
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 15 June 2015
Sec. Cytokines and Soluble Mediators in Immunity
Volume 6 - 2015 | https://doi.org/10.3389/fimmu.2015.00308
This article is part of the Research Topic The CXCR4 ligand/receptor family and the DPP4 protease in high-risk cardiovascular patients View all 14 articles
 Martine C. Morrison1*
Martine C. Morrison1* Robert Kleemann1,2
Robert Kleemann1,2
Obesity is associated with a chronic low-grade inflammatory state that drives the development of obesity-related co-morbidities such as insulin resistance/type 2 diabetes, non-alcoholic fatty liver disease (NAFLD), and cardiovascular disease. This metabolic inflammation is thought to originate in the adipose tissue, which becomes inflamed and insulin resistant when it is no longer able to expand in response to excess caloric and nutrient intake. The production of inflammatory mediators by dysfunctional adipose tissue is thought to drive the development of more complex forms of disease such as type 2 diabetes and NAFLD. An important factor that may contribute to metabolic inflammation is the cytokine macrophage migration inhibitory factor (MIF). Increasing evidence suggests that MIF is released by adipose tissue in obesity and that it is also involved in metabolic and inflammatory processes that underlie the development of obesity-related pathologies. This review provides a comprehensive summary of our current knowledge on the role of MIF in obesity, its production by adipose tissue, and its involvement in the development of insulin resistance, type 2 diabetes, and NAFLD. We discuss the main findings from recent clinical studies in obese subjects and weight-loss intervention studies as well as results from clinical studies in patients with insulin resistance and type 2 diabetes. Furthermore, we summarize findings from experimental disease models studying the contribution of MIF in obesity and insulin resistance, type 2 diabetes, and hepatic lipid accumulation and fibrosis. Although many of the findings support a pro-inflammatory role of MIF in disease development, recent reports also provide indications that MIF may exert protective effects under certain conditions.
The development of obesity-associated co-morbidities such as type 2 diabetes (T2D), non-alcoholic fatty liver disease (NAFLD), and cardiovascular disease (CVD) is considered to be driven by the chronic low-grade inflammatory state that characterizes obesity (1). During obesity, excess caloric and nutrient intake, i.e., metabolic overload, leads to adipose tissue expansion and visceral adiposity. When the maximal expandability of an adipose tissue depot is reached (when adipocytes have maximally expanded) this leads to adipose tissue dysfunction and infiltration of immune cells, which is associated with insulin resistance of the adipose tissue itself (2, 3). Furthermore, adipocytes and infiltrated immune cells of this inflamed adipose tissue produce inflammatory mediators that can be released into the circulation (4). Thus, the adipose tissue becomes a source of inflammation that can drive pathogenesis in other tissues, leading to the development of T2D, NAFLD, and CVD (Figure 1).
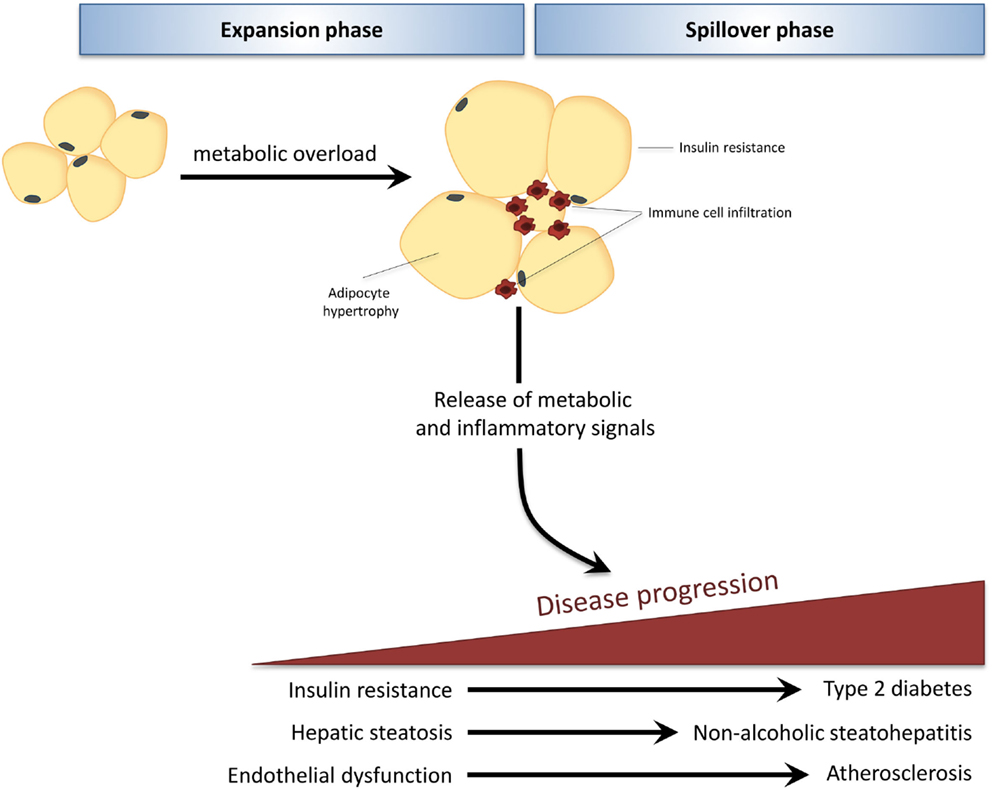
Figure 1. In obesity, adipose tissue can become a source of inflammation that drives disease development in distant organs. Caloric and nutrient excess, i.e., metabolic overload, leads to expansion of adipose tissue depots (expansion phase). When the maximal expandability of an adipose tissue depot is reached due to prolonged metabolic overload, the adipose tissue becomes dysfunctional, is infiltrated by immune cells, and becomes insulin resistant. Adipocytes and infiltrated immune cells produce inflammatory mediators that spill over into the circulation (spillover phase) where they drive the progression of obesity-related co-morbidities in distant organs, such as type 2 diabetes, non-alcoholic fatty liver disease, and atherosclerosis.
Macrophage migration inhibitory factor (MIF) is a cytokine that is ubiquitously expressed, both by immune and non-immune cells. It is well known for its pro-inflammatory effects and is recognized as a negative regulator of the immunosuppressive actions of glucocorticoids (5). In line with this, MIF has been implicated in the development of many acute inflammatory and auto-immune diseases (6, 7), as well as chronic inflammatory metabolic disorders (8–10). MIF can act via its receptor CD74 (11), and controls the recruitment of inflammatory cells via CXCR2 and CXCR4 signaling (8). Furthermore, MIF can exert pro-inflammatory effects through its enzymatic tautomerase and oxidoreductase activity (12, 13). Several lines of evidence provide indications that MIF, besides regulating inflammation, may also be linked to energy metabolism. It is expressed in metabolically active tissues such as the adipose tissue and the liver (9, 14). Its expression by adipocytes is regulated by glucose and insulin (15) and it has been shown to have catabolic effects in muscle (16). Furthermore, it co-localizes with insulin within the secretory granules of pancreatic beta cells and is a modulator of insulin release (17). Increasing evidence suggests that MIF may also control inflammatory and metabolic processes in the pathogenesis of obesity and associated disorders including insulin resistance, T2D, and NAFLD. Here, we provide a comprehensive review of the current knowledge on the role of MIF in obesity, MIF production by adipose tissue, and its role in the development of insulin resistance, type 2 diabetes, and NAFLD. Since epidemiological studies have shown both insulin resistance (18, 19) and NAFLD (20, 21) to be associated with future CVD risk, this may also have relevance for the role of MIF in vascular disease, which has been described elsewhere for rodents (22, 23) and humans (24, 25), and will not be addressed here. This survey aims to advance our understanding on the functions of MIF in non-pathological and pathological processes connected to obesity and glucose homeostasis, with particular emphasis on adipose tissue and liver.
All values are mean ± SEM. Where the original article stated mean ± SD, SEM was calculated from SD by dividing SD by the square root of n.
Several studies provide evidence for a positive association between obesity and circulating MIF levels (Table 1). Dandona and co-workers (26) reported that obese individuals (BMI 37.5 ± 4.9 kg/m2) had significantly higher plasma MIF levels (2.8 ± 2.0 ng/ml) than lean individuals (BMI 22.6 ± 3.4 kg/m2; plasma MIF 1.2 ± 0.6 ng/ml) and that there was a highly significant positive correlation between MIF levels and BMI. Results from another study by the same group (27) confirmed these results, reporting MIF levels of 3.3 ± 2.4 ng/ml in obese subjects (BMI 40.0 ± 4.4 kg/m2), which were significantly higher than the 1.3 ± 0.8 ng/ml observed in the lean controls (BMI 22.6 ± 1.9 kg/m2). Again, there was a positive correlation (p = 0.10) between MIF levels and BMI. Others have reported similar findings of increased circulating MIF in obese individuals compared with healthy lean controls (28–31), and showed associations with % body fat and % truncal fat (31). Studies in pre-pubescent schoolchildren (age 5–13 years) and male adolescents (age 13–17 years) demonstrate that these associations are not only observed in adults, reporting increased MIF levels in overweight and obesity in childhood and adolescence (32, 33), and showing positive correlations with waist circumference and BMI (33) in this study population as well.
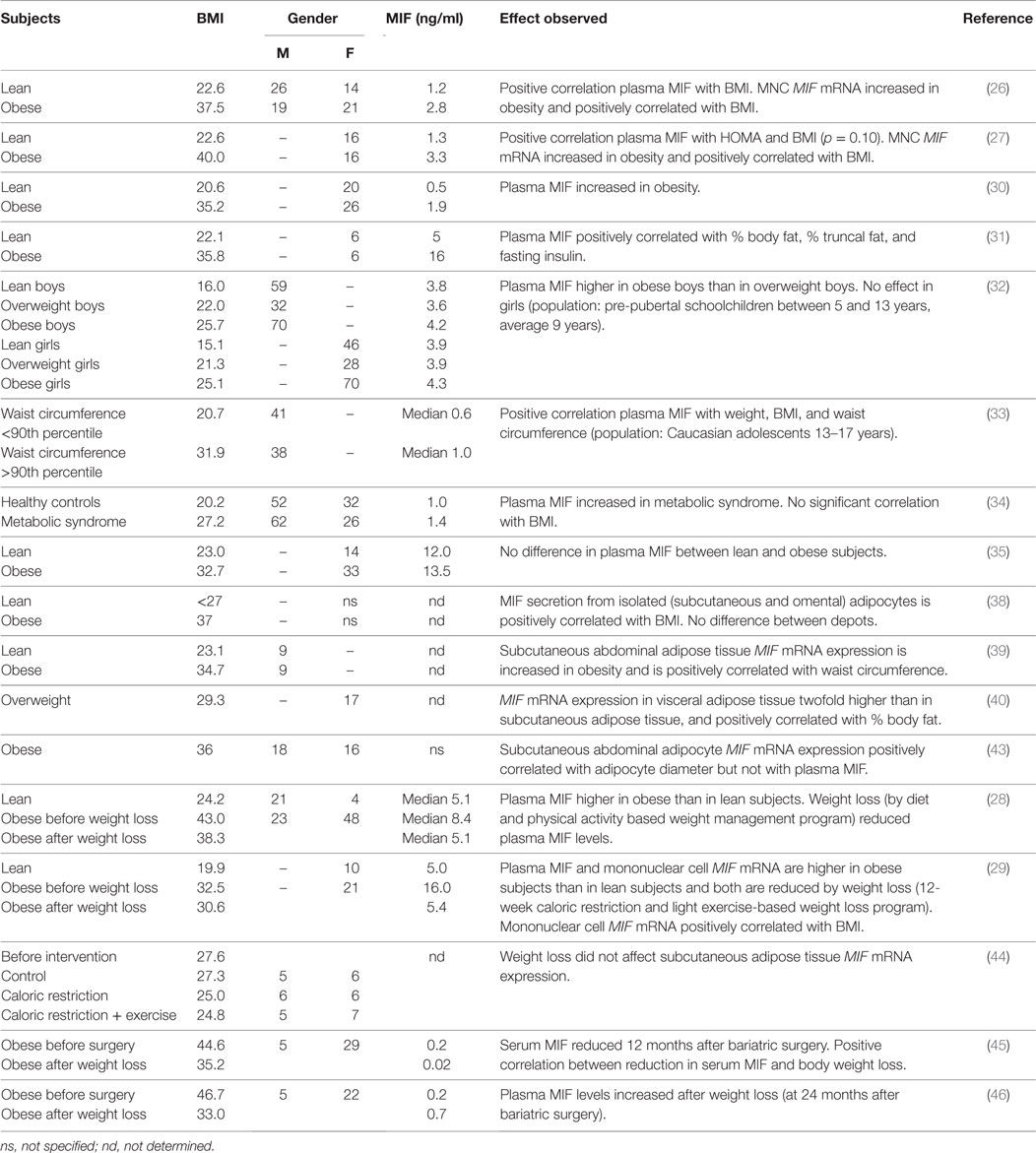
Table 1. Relationship between (circulating) MIF and obesity and effects of weight loss thereupon.
A study by Kim et al. (34) compared healthy Korean subjects (BMI 20.2 kg/m2) without metabolic syndrome with patients with metabolic syndrome (BMI 27.2 kg/m2) and found higher levels in patients than controls (1.4 ± 0.1 and 1.0 ± 0.1 ng/ml, respectively), which was significant in women but not in men. N.B. there was no correlation between circulating MIF and BMI in this study, suggesting that other MIF-inducing factors play a role when overweight/obesity progresses toward a phenotype of metabolic disease.
Of note, plasma MIF concentrations tend to be higher in males than in females (26, 34), suggesting an inducing effect of male sex hormones. Indeed, circulating MIF levels are two to threefold higher in women with polycystic ovary syndrome (PCOS), an endocrine disorder characterized by elevated levels of androgens (31, 35). Within PCOS patients, the effect of obesity is not significant due to large variation within this population, and comparable plasma MIF levels were found in non-obese and obese patients [~35 and ~55 ng/ml, respectively, in Ref. (31); 35.2 and 48.6 ng/ml, respectively, in Ref. (35)].
Remarkably, there is a large variation in the absolute circulating MIF values in documented obesity studies, indicating that there may be underlying methodological differences causing these diverse values. A part of the variation may be explained by different analytical methods (e.g., different ELISA kits, multiplex technology) but even studies employing the same analytical tools report substantial differences [e.g., Ref. (26, 31)]. Hence, other technical or methodological issues may be responsible for these discrepancies. The MIF molecule itself and its oligomers are very hydrophobic (36, 37) and tend to precipitate or stick to plastic material and differences in sample preparation and/or pipetting can result in differences as reported in the human studies. Therefore, it is difficult to make comparisons between different studies. However, the direction of the effect (obese higher than lean) is mostly consistent and overall, these studies demonstrate a well-established association between obesity and circulating MIF that seems to transcend differences in age, sex, and ethnicity. The observed positive correlations with BMI and % body fat provide indication that the observed association may be related to the increased adipose tissue mass that is inherent to obesity and hint at the adipose tissue as a contributor to the observed association.
Several groups have investigated adipose tissue as a potential source of circulating MIF in obesity (Table 1). Skurk and co-workers (38) demonstrated that primary human pre-adipocytes and mature adipocytes have the capacity to produce and secrete MIF and the secretion of MIF was found to increase in parallel with the (in vitro) differentiation of pre-adipocytes. There was a close positive correlation between MIF release from mature adipocytes and donor BMI, which was observed in both visceral and omental adipocytes, with no differences between these two depots. Others have shown similar effects on the gene expression level. González-Muniesa and colleagues (39) showed that MIF mRNA expression in subcutaneous abdominal adipose tissue is increased in obese individuals and correlates with waist circumference. A positive correlation was also reported by Alvehus and co-workers (40) for the visceral adipose tissue depot, which is an important source of inflammation and, in contrast to subcutaneous adipose tissue, is associated with inflammatory metabolic disorders such as NAFLD and CVD (41, 42). Alvehus et al. (40) showed that MIF mRNA expression in the visceral adipose tissue was higher than in the subcutaneous adipose tissue (~2-fold) and was positively correlated with body fat percentage. Adipose tissue inflammation is considered to be a consequence of adipocyte hypertrophy and is thought to be triggered by maximal expansion of adipocytes (2, 3). In line with this notion, subcutaneous abdominal adipocyte diameter correlates with MIF mRNA expression (43).
If indeed the increased levels of circulating MIF in obesity are primarily the result of increased adipose tissue mass and adipocyte expansion, one would logically expect plasma MIF levels to decrease when obese subjects lose weight. Indeed, reduced MIF levels have been reported upon weight loss. Church and colleagues (28), for instance, observed significant reductions in plasma MIF levels in obese individuals after a diet and physical activity based weight-loss program. Participants’ BMI after this 8.5-month program was reduced from 43.0 ± 8.6 to 38.3 ± 7.6 kg/m2 (reflecting an average weight loss of 14.4 kg), which resulted in completely normalized plasma MIF levels comparable to those of lean controls (from median 8.4 to 5.1 ng/ml). Similar results were reported in a different study on the effects of weight loss (29) in which obese subjects participated in a 3.5-month diet and light exercise-induced weight-loss program. A reduction in BMI from 32.5 ± 1.2 to BMI 30.6 ± 1.6 kg/m2 (reflecting a weight loss of 4.0 ± 0.4 kg) was accompanied by a reduction in serum MIF from 16.0 ± 4.0 to 5.4 ± 0.4 ng/ml, which again was comparable to lean control subjects. Remarkably, the observed weight loss in both these studies in obese subjects resulted in completely normalized MIF levels comparable to those observed in lean subjects, even though BMI was still considerably higher in the obese subjects (and subjects were still obese even after weight loss), thus indicating that other factors rather than fat mass per se (e.g., insulin sensitivity, hormonal changes) may determine circulating MIF levels, or that weight loss may normalize the state of a particular adipose tissue depot that is primarily responsible for MIF production and secretion. Furthermore, a relatively small reduction in adipocyte size may already be sufficient to prevent further adipocyte damage and thereby attenuate the inflammatory process in adipose tissue.
Only a few studies have examined whether a specific adipose tissue depot is responsible for the release of MIF into the circulation. Support for the view that circulating MIF must be produced by adipose tissue depots other than the subcutaneous adipose tissue comes from studies by Koska et al. (43) who showed that subcutaneous adipose tissue MIF mRNA does not correlate with circulating MIF levels, and Tam et al. (44) who found no effect of weight loss on subcutaneous adipose tissue MIF mRNA expression. Identification of the underlying factors that determine circulating MIF in obesity warrants further investigation.
Two studies on the effects of bariatric surgery-induced weight loss in morbidly obese subjects report conflicting results in this specific population. Fenske et al. (45) showed that weight loss at 12 months after bariatric surgery (BMI from 44.6 ± 0.9 to 35.2 kg/m2) resulted in reductions in serum MIF levels from 0.2 ng/ml before surgery to 0.02 ng/ml at 12 months after surgery. Van Dielen and colleagues (46), on the other hand, reported similarly low-plasma MIF levels in morbidly obese subjects before surgery (BMI 46.7 ± 1.1 kg/m2, plasma MIF 0.2 ng/ml) but observed increased MIF levels at 24 months after bariatric surgery (weight loss to BMI 33.0 ± 0.9 kg/m2, plasma MIF 0.7 ng/ml). To gain insight into the processes that determine circulating MIF levels in these patients, longitudinal analyses after bariatric surgery are needed and it is possible that co-variables such as impaired insulin secretion and diabetes may determine plasma MIF levels in this extreme group of metabolically deregulated subjects.
Overall, the above studies indicate that whole-body adiposity is not a key determinant of circulating MIF and suggest that MIF is produced by specific adipose tissue depots during a particular period of depot growth (i.e., adipocyte expansion within the depot) and resulting adipose tissue inflammation. Of note, circulating MIF may also be produced by other tissues and cells in obesity. Besides adipose tissue as a possible source of circulating MIF in obesity, there are some reports on MIF expression by mononuclear cells (MNCs), with reports of increased MIF mRNA expression by MNC from obese subjects (26, 27, 29). Although MNC MIF expression did not correlate with plasma MIF levels (26), a correlation with BMI was reported in two of these studies (26, 27) and weight loss (BMI from 32.5 ± 1.2 to 30.6 ± 1.6 kg/m2) significantly reduced MNC MIF mRNA expression (29).
Results from observational studies have shown increased levels of circulating MIF in insulin resistant and T2D subjects (Table 2). Yabunaka et al. (47) reported increased serum MIF levels in Japanese type 2 diabetic subjects (20.7 ± 1.5 ng/ml) compared with healthy control subjects (5.2 ± 0.3 ng/ml); however, there was no correlation between MIF and fasting plasma glucose, HbA1c, or diabetes duration. Interestingly, BMI was comparable between T2D patients and controls in this study (23.9 ± 0.3 kg/m2 in T2D vs. 23.8 ± 0.4 kg/m2 in controls) indicating that the observed difference was not attributable to a higher BMI in the diabetic subjects. (N.B. as the study was conducted in an Asian population absolute BMI values are lower than in Caucasian diabetes patients.) Similarly, Yu and colleagues (48) showed increased MIF levels in Chinese T2D patients (2.6 ± 0.1 ng/ml) compared with healthy controls (2.1 ± 0.1 ng/ml), which again were independent of BMI (23.9 ± 0.1 kg/m2 in T2D vs. 22.9 ± 0.3 kg/m2 in controls). In line with these findings, Sanchez-Zamora et al. reported increased MIF levels (~0.2 vs. ~0.05 ng/ml) in Mexican T2D patients relative to healthy controls (49), although BMI for the different groups was not stated in this study.
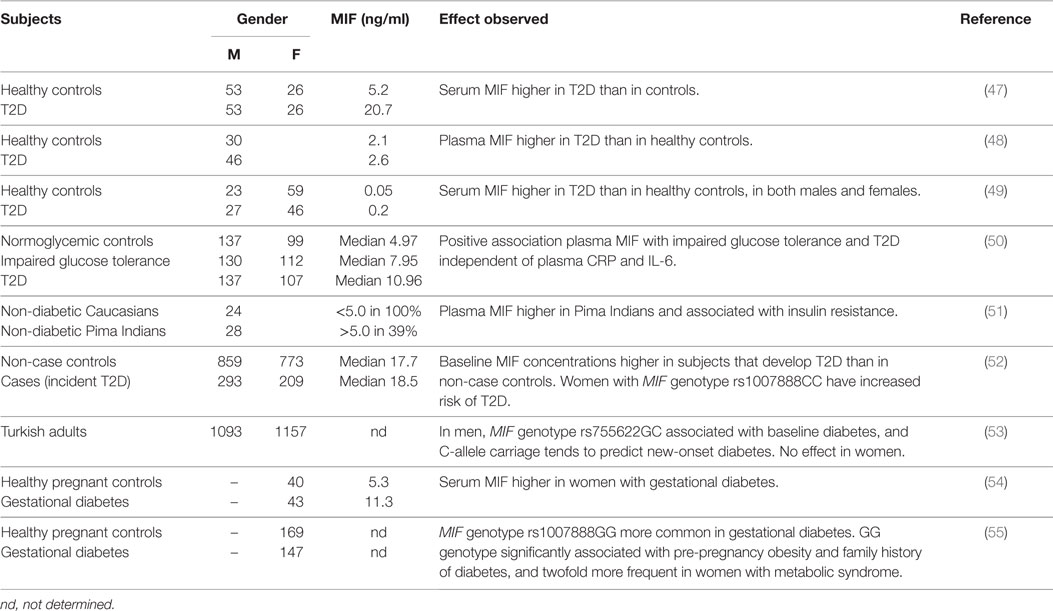
Table 2. Relationship between (plasma) MIF and insulin resistance/T2D.
These associations with MIF have also been reported in earlier stages of T2D development (impaired glucose tolerance, insulin resistance), before overt T2D is present. Herder et al. (50) reported increased serum MIF concentrations in German individuals with impaired glucose tolerance (median MIF 7.95 vs. 4.97 ng/ml in healthy controls) and further increased levels in individuals with T2D (median MIF 10.96 ng/ml), suggesting a gradual increase of MIF with severity of the disease. In this study, BMI was higher in the insulin resistant and T2D subjects than in the healthy controls. In line with these results, a positive association between plasma MIF levels and insulin resistance was shown in a study in Pima Indians (51) and correlations with homeostatic model assessment (HOMA; a method used to quantify insulin resistance) (26, 27) and β-cell dysfunction (28) have also been reported. Furthermore, MIF mRNA expression in freshly isolated subcutaneous abdominal adipocytes was found to be negatively associated with donor peripheral and hepatic insulin action in another study in Pima Indians (43).
In a prospective case-cohort study (52), baseline levels of serum MIF were found to be higher in incident T2D cases than in non-case controls (mean follow-up 10.1 ± 0.1 years, only significant in women) and this association between elevated MIF serum levels and T2D risk was stronger in obese than in non-obese women. Furthermore, this study reported significant associations between serum MIF levels in women and MIF genotype rs1007888CC, and carriers of this allele had a 1.7-fold increased risk of T2D. A different MIF genotype (rs755622GC) was shown to be associated with baseline diabetes in men but not in women in a population-based cohort study in Turkish subjects, and carriage of the C-allele tended to predict new-onset diabetes in this population (53).
MIF has also been implicated in gestational diabetes, with higher serum MIF in women with gestational diabetes (11.2 ± 0.75 ng/ml) than in healthy pregnant controls (5.31 ± 0.64 ng/ml) (54). Furthermore, associations between MIF genotype Rs1007888GG and gestational diabetes as well as pre-pregnancy obesity and family history of diabetes have been reported (55). This specific genotype was also found to be twofold more frequent in women with metabolic syndrome (55).
Compared with the overweight/obesity studies, there are relatively few studies in insulin resistant/T2D subjects, and these have been conducted in very specific populations (e.g., Asians, Pima Indians). While there were intervention studies (weight loss, bariatric surgery) in overweight/obese subjects, no such reports exist in the case of T2D. Therefore, conclusions should be drawn carefully. Overall, circulating MIF seems to be associated with insulin resistance and overt T2D, and MIF levels rise with the severity of disease. This suggests that MIF-inducing processes unrelated to obesity/adiposity and adipose tissue expression may contribute to circulating MIF levels as overweight/obesity progresses to T2D. Inflammatory processes in other tissues or in circulating immune cells may be of importance in this respect. The involvement of sources of inflammation other than adipose tissue may explain that the associations of MIF and metabolic disease (metabolic syndrome, insulin resistance, T2D) appear to be independent of obesity with increasing complexity of disease (Figure 2).
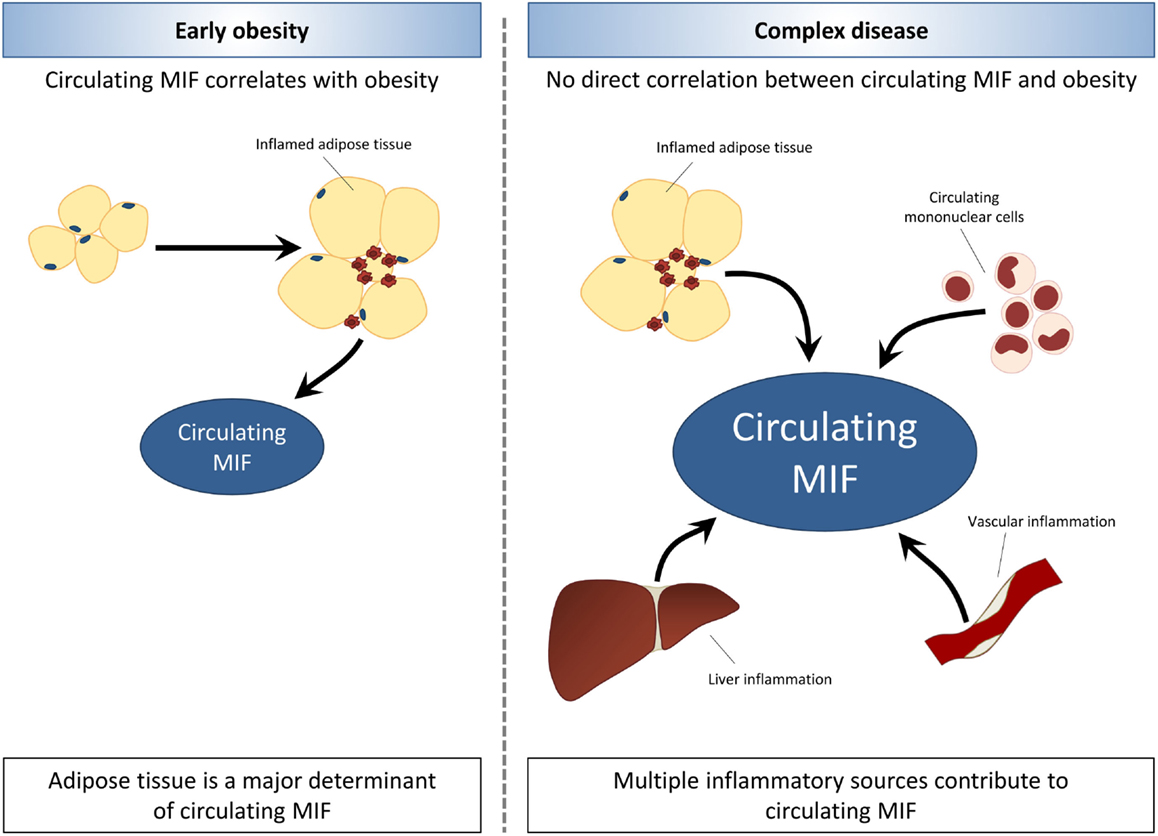
Figure 2. Associations between MIF and obesity are lost with increasing disease complexity. In early obesity, circulating MIF levels are increased in association with expansion and inflammation of adipose tissue. The development of obesity-related co-morbidities is related to elevated MIF levels, which rise with increasing severity of disease. In later stages of obesity-associated disease, MIF levels appear to be elevated independent of obesity and adiposity. Thus, indicating that other sources of inflammation such as circulating mononuclear cells, liver inflammation, and vascular inflammation may contribute to circulating MIF levels with increasing disease complexity.
To date, only one study has examined the role of MIF in NALFD patients. Akyildiz et al. (56) investigated the effect of MIF-173 G/C gene polymorphism in NAFLD and found no difference in genotype or C-allele frequency between healthy controls (n = 104) and NAFLD patients (n = 91). Furthermore, they did not find any differences in genotype or C-allele frequency within the NAFLD patient group when subjects were stratified for NAFLD score (steatosis/borderline NASH/NASH) or fibrosis stage. Immunohistological investigation of MIF expression in liver biopsies from the NAFLD patients in this study showed that MIF staining was increased in both hepatocytes and MNCs in NASH patients compared to those with only steatosis. While hepatocyte MIF expression was not associated with fibrosis stage, fibrosis stage was significantly higher in presence of MIF staining in MNCs and positive MNC MIF staining was associated with a 3.6-fold increased risk of fibrosis.
Several studies in genetically engineered mouse models that lack the Mif gene have aimed to unravel the role of MIF in both non-pathological glucose homeostasis and in the obese/insulin resistant state. The results of these studies have been summarized in Table 3.
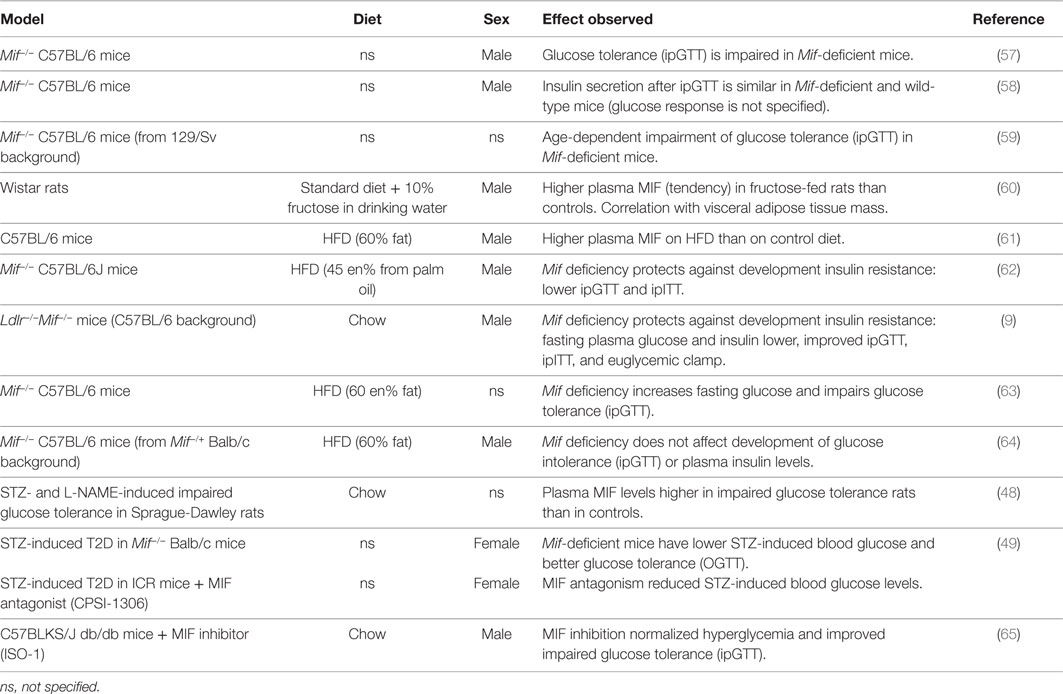
Table 3. MIF in experimental models of obesity and insulin resistance/T2D.
Nikolic et al. (57) demonstrated that Mif-deficient mice (male, 12 weeks old, diet not specified, C57BL/6 background) have impaired insulin signaling (AKT phosphorylation) in liver and adipose tissue. This was reflected in their response to an intraperitoneal glucose tolerance test (ipGTT), during which Mif−/− mice had a similar insulin response to wild-type mice, but showed impaired clearance of glucose. In this experiment, glucocorticoid receptor inhibition rescued the observed impairment of insulin signaling in liver and adipose tissue, and enhanced glucose-stimulated insulin release and glucose clearance during the ipGTT. A second study by the same group (58) confirmed that Mif−/− mice (male, 8–12 weeks old, diet not specified, C57BL/6 background) show a comparable insulin response to an ipGTT to wild-type mice (however, the glucose response was not shown in this study). A rationale for these observations was provided by results from in vitro experiments (58) that showed that conditioned medium containing insulin secreted from Mif−/− pancreatic islets had lower activity than conditioned medium with insulin produced by wild-type islets, as demonstrated by reduced insulin signaling (AKT phosphorylation) and glucose uptake in a human liver carcinoma cell line (Hep G2 cells). Furthermore, this study revealed that MIF physically interacts and binds with insulin in wild-type islets and affects the conformation of insulin, reducing the concentration of monomer/dimer insulin and increasing hexamer formation of insulin, which may contribute to enabling insulin’s full functionality. These effects were found to be independent of MIF’s tautomerase activity.
Serre-Beinier and colleagues (59) investigated the role of MIF in carbohydrate homeostasis over time with aging. For this they used Mif−/− C57BL/6 mice from a 129/Sv background (diet and sex not specified) and examined glucose and insulin tolerance over time. Mif−/− mice had lower (13%) body weight at birth, but gained weight faster than wild-type mice. By 4 months of age, body weights were similar to those of wild-type mice, and at 12 months of age, they were 17% higher than in wild-type mice. While fasting blood glucose remained stable over time in both Mif-deficient and wild-type mice, fasting insulin increased over time in Mif−/− mice but remained stable in wild-type mice. IpGTTs showed that Mif−/− mice cleared glucose more rapidly than wild-type mice, but insulin levels were higher in the Mif-deficient mice, indicating that they required more insulin to achieve this effect. At 12 months of age, glucose clearance was impaired relative to wild-type mice, with comparable insulin levels. These results are in line with findings by Nikolic et al. (57) and Vujicic et al. (58) described above, which suggest that insulin produced in absence of MIF may have reduced activity/functionality. The response to exogenous insulin (during intraperitoneal insulin tolerance tests; ipITTs, and euglycemic–hyperinsulinemic clamps) was similar between genotypes at 4 and 12 months of age and there were no differences in insulin-stimulated AKT phosphorylation in muscle, indicating that insulin sensitivity of this peripheral tissue is not affected by MIF.
While these studies indicate that MIF may contribute to insulin function in non-pathological glucose homeostasis (i.e., in absence of obesity or disease), the associations observed in human studies indicate that it may have a detrimental role in obesity and the development of obesity-associated insulin resistance and T2D, i.e. conditions of metabolic stress and disturbed metabolic homeostasis. Several groups have addressed this using experimental models of obesity and insulin resistance/T2D.
Several groups have investigated the role of MIF in obesity and associated development of insulin resistance/T2D using high-fat diet (HFD)-induced rodent models of disease (Table 3). Velickovic and colleagues (60) reported higher plasma MIF levels in fructose-fed Wistar rats than in controls (13.6 ± 1.7 vs. 11.0 ± 0.9 ng/ ml), and showed that MIF levels correlated with visceral adipose tissue mass, consistent with observations made in humans. In line with this, Saksida et al. (61) showed that HFD feeding in C57BL/6 mice resulted in an obese phenotype, with increased fasting glycemia and serum insulin, which was accompanied by increased serum MIF levels (~7 ng/ml compared with ~0 ng/ml in control diet-fed mice). This study also showed that Mif mRNA expression in pancreatic islets was increased in HFD-fed mice. Furthermore, they showed that in wild-type pancreatic islets, palmitic acid (a saturated fatty acid which is present in human diets as well as in experimental HFDs) induces MIF production and apoptosis, while pancreatic islets from Mif-deficient mice were entirely resistant to palmitic acid-induced apoptosis, which may be of importance in β-cell apoptosis in late-stage obesity-associated T2D.
Finucane and colleagues (62) also described protective effects of Mif deficiency in HFD-induced obesity/insulin resistance. In this study, Mif−/− mice (male, 8–9 weeks old, C57BL/6J background) were fed a palm oil based (rich in palmitic acid) HFD (45 kcal% fat) for 16 weeks. Mif deficiency reduced HFD-induced body weight gain and fat mass and improved the response to ipGTT and ipITT. Glucose clearance during the ipGTT was improved compared with wild-type mice, with a reduced insulin response and similarly, the Mif−/− mice showed a more efficient glucose clearance in the ipITT. Together these glucose and insulin tolerance tests show that the response to both endogenous insulin (produced during ipGTT) and exogenous insulin (injected in ipITT) is improved in absence of MIF, thus suggesting an improvement in peripheral insulin signaling in obese Mif−/− mice. These effects were also observed when mice were weight-matched to exclude effects of body weight differences, suggesting that Mif deficiency improves insulin signaling in peripheral tissues, such as white adipose tissue.
Support for this notion comes from studies that analyzed the effects of MIF on insulin signaling in adipose tissue. Finucane et al. (62) further showed that in wild-type mice, obesity increased MIF protein expression in the epididymal adipose tissue, for which the stromal vascular fraction rather than the adipocyte fraction was the primary cellular source. Mif−/− mice were found to have reduced adipose tissue inflammation (reduced M1 macrophage infiltration and reduced ex vivo cytokine secretion) and this was accompanied by improved adipose tissue insulin signaling as demonstrated by increased insulin-stimulated AKT phosphorylation and Glut4 expression in vivo, as well as increased insulin-stimulated glucose uptake into adipose tissue explants. Similarly, Verschuren et al. (9) used the hyperlipidemic Ldlr−/− background to induce obesity and insulin resistance (male mice up to 52 weeks old, chow, C57BL/6 background) and showed that Mif deficiency protects against the development of insulin resistance. Mif-deficient mice had lower fasting glucose and insulin levels and showed improved glucose tolerance (assessed by ipGTT, with comparable insulin levels during the test) and improved insulin sensitivity (assessed by ipITT). Furthermore, a hyperinsulinemic–euglycemic clamp showed that insulin resistance was reduced in Mif-deficient mice. In line with the findings by Finucane et al. (62), analysis of adipose tissue in this study showed improvement of peripheral insulin signaling. This study also revealed improvements in adipose tissue inflammation in absence of MIF, with smaller adipocytes, reduced macrophage infiltration and crown-like structure formation and a lower M1/M2 macrophage ratio. This was accompanied by improved insulin signaling in adipose tissue (increased insulin-stimulated Pi3-Kinase activity and AKT phosphorylation), which further corroborates the observed improvements in glucose handling and insulin sensitivity. In both these studies, Mif deficiency improved the inflammatory milieu in adipose tissue and resulted in improvement of insulin signaling.
While above studies used HFDs with a moderate fat content (translational to the human situation), two studies that used diets with a supraphysiological fat content (60 kcal%), made different observations; Heinrichs et al. (63) reported that Mif deficiency in HFD-fed C57BL/6 mice (age and sex not specified) promotes weight gain and has detrimental effects on glucose tolerance (impaired glucose clearance during ipGTT). Remarkably, adipose tissue inflammation was reduced in absence of Mif in this study, in line with observations in the more physiological diet-induced obesity studies. A study by Conine and co-workers (64) showed no difference between Mif−/− mice (male, 6–8 weeks old, C57BL/6 mice from Mif−/+ Balb/c background) and wild-type mice after 15 weeks of HFD feeding. In this study, body weight gain and adipose tissue inflammation were similar between genotypes, and there was no difference in glucose intolerance (assessed by ipGTT) or fasting blood glucose and insulin levels.
Others have used streptozocin (STZ; a toxic glucose analog that preferentially accumulates in pancreatic beta cells but that can also be harmful to other tissues) to induce a (pre-)diabetic phenotype (severity depends on STZ dosage and regimen). Yu et al. (48) showed that plasma MIF levels were higher in a pre-diabetic rat model of impaired glucose tolerance (HFD-fed and STZ- and L-NAME-treated Sprague-Dawley rats) than in chow-fed controls (32.5 ± 1.9 vs. 24.8 ± 1.6 ng/ml). Sanchez-Zamora and co-workers (49) showed that Mif deficiency in Balb/c mice (female, age and diet not specified) lowered STZ-induced blood glucose levels and improved glucose tolerance in an oral glucose tolerance test (OGTT) without affecting insulin production by pancreatic β-cells. Similarly, MIF antagonism (with small molecule inhibitor CPSI-1306) reduced STZ-induced blood glucose levels in ICR outbred mice in the same study (49). Together, these studies in STZ-induced diabetes suggest an improvement of peripheral insulin sensitivity and/or more active insulin in Mif-deficient mice. The latter, however, would contradict findings by Nikolic et al. (57) and Vujicic et al. (58) discussed above.
Overall, these STZ studies lend further support to the notion that Mif deficiency protects from (pre)diabetes and are thus in line with most of the observations made in diet-induced models of obesity and metabolic disease. While in normal physiology, MIFs effects in glucose homeostasis were found to be independent of MIFs tautomerase activity (58), results from a study by Wang et al. (65) suggest that this activity may play a role under pathophysiological conditions. The authors showed that in diabetic db/db mice (male, 8 weeks old, chow, C57BLKS/J background), which have an autosomal recessive mutation in the leptin receptor, treatment with the MIF tautomerase activity inhibitor ISO-1 reduced fasting glucose levels to those observed in non-diabetic controls (db/m mice). These effects were further supported by results from an ipGTT, which revealed significant improvement in glucose handling in ISO-1 treated mice. In the lean, non-diabetic control mice, ISO-1 treatment did not affect fasting blood glucose levels or ipGTT responses, consistent with the discussed observations made by Vujijic et al. (58).
Altogether, these studies indicate that while MIF seems to have a function for insulin conformation and functionality in non-pathological glucose homeostasis, its role is less clear when metabolic stress and pathological processes come into play. Under these conditions, Mif deficiency appears to be protective against white adipose tissue inflammation and may improve glucose metabolism and peripheral insulin resistance although results are partly conflicting. An important explanation for these discrepancies is likely to be the choice of rodent model in combination with the experimental conditions employed. Inbred mouse strains are known to differ greatly in their susceptibility to obesity as well as in glucose metabolism and insulin resistance (66–68). Differences in the genetic background of knockout mice (as well as successfulness of backcrossing onto a different strain) are therefore likely to have a large impact on study outcomes. A second factor that may influence study results is the choice of diet (i.e., percentage of fat, type of fat, palmitic acid content) and the age that mice are started on this dietary treatment [for instance, older Ldlr−/− mice show a more aggravated disease phenotype in response to HFD feeding than younger mice (69)]. Although information on the mouse strain and dietary conditions used are critical for the interpretation and comparison of different studies, they are often inadequately described. Overall, the prevailing finding is that Mif deficiency is protective in insulin resistance, glucose tolerance, and STZ-induced diabetes, and reduced adipose tissue inflammation and improved insulin signaling in adipose tissue may play a role in these beneficial effects, in line with observations made in human studies. Besides driving the development of insulin resistance and T2D in obesity, metabolic inflammation is also believed to drive the development of NAFLD (Figure 1).
Several studies provide indication that MIF may also play a role in the development of hepatic steatosis, inflammation and fibrosis (Table 4). Finucane and colleagues (62) showed that Mif deficiency alleviates HFD-induced hepatic steatosis (male, 8–9 weeks old, 16 weeks HFD 45 kcal% fat from palm oil, C57BL/6J background). In Mif−/− mice, liver weight was reduced, and hepatic triglyceride content (as well as histologically observed steatosis) was reduced. Mif-deficient mice also had lower plasma alanine transaminase (ALAT) levels. Hepatic gene expression analyses revealed that lipogenic gene expression (i.e., Cd36, Dgat-1, Fasn, Srebp1-c, Pgc-a1, Lpl, Ppary) was significantly lower in absence of MIF, providing a possible rationale for the observed reduction in hepatic steatosis. In line with these results, Heinrichs et al. (63) observed that hepatic steatosis was reduced in absence of MIF in both HFD-fed (60 kcal% fat, 16 weeks) and methionine and choline-deficient (MCD) diet-fed (8 weeks) Mif−/− mice (sex and age not specified, C57BL/6 background). In the HFD-fed mice, Mif deficiency increased liver weight while liver damage markers ALAT and aspartate transaminase (ASAT) were slightly reduced. The observed reduction in hepatic steatosis (analyzed by histological Oil Red O staining and biochemical measurement of hepatic triglycerides) was accompanied by increased lipogenic gene expression (i.e., transcription factors Lxra and Srebp-1, and enzymes Acc, Fas, Dgat1, Dgat2), which was also observed in the MCD diet-fed mice. Furthermore, HFD-feeding resulted in increased hepatic inflammatory cell infiltration (1.6-fold increase in F4/80+ macrophages) in Mif−/− mice, which were skewed toward a M2 polarization. Mechanistic in vitro experiments in oleic acid/IL-β stimulated mouse hepatoma cells Hepa1-6 and primary murine hepatocytes showed that recombinant MIF reduced hepatocyte lipid content through a CD74 and AMPK-mediated pathway. In contrast to the above studies, Conine et al. (64) observed no difference in HFD-induced (60 kcal% fat) hepatic lipid accumulation in presence or absence of Mif (male, 6–8 weeks old, C57BL/6 mice from Mif−/+ Balb/c background). It is not clear whether absence of phenotypical differences in this study may be related to the relatively young age of the mice at the start of the study, the extreme (supraphysiological) dietary fat content or the genetic background of the mice.
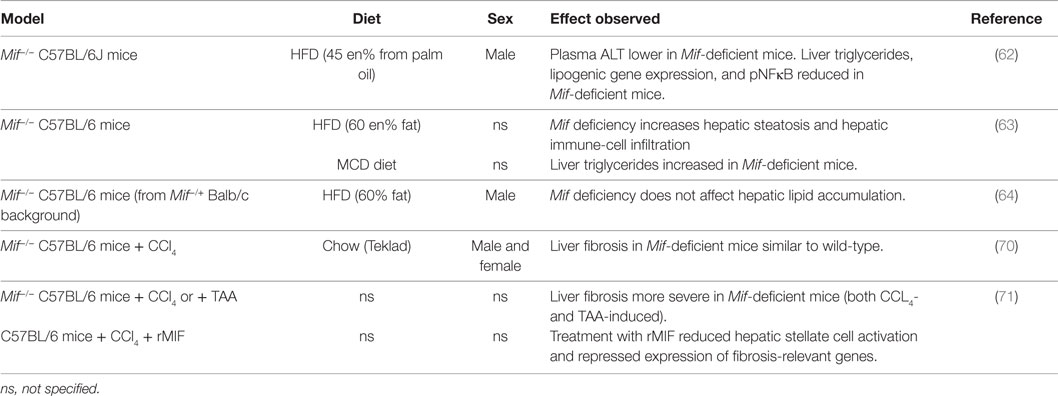
Table 4. MIF in experimental models of NAFLD/liver fibrosis.
While the above studies focused mainly on the lipid accumulation that is observed in NAFLD, others have investigated the role of MIF in the development of hepatic fibrosis, a characteristic of progressive NASH. Barnes and co-workers (70) studied effects of MIF in chemically induced liver fibrosis (male and female, 10–12 weeks old, C57BL/6 Mif−/− mice). In wild-type mice, a single dose of CCl4-induced mRNA expression of Mif and its cognate receptor Cd74 (peaking between 4 and 8 h and resolving by 18 h), which was also observed in plasma with MIF protein levels increased as early as 2 h after injection (indicating a release of preformed pools of MIF). Circulating ALAT and ASAT were similarly increased in Mif−/− and wild-type mice indicating comparative liver damage between genotypes. Assessment of hepatic stellate cell activation upon chronic CCl4 treatment (2 i.p. injections per week for 5 weeks) revealed increased mRNA expression of Acta2 (α-SMA) and Col1a1 (Collagen type I, alpha 1) in male Mif−/− mice compared with wild-type, while expression of these genes was reduced in female Mif−/− mice compared with wild-type mice. Despite these effects of Mif deficiency on hepatic stellate cell activation, hepatic collagen deposition (assessed by quantification of Sirius Red staining and hydroxyproline measurement) was comparable to wild-type mice in both male and female Mif−/− mice. Authors then continued their studies in female mice only, to investigate a potential role of MIF on the progression of fibrosis via the regulation of extracellular matrix (ECM) degradation. They observed reduced infiltration of restorative macrophages in Mif−/− mice, which was accompanied by reduced expression (mRNA, protein, and enzymatic activity) of the ECM-degrading metalloproteinase MMP13, suggesting a possible proresolution role for MIF during fibrosis.
Heinrichs and colleagues (71) also investigated the effects of Mif deficiency (age and sex not specified, C57BL/6 Mif−/− mice) in two different models of chemically induced liver fibrosis. In this study, hepatic fibrosis (assessed histologically and by hydroxyproline measurement) was increased in absence of MIF in both CCl4- and TAA-induced liver fibrosis. In line with this, Mif-deficient mice had increased mRNA expression of Col1a1, Timp1, Mmp2, and Tgfb1, as well as increased α-SMA protein expression. FACS analysis revealed no differences between genotypes in immune-cell subset infiltration, suggesting that differences in fibrosis development may be explained by effects on hepatic stellate cell activation rather than by effects on the intrahepatic immune response. In vitro studies in murine hepatic stellate cells showed that treatment with recombinant MIF inhibited PDGF-induced hepatic stellate cell activation, which was found to be mediated through CD74 and AMPK signaling. In line with these in vitro results, follow-up in vivo studies showed that CCl4-induced hepatic fibrosis was increased in Cd74−/− mice. Deficiency of Cxcr4, however, another MIF receptor expressed by hepatic stellate cells, did not affect fibrosis development. Therapeutic application of recombinant MIF in CCl4-treated wild-type mice reduced hepatic stellate cell activation and repressed expression of fibrosis-relevant genes. However, the effects of this recombinant MIF treatment on fibrosis development are not mentioned.
Overall, very few studies to date have investigated the effects of MIF in the development of hepatic co-morbidities. MIF appears to have detrimental effects in HFD-induced hepatic lipid accumulation through effects on lipogenic gene expression, providing further support for the notion that MIF may indeed not only have pro-inflammatory effects but may also be linked to metabolism. In chemically-induced liver fibrosis, it seems that MIF may have protective effects, possibly through an effect on the resolution of fibrosis. However, results from a clinical study in NAFLD patients indicate the opposite, showing that increased MIF expression in liver is associated with increased risk of fibrosis. This discrepancy may be explained by differences in disease etiology in chemically induced liver damage used in the experimental disease models, compared with the metabolically induced liver damage that is typical for human NAFLD development.
In early obesity, circulating MIF levels are increased in association with expansion and inflammation of adipose tissue. The development of insulin resistance and T2D in obesity is related to elevated MIF levels, which rise with increasing severity of disease. In later stages of obesity-associated T2D, MIF levels appear to be elevated independent of obesity and adiposity, indicating that with increasing disease complexity, other sources of inflammation may contribute to circulating MIF levels.
Mechanistic studies in experimental models of disease have shown that MIF contributes to insulin function in non-pathological glucose homeostasis (i.e., in absence of obesity or disease) but its role is less clear when metabolic stress and pathological processes come into play. Under these conditions, Mif deficiency appears to be protective against adipose tissue inflammation and may improve insulin resistance and T2D in both HFD- and STZ-induced models. Furthermore, there are some first indications that MIF may contribute to development of diet-induced hepatic steatosis in NAFLD, while observations in advanced liver disease are conflicting, describing both pro- and anti-fibrotic effects of MIF.
To fully understand the intricacies of MIFs contribution to obesity-related disease development, future work is needed to decipher temporal (non-pathological – early disease – advanced complex disease) and spatial (tissue-specific) effects of MIF, as well as interorgan crosstalk that may be mediated by circulating MIF. For the interpretation of such studies, it is of paramount importance that they would be performed in translational, physiological (diet-induced) models under well-described experimental conditions. In addition, future studies assessing the complex interplay between metabolic and inflammatory balances will greatly enhance our understanding of the development of obesity-related diseases, and the role of MIF therein. Although most published data on MIF point toward a detrimental role in advanced stages of disease, the potential protective effects of this mediator under certain conditions deserve further investigation.
MM and RK had substantial contributions to the conception and design of the work, drafted and revised the manuscript, had final approval of the version to be published and agree to be accountable for all aspects of the work.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
This work was funded by the TNO research programs “Nutrition and Health” and “Predictive Health Technologies” (RK and MM) as well as by TI Food and Nutrition, a public–private partnership on pre-competitive research in food and nutrition (to MM).
ALAT, alanine transaminase; ASAT, aspartate transaminase; HFD, high-fat diet; HOMA, homeostatic model assessment; ipGTT, intraperitoneal glucose tolerance test; ipITT, intraperitoneal insulin tolerance test; MCD, methionine and choline deficient; MIF, macrophage migration inhibitory factor; MNC, mononuclear cell; NAFLD, non-alcoholic fatty liver disease; NASH, non-alcoholic steatohepatitis; T2D, type 2 diabetes.
1. Lumeng CN, Saltiel AR. Inflammatory links between obesity and metabolic disease. J Clin Invest (2011) 121(6):2111–7. doi: 10.1172/JCI57132
2. Liang W, Tonini G, Mulder P, Kelder T, van Erk M, van den Hoek AM, et al. Coordinated and interactive expression of genes of lipid metabolism and inflammation in adipose tissue and liver during metabolic overload. PLoS One (2013) 8(9):e75290. doi:10.1371/journal.pone.0075290
3. Lafontan M. Adipose tissue and adipocyte dysregulation. Diabetes Metab (2014) 40(1):16–28. doi:10.1016/j.diabet.2013.08.002
4. Gregor MF, Hotamisligil GS. Inflammatory mechanisms in obesity. Annu Rev Immunol (2011) 29:415–45. doi:10.1146/annurev-immunol-031210-101322
5. Flaster H, Bernhagen J, Calandra T, Bucala R. The macrophage migration inhibitory factor-glucocorticoid dyad: regulation of inflammation and immunity. Mol Endocrinol (2007) 21(6):1267–80. doi:10.1210/me.2007-0065
6. Renner P, Roger T, Calandra T. Macrophage migration inhibitory factor: gene polymorphisms and susceptibility to inflammatory diseases. Clin Infect Dis (2005) 41(Suppl 7):S513–9. doi:10.1086/432009
7. Hoi AY, Iskander MN, Morand EF. Macrophage migration inhibitory factor: a therapeutic target across inflammatory diseases. Inflamm Allergy Drug Targets (2007) 6(3):183–90. doi:10.2174/187152807781696455
8. Bernhagen J, Krohn R, Lue H, Gregory JL, Zernecke A, Koenen RR, et al. MIF is a noncognate ligand of CXC chemokine receptors in inflammatory and atherogenic cell recruitment. Nat Med (2007) 13(5):587–96. doi:10.1038/nm1567
9. Verschuren L, Kooistra T, Bernhagen J, Voshol PJ, Ouwens DM, van Erk M, et al. MIF deficiency reduces chronic inflammation in white adipose tissue and impairs the development of insulin resistance, glucose intolerance, and associated atherosclerotic disease. Circ Res (2009) 105(1):99–107. doi:10.1161/CIRCRESAHA.109.199166
10. Noels H, Bernhagen J, Weber C. Macrophage migration inhibitory factor: a noncanonical chemokine important in atherosclerosis. Trends Cardiovasc Med (2009) 19(3):76–86. doi:10.1016/j.tcm.2009.05.002
11. Bucala R, Shachar I. The integral role of CD74 in antigen presentation, MIF signal transduction, and B cell survival and homeostasis. Mini Rev Med Chem (2014) 14(14):1132–8.
12. Rosengren E, Aman P, Thelin S, Hansson C, Ahlfors S, Bjork P, et al. The macrophage migration inhibitory factor MIF is a phenylpyruvate tautomerase. FEBS Lett (1997) 417(1):85–8. doi:10.1016/S0014-5793(97)01261-1
13. Kleemann R, Kapurniotu A, Frank RW, Gessner A, Mischke R, Flieger O, et al. Disulfide analysis reveals a role for macrophage migration inhibitory factor (MIF) as thiol-protein oxidoreductase. J Mol Biol (1998) 280(1):85–102. doi:10.1006/jmbi.1998.1864
14. Hirokawa J, Sakaue S, Tagami S, Kawakami Y, Sakai M, Nishi S, et al. Identification of macrophage migration inhibitory factor in adipose tissue and its induction by tumor necrosis factor-alpha. Biochem Biophys Res Commun (1997) 235(1):94–8. doi:10.1006/bbrc.1997.6745
15. Sakaue S, Nishihira J, Hirokawa J, Yoshimura H, Honda T, Aoki K, et al. Regulation of macrophage migration inhibitory factor (MIF) expression by glucose and insulin in adipocytes in vitro. Mol Med (1999) 5(6):361–71.
16. Benigni F, Atsumi T, Calandra T, Metz C, Echtenacher B, Peng T, et al. The proinflammatory mediator macrophage migration inhibitory factor induces glucose catabolism in muscle. J Clin Invest (2000) 106(10):1291–300. doi:10.1172/JCI9900
17. Waeber G, Calandra T, Roduit R, Haefliger JA, Bonny C, Thompson N, et al. Insulin secretion is regulated by the glucose-dependent production of islet beta cell macrophage migration inhibitory factor. Proc Natl Acad Sci U S A (1997) 94(9):4782–7. doi:10.1073/pnas.94.9.4782
18. Hanley AJ, Williams K, Stern MP, Haffner SM. Homeostasis model assessment of insulin resistance in relation to the incidence of cardiovascular disease: the San Antonio heart study. Diabetes Care (2002) 25(7):1177–84. doi:10.2337/diacare.25.7.1177
19. Bonora E, Kiechl S, Willeit J, Oberhollenzer F, Egger G, Meigs JB, et al. Insulin resistance as estimated by homeostasis model assessment predicts incident symptomatic cardiovascular disease in Caucasian subjects from the general population: the Bruneck study. Diabetes Care (2007) 30(2):318–24. doi:10.2337/dc06-0919
20. Edens MA, Kuipers F, Stolk RP. Non-alcoholic fatty liver disease is associated with cardiovascular disease risk markers. Obes Rev (2009) 10(4):412–9. doi:10.1111/j.1467-789X.2009.00594.x
21. Chiang CH, Huang CC, Chan WL, Chen JW, Leu HB. The severity of non-alcoholic fatty liver disease correlates with high sensitivity C-reactive protein value and is independently associated with increased cardiovascular risk in healthy population. Clin Biochem (2010) 43(18):1399–404. doi:10.1016/j.clinbiochem.2010.09.003
22. Kleemann R, Zadelaar S, Kooistra T. Cytokines and atherosclerosis: a comprehensive review of studies in mice. Cardiovasc Res (2008) 79(3):360–76. doi:10.1093/cvr/cvn120
23. Asare Y, Schmitt M, Bernhagen J. The vascular biology of macrophage migration inhibitory factor (MIF). expression and effects in inflammation, atherogenesis and angiogenesis. Thromb Haemost (2013) 109(3):391–8. doi:10.1160/TH12-11-0831
24. Burger-Kentischer A, Goebel H, Seiler R, Fraedrich G, Schaefer HE, Dimmeler S, et al. Expression of macrophage migration inhibitory factor in different stages of human atherosclerosis. Circulation (2002) 105(13):1561–6. doi:10.1161/01.CIR.0000012942.49244.82
25. Verschuren L, Lindeman JH, van Bockel JH, Abdul-Hussien H, Kooistra T, Kleemann R. Up-regulation and coexpression of MIF and matrix metalloproteinases in human abdominal aortic aneurysms. Antioxid Redox Signal (2005) 7(9–10):1195–202. doi:10.1089/ars.2005.7.1195
26. Dandona P, Aljada A, Ghanim H, Mohanty P, Tripathy C, Hofmeyer D, et al. Increased plasma concentration of macrophage migration inhibitory factor (MIF) and MIF mRNA in mononuclear cells in the obese and the suppressive action of metformin. J Clin Endocrinol Metab (2004) 89(10):5043–7. doi:10.1210/jc.2004-0436
27. Ghanim H, Aljada A, Hofmeyer D, Syed T, Mohanty P, Dandona P. Circulating mononuclear cells in the obese are in a proinflammatory state. Circulation (2004) 110(12):1564–71. doi:10.1161/01.CIR.0000142055.53122.FA
28. Church TS, Willis MS, Priest EL, Lamonte MJ, Earnest CP, Wilkinson WJ, et al. Obesity, macrophage migration inhibitory factor, and weight loss. Int J Obes (Lond) (2005) 29(6):675–81. doi:10.1038/sj.ijo.0802942
29. Sheu WH, Chang TM, Lee WJ, Ou HC, Wu CM, Tseng LN, et al. Effect of weight loss on proinflammatory state of mononuclear cells in obese women. Obesity (Silver Spring) (2008) 16(5):1033–8. doi:10.1038/oby.2008.37
30. Sumarac-Dumanovic M, Stevanovic D, Ljubic A, Jorga J, Simic M, Stamenkovic-Pejkovic D, et al. Increased activity of interleukin-23/interleukin-17 proinflammatory axis in obese women. Int J Obes (Lond) (2009) 33(1):151–6. doi:10.1038/ijo.2008.216
31. Gonzalez F, Rote NS, Minium J, Weaver AL, Kirwan JP. Elevated circulating levels of macrophage migration inhibitory factor in polycystic ovary syndrome. Cytokine (2010) 51(3):240–4. doi:10.1016/j.cyto.2010.06.008
32. Murdolo G, Nowotny B, Celi F, Donati M, Bini V, Papi F, et al. Inflammatory adipokines, high molecular weight adiponectin, and insulin resistance: a population-based survey in prepubertal schoolchildren. PLoS One (2011) 6(2):e17264. doi:10.1371/journal.pone.0017264
33. Kamchybekov U, Figulla HR, Gerdes N, Jung C. Macrophage migration inhibitory factor is elevated in obese adolescents. Arch Physiol Biochem (2012) 118(4):204–9. doi:10.3109/13813455.2012.693089
34. Kim H, Lee S, Kim HJ, Kong MH, Kim YR, Kang SH, et al. Elevated levels of macrophage migration inhibitory factor in women with metabolic syndrome. Horm Metab Res (2011) 43(9):642–5. doi:10.1055/s-0031-1283150
35. Mejia-Montilla J, Alvarez-Mon M, Reyna-Villasmil E, Torres-Cepeda D, Santos-Bolivar J, Reyna-Villasmil N, et al. Macrophage migration inhibitory factor in obese and non obese women with polycystic ovary syndrome. Endocrinol Nutr (2015) 62(1):31–7. doi:10.1016/j.endonu.2014.09.005
36. Mischke R, Gessner A, Kapurniotu A, Juttner S, Kleemann R, Brunner H, et al. Structure activity studies of the cytokine macrophage migration inhibitory factor (MIF) reveal a critical role for its carboxy terminus. FEBS Lett (1997) 414(2):226–32. doi:10.1016/S0014-5793(97)01039-9
37. Mischke R, Kleemann R, Brunner H, Bernhagen J. Cross-linking and mutational analysis of the oligomerization state of the cytokine macrophage migration inhibitory factor (MIF). FEBS Lett (1998) 427(1):85–90. doi:10.1016/S0014-5793(98)00400-1
38. Skurk T, Herder C, Kraft I, Muller-Scholze S, Hauner H, Kolb H. Production and release of macrophage migration inhibitory factor from human adipocytes. Endocrinology (2005) 146(3):1006–11. doi:10.1210/en.2004-0924
39. González-Muniesa P, Marrades MP, Martinez JA, Moreno-Aliaga MJ. Differential proinflammatory and oxidative stress response and vulnerability to metabolic syndrome in habitual high-fat young male consumers putatively predisposed by their genetic background. Int J Mol Sci (2013) 14(9):17238–55. doi:10.3390/ijms140917238
40. Alvehus M, Buren J, Sjostrom M, Goedecke J, Olsson T. The human visceral fat depot has a unique inflammatory profile. Obesity (Silver Spring) (2010) 18(5):879–83. doi:10.1038/oby.2010.22
41. van der Poorten D, Milner KL, Hui J, Hodge A, Trenell MI, Kench JG, et al. Visceral fat: a key mediator of steatohepatitis in metabolic liver disease. Hepatology (2008) 48(2):449–57. doi:10.1002/hep.22350
42. Britton KA, Massaro JM, Murabito JM, Kreger BE, Hoffmann U, Fox CS. Body fat distribution, incident cardiovascular disease, cancer, and all-cause mortality. J Am Coll Cardiol (2013) 62(10):921–5. doi:10.1016/j.jacc.2013.06.027
43. Koska J, Stefan N, Dubois S, Trinidad C, Considine RV, Funahashi T, et al. mRNA concentrations of MIF in subcutaneous abdominal adipose cells are associated with adipocyte size and insulin action. Int J Obes (Lond) (2009) 33(8):842–50. doi:10.1038/ijo.2009.106
44. Tam CS, Covington JD, Ravussin E, Redman LM; Pennington CALERIE Team. Little evidence of systemic and adipose tissue inflammation in overweight individuals. Front Genet (2012) 3:58. doi:10.3389/fgene.2012.00058
45. Fenske WK, Dubb S, Bueter M, Seyfried F, Patel K, Tam FW, et al. Effect of bariatric surgery-induced weight loss on renal and systemic inflammation and blood pressure: a 12-month prospective study. Surg Obes Relat Dis (2013) 9(4):559–68. doi:10.1016/j.soard.2012.03.009
46. van Dielen FM, Buurman WA, Hadfoune M, Nijhuis J, Greve JW. Macrophage inhibitory factor, plasminogen activator inhibitor-1, other acute phase proteins, and inflammatory mediators normalize as a result of weight loss in morbidly obese subjects treated with gastric restrictive surgery. J Clin Endocrinol Metab (2004) 89(8):4062–8. doi:10.1210/jc.2003-032125
47. Yabunaka N, Nishihira J, Mizue Y, Tsuji M, Kumagai M, Ohtsuka Y, et al. Elevated serum content of macrophage migration inhibitory factor in patients with type 2 diabetes. Diabetes Care (2000) 23(2):256–8. doi:10.2337/diacare.23.2.256
48. Yu XY, Chen HM, Liang JL, Lin QX, Tan HH, Fu YH, et al. Hyperglycemic myocardial damage is mediated by proinflammatory cytokine: macrophage migration inhibitory factor. PLoS One (2011) 6(1):e16239. doi:10.1371/journal.pone.0016239
49. Sanchez-Zamora Y, Terrazas LI, Vilches-Flores A, Leal E, Juarez I, Whitacre C, et al. Macrophage migration inhibitory factor is a therapeutic target in treatment of non-insulin-dependent diabetes mellitus. FASEB J (2010) 24(7):2583–90. doi:10.1096/fj.09-147066
50. Herder C, Kolb H, Koenig W, Haastert B, Muller-Scholze S, Rathmann W, et al. Association of systemic concentrations of macrophage migration inhibitory factor with impaired glucose tolerance and type 2 diabetes: results from the cooperative health research in the region of Augsburg, survey 4 (KORA S4). Diabetes Care (2006) 29(2):368–71. doi:10.2337/diacare.29.02.06.dc05-1474
51. Vozarova B, Stefan N, Hanson R, Lindsay RS, Bogardus C, Tataranni PA, et al. Plasma concentrations of macrophage migration inhibitory factor are elevated in Pima Indians compared to Caucasians and are associated with insulin resistance. Diabetologia (2002) 45(12):1739–41. doi:10.1007/s00125-002-0896-4
52. Herder C, Klopp N, Baumert J, Muller M, Khuseyinova N, Meisinger C, et al. Effect of macrophage migration inhibitory factor (MIF) gene variants and MIF serum concentrations on the risk of type 2 diabetes: results from the MONICA/KORA Augsburg case-cohort study, 1984-2002. Diabetologia (2008) 51(2):276–84. doi:10.1007/s00125-007-0800-3
53. Coban N, Onat A, Yildirim O, Can G, Erginel-Unaltuna N. Oxidative stress-mediated (sex-specific) loss of protection against type-2 diabetes by macrophage migration inhibitory factor (MIF)-173G/C polymorphism. Clin Chim Acta (2015) 438:1–6. doi:10.1016/j.cca.2014.07.037
54. Yilmaz O, Kucuk M, Kebapcilar L, Altindag T, Yuksel A, Yuvanc HO, et al. Macrophage migration-inhibitory factor is elevated in pregnant women with gestational diabetes mellitus. Gynecol Endocrinol (2012) 28(1):76–9. doi:10.3109/09513590.2011.588757
55. Aslani S, Hossein-nezhad A, Maghbooli Z, Mirzaei K, Karimi F. Genetic variation in macrophage migration inhibitory factor associated with gestational diabetes mellitus and metabolic syndrome. Horm Metab Res (2011) 43(8):557–61. doi:10.1055/s-0031-1275706
56. Akyildiz M, Gunsar F, Nart D, Sahin O, Yilmaz F, Akay S, et al. Macrophage migration inhibitory factor expression and MIF gene -173 G/C polymorphism in nonalcoholic fatty liver disease. Eur J Gastroenterol Hepatol (2010) 22(2):192–8. doi:10.1097/MEG.0b013e328331a596
57. Nikolic I, Vujicic M, Saksida T, Berki T, Stosic-Grujicic S, Stojanovic I. The role of endogenous glucocorticoids in glucose metabolism and immune status of MIF-deficient mice. Eur J Pharmacol (2013) 714(1–3):498–506. doi:10.1016/j.ejphar.2013.06.037
58. Vujicic M, Senerovic L, Nikolic I, Saksida T, Stosic-Grujicic S, Stojanovic I. The critical role of macrophage migration inhibitory factor in insulin activity. Cytokine (2014) 69(1):39–46. doi:10.1016/j.cyto.2014.05.013
59. Serre-Beinier V, Toso C, Morel P, Gonelle-Gispert C, Veyrat-Durebex C, Rohner-Jeanrenaud F, et al. Macrophage migration inhibitory factor deficiency leads to age-dependent impairment of glucose homeostasis in mice. J Endocrinol (2010) 206(3):297–306. doi:10.1677/JOE-09-0342
60. Velickovic N, Djordjevic A, Vasiljevic A, Bursac B, Milutinovic DV, Matic G. Tissue-specific regulation of inflammation by macrophage migration inhibitory factor and glucocorticoids in fructose-fed wistar rats. Br J Nutr (2013) 110(3):456–65. doi:10.1017/S0007114512005193
61. Saksida T, Stosic-Grujicic S, Timotijevic G, Sandler S, Stojanovic I. Macrophage migration inhibitory factor deficiency protects pancreatic islets from palmitic acid-induced apoptosis. Immunol Cell Biol (2012) 90(7):688–98. doi:10.1038/icb.2011.89
62. Finucane OM, Reynolds CM, McGillicuddy FC, Harford KA, Morrison M, Baugh J, et al. Macrophage migration inhibitory factor deficiency ameliorates high-fat diet induced insulin resistance in mice with reduced adipose inflammation and hepatic steatosis. PLoS One (2014) 9(11):e113369. doi:10.1371/journal.pone.0113369
63. Heinrichs D, Berres ML, Coeuru M, Knauel M, Nellen A, Fischer P, et al. Protective role of macrophage migration inhibitory factor in nonalcoholic steatohepatitis. FASEB J (2014) 28(12):5136–47. doi:10.1096/fj.14-256776
64. Conine SJ, Cross JV. MIF deficiency does not alter glucose homeostasis or adipose tissue inflammatory cell infiltrates during diet-induced obesity. Obesity (Silver Spring) (2014) 22(2):418–25. doi:10.1002/oby.20555
65. Wang Z, Wei M, Wang M, Chen L, Liu H, Ren Y, et al. Inhibition of macrophage migration inhibitory factor reduces diabetic nephropathy in type II diabetes mice. Inflammation (2014) 37(6):2020–9. doi:10.1007/s10753-014-9934-x
66. West DB, Boozer CN, Moody DL, Atkinson RL. Dietary obesity in nine inbred mouse strains. Am J Physiol (1992) 262(6 Pt 2):R1025–32.
67. Berglund ED, Li CY, Poffenberger G, Ayala JE, Fueger PT, Willis SE, et al. Glucose metabolism in vivo in four commonly used inbred mouse strains. Diabetes (2008) 57(7):1790–9. doi:10.2337/db07-1615
68. Parks BW, Sallam T, Mehrabian M, Psychogios N, Hui ST, Norheim F, et al. Genetic architecture of insulin resistance in the mouse. Cell Metab (2015) 21(2):334–46. doi:10.1016/j.cmet.2015.01.002
69. Gupte AA, Liu JZ, Ren Y, Minze LJ, Wiles JR, Collins AR, et al. Rosiglitazone attenuates age- and diet-associated nonalcoholic steatohepatitis in male low-density lipoprotein receptor knockout mice. Hepatology (2010) 52(6):2001–11. doi:10.1002/hep.23941
70. Barnes MA, McMullen MR, Roychowdhury S, Madhun NZ, Niese K, Olman MA, et al. Macrophage migration inhibitory factor is required for recruitment of scar-associated macrophages during liver fibrosis. J Leukoc Biol (2015) 97(1):161–9. doi:10.1189/jlb.3A0614-280R
Keywords: MIF, obesity, adipose tissue, insulin resistance, type 2 diabetes, non-alcoholic fatty liver disease
Citation: Morrison MC and Kleemann R (2015) Role of macrophage migration inhibitory factor in obesity, insulin resistance, type 2 diabetes, and associated hepatic co-morbidities: a comprehensive review of human and rodent studies. Front. Immunol. 6:308. doi: 10.3389/fimmu.2015.00308
Received: 01 May 2015; Accepted: 29 May 2015;
Published: 15 June 2015
Edited by:
Jürgen Bernhagen, Rhine-Westphalia Institute of Technology Aachen University, GermanyReviewed by:
Bong-Sung Kim, Rhine-Westphalia Institute of Technology Aachen University, GermanyCopyright: © 2015 Morrison and Kleemann. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Martine C. Morrison, Department of Metabolic Health Research, Netherlands Organization for Applied Scientific Research (TNO), Zernikedreef 9, Leiden 2333 CK, Netherlands,bWFydGluZS5tb3JyaXNvbkB0bm8ubmw=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.