stinissen P. Memory CD4+CD127high T cells from patients with multiple sclerosis produce IL-17 in response to myelin antigens. J Neuroimmunol (2010) 226:185–91. doi:10.1016/j.jneuroim.2010.05.025
100. Schlapbach C, Gehad A, Yang C, Watanabe R, Guenova E, Teague JE, et al. Human TH9 cells are skin-tropic and have autocrine and paracrine proinflammatory capacity. Sci Transl Med (2014) 6:219ra8. doi:10.1126/scitranslmed.3007828
101. Jager A, Dardalhon V, Sobel RA, Bettelli E, Kuchroo VK. Th1, Th17, and Th9 effector cells induce experimental autoimmune encephalomyelitis with different pathological phenotypes. J Immunol (2009) 183:7169–77. doi:10.4049/jimmunol.0901906
102. Eyerich S, Eyerich K, Pennino D, Carbone T, Nasorri F, Pallotta S, et al. Th22 cells represent a distinct human T cell subset involved in epidermal immunity and remodeling. J Clin Invest (2009) 119:3573–85. doi:10.1172/JCI40202
103. Duhen T, Geiger R, Jarrossay D, Lanzavecchia A, Sallusto F. Production of interleukin 22 but not interleukin 17 by a subset of human skin-homing memory T cells. Nat Immunol (2009) 10:857–63. doi:10.1038/ni.1767
104. Rolla S, Bardina V, De Mercanti S, Quaglino P, De Palma R, Gned D, et al. Th22 cells are expanded in multiple sclerosis and are resistant to IFN-β. J Leukoc Biol (2014) 96:1155–64. doi:10.1189/jlb.5A0813-463RR
105. Peelen E, Damoiseaux J, Smolders J, Knippenberg S, Menheere P, Tervaert JW, et al. Th17 expansion in MS patients is counterbalanced by an expanded CD39+ regulatory T cell population during remission but not during relapse. J Neuroimmunol (2011) 240-241:97–103. doi:10.1016/j.jneuroim.2011.09.013
106. Viglietta V, Baecher-Allan C, Weiner HL, Hafler DA. Loss of functional suppression by CD4+CD25+ regulatory T cells in patients with multiple sclerosis. J Exp Med (2004) 199:971–9. doi:10.1084/jem.20031579
107. Haas J, Hug A, Viehöver A, Fritzsching B, Falk CS, Filser A, et al. Reduced suppressive effect of CD4+CD25high regulatory T cells on the T cell immune response against myelin oligodendrocyte glycoprotein in patients with multiple sclerosis. Eur J Immunol (2005) 35:3343–52. doi:10.1002/eji.200526065
108. Feger U, Luther C, Poeschel S, Melms A, Tolosa E, Wiendl H. Increased frequency of CD4 + CD25 + regulatory T cells in the cerebrospinal fluid but not in the blood of multiple sclerosis patients. Clin Exp Immunol (2007) 147:412–8. doi:10.1111/j.1365-2249.2006.03271.x
109. Venken K, Hellings N, Hensen K, Rummens J, Medaer R, D’hooghe MB, et al. Secondary progressive in contrast to relapsing-remitting multiple sclerosis patients show a normal CD4 + CD25 + regulatory T-cell function and FOXP3 expression. J Neurosci Res (2006) 83:1432–46. doi:10.1002/jnr.20852
110. Michel L, Berthelot L, Pettré S, Wiertlewski S, Lefrère F, Braudeau C, et al. Patients with relapsing-remitting multiple sclerosis have normal Treg function when cells expressing IL-7 receptor α-chain are excluded from the analysis. J Clin Invest (2008) 118:3411–9. doi:10.1172/JCI35365
111. Frisullo G, Nociti V, Iorio R, Patanella AK, Caggiula M, Marti A, et al. Regulatory T cells fail to suppress CD4 + T-bet + T cells in relapsing multiple sclerosis patients. Immunology (2009) 127:418–28. doi:10.1111/j.1365-2567.2008.02963.x
112. Kumar M, Putzki N, Limmroth V, Remus R, Lindemann M, Knop D, et al. CD4+CD25+FoxP3+ T lymphocytes fail to suppress myelin basic protein-induced proliferation in patients with multiple sclerosis. J Neuroimmunol (2006) 180:178–84. doi:10.1016/j.jneuroim.2006.08.003
113. Venken K, Hellings N, Broekmans T, Hensen K, Rummens J, Stinissen P. Natural Naive CD4+CD25+CD127low regulatory T cell (Treg) development and function are disturbed in multiple sclerosis patients: recovery of memory Treg homeostasis during disease progression. J Immunol (2008) 180:6411–20. doi:10.4049/jimmunol.180.9.6411
114. Haas J, Fritzsching B, Trubswetter P, Korporal M, Milkova L, Fritz B, et al. Prevalence of newly generated naive regulatory T cells (Treg) is critical for Treg suppressive function and determines Treg dysfunction in multiple sclerosis. J Immunol (2007) 179:1322–30. doi:10.4049/jimmunol.179.2.1322
115. Hug A, Korporal M, Schroder I, Haas J, Glatz K, Storch-Hagenlocher B, et al. Thymic export function and T cell homeostasis in patients with relapsing remitting multiple sclerosis. J Immunol (2003) 171:432–7. doi:10.4049/jimmunol.171.1.432
116. Fletcher JM, Lonergan R, Costelloe L, Kinsella K, Moran B, O’Farrelly C, et al. CD39+Foxp3+ regulatory T cells suppress pathogenic Th17 cells and are impaired in multiple sclerosis. J Immunol (2009) 183:7602–10. doi:10.4049/jimmunol.0901881
117. Korn T, Reddy J, Gao W, Bettelli E, Awasthi A, Petersen TR, et al. Myelin-specific regulatory T cells accumulate in the CNS but fail to control autoimmune inflammation. Nat Med (2007) 13:423–31. doi:10.1038/nm1564
118. Gurevich M, Achiron A. The switch between relapse and remission in multiple sclerosis: continuous inflammatory response balanced by Th1 suppression and neurotrophic factors. J Neuroimmunol (2012) 252:83–8. doi:10.1016/j.jneuroim.2012.07.014
119. Herrmann MM, Gaertner S, Stadelmann C, van den Brandt J, Böscke R, Budach W, et al. Tolerance induction by bone marrow transplantation in a multiple sclerosis model. Blood (2005) 106:1875–83. doi:10.1182/blood-2004-12-4607
120. Babbe H, Roers A, Waisman A, Lassmann H, Goebels N, Hohlfeld R, et al. Clonal expansions of CD8(+) T cells dominate the T cell infiltrate in active multiple sclerosis lesions as shown by micromanipulation and single cell polymerase chain reaction. J Exp Med (2000) 192:393–404. doi:10.1084/jem.192.3.393
121. Skulina C, Schmidt S, Dornmair K, Babbe H, Roers A, Rajewsky K, et al. Multiple sclerosis: brain-infiltrating CD8+ T cells persist as clonal expansions in the cerebrospinal fluid and blood. Proc Natl Acad Sci U S A (2004) 101:2428–33. doi:10.1073/pnas.0308689100
122. Sauer BM, Schmalstieg WF, Howe CL. Axons are injured by antigen-specific CD8+ T cells through a MHC class I- and granzyme B-dependent mechanism. Neurobiol Dis (2013) 59:194–205. doi:10.1016/j.nbd.2013.07.010
123. Zang YCQ, Li S, Rivera VM, Hong J, Robinson RR, Breitbach WT, et al. Increased CD8+ cytotoxic T cell responses to myelin basic protein in multiple sclerosis. J Immunol (2004) 172:5120–7. doi:10.4049/jimmunol.172.8.5120
124. Liu G, Fang L, Hjelmstrom P, Gao X. Increased CD8+ central memory T cells in patients with multiple sclerosis. Mult Scler (2007) 13:149–55. doi:10.1177/1352458506069246
125. Jacobsen M, Cepok S, Quak E, Happel M, Gaber R, Ziegler A, et al. Oligoclonal expansion of memory CD8+ T cells in cerebrospinal fluid from multiple sclerosis patients. Brain (2002) 125:538–50. doi:10.1093/brain/awf059
126. Malmestrom C, Lycke J, Haghighi S, Andersen O, Carlsson L, Wadenvik H, et al. Relapses in multiple sclerosis are associated with increased CD8+ T-cell mediated cytotoxicity in CSF. J Neuroimmunol (2008) 196:159–65. doi:10.1016/j.jneuroim.2008.03.001
127. Huseby ES, Liggitt D, Brabb T, Schnabel B, Ohlén C, Goverman J. A pathogenic role for myelin-specific CD8(+) T cells in a model for multiple sclerosis. J Exp Med (2001) 194:669–76. doi:10.1084/jem.194.5.669
128. Cunnusamy K, Baughman EJ, Franco J, Ortega SB, Sinha S, Chaudhary P, et al. Disease exacerbation of multiple sclerosis is characterized by loss of terminally differentiated autoregulatory CD8+ T cells. Clin Immunol (2014) 152:115–26. doi:10.1016/j.clim.2014.03.005
129. Baughman EJ, Mendoza JP, Ortega SB, Ayers CL, Greenberg BM, Frohman EM, et al. Neuroantigen-specific CD8+ regulatory T-cell function is deficient during acute exacerbation of multiple sclerosis. J Autoimmun (2011) 36:115–24. doi:10.1016/j.jaut.2010.12.003
130. Correale J, Villa A. Role of CD8+ CD25+ Foxp3+ regulatory T cells in multiple sclerosis. Ann Neurol (2009) 67:625–38. doi:10.1002/ana.21944
131. Hauser SL, Waubant E, Arnold DL, Vollmer T, Antel J, Fox RJ, et al. B-cell depletion with rituximab in relapsing-remitting multiple sclerosis. N Engl J Med (2008) 358:676–88. doi:10.1056/NEJMoa0706383
132. Link H, Huang Y. Oligoclonal bands in multiple sclerosis cerebrospinal fluid: an update on methodology and clinical usefulness. J Neuroimmunol (2006) 180:17–28. doi:10.1016/j.jneuroim.2006.07.006
133. Genain CP, Cannella B, Hauser SL, Raine CS. Identification of autoantibodies associated with myelin damage in multiple sclerosis. Nat Med (1999) 5:170–5. doi:10.1038/5532
134. O’Connor KC, Appel H, Bregoli L, Call ME, Catz I, Chan JA, et al. Antibodies from inflamed central nervous system tissue recognize myelin oligodendrocyte glycoprotein. J Immunol (2005) 175:1974–82. doi:10.4049/jimmunol.175.3.1974
135. Bar-Or A, Fawaz L, Fan B, Darlington PJ, Rieger A, Ghorayeb C, et al. Abnormal B-cell cytokine responses a trigger of T-cell-mediated disease in MS? Ann Neurol (2010) 67:452–61. doi:10.1002/ana.21939
136. Lanzavecchia A. Antigen-specific interaction between T and B cells. Nature (1985) 314:537–9. doi:10.1038/314537a0
137. Molnarfi N, Schulze-Topphoff U, Weber MS, Patarroyo JC, Prod’homme T, Varrin-Doyer M, et al. MHC class II-dependent B cell APC function is required for induction of CNS autoimmunity independent of myelin-specific antibodies. J Exp Med (2013) 210:2921–37. doi:10.1084/jem.20130699
138. Harp CT, Ireland S, Davis LS, Remington G, Cassidy B, Cravens PD, et al. Memory B cells from a subset of treatment-naïve relapsing-remitting multiple sclerosis patients elicit CD4+ T-cell proliferation and IFN-γ production in response to myelin basic protein and myelin oligodendrocyte glycoprotein. Eur J Immunol (2010) 40:2942–56. doi:10.1002/eji.201040516
139. Monson NL, Brezinschek H, Brezinschek RI, Mobley A, Vaughan GK, Frohman EM, et al. Receptor revision and atypical mutational characteristics in clonally expanded B cells from the cerebrospinal fluid of recently diagnosed multiple sclerosis patients. J Neuroimmunol (2005) 158:170–81. doi:10.1016/j.jneuroim.2004.04.022
140. Delves PJ, Roitt IM. The immune system. Second of two parts. N Engl J Med (2000) 343:108–17. doi:10.1056/NEJM200007133430207
141. Kinnunen T, Chamberlain N, Morbach H, Cantaert T, Lynch M, Preston-Hurlburt P, et al. Specific peripheral B cell tolerance defects in patients with multiple sclerosis. J Clin Invest (2013) 123:2737–41. doi:10.1172/JCI68775
142. Iwata Y, Matsushita T, Horikawa M, DiLillo DJ, Yanaba K, Venturi GM, et al. Characterization of a rare IL-10-competent B-cell subset in humans that parallels mouse regulatory B10 cells. Blood (2011) 117:530–41. doi:10.1182/blood-2010-07-294249
143. Yoshizaki A, Miyagaki T, DiLillo DJ, Matsushita T, Horikawa M, Kountikov EI, et al. Regulatory B cells control T-cell autoimmunity through IL-21-dependent cognate interactions. Nature (2012) 491:264–8. doi:10.1038/nature11501
144. Knippenberg S, Peelen E, Smolders J, Thewissen M, Menheere P, Cohen Tervaert JW, et al. Reduction in IL-10 producing B cells (Breg) in multiple sclerosis is accompanied by a reduced naïve/memory Breg ratio during a relapse but not in remission. J Neuroimmunol (2011) 239:80–6. doi:10.1016/j.jneuroim.2011.08.019
145. Michel L, Chesneau M, Manceau P, Genty A, Garcia A, Salou M, et al. Unaltered regulatory B-cell frequency and function in patients with multiple sclerosis. Clin Immunol (2014) 155:198–208. doi:10.1016/j.clim.2014.09.011
146. Duddy M, Niino M, Adatia F, Hebert S, Freedman M, Atkins H, et al. Distinct effector cytokine profiles of memory and naive human B cell subsets and implication in multiple sclerosis. J Immunol (2007) 178:6092–9. doi:10.4049/jimmunol.178.10.6092
147. Correale J, Farez M, Razzitte G. Helminth infections associated with multiple sclerosis induce regulatory B cells. Ann Neurol (2008) 64:187–99. doi:10.1002/ana.21438
148. Cohn L, Delamarre L. Dendritic cell-targeted vaccines. Front Immunol (2014) 5:255. doi:10.3389/fimmu.2014.00255
149. Burster T, Beck A, Tolosa E, Schnorrer P, Weissert R, Reich M, et al. Differential processing of autoantigens in lysosomes from human monocyte-derived and peripheral blood dendritic cells. J Immunol (2005) 175:5940–9. doi:10.4049/jimmunol.175.9.5940
150. Delamarre L, Mellman I. Harnessing dendritic cells for immunotherapy. Semin Immunol (2011) 23:2–11. doi:10.1016/j.smim.2011.02.001
151. Hertz CJ, Kiertscher SM, Godowski PJ, Bouis DA, Norgard MV, Roth MD, et al. Microbial lipopeptides stimulate dendritic cell maturation via toll-like receptor 2. J Immunol (2001) 166:2444–50. doi:10.4049/jimmunol.166.4.2444
152. Mehta-Damani A, Markowicz S, Engleman EG. Generation of antigen-specific CD4+ T cell lines from naive precursors. Eur J Immunol (1995) 25:1206–11. doi:10.1002/eji.1830250511
153. Hildner K, Edelson BT, Purtha WE, Diamond M, Matsushita H, Kohyama M, et al. Batf3 deficiency reveals a critical role for CD8alpha+ dendritic cells in cytotoxic T cell immunity. Science (2008) 322:1097–100. doi:10.1126/science.1164206
154. Karni A, Abraham M, Monsonego A, Cai G, Freeman GJ, Hafler D, et al. Innate immunity in multiple sclerosis: myeloid dendritic cells in secondary progressive multiple sclerosis are activated and drive a proinflammatory immune response. J Immunol (2006) 177:4196–202. doi:10.4049/jimmunol.177.6.4196
155. van Zwam M, Samsom JN, Nieuwenhuis EE, Melief M, Wierenga-Wolf AF, Dijke IE, et al. Myelin ingestion alters macrophage antigen-presenting function in vitro and in vivo. J Leukoc Biol (2011) 90:123–32. doi:10.1189/jlb.1209813
156. Pesic M, Bartholomäus I, Kyratsous NI, Heissmeyer V, Wekerle H, Kawakami N. 2-photon imaging of phagocyte-mediated T cell activation in the CNS. J Clin Invest (2013) 123:1192–201. doi:10.1172/JCI67233
157. Fabriek BO, Van Haastert ES, Galea I, Polfliet MM, Döpp ED, Van Den Heuvel MM, et al. CD163-positive perivascular macrophages in the human CNS express molecules for antigen recognition and presentation. Glia (2005) 51:297–305. doi:10.1002/glia.20208
158. Jack C, Ruffini F, Bar-Or A, Antel JP. Microglia and multiple sclerosis. J Neurosci Res (2005) 81:363–73. doi:10.1002/jnr.20482
159. Woodroofe MN, Bellamy AS, Feldmann M, Davison AN, Cuzner ML. Immunocytochemical characterisation of the immune reaction in the central nervous system in multiple sclerosis. Possible role for microglia in lesion growth. J Neurol Sci (1986) 74:135–52. doi:10.1016/0022-510X(86)90100-0
160. Bö L, Mörk S, Kong PA, Nyland H, Pardo CA, Trapp BD. Detection of MHC class II-antigens on macrophages and microglia, but not on astrocytes and endothelia in active multiple sclerosis lesions. J Neuroimmunol (1994) 51:135–46. doi:10.1016/0165-5728(94)90075-2
161. Kort JJ, Kawamura K, Fugger L, Weissert R, Forsthuber TG. Efficient presentation of myelin oligodendrocyte glycoprotein peptides but not protein by astrocytes from HLA-DR2 and HLA-DR4 transgenic mice. J Neuroimmunol (2006) 173:23–34. doi:10.1016/j.jneuroim.2005.11.014
162. Ebers GC, Bulman DE, Sadovnick AD, Paty DW, Warren S, Hader W, et al. A population-based study of multiple sclerosis in twins. N Engl J Med (1986) 315:1638–42. doi:10.1056/NEJM198612253152603
163. Hedstrom AK, Sundqvist E, Baarnhielm M, Nordin N, Hillert J, Kockum I, et al. Smoking and two human leukocyte antigen genes interact to increase the risk for multiple sclerosis. Brain (2011) 134:653–64. doi:10.1093/brain/awq371
164. Beecham AH, Patsopoulos NA, Xifara DK, Davis MF, Kemppinen A, Cotsapas C, et al. Analysis of immune-related loci identifies 48 new susceptibility variants for multiple sclerosis. Nat Genet (2013) 45:1353–60. doi:10.1038/ng.2770
165. Fugger L, Friese MA, Bell JI. From genes to function: the next challenge to understanding multiple sclerosis. Nat Rev Immunol (2009) 9:408–17. doi:10.1038/nri2554
166. Hensiek AE, Sawcer SJ, Feakes R, Deans J, Mander A, Akesson E, et al. HLA-DR 15 is associated with female sex and younger age at diagnosis in multiple sclerosis. J Neurol Neurosurg Psychiatry (2002) 72:184–7. doi:10.1136/jnnp.72.2.184
167. Friese MA, Jakobsen KB, Friis L, Etzensperger R, Craner MJ, McMahon RM, et al. Opposing effects of HLA class I molecules in tuning autoreactive CD8+ T cells in multiple sclerosis. Nat Med (2008) 14:1227–35. doi:10.1038/nm.1881
168. Burton PR, Clayton DG, Cardon LR, Craddock N, Deloukas P, Duncanson A, et al. Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature (2007) 447:661–78. doi:10.1038/nature05911
169. Gregersen PK, Kosoy R, Lee AT, Lamb J, Sussman J, McKee D, et al. Risk for myasthenia gravis maps to a 151 Pro→Ala change in TNIP1 and to human leukocyte antigen-B*08. Ann Neurol (2012) 72:927–35. doi:10.1002/ana.23691
170. Gregersen PK, Silver J, Winchester RJ. The shared epitope hypothesis. An approach to understanding the molecular genetics of susceptibility to rheumatoid arthritis. Arthritis Rheum (1987) 30:1205–13. doi:10.1002/art.1780301102
171. Jones EY, Fugger L, Strominger JL, Siebold C. MHC class II proteins and disease: a structural perspective. Nat Rev Immunol (2006) 6:271–82. doi:10.1038/nri1805
172. Mohme M, Hotz C, Stevanovic S, Binder T, Lee J, Okoniewski M, et al. HLA-DR15-derived self-peptides are involved in increased autologous T cell proliferation in multiple sclerosis. Brain (2013) 136:1783–98. doi:10.1093/brain/awt108
173. Kirschmann DA, Duffin KL, Smith CE, Welply JK, Howard SC, Schwartz BD, et al. Naturally processed peptides from rheumatoid arthritis associated and non-associated HLA-DR alleles. J Immunol (1995) 155:5655–62.
174. Moldovan IR, Rudick RA, Cotleur AC, Born SE, Lee J, Karafa MT, et al. Interferon gamma responses to myelin peptides in multiple sclerosis correlate with a new clinical measure of disease progression. J Neuroimmunol (2003) 141:132–40. doi:10.1016/S0165-5728(03)00221-2
175. Olsson T, Sun J, Hillert J, Höjeberg B, Ekre HP, Andersson G, et al. Increased numbers of T cells recognizing multiple myelin basic protein epitopes in multiple sclerosis. Eur J Immunol (1992) 22:1083–7. doi:10.1002/eji.1830220431
176. Hedegaard CJ, Krakauer M, Bendtzen K, Lund H, Sellebjerg F, Nielsen CH. T helper cell type 1 (Th1), Th2 and Th17 responses to myelin basic protein and disease activity in multiple sclerosis. Immunology (2008) 125:161–9. doi:10.1111/j.1365-2567.2008.02837.x
177. Valli A, Sette A, Kappos L, Oseroff C, Sidney J, Miescher G, et al. Binding of myelin basic protein peptides to human histocompatibility leukocyte antigen class II molecules and their recognition by T cells from multiple sclerosis patients. J Clin Invest (1993) 91:616–28. doi:10.1172/JCI116242
178. Martin R, Howell MD, Jaraquemada D, Flerlage M, Richert J, Brostoff S, et al. A myelin basic protein peptide is recognized by cytotoxic T cells in the context of four HLA-DR types associated with multiple sclerosis. J Exp Med (1991) 173:19–24. doi:10.1084/jem.173.1.19
179. Greer JM, Csurhes PA, Pender MP, McCombe PA. Effect of gender on T-cell proliferative responses to myelin proteolipid protein antigens in patients with multiple sclerosis and controls. J Autoimmun (2004) 22:345–52. doi:10.1016/j.jaut.2004.03.004
180. Pender MP, Csurhes PA, Greer JM, Mowat PD, Henderson RD, Cameron KD, et al. Surges of increased T cell reactivity to an encephalitogenic region of myelin proteolipid protein occur more often in patients with multiple sclerosis than in healthy subjects. J Immunol (2000) 165:5322–31. doi:10.4049/jimmunol.165.9.5322
181. Greer JM, Csurhes PA, Cameron KD, McCombe PA, Good MF, Pender MP. Increased immunoreactivity to two overlapping peptides of myelin proteolipid protein in multiple sclerosis. Brain (1997) 120:1447–60. doi:10.1093/brain/120.8.1447
182. Krogsgaard M, Wucherpfennig KW, Cannella B, Hansen BE, Svejgaard A, Pyrdol J, et al. Visualization of myelin basic protein (MBP) T cell epitopes in multiple sclerosis lesions using a monoclonal antibody specific for the human histocompatibility leukocyte antigen (HLA)-DR2-MBP 85-99 complex. J Exp Med (2000) 191:1395–412. doi:10.1084/jem.191.8.1395
183. Kappos L, Comi G, Panitch H, Oger J, Antel J, Conlon P, et al. Induction of a non-encephalitogenic type 2 T helper-cell autoimmune response in multiple sclerosis after administration of an altered peptide ligand in a placebo-controlled, randomized phase II trial. The altered peptide ligand in relapsing MS study group. Nat Med (2000) 6:1176–82. doi:10.1038/80525
184. Vogt AB, Kropshofer H, Kalbacher H, Kalbus M, Rammensee HG, Coligan JE, et al. Ligand motifs of HLA-DRB5*0101 and DRB1*1501 molecules delineated from self-peptides. J Immunol (1994) 153:1665–73.
185. Vergelli M, Kalbus M, Rojo SC, Hemmer B, Kalbacher H, Tranquill L, et al. T cell response to myelin basic protein in the context of the multiple sclerosis-associated HLA-DR15 haplotype: peptide binding, immunodominance and effector functions of T cells. J Neuroimmunol (1997) 77:195–203. doi:10.1016/S0165-5728(97)00075-1
186. Belogurov A, Kudriaeva A, Kuzina E, Smirnov I, Bobik T, Ponomarenko N, et al. Multiple sclerosis autoantigen myelin basic protein escapes control by ubiquitination during proteasomal degradation. J Biol Chem (2014) 289:17758–66. doi:10.1074/jbc.M113.544247
187. Pelfrey CM, Trotter JL, Tranquill LR, McFarland HF. Identification of a novel T cell epitope of human proteolipid protein (residues 40-60) recognized by proliferative and cytolytic CD4+ T cells from multiple sclerosis patients. J Neuroimmunol (1993) 46:33–42. doi:10.1016/0165-5728(93)90231-M
188. Pelfrey CM, Tranquill LR, Vogt AB, McFarland HF. T cell response to two immunodominant proteolipid protein (PLP) peptides in multiple sclerosis patients and healthy controls. Mult Scler (1996) 1:270–8.
189. Trotter JL, Pelfrey CM, Trotter AL, Selvidge JA, Gushleff KC, Mohanakumar T, et al. T cell recognition of myelin proteolipid protein and myelin proteolipid protein peptides in the peripheral blood of multiple sclerosis and control subjects. J Neuroimmunol (1998) 84:172–8. doi:10.1016/S0165-5728(97)00260-9
190. Markovic-Plese S, Fukaura H, Zhang J, al-Sabbagh A, Southwood S, Sette A, et al. T cell recognition of immunodominant and cryptic proteolipid protein epitopes in humans. J Immunol (1995) 155:982–92.
191. Trotter JL, Hickey WF, van der Veen RC, Sulze L. Peripheral blood mononuclear cells from multiple sclerosis patients recognize myelin proteolipid protein and selected peptides. J Neuroimmunol (1991) 33:55–62. doi:10.1016/0165-5728(91)90034-5
192. Sun J, Link H, Olsson T, Xiao BG, Andersson G, Ekre HP, et al. T and B cell responses to myelin-oligodendrocyte glycoprotein in multiple sclerosis. J Immunol (1991) 146:1490–5.
193. Wallström E, Khademi M, Andersson M, Weissert R, Linington C, Olsson T. Increased reactivity to myelin oligodendrocyte glycoprotein peptides and epitope mapping in HLA DR2(15)+ multiple sclerosis. Eur J Immunol (1998) 28:3329–35. doi:10.1002/(SICI)1521-4141(199810)28:10<3329::AID-IMMU3329>3.0.CO;2-B
194. Raddassi K, Kent SC, Yang J, Bourcier K, Bradshaw EM, Seyfert-Margolis V, et al. Increased frequencies of myelin oligodendrocyte glycoprotein/MHC class II-binding CD4 cells in patients with multiple sclerosis. J Immunol (2011) 187:1039–46. doi:10.4049/jimmunol.1001543
195. Kerlero de Rosbo N, Milo R, Lees MB, Burger D, Bernard CC, Ben-Nun A. Reactivity to myelin antigens in multiple sclerosis. Peripheral blood lymphocytes respond predominantly to myelin oligodendrocyte glycoprotein. J Clin Invest (1993) 92:2602–8. doi:10.1172/JCI116875
196. Varrin-Doyer M, Shetty A, Spencer CM, Schulze-Topphoff U, Weber MS, Bernard CC, et al. MOG transmembrane and cytoplasmic domains contain highly stimulatory T-cell epitopes in MS. Neurol Neuroimmunol Neuroinflamm (2014) 1:e20. doi:10.1212/NXI.0000000000000020
197. Kennel De March A, De Bouwerie M, Kolopp-Sarda MN, Faure GC, Béné MC, Bernard CC. Anti-myelin oligodendrocyte glycoprotein B-cell responses in multiple sclerosis. J Neuroimmunol (2003) 135:117–25. doi:10.1016/S0165-5728(02)00434-4
198. Khalil M, Reindl M, Lutterotti A, Kuenz B, Ehling R, Gneiss C, et al. Epitope specificity of serum antibodies directed against the extracellular domain of myelin oligodendrocyte glycoprotein: influence of relapses and immunomodulatory treatments. J Neuroimmunol (2006) 174:147–56. doi:10.1016/j.jneuroim.2006.01.012
199. Lindert RB, Haase CG, Brehm U, Linington C, Wekerle H, Hohlfeld R. Multiple sclerosis: B- and T-cell responses to the extracellular domain of the myelin oligodendrocyte glycoprotein. Brain (1999) 122:2089–100. doi:10.1093/brain/122.11.2089
200. Zhou D, Srivastava R, Nessler S, Grummel V, Sommer N, Bruck W, et al. Identification of a pathogenic antibody response to native myelin oligodendrocyte glycoprotein in multiple sclerosis. Proc Natl Acad Sci U S A (2006) 103:19057–62. doi:10.1073/pnas.0607242103
201. Lalive PH, Menge T, Delarasse C, Della Gaspera B, Pham-Dinh D, Villoslada P, et al. Antibodies to native myelin oligodendrocyte glycoprotein are serologic markers of early inflammation in multiple sclerosis. Proc Natl Acad Sci U S A (2006) 103:2280–5. doi:10.1073/pnas.0510672103
202. Probstel AK, Dornmair K, Bittner R, Sperl P, Jenne D, Magalhaes S, et al. Antibodies to MOG are transient in childhood acute disseminated encephalomyelitis. Neurology (2011) 77:580–8. doi:10.1212/WNL.0b013e318228c0b1
203. Gaertner S, de Graaf KL, Greve B, Weissert R. Antibodies against glycosylated native MOG are elevated in patients with multiple sclerosis. Neurology (2004) 63:2381–3. doi:10.1212/01.WNL.0000147259.34163.33
204. Klawiter EC, Piccio L, Lyons J, Mikesell R, O’Connor KC, Cross AH. Elevated intrathecal myelin oligodendrocyte glycoprotein antibodies in multiple sclerosis. Arch Neurol (2010) 67:1102–8. doi:10.1001/archneurol.2010.197
205. Karni A, Bakimer-Kleiner R, Abramsky O, Ben-Nun A. Elevated levels of antibody to myelin oligodendrocyte glycoprotein is not specific for patients with multiple sclerosis. Arch Neurol (1999) 56:311–5. doi:10.1001/archneur.56.3.311
206. Tomassini V, de Giglio L, Reindl M, Russo P, Pestalozza I, Pantano P, et al. Anti-myelin antibodies predict the clinical outcome after a first episode suggestive of MS. Mult Scler (2007) 13:1086–94. doi:10.1177/1352458507077622
207. Pelayo R, Tintoré M, Montalban X, Rovira A, Espejo C, Reindl M, et al. Antimyelin antibodies with no progression to multiple sclerosis. N Engl J Med (2007) 356:426–8. doi:10.1056/NEJMc062467
208. Berger T, Rubner P, Schautzer F, Egg R, Ulmer H, Mayringer I, et al. Antimyelin antibodies as a predictor of clinically definite multiple sclerosis after a first demyelinating event. N Engl J Med (2003) 349:139–45. doi:10.1056/NEJMoa022328
209. Kuhle J, Pohl C, Mehling M, Edan G, Freedman MS, Hartung H, et al. Lack of association between antimyelin antibodies and progression to multiple sclerosis. N Engl J Med (2007) 356:371–8. doi:10.1056/NEJMoa063602
210. Lim ET, Berger T, Reindl M, Dalton CM, Fernando K, Keir G, et al. Anti-myelin antibodies do not allow earlier diagnosis of multiple sclerosis. Mult Scler (2005) 11:492–4. doi:10.1191/1352458505ms1187sr
211. Haase CG, Guggenmos J, Brehm U, Andersson M, Olsson T, Reindl M, et al. The fine specificity of the myelin oligodendrocyte glycoprotein autoantibody response in patients with multiple sclerosis and normal healthy controls. J Neuroimmunol (2001) 114:220–5. doi:10.1016/S0165-5728(00)00462-8
212. Koehler NK, Genain CP, Giesser B, Hauser SL. The human T cell response to myelin oligodendrocyte glycoprotein: a multiple sclerosis family-based study. J Immunol (2002) 168:5920–7. doi:10.4049/jimmunol.168.11.5920
213. Weissert R, Kuhle J, de Graaf KL, Wienhold W, Herrmann MM, Müller C, et al. High immunogenicity of intracellular myelin oligodendrocyte glycoprotein epitopes. J Immunol (2002) 169:548–56. doi:10.4049/jimmunol.169.1.548
214. Garcia-Vallejo JJ, Ilarregui JM, Kalay H, Chamorro S, Koning N, Unger WW, et al. CNS myelin induces regulatory functions of DC-SIGN-expressing, antigen-presenting cells via cognate interaction with MOG. J Exp Med (2014) 211:1465–83. doi:10.1084/jem.20122192
215. Van Noort JM, van Sechel AC, Bajramovic JJ, el Ouagmiri M, Polman CH, Lassmann H, et al. The small heat-shock protein alpha B-crystallin as candidate autoantigen in multiple sclerosis. Nature (1995) 375:798–801. doi:10.1038/375798a0
216. Rothbard JB, Zhao X, Sharpe O, Strohman MJ, Kurnellas M, Mellins ED, et al. Chaperone activity of α B-crystallin is responsible for its incorrect assignment as an autoantigen in multiple sclerosis. J Immunol (2011) 186:4263–8. doi:10.4049/jimmunol.1003934
217. Fissolo N, Haag S, de Graaf KL, Drews O, Stevanovic S, Rammensee HG, et al. Naturally presented peptides on major histocompatibility complex I and II molecules eluted from central nervous system of multiple sclerosis patients. Mol Cell Proteomics (2009) 8:2090–101. doi:10.1074/mcp.M900001-MCP200
218. Vincent A. Unravelling the pathogenesis of myasthenia gravis. Nat Rev Immunol (2002) 2:797–804. doi:10.1038/nri916
219. Almon RR, Andrew CG, Appel SH. Serum globulin in myasthenia gravis: inhibition of alpha-bungarotoxin binding to acetylcholine receptors. Science (1974) 186:55–7. doi:10.1126/science.186.4158.55
220. Lindstrom JM, Seybold ME, Lennon VA, Whittingham S, Duane DD. Antibody to acetylcholine receptor in myasthenia gravis. Prevalence, clinical correlates, and diagnostic value. Neurology (1976) 26:1054–9. doi:10.1212/WNL.26.11.1054
221. Toyka KV, Brachman DB, Pestronk A, Kao I. Myasthenia gravis: passive transfer from man to mouse. Science (1975) 190:397–9. doi:10.1126/science.1179220
222. Hoch W, McConville J, Helms S, Newsom-Davis J, Melms A, Vincent A. Auto-antibodies to the receptor tyrosine kinase MuSK in patients with myasthenia gravis without acetylcholine receptor antibodies. Nat Med (2001) 7:365–8. doi:10.1038/85520
223. Leite MI, Jacob S, Viegas S, Cossins J, Clover L, Morgan BP, et al. IgG1 antibodies to acetylcholine receptors in ‘seronegative’ myasthenia gravis. Brain (2008) 131:1940–52. doi:10.1093/brain/awn092
224. Higuchi O, Hamuro J, Motomura M, Yamanashi Y. Autoantibodies to low-density lipoprotein receptor-related protein 4 in myasthenia gravis. Ann Neurol (2011) 69:418–22. doi:10.1002/ana.22312
225. Hohlfeld R, Toyka KV, Heininger K, Grosse-Wilde H, Kalies I. Autoimmune human T lymphocytes specific for acetylcholine receptor. Nature (1984) 310:244–6. doi:10.1038/310244a0
226. Balandina A, Lécart S, Dartevelle P, Saoudi A, Berrih-Aknin S. Functional defect of regulatory CD4+CD25+ T cells in the thymus of patients with autoimmune myasthenia gravis. Blood (2005) 105:735–41. doi:10.1182/blood-2003-11-3900
227. Hohlfeld R, Michels M, Tesch H, Fahsbender A, Heininger K, Conti-Tronconi BM, et al. Epstein-Barr virus-transformed B cells can present acetylcholine receptor to autologous autoreactive T cells. Immunol Lett (1986) 12:171–4. doi:10.1016/0165-2478(86)90101-X
228. Wingerchuk DM, Lennon VA, Pittock SJ, Lucchinetti CF, Weinshenker BG. Revised diagnostic criteria for neuromyelitis optica. Neurology (2006) 66:1485–9. doi:10.1212/01.wnl.0000216139.44259.74
229. Lennon VA, Wingerchuk DM, Kryzer TJ, Pittock SJ, Lucchinetti CF, Fujihara K, et al. A serum autoantibody marker of neuromyelitis optica: distinction from multiple sclerosis. Lancet (2004) 364:2106–12. doi:10.1016/S0140-6736(04)17551-X
230. Lennon VA, Kryzer TJ, Pittock SJ, Verkman AS, Hinson SR. IgG marker of optic-spinal multiple sclerosis binds to the aquaporin-4 water channel. J Exp Med (2005) 202:473–7. doi:10.1084/jem.20050304
231. Weinshenker BG, Wingerchuk DM, Vukusic S, Linbo L, Pittock SJ, Lucchinetti CF, et al. Neuromyelitis optica IgG predicts relapse after longitudinally extensive transverse myelitis. Ann Neurol (2006) 59:566–9. doi:10.1002/ana.20770
232. Roemer SF, Parisi JE, Lennon VA, Benarroch EE, Lassmann H, Bruck W, et al. Pattern-specific loss of aquaporin-4 immunoreactivity distinguishes neuromyelitis optica from multiple sclerosis. Brain (2007) 130:1194–205. doi:10.1093/brain/awl371
233. Sabater L, Giralt A, Boronat A, Hankiewicz K, Blanco Y, Llufriu S, et al. Cytotoxic effect of neuromyelitis optica antibody (NMO-IgG) to astrocytes: an in vitro study. J Neuroimmunol (2009) 215:31–5. doi:10.1016/j.jneuroim.2009.07.014
234. Brück W, Popescu B, Lucchinetti CF, Markovic-Plese S, Gold R, Thal DR, et al. Neuromyelitis optica lesions may inform multiple sclerosis heterogeneity debate. Ann Neurol (2012) 72:385–94. doi:10.1002/ana.23621
235. Vincent T, Saikali P, Cayrol R, Roth AD, Bar-Or A, Prat A, et al. Functional consequences of neuromyelitis optica-IgG astrocyte interactions on blood-brain barrier permeability and granulocyte recruitment. J Immunol (2008) 181:5730–7. doi:10.4049/jimmunol.181.8.5730
236. Bennett JL, Lam C, Kalluri SR, Saikali P, Bautista K, Dupree C, et al. Intrathecal pathogenic anti-aquaporin-4 antibodies in early neuromyelitis optica. Ann Neurol (2009) 66:617–29. doi:10.1002/ana.21802
237. Bradl M, Misu T, Takahashi T, Watanabe M, Mader S, Reindl M, et al. Neuromyelitis optica: pathogenicity of patient immunoglobulin in vivo. Ann Neurol (2009) 66:630–43. doi:10.1002/ana.21837
238. Sato DK, Callegaro D, Lana-Peixoto MA, Waters PJ, de Haidar Jorge FM, Takahashi T, et al. Distinction between MOG antibody-positive and AQP4 antibody-positive NMO spectrum disorders. Neurology (2014) 82:474–81. doi:10.1212/WNL.0000000000000101
239. Kitley J, Waters P, Woodhall M, Leite MI, Murchison A, George J, et al. Neuromyelitis optica spectrum disorders with aquaporin-4 and myelin-oligodendrocyte glycoprotein antibodies: a comparative study. JAMA Neurol (2014) 71:276–83. doi:10.1001/jamaneurol.2013.5857
240. Saadoun S, Waters P, Bell BA, Vincent A, Verkman AS, Papadopoulos MC. Intra-cerebral injection of neuromyelitis optica immunoglobulin G and human complement produces neuromyelitis optica lesions in mice. Brain (2010) 133:349–61. doi:10.1093/brain/awp309
241. Asavapanumas N, Verkman AS. Neuromyelitis optica pathology in rats following intraperitoneal injection of NMO-IgG and intracerebral needle injury. Acta Neuropathol Commun (2014) 2:48. doi:10.1186/2051-5960-2-48
242. Linhares UC, Schiavoni PB, Barros PO, Kasahara TM, Teixeira B, Ferreira TB, et al. The ex vivo production of IL-6 and IL-21 by CD4+ T cells is directly associated with neurological disability in neuromyelitis optica patients. J Clin Immunol (2013) 33:179–89. doi:10.1007/s10875-012-9780-2
243. Matsuya N, Komori M, Nomura K, Nakane S, Fukudome T, Goto H, et al. Increased T-cell immunity against aquaporin-4 and proteolipid protein in neuromyelitis optica. Int Immunol (2011) 23:565–73. doi:10.1093/intimm/dxr056
244. Van Belle TL, Coppieters KT, von Herrath MG. Type 1 diabetes: etiology, immunology, and therapeutic strategies. Physiol Rev (2011) 91:79–118. doi:10.1152/physrev.00003.2010
245. Arif S, Tree TI, Astill TP, Tremble JM, Bishop AJ, Dayan CM, et al. Autoreactive T cell responses show proinflammatory polarization in diabetes but a regulatory phenotype in health. J Clin Invest (2004) 113(3):451–63. doi:10.1172/JCI19585
246. Marwaha AK, Crome SQ, Panagiotopoulos C, Berg KB, Qin H, Ouyang Q, et al. Cutting edge: increased IL-17-secreting T cells in children with new-onset type 1 diabetes. J Immunol (2010) 185:3814–8. doi:10.4049/jimmunol.1001860
247. Honkanen J, Nieminen JK, Gao R, Luopajarvi K, Salo HM, Ilonen J, et al. IL-17 immunity in human type 1 diabetes. J Immunol (2010) 185:1959–67. doi:10.4049/jimmunol.1000788
248. Ferraro A, Socci C, Stabilini A, Valle A, Monti P, Piemonti L, et al. Expansion of Th17 cells and functional defects in T regulatory cells are key features of the pancreatic lymph nodes in patients with type 1 diabetes. Diabetes (2011) 60:2903–13. doi:10.2337/db11-0090
249. Pinkse GG, Tysma OH, Bergen CA, Kester MG, Ossendorp F, van Veelen PA, et al. Autoreactive CD8 T cells associated with cell destruction in type 1 diabetes. Proc Natl Acad Sci U S A (2005) 102:18425–30. doi:10.1073/pnas.0508621102
250. Lindley S, Dayan CM, Bishop A, Roep BO, Peakman M, Tree TI. Defective suppressor function in CD4(+)CD25(+) T-cells from patients with type 1 diabetes. Diabetes (2005) 54:92–9. doi:10.2337/diabetes.54.1.92
251. Schneider A, Rieck M, Sanda S, Pihoker C, Greenbaum C, Buckner JH. The effector T cells of diabetic subjects are resistant to regulation via CD4+ FOXP3+ regulatory T cells. J Immunol (2008) 181:7350–5. doi:10.4049/jimmunol.181.10.7350
252. Brooks-Worrell B, Gersuk VH, Greenbaum C, Palmer JP. Intermolecular antigen spreading occurs during the preclinical period of human type 1 diabetes. J Immunol (2001) 166:5265–70. doi:10.4049/jimmunol.166.8.5265
253. Palmer JP, Asplin CM, Clemons P, Lyen K, Tatpati O, Raghu PK, et al. Insulin antibodies in insulin-dependent diabetics before insulin treatment. Science (1983) 222:1337–9. doi:10.1126/science.6362005
254. Toma A, Haddouk S, Briand J, Camoin L, Gahery H, Connan F, et al. Recognition of a subregion of human proinsulin by class I-restricted T cells in type 1 diabetic patients. Proc Natl Acad Sci U S A (2005) 102:10581–6. doi:10.1073/pnas.0504230102
255. Bonifacio E, Bingley PJ, Shattock M, Dean BM, Dunger D, Gale EA, et al. Quantification of islet-cell antibodies and prediction of insulin-dependent diabetes. Lancet (1990) 335:147–9. doi:10.1016/0140-6736(90)90013-U
256. Baekkeskov S, Aanstoot HJ, Christgau S, Reetz A, Solimena M, Cascalho M, et al. Identification of the 64K autoantigen in insulin-dependent diabetes as the GABA-synthesizing enzyme glutamic acid decarboxylase. Nature (1990) 347:151–6. doi:10.1038/347151a0
257. Bonifacio E, Lampasona V, Genovese S, Ferrari M, Bosi E. Identification of protein tyrosine phosphatase-like IA2 (islet cell antigen 512) as the insulin-dependent diabetes-related 37/40K autoantigen and a target of islet-cell antibodies. J Immunol (1995) 155:5419–26.
258. Wenzlau JM, Juhl K, Yu L, Moua O, Sarkar SA, Gottlieb P, et al. The cation efflux transporter ZnT8 (Slc30A8) is a major autoantigen in human type 1 diabetes. Proc Natl Acad Sci U S A (2007) 104:17040–5. doi:10.1073/pnas.0705894104
259. Peakman M, Stevens EJ, Lohmann T, Narendran P, Dromey J, Alexander A, et al. Naturally processed and presented epitopes of the islet cell autoantigen IA-2 eluted from HLA-DR4. J Clin Invest (1999) 104:1449–57. doi:10.1172/JCI7936
260. Nepom GT, Lippolis JD, White FM, Masewicz S, Marto JA, Herman A, et al. Identification and modulation of a naturally processed T cell epitope from the diabetes-associated autoantigen human glutamic acid decarboxylase 65 (hGAD65). Proc Natl Acad Sci U S A (2001) 98:1763–8. doi:10.1073/pnas.98.4.1763
261. Honeyman MC, Cram DS, Harrison LC. Glutamic acid decarboxylase 67-reactive T cells: a marker of insulin-dependent diabetes. J Exp Med (1993) 177:535–40. doi:10.1084/jem.177.2.535
262. Reijonen H, Mallone R, Heninger A, Laughlin EM, Kochik SA, Falk B, et al. GAD65-specific CD4+ T-cells with high antigen avidity are prevalent in peripheral blood of patients with type 1 diabetes. Diabetes (2004) 53:1987–94. doi:10.2337/diabetes.53.8.1987
263. Dang M, Rockell J, Wagner R, Wenzlau JM, Yu L, Hutton JC, et al. Human type 1 diabetes is associated with T cell autoimmunity to zinc transporter 8. J Immunol (2011) 186:6056–63. doi:10.4049/jimmunol.1003815
264. Jarchum I, Nichol L, Trucco M, Santamaria P, DiLorenzo TP. Identification of novel IGRP epitopes targeted in type 1 diabetes patients. Clin Immunol (2008) 127:359–65. doi:10.1016/j.clim.2008.01.015
265. Gottlieb PA, Delong T, Baker RL, Fitzgerald-Miller L, Wagner R, Cook G, et al. Chromogranin A is a T cell antigen in human type 1 diabetes. J Autoimmun (2014) 50:38–41. doi:10.1016/j.jaut.2013.10.003
266. von Herrath M, Sanda S, Herold K. Type 1 diabetes as a relapsing-remitting disease? Nat Rev Immunol (2007) 7:988–94. doi:10.1038/nri2192
267. Takemura S, Klimiuk PA, Braun A, Goronzy JJ, Weyand CM. T cell activation in rheumatoid synovium is B cell dependent. J Immunol (2001) 167:4710–8. doi:10.4049/jimmunol.167.8.4710
268. Shiokawa S, Matsumato N, Nishimura J. Clonal analysis of B cells in the synovial membrane of patients with rheumatoid arthritis. Scand J Rheumatol (2003) 32:12–8. doi:10.1080/03009740310000346
269. Yamada H, Nakashima Y, Okazaki K, Mawatari T, Fukushi J, Kaibara N, et al. Th1 but not Th17 cells predominate in the joints of patients with rheumatoid arthritis. Ann Rheum Dis (2007) 67:1299–304. doi:10.1136/ard.2007.080341
270. Leipe J, Grunke M, Dechant C, Reindl C, Kerzendorf U, Schulze-Koops H, et al. Role of Th17 cells in human autoimmune arthritis. Arthritis Rheum (2010) 62:2876–85. doi:10.1002/art.27622
271. Jovanovic DV, Di Battista JA, Martel-Pelletier J, Jolicoeur FC, He Y, Zhang M, et al. IL-17 stimulates the production and expression of proinflammatory cytokines, IL-beta and TNF-alpha, by human macrophages. J Immunol (1998) 160:3513–21.
272. Van Hamburg JP, Asmawidjaja PS, Davelaar N, Mus AM, Colin EM, Hazes JM, et al. Th17 cells, but not Th1 cells, from patients with early rheumatoid arthritis are potent inducers of matrix metalloproteinases and proinflammatory cytokines upon synovial fibroblast interaction, including autocrine interleukin-17A production. Arthritis Rheum (2011) 63:73–83. doi:10.1002/art.30093
273. Ehrenstein MR, Evans JG, Singh A, Moore S, Warnes G, Isenberg DA, et al. Compromised function of regulatory T cells in rheumatoid arthritis and reversal by anti-TNFalpha therapy. J Exp Med (2004) 200:277–85. doi:10.1084/jem.20040165
274. Edwards JCW, Szczepanski L, Szechinski J, Filipowicz-Sosnowska A, Emery P, Close DR, et al. Efficacy of B-cell-targeted therapy with rituximab in patients with rheumatoid arthritis. N Engl J Med (2004) 350:2572–81. doi:10.1056/NEJMoa032534
275. Verheijden GF, Rijnders AW, Bos E, Coenen-de Roo CJ, van Staveren CJ, Miltenburg AM, et al. Human cartilage glycoprotein-39 as a candidate autoantigen in rheumatoid arthritis. Arthritis Rheum (1997) 40:1115–25. doi:10.1002/art.1780400616
276. Li NL, Zhang DQ, Zhou KY, Cartman A, Leroux JY, Poole AR, et al. Isolation and characteristics of autoreactive T cells specific to aggrecan G1 domain from rheumatoid arthritis patients. Cell Res (2000) 10:39–49. doi:10.1038/sj.cr.7290034
277. Franklin EC, Holman HR, Muller-Eberhard HJ, Kunkel HG. An unusual protein component of high molecular weight in the serum of certain patients with rheumatoid arthritis. J Exp Med (1957) 105:425–38. doi:10.1084/jem.105.5.425
278. Van Zeben D, Hazes JM, Zwinderman AH, Cats A, van der Voort EA, Breedveld FC. Clinical significance of rheumatoid factors in early rheumatoid arthritis: results of a follow up study. Ann Rheum Dis (1992) 51:1029–35. doi:10.1136/ard.51.9.1029
279. Oda A, Miyata M, Kodama E, Satoh H, Sato Y, Nishimaki T, et al. Antibodies to 65Kd heat-shock protein were elevated in rheumatoid arthritis. Clin Rheumatol (1994) 13:261–4.
280. Trouw LA, Huizinga TW, Toes RE. Autoimmunity in rheumatoid arthritis: different antigens – common principles. Ann Rheum Dis (2013) 72(Suppl 2):ii132. doi:10.1136/annrheumdis-2012-202349
281. Brink M, Hansson M, Mathsson L, Jakobsson P, Holmdahl R, Hallmans G, et al. Multiplex analyses of antibodies against citrullinated peptides in individuals prior to development of rheumatoid arthritis. Arthritis Rheum (2013) 65:899–910. doi:10.1002/art.37835
282. Amara K, Steen J, Murray F, Morbach H, Fernandez-Rodriguez BM, Joshua V, et al. Monoclonal IgG antibodies generated from joint-derived B cells of RA patients have a strong bias toward citrullinated autoantigen recognition. J Exp Med (2013) 210:445–55. doi:10.1084/jem.20121486
283. Van der Woude D, Rantapaa-Dahlqvist S, Ioan-Facsinay A, Onnekink C, Schwarte CM, Verpoort KN, et al. Epitope spreading of the anti-citrullinated protein antibody response occurs before disease onset and is associated with the disease course of early arthritis. Ann Rheum Dis (2010) 69:1554–61. doi:10.1136/ard.2009.124537
284. Kuhn KA, Kulik L, Tomooka B, Braschler KJ, Arend WP, Robinson WH, et al. Antibodies against citrullinated proteins enhance tissue injury in experimental autoimmune arthritis. J Clin Invest (2006) 116:961–73. doi:10.1172/JCI25422
285. Trouw LA, Haisma EM, Levarht EW, van der Woude D, Ioan-Facsinay A, Daha MR, et al. Anti-cyclic citrullinated peptide antibodies from rheumatoid arthritis patients activate complement via both the classical and alternative pathways. Arthritis Rheum (2009) 60:1923–31. doi:10.1002/art.24622
286. Shi J, Knevel R, Suwannalai P, van der Linden MP, Janssen GM, van Veelen PA, et al. Autoantibodies recognizing carbamylated proteins are present in sera of patients with rheumatoid arthritis and predict joint damage. Proc Natl Acad Sci U S A (2011) 108:17372–7. doi:10.1073/pnas.1114465108
287. Strollo R, Ponchel F, Malmström V, Rizzo P, Bombardieri M, Wenham CY, et al. Autoantibodies to posttranslationally modified type II collagen as potential biomarkers for rheumatoid arthritis. Arthritis Rheum (2013) 65:1702–12. doi:10.1002/art.37964
288. Rowley M, Tait B, Mackay IR, Cunningham T, Phillips B. Collagen antibodies in rheumatoid arthritis. Significance of antibodies to denatured collagen and their association with HLA-DR4. Arthritis Rheum (1986) 29:174–84. doi:10.1002/art.1780290204
289. Vincent A, Bien CG, Irani SR, Waters P. Autoantibodies associated with diseases of the CNS: new developments and future challenges. Lancet Neurol (2011) 10:759–72. doi:10.1016/S1474-4422(11)70096-5
290. Moscato EH, Peng X, Jain A, Parsons TD, Dalmau J, Balice-Gordon RJ. Acute mechanisms underlying antibody effects in anti-N-methyl-D-aspartate receptor encephalitis. Ann Neurol (2014) 76:108–19. doi:10.1002/ana.24195
291. Dalmau J, Tüzün E, Wu H, Masjuan J, Rossi JE, Voloschin A, et al. Paraneoplastic anti-N-methyl-D-aspartate receptor encephalitis associated with ovarian teratoma. Ann Neurol (2007) 61:25–36. doi:10.1002/ana.21050
292. Tüzün E, Zhou L, Baehring JM, Bannykh S, Rosenfeld MR, Dalmau J. Evidence for antibody-mediated pathogenesis in anti-NMDAR encephalitis associated with ovarian teratoma. Acta Neuropathol (2009) 118:737–43. doi:10.1007/s00401-009-0582-4
293. Voltz R, Dalmau J, Posner JB, Rosenfeld MR. T-cell receptor analysis in anti-Hu associated paraneoplastic encephalomyelitis. Neurology (1998) 51:1146–50. doi:10.1212/WNL.51.4.1146
294. Roberts WK, Deluca IJ, Thomas A, Fak J, Williams T, Buckley N, et al. Patients with lung cancer and paraneoplastic Hu syndrome harbor HuD-specific type 2 CD8+ T cells. J Clin Invest (2009) 119:2042–51. doi:10.1172/JCI36131
295. Nakanishi K, Komatsu Y, Kogawa N, Matsushita H. Analysis of eluted peptides from type 1 diabetes-susceptible HLA class II molecules identified novel islet protein, heparin/heparan sulfate-interacting protein. Biochem Biophys Res Commun (2005) 329:356–61. doi:10.1016/j.bbrc.2005.01.144
296. De Graaf KL, Albert M, Weissert R. Autoantigen conformation influences both B- and T-cell responses and encephalitogenicity. J Biol Chem (2012) 287:17206–13. doi:10.1074/jbc.M111.304246
297. Wordsworth BP, Lanchbury JS, Sakkas LI, Welsh KI, Panayi GS, Bell JI. HLA-DR4 subtype frequencies in rheumatoid arthritis indicate that DRB1 is the major susceptibility locus within the HLA class II region. Proc Natl Acad Sci U S A (1989) 86:10049–53. doi:10.1073/pnas.86.24.10049
298. Nepom GT, Byers P, Seyfried C, Healey LA, Wilske KR, Stage D, et al. HLA genes associated with rheumatoid arthritis. Identification of susceptibility alleles using specific oligonucleotide probes. Arthritis Rheum (1989) 32:15–21. doi:10.1002/anr.1780320104
299. Carthy D, Ollier W, Papasteriades C, Pappas H, Thomson W. A shared HLA-DRB1 sequence confers RA susceptibility in Greeks. Eur J Immunogenet (1993) 20:391–8. doi:10.1111/j.1744-313X.1993.tb00158.x
300. Begovich AB, Carlton VE, Honigberg LA, Schrodi SJ, Chokkalingam AP, Alexander HC, et al. A missense single-nucleotide polymorphism in a gene encoding a protein tyrosine phosphatase (PTPN22) is associated with rheumatoid arthritis. Am J Hum Genet (2004) 75:330–7. doi:10.1086/422827
301. Cucca F, Lampis R, Congia M, Angius E, Nutland S, Bain SC, et al. A correlation between the relative predisposition of MHC class II alleles to type 1 diabetes and the structure of their proteins. Hum Mol Genet (2001) 10:2025–37. doi:10.1093/hmg/10.19.2025
302. Todd JA, Bell JI, McDevitt HO. HLA-DQ beta gene contributes to susceptibility and resistance to insulin-dependent diabetes mellitus. Nature (1987) 329:599–604. doi:10.1038/329599a0
303. Bottini N, Musumeci L, Alonso A, Rahmouni S, Nika K, Rostamkhani M, et al. A functional variant of lymphoid tyrosine phosphatase is associated with type I diabetes. Nat Genet (2004) 36:337–8. doi:10.1038/ng1323
304. Vella A, Cooper JD, Lowe CE, Walker N, Nutland S, Widmer B, et al. Localization of a type 1 diabetes locus in the IL2RA/CD25 region by use of tag single-nucleotide polymorphisms. Am J Hum Genet (2005) 76:773–9. doi:10.1086/429843
305. Nisticò L, Buzzetti R, Pritchard LE, Van der Auwera B, Giovannini C, Bosi E, et al. The CTLA-4 gene region of chromosome 2q33 is linked to, and associated with, type 1 diabetes. Belgian diabetes registry. Hum Mol Genet (1996) 5:1075–80. doi:10.1093/hmg/5.7.1075
306. Smyth DJ, Cooper JD, Bailey R, Field S, Burren O, Smink LJ, et al. A genome-wide association study of nonsynonymous SNPs identifies a type 1 diabetes locus in the interferon-induced helicase (IFIH1) region. Nat Genet (2006) 38:617–9. doi:10.1038/ng1800
307. Todd JA, Walker NM, Cooper JD, Smyth DJ, Downes K, Plagnol V, et al. Robust associations of four new chromosome regions from genome-wide analyses of type 1 diabetes. Nat Genet (2007) 39:857–64. doi:10.1038/ng2068
308. Barratt BJ, Payne F, Lowe CE, Hermann R, Healy BC, Harold D, et al. Remapping the insulin gene/IDDM2 locus in type 1 diabetes. Diabetes (2004) 53:1884–9. doi:10.2337/diabetes.53.7.1884
309. Matsushita T, Matsuoka T, Isobe N, Kawano Y, Minohara M, Shi N, et al. Association of the HLA-DPB1*0501 allele with anti-aquaporin-4 antibody positivity in Japanese patients with idiopathic central nervous system demyelinating disorders. Tissue Antigens (2009) 73:171–6. doi:10.1111/j.1399-0039.2008.01172.x
310. Zephir H, Fajardy I, Outteryck O, Blanc F, Roger N, Fleury M, et al. Is neuromyelitis optica associated with human leukocyte antigen? Mult Scler (2009) 15:571–9. doi:10.1177/1352458508102085
311. Quintana FJ, Farez MF, Izquierdo G, Lucas M, Cohen IR, Weiner HL. Antigen microarrays identify CNS-produced autoantibodies in RRMS. Neurology (2012) 78:532–9. doi:10.1212/WNL.0b013e318247f9f3
312. Andersson M, Yu M, Söderström M, Weerth S, Baig S, Solders G, et al. Multiple MAG peptides are recognized by circulating T and B lymphocytes in polyneuropathy and multiple sclerosis. Eur J Neurol (2002) 9:243–51. doi:10.1046/j.1468-1331.2002.00391.x
313. Kerlero de Rosbo N, Kaye JF, Eisenstein M, Mendel I, Hoeftberger R, Lassmann H, et al. The myelin-associated oligodendrocytic basic protein region MOBP15-36 encompasses the immunodominant major encephalitogenic epitope(s) for SJL/J mice and predicted epitope(s) for multiple sclerosis-associated HLA-DRB1*1501. J Immunol (2004) 173:1426–35. doi:10.4049/jimmunol.173.2.1426
314. Muraro PA, Kalbus M, Afshar G, McFarland HF, Martin R. T cell response to 2’,3’-cyclic nucleotide 3’-phosphodiesterase (CNPase) in multiple sclerosis patients. J Neuroimmunol (2002) 130:233–42. doi:10.1016/S0165-5728(02)00229-1
315. Schmidt S, Linington C, Zipp F, Sotgiu S, de Waal Malefyt R, Wekerle H, et al. Multiple sclerosis: comparison of the human T-cell response to S100 beta and myelin basic protein reveals parallels to rat experimental autoimmune panencephalitis. Brain (1997) 120:1437–45. doi:10.1093/brain/120.8.1437
316. Banki K, Colombo E, Sia F, Halladay D, Mattson DH, Tatum AH, et al. Oligodendrocyte-specific expression and autoantigenicity of transaldolase in multiple sclerosis. J Exp Med (1994) 180:1649–63. doi:10.1084/jem.180.5.1649
317. Pevzner A, Schoser B, Peters K, Cosma N, Karakatsani A, Schalke B, et al. Anti-LRP4 autoantibodies in AChR- and MuSK-antibody-negative myasthenia gravis. J Neurol (2012) 259:427–35. doi:10.1007/s00415-011-6194-7
318. Petit-Pedrol M, Armangue T, Peng X, Bataller L, Cellucci T, Davis R, et al. Encephalitis with refractory seizures, status epilepticus, and antibodies to the GABAA receptor: a case series, characterisation of the antigen, and analysis of the effects of antibodies. Lancet Neurol (2014) 13:276–86. doi:10.1016/S1474-4422(13)70299-0
319. Boronat A, Gelfand JM, Gresa-Arribas N, Jeong H, Walsh M, Roberts K, et al. Encephalitis and antibodies to dipeptidyl-peptidase-like protein-6, a subunit of Kv4.2 potassium channels. Ann Neurol (2013) 73:120–8. doi:10.1002/ana.23756
320. Lancaster E, Martinez-Hernandez E, Titulaer MJ, Boulos M, Weaver S, Antoine J, et al. Antibodies to metabotropic glutamate receptor 5 in the Ophelia syndrome. Neurology (2011) 77:1698–701. doi:10.1212/WNL.0b013e3182364a44
321. Dalmau J, Graus F, Rosenblum MK, Posner JB. Anti-Hu – associated paraneoplastic encephalomyelitis/sensory neuronopathy. A clinical study of 71 patients. Medicine (1992) 71:59–72. doi:10.1097/00005792-199203000-00001
322. Tanaka M, Maruyama Y, Sugie M, Motizuki H, Kamakura K, Tanaka K. Cytotoxic T cell activity against peptides of Hu protein in anti-Hu syndrome. J Neurol Sci (2002) 201:9–12. doi:10.1016/S0022-510X(02)00157-0
323. Greenlee JE, Brashear HR. Antibodies to cerebellar Purkinje cells in patients with paraneoplastic cerebellar degeneration and ovarian carcinoma. Ann Neurol (1983) 14:609–13. doi:10.1002/ana.410140603
324. Jaeckle KA, Graus F, Houghton A, Cardon-Cardo C, Nielsen SL, Posner JB. Autoimmune response of patients with paraneoplastic cerebellar degeneration to a Purkinje cell cytoplasmic protein antigen. Ann Neurol (1985) 18:592–600. doi:10.1002/ana.410180513
325. Luque FA, Furneaux HM, Ferziger R, Rosenblum MK, Wray SH, Schold SC, et al. Anti-Ri: an antibody associated with paraneoplastic opsoclonus and breast cancer. Ann Neurol (1991) 29:241–51. doi:10.1002/ana.410290303
326. Dalmau J, Gultekin SH, Voltz R, Hoard R, DesChamps T, Balmaceda C, et al. Ma1, a novel neuron- and testis-specific protein, is recognized by the serum of patients with paraneoplastic neurological disorders. Brain (1999) 122:27–39. doi:10.1093/brain/122.1.27
327. Dalmau J, Graus F, Villarejo A, Posner JB, Blumenthal D, Thiessen B, et al. Clinical analysis of anti-Ma2-associated encephalitis. Brain (2004) 127:1831–44. doi:10.1093/brain/awh203
328. Bataller L, Wade DF, Graus F, Stacey HD, Rosenfeld MR, Dalmau J. Antibodies to Zic4 in paraneoplastic neurologic disorders and small-cell lung cancer. Neurology (2004) 62:778–82. doi:10.1212/01.WNL.0000113749.77217.01
329. Kim J, Namchuk M, Bugawan T, Fu Q, Jaffe M, Shi Y, et al. Higher autoantibody levels and recognition of a linear NH2-terminal epitope in the autoantigen GAD65, distinguish stiff-man syndrome from insulin-dependent diabetes mellitus. J Exp Med (1994) 180:595–606. doi:10.1084/jem.180.2.595
330. Monstad SE, Nøstbakken JK, Vedeler CA. CRMP5 antibodies found in a patient with limbic encephalitis and myasthenia gravis. J Neurol Neurosurg Psychiatry (2009) 80:241–2. doi:10.1136/jnnp.2008.149336
331. Antoine JC, Absi L, Honnorat J, Boulesteix JM, de Brouker T, Vial C, et al. Antiamphiphysin antibodies are associated with various paraneoplastic neurological syndromes and tumors. Arch Neurol (1999) 56:172–7. doi:10.1001/archneur.56.2.172
332. Mosmann TR, Cherwinski H, Bond MW, Giedlin MA, Coffman RL. Two types of murine helper T cell clone. I. Definition according to profiles of lymphokine activities and secreted proteins. J Immunol (1986) 136:2348–57.
333. Sakaguchi S. Regulatory T cells: key controllers of immunologic self-tolerance. Cell (2000) 101:455–8. doi:10.1016/S0092-8674(00)80856-9
334. Szabo SJ, Sullivan BM, Stemmann C, Satoskar AR, Sleckman BP, Glimcher LH. Distinct effects of T-bet in TH1 lineage commitment and IFN-gamma production in CD4 and CD8 T cells. Science (2002) 295:338–42. doi:10.1126/science.1065543
335. Delves PJ, Roitt IM. The immune system. First of two parts. N Engl J Med (2000) 343:37–49. doi:10.1056/NEJM200007063430107
336. Zheng W, Flavell RA. The transcription factor GATA-3 is necessary and sufficient for Th2 cytokine gene expression in CD4 T cells. Cell (1997) 89:587–96. doi:10.1016/S0092-8674(00)80240-8
337. Bettelli E, Korn T, Oukka M, Kuchroo VK. Induction and effector functions of T(H)17 cells. Nature (2008) 453:1051–7. doi:10.1038/nature07036
338. Ivanov II, McKenzie BS, Zhou L, Tadokoro CE, Lepelley A, Lafaille JJ, et al. The orphan nuclear receptor RORγt directs the differentiation program of proinflammatory IL-17+ T helper cells. Cell (2006) 126:1121–33. doi:10.1016/j.cell.2006.07.035
339. Hori S, Nomura T, Sakaguchi S. Control of regulatory T cell development by the transcription factor Foxp3. Science (2003) 299:1057–61. doi:10.1126/science.1079490
340. Dardalhon V, Awasthi A, Kwon H, Galileos G, Gao W, Sobel RA, et al. IL-4 inhibits TGF-β-induced Foxp3+ T cells and, together with TGF-β, generates IL-9+ IL-10+ Foxp3- effector T cells. Nat Immunol (2008) 9:1347–55. doi:10.1038/ni.1677
341. Chang H, Sehra S, Goswami R, Yao W, Yu Q, Stritesky GL, et al. The transcription factor PU.1 is required for the development of IL-9-producing T cells and allergic inflammation. Nat Immunol (2010) 11:527–34. doi:10.1038/ni.1867
342. Veldhoen M, Uyttenhove C, van Snick J, Helmby H, Westendorf A, Buer J, et al. Transforming growth factor-β ‘reprograms’ the differentiation of T helper 2 cells and promotes an interleukin 9–producing subset. Nat Immunol (2008) 9:1341–6. doi:10.1038/ni.1659
343. Kaplan MH, Hufford MM, Olson MR. The development and in vivo function of T helper 9 cells. Nat Rev Immunol (2015) 15:295–307. doi:10.1038/nri3824
Keywords: autoimmune disease, multiple sclerosis, antigen presentation, autoantigen, T cell, B cell, HLA, MHC
Citation: Riedhammer C and Weissert R (2015) Antigen presentation, autoantigens, and immune regulation in multiple sclerosis and other autoimmune diseases. Front. Immunol. 6:322. doi: 10.3389/fimmu.2015.00322
Received: 24 March 2015; Accepted: 03 June 2015;
Published: 17 June 2015
Edited by:
Hans-Peter Hartung, Heinrich-Heine University of Düsseldorf, GermanyReviewed by:
Jorge Correale, Fundación para la Lucha contra las Enfermedades Neurológicas de la Infancia, ArgentinaCopyright: © 2015 Riedhammer and Weissert. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Robert Weissert, Department of Neurology, Neuroimmunology, University of Regensburg, Universitaetsstrasse 84, Regensburg 93053, Germany,cm9iZXJ0LndlaXNzZXJ0QHVrci5kZQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.